

Abstract
Purpose:
Clinical evidence indicates that treatment with estrogens elicits anti-cancer effects in ~30% of patients with advanced endocrine-resistant estrogen receptor alpha (ER)-positive breast cancer. Despite the proven efficacy of estrogen therapy, its mechanism of action is unclear and this treatment remains under-utilized. Mechanistic understanding may offer strategies to enhance therapeutic efficacy.
Experimental Design:
We performed genome-wide CRISPR/Cas9 screening and transcriptomic profiling in long-term estrogen-deprived (LTED) ER+ breast cancer cells to identify pathways required for therapeutic response to the estrogen 17β-estradiol (E2). We validated findings in cell lines, patient-derived xenografts (PDXs), and patient samples, and developed a novel combination treatment through testing in cell lines and PDX models.
Results:
Cells treated with E2 exhibited replication-dependent markers of DNA damage and the DNA damage response prior to apoptosis. Such DNA damage was partially driven by the formation of DNA:RNA hybrids (R-loops). Pharmacological suppression of the DNA damage response via poly(ADP-ribose) polymerase (PARP) inhibition with olaparib enhanced E2-induced DNA damage. PARP inhibition synergized with E2 to suppress growth and prevent tumor recurrence in BRCA1/2-mutant and BRCA1/2-wild-type cell line and PDX models.
Conclusions:
E2-induced ER activity drives DNA damage and growth inhibition in endocrine-resistant breast cancer cells. Inhibition of the DNA damage response using drugs such as PARP inhibitors can enhance therapeutic response to E2. These findings warrant clinical exploration of the combination of E2 with DNA damage response inhibitors in advanced ER+ breast cancer, and suggest that PARP inhibitors may synergize with therapeutics that exacerbate transcriptional stress.
Keywords: breast cancer, endocrine resistance, estrogen, PARP inhibitor, DNA damage
Introduction
The majority of breast tumors express estrogen receptor α (ER), which typically reflects a degree of dependence upon estrogens for tumor growth. ER+/HER2− breast cancer is commonly treated with anti-estrogens that antagonize ER (e.g., tamoxifen, fulvestrant) or aromatase inhibitors that suppress estrogen biosynthesis. Although such endocrine therapies have improved outcomes for patients overall, endocrine resistance remains a clinical problem: approximately 20-30% of early-stage ER+ breast cancer patients experience disease recurrence. Despite the acquisition of resistance to anti-estrogens, loss of ER expression is rare in recurrent ER+ breast cancer and occurs in <10% of cases (1). Within the past 15 years, advances have been made in developing tumor-targeted therapies for endocrine-resistant disease (e.g., inhibitors of mTOR, CDK4/6, and PI3K). Since recurrent ER+ breast tumors remain at least partially dependent upon ER activity (2), approved tumor-targeted therapies are often administered in combination with an endocrine agent. Although these therapies have increased progression-free survival in patients, metastatic disease is typically fatal, and there remains a pressing need for new treatment options.
Decades of clinical evidence have demonstrated therapeutic efficacy of estrogen treatments in a subset of breast cancer patients (3–6). In the setting of endocrine-resistant advanced/metastatic ER+ disease, estrogens elicit anti-cancer effects in ~30% of patients, translating into thousands of patients worldwide who could benefit from these treatments (7–11). Despite robust clinical evidence of efficacy, estrogen treatments remain under-utilized due in part to their unknown and seemingly paradoxical anti-cancer mechanism(s) of action. We previously demonstrated that therapeutic response to the natural estrogen 17β-estradiol (E2) requires ER, and hyperactivation of ER transcriptional activity through high levels of receptor expression and acute stimulation with ligand elicits anti-cancer effects (12). Herein, we demonstrate that estrogen therapy-induced apoptosis is dependent upon cell cycle progression. E2 induces ER-dependent DNA damage requiring R-loop formation and S-phase DNA replication, which can be exploited therapeutically through poly (ADP-ribose) polymerase (PARP) inhibition to enhance efficacy.
Materials and Methods
Cell culture
HCC-1428 cells were obtained from American Type Culture Collection (ATCC) and cultured in DMEM with 10% FBS (HyClone Laboratories). T47D/pInd20-ESR1 cells (12) were maintained in DMEM + 10% FBS. HCC-1428/LTED cells were generated through long-term (~1 yr) culture in hormone-depleted conditions (HD) via phenol red-free DMEM with 10% dextran/charcoal-treated FBS (DCC-FBS; Hyclone Laboratories) as described (13). Cells were stably transfected with lentiviral vectors encoding luciferase, FLAG-tagged ER (FLAG-ESR1), doxycycline (dox)-inducible ER (pInducer20-ESR1), dox-inducible shRNA targeting ER (ESR1), or non-targeting shControl (12). Cells stably overexpressing ESR1 cells were maintained in HD medium as described (12). Cells were transiently transfected with plasmids encoding RNase H1 or vector control. Cell lines were confirmed to be mycoplasma-free (Universal Mycoplasma Detection Kit; ATCC) and authenticated by STR genotyping at the University of Vermont Cancer Center DNA Analysis Facility.
Human Tissue Specimens
Tumor tissue specimens obtained from 2 patients were molecularly analyzed herein. Those patients were enrolled in a clinical study (14) approved by the Dartmouth Health Human Research Protection Program, registered on clinicaltrials.gov (NCT02188745), and conducted in accordance with ICH Good Clinical Practice. The study was monitored by the Dartmouth Cancer Center Data, Safety Monitoring, and Accrual Committee. Written informed consent was obtained from participants prior to performing study-related procedures. All procedures performed involving human participants were in accordance with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Patients underwent clinically indicated baseline (pre-treatment) tumor biopsies, and research biopsies of the same tumor lesions on Day 14 of treatment with E2 (2 mg taken orally T.I.D.). Tissue specimens were formalin-fixed and paraffin-embedded.
Immunohistochemistry
Five-micron sections of FFPE tissue were mounted on slides, deparaffinized in xylene, and rehydrated in a graded ethanol series. For IHC, antigen retrieval was performed in citrate buffer, pH 6 (VWR), and sections were permeabilized with 0.2% Triton X-100 in PBS. Sections were blocked in 5% goat serum in PBS for 30 min, then incubated overnight in blocking solution with primary antibody at 4°C. Primary antibodies included: phospho-histone H2AXS139 (Cell Signaling Technology #9718); S9.6 (Millipore Sigma #MABE1095), cleaved caspase 3 (Cell Signaling Technology #9664); phospho-ATMS1981 (Abcam #81292); phospho-Chk2T68 (Cell Signaling Technology #2917); progesterone receptor A/B (Cell Signaling Technology #3153). Sections were washed and treated with 0.3% hydrogen peroxide, and signal was developed using the VectaStain Elite ABC-HRP kit with DAB substrate (Vector Laboratories). Sections were counterstained with hematoxylin. IHC staining of PDX tumors was quantified in 3 representative microscopic fields/tumor at 200x magnification using Halo software (Indica Labs).
Immunofluorescence
Tumors:
Slide-mounted 5-micron sections of FFPE tissue were processed as above through the permeabilization step, and blocked in 1% BSA in PBS for 1 h. Sections were incubated with primary antibody in blocking solution overnight at 4°C, washed 3 times, incubated with a fluorescently labeled secondary antibody for 1 h at room temp., washed 3 times, and mounted in ProLong Gold with DAPI (ThermoFisher Scientific). Sections were imaged at 100x magnification, and fluorescent intensity was quantified using CellProfiler software (RRID: SCR_007358).
Cell lines:
Cells were grown on glass coverslips, treated as indicated in figure legends, and fixed in 1.85% formaldehyde. Cells were permeabilized with 0.1% Triton X-100 in PBS, and blocked in 1% BSA in PBS for 1 h. Cells were incubated with primary antibody in blocking solution overnight at 4°C, wash 3 times, and incubated with a fluorescently labeled secondary antibody at 1 h at room temp. Coverslips were mounted in ProLong Gold with DAPI (ThermoFisher Scientific). Cells were imaged at 400x magnification. Staining was quantified by manually counting γH2AX, 53BP1, and PCNA foci per nucleus, and fluorescent intensity was quantified in S9.6-immunostained cells using CellProfiler software (RRID: SCR_007358). Staining was quantified in ≥100 cells per treatment group.
Primary antibodies included:
53BP1 (Cell Signaling Technology #4937); phospho-histone H2AXS139 (Cell Signaling Technology #9718); S9.6 (Millipore Sigma #MABE1095); estrogen receptor alpha (Santa Cruz Biotechnology #sc-8002); PCNA (Cell Signaling Technology #8580). Secondary antibodies included: AlexaFluor 488 goat anti-rabbit IgG (Life Technologies #A11034); AlexaFluor 647 goat anti-mouse IgG Fab2 (Cell Signaling Technology #4410).
Immunoblotting
All chemicals were purchased from Sigma unless otherwise noted. Cells were lysed in RIPA buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40, 10% glycerol, 1 mM EDTA, 1 mM EGTA, 5 mM NaPPi, 50 mM NaF, 10 mM Na β-glycerophosphate) plus fresh HALT protease inhibitor cocktail (Pierce) and 1 mM Na3VO4 (New England Biolabs). Frozen tumor fragments were thawed, homogenized, and lysed in the same solution. Cell/tumor lysates were sonicated at 30% power for 15 s, centrifuged at 17,000 x g for 10 min at 4°C, and protein content in supernatant was quantified by BCA Assay (Pierce). Protein extracts were reduced and denatured in LDS sample buffer (GenScript) plus 1.25% β-mercaptoethanol. Proteins were separated by SDS-PAGE, transferred to nitrocellulose, and stained with Ponceau S to visually confirm protein loading and transfer. Blots were probed with primary antibodies against ER (Santa Cruz Biotechnology; cat.# sc-8002), vinculin (Cell Signaling Technology; cat.# 13901), PR (Cell Signaling Technology; cat.#8757), poly/mono-ADP ribose (Cell Signaling Technology; cat.#83732), Rb (Cell Signaling Technology; cat.#9309), phospho-RbS780 (Cell Signaling Technology; cat.#9307), and FLAG (Millipore Sigma; cat.# F3165). Signal was detected with DyLight conjugated secondary antibodies (Cell Signaling Technology) using the LI-COR Odyssey system (LI-COR Biosciences).
Mouse tumor growth studies
Studies were approved by the Dartmouth College IACUC. The WHIM16 tumor model was obtained from the Washington University HAMLET core (15). The CTG-3346 tumor model was developed from recurrent breast tumor tissue obtained by ultrasound-guided biopsy at Dartmouth-Hitchcock Medical Center: fresh tissue cores were placed into transport medium, stored at 4°C, and sent overnight to Champion Oncology. A tissue core was implanted s.c. into two female NOD-scid/IL2Rγ−/− (NSG) mice (without supplementation with exogenous E2). Tumor growth was monitored by palpation. After ~8 months, P1 tumors were harvested and cut into fragments that were serially transplanted into P2 ovariectomized (ovx) mice. P4 and P5 tumors were used for treatment studies herein. The CTG-3346 PDX model is available upon request.
Ovariectomized (ovx) NSG mice were obtained from the Dartmouth Cancer Center Mouse Modeling Shared Resource. Tumor fragments (~8 mm3) were implanted subcutaneously (s.c.) into ovx mice aged 4-6 wk. Tumor dimension were measured twice weekly with calipers, and tumor volume was calculated as: length x width2/2. Mice were randomized to treatment groups when tumor volume reached ~200 mm3. E2 was administered by s.c. beeswax pellet containing 1 mg E2 as described in ref. (16), which were replaced every 30 d. Olaparib (MedChemExpress) was dissolved in 10% (2-hydroxypropyl)-β-cyclodextrin (Sigma-Aldrich) in PBS. Olaparib (50 mg/kg/d) or vehicle was delivered by daily intraperitoneal (i.p.) injection. For molecular analysis, tumors were harvested at the indicated time points and either snap frozen, or formalin-fixed and paraffin-embedded.
CRISPR/Cas9 knockout screen
HCC-1428/LTED cells stably expressing Cas9-Blast were generated by lentiviral transduction of the lentiCas9-Blast plasmid (gift from Feng Zhang, Broad Institute; obtained from Addgene #52962). Two days following viral transduction, cells were selected with 5 μg/mL blasticidin for 3 wk, and Cas9 expression was confirmed by RT-qPCR. 120 million cells stably expressing Cas9 were lentivirally transduced with the human Brunello CRISPR knockout pooled library (gift from David Root and John Doench, Broad Institute; obtained from Addgene #73178) at an MOI of 0.3. Two days following viral transduction, cells were treated with 1 μg/mL puromycin for 7 d. Fourteen days following transduction, cells were trypsinized and split into 3 treatment groups at 35 million cells per group. Group A was immediately flash-frozen to represent baseline. Group B was treated with HD medium for 3 wk. Group C was treated with HD medium + 1 nM E2 for 3 wk. Cells in Groups B and C were then trypsinized and flash-frozen. Genomic DNA was collected from the 3 groups using the Blood DNA Maxi Kit (Qiagen). Samples were prepared by PCR amplification of the genomically integrated sgRNA sequences: 100 μg of genomic DNA per sample was split into 100 PCR reactions, using 30 PCR cycles, followed by pooling of PCR products. PCR products were purified with the QIAquick PCR Purification Kit (Qiagen). Amplicon size was confirmed by gel electrophoresis. A secondary PCR was performed with 100 ng per sample for 10 cycles to attach Illumina adapters. PCR products were purified by adding 0.8X and subsequently 2X KAPA Pure beads. Libraries were sequenced using an Illumina NextSeq 500 (2 x 75 bp) on High Output setting. The 5’ ends of reads were trimmed to 5’-CACCG-3’ using Cutadapt (17). MAGeCK (v.0.5.9) was used to extract read counts for each sgRNA using the count function (18). The mle function was used to compare read counts from cells treated ± E2 after controlling for baseline counts and library size. The output included beta scores. A differential beta score was calculated by taking the difference in beta scores between the E2-treated and HD conditions. To determine significant pathway enrichment, gene set enrichment analysis for Hallmarks pathways was performed using the Molecular Signatures Database (19).
RNA-seq analysis
HCC-1428 and HCC-1428/LTED cells were treated with HD medium for 3 d, and then treated ± 1 nM E2 x 24 h in triplicate in 100-mm dishes. RNA was extracted using RNeasy Plus Mini Kit (Qiagen catalog # 1062832). RNA quality was assessed on a fragment analyzer (Advanced Analytical Technologies, Agilent), and RNA was quantified by Qubit. In preparation for RNA sequencing (RNA-seq), ribo-depleted libraries were prepared from 2.5 μg of total RNA using the Globin-Zero Gold (Illumina catalog # GZG1206) and TruSeq Stranded Total RNA (Illumina catalog # RS-122-2201) workflows according to manufacturer’s instructions. Each library was uniquely barcoded, quantified by qPCR (Kapa Biosystems catalog # KK4824), and pooled for sequencing on an Illumina NextSeq 500 (2 × 75-bp). Reads were checked for quality using fastqc (RRID:SCR_014583) and if necessary were trimmed using Trimmomatic (RRID:SCR_011848) to trim regions with phred Q > 30 (RRID:SCR_001017). High-quality reads were then aligned to reference genome hg19 using STAR (20). Gene counts were normalized by frequency per kilobase million (20). Differential expression of genes was determined using the limma (RRID:SCR_010943) and DESeq2 (RRID:SCR_015687) packages in the R environment (RRID: SCR_001905), and multiple testing correction was performed using the FDR Benjamini–Hochberg method.
Principal components analysis (PCA) was used to visualize variation between samples based on expression of the top 2,000 most variably expressed genes within each cell line. One replicate sample from control-treated HCC-1428/LTED cells (“SH50”) that was sequenced in a separate batch proved to be an outlier by PCA and hierarchical clustering; this sample was excluded from further analysis.
To determine significant gene expression pathway enrichment between time points, we conducted single-sample gene set enrichment analysis (ssGSEA) (21) for Hallmarks pathways in GenePattern (22) using default arguments. Gene set enrichment scores were normalized based on numbers of genes within each Hallmarks gene set. Normalized enrichment scores (NES) were compared between treatment groups by Bonferroni multiple comparison-adjusted posthoc test. RNA-seq data were deposited at NCBI Sequence Read Archive (SRA) under accession # PRJNA816494.
Growth assays
Parental cells were HD for 3 d prior to seeding. Cells were seeded in triplicate at 12,000 cells/well in 12-well plates, then treated as indicated for 4 wk. Cells were fixed and stained with 0.5% crystal violet in 20% methanol for 10 min. Excess dye was rinsed out with water. Plates were scanned, and area fraction of staining in each well was determined using ImageJ (RRID:SCR_003070).
Statistical analysis
Cell growth data and IHC scores were analyzed by t-test (for 2-group experiments) or ANOVA followed by Bonferroni multiple comparison-adjusted post hoc testing between groups (for experiments with ≥3 groups). Pairwise comparisons of immunofluorescence scores used the Cramer-von Mises nonparametric test. Cell culture experiments were repeated on 3 independent occasions, and the data met the assumptions of all tests. Tumor growth data were analyzed using the following linear mixed model : Log10(tumor volumeit) = ai + b * t + eit, where i represents the ith mouse, t represents time of tumor volume measurement, ai represents the mouse-specific log tumor volume at t = 0, b represents the rate of tumor volume growth, and eit represents deviation of measurements from the model over time (23,24). Mouse heterogeneity (the baseline tumor volume) is represented by variance of ai, and b*loge(10) * 100 indicates tumor volume increase (%) per week. Treatment groups were compared using a z-test for slopes with standard error derived from the output of the function lme from the library nlme in R. Synergy was determined as described in ref. (25).
Mouse tumor growth data were analyzed using the following linear mixed model: Log10(tumor volumeit) = ai + b * t + eit, where i represents the ith mouse, t represents time of tumor volume measurement, ai represents the mouse-specific log tumor volume at t = 0, b represents the rate of tumor volume growth, and eit represents deviation of measurements from the model over time (26,27). Mouse heterogeneity (the baseline tumor volume) is represented by variance of ai, and b * loge[10]* 100 indicates tumor volume increase (%) per week. Treatment groups were compared using a z-test for slopes with standard error derived from the output of the function lme from the library nlme in R. Synergy was determined as described in ref. (25). Comparison of tumor recurrence curves was performed by log-rank test.
Data availability
Data generated in this study are available upon request from the corresponding authors. RNA sequencing data are available on NCBI SRA (# PRJNA816494).
Results
Functional genomic screening suggests roles for cell cycle progression and DNA damage response in anti-cancer effects of 17β-estradiol
Parental ER+/HER2- HCC-1428 breast cancer cells are dependent upon E2 for growth (13). In contrast, their long-term estrogen-deprived (LTED) derivatives exhibit acquired resistance to hormone deprivation (HD) and growth is inhibited by re-treatment with 1 nM E2 [Fig. 1A and ref. (12)]. This concentration of E2 is within the pre-menopausal range in humans, and can be achieved pharmacologically in patients treated with E2 therapy (7).
Figure 1. 17β-estradiol induces DNA damage in long-term estrogen-deprived cells that therapeutically respond to E2.
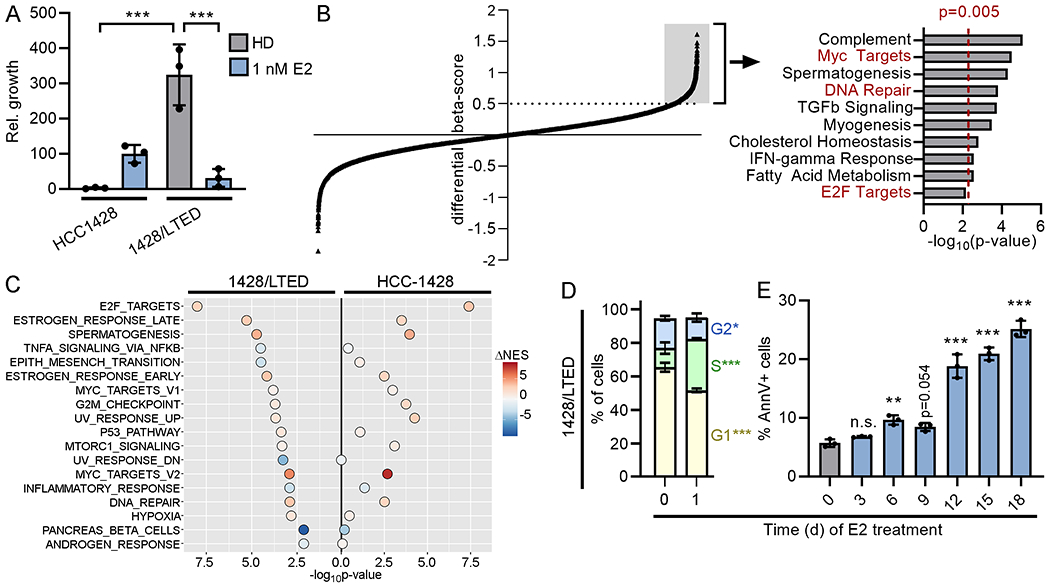
(A) Parental cells were HD for 3 d prior to seeding. Cells were treated as indicated for 4 wk and relative growth was measured. (B) 1428/LTED cells stably expressing Cas9 were transduced with a sgRNA library. Cells were treated ± 1 nM E2 for 3 wk, and beta-scores were calculated (18). Each point represents one gene. Genes with a differential beta-score ≥0.5 (n=1194, gray box) were analyzed for enrichment with Hallmarks gene sets (right). Cell cycle and DNA repair pathways are highlighted in red font. (C) HCC-1428 (HD for 3 d) and 1428/LTED cells were treated ± 1 nM E2 for 24 h, and RNA was harvested for sequencing. Single-sample gene set enrichment analysis (ssGSEA) for Hallmarks pathways was performed for each replicate sample, and normalized gene set enrichment scores (NES) were compared between treatment groups. Gene sets significantly (p<0.05) altered in 1428/LTED cells by E2 treatment are shown, and p-values and differences in mean NES induced by E2 treatment are indicated for each cell line. (D) Cells treated ± 1 nM E2 x 24 h were fixed and stained with propidium iodide (PI). DNA content was measured by flow cytometry. Sub-G1 cells were excluded from plots. Proportions of cells in each phase were compared. (E) Cells were treated ± 1 nM E2 as indicated. Three days after last medium change, cells were harvested, stained with FITC-tagged Annexin V (AnnV), and analyzed by flow cytometry. In (A/D/E), data are shown as mean of triplicates ± SD. *p<0.05, **p<0.005, ***p<0.0005, n.s. = not significant compared to control unless otherwise indicated.
To identify candidate mechanisms underlying therapeutic response to E2, we performed a genome-wide CRISPR/Cas9 knockout screen using 1428/LTED cells. Beta-scores were calculated as a measure of gene essentiality in the presence and absence of E2. We then calculated the difference in beta-scores between E2-treated and HD conditions to identify genes that were required for the growth-inhibitory effect of E2 (i.e., genes that, when lost, rescued from E2; differential beta-score ≥0.5, Fig. 1B). Pathways analysis of this subset of genes revealed enrichment for pathways involved in DNA repair and cell cycle progression (i.e., E2F and Myc targets). Additionally, we observed that knockout of multiple CDKs (CDK4, CDK9, CDK7, CDK2) each rescued 1428/LTED cells from the growth inhibitory effects of E2 (Supplementary Table S1). Gene set enrichment analysis of transcriptomic profiles of 1428/LTED cells showed E2-induced engagement of pathways involved in cell cycle (E2F and Myc targets) and DNA damage response (G2/M checkpoint, p53 pathway, UV response, DNA repair), some of which were not detected in parental HCC-1428 cells (Fig. 1C and Supplementary Table S2). In addition, the magnitude of gene expression changes induced by E2 was often greater in 1428/LTED cells than HCC-1428 cells (Supplementary Figure S1). Consistent with these results, 1428/LTED cells exhibited an accumulation of cells in S-phase following 1 d of E2 treatment, which preceded the onset of apoptosis (Fig. 1D/E). Based on these collective data, we hypothesized that estrogen therapy induces DNA damage during replication.
17β-estradiol treatment induces ER- and cell cycle-dependent DNA damage
Cellular response to DNA damage was assessed in HCC-1428 and 1428/LTED cells by immunofluorescent staining for phospho-histone H2AXSer139 (γH2AX), a marker of cellular response to double-strand DNA breaks. 1428/LTED cells exhibited significant increases in γH2AX foci following both 1 and 24 h of E2 treatment, while parental HCC-1428 cells did not (Fig. 2A/B). As another marker of response to DNA breaks, 1428/LTED cells also exhibited an increase in p53 binding protein (53BP1) foci upon treatment with E2 (Supplementary Figure S2A). BrdU pulse labeling revealed that the majority of cells incurring E2-induced DNA damage (γH2AX+) were in S-phase (BrdU+), and blocking G1-to-S cell cycle progression through treatment with the CDK4/6-selective inhibitor abemaciclib prevented DNA damage induced by E2 (Fig. 2C/D and Supplementary Figure 3A).
Figure 2. 17β-estradiol-induced DNA damage is dependent upon overexpression of ER.
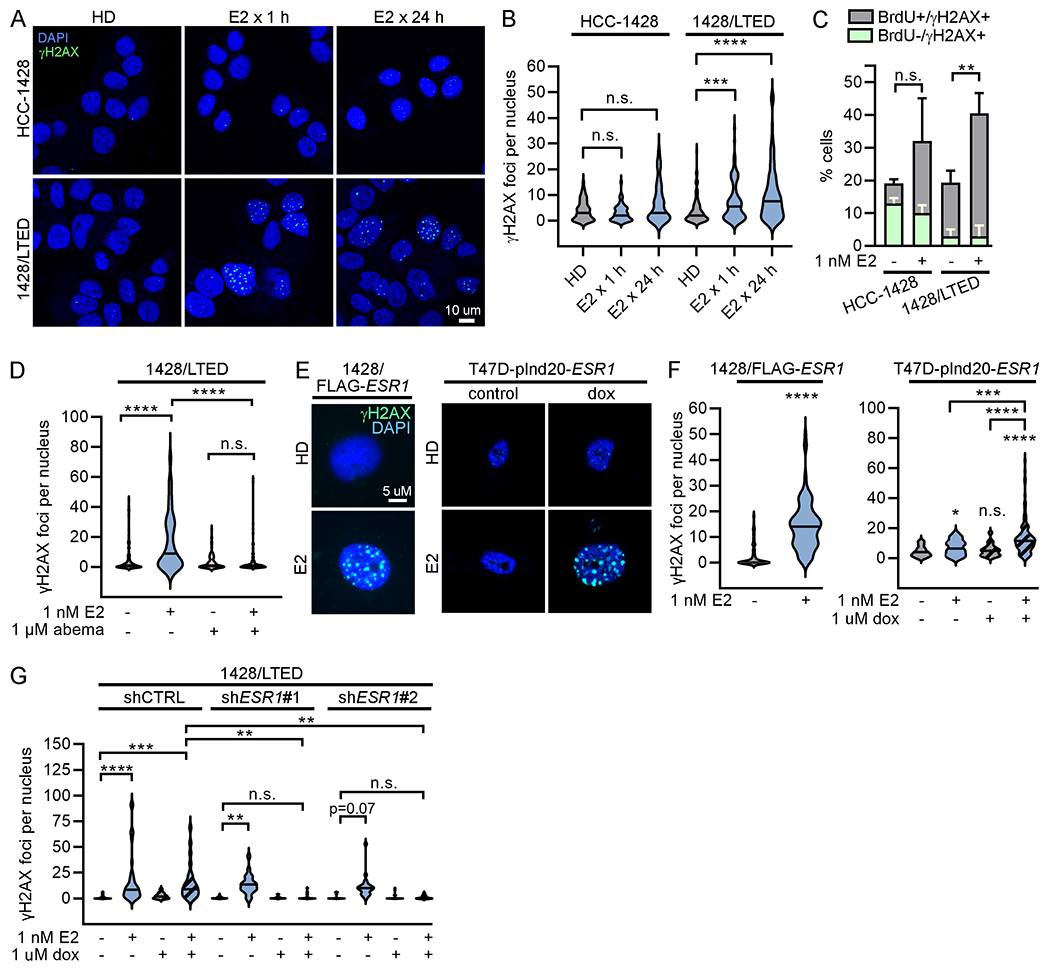
(A/B) HCC-1428 (HD x 3 d) and 1428/LTED cells were treated ± 1 nM E2. Cells were fixed and stained for γH2AX (green) and DAPI (blue). γH2AX foci were counted in ≥100 nuclei/group. (C) HCC-1428 (HD x 3 d) and 1428/LTED cells were treated ± E2 for 21 h, and labeled with BrdU for another 3 h. Cells were stained for γH2AX, BrdU, and cleaved PARP for flow cytometry analysis. Cleaved PARP-positive cells were excluded from analysis. Proportions of cells with DNA breaks (γH2AX-positive) that were or were not in S-phase (i.e., did or did not incorporate BrdU) were plotted. Proportions of γH2AX+/BrdU+ cells were statistically compared. Data are shown as mean of triplicates + SD. (D) Cells were treated ± E2 ± abemaciclib x 24 h and analyzed as in (B). (E/F) T47D/pInd20-ESR1 cells were pretreated with HD x 7 d, and then treated with HD ± dox x 14 d prior to seeding. All cells were then treated as indicated x 24 h and analyzed as in (B). (G) 1428/LTED cell lines expressing dox-inducible shRNA targeting ESR1 (two independent constructs) or non-silencing control were treated ± dox for 2 d, and then treated ± E2 x 24 h and analyzed as in (B). In (A/E), representative images are shown. *p<0.05, **p<0.005, ***p<0.0005, ****p<0.0001, n.s. = not significant.
1428/LTED cells express increased ER levels compared to parental cells, which we previously demonstrated drives therapeutic response to E2. Forced overexpression of ER (ESR1) in HCC-1428 or MDA-MB-415 ER+/HER2- breast cancer cells, and doxycycline (dox)-inducible overexpression of ER in T47D cells drive hormone-independent growth and convert E2 from a growth promoter to a growth suppressor [Supplementary Figure S4A/B and ref. (12)]. Similar to 1428/LTED cells, exogenous ER overexpression increased E2-induced DNA damage as measured by γH2AX and 53BP1 foci (Fig. 2E/F and Supplementary Figure S2B/C). Conversely, doxycycline-induced RNAi-mediated knock-down of ER blocked the ability of E2 to induce DNA damage in 1428/LTED cells (Fig. 2G and Supplementary Figure S4C). ER-negative BT-20 cells did not exhibit an increase in DNA damage following E2 treatment (Supplementary Figure S5A). These data show that ER drives estrogen-inducible DNA damage in ER+ breast cancer cells. Similar to 1428/LTED cells, E2 induced DNA damage in ER-overexpressing cells mainly during S-phase, and treatment with abemaciclib prevents DNA damage upon E2 treatment (Supplementary Figure S3). Although MCF-7/LTED cells are also growth-inhibited by E2 (28), a DNA damage response was not consistently observed (Supplementary Figure S5B), suggesting that this model responds to E2 via a different mechanism. Together, these results indicate that E2 treatment induces DNA damage during replication in ER-overexpressing models that are growth-inhibited by E2.
Estrogen therapy induces a DNA damage response in human tumors and PDX models
Paired baseline and on-treatment metastatic tumor biopsy specimens were obtained from 2 subjects in clinical trial NCT02188745, which evaluated the therapeutic effects of E2 in patients with endocrine-resistant breast cancer. Both tumors were ER+/PR+/HER2- by IHC (Supplementary Figure S6). Following 2 wk of treatment with E2 (2 mg p.o. TID), γH2AX staining significantly increased (Fig. 3A and Supplementary Figure S7). ER levels decreased during E2 treatment (Fig. 3A), consistent with ER protein turnover following estrogen-induced ER activation (29). These data suggest that estrogen therapy induces DNA damage in human ER+/HER2− breast tumors.
Figure 3. 17β-estradiol therapy induces DNA damage in endocrine-resistant tumors.
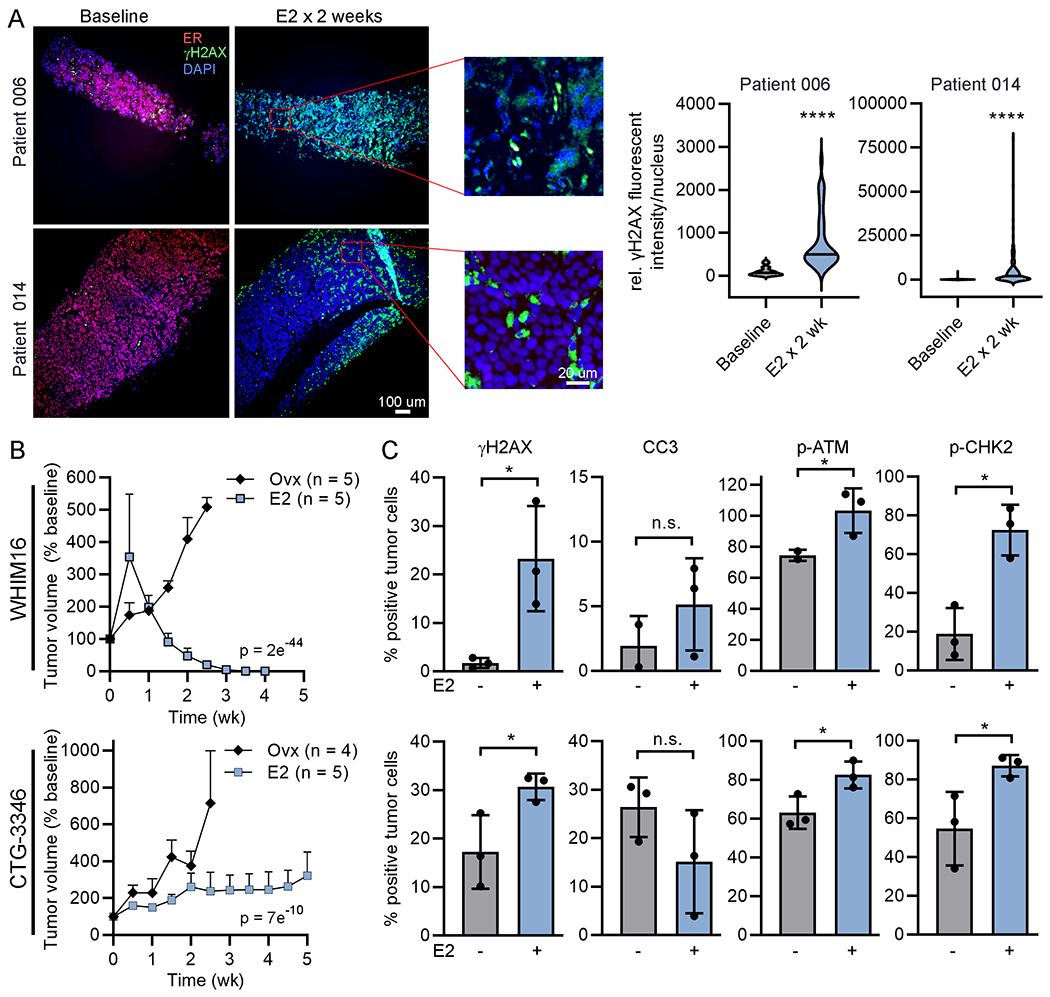
(A) Biopsy samples of advanced ER+ breast tumors were obtained from 2 patients before and 2 wk after treatment with E2. FFPE tumor sections were stained for ER (red), γH2AX (green), and DAPI (blue). γH2AX intensity was quantified in ≥100 nuclei/specimen. Representative exposure-matched image pairs are shown. (B) Ovx mice bearing tumors ~200 mm3 were randomized to treatment ± E2. Tumor volumes were serially measured. Data are shown as mean + SEM, and were analyzed by nonlinear mixed modeling. (C) Tumors (n=3/group) were harvested from ovx mice treated ± E2 for 24 h. FFPE sections were analyzed by IHC. Data are shown as mean ± SD. *p<0.05, ****p<0.0001, n.s. = not significant.
The ability of E2 to induce a DNA damage response was evaluated in 2 estrogen-independent PDX models that are growth-inhibited by E2. The ER+/PR+/HER2- WHIM16 PDX model grows in ovariectomized (ovx) mice, modeling resistance to aromatase inhibitor-induced estrogen deprivation in patients, and regresses upon treatment with E2 [Fig. 3B, Supplementary Figure S8, and refs. (12,15,28)]. CTG-3346 is a novel PDX model that grows in ovx mice and is growth-inhibited by treatment with E2 (Fig. 3B and Supplementary Figure S8). CTG-3346 was derived from a patient treated as described as in Supplementary Figure S9A, and this model retains the ER+/PR+/HER2- status of the patient’s recurrent tumor (Supplementary Figure S9B). CTG-3346 tumors exhibit mutation and copy number loss of RB1, and consequent loss of Rb protein as a major tumor suppressor and effector of CDK4/6 (Supplementary Figure S9C), which is consistent with the patient’s tumor resistance to the CDK4/6 inhibitor palbociclib [Supplementary Figure S9A and ref. (30)]. Despite prior treatment of the patient with multiple lines of endocrine therapies, CTG-3346 retains functional ER, as measured by an increase in mRNA expression of the ER target gene TFF1 upon treatment of mice with E2 (Supplementary Figure S9D).
E2 treatment for 24 h significantly increased γH2AX in both PDX models growing in ovx mice (Fig. 3C and Supplementary Figure S10). However, E2 did not alter cleaved caspase-3 positivity, indicating that the DNA damage reflected by γH2AX is not related to apoptosis. E2 treatment also significantly increased proportions of cells expressing phospho-ATMS1981 and phospho-CHK2T68, which are markers of an activated DNA damage response (Fig. 3C and Supplementary Figure S10).
Estrogen therapy induces transcriptional stress leading to DNA damage
Since E2 induced an increase in S-phase cells, E2-induced DNA damage was dependent upon G1-to-S progression (Figs. 1D and 2D, and Supplementary Figure S3), and LTED and ER-overexpressing cells exhibit E2-induced hyperactivation of ER transcriptional activity (12,28), we hypothesized that estrogen-induced DNA damage results from ER transcription-driven replication stress via R-loops. While E2 increased replication stress in 1428/LTED and ER-overexpressing cells as shown by proliferating cell nuclear antigen (PCNA) focus formation (31) (Supplementary Figure S11), cells stimulated to enter S-phase may be expected to undergo replication stress. We there evaluated the contribution of E2-induced transcription to DNA damage.
R-loops are 3-stranded DNA:RNA hybrid structures that form when nascent mRNA re-anneals to the template strand of DNA, impairing re-annealing of complementary DNA strands. While R-loops play roles in transcriptional regulation (32–34), these structures can induce genome instability and DNA breaks, potentially through collision with replication forks (35). Furthermore, ER transcriptional activation has been linked to R-loop formation in MCF-7 cells (36). Using the R-loop structure-specific S9.6 antibody, we observed E2-induced increases in nuclear R-loop formation in 1428/LTED cells that were significantly higher than those detected in parental HCC-1428 cells (Fig. 4A/B). Similarly, 1428/FLAG-ESR1 and T47D/pInd20-ESR1 cells showed E2-induced R-loop formation, which was significantly higher upon dox-induced ER overexpression in the T47D model (Supplementary Figure S12A/B). In addition, IHC analysis of PDX models showed increased R-loops after 24 h of E2 treatment in vivo (Fig. 4C). RNase H1 can degrade the RNA specifically within RNA-DNA hybrids. Transient ectopic expression of RNase H1 suppressed E2-induced R-loop formation and prevented DNA damage (Fig. 4D/E and Supplementary Figure S12C), supporting a causative role for R-loops in DNA damage incurred from E2.
Figure 4. 17β-estradiol-induced R-loop formation drives DNA damage.
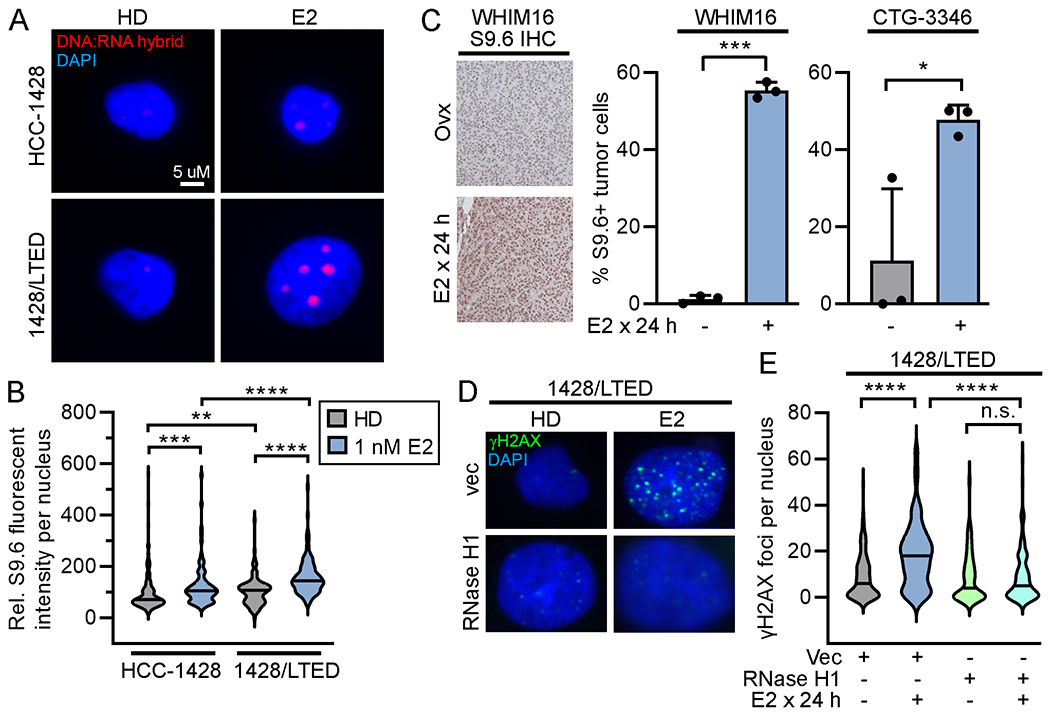
(A/B) Cells were treated ± 1 nM E2 x 24 h, fixed, and stained for DNA/RNA hybrids (S9.6 antibody) and with DAPI. Fluorescence intensity was quantified in ≥100 nuclei/group. (C) Tumors were harvested from ovx mice treated ± E2 (n=3/group). FFPE sections were stained for DNA/RNA hybrids. Proportions of positively staining nuclei were calculated. Data are shown as mean + SD. (D/E) Cells were transiently transfected with plasmids encoding RNase H1 or vector control. Two days later, cells were treated ± 1 nM E2 x 24 h, then fixed and stained for γH2AX and DAPI. γH2AX foci were counted in ≥100 nuclei/group. In (A/D), representative images are shown. *p<0.05, **p<0.005, ***p<0.0005, ****p<0.0001, n.s. = not significant.
PARP inhibition enhances 17β-estradiol/ER-induced DNA damage and synergistically inhibits growth
We next sought to determine whether inhibiting repair of DNA damage could enhance the therapeutic effects of E2. PARP1/2 are involved in DNA damage repair, and PARP1 play a role in the repair of R-loops and R-loop-associated DNA damage (37,38). We thus hypothesized that inhibition of PARP would enhance E2/ER-induced DNA damage. Treatment with the PARP1/2 inhibitor olaparib synergized with E2 to suppress the growth of 1428/LTED and dox-induced T47/pInd20-ESR1 cells (Fig. 5A). Accordingly, olaparib potentiated the DNA damage induced by E2 (Fig. 5B/C). These effects were observed in both the BRCA1/2-wild-type T47D model and the BRCA2-altered HCC-1428 models. Olaparib also enhanced R-loop formation when combined with E2 in 1428/LTED and 1428/FLAG-ESR1 cells (Supplementary Figure S13).
Figure 5. PARP inhibition synergizes with 17β-estradiol to induce DNA damage and inhibit growth in vitro.
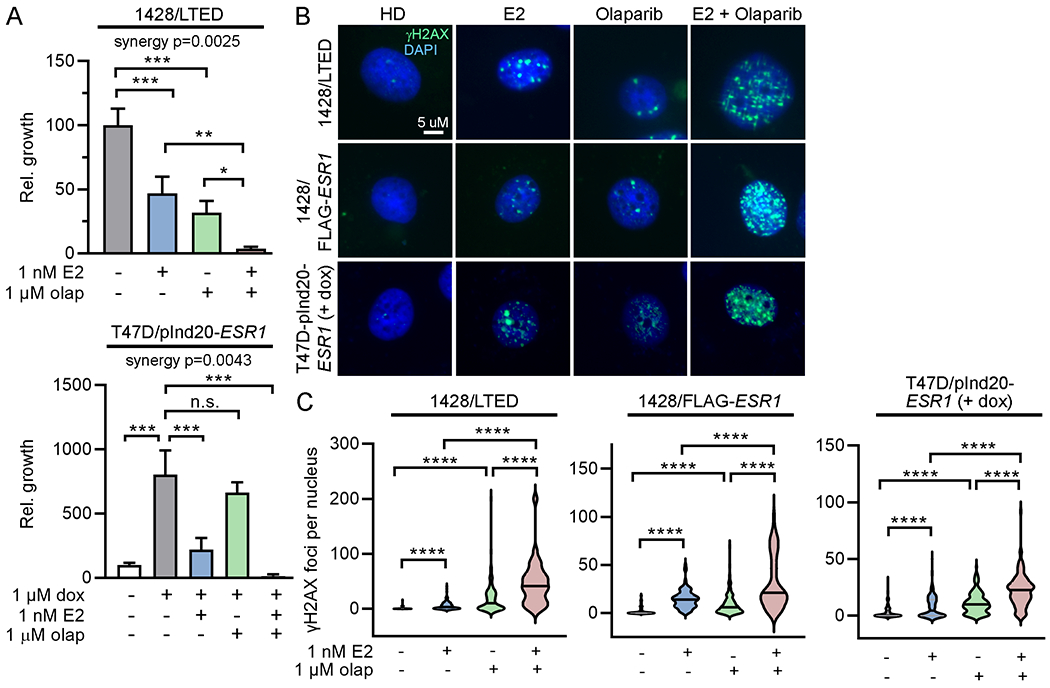
(A) T47D/pInd20-ESR1 cells were HD x 7 d, and pretreated with HD + dox x 14 d prior to seeding. Both lines were then treated ± olaparib for 2 d, followed by treatment ± olaparib ± E2 for 4 wk, and relative growth was measured. Data are shown as mean of triplicates + SD. (B/C) T47D/pInd20-ESR1 cells were pretreated as in (A) before seeding. All lines were then treated ± olaparib for 2 d, followed by treatment ± E2 ± olaparib for 24 h. Cells were fixed and stained for γH2AX and DAPI. Representative images are shown. γH2AX foci were counted in ≥100 nuclei/group. *p<0.05, **p<0.005, ***p<0.0005, ****p<0.0001, n.s. = not significant.
Currently, PARP inhibitors are FDA-approved for the treatment of breast cancer patients with germline genetic alterations in BRCA1 or BRCA2. BRCA1/2 encode proteins involved in homologous recombination repair of DNA (39,40) and are postulated to be synthetically lethal with PARP inhibition (41,42). In accordance with the BRCA1/2 mutation status of these cell lines, HCC-1428 derivatives responded to olaparib treatment as a monotherapy, while T47D derivatives did not (Fig. 5A). However, the synergistic effect of olaparib and E2 in both models indicates that this drug combination is effective regardless of BRCA1/2 status.
PARP inhibition synergizes with 17β-estradiol to inhibit to tumor growth and prevent recurrence
Ovx mice bearing WHIM16 or CTG-3346 tumors were randomized to treatment with vehicle, olaparib, E2, or the combination of E2 and olaparib. E2 was administered continuously, and olaparib was administered for 28 d (gray shading in Fig. 6A/B). Neither PDX model responded to olaparib monotherapy (Fig. 6A/B and Supplementary Figure S14). However, olaparib synergized with E2 to delay recurrence of WHIM16 tumors (Fig. 6A/C) and prevent growth of CTG-3346 tumors (Fig. 6B). Immunoblot analysis of lysates of tumors harvested from mice at 4 h after drug treatment on Day 2 (i.e., 28 h post-treatment initiation) confirmed drug target engagement: E2 induced ER transcriptional activity as evidenced by increased expression of progesterone receptor (PR), which is encoded by the ER-inducible gene PR, and decreased ER levels as described (12); olaparib decreased levels of ADP-ribosylation of proteins (Fig. 6D).
Figure 6. PARP inhibition synergizes with 17β-estradiol against tumors in vivo and prevents recurrence.
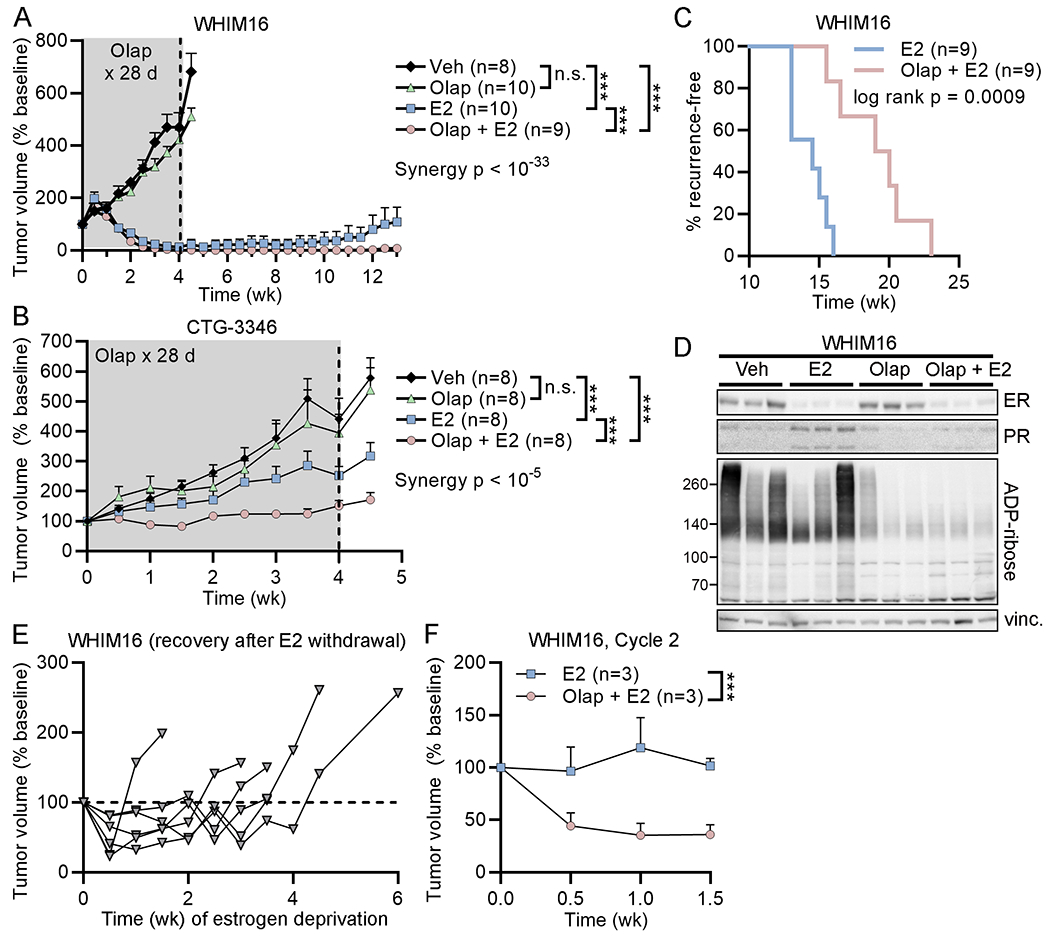
(A/B) Ovx mice bearing tumors ~200 mm3 were randomized to treatment as indicated. E2 was delivered continuously, and olaparib was administered daily for 28 d (gray shading). Data are shown as mean + SEM. (C) After 10 wk of treatment in (A), mice without palpable tumors were monitored for recurrence. Time to recurrence was calculated as time from treatment start until tumors re-grew to baseline volume. Proportions of mice that were recurrence-free over time are shown. (D) Ovx mice bearing WHIM16 tumors were treated as in (A), and tumors were harvested 4 h after drug treatment on Day 2. Tumor lysates were analyzed by immunoblot. vinc.- vinculin loading control. (E) Mice bearing tumors that recurred during E2 monotherapy in (C) were treated with estrogen deprivation starting on Day 0, which stunted tumor growth. Each line represents one mouse. Tumors eventually resumed growth, which was defined as two consecutive biweekly volume measurements above baseline. (F) Mice with tumors that resumed estrogen-independent growth in (D) were randomized to treatment with a second cycle of E2 ± olaparib. Data are shown as mean + SEM. ***p<0.0005, n.s. = not significant.
We previously demonstrated that WHIM16 tumors that acquire resistance to E2 treatment are sensitive to estrogen deprivation, and such tumors ultimately regain estrogen-independent growth and re-sensitization to E2 therapy (12). Following recurrence on E2 monotherapy, mice bearing WHIM16 tumors were estrogen-deprived until tumors resumed growth (Fig. 6E). These tumor-bearing mice were then randomized to a second round of treatment with E2 ± olaparib. The addition of olaparib significantly improved the anti-tumor effects of E2 (Fig. 6F and Supplementary Figure S15). These data collectively suggest that PARP inhibition enhanced the therapeutic effects of E2 by increasing R-loops and DNA damage, and that this synergistic effect can occur in the absence of BRCA1/2 alterations.
Discussion
Prior work supported a link between overexpression/amplification of ER, hyperactivation of ER-driven transcription, and therapeutic response to E2 (12,15,28,43). However, the molecular mechanism underlying these effects remained unclear. Herein, we demonstrate that E2 induces ER-dependent S-phase-specific DNA damage and R-loop accumulation that is exacerbated by ER overexpression and adaptation to growth in hormone-depleted conditions. While estrogens have been shown to induce DNA damage and genotoxic stress through several mechanisms, we present evidence of E2-induced DNA damage specifically associated with ER activation and a downstream growth-inhibitory response. This mechanism led us to develop a novel combination therapy with PARP inhibitors and E2, which synergize to enhance therapeutic response in models of endocrine-resistant ER+ breast cancer.
Estrogens can induce DNA damage through both ER-dependent and -independent mechanisms. Estrogens and metabolites have been shown to directly induce genotoxic stress in the absence of ER, including through the formation of DNA adducts (44–46). In our models, E2 induced DNA damage most prominently in LTED and ER-overexpressing models, suggesting an ER-dependent mechanism of DNA damage (Figs. 1B/F/G and 3C/D, and Supplementary Figs. S2 and S5). ER-initiated transcription can engage topoisomerase IIβ to induce transient double-stranded DNA breaks (47), which are postulated to relieve topological constraints to improve accessibility of DNA for transcription. E2-activated ER can also induce the formation of R-loops leading to DNA breaks at ER-responsive genes (36), but this effect had not previously been associated with a growth-inhibitory response. It is conceivable that the phenotypes resulting from estrogen-induced DNA damage depend upon doses of ligand and receptor: long-term exposure to relatively lower (endogenous) levels of estrogens may drive genomic instability and increase cancer risk (36,48); acute exposure to higher (pharmacological) levels of estrogens, particularly in the context of ER amplification/overexpression, elicits cellular toxicity [ref. (12) and data shown herein].
Similar to estrogen therapy for endocrine-resistant breast cancer, a subset of patients with castration-resistant prostate cancer benefit from treatment with androgens, and this effect may be dependent upon androgen receptor overexpression or amplification. Androgen treatment induces DNA damage, which is exacerbated in BRCA2-deficient cells (49,50). A recent clinical study showed that 16/36 (44%) patients with metastatic castration-resistant prostate cancer experienced a PSA50 response (i.e., a decline ≥50% from baseline in plasma prostate-specific antigen levels) upon treatment with bipolar androgen therapy (BAT; testosterone cypionate/enanthate administered every 28 d) in combination with olaparib regardless of tumor BRCA1/2 status (51). Our results do not indicate that BRCA1/2 mutations are a requirement for therapeutic response to E2; however, BRCA1/2 mutations or other defects in homologous recombination-mediated DNA repair may increase the likelihood that tumors therapeutically respond to estrogen by preventing repair of estrogen/ER-induced DNA damage. In this study, we leveraged combination treatment with a PARP inhibitor due to the roles of PARP1/2 in repair of R-loops and DNA damage. However, other inhibitors of the DNA damage response may also synergize with estrogen therapy; as new inhibitors enter clinic development (e.g., agents targeting WEE1, ATR, etc.), this concept will warrant further investigation.
This work raises several points surrounding clinical implementation of estrogen therapy. Firstly, these findings offer the possibility of expanding the use of PARP inhibitors to a larger patient population. Currently, PARP inhibitors are FDA-approved for breast cancer only in the setting of germline BRCA1/2 genetic alterations, which constitute ~5% of breast cancer cases (52). In our studies, olaparib synergized with E2 in both BRCA1/2-mutant and -wild-type models, and synergy was seen even in models that were non-responsive to olaparib monotherapy (Figs. 5A and 6A/B). A clinical indication for E2/PARP inhibitor combination therapy in advanced ER+/HER2- disease regardless of BRCA1/2 status would substantially increase the number of patients who could benefit from a repurposed PARP inhibitor.
We previously demonstrated that p53 knockdown by siRNA prevented apoptosis induction by E2 in MCF-7/LTED cells (28). Pathways analysis of transcriptomic profiles showed significant E2-induced enrichment for “P53_Pathway” in 1428/LTED cells but not parental HCC-1428 cells (Fig. 1C). However, we observed anti-cancer effects of E2 herein in both p53-wild-type (HCC-1428-derived) and p53-mutant (T47D-derived, WHIM16, CTG-3346) models. Furthermore, the patient who experienced the longest duration of clinical benefit from cycling treatment with E2 (8 wk) and an aromatase inhibitor (16 wk) in our recently completely POLLY clinical study had p53-mutant ER+ disease (14). These observations collectively suggest that p53 function is not required for therapeutic response to E2, but p53 may be engaged when present.
Another point raised by this work is the efficacy of E2 therapy in the landscape of CDK4/6 inhibitors. Treatment with CDK4/6 inhibitors has become a standard of care in advanced ER+ breast cancer, and acquired resistance to CDK4/6 inhibitors is an emerging clinical problem. Since CDK4/6 inhibitors are a relatively recent development in the field, strategies for the treatment of ER+ breast cancer following progression on CDK4/6 inhibitors remain underexplored. We developed the CTG-3346 PDX tumor from a patient with acquired resistance to the CDK4/6 inhibitor palbociclib. CTG-3346 exhibits loss/mutation of Rb (Supplementary Figure S9A/C), consistent with acquired resistance to CDK4/6 inhibition (30). The growth-inhibitory effects of E2 in this model (Fig. 3B) indicate that E2 therapy remains an effective treatment option in the post-CDK4/6 inhibitor setting.
Although E2 therapy may remain effective despite resistance to CDK4/6 inhibitors, this work cautions against combining E2 with CDK4/6 inhibitors. Our data indicate that cell cycle progression and DNA replication are required for E2-induced DNA damage, and CDK4/6 inhibition antagonized the DNA-damaging effects of E2 (Fig. 2D and Supplementary Figure S3C). A similar antagonistic effect has been reported with CDK4/6 inhibitors co-administered with cytotoxic chemotherapies that target S-phase and mitotic cells (53,54). However, the antagonistic effects of these drug combinations may be dependent upon the order and timing with which they are administered. Several studies have suggested that pre-treatment with cytotoxic chemotherapies followed by treatment with CDK4/6 inhibitors elicits synergistic effects. Such synergy may be due to the repressed transcription of DNA repair genes by CDK4/6 inhibitors, which could impede DNA repair following treatment with cytotoxic chemotherapy (55,56). If E2 is considered a DNA-damaging agent in endocrine-resistant ER+ breast cancer, then strategically timed CDK4/6 inhibition following E2 treatment may convert these seemingly antagonistic drugs to synergistic; this concept warrants further study.
Supplementary Material
Statement of Translational Relevance.
A subset of patients with endocrine-resistant estrogen receptor alpha (ER)-positive breast cancer benefit from treatment with estrogens. However, the molecular effects and anti-cancer mechanism of action of estrogen therapy are unclear, which has limited the clinical use of this seemingly paradoxical treatment. We show that therapeutic response to the estrogen 17β-estradiol is dependent upon re-engagement of ER, and that 17β-estradiol treatment induces ER-dependent DNA damage in cells adapted to growth without estrogens. Pharmacological inhibition of poly(ADP-ribose) polymerase (PARP) synergizes with 17β-estradiol to enhance DNA-damage and therapeutic response. Importantly, this synergistic effect was observed regardless of BRCA1/2 mutation status. These findings collectively offer 17β-estradiol and PARP inhibitor combination treatment as a novel therapeutic strategy for patients with advanced ER+ breast cancer. Moreover, these data indicate that PARP inhibitors may have applications beyond homologous recombination-deficient tumors.
Acknowledgements
This work was supported by Susan G. Komen (CCR1533084 to TWM) and NIH (R01CA200994, R01CA267691, R01CA262232, and R01CA211869 to TWM; F31CA243409 to NAT; the Center for Quantitative Biology at Dartmouth P20GM130454; Dartmouth College Cancer Center Support Grant P30CA023108; S10OD030242). We thank Alan Eastman, Chris Shoemaker, Scott Kaufmann, and Matthew Goetz for helpful discussions, and the following Dartmouth Cancer Center Shared Resources for their support: Mouse Modeling; Pathology (RRID: SCR_023479); Biostatistics; Bioinformatics; Microscopy; Genomics & Molecular Biology (RRID: SCR_021293).
Footnotes
Conflicts of Interest Disclosure Statement: PH and YM are employees of Champions Oncology. All other authors declare no conflicts of interest.
References Cited
- 1.Shiino S, Kinoshita T, Yoshida M, Jimbo K, Asaga S, Takayama S, et al. Prognostic Impact of Discordance in Hormone Receptor Status Between Primary and Recurrent Sites in Patients With Recurrent Breast Cancer. Clinical breast cancer 2016;16(4):e133–40. [DOI] [PubMed] [Google Scholar]
- 2.Osborne CK, Pippen J, Jones SE, Parker LM, Ellis M, Come S, et al. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: results of a North American trial. J Clin Oncol 2002;20(16):3386–95. [DOI] [PubMed] [Google Scholar]
- 3.Haddow A, Watkinson JM, Paterson E, Koller PC. Influence of Synthetic Oestrogens on Advanced Malignant Disease. British medical journal 1944;2(4368):393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ingle JN, Ahmann DL, Green SJ, Edmonson JH, Bisel HF, Kvols LK, et al. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. The New England journal of medicine 1981;304(1):16–21. [DOI] [PubMed] [Google Scholar]
- 5.Peethambaram PP, Ingle JN, Suman VJ, Hartmann LC, Loprinzi CL. Randomized trial of diethylstilbestrol vs. tamoxifen in postmenopausal women with metastatic breast cancer. An updated analysis. Breast Cancer Res Treat 1999;54(2):117–22. [DOI] [PubMed] [Google Scholar]
- 6.Kennedy BJ. Systemic Effects of Androgenic and Estrogenic Hormones in Advanced Breast Cancer. J Am Geriatr Soc 1965;13:230–5. [DOI] [PubMed] [Google Scholar]
- 7.Ellis MJ, Gao F, Dehdashti F, Jeffe DB, Marcom PK, Carey LA, et al. Lower-dose vs high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: a phase 2 randomized study. Jama 2009;302(7):774–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwase H, Yamamoto Y, Yamamoto-Ibusuki M, Murakami KI, Okumura Y, Tomita S, et al. Ethinylestradiol is beneficial for postmenopausal patients with heavily pre-treated metastatic breast cancer after prior aromatase inhibitor treatment: a prospective study. British journal of cancer 2013;109(6):1537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zucchini G, Armstrong AC, Wardley AM, Wilson G, Misra V, Seif M, et al. A phase II trial of low-dose estradiol in postmenopausal women with advanced breast cancer and acquired resistance to aromatase inhibition. European journal of cancer 2015;51(18):2725–31. [DOI] [PubMed] [Google Scholar]
- 10.Lonning PE, Taylor PD, Anker G, Iddon J, Wie L, Jorgensen LM, et al. High-dose estrogen treatment in postmenopausal breast cancer patients heavily exposed to endocrine therapy. Breast Cancer Res Treat 2001;67(2):111–6. [DOI] [PubMed] [Google Scholar]
- 11.Agrawal A, Robertson JF, Cheung KL. Efficacy and tolerability of high dose “ethinylestradiol” in post-menopausal advanced breast cancer patients heavily pre-treated with endocrine agents. World journal of surgical oncology 2006;4:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Traphagen NA, Hosford SR, Jiang A, Marotti JD, Brauer BL, Demidenko E, et al. High estrogen receptor alpha activation confers resistance to estrogen deprivation and is required for therapeutic response to estrogen in breast cancer. Oncogene 2021;40(19):3408–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller TW, Hennessy BT, Gonzalez-Angulo AM, Fox EM, Mills GB, Chen H, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest 2010;120(7):2406–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwartz GN, Kaufman PA, Giridhar KV, Marotti JD, Chamberlin MD, Arrick BA, et al. Alternating 17beta-estradiol and aromatase inhibitor therapies is efficacious in post-menopausal women with advanced endocrine-resistant ER+ breast cancer. Clin Cancer Res 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell reports 2013;4(6):1116–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeRose YS, Gligorich KM, Wang G, Georgelas A, Bowman P, Courdy SJ, et al. Patient-derived models of human breast cancer: protocols for in vitro and in vivo applications in tumor biology and translational medicine. Curr Protoc Pharmacol 2013;Chapter 14:Unit14 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 2011;17(1):3. [Google Scholar]
- 18.Li W, Koster J, Xu H, Chen CH, Xiao T, Liu JS, et al. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome biology 2015;16:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462(7269):108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nature genetics 2006;38(5):500–1. [DOI] [PubMed] [Google Scholar]
- 23.Demidenko E Mixed models : theory and applications with R. Hoboken, New Jersey: Wiley; 2013. xxvii, 717 pages p. [Google Scholar]
- 24.Demidenko E Advanced statistics with applications in R. Wiley series in probability and statistics. Hoboken, NJ: Wiley,; 2020. p 1 online resource. [Google Scholar]
- 25.Demidenko E, Miller TW. Statistical determination of synergy based on Bliss definition of drugs independence. Plos One 2019;14(11):e0224137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Demidenko E Mixed Models: Theory and Applications with R. Hoboken, NJ: Wiley; 2013. [Google Scholar]
- 27.Demidenko E Advanced Statistics with Applications in R. Wiley; (Hoboken, NJ: ). 2020. [Google Scholar]
- 28.Hosford SR, Shee K, Wells JD, Traphagen NA, Fields JL, Hampsch RA, et al. Estrogen therapy induces an unfolded protein response to drive cell death in ER+ breast cancer. Molecular oncology 2019;13(8):1778–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pakdel F, Le Goff P, Katzenellenbogen BS. An assessment of the role of domain F and PEST sequences in estrogen receptor half-life and bioactivity. The Journal of steroid biochemistry and molecular biology 1993;46(6):663–72. [DOI] [PubMed] [Google Scholar]
- 30.Kumarasamy V, Vail P, Nambiar R, Witkiewicz AK, Knudsen ES. Functional Determinants of Cell Cycle Plasticity and Sensitivity to CDK4/6 Inhibition. Cancer Res 2021;81(5):1347–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mailand N, Gibbs-Seymour I, Bekker-Jensen S. Regulation of PCNA-protein interactions for genome stability. Nature reviews Molecular cell biology 2013;14(5):269–82. [DOI] [PubMed] [Google Scholar]
- 32.Ginno PA, Lott PL, Christensen HC, Korf I, Chedin F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell 2012;45(6):814–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skourti-Stathaki K, Proudfoot NJ, Gromak N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 2011;42(6):794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Y, McBride KM, Hensley S, Lu Y, Chedin F, Bedford MT. Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Mol Cell 2014;53(3):484–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamperl S, Bocek MJ, Saldivar JC, Swigut T, Cimprich KA. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017;170(4):774–86 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stork CT, Bocek M, Crossley MP, Sollier J, Sanz LA, Chedin F, et al. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. eLife 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye BJ, Kang HJ, Lee-Kwon W, Kwon HM, Choi SY. PARP1-mediated PARylation of TonEBP prevents R-loop-associated DNA damage. DNA Repair (Amst) 2021;104:103132. [DOI] [PubMed] [Google Scholar]
- 38.Cristini A, Groh M, Kristiansen MS, Gromak N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell reports 2018;23(6):1891–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell 1999;4(4):511–8. [DOI] [PubMed] [Google Scholar]
- 40.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell 2001;7(2):263–72. [DOI] [PubMed] [Google Scholar]
- 41.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434(7035):917–21. [DOI] [PubMed] [Google Scholar]
- 42.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434(7035):913–7. [DOI] [PubMed] [Google Scholar]
- 43.Kota K, Brufsky A, Oesterreich S, Lee A. Estradiol as a Targeted, Late-Line Therapy in Metastatic Breast Cancer with Estrogen Receptor Amplification. Cureus 2017;9(7):e1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Savage KI, Matchett KB, Barros EM, Cooper KM, Irwin GW, Gorski JJ, et al. BRCA1 deficiency exacerbates estrogen-induced DNA damage and genomic instability. Cancer Res 2014;74(10):2773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yue W, Wang JP, Li Y, Fan P, Liu G, Zhang N, et al. Effects of estrogen on breast cancer development: Role of estrogen receptor independent mechanisms. Int J Cancer 2010;127(8):1748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tripathi K, Mani C, Somasagara RR, Clark DW, Ananthapur V, Vinaya K, et al. Detection and evaluation of estrogen DNA-adducts and their carcinogenic effects in cultured human cells using biotinylated estradiol. Mol Carcinog 2017;56(3):1010–20. [DOI] [PubMed] [Google Scholar]
- 47.Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, et al. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006;312(5781):1798–802. [DOI] [PubMed] [Google Scholar]
- 48.Bhardwaj P, Iyengar NM, Zahid H, Carter KM, Byun DJ, Choi MH, et al. Obesity promotes breast epithelium DNA damage in women carrying a germline mutation in BRCA1 or BRCA2. Science translational medicine 2023;15(684):eade1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chatterjee P, Schweizer MT, Lucas JM, Coleman I, Nyquist MD, Frank SB, et al. Supraphysiological androgens suppress prostate cancer growth through androgen receptor-mediated DNA damage. J Clin Invest 2019;130:4245–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schweizer MT, Antonarakis ES, Wang H, Ajiboye AS, Spitz A, Cao H, et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: results from a pilot clinical study. Sci Transl Med 2015;7(269):269ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schweizer MT, Gulati R, Yezefski T, Cheng HH, Mostaghel E, Haffner MC, et al. Bipolar androgen therapy plus olaparib in men with metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rebbeck TR, Mitra N, Wan F, Sinilnikova OM, Healey S, McGuffog L, et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA 2015;313(13):1347–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dean JL, McClendon AK, Knudsen ES. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J Biol Chem 2012;287(34):29075–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pikman Y, Alexe G, Roti G, Conway AS, Furman A, Lee ES, et al. Synergistic Drug Combinations with a CDK4/6 Inhibitor in T-cell Acute Lymphoblastic Leukemia. Clin Cancer Res 2017;23(4):1012–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cao J, Zhu Z, Wang H, Nichols TC, Lui GYL, Deng S, et al. Combining CDK4/6 inhibition with taxanes enhances anti-tumor efficacy by sustained impairment of pRB-E2F pathways in squamous cell lung cancer. Oncogene 2019;38(21):4125–41. [DOI] [PubMed] [Google Scholar]
- 56.Salvador-Barbero B, Alvarez-Fernandez M, Zapatero-Solana E, El Bakkali A, Menendez MDC, Lopez-Casas PP, et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 2020;38(4):584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated in this study are available upon request from the corresponding authors. RNA sequencing data are available on NCBI SRA (# PRJNA816494).