

Abstract
Microglia are dynamic resident immune cells of the central nervous system (CNS) that sense, survey, and respond to changes in their environment. In disease states, microglia transform from homeostatic to diverse molecular phenotypic states that play complex and causal roles in neurologic disease pathogenesis, as evidenced by the identification of microglial genes as genetic risk factors for neurodegenerative disease. While advances in transcriptomic profiling of microglia from the central nervous system of humans and animal models have provided transformative insights, the transcriptome is only modestly reflective of the proteome. Proteomic profiling of microglia is therefore more likely to provide functionally and therapeutically relevant targets. In this review, we discuss molecular insights gained from transcriptomic studies of microglia in the context of Alzheimer’s disease as a prototypic neurodegenerative disease, and highlight existing and emerging approaches for proteomic profiling of microglia derived from in vivo model systems and human brain.
Keywords: inflammation, microglia, neurodegeneration, proteomics, signaling
1. INTRODUCTION
Microglia are the predominant resident myeloid cells of the central nervous system (CNS). Under homeostatic conditions, they survey microenvironments in the brain to phagocytose pathogens, dead cells, and cellular debris [1,2]. During development, they are critical to refining synaptic architecture by pruning supernumerary synapses. When microglia respond to activating signals or triggers, they dramatically change their morphology from highly ramified to ameboid states, proliferate, produce cytokines, and upregulate phagosome machinery [3–7]. Microglia can also contribute to CNS injury by producing neurotoxic factors, and potentially engulfing healthy synapses. Despite their critical roles in homeostasis, development, and disease, they have been historically difficult to study. Microglia comprise 10–12% of total brain cells with regional variation[8,9]. They do not communicate with action potentials as neurons do, and unlike other brain cell types, they derive from myeloid lineage [10]. Historically, microglia’s minority status in the brain along with the lack of surface markers suitable to distinguish microglia from other myeloid and brain cells have posed technical obstacles in isolation and molecular characterization. Additionally, microglia evade viral infection, thus making viral direction of genetic vectors exceedingly challenging. Despite these traditional technical barriers to genetically manipulating and isolating microglia from brain, there have been clues that microglia are causal mediators of autoimmune, aged, inflammatory, and neurodegenerative conditions [11,12].
Genome wide association studies (GWAS) of neurodegenerative diseases including Alzheimer’s disease (AD) have identified susceptibility loci involved in the innate immune system [13,14]. Recent transcriptomic advancements have revealed molecular heterogeneity in microglia under homeostatic and disease associated conditions. In the context of amyloid beta (Aβ) pathology in AD, microglia transform from homeostatic to disease associated microglia (DAM) phenotypes, which may mediate pro-inflammatory, disease-promoting as well as protective responses [15,16]. While transcriptome-level insights have been transformative, protein-level investigation of microglia particularly in their native state in the brain are necessary to guide prioritization of functionally-relevant and druggable targets for disease modification. We review the current literature on the role of microglia in AD as a prototypic neurodegenerative disease, and focus on existing and emerging approaches for proteomic profiling of microglia in in vivo model systems and human brain.
2. GENOMIC AND TRANSCRIPTOMIC INSIGHTS INTO MICROGLIAL HETEROGENEITY IN ALZHEIMER’S DISEASE.
A series of GWAS in late-onset Alzheimer’s disease (LOAD) patient populations showed that microglia are important mediators of AD [14,17–22]. While only comprising 10–12% of the total brain cell population, microglia express several risk genes (eg. TREM2, CD33, APOE) associated with LOAD [8,23,24]. Apolipoprotein E (APOE) is one of the strongest and common genetic risk factors for LOAD, and is highly expressed at the transcript level in both astrocytes and microglia, and several studies have identified immune regulatory roles for APOE in animal models of AD pathology [25,26]. The APOE4 allele [27], as compared to APOE3, increases the risk for LOAD by threefold in heterozygous E3/E4 carriers and 15-fold in homozygous (E4/E4) carriers [28–30]. Additionally, microglia highly express triggering receptor expressed on myeloid cells 2 (TREM2), a gene which encodes a protein critical for microglial survival and proliferation, immune signaling, phagocytosis and recruitment to Aβ plaques [31–33]. GWAS revealed that partial loss-of-function mutations in TREM2 significantly increased the risk of late onset AD by 3–5-fold [32,34,35]. Stemming from these seminal human genetic studies, selective genetic modulation of these AD risk-genes specifically in microglia using conditional genetic approaches in animal models have advanced our understanding of how these microglial genes contribute to AD pathogenesis [31,36–39].
Advancements in single-cell RNA sequencing (scRNA-seq) of brain myeloid cells from animal models of AD pathology and post-mortem human brain from AD subjects have refined our understanding of the complex and diverse molecular transformations of microglia. A landmark study conducted in 2017 used scRNA-seq to isolate transcriptomes of microglia from differentially aged wild-type and transgenic 5xFAD mice, a model of Aβ pathology, and identified a unique population of DAM [40]. Within 5xFAD mice, microglia transition from a homeostatic state to a DAM state between the ages of 3–8 months [40]. DAM transcriptomic signatures included high expression of previously identified human AD risk genes (APOE and TREM2) and included a repression of homeostatic genes (Cd33, Csf1r, Cst3, Cx3cr1, P2ry12, P2ry13, Tmem119). DAM also exhibited signatures of increased lipid metabolism, phagocytosis, and lysosomal activity. Genetic loss of Trem2 in 5xFAD mice further resolved two sequential phases within the DAM activation cascade. The first Trem2-independent phase represses homeostatic factors, whereas the second Trem2-dependent phase prepares microglia for increased phagocytic activity [40]. Since this study was published, a multitude of microglial scRNA-seq investigations have been conducted in a wide variety of mouse models of neurodegeneration in and beyond amyloid-based models [15]. Converging evidence now supports wide prevalence of DAM across various models of CNS injury, inflammation, and aging [16,41,42], although Aβ pathology appears to be one of the strongest inducers of the DAM phenotype. Taken together, it is possible that DAM phenotypes may be a universal microglial response to injury [16,41,42]. The immense complexity of microglial transcriptomic phenotypes in the brain have also necessitated standardization in definitions of microglial states and nomenclature [15].
Transcriptomic signatures of human microglia are also known to diverge significantly from those of rodent microglia in homeostatic and neurodegenerative conditions [23,43–47]. ScRNA-seq studies on microglia isolated from postmortem human brain regions (prefrontal cortices, frontal cortices, and parietal lobes) demonstrate divergence in human AD gene-signatures distinct from mouse models though APOE emerges as a common differentially enriched gene in mice and humans [43,45,48]. Consistent with mouse models, TREM2 loss-of-function mutations in humans weakens microglial activation in humans [44]. In a large human microglia single-cell analysis of over 16,000 microglia from human AD and nonpathological control cases [47], Olah et al., 2020 identified nine transcriptional clusters with 80% of the microglia clustering into 2 homeostatic clusters [47]. The only cluster containing genes significantly altered with pathologic diagnosis of AD in the human dorsolateral prefrontal cortex overlapped considerably (>85%) with the murine DAM expression profile, with enrichment in genes involved with inflammatory demyelination, ischemia, and AD [47]. However, this cluster significantly decreased its expression in AD with histological validation of reduced frequency of microglia associated with this cluster in AD human brain.
The reported transcriptomic discord between human and rodent microglia are likely driven by a combination of biological and experimental parameters. Importantly, rodent models are maintained in well-controlled environments with sterilized food, water, bedding, and cages [49]. These environments are designed to reduce incidence of pathogen and contaminant exposure over the animal’s lifetime and therefore may impact immune activation, which could confound interpretations comparing human and rodent studies. ScRNA-seq studies on fresh rodent brain are more amenable to intact-isolation of microglia as compared to human tissues which are typically frozen [50]. Studies using archived (frozen) postmortem human brain samples are also confounded by on post-mortem interval, comorbidities, and independent effects of aging. Furthermore, human brain samples of advanced neurodegeneration introduce a sampling bias towards terminal disease stage [50]. Alternatively, animal models lend experimental control over selecting final age end-points which can reflect initial, moderate, and advanced disease states. Given these caveats, the level of discordance in transcriptomic characteristics and immune responses between mouse and human microglia still remains to be clarified. Given these apparent discordances, it is critical to assess microglial transcriptomic signatures directly from human brain samples to verify observations made in mouse models [51].
3. SIGNALING MECHANISMS IN MICROGLIA AND THEIR IMPACT ON AD PATHOGENESIS.
Despite advancements in our understanding of the transcriptomic diversity across sub-populations of microglia in disease, proteomic studies are critical to reveal cell signaling mechanisms. Proteins, not transcripts, are the effector molecules of signaling cascades that give rise to microglial phenotypes and coordinate specific biological functions [52]. Microglia depend on intra and inter cellular signaling to sense injury, transition to activated states, mobilize to the sites of pathological insults, phagocytose debris, and prune synapses [53]. These functions are carried out by complex signaling cascades, some of which are highlighted in Figure 1. Microglia use the fractalkine receptor (CX3CR1) to bind and promote phagocytosis of microtubule-associated protein tau (MAPT) [54,55]. TAU can interact and promote the seeding of Aβ plaques and are more readily phosphorylated in the presence of Aβ [56]. Microglia rely on TREM2 signaling to detect environmental cues (e.g., APOE, Aβ, phospholipids, and apoptotic cells), direct motility to injury sites, stimulate proliferation, and induce the DAM program [57–60]. TREM2 decrease reduces microglia activation observed by impaired phagocytic capacity of injured neurons and decreased inflammatory responses [61]. Cell surface presence of TREM2 requires adaptor protein DNAX-activation protein 12 (DAP12) to initiate intracellular signaling via the recruitment of the tyrosine protein kinase SYK [62,63]. Mice lacking SYK showed exacerbated Aβ neuropathology and worse behavioral deficits [64]. TREM2 also associates with DAP10 to promote the recruitment of phosphatidylinositol 3-kinase (PI3K) [65]. TREM2-DAP12-DAP10 signaling primes downstream protein and lipid phosphorylation events which initiate a myriad of signaling cascades supporting diverse cellular responses. These responses include cytoskeletal reorganization and Ca2+ signaling which both in turn influence microglial motility and process extension, mechanistic target of rapamycin (mTOR) signaling which promotes proliferation and differentiation of immune cells, autophagy, and apoptosis, and mitogen-activated protein kinase (MAPK) activation [32,66]. In turn, MAPK cascades, including the extracellular signal-regulated kinases1 and 2 pathway (ERK1/2) regulates gene transcription to promote cell proliferation, migration and immune function [67]. Studies found that the MAPK/ERK pathway is a central mechanism of AD and is strongly correlated with AD pathology and cognitive decline[68–71]. Notably, the strong association between MAPKs (ERK1 and 2) and human AD pathology was identified at the proteomic, but not at the transcriptomic level [71]. Because post-transcriptional and post-translational regulation is important for some proteins, jointly measuring both RNA and protein can reveal disease-relevant regulatory mechanisms [72–75]. Joint measurement of RNA and protein can also reveal changes at the RNA-level not reflected at the protein-level and vice-versa. In mouse models of AD pathology, microglia are characterized by elevated ERK signaling, and ERK inhibition reduces microglial activation and proinflammatory immune responses [68]. Taken together, TREM2 is a critical surveillance sensor that enables microglia to detect pathological cues in the environment, induce activation programs via an orchestration of signaling cascades (e.g., Ca2+, mTOR, MAPK/ERK) that enable microglia to proliferate, mobilize to damage-sites, and clear cellular debris and pathological proteins.
Figure 1. Microglial responses in Alzheimer’s Disease.
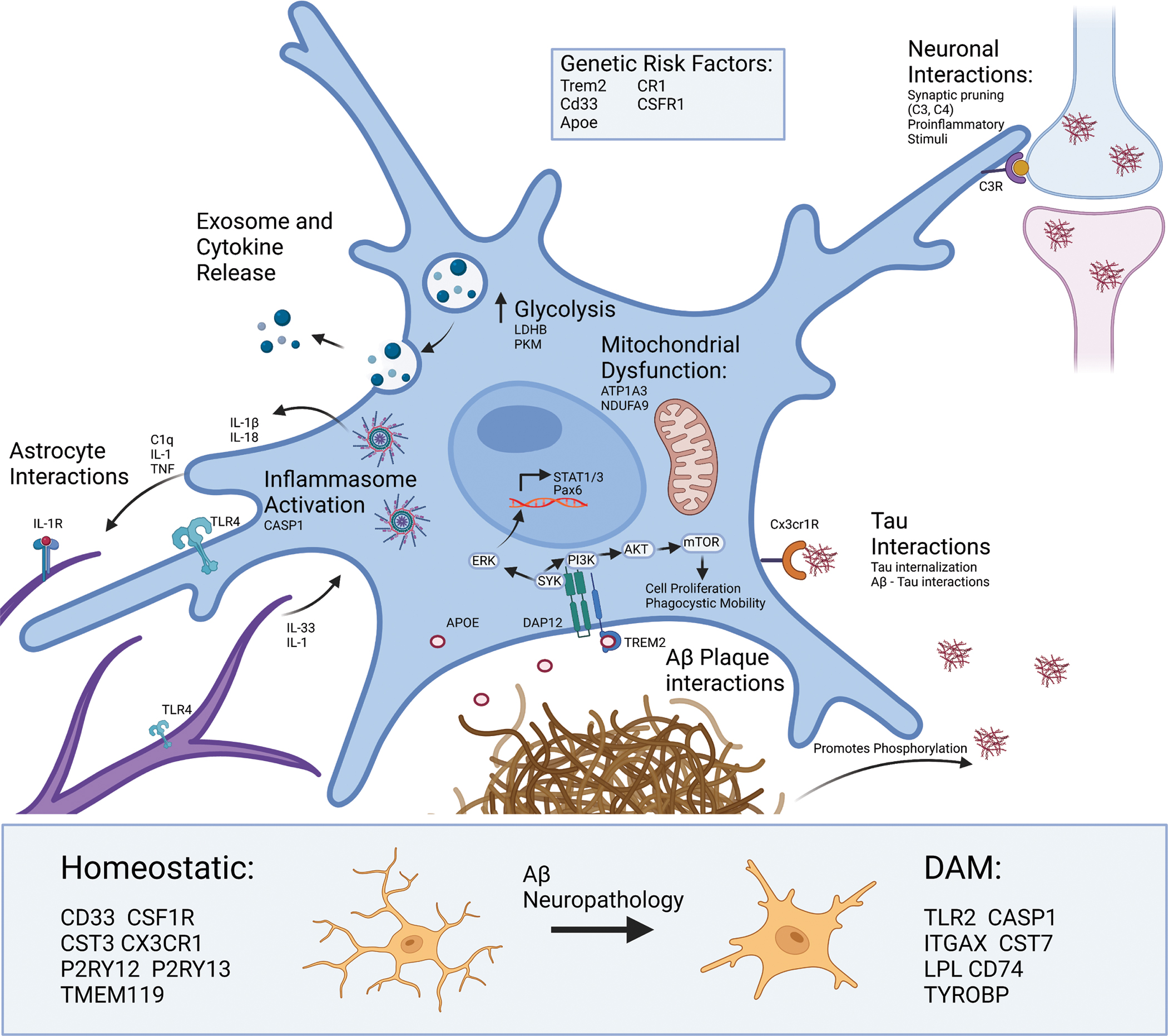
Several genetic risk loci for AD (TREM2, CD33, CSF1R, CR1, APOE) encode proteins involved in signaling cascades that support broad phenotypic shifts in motility, metabolism, phagocytosis, proliferation, cytokine production, exosome production and release, and apoptosis. Microglia use complement signaling to identify both healthy and dead neurons for degradation. Microglia can use signaling cascades to detect and recruit to pathological proteins including TAU and Aβ. For example, fractalkine signaling (via fractalkine receptor CX3CR1) allows microglia to detect and phagocytose Tau [54,216,217]. Microglial interactions with Aβ plaques are distinct, where microglia actively surround Aβ plaques and interact with them via mechanisms involving APOE, TREM2 and its receptor. This receptor is responsible for the TREM2-dependent signaling pathway which results in activation of SYK and ERK [218,219]. ERK then crosses the nucleus to allow for transcription of key inflammatory signaling molecules such as STAT1. Whereas activation of the mTOR pathway leads to further phagocytosis and cell proliferation. TAU and Aβ can also directly interact with each other, where tau tangles can act as a seed for Aβ plaque accumulation and Aβ can promote the phosphorylation of tau necessary for tau fibrillization. Cross-talk between microglia and astrocytes is largely responsible for activation of the TLR4 and IL1R on both cell types which can shift astrocytes and microglia toward more proinflammatory cytokine release [77]. Proteomic studies evidence an increase in CASP1 as well as IL-1β indicating formation and activation of the NLRP3 inflammasome which cleaves proIL-1β and proIL-18 prior to release from microglia. These signaling events also result in increased production and release of exosomes that contain key signaling proteins like TNF, and other cargo. This corresponds with metabolic reprograming in microglia, where there is an increased movement of glucose to glycolysis but a reduction in mitochondrial activity, which is in turn sustains ATP production and calcium-dependent mechanisms necessary for DAM function. Aβ and other neuropathologies also transform microglia from homeostatic and disease-associated microglia (DAM) states, and key markers of these states also shown below. [16,220]
In addition to the signaling cascades which enable microglia to sense and respond to the extracellular environment, activated microglia produce cytokines which in turn change the phenotypes other brain cells, such as astrocytes (Figure 1). For in-depth reviews on astrocyte-microglial crosstalk, see Matejuk & Ransohoff., 2020; and Vainchtein & Molofsky., 2020 [76,77]. Briefly, media derived from LPS-activated microglia containing elevated levels of IL-1α, TNFα, and C1Q induce an activated neurotoxic phenotype in astrocytes which contribute to neuronal death [78]. Furthermore, studies show that microglia-astrocyte crosstalk is necessary for the astrocytic Toll-like receptor (TLR) induced inflammatory response [79,80]. Although studies have shed light on the molecular cross-talk between microglia and astrocytes, future work that integrates proteomic approaches will uncover novel mechanisms of microglial-astrocytic crosstalk and contributions to neurodegeneration.
Another class of key microglial signaling effectors in development and pathology includes complement proteins. The complement cascade is a critical effector mechanism of the CNS and peripheral immune response that comprises of over 40 proteins [81]. Neurons and their synapses express complement activators “eat me signals” as well as complement inhibitors “don’t eat me signals,” which evolved in the mammalian CNS to support the refinement of synaptic architecture by microglia during critical developmental periods [1,82,83]. In neurodegenerative contexts, injurious stimuli (e.g., apoptotic cells, pathological proteins such as Aβ and TAU) activate the C1 complex (C1Q, C1R2, and C1S2) [81]. Following C1 complex activation, complement proteins C2 and C4 combine to create the C3 convertase, C4B2B. C4B2B cleaves C3 to form C3A, which promotes chemotaxis and microglial activation via the C3A receptor, C3AR. When C1 binding initiates complement activation on synapses, C3 tags them for elimination via microglial phagocytosis [1,84]. In AD, the complement pathway contributes to synaptic loss and neuronal damage [85–87]. One recent large scale proteomics study conducted by Bai et al., 2020 used differentially staged post-mortem human AD brain to identify complement proteins as a key hallmark denoting pathological transition from mild cognitive impairment to AD [88]. While we discussed TREM2 and complement mediated signaling as key AD-related pathways in microglia, several other receptor-mediated signaling pathways exist in microglia based on the diverse array of surface receptors expressed by these cells [53,89,90]. Taken together, microglia employ complex signaling cascades to support a wide variety of phenotypic changes in neurodegeneration. Signaling cascades and disease mechanisms rely in large part on the tight regulation of post-transcriptional and post-translational changes which transcriptomics cannot resolve, proteomic investigations are needed. Additionally, since proteomic changes are modestly reflected by transcriptomic-level changes, studies focused on proteomic profiling of microglia are critical to expand our understanding of microglial contributions to disease [91,92].
4. INSIGHTS INTO PROTEOMIC PHENOTYPES OF MICROGLIA FROM BULK BRAIN “OMICS”
As compared to the transcriptome, the proteome of microglia is orders of magnitude more complex and is more proximate to biological functions. Poor concordance between the transcriptome and proteome particularly in the CNS, has been observed [93,94]. Transcriptomic analyses are unable to capture post-translational modifications (PTMs), many of which regulate protein function and localization within the cell. PTMs are more likely to represent druggable targets for disease-modification as well as potential biomarkers in neurodegeneration. Therefore, complimentary assessments of the transcriptome and proteome of bulk tissue and of individual CNS cell types can provide a comprehensive understanding of cellular functions and disease processes. Quantitative mass spectrometry (MS) has been successfully applied to analyze postmortem human and mouse brain. These MS-based studies of bulk tissue are coupled with bioinformatic approaches to resolve several groups (or modules) of co-expressed proteins, which may represent shared biological functions, cellular endophenotypes, subcellular compartments, or shared upstream regulation [71,95]. These network-based approaches shaped the current understanding of the proteomic transformations that occur in the brain in neurodegeneration. While bulk tissue proteomics do not directly resolve cell type-specific biology, it is possible to indirectly infer cellular mechanisms by deconvoluting the bulk proteome using reference gene/protein markers of individual brain cell types. In this manner, postmortem human bulk tissue proteomics provide excellent opportunities to study immune and glial mechanisms of AD, with potential inferences related to microglia. We next highlight insights into metabolic, inflammasome and complement related immune protein alterations that occur in AD brain, as revealed by recent bulk tissue MS studies.
4.1. Metabolic re-programing in glial cells in AD brain
In a large-scale label-free quantification (LFQ)-MS analysis of over 2000 post-mortem brain samples, 453 of which were derived from asymptomatic/prodromal AD (AsymAD) or AD cases, a module of proteins (M4) enriched with microglial markers and sugar metabolism proteins, emerged as the module with the strongest correlation to AD traits including cognition, Aβ deposition, neurofibrillary tangles, and overall functional status [96]. M4 proteins increased significantly in AD cases compared to controls. In addition to containing several AD genetic risk factors, the hub proteins associated with the M4 module include MSN, PLEC, ITGB1, PRDX1, and CD44. Specifically, glycolytic proteins including LDHB, PKM, and GAPDH and proteins associated with glycolytic flux including PRDX1, DDAH and PARK7 were also representative of the M4 module, along with glial proteins CASP1, SPP1, and MAPK1 [96]. Conversely, another module (M3) enriched in mitochondrial proteins, including electron transport activity (ATP1A3) and NADH dehydrogenase activity (NDUFA9 and NDUFA10), was decreased in AD, independent of aging [96]. The simultaneous decrease in mitochondrial proteins and increase in glycolysis proteins in the brain exhibits the metabolic reprogramming known as the Warburg effect [97–99]. Since M4 was disproportionately enriched in microglial and astrocyte protein markers, it is likely that glial metabolic reprograming towards glycolysis is a major pathogenic phenotype of AD pathology. As homeostatic microglia transform to DAM, they shift from aerobic respiration with oxidative phosphorylation produced ATP, to utilizing glycolysis for ATP production. These proteomic findings are also consistent with literature describing a state of insulin resistance in AD brain [100]. Lipid peroxidation, resulting in lipid degradation via 4-hydroxynonenal also increases with the severity of AD [101]. Increases in glycogen-synthase kinase 3β in AD patients further promotes oxidative stress within the brain [101]. In a following deeper proteomic study of control and AD post-mortem brains, the parent M4 module was further sub-divided into two modules: M7 (enriched in MAPK signaling proteins) and M11 (Cell-Extracellular-Matrix [ECM] interactions) [71], both of which were strongly associated with cognitive decline, even after adjusting for neuropathological AD hallmarks [71]. Taken together, these studies demonstrate that microglial metabolic shifts (metabolic reprograming) associate with cognitive decline independent of AD neuropathology. Bulk brain proteomes suggest that microglial metabolic reprogramming is a key driver of AD etiology, thus the relationship between metabolism and neuroinflammation warrants further investigation. Whether the glial, metabolic, and immune protein changes captured in M4 in AD are protective or detrimental, is unclear and requires direct investigation in appropriate model systems.
4.2. Evidence for increased inflammasome activation in AD brain.
Inflammasomes are critical mechanistic components of the innate immune system’s ability to detect pathogens and mount a concerted cellular response against them [102]. Inflammasomes are made up of multi-protein complex with specialized sensory, adaptor and effector caspase components [102,103]. The activation of the NOD-, LRR-, and pyrin domain containing 3 (NLRP3) inflammasome, specifically, is associated both with human post-mortem AD brain and transgenic mouse models of AD [104–108]. Caspase-1 (CASP1), a key component of the NLRP3 inflammasome, is also upregulated in AD brain and is a member of module M4, that contains proteins upregulated in AD several of which are enriched in microglia and involved sugar/carbohydrate metabolism [109]. The NLRP3 inflammasome is a multiprotein complex heavily involved in inflammation, particularly in the cleavage and release of the interleukin (IL)-1β and IL-18 proinflammatory cytokines [110]. Proinflammatory responses in microglia depend heavily on bioenergetic shifts. A strong component of the proinflammatory response and bioenergetic shifts, the NLRP3 inflammasome is also activated in frontotemporal dementia (FTD) patients [105]. Ising et al found an increase in cleavage of caspase 1, mature IL-1β, and apoptosis associated speck like proteins (ASC) in AD and FTD patients [105]. This was also highlighted in Tau22 mice, which overexpress Tau through IHC and western blots [105]. These Tau 22 mice also show higher Casp1 and Il1b gene expression [105]. When crossing Tau22 mice with Pycard or Cias1 deficient mice (Tau22/Asc−/− or Tau22/Nlrp3−/−), there was a reduction in cleaved CASP1 and IL-1β as well as a reduction in special memory showed through a Morris water maze [105]. Westerns of Tau22/Asc−/− and Tau22/Nlrp3−/− mice show a decrease in PP2A phosphatase activity and 18 genes, including Ccl3 [105]. Injection of APP/PS1 into inflammasome KO mice shows an increase in IL-1β and phosphorylated TAU [105]. Meissner et al highlights the strong connection between metabolism and the NLRP3 inflammasome through evaluating changes in superoxide dismutase 1 (SOD1) influence on the NLRP3 inflammasome [111]. In vitro, SOD-1 deficient macrophages show a reduction in cleaved IL-1β and IL-18, while inflammasome independent cytokines such as TNF-α and KC remain unaffected [111]. SOD-1 deficient macrophages have increased reactive oxygen species (ROS) due to the lack of superoxide presence[111]. In vivo in SOD-1 KO mice that received LPS and ATP stimulation to induce inflammasome formation, reduced glutathionylation was observed, further inhibiting CASP1 activity as shown by a reduction NLRP3 inflammasome cleavage of IL-1β [111]. Recently, therapeutic targets for the inflammasome are being developed for treatment of AD. One such therapeutic agent is JC124, a sulfonamide-based selective inhibitor of the NLRP3 inflammasome. APP/PS1 mice treated with JC124 showed improvements in behavioral contexts such as fear conditioning [112]. JC124 treatment also reduced microglial and astrocyte activation as measured by a reduction in IBA1 and GFAP positivity, while potentially increasing synaptic plasticity through measurable increases in synapsin 1, synaptophysin, and PSD-95 [112]. JC124 also reduced the Aβ plaque load and IL-1β and CASP1 [112]. This positions the NLRP3 inflammasome as a potential target for regulating microglial activity and attenuating the detrimental effects of AD pathology.
4.3. Implication of complement signaling in transition from mild cognitive impairment to AD dementia.
Several proteomic studies on differentially-staged AD postmortem human brain implicate complement signaling in the transition from mild cognitive impairment to AD. Under homeostatic conditions, neurons express complement activators and inhibitors to mark their synapses for pruning by microglia (see Section 3 for comprehensive review of complement signaling). This role is critical to the refinement of synaptic architecture [1,82,113,114].
In the context of AD etiology, complement signaling is necessary for synaptic toxicity of amyloid in mouse models [86] and expression of complement components C3 and C3A receptor (C4AR1) correlate positively with cognitive decline and Braak staging in human AD [115]. Additionally, C3AR-deficiency in PS19 mice rescues tau pathology and attenuates neuroinflammation, synaptic deficits, and neurodegeneration [115], C3-deficient C57BL/6 mice exhibit resilience against age-dependent CA3 neuronal loss and enhanced synaptic plasticity at 12 months [116], and aged C3-deficient APP/PS1 mice perform significantly better on learning and memory tests than APP/PS1 mice [84]. In human bulk-brain proteomic studies, complement proteins significantly increase with disease stage [88,109,117]. In one human brain proteomic study, Bai et. al., 2020 used TMT-MS to analyze the proteomes of 90 human frontal cortical samples from varying disease stages including mild cognitive impairment (MCI) and later-stage AD [118]. The protein cluster associated with the transition from MCI to AD included complement proteins (CR1, C1S, C3, C4A and C4B), which were consistent with high abundance of complement proteins C4A and C4B also contained in the M4 module in the independent analysis discussed above [96]. It is possible that chronic activation of microglia by sustained Aβ and TAU deposition up-regulates microglial complement signaling; increasing microglial-mediated synaptic pruning. Given that synaptic loss, not Aβ nor TAU accumulation, is the strongest pathological correlate to cognitive decline, [119,120] it is possible that chronically activated microglia are aberrantly pruning synapses, advancing the transition from MCI to AD in human patients. Furthermore, in addition to generating human MCI and AD bulk brain proteomes, Bai et al., 2020 also generated bulk cortical proteomes deriving from differentially aged wildtype and 5xFAD mice (3, 6, and 12 months) [88]. The analysis compared differentially abundant proteins (DAPs) from 5xFAD mice with human MCI and AD patients and found that the proteomes from older 5xFAD mice (12 months) were most reflective of human AD pathology, sharing 89 and 169 protein alterations with human MCI and AD brain, respectively. The analysis identified 37 human-specific and 69 5xFAD mouse specific DAPs. Uniquely in humans, AD pathology significantly reduces the abundance of specific synaptic proteins (CAMK2 and NPTX2) and neurotrophic factors (BDNF and VGF) [88], a trend which deviates from 5xFAD mouse models in this study. While synaptic loss in 5xFAD mice is well documented; several studies converge to show significant synaptic loss only beyond 4 months of age, none of these studies herein referenced note a decrease in the proteins CAMK2 or NPTX2 specifically [86,121–125]. Due to the limitations of bulk-brain proteomics, the microglial-specific proteomic changes taking place during the transition from MCI to AD in humans or in mouse models of neurodegeneration are currently not known, despite many studies linking complement signaling with synaptic loss and cognitive decline in both mouse and human studies.
The divergence of vulnerability of sub-classes of neuronal and synaptic loss between human AD brain and brains derived from mouse models of neurodegeneration highlight the importance of caution when interpreting the physiological significance of neurodegenerative pathology exhibited by mouse models of AD. There is currently no mouse model which displays the full spectrum of human AD etiology [126]. The 5xFAD mouse model specifically drives the over-expression of high levels of mutant APP under the Thy1 promoter [127]. The rapid and aggressive Aβ pathology exhibited by the 5xFAD mouse models must be taken into consideration. Whether or not proteomic differences between human AD and murine 5xFAD mouse models are due to differences in microglial cells specifically are difficult to assess in analyses deriving from bulk brain tissue. The inability to resolve cell-type specific changes from bulk tissue lends necessity for isolation-based approaches.
5. INSIGHTS FROM PROTEOMICS OF MICROGLIA USING ISOLATION-BASED METHODS
5.1. Isolation-based methods for microglial proteomics
With the advent of fluorescence-activated cell sorting (FACS), magnetic activated cell sorting (MACS) and immunopanning approaches, it is possible to distinguish microglia from other brain myeloid populations (infiltrating, perivascular and border-associated macrophages) with high confidence, and subsequently isolate relatively pure microglia from adult mammalian brain for down-stream proteomics studies (Figure 2) [53,95,128–132]. These methods utilize antibodies against microglial surface proteins to positively identify and then purify these cells. FACS, MACS and immunopanning result in high microglial yields from both rodent and human brain tissue prepared using either enzymatic or mechanical dissociation methods. In our hands, CD11B+ bead-based MACS isolation typically yields 100,000–200,000 live microglia from one adult mouse brain while FACS-based purification yields 50,000–100,000 CD11B+ CD45 intermediate microglia from one mouse brain [95,128,130]. Additional purity of microglia can be obtained by using additional microglial markers such as TMEM119. These numbers are compatible with bulk proteomics studies which can investigate molecular characteristics of microglia as a whole population. The ability to use cell sorting methods (FACS or MACS) and immunopanning to isolate microglia from both human brain and rodents enables us to directly compare molecular transformations in postmortem human brain tissue with mouse models of aging and neurodegeneration. FACS-based microglial isolation from post-mortem human brain tissue is possible, and can be accomplished using a combination of markers such as CD11B CD45, along with other lymphocytes, monocyte, and neutrophil markers. However, unlike mouse tissue where adequate cardiac perfusion can remove unwanted blood-derived monocytes, human post-mortem tissues typically contain non-microglial myeloid cells is variable proportions which are more difficult to exclude during purification [133].
Figure 2. Isolation-dependent approaches for microglial proteomics.
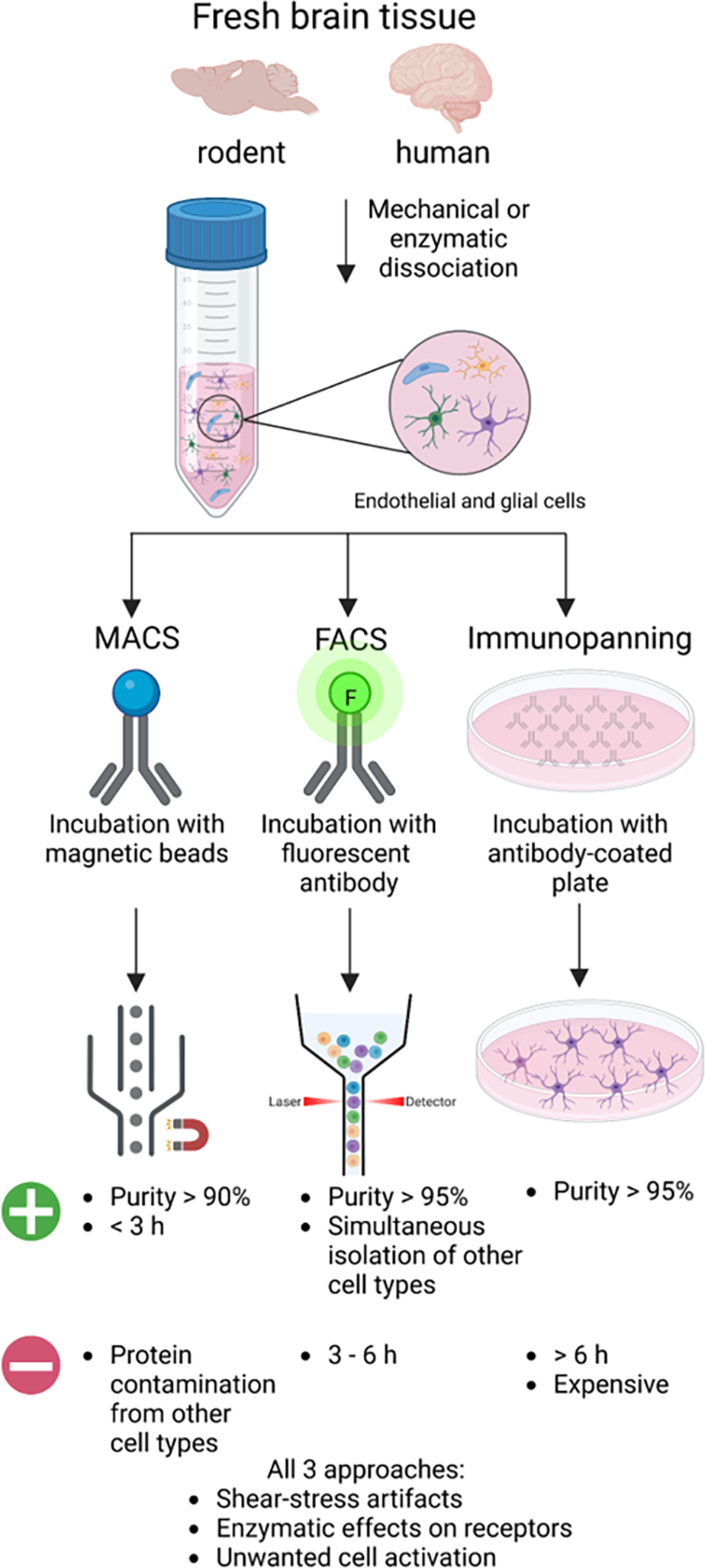
Acute isolation of microglia and other brain cell types requires fresh brain tissue that undergoes mechanical or enzymatic dissociation to generate a heterogenous single-cell suspension that can be prepared for MACS, FACS, or immunopanning methods. For MACS, the single-cell suspension is incubated with a magnetic microbead conjugated to an antibody that binds a cell surface receptor. Then, a magnet is used to select for the desired cell type and the unbound cells are washed away. The magnetically bound cells are released and collected for downstream analysis. For FACS, the single-cell suspension is incubated with a fluorophore-conjugated antibody specific to a cell surface receptor. Following, the desired cell type is sorted based on size and fluorescent signal. The sorted cells are collected for downstream analysis. For immunopanning, single-cell suspensions are plated on a cell culture dish coated with an antibody specific to a cell surface receptor. The unbound cells are washed away, and the bound cells are collected for downstream analysis.
One long-standing challenge inherent to cell-sorting based isolation methods of microglia relies on the detection and validation of microglial markers. Because microglia derive from myeloid lineage, and must undergo developmental transformation as microglial progenitor cells, it is exceedingly challenging to distinguish microglia from other macrophages residing in the choroid plexus, meninges, and perivascular spaces, especially in the setting of brain injury or established neuropathology [10]. This is because some monocyte markers are upregulated (eg. CD45-high) while canonical microglial markers (eg. Tmem119) are decreased with progressive pathology [134]. Under homeostatic conditions, mouse microglia typically express high CD11B-positive (+), low to intermediate levels of CD45 and low Ly6c on the cell surface, whereas non-microglial macrophages express high levels of CD45 and Ly6c under homeostatic conditions [135–137]. In mouse models of AD, mononuclear phagocytes (MP) in the central nervous system have a higher proportion of CD45high cells, and CD45high surround and phagocytose Aβ, and demonstrate persistently elevated phagocytic capacity for fibrillar Aβ42 with age [138]. While FACS can purify microglia using both positive and negative selection using a combination of markers, MACS typically relies only on CD11B as a positive selection marker. Therefore, MACS and FACS purified microglia result in different levels of contamination by non-microglial myeloid cells, a factor that must be considered in experimental design.
There are notable assay-level differences in microglial proteomes prepared between FACS and MACS. MACS has been shown to have a comparatively higher yield than FACS, with a cell viability exceeding 85% for both [139]. Additional considerations include the longer processing time for FACS, and molecular alterations associated with FACS protocol and the difficulty of using multiple marker profiles, and inability to perform live/dead gating with MACS [139,140]. Additionally, one study compared the proteomic differences between microglia isolated via FACS or MACS from 4-month-old C57/Bl6J wildtype mice [130]. MACS-isolated microglial proteomes showed high abundance of non-microglial synaptic and neuron-projection proteins despite >90% microglial purity, whereas FACS-isolated microglial proteomes showed enrichment of immune function proteins. Another advantage of FACS is the ability to isolate other brain cell types in parallel (eg. macrophages, endothelial cells, astrocytes, and oligodendrocytes while MACS only allows one cell type at a time [141]. On the other hand, MACS is much faster than FACS, while FACS is generally associated with higher cell loss [139,142]. Despite the differences in proteomic purity, both methods yielded a consensus set of 203 core microglial proteins, including IBA1, COTL1, and AD-relevant proteins such as potassium channel tetramerization domain-containing protein 2 (KCTD2), bridging integrator 1 (BIN1), and Moesin (MSN) [129,130].
5.2. What have we learned from isolation-based microglial proteomics in AD pathology?
Advanced age is the strongest risk factor for AD [143]. It is important to isolate the impact of advanced aging on microglial phenotype to contextualize studies of microglial changes with age and AD etiology. The first study to isolate the impact of age on microglial proteomes, without the context of disease, compared primary microglia of 3–5-month-old mice to those of 20–24 months [144]. They identified 156 DAPs involved with inflammatory signaling, mitochondrial function, and cellular metabolism with age. Notably, this study identifies an age-dependent decrease in mTORC2; contributing to a pro-inflammatory phenotype. mTORC2 promotes protein synthesis in response to growth factors and in-turn regulates cellular metabolism and macrophage polarization [145,146]. With age, microglia lose their ability to sense and respond to growth factors in the environment, via the reduction of mTORC2. Microglia in turn lose their plasticity, or their ability to polarize between inflammatory and homeostatic states[145]. These results are consistent with a human AD proteomic study identifying showing a decrease in both the transcriptional regulation of inflammatory signaling and a decrease in polarization with age, and an increase in mitochondrial proteins using fatty acids and ketone bodies as energy substrates [96]. Both human and mouse proteomic studies converge to show a unique aged microglial phenotype characterized by shifts in transcriptional regulation of inflammatory signaling, a metabolic re-programming shifting from glucose to fatty acids and ketones to generate energy, and a loss of plasticity. These proteomic studies on aged microglia are consistent with other studies showing that aged microglia mount a larger inflammatory response to acute inflammatory stimuli compared with younger mice [147,148], and morphologically shift to amoeboid, less ramified states characterized by cytoplasmic hypertrophy [149–151]. The age-driven changes in microglial metabolism and plasticity are important to take in consideration with studies assessing the impact of inflammation or AD etiology on the isolated microglial proteome.
Neuroinflammation, as characterized by activation of microglia and astrocytes and an increased expression of pro-inflammatory cytokines, is a central component of age and a hallmark of post-mortem human AD brain [152–157]. To distinguish the relative impact of inflammation and Aβ pathology on the microglial proteome in aged mice (6–7 months), Rangaraju et al., 2018 performed a quantitative MS study on purified CD11B+ microglia from wildtype mice, wildtype mice treated with Lipopolysaccharide (LPS), and 5xFAD mice [129]. LPS suppressed mitochondrial oxidative phosphorylation with concurrent increases in stress-response and glycolytic proteins [129]. When comparing DAPs enriched in 5xFAD brain or LPS-treated mice, this study identified several common proinflammatory proteins (top 5 including CLU, NUDT2, GLIPR2, DIABLO, and CSTF) and 30 proteins which were decreased both in 5xFAD and LPS treated mice (top 5 including BCORL1, PLEKHG1, RASGEF1A, IPO4, and BICD1). The direction of change of common proteins between 5xFAD and LPS were largely concordant (r=0.47), suggesting that Aβ pathology induces a pro-inflammatory state in microglia. Pleckstrin homology and RhoGEF domain containing G1 (PLEKHG1) was decreased in microglia derived from both LPS-treated mice and 5xFAD mice has been associated with human AD as well. For example, a recent study integrated single-nuclei RNA-seq (snRNA-seq) and proteomic datasets from human AD cortical and serum samples, and identified PLEKHG1 as a novel candidate biomarker. PLEKHG1 shows a decreased gene expression in human AD, though no studies have yet reported the role of PLEKHG1 in the nervous system [158].
Rangaraju et al., 2018 then identified specific proteomic changes in murine 5xFAD microglia consistent with proteomic datasets derived from frontal cortices of human post-mortem AD tissue. They identified 11 proteins similarly increased with Alzheimer’s disease and 5xFAD microglia (CLU, COTL1, HTRA1, APOE, APP) and 23 proteins decreased with AD and 5xFAD (including VGF, RTN, ALPL, SCN3A, and CAMK4). Notably, Clusterin (CLU) is a common protein which is significantly increased in microglia in response to acute inflammation (LPS), in microglia in murine models of amyloid pathology (5xFAD), and in human AD brain. CLU is an apolipoprotein that binds to TREM2 and facilitates microglial uptake of amyloid-beta (Aβ), and has long been associated with AD genetic risk [20,58,159]. Neuroinflammation induced by repeated LPS has been shown to induce memory impairment, increased activity of β and γ secretases, and increase generation of toxic species of Aβ 1–42[160–163]. The generation of toxic Aβ is a common etiology across LPS administration, 5xFAD pathology, and human AD, and the resulting increase in microglial-derived CLU emerges as a conserved response to facilitate microglial engulfment of these toxic amyloid species.
To understand microglial proteomic changes preceding Aβ plaques, Boza-Serrano et al., 2018 used MACS to isolate CD11B+ primary microglia from WT and 5xFAD aged to time points before plaque deposition (2 weeks and 6 weeks), and after plaque deposition (10 weeks), for downstream MS analysis [164]. Their results showed a significant increase in inflammatory pathways in 5xFAD mice before the onset of plaque deposition. One hypothesis posits that neuronal release of soluble Aβ at 6 weeks could be activating microglia prior to the presence of insoluble plaque deposition. A comprehensive proteomic study of acutely isolated microglia from later stages of 5xFAD mice showed that APOE expression is enriched in 5xFAD mice and increases with age in both wildtype and 5xFAD mice [165]. The histological characterizations of this study did not detect APOE in ramified microglia whereas plaque-associated microglia contained APOE and Aβ, suggesting that both may be phagocytosed by plaque-associated microglia.
A recent investigation used high resolution MS to generate ex vivo human microglial maps isolated from 5 amygdalohippocampectomy samples to compare with proteomes generated from primary mouse microglia and BV2 cell lines [166]. This comprehensive study identified 9,456 human microglial proteins and 9,629 mouse microglial proteins. Comparing proteomes generated from ex vivo mouse and human microglia showed a significant enrichment in FC receptor activation, phagocytosis and inflammatory responses in human microglia [166]. Human microglia also showed a significant enrichment in proteins associated with key AD risk loci (APOE, CLU, and SORL1) and inflammatory cytokines IL-18 and immune cell marker CD45. Comparisons between in vitro and ex vivo microglia altogether revealed a higher prevalence of homeostatic markers in ex vivo microglia; with notable absence of homeostatic microglial markers P2RY12, TMEM119 and CX3CR1 in mouse BV2 microglial cell lines [166]. These findings have critical implications on the divergent identities of homeostatic and activated microglia between human and murine microglia, and between in vivo and in vitro culture models.
6. EMERGING ISOLATION-INDEPENDENT APPROACHES FOR MICROGLIAL PROTEOMICS
Traditional methods to isolate microglia from adult brain rely on harsh reagents or mechanical dissociation and require fresh unfrozen brain tissue. The isolation process alters the microglia transcriptome and likely the proteome, introducing artifacts into datasets, likely confounding the ability to confidently determine the microglial proteome in their native state [167]. Furthermore, the harsh conditions of the isolation process introduce a sampling bias to isolating healthy microglia; limiting our ability to effectively capture disease-associated proteomic changes. The unmet need to capture the native proteomic signatures of microglia and other neural- and glial-cell types, in homeostatic and disease-associated states, has motivated the development of in vivo protein-labeling methodologies, which can be combined with affinity purification and MS (Figure 3). Broadly, in vivo proteomic labeling can be achieved via metabolic labeling of nascent proteins using bio-orthogonal non-canonical amino acid tagging (BONCAT) as well as by proximity-dependent biotinylation methods, including cell type-specific in vivo biotinylation of proteins (CIBOP).
Figure 3. Isolation-independent methods for in vivo proteomic labeling of microglia.
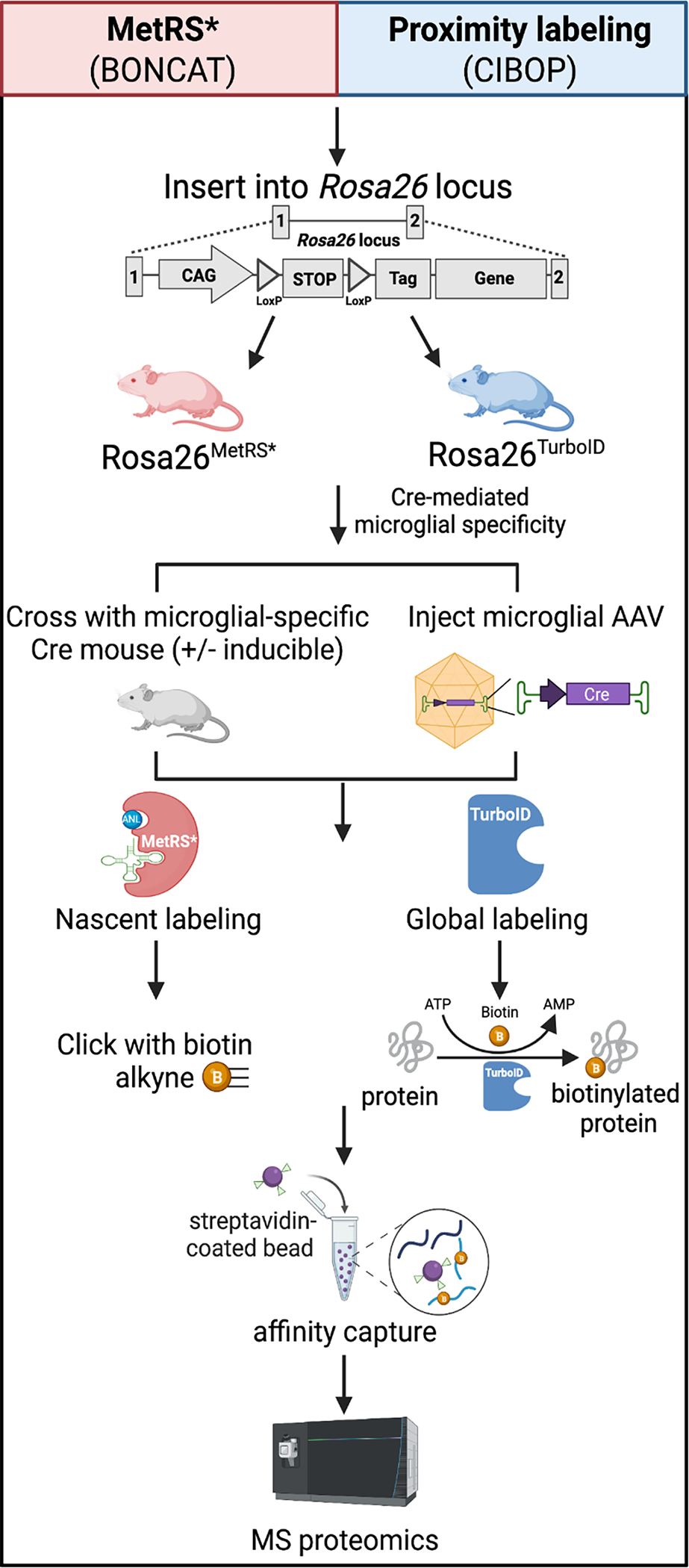
In vivo biorthogonal amino acid tagging (BONCAT) of proteins (red panel) and cell type-specific in vivo biotinylation of proteins (CIBOP) (blue panel) is achieved by inserting mutant MetRS (L274G) or MetRS*, or TurboID, into the Rosa26 locus to generate in vivo proteomic labeling in mouse models. Following, microglia specificity can be achieved by either breeding the MetRS* or TurboID models with a microglia-specific Cre mouse, with or without inducibility, or by injecting a microglial-specific adeno-associated virus (AAV) to deliver Cre. MetRS* contains a mutation (L247G) in the amino acid binding site which tags nascent proteins with an azide tagged methionine analog, azidonorleucine (ANL). The azide residue of ANL can undergo “click” chemistry in which ANL-tagged proteins residues are “clicked” with a PEG-biotin-alkyne. Proximity labeling using CIBOP is achieved by the biotin ligase, TurboID, that biotinylates endogenous proteins in close proximity. After, MetRS* or TurboID tagged proteins can undergo biotin affinity capture using streptavidin-coated beads and processed for downstream mass MS-based proteomics.
6.1. Nascent proteomic in vivo labeling using bio-orthogonal chemistry
The BONCAT strategy is a method of labeling newly synthesized proteins with a methionine (Met) analogue using a mutated Met tRNA synthetase (MetRS, mutation L274G) [168]. In the presence of the mutant MetRS, newly-synthesized or nascent proteins incorporate azidonorleucine (ANL), a methionine analogue with a “Click”-able azide moiety, in place of Met. Using the conditional MetRS-L274G mouse model (Strain Jackson Labs mouse strain #028071) combined with a Cre driver line (CamK2a promoter for excitatory neurons and Gad2 promoter for inhibitor neurons), the proteome of excitatory and inhibitory neurons in vivo were labeled and characterized by MS [169]. After supplying ANL into the drinking water for 3 weeks, the mice labeled newly synthesized proteins with the clickable methionine analogue. After conjugating ANL-tagged proteins to biotin alkyne, the labeled peptides can be purified by means of streptavidin-based affinity capture. Because the BONCAT method only labels newly synthesized proteins, there may be challenges with achieving a desired depth of proteome, however, this feature may be an asset for studying event-related or stimuli-related changes to the proteome. Recently, BONCAT has been successfully applied to astrocytes as well [170]. The extension of the BONCAT method to label and profile microglia, other brain immune cells and glial subtypes has not been reported thus far.
6.2. Proximity labeling methods for in vivo proteomic labeling
As a complimentary approach to BONCAT, which labels the nascent proteome, more global cellular proteomic labeling can be achieved using proximity labeling methods. Several biotin ligases with varying degrees of promiscuity, labeling-radius, and efficiency have been developed (including BioID and TurboID) [171–173]. Among these, TurboID is a proximity-labeling biotin ligase engineered to promiscuously biotinylate proteins within minutes and within a 10 nm radius [174,175]. Cytosolic proteomic labeling of several thousand proteins in mammalian cells can be achieved if TurboID is fused to a nuclear export sequence (TurboID-NES). TurboID-NES was recently engineered into the safe-harbor chromosomal Rosa26 locus of mice, for conditional TurboID expression in cell type or tissues of interest in vivo [176]. By expressing Cre-recombinase under astrocyte-specific and neuron-specific promoters (Aldh1 and Camk2a, respectively), using a combination of AAV-based or transgenic Cre recombinase expression, TurboID-NES was expressed in CAMK2A excitatory neurons and ALDH1l1 positive astrocytes in the adult mouse brain. This study then used streptavidin enrichment of biotinylated proteins and MS, and quantified 1,380 proteins labeled and enriched in both neurons and astrocytes, each with distinct cell type proteomic signatures, and furthermore, unique regional proteomic differences in the mouse brain [176]. To date, the CIBOP approach has been applied to neurons and astrocytes, and within in vitro BV2 and N2A cell lines, although extensions to other glial cell types in vivo including microglia are anticipated [177,178]. The CIBOP approach paired with MS-based proteomics may provide much needed proteomic insights into glial contributions to neurological disease pathogenesis.
Another proximity labeling method for native-state proteomics of neurons and glia in the brain utilizes ascorbate peroxidase (APEX). APEX rapidly biotinylates proteins within <20 nm radius of the peroxidase enzyme[179–181]. In the presence of hydrogen peroxide, APEX oxidizes biotin-phenols to phenoxyl radicals which in turn biotinylate nucleophilic sites on amino acids[181]. Since its inception, APEX has been used for several applications, ranging from mapping protein-protein interactomes [182], purifying proteomes of subcellular organelles by means of fusing APEX to specific site of interest [183], to transcriptomic profiling [184,185]. Unlike TurboID-based CIBOP where biotinylation can occur in vivo, APEX-based labeling requires hydrogen peroxide for biotinylation, which can only be performed ex vivo after tissue isolation or dissection. Chen et al., in 2015 successfully applied the technology to live Drosophila tissue [186] and recently APEX has been successfully used for cell type-specific ex vivo labeling of mouse brain tissues to resolve subcellular proteomics of neuronal subtypes [187,188].
6.3. Adeno-associated virus (AAVs) based labeling of microglia
Methods to detect and label microglia in the brain have relied on germline transgenic mouse models that introduce genetic manipulations, or the addition of transgenes. However, generating transgenic animal models can be expensive, time-consuming, require technical expertise in animal husbandry and genetics and can suffer from potential unwanted consequences of genetic manipulations. Given that BONCAT and CIBOP represent viable options for in vivo proteomic labeling of neurons and glial cells, AAV-based delivery of TurboID or MetRS or Cre recombinase constructs may dramatically reduce the complexity of in vivo studies especially with cell-type-specific proteomics applied to genetic models of neurological diseases. For these to succeed, microglia-specific AAVs are necessary. Viral vectors circumvent the limitations of transgenic animal models and have been used for delivery of Cre recombinase in synapsin-positive hippocampal neurons in TurboID mice for proteomic labeling in vivo [176]. Though, until recently, in vivo viral transduction of microglia has been challenging due to immune activation and poor transduction efficiency [189]. Lin and colleagues report the development of highly efficient AAV variants (AAV-cMG and AAV-MG) to specifically target microglia in vitro and in vivo [190], opening an exciting new avenue to explore microglial proteomic signatures in the brain. The use of these highly efficient microglia-specific AAV variants may be used to target Cre recombinase expression to microglia in MetRS and TurboID mouse models to reliably label the microglial proteome using BONCAT or CIBOP approaches. Subsequent TurboID-enriched proteomes can then be quantified using MS-based proteomics. These methods have been demonstrated in the study of neuronal and astrocytic proteomes, but microglia-specific applications in vivo are eagerly anticipated. The recent breakthroughs in specific AAV serotypes to target microglia in the mouse brain provide exciting opportunities for efficient proteomic labeling of microglia in animal models, obviating needs to complex breeding schema.
7. AVENUES FOR PROTEOMICS OF MICROGLIA-DERIVED EXOSOMES
One potential mechanism by which microglia mediate CNS diseases is exosome release. Exosomes are small extracellular vesicles of endocytic origin and are composed of proteins, nucleic acids, and lipids. Exosomes are critical to the transport of cargo containing proteins, messenger mRNAs (mRNAs), and microRNAs (miRNAs) between cells to facilitate intercellular communication and influence downstream signaling events [191]. Exosomes can interact with recipient cells through endocytosis, fusion with the plasma membrane, or ligand receptor interaction, resulting in cellular alterations of recipient cells [192]. Microglia-derived exosomes can transfer antigens and other proteins to recipient cells, including neurons and glia to deliver proinflammatory cytokines or pathologic TAU, thus positioning exosomes as key mediators in neurodegenerative etiologies [193,194]. Microglial depletion and suppression of exosome biogenesis in a tau mouse model of AD pathology significantly suppresses pathologic tau propagation [194]. Current cell isolation approaches pose significant limitations that impact proteomic characterization of microglia-derived exosomes in vivo [95,129,130]. Proteomic analyses of microglial-derived exosomes are currently limited by very low protein yield for MS, and lack of definitive verification of their cell of origin. Despite these challenges, several in vitro proteomic studies on microglial-derived exosomes released in response to inflammatory stimuli have been published [195]. Proteomic analysis of exosomes derived from the N9 microglial cell line identified the composition of microglia-derived exosomes from cell culture medium [196]. Of particular interest, was the aminopeptidase N or CD13 found in microglial exosomal proteins but not in exosomes derived from B cells and dendritic cells. Functional assays looking at aminopeptidase activity, revealed that microglia exosomal CD13 is active in neuropeptide degradation. Yang et al. characterized microglial extracellular vesicular (EV) protein cargo following LPS and tumor necrosis factor (TNF) inhibitor treatment in BV2 cell lines [197]. Following LPS, EVs contained high levels of proinflammatory cytokines TNF and IL-10, seen by ELISA. Furthermore, inhibition of the TNF signaling pathway resulted in a reduction of EVs released from LPS treated microglia. MS experiments identified 49 LPS-induced DAPs contained within EVs largely associated with transcription and translation. This study provides proteomic evidence for unique exchange of cargo through EV release in inflammatory contexts. Verderio et al., 2012 found that microvesicles and exosomes derived from LPS-preactivated cultured microglia, induced a dose-dependent activation of astrocytes and microglia [198]. The findings of this study suggest that the cargo from microvesicles and exosomes can transfer an inflammatory signal to recipient cells, thus potentially exacerbating neuroinflammatory conditions. Currently, there is a critical gap in our understanding of the specific proteins within microglia-derived exosomes that lead to the perpetuation of AD pathology. Proteomic studies of microglia-derived exosomes from bulk brain tissue as well as biofluids such as plasma and cerebrospinal fluid, could further delineate homeostatic-exosomes from DAM-derived exosomes and contribute to the nomination of novel neurodegenerative disease biomarkers. To this end, the development of protein-labeling methodologies using bio-orthogonal chemistry or proximity labeling, microglia-specific AAVs for in vivo applications, as well as exosome directed genetic tools (such exosome reporter CD63-GFPflox/flox mice) have all recently emerged as a promising toolkits to characterize the proteome of microglia-derived exosomes for the first time in vivo in the context of AD pathology [169,176,199].
8. SINGLE-CELL MICROGLIAL PROTEOMICS: ARE WE THERE YET?
Single-cell transcriptomics of human and mouse microglia and other glial cells have revealed immense heterogeneity, with unique signatures observed during development, aging, and disease. Given that the proteome is several orders of magnitude more complex than the mRNA transcriptome [200,201], it is likely that additional disease-relevant and therapeutically-meaningful cellular heterogeneity can be revealed if microglia can be studied using single-cell proteomics methods. Several recent technical advances in proteomics methodologies now make single-microglial proteomics feasible [136]. Methods to quantify proteins in single mammalian cells have evolved over the last decade, ranging from single cell Western blots, multi-parameter flow cytometry, antibody-based multiplexed approaches, to more recently, high-throughput MS-based methods [202]. Among single-cell proteomics methods, multi-parameter flow cytometry and mass cytometry based on time of flight (CyTOF) can measure 40–50 proteins of interest using antibody-based methods [203]. Flow cytometry uses physical characteristics (size and complexity) and fluorophore-conjugated antibodies to characterize cells using pre-selected cell surface and intracellular proteins of interest. CyTOF is a single-cell proteomics technique which uses rare metal isotopes conjugated to antibodies. Since the overlap between metal isotopes is minimal, over 40 distinct protein targets can be measured in a multiplexed manner. CyTOF has been successfully applied to study freshly-isolated as well as cryopreserved post-mortem human microglia to detect 55 extra-cellular and intra-cellular protein markers, revealing regional phenotypic signatures of human microglia in the brain which can be captured using a panel of 4 markers (CD45, CD64, CD68 and HLA-DR) [27]. Advances in multi-parameter flow cytometry are also able to approach the level of high-dimensionality achievable by CyTOF [204]. A limitation of both multi-parameter flow cytometry and CyTOF is the dependency on well-validated antibodies. While multi-parameter flow cytometry-based sorting can purify populations of interest for other omics approaches, CyTOF is not suitable for this. Flow cytometry and CyTOF remain powerful methods to study microglia and other glial cells in various contexts, especially when targets have been identified a-priori. For less-biased and high-throughput phenotyping of microglia, MS-based methods are more suitable to measure hundreds to thousands of proteins from a single cell.
Nanodroplet processing in one pot for trace samples (NanoPOTS) was developed in 2018 and used robotic handling and miniaturization to nanoliter scale to quantify nearly 3,000 proteins from pools on 10–14 cells per sample [205,206]. NanoPOTS was successfully applied for shotgun MS characterization of single cells and pools of 3–20 cells from day 15 chicken embryos, although only 200 unique peptides were identified from single cells while cell pools yielded deeper proteomes by MS [207]. Downstream of laser capture microdissection (LCM), NanoPOTS can spatially-resolve proteomes in rodent mouse brain, recovering an average proteomic depth of 2,000 proteins per 100 μm - diameter section in label-free contexts [208]. One important limitation to NanoPOTS includes the use of nanoliter-scale volumes which limits compatibility with traditionally automated LC-MS systems, and reliance on autosampler and TMT labeling to increase throughput [206]. Single Cell ProtEomics by Mass Spectrometry (SCoPE-MS) was pioneered by Nikolai Slavov’s laboratory in 2018, as the first MS-based single-cell proteomics approach that was able to quantify over a thousand proteins in single mammalian cells [202]. SCoPE-MS minimizes losses at several steps of the MS pipeline by an innovative use of mechanical lysis of single cells via acoustic sonication, eliminating the need for chemical lysis steps, and mixing of labeled carrier peptides with labeled peptides from single cells prior to ionization and MS analysis. SCoPE-MS also utilizes multiplexed TMT technology to maximize sensitivity and throughput. This method was able to quantify over a thousand proteins per cell, which were primarily highly-abundant proteins (>105 copies/cell) but not proteins with lower copy numbers (<104 copies per cell). Importantly, SCoPE-MS differentiated different cellular states of embryonic cell differentiation in vitro, although this method has yet to be applied to cells isolated from the brain. A next-generation version of SCoPE-MS (SCoPE2) was then developed to improve upon miniaturization, protein quantification, throughput, and scalability [209,210]. In the original description of the method, SCoPE2 quantified >2,500 peptides and >1,000 proteins per cell and was able to distinguish monocytes from differentiated macrophages in vitro, including intermediary differentiation states. Although yet to be applied to brain cell types or microglia, SCoPE2 is a promising single-cell proteomics method that may be applicable to the study of microglia. SCoPE2 workflow is compatible with cells purified via manual picking, FACS-purification, or other droplet-based microfluidics methods. For additional technical details of labeling-based SCoPE-MS and SCoPE2 methods and their challenges, readers are referred to two review papers [75,211]. Chip-based systems capable of cell isolation, counting, imaging and processing for proteomics have also been developed for single-cell applications (SciProChip) and small cell pools (iProChip), allowing quantification of over 1,400 proteins from single cells (PC-9 cells) by LC-MS/MS analysis, potentially representing a highly-sensitive labeling-independent approach in contrast to NanoPOTS and SCOPE2 methods [212].
More recently, unlabeled approaches using highly-miniaturized and ultrasensitive pico-liter scale methods have been pioneered by Matthias Mann’s group. These methods couple the Ion Mobility Spectrometry (IMS) technique with MS methods, and apply parallel accumulation-serial fragmentation (PASEF) and ultrasensitive MS instrumentation. PASEF is a MS approach in which ionized peptides are first retained in a trapped ion mobility spectrometry (TIMS) chamber of a TIMS-QTOF instrument, after which they are released into a vacuum system based on their ion mobility for precursor fragmentation, in either data-independent acquisition (diaPASEF) or data-dependent acquisition (ddaPASEF) modes [213,214]. PASEF allows for selection of multiple precursor ions for fragmentation as opposed to sequential selection, which increases throughput and quantitation. Using the diaPASEF mode and a new MS instrument with optimized technical parameters (Bruker timsTOF SCP), FACS-purified single HeLa cells were analyzed by MS, yielding single-cell proteomes of HeLa cells in which over 2,000 proteins were quantified per cell, and proteomic alterations during the HeLa cell cycle were resolved. 430 single-cell HeLa proteomes were also contrasted with single-cell RNA sequencing data showing that the core proteome of HeLa cells is more stable than the transcriptome [215].
To summarize, rapid and unprecedented technical advances have occurred in the field of single-cell proteomics. A high-level summary of the single-cell proteomic techniques discussed in this paper is provided in Table 1. These advances position researchers to investigate the proteome of mammalian cells in health and disease model systems and human tissues, with a depth of coverage that is beginning to approach single cell transcriptomics methods. While scalability, cost, and efficiency still need to be further optimized, these methods are capable of resolving cellular proteomes with depth in the 1,000–2,000 proteins/cell range which are several orders of magnitude higher than lower-throughput antibody or probe-based methods. Extension of single-cell proteomics methods to spatial proteomic profiling of single cells is also anticipated. While none of these methods have been extended beyond cell lines in vitro, it is only a matter of time that these single cell proteomics pipelines will be applied to the study of single cells isolated from tissues including the brain. Since microglia are one of the most accessible cell types from adult mammalian brain, single cell microglial proteomics studies of fresh human post-mortem brain and animal models are eagerly anticipated.
Table 1.
Single Cell Proteomic Methodologies
| Technique | Proteomic Depth (Number of proteins) | Cellular Isolation Mechanism | Published Applications | Advantages | Limitations |
|---|---|---|---|---|---|
| Mass Cytometry (CYTOF) | 40–50 / cell.[203] | Mass cytometry uses rare metal isotope conjugated antibodies.[203] | Cultured mammalian cells.[221] Human microglia from fresh & cryopreserved brain.[27] |
Compatible with frozen brain tissue.[27] Detection of intracellular and extracellular proteins.[27] |
Dependent on well-validated antibodies and targets. Inability to sort cells for other applications. |
| Nanodroplet Processing in One pot for Trace Samples (NanoPOTS) | ~1500–3000 from ~10–100 cultured cells.[205] ~2,000 proteins / 100 μm diameter brain section. [208,222] |
Flow cytometry of cultured cells.[205,223] Laser Capture Microdissection from brain tissue. |
Cultured mammalian cells [223] & rodent brain.[208,222] | Spatial resolution of proteomic signatures in rodent brain. Compatible with chemical multiplexing. [224] |
Nanoliter-scale volumes limit compatibility with automated LC-MS. [206] |
| Single Cell ProtEomics by Mass Spectrometry (SCoPE-MS / SCoPE2) |
>1,000 / cell. [202] | Acoustic sonication.[202] | Cultured mammalian cells.[202,209] | Obviate dependence on chemical lysing. Accurate quantification of protein changes across single cells.[211] |
Lower accuracy of comparing abundances of different proteins. [211] Limited capability in quantifying lower-abundance proteins.[211] |
| Single cell integrated Proteomics Chip (SciProChip) | >1,400 / cell. [225] | Size-based digital microfluidic isolation.[226] | Cultured mammalian cells. [225] | High label-free sensitivity.[225] Integrated isolation, lysis, counting, imaging, and processing.[225] |
Size-based isolation alone limits analyses of sub-populations of cells.[225] Sample loss with manual peptide transfer to autosampler.[225] |
| Parallel Accumulation-Serial Fragmentation (PASEF) | >2,000 / cell. [215] | Flow cytometry of cultured cells.[215] | Cultured mammalian cells.[215] | High label-free sensitivity.[215] Fast Scanning Speed.[227] |
Limited throughput of 20–40 single cells / day. [227] |
9. CONCLUDING REMARKS
In conclusion, understanding molecular alterations occurring in microglia in states of homeostasis and neurodegenerative disease pathology, can provide critical biological and translational insights. Since microglia are causal mediators of pathology in AD and other neurodegenerative diseases, understanding their molecular transformations, while considering the effects of aging and other genetic and environmental factors, can identify targets for disease-modification and potential biomarkers. Rapid advances in microglial transcriptomics using bulk and single cell methods have dominated the field of microglial biology. Given the discordance between transcript and protein levels in the brain, and the immense complexity of the proteome that extends to post-translational modifications, proteomic studies of microglia are likely to provide transformative insights that cannot be captured by microglial transcriptomes. Bulk tissue proteomics of brain tissues tend to under-sample microglial proteins and cannot directly resolve microglia-specific changes occurring in the brain. Advances in isolation-based approaches for microglial proteomics along with technological improvements in sensitivity and throughput of MS instruments and workflow pipelines, now allow microglia-specific proteomics from human post-mortem samples as well as from mouse brain tissue with proteomic coverage exceeding 3,000–5,000 proteins per sample. To overcome the potential biases of isolation-based approaches, isolation-independent in vivo proteomic labeling approaches such as BONCAT and CIBOP represent promising methods that could be applied for native state microglia-specific proteomics in animal models of neurodegeneration, to compliment isolation-based methods. Recent advances in single cell proteomics technologies have also provided the foundation for future microglia-specific proteomics studies in humans and animal models. The field of microglial biology therefore eagerly anticipates the next wave of studies using microglial proteomics approaches.
FUNDING AND ACKNOWLEDGEMENTS
The funding which supports the authors of this publication derive from the National Institute of Health under the following award numbers: F31AG071319 (S. Sunna), F31AG074665-01 (C. Bowen), 1F31NS127530 (J. Santiago) & R01AG075820, R01NS114130, RF1AG071587 (S. Rangaraju). Figures and illustrations were created using BioRender.com.
Abbreviations:
- 5xFAD
five familial Alzheimer’s disease
- AAV
adeno-associated virus
- Aβ
amyloid beta
- AD
Alzheimer’s disease
- ANL
azidonorleucine
- APEX
engineered ascorbate peroxidase
- AsymAD
asymptomatic Alzheimer’s disease
- BONCAT
bio-orthogonal non-canonical amino acid tagging
- CIBOP
cell type specific biotinylation of proteins
- CYTOF
cytometry by time of flight
- DAM
disease associated microglia
- DAP
differentially abundant proteins
- DDAPASEF
data dependent acquisition parallel accumulation-serial fragmentation
- DIAPASEF
data independent acquisition parallel accumulation-serial fragmentation
- ECM
extracellular matrix
- EV
extracellular vesicle
- FACS
fluorescence activated cell sorting
- GWAS
genome wide association studies
- IMS
ion mobility spectrometry
- iProChip
integrated proteomics chip
- LFQ
label free quantification
- LOAD
late onset Alzheimer’s disease
- LPS
lipopolysaccharide
- MCI
mild cognitive impairment
- MetRS
methionyl-tRNA-synthetase
- MP
mononuclear phagocytes
- PASEF
parallel accumulation-serial fragmentation
- SciProChip
single-cell iProChip
- SCOPE-MS
single-cell proteomics by mass spectrometry
- scRNASeq
single-cell RNA-seq
- TIMSTOF SCP
trapped ion mobility spectrometry
Biographies

Sydney Sunna is a 5th year graduate student in Emory University’s Neuroscience Doctoral Program, jointly mentored by Dr. Rangaraju and Dr. Seyfried. She received her B.Sc. degree in Neuroscience and Cognitive Science and Molecular and Cellular Biology with dean’s list distinction from the University of Arizona in 2017. Sydney’s doctoral research focuses on characterizing glial and neuronal proteomes labeled with the biotin ligase TurboID, and using TurboID-mediated proximity labeling to purify cell-specific proteomes from brain homogenate under inflammatory and homeostatic conditions. She is currently a F31 NRSA recipient.

Christine Bowen is a graduate student in the Seyfried and Rangaraju labs. She studies how microglial Kv1.3 potassium channel regulate neuroinflammation in Alzheimer’s disease and stroke models. Christine utilizes in vivo mouse models, proteomics, and transcriptomics on her project. She completed her bachelors of science in cellular and molecular biology at Appalachian State University. Before starting graduate school, Christine was a Post-baccalaureate IRTA fellow in the National Toxicology Program at the National Institute of Environmental Health Sciences. Her current position is supported by the NRSA F31 Fellowship.

Christina Ramelow is a 3rd year PhD Candidate in the Neuroscience Graduate Program at Emory University, mentored by Dr. Rangaraju. Her research is investigating astrocytic proteomic and transcriptomic contributions to aging and Alzheimer’s disease pathogenesis. Christina obtained a Bachelor of Science and Master of Science in Biology from Eastern Washington University. As an undergrad, she participated in the TRIO Ronald E. McNair Post Baccalaureate Achievement Program and the NSF S-STEM Scholar Program where she found a passion for biomedical research, outreach, and mentoring.

Juliet Santiago is a 4th year PhD Candidate in the Neuroscience Graduate Program at Emory University. Juliet is mentored by Dr. Rangaraju. Her research focuses on understanding the role of microglia-dervied extracellular vesicles in neuroinflammation and neurodegenerative disease. Juliet attended the University of Florida for her undergraduate education where she majored in Microbiology and Cell Science. Juliet is currently a F31 NRSA recipient for her graduate work.

Dr. Kumar is an instructor at Emory University. His research focuses on understanding the role of central and peripheral immune cell’s impact on the progression neurological disorders, including Alzheimer’s disease. Dr. Kumar uses both transcriptomics and proteomics-based approach to investigate mechanisms of neurodegeneration in Alzheimer’s disease to find novel therapeutic strategies.

Dr. Rangaraju is a board-certified neurologist and physician scientist with expertise in vascular neurology, microglial biology, mechanisms of neurodegeneration and proteomics methodology. His laboratory investigates mechanisms of neurodegeneration in Alzheimer’s disease and stroke, with a focus on microglia as causal mediators. His laboratory has optimized protocols for mass spectrometry-based phenotyping of microglia from the adult brain, developed tools for in vivo proteomic labeling for cell type-specific native-state proteomics, and has identified immune targets such as the Kv1.3 channel as a therapeutic target for neuro-immune modulation in Alzheimer’s disease pathology and stroke.
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors do not have anything to disclose.
REFERENCES
- [1].Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, … Barres BA (2007). The classical complement cascade mediates CNS synapse elimination. Cell, 131(6), 1164–1178. doi: 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- [2].Crapser JD, Arreola MA, Tsourmas KI, & Green KN (2021). Microglia as hackers of the matrix: sculpting synapses and the extracellular space. Cell Mol Immunol, 18(11), 2472–2488. doi: 10.1038/s41423-021-00751-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Penfield W (1932). Cytology & Cellular Pathology of the Nervous System (Vol. 2): PB Hoeber, Incorporated. [Google Scholar]
- [4].Ling EA, Ng YK, Wu CH, & Kaur C (2001). Microglia: its development and role as a neuropathology sensor. Prog Brain Res, 132, 61–79. doi: 10.1016/s0079-6123(01)32066-6 [DOI] [PubMed] [Google Scholar]
- [5].Hanisch UK (2002). Microglia as a source and target of cytokines. Glia, 40(2), 140–155. doi: 10.1002/glia.10161 [DOI] [PubMed] [Google Scholar]
- [6].Heppner FL, Prinz M, & Aguzzi A (2001). Pathogenesis of prion diseases: possible implications of microglial cells Progress in Brain Research (Vol. 132, pp. 737–750): Elsevier. [DOI] [PubMed] [Google Scholar]
- [7].Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, … Amit I (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell, 169(7), 1276–1290.e1217. doi: 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- [8].Lawson LJ, Perry VH, & Gordon S (1992). Turnover of resident microglia in the normal adult mouse brain. Neuroscience, 48(2), 405–415. doi: 10.1016/0306-4522(92)90500-2 [DOI] [PubMed] [Google Scholar]
- [9].Lawson LJ, Perry VH, Dri P, & Gordon S (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience, 39(1), 151–170. doi: 10.1016/0306-4522(90)90229-w [DOI] [PubMed] [Google Scholar]
- [10].Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, … Merad M (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science, 330(6005), 841–845. doi: 10.1126/science.1194637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hickman S, Izzy S, Sen P, Morsett L, & El Khoury J (2018). Microglia in neurodegeneration. Nat Neurosci, 21(10), 1359–1369. doi: 10.1038/s41593-018-0242-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Luo XG, Ding JQ, & Chen SD (2010). Microglia in the aging brain: relevance to neurodegeneration. Mol Neurodegener, 5, 12. doi: 10.1186/1750-1326-5-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. (2019). Science, 365(6460). doi: 10.1126/science.aav7188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Efthymiou AG, & Goate AM (2017). Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener, 12(1), 43. doi: 10.1186/s13024-017-0184-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Paolicelli RC, Sierra A, Stevens B, Tremblay ME, Aguzzi A, Ajami B, … Wyss-Coray T (2022). Microglia states and nomenclature: A field at its crossroads. Neuron, 110(21), 3458–3483. doi: 10.1016/j.neuron.2022.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, & Amit I (2018). Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell, 173(5), 1073–1081. doi: 10.1016/j.cell.2018.05.003 [DOI] [PubMed] [Google Scholar]
- [17].Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, … Amouyel P (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet, 45(12), 1452–1458. doi: 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, … Amouyel P (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet, 41(10), 1094–1099. doi: 10.1038/ng.439 [DOI] [PubMed] [Google Scholar]
- [19].Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, … Williams J (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet, 43(5), 429–435. doi: 10.1038/ng.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, … Williams J (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet, 41(10), 1088–1093. doi: 10.1038/ng.440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, … Posthuma D (2019). Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet, 51(3), 404–413. doi: 10.1038/s41588-018-0311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, … Schellenberg GD (2011). Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet, 43(5), 436–441. doi: 10.1038/ng.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Masuda T, Sankowski R, Staszewski O, & Prinz M (2020). Microglia Heterogeneity in the Single-Cell Era. Cell Rep, 30(5), 1271–1281. doi: 10.1016/j.celrep.2020.01.010 [DOI] [PubMed] [Google Scholar]
- [24].Mittelbronn M, Dietz K, Schluesener HJ, & Meyermann R (2001). Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol, 101(3), 249–255. doi: 10.1007/s004010000284 [DOI] [PubMed] [Google Scholar]
- [25].Kloske CM, & Wilcock DM (2020). The Important Interface Between Apolipoprotein E and Neuroinflammation in Alzheimer’s Disease. Front Immunol, 11, 754. doi: 10.3389/fimmu.2020.00754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shi Y, & Holtzman DM (2018). Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol, 18(12), 759–772. doi: 10.1038/s41577-018-0051-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bottcher C, Schlickeiser S, Sneeboer MAM, Kunkel D, Knop A, Paza E, … Priller J (2019). Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat Neurosci, 22(1), 78–90. doi: 10.1038/s41593-018-0290-2 [DOI] [PubMed] [Google Scholar]
- [28].Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, … Pericak-Vance MA (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science, 261(5123), 921–923. doi: 10.1126/science.8346443 [DOI] [PubMed] [Google Scholar]
- [29].Guerreiro RJ, Gustafson DR, & Hardy J (2012). The genetic architecture of Alzheimer’s disease: beyond APP, PSENs and APOE. Neurobiol Aging, 33(3), 437–456. doi: 10.1016/j.neurobiolaging.2010.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Karch CM, & Goate AM (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry, 77(1), 43–51. doi: 10.1016/j.biopsych.2014.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gratuze M, Leyns CEG, & Holtzman DM (2018). New insights into the role of TREM2 in Alzheimer’s disease. Mol Neurodegener, 13(1), 66. doi: 10.1186/s13024-018-0298-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, … Colonna M (2017). TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell, 170(4), 649–663 e613. doi: 10.1016/j.cell.2017.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ulrich JD, Ulland TK, Colonna M, & Holtzman DM (2017). Elucidating the Role of TREM2 in Alzheimer’s Disease. Neuron, 94(2), 237–248. doi: 10.1016/j.neuron.2017.02.042 [DOI] [PubMed] [Google Scholar]
- [34].Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, … Alzheimer Genetic Analysis, G. (2013). TREM2 variants in Alzheimer’s disease. N Engl J Med, 368(2), 117–127. doi: 10.1056/NEJMoa1211851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, … Stefansson K (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med, 368(2), 107–116. doi: 10.1056/NEJMoa1211103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, … Butovsky O (2017). The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity, 47(3), 566–581 e569. doi: 10.1016/j.immuni.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, … Colonna M (2016). TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med, 213(5), 667–675. doi: 10.1084/jem.20151948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhu B, Liu Y, Hwang S, Archuleta K, Huang H, Campos A, … Huang TY (2022). Trem2 deletion enhances tau dispersion and pathology through microglia exosomes. Mol Neurodegener, 17(1), 58. doi: 10.1186/s13024-022-00562-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fitz NF, Nam KN, Wolfe CM, Letronne F, Playso BE, Iordanova BE, … Koldamova R (2021). Phospholipids of APOE lipoproteins activate microglia in an isoform-specific manner in preclinical models of Alzheimer’s disease. Nat Commun, 12(1), 3416. doi: 10.1038/s41467-021-23762-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, … Amit I (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell, 169(7), 1276–1290.e1217. doi: 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- [41].Chen Y, & Colonna M (2021). Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? J Exp Med, 218(9). doi: 10.1084/jem.20202717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Spurgat MS, & Tang SJ (2022). Single-Cell RNA-Sequencing: Astrocyte and Microglial Heterogeneity in Health and Disease. Cells, 11(13). doi: 10.3390/cells11132021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, … Tsai LH (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature, 570(7761), 332–337. doi: 10.1038/s41586-019-1195-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, … Colonna M (2020). Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med, 26(1), 131–142. doi: 10.1038/s41591-019-0695-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Del-Aguila JL, Li Z, Dube U, Mihindukulasuriya KA, Budde JP, Fernandez MV, … Harari O (2019). A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res Ther, 11(1), 71. doi: 10.1186/s13195-019-0524-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Geirsdottir L, David E, Keren-Shaul H, Weiner A, Bohlen SC, Neuber J, … Prinz M (2020). Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell, 181(3), 746. doi: 10.1016/j.cell.2020.04.002 [DOI] [PubMed] [Google Scholar]
- [47].Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, … De Jager PL (2020). Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun, 11(1), 6129. doi: 10.1038/s41467-020-19737-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Srinivasan K, Friedman BA, Etxeberria A, Huntley MA, van der Brug MP, Foreman O, … Hansen DV (2020). Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep, 31(13), 107843. doi: 10.1016/j.celrep.2020.107843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Council NR (2011). Guide for the Care and Use of Laboratory Animals: Eighth Edition. Washington, DC: The National Academies Press. [Google Scholar]
- [50].Thrupp N, Sala Frigerio C, Wolfs L, Skene NG, Fattorelli N, Poovathingal S, … Fiers M (2020). Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep, 32(13), 108189. doi: 10.1016/j.celrep.2020.108189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Geirsdottir L, David E, Keren-Shaul H, Weiner A, Bohlen SC, Neuber J, … Prinz M (2019). Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell, 179(7), 1609–1622.e1616. doi: 10.1016/j.cell.2019.11.010 [DOI] [PubMed] [Google Scholar]
- [52].Graves PR, & Haystead TAJ (2002). Molecular Biologist’s Guide to Proteomics. Microbiology and Molecular Biology Reviews, 66(1), 39–63. doi: doi: 10.1128/MMBR.66.1.39-63.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, & El Khoury J (2013). The microglial sensome revealed by direct RNA sequencing. Nat Neurosci, 16(12), 1896–1905. doi: 10.1038/nn.3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chidambaram H, Das R, & Chinnathambi S (2020). Interaction of Tau with the chemokine receptor, CX3CR1 and its effect on microglial activation, migration and proliferation. Cell & Bioscience, 10(1), 109. doi: 10.1186/s13578-020-00474-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bolós M, Llorens-Martín M, Perea JR, Jurado-Arjona J, Rábano A, Hernández F, & Avila J (2017). Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol Neurodegener, 12(1), 59. doi: 10.1186/s13024-017-0200-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang Y, Sloan Steven A., Clarke Laura E., Caneda C, Plaza Colton A., Blumenthal Paul D., … Barres Ben A. (2016). Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron, 89(1), 37–53. doi: 10.1016/j.neuron.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, … Colonna M (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell, 160(6), 1061–1071. doi: 10.1016/j.cell.2015.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yeh FL, Wang Y, Tom I, Gonzalez LC, & Sheng M (2016). TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron, 91(2), 328–340. doi: 10.1016/j.neuron.2016.06.015 [DOI] [PubMed] [Google Scholar]
- [59].Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, … Bu G (2015). Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J Biol Chem, 290(43), 26043–26050. doi: 10.1074/jbc.M115.679043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, … Xu H (2018). TREM2 Is a Receptor for beta-Amyloid that Mediates Microglial Function. Neuron, 97(5), 1023–1031 e1027. doi: 10.1016/j.neuron.2018.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Takahashi K, Rochford CD, & Neumann H (2005). Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med, 201(4), 647–657. doi: 10.1084/jem.20041611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Konishi H, & Kiyama H (2018). Microglial TREM2/DAP12 Signaling: A Double-Edged Sword in Neural Diseases. Front Cell Neurosci, 12, 206. doi: 10.3389/fncel.2018.00206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Haure-Mirande JV, Audrain M, Ehrlich ME, & Gandy S (2022). Microglial TYROBP/DAP12 in Alzheimer’s disease: Transduction of physiological and pathological signals across TREM2. Mol Neurodegener, 17(1), 55. doi: 10.1186/s13024-022-00552-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wang S, Sudan R, Peng V, Zhou Y, Du S, Yuede CM, … Colonna M (2022). TREM2 drives microglia response to amyloid-β via SYK-dependent and -independent pathways. Cell, 185(22), 4153–4169.e4119. doi: 10.1016/j.cell.2022.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, & Humphrey MB (2010). TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal, 3(122), ra38. doi: 10.1126/scisignal.2000500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zou Z, Tao T, Li H, & Zhu X (2020). mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell & Bioscience, 10(1), 31. doi: 10.1186/s13578-020-00396-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kim EK, & Choi EJ (2010). Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta, 1802(4), 396–405. doi: 10.1016/j.bbadis.2009.12.009 [DOI] [PubMed] [Google Scholar]
- [68].Chen MJ, Ramesha S, Weinstock LD, Gao T, Ping L, Xiao H, … Rangaraju S (2021). Extracellular signal-regulated kinase regulates microglial immune responses in Alzheimer’s disease. J Neurosci Res, 99(6), 1704–1721. doi: 10.1002/jnr.24829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Johnson ECB, Carter EK, Dammer EB, Duong DM, Gerasimov ES, Liu Y, … Seyfried NT (2022). Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat Neurosci, 25(2), 213–225. doi: 10.1038/s41593-021-00999-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, … Levey AI (2017). A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst, 4(1), 60–72 e64. doi: 10.1016/j.cels.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Johnson ECB, Carter EK, Dammer EB, Duong DM, Gerasimov ES, Liu Y, … Seyfried NT (2022). Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nature Neuroscience, 25(2), 213–225. doi: 10.1038/s41593-021-00999-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Mahdessian D, Cesnik AJ, Gnann C, Danielsson F, Stenström L, Arif M, … Lundberg E (2021). Spatiotemporal dissection of the cell cycle with single-cell proteogenomics. Nature, 590(7847), 649–654. doi: 10.1038/s41586-021-03232-9 [DOI] [PubMed] [Google Scholar]
- [73].Specht H, Emmott E, Petelski AA, Huffman RG, Perlman DH, Serra M, … Slavov N (2021). Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biology, 22(1), 50. doi: 10.1186/s13059-021-02267-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Buccitelli C, & Selbach M (2020). mRNAs, proteins and the emerging principles of gene expression control. Nature Reviews Genetics, 21(10), 630–644. doi: 10.1038/s41576-020-0258-4 [DOI] [PubMed] [Google Scholar]
- [75].Slavov N (2022). Scaling Up Single-Cell Proteomics. Mol Cell Proteomics, 21(1), 100179. doi: 10.1016/j.mcpro.2021.100179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Matejuk A, & Ransohoff RM (2020). Crosstalk Between Astrocytes and Microglia: An Overview. Frontiers in Immunology, 11. doi: 10.3389/fimmu.2020.01416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Vainchtein ID, & Molofsky AV (2020). Astrocytes and Microglia: In Sickness and in Health. Trends in Neurosciences, 43(3), 144–154. doi: 10.1016/j.tins.2020.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, … Barres BA (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541(7638), 481–487. doi: 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Barbierato M, Facci L, Argentini C, Marinelli C, Skaper SD, & Giusti P (2013). Astrocyte-microglia cooperation in the expression of a pro-inflammatory phenotype. CNS Neurol Disord Drug Targets, 12(5), 608–618. doi: 10.2174/18715273113129990064 [DOI] [PubMed] [Google Scholar]
- [80].Facci L, Barbierato M, Marinelli C, Argentini C, Skaper SD, & Giusti P (2014). Toll-like receptors 2, −3 and −4 prime microglia but not astrocytes across central nervous system regions for ATP-dependent interleukin-1β release. Sci Rep, 4, 6824. doi: 10.1038/srep06824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Schartz ND, & Tenner AJ (2020). The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J Neuroinflammation, 17(1), 354. doi: 10.1186/s12974-020-02024-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, … Stevens B (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron, 74(4), 691–705. doi: 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, … Stevens B (2018). CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron, 100(1), 120–134.e126. doi: 10.1016/j.neuron.2018.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, … Lemere CA (2017). Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med, 9(392). doi: 10.1126/scitranslmed.aaf6295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Dejanovic B, Huntley MA, De Maziere A, Meilandt WJ, Wu T, Srinivasan K, … Sheng M (2018). Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron, 100(6), 1322–1336 e1327. doi: 10.1016/j.neuron.2018.10.014 [DOI] [PubMed] [Google Scholar]
- [86].Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, … Stevens B (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science, 352(6286), 712–716. doi: 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S, … Hanson JE (2019). Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep, 28(8), 2111–2123.e2116. doi: 10.1016/j.celrep.2019.07.060 [DOI] [PubMed] [Google Scholar]
- [88].Bai B, Wang X, Li Y, Chen PC, Yu K, Dey KK, … Peng J (2020). Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron, 105(6), 975–991.e977. doi: 10.1016/j.neuron.2019.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Doens D, & Fernandez PL (2014). Microglia receptors and their implications in the response to amyloid beta for Alzheimer’s disease pathogenesis. J Neuroinflammation, 11, 48. doi: 10.1186/1742-2094-11-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Rangaraju S, Dammer EB, Raza SA, Rathakrishnan P, Xiao H, Gao T, … Levey AI (2018). Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegener, 13(1), 24. doi: 10.1186/s13024-018-0254-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, … Levey AI (2017). A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Systems, 4(1), 60–72.e64. doi: 10.1016/j.cels.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Higginbotham L, Ping L, Dammer EB, Duong DM, Zhou M, Gearing M, … Seyfried NT (2020). Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Sci Adv, 6(43). doi: 10.1126/sciadv.aaz9360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Vogel C, & Marcotte EM (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet, 13(4), 227–232. doi: 10.1038/nrg3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, … Simons M (2015). Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci, 18(12), 1819–1831. doi: 10.1038/nn.4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Rayaprolu S, Higginbotham L, Bagchi P, Watson CM, Zhang T, Levey AI, … Seyfried NT (2021). Systems-based proteomics to resolve the biology of Alzheimer’s disease beyond amyloid and tau. Neuropsychopharmacology, 46(1), 98–115. doi: 10.1038/s41386-020-00840-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, … Seyfried NT (2020). Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med, 26(5), 769–780. doi: 10.1038/s41591-020-0815-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Warburg O (1956). On respiratory impairment in cancer cells. Science, 124(3215), 269–270. [PubMed] [Google Scholar]
- [98].Vander Heiden MG, Cantley LC, & Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science, 324(5930), 1029–1033. doi: 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Griffin JL, & Shockcor JP (2004). Metabolic profiles of cancer cells. Nature reviews cancer, 4(7), 551–561. [DOI] [PubMed] [Google Scholar]
- [100].Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang HY, Ahima RS, … Nathan DM (2018). Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol, 14(3), 168–181. doi: 10.1038/nrneurol.2017.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Lee S, Tong M, Hang S, Deochand C, & de la Monte S (2013). CSF and Brain Indices of Insulin Resistance, Oxidative Stress and Neuro-Inflammation in Early versus Late Alzheimer’s Disease. J Alzheimers Dis Parkinsonism, 3, 128. doi: 10.4172/2161-0460.1000128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Moloudizargari M, Moradkhani F, Asghari N, Fallah M, Asghari MH, Moghadamnia AA, & Abdollahi M (2019). NLRP inflammasome as a key role player in the pathogenesis of environmental toxicants. Life Sciences, 231, 116585. doi: 10.1016/j.lfs.2019.116585 [DOI] [PubMed] [Google Scholar]
- [103].Hanslik KL, & Ulland TK (2020). The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front Neurol, 11, 570711. doi: 10.3389/fneur.2020.570711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, … Heneka MT (2017). Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature, 552(7685), 355–361. doi: 10.1038/nature25158 [DOI] [PubMed] [Google Scholar]
- [105].Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, … Heneka MT (2019). NLRP3 inflammasome activation drives tau pathology. Nature, 575(7784), 669–673. doi: 10.1038/s41586-019-1769-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Stancu IC, Cremers N, Vanrusselt H, Couturier J, Vanoosthuyse A, Kessels S, … Dewachter I (2019). Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol, 137(4), 599–617. doi: 10.1007/s00401-018-01957-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Tan MS, Tan L, Jiang T, Zhu XC, Wang HF, Jia CD, & Yu JT (2014). Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis, 5(8), e1382. doi: 10.1038/cddis.2014.348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Denes A, Coutts G, Lénárt N, Cruickshank SM, Pelegrin P, Skinner J, … Brough D (2015). AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc Natl Acad Sci U S A, 112(13), 4050–4055. doi: 10.1073/pnas.1419090112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, … Seyfried NT (2020). Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine, 26(5), 769–780. doi: 10.1038/s41591-020-0815-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Swanson KV, Deng M, & Ting JPY (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nature Reviews Immunology, 19(8), 477–489. doi: 10.1038/s41577-019-0165-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Meissner F, Molawi K, & Zychlinsky A (2008). Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat Immunol, 9(8), 866–872. doi: 10.1038/ni.1633 [DOI] [PubMed] [Google Scholar]
- [112].Kuwar R, Rolfe A, Di L, Blevins H, Xu Y, Sun X, … Sun D (2021). A Novel Inhibitor Targeting NLRP3 Inflammasome Reduces Neuropathology and Improves Cognitive Function in Alzheimer’s Disease Transgenic Mice. J Alzheimers Dis, 82(4), 1769–1783. doi: 10.3233/jad-210400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Magdalon J, Mansur F, Teles e Silva AL, de Goes VA, Reiner O, & Sertié AL (2020). Complement System in Brain Architecture and Neurodevelopmental Disorders. Frontiers in Neuroscience, 14. doi: 10.3389/fnins.2020.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Andoh M, & Koyama R (2021). Microglia regulate synaptic development and plasticity. Developmental Neurobiology, 81(5), 568–590. doi: 10.1002/dneu.22814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Litvinchuk A, Wan YW, Swartzlander DB, Chen F, Cole A, Propson NE, … Zheng H (2018). Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron, 100(6), 1337–1353.e1335. doi: 10.1016/j.neuron.2018.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, … Lemere CA (2015). Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J Neurosci, 35(38), 13029–13042. doi: 10.1523/jneurosci.1698-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Bai B, Hales CM, Chen P-C, Gozal Y, Dammer EB, Fritz JJ, … Peng J (2013). U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proceedings of the National Academy of Sciences, 110(41), 16562–16567. doi: 10.1073/pnas.1310249110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Bai B, Wang X, Li Y, Chen PC, Yu K, Dey KK, … Peng J (2020). Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron, 106(4), 700. doi: 10.1016/j.neuron.2020.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Subramanian J, Savage JC, & Tremblay M-È (2020). Synaptic Loss in Alzheimer’s Disease: Mechanistic Insights Provided by Two-Photon in vivo Imaging of Transgenic Mouse Models. Frontiers in Cellular Neuroscience, 14. doi: 10.3389/fncel.2020.592607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].de Wilde MC, Overk CR, Sijben JW, & Masliah E (2016). Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimer’s & Dementia, 12(6), 633–644. doi: 10.1016/j.jalz.2015.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, … Vassar R (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci, 26(40), 10129–10140. doi: 10.1523/jneurosci.1202-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Neuman KM, Molina-Campos E, Musial TF, Price AL, Oh KJ, Wolke ML, … Nicholson DA (2015). Evidence for Alzheimer’s disease-linked synapse loss and compensation in mouse and human hippocampal CA1 pyramidal neurons. Brain Struct Funct, 220(6), 3143–3165. doi: 10.1007/s00429-014-0848-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Buskila Y, Crowe SE, & Ellis-Davies GC (2013). Synaptic deficits in layer 5 neurons precede overt structural decay in 5xFAD mice. Neuroscience, 254, 152–159. doi: 10.1016/j.neuroscience.2013.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Eimer WA, & Vassar R (2013). Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Aβ42 accumulation and Caspase-3 activation. Molecular Neurodegeneration, 8(1), 2. doi: 10.1186/1750-1326-8-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Seo NY, Kim GH, Noh JE, Shin JW, Lee CH, & Lee KJ (2021). Selective Regional Loss of Cortical Synapses Lacking Presynaptic Mitochondria in the 5xFAD Mouse Model. Front Neuroanat, 15, 690168. doi: 10.3389/fnana.2021.690168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Oblak AL, Lin PB, Kotredes KP, Pandey RS, Garceau D, Williams HM, … Lamb BT (2021). Comprehensive Evaluation of the 5XFAD Mouse Model for Preclinical Testing Applications: A MODEL-AD Study. Frontiers in Aging Neuroscience, 13. doi: 10.3389/fnagi.2021.713726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Moechars D, Lorent K, De Strooper B, Dewachter I, & Van Leuven F (1996). Expression in brain of amyloid precursor protein mutated in the alpha-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. The EMBO Journal, 15(6), 1265–1274. doi: 10.1002/j.1460-2075.1996.tb00468.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Ramesha S, Rayaprolu S, & Rangaraju S (2020). Flow Cytometry Approach to Characterize Phagocytic Properties of Acutely-Isolated Adult Microglia and Brain Macrophages In Vitro. J Vis Exp(160). doi: 10.3791/61467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Rangaraju S, Dammer EB, Raza SA, Gao T, Xiao H, Betarbet R, … Seyfried NT (2018). Quantitative proteomics of acutely-isolated mouse microglia identifies novel immune Alzheimer’s disease-related proteins. Molecular Neurodegeneration, 13(1), 34. doi: 10.1186/s13024-018-0266-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Rayaprolu S, Gao T, Xiao H, Ramesha S, Weinstock LD, Shah J, … Rangaraju S (2020). Flow-cytometric microglial sorting coupled with quantitative proteomics identifies moesin as a highly-abundant microglial protein with relevance to Alzheimer’s disease. Mol Neurodegener, 15(1), 28. doi: 10.1186/s13024-020-00377-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Bohlen CJ, Bennett FC, & Bennett ML (2019). Isolation and Culture of Microglia. Curr Protoc Immunol, 125(1), e70. doi: 10.1002/cpim.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Nolle A, van Dijken I, Waelti CM, Calini D, Bryois J, Lezan E, … Hoozemans JJM (2021). Enrichment of Glial Cells From Human Post-mortem Tissue for Transcriptome and Proteome Analysis Using Immunopanning. Front Cell Neurosci, 15, 772011. doi: 10.3389/fncel.2021.772011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Mizee MR, Miedema SSM, van der Poel, Adelia M, Schuurman KG, van Strien ME, … Huitinga I (2017). Isolation of primary microglia from the human post-mortem brain: effects of ante- and post-mortem variables. Acta Neuropathologica Communications, 5(1), 16. doi: 10.1186/s40478-017-0418-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Crotti A, & Ransohoff RM (2016). Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity, 44(3), 505–515. doi: 10.1016/j.immuni.2016.02.013 [DOI] [PubMed] [Google Scholar]
- [135].Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang L. c., Means TK, & El Khoury J (2013). The microglial sensome revealed by direct RNA sequencing. Nature Neuroscience, 16(12), 1896–1905. doi: 10.1038/nn.3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, … Barres BA (2016). New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences, 113(12), E1738–E1746. doi: 10.1073/pnas.1525528113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, & Luster AD (2007). Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nature Medicine, 13(4), 432–438. doi: 10.1038/nm1555 [DOI] [PubMed] [Google Scholar]
- [138].Rangaraju S, Raza SA, Li NXA, Betarbet R, Dammer EB, Duong D, … Levey AI (2018). Differential Phagocytic Properties of CD45low Microglia and CD45high Brain Mononuclear Phagocytes—Activation and Age-Related Effects. Frontiers in Immunology, 9. doi: 10.3389/fimmu.2018.00405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Pan J, & Wan J (2020). Methodological comparison of FACS and MACS isolation of enriched microglia and astrocytes from mouse brain. Journal of Immunological Methods, 486, 112834. doi: 10.1016/j.jim.2020.112834 [DOI] [PubMed] [Google Scholar]
- [140].Kang SS, Ebbert MTW, Baker KE, Cook C, Wang X, Sens JP, … Fryer JD (2018). Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J Exp Med, 215(9), 2235–2245. doi: 10.1084/jem.20180653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Swartzlander DB, Propson NE, Roy ER, Saito T, Saido T, Wang B, & Zheng H (2018). Concurrent cell type-specific isolation and profiling of mouse brains in inflammation and Alzheimer’s disease. JCI Insight, 3(13). doi: 10.1172/jci.insight.121109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Sutermaster BA, & Darling EM (2019). Considerations for high-yield, high-throughput cell enrichment: fluorescence versus magnetic sorting. Scientific Reports, 9(1), 227. doi: 10.1038/s41598-018-36698-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Guerreiro R, & Bras J (2015). The age factor in Alzheimer’s disease. Genome Med, 7, 106. doi: 10.1186/s13073-015-0232-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Flowers A, Bell-Temin H, Jalloh A, Stevens SM Jr., & Bickford PC (2017). Proteomic anaysis of aged microglia: shifts in transcription, bioenergetics, and nutrient response. J Neuroinflammation, 14(1), 96. doi: 10.1186/s12974-017-0840-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Byles V, Covarrubias AJ, Ben-Sahra I, Lamming DW, Sabatini DM, Manning BD, & Horng T (2013). The TSC-mTOR pathway regulates macrophage polarization. Nature Communications, 4(1), 2834. doi: 10.1038/ncomms3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Bracho-Valdés I, Moreno-Alvarez P, Valencia-Martínez I, Robles-Molina E, Chávez-Vargas L, & Vázquez-Prado J (2011). mTORC1- and mTORC2-interacting proteins keep their multifunctional partners focused. IUBMB Life, 63(10), 896–914. doi: 10.1002/iub.558 [DOI] [PubMed] [Google Scholar]
- [147].Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, & Johnson RW (2005). Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. Faseb j, 19(10), 1329–1331. doi: 10.1096/fj.05-3776fje [DOI] [PubMed] [Google Scholar]
- [148].Sierra A, Gottfried-Blackmore AC, McEwen BS, & Bulloch K (2007). Microglia derived from aging mice exhibit an altered inflammatory profile. Glia, 55(4), 412–424. doi: 10.1002/glia.20468 [DOI] [PubMed] [Google Scholar]
- [149].Sheng JG, Mrak RE, & Griffin W (1998). Enlarged and phagocytic, but not primed, interleukin-1α-immunoreactive microglia increase with age in normal human brain. Acta neuropathologica, 95, 229–234. [DOI] [PubMed] [Google Scholar]
- [150].Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, & Wong WT (2011). Age-related alterations in the dynamic behavior of microglia. Aging cell, 10(2), 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Tremblay MÈ, Zettel ML, Ison JR, Allen PD, & Majewska AK (2012). Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia, 60(4), 541–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ, & Cotman CW (2012). Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. Journal of neuroinflammation, 9(1), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Gomez-Nicola D, & Boche D (2015). Post-mortem analysis of neuroinflammatory changes in human Alzheimer’s disease. Alzheimer’s research & therapy, 7, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Sudduth TL, Schmitt FA, Nelson PT, & Wilcock DM (2013). Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiology of aging, 34(4), 1051–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Knezevic D, & Mizrahi R (2018). Molecular imaging of neuroinflammation in Alzheimer’s disease and mild cognitive impairment. Progress in Neuro-Psychopharmacology and Biological Psychiatry, 80, 123–131. [DOI] [PubMed] [Google Scholar]
- [156].Lagarde J, Sarazin M, & Bottlaender M (2018). In vivo PET imaging of neuroinflammation in Alzheimer’s disease. Journal of Neural Transmission, 125, 847–867. [DOI] [PubMed] [Google Scholar]
- [157].Zimmer ER, Leuzy A, Benedet AL, Breitner J, Gauthier S, & Rosa-Neto P (2014). Tracking neuroinflammation in Alzheimer’s disease: the role of positron emission tomography imaging. Journal of neuroinflammation, 11(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Yu QS, Feng WQ, Shi LL, Niu RZ, & Liu J (2022). Integrated Analysis of Cortex Single-Cell Transcriptome and Serum Proteome Reveals the Novel Biomarkers in Alzheimer’s Disease. Brain Sci, 12(8). doi: 10.3390/brainsci12081022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, … Amouyel P (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet, 41(10), 1094–1099. doi: 10.1038/ng.439 [DOI] [PubMed] [Google Scholar]
- [160].Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, & Hong JT (2008). Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. Journal of neuroinflammation, 5(1), 37. doi: 10.1186/1742-2094-5-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Wang LM, Wu Q, Kirk RA, Horn KP, Ebada Salem AH, Hoffman JM, … Morton KA (2018). Lipopolysaccharide endotoxemia induces amyloid-β and p-tau formation in the rat brain. Am J Nucl Med Mol Imaging, 8(2), 86–99. [PMC free article] [PubMed] [Google Scholar]
- [162].Kahn MS, Kranjac D, Alonzo CA, Haase JH, Cedillos RO, McLinden KA, … Chumley MJ (2012). Prolonged elevation in hippocampal Aβ and cognitive deficits following repeated endotoxin exposure in the mouse. Behavioural brain research, 229(1), 176–184. [DOI] [PubMed] [Google Scholar]
- [163].Tejera D, Mercan D, Sanchez-Caro JM, Hanan M, Greenberg D, Soreq H, … Heneka MT (2019). Systemic inflammation impairs microglial Aβ clearance through NLRP 3 inflammasome. The EMBO Journal, 38(17), e101064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Boza-Serrano A, Yang Y, Paulus A, & Deierborg T (2018). Innate immune alterations are elicited in microglial cells before plaque deposition in the Alzheimer’s disease mouse model 5xFAD. Sci Rep, 8(1), 1550. doi: 10.1038/s41598-018-19699-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Rangaraju S, Dammer EB, Raza SA, Rathakrishnan P, Xiao H, Gao T, … Levey AI (2018). Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Molecular Neurodegeneration, 13(1), 24. doi: 10.1186/s13024-018-0254-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Lloyd AF, Martinez-Muriana A, Hou P, Davis E, Mancuso R, Brenes AJ, … Howden AJM (2022). Deep proteomic analysis of human microglia and model systems reveal fundamental biological differences of <em>in vitro</em> and <em>ex vivo</em> cells. bioRxiv, 2022.2007.2007.498804. doi: 10.1101/2022.07.07.498804 [DOI] [Google Scholar]
- [167].Ocañas SR, Pham KD, Blankenship HE, Machalinski AH, Chucair-Elliott AJ, & Freeman WM (2022). Minimizing the <em>Ex Vivo</em> Confounds of Cell-Isolation Techniques on Transcriptomic and Translatomic Profiles of Purified Microglia. eneuro, 9(2), ENEURO.0348–0321.2022. doi: 10.1523/ENEURO.0348-21.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Dieterich DC, Link AJ, Graumann J, Tirrell DA, & Schuman EM (2006). Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc Natl Acad Sci U S A, 103(25), 9482–9487. doi: 10.1073/pnas.0601637103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Alvarez-Castelao B, Schanzenbächer CT, Hanus C, Glock C, tom Dieck S, Dörrbaum AR, … Schuman EM (2017). Cell-type-specific metabolic labeling of nascent proteomes in vivo. Nature Biotechnology, 35(12), 1196–1201. doi: 10.1038/nbt.4016 [DOI] [PubMed] [Google Scholar]
- [170].Prabhakar P, Pielot R, Landgraf P, Wissing J, Bayrhammer A, van Ham M, … Müller A (2022). Monitoring regional astrocyte diversity by cell-type specific proteomic labeling in vivo. bioRxiv, 2022.2006.2007.495069. doi: 10.1101/2022.06.07.495069 [DOI] [PubMed] [Google Scholar]
- [171].Mathew B, Bathla S, Williams KR, & Nairn AC (2022). Deciphering Spatial Protein-Protein Interactions in Brain Using Proximity Labeling. Mol Cell Proteomics, 21(11), 100422. doi: 10.1016/j.mcpro.2022.100422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Qin W, Cho KF, Cavanagh PE, & Ting AY (2021). Deciphering molecular interactions by proximity labeling. Nat Methods, 18(2), 133–143. doi: 10.1038/s41592-020-01010-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Roux KJ, Kim DI, Raida M, & Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol, 196(6), 801–810. doi: 10.1083/jcb.201112098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Cho KF, Branon TC, Udeshi ND, Myers SA, Carr SA, & Ting AY (2020). Proximity labeling in mammalian cells with TurboID and split-TurboID. Nat Protoc, 15(12), 3971–3999. doi: 10.1038/s41596-020-0399-0 [DOI] [PubMed] [Google Scholar]
- [175].Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, … Ting AY (2018). Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol, 36(9), 880–887. doi: 10.1038/nbt.4201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Rayaprolu S, Bitarafan S, Santiago JV, Betarbet R, Sunna S, Cheng L, … Rangaraju S (2022). Cell type-specific biotin labeling in vivo resolves regional neuronal and astrocyte proteomic differences in mouse brain. Nat Commun, 13(1), 2927. doi: 10.1038/s41467-022-30623-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Sunna S, Bowen C, Zeng H, Rayaprolu S, Kumar P, Bagchi P, … Rangaraju S (2022). Cellular proteomic profiling using proximity labelling by TurboID-NES in microglial and neuronal cell lines. bioRxiv, 2022.2009.2027.509765. doi: 10.1101/2022.09.27.509765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Rayaprolu S, Bitarafan S, Santiago JV, Betarbet R, Sunna S, Cheng L, … Rangaraju S (2022). Cell type-specific biotin labeling in vivo resolves regional neuronal and astrocyte proteomic differences in mouse brain. Nature Communications, 13(1), 2927. doi: 10.1038/s41467-022-30623-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, … Ting AY (2012). Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol, 30(11), 1143–1148. doi: 10.1038/nbt.2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, & Ting AY (2013). Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science, 339(6125), 1328–1331. doi: 10.1126/science.1230593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Nguyen TMT, Kim J, Doan TT, Lee MW, & Lee M (2020). APEX Proximity Labeling as a Versatile Tool for Biological Research. Biochemistry, 59(3), 260–269. doi: 10.1021/acs.biochem.9b00791 [DOI] [PubMed] [Google Scholar]
- [182].Mannix KM, Starble RM, Kaufman RS, & Cooley L (2019). Proximity labeling reveals novel interactomes in live Drosophila tissue. Development, 146(14). doi: 10.1242/dev.176644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Bersuker K, Peterson CWH, To M, Sahl SJ, Savikhin V, Grossman EA, … Olzmann JA (2018). A Proximity Labeling Strategy Provides Insights into the Composition and Dynamics of Lipid Droplet Proteomes. Dev Cell, 44(1), 97–112 e117. doi: 10.1016/j.devcel.2017.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Kaewsapsak P, Shechner DM, Mallard W, Rinn JL, & Ting AY (2017). Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. Elife, 6. doi: 10.7554/eLife.29224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Fazal FM, Han S, Parker KR, Kaewsapsak P, Xu J, Boettiger AN, … Ting AY (2019). Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell, 178(2), 473–490 e426. doi: 10.1016/j.cell.2019.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Chen CL, Hu Y, Udeshi ND, Lau TY, Wirtz-Peitz F, He L, … Perrimon N (2015). Proteomic mapping in live Drosophila tissues using an engineered ascorbate peroxidase. Proc Natl Acad Sci U S A, 112(39), 12093–12098. doi: 10.1073/pnas.1515623112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Dumrongprechachan V, Salisbury RB, Soto G, Kumar M, MacDonald ML, & Kozorovitskiy Y (2021). Cell-type and subcellular compartment-specific APEX2 proximity labeling reveals activity-dependent nuclear proteome dynamics in the striatum. Nat Commun, 12(1), 4855. doi: 10.1038/s41467-021-25144-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Hobson BD, Choi SJ, Mosharov EV, Soni RK, Sulzer D, & Sims PA (2022). Subcellular proteomics of dopamine neurons in the mouse brain. eLife, 11, e70921. doi: 10.7554/eLife.70921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Maes ME, Colombo G, Schulz R, & Siegert S (2019). Targeting microglia with lentivirus and AAV: Recent advances and remaining challenges. Neurosci Lett, 707, 134310. doi: 10.1016/j.neulet.2019.134310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Lin R, Zhou Y, Yan T, Wang R, Li H, Wu Z, … Luo M (2022). Directed evolution of adeno-associated virus for efficient gene delivery to microglia. Nat Methods, 19(8), 976–985. doi: 10.1038/s41592-022-01547-7 [DOI] [PubMed] [Google Scholar]
- [191].Thery C, Zitvogel L, & Amigorena S (2002). Exosomes: composition, biogenesis and function. Nat Rev Immunol, 2(8), 569–579. doi: 10.1038/nri855 [DOI] [PubMed] [Google Scholar]
- [192].Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, & Ratajczak MZ (2006). Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia, 20(9), 1487–1495. doi: 10.1038/sj.leu.2404296 [DOI] [PubMed] [Google Scholar]
- [193].Zheng Y, He R, Wang P, Shi Y, Zhao L, & Liang J (2019). Exosomes from LPS-stimulated macrophages induce neuroprotection and functional improvement after ischemic stroke by modulating microglial polarization. Biomater Sci, 7(5), 2037–2049. doi: 10.1039/c8bm01449c [DOI] [PubMed] [Google Scholar]
- [194].Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, … Ikezu T (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci, 18(11), 1584–1593. doi: 10.1038/nn.4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Ceccarelli L, Marchetti L, Rizzo M, Moscardini A, Cappello V, Da Pozzo E, … Martini C (2022). Human Microglia Extracellular Vesicles Derived from Different Microglia Cell Lines: Similarities and Differences. ACS Omega, 7(27), 23127–23137. doi: 10.1021/acsomega.2c00816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Potolicchio I, Carven GJ, Xu X, Stipp C, Riese RJ, Stern LJ, & Santambrogio L (2005). Proteomic analysis of microglia-derived exosomes: metabolic role of the aminopeptidase CD13 in neuropeptide catabolism. J Immunol, 175(4), 2237–2243. doi: 10.4049/jimmunol.175.4.2237 [DOI] [PubMed] [Google Scholar]
- [197].Yang Y, Boza-Serrano A, Dunning CJR, Clausen BH, Lambertsen KL, & Deierborg T (2018). Inflammation leads to distinct populations of extracellular vesicles from microglia. J Neuroinflammation, 15(1), 168. doi: 10.1186/s12974-018-1204-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Verderio C, Muzio L, Turola E, Bergami A, Novellino L, Ruffini F, … Furlan R (2012). Myeloid microvesicles are a marker and therapeutic target for neuroinflammation. Ann Neurol, 72(4), 610–624. doi: 10.1002/ana.23627 [DOI] [PubMed] [Google Scholar]
- [199].Men Y, Yelick J, Jin S, Tian Y, Chiang MSR, Higashimori H, … Yang Y (2019). Exosome reporter mice reveal the involvement of exosomes in mediating neuron to astroglia communication in the CNS. Nat Commun, 10(1), 4136. doi: 10.1038/s41467-019-11534-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Manzoni C, Kia DA, Vandrovcova J, Hardy J, Wood NW, Lewis PA, & Ferrari R (2018). Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences. Brief Bioinform, 19(2), 286–302. doi: 10.1093/bib/bbw114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Ghazalpour A, Bennett B, Petyuk VA, Orozco L, Hagopian R, Mungrue IN, … Lusis AJ (2011). Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet, 7(6), e1001393. doi: 10.1371/journal.pgen.1001393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Budnik B, Levy E, Harmange G, & Slavov N (2018). SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol, 19(1), 161. doi: 10.1186/s13059-018-1547-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Korin B, Dubovik T, & Rolls A (2018). Mass cytometry analysis of immune cells in the brain. Nat Protoc, 13(2), 377–391. doi: 10.1038/nprot.2017.155 [DOI] [PubMed] [Google Scholar]
- [204].Spiteri AG, Terry RL, Wishart CL, Ashhurst TM, Campbell IL, Hofer MJ, & King NJC (2021). High-parameter cytometry unmasks microglial cell spatio-temporal response kinetics in severe neuroinflammatory disease. J Neuroinflammation, 18(1), 166. doi: 10.1186/s12974-021-02214-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Zhu Y, Piehowski PD, Zhao R, Chen J, Shen Y, Moore RJ, … Kelly RT (2018). Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat Commun, 9(1), 882. doi: 10.1038/s41467-018-03367-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Zhou M, Uwugiaren N, Williams SM, Moore RJ, Zhao R, Goodlett D, … Zhu Y (2020). Sensitive Top-Down Proteomics Analysis of a Low Number of Mammalian Cells Using a Nanodroplet Sample Processing Platform. Analytical chemistry, 92(10), 7087–7095. doi: 10.1021/acs.analchem.0c00467 [DOI] [PubMed] [Google Scholar]
- [207].Zhu Y, Scheibinger M, Ellwanger DC, Krey JF, Choi D, Kelly RT, … Barr-Gillespie PG (2019). Single-cell proteomics reveals changes in expression during hair-cell development. Elife, 8. doi: 10.7554/eLife.50777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Piehowski PD, Zhu Y, Bramer LM, Stratton KG, Zhao R, Orton DJ, … Burnum-Johnson KE (2020). Automated mass spectrometry imaging of over 2000 proteins from tissue sections at 100-μm spatial resolution. Nature Communications, 11(1), 8. doi: 10.1038/s41467-019-13858-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Petelski AA, Emmott E, Leduc A, Huffman RG, Specht H, Perlman DH, & Slavov N (2021). Multiplexed single-cell proteomics using SCoPE2. Nat Protoc, 16(12), 5398–5425. doi: 10.1038/s41596-021-00616-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Specht H, Emmott E, Petelski AA, Huffman RG, Perlman DH, Serra M, … Slavov N (2021). Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol, 22(1), 50. doi: 10.1186/s13059-021-02267-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Slavov N (2021). Single-cell protein analysis by mass spectrometry. Curr Opin Chem Biol, 60, 1–9. doi: 10.1016/j.cbpa.2020.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Gebreyesus ST, Siyal AA, Kitata RB, Chen ES, Enkhbayar B, Angata T, … Tu HL (2022). Streamlined single-cell proteomics by an integrated microfluidic chip and data-independent acquisition mass spectrometry. Nat Commun, 13(1), 37. doi: 10.1038/s41467-021-27778-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Meier F, Kohler ND, Brunner AD, Wanka JH, Voytik E, Strauss MT, … Mann M (2021). Deep learning the collisional cross sections of the peptide universe from a million experimental values. Nat Commun, 12(1), 1185. doi: 10.1038/s41467-021-21352-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Meier F, Beck S, Grassl N, Lubeck M, Park MA, Raether O, & Mann M (2015). Parallel Accumulation-Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device. J Proteome Res, 14(12), 5378–5387. doi: 10.1021/acs.jproteome.5b00932 [DOI] [PubMed] [Google Scholar]
- [215].Brunner AD, Thielert M, Vasilopoulou C, Ammar C, Coscia F, Mund A, … Mann M (2022). Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. Mol Syst Biol, 18(3), e10798. doi: 10.15252/msb.202110798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Sierksma A, Lu A, Mancuso R, Fattorelli N, Thrupp N, Salta E, … Fiers M (2020). Novel Alzheimer risk genes determine the microglia response to amyloid-β but not to TAU pathology. EMBO Molecular Medicine, 12(3), e10606. doi: 10.15252/emmm.201910606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Arnoux I, & Audinat E (2015). Fractalkine Signaling and Microglia Functions in the Developing Brain. Neural Plast, 2015, 689404. doi: 10.1155/2015/689404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Konishi H, & Kiyama H (2018). Microglial TREM2/DAP12 Signaling: A Double-Edged Sword in Neural Diseases. Frontiers in Cellular Neuroscience, 12. doi: 10.3389/fncel.2018.00206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Turnbull IR, & Colonna M (2007). Activating and inhibitory functions of DAP12. Nature Reviews Immunology, 7(2), 155–161. doi: 10.1038/nri2014 [DOI] [PubMed] [Google Scholar]
- [220].Sobue A, Komine O, Hara Y, Endo F, Mizoguchi H, Watanabe S, … Yamanaka K (2021). Microglial gene signature reveals loss of homeostatic microglia associated with neurodegeneration of Alzheimer’s disease. Acta Neuropathologica Communications, 9(1), 1. doi: 10.1186/s40478-020-01099-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Lin D, & Maecker HT (2018). Mass Cytometry Assays for Antigen-Specific T Cells Using CyTOF. Methods Mol Biol, 1678, 37–47. doi: 10.1007/978-1-4939-7346-0_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Zhu Y, Dou M, Piehowski PD, Liang Y, Wang F, Chu RK, … Kelly RT (2018). Spatially Resolved Proteome Mapping of Laser Capture Microdissected Tissue with Automated Sample Transfer to Nanodroplets. Mol Cell Proteomics, 17(9), 1864–1874. doi: 10.1074/mcp.TIR118.000686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Zhu Y, Scheibinger M, Ellwanger DC, Krey JF, Choi D, Kelly RT, … Barr-Gillespie PG (2019). Single-cell proteomics reveals changes in expression during hair-cell development. Elife, 8, e50777. doi: 10.7554/eLife.50777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Williams SM, Liyu AV, Tsai C-F, Moore RJ, Orton DJ, Chrisler WB, … Zhu Y (2020). Automated Coupling of Nanodroplet Sample Preparation with Liquid Chromatography–Mass Spectrometry for High-Throughput Single-Cell Proteomics. Analytical chemistry, 92(15), 10588–10596. doi: 10.1021/acs.analchem.0c01551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Gebreyesus ST, Siyal AA, Kitata RB, Chen ES-W, Enkhbayar B, Angata T, … Tu H-L (2022). Streamlined single-cell proteomics by an integrated microfluidic chip and data-independent acquisition mass spectrometry. Nature Communications, 13(1), 37. doi: 10.1038/s41467-021-27778-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Lamanna J, Scott EY, Edwards HS, Chamberlain MD, Dryden MDM, Peng J, … Wheeler AR (2020). Digital microfluidic isolation of single cells for -Omics. Nat Commun, 11(1), 5632. doi: 10.1038/s41467-020-19394-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Petrosius V, & Schoof EM (2023). Recent advances in the field of single-cell proteomics. Transl Oncol, 27, 101556. doi: 10.1016/j.tranon.2022.101556 [DOI] [PMC free article] [PubMed] [Google Scholar]