

Graphical Abstract
In this review, Lobel et al. summarize the current understanding of the metabolic requirements for the functions of multiple types of cells in the tumor microenvironment, including tumor, immune, stromal, and endothelial cells; as well as the metabolic interplay between these diverse cell types.
Summary
Over the last two decades the rapidly expanding field of tumor metabolism has enhanced our knowledge of the impact of nutrient availability on metabolic reprogramming in cancer. Apart from established roles in cancer cells themselves, various nutrients, metabolic enzymes, and stress responses are key to the activities of tumor microenvironmental immune, fibroblastic, endothelial, and other cell types that support malignant transformation. In this article, we review our current understanding of how nutrient availability affects metabolic pathways and responses in both cancer and “stromal” cells, by dissecting major examples and their regulation of cellular activity. Understanding the relationship of nutrient availability to cellular behaviors in the tumor ecosystem will broaden the horizon of exploiting novel therapeutic vulnerabilities in cancer.
Introduction
Metabolism encompasses a large array of chemical reactions devoted to energetics that occur in living cells, tissues, and organisms. Multiple integrated steps catalyzed by specific enzymes form complex metabolic pathways, providing metabolites and energy that support cellular activities central to life. As such, these pathways are highly conserved across species. Nevertheless, metabolic pathways are dynamically regulated in a complex, context-dependent manner to balance the anabolic and catabolic needs within a cell. It is increasingly appreciated that metabolic fluxes could be rewired to enable cellular adaptions to nutritional fluctuation. Altered metabolism, previously described in a variety of cancer cells, allows them to thrive through environmental challenges, including nutrient-scarce conditions, limited oxygen supply, immune attack, and/or clinical interventions.
Metabolic changes in both tumor and adjacent stromal cells (heterogeneous immune cell types, cancer associated fibroblast, endothelial cells, neuronal cells) are achieved not only by modulation of conventional activities of key enzymes, but also external nutrient sources. Moreover, multiple metabolic enzymes exhibit unexpected activities apart from their canonical functions, such as in the regulation of gene transcription, DNA damage repair, and cell fate decisions. Accumulating evidence supports an emerging paradigm that nutrients, metabolic enzymes and related metabolites provide multi-functional hubs to ensure the plasticity and adaptation of both normal and cancer cells in complex multicellular ecosystems within solid tumors.
In this review, we will summarize current knowledge of how external and internal nutrients influence metabolic pathways and stress responses which have been identified and elucidated in the context of cancer. We will discuss how these relationships reshape cancer cell status and modulate the tumor microenvironment. The article is organized by methodically examining principal nutrients and related intracellular pathways. We believe a deeper understanding of various responses to tumor microenvironmental nutrients will identify potential new targets for precision cancer therapeutics.
Glucose
Glucose is a key source of energy and biosynthetic material for all mammalian cells. It is metabolized via glycolysis into pyruvate, which can then either be shuttled into the mitochondria to supply carbon to the tricarboxylic acid (TCA) cycle and generate adenosine triphosphate (ATP) via oxygen-dependent oxidative phosphorylation (OXPHOS), or be reduced to lactate via lactate dehydrogenase (LDH).1 Most mammalian cells at rest generate almost all ATP via OXPHOS in the presence of oxygen.2 Malignant cells, instead, primarily generate ATP via aerobic glycolysis and lactate secretion despite the availability of oxygen, a phenomenon termed the “Warburg effect”.3 Aerobic glycolysis, while less energetically efficient than OXPHOS, supports the generation of metabolites such as lipids, nucleotides, and amino acids that are required for cell growth and proliferation.4,5 As such, it is not surprising that cancer cell proliferation and tumor formation are impaired by glucose restriction, inhibition of glycolysis, or inhibition of lactate production.6–8 Growth factor signaling pathways such as the Kras pathway and the phosphoinositide 3-kinase (PI3K) pathway, which are overactivated in many malignancies, also orchestrate metabolic reprogramming to promote glucose uptake and metabolism.9,10
While glucose metabolism is critical for tumor cells, many other cells in the TME are also dependent on glucose metabolism for their metabolic functions, particularly T cells.4 Resting T cells are largely quiescent and generate the majority of their ATP from glucose metabolized by OXPHOS or via fatty acid oxidation, but are poised to rapidly produce enzymes necessary for glycolysis after activation.11,12 T cell receptor signaling-mediated activation leads to rapid metabolic reprogramming to support proliferation and effector T effector cell functions, with increased uptake of glucose and amino acids. Knockdown of the glucose uptake transporter Glut1 selectively impairs glycolysis in activated T effector cells, as well as their growth, proliferation, and effector functions. Immunosuppressive regulatory T cells (Tregs), in contrast, were unaffected by Glut1 loss.13 T cell activation also induces growth factor signaling pathways such as Myc and the PI3K pathway, which both regulate activated T cell growth and proliferation in similar ways to cancer cells, and promote glucose uptake and glycolysis.14,15 Reciprocally, LDH activity, which maintains flux through aerobic glycolysis, promotes PI3K signaling, T helper type 1 (Th1) differentiation, and interferon-gamma (IFNg) expression in activated T cells.16,17
Dendritic cells (DCs), specialized antigen-presenting cells that potently stimulate T cell responses, are present in the TME in an immature and tolerogenic state until activation by pattern recognition receptor signaling in combination with uptake of antigen. Tumor-infiltrating DC metabolism has been recently reviewed in depth.18 Once activated, DCs upregulate the chemokine receptor CCR7, and migrate out of the TME to lymph nodes in order to present antigen to T cells. This activation process rapidly induces a metabolic switch from OXPHOS to glycolysis for ATP production, and a lack of glucose or blockade of glycolysis with 2-deoxyglycose (2-DG) inhibited expression of activation markers on DCs derived in vitro.19,20 2-DG treatment also inhibited expression of CCR7 and DC migration in vivo.21 Similar to tumor cells or T cells, this shift to a primarily glycolytic metabolic program supports the generation of anabolic materials necessary for the stressful process of migration, especially through de novo lipid synthesis.20,22
Glucose plays a central role in the functioning of both malignant and immune cells, but its high rate of uptake by cancer cells may limit its availability to other cells in the TME.3 A study quantifying metabolites in pancreatic ductal adenocarcinoma (PDAC) tumor interstitial fluid (TIF) compared to plasma found that glucose was present at lower levels in TIF than in plasma, though to a small degree.23 Several studies have shown that oncogenic cells compete for glucose with immune cells in the tumor microenvironment, and that this competition leads to a reduction of tumor suppressive T cell activity.24,25 Oncogenic expression of glycolytic genes has been shown to correlate with resistance to adoptive T cell therapy, potentially via decreased production of IRF1 and CXCL10, though this may also be due to increased recruitment of immunosuppressive myeloid-derived suppressor cells (MDSCs).26,27 Expression of GLUT1 in human PDAC tumor sections correlated with disease prognosis as well as the expression of the immune checkpoint programmed cell death protein 1 (PD-1) on tumor-infiltrating T cells, which could be reversed in a model of PDAC by tumor-intrinsic knockdown of the glycolytic enzyme phosphofructokinase.28
Collectively, these studies suggest that tumor cell glucose uptake and metabolism is a limiting factor for the activity of tumor-infiltrating immune cells. However, somewhat contrary to this conclusion, a recent study has shown that most of the glucose in the TME is taken up not by tumor cells, but by tumor-infiltrating myeloid cells.29 This study may challenge the idea that tumor cell glucose uptake limits glucose uptake by intratumoral T cells or drives T cell dysfunction. Instead, T cell functions and metabolic fitness may be hindered by other unknown factors present in the TME. T cells isolated from both human and murine tumors had reduced mitochondrial mass and function related only to activation in the TME.30 A similar study found that CD8 T cells from clear cell renal cell carcinoma (ccRCC) tumors were functionally defective and unable to effectively take up and metabolize glucose, likely also due to their small and fragmented mitochondria.31 A follow up study demonstrated that intratumoral T cell effector functions could be rescued by costimulation with CD28 via restored T cell glycolytic capacity.32 Glucose uptake also regulates the functions of myeloid cells, as loss of GLUT1 inhibits macrophage phagocytosis.33
Although inhibition of glycolysis could represent a promising avenue to target tumor metabolism, it is possible that the importance of glucose in tumor-infiltrating immune populations may limit its effectiveness. The requirements for glucose in immune cell functions, however, are nuanced. While T short-term effector functions of T cells are supported by glucose metabolism, formation of long-lived memory T cells appears to be inhibited by flux through glycolysis.34 Overexpression of the glycolytic enzyme phosphoglycerate mutase-1 in CD8 T cells adoptively transferred into mice impaired their survival and proliferation in vivo. In the same model, treatment with the glycolysis inhibitor 2-DG during ex vivo activation of CD8 T cells increased the formation of memory cells, as well as their antitumor effectiveness.34 Inhibition of the signaling and metabolic regulator AKT also led to increased persistence and antitumor effectiveness of T cells after transfer.35 Inhibition of glycolysis may also modulate myeloid cells in the TME in beneficial ways. Treatment with 2-DG was found in a separate study to limit the recruitment MDSCs via decreased tumor cell production of the cytokines granulocyte colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF).27 One alternative strategy might be to solely target tumor cell glycolytic metabolism. Using nanoparticle-based delivery of a prodrug of the pyruvate dehydrogenase kinase inhibitor DCA, selective inhibition of glycolysis in oncogenic cells in combination with anti-PD1 therapy was shown to increase the number of tumor-infiltrating lymphocytes in a murine model of colon cancer.36
Lactate
The majority of glucose taken up by cancer cells is converted to lactate via lactate dehydrogenase (LDH) and then secreted.3 This process, which regenerates nicotinamide adenine dinucleotide (NAD+) from its reduced form, NADH, is required to allow glycolysis to proceed unimpeded.37 The primary lactate transporters expressed on cancer cells are monocarboxylate transporters (MCT) 1, 2, and 4, with MCT4 being the main transporter by which highly glycolytic cells export lactate.38,39 The high level of lactate secreted from oncogenic cells into the TME has many beneficial effects for tumors, both through effects mediated by lactate itself and those mediated by intratumoral pH. While we will focus on lactate’s direct functions, the effects of intratumoral acidosis are exceptionally important and have been previously reviewed.40 Lactate can activate signaling or metabolic pathways as a ligand for the G-protein coupled receptors GPR81 and GPR132.41 GPR81 is critical for the survival of PDAC cells and upregulates the expression of both MCT1 and 4, as well as CD147, which is required for their trafficking to the plasma membrane.42 Autocrine activation via lactate of GPR81 in breast cancer cells also upregulates programmed death-ligand 1 (PD-L1) to suppress anti-tumor immune activity.43
Lactate has broad effects on both myeloid and lymphoid immune populations, which have recently been extensively reviewed (Figure 1).41,44 Tumor-derived lactate inhibits macrophage inflammatory cytokine production and polarizes TAMs towards an immunosuppressive phenotype via multiple signaling pathways, including but not limited to HIF1a, ERK/STAT3, the odorant receptor OLFR78, and GPR132.45–49 Administration of lactate has been shown to reduce inflammation in models of pancreatitis, hepatitis, acute liver injury, and intestinal inflammation via the modulation of macrophage activity.50,51 It has recently been discovered that lactate can regulate chromatin accessibility and gene expression via post-translational addition of lactate to histones, a process known as lactylation. In M1-polarized macrophages, histone lactylation increases the expression of genes classically expressed by immunosuppressive macrophages, as well as genes associated with wound healing.52 High concentrations of lactate shift monocyte metabolism towards OXPHOS and an anti-inflammatory phenotype, inhibit inflammatory cytokine secretion after LPS stimulation, and inhibit monocyte migration while promoting the motility of tumor cells.53,54 Tumor cell lactate production via LDH supports the generation of immunosuppressive MDSCs while also inhibiting the cytolytic effects of natural killer (NK) cells.55 In a model of pancreatic cancer, radiation-induced tumor cell lactate production promoted anti-inflammatory MDSC activity via HIF1α and GPR81 signaling.56 GPR81 signaling has also been implicated in lactate’s suppressive effects on antigen presentation by tumor-infilitrating dendritic cells.57,58 Lactate exposure tends to inhibit the functions of antitumor T cells. Lactate inhibits CD4 and CD8 T cell migration as well as CD8 T-cell mediated cytolysis.59 Tregs, by contrast, are able to function in environments with lactate levels due to FOXP3-mediated suppression of both glycolysis and Myc expression.60 Lactate also polarizes CD4 T cells towards Th17 differentiation, and can additionally induce a Treg phenotype in Th17 cells.61,62
Figure 1:
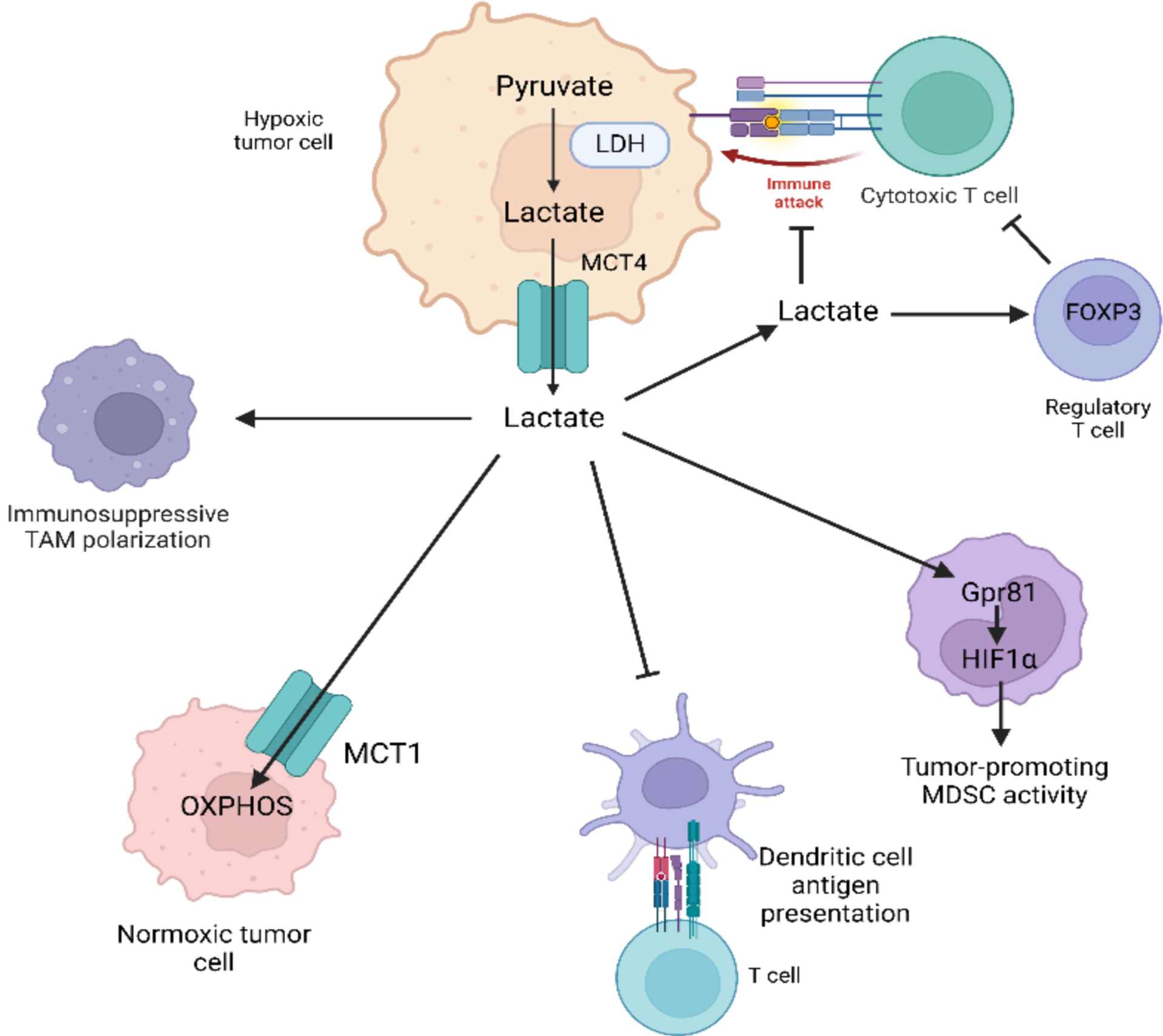
Lactate broadly generates a more immunosuppressive TME by reducing anti-tumor immune activity and promoting MDSC and Treg activity.
Although long treated as a mere waste product that contributes to intratumoral acidosis, lactate also represents an important source of fuel for both malignant and non-malignant cells (Figure 1). Lactate can be shuttled between hypoxic cancer cells in the “core” of a tumor, which secrete it via MCT4, and tumor cells in regions with more abundant oxygen that take it up via MCT1 and utilize it to fuel OXPHOS.63,64 A study using in vivo administration of [U-13C]-labeled lactate demonstrated that both human and murine non-small cell lung cancer tumors utilize lactate as a primary source of fuel for the TCA cycle.65 This phenomenon also occurs in prostate tumor cells, which take up lactate supplied primarily by cancer-associated fibroblasts (CAFs) and proceed to increase lipid uptake into lipid droplets, as well as the expression of genes relating to lipid metabolism and storage.66 A separate study found that contact with prostate cancer cells induced CAFs to increase their expression of glucose transporters, engage in aerobic glycolysis, and secrete high levels of lactate.67 Reciprocally, prostate cancer cells take up decreased levels of glucose and increased levels of lactate via MCT1 after contact with CAFs. Tumor cell secretion of lactate can also stimulate angiogenesis via increased reactive oxygen species (ROS) in endothelial cells that take lactate up.68 It should be noted that non-tumor tissues can also utilize lactate as a source of energy. Using [U-13C] lactate infusion, Hui et al. found that while lung and pancreatic tumors utilize more lactate than glucose to generate TCA cycle intermediates, non-tumor-bearing mice infused with [U-13C] lactate turn it over at the highest rate of any metabolite studied, with high levels of labeling of TCA cycle metabolites. During fasting, lactate was found to contribute to TCA cycle metabolite pools in all tissues aside from the brain.69
Given the many advantages that tumor cells gain from either importing or exporting lactate, inhibition of MCT1 and/or MCT4 represents a potentially potent therapeutic strategy against multiple types of cancer. Inhibiting MCT1 in a model of squamous cervical cancer led to reduced tumor growth due to the inability of cancer cells to take up lactate to power OXPHOS.70 MCT1 inhibition was also shown to increase oxidative metabolism in models of colon cancer and lymphoma, and could be combined with the electron transport chain complex I inhibitor metformin to reduce this metabolism and impede tumor growth in vivo.71 Dual inhibition of MCT1 and MCT4 was also found to synergize with metformin treatment to reduce tumor growth in multiple models of cancer.72,73
Glutamine
Glutamine, the most abundant plasma amino acid, is taken up avidly by proliferating cells and used as a source of nitrogen for biosynthetic processes, as well as a source of carbon for anabolic metabolites. The full extent of glutamine metabolic pathways and their relevance to cancer have been extensively reviewed previously, but will be briefly summarized here (Figure 2).74,75 The most well-studied glutamine metabolic pathway is glutaminolysis, in which glutamine is trafficked into the mitochondria and converted into glutamate by the rate-limiting enzyme glutaminase (GLS). This glutamate can be utilized to produce glutathione via glutathione synthetase, generate aspartate or alanine via GOT1 and GPT1 respectively, or be converted into alpha-ketoglutarate (α-KG) via glutamate dehydrogenase (GDH) and enter into the TCA cycle.75 The process of glutamine contributing to TCA cycle metabolites, known as TCA cycle anapleurosis, serves to maintain metabolite pools in highly proliferative cells, as metabolites such as citrate and aspartate (via oxaloacetate) can be trafficked out of the mitochondria and utilized to produce fatty acids and nucleotides, respectively.75 Glutamine also acts as a source of nitrogen to contribute to O- and N-linked glycosylation of proteins, in combination with products of glycolysis, via the hexosamine biosynthetic pathway (HBP), as well as to the de novo production of nucleotides.76,77 Lastly, glutamine promotes molecular target of rapamycin complex 1 (mTORC1) signaling via both leucine-dependent and leucine-independent mechanisms.78–80 These varied metabolic functions render glutamine “conditionally essential” for highly proliferative cells, including both immune and cancerous cells.81
Figure 2.
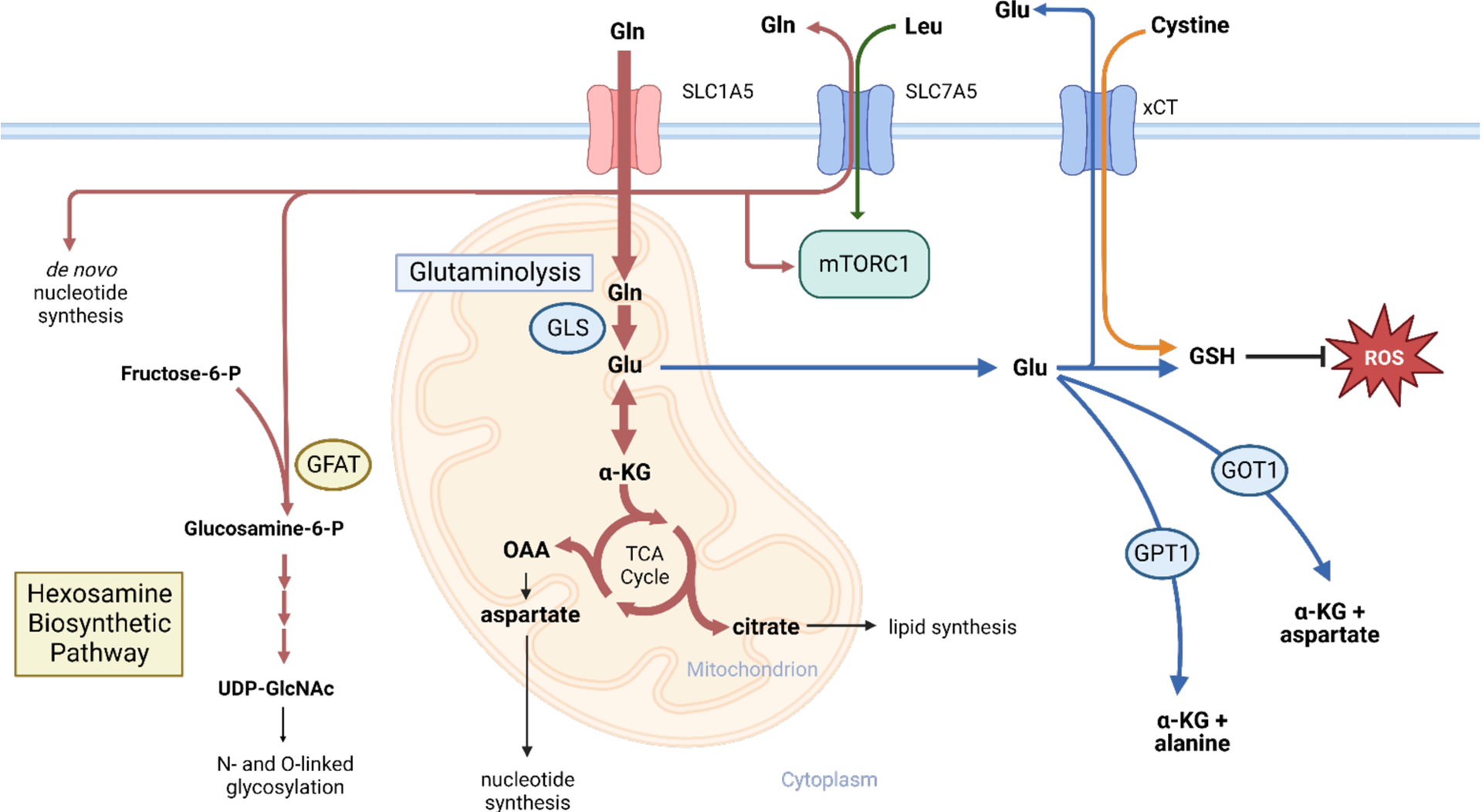
Glutamine metabolic pathways. Glutamine supports many pro-growth metabolic or signaling pathways in highly proliferative oncogenic or normal cells.
Perhaps due to this key role in supporting cell proliferation, glutamine uptake and metabolism are promoted by several oncogenes. MYC is well known to promote the expression of glutamine transporters such as SLC1A5 (also known as ASCT2), the main glutamine transporter expressed in cancer cells, and SLC7A5, a glutamine-leucine antiporter involved in mTORC1 activation, as well as critical glutamine metabolic enzymes such as GLS.82,83 Oncogenic KRAS promotes glutamine metabolism and transport; tumor cells expressing high levels of KRAS require flux through SLC7A5 for growth, and KRAS-mutant tumors that lack KEAP1 expression utilize glutamine as the main carbon source for the TCA cycle.84–86 HIF1α and mutant P53 have also been implicated in promoting glutamine uptake and metabolism.87,88 EGFR activation drives flux through glutaminolysis via the MEK/ERK pathway, which mediates its effects on glutaminolysis via GDH.89 Additionally, the tumor suppressor protein RB downregulates the expression of SLC1A5.90
Tumor cells rely largely on glutamine supplied from plasma, but cells in the TME contribute to these pools as well. In a murine model of ovarian cancer, glutamine generated by CAFs via glutamine synthetase (GLNS) and secreted into the TME was found to be critical for the growth of tumor cells. Nanoparticle-targeted siRNA knockdown of both CAF Glns and tumor cell Gls generated a “synthetic lethal” phenotype that significantly decreased tumor growth.91
While glutamine is an essential metabolite to support tumor cell growth, it has many roles in immune cells as well. Resting T cells, as mentioned previously, are largely quiescent with low rates of metabolism and glutamine uptake, but reprogram their metabolism after activation to support rapid proliferation, cytokine production, and effector functions.12 Similarly to oncogenic cells, activated T cells significantly upregulate C-MYC expression and MAPK/ERK signaling, leading to increased glutamine uptake and metabolism.15,92 Glutamine deprivation was found to impair activation-related growth and proliferation, but these functions could be partially restored by adding back nucleotides or polyamines.15 Additionally, along with products of glycolysis, glutamine is necessary to fuel flux through the hexosamine biosynthetic pathway, which produces the substrates for O- or N-linked glycosylation of proteins.77 O-linked glycosylation in particular has proven critical in the regulation of T cell signaling and cytokine production.93,94 Glutamine metabolism also regulates the differentiation and functions of T cells after activation. Inhibition of glutaminase prevents Th17 differentiation while promoting Th1 and CD8 T cell differentiation and effector functions via upregulation of Tbet.81
Given the high levels of glutamine uptake by cancer cells, as well as the requirements for glutamine in T-cells, competition for glutamine between cancer and T cells in the TME may exist. Unlike glucose, the majority of glutamine that enters the TME appears to be taken up by cancer cells.29 One recent study found that effective treatment of a model of lung cancer with immune checkpoint blockade required glutamate release into the TME for CD8 T cell activation, and that this activation was impaired by treatment with an inhibitor of glutamine metabolism.95 A separate study showed that knockout of tumor cell glutaminase in triple negative breast cancer tumors increases both the concentration of glutamine in tumor interstitial fluid (TIF) and levels of activation of cytotoxic T cells, leading the authors to propose a “glutamine steal” hypothesis, wherein low glutamine availability in TIF due to tumor cell uptake of glutamine impairs anti-tumor immunity.96 However, glutamine levels in TIF across tumors and types of cancer are likely highly variable and context-dependent. In PDAC tumor TIF, glutamine concentrations were almost identical to those found in plasma, indicating that it may not limit immune cell function.23 Additionally, glutamine deprivation may serve to polarize tumor-infiltrating myeloid cells towards a more anti-tumor phenotype. Macrophages, in particular, when cultured in glutamine-deprived media or with inhibitors of glutaminolysis, were polarized towards an M1-like phenotype.97,98 Targeting macrophage glutamine synthetase via pharmacologic or genetic means polarized macrophages towards a more immune-suppressive phenotype and augmented their ability to recruit T cells.99 It is clear that more study is required into the intratumoral availability of glutamine and its effects on tumor-infiltrating immune cells across many tumor types.
The central role of glutamine in supporting tumor cell growth and proliferation represents a potential metabolic vulnerability in cancer cells, and so several inhibitors of glutamine metabolism and uptake have been developed to target this vulnerability.
The pan-glutamine antagonist 6-Diazo-5-oxo-L-norleucine (DON) potently and irreversibly inhibits all glutamine-utilizing enzymes and has long been noted to be effective against preclinical models of cancer, but failed several clinical trials due to its harsh side effect profile.100 The next compound to be tested clinically was CB-839, a selective GLS inhibitor that potently inhibits the growth of several types of cancer in vitro, but has had mixed results in in vivo models of cancer.101–105 While initially promising, it has had mixed results in clinical trials in combination with mTOR inhibitors or conventional chemotherapeutic agents.106,107 This relative lack of in vivo efficacy has been attributed to the ability of oncogenic cells to rewire their metabolism to depend less on the products of glutaminolysis, and has thus led to refocused interest in compounds that broadly target glutamine metabolism or uptake.104
In order to broadly target glutamine metabolism while minimizing toxicity, prodrugs of DON have been developed that contain moieties designed to be selectively cleaved by tumor cells or in the tumor microenvironment. JHU-083, the first of these compounds to be tested extensively, inhibits the growth of several in vivo models of cancer, while also polarizing the immune microenvironment to a more pro-inflammatory state. Despite the long-noted requirement for glutamine in activated T cells, JHU-083 counterintuitively does not impede and in fact stimulates anti-tumor T cell activity.108 This unexpected benefit is due to the ability of activated T cells to reprogram their metabolism to utilize acetate as fuel for the TCA cycle, in contrast to tumor cells, which also leads to synergy between JHU-083 and anti-PD1 immune checkpoint blockade.108 JHU-083 was also shown to reduce the recruitment of immune suppressive MDSCs to the tumor microenvironment via reduced tumor production of CSF1, and additionally increased MHCII expression in macrophages due to reduced tumor indoleamine-2,3-dioxygenase (IDO) activity and lower production of the immunosuppressive metabolite kynurenine.109 A second prodrug of DON, DRP-104, has only recently been developed but is already in human trials. Like JHU-083, DRP-104 is selectively converted to DON by oncogenic cells, induces the generation of a more inflammatory tumor immune microenvironment, inhibits the growth of several in vivo cancer models, and synergizes with immune checkpoint blockade in those models.110
The second class of compounds developed to more broadly target glutamine metabolism are amino acid transporter blockers, the most well-studied of which is V9302. V9302 selectively and potently inhibits SLC1A5, the glutamine transporter most highly expressed in oncogenic cells.111 In contrast to DON and its prodrugs, V9302 does not inhibit glutaminergic metabolic pathways or the intracellular production of glutamine via glutamine synthetase.100,111 V9302 has been shown to effectively inhibit in vivo tumor growth as a monotherapy and to synergize with immune checkpoint blockade, tyrosine kinase inhibitors, as well as cytotoxic chemotherapy.96,111–116 In multiple myeloma, V9302 was shown to synergize with proteasome inhibitors.117 Jin et al showed that V9302 in combination with CB-839 potently inhibited the growth of a model of liver cancer to a greater extent than either compound alone by reducing glutathione and increasing ROS, indicating that there may be merit to combining multiple approaches that target glutamine.118
Leveraging Amino acid dependency for therapeutic purposes
Amino acids (AAs), structural units of proteins, constitute an important energy source for cancer cells. There are non-essential AAs (NEAAs) such as alanine, arginine, serine, glutamine etc., which are produced in healthy cells. There are also dietarily obtained essential AAs (EAAs) including methionine and tryptophan, as well as three branched chain amino acids (BCAAs): leucine, isoleucine, and valine. To meet the increased demand for AAs, cancer cells need to coordinate AA uptake, biosynthesis and catabolic processes.119 Cancer cells develop a dependency on both NEAAs and EAAs.
Cancer cells can have selective dependency on amino acids, making some better targets than others.120 Three main approaches to target amino acid metabolism are available: inhibiting AA transporters, targeting biosynthesis and depleting AAs directly. Targeting AA transporters is often not the best approach due to redundancies in the transporter family. Previous studies showed that phosphoglycerate dehydrogenase (PHGDH), the enzyme involved in serine biosynthesis, is commonly amplified in various cancers such as melanomas and breast cancers.121 PHGDH inhibitors were recently developed and have been shown to be efficacious in reducing breast cancer cell and xenograft growth.122,123 To deplete AAs directly, asparaginase (ASNS), which breaks down asparagine from aspartate, has become an essential component in acute lymphoblastic leukemia (ALL) therapy.124 Despite distinct dependencies among cancer types, regions within one tumor can also have distinct metabolic profiles that could sensitize them to novel therapeutic interventions. Pan et al. showed that tumor “cores” are much lower in many amino acids including arginine, asparagine, glutamine, serine, and aspartic acid compared to the tumor periphery.125 They found that low glutamine in the tumor core leads to BRAF inhibitor (BRAFi) resistance. H3K27 hypermethylation mediates the low glutamine-induced resistance. EPZ005687, an inhibitor of H3K27me3, mitigates BRAFi resistance and reduces overall tumor volume.
Surrounding cell types provide amino acids to cancer cells
Other cell types in the TME are important sources of AAs for tumor cells.126 Resistance mechanisms have been reported following ASNS therapy for ALL treatment.127,128 Bone marrow-derived mesenchymal cells express ASNS at levels almost 20-fold higher than ALL cells, secreting asparagine to cancer cells and sparing them from ASNS cytotoxicity.128 ASNS also hydrolyzes glutamine to glutamate, leading to a decreased level of glutamine, which is also a dependency for ALL cells.127 A study by Ehsanipour et al. showed that adipocytes protect leukemic cells from ASNS cytotoxicity by glutamine secretion, and obesity significantly impairs ASNS therapy in mice with ALL implant.127 Pancreatic cancer is characterized by dense desmoplasia and represents another classic example of obtaining AAs from surrounding CAFs. Pancreatic stellate cells (PSCs), a major type of pancreatic CAF, secrete alanine to fuel PDAC growth by serving as a major carbon source for biosynthesis.126 Interestingly, alanine secretion is dependent on PSC autophagy, as autophagy inhibition in PSCs significantly decreases alanine secretion and tumor growth in cancer cell-PSC co-injection models. A recent study from the same group identified a selective alanine transporter, SLC38A2, mediating PDAC-PSC crosstalk.129 PDAC cells lacking this transporter failed to concentrate intracellular alanine, leading to metabolic crisis and profound tumor regression. In addition to directly secreting AAs, CAFs can secrete branched chain keto acids (BCKAs) to fuel PDAC growth.130 CAF-secreted BCKAs act as substrates for de novo synthesis of BCAAs via the enzyme BCAT2, and newly synthesized BCAAs contribute to protein synthesis and cancer growth. CAFs are an important player in the ovarian cancer TME, as they facilitate ovarian tumor growth, proliferation, and metastasis.131 As mentioned above, in high grade ovarian cancer, stromal CAFs significantly upregulate glutamine anabolic pathways, and secrete glutamine to promote cancer cell growth.91
In addition to CAFs, other cell types such as peripheral neurons can provide key amino acids to cancer cells, as noted by Banh et al.132 In that study, neurons were shown to upregulate serine secretion to PDAC cells under serine depleted conditions. While axons might be in nutrient-poor tumors, neuronal cell bodies can obtain abundant nutrients from circulation. Axons release serine to support the survival of a subset of exogenous serine dependent human PDAC cells under serine/glycine deprivation. Serine deprivation results in an inhibited mRNA translation on two serine codons (TCC and TCT), but also leads to a selective translation and secretion of nerve growth factor (NGF) to promote nerve innervation around tumors.
Macropinocytosis and autophagy contribute to intracellular amino acid pools
In addition to uptaking a specific AA, cancer cells evolve mechanisms to import proteins in a non-selective manner known as macropinocytosis, providing AA after protein breakdown.133,134 Macropinocytosis is a highly conserved endocytic process wherein fluid and other nutrients are taken up through macropinosomes.133 Macropinosomes are formed in an actin-dependent process which pushes membranes inward to form membrane vesicles. Commisso et al. showed that Ras-transformed cells use macropinocytosis to transport extracellular proteins such as albumin into the cells.133 The subsequent degradation of albumin supplies AAs such as glutamine to support tumor biosynthesis. Macropinocytosis inhibition using EIPA leads to tumor regression in RAS-mutant PDAC xenografts but not RAS-WT cell implants.133 A recent study showed that macropinocytosis renders PTEN-null PDAC cells resistant to mTOR inhibition.135 Macropinocytosis-mediated protein scavenging restores phosphorylation of AKT, leading to recovered proliferation under mTOR inhibition. A separate paper suggested that macropinocytosis is also employed by non-RAS-transformed cancers such as hepatocellular carcinoma (HCC) under hypoxia.136 Under hypoxia, HIF1 induces HCC cell macropinocytosis by activating EHD2, a protein important in actin remodeling and membrane ruffling.136 Not only is macropinocytosis used by cancer cells, it is also used by CAFs.137 It was shown that under glutamine deficiency, macropinocytosis was upregulated in CAFs through CaMKK2-AMPK signaling. Macropinocytosis enhances CAF fitness, rendering them better at secreting amino acids to promote tumor survival. CAFs deficient in macropinocytosis fail to support tumor growth in a co-injection model.137
Autophagy is a physiological cellular process for degrading cytoplasmic constituents by delivering them to lysosomes. It is another adaptive mechanism used by several types of cancers under metabolic stress, although it is reported to have both tumor-promoting or tumor-restraining roles.138 Nutrient starvation is the most typical trigger for autophagy, although sensitivity can vary depending on which AA is deficient and the type of tissue.139 Pancreatic cancer is one of the cancers which relies on autophagy for survival. Inhibition of autophagy using chloroquine decreases PDAC tumor burden. In addition, combination of ERK and autophagy inhibition have synergistic effects on slowing tumor growth.140 Autophagy inhibition can lead to impaired mitochondrial functions, increased reactive oxygen species, and DNA damage.141 Recent studies have started to shed light on the metabolic substrates autophagy provides to PDAC. Autophagy was shown to maintain cysteine homeostasis by regulating the localization of SLC7A11.142 It was shown that under cysteine deficiency, SLC7A11 localizes to the plasma membrane via associating with LC3, which is essential to transport SLC7A11 along microtubules. Under autophagy inhibition, SLC7A11 is phosphorylated and translocated to lysosomes. Another recent paper showed that autophagy can select ferritin for degradation via nuclear receptor coactivator 4 (NCOA4), releasing iron to fuel PDAC growth.143 Ferritin targeted autophagy, also called ferritinophagy, supports iron-sulfur cluster protein synthesis to maintain mitochondrial homeostasis in pancreatic tumors. Ncoa4 knockout improves survival of PDAC-bearing GEMM, and enhanced ferritinophagy via Ncoa4 overexpression accelerates PDAC initiation.
Lipid dysregulation and alternative sources for lipid homeostasis
Lipids represent a complex family of hydrophobic biomolecules with significant impact on signaling, as well as energy storage and production. In addition, they are also the building blocks of cellular membranes. Major classes of lipids include sterols, monoglycerides, diacylglycerides (DGs), triglycerides (TGs), phospholipids, and glycolipids. The basic components of most lipids are fatty acids (FAs) and a glycerol backbone. FAs contain one or more hydrocarbon chain(s) that vary in the presence of double bonds and the number of carbon atoms, and can be saturated, monounsaturated, and polyunsaturated.144 Polyunsaturated fatty acids (PUFAs), containing two or more double bonds, are essential and must be dietarily obtained. In contrast to PUFAs, monounsaturated fatty acids (MUFAs) can be made by humans via de novo synthesis or incorporation from diet.
De novo lipogenesis in adults is largely restricted to the liver, adipose tissues, and lactating breast. However, cancer cells reactivate FA biosynthesis as a potential adaptive mechanism to cope with limited availability of serum lipids in the TME.145 Multiple studies since the 1970s have shown that increased lipogenesis is essential for tumor growth.145 Elevated expression of fatty acid synthase (FASN) was correlated with poor prognosis in cancers such as colon, lung, prostate, and others.146,147,148 The FASN inhibitor TVB-2640 is currently in phase I trials in solid tumors, with promising results either as a monotherapy or in combination with paclitaxel. Responses have been observed in multiple cancer types, including ovarian cancer, breast cancer and KRASmut NSCLC.149
Normal cells use glucose as the main substrate for acetyl-CoA and subsequent lipid synthesis. However, in cancer cells, glucose is shunted away from entering the TCA cycle, inhibiting glucose-based acetyl-CoA generation.150,151 To circumvent this challenge, cancer cells rely on glutamine and acetate as alternative sources for lipid synthesis.152,153 Cancer cells live in metabolically stressed conditions, and studies have shown that in vivo conditions are best mimicked by culturing cells under low serum and hypoxia.154,155 An siRNA screen of lipid metabolism targets found acetyl-CoA synthetase 2 (ACSS2) as one of the top hits under low serum and hypoxia.154 Upregulation of ACSS2 is observed in breast and prostate cancers and correlated with a poor prognosis. ACSS2 upregulation enhances the ability of cancer cells to utilize acetate, which was shown to be an important source of lipid synthesis under hypoxia.154,156
A significant heterogeneity in metabolic profiles exists in solid tumors. More or less proximity to vasculature might contribute to oxygen and nutrient gradients in different tumor regions.157 Hypoxic cancer cells are dependent on exogenous lipids for proliferation and survival,158,159 and several possibilities were proposed to understand the reason behind their lipid auxotrophy. Hypoxia could directly limit substrates for desaturation reactions. Stearoyl CoA desaturase 1 (SCD1), a rate limiting enzyme for MUFA production, requires NADPH and molecular oxygen to function. MUFAs significantly rescue hypoxic cancer cell death when deprived of exogenous lipids.155 A recent study suggested that hypoxia reduces NAD+ regeneration via the electron transport chain (ETC), leading to decreased lipid biogenesis.156 They found that lipid synthesis has a significantly higher NAD+ consumption than the production of other biomolecules, and the limitation of NAD+ regeneration is a bottle neck for lipid synthesis under hypoxia. ETC inhibition under normoxia mimics the effect of hypoxia on cancer cell proliferation and lipid synthesis. Acetate also appears to bypass NAD+ requirements and rescue proliferation in hypoxic cells.
Despite an overall decrease in lipid synthesis under hypoxia, SCD1 expression is frequently dysregulated in various human cancers, and correlated with cancer aggressiveness, stem-like features and chemoresistance.160, 161, 162 The tumor promoting function of SCD1 was thought to be unsaturated FA-dependent.9 A recent study by Lien et al. showed that caloric restriction slows tumor growth via impairing SCD activity and causing lipid imbalance.163 SCD activity is required for cancer cells under exogenous lipid limitation, and reinforced expression of SCD diminishes the beneficial effects of caloric restriction on controlling tumor growth. However, some cancer cells are not sensitive to SCD inhibition under lipid deprivation. It was recently shown that fatty acid desaturase 2 (FADS2) is upregulated and becomes a dependency in liver and lung cancer lines under SCD inhibition.164 FADS2 synthesizes sapienate, which contains a double bond at the Δ6 position in contrast to the Δ9 position in palmitoleic acid produced by SCD.144 Sapienate and its elongation product cis-8-octadecenoate are used for membrane synthesis, rescuing SCD-independent cell death under lipid deprivation.164
Lipid scavenging from CAFs and adipocytes
For cancer cells that are not sensitive to SCDi, lipid uptake from exogenous sources is another adaptive mechanism they employ. Oncogenic Ras transformation in immortalized baby mouse kidney (iBMK) cells mimics the effects of hypoxia in terms of FA scavenging, elevating lysophosphatidylcholines (LPCs), especially LPCs with unsaturated fatty acyl chains.165
CAFs may be one of the major sources of exogenous lipids for cancer cells. It was shown that LPCs secreted from activated pancreatic stellate cells are used by PDAC cells to support membrane synthesis.166 In addition, LPC can be converted to lysophosphatidic acids (LPA) in the extracellular space by the secreted enzyme autotaxin. LPA can serve as a potent pro-growth and pro-migration signaling molecules in cancers.166,167 In colon cancer, Gong et al. showed that CAFs promote colorectal cancer cell (CRC) migration through lipid secretion.168 Co-culturing CAFs with CRCs reshaped CRC lipidome by increasing the overall abundance of DGs, TGs, phosphatidylcholines (PCs) and phosphatidylethanolamine (PEs). In addition, long chain poly-unsaturated fatty acyls are preferentially taken up by the CRCs. FASN inhibition in CAFs and inhibition of CD36, one of the most abundant fatty acid transporters, decreased CRC migration. Furthermore, another study also showed that PCs with unsaturated acyl chains are secreted by CAFs, and they support CRC growth metastasis by enhancing membrane fluidity.169
In addition to CAFs, cancer-associated adipocytes (CAAs) could also support tumor growth by secreting fatty acids and growth factors.170,171,172 Adipocytes near cancer cells are usually observed with smaller sizes compared to more distal adipocytes, with a decrease in TG stores.173 Cancer cells can trigger lipolysis in intra-or peri-tumoral adipocytes and even a global lipolysis program to induce muscular atrophy (cachexia syndrome).174 Co-culture of omental adipocytes with ovarian cancer cells stimulates fatty acid uptake through upregulating CD36. CD36 inhibition reduces ovarian cancer tumor burden and metastasis by potentially reducing FA and cholesterol supply.175 A study by Tabe et al. showed that bone marrow (BM) adipocytes support survival of acute monocytic leukemia (AMoL), a subtype of acute myeloid leukemia. BM adipocytes prevent the spontaneous apoptosis of AMoL cells by providing free FAs which are transported by CD36 and used as signaling molecules to activate fatty acid oxidation (FAO) and anti-apoptotic programs.176 In addition to getting free FAs from stromal cells, it was shown that cancer cells secrete lipoprotein lipase (LPL) to hydrolyze extracellular triglycerides in lipoproteins circulating in the bloodstream. LPL, in the presence of lipoproteins, stimulates cancer cell growth.177
We provide a list of aforementioned nutrient-dependencies in Table 1.
Table 1:
A list of cellular nutrient dependencies under various settings in human and mouse.
| Nutrient | Setting | Cell Type/Crosstalk | Species | References |
|---|---|---|---|---|
| Glucose | Lung cancer | Kras-driven cancer cells | Human, mouse | 7 |
| PDAC | Kras-driven cancer cells | Mouse | 9 | |
| Breast cancer | ErbB2-driven breast cancer cells | Human, mouse | 7 | |
| Cell culture | Activated T cells | Human, mouse | 11, 13, 15–17 | |
| Cell culture | Dendritic cells | Mouse | 19, 20 | |
| Melanoma | Tumor cell - T cell competition | Mouse | 24 | |
| Sarcoma | Tumor cell - T cell competition | Mouse | 25 | |
| Lactate | PDAC | Tumor cells | Human, Mouse | 42 |
| Lung tumors | Tumor cells | Human | 65 | |
| Lewis Lung Carcinoma, B16- F1 Melanoma | Tumor-associated macrophages | Mouse | 45 | |
| PDAC | MDSCs | Mouse | 55 | |
| PDAC | NK cells | Mouse | 55 | |
| Cell culture | CD4 and CD8 T cells | Human, mouse | 59 | |
| Cell culture | Tregs | Mouse | 60 | |
| Glutamine | Cultured Glioma cells, MEFs | Tumor cells, MEFs | Human, Mouse | 83 |
| Cell culture | Activated T cells | Mouse | 15, 92 | |
| Triple-negative breast cancer | Tumor cell - T cell competition | Mouse | 96 | |
| PDAC | Tumor cell - T cell competition | Mouse | 23 | |
| Cell culture | Macrophages | Mouse | 97, 98 | |
| Ovarian cancer | CAF and cancer cell crosstalk | Human, mouse | 91 | |
| Serine | Breast cancer | PHGDH dependent cancer cells | Human | 122,123 |
| PDAC | Neuron and cancer cell crosstalk | Human | 132 | |
| Alanine | PDAC | PSC and cancer cell crosstalk | Human, mouse | 126 |
| BCKA | PDAC | CAF and cancer cell crosstalk | Human | 130 |
| Albumin | PDAC, macropinocytosis | RAS-driven cancer cells | Human, mouse | 133 |
| HCC, macropinocytosis | Non-RAS driven cancer cells under hypoxia | Human, mouse | 136 | |
| PDAC, macropinocytosis | Pancreatic CAFs | Human, mouse | 137 | |
| Cysteine | PDAC, autophagy | Pancreatic cancer cells | Human, mouse | 142 |
| Lysophospholipids | Lung cancer, Ras transformed cells | Hypoxic and RAS-driven cancer cells | Human, mouse | 165 |
| PDAC | PSC and cancer cell crosstalk | Human, mouse | 166 | |
| Lipids | CRC | CAF and cancer cell crosstalk | Human | 168, 169 |
| Ovarian cancer | Adipocyte and cancer cell crosstalk | Human | 175 | |
| AML subtype | Adipocyte and cancer cell crosstalk | Human | 176 |
Targeting ER stress responses and integrated stress responses in cancers
Multiple stressors in the tumor microenvironment, either cell intrinsic or extrinsic, can trigger stress responses. Endoplasmic reticulum (ER) stress responses, also called unfolded protein responses (UPR), can be stimulated by hypoxia, nutrient deprivation, acidic pH, upregulated protein anabolism and secretory activity.178 The UPR is mediated by three major sensors: PKR-like ER kinase (PERK), inositol-requiring enzyme-1α (IRE1α) and activating transcription factor 6α (ATF6α). These three sensors reside in the ER membrane and can detect membrane composition changes and misfolded proteins. Pathways mediated by PERK, IRE1α and ATF6α are non-redundant and coordinate well-controlled molecular events in cells. It was previously reported that only severe oxygen deprivation (<1%) activates UPR,179 raising the possibility that ER stress elevation is spatially controlled in tumor cells with a different oxygen gradient.178 In addition, the magnitude of ER stress can have differential outcomes on the malignant cells. Moderate ER stress fueled by oncogenic changes and TMEs can promote cancer cell proliferation,180,181,182,183,184,185metastasis,186,187 and chemoresistance.188 If this stress is unresolved, apoptotic programs under ER stress pathways will be activated.155,189 The integrated stress response (ISR) is an evolutionally conserved signaling network to maintain cell/tissue homeostasis with four kinase sensors: PERK, general control nonderepressible 2 (GCN2), protein kinase R (PKR) and eIF2α kinase heme-regulated inhibitor (HRI). These sensors respond to environmental stimuli such as protein homeostasis defects, nutrient deprivation, viral infection, and oxidative stress. All these stress sources converge onto a single control machinery: phosphorylation of translation initiation factor eIF2α.190 Global translation is attenuated resulting from eIF2α phosphorylation. Similar to the UPR, the ISR has a dual role in physiological and various cancer conditions: promoting cancer growth by balancing protein synthesis or causing apoptosis when proteotoxic stress cannot be tolerated.191,192,193 Because the functions of the UPR and ISR are context dependent, generalization and extrapolation need to be carefully applied. When they are promoting cancer cell fitness, the UPR and ISR can be easily targeted by small molecule drugs due to well-characterized functional domains and dynamics. We provide an illustration of the drugs targeting different arms of the UPR and ISR (Figure 4) and a table of their applications in various cancers (Table 2).
Figure 4:
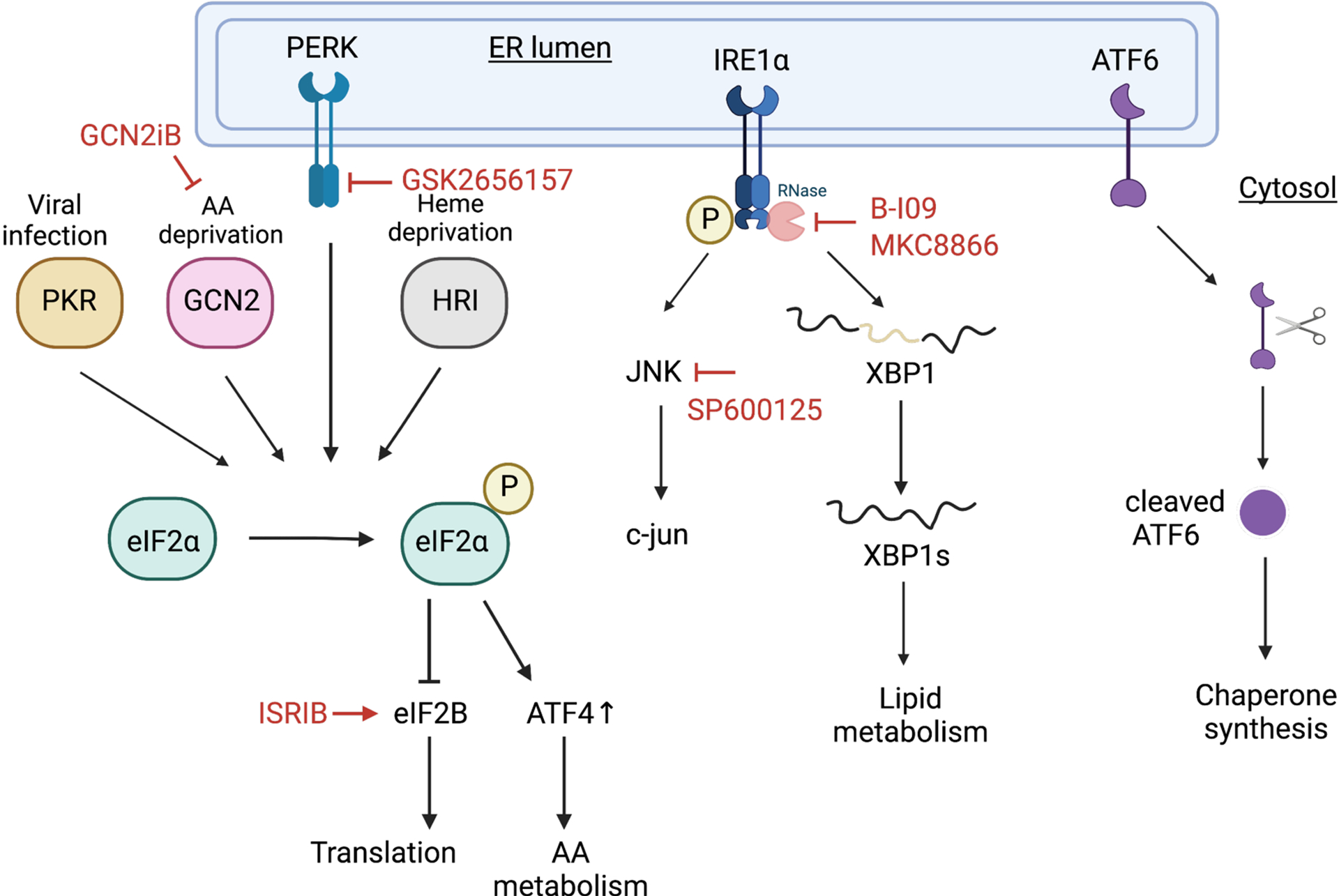
Unfolded protein response and integrated stress response pathways have druggable targets. Red lines highlight current druggable targets in cancer applications.
Table 2:
A non-exhaustive list of drugs targeting ER stress responses and their cancer applications.
| Drug | Target | Application | References |
|---|---|---|---|
| B-I09 | IRElα - XBPls | B cell lymphoma, neuroblastoma, CLL, MM and ARID1A- mutant ovarian cancers | 182, 201–203 |
| MKC8866 | IRElα - XBP1s | Prostate cancer | 204 |
| ISRIB | eIF2B | Prostate cancer | 191 |
| SP600125 | JNK | Pancreatic cancer | 185 |
| GSK2656157 | PERK | Pancreatic cancer | 181 |
| GCN2iB | GCN2 | Liver cancer under arginine restriction | 209 |
Lipid imbalance and ER stress responses
Hypoxia is a common TME feature and a strong stimulus for the UPR. Post-translational disulfide bond formation is oxygen-dependent, and the disruption of which under hypoxia increases misfolded proteins, activating a UPR.194 Moreover, molecular oxygen is required for the activity of the desaturase SCD1.195 Young et al. showed that hypoxia decreases unsaturated oleic acids and linoleic acids, leading to toxic saturated fatty acid buildup in malignant cells. Lipid saturation under hypoxia activates ER stress responses and causes a significant decrease in cell viability partially through the IRE1α pathway.155 Lipid droplets (LD), containing TGs and cholesterol esters, are an important feature for clear cell renal cell carcinoma (ccRCC). Qiu et al. showed that PLIN2, the gene encoding for the LD coating protein perilipin 2, is important for maintaining ccRCC cell viability by regulating ER homeostasis. PLIN2 knockdown triggers cytotoxic ER stress responses, mainly through the IRE1α and ATF6α pathway.189 Under hypoxic conditions, saturated FAs are incorporated into TGs, but unsaturated FAs are released into the phospholipid pool, supporting ccRCC cell viability. The disruption of TG synthesis via DGAT knockdown impairs the buffering ability of TGs, leading to toxic fatty acid buildup.196 The above studies suggest lipid imbalance is important for cellular and ER homeostasis. Volmer et al. showed that membrane saturation is directly sensed by the transmembrane domain of IRE1α and PERK to activate ER stress.197 Griffiths et al. showed that ER stress can also be indirectly caused by reactive oxygen species (ROS) accumulation as a result from lipid composition changes.198 Sterol regulatory element binding protein (SREBP) ablation leads to a loss of mono- and poly- unsaturated fatty acids, which results in ROS and ROS-induced ER stress activation.
ER stress response pathways as cancer cell dependencies
Though similarly experiencing hypoxic stress,199 triple-negative breast cancer employ IRE1α- XBP1 pathway as a survival mechanism. XBP1 activates a HIF1α transcriptional program, promoting adaptive responses of basal breast tumors within a cytotoxic solid tumor microenvironment.183 IRE1α has RNase activity after activation through dimerization and autophosphorylation. IRE1α removes 26 nucleotides from unspliced XBP1 to form a functional transcription factor XBP1s, which plays a crucial role in lipid metabolism and maintaining ER homeostasis.200 IRE1α-XBP1s plays an essential role in MYC-driven cancers such as neuroblastoma and B-cell lymphoma (BL).182 Pharmacological depletion of XBP1s using B-I09 suppresses BL growth via SCD1 activity.182 B-I09 was also shown to be efficacious in chronic lymphocytic leukemia (CLL),201 multiple myeloma (MM),202 and ARID1A-mutant ovarian cancers.203 Another IRE1α RNase inhibitor, MKC8866, has also been shown to inhibit prostate cancer growth.204
Synthetic lethality between amino acid deficiency and the ISR
AA deficiency leads to a build-up of uncharged tRNAs, which is sensed by GCN2. GCN2 binding to uncharged tRNAs results in activation of ISR and translational silencing.205 However, when GCN2 is activated, stress-related programs regulated by ATF4 are selectively translated to promote AA transport and synthesis as an adaptation. If this adaptation is unsuccessful, ATF4-CHOP will activate apoptosis.206,207 As previously noted, metabolites in TIF isolated from murine PDAC have depleted levels of AAs such as arginine relative to plasma.23 Previously, we mentioned that ASNS has been used as a targeted treatment for ALL, because it breaks down arginine that the cancer cells depend on.124 ASNS treatment activates phosphorylation of GCN2 and eIF2α, and sensitizes ALL and some PDAC tumors to GCN2 inhibitors.208 A recent study showed that hepatocellular carcinoma (HCC) depends on exogenous arginine due to suppressed urea cycle enzymes including argininosuccinate synthetase (ASS1) and argininosuccinate lyase (ASL) compared to healthy liver.209 Arginine is synthesized through the combined functions of ASS1 and ASL, and the deficiency in these enzymes renders HCC cells reliant on exogenous arginine uptake. Arginine restriction induces GCN2-dependent cell-cycle arrest through p21, and GCN2 inhibition induces senescence in HCC cells, rendering HCC cells sensitive to senolytic treatment.
Synthetic lethality between proteotoxicity and the ISR
Activation of oncogenes and loss of tumor suppressors can often lead to hyperactivated protein synthesis. If protein synthesis exceeds lipid synthesis homeostasis, ISR and UPR are activated.155 Dysregulated mTORC1 signaling via Tsc2 knockout induces cell death under low serum and low oxygen (SO) conditions.155 SO conditions increase membrane saturation, disrupting ER membrane homeostasis. Tsc2 knockout cells undergo cell death because they are unable to balance protein and lipid synthesis, which triggers “lipotoxicity” through the cytotoxic UPR.
It is also important to balance proteostasis to ensure the protein translational rate matches with tumor growth. C-MYC overexpression often leads to an anabolic program and cellular proliferation, which could disrupt proteostasis and induce the ISR. MYC increases the level of uncharged tRNAs, upregulating ATF4 by GCN2 activation.184 ATF4 inhibition is synthetically lethal with MYC activation, delaying MYC-driven lymphoma. A recent study by Nguyen et al. similarly suggested that protein synthesis needs to be fine-tuned for optimal tumor growth.191 Myc amplification in combination with Pten loss accelerates prostate cancer (PCa) progression compared to the Myc overexpression or Pten loss alone in a transgenic mouse model. Paradoxically, the coexistence of two mutations leads to a decrease in global protein synthesis even though the two mutations alone independently enhance proteins synthesis. They also found that the level of eIF2α phosphorylation correlates with tumor progression and is significantly upregulated in Myc and Pten double mutants. ISRIB is a compound that blocks the ISR through replenishing eIF2B.190 eIF2B is inhibited by eIF2α phosphorylation,190 and ISRIB reverses p-eIF2α’s function in translational silencing.191 ISRIB greatly reduces prostate tumor burden in Ptenfl/fl and Myctg mice, extending survival of a humanized model of metastatic PCa.
Concluding Remarks
Accumulating evidence supports the notion that external nutrient availability has a significant impact on cancer initiation and progression4, 5. In this review, we focused on key roles of nutrients and related metabolic pathways and stress responses from carbon and nitrogen metabolism and their connections to cancer. We speculate that at least two general mechanisms exist that confer nutrients with unexpected functions: 1) Subcellular compartmentalization of nutrients, metabolites, and metabolic enzymes: some enzymes may directly serve as transcription factors or regulators after nuclear translocation, whereas re-distribution of nutrients may establish novel protein-protein interactions, leading to functional alterations of signaling pathways; 2) Shifted enzymatic activities based on the availability of particular substrates. We speculate that these nutrients like glucose, amino acids, and lipids may be essential for orchestrating cellular metabolism, and metabolic crosstalk with signaling effectors that are prerequisite for disease progression. Recently, a group developed a theoretical model to elucidate cancer–associated metabolic disorders based on redox balance and electron transfer, which may provide a central platform to integrate nutrient availability with conventional and “moonlighting” functions of metabolic enzymes, especially in the context of cancer (Liu et al., 2020; Yang et al., 2021). Future work will continue to identify underappreciated relationships between nutrients, metabolic enzymes, and intracellular stress responses, with the hope of developing new therapeutics against human malignancies.
Figure 3:
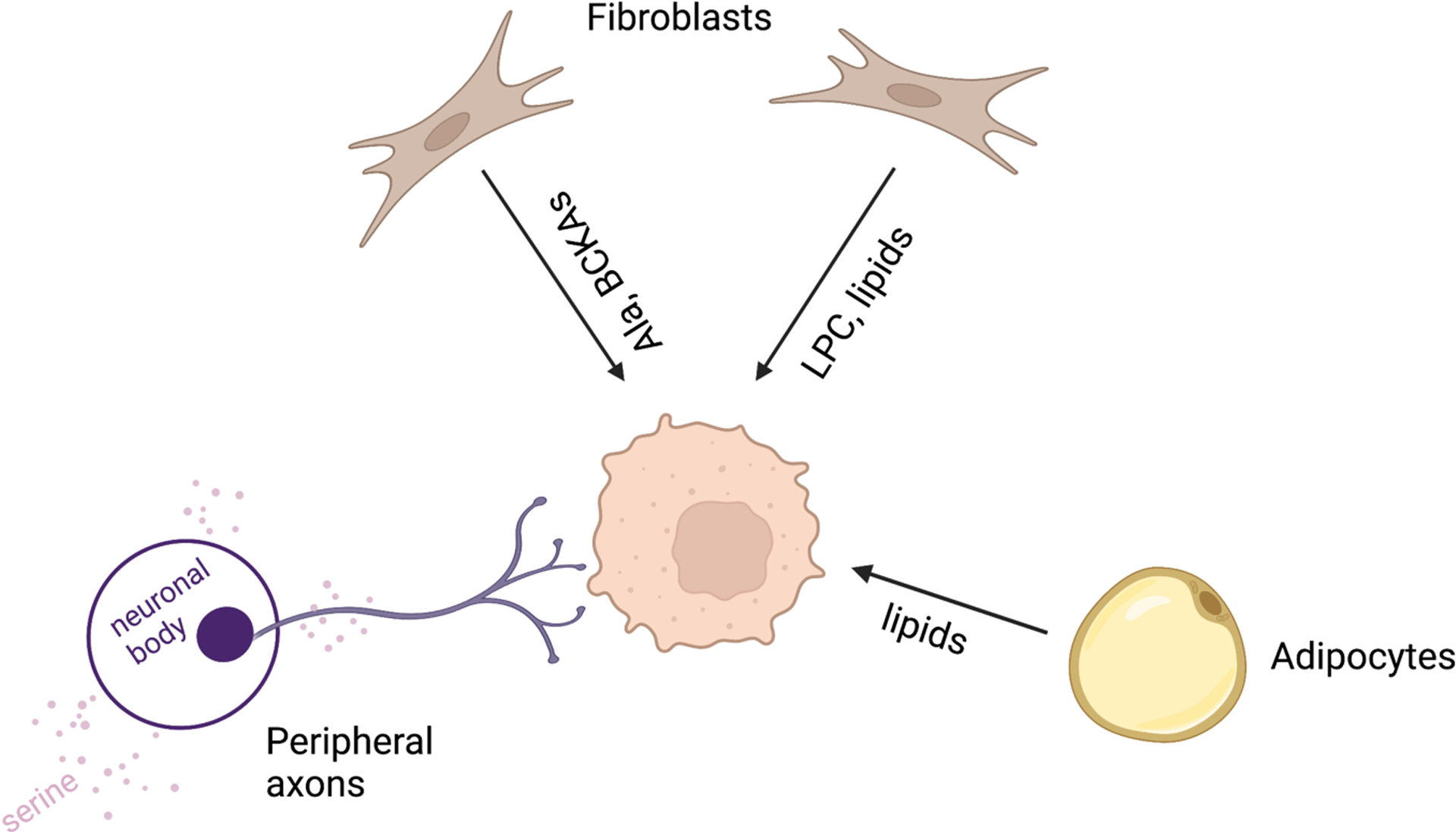
Tumor cells obtain nutrients from surrounding cell types. Pancreatic CAFs feed alanine (Ala), BCKA and LPCs to the tumor cells. Adipocytes secrete lipids to support ovarian cancer and acute myeloid leukemia. Peripheral axons release serine to support mRNA translation in pancreatic cancer.
Acknowledgements
This work was supported by the National Cancer Institute (P01 CA104838, R01 CA158301, and R35 CA220483 to M.C.S.). We apologize to those whose work was not cited because of space limitations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lunt SY, and Vander Heiden MG (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27, 441–464. 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 2.Rolfe DF, and Brown GC (1997). Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiological Reviews 77, 731–758. 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- 3.Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 324, 1029–1033. 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reinfeld BI, Rathmell WK, Kim TK, and Rathmell JC (2022). The therapeutic implications of immunosuppressive tumor aerobic glycolysis. Cell Mol Immunol 19, 46–58. 10.1038/s41423-021-00727-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeBerardinis RJ, and Chandel NS (2020). We need to talk about the Warburg effect. Nat Metab 2, 127–129. 10.1038/s42255-020-0172-2. [DOI] [PubMed] [Google Scholar]
- 6.Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, and Thompson CB (2001). Growth Factors Can Influence Cell Growth and Survival through Effects on Glucose Metabolism. Molecular and Cellular Biology 21, 5899–5912. 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, et al. (2013). Hexokinase 2 Is Required for Tumor Initiation and Maintenance and Its Systemic Deletion Is Therapeutic in Mouse Models of Cancer. Cancer Cell 24, 213–228. 10.1016/j.ccr.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fantin VR, St-Pierre J, and Leder P (2006). Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9, 425–434. 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 9.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670. 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buzzai M, Bauer DE, Jones RG, DeBerardinis RJ, Hatzivassiliou G, Elstrom RL, and Thompson CB (2005). The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid β-oxidation. Oncogene 24, 4165–4173. 10.1038/sj.onc.1208622. [DOI] [PubMed] [Google Scholar]
- 11.Ricciardi S, Manfrini N, Alfieri R, Calamita P, Crosti MC, Gallo S, Müller R, Pagani M, Abrignani S, and Biffo S (2018). The Translational Machinery of Human CD4+ T Cells Is Poised for Activation and Controls the Switch from Quiescence to Metabolic Remodeling. Cell Metabolism 28, 895–906.e5. 10.1016/j.cmet.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buck MD, O’Sullivan D, and Pearce EL (2015). T cell metabolism drives immunity. Journal of Experimental Medicine 212, 1345–1360. 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, et al. (2014). The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab 20, 61–72. 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wofford JA, Wieman HL, Jacobs SR, Zhao Y, and Rathmell JC (2008). IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood 111, 2101–2111. 10.1182/blood-2007-06-096297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, et al. (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882. 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, and Li MO (2016). Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484. 10.1126/science.aaf6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu K, Yin N, Peng M, Stamatiades EG, Shyu A, Li P, Zhang X, Do MH, Wang Z, Capistrano KJ, et al. (2021). Glycolysis fuels phosphoinositide 3-kinase signaling to bolster T cell immunity. Science 371, 405–410. 10.1126/science.abb2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giovanelli P, Sandoval TA, and Cubillos-Ruiz JR (2019). Dendritic Cell Metabolism and Function in Tumors. Trends in Immunology 40, 699–718. 10.1016/j.it.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, et al. (2010). Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749. 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Everts B, Amiel E, Huang SC-C, Smith AM, Chang C-H, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJW, et al. (2014). TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nature Immunology 15, 323–332. 10.1038/ni.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guak H, Al Habyan S, Ma EH, Aldossary H, Al-Masri M, Won SY, Ying T, Fixman ED, Jones RG, McCaffrey LM, et al. (2018). Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat Commun 9, 2463. 10.1038/s41467-018-04804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gotoh K, Morisaki T, Setoyama D, Sasaki K, Yagi M, Igami K, Mizuguchi S, Uchiumi T, Fukui Y, and Kang D (2018). Mitochondrial p32/C1qbp Is a Critical Regulator of Dendritic Cell Metabolism and Maturation. Cell Reports 25, 1800–1815.e4. 10.1016/j.celrep.2018.10.057. [DOI] [PubMed] [Google Scholar]
- 23.Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Vander Heiden MG, and Muir A (2019). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 8, e44235. 10.7554/eLife.44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho P-C, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui Y-C, Cui G, Micevic G, Perales JC, et al. (2015). Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162, 1217–1228. 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJW, et al. (2015). Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162, 1229–1241. 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, Williams LJ, Wang Z, Bristow CA, Carugo A, et al. (2018). Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metabolism 27, 977–987.e4. 10.1016/j.cmet.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, Wei S, Zhao L, Vatan L, Wen B, et al. (2018). Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab 28, 87–103.e6. 10.1016/j.cmet.2018.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cortese N, Capretti G, Barbagallo M, Rigamonti A, Takis PG, Castino GF, Vignali D, Maggi G, Gavazzi F, Ridolfi C, et al. (2020). Metabolome of Pancreatic Juice Delineates Distinct Clinical Profiles of Pancreatic Cancer and Reveals a Link between Glucose Metabolism and PD-1+ Cells. Cancer Immunol Res 8, 493–505. 10.1158/2326-6066.CIR-19-0403. [DOI] [PubMed] [Google Scholar]
- 29.Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, Sugiura A, Cohen AS, Ali A, Do BT, et al. (2021). Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282–288. 10.1038/s41586-021-03442-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, and Delgoffe GM (2016). The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 45, 374–388. 10.1016/j.immuni.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, Chiang Y-CJ, Corona AL, Gemta LF, Vincent BG, et al. (2017). Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2. 10.1172/jci.insight.93411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beckermann KE, Hongo R, Ye X, Young K, Carbonell K, Healey DCC, Siska PJ, Barone S, Roe CE, Smith CC, et al. (2020). CD28 costimulation drives tumor-infiltrating T cell glycolysis to promote inflammation. JCI Insight 5. 10.1172/jci.insight.138729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Freemerman AJ, Zhao L, Pingili AK, Teng B, Cozzo AJ, Fuller AM, Johnson AR, Milner JJ, Lim MF, Galanko JA, et al. (2019). Myeloid Slc2a1-Deficient Murine Model Revealed Macrophage Activation and Metabolic Phenotype Are Fueled by GLUT1. J Immunol 202, 1265–1286. 10.4049/jimmunol.1800002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, et al. (2013). Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 123, 4479–4488. 10.1172/JCI69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, Tran E, Hanada K-I, Yu Z, Palmer DC, et al. (2015). Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res 75, 296–305. 10.1158/0008-5472.CAN-14-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolb D, Kolishetti N, Surnar B, Sarkar S, Guin S, Shah AS, and Dhar S (2020). Metabolic Modulation of the Tumor Microenvironment Leads to Multiple Checkpoint Inhibition and Immune Cell Infiltration. ACS Nano 14, 11055–11066. 10.1021/acsnano.9b10037. [DOI] [PubMed] [Google Scholar]
- 37.Elia I, and Haigis MC (2021). Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nat Metab 3, 21–32. 10.1038/s42255-020-00317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pinheiro C, Longatto-Filho A, Azevedo-Silva J, Casal M, Schmitt FC, and Baltazar F (2012). Role of monocarboxylate transporters in human cancers: state of the art. J Bioenerg Biomembr 44, 127–139. 10.1007/s10863-012-9428-1. [DOI] [PubMed] [Google Scholar]
- 39.Fisel P, Schaeffeler E, and Schwab M (2018). Clinical and Functional Relevance of the Monocarboxylate Transporter Family in Disease Pathophysiology and Drug Therapy. Clinical and Translational Science 11, 352–364. 10.1111/cts.12551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corbet C, and Feron O (2017). Tumour acidosis: from the passenger to the driver’s seat. Nat Rev Cancer 17, 577–593. 10.1038/nrc.2017.77. [DOI] [PubMed] [Google Scholar]
- 41.Certo M, Llibre A, Lee W, and Mauro C (2022). Understanding lactate sensing and signalling. Trends in Endocrinology & Metabolism 33, 722–735. 10.1016/j.tem.2022.07.004. [DOI] [PubMed] [Google Scholar]
- 42.Roland CL, Arumugam T, Deng D, Liu SH, Philip B, Gomez S, Burns WR, Ramachandran V, Wang H, Cruz-Monserrate Z, et al. (2014). Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res 74, 5301–5310. 10.1158/0008-5472.CAN-14-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng J, Yang H, Zhang Y, Wei H, Zhu Z, Zhu B, Yang M, Cao W, Wang L, and Wu Z (2017). Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene 36, 5829–5839. 10.1038/onc.2017.188. [DOI] [PubMed] [Google Scholar]
- 44.Luo Y, Li L, Chen X, Gou H, Yan K, and Xu Y (2022). Effects of lactate in immunosuppression and inflammation: Progress and prospects. International Reviews of Immunology 41, 19–29. 10.1080/08830185.2021.1974856. [DOI] [PubMed] [Google Scholar]
- 45.Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563. 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, Yang J, Pan J, Hu S, Zhang C, et al. (2018). Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 17, 428–438. 10.1080/15384101.2018.1444305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vadevoo SMP, Gunassekaran GR, Lee C, Lee N, Lee J, Chae S, Park J-Y, Koo J, and Lee B (2021). The macrophage odorant receptor Olfr78 mediates the lactate-induced M2 phenotype of tumor-associated macrophages. Proc Natl Acad Sci U S A 118, e2102434118. 10.1073/pnas.2102434118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen P, Zuo H, Xiong H, Kolar MJ, Chu Q, Saghatelian A, Siegwart DJ, and Wan Y (2017). Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci U S A 114, 580–585. 10.1073/pnas.1614035114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang K, Xu J, Fan M, Tu F, Wang X, Ha T, Williams DL, and Li C (2020). Lactate Suppresses Macrophage Pro-Inflammatory Response to LPS Stimulation by Inhibition of YAP and NF-κB Activation via GPR81-Mediated Signaling. Frontiers in Immunology 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoque R, Farooq A, Ghani A, Gorelick F, and Mehal WZ (2014). Lactate Reduces Liver and Pancreatic Injury in Toll-Like Receptor– and Inflammasome-Mediated Inflammation via GPR81-Mediated Suppression of Innate Immunity. Gastroenterology 146, 1763–1774. 10.1053/j.gastro.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou H-C, Yu W-W, Yan X-Y, Liang X-Q, Ma X-F, Long J-P, Du X-Y, Mao H-Y, and Liu H-B (2022). Lactate-driven macrophage polarization in the inflammatory microenvironment alleviates intestinal inflammation. Front Immunol 13, 1013686. 10.3389/fimmu.2022.1013686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al. (2019). Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580. 10.1038/s41586-019-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ratter JM, Rooijackers HMM, Hooiveld GJ, Hijmans AGM, de Galan BE, Tack CJ, and Stienstra R (2018). In vitro and in vivo Effects of Lactate on Metabolism and Cytokine Production of Human Primary PBMCs and Monocytes. Frontiers in Immunology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goetze K, Walenta S, Ksiazkiewicz M, Kunz-Schughart LA, and Mueller-Klieser W (2011). Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. International Journal of Oncology 39, 453–463. 10.3892/ijo.2011.1055. [DOI] [PubMed] [Google Scholar]
- 55.Husain Z, Huang Y, Seth P, and Sukhatme VP (2013). Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol 191, 1486–1495. 10.4049/jimmunol.1202702. [DOI] [PubMed] [Google Scholar]
- 56.Yang X, Lu Y, Hang J, Zhang J, Zhang T, Huo Y, Liu J, Lai S, Luo D, Wang L, et al. (2020). Lactate-Modulated Immunosuppression of Myeloid-Derived Suppressor Cells Contributes to the Radioresistance of Pancreatic Cancer. Cancer Immunol Res 8, 1440–1451. 10.1158/2326-6066.CIR-20-0111. [DOI] [PubMed] [Google Scholar]
- 57.Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, Mackensen A, and Kreutz M (2006). Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107, 2013–2021. 10.1182/blood-2005-05-1795. [DOI] [PubMed] [Google Scholar]
- 58.Brown TP, Bhattacharjee P, Ramachandran S, Sivaprakasam S, Ristic B, Sikder MOF, and Ganapathy V (2020). The lactate receptor GPR81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene 39, 3292–3304. 10.1038/s41388-020-1216-5. [DOI] [PubMed] [Google Scholar]
- 59.Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D’Acquisto F, Bland EJ, Bombardieri M, Pitzalis C, Perretti M, et al. (2015). Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLOS Biology 13, e1002202. 10.1371/journal.pbio.1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ, Kopinski PK, Wang L, et al. (2017). Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metabolism 25, 1282–1293.e7. 10.1016/j.cmet.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pucino V, Certo M, Bulusu V, Cucchi D, Goldmann K, Pontarini E, Haas R, Smith J, Headland SE, Blighe K, et al. (2019). Lactate Buildup at the Site of Chronic Inflammation Promotes Disease by Inducing CD4+ T Cell Metabolic Rewiring. Cell Metabolism 30, 1055–1074.e8. 10.1016/j.cmet.2019.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lopez Krol A, Nehring HP, Krause FF, Wempe A, Raifer H, Nist A, Stiewe T, Bertrams W, Schmeck B, Luu M, et al. (2022). Lactate induces metabolic and epigenetic reprogramming of pro-inflammatory Th17 cells. EMBO Rep 23, e54685. 10.15252/embr.202254685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guillaumond F, Leca J, Olivares O, Lavaut M-N, Vidal N, Berthezène P, Dusetti NJ, Loncle C, Calvo E, Turrini O, et al. (2013). Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A 110, 3919–3924. 10.1073/pnas.1219555110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lyssiotis CA, and Kimmelman AC (2017). Metabolic Interactions in the Tumor Microenvironment. Trends in Cell Biology 27, 863–875. 10.1016/j.tcb.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371.e9. 10.1016/j.cell.2017.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ippolito L, Comito G, Parri M, Iozzo M, Duatti A, Virgilio F, Lorito N, Bacci M, Pardella E, Sandrini G, et al. (2022). Lactate Rewires Lipid Metabolism and Sustains a Metabolic–Epigenetic Axis in Prostate Cancer. Cancer Research 82, 1267–1282. 10.1158/0008-5472.CAN-21-0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, Lanciotti M, Serni S, Cirri P, and Chiarugi P (2012). Reciprocal Metabolic Reprogramming through Lactate Shuttle Coordinately Influences Tumor-Stroma Interplay. Cancer Research 72, 5130–5140. 10.1158/0008-5472.CAN-12-1949. [DOI] [PubMed] [Google Scholar]
- 68.Végran F, Boidot R, Michiels C, Sonveaux P, and Feron O (2011). Lactate Influx through the Endothelial Cell Monocarboxylate Transporter MCT1 Supports an NF-κB/IL-8 Pathway that Drives Tumor Angiogenesis. Cancer Research 71, 2550–2560. 10.1158/0008-5472.CAN-10-2828. [DOI] [PubMed] [Google Scholar]
- 69.Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Zhan, Yanxiang Guo J, et al. (2017). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. 10.1038/nature24057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, et al. (2008). Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 118, 3930–3942. 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beloueche-Babari M, Wantuch S, Casals Galobart T, Koniordou M, Parkes HG, Arunan V, Chung Y-L, Eykyn TR, Smith PD, and Leach MO (2017). MCT1 inhibitor AZD3965 increases mitochondrial metabolism, facilitating combination therapy and non-invasive magnetic resonance spectroscopy. Cancer Res 77, 5913–5924. 10.1158/0008-5472.CAN-16-2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Benjamin D, Robay D, Hindupur SK, Pohlmann J, Colombi M, El-Shemerly MY, Maira S-M, Moroni C, Lane HA, and Hall MN (2018). Dual Inhibition of the Lactate Transporters MCT1 and MCT4 Is Synthetic Lethal with Metformin due to NAD+ Depletion in Cancer Cells. Cell Rep 25, 3047–3058.e4. 10.1016/j.celrep.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bonglack EN, Messinger JE, Cable JM, Ch’ng J, Parnell KM, Reinoso-Vizcaíno NM, Barry AP, Russell VS, Dave SS, Christofk HR, et al. (2021). Monocarboxylate transporter antagonism reveals metabolic vulnerabilities of viral-driven lymphomas. Proc Natl Acad Sci U S A 118, e2022495118. 10.1073/pnas.2022495118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.DeBerardinis RJ, and Cheng T (2010). Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29, 313–324. 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Altman BJ, Stine ZE, and Dang CV (2016). From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 16, 619–634. 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Akella NM, Ciraku L, and Reginato MJ (2019). Fueling the fire: emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biology 17, 52. 10.1186/s12915-019-0671-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chiaradonna F, Ricciardiello F, and Palorini R (2018). The Nutrient-Sensing Hexosamine Biosynthetic Pathway as the Hub of Cancer Metabolic Rewiring. Cells 7, 53. 10.3390/cells7060053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al. (2009). Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534. 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jewell JL, Kim YC, Russell RC, Yu F-X, Park HW, Plouffe SW, Tagliabracci VS, and Guan K-L (2015). Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347, 194–198. 10.1126/science.1259472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meng D, Yang Q, Wang H, Melick CH, Navlani R, Frank AR, and Jewell JL (2020). Glutamine and asparagine activate mTORC1 independently of Rag GTPases. J Biol Chem 295, 2890–2899. 10.1074/jbc.AC119.011578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, Maseda D, Liberti MV, Paz K, Kishton RJ, et al. (2018). Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 175, 1780–1795.e19. 10.1016/j.cell.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gao P, Tchernyshyov I, Chang T-C, Lee Y-S, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. (2009). c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765. 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang X-Y, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, et al. (2008). Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences 105, 18782–18787. 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sánchez-Rivera FJ, et al. (2017). Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 23, 1362–1368. 10.1038/nm.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Watanabe T, Kobunai T, Yamamoto Y, Matsuda K, Ishihara S, Nozawa K, Iinuma H, Ikeuchi H, and Eshima K (2011). Differential gene expression signatures between colorectal cancers with and without KRAS mutations: Crosstalk between the KRAS pathway and other signalling pathways. European Journal of Cancer 47, 1946–1954. 10.1016/j.ejca.2011.03.029. [DOI] [PubMed] [Google Scholar]
- 86.Najumudeen AK, Ceteci F, Fey SK, Hamm G, Steven RT, Hall H, Nikula CJ, Dexter A, Murta T, Race AM, et al. (2021). The amino acid transporter SLC7A5 is required for efficient growth of KRAS-mutant colorectal cancer. Nat Genet 53, 16–26. 10.1038/s41588-020-00753-3. [DOI] [PubMed] [Google Scholar]
- 87.Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, Mamer OA, Avizonis D, Shackelford DB, Shaw RJ, et al. (2014). Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proceedings of the National Academy of Sciences 111, 2554–2559. 10.1073/pnas.1312570111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tran TQ, Lowman XH, Reid MA, Mendez-Dorantes C, Pan M, Yang Y, and Kong M (2017). Tumor-associated mutant p53 promotes cancer cell survival upon glutamine deprivation through p21 induction. Oncogene 36, 1991–2001. 10.1038/onc.2016.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang R, Li X, Wu Y, Zhang G, Liu X, Li Y, Bao Y, Yang W, and Cui H (2020). EGFR activates GDH1 transcription to promote glutamine metabolism through MEK/ERK/ELK1 pathway in glioblastoma. Oncogene 39, 2975–2986. 10.1038/s41388-020-1199-2. [DOI] [PubMed] [Google Scholar]
- 90.Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG, Dean DC, and Clem BF (2014). Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 33, 556–566. 10.1038/onc.2012.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang L, Achreja A, Yeung T-L, Mangala LS, Jiang D, Han C, Baddour J, Marini JC, Ni J, Nakahara R, et al. (2016). Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metabolism 24, 685–700. 10.1016/j.cmet.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, Turay AM, and Frauwirth KA (2010). Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. Journal of Immunology 185, 1037–1044. 10.4049/jimmunol.0903586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lund PJ, Elias JE, and Davis MM (2016). Global Analysis of O-GlcNAc Glycoproteins in Activated Human T Cells. The Journal of Immunology 197, 3086–3098. 10.4049/jimmunol.1502031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, van Aalten DMF, and Cantrell DA (2016). Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat Immunol 17, 712–720. 10.1038/ni.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Best SA, Gubser PM, Sethumadhavan S, Kersbergen A, Abril YLN, Goldford J, Sellers K, Abeysekera W, Garnham AL, McDonald JA, et al. (2022). Glutaminase inhibition impairs CD8 T cell activation in STK11-/Lkb1-deficient lung cancer. Cell Metabolism 34, 874–887.e6. 10.1016/j.cmet.2022.04.003. [DOI] [PubMed] [Google Scholar]
- 96.Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, Cho SH, Paik Y, Wang Q, Zhang S, et al. (2021). Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest 131. 10.1172/JCI140100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jha AK, Huang SC-C, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, et al. (2015). Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunity 42, 419–430. 10.1016/j.immuni.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 98.Liu P-S, Wang H, Li X, Chao T, Teav T, Christen S, Di Conza G, Cheng W-C, Chou C-H, Vavakova M, et al. (2017). α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol 18, 985–994. 10.1038/ni.3796. [DOI] [PubMed] [Google Scholar]
- 99.Palmieri EM, Menga A, Martín-Pérez R, Quinto A, Riera-Domingo C, De Tullio G, Hooper DC, Lamers WH, Ghesquière B, McVicar DW, et al. (2017). Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep 20, 1654–1666. 10.1016/j.celrep.2017.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lemberg KM, Vornov JJ, Rais R, and Slusher BS (2018). We’re Not “DON” Yet: Optimal Dosing and Prodrug Delivery of 6-Diazo-5-oxo-L-norleucine. Mol. Cancer Ther 17, 1824–1832. 10.1158/1535-7163.MCT-17-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, et al. (2014). Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol Cancer Ther 13, 890–901. 10.1158/1535-7163.MCT-13-0870. [DOI] [PubMed] [Google Scholar]
- 102.Thompson RM, Dytfeld D, Reyes L, Robinson RM, Smith B, Manevich Y, Jakubowiak A, Komarnicki M, Przybylowicz-Chalecka A, Szczepaniak T, et al. (2017). Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 8, 35863–35876. 10.18632/oncotarget.16262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, et al. (2016). Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metabolism 23, 517–528. 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Biancur DE, Paulo JA, Małachowska B, Quiles Del Rey M, Sousa CM, Wang X, Sohn ASW, Chu GC, Gygi SP, Harper JW, et al. (2017). Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nature Communications 8, 1–15. 10.1038/ncomms15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee P, Malik D, Perkons N, Huangyang P, Khare S, Rhoades S, Gong Y-Y, Burrows M, Finan JM, Nissim I, et al. (2020). Targeting glutamine metabolism slows soft tissue sarcoma growth. Nature Communications 11, 1–15. 10.1038/s41467-020-14374-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee C-H, Motzer R, Emamekhoo H, Matrana M, Percent I, Hsieh JJ, Hussain A, Vaishampayan U, Liu S, McCune S, et al. (2022). Telaglenastat plus Everolimus in Advanced Renal Cell Carcinoma: A Randomized, Double-Blinded, Placebo-Controlled, Phase II ENTRATA Trial. Clin Cancer Res 28, 3248–3255. 10.1158/1078-0432.CCR-22-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tannir NM, Agarwal N, Porta C, Lawrence NJ, Motzer R, McGregor B, Lee RJ, Jain RK, Davis N, Appleman LJ, et al. (2022). Efficacy and Safety of Telaglenastat Plus Cabozantinib vs Placebo Plus Cabozantinib in Patients With Advanced Renal Cell Carcinoma: The CANTATA Randomized Clinical Trial. JAMA Oncol. 10.1001/jamaoncol.2022.3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Leone RD, Zhao L, Englert JM, Sun I-M, Oh M-H, Sun I-H, Arwood ML, Bettencourt IA, Patel CH, Wen J, et al. (2019). Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021. 10.1126/science.aav2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Oh M-H, Sun I-H, Zhao L, Leone RD, Sun I-M, Xu W, Collins SL, Tam AJ, Blosser RL, Patel CH, et al. (2020). Targeting glutamine metabolism enhances tumor specific immunity by modulating suppressive myeloid cells. J Clin Invest. 10.1172/JCI131859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yokoyama Y, Estok TM, and Wild R (2022). Sirpiglenastat (DRP-104) Induces Antitumor Efficacy through Direct, Broad Antagonism of Glutamine Metabolism and Stimulation of the Innate and Adaptive Immune Systems. Molecular Cancer Therapeutics 21, 1561–1572. 10.1158/1535-7163.MCT-22-0282. [DOI] [PubMed] [Google Scholar]
- 111.Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, Kondo J, Coffey RJ, Johnson MO, Rathmell JC, et al. (2018). Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med 24, 194–202. 10.1038/nm.4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Byun J-K, Park M, Lee S, Yun JW, Lee J, Kim JS, Cho SJ, Jeon H-J, Lee I-K, Choi Y-K, et al. (2020). Inhibition of Glutamine Utilization Synergizes with Immune Checkpoint Inhibitor to Promote Antitumor Immunity. Molecular Cell 80, 592–606.e8. 10.1016/j.molcel.2020.10.015. [DOI] [PubMed] [Google Scholar]
- 113.Li Q, Zhong X, Yao W, Yu J, Wang C, Li Z, Lai S, Qu F, Fu X, Huang X, et al. (2022). Inhibitor of glutamine metabolism V9302 promotes ROS-induced autophagic degradation of B7H3 to enhance antitumor immunity. J Biol Chem 298, 101753. 10.1016/j.jbc.2022.101753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tang Y, Wang S, Li Y, Yuan C, Zhang J, Xu Z, Hu Y, Shi H, and Wang S (2022). Simultaneous glutamine metabolism and PD-L1 inhibition to enhance suppression of triple-negative breast cancer. J Nanobiotechnology 20, 216. 10.1186/s12951-022-01424-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu Y, Ge X, Pang J, Zhang Y, Zhang H, Wu H, Fan F, and Liu H (2021). Restricting Glutamine Uptake Enhances NSCLC Sensitivity to Third-Generation EGFR-TKI Almonertinib. Front Pharmacol 12, 671328. 10.3389/fphar.2021.671328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim G, Jang S-K, Kim YJ, Jin H-O, Bae S, Hong J, Park I-C, and Lee JH (2022). Inhibition of Glutamine Uptake Resensitizes Paclitaxel Resistance in SKOV3-TR Ovarian Cancer Cell via mTORC1/S6K Signaling Pathway. Int J Mol Sci 23, 8761. 10.3390/ijms23158761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Prelowska MK, Mehlich D, Ugurlu MT, Kedzierska H, Cwiek A, Kosnik A, Kaminska K, Marusiak AA, and Nowis D (2021). Inhibition of the ʟ-glutamine transporter ASCT2 sensitizes plasma cell myeloma cells to proteasome inhibitors. Cancer Letters 507, 13–25. 10.1016/j.canlet.2021.02.020. [DOI] [PubMed] [Google Scholar]
- 118.Jin H, Wang S, Zaal EA, Wang C, Wu H, Bosma A, Jochems F, Isima N, Jin G, Lieftink C, et al. (2020). A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. Elife 9, e56749. 10.7554/eLife.56749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lieu EL, Nguyen T, Rhyne S, and Kim J (2020). Amino acids in cancer. Exp Mol Med 52, 15–30. 10.1038/s12276-020-0375-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim J, and DeBerardinis RJ (2019). Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metabolism 30, 434–446. 10.1016/j.cmet.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li AM, and Ye J (2020). The PHGDH enigma: Do cancer cells only need serine or also a redox modulator? Cancer Letters 476, 97–105. 10.1016/j.canlet.2020.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pacold ME, Brimacombe KR, Chan SH, Rohde JM, Lewis CA, Swier LJYM, Possemato R, Chen WW, Sullivan LB, Fiske BP, et al. (2016). A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nature Chemical Biology 12, 452–458. 10.1038/nchembio.2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mullarky E, Lucki NC, Zavareh RB, Anglin JL, Gomes AP, Nicolay BN, Wong JCY, Christen S, Takahashi H, Singh PK, et al. (2016). Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proceedings of the National Academy of Sciences of the United States of America 113, 1778–783. 10.1073/pnas.1521548113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pieters R, Hunger SP, Boos J, Rizzari C, Silverman L, Baruchel A, Goekbuget N, Schrappe M, and Pui C-H (2011). L-asparaginase treatment in acute lymphoblastic leukemia: A focus on Erwinia asparaginase. Cancer 117, 238–249. 10.1002/cncr.25489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, Yang Y, Hernandez-Davies JE, Rosales KK, Li H, et al. (2016). Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol 18, 1090–1101. 10.1038/ncb3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. 10.1038/nature19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ehsanipour EA, Sheng X, Behan JW, Wang X, Butturini A, Avramis VI, and Mittelman SD (2013). Adipocytes cause leukemia cell resistance to L-asparaginase via release of glutamine. Cancer Res 73, 2998–3006. 10.1158/0008-5472.CAN-12-4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Iwamoto S, Mihara K, Downing JR, Pui C-H, and Campana D (2007). Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J Clin Invest 117, 1049–1057. 10.1172/JCI30235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Parker SJ, Amendola CR, Hollinshead KER, Yu Q, Yamamoto K, Encarnación-Rosado J, Rose RE, LaRue MM, Sohn ASW, Biancur DE, et al. (2020). Selective Alanine Transporter Utilization Creates a Targetable Metabolic Niche in Pancreatic Cancer. Cancer Discovery 10, 1018–1037. 10.1158/2159-8290.CD-19-0959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhu Z, Achreja A, Meurs N, Animasahun O, Owen S, Mittal A, Parikh P, Lo T-W, Franco-Barraza J, Shi J, et al. (2020). Tumour-reprogrammed stromal BCAT1 fuels branched-chain ketoacid dependency in stromal-rich PDAC tumours. Nat Metab 2, 775–792. 10.1038/s42255-020-0226-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yeung T-L, Leung CS, Wong K-K, Samimi G, Thompson MS, Liu J, Zaid TM, Ghosh S, Birrer MJ, and Mok SC (2013). TGF-β modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res 73, 5016–5028. 10.1158/0008-5472.CAN-13-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Banh RS, Biancur DE, Yamamoto K, Sohn ASW, Walters B, Kuljanin M, Gikandi A, Wang H, Mancias JD, Schneider RJ, et al. (2020). Neurons Release Serine to Support mRNA Translation in Pancreatic Cancer. Cell 183, 1202–1218.e25. 10.1016/j.cell.2020.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Commisso C, and Debnath J (2018). Macropinocytosis Fuels Prostate Cancer. Cancer Discovery 8, 800–802. 10.1158/2159-8290.CD-18-0513. [DOI] [PubMed] [Google Scholar]
- 135.Michalopoulou E, Auciello FR, Bulusu V, Strachan D, Campbell AD, Tait-Mulder J, Karim SA, Morton JP, Sansom OJ, and Kamphorst JJ (2020). Macropinocytosis Renders a Subset of Pancreatic Tumor Cells Resistant to mTOR Inhibition. Cell Rep 30, 2729–2742.e4. 10.1016/j.celrep.2020.01.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang MS, Cui JD, Lee D, Yuen VW-H, Chiu DK-C, Goh CC, Cheu JW-S, Tse AP-W, Bao MH-R, Wong BPY, et al. (2022). Hypoxia-induced macropinocytosis represents a metabolic route for liver cancer. Nat Commun 13, 954. 10.1038/s41467-022-28618-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang Y, Recouvreux MV, Jung M, Galenkamp KMO, Li Y, Zagnitko O, Scott DA, Lowy AM, and Commisso C (2021). Macropinocytosis in Cancer-Associated Fibroblasts Is Dependent on CaMKK2/ARHGEF2 Signaling and Functions to Support Tumor and Stromal Cell Fitness. Cancer Discovery 11, 1808–1825. 10.1158/2159-8290.CD-20-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chavez-Dominguez R, Perez-Medina M, Lopez-Gonzalez JS, Galicia-Velasco M, and Aguilar-Cazares D (2020). The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Front. Oncol 10, 578418. 10.3389/fonc.2020.578418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mizushima N (2007). Autophagy: process and function. Genes Dev. 21, 2861–2873. 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 140.Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD, et al. (2019). Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med 25, 628–640. 10.1038/s41591-019-0368-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, Deng J, Hai J, Yang S, Wong KK, et al. (2018). Autophagy Sustains Pancreatic Cancer Growth through Both Cell-Autonomous and Nonautonomous Mechanisms. Cancer Discovery 8, 276–287. 10.1158/2159-8290.CD-17-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mukhopadhyay S, Biancur DE, Parker SJ, Yamamoto K, Banh RS, Paulo JA, Mancias JD, and Kimmelman AC (2021). Autophagy is required for proper cysteine homeostasis in pancreatic cancer through regulation of SLC7A11. Proc. Natl. Acad. Sci. U.S.A 118, e2021475118. 10.1073/pnas.2021475118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Santana-Codina N, del Rey MQ, Kapner KS, Zhang H, Gikandi A, Malcolm C, Poupault C, Kuljanin M, John KM, Biancur DE, et al. (2022). NCOA4-Mediated Ferritinophagy Is a Pancreatic Cancer Dependency via Maintenance of Iron Bioavailability for Iron–Sulfur Cluster Proteins. Cancer Discovery 12, 2180–2197. 10.1158/2159-8290.CD-22-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Snaebjornsson MT, Janaki-Raman S, and Schulze A (2020). Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metabolism 31, 62–76. 10.1016/j.cmet.2019.11.010. [DOI] [PubMed] [Google Scholar]
- 145.Röhrig F, and Schulze A (2016). The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 16, 732–749. 10.1038/nrc.2016.89. [DOI] [PubMed] [Google Scholar]
- 146.Ogino S, Nosho K, Meyerhardt JA, Kirkner GJ, Chan AT, Kawasaki T, Giovannucci EL, Loda M, and Fuchs CS (2008). Cohort Study of Fatty Acid Synthase Expression and Patient Survival in Colon Cancer. JCO 26, 5713–5720. 10.1200/JCO.2008.18.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rossi S, Graner E, Febbo P, Weinstein L, Bhattacharya N, Onody T, Bubley G, Balk S, and Loda M (2003). Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol Cancer Res 1, 707–715. [PubMed] [Google Scholar]
- 148.Visca P, Sebastiani V, Botti C, Diodoro MG, Lasagni RP, Romagnoli F, Brenna A, De Joannon BC, Donnorso RP, Lombardi G, et al. (2004). Fatty acid synthase (FAS) is a marker of increased risk of recurrence in lung carcinoma. Anticancer Res 24, 4169–4173. [PubMed] [Google Scholar]
- 149.Falchook G, Infante J, Arkenau H-T, Patel MR, Dean E, Borazanci E, Brenner A, Cook N, Lopez J, Pant S, et al. (2021). First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine 34, 100797. 10.1016/j.eclinm.2021.100797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kim J, Tchernyshyov I, Semenza GL, and Dang CV (2006). HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metabolism 3, 177–185. 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 151.Papandreou I, Cairns RA, Fontana L, Lim AL, and Denko NC (2006). HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metabolism 3, 187–197. 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 152.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. (2012). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384. 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kamphorst JJ, Chung MK, Fan J, and Rabinowitz JD (2014). Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer & Metabolism 2, 23. 10.1186/2049-3002-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, et al. (2015). Acetyl-CoA Synthetase 2 Promotes Acetate Utilization and Maintains Cancer Cell Growth under Metabolic Stress. Cancer Cell 27, 57–71. 10.1016/j.ccell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. http://genesdev.cshlp.org/content/27/10/1115. [DOI] [PMC free article] [PubMed]
- 156.Li Z, Ji BW, Dixit PD, Tchourine K, Lien EC, Hosios AM, Abbott KL, Rutter JC, Westermark AM, Gorodetsky EF, et al. (2022). Cancer cells depend on environmental lipids for proliferation when electron acceptors are limited. Nat Metab 4, 711–723. 10.1038/s42255-022-00588-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li F, and Simon MC (2020). Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Developmental Cell 54, 183–195. 10.1016/j.devcel.2020.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jain IH, Calvo SE, Markhard AL, Skinner OS, To T-L, Ast T, and Mootha VK (2020). Genetic Screen for Cell Fitness in High or Low Oxygen Highlights Mitochondrial and Lipid Metabolism. Cell 181, 716–727.e11. 10.1016/j.cell.2020.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li J-L, et al. (2014). Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep 9, 349–365. 10.1016/j.celrep.2014.08.056. [DOI] [PubMed] [Google Scholar]
- 160.Wang C, Shi M, Ji J, Cai Q, Zhao Q, Jiang J, Liu J, Zhang H, Zhu Z, and Zhang J (2020). Stearoyl-CoA desaturase 1 (SCD1) facilitates the growth and anti-ferroptosis of gastric cancer cells and predicts poor prognosis of gastric cancer. Aging 12, 15374–15391. 10.18632/aging.103598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bansal S, Berk M, Alkhouri N, Partrick DA, Fung JJ, and Feldstein A (2014). Stearoyl-CoA desaturase plays an important role in proliferation and chemoresistance in human hepatocellular carcinoma. J Surg Res 186, 29–38. 10.1016/j.jss.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Noto A, De Vitis C, Pisanu ME, Roscilli G, Ricci G, Catizone A, Sorrentino G, Chianese G, Taglialatela-Scafati O, Trisciuoglio D, et al. (2017). Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 36, 4671–4672. 10.1038/onc.2017.212. [DOI] [PubMed] [Google Scholar]
- 163.Lien EC, Westermark AM, Zhang Y, Yuan C, Li Z, Lau AN, Sapp KM, Wolpin BM, and Vander Heiden MG (2021). Low glycaemic diets alter lipid metabolism to influence tumour growth. Nature 599, 302–307. 10.1038/s41586-021-04049-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Vriens K, Christen S, Parik S, Broekaert D, Yoshinaga K, Talebi A, Dehairs J, Escalona-Noguero C, Schmieder R, Cornfield T, et al. (2019). Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 566, 403–406. 10.1038/s41586-019-0904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids | PNAS. https://www.pnas.org/doi/10.1073/pnas.1307237110. [DOI] [PMC free article] [PubMed]
- 166.Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, Tumanov S, Allen-Petersen BL, Link J, Kendsersky ND, et al. (2019). A stromal lysolipid-autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discov 9, 617–627. 10.1158/2159-8290.CD-18-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mills GB, and Moolenaar WH (2003). The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer 3, 582–591. 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 168.Gong J, Lin Y, Zhang H, Liu C, Cheng Z, Yang X, Zhang J, Xiao Y, Sang N, Qian X, et al. (2020). Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis 11, 267. 10.1038/s41419-020-2434-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Peng S, Li Y, Huang M, Tang G, Xie Y, Chen D, Hu Y, Yu T, Cai J, Yuan Z, et al. (2022). Metabolomics reveals that CAF-derived lipids promote colorectal cancer peritoneal metastasis by enhancing membrane fluidity. Int J Biol Sci 18, 1912–1932. 10.7150/ijbs.68484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, Shtein HC, Cairns R, Thomas KC, Fazakerley DJ, Grewal T, et al. (2017). Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer & Metabolism 5, 1. 10.1186/s40170-016-0163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2 | Science Translational Medicine. https://www.science.org/doi/10.1126/scitranslmed.aag0945. [DOI] [PMC free article] [PubMed]
- 172.Laurent V, Guérard A, Mazerolles C, Le Gonidec S, Toulet A, Nieto L, Zaidi F, Majed B, Garandeau D, Socrier Y, et al. (2016). Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat Commun 7, 10230. 10.1038/ncomms10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, Wang YY, Meulle A, Salles B, Le Gonidec S, et al. (2011). Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res 71, 2455–2465. 10.1158/0008-5472.CAN-10-3323. [DOI] [PubMed] [Google Scholar]
- 174.Cao Y Adipocyte and lipid metabolism in cancer drug resistance. J Clin Invest 129, 3006–3017. 10.1172/JCI127201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, Nieman KM, Pascual G, Benitah SA, Montag A, et al. (2018). Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 37, 2285–2301. 10.1038/s41388-017-0093-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tabe Y, Yamamoto S, Saitoh K, Sekihara K, Monma N, Ikeo K, Mogushi K, Shikami M, Ruvolo V, Ishizawa J, et al. (2017). Bone Marrow Adipocytes Facilitate Fatty Acid Oxidation Activating AMPK and a Transcriptional Network Supporting Survival of Acute Monocytic Leukemia Cells. Cancer Research 77, 1453–1464. 10.1158/0008-5472.CAN-16-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kuemmerle NB, Rysman E, Lombardo PS, Flanagan AJ, Lipe BC, Wells WA, Pettus JR, Froehlich HM, Memoli VA, Morganelli PM, et al. (2011). Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol Cancer Ther 10, 427–436. 10.1158/1535-7163.MCT-10-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chen X, and Cubillos-Ruiz JR (2021). Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer 21, 71–88. 10.1038/s41568-020-00312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Koumenis C, and Wouters BG (2006). “Translating” Tumor Hypoxia: Unfolded Protein Response (UPR)–Dependent and UPR-Independent Pathways. Molecular Cancer Research 4, 423–436. 10.1158/1541-7786.MCR-06-0150. [DOI] [PubMed] [Google Scholar]
- 180.Zhao N, Cao J, Xu L, Tang Q, Dobrolecki LE, Lv X, Talukdar M, Lu Y, Wang X, Hu DZ, et al. (2018). Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. Journal of Clinical Investigation 128, 1283–1299. 10.1172/JCI95873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Atkins C, Liu Q, Minthorn E, Zhang S-Y, Figueroa DJ, Moss K, Stanley TB, Sanders B, Goetz A, Gaul N, et al. (2013). Characterization of a Novel PERK Kinase Inhibitor with Antitumor and Antiangiogenic Activity. Cancer Research 73, 1993–2002. 10.1158/0008-5472.CAN-12-3109. [DOI] [PubMed] [Google Scholar]
- 182.Xie H, Tang C-HA, Song JH, Mancuso A, Del Valle JR, Cao J, Xiang Y, Dang CV, Lan R, Sanchez DJ, et al. (2018). IRE1α RNase–dependent lipid homeostasis promotes survival in Myc-transformed cancers. Journal of Clinical Investigation 128, 1300–1316. 10.1172/JCI95864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, et al. (2014). XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 508, 103–107. 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tameire F, Verginadis II, Leli NM, Polte C, Conn CS, Ojha R, Salas Salinas C, Chinga F, Monroy Alexandra. M, Fu W, et al. (2019). ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat Cell Biol 21, 889–899. 10.1038/s41556-019-0347-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Genovese G, Carugo A, Tepper J, Robinson FS, Li L, Svelto M, Nezi L, Corti D, Minelli R, Pettazzoni P, et al. (2017). Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature 542, 362–366. 10.1038/nature21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Feng Y, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JHL, Proia TA, Jin DX, Reinhardt F, Ploegh HL, Wang Q, et al. (2014). Epithelial-to-Mesenchymal Transition Activates PERK–eIF2α and Sensitizes Cells to Endoplasmic Reticulum Stress. Cancer Discovery 4, 702–715. 10.1158/2159-8290.CD-13-0945. [DOI] [PubMed] [Google Scholar]
- 187.Dey S, Sayers CM, Verginadis II, Lehman SL, Cheng Y, Cerniglia GJ, Tuttle SW, Feldman MD, Zhang PJL, Fuchs SY, et al. (2015). ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Invest 125, 2592–2608. 10.1172/JCI78031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Avril T, Vauléon E, and Chevet E (2017). Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 6, e373–e373. 10.1038/oncsis.2017.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Qiu B, Ackerman D, Sanchez DJ, Li B, Ochocki JD, Grazioli A, Bobrovnikova-Marjon E, Diehl JA, Keith B, and Simon MC (2015). HIF-2α dependent lipid storage promotes endoplasmic reticulum homeostasis in clear cell renal cell carcinoma. Cancer Discov 5, 652–667. 10.1158/2159-8290.CD-14-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Costa-Mattioli M, and Walter P (2020). The integrated stress response: From mechanism to disease. Science 368, eaat5314. 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Nguyen HG, Conn CS, Kye Y, Xue L, Forester CM, Cowan JE, Hsieh AC, Cunningham JT, Truillet C, Tameire F, et al. (2018). Development of a stress response therapy targeting aggressive prostate cancer. Sci. Transl. Med 10, eaar2036. 10.1126/scitranslmed.aar2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Darini C, Ghaddar N, Chabot C, Assaker G, Sabri S, Wang S, Krishnamoorthy J, Buchanan M, Aguilar-Mahecha A, Abdulkarim B, et al. (2019). An integrated stress response via PKR suppresses HER2+ cancers and improves trastuzumab therapy. Nat Commun 10, 2139. 10.1038/s41467-019-10138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Stone S, Ho Y, Li X, Jamison S, Harding HP, Ron D, and Lin W (2016). Dual role of the integrated stress response in medulloblastoma tumorigenesis. Oncotarget 7, 64124–64135. 10.18632/oncotarget.11873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.May D, Itin A, Gal O, Kalinski H, Feinstein E, and Keshet E (2005). Ero1-L alpha plays a key role in a HIF-1-mediated pathway to improve disulfide bond formation and VEGF secretion under hypoxia: implication for cancer. Oncogene 24, 1011–1020. 10.1038/sj.onc.1208325. [DOI] [PubMed] [Google Scholar]
- 195.Ackerman D, and Simon MC (2014). Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends in Cell Biology 24, 472–478. 10.1016/j.tcb.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ackerman D, Tumanov S, Qiu B, Michalopoulou E, Spata M, Azzam A, Xie H, Simon MC, and Kamphorst JJ (2018). Triglycerides Promote Lipid Homeostasis during Hypoxic Stress by Balancing Fatty Acid Saturation. Cell Rep 24, 2596–2605.e5. 10.1016/j.celrep.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Volmer R, van der Ploeg K, and Ron D (2013). Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proceedings of the National Academy of Sciences 110, 4628–4633. 10.1073/pnas.1217611110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Griffiths B, Lewis CA, Bensaad K, Ros S, Zhang Q, Ferber EC, Konisti S, Peck B, Miess H, East P, et al. (2013). Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer & Metabolism 1, 3. 10.1186/2049-3002-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Carroll CP, Bolland H, Vancauwenberghe E, Collier P, Ritchie AA, Clarke PA, Grabowska AM, Harris AL, and McIntyre A (2022). Targeting hypoxia regulated sodium driven bicarbonate transporters reduces triple negative breast cancer metastasis. Neoplasia 25, 41–52. 10.1016/j.neo.2022.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Chen Y, and Brandizzi F (2013). IRE1: ER stress sensor and cell fate executor. Trends in Cell Biology 23, 547–555. 10.1016/j.tcb.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Tang C-HA, Ranatunga S, Kriss CL, Cubitt CL, Tao J, Pinilla-Ibarz JA, Del Valle JR, and Hu C-CA (2014). Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J Clin Invest 124, 2585–2598. 10.1172/JCI73448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Shao A, Xu Q, Spalek WT, Cain CF, Kang CW, Tang C-HA, Del Valle JR, and Hu C-CA (2020). Development of Tumor-Targeting IRE-1 Inhibitors for B-cell Cancer Therapy. Molecular Cancer Therapeutics 19, 2432–2444. 10.1158/1535-7163.MCT-20-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zundell JA, Fukumoto T, Lin J, Fatkhudinov N, Nacarelli T, Kossenkov AV, Liu Q, Cassel J, Hu C-CA, Wu S, et al. (2021). Targeting the IRE1α/XBP1 Endoplasmic Reticulum Stress Response Pathway in ARID1A-Mutant Ovarian Cancers. Cancer Res 81, 5325–5335. 10.1158/0008-5472.CAN-21-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sheng X, Nenseth HZ, Qu S, Kuzu OF, Frahnow T, Simon L, Greene S, Zeng Q, Fazli L, Rennie PS, et al. (2019). IRE1α-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat Commun 10, 323. 10.1038/s41467-018-08152-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J, and Anderson P (2002). Evidence That Ternary Complex (eIF2-GTP-tRNAi Met)–Deficient Preinitiation Complexes Are Core Constituents of Mammalian Stress Granules. MBoC 13, 195–210. 10.1091/mbc.01-05-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kilberg MS, Shan J, and Su N (2009). ATF4-DEPENDENT TRANSCRIPTION MEDIATES SIGNALING OF AMINO ACID LIMITATION. Trends Endocrinol Metab 20, 436–443. 10.1016/j.tem.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, Kim Y-J, Lee H-K, Cortese JF, Wirth DF, et al. (2012). Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol 8, 311–317. 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Nakamura A, Nambu T, Ebara S, Hasegawa Y, Toyoshima K, Tsuchiya Y, Tomita D, Fujimoto J, Kurasawa O, Takahara C, et al. (2018). Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proceedings of the National Academy of Sciences 115, E7776–E7785. 10.1073/pnas.1805523115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Missiaen R, Anderson NM, Kim LC, Nance B, Burrows M, Skuli N, Carens M, Riscal R, Steensels A, Li F, et al. (2022). GCN2 inhibition sensitizes arginine-deprived hepatocellular carcinoma cells to senolytic treatment. Cell Metab 34, 1151–1167.e7. 10.1016/j.cmet.2022.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]