

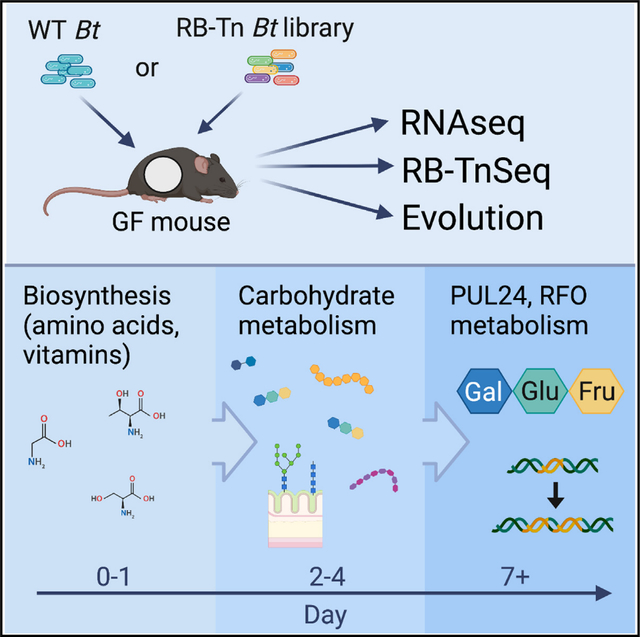
SUMMARY
To understand how a bacterium ultimately succeeds or fails in adapting to a new host, it is essential to assess the temporal dynamics of its fitness over the course of colonization. Here, we introduce a human-derived commensal organism, Bacteroides thetaiotaomicron (Bt), into the guts of germ-free mice to determine whether and how the genetic requirements for colonization shift over time. Combining a high-throughput functional genetics assay and transcriptomics, we find that gene usage changes drastically during the first days of colonization, shifting from high expression of amino acid biosynthesis genes to broad upregulation of diverse polysaccharide utilization loci. Within the first week, metabolism becomes centered around utilization of a predominant dietary oligosaccharide, and these changes are largely sustained through 6 weeks of colonization. Spontaneous mutations in wild-type Bt also evolve around this locus. These findings highlight the importance of considering temporal colonization dynamics in developing more effective microbiome-based therapies.
Graphical Abstract
In brief
Kennedy et al. evaluate temporal dynamics of Bacteroides thetaiotaomicron (Bt) adaptation to a new host. Bt metabolic priorities shift over the course of colonization, culminating in strong selective pressure to efficiently consume dietary resources. This work highlights the importance of considering temporal dynamics in developing better microbiome-based therapies.
INTRODUCTION
Rapid adaptation is paramount to the survival of any species undergoing an environmental transition. For microbial taxa, which are frequently and rapidly dispersed across dramatically different habitats and microenvironments, processes of local adaptation may arise as primary determinants of microbial colonization success and resulting biogeography.1 The mammalian gut is an environment regularly bombarded with a diverse array of exogenous microorganisms. As such, it represents a biologically and clinically relevant system to explore rapid microbial adaptational processes.
An emerging body of literature has begun probing the evolutionary and selective dynamics of exogenous microbes during colonization of the mammalian gut but has largely neglected early-time-point or transient dynamics en route to long-term persistence. For instance, several studies using transposon sequencing (TnSeq)-based approaches have evaluated the genetic requirements for colonization across various microbes in the guts of conventionally raised, gnotobiotic, and germ-free mice, but have assessed fitness only at a single time point after introduction.2,3 Wu et al. evaluated TnSeq mutant abundances of four Bacteroides strains over a 16-day time course but performed only broad characterization of population-level shifts at intermediate time points, restricting more rigorous gene-level functional analyses to a single endpoint.4 Several recent evolution studies have demonstrated the extent to which carbon limitation and metabolic demands drive the evolutionary trajectories of commensal and probiotic strains such as Bacteroides thetaiotaomicron (Bt) or Escherichia coli in the gut.5–8 However, despite dense time-course sampling, these studies do not identify genes that are important for fitness specifically at earlier stages of colonization. On shorter adaptive timescales, transcriptomics analyses of commensal microbes such as Bt in vivo and in vitro have demonstrated that gene expression profiles adapt quickly to factors such as diet,9 community membership,10,11 immune activation,12 or spatial localization within the gut.13 However, no studies have yet comprehensively evaluated the temporal transcriptional profile of a commensal microbe over the course of colonization.
In the following experiments, we introduce a human-derived commensal organism, Bt, into the guts of germ-free C57Bl/6 mice to determine whether the genetic requirements for colonization shift over time and, if so, to characterize the biological functions required for microbial survival at different stages of colonization and persistence. Use of a germ-free monocolonization model allows us to reduce the staggering complexity of the gut microbial ecosystem into experimentally tractable and readily interpretable components: here, we rigorously outline population-level microbial colonization dynamics for a widely used model organism and the host-microbe interactions that drive them. Using this germ-free model as a baseline, future work can evaluate the distinct contributions of microbe-microbe interactions and other emergent community-level properties to community assembly and colonization dynamics.
To identify the microbial genes important for fitness in this context, we combine two complementary unbiased approaches: transcriptomics (RNA sequencing), which reveals global gene usage patterns, and a functional genetics approach (BarSeq3,14 [bar-coded anatomy resolved by sequencing]) to assess fitness consequences of gene disruptions at a global scale over the course of gut colonization. We validate these results with both in vivo metabolomics analysis and in vitro microbial growth experiments. Finally, we evaluate spontaneous evolution of wild-type (WT) Bt in the gut to survey natural population-level fitness dynamics. Our results indicate that adaptation to the host gut occurs in distinct stages. During the earliest stage of colonization, genes involved in amino acid and vitamin biosynthesis are upregulated and, in some cases, play essential roles in survival of Bt. By colonization days 2–4, Bt shifts toward upregulation of a diverse array of carbohydrate metabolism genes. This broad survey of available resources continues through day 7 before ultimately centering on upregulation of a polysaccharide utilization locus (PUL) responsible for the degradation of raffinose-family oligosaccharides (RFOs) rich in the standard chow diet fed to our mice. This metabolic shift accompanies a change in Bt localization from the mucus toward the lumen, where the expression profile remains largely consistent through at least 6 weeks of colonization. Spontaneous mutations in WT Bt also evolve around the same PUL, highlighting the importance of efficient carbohydrate metabolism for long-term persistence.
These experiments lay the groundwork for future delineation of shifting selective pressures in various host backgrounds and microbiome compositions. We expect that these insights into the temporally dynamic stresses that microbes must overcome to colonize and persist in the gut will prove invaluable to our understanding of microbial adaptation and the development of microbiome-based therapies.
RESULTS
Both transcriptional and genetic fitness determinants shift over the course of Bt colonization and persistence
To evaluate global transcription during colonization, we introduced WT Bt into germ-free (GF) C57Bl/6 mice and collected cecal contents at days 0.5, 1, 2, 4, 7, 14, and 42 after colonization (Figure 1A, n = 3–4 mice per time point). A control cohort of GF mice was gavaged with sterile phosphate-buffered saline, and in vitro control samples were collected from n = 4 Bt cultures at mid-log phase in brain heart infusion supplemented medium. After rRNA and host RNA depletion, the bacterial RNA samples were sequenced and compared across time points. In parallel, to assess functional genetic requirements during colonization, we introduced a rich library of randomly barcoded Tn insertion (RB-Tn) mutants of Bt into four different cohorts of GF C57Bl/6 mice (n = 3–5 mice/cohort) and collected near-daily fecal samples (Figure 1A). Amplification and sequencing of the transposon barcodes reveals the relative abundance of each mutant in the library at each time point.14 RB-Tn experiments were carried out according to two slightly different protocols, with adjustments made to optimize experimental logistics and reduce bottleneck effects (Figure S1 and STAR Methods).
Figure 1. Bt gene expression and genetic fitness determinants shift over the course of colonization and persistence.
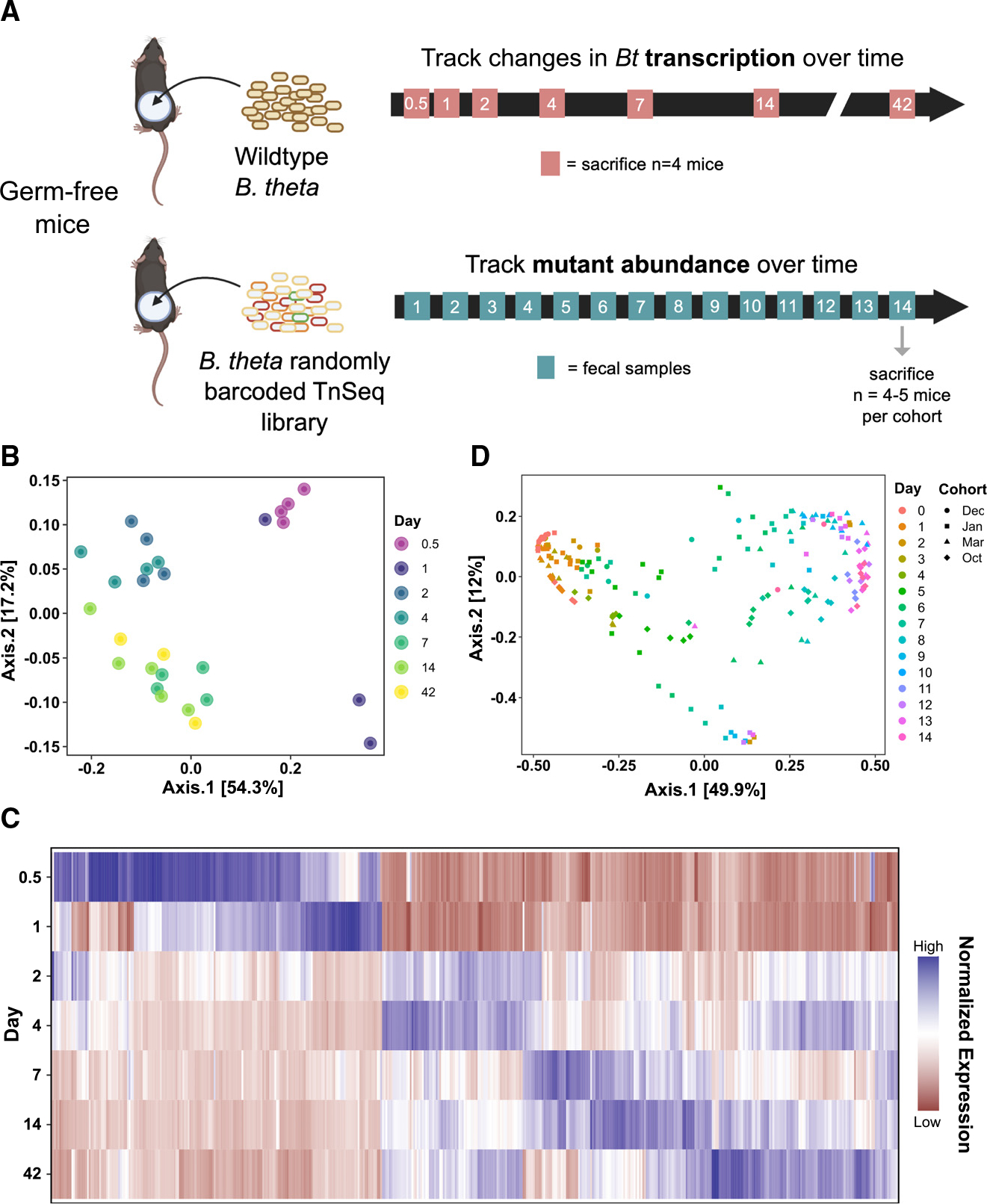
(A) Experimental scheme of transcriptomics (top) and functional genetics (bottom) experiments in germ-free (GF) mice.
(B) Principal coordinates analysis (PCoA) using Bray-Curtis dissimilarity on the global gene expression profile of WT Bt at 0.5, 1, 2, 4, 7, 14, and 42 days of colonization (n = 4–7 per group).
(C) Heatmap visualizing all genes that were significantly differentially expressed at any time point relative to day 1 (criteria: logFDR < −3, |log2FC| > 2, base mean >50 RPM). Each column represents an individual gene (Table S5); data are normalized across all time points for each gene.
(D) PCoA using Bray-Curtis dissimilarity on the abundances of RB-Tn mutant strains in fecal samples, colored by experimental day, shaped by experimental cohort. Four different cohorts of mice (n = 3–5 per cohort) are represented.
First, we asked whether there are differences in Bt gene expression at different times after introduction into the mouse gut. We first performed principal coordinates analysis (PCoA) using Bray-Curtis dissimilarity on all gene expression data (Figure 1B). We performed PERMANOVA analysis including both experimental day and cohort as explanatory factors, and found that only experimental day significantly contributed to clustering, explaining 79% of the variation in the dataset (Table S3).
We next performed post hoc analyses to evaluate clustering by experimental day. We confirmed that the in vitro expression profile of Bt clustered distinctly from all other time points (adjusted p < 0.05 for all comparisons, Table S3) and excluded in vitro samples from further analysis. Examining in vivo results, we found that day 0.5 and day 1 were statistically indistinguishable, as were day 2 and day 4, and day 14 and day 42, but that all other time points formed significantly distinct clusters (Figure 1B and Table S3). Therefore, for all further analyses, day 0.5 and day 1 data were combined (“D0.5/D1”), day 2 and day 4 data were combined (“D2/D4”), and day 14 and day 42 data were combined (“D14/D42”). The largest changes in gene expression, in which samples traverse principal coordinate 1 (PC1), occurred within 2 days after introduction to the gut. Although day 7 and D14/D42 were statistically distinct, these clusters spanned a smaller and overlapping range within PCoA space, and therefore reflect relatively minor changes in global gene expression profile. To further assess overarching gene expression changes in Bt over 6 weeks of gut colonization, all genes that met stringent criteria of differential expression (log false discovery rate [FDR] < −3, |log2fold change [FC]| > 2, and base mean >50 reads per million [RPM]) at any time point relative to day 1 were displayed in a heatmap (Figure 1C and Table S5). Even by these stringent criteria, we identified 489 differentially expressed genes (DEGs), which show a temporal pattern of relative peaks in expression at different time points.
Consistent with these global changes in gene expression over time, PCoA ordination using the relative abundance profiles of the RB-Tn mutant strains within each mouse in our functional genetics experiment reveals that the mutant pool composition shifted across time in a replicable pattern (Figure 1D) across four independent cohorts of this experiment, despite protocol adjustments. PERMANOVA showed that the data cluster significantly by experimental day (p = 0.0001, R2 = 0.457), with less substantial but significant contributions by individual mouse ID (p = 0.0001, R2 = 0.135) and experimental cohort (p = 0.0001, R2 = 0.092). The largest shifts across PC1 occurred between day 1 and day 7, with the mutant pool changing less dramatically between day 7 and day 14. Together, these data suggest that different sets of genes mediate colonization and growth immediately upon gut entry and later in the adaptational process.
Amino acid and vitamin biosynthesis are transcriptionally upregulated and functionally significant during early colonization of the gut
To gain a more comprehensive understanding of the gene pathways expressed during the acute phase of adaptation to the gut, we performed pairwise comparisons of gene expression across sequential time points. We identified extensive shifts in the Bt transcriptome between D0.5/D1 and D2/D4, with expression of 85 genes relatively enriched on D0.5/D1 and expression of 192 genes enriched at D2/D4 (Figure 2A). The pathways enriched at D0.5/D1 comprised a largely unique set of genes from those that dominate the Bt expression profile in vitro (Table S5). To functionally characterize these DEGs, we mapped them to the Kyoto Encyclopedia of Genes and Genomes (KEGG) catalog (Figure 2B). Although fewer DEGs were upregulated at D0.5/D1 compared to D2/D4, more of these genes mapped to known KEGG pathways, spanning a variety of functions, many of which center around metabolism. In particular, 30 DEGs upregulated at D0.5/D1 mapped to amino acid metabolism functions compared to ten amino acid genes upregulated at D2/D4.
Figure 2. The Bt transcriptome undergoes dramatic remodeling during the first week after introduction to the murine gut.
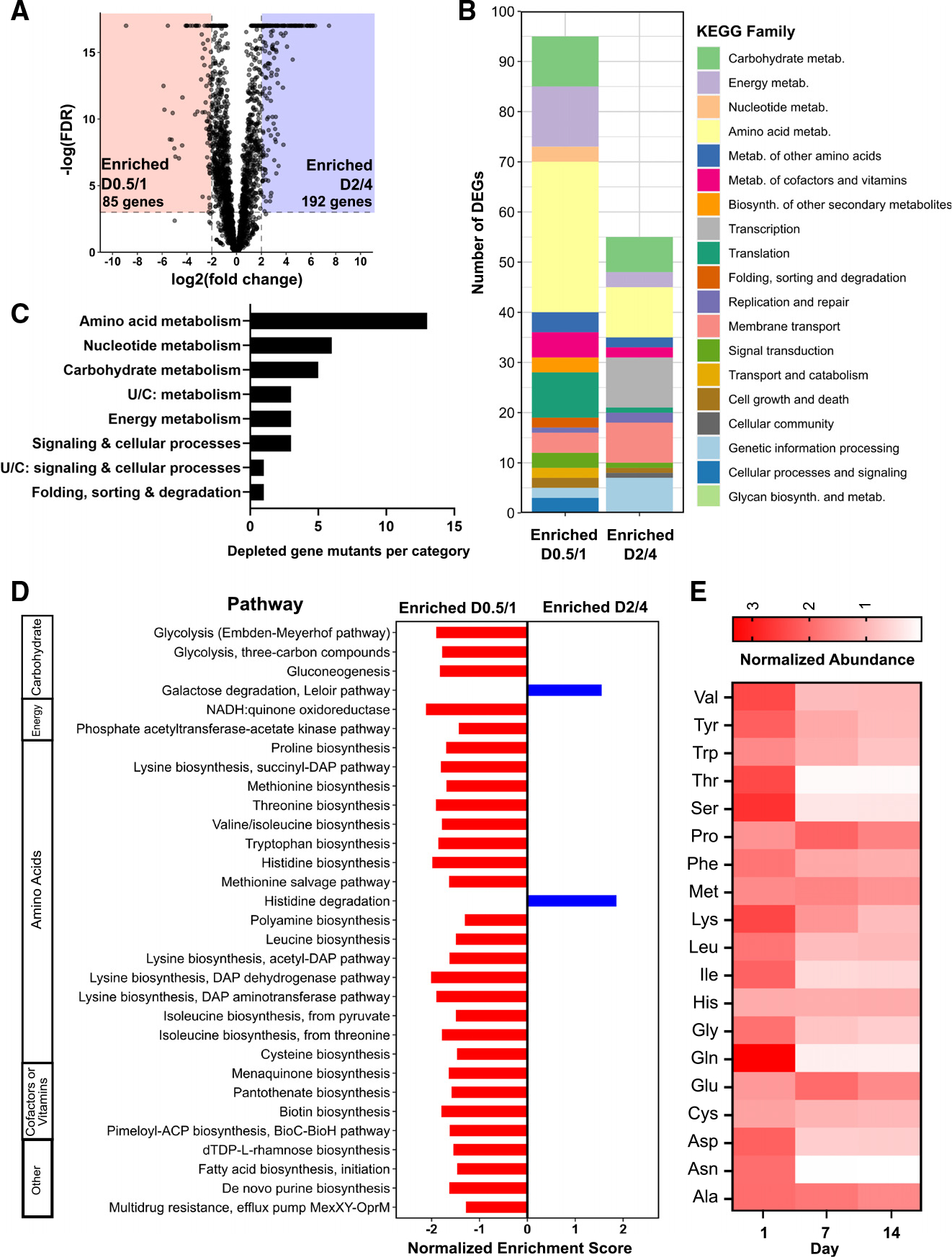
(A) Volcano plot of significant transcriptional differences between day 0.5 (D0.5)/D1 and D2/D4. Eighty-five genes had significantly increased expression on D0.5/D1 (red), while 192 had significantly increased expression on D2/D4 (blue) (cutoff: log(FDR-adjusted p value) < −3, |log2FC| > 2, maximum group mean >50 RPM).
(B) DEGs from (A) colored by KEGG family. Genes with no KEGG annotations were excluded.
(C) Number of gene mutants significantly depleted in the RB-Tn experiment (t statistic < −3σ) on day 1 of the experiment, plotted by KEGG family.
(D) GSEA using transcriptomics data for all KEGG metabolic modules, comparing D0.5/D1 to D2/D4. Only pathways with significant differential expression (p < 0.05) are shown.
(E) Abundances of amino acids in the ceca of ex-GF mice at day 1, day 7, and day 14 of colonization measured using gas chromatography-mass spectrometry (GCMS) and normalized to internal standards and to respective GF day-0 controls (n = 5 mice for GF day 0, n = 4 for post-colonization samples). See also Table S5.
To further explore the metabolic functions characteristic of the early phase of colonization, we performed gene set enrichment analysis (GSEA) for all KEGG metabolism modules across D0.5/D1 and D2/D4 (Figure 2D). Expression of pathways corresponding to the biosynthesis of many amino acids was enriched specifically at the D0.5/D1 time point. Expression of genes involved in the biosynthesis of biotin, a cofactor required for amino acid biosynthesis, was concurrently upregulated at D0.5/D1. These results are supported by RB-TnSeq analysis: by mapping the significantly depleted gene mutants (t statistic< −3σ) on the first day of the functional genetics assay using KEGG, we identified amino acid metabolism as the KEGG family with the largest number of significantly depleted mutants (Figure 2C). These genes are identified by gold stars on the amino acid biosynthesis pathway map in Figure S2, which illustrates reactions with upregulated gene expression at D0.5/D1 compared to D2/D4. We observe that biosynthesis of most amino acids involves multiple reactions that are transcriptionally enriched at D0.5/D1, or one step that is functionally essential.
Not only do both our transcriptomics analysis and functional genetics screen support a key role for amino acid biosynthesis early in colonization, but metabolomic analysis of the cecal contents corroborates this finding. We measured the levels of specific amino acids in the cecum of GF mice and compared those to cecal levels in ex-GF mice at day 1, day 7, and day 14 after colonization with WT Bt, and found that amino acid levels were generally higher on day 1 than on day 7 or day 14 (Figure 2E), as shown previously in an E. coli colonization model.8 This difference was especially profound and statistically significant for amino acids in the glycine-serine-threonine pathway and for asparagine (Table S4). By contrast, glutamate and proline reach their highest levels at day 7 before being depleted in the second week.
Although upregulation of amino acid and vitamin biosynthesis at D0.5/D1 is reminiscent of the stringent response (SR), in which growth is inhibited under conditions of nutrient limitation to prioritize biosynthesis of essential nutrients,15 further analysis suggests that early colonization is a distinct transcriptional program. Using a list of Bt-specific (p)ppGpp-mediated SR pathways manually curated from transcriptomic analyses by Schofield et al.,16 we performed GSEA to determine whether SR transcriptional patterns were better represented at D0.5/D1 or D2/D4. Almost all pathways expected to be downregulated during SR were downregulated at D2/D4 rather than D0.5/D1, but pathways expected to be upregulated during SR were generally upregulated at D0.5/D1 (Table S5). Thus, although some pathway targets of the (p)ppGpp-mediated SR transcriptional program may be differentially expressed during gut colonization, these likely represent a distinct but overlapping response to the shifting selective pressures of the gut resource environment.
A shift toward enhanced expression of diverse sugar metabolism genes occurs during the first week of gut colonization
Of the 192 DEGs upregulated at D2/D4 compared to D0.5/D1, only 50 mapped to known KEGG orthologs (Figure 2B and Table S5). Among these, we noted specific enrichment of genes involved in transcription, membrane transport, and genetic information processing, which represents Bt’s seven identical insertion sequence 3 (IS3)-family transposases. In the functional genetics experiment, we found that by day 2, amino acid metabolism had been surpassed by carbohydrate metabolism as the KEGG family with the largest number of depleted gene mutants (Figure 3A). This suggests that amino acid metabolism becomes less functionally essential after day 1 and that efficient carbohydrate metabolism becomes more essential.
Figure 3. A shift toward greater expression of diverse sugar metabolism genes occurs during the first week of gut colonization.
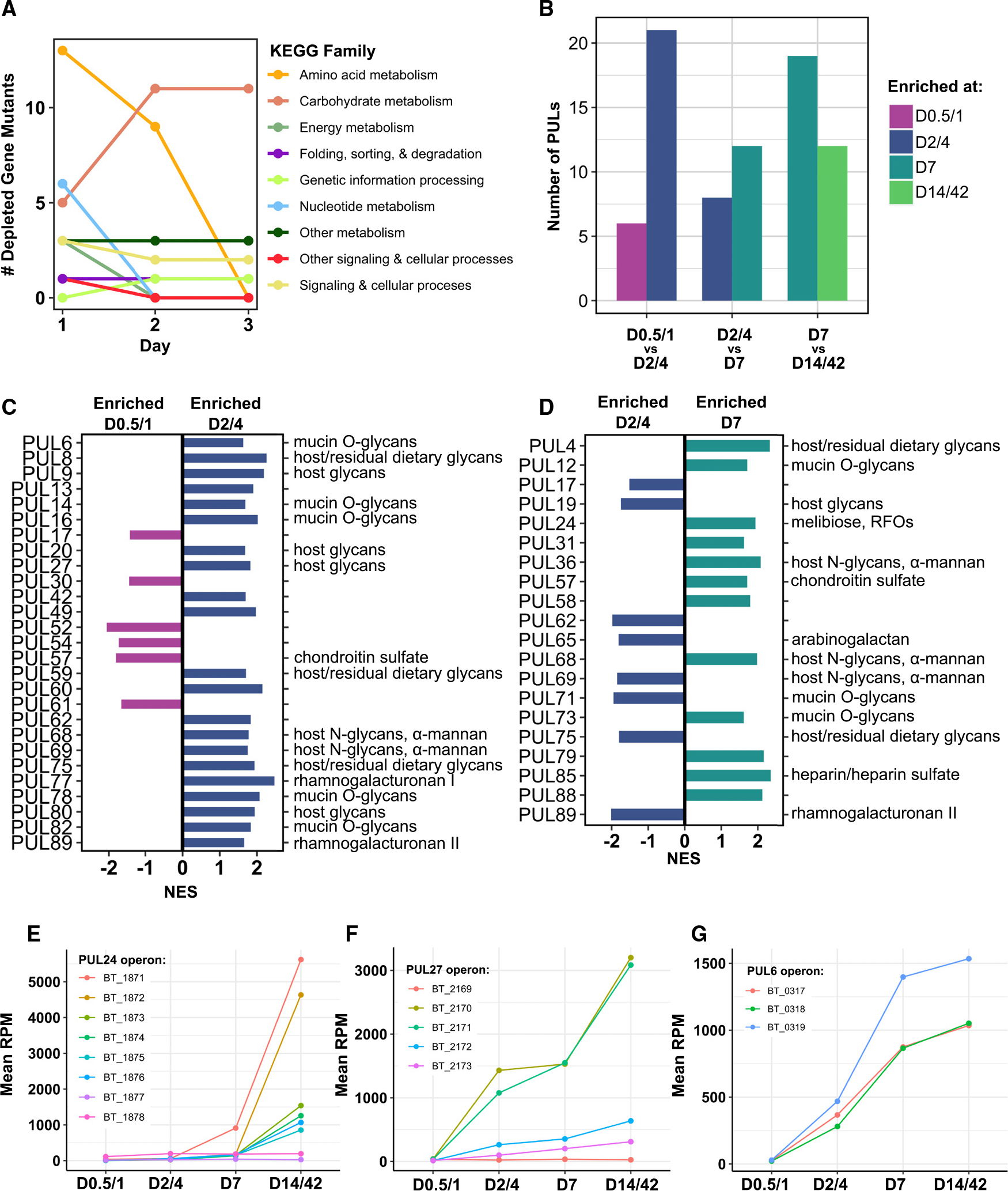
(A) Number of gene mutants significantly depleted from day 1 to day 3 of the RB-Tn experiment (t statistic < −3σ), colored by KEGG family.
(B–G) Number of PULs significantly differentially expressed (adjusted p < 0.05) across sequential pairwise comparisons identified via GSEA using transcriptomics data. Specific PULs differentially expressed across (C) day 0.5 (D0.5)/D1 vs. D2/D4 and (D) D2/D4 vs. D7 identified via GSEA. Predicted PUL substrates are listed on the right. Mean expression levels (RPM) for all genes in the (E) PUL24, (F) PUL27, and (G) PUL6 operons. Error bars excluded for visual clarity; statistical comparisons done via GSEA.
See also Table S5.
Members of the genus Bacteroides are well known for their ability to digest a wide variety of polysaccharides.17–19 According to the CAZy database, Bt possesses 359 glycoside hydrolases, 87 glycosyl transferases, 15 polysaccharide lyases, and 19 carbohydrate esterases.20 To further probe carbohydrate utilization, we performed GSEA on all PULs in the Bt genome in pairwise comparisons across sequential time points. Across all time points, 49 unique PULs were significantly differentially expressed. We find that from D0.5/D1 to D2/D4, expression of 21 PULs was upregulated compared to only six that were downregulated (Figures 3B and 3C). From D2/D4 to day 7, 12 PULs were upregulated compared to eight that were downregulated (Figures 3B and 3D), and from day 7 to D14/D42, more PULs were downregulated than upregulated (Figure 3B). Collectively, these data show that over the first week of colonization, Bt increases expression of a broad array of PULs, but expression of many of these PULs is reduced as time goes on, perhaps to optimize utilization of available resources.
To identify specific PULs that may be particularly central to efficient resource utilization, we searched for PUL genes whose mean expression across mice increased monotonically over all time points, and found genes belonging to 21 PULs, including PUL24 and PUL59, both of which are adjacent to IS3-family transposases (Table S5). Of these 21 PULs, only PUL24, PUL27, and PUL6 had expression patterns that were broadly consistent across all genes in the PUL (Figures 3E and 3F). These PULs moreover reached substantially higher levels of expression (~1,500–6,000 RPM) than any of the other 18 PULs (~200–400 RPM). PUL27 and PUL6 are predicted to enable degradation of host mucosal glycans, suggesting that Bt may be foraging for sugars through degradation of the mucus layer.17 The last gene of PUL24 encodes an α-galactosidase (BT1871), which is predicted to confer the ability to hydrolyze the α−1,6 glycosidic linkage in RFOs, a major component of the fiber-rich diet fed to our mice, as well as many standard mouse chows.5 Although this study focuses on understanding the shifting genetic determinants of colonization over time, we recognize that the effect of diet is intrinsic to the results of any gut microbiome study.
Global Bt gene expression stabilizes after 1 week of colonization
In contrast to the 277 DEGs identified between D0.5/D1 and D2/D4, we found only 21 DEGs that met our criteria between D2/D4 and day 7, and only seven between day 7 and D14/D42 (Table S5). Of the DEGs between D2/D4 and day 7, the only one that mapped to any KEGG pathway was in the PUL24 operon, and similarly, five of the seven DEGs between day 7 and D14/D42 fell into the PUL24 operon. Thus, we infer that Bt’s transcriptional profile has largely stabilized within the first week of colonization and is maintained thereafter, excepting the continued upregulation of certain PUL genes.
Upregulation of α-galactosidase activity confers a significant growth advantage to Bt in GF mice fed a standard RFO-rich diet
Our functional genetics screen confirmed the importance of PUL24: around 4 days after introduction of the RB-Tn mutant library into GF mice, the diversity of the community begins to collapse due to strong positive selection for a small pool of mutants that seemed to have gained fitness from the Tn insertion (Figure 4A). This could be caused by RB-Tn insertions in regulatory elements or by polar effects on downstream genes driven by readthrough from the antibiotic resistance promoter of the RB-Tn insertion.3 RB-Tn mutants with insertions in PUL24 were among the most positively selected mutants at the end of the 2-week experiment, exhibiting growth that exceeded a neutral expectation that final abundances would simply reflect initial abundances in the inoculum (Figure 4B). Furthermore, most of the hyperfit mutants had Tn insertions in one of three operons: PUL24 (BT1871–1877), PUL39 (BT2851–2860), or BT3130–3134, all of which encode at least one α-galactosidase, situated at the tail end of the operon (Figure 4C). At the end of the RB-Tn selection experiments, the populations were overtaken by mutants carrying Tn insertions upstream of these α-galactosidase genes (Figure 4D). This was true for all mice across four independent experimental cohorts, which were carried out months apart from one another (Figure 4E).
Figure 4. Colonization of a complex Bt mutant library within GF mice selects for disruptions upstream of α-galactosidase genes.
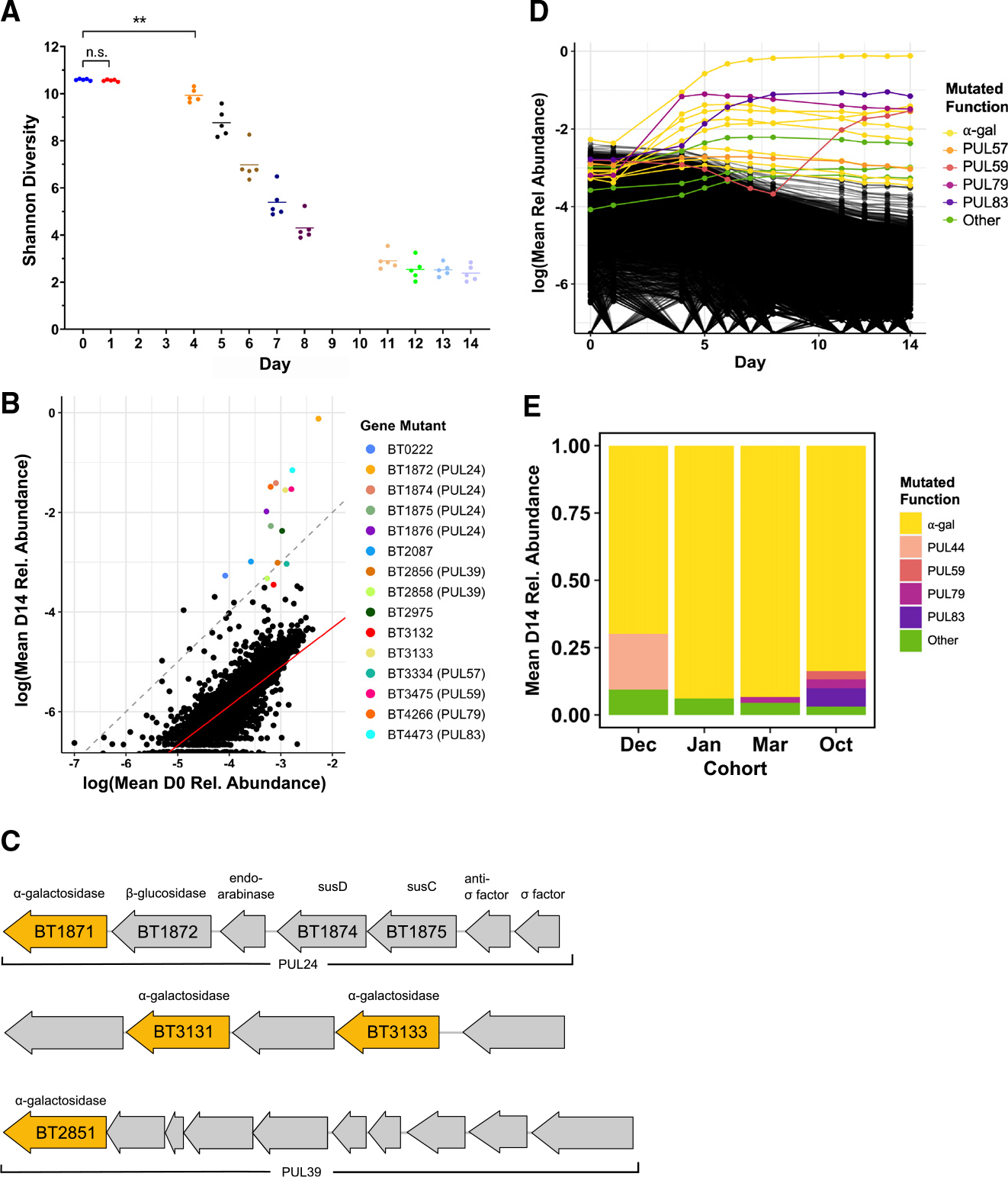
A–D) (A, B, and D) Data from a single experimental run (October). (A) Shannon diversity of the RB-Tn mutant pool in mice over time. Each point represents the RB-Tn pool within a single mouse on the indicated day (**p < 0.01, t test, n = 5/time point). (B) Initial (inoculum, day 0) vs. final (day 14) relative abundance of RB-Tn mutant strains. Each point represents the summed relative abundance of all mutant strains that mapped to given gene, averaged across mice. Dashed gray line designates a 1:1 relationship between starting and final abundance; red line represents a linear regression best-fit line generated from the log-transformed data (p < 2e−16, R2 = 0.6461; STAR Methods). (C) Organization of PUL 24 (BT1871–1877), an unnamed PUL containing BT3130–3134, and PUL 39 (BT2851–2860). Predicted α-galactosidases are colored yellow. (D) Relative abundance of RB-Tn mutant strains over time. Each line represents the summed relative abundance of all mutant strains that mapped to a given gene, averaged across mice. Top 15 most abundant gene mutants at day 14 are colored by the PUL to which they mapped; all others are black.
(E) Day-14 relative abundance of gene mutants mapping to operons that encode α-galactosidase functions (yellow), known PULs, or other gene functions averaged across mice for each experimental run.
This RB-Tn pool has previously been assayed in over 300 different conditions including distinct carbon or nitrogen sources and specific stress conditions.3 The mutants that we identified as hyperfit in GF mice exhibit a phenotype significantly deviant from WT in only two conditions: within GF mice and in defined culture with melibiose—a disaccharide of glucose and galactose, and a breakdown product of the trisaccharide raffinose (Figure S3A)—as the sole carbon source. In both cases, these mutants exhibit a growth advantage. Indeed, when we isolated the most abundant strains from day 14 of the RB-Tn experiment, we found that their growth rate was significantly higher than WT when melibiose is the sole carbon source (Figure 5A and Table S6). In this condition, Bt must hydrolyze the α−1,6 glycosidic bond to harvest and metabolize the monosaccharide sugars. The competitive advantage of these mutants cannot be attributed to either of the monosaccharides or to hydrolysis of α−1,2 glycosidic bonds, as the mutant growth rates on these carbon substrates (glucose, galactose, and sucrose) are indistinguishable from WT (Figure S3B). In raffinose medium, which contains both α−1,2 and α−1,6 linkages, mutants exhibit an attenuated but still significant growth advantage (Figure S3B, p = 3.9e−05, t test, n = 4–6).
Figure 5. Upregulation of α-galactosidase activity confers a significant growth advantage to Bt in GF mice fed a standard RFO-rich diet.
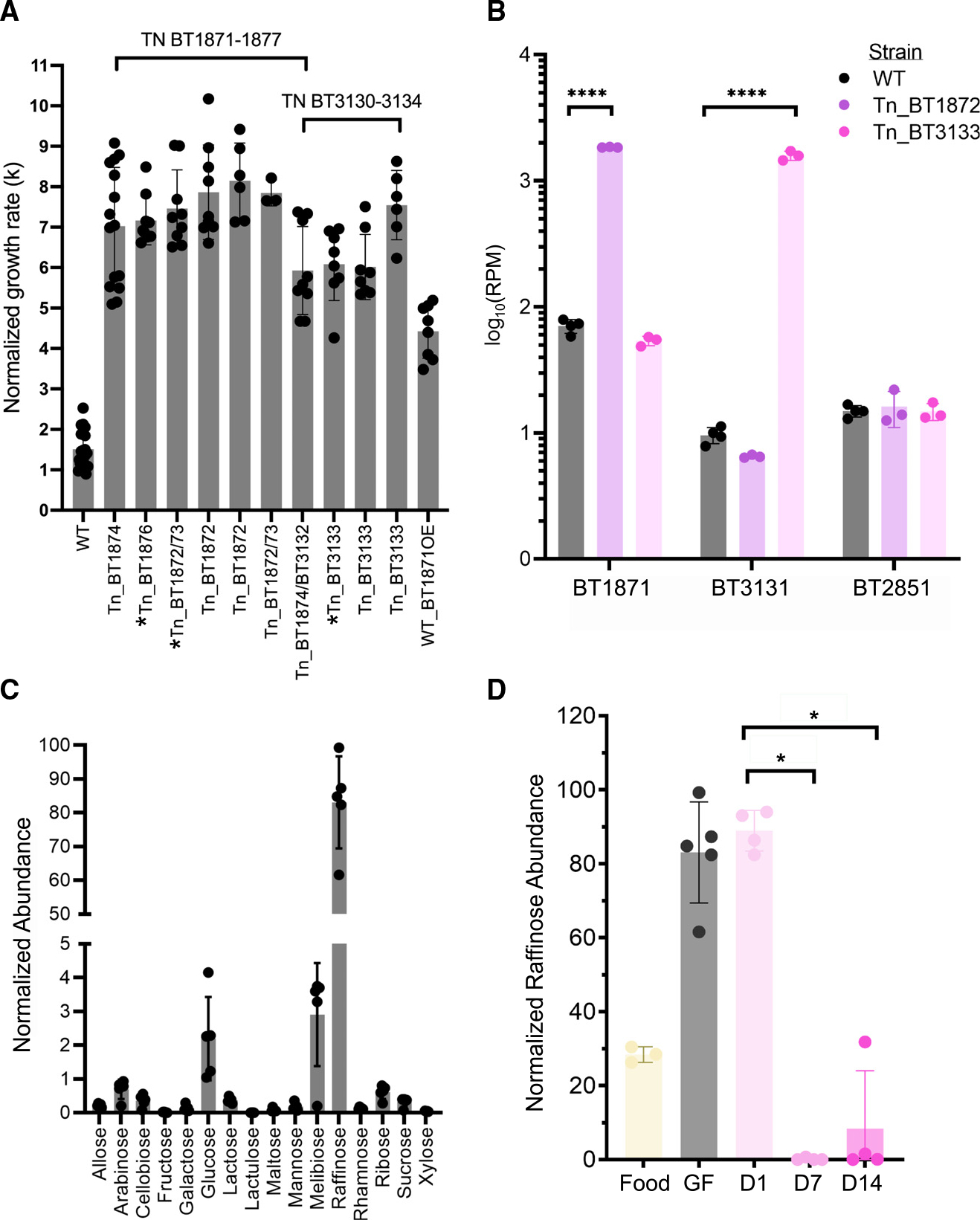
(A) Log-phase growth rates (k) of the most abundant RB-Tn mutant strains isolated from mice and grown in melibiose minimal medium. Strains with asterisks carry other mutations in addition to the transposon insertion. The WT + BT1871OE strain carries a plasmid copy of BT1871 expressed from the rpoD promoter that was integrated into the WT genome at the attN1 site. All mutant strains grew significantly faster than WT (p < 0.0001).
(B) Normalized abundance of α-galactosidase mRNA (RPM) measured in WT and hyperfit RB-Tn mutants isolated from mice and grown in Varel-Bryant defined medium with 20 mM melibiose as the sole carbon substrate.21
(C) Abundance of various sugars in the GF mouse cecum as measured using GCMS and normalized to internal standards.
(D) Abundance of raffinose in the standard chow fed to GF mice, within GF ceca before colonization, or 1, 7, or 14 days post colonization.
*p < 0.05; **p < 0.01; ***p < 0.0001, ****p < 0.00001.
See also Table S6.
Given that release of the monosaccharide sugars of melibiose depends on hydrolysis of an α−1,6 glycosidic bond, we hypothesized that the fitness phenotypes both in mice and in vitro did in fact depend on overexpression of the α-galactosidase gene downstream of the Tn insertion site. To test the hypothesis that the Tn insertion enhanced expression of downstream genes, we grew WT and hyperfit mutants in melibiose medium and measured the expression of α-galactosidase genes. For this experiment we used two mutants, one carrying an insertion in PUL24 and another carrying an insertion in the BT3130–3134 operon. For both mutants, we found >10-fold overexpression of α-galactosidase downstream of the insertion but within the same operon (Figure 5B, BT1871: p < 1e−6, t test; BT3131: p = 4e−6, n = 3–4/group). Meanwhile, expression of an α-galactosidase (BT2851) located outside the operons that carry the insertion was similar between WT and the mutants.
We then overexpressed BT1871 from a strong, constitutively active promoter (PrpoD) in WT Bt and observed a 3-fold increase in log-phase growth rate relative to WT when melibiose was the sole carbon source (Figure 5A and Table S6). We note that even this constitutive-expression mutant did not exhibit as much growth advantage as the RB-Tn mutant strains. It is possible that the RB-Tn strains may have evolved additional mutations that further promote growth on melibiose media, although we identified no new junctions suggesting genomic rearrangements and no changes in coverage suggesting duplications in whole-genome short-read sequencing of RB-Tn isolates. Alternatively, the promoter used in front of the erythromycin resistance cassette in the RB-Tn mutants may simply be stronger than the PrpoD promoter we used here.
Metabolomic measurements of a carbohydrate panel confirm that raffinose and its constituent sugars are among the most abundant carbohydrate substrates within the ceca of GF mice fed a standard chow (Figure 5C). Although mammals can catalyze hydrolysis at the α−1,2 linkages in raffinose and sucrose, the α−1,6 linkages in raffinose and melibiose can only be hydrolyzed by gut microbes (Figure S3A).22 Accordingly, raffinose and melibiose build up in the cecum of our GF mice at high concentrations until Bt is introduced. After 7 days, during which time Bt has initiated overexpression of α-galactosidase genes, these sugars are depleted (Figure 5D, p = 0.02857, Wilcoxon test, n = 4; Figure S3C, p = 0.006193, t test, n = 4). In contrast, sucrose is consumed by the host and no significant changes are observed in sucrose concentration following Bt colonization (Figure S3D).
Together, we find that when mice are fed standard high-fiber chow, the high concentration of RFOs accumulating in the lower gastrointestinal (GI) tract creates an environment that strongly selects for Bt strains that can make efficient use of these sugars through increased expression of α-galactosidases. We infer that efficient carbohydrate metabolism—particularly for abundant dietary fibers—is a major determinant of population-level selective dynamics during the persistence phase of Bt engraftment within the gut.
Changes in resource-use strategies coincide with shifts in Bt localization from the mucus toward the lumen
We next sought to determine whether Bt localization changes over the course of colonization. To assess Bt localization at days 1, 2, 4, 7, and 42, we labeled fixed colonic cross-sections with antibodies to MUC2, the predominant colonic mucin,23 and the DNA stain 4′,6-diamidino-2-phenylindole (DAPI), and imaged them with a laser-scanning confocal microscope. We then quantified the average epithelial proximity of Bt by measuring the distance from the epithelial surface along perpendicular tracts to the nearest bacterial cell (STAR Methods).
At days 1 and 2, Bt was deeply embedded within a largely unstructured mucus layer, often directly adjacent to the epithelium (Figures 6A and 6B). By day 4, an observable but thin and patchy inner mucus layer began to form (Figure 6C), which expanded in thickness by day 7 (Figure 6D). This layer still allowed substantial penetration by Bt, although mean distance from Bt to the epithelium was significantly increased compared to day 1 (Figure 6F, p = 0.037, t test, n = 4/group). At day 42, a defined inner mucus layer covered the epithelial surface of each cross-section, effectively excluding Bt, which localized primarily to the shedding outer mucus layer and luminal space (Figures 6E and 6F, p = 0.024, t test, n = 3–4/group). Thus, as the impermeable mucus layer forms, we identify a shift in Bt spatial localization from deeply embedded in the mucus layer early in colonization toward the luminal space by day 7 and onward.
Figure 6. Bt localization shifts from the mucus toward the luminal space over colonization.
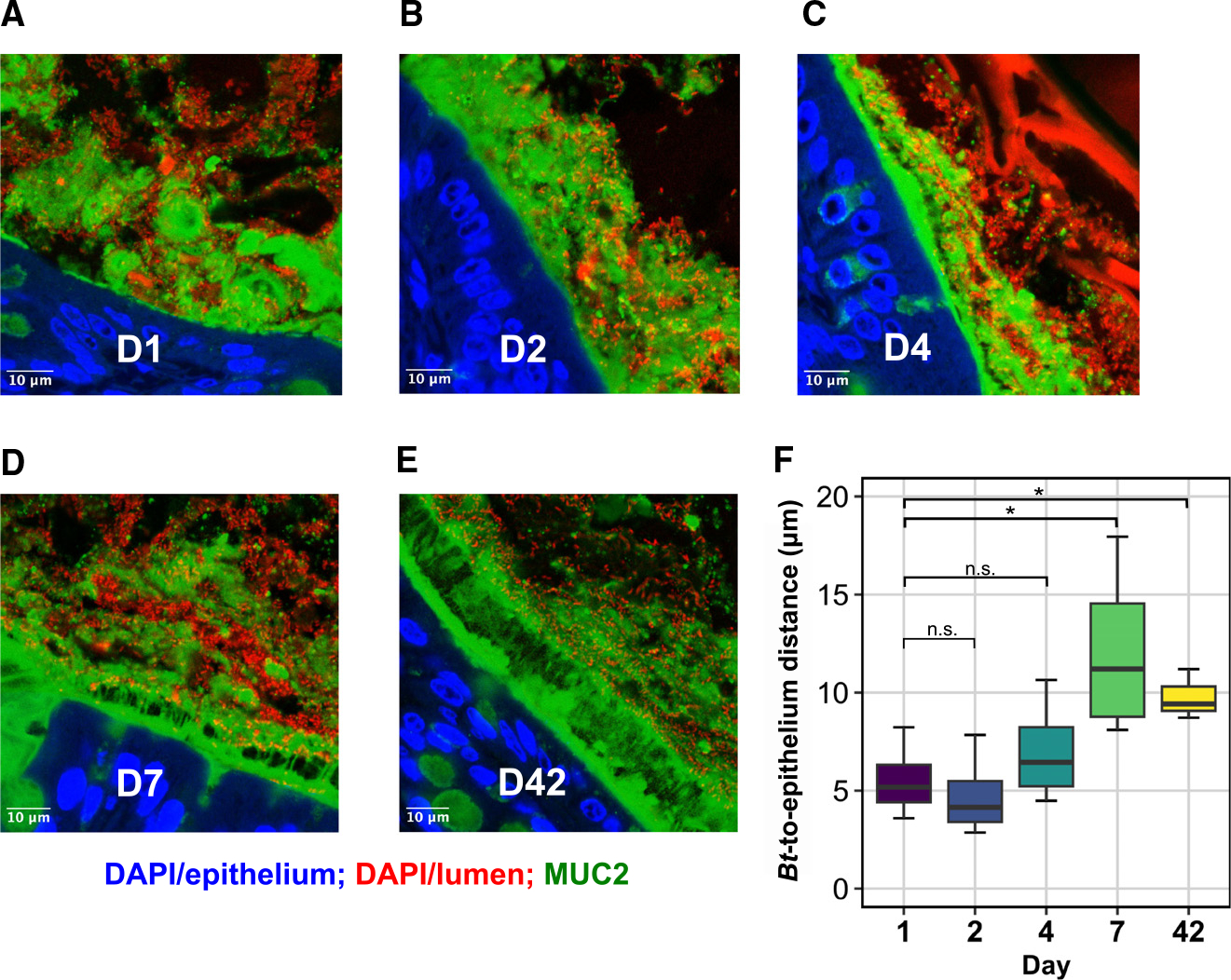
(A–E) Representative cross-sections of distal mouse colon fixed in Carnoy’s solution at (A) day 1 (D1), (B) D2, (C) D4, (D) D7, and (E) D42 of colonization by Bt. Blue, DAPI staining in the epithelium; red, DAPI staining in the lumen of gut, including bacteria, debris, and shed host nuclei; green, antibody staining for MUC2. Scale bars, 10 μm.
(F) Mean Bt epithelial proximity. n = 3–4/group, t test, *p < 0.05.
Our RNA-sequencing data indicated that Bt undergoes a metabolic shift around day 7 of colonization, switching from broadly upregulated PUL metabolism, including many loci predicted to encode mucosal glycans, to a metabolic profile strongly centered around dietary RFO consumption. Here, we find that Bt spatial localization similarly shifts around day 7 of colonization from the mucus into the lumen. We have therefore identified a loose correlation between the timing of shifts in Bt resource-use strategies and its spatial localization with respect to the mucus layer and luminal space.
Strong selection for efficient RFO metabolism leads to emergence of spontaneous Bt mutants with duplicates of the BT1871 locus through an IS3-family transposable element
The transposon mutants in our functional genetics experiment were able to gain ~10-fold increase in α-galactosidase expression due to readthrough from an extremely strong, synthetic promoter. We wondered whether the selective pressure for elevated α-galactosidase activity to utilize the α−1,6-linked sugars abundant in the diet would drive evolution of a spontaneous mutant with enhanced α-galactosidase activity. One week after the initial colonization of GF mice with WT Bt in an ongoing transcriptomics experiment, three mice from two cages were separated into individual cages for 6 weeks of observation, a length of time predicted to be sufficient for spontaneous mutations to arise and stabilize.7 At the end of 6 weeks, we performed shotgun sequencing on fecal material from all three mice. Assembly of the short-read sequences revealed that there was at least 2× coverage of the BT1871 locus for all samples (Figure 7A). A dip to 1× coverage in the middle of this region mapped to an IS3-family transposase, of which there are six other identical copies in the genome; hence, the corresponding coverage was diluted among the different copies. BreSeq analysis identified three new junctions in the Bt genome indicative of genome rearrangements (Figure 7B). Notably, samples from mouse A and mouse C, which were co-housed during the first week of the experiment, shared an identical junction, suggesting that this mutation arose in Bt within the first week of colonization and was transmitted across mice. Samples from mouse B and mouse C each possessed unique junctions, indicating that these junctions arose independently. In all, three independent junctions spontaneously evolved in this experiment, and all three junctions were formed between either BT1872 or BT1873 and a locus downstream of BT1871, which encodes for rRNAs, tRNAs, and a ribosome recycling factor. All three new junctions occupied a significant portion of the sequencing reads (60%–73%), suggesting that the mutants carrying these junctions were competitively dominant in the gut environment.
Figure 7. Under strong selective pressure, Bt duplicates the BT1871 locus with the help of a transposable element.
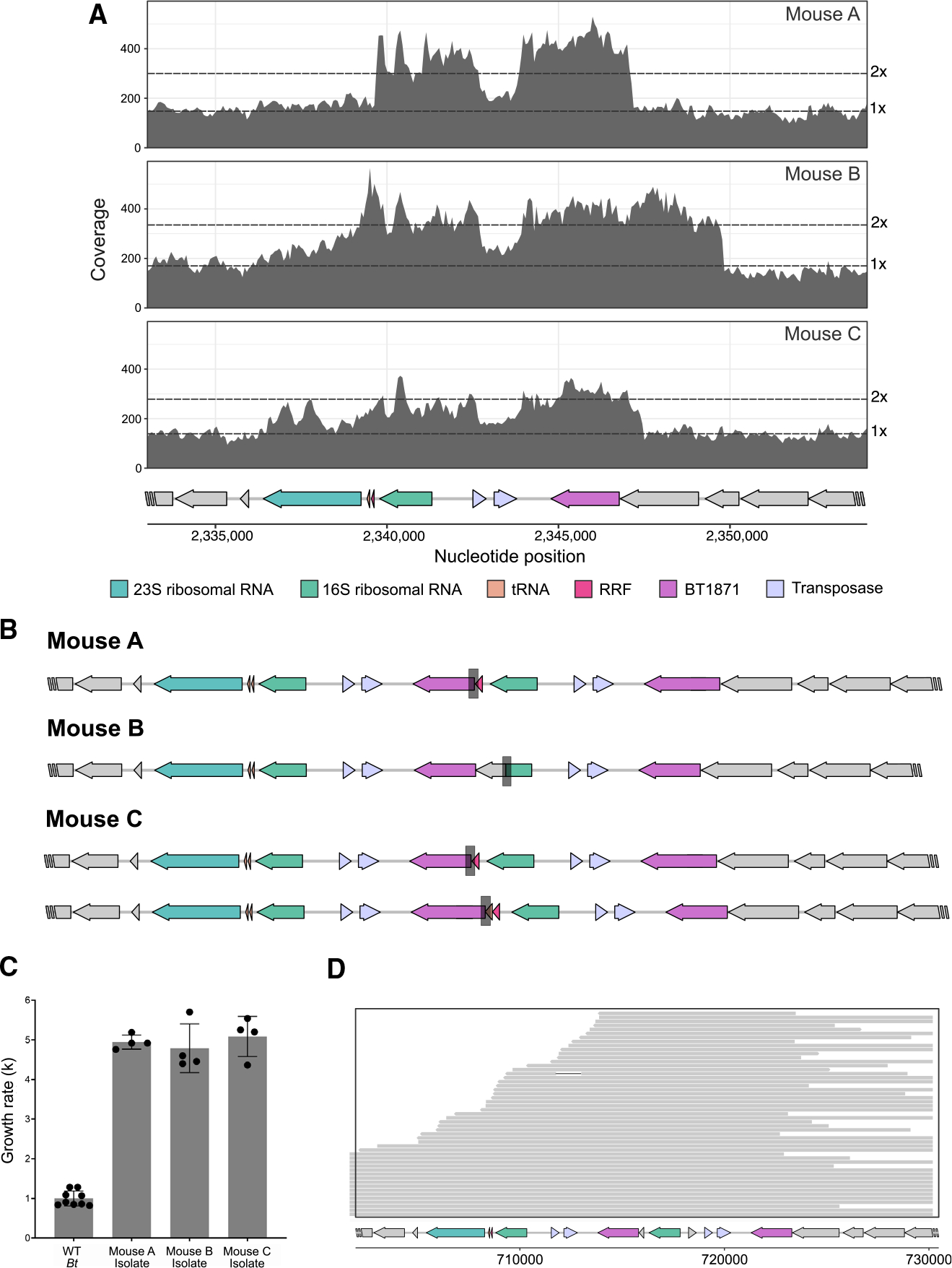
(A) Shotgun sequencing of bulk fecal sample from three mice inoculated with WT Bt shows at least 2× coverage of the BT1871 locus.
(B) New Bt genomic junctions identified via shotgun sequencing of bulk fecal material from each mouse.
(C) Log-phase growth rates (k) of three mutant strains isolated from feces of mice colonized with WT Bt for 6 weeks compared to growth rate of the ancestral WT Bt in defined medium supplemented with melibiose (n = 4 technical replicates per strain). p < 0.05.
(D) Long-read MinION sequencing of an abundant mutant in mouse C (MZ65) reveals the duplication of a long segment including the BT1871 gene, which generates a tandem repeat of the locus. Each gray bar represents a single long read, which together span the region containing the tandem repeat.
IS3-family transposases are known to act by a copy-and-paste mechanism.24 We wondered whether this transposase, which we had identified as significantly upregulated from D0.5/D1 to D2/D4 (Table S5), could have created multiple copies of the BT1871 locus. We isolated individual strains likely to contain these mutations by plating bulk fecal material and then selecting a single strain from each mouse that exhibited a growth advantage compared to WT on melibiose medium (Figure 7C). We then performed long-read sequencing of each strain with MinION. In one of these isolates, we identified a tandem repeat of the BT1871 locus (Figure 7D), which places the original copy of BT1871 downstream of a strong rRNA promoter. Interestingly, the six identical copies of this IS3-family transposase (NCBI: 60924995) are often found adjacent to PULs in the WT Bt genome. This suggests that these transposable elements confer a mechanism to modulate gene expression that may be advantageous in adapting to new metabolic landscapes.
Interactions with the host are relatively consistent throughout colonization
Although the host response to monocolonization is not the focus of this paper and has been thoroughly cataloged elsewhere,25–28 we assessed levels of cecal immunoglobulin A (IgA) and serum IgG, as well as several pro-inflammatory cytokines from colonic mucosal scrapings. We identified no significant or consistent trends in any of these metrics (Figure S4) Previous work has outlined how IgA binding to commensal bacteria such as Bt may impact gene expression, in some cases affecting dietary polysaccharide usage and microbial localization.27,29–31 We could not recapitulate these specific changes in our Bt gene expression time course. This may reflect differences in IgA assay protocols,29 or it may suggest that Bt’s response to IgA binding is sensitive to the specific mouse chow composition and availability of dietary resources.
We then performed targeted GSEA on expression of Bt’s capsular polysaccharide (CPS) loci, which encode outer membrane proteins that mediate interaction between Bacteroides genera and the host immune system32 and have been shown previously to play a critical role in Bacteroides gut colonization.2,33,34 We found that CPS loci 1 and 3, and to a lesser extent 2, 4, and 7, were highly expressed early in colonization and then downregulated, whereas CPS5 and CPS6 had increasing relative expression from day 7 onward (Figures S5A–S5D). Only CPS4 mutants showed severe fitness defects during the early days of the functional genetics assay, in line with previous reports (Figure S5E).2 These data indicate that Bt modifies its outer membrane upon entry into the gut and over the course of persistence, which may reflect adaptation to the host immune system, perhaps in parallel to shifts in localization.
DISCUSSION
Microbial adaptation during colonization is a dynamic process
Bacteroides species are dominant and prevalent in the guts of many human populations.35,36 Instrumental to their success is their ability not only to tolerate stress but to quickly adapt in order to grow under a range of conditions.37,38 Although Bt has been used as a model organism to understand the genetic drivers of gut colonization for commensal organisms,2,4,39 a single representative time point has been assessed in most previous investigations, which fail to consider the dynamism of the adaptational process. For example, whereas previous work based on in vivo Bt gene expression at a single time point characterized only one metabolic phase that prioritized carbohydrate transport and metabolism,9 our time-course data suggest that Bt undergoes a temporally dynamic adaptational program. We show that Bt prioritizes biosynthesis of amino acids and other essential compounds early in colonization, before ramping up expression of a diverse array of PULs and ultimately centering metabolism around degradation of a single abundant dietary carbohydrate. This may reflect a transition from an initial stress response during establishment in the gut to a more growth-focused strategy that surveys and optimizes utilization of available environmental resources. We find that the set of genes required for Bt to initially establish colonization in the gut are distinct from those required to persist (Figure 1). The results presented here suggest that because of the rapidly shifting adaptational profile exhibited by microbial populations over the course of colonization, investigators must take care in selecting the appropriate time points to address their experimental questions.
Early colonization is a distinct adaptational phase
The process of colonizing a new host presents a diverse array of environmental changes that require physiological adaptation. The largest transcriptomic changes that we observed across 6 weeks of Bt colonization occurred in the first week (Figure 1B), as did the largest shifts in RB-Tn mutant abundances (Figure 1C). Our findings indicate that Bt upregulates a broad swath of metabolic processes upon arrival in the lower GI tract during the earliest stage of colonization (Figure 2B). Work from Watson et al. examining the qualities of microbes that successfully colonize the gut after fecal microbial transplant suggests that microbes that can be metabolically “self-sufficient” may be better suited to engraft and persist in a new host environment.40 We show that, similarly, Bt casts a wide metabolic net upon gut entry, upregulating carbohydrate metabolism, energy metabolism, vitamin and cofactor biosynthesis, and especially amino acid biosynthesis. We like-wise see that Bt upregulates a diverse array of PULs during early colonization (Figure 3). This may better enable Bt to survive conditions of resource scarcity until a more energetically efficient transcriptional program can be fine-tuned to the particulars of its new resource landscape.
Selection for efficient carbohydrate metabolism drives long-term persistence
During the later stages of colonization and persistence, Bt re-centers its metabolism around carbohydrate utilization. These results add to a growing body of work identifying metabolic necessity in general, and carbohydrate metabolism in particular, as powerful drivers of microbial fitness and evolution in the gut. For instance, one study showed that E. coli evolves mutations to modulate carbohydrate utilization depending on dietary conditions,6 while another identified selection for mutations in either amino acid or carbohydrate metabolism depending on the competitive context.8 Research using Bt has demonstrated rapid evolution of divergent metabolic strategies depending on mouse diet.7 One study even showed how microbial competitive fitness in the mouse gut can be promoted by engineering a unique PUL into Bt and introducing the strain in combination with its associated carbohydrate substrate.41 Although microbes mediate competition in a number of ways ranging from direct attack to occupation of physical space to indirect interactions via the host,42 these findings collectively suggest that competition for scarce metabolic resources is a primary evolutionary battleground within and among microbial species in the gut ecosystem.
In these experiments, we identified strong selective pressure for improved utilization of a specific carbohydrate resource. However, previous work shows that genetic drift and transmission of microbes between hosts can significantly impact the evolution and spread of mutant populations.5 Therefore, it is important to consider how transmission between hosts may have impacted our results. In our RB-TnSeq experiments, mice were co-housed, facilitating intra-cage transmission. This may have artificially inflated the replicability of our results within cages, although we still saw near-identical trends emerge across four independent experiments and eight independent cages.
Changes in microbial localization and metabolic gene expression profile correlate
We found that changes in Bt resource use over the course of colonization paralleled shifts in its localization from the inner mucus layer toward the luminal space (Figure 6). This aligns with previous work showing that the mucus of GF mice is permeable to bacteria and takes up to 6 weeks of conventionalization to fully mature into the impermeable protective state that excludes Bt and most other gut commensals.43,44 Although we provide only correlational evidence here and causal experimentation is beyond the scope of this work, we hypothesize that the local microenvironment may dictate the selective pressures that ultimately drive Bt gene expression and evolution: Bt is known to use mucosal glycans only as a carbohydrate resource of last resort.45 However, Bt that gets trapped in the permeable immature mucus layer early in colonization may not have direct access to luminal dietary resources. Under these resource-limited conditions, the population of mucus-embedded Bt may upregulate various PULs for consumption of mucosal glycans, as has been demonstrated in other in vivo mouse studies of Bt under conditions of dietary resource deficiencies.9,45–48 As the mucus layer matures and Bt gradually moves into the lumen, its metabolism shifts from broad mucosal glycan foraging while embedded (D2/D4) toward hyperutilization of a single abundant dietary resource while excluded (days 7–42).
Although more rigorous follow-up is needed to establish a causal relationship between localization and resource use, the correlation reported here provides preliminary support for the notion that the spatial localization and metabolic profile of Bt are deeply intertwined, flexible, and highly responsive to the specific balance of selective pressures such as resource availability and energetic efficiency. These factors will doubtless be heavily impacted by the presence of additional community members. Nevertheless, we conclude that resource-use profiling and localization may emerge as particularly informative descriptors of available niche space in the gut.
IS3 transposable elements: A novel mechanism to modulate expression of specific CAZymes?
In GF mice fed a standard RFO-rich diet, the selective pressure for mutants with increased α-galactosidase activity is intense. Mutations in the BT1871 locus were consistently positively selected when WT Bt evolved in mice for 6 weeks. In all three mutant populations, the downstream side of the duplicated region ends midway through a ribosomal gene, meaning that there are no known transcriptional terminators between the ribosomal promoter and the α-galactosidase gene, BT1871. It is likely that these mutants increased α-galactosidase activity due to readthrough from the strong ribosomal promoter in the upstream copy of the locus. Although increased α-galactosidase expression could be simply due to the presence of two copies of BT1871, the growth phenotype that these mutants exhibit, in which log-phase growth is five times faster than in WT, is on par with that of the RB-Tn mutants, for which α-galactosidase expression was more than ten times greater than in WT.
In Bt, identical copies of this IS3 transposable element occur in seven locations, often adjacent to PULs and almost always paired with a ribosomal gene. These genes were upregulated at D2/D4 of our transcriptomics experiment, shortly before we saw extensive upregulation of PUL24 genes. It is well known in both mammals and prokaryotes that transposable elements can modulate the expression of nearby and distant genes.49 Transposable elements may be selected for because the genetic “cargo” that they shuttle along (such as an antibiotic cassette) is beneficial to the organism or because the position where the insertion occurs results in some downstream effect that is beneficial to fitness. Both reasons may contribute to the selection of the tandem duplication of the BT1871 locus. Future studies will be necessary to investigate whether other IS3 elements in Bt perform similar functions in modulating expression of nearby genes and whether such a mechanism can be found in other families of bacteria.
Limitations of the study
One limitation of this study is that many microbial genes are poorly annotated. At most of our transcriptomic time points, only a minority of the DEGs have known annotations, but those unannotated genes may still serve meaningful adaptive functions. These genes (Table S5B) would be excellent candidates for continued functional characterization through controlled in vitro and in vivo experiments. We also note the key limitation that this study was performed in a highly simplified monocolonization model. While these results provide evidence of general eco-evolutionary principles and highlight important considerations for understanding microbial GI colonization more broadly—e.g., the dynamism of the adaptational process and the importance of choosing time points carefully, the remarkable genetic mutability of bacteria, and the feedback cycles between a microbe and its resource environment—the specific results presented here may not directly apply to scenarios with more complex microbial communities. However, by eventually comparing these results to those from more complex communities, this dataset can provide even greater insight into how host-microbe interactions and inter- and intraspecific microbial interactions each contribute to the adaptational process.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents may be directed to and will be fulfilled by the lead contact, Eugene Chang (echang@medicine.bsd.uchicago.edu).
Materials availability
Bacterial strains obtained in this study will be made available upon request addressed to the lead contact.
Data and code availability
All sequencing data, including DNA and RNA datasets, have been deposited at NCBI Sequence Read Archive and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Goat polyclonal anti-IgA | Southern Biotech | Cat#OB1040–01 |
| Goat anti-mouse IgA:HRP | BioRad | Cat#STAR137P |
| Rabbit polyclonal anti-mouse Muc2 | Santa Cruz Biotechnology | Cat#sc-15334 |
| Alexa Fluor 488 goat anti-rabbit IgG | Invitrogen | Cat#A-11008 |
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
| Bacteroides thetaiotaomicron VPI-5482 wildtype | Gift from Deutschbauer lab | AMD595 |
| RB-Tn mutant isolated from monocolonized mice with barcoded transposon insertion in BT1876 | Gift from Deutschbauer lab | AMD595-derived RB-Tn library3 |
| RB-Tn mutant isolated from monocolonized mice with barcoded transposon insertion in BT1874 | This work | Tn_BT1874 |
| RB-Tn mutant isolated from monocolonized mice with barcoded transposon insertion in BT1876 | This work | Tn_BT1876 |
| RB-Tn mutant isolated from monocolonized mice with barcoded transposon insertion in the intergenic region between BT1872 and BT1873 | This work | Tn_BT1872–83 |
| RB-Tn mutant isolated from monocolonized mice with barcoded transposon insertion in BT1872 | This work | Tn_BT1872 |
| RB-Tn mutant isolated from monocolonized mice with barcoded transposon insertion in BT1874 and BT3132 | This work | Tn_BT1874/BT3132 |
| RB-Tn mutant isolated from monocolonized mice with barcoded transposon insertion in BT3133 | This work | Tn_BT3133 |
| WT Bt with a copy of BT1871 integrated into the Bt genome at the attN1 site under the BT1311 (rpoD) constitutive promoter. | This work | WT + BT1871OE |
| Bt mutant isolated from feces after 6-week monoassociation of Bt; carries a tandem repeat of the BT1871 locus (see Figure 7). | This work | MZ65 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Brain Heart Infusion Broth | Fisher | Cat#DF0037178 |
| Agar | Fisher | Cat#DF0145–17-0 |
| Hemin | Fisher | Cat#51280–5G |
| Glucose | Sigma | Cat#158968–500G |
| Galactose | Sigma | Cat#G0750–500G |
| Sucrose | Fisher | Cat#AAJ64270A1 |
| Raffinose | Fisher | Cat#AC195670250 |
| Melibiose | Sigma | Cat#M5500–100G |
| Erythromycin | Fisher | Cat#AC227330050 |
| Phosphate Buffered Saline | Sigma | Cat#P3813–10PAK |
| Glycerol | Fisher | Cat#BP2291 |
| Ambion TRIzol Reagent | Fisher | Cat#15–596-018 |
| Chloroform | Fisher | Cat#AC390760010 |
| Isopropanol | Fisher | Cat#BP2618500 |
| Ethanol | Fisher | Cat#BP2818500 |
| DNAse | Fisher | Cat#18068015 |
| Tris | Fisher | Cat#BP152–10 |
| EDTA | Fisher | Cat#PR-V4231 |
| NaCl | Fisher | Cat#S640–10 |
| SDS | Sigma | Cat#L3771–25G |
| Phenol:Chloroform:Isoamyl Alcohol | Fisher | Cat#AM9732 |
| Proteinase K | Sigma | Cat#3115852001 |
| HCl | Sigma | Cat#A144–212 |
| Methanol | Fisher | Cat#A452SK-4 |
| Methoxyamine | Sigma | Cat#226904 |
| Pyridine | Sigma | Cat#270970 |
| Derivatizing reagent (BSTFA +1% TMCS) | Sigma | Cat#B-023 |
| Ethyl acetate | Sigma | Cat#650528 |
| ELISA diluent | R&D Systems | Cat#DY995 |
| Acetic acid | Fisher | Cat#A38–500 |
| Xylene | Fisher | Cat#X3S-4 |
| Lysozyme | Fisher | Cat#89833 |
| Sodium citrate | Fisher | Cat#S279–500 |
| DAPI | Sigma | Cat#D8417–5MG |
| ProLong Gold Anti-Fade mounting medium | Fisher | Cat#P10144 |
| iTaq Universal SYBR Green Supermix | BioRad | Cat#1725124 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| DNeasy PowerSoil Kit. | Qiagen | Cat#47016 |
| Rapid Barcoding Kit | Oxford Nanopore Technologies | Cat#SQK-RBK004 |
| Transcriptor First Strand cDNA Synthesis Kit | Roche | Cat#35081963001 |
| Invitrogen IgG (Total) Mouse Uncoated ELISA Kit | Invitrogen | Cat#88–50400-88 |
|
| ||
| Deposited Data | ||
|
| ||
| RNAseq raw data from Bt at different time points | This work | https://www.ncbi.nlm.nih.gov/sra (accession: PRJNA797447) |
| Shotgun sequencing raw data from evolution experiment fecal samples | This work | https://www.ncbi.nlm.nih.gov/sra (accession: PRJNA797447) |
| Long-read sequencing raw data from isolate MZ65 | This work | https://www.ncbi.nlm.nih.gov/sra (accession: PRJNA797447) |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| Mouse: germ-free wild-type C57BL/6J | University of Chicago Gnotobiotic Core Facility |
N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Custom Ribo-Zero Plus Probes | SeqCenter | See Table S1A |
| qPCR primers | Miyoshi et al., 201750 | See Table S1B |
|
| ||
| Software and Algorithms | ||
|
| ||
| bcl2fastq | Illumina | Version 2.20.0.422 |
| MinKNOW | Oxford Nanopore Technologies | Version 4.3.4 |
| Guppy | Oxford Nanopore Technologies | Version 5.0.11 |
| Flye | https://github.com/fenderglass/Flye | Version 2.651 |
| Pilon | https://github.com/broadinstitute/pilon | Version 1.2352 |
| MassHunter Quantitative Analysis software |
Agilent Technologies | Version B.10 |
| R software | https://www.r-project.org/ | Version 4.2.153 |
| vegan R software | https://cran.r-project.org/web/packages/vegan/index.html | Version 2.6–454 |
| pairwiseAdonis R software | https://github.com/pmartinezarbizu/pairwiseAdonis | Version 0.4 |
| DESeq2 R software | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | Version 1.36.055 |
| anvi’o | https://anvio.org/ | Version 7.156 |
| fgsea R package | https://bioconductor.org/packages/release/bioc/html/fgsea.html | Version 3.1757 |
| Graphpad Prism | https://www.graphpad.com/features | Version 9 |
| Prodigal | https://github.com/hyattpd/Prodigal | Version 2.6.358 |
| bowtie2 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml | Version v2.3.5.159 |
| samtools | http://www.htslib.org/ | Version 1.1160 |
| gggenes | https://github.com/wilkox/gggenes/tree/master | Version 0.4.1 |
| blastn | https://blast.ncbi.nlm.nih.gov/Blast.cgi | Version 2.5.0 |
| minimap2 | https://github.com/lh3/minimap2 | Version 2.1761 |
| IGV | https://igv.org/ | Version 2.11.162 |
| LAS_X Leica | Leica | N/A |
|
| ||
| Other | ||
|
| ||
| Standard mouse chow | LabDiets | 5K67 |
| Anaerobic chamber | Coy Laboratory Products | N/A |
| GENSYS 40-Vis spectrophotometer | Thermo Scientific | N/A |
| Mini-BeadBeater-96 | BioSpec Products | N/A |
| BioAnalyzer | Agilent | N/A |
| NextSeq2000 | Illumina | N/A |
| IsoCage P Bioexclusion cages | Tecniplast | N/A |
| Agencourt AMPure XP beads | Beckman Coulter | Cat#A63882 |
| Flow Cell (R9.4.1) | Oxford Nanopore Technologies | Cat#FLO-MIN106D |
| MinION | Oxford Nanopore Technologies | N/A |
| Beadruptor tubes | Fisher | Cat#15–340-154 |
| Bead Mill 24 Homogenizer | Fisher | Cat#15–340-163 |
| Mass spectrometry autosampler vial | Microliter | Cat#09–1200 |
| Biotage SPE Dry 96 Dual | Biotage | Cat#3579M |
| Thermomixer C | Eppendorf | Cat#2231001005 |
| Agilent 7890A GC system | Agilent | N/A |
| Agilent 5975C MS detector | Agilent | N/A |
| HP-5MSUI column | Agilent | Cat#19091S-433UI |
| CFX384 Real-Time System | BioRad | N/A |
| Leica SP8 laser scanning confocal microscope | Leica | N/A |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
Female 8–12 week-old C57Bl/6J germ-free mice were bred and maintained in plastic gnotobiotic isolators or bioexclusion racks within the University of Chicago Gnotobiotic Core Facility and fed ad libitum autoclaved standard chow diet (LabDiets 5K67). Littermates were randomly assigned to treatment groups. All murine experimental procedures were institutionally approved.
Bacterial strains and growth conditions
The bacterial strains used in this study, including the RB-TnSeq library, are listed in the Key Resources Table. Bacteroides thetaiotaomicron VPI-5482 [AMD595], and all its derivatives were cultured anaerobically at 37°C in liquid Brain Heart Infusion Supplemented (BHI-S) medium or defined Varel-Bryant medium as described in Liu et al.,3 and Varel and Bryant.21 Varel-Bryant medium with no carbon source was supplemented with glucose, galactose, sucrose, raffinose, or melibiose to a final concentration of 20 mM. An anaerobic chamber (Coy Laboratory Products) filled from tanks containing 10% CO2, 7.5% H2, and 82.5% N2 was used for all anaerobic microbiology procedures, with working conditions near 2–3% H2.
For growth rate measurements, colonies of Bt were inoculated into 3 mL of BHI-S in plastic culture tubes and grown overnight at 37°C, for a total of 6 biological replicates. Immediately before inoculating with the cells, sealed Hungate tubes containing 10 mL of VB medium and a 20 mL headspace were inoculated via syringe with autoclaved sugar solution and hemin solution for a final concentration of 20 mM sugar and 5 μg/mL hemin. Overnight cultures were diluted to OD600 = 1, and 100 μL of the diluted culture was inoculated into the prepared media tubes via syringe. Anaerobically sealed cultures were grown outside of the chamber in a 37°C incubator with no shaking. Every 45 min, the cultures were taken from the incubator, cells resuspended by shaking, and their OD600 readings measured by a GENSYS 40-Vis spectrophotometer.
METHOD DETAILS
In vivo transcriptomic experiments
Mice were inoculated with a single dose of wildtype Bt VPI-5482 at 106 – 108 CFU/200 μL, and were then sacrificed at 0.5, 1, 2, 4, 7, 14, and 42 days post-colonization. Luminal contents of the mice cecum were immediately snap-frozen in liquid N2 and stored at −80°C. About 50 mg of the contents were transferred into 2 mL screw-cap tubes, followed by the addition of 1 mL TRIzol reagent for the isolation of RNA. The samples were homogenized by beadbeating with 0.1 mm glass beads in a Mini-BeadBeater-96 for 2 min. Total RNA isolation and purification were performed using the TRI reagent protocol and quality checked by BioAnalyzer. All library preparation and sequencing work was performed by SeqCenter (Pittsburgh, PA). Initial DNAse treatment is performed with Invitrogen DNAse (RNAse free). Library preparation is performed using Illumina’s Stranded Total RNA Prep Ligation with Ribo-Zero plus. Custom Ribo-Zero probes were designed for Bt and supplemented alongside the standard probe set. Custom probe sequences can be found in Table S1. Sequencing was performed on a NextSeq2000 giving 2×50bp reads. Post sequencing, bcl2fastq (v2.20.0.422) was used to demultiplex and trim adaptors.
In vivo genome-wide mutant fitness assays
Four individual cohorts of mice were used, indicated by the month of the experiment. For each cohort, mice were housed in cages of 2–3 animals either within a gnotobiotic isolator (Dec, Jan) or in hermetically sealed Tecniplast IsoCage P Bioexclusion cages on a rack system (Mar, Oct), and allowed to acclimate for 3 days prior to colonization (Figure S1A). Mice were inoculated with a single dose of the Bt RB-TnSeq mutant library at 106 – 108 CFU/200 μL. The inoculum was prepared by one of two methods: 1) frozen 2 mL aliquots of the Bt RB-TnSeq library were thawed and gavaged directly into mice (Dec, Jan), or 2) a thawed 2 mL aliquot of the Bt RB-TnSeq library was grown in 150 mL BHI-S medium overnight with 20 μg/mL erythromycin (16 h), and backdiluted to OD600 = 0.05 the next morning to allow for fresh cells to reach mid-log phase (3 h) by the time of inoculation (Mar, Oct). For each experiment, at least 3 cell pellets of the inoculum were collected as Time = 0 references. Each mouse was colonized by oral gavage with 200 μL of the Bt transposon library. Stool samples were collected daily (excluding weekends), up to 14 days post-colonization to assess longitudinal shifts in mutant abundance. Mice were monitored and weighed daily. Genomic DNA was extracted using the DNeasy PowerSoil Kit.
The IsoCage P rack system facilitated sample collection while maintaining gnotobiotic conditions, and although the fresh and frozen inocula exhibited nearly identical coverage of the Bt genome (Figure S1B) and harbored statistically indistinguishable levels of diversity (Figures S1C and S1D), use of fresh rather than frozen inoculum substantially reduced bottleneck effects, allowing for low-abundance mutants to reach the gut and persist at a much higher rate through at least D1 (Figure S1E), with diversity only beginning to decrease at D4 (Figure 4A). Because of the bottleneck effect in the frozen inoculum cohorts, analysis of early time points in the functional genetics experiments is restricted to experiments that used fresh inoculum (Mar, Oct). Results from later time points converged across all four runs of the experiment, in spite of differences in protocol (Figures 1C and 4E).
Isolation of mutants from fecal matter
Mouse feces were collected and immediately homogenized in 500 mL 25% glycerol solution and stored at −80°C. Prior to isolation, glycerol stocks were allowed to thaw on the benchtop for 10 min, centrifuged for 30 s at 2,000 RPM. On each 150 mm BHI-S plate, 100 μL of 10−3, 10−4, or 10−5 dilution of the glycerol stock was spread using 4.5 mm glass beads. The plates were incubated anaerobically at 37°C for two days. Individual colonies were picked into 1 mL 96-well plates containing 750 μL BHI-S in each well. After 16 h of growth, glycerol was added to a final concentration of 20% and the isolates were stored. The isolate stocks were used as the template in PCR amplifying the barcoded region of the mutants. The PCR products were sent for Sanger sequencing.
Host-associated evolution of spontaneous Bt mutants
Three female mice 8–12 wk-old C57Bl/6J GF mice were co-housed in the same gnotobiotic isolator and fed standard chow diet ad libitum, and were then given a single dose of wildtype Bt VPI-5482 at 106 – 108 CFU/200 μL. One week post inoculation of Bt, the three mice were separated into individual cages. A fecal sample was taken six weeks post inoculation. DNA was extracted from the fecal pellets by the phenol-chloroform method, followed by ethanol precipitation, and sent for shotgun sequencing. Individual isolates from each fecal pellet were cultured from the bulk material as outlined in Isolation of mutants from fecal matter. We assayed for isolates with increased growth in VB-melibiose and selected one isolate from each mouse for MinION long-read sequencing.
Isolate genome sequencing, assembly, and polishing
To provide greater context for the delineation of the complex chromosomal rearrangements associated with the BT1872/BT1873 operon, a long-read sequencing strategy was employed. The isolate genomes assessed were wildtype Bt, the strain used for the mouse experiments, and three spontaneous mutant cultivars (MZ55, MZ58 and MZ65), which demonstrated enhanced growth rates in the presence of melibiose, recovered from the feces of mice six weeks after initial inoculation. Total genomic HMW DNA was extracted by a standard phenol chloroform protocol on overnight 25 mL BHIS broth cultures.63 DNA was resuspended in 0.1 mL 10 mM Tris-HCl, pH 8.5.
Slow pipetting, wide bore pipette tips and steps to minimize velocity gradients were implemented throughout to avoid further shearing of DNA molecules. Libraries were prepared with the Rapid Barcoding Kit (SQK-RBK004) and the standard protocols from Oxford Nanopore Technologies were used with the following modifications. DNA fragmentation was performed on 10 μg DNA using 10 passes through a 22G needle in a 250 μL volume before purification using 0.5% Agencourt AMPure XP beads (A63882, Beckman Coulter). Each elution step of the AMPureXP beads was performed using 10 mM Tris-Cl pH 8.5 instead of water, at 37°C for 5 min. The gDNA inputs into library preparation ranged between 0.5 μg and 1.2 μg (Table S2), based on sample availability in a standard 8.5 μL volume, with 1.5 μL Fragmentation mix added to each sample. Barcoded libraries were pooled so each sample contributed an equal input mass (~0.5 μg). Using MinKNOW v4.3.4, a single R9.4/FLO-MIN106 flow cell (Oxford Nanopore Technologies) sequenced the final prepared library with a starting voltage of −180 mV and a run time of 72 h. Guppy v5.0.11 and the sup model were used for post-run basecalling, sample de-multiplexing and the conversion of raw FAST5 files to FASTQ files. For downstream analyses, we only used reads with a minimum quality score of 7. We assembled long-reads contigs with Flye.51 Additional DNA extractions were carried out for every isolate using a standard phenol-chloroform extraction and send for short-read sequencing. We then used the short-reads to polish the long-read assemblies using Pilon v1.23.52
Metabolite extraction from cecal material
Metabolites were extracted with the addition of extraction solvent (80% methanol spiked with internal standards and stored at −80°C, Table S4) to pre-weighed fecal/cecal samples at a ratio of 100 mg of material per mL of extraction solvent in beadruptor tubes (Fisherbrand; 15-340-154). Samples were homogenized at 4°C on a Bead Mill 24 Homogenizer (Fisher; 15-340-163), set at 1.6 m/s with 6 30-s cycles, 5 s off per cycle. Samples were then centrifuged at −10°C, 20,000 × g for 15 min and the supernatant was used for subsequent metabolomic analysis.
Metabolite analysis using GC-EI-MS and methoxyamine and TMS derivatization
Metabolites were analyzed using gas chromatography-mass spectrometry (GCMS) with electron impact ionization. To a mass spectrometry autosampler vial (Microliter; 09–1200), 100 μL of metabolite extract was added and dried down completely under a nitrogen stream at 30 L/min (top) and 1 L/min (bottom) at 30°C (Biotage SPE Dry 96 Dual; 3579M). To dried samples, 50 μL of freshly prepared 20 mg/mL methoxyamine (Sigma; 226904) in pyridine (Sigma; 270970) was added and incubated in a thermomixer C (Eppendorf) for 90 min at 30°C and 1400 rpm. After samples are cooled to room temperature, 80 μL of derivatizing reagent (BSTFA +1% TMCS; Sigma; B-023) and 70 μL of ethyl acetate (Sigma; 439169) were added and samples were incubated in a thermomixer at 70°C for 1 h and 1400 rpm. Samples were cooled to RT and 400 μL of Ethyl Acetate was added to dilute samples. Turbid samples were transferred to microcentrifuge tubes and centrifuged at 4°C, 20,000 × g for 15 min. Supernatants were then added to mass spec vials for GCMS analysis. Samples were analyzed using a GC-MS (Agilent 7890A GC system, Agilent 5975C MS detector) operating in electron impact ionization mode, using an HP-5MSUI column (30 m × 0.25 mm, 0.25 μm; Agilent Technologies 19091S-433UI) and 1 μL injection. Oven ramp parameters: 1 min hold at 60°C, 16°C per min up to 300°C with a 7 min hold at 300°C. Inlet temperature was 280°C and transfer line was 300°C. Data analysis was performed using MassHunter Quantitative Analysis software (version B.10, Agilent Technologies) and confirmed by comparison to authentic standards. Normalized peak areas were calculated by dividing raw peak areas of targeted analytes by averaged raw peak areas of internal standards.
RT-qPCR
Total messenger RNA was isolated from colonic mucosal scrapings with TRIzol reagent according to the same protocol used for cecal RNA isolation. Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics Corporation) was used to obtain cDNA. Real-time qPCR was performed using iTaq Universal SYBR Green Supermix with CFX384 Real-Time System (Bio-Rad). Primers are listed in Table S1.
ELISA
For cecal IgA ELISA, frozen cecal samples were resuspended in 1 mL of ELISA diluent per 100mg of cecal contents and homogenized by bead beating for 1 min. ELISA was performed using goat polyclonal anti-IgA antibody (capture antibody) (Southern Biotech: 1040–01) and goat anti-IgA antibody labeled with HRP (secondary antibody) (Bio-Rad: STAR137P). IgG ELISA was performed on frozen serum samples using Invitrogen IgG (Total) Mouse Uncoated ELISA Kit.
Histological procedures
During sacrifice of mice colonized with WT Bt for transcriptomics experiments, distal colonic cross-sections were collected and placed in cassettes. Tissues were fixed by immersion in Carnoy’s solution (60% ethanol, 30% chloroform, 10% acetic acid) for 3 h and were stored in 70% ethanol until tissue sectioning. When ready to process, samples were dehydrated by two successive washes each in methanol for 35 min, ethanol for 30 min, and xylene for 25 min. Tissue samples within cassettes were then submerged in melted paraffin at 68°C for 1hr, removed, and kept at room temperature until sectioning. Paraffin blocks were cut into 4-μm-thick sections and deparaffinized for immunofluorescence.
Immunostaining and imaging
After deparaffinization and rehydration, slides were incubated in lysozyme solution at 37°C for 20 min, and then in antigen retrieval solution (10 mM sodium citrate [pH 6.0]) at 90°C for 10 min. For mucus visualization, a polyclonal rabbit anti-mouse Muc2-specific antibody (Santa Cruz Biotechnology) was diluted 1:100 in blocking buffer (Dako), applied to the slide, and incubated for 2 h in the dark at room temperature. Slides were washed gently three times in TBS-T. The secondary antibody (Alexa Fluor 488 goat anti-rabbit IgG, Invitrogen) was diluted 1:100 in blocking buffer, applied to the slide, and incubated for 30 min in the dark at room temperature. Slides were again washed gently three times in TBS-T, and were then stained with DAPI 10 μg/mL (Sigma), incubated for 1 min, and washed three times in PBS. Slides were then dried, mounted with ProLong Gold Anti-Fade mounting medium (Invitrogen) and stored at room temperature in the dark until imaging. Images were acquired on a Leica SP8 laser scanning confocal microscope with the LAS_X Leica software (Leica). All samples were imaged with a 40× oil-immersion objective. Images were acquired at a frame size of 1024 × 1024 with 16-bit depth.
QUANTIFICATION AND STATISTICAL ANALYSIS
PCoA, differential expression analysis, and Gene Set Enrichment Analysis (GSEA) on transcriptomic data
The R53 package ‘phyloseq’64 was used to calculate Bray-Curtis dissimilarity for all pairwise combinations of transcriptome samples, which was then ordinated to create PCoA plots. PERMANOVA analysis using the R package ‘vegan’ was performed to evaluate sample clustering by experimental day with significance criteria set at p < 0.05. Post-hoc pairwise PERMANOVA analyses using FDR method to correct for multiple testing (‘pairwiseAdonis’) were then performed to evaluate significant pairwise differences in clustering (Table S3).
Gene calls were mapped to KEGG ortholog (Kofam) annotations in R. Differential expression analysis of each gene call was performed by pairwise comparisons with ‘deseq2′,55 with the significance criteria log(FDR-adjusted p value) < −3, |log2(fold change)| > 2, and max group mean >50 RPM. Metabolism for the Bt genome was estimated using the anvi’o v7.1 program ‘anvi-estimate-genome.’ Gene calls for each metabolic pathway found within the Bt genome were then transferred into a unique GSEA pathway query list in R (‘fgsea’),57 and pathway enrichment was then calculated using the differential expression statistics calculated with ‘deseq2’.55 Pathways were filtered for padj < 0.05 and the normalized enrichment scores (NES) was plotted (PRISMv9). Other custom gene lists were created to calculate the enrichment of other gene sets including the polysaccharide utilization loci (PULs),20 capsular polysaccharide loci (CPSs),65 and genes associated with the Bt stringent response.16 Statistical results from these RNAseq analyses are presented in Table S5.
Pipeline for measuring relative abundance and fitness scores of RB-Tn mutants
RB-TnSeq strain and gene fitness scores were calculated as described previously from strain-level count data (significance threshold: t-statistic < −3σ).14 For temporal abundance analyses of TnSeq mutants for each cohort, we first created a feature table with raw counts of strain-level mutants across each sample. This table was filtered to remove strains with counts of 1, as these are likely produced by sequencing error. Next, counts were normalized by the count of a synthetic spike-in barcode that was introduced at 20 p.m. into each sample during PCR amplification of the barcodes. Samples where the spike-in represented >30% of total reads were discarded. The synthetic spike-in barcode was subsequently removed as a feature from the table, and the resulting tables were used for alpha diversity analyses via the R package ‘vegan’.54 For all other relative abundance analyses, strains were assigned to genes based on previous mapping by Liu et al.,3 as well as manual mapping performed for this experiment. Strains that had not been mapped here or elsewhere were binned together as “non-mapping” strains, and strain-level counts were then summed for each gene. For all subsequent analyses, we further filtered out genes that mapped to Bt plasmids in order to focus on chromosomal gene fitness patterns. The R package ‘phyloseq’ was used to calculate Bray-Curtis dissimilarity for all pairwise combinations of samples, which was then used to create PCoA plots.64 PERMANOVA analysis using the R package ‘vegan’ was performed to evaluate sample clustering by experimental day with significance criteria set at p < 0.05 (Table S3). Finally, filtered count tables were adjusted to relative abundance based on the total remaining counts, and used to track gene-level relative abundance over time. For linear regression of initial vs. final relative abundance of gene mutants, genes with zero-counts were re-assigned a relative abundance of 1×10−7 to perform log-transformation of the data.
Metabolomics statistics
As amino acid abundances (Figure 2E) were normalized within compounds and cannot be compared across compounds, for each metabolite, we performed one-way ANOVA across all time points, with post-hoc follow-up tests and FDR correction (Table S4). Carbohydrate metabolite abundances (Figures 5D, S3C and S3D) were compared within compounds across time points using one-way ANOVA with post-hoc follow-up tests and FDR correction (Table S6).
Bacterial growth rate statistics
Growth rates of different Tn mutant isolates (Figures 5A and S3B) were compared by one-way ANOVA with post-hoc follow-up tests and FDR correction (Table S6).
Genome mapping and coverage visualization
We used anvi’o v7.156 and the pangenomic workflow to compute and visualize genomic coverage for each isolate genome from the evolution experiments. Briefly, the workflow uses (1) Prodigal v2.6.358 to identify open-reading frames (ORFs), (2) ‘anvi-run-hmm’ to identify single copy core genes from bacteria (n = 71)66 and ribosomal RNAs (n = 12, modified from1) using HMMER v3.3,67 (3) ‘anvi run-ncbi-cogs’ and ‘anvi-run-kegg-kofams’ to annotate ORFs with the NCBI’s Clusters of Orthologous Groups (COGs),68 and the KOfam HMM database of KEGG orthologs (KOs)69,70 respectively. We used bowtie2 v2.3.5.159 to recruit short-reads to the contigs, and samtools v1.1160 to convert SAM files to BAM files. We profiled the resulting BAM files with ‘anvi-profile’ and used the program ‘anvi-merge’ to combine all single profiles into a merged profile for downstream visualization. We used ‘anvi-get-split-coverages’ and ‘anvi-script-visualize-split-coverages’ to generate coverage plots. We used ‘anvi-export-gene-calls’ and gggenes v0.4.1 to visualize the genomic context around BT1871.
BT1871 copy number
We used blast71 to compute the number of long-reads with two copies of the BT1871 locus in the MZ65 isolate. We extracted the gene sequence (1989 bp) from the initial Bt genome (AMD595) using anvi’o interactive interface and used blastn v2.5.0 to blast the long-reads from MZ65. Blast hits with an alignment length >180% of the gene length were flagged as “two copies” and hits with alignment length between 80% and 105% were flagged as “one copy”. To visualize long-reads with two copies of BT1871, we used minimap2 v2.1761 to map the MZ65 long-reads to the MZ65 genome, which had two copies of the BT1871 region. We used samtools v1.11 to extract the reads with two copies as identified above and used IGV v2.11.162 to visualize the mapping and generate a figure.
Image quantification
For each mouse, a single colonic cross-section was used to generate 6 representative images. Images were masked to split the DAPI signal into an epithelial channel (blue) and a luminal channel (red). The MUC2 channel was green. To assess proximity of Bt to the epithelium, at 10 evenly-spaced points along the epithelium in each frame, a perpendicular line was drawn until it intersected the nearest bacterial cell, and this distance was measured. These measurements were averaged across all frames for each mouse. Average measurements of n = 3–4 mice per time point were used to compare Bt epithelial proximity across experimental days by performing one-way ANOVA and then post-hoc follow-up tests with FDR correction to compare all time points to D1. Differences were considered significant at q < 0.05.
Supplementary Material
Highlights.
B. thetaiotaomicron adaptation to the germ-free mouse gut is a dynamic process
Early selective pressures are distinct from intermediate and long-term pressures
Selective pressure for efficient dietary resource use dominates in the long term
Changes in Bt spatial localization parallel changes in resource use
ACKNOWLEDGMENTS
We thank Hualan Liu for invaluable experimental help with the RB-TnSeq libraries. We thank David Hershey and A. Murat Eren for helpful scientific input. We also thank the Chang lab members for scientific support received. The graphical abstract was created using BioRender. This work was performed with support from NIH T32DK007074 (M.Z., M.S.K.), NIH RC2DK122394 (E.B.C.), NIH T32GM007281 (M.S.K.), and the Host-Microbe and Tissue and Cell Engineering cores of the UChicago DDRCC, Center for Interdisciplinary Study of Inflammatory Intestinal Disorders (C-IID) (NIDDK P30 DK042086).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113009.
REFERENCES
- 1.O’Malley MA (2007). The nineteenth century roots of “everything is everywhere. Nat. Rev. Microbiol. 5, 647–651. 10.1038/nrmicro1711. [DOI] [PubMed] [Google Scholar]
- 2.Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, and Gordon JI (2009). Identifying Genetic Determinants Needed to Establish a Human Gut Symbiont in Its Habitat. Cell Host Microbe 6, 279–289. 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu H, Shiver AL, Price MN, Carlson HK, Trotter VV, Chen Y, Escalante V, Ray J, Hern KE, Petzold CJ, et al. (2021). Functional genetics of human gut commensal Bacteroides thetaiotaomicron reveals metabolic requirements for growth across environments. Cell Rep. 34, 108789. 10.1016/j.celrep.2021.108789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu M, McNulty NP, Rodionov DA, Khoroshkin MS, Griffin NW, Cheng J, Latreille P, Kerstetter RA, Terrapon N, Henrissat B, et al. (2015). Genetic determinants of in vivo fitness and diet responsiveness in multiple human gut Bacteroides. Science 350, aac5992. 10.1126/SCIENCE.AAC5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vasquez KS, Willis L, Cira NJ, Ng KM, Pedro MF, Aranda-Díaz A, Rajendram M, Yu FB, Higginbottom SK, Neff N, et al. (2021). Quantifying rapid bacterial evolution and transmission within the mouse intestine. Cell Host Microbe 29, 1454–1468.e4. 10.1016/j.chom.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crook N, Ferreiro A, Gasparrini AJ, Pesesky MW, Gibson MK, Wang B, Sun X, Condiotte Z, Dobrowolski S, Peterson D, and Dantas G (2019). Adaptive strategies of the candidate probiotic E. coli Nissle in the mammalian gut. Cell Host Microbe 25, 499–512.e8. 10.1016/J.CHOM.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dapa T, Ramiro RS, Pedro MF, Gordo I, and Xavier KB (2022). Diet leaves a genetic signature in a keystone member of the gut microbiota. Cell Host Microbe 30, 183–199.e10. 10.1016/J.CHOM.2022.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Barroso-Batista J, Pedro MF, Sales-Dias J, Pinto CJG, Thompson JA, Pereira H, Demengeot J, Gordo I, and Xavier KB (2020). Specific Eco-evolutionary Contexts in the Mouse Gut Reveal Escherichia coli Metabolic Versatility. Curr. Biol. 30, 1049–1062.e7. 10.1016/j.cub.2020.01.050. [DOI] [PubMed] [Google Scholar]
- 9.Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, Buhler JD, and Gordon JI (2005). Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science (New York, N.Y.) 307, 1955–1959. 10.1126/SCIENCE.1109051. [DOI] [PubMed] [Google Scholar]
- 10.Sonnenburg JL, Chen CTL, and Gordon JI (2006). Genomic and Metabolic Studies of the Impact of Probiotics on a Model Gut Symbiont and Host. PLoS Biol. 4, e413. 10.1371/journal.pbio.0040413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, Shah N, Wang C, Magrini V, Wilson RK, et al. (2009). Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc. Natl. Acad. Sci. USA 106, 5859–5864. 10.1073/pnas.0901529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becattini S, Sorbara MT, Kim SG, Littmann EL, Dong Q, Walsh G, Wright R, Amoretti L, Fontana E, Hohl TM, and Pamer EG (2021). Rapid transcriptional and metabolic adaptation of intestinal microbes to host immune activation. Cell Host Microbe 29, 378–393.e5. 10.1016/j.chom.2021.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donaldson GP, Chou WC, Manson AL, Rogov P, Abeel T, Bochicchio J, Ciulla D, Melnikov A, Ernst PB, Chu H, et al. (2020). Spatially distinct physiology of Bacteroides fragilis within the proximal colon of gnotobiotic mice. Nat. Microbiol. 5, 746–756. 10.1038/s41564-020-0683-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wetmore KM, Price MN, Waters RJ, Lamson JS, He J, Hoover CA, Blow MJ, Bristow J, Butland G, Arkin AP, and Deutschbauer A (2015). Rapid Quantification of Mutant Fitness in Diverse Bacteria by Sequencing Randomly Bar-Coded Transposons. mBio 6, e00306–e00315. 10.1128/MBIO.00306-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irving SE, Choudhury NR, and Corrigan RM (2021). The stringent response and physiological roles of (pp)pGpp in bacteria. Nat. Rev. Microbiol. 19, 256–271. 10.1038/s41579-020-00470-y. [DOI] [PubMed] [Google Scholar]
- 16.Schofield WB, Zimmermann-Kogadeeva M, Zimmermann M, Barry NA, and Goodman AL (2018). The Stringent Response Determines the Ability of a Commensal Bacterium to Survive Starvation and to Persist in the Gut. Cell Host Microbe 24, 120–132.e6. 10.1016/J.CHOM.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martens EC, Chiang HC, and Gordon JI (2008). Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4, 447–457. 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wexler AG, and Goodman AL (2017). An insider’s perspective: Bacteroides as a window into the microbiome. Nat. Microbiol. 2, 17026. 10.1038/nmicrobiol.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El Kaoutari A, Armougom F, Gordon JI, Raoult D, and Henrissat B (2013). The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 11, 497–504. 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- 20.Drula E, Garron M-L, Dogan S, Lombard V, Henrissat B, and Terrapon N (2022). The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50, D571–D577. 10.1093/nar/gkab1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varel VH, and Bryant MP (1974). Nutritional Features of Bacteroides fragilis subsp. fragilis. Appl. Microbiol. 28, 251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adamberg K, Adamberg S, Ernits K, Larionova A, Voor T, Jaagura M, Visnapuu T, and Alamäe T (2018). Composition and metabolism of fecal microbiota from normal and overweight children are differentially affected by melibiose, raffinose and raffinose-derived fructans. Anaerobe 52, 100–110. 10.1016/j.anaerobe.2018.06.009. [DOI] [PubMed] [Google Scholar]
- 23.Johansson MEV, Sjövall H, and Hansson GC (2013). The gastrointestinal mucus system in health and disease. Nat. Rev. Gastroenterol. Hepatol. 10, 352–361. 10.1038/nrgastro.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandler M, Fayet O, Rousseau P, Ton Hoang B, and Duval-Valentin G (2015). Copy-out-Paste-in Transposition of IS911: A Major Transposition Pathway. Microbiol. Spectr 3. 10.1128/MICROBIOLSPEC.MDNA3-0031-2014. [DOI] [PubMed] [Google Scholar]
- 25.Macpherson AJ, and Harris NL (2004). Interactions between commensal intestinal bacteria and the immune system. Nat. Rev. Immunol. 4, 478–485. 10.1038/nri1373. [DOI] [PubMed] [Google Scholar]
- 26.Bunker JJ, Erickson SA, Flynn TM, Henry C, Koval JC, Meisel M, Jabri B, Antonopoulos DA, Wilson PC, and Bendelac A (2017). Natural polyreactive IgA antibodies coat the intestinal microbiota. Science 358, eaan6619. 10.1126/science.aan6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peterson DA, McNulty NP, Guruge JL, and Gordon JI (2007). IgA Response to Symbiotic Bacteria as a Mediator of Gut Homeostasis. Cell Host Microbe 2, 328–339. 10.1016/j.chom.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 28.Hapfelmeier S, Lawson MAE, Slack E, Kirundi JK, Stoel M, Heikenwalder M, Cahenzli J, Velykoredko Y, Balmer ML, Endt K, et al. (2010). Reversible Microbial Colonization of Germ-Free Mice Reveals the Dynamics of IgA Immune Responses. Science 328, 1705–1709. 10.1126/science.1188454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joglekar P, Ding H, Canales-Herrerias P, Pasricha PJ, Sonnenburg JL, and Peterson DA (2019). Intestinal IgA Regulates Expression of a Fructan Polysaccharide Utilization Locus in Colonizing Gut Commensal Bacteroides thetaiotaomicron. mBio 10, 023244–19–e2419. 10.1128/mBio.02324-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakajima A, Vogelzang A, Maruya M, Miyajima M, Murata M, Son A, Kuwahara T, Tsuruyama T, Yamada S, Matsuura M, et al. (2018). IgA regulates the composition and metabolic function of gut microbiota by promoting symbiosis between bacteria. J. Exp. Med. 215, 2019–2034. 10.1084/jem.20180427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huus KE, Bauer KC, Brown EM, Bozorgmehr T, Woodward SE, Serapio-Palacios A, Boutin RCT, Petersen C, and Finlay BB (2020). Commensal Bacteria Modulate Immunoglobulin A Binding in Response to Host Nutrition. Cell Host Microbe 27, 909–921.e5. 10.1016/j.chom.2020.03.012. [DOI] [PubMed] [Google Scholar]
- 32.Mazmanian SK, Round JL, and Kasper DL (2008). A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625. 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 33.Coyne MJ, Chatzidaki-Livanis M, Paoletti LC, and Comstock LE (2008). Role of glycan synthesis in colonization of the mammalian gut by the bacterial symbiont Bacteroides fragilis. Proc. Natl. Acad. Sci. USA 105, 13099–13104. 10.1073/pnas.0804220105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Porter NT, Canales P, Peterson DA, and Martens EC (2017). A Subset of Polysaccharide Capsules in the Human Symbiont Bacteroides thetaiotaomicron Promote Increased Competitive Fitness in the Mouse Gut. Cell Host Microbe 22, 494–506.e8. 10.1016/J.CHOM.2017.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. (2010). A human gut microbial gene catalog established by metagenomic sequencing. Nature 464, 59–65. 10.1038/NATURE08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuwahara T, Yamashita A, Hirakawa H, Nakayama H, Toh H, Okada N, Kuhara S, Hattori M, Hayashi T, and Ohnishi Y (2004). Genomic analysis of Bacteroides fragilis reveals extensive DNA inversions regulating cell surface adaptation. Proc. Natl. Acad. Sci. USA 101, 14919–14924. 10.1073/pnas.0404172101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo Y, Kitamoto S, and Kamada N (2020). Microbial adaptation to the healthy and inflamed gut environments. Gut Microb. 12, 1857505. 10.1080/19490976.2020.1857505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Townsend GE, Han W, Schwalm ND, Hong X, Bencivenga-Barry NA, Goodman AL, and Groisman EA (2020). A Master Regulator of Bacteroides thetaiotaomicron Gut Colonization Controls Carbohydrate Utilization and an Alternative Protein Synthesis Factor. mBio 11, e03221–19. 10.1128/MBIO.03221-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watson AR, Füssel J, Veseli I, DeLongchamp JZ, Silva M, Trigodet F, Lolans K, Shaiber A, Fogarty E, Runde JM, et al. (2023). Metabolic independence drives gut microbial colonization and resilience in health and disease. Genome Biol. 24, 78. 10.1186/s13059-023-02924-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shepherd ES, Deloache WC, Pruss KM, Whitaker WR, and Sonnenburg JL (2018). An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature 557, 434–438. 10.1038/s41586-018-0092-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hibbing ME, Fuqua C, Parsek MR, and Peterson SB (2010). Bacterial competition: surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 8, 15–25. 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johansson MEV, Jakobsson HE, Holmén-Larsson J, Schütte A, Ermund A, Rodríguez-Piñeiro AM, Arike L, Wising C, Svensson F, Bäckhed F, and Hansson GC (2015). Normalization of Host Intestinal Mucus Layers Requires Long-Term Microbial Colonization. Cell Host Microbe 18, 582–592. 10.1016/j.chom.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johansson MEV, Phillipson M, Petersson J, Velcich A, Holm L, and Hansson GC (2008). The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 105, 15064–15069. 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, et al. (2016). A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 167, 1339–1353.e21. 10.1016/j.cell.2016.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Earle KA, Billings G, Sigal M, Lichtman JS, Hansson GC, Elias JE, Amieva MR, Huang KC, and Sonnenburg JL (2015). Quantitative Imaging of Gut Microbiota Spatial Organization. Cell Host Microbe 18, 478–488. 10.1016/J.CHOM.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hayase E, Hayase T, Jamal MA, Miyama T, Chang C-C, Ortega MR, Ahmed SS, Karmouch JL, Sanchez CA, Brown AN, et al. (2022). Mucus-degrading Bacteroides link carbapenems to aggravated graft-versus-host disease. Cell 185, 3705–3719.e14. 10.1016/j.cell.2022.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sonnenburg ED, and Sonnenburg JL (2014). Starving our Microbial Self: The Deleterious Consequences of a Diet Deficient in Microbiota-Accessible Carbohydrates. Cell Metabol. 20, 779–786. 10.1016/j.cmet.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siguier P, Filée J, and Chandler M (2006). Insertion sequences in prokaryotic genomes. Curr. Opin. Microbiol. 9, 526–531. 10.1016/j.mib.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 50.Miyoshi J, Bobe AM, Miyoshi S, Huang Y, Hubert N, Delmont TO, Eren AM, Leone V, and Chang EB (2017). Peripartum Antibiotics Promote Gut Dysbiosis, Loss of Immune Tolerance, and Inflammatory Bowel Disease in Genetically Prone Offspring. Cell Rep 20, 491–504. 10.1016/j.celrep.2017.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kolmogorov M, Yuan J, Lin Y, and Pevzner PA (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- 52.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, and Earl AM (2014). Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS One 9, e112963. 10.1371/JOURNAL.PONE.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.R Core Team R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. [Google Scholar]
- 54.Oksanen J, Simpson G, Blanchet F, Kindt R, Legendre P, Minchin P, O’Hara R, Solymos P, Stevens M, Szoecs E, et al. (2022). Vegan: Community Ecology Package.
- 55.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eren AM, Kiefl E, Shaiber A, Veseli I, Miller SE, Schechter MS, Fink I, Pan JN, Yousef M, Fogarty EC, et al. (2021). Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 6, 3–6. 10.1038/s41564-020-00834-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A (2021). Fast Gene Set Enrichment Analysis. BioRxiv, 060012. 10.1101/060012. [DOI] [Google Scholar]
- 58.Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, and Hauser LJ (2010). Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11, 119–211. 10.1186/1471-2105-11-119/TABLES/5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/BIOINFORMAT-ICS/BTP352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. 10.1093/BIOINFORMATICS/BTY191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative Genomics Viewer. Nat. Biotechnol. 29, 24–26. 10.1038/NBT.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trigodet F, Lolans K, Fogarty E, Shaiber A, Morrison HG, Barreiro L, Jabri B, and Eren AM (2022). High molecular weight DNA extraction strategies for long-read sequencing of complex metagenomes. Mol. Ecol. Resour. 22, 1786–1802. 10.1111/1755-0998.13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McMurdie PJ, and Holmes S (2013). phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS One 8, e61217. 10.1371/JOURNAL.PONE.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mazmanian SK (2008). Capsular Polysaccharides of Symbiotic Bacteria Modulate Immune Responses During Experimental Colitis (2008). J. Pediatr. Gastroenterol. Nutr. 46, E11–E12. 10.1097/01.mpg.0000313824.70971.a7. [DOI] [PubMed] [Google Scholar]
- 66.Lee MD (2019). GToTree: a user-friendly workflow for phylogenomics. Bioinformatics 35, 4162–4164. 10.1093/BIOINFORMATICS/BTZ188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eddy SR (2011). Accelerated Profile HMM Searches. PLoS Comput. Biol. 7, e1002195. 10.1371/JOURNAL.PCBI.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinf. 4, 41. 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kanehisa M, and Goto S (2000). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30. 10.1093/NAR/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, and Ogata H (2020). KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252. 10.1093/BIOINFORMATICS/BTZ859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data, including DNA and RNA datasets, have been deposited at NCBI Sequence Read Archive and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Goat polyclonal anti-IgA | Southern Biotech | Cat#OB1040–01 |
| Goat anti-mouse IgA:HRP | BioRad | Cat#STAR137P |
| Rabbit polyclonal anti-mouse Muc2 | Santa Cruz Biotechnology | Cat#sc-15334 |
| Alexa Fluor 488 goat anti-rabbit IgG | Invitrogen | Cat#A-11008 |
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
| Bacteroides thetaiotaomicron VPI-5482 wildtype | Gift from Deutschbauer lab | AMD595 |
| RB-Tn mutant isolated from monocolonized mice with barcoded transposon insertion in BT1876 | Gift from Deutschbauer lab | AMD595-derived RB-Tn library3 |
| RB-Tn mutant isolated from monocolonized mice with barcoded transposon insertion in BT1874 | This work | Tn_BT1874 |
| RB-Tn mutant isolated from monocolonized mice with barcoded transposon insertion in BT1876 | This work | Tn_BT1876 |
| RB-Tn mutant isolated from monocolonized mice with barcoded transposon insertion in the intergenic region between BT1872 and BT1873 | This work | Tn_BT1872–83 |
| RB-Tn mutant isolated from monocolonized mice with barcoded transposon insertion in BT1872 | This work | Tn_BT1872 |
| RB-Tn mutant isolated from monocolonized mice with barcoded transposon insertion in BT1874 and BT3132 | This work | Tn_BT1874/BT3132 |
| RB-Tn mutant isolated from monocolonized mice with barcoded transposon insertion in BT3133 | This work | Tn_BT3133 |
| WT Bt with a copy of BT1871 integrated into the Bt genome at the attN1 site under the BT1311 (rpoD) constitutive promoter. | This work | WT + BT1871OE |
| Bt mutant isolated from feces after 6-week monoassociation of Bt; carries a tandem repeat of the BT1871 locus (see Figure 7). | This work | MZ65 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Brain Heart Infusion Broth | Fisher | Cat#DF0037178 |
| Agar | Fisher | Cat#DF0145–17-0 |
| Hemin | Fisher | Cat#51280–5G |
| Glucose | Sigma | Cat#158968–500G |
| Galactose | Sigma | Cat#G0750–500G |
| Sucrose | Fisher | Cat#AAJ64270A1 |
| Raffinose | Fisher | Cat#AC195670250 |
| Melibiose | Sigma | Cat#M5500–100G |
| Erythromycin | Fisher | Cat#AC227330050 |
| Phosphate Buffered Saline | Sigma | Cat#P3813–10PAK |
| Glycerol | Fisher | Cat#BP2291 |
| Ambion TRIzol Reagent | Fisher | Cat#15–596-018 |
| Chloroform | Fisher | Cat#AC390760010 |
| Isopropanol | Fisher | Cat#BP2618500 |
| Ethanol | Fisher | Cat#BP2818500 |
| DNAse | Fisher | Cat#18068015 |
| Tris | Fisher | Cat#BP152–10 |
| EDTA | Fisher | Cat#PR-V4231 |
| NaCl | Fisher | Cat#S640–10 |
| SDS | Sigma | Cat#L3771–25G |
| Phenol:Chloroform:Isoamyl Alcohol | Fisher | Cat#AM9732 |
| Proteinase K | Sigma | Cat#3115852001 |
| HCl | Sigma | Cat#A144–212 |
| Methanol | Fisher | Cat#A452SK-4 |
| Methoxyamine | Sigma | Cat#226904 |
| Pyridine | Sigma | Cat#270970 |
| Derivatizing reagent (BSTFA +1% TMCS) | Sigma | Cat#B-023 |
| Ethyl acetate | Sigma | Cat#650528 |
| ELISA diluent | R&D Systems | Cat#DY995 |
| Acetic acid | Fisher | Cat#A38–500 |
| Xylene | Fisher | Cat#X3S-4 |
| Lysozyme | Fisher | Cat#89833 |
| Sodium citrate | Fisher | Cat#S279–500 |
| DAPI | Sigma | Cat#D8417–5MG |
| ProLong Gold Anti-Fade mounting medium | Fisher | Cat#P10144 |
| iTaq Universal SYBR Green Supermix | BioRad | Cat#1725124 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| DNeasy PowerSoil Kit. | Qiagen | Cat#47016 |
| Rapid Barcoding Kit | Oxford Nanopore Technologies | Cat#SQK-RBK004 |
| Transcriptor First Strand cDNA Synthesis Kit | Roche | Cat#35081963001 |
| Invitrogen IgG (Total) Mouse Uncoated ELISA Kit | Invitrogen | Cat#88–50400-88 |
|
| ||
| Deposited Data | ||
|
| ||
| RNAseq raw data from Bt at different time points | This work | https://www.ncbi.nlm.nih.gov/sra (accession: PRJNA797447) |
| Shotgun sequencing raw data from evolution experiment fecal samples | This work | https://www.ncbi.nlm.nih.gov/sra (accession: PRJNA797447) |
| Long-read sequencing raw data from isolate MZ65 | This work | https://www.ncbi.nlm.nih.gov/sra (accession: PRJNA797447) |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| Mouse: germ-free wild-type C57BL/6J | University of Chicago Gnotobiotic Core Facility |
N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Custom Ribo-Zero Plus Probes | SeqCenter | See Table S1A |
| qPCR primers | Miyoshi et al., 201750 | See Table S1B |
|
| ||
| Software and Algorithms | ||
|
| ||
| bcl2fastq | Illumina | Version 2.20.0.422 |
| MinKNOW | Oxford Nanopore Technologies | Version 4.3.4 |
| Guppy | Oxford Nanopore Technologies | Version 5.0.11 |
| Flye | https://github.com/fenderglass/Flye | Version 2.651 |
| Pilon | https://github.com/broadinstitute/pilon | Version 1.2352 |
| MassHunter Quantitative Analysis software |
Agilent Technologies | Version B.10 |
| R software | https://www.r-project.org/ | Version 4.2.153 |
| vegan R software | https://cran.r-project.org/web/packages/vegan/index.html | Version 2.6–454 |
| pairwiseAdonis R software | https://github.com/pmartinezarbizu/pairwiseAdonis | Version 0.4 |
| DESeq2 R software | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | Version 1.36.055 |
| anvi’o | https://anvio.org/ | Version 7.156 |
| fgsea R package | https://bioconductor.org/packages/release/bioc/html/fgsea.html | Version 3.1757 |
| Graphpad Prism | https://www.graphpad.com/features | Version 9 |
| Prodigal | https://github.com/hyattpd/Prodigal | Version 2.6.358 |
| bowtie2 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml | Version v2.3.5.159 |
| samtools | http://www.htslib.org/ | Version 1.1160 |
| gggenes | https://github.com/wilkox/gggenes/tree/master | Version 0.4.1 |
| blastn | https://blast.ncbi.nlm.nih.gov/Blast.cgi | Version 2.5.0 |
| minimap2 | https://github.com/lh3/minimap2 | Version 2.1761 |
| IGV | https://igv.org/ | Version 2.11.162 |
| LAS_X Leica | Leica | N/A |
|
| ||
| Other | ||
|
| ||
| Standard mouse chow | LabDiets | 5K67 |
| Anaerobic chamber | Coy Laboratory Products | N/A |
| GENSYS 40-Vis spectrophotometer | Thermo Scientific | N/A |
| Mini-BeadBeater-96 | BioSpec Products | N/A |
| BioAnalyzer | Agilent | N/A |
| NextSeq2000 | Illumina | N/A |
| IsoCage P Bioexclusion cages | Tecniplast | N/A |
| Agencourt AMPure XP beads | Beckman Coulter | Cat#A63882 |
| Flow Cell (R9.4.1) | Oxford Nanopore Technologies | Cat#FLO-MIN106D |
| MinION | Oxford Nanopore Technologies | N/A |
| Beadruptor tubes | Fisher | Cat#15–340-154 |
| Bead Mill 24 Homogenizer | Fisher | Cat#15–340-163 |
| Mass spectrometry autosampler vial | Microliter | Cat#09–1200 |
| Biotage SPE Dry 96 Dual | Biotage | Cat#3579M |
| Thermomixer C | Eppendorf | Cat#2231001005 |
| Agilent 7890A GC system | Agilent | N/A |
| Agilent 5975C MS detector | Agilent | N/A |
| HP-5MSUI column | Agilent | Cat#19091S-433UI |
| CFX384 Real-Time System | BioRad | N/A |
| Leica SP8 laser scanning confocal microscope | Leica | N/A |