

Abstract
MRTX1133 is the first non-covalent inhibitor against the KRASG12D mutant that demonstrated specificity and potency in pre-clinical tumor models. Here, we used isogenic cell lines expressing a single Ras allele to evaluate the selectivity of this compound. In addition to KRASG12D, MRTX1133 showed significant activity against several other KRAS mutants as well as wildtype KRAS protein. In contrast, MRTX1133 exhibited no activity against both G12D and wildtype forms of HRAS and NRAS proteins. Functional analysis revealed that the selectivity of MRTX1133 towards KRAS is associated with its binding to H95 on KRAS, a residue that is not conserved in HRAS and NRAS. Reciprocal mutation of amino acid 95 among the three Ras paralogs resulted in reciprocal change in their sensitivity towards MRTX1133. Thus, H95 is an essential selectivity handle for MRTX1133 towards KRAS. Amino acid diversity at residue 95 could facilitate the discovery of pan-KRAS inhibitors as well as HRAS and NRAS paralog-selective inhibitors.
INTRODUCTION
The discovery(1–3) and clinical development(4,5) of mutant-selective inhibitors against the small GTPase KRAS is a major breakthrough in targeted therapy. All three family members of the Ras small GTPases, KRAS, HRAS, and NRAS, are oncogenes in human cancer. Oncogenic mutations in Ras protein attenuate its GTPase activity and increase the level of GTP-bound, active form of Ras in the cell. The MAP kinase (MAPK) pathway, which consists of the RAF/MEK/ERK kinase cascade, is a major downstream effector of Ras: it mediates mitogenic signaling from Ras and is commonly used to assess Ras activity in cells(6). The first KRAS mutant to be successfully targeted was the KRASG12C mutant. KRASG12C inhibitors rely on their ability to covalently cross-link to the mutant C12 residue on KRAS to achieve their exquisite mutant selectivity(1–3). Recently, MRTX1133 was disclosed as the first non-covalent inhibitor with selectivity toward the KRASG12D mutant(7–9). MRTX1133 was evolved extensively from the scaffold of adagrasib by introducing selectivity towards the D12 residue and by optimizing affinity toward the KRAS switch II pocket(7). The discovery of MRTX1133 proves that KRAS mutant without a cross-linkable residue can be successfully targeted with non-covalent inhibitors.
METHODS
Cell culture and reagents.
Human cancer cell lines were grown in RPMI-1640 medium (Lonza #12-702F) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific #10438026) and 100 units/mL of penicillin and 100 ug/mL of streptomycin (Thermo Fisher #15140). Cell lines were authenticated by short tandem repeat DNA profiling (LabCorp) and tested negative for mycoplasma using the MycoAlert PLUS Detection Kit (Lonza #LT07-710) and positive control (Lonza #LT07-518) (Supplementary Table 1). Rasless MEF cells expressing different Ras alleles were generated by and obtained from the National Cancer Institute (NCI) Ras Initiative (https://www.cancer.gov/research/key-initiatives/ras/ras-central/blog/2017/rasless-mefs-drug-screens), and were grown in DMEM (Lonza #12-604F) supplement with 10% fetal bovine serum (Thermo Fisher Scientific #10438026) and 100 units/mL of penicillin and 100 ug/mL of streptomycin (Thermo Fisher #15140). Puromycin (Thermo Fisher Scientific #A1113803) was added in the culture media to a final concentration of 2.5 μg/ml to maintain the stable expression of wild-type HRAS in Rasless MEF cells. Blasticidin (Thermo Fisher Scientific #A1113903) was added in the culture media to a final concentration of 4 μg/ml to maintain the stable expression of all the other wild-type or mutant Ras. All the cells were cultured at 37 °C in a humidified 5% CO2 incubator. MRTX1133 (ChemGood #C-1420) and Trametinib (Selleckchem #S2673) were dissolved in DMSO at the stock concentration of 10 mM.
Cell viability assay.
Cell viability assay was performed using CellTiter-Glo One reagent (Promega #G8462) following the manufacturer’s instructions. Briefly, human cancer cells (2000 cells per well) or Rasless MEF cells (3000 cells per well) were plated and cultured in black-walled clear bottom 96-well tissue culture-treated plates (Corning #3603) for 24 hours. Next day, equal volume of media containing 2X MRTX1133 or trametinib was added into wells such that the final concentrations of the drug reached the desired concentration in culture. Cells were treated for 3 days, and CellTiter-Glo assay was performed at the end point. Luminescence signal was detected on a TECAN Infinite M200 plate reader (Tecan Trading AG, Switzerland), and cell viability was calculated by normalizing the luminescence signal to non-treated conditions. Dose-response curve was fitted and plotted using GraphPad Prism 9.0 (GraphPad Software, LLC).
Immunoblotting and antibodies.
To collect whole cell extracts for immunoblotting, cells were kept on ice and washed with ice-cold DPBS (Corning #21-031-CV) twice then lysed in RIPA lysis buffer (50 mM Tris-HCl pH7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% sodium deoxycholate, 2 mM Na3VO4, 20 mM sodium pyrophosphate, 20 mM NaF, 20 mM β-glycerophosphate) containing additional protease inhibitors (Sigma-Aldrich #11836170001) and phosphatase inhibitors (Sigma-Aldrich # 4906837001). Total protein concentration was determined by BCA assay (Thermo Fisher Scientific #23225). Cell lysate was mixed with Laemmli Sample Buffer (Bio-Rad #1610747) and denatured at 95°C for 10 minutes. Samples were separated by SDS-PAGE in an 8–16% gradient Tris/Glycine gel (Bio-Rad #5671105) then transferred onto a nitrocellulose membrane by using the Trans-Blot Turbo RTA transfer kit (Bio-Rad #1704271). To detect protein abundance, membranes were first blocked in 5% blocking milk (Bio-Rad #1706404) in TBS (Quality Biological #351-086-131) containing 0.1% Tween-20 (Sigma #P1379) for 30 minutes, then hybridized with primary antibodies (Supplementary Table 2) following manufacturers’ instructions. After primary antibody hybridization, membranes were washed five minutes in TBS containing 0.1% Tween-20 for three times then hybridized with horseradish peroxidase (HRP)-conjugated secondary antibodies (Supplementary Table 2) in TBS containing 0.1% Tween-20 for one hour. After secondary antibody hybridization, membranes were washed 10 minutes in TBS containing 0.1% Tween-20 for three times. HRP signal was detected by using the Immobilon Forte Western HRP Substrate solution (Sigma #WBLUF0100) and acquired by using a ChemiDoc Touch Imaging System (Bio-Rad Laboratories, Inc). Chemiluminescence signal was quantified using the Image Lab Software (Bio-Rad Laboratories, Inc).
Construction of mutant Ras expression vectors.
Plasmids harboring WT and G12D mutant KRAS, HRAS, and NRAS cDNA were obtained from the NCI Ras Initiative. Ras double mutants with point-mutations in the 95th and 96th amino acid residues were generated by site-directed mutagenesis (Agilent Technologies #200521) using the primers listed in Supplementary Table 3 per manufacturer’s recommended PCR conditions. The coding region of the Ras cDNA and point mutations were verified by Sanger Sequencing. To construct N-terminal HA-tagged Ras-expressing plasmids, the coding region of Ras cDNA was first PCR-amplified from the plasmids above by using CloneAmp HiFi PCR Premix (Takara #639298) and the primers listed in Supplementary Table 4 using manufacturer’s recommended PCR conditions. The PCR products were purified by gel purification (Qiagen #28706) then digested with the restriction enzymes NotI (New England Biolabs #R3189) and BamHI (New England Biolabs #R3136). Digested PCR fragments were gel-purified (Qiagen #28104) and ligated into the pLVX-Puro vector (Takara #632164) between the PspOMI and BamHI restriction sites by using T4 DNA ligase (New England Biolabs #M0202). Ligated plasmids were transformed into NEB Stable Competent E. coli (New England Biolabs #C3040) following manufacturer’s instructions, and single colonies were isolated and grown for plasmid preparation (Qiagen #28115). The entire coding region of the HA-Ras cDNA were verified by Sanger Sequencing.
Lentivirus production and generation of HA-Ras-expressing stable cells.
To produce lentivirus, 293FT cells were co-transfected with 1 μg of the pLVX-Puro vectors carrying the cDNAs expressing HA-tagged Ras variants, 0.5 μg of pMD2.G (Addgene #12259), 0.75 μg of psPAX2 (Addgene #12260) in 6-well plates by using TransIT-293 transfection reagent (Mirus #MIR2705) following manufacturer’s instructions. Sixty hours after transfection, virus-containing media was collected, aliquoted, and frozen at −80°C. To generate cell lines stably expressing HA-Ras variants, cells were plated and grown overnight. Culture medium was replaced with lentivirus-containing medium at the ratio of 100 μL virus-containing medium per 25,000 cells seeded in the presence of 6 μg/mL polybrene (Thermo Fisher Scientific #TR1003G), followed by centrifugation at 2,000 rpm in room temperature for 30 minutes. Cells were cultured and incubated with the lentiviral media at 37°C in a humidified 5% CO2 incubator for 24 hours, and then cultured in normal culture medium for another 24 hours. Cells stably expressing the transduced genes were selected with 5 μg/ml puromycin for 3 days and subsequently maintained in medium containing 2.5 μg/ml puromycin. HA-Ras expression was verified by immunoblotting.
G-LISA and RBD pull down assays.
To determine the inhibitory effect of MRTX1133 on the level of active Ras proteins in Rasless MEFs expressing a single Ras allele, a colorimetric-based Ras G-LISA activation assay was used (Cytoskeleton #BK131). Briefly, cells were treated with MRTX1133 or DMSO control for 3 days, and cell lysates were collected on ice, aliquoted, and frozen in liquid nitrogen. Total protein concentration in the lysates were determined by using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific #23225). Briefly, 25 μg of totally lysates were subjected to G-LISA assay in duplicated wells following the manufacturer’s instructions, and another 15 ug of total lysates were subjected to immunoblotting to determine Ras expression level as the input. Colorimetric signal in each well was quantified using a TECAN plate reader, and a background signal from wells containing no lysate was subtracted to yield a specific signal. If the signal of a sample well was below that of the back background signal, the specific signal of that well was treated as being zero (i.e. below detection limit). Relative level of active Ras protein was calculated by normalizing against DMSO-treated samples and adjusted by the input level from the corresponding immunoblot. To determine the inhibitory effect of MRTX1133 on HA-tagged Ras with double mutations, a RBD pull-down assay was used (Cytoskeleton #BK008). Briefly, KRASG12D Rasless MEFs stably expressing HA-Ras double mutants was treated with MRTX1133 or DMSO for 24 hours. Cell lysate was collected on ice, aliquoted, and frozen in liquid nitrogen. Total protein concentration in the lysate was determined with BCA assay, and 300 μg of totally lysate was subjected to RBD pull-down assay following the manufacturer’s instruction. The level of active Ras protein in the pull down was analyzed by immunoblotting using Ras antibody (Sigma-Aldrich #OP40). Total was lysate was also subjected to immunoblotting to determine Ras expression level as the input.
Data Availability.
The data generated by the listed authors in this study are available upon request from the corresponding authors.
RESULTS
We first tested the activity of MRTX1133 in a panel of human cancer cell lines harboring different KRAS mutations. Consistent with the initial reports(7,8), two KRASG12D mutant cancer cell lines, Panc10.05 and AsPC-1, exhibited exquisite sensitivity to MRTX1133 (Figure 1A). MRTX1133 at concentration up to 1 μM showed no toxicity in a BRAF V600E mutant cell line, although non-specific toxicity was apparent at 10 μM (Figure 1B). Cancer cell lines with other KRAS mutant alleles, including H358 (KRASG12C) and SW620 (KRASG12V), also showed partial sensitivity to MRTX1133 (Figure 1C). In sensitive cell lines, MRTX1133 caused a corresponding decrease in the level of phosphor-ERK (pERK) (Figure 1D). Thus, MRTX1133 has inhibitory activities, albeit less potent, towards non-G12D KRAS mutants. We next tested an NRASG12D mutant multiple myeloma cell line, INA-6, that was previous shown to be dependent on the NRAS oncogene by CRISPR KO(10) (Figure S1). We were surprised that it was insensitive to MRTX1133 despite being highly sensitive to the MEK inhibitor trametinib (Figure 1E&F). This indicates that MRTX1133 may have strong paralog selectivity towards KRAS.
Figure 1. Selectivity of MRTX1133 in Ras mutant human cancer cell lines.
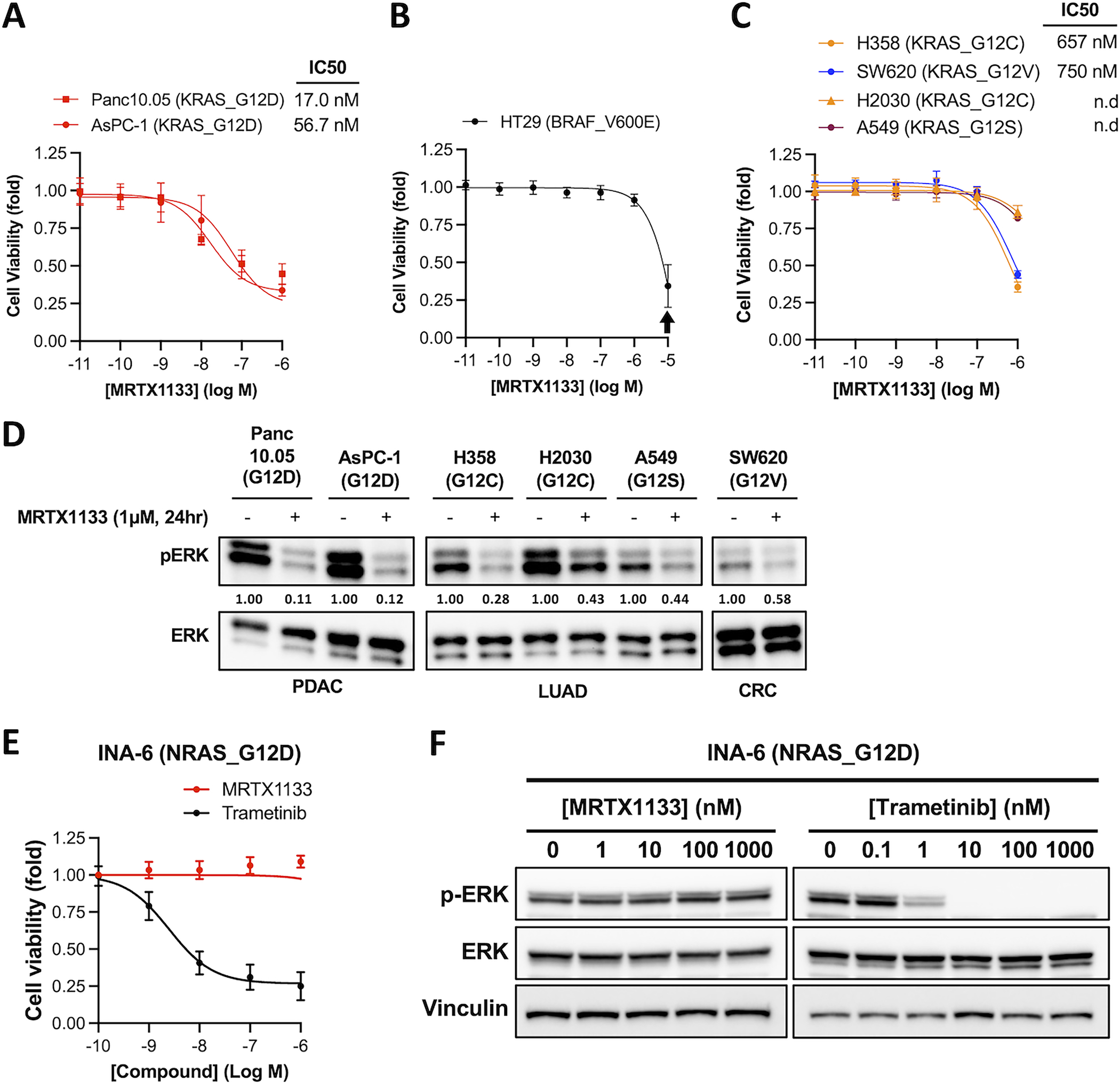
A-C. Human cancer cell lines harboring KRAS or BRAF mutations were treated with MRTX1133 for 3 days. Cell viability was determined using the CellTiter-Glo assay. IC50 values was estimated from curve fitting using three independent repeats (error bars represent S.D. of 3 independent experiments; n.d., not determined). Arrow indicates concentration of MRTX1133 (10 μM) that causes non-specific toxicity.
D. Human cancer cell lines harboring KRAS or BRAF mutations were treated with 1 μM MRTX1133 (+) or DMSO (−) for one day. Cell lysates were harvested and immunoblotted for pERK and total ERK. Changes in the level of pERK relative to untreated sample was quantified and shown under the pERK blots (a representative of two independent experiments is shown).
E. INA-6 cells were treated with MRTX1133 or Trametinib for 3 days. Cell viability assay was performed analogously to that in panel A (error bars represent S.D. of 3 independent experiments).
F. INA-6 cells were treated with different concentrations of MRTX1133 or trametinib for one day. Cell lysates were harvested and immunoblotted for pERK and total ERK (a representative of two independent experiments is shown).
The sensitivity of human cancer cell lines towards MRTX1133 could be confounded by other co-existing mutations in the cell and by the variable expression levels of wildtype (WT) KRAS, HRAS and NRAS alleles that could transmit Ras signaling(11). We therefore examined the activity of MRTX1133 in a panel of isogenic “Rasless” mouse embryonic fibroblasts (MEFs) that express only a single Ras allele. Rasless MEFs were previously generated by deleting all three Ras genes, and this led to growth arrest(12). The NCI Ras Initiative stably re-expressed WT and mutant Ras cDNAs in Rasless MEFs, and this rescued their proliferation(13). Thus, in the re-constituted Rasless MEFs, proliferation is strictly dependent on the sole Ras allele being expressed. This isogenic system is uniquely useful for evaluating the selectivity of a Ras inhibitor.
Consistent with human cancer cell lines, KRASG12D Rasless MEFs were highly sensitive to MRTX1133 with an IC50 of 70 nM (Figure 2A). As a control for its specificity, we found that MRTX1133 had no impact on the viability of BRAF V600E Rasless MEFs at concentration up to 2 μM, and non-specific toxicity was only evident at 10 μM (Figure 2A). KRASG12C, KRASG12V, and KRASG13D Rasless MEFs exhibited partial sensitivity to MRTX1133, whereas KRASG12R, KRASQ61L and KRASQ61R mutants were insensitive (Figure 2B). Concordantly, MRTX1133 significantly decreased pERK level in sensitive cell lines but had minimal impact on pERK level in insensitive cell lines (Figure 2C&D).
Figure 2. The selectivity of MRTX1133 in Rasless MEFs expressing different Ras alleles.
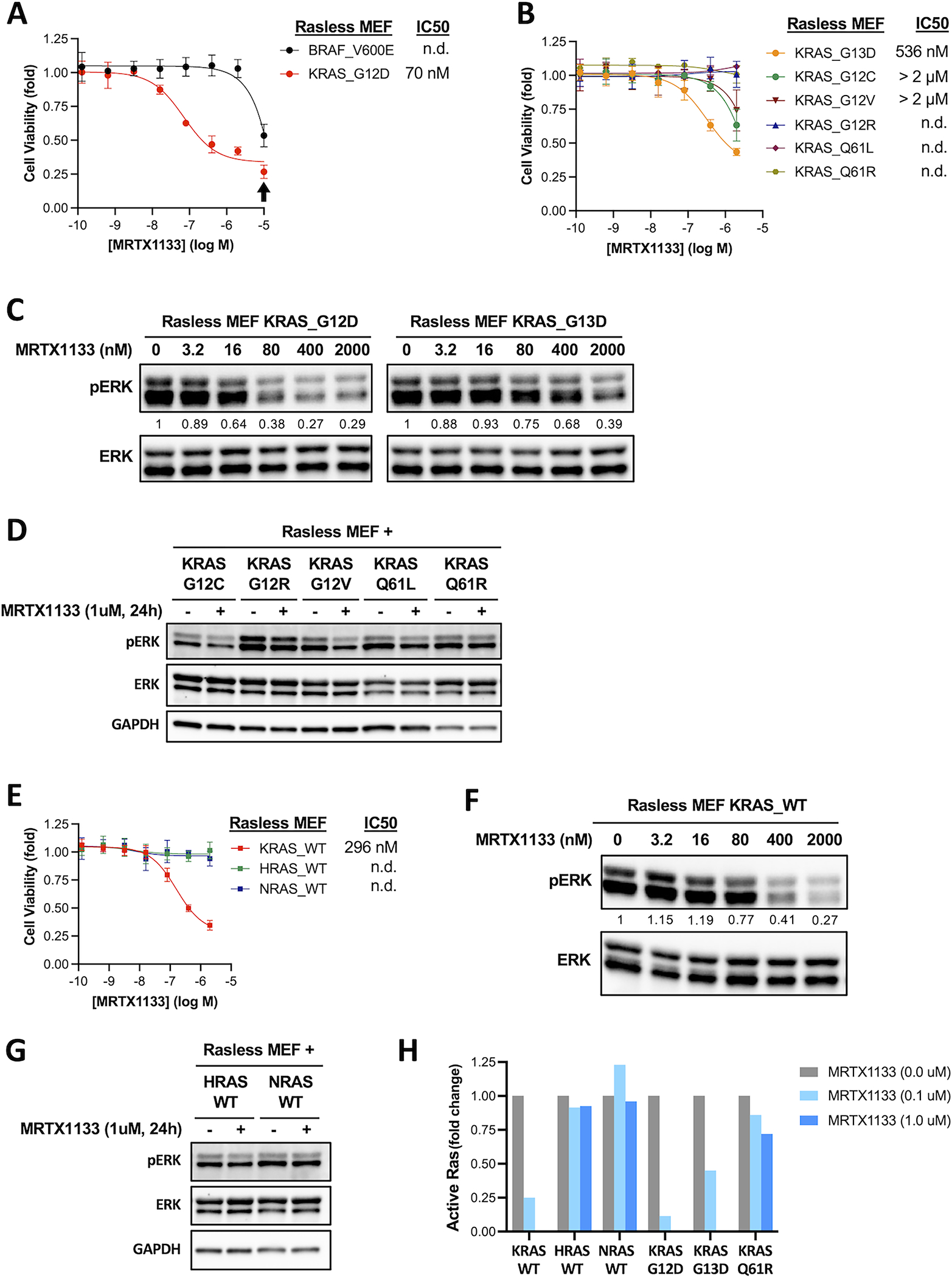
A-B. Isogenic Rasless MEFs expressing human KRAS hotspot mutations or BRAF V600E were treated with MRTX1133 for 3 days. Cell viability was determined using the CellTiter-Glo assay. IC50 values was estimated from curve fitting from three independent repeats (error bars represent S.D.; n.d., not determined). Arrow indicates concentration of MRTX1133 (10 μM) that causes non-specific toxicity.
C. Rasless MEFs expressing human KRASG12D (left panels) or KRASG13D (right panels) were treated with different concentrations of MRTX1133 for one day. Cell lysates were harvested and immunoblotted for phosphorylated ERK (pERK) and total ERK. Changes in the level of pERK relative to untreated sample was quantified and shown under the pERK blots (a representative experiment from two independent repeats is shown).
D. Rasless MEFs expressing G12C, G12R, G12V, Q61L, or Q61R mutant of human KRAS were treated with 1 μM MRTX1133 (+) or DMSO (−) for one day. Cell lysates were harvested and immunoblotted for pERK, total ERK, and GAPDH (a representative experiment from two independent repeats is shown).
E. Cell viability dose-response curves of isogenic Rasless MEFs expressing WT human KRAS, HRAS or NRAS protein. Experiments were performed analogously to those in panel A. IC50 values was estimated from curve fitting from three independent repeats (error bars represent S.D.; n.d., not determined).
F. Rasless MEFs expressing WT human KRAS were treated with different concentrations of MRTX1133 for one day and the level of pERK was analyzed by immunoblotting (a representative experiment from two independent repeats is shown).
G. Rasless MEFs expressing WT human HRAS or NRAS protein were treated with 1 μM MRTX1133 (+) or DMSO (−) for one day and the level of pERK was analyzed by immunoblotting (a representative experiment from two independent repeats is shown).
H. Rasless MEFs expressing different wild-type or mutant Ras alleles were treated with MRTX1133 for one day. Cell lysates were harvested, and the level of active Ras protein was measured by G-LISA assay. Background-corrected signals were normalized against non-treated controls within each cell line. Data represent average of two independent repeats (the level of active KRAS protein in KRASG12D, KRASG13D and KRASWT Rasless MEFs treated with 1 μM MRTX1133 was below detection limit).
Next, we tested the sensitivity of KRASWT, HRASWT or NRASWT Rasless MEFs. Unlike KRASWT human cancer cell lines that showed insensitivity to MRTX1133(8), KRASWT Rasless MEFs were sensitive to MRTX1133, with an IC50 of 296 nM that was approximately 4-fold higher than that of KRASG12D Rasless MEFs (Figure 2E). Thus, the mutant D12 residue is important, but not essential, for MRTX1133 binding. In contrast, HRASWT and NRASWT Rasless MEFs were completely insensitive to MRTX1133 (Figure 2E). Concordantly, MRTX1133 caused a dose-dependent decrease in pERK level in KRASWT Rasless MEFs but not in HRASWT and NRASWT Rasless MEFs (Figure 2F&G). To directly evaluate the impact of MRTX1133 on Ras activity, we measured the level of active Ras in cells with a Ras-binding domain (RBD) binding assay. We found that MRTX1133 was most effective at reducing the level of active KRASG12D and KRASWT protein in the cells, it also reduced the level of active KRASG13D protein to a lesser extent. In contrast, MRTX1133 had little impact on the level of active KRASQ61R and that of HRASWT and NRASWT protein (Figure 2H). These results support the notion that the activity of MRTX1133 is not confined to KRASG12D, and it can inhibit KRASWT as well as other KRAS mutants including G13D and G12C, albeit at lower potency.
The striking difference in the selectivity of MRTX1133 for KRAS over HRAS and NRAS prompted us to investigate the underlying mechanism. Prior structural studies on MRTX1133 showed that the compound makes multiple contacts throughout the G-domain of KRAS(8). All amino acid residues in KRASG12D that made direct interaction with MRTX1133 are conserved among the three Ras paralogs except H95 (Figure S2A). The imidazole side chain of H95 on KRAS makes at least three important contributions to drug binding: it forms a direct hydrogen bond with the MRTX1133 pyrimidine ring; it forms a tripartite hydrogen bonding interaction with both the MRTX1133 pyrimidine ring and the Y64 phenol hydroxyl group; and it forms a T-shaped interaction with the Y96 phenol ring to stabilize the hydrogen bonding between Y96 and MRTX1133 (Figure S2B). In HRAS and NRAS, the corresponding residues are Q95 and L95, respectively (Figure S2A). Thus, we hypothesized that the non-conserved H95 on KRAS provides a critical selectivity handle for MRTX1133, and substitution of H95 with Q and L, as seen in HRAS and NRAS, respectively, could disrupt MRTX1133 binding.
To test this hypothesis, we generated reciprocal mutations in G12D mutant of KRAS, HRAS and NRAS where residue 95 was “swapped” among these paralogs. We stably expressed Ha-tagged cDNAs of these double mutants in the parental KRASG12D Rasless MEFs so we could directly compare their sensitivity against KRASG12D single mutant in the same cells (Figure S2C). Expression of the HA-KRASG12D/H95Q and the HA-KRASG12D/H95L double mutants rendered cells fully insensitive to MRTX1133 (Figure 3A). Using RBD binding assay to directly measure the level of active Ras protein, we found that MRTX1133 was ineffective at reducing the level of active KRAS double mutants (Figure 3B). Consequently, expression of these double mutants significantly attenuated the downregulation of pERK by MRTX1133 (Figure 3C). Consistent with the notion that MRTX1133 is unable to inhibit HRAS and NRAS, expression of HA-HRASG12D and HA-NRASG12D single mutant rendered cells insensitive to MRTX1133. In contrast, cells expressing the HA-HRASG12D/Q95H and the HA-NRASG12D/L95H double mutants remain fully sensitive to MRTX1133 (Figure 3D). Concordantly, the level of active HA-HRASG12D and HA-NRASG12D single mutants remained unchanged with MRTX1133 treatment, whereas the level of active HA-HRASG12D/Q95H and the HA-NRASG12D/L95H double mutant was effectively reduced by MRTX1133 (Figure 3E). This differential sensitivity was also reflected by corresponding changes in pERK level in the cell (Figure 3F).
Figure 3. Histidine 95 on KRAS determines the paralog selectivity of MRTX1133.
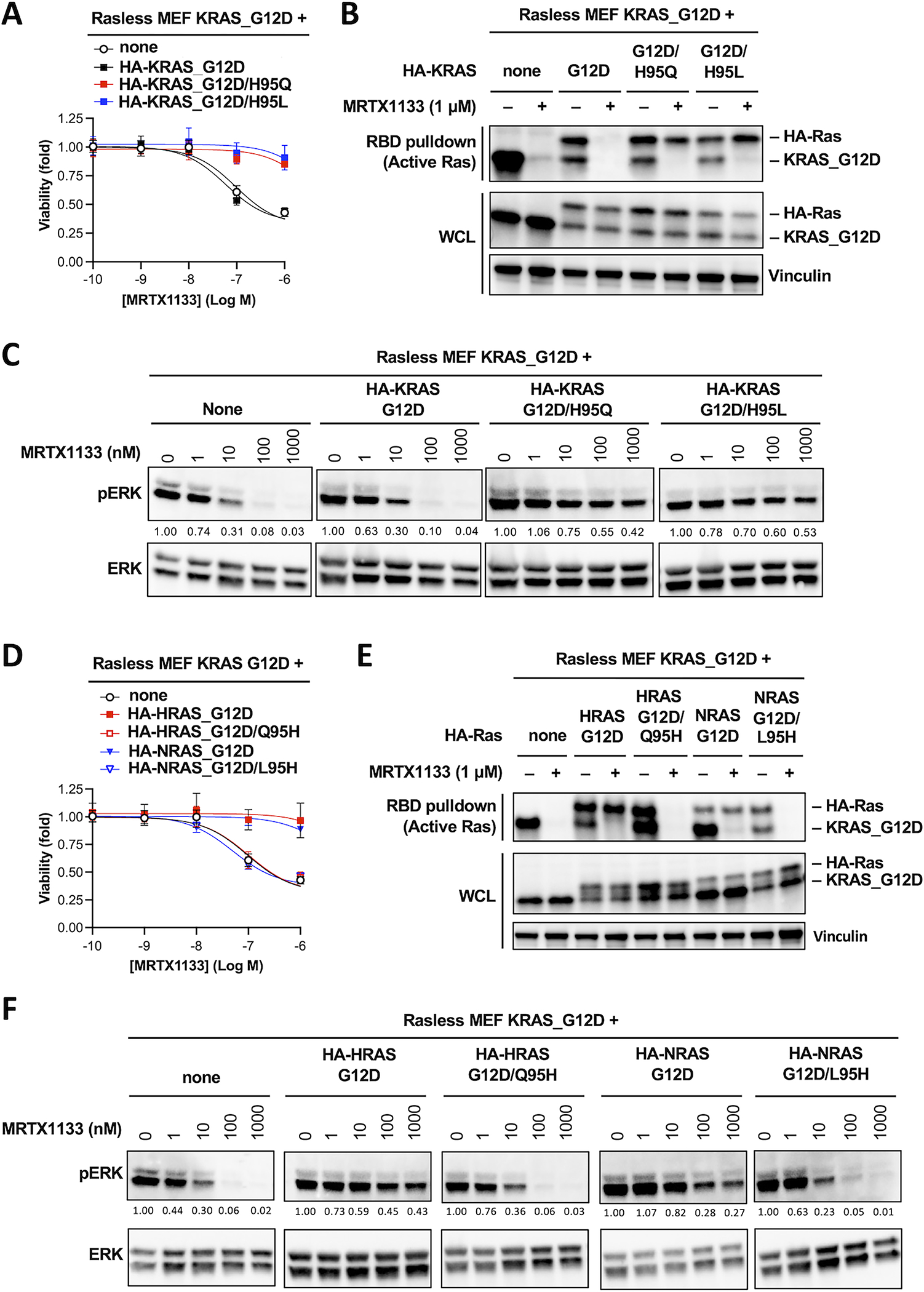
A. KRASG12D Rasless MEFs were stably transduced with cDNAs expressing HA-tagged KRASG12D, KRASG12D/H95Q or KRASG12D/H95L. Resulting cells were treated with MRTX1133 for 3 days. Cell viability was determined using the CellTiter-Glo assay. Data shown as average of three independent repeats (Error bars represent S.D. Data in Figure 3A, 3D and 4A are from the same series of experiments, and they share the same “none” and “HA-KRAS_G12D” controls. Data from these two controls are therefore duplicated in these panels).
B. Cell lines used in panel A were treated with 1 μM MRTX1133 (+) or DMSO (−) for one day. Whole cell lysates were harvested and the level of active KRAS was measured using RBD pulldown assay. Same whole cell lysates were immunoblotted to determine the input levels (WCL). Untagged KRASG12D and HA-tagged KRAS were detected with a pan-Ras antibody (a representative experiment from two independent repeats is shown).
C. Cell lines used in panel A were treated with MRTX1133 for one day. Cell lysates were immunoblotted for pERK and total ERK. Changes in the level of pERK relative to untreated sample was quantified and shown under the pERK blot (a representative experiment from two independent repeats is shown. Data in Figure 3C and 4C are from the same series of experiments, and they share the same “none” and “HA-KRAS_G12D” controls. Blots from these two controls are therefore duplicated in these panels).
D-F. Analogous experiments as in panels A-C were performed in KRASG12D Rasless MEFs transduced with cDNAs expressing HA-tagged HRASG12D, HRASG12D/Q95H, NRASG12D, or NRASG12D/L95H mutants (Data in Figure 3A, 3D and 4A are from the same series of experiments, and they share the same “none” and “HA-KRAS_G12D” controls. Data from these two controls are therefore duplicated in these panels).
In patients treated with the KRASG12C inhibitor adagrasib, acquired resistance can arise via secondary mutation at H95 and Y96 because these residues form hydrogen bonds with the covalently bound ligand(14),(15,16) (Figure S2D). We therefore tested whether secondary mutation at Y96 could confer resistance to MRTX1133. Expression of a HA-KRASG12D/Y96D double mutant rendered KRASG12D Rasless MEFs fully resistant to MRTX1133 (Figure 4A). MRTX1133 was unable to reduce the level of active HA-KRASG12D/Y96D mutant (Figure 4B), and the level of pERK in cells expressing the double mutant was resistant to MRTX1133 (Figure 4C). To further corroborate these findings, we tested the effect of HA-tagged KRASG12D/H95Q, KRASG12D/H95L and KRASG12D/Y96D double mutants in three human pancreatic cancer cell lines Panc10.05, KP4 and SW1990. These cell lines harbor endogenous KRASG12D mutation and are sensitive to MRTX1133. Expression of these KRAS double mutants conferred functional resistance to MRTX1133 and preserved pERK level to various degrees in the presence of MRTX1133 (Figure 4D&E, Figure S3). In conclusion, our results indicate that the non-conserved H95 residue in KRAS presents an essential selectivity handle for MRTX1133 to effectively inhibit KRAS while sparing HRAS and NRAS protein.
Figure 4. Mutation in H95 and Y96 on KRAS as a potential mechanism for acquired resistance to MRTX1133.
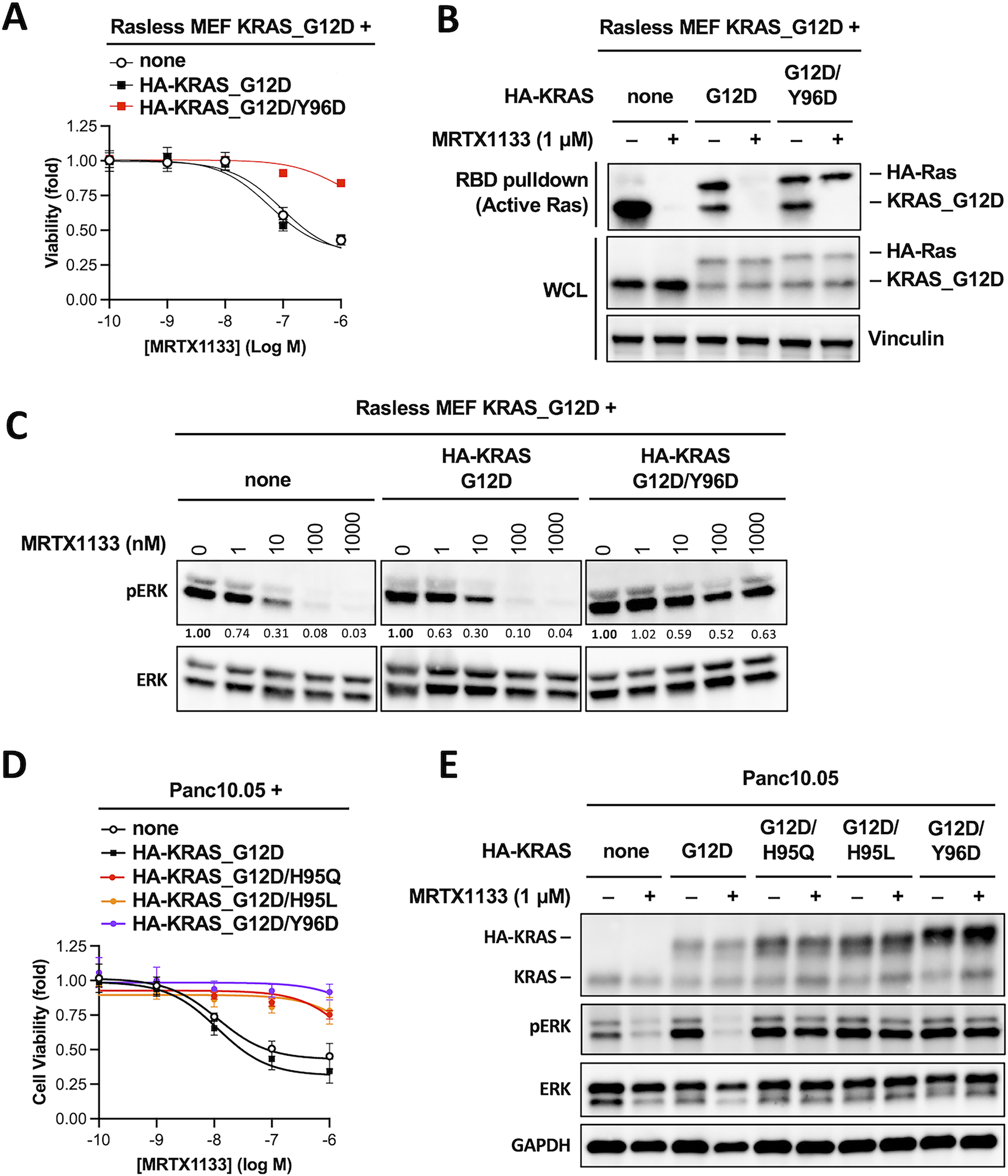
A. KRASG12D Rasless MEFs were stably transduced with HA-tagged KRASG12D or KRASG12D/Y96D. Cells were treated with MRTX1133 for 3 days and cell viability was determined using CellTiter-Glo assay. Dose-response curves were fitted from three independent repeats (Error bars represent S.D. (Data in Figure 3A, 3D and 4A are from the same series of experiments, and they share the same “none” and “HA-KRAS_G12D” controls. Data from these two controls are therefore duplicated in these panels).
B. Cells used in panel A were treated with 1 μM MRTX1133 (+) or DMSO (−) for one day. Whole cell lysates were harvested and subjected to RBD pulldown assay to determine active Ras levels. The same whole cell lysates were also subjected to immunoblotting to determine the input levels (WCL). Shown is a representative of two independent experiments.
C. Cells used in panel A were treated with different concentrations of MRTX1133 for one day. Cell lysates were harvested and immunoblotted for pERK and total ERK. Changes in the level of pERK relative to untreated sample was quantified and shown under the pERK blots (a representative of two independent experiments is shown. Data in Figure 3C and 4C are from the same series of experiments, and they share the same “none” and “HA-KRAS_G12D” controls. Blots from these two controls are therefore duplicated in these panels).
D. Human pancreatic cancer cell line Panc10.05 were stably transduced with HA-tagged KRAS mutants as indicated. Cells were treated with MRTX1133 for 3 days and cell viability was determined using CellTiter-Glo assay. Dose-response curves were fitted from three independent repeats (error bars represent S.D.).
E, Cells used in panel D were treated with or without 1 μM MRTX1133 (+) or DMSO (−) for one day. Cell lysates were harvested and immunoblotted for pERK and total ERK (a representative of two independent experiments is shown).
DISCUSSION
In conclusion, our results indicate that the non-conserved H95 residue in KRAS presents an essential selectivity handle for MRTX1133 to effectively inhibit KRAS while sparing HRAS and NRAS protein. Amino acid diversity at residue 95 can be exploited for the development of pan-KRAS inhibitors as well as HRAS and NRAS paralog-selective inhibitors. Additional implications of our finding are discussed in the Supplementary Discussion section.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
The non-conserved H95 residue on KRAS is required for the selectivity of the KRASG12D inhibitor MRTX1133 and can be exploited for the development of pan-KRAS inhibitors.
ACKNOWLEDGEMENTS
We would like to thank the National Cancer Institute Ras Initiative for generously providing the panel of Rasless MEF cell lines and human Ras cDNA plasmids for cloning, the CCR Genomics Core facility for sequencing support, and Dr. John S Schneekloth at the NCI for helpful discussion.
FUNDING
This research was supported by the Intramural Research Program (ZIABC011732) of the National Cancer Institute, NIH, to JL.
Footnotes
Conflict of interest disclosure statement: The authors declare no conflict of interest.
REFERENCES
- 1.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013;503:548–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575:217–23 [DOI] [PubMed] [Google Scholar]
- 3.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 2020;10:54–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med 2021;384:2371–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janne PA, Riely GJ, Gadgeel SM, Heist RS, Ou SI, Pacheco JM, et al. Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRAS(G12C) Mutation. N Engl J Med 2022;387:120–31 [DOI] [PubMed] [Google Scholar]
- 6.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 2011;11:761–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J Med Chem 2021 [DOI] [PubMed] [Google Scholar]
- 8.Hallin J, Bowcut V, Calinisan A, Briere DM, Hargis L, Engstrom LD, et al. Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat Med 2022;28:2171–82 [DOI] [PubMed] [Google Scholar]
- 9.Kemp SB, Cheng N, Markosyan N, Sor R, Kim IK, Hallin J, et al. Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov 2023;13:298–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y, Bolomsky A, Oellerich T, Chen P, Ceribelli M, Haupl B, et al. Oncogenic RAS commandeers amino acid sensing machinery to aberrantly activate mTORC1 in multiple myeloma. Nat Commun 2022;13:5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Young A, Lou D, McCormick F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov 2013;3:112–23 [DOI] [PubMed] [Google Scholar]
- 12.Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, et al. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J 2010;29:1091–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.A Panel of Isogenic RAS-Dependent Cell Lines Developed at the Frederick National Laboratory. <https://www.cancer.gov/research/key-initiatives/ras/ras-central/blog/2017/rasless-mefs-drug-screens>.
- 14.Fell JB, Fischer JP, Baer BR, Blake JF, Bouhana K, Briere DM, et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRAS(G12C) Inhibitor for the Treatment of Cancer. J Med Chem 2020;63:6679–93 [DOI] [PubMed] [Google Scholar]
- 15.Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, et al. Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N Engl J Med 2021;384:2382–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated by the listed authors in this study are available upon request from the corresponding authors.