

Abstract
Sensorimotor gating disruptions have been noted in several psychiatric and neurodegenerative disorders. However, the involvement of sensorimotor gating processes in eating disorders has not been well characterized. Our objective was to examine the sensorimotor gating of the acoustic startle response following dietary-induced binge eating and high-fat diet (HFD) induced weight gain in male C57B/6J mice. Acute administration of the norepinephrine reuptake inhibitor, nisoxetine (0.5 and 5 mg/kg), and a dopamine reuptake inhibitor, GBR 12783 (1.6 and 16mg/kg), were either given alone or in combination to assess norepinephrine and dopamine alterations, respectively. Male mice with repeated bouts of calorie restriction (Restrict) and with limited access to a sweetened fat food (Binge) demonstrated an escalation of intake over 2.5 weeks under standard chow conditions. Restrict Binge (RB) mice had a reduced startle response to the startle pulse (110 dB) compared with the Naive control group at 5 mg/kg nisoxetine. There was an overall effect of nisoxetine (0.5 and 5 mg/kg) to increase percent inhibition at pre-pulse (74 dB), %PP74. Under HFD conditions, the RB group did not demonstrate a binge-like eating phenotype. The RB group on HFD had a higher response to 74 dB with nisoxetine (5.0 mg/kg) compared with a combinational dose of nisoxetine (5.0 mg/kg) and GBR 12783 (1.6 mg/kg). These findings suggest that dietary conditions that promote binge-like eating can influence the central noradrenergic and dopaminergic controls of the acoustic startle response and potentially influence sensorimotor gating.
Keywords: eating pathologies, rodent models bulimia nervosa, binge eating, eating disorders, pre-attentive processing, sensory motor integration
Introduction
Sensorimotor gating is the neural process by which sensory information is integrated and transmitted to motor output. Sensorimotor gating paradigms have been used to uncover disease-related neural impairments, as well as screen for novel pharmacotherapies (Mansbach, Geyer, and Braff 1988; Peres et al. 2016). Disruption in sensorimotor gating has been demonstrated with weight gain and is associated with psychiatric disorders including obsessive compulsive disorder, post-traumatic stress disorder, and most notably, schizophrenia (Ahmari et al. 2012; Bakshi et al. 2012; Braff, Callaway, and Naylor 1977). Sensorimotor gating can be behaviorally assessed by pre-pulse inhibition (PPI) of the startle reflex (Geyer et al. 2001; Labouesse et al. 2013; Li, Kshatriya, and Bello 2022; Ouagazzal, Jenck, and Moreau 2001). PPI is quantified as a reduced response to a startle stimulus (pulse) when preceded by another stimulus of lesser intensity (pre-pulse) (Graham 1975; Hoffman and Searle 1968). As such, sensorimotor gating methods using PPI have clinically relevant outcomes in rodent models (Geyer et al. 2001; Ouagazzal, Jenck, and Moreau 2001).
Diet-induced weight gain by high-fat diet (HFD) consumption is associated with metabolic dysfunction (Borkman et al. 1991; Bray 1991). Animal and human studies demonstrate that HFD also impairs cognitive function by acting on the hippocampus and prefrontal cortex (Francis and Stevenson 2013; Morris et al. 2004; Morrison et al. 2010). Owing to the effect that HFD has on the brain, evidence suggests that HFD also contributes to neurodegenerative diseases including Alzheimer’s and Parkinson’s disease (Johnson et al. 1999; Morris et al. 2003). Along these lines, disruption in PPI has been demonstrated with diet-induced weight gain (Labouesse et al. 2013; Harrington and Coscina 1983). It has been reported that startle response positively correlates with weight gain following 8 days of exposure to high-fat food in adult rats (Harrington and Coscina 1983). Additionally, chronic consumption of HFD during different developmental windows impairs the startle response in mice (Labouesse et al. 2013). Despite these findings, the influences of specific dietary conditions, such as highly palatable food access and caloric restriction, on sensorimotor outcomes have not been investigated.
Impairments in sensorimotor gating have been associated with repetitive behaviors and symptoms associated psychiatric disorders, but the relationship is not clear (Ahmari et al. 2012; Steinman et al. 2016). Previous investigations with rodents have utilized recurrent bouts of intermittent access to sweetened fat food and calorie restriction to promote binge-like eating (Sachdeo et al. 2019; Bello, Yeh, and James 2019; Corwin, Avena, and Boggiano 2011; Cottone et al. 2008). Recurrent binge eating is a central eating pathology of clinically defined eating disorders, such as bulimia nervosa (BN) and binge eating disorder (BED) (Association 2013). Although clinical binge eating is accompanied by “a sense of a loss of control,” rodent models can provide insight into the consequences of dietary constraints that promote the driven nature of disordered eating (Association 2013; Corwin, Avena, and Boggiano 2011; Hildebrandt and Ahmari 2021; Heal and Gosden 2022). The overall hypothesis of these experiments is that recurrent dietary conditions that promote binge eating lead to impairments in sensorimotor gating.
A major contributor to binge-like eating is that binge foods are highly palatable, typically with a high fat and sugar content (Elmore and de Castro 1991; Gibson 2006; Greeno and Wing 1994). Repeated consumption of highly palatable food has reinforcing and anxiolytic effects, which contribute to the motivational saliency of these foods (Foster et al. 2009; Pecoraro et al. 2004; George et al. 1990; Kaye et al. 1990; Rinaman 2011; Ritter, Pelzer, and Ritter 1978; Valentino et al. 1998). Notably, alterations in central monoamines, such as dopamine and norepinephrine, have been implicated as the underlining neural substrate for motivational saliency of highly palatable foods (Billes and Cowley 2007; Manning et al. 2021; Nobrega and Coscina 1986; Cannon et al. 2004). One unexplored way to assess the neural consequences of palatable food could be to use a sensorimotor gating paradigm. Specifically, it has been shown that disruptions in both central dopamine and norepinephrine targets have impaired sensorimotor gating responses in rodents (Alsene et al. 2011; Manning et al. 2021). In addition, selective norepinephrine reuptake inhibitors (e.g., atomoxetine) and non-selective norepinephrine/dopamine reuptake inhibitors (e.g., methylphenidate) restored drug-induced sensorimotor gating deficits (Woo et al. 2014).
The objective of this novel study was to investigate the influence of dietary-induced binge eating and high-fat diet feeding on the sensorimotor gating of an acoustic startle response. We also examined whether we could influence alterations in the sensorimotor gating responses. This was done by the acute administration of a central-acting norepinephrine reuptake inhibitor, nisoxetine, and a central-acting dopamine reuptake inhibitor, GBR 12783, alone or in combination.
Methods
Subjects
A total of 40 C57BL/6J male mice (Jackson Laboratories, Bar Harbor, ME; Cat. No. 000664) were obtained at 6 weeks of age and acclimated for one week. Mice were single-housed and maintained on a 12-hour (0500 h: lights on, 1700 h: lights off) light-dark cycle throughout the entire study. Mice received ad libitum standard rodent chow (LabDiet 5001; 13.5% fat, 28.5% protein; 58% carbohydrates; 3.36 Kcal/g; St. Louis, MO) unless otherwise specified, and water was available ad libitum throughout. All procedures performed on mice were approved by the Rutgers University IACUC.
Pre-pulse Inhibition (PPI)
All mice underwent multiple PPI testing. PPI testing was conducted in four fan-ventilated, sound-attenuated test chambers (50.8-cm W × 33-cm H × 30.5-cm D) (Med Associates Inc., St. Albans, VT). Each chamber housed an animal holder (6.025-cm L × 6.19-cm W × 4.8-cm H) situated atop a piezoelectric accelerometric platform table. The mice were exposed to six different types of stimuli: (1) null: background noise only (200 ms), (2) P74: low pre-pulse (74 dB/20 ms) given alone, (3) P90: high pre-pulse (90 dB/20 ms) given alone, (4) PP74: P74 given 100 ms prior to the onset of the startle stimuli, (5) PP90: P90 given 100 ms prior to the onset of the startle stimuli, (6) and the startle stimulus (110 dB/ 40 ms). All trials were applied ten times and presented in random order (P74 and P90 were only given five times) and the average inter-trial interval was 15 s (10–20 s) per session. Percent pre-pulse inhibition with pre-pulse intensities of 74 (%PP74) and 90 db (%PP90). Percent pre-pulse inhibition was calculated as: [(pre-pulse preceding startle stimulus-startle stimulus alone) / (startle stimulus alone)] * 100. The PPI testing protocol has been previously described (Ouagazzal, Jenck, and Moreau 2001; Kshatriya et al. 2020).
Dietary-induced binge eating on standard chow.
At 7 weeks of age, all mice underwent an initial, baseline PPI testing. Seven days later, all mice were subjected to a 24-hour pre-exposure to ~5g of sweetened fat (8.6 kcal/g) comprised of vegetable shortening (Crisco, The J.M. Smucker Company, Orrville, OH) and 10% sucrose (Domino Sugar; American Sugar Refining) as well as ~5g of standard chow. At 9 weeks of age, mice were assigned to four feeding groups. Feeding groups were based on two independent dietary conditions, intermittent calorie deprivation (24 h) and intermittent sweetened fat access (30 min). Intermittent calorie deprivation (24 h) occurred on Days 2 and 5 at 1430 h, while the refeeding with standard chow and 30-min access to the sweetened fat (i.e., “Binge”) occurred at 1430 h on Days 3 and 6 of the 7-day feeding schedule. Days 1, 4, and 7 were ad libitum standard chow access with no calorie deprivation for all groups (Sachdeo et al. 2019). The feeding groups protocol were as follows: Restrict Binge (RB) group had both intermittent calorie restriction (Days 2 and 5) and sweetened fat access (Days 3 and 6). Binge (B) group had sweetened fat access (Days 3 and 6) without any calorie restriction. Restrict (R) group had intermittent calorie restriction (Days 2 and 5) without any repeated access to the sweetened fat. Naive (N) group had neither repeated access to sweetened fat nor calorie restriction, see Table 1. Body weights and food intakes were weighed to the nearest 0.01 g. After 2.5 weeks, all mice underwent a second PPI testing on day 6 of the feeding protocol. The 2.5 week time point was chosen because we previously observed an escalation intake with this dietary binge eating paradigm in mice (Sachdeo et al. 2019) and rats (Bello, Patinkin, and Moran 2011; Bello, Walters, et al. 2014; Bello, Yeh, and James 2019; Bello, Yeh, et al. 2014). The 7-day feeding schedule was repeated weekly unless otherwise noted.
Table 1. Dietary-induced binge eating feeding protocols.
Seven-day feeding protocol for mice maintained on standard chow or high-fat diet (45% fat). Days 1, 4, and 7 were ad libitum feeding for all groups. Sweetened fat was vegetable shortening blended with 10% sucrose (8.6 kcal/g). All groups were n = 10.
| Groups | Calorie Restriction (Days 2, 5) | Sweetened Fat (Days 3, 6) |
|---|---|---|
| Restrict Binge (RB) | Intermittent (24 h) | 30 min |
| Binge (B) | None | 30 min |
| Restrict (R) | Intermittent (24 h) | None |
| Naive (N) | None | None |
Nisoxetine and GBR 12783 testing under standard chow conditions.
Following this within subject design, mice continued their respective feeding protocol and had PPI testing every 10 days. Testing occurred on Day 3 or Day 6 of the feeding protocol at 1-h post-intraperitoneal (IP) injection. All animals received seven different treatments as follows in order of injection: (1) saline; (2) nisoxetine (NIS) (Tocris, Cat. No. 1025, Bristol, United Kingdom) [0.5 mg/kg]; (3) NIS [5 mg/kg]; (4) GBR 12783 dihydrochloride (GBR) (Tocris, Cat. No. 0513, Bristol, United Kingdom) [1.6 mg/kg] in 10% DMSO saline; (5) GBR [16 mg/kg] in 10% DMSO saline; (6) 10% DMSO saline; and (7) combination of NIS [5 mg/kg] and GBR [1.6 mg/kg] in 10% DMSO saline, see Figure 1A.
Figure 1. Design of PPI schedule and dosing experiments.

Each box represents a sensorimotor gating session of an acoustic startle. PPI sessions were performed on Days 3 and 6 of the feeding protocols, see Table 1. Notably, RB and B mice did not receive sweetened fat on PPI days. Acute dosing occurred every 10 days to provide an intervening binge episode and to provide a sufficient washout period. A: Mice were maintained on a standard diet and exposed to one of the four feeding protocols. The 7-day feeding protocol was repeated weekly. After the 2.5-week session, each mice received an IP injection of vehicle, nisoxetine, GBR, or their combination, 1 h before the designated PPI session. After the completion of the series, mice were fed a high-fat diet (HFD; 45 % fat) ad libitum for 7 weeks not on their respectively feeding protocols. B: Mice were exposed to their respective feeding protocols while being maintained on HFD. The 7-day feeding protocol was repeated weekly. Each mouse received an IP injection of vehicle, nisoxetine, GBR, and combination series,1 h before the designated PPI session. NISL = nisoxetine 0.5 mg/k; NISH = nisoxetine 5 mg/kg; GBRL = 1.6 mg/kg GBR 12783; GBRH = 16 mg/kg GBR 12783,
Dietary-induced binge eating on high fat diet.
After completion of all PPI testing while on standard chow, all mice that were placed on the ad libitum high-fat diet (HFD; Research Diets D12451; 4.73 kcal/g, 45% fat, 20% protein, 35% carbohydrate; New Brunswick, NJ) for 7 weeks underwent baseline PPI on HFD, see Figure 1B. The 7-week time point on high fat diet was used because we observed an increase in body weight after 6 weeks on HFD(Li, Kshatriya, and Bello 2022). On week 8, mice were given access to ~5g sweetened fat and ~5g HFD for 24 h. One week afterward, on week 9 of HFD, mice began the binge feeding protocols with the same group assignments while maintained on HFD, see Table 1. After 2.5 weeks on the feeding protocol, mice underwent a second round of PPI, see Figure 1B.
Nisoxetine and GBR 12783 testing under high-fat diet conditions.
While remaining on the feeding protocol and HFD, mice were then subjected to PPI testing every 10 days, 1 hour after receiving IP injections. Four treatments were given as follows: (1) 10% DMSO saline; (2) NIS [5 mg/kg]; (3) GBR [16 mg/kg] in 10% DMSO saline; and (4) a combination of NIS [5 mg/kg] and GBR [1.6 mg/kg] in 10% DMSO saline, see Figure 1B.
Statistical Analysis
All statistical analysis was performed on Statistica (TIBCO Software, Inc.). Data are represented as mean ± SEM. Body weight data were analyzed by one-way analysis of variance (ANOVA). Separate ANOVA with repeated measures were used to analyze food intake and PPI under conditions of baseline, vehicle, nisoxetine, GBR, or the combination. Treatment was a repeated measure. Tukey’s post hoc analyses were performed when justified. In all analyses, significance was p < 0.05. A multivariant ANOVA was also performed in which pulse and treatment were repeated measures. For all significant main effects and interactions, degrees of freedom and F- values were included.
Results
Caloric intakes during the 30-minute binge period over 2.5 weeks on the feeding protocols on standard chow.
For total 30-min kcal intake over time, there were overall group [F (3, 36) = 270.1, p< 0.001], time [F (4, 144) = 7.5, p < 0.0001], and group × time [F (12, 144) = 7.7, p < 0.00001] effects. The group effect revealed significant differences between all groups (p < 0.001). The time × group interaction revealed that RB animals had more intake at all respective time points compared to all groups (p < 0.0001). RB was the only group that demonstrated an escalation of intake over time, that is, all binge episodes were greater than Binge 1 (p < 0.005), see Figure 2A. An examination of the different dietary components contributing to the total kcal intake from Binge 1 and Binge 5 revealed that there was an effect in groups receiving sweetened fat [F (1, 18) = 43.5, p < 0.0001], a time effect [F (1, 18) =36.7, p < 0.0001], and group × time effect [F (1, 18) = 13.9, p < 0.01]. Sweetened fat consumption for RB animals was greater at Binge 5 compared with Binge 1 (p < 0.001). Comparison of standard chow consumption from Binge 1 to the last Binge 5 revealed a group effect [F (3, 36) = 40.2, p < 0.0005] and a time × group effect [F (3, 36) = 2.9, p < 0.05], see Figure 2B. However, no group consumed more standard chow from Binge 1 to Binge 5. At the completion of the 2.5 weeks, body weights were 25.60 ± 0.43 g for N, 25.98 ± 0.42 g for B, 26.43 ± 0.56 g for R, and 25.49 ± 0.39 g for RB. Compared to baseline, all groups had a greater body weight following 2.5 weeks on the standard chow feeding protocol, but no differences between groups [F (3, 36) = 4.52, p<0.01)] (Figure 1S).
Figure 2. Standard chow dietary-induced binge eating intakes during the 30-min access periods.
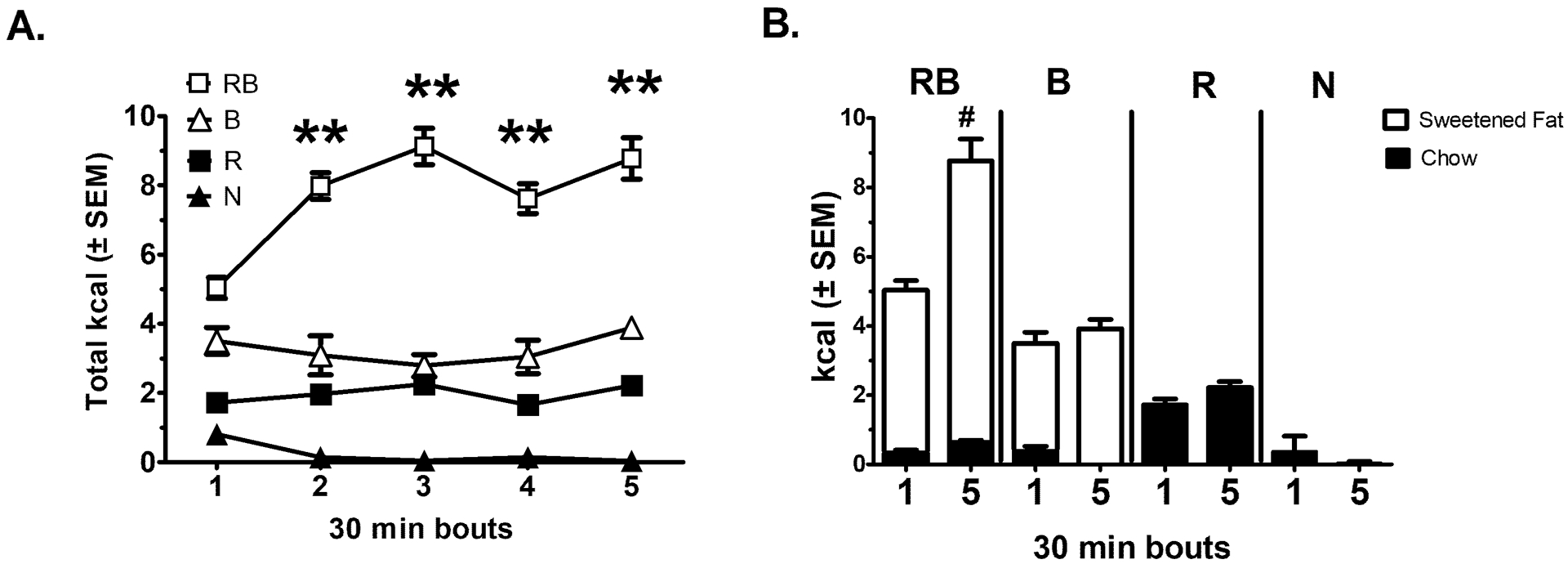
As illustrated in Table 1, groups represent Restricted Binge (RB), Binge (B), Restrict (R), and Naive (N). All mice had a pre-exposure to the sweetened fat (vegetable shortening with 10% sucrose) 1 week prior to starting feeding protocols. All groups n = 10. A: Represents total kcal intake during the 30 min access period. RB demonstrated an increase in total kcal intake from Binge 1. ** indicates p < 0.005. B. Com-parison of chow intake (black bars) and sweetened fat intake (white bars) from Binge 1 to Binge 5. # indicates p < 0.001 from sweetened fat intake for RB.
Startle response and PPI before and after dietary-induced binge-like eating and feeding protocol on standard chow.
One week before the pre-exposure to the sweetened fat and 2 weeks prior to starting the feeding protocols, all mice underwent a baseline PPI session. After the 2.5 week feeding protocol, all mice underwent another PPI (2.5 weeks, Figure 3A). Due to a technical error, for the PPI sessions, data are reported for the B and N groups with n = 5, and RB and R groups had n = 10, see figure 3A.
Figure 3. Startle response and pre-pulse inhibition before and after feeding protocols on standard chow.

Mice were exposed to the acoustic startle response session before (baseline) or after the feeding protocols (2.5 weeks), RB (n = 10), B (n = 5), R (n= 10), N (n = 5). A: Startle response from background noise only (Null), low pre-pulse (74 dB/20 ms) given alone (P74), high pre-pulse (90 dB/20 ms) given alone (P90), and startle stimulus pulse (110 dB/ 40 ms)(Startle). * indicates p < 0.01 from baseline response. B: Percent pre-pulse inhibition of PP74 is the P74 given 100 ms prior to the onset of the startle stimuli. PP90 is P90 given 100 ms prior to the onset of the startle stimuli. % pre-pulse inhibition was calculated as: [(pre-pulse preceding startle stimulus-startle stimulus alone) / (startle stimulus alone)] * 100. ** indicates p < 0.005 from baseline %PP74.
For P74, there was an overall time effect [F (1, 26) = 12.9, p < 0.005] with mice demonstrating a higher response to P74 after 2.5 weeks (P < 0.01), see Figure 3A.
For the startle pulse (110 dB), there was an overall group effect [F (3, 26) = 4.3, p < 0.05], with RB having a lower response compared with N (p < 0.01), a time effect: [F (1, 26) = 9.0, p < 0.005], with an overall increased response to the startle pulse after 2.5 weeks (p < 0.01). The group × time effect approached significance [F (3, 26) = 2.8, p = 0.06] with N having a higher startle response than RB at 2.5 weeks (p < 0.05; planned comparison), see Figure 3A.
For %PP74, there was an overall time effect [F (1, 26) = 8.2, p < 0.01] with a higher %PP74 at 2.5 weeks (p < 0.005), see Figure 3B.
For our multivariant ANOVA, we did not observe a pulse × treatment × group effect, see Supplemental statistical analysis.
Nisoxetine effects on startle response and PPI in mice exposed to dietary-induced binge-like eating on standard chow.
All mice received a within design dosing scheme of acute nisoxetine. Due to a technical error, for the PPI sessions, data reported for the B and N groups were n = 5 each, and RB and R groups were n = 10 each. The response for the null stimulus following saline, nisoxetine- low (Nis L; 0.5 mg/kg), and nisoxetine-high (Nis H ; 5 mg/kg) treatments showed there was an overall dose effect [F (2, 52) = 3.3, p < 0.05]. Post hoc testing revealed the response following the Nis H dose was less saline treatment (p < 0.05), see Figure 4A.
Figure 4. Dose-dependent nisoxetine effects of startle response and pre-pulse inhibition on standard chow.

Mice were exposed to the acoustic startle response session while maintained on the feeding protocol. Each mouse received an IP dose of vehicle (saline), nisoxetine 0.5 mg/kg (NISL), and nisoxetine 5 mg/kg (NISH) with a 10-day washout period between doses, RB (n = 10), B (n = 5), R (n= 10), N (n = 5). A: Startle response for Null, P74, P90, and Startle. * indicates P < 0.05 from saline dose. # indicates p < 0.05 from Naive NISH dose. B: Percent pre-pulse inhibition of PP74 and PP90. * indicates p < 0.05 from saline %PP74, ** indicates p < 0.005 from saline %PP74.
Similar for P90, there was an overall dose effect [F (2, 52) = 8.1, p < 0.005] and the P90 response was lower following the Nis H dose compared with saline (p < 0.05), see Figure 4A.
For startle pulse, there was a group effect [F (3, 26) = 4.5, p < 0.05] and group x dose effect [F (6, 52) = 3.4, p < 0.01]. Post hoc testing revealed that RB was lower than N at Nis H (p < 0.05), see Figure 4A.
For %PP74, there was an overall dose effect [F (2, 52) = 8.5, p < 0.001]. Post hoc testing indicated significantly higher inhibition than saline for Nis L (p < 0.05) and Nis H (p < 0.005), see Figure 4B.
For our multivariant ANOVA, we did not observe a pulse × treatment × group effect, see Supplemental statistical analysis.
Dose-dependent GBR 12783 alone or in combination with nisoxetine effects on startle response and PPI in mice exposed to dietary-induced binge-like eating on standard chow.
Following the nisoxetine dosing, all mice received a within design dosing scheme of acute GBR 12783 alone or in combination with nisoxetine (5 mg/kg). All groups were n = 10. Comparisons were performed of the startle response following 10% DMSO (vehicle), GBR 12783 low (GBR L; 1.6mg/kg), GBR 12783 high (GBR H; 16 mg/kg), GBR L with Nis H (GBR L: Nis H), see Figure 5A.
Figure 5. Dose-dependent effects of GBR 12783 (alone) or in combination with high dose nisoxetine on startle response and pre-pulse inhibition on standard chow.

Mice were exposed to the acoustic startle response session while maintained on their respective feeding protocols. Each mouse received an IP dose of vehicle (10% DMSO), GBR 12783 1.6 mg/kg (GBRL), GBR 12783 16 mg/kg (GBRH), and GBRL with nisoxetine 5 mg/kg (GBRL:NISH) with a 10-day washout period between doses. All groups were n = 10. A: Startle response from Null, P74, P90, and Startle pulse. & indicates p < 0.05 from all other doses and DMSO. ** indicates p < 0.005 from other GBR and combinational dose. B: Percent pre-pulse inhibition of PP74 and PP90. # indicates p < 0.05 from B group at GBRH.
For the null stimulus, there was an overall group effect [F (3, 33) = 7.45, p < 0.001] and overall dose effect [F (3, 99) = 6.1, p < 0.001]. Post hoc testing for the group effect indicated that N response was significantly less than RB (p < 0.05) and R (p < 0.005). Post hoc testing revealed that GBR H dose produced a greater response than GBR L and GBR L: Nis H (p < 0.05 for both), see figure 5A.
For P74, there was an overall dose effect [F (3, 99) = 5.7, p< 0.005]. Post hoc testing indicated the P74 response to GBR L was significantly higher from all other doses and DMSO (p < 0.05 for all), see Figure 5A.
For P90, there was an overall dose effect [F (3, 99) = 12.0, p < 0.0001]. Post hoc testing of the dose effect for P90 showed the response to GBR L was highest compared with all other doses and DMSO (p < 0.005), see Figure 5A.
For the startle stimulus, there was an overall group effect [F (3, 33) = 5.3, p < 0.005] and overall dose effect [F (3, 99) = 26.8, p< 0.005]. Post hoc testing of the group effect revealed the RB and B had lower startle responses compared with N (p < 0.05 for both). For dose effect, post hoc testing revealed the GBR L response was higher than all other doses and DMSO (p < 0.05), see Figure 5A.
For %PP74, there was an overall group effect [F (3, 33) = 4.1, p < 0.05], overall dose effect [F (3, 99) = 13.4, p < 0.0005] and group × dose effect [F (9, 99) = 2.5, p < 0.05]. Post hoc testing revealed at GBR H, RB and R had lower %PP74 than B (p < 0.05 for both), see Figure 5B.
For %PP90, there was a dose × group effect [F (9, 99) = 2.9, p < 0.005]. Post hoc testing did not reveal group differences at any dose, see Figure 5B.
For our multivariant ANOVA, we did not observe a pulse × treatment × group effect, see Supplemental statistical analysis.
Caloric intakes during the 30-minute binge period over 2.5 weeks on the feeding protocols on high-fat diet.
Following the 7 weeks of HFD, mice underwent a baseline PPI and were exposed to their respective feeding protocols for 2.5 weeks, see Table 1. All groups were n =10. For 30-min total kcal intake over time, there was an overall group effect [F (3, 36) = 36.2, p < 0.0001]. RB and B consumed more total kcal during the 30-min compared with R and N (p < 0.005), see Figure 6A. There were no differences in the dietary components contributing to the total kcal intake from Binge 1 and Binge 5, see Figure 6B. At the completion of the 2.5-week feeding protocol on HFD, there was a difference in body weight [F (3, 36) = 3.5, p < 0.05]. Post hoc testing revealed B had higher body weights than R (p < 0.05). Body weights were 48.9 ± 1.24 g for N, 49.7 ± 0.7 g for B, 45.53 ± 0.8 g for R, and 47.26 ± 0.68 g for RB.
Figure 6. High-fat diet dietary-induced binge eating intakes during the 30-min access periods.

As illustrated in Table 1, groups represent Restrict Binge (RB), Binge (B), Restrict (R), and Naive (N). All groups had exposure to the sweetened fat (vegetable shortening with 10% sucrose) for 24 h prior to restarting the feeding protocols on HFD. All groups were n = 10. A: Represents total kcal intake during the 30 min access period. B. Comparison of HFD intake (black bars) and sweetened fat intake (white bars) from Binge 1 to Binge 5.
Startle response and PPI before and after dietary-induced binge-like eating and feeding protocol on high-fat diet.
Baseline PPI was performed one week before the 24 h exposure to the sweetened fat and 2 weeks prior to restarting the feeding protocols. All mice underwent a PPI session after being re-exposed to their respective feeding protocol for 2.5 weeks under HFD. All groups were n = 10, see Figure 7A.
Figure 7. Startle response and pre-pulse inhibition before and after feeding protocols on high-fat diet.

Mice were exposed to the acoustic startle response session before (baseline) or after the feeding protocols (2.5 weeks). All groups were n = 10. A: Startle response from background noise only (Null), low pre-pulse (74 dB/20 ms) given alone (P74), high pre-pulse (90 dB/20 ms) given alone (P90), and startle stimulus pulse (110 dB/ 40 ms)(Startle). * indicates p < 0.05 from baseline response. B: Percent pre-pulse inhibition of PP74 is the P74 given 100 ms prior to the onset of the startle stimuli (%PP74). Percent pre-pulse inhibition PP90 is the P90 given 100 ms prior to the onset of the startle stimuli (%PP90). * indicates p < 0.05 from baseline %PP74.
For P90, there was an overall time effect [F (1, 36) = 5.39, p < 0.05] and an overall group effect [F (3, 36) =3.7, p< 0.05]. Post hoc testing indicated the response to P90 was lower after the 2.5 weeks (p < 0.05). The B group had a higher response to P90 compared with R (p< 0.05), see Figure 7A.
For the startle pulse, there was an overall time effect [F (1, 36) = 26.7, p < 0.001] and overall group effect [F (1, 36) = 26.8, p < 0.00001]. There was a lower response to startle stimulus after 2.5 weeks (p < 0.05). For group, RB group response was higher than N (p < 0.05), see Figure 7A.
For %PP74, there was a time effect [F (1, 36) = 11.2, p< 0.005] with lower %PP74 after 2.5 weeks on HFD (p < 0.05), see Figure 7B.
For our multivariant ANOVA, we did not observe a pulse × treatment × group effect, see Supplemental statistical analysis.
Nisoxetine and GBR 12783 alone or in combination effects on startle response and PPI on feeding protocols on high-fat diet.
Following the 2.5 weeks PPI, all mice were continued on the feeding protocols and received a within dose scheme of 10% DMSO (vehicle), nisoxetine high (Nis H; 5.0 mg/kg) GBR 12783 high (GBR H; 16 mg/kg), and combination of GBR 12783 low (GBR L; 1.6 mg/kg) and nisoxe-tine high (GBR L: Nis H), see Figure 8A.
Figure 8. Nisoxetine and GBR 12783 alone or in combination effects of startle response and pre-pulse inhibition on high-fat diet.

Mice were exposed to the acoustic startle response session while maintained on their respective feeding protocols on HFD. Each mouse received an IP dose of vehicle (10% DMSO), nisoxetine 5 mg/kg (NISH), GBR 12783 16 mg/kg (GBRH), and GBR 12783 1.6 mg/kg and NISH (GBRL: NISH) with at least a 10-day washout period between doses. All groups were n = 10. A: Startle response from Null, P74, P90, and Startle. & indicates p< 0.05 from all other doses and DMSO. @ indicates p < 0.05 from NisH and GBRL:NISH. $ indicates p < 0.05 from RB NISH P74 response. * indicates p < 0.05 from DMSO startle. B: Percent pre-pulse inhibition of PP74 (%PP74) and PP90 (%PP90).
For null stimulus, there was an overall dose effect [F (3, 108) = 23.3, p< 0.00001], with the GBR H dose having a higher response than all other doses (p < 0.05 for all), see Figure 8A.
For P74, there was an overall dose response [F (3, 108) = 3.7, p < 0.05] with a higher response to GBR H compared with Nis H and GBR L:Nis H (p < 0.05 for both). For P74, there was a group × dose effect [F (9, 108) = 2.6, p < 0.01]; the RB group had a higher response with NIS H than with GBR L:Nis H (p < 0.005 for both), see Figure 8A.
For P90, there was an overall dose effect [F (3, 108) = 4.0, p< 0.01]; GBR H was higher than NIS H and GBR L: Nis H (p < 0.05 for both), see Figure 8A.
For the startle pulse, there was an overall dose effect [F (3, 108) = 8.0, p < 0.0005] with a higher response to GBR H compared with Nis H and GBR L: Nis H (p < 0.05 for both). In addition, the response to NISH + GBR L was less than DMSO (p < 0.05), see Figure 8A.
For %PP74 and %PP90, there were no significant differences, see Figure 8B.
For our multivariant ANOVA, we did not observe a pulse × treatment × group effect, see Supplemental statistical analysis.
Discussion
The present study sought to investigate the effect of dietary-induced binge-like eating and the influence of high-fat diet (HFD) feeding on sensorimotor gating of the acoustic startle response. We also investigated whether there were differential responses to acute systemic administration of a selective norepinephrine reuptake inhibitor, nisoxetine, and a selective dopamine reuptake inhibitor, GBR 12783, alone and in combination. Our overall hypothesis was that intermittent recurrent dietary conditions that promote binge eating lead to impairments in sensorimotor gating. Our observed effects from the dietary conditions were more pronounced with use of selective norepinephrine or dopamine reuptake inhibitors. Alterations in the noradrenergic system have been implicated in binge eating pathologies (Pruccoli et al. 2021). One of our major findings of this study was that a high dose of nisoxetine (5 mg/kg) resulted in a diminished response to the startle pulse (110 dB) in the restrict binge (RB) group compared with the control (N) group under standard chow conditions. Because the RB demonstrated binge-like eating, these findings suggest either the overconsumption behavior or the combination of intermittent calorie restriction with sweetened fat access reduced the nisoxetine-induced increase in the acoustic startle response.
Surprisingly, we did not observe a differential pre-pulse inhibition (PPI) based on dietary conditions, we did observe that systemic nisoxetine (0.5 mg/kg and 5 mg/kg) increased pre-pulse inhibition at P74 (%PP74). Systemic nisoxetine (10 mg/kg or 30 mg/kg) has been demonstrated to normalize deficits of acoustic startle pre-pulse inhibition (PPI) in dopamine transporter (DAT) KO mice (Yamashita et al. 2006). A similar normalization of a PPI was observed with the selective serotonin reuptake inhibitor (SSRI) fluoxetine (30 mg/kg), but not with another SSRI, citalopram (30 or 100 mg/kg)(Yamashita et al. 2006). Compared with citalopram and other SSRI compounds, fluoxetine has been shown to increase norepinephrine concentrations in the prefrontal cortex of rats (Bymaster et al. 2002). Together, these findings suggest that compounds that increase central norepinephrine can impact sensorimotor gating.
On standard chow, we observed that mice exposed to repeated bouts of intermittent calorie restriction and sweetened fat access resulted in increased caloric intake over the 2.5-week period in the Restrict Binge (RB) group. The escalation in caloric intake by the RB group was attributed to the increased intake of sweetened fat in the 30 min access period. We have reported similar effects using this rodent model of dietary-induced binge eating (Bello, Patinkin, and Moran 2011; Bello, Walters, et al. 2014; Bello, Yeh, et al. 2014; Sachdeo et al. 2019). Although cumulative caloric intakes were not measured in this study, it has been demonstrated by several laboratories intermittent limited access to a highly palatable food produces a cyclical feeding pattern, whereby rodents increase caloric intake during highly palatable food access periods and demonstrate a compensatory reduction in caloric intake during periods without highly palatable food access (Babbs, Wojnicki, and Corwin 2012; Bello, Yeh, et al. 2014; Corwin, Avena, and Boggiano 2011; Cottone et al. 2008; Kinzig, Hargrave, and Honors 2008; Sachdeo et al. 2019). This cyclical feeding pattern is the reason there were no differences in body weights between groups. For this study, mice were placed on ad libitum HFD for 7 weeks and we re-initiated the dietary-induced binge eating-like feeding protocol. When on the HFD, the RB group did not exhibit binge-like eating during the 30-min access period. Nonetheless, both groups (RB and Binge; B) that had access to sweetened fat consumed more calories during the 30-min access period.
Dopaminergic involvements in the acoustic startle response and PPI have been well-characterized (Mansbach, Geyer, and Braff 1988; Zhang et al. 2000). Specific involvement of the dopamine D2 receptors (D2R) in modulating PPI, as well as a reduced PPI in DAT KO mice have strongly implicated the dopaminergic systems in sensorimotor gating (Zhang et al. 2000; Ouagazzal, Jenck, and Moreau 2001; Yamashita et al. 2006). In the present study, there was a dose-dependent response in the acoustic startle to GBR 12783, whereby the low dose (1.6 mg/kg) enhanced and the high dose (16 mg/kg) diminished startle responses. Also, with the 16 mg/kg dose, there was a reduction in the %PP74 in the RB and R groups compared with B group. Because we observed an enhanced acoustic startle response with the high-dose nisoxetine (5 mg/kg) and low-dose GBR 12783 (1.6 mg/kg), we speculated that a combination of high-dose nisoxetine with a low dose of GBR 12783 would produce a differential startle response in the feeding groups. Essentially, when high-dose nisoxetine was combined with low-dose GBR12783 the acoustic responses were similar to the vehicle treatment when fed standard chow. We did, however, observe that the RB group on HFD had a lower to response to p74 with a combinational dose of nisoxetine (5.0 mg/kg) and GBR 12783 (1.6 mg/kg) than compared with nisoxetine (5.0 mg/kg) alone. Notably, the RB group on HFD did not have a binge-like phenotype but were exposed to intermittent bouts of calorie restriction and sweetened fat access. Further experiments are needed to understand the importance of this acoustic startle response to the combinational effects of targeting the central noradrenergic and dopaminergic systems under high-fat diet feeding conditions.
Several limitations are evident in the present experiments. First, the use of a within subject design was meaningful for determining the consequences of dietary-induced binge eating on sensorimotor gating over time. The findings with a within approach need to be interpreted with caution since there is an observed habituation over time (Ouagazzal, Jenck, and Moreau 2001; Dulawa and Geyer 2000). Notably, the mouse strain used in the present study was C57BL6/J, which reportedly has the least amount of startle habituation over time (Ouagazzal, Jenck, and Moreau 2001; Plappert, Pilz, and Schnitzler 2004). In addition, we did not find any differences at %PP90, this could suggest that the 90dB could have evoked a startle response and interfered with the pre-pulse inhibition (Dulawa and Geyer 2000). An additional related limitation is interpreting the age-related differences in acoustic startle response (Parham and Willott 1988). For this study, mice began the feeding paradigm and PPI paradigm at 7 weeks of age and completed the study when the mice were 9 months of age. As a result, we did not directly compare the startle response on standard chow with startle response on HFD. Nonetheless, chronic exposure to HFD has been shown to impair PPI, and this HFD-reduction in PPI can be normalized with the antipsychotic haloperidol (Labouesse et al. 2013). Even though there is an apparent reduction with HFD in our study, comparisons were only performed within the individual diet conditions because we could not account for the possible confound of age. Age-related impairments in acoustic responses in C57Bl6/J have been attributed to a progressive hearing loss occurring during the first 12 months of age (Parham and Willott 1988). Prolonged metabolic impairments, such as poorly controlled type II diabetes mellitus, have been noted to impair the evoked otoacoustic emissions in human subjects (Sasso et al. 1999). Interesting, there were no differences in the otoacoustic emissions in diabetic and control subjects during an acute hyperglycemic clamping procedure, suggesting that long term poorly controlled glycemic levels influence acoustic sensorineural signaling (Sasso et al. 1999). Given our current data, there is no way to confidently attribute the apparent reduction in acoustic startle response to the age of the mice or HFD exposure or some other relationship between age and diet factors. Another limitation in generalizing our findings is that we only used male mice for this study. The reason for using this sex only was because the male mouse sensorimotor gating responses have been well-characterized in a large meta-analysis study (Shoji and Miyakawa 2018). Another possible interpretive limitation was that there was an increase in background (Null) activity with high doses of nisoxetine (5.0 mg/kg) and GBR 12783 (16 mg/kg). Although the calculated startle response accounts for the response during the null period, this could be an additional source of variation. A large meta-analysis of acoustic startle responses in C57Bl/6J male mice (n = 1363) found that while there was no significant association between acoustic startle response and PPI, basal startle reactivity influenced PPI (Shoji and Miyakawa 2018). This limitation could be addressed by separating mice based on high and low basal startle reactivity. However, the sample sizes of the current study were not powered for that analysis.
Previous work from our laboratory has suggested a dampening of the central noradrenergic system response in rats exposed to a dietary-induced binge eating protocol (Bello, Walters, et al. 2014; Bello, Yeh, and James 2019; Bello, Yeh, et al. 2014). Our present findings suggest that dietary-induced binge eating disrupts the central norepinephrine controls of an acoustic startle response in male mice. The importance of these findings could represent an alteration in responses to altering stimuli associated with disordered eating. Women have a higher prevalence of eating disorders than men (Association 2013). One critical element in understanding the importance of these findings is to determine whether our findings are sex dependent. Previous studies with acoustic sensorimotor gating with eating disorders populations were performed in women with anorexia nervosa (n =20) (Steinman et al. 2016). In that study there were no differences in the level of pre-pulse inhibition between women with anorexia nervosa and healthy controls. More work is needed to understand the underlying mechanism of sensorimotor integration and whether impairment of these neural processes contributes to binge eating pathologies in clinical populations.
Supplementary Material
Acknowledgments
The authors would like to thank Rifke Anolik and Joshua Corris for their technical assistance and Dr. Lihong Hao for statistical assistance. Additionally, the authors would like to thank Stacey Pontoriero for editorial assistance.
Funding:
This research was funded by USDA-NIFA, NJ06162 and NJ06280 and NCCIH of the NIH under Award Number R01AT008933.
Footnotes
Conflicts of Interests and Disclaimers: None.
References
- Ahmari SE, Risbrough VB, Geyer MA, and Simpson HB. 2012. ‘Impaired sensorimotor gating in unmedicated adults with obsessive-compulsive disorder’, Neuropsychopharmacology, 37: 1216–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsene KM, Rajbhandari AK, Ramaker MJ, and Bakshi VP. 2011. ‘Discrete forebrain neuronal networks supporting noradrenergic regulation of sensorimotor gating’, Neuropsychopharmacology, 36: 1003–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Association, American Psychiatric. 2013. Diagnostic and statistical manual of mental disorders: DSM-V (American Psychiatric Association: Washington, DC: ). [Google Scholar]
- Babbs RK, Wojnicki FH, and Corwin RL. 2012. ‘Assessing binge eating. An analysis of data previously collected in bingeing rats’, Appetite, 59: 478–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakshi VP, Alsene KM, Roseboom PH, and Connors EE. 2012. ‘Enduring sensorimotor gating abnormalities following predator exposure or corticotropin-releasing factor in rats: a model for PTSD-like information-processing deficits?’, Neuropharmacology, 62: 737–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello NT, Patinkin ZW, and Moran TH. 2011. ‘Opioidergic consequences of dietary-induced binge eating’, Physiol Behav, 104: 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello NT, Walters AL, Verpeut JL, and Caverly J. 2014. ‘Dietary-induced binge eating increases prefrontal cortex neural activation to restraint stress and increases binge food consumption following chronic guanfacine’, Pharmacol Biochem Behav, 125: 21–28. [DOI] [PubMed] [Google Scholar]
- Bello NT, Yeh CY, and James MH. 2019. ‘Reduced Sensory-Evoked Locus Coeruleus-Norepinephrine Neural Activity in Female Rats With a History of Dietary-Induced Binge Eating’, Front Psychol, 10: 1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello NT, Yeh CY, Verpeut JL, and Walters AL. 2014. ‘Binge-like eating attenuates nisoxetine feeding suppression, stress activation, and brain norepinephrine activity’, PLoS One, 9: e93610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billes SK, and Cowley MA. 2007. ‘Inhibition of dopamine and norepinephrine reuptake produces additive effects on energy balance in lean and obese mice’, Neuropsychopharmacology, 32: 822–34. [DOI] [PubMed] [Google Scholar]
- Borkman M, Campbell LV, Chisholm DJ, and Storlien LH. 1991. ‘Comparison of the effects on insulin sensitivity of high carbohydrate and high fat diets in normal subjects’, J Clin Endocrinol Metab, 72: 432–7. [DOI] [PubMed] [Google Scholar]
- Braff DL, Callaway E, and Naylor H. 1977. ‘Very short-term memory dysfunction in schizophrenia. Defective short time constant information processing in schizophrenia’, Arch Gen Psychiatry, 34: 25–30. [DOI] [PubMed] [Google Scholar]
- Bray GA 1991. ‘Obesity, a disorder of nutrient partitioning: the MONA LISA hypothesis’, J Nutr, 121: 1146–62. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Zhang W, Carter PA, Shaw J, Chernet E, Phebus L, Wong DT, and Perry KW. 2002. ‘Fluoxetine, but not other selective serotonin uptake inhibitors, increases norepinephrine and dopamine extracellular levels in prefrontal cortex’, Psychopharmacology (Berl), 160: 353–61. [DOI] [PubMed] [Google Scholar]
- Cannon CM, Abdallah L, Tecott LH, During MJ, and Palmiter RD. 2004. ‘Dysregulation of striatal dopamine signaling by amphetamine inhibits feeding by hungry mice’, Neuron, 44: 509–20. [DOI] [PubMed] [Google Scholar]
- Corwin RL, Avena NM, and Boggiano MM. 2011. ‘Feeding and reward: perspectives from three rat models of binge eating’, Physiol Behav, 104: 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottone P, Sabino V, Steardo L, and Zorrilla EP. 2008. ‘Intermittent access to preferred food reduces the reinforcing efficacy of chow in rats’, Am J Physiol Regul Integr Comp Physiol, 295: R1066–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulawa SC, and Geyer MA. 2000. ‘Effects of strain and serotonergic agents on prepulse inhibition and habituation in mice’, Neuropharmacology, 39: 2170–9. [DOI] [PubMed] [Google Scholar]
- Elmore DK, and de Castro JM. 1991. ‘Meal patterns of normal, untreated bulimia nervosa and recovered bulimic women’, Physiol Behav, 49: 99–105. [DOI] [PubMed] [Google Scholar]
- Foster MT, Warne JP, Ginsberg AB, Horneman HF, Pecoraro NC, Akana SF, and Dallman MF. 2009. ‘Palatable foods, stress, and energy stores sculpt corticotropin-releasing factor, adrenocorticotropin, and corticosterone concentrations after restraint’, Endocrinology, 150: 2325–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis H, and Stevenson R. 2013. ‘The longer-term impacts of Western diet on human cognition and the brain’, Appetite, 63: 119–28. [DOI] [PubMed] [Google Scholar]
- George DT, Kaye WH, Goldstein DS, Brewerton TD, and Jimerson DC. 1990. ‘Altered norepinephrine regulation in bulimia: effects of pharmacological challenge with isoproterenol’, Psychiatry Res, 33: 1–10. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Krebs-Thomson K, Braff DL, and Swerdlow NR. 2001. ‘Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review’, Psychopharmacology (Berl), 156: 117–54. [DOI] [PubMed] [Google Scholar]
- Gibson EL 2006. ‘Emotional influences on food choice: sensory, physiological and psychological pathways’, Physiol Behav, 89: 53–61. [DOI] [PubMed] [Google Scholar]
- Graham FK 1975. ‘Presidential Address, 1974. The more or less startling effects of weak prestimulation’, Psychophysiology, 12: 238–48. [DOI] [PubMed] [Google Scholar]
- Greeno CG, and Wing RR. 1994. ‘Stress-induced eating’, Psychol Bull, 115: 444–64. [DOI] [PubMed] [Google Scholar]
- Harrington ME, and Coscina DV. 1983. ‘Early weight gain and behavioral responsivity as predictors of dietary obesity in rats’, Physiol Behav, 30: 763–70. [DOI] [PubMed] [Google Scholar]
- Heal DJ, and Gosden J. 2022. ‘What pharmacological interventions are effective in binge-eating disorder? Insights from a critical evaluation of the evidence from clinical trials’, Int J Obes (Lond), 46: 677–95. [DOI] [PubMed] [Google Scholar]
- Hildebrandt BA, and Ahmari SE. 2021. ‘Breaking It Down: Investigation of Binge Eating Components in Animal Models to Enhance Translation’, Front Psychiatry, 12: 728535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman HS, and Searle JL. 1968. ‘Acoustic and temporal factors in the evocation of startle’, J Acoust Soc Am, 43: 269–82. [DOI] [PubMed] [Google Scholar]
- Johnson CC, Gorell JM, Rybicki BA, Sanders K, and Peterson EL. 1999. ‘Adult nutrient intake as a risk factor for Parkinson’s disease’, Int J Epidemiol, 28: 1102–9. [DOI] [PubMed] [Google Scholar]
- Kaye WH, Ballenger JC, Lydiard RB, Stuart GW, Laraia MT, O’Neil P, Fossey MD, Stevens V, Lesser S, and Hsu G. 1990. ‘CSF monoamine levels in normal-weight bulimia: evidence for abnormal noradrenergic activity’, Am J Psychiatry, 147: 225–9. [DOI] [PubMed] [Google Scholar]
- Kinzig KP, Hargrave SL, and Honors MA. 2008. ‘Binge-type eating attenuates corticosterone and hypophagic responses to restraint stress’, Physiol Behav, 95: 108–13. [DOI] [PubMed] [Google Scholar]
- Kshatriya D, Hao L, Li X, and Bello NT. 2020. ‘Raspberry Ketone [4-(4-Hydroxyphenyl)-2-Butanone] Differentially Effects Meal Patterns and Cardiovascular Parameters in Mice’, Nutrients, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labouesse MA, Stadlbauer U, Langhans W, and Meyer U. 2013. ‘Chronic high fat diet consumption impairs sensorimotor gating in mice’, Psychoneuroendocrinology, 38: 2562–74. [DOI] [PubMed] [Google Scholar]
- Li X, Kshatriya D, and Bello NT. 2022. ‘Weight-gain propensity and morphine withdrawal alters locomotor behavior and regional norepinephrine-related gene expression in male and female mice’, Pharmacol Biochem Behav, 213: 173329. [DOI] [PubMed] [Google Scholar]
- Manning EE, Wang AY, Saikali LM, Winner AS, and Ahmari SE. 2021. ‘Disruption of prepulse inhibition is associated with compulsive behavior severity and nucleus accumbens dopamine receptor changes in Sapap3 knockout mice’, Sci Rep, 11: 9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansbach RS, Geyer MA, and Braff DL. 1988. ‘Dopaminergic stimulation disrupts sensorimotor gating in the rat’, Psychopharmacology (Berl), 94: 507–14. [DOI] [PubMed] [Google Scholar]
- Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Aggarwal N, Schneider J, and Wilson RS. 2003. ‘Dietary fats and the risk of incident Alzheimer disease’, Arch Neurol, 60: 194–200. [DOI] [PubMed] [Google Scholar]
- Morris MC, Evans DA, Bienias JL, Tangney CC, and Wilson RS. 2004. ‘Dietary fat intake and 6-year cognitive change in an older biracial community population’, Neurology, 62: 1573–9. [DOI] [PubMed] [Google Scholar]
- Morrison CD, Pistell PJ, Ingram DK, Johnson WD, Liu Y, Fernandez-Kim SO, White CL, Purpera MN, Uranga RM, Bruce-Keller AJ, and Keller JN. 2010. ‘High fat diet increases hippocampal oxidative stress and cognitive impairment in aged mice: implications for decreased Nrf2 signaling’, J Neurochem, 114: 1581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobrega JN, and Coscina DV. 1986. ‘Dopamine-norepinephrine interactions in the development of hyperphagia and obesity following medial hypothalamic lesions’, Pharmacol Biochem Behav, 25: 401–9. [DOI] [PubMed] [Google Scholar]
- Ouagazzal AM, Jenck F, and Moreau JL. 2001. ‘Drug-induced potentiation of prepulse inhibition of acoustic startle reflex in mice: a model for detecting antipsychotic activity?’, Psychopharmacology (Berl), 156: 273–83. [DOI] [PubMed] [Google Scholar]
- Parham K, and Willott JF. 1988. ‘Acoustic startle response in young and aging C57BL/6J and CBA/J mice’, Behav Neurosci, 102: 881–6. [DOI] [PubMed] [Google Scholar]
- Pecoraro N, Reyes F, Gomez F, Bhargava A, and Dallman MF. 2004. ‘Chronic stress promotes palatable feeding, which reduces signs of stress: feedforward and feedback effects of chronic stress’, Endocrinology, 145: 3754–62. [DOI] [PubMed] [Google Scholar]
- Peres FF, Levin R, Almeida V, Zuardi AW, Hallak JE, Crippa JA, and Abilio VC. 2016. ‘Cannabidiol, among Other Cannabinoid Drugs, Modulates Prepulse Inhibition of Startle in the SHR Animal Model: Implications for Schizophrenia Pharmacotherapy’, Front Pharmacol, 7: 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plappert CF, Pilz PK, and Schnitzler HU. 2004. ‘Factors governing prepulse inhibition and prepulse facilitation of the acoustic startle response in mice’, Behav Brain Res, 152: 403–12. [DOI] [PubMed] [Google Scholar]
- Pruccoli J, Parmeggiani A, Cordelli DM, and Lanari M. 2021. ‘The Role of the Noradrenergic System in Eating Disorders: A Systematic Review’, Int J Mol Sci, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaman L 2011. ‘Hindbrain noradrenergic A2 neurons: diverse roles in autonomic, endocrine, cognitive, and behavioral functions’, Am J Physiol Regul Integr Comp Physiol, 300: R222–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter S, Pelzer NL, and Ritter RC. 1978. ‘Absence of glucoprivic feeding after stress suggest impairment of noradrenergic neuron function’, Brain Res, 149: 399–411. [DOI] [PubMed] [Google Scholar]
- Sachdeo BLY, Yu L, Giunta GM, and Bello NT. 2019. ‘Binge-Like Eating Is Not Influenced by the Murine Model of OPRM1 A118G Polymorphism’, Front Psychol, 10: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasso FC, Salvatore T, Tranchino G, Cozzolino D, Caruso AA, Persico M, Gentile S, Torella D, and Torella R. 1999. ‘Cochlear dysfunction in type 2 diabetes: a complication independent of neuropathy and acute hyperglycemia’, Metabolism, 48: 1346–50. [DOI] [PubMed] [Google Scholar]
- Shoji H, and Miyakawa T. 2018. ‘Relationships between the acoustic startle response and prepulse inhibition in C57BL/6J mice: a large-scale meta-analytic study’, Mol Brain, 11: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman SA, Ahmari SE, Choo T, Kimeldorf MB, Feit R, Loh S, Risbrough V, Geyer MA, Steinglass JE, Wall M, Schneier FR, Fyer AJ, and Simpson HB. 2016. ‘Prepulse Inhibition Deficits Only in Females with Obsessive-Compulsive Disorder’, Depress Anxiety, 33: 238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentino RJ, Curtis AL, Page ME, Pavcovich LA, and Florin-Lechner SM. 1998. ‘Activation of the locus ceruleus brain noradrenergic system during stress: circuitry, consequences, and regulation’, Adv Pharmacol, 42: 781–4. [DOI] [PubMed] [Google Scholar]
- Woo H, Park SJ, Lee Y, Kwon G, Gao Q, Lee HE, Ahn YJ, Shin CY, Cheong JH, and Ryu JH. 2014. ‘The effects of atomoxetine and methylphenidate on the prepulse inhibition of the acoustic startle response in mice’, Prog Neuropsychopharmacol Biol Psychiatry, 54: 206–15. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Fukushima S, Shen HW, Hall FS, Uhl GR, Numachi Y, Kobayashi H, and Sora I. 2006. ‘Norepinephrine transporter blockade can normalize the prepulse inhibition deficits found in dopamine transporter knockout mice’, Neuropsychopharmacology, 31: 2132–9. [DOI] [PubMed] [Google Scholar]
- Zhang J, Forkstam C, Engel JA, and Svensson L. 2000. ‘Role of dopamine in prepulse inhibition of acoustic startle’, Psychopharmacology (Berl), 149: 181–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.