

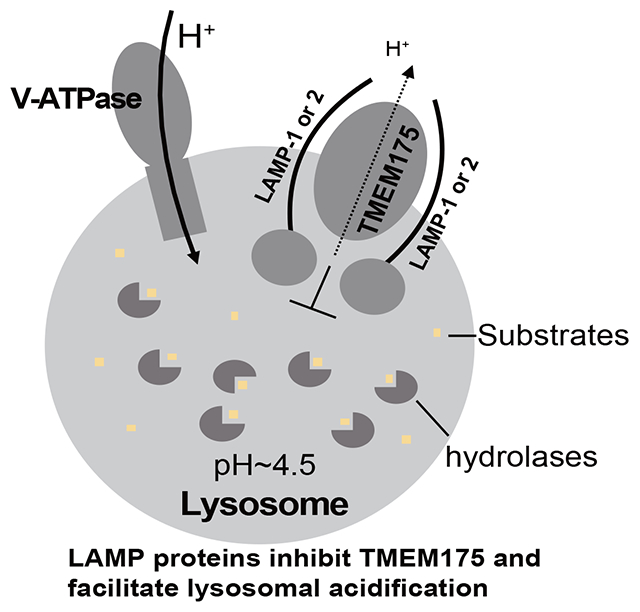
Summary
Maintaining a highly acidic lysosomal pH is central to cellular physiology. Here we use functional proteomics, single particle cryo-EM, electrophysiology, and in vivo imaging to unravel a key biological function of human lysosome-associated membrane proteins (LAMP-1 and LAMP-2) in regulating lysosomal pH homeostasis. Despite being widely used as a lysosomal marker, the physiological functions of the LAMP proteins have long been overlooked. We show that LAMP-1 and LAMP-2 directly interact with and inhibit the activity of the lysosomal cation channel TMEM175, a key player in lysosomal pH homeostasis implicated in Parkinson’s disease. This LAMP inhibition mitigates the proton conduction of TMEM175 and facilitates lysosomal acidification to a lower pH environment crucial for optimal hydrolase activity. Disrupting the LAMP-TMEM175 interaction alkalinizes the lysosomal pH and compromises the lysosomal hydrolytic function. In light of the ever-increasing importance of lysosomes to cellular physiology and diseases, our data have widespread implications for lysosomal biology.
Keywords: lysosomal LAMP proteins, LAMP-1 and LAMP-2, TMEM175, lysosomal pH homeostasis lysosome acidification, lysosomal hydrolytic function, risk factor for Parkinson’s disease
Graphical Abstract
eTOC Blurb:
Zhang et al. utilized a multidisciplinary approach to unravel a key functional role of LAMP proteins in regulating lysosomal pH homeostasis. They demonstrated that LAMP-1 and 2 directly interact with TMEM175 and inhibit its channel activity. This LAMP inhibition facilitates the lysosomal acidification to a lower pH optimal for the lysosomal hydrolytic activity.
Introduction
Lysosomes are acidic membrane-bounded organelles serving as the degradation and recycling center of the cell1,2. Lysosomes also function as a signaling hub and participate in multiple key biological processes including autophagy, endocytosis, exocytosis, Ca2+ signaling, and nutrient sensing3–8. Containing over 60 different types of hydrolases, lysosomes are capable of degrading essentially all cellular macromolecules. The lysosomal hydrolytic function requires a highly acidic luminal pH maintained by a dynamic equilibrium of proton influx and efflux as well as the counter-ion movement across the lysosomal membrane9–12. The lysosomal acidification (proton influx) is achieved by the action of V-ATPase which utilizes the free energy of ATP hydrolysis to pump protons into the lysosome13. The counter-ion movement is necessary to dissipate the membrane potential generated by the proton influx from the V-ATPase action10,14,15. The lysosomal pH quickly alkalizes upon V-ATPase inhibition16, indicating the presence of proton leakage which is important for lysosomal pH homeostasis17. The molecular identity of the lysosomal protein responsible for the proton leak has been elusive until a recent study reported that the transmembrane protein 175 (TMEM175) is the bona fide lysosomal proton leak channel18.
TMEM175, a potential risk factor for Parkinson’s disease (PD) from the genome-wide association studies19–21, was initially characterized as an endo/lysosomal K+ channel, whose function is important for setting the lysosomal membrane potential, maintaining pH stability under starved conditions, and regulating autophagosome/lysosome fusion22. However, compared to the canonical 6-TM K+ channels, TMEM175 bears no sequence homology and adopts a distinct structure and thereby utilizes a completely different mechanism to achieve K+ selectivity23–26. Intriguingly, two recent studies demonstrated that TMEM175 exhibits a unique pH-dependent dual permeability property18,25. While TMEM175 can selectively conduct K+ at neutral pH, acidic luminal pH converts it to a highly proton-selective channel. Thus, under physiological conditions, TMEM175 functions as a low pH-activated proton channel in the lysosome whose proton efflux activity balances the proton-influx from V-ATPase and maintains the lysosomal pH homeostasis. TMEM175 knockout causes over-acidification of lysosomes which in turn impairs the lysosomal hydrolytic activity18,22,27.
To maintain a steady-state lysosomal pH, only a small rate of lysosomal proton leak is needed to offset the proton influx from V-ATPase12 and the acidic pH-elicited proton currents from TMEM175 appear to be too potent for this proton relief function. Furthermore, lysosome exhibits a more acidic pH than endosome partly due to a slower rate of proton leak17, indicating a tighter control of proton leakage in the lysosome. In this study, we discovered that the ubiquitous lysosome-associated membrane proteins (LAMP-1 and LAMP-2) play an integral role in lysosomal pH homeostasis by controlling the TMEM175 channel activity. The LAMP proteins constitute about 50% of lysosomal membrane proteins and have been widely used as standard markers for the lysosomal compartment28–35, yet their biological functions have long been overlooked. They are type I transmembrane proteins with a large, heavily glycosylated luminal domain, a single transmembrane domain, and a short cytoplasmic tail important for lysosomal targeting31,36,37. Because of their high abundance and heavy glycosylation, LAMP proteins were originally thought to be structural proteins protecting the lysosomal membrane from the hydrolytic action of the luminal hydrolases38,39. However, lysosomal integrity does not appear to be compromised in cells with both LAMP-1 and 2 genes knocked out40. Studies of LAMP-1 and /or LAMP-2 deficient mice and cells indicated that LAMP proteins are important for the fusion of lysosomes with phagosomes and autophagosomes34,41,42. In humans, LAMP-2 deficiency leads to Danon disease, a genetic disorder characterized by myopathy, cardiomyopathy, and mental retardation43. LAMP-2 was also implicated in cholesterol export from lysosomes40,44, chaperone-mediated autophagy45, and anchoring the tuberous sclerosis complex (TSC) to the cytoplasmic surface of lysosomes to suppress mTORC1 signaling46 All these studies demonstrated that LAMP proteins play some important role in lysosomal activity more than just lysosomal structural proteins. To this end, we utilized a multidisciplinary approach to unravel a key functional role of LAMP proteins in regulating lysosomal pH homeostasis. We demonstrate that LAMP-1 and 2 directly interact with TMEM175 and inhibit its channel activity. This LAMP inhibition facilitates the lysosomal acidification to a lower pH optimal for the enzymatic activities of hydrolases.
Results
Identification of LAMP-1 and LAMP-2 as the partners of TMEM175
We performed a proteomic screening to identify potential candidates of TMEM175-interacting proteins that may regulate its channel function in lysosome using HEK293 and SH-SY5Y cells which have been previously used for functional characterization of TMEM17522,25,27. FLAG-tagged TMEM175 was transiently expressed in these two cell lines and the workflow of the proteomic analysis is summarized in Figure 1A. A total of 21 proteins from SH-SY5Y and 19 proteins from HEK293 were identified as putative TMEM175-interacting proteins in our proteomic analysis (Figure 1B, Figure S1, Table S1, and STAR Methods). Of all the candidates, LAMP-1 and LAMP-2 stand out to be among the most abundant proteins from our proteomic analysis of both SH-SY5Y and HEK293 cells; they are also the only two overlapping candidates that were identified from the proteomic screening of both cell lines (Figure 1B and Figure S1A).
Figure 1. Identification of LAMP-1 and LAMP-2 as TMEM175-interacting proteins.
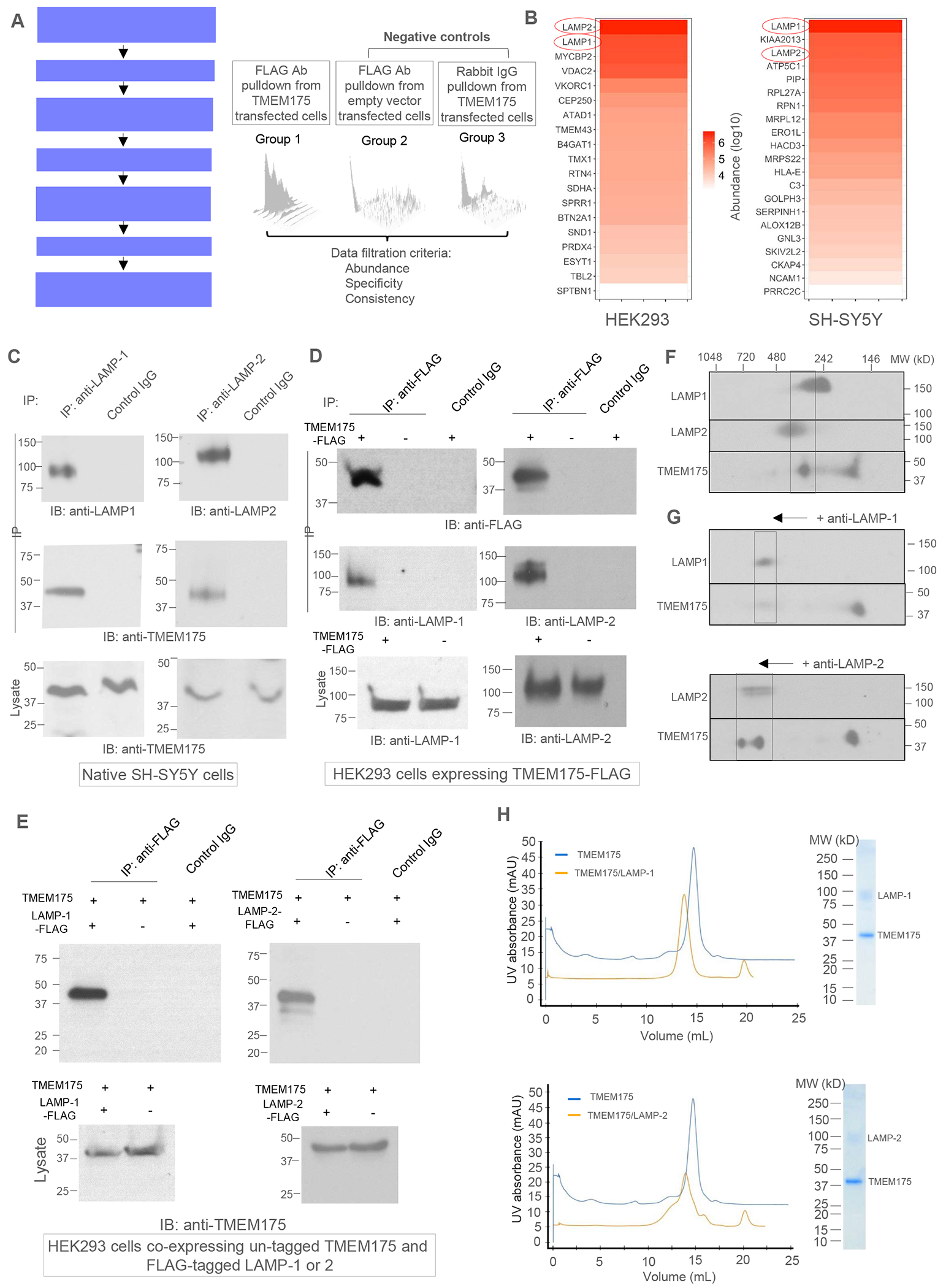
(A) Workflow of the proteomic screening of TMEM175-interacting proteins.
(B) Lists of putative TMEM175-interacting proteins identified from the pull-down samples from HEK293 and SH-SY5Y cells.
(C) Co-immunoprecipitation (Co-IP) of TMEM175 and LAMP proteins in native SH-SY5Y cells. LAMP-1 and 2 were immunoprecipitated by respective antibodies and TMEM175 was detected in the IP samples by immunoblotting (IB). Numbers beside each gel mark the molecular weights in kDa.
(D) Co-IP of TMEM175 and LAMP proteins in HEK293 cells expressing FLAG-tagged TMEM175. TMEM175 was immunoprecipitated by anti-FLAG antibodies and LAMP-1 and 2 were detected in the IP sample.
(E) Co-IP of TMEM175 and LAMP proteins in HEK293 cells co-expressing untagged TMEM175 and FLAG-tagged LAMP-1 or 2. Overexpressed LAMP-1 or 2 were immunoprecipitated by anti-FLAG antibodies and TMEM175 was detected in the IP samples.
(F) 2-D gel electrophoresis (BN-PAGE and SDS-PAGE) of SH-SY5Y cell lysate. Anti-LAMP-1, LAMP-2, and TMEM175 antibodies were used for immunoblotting.
(G) 2-D gel electrophoresis of SH-SY5Y cell lysate after mobility shift by anti-LAMP-1 or LAMP-2 antibodies.
(H) Gel filtration profiles and SDS-PAGEs of purified TMEM175/LAMP-1 (upper panel) and TMEM175/LAMP-2 (lower panel) complexes. The profiles of TMEM175 (blue) are also shown for comparison.
To verify the interaction between TMEM175 and LAMP proteins, we performed the co-immunoprecipitation (Co-IP) experiment using the lysates of native SH-SY5Y cells without transfection. Indeed, TMEM175 was immunoprecipitated with both LAMP-1 and LAMP-2, suggesting complex formation between TMEM175 and LAMP proteins (Figure 1C). The amount of endogenous TMEM175 in native HEK293 cells appears to be too low to be detected by the anti-TMEM175 antibodies used in our study. We therefore transiently expressed FLAG-tagged TMEM175 in HEK293 cells and validated its interaction with LAMP proteins by Co-IP experiment (Figure 1D). The endogenous LAMP proteins were detected with the over-expressed TMEM175-FLAG when immunoprecipitated by anti-FLAG antibodies. Furthermore, when un-tagged TMEM175 was co-expressed with FLAG-tagged LAMP-1 or LAMP-2, it was detected with the LAMP proteins immunoprecipitated by anti-flag antibodies (Figure 1E).
We also analyzed the cell lysates from native SH-SY5Y cells using two-dimensional BN-PAGE (blue-native polyacrylamide gel electrophoresis) and SDS-PAGE to verify the complex formation between TMEM175 and LAMP proteins (Figure 1 F & 1G). After 2nd dimensional SDS-PAGE and immunoblotting with their respective antibodies, a significant portion of TMEM175 was detected at a higher molecular weight (MW) between 242 and 480 kDa on BN-PAGE, overlapping with LAMP-1 and LAMP-2 (Figure 1F). To confirm the co-migration of TMEM175 and LAMP in the same complex, we also performed a mobility-shift assay by adding antibodies against LAMP-1 or LAMP-2 to the cell lysates before 2-D gel electrophoresis. The bound antibodies shift LAMP-1 and LAMP-2 to a higher MW position (> 480 kDa) and TMEM175 shifts to the same size as the LAMP proteins (Figure 1G). Similar results were also observed in the 2-D gel electrophoresis analysis of cell lysates from HEK293 cells co-expressing TMEM175 and FLAG-tagged LAMP-1 or LAMP-2 (Figure S2). Thus, both Co-IP and 2-D gel electrophoresis experiments performed under native or overexpressed conditions demonstrated that TMEM175 interacts with LAMP proteins in vivo.
Biochemical purification of TMEM 175/LAMP complexes
To biochemically demonstrate that TMEM175 and LAMP proteins form a stable complex through direct interaction, we co-expressed His-tagged TMEM175 with FLAG-tagged LAMP-1 or LAMP-2 in HEK293F cells using the BacMam system (STAR Methods). Overexpressed proteins were solubilized in DDM detergent and purified using two steps of affinity chromatography – Ni-NTA affinity column followed by anti-FLAG M2 affinity column (STAR Methods). The purified complex elutes as a mono-dispersed peak with a size larger than TMEM175 alone on size-exclusion chromatography and consists of TMEM175 and LAMP-1 or LAMP-2 as indicated by SDS-PAGE, confirming the formation of a stable complex between TMEM175 and LAMP-1 or LAMP-2 (Figure 1H). Due to heavy glycosylation, LAMP proteins migrate as a smeary band with higher molecular weight (~100 kDa) on SDS-PAGE. The complex has a 2:2 subunit stoichiometry as demonstrated later in the structural determination of the purified complex using single-particle cryo-EM.
LAMP proteins inhibit the channel activity of TMEM175
TMEM175 is a K+-selective channel at neutral pH but becomes a highly proton-selective channel at lower luminal pH18,22,25. Two approaches were taken to measure the effect of LAMP-binding on both K+- and proton-conducting activities of TMEM175: one is the electrophysiological recording of HEK293 cells co-expressing TMEM175 with LAMP-1 or LAMP-2 and the other is in vitro K+ flux assay using proteoliposomes containing the purified TMEM175/LAMP complexes.
When over-expressed in HEK293 cells, both TMEM175 and LAMP proteins can be trafficked to the plasma membrane, allowing for direct measurement of TMEM175 channel activity with or without LAMP co-expression by patching the plasma membrane22,23,47–49 (Figure S3A). In this setting, the extracellular side is equivalent to the luminal side of the channel in lysosome. We chose Cs+ as the permeating ion in most of our electrophysiological recordings at neutral pH because TMEM175 conducts Cs+ better than K+ and Cs+ also reduces background currents by blocking the endogenous K+ channels23. Under bi-ionic conditions with Na+ in the bath (extracellular) and Cs+ in the pipette (intracellular), highly Cs+ selective outward currents were recorded at symmetrical pH of 7.4 in the whole cell patch of HEK293 expressing TMEM175 alone (Figure 2A & 2B). Upon changing the bath pH to acidic conditions, TMEM175 becomes a proton-activated proton channel conducting a higher inward proton current at lower pH (Figure 2C). This pH-dependent change of ion selectivity and channel activity of TMEM175 was also demonstrated in two recent studies18,25. However, when TMEM175 was co-expressed with LAMP-1 or 2, its outward Cs+ currents at neutral luminal pH (Figure 2A & 2B) and inward proton currents at acidic luminal pH (Figure 2D–2F) were both significantly reduced, indicating that LAMP proteins inhibit TMEM175 channel activity regardless of luminal pH. To confirm that the observed current reduction upon LAMP-1 or 2 co-expression is not caused by a decreased surface expression of TMEM175, we performed a surface protein pulldown assay demonstrating equivalent expression of TMEM175 in the plasma membrane with or without LAMP co-expression (STAR Methods and Figure S3B).
Figure 2. LAMP binding inhibits the channel activity of TMEM175.
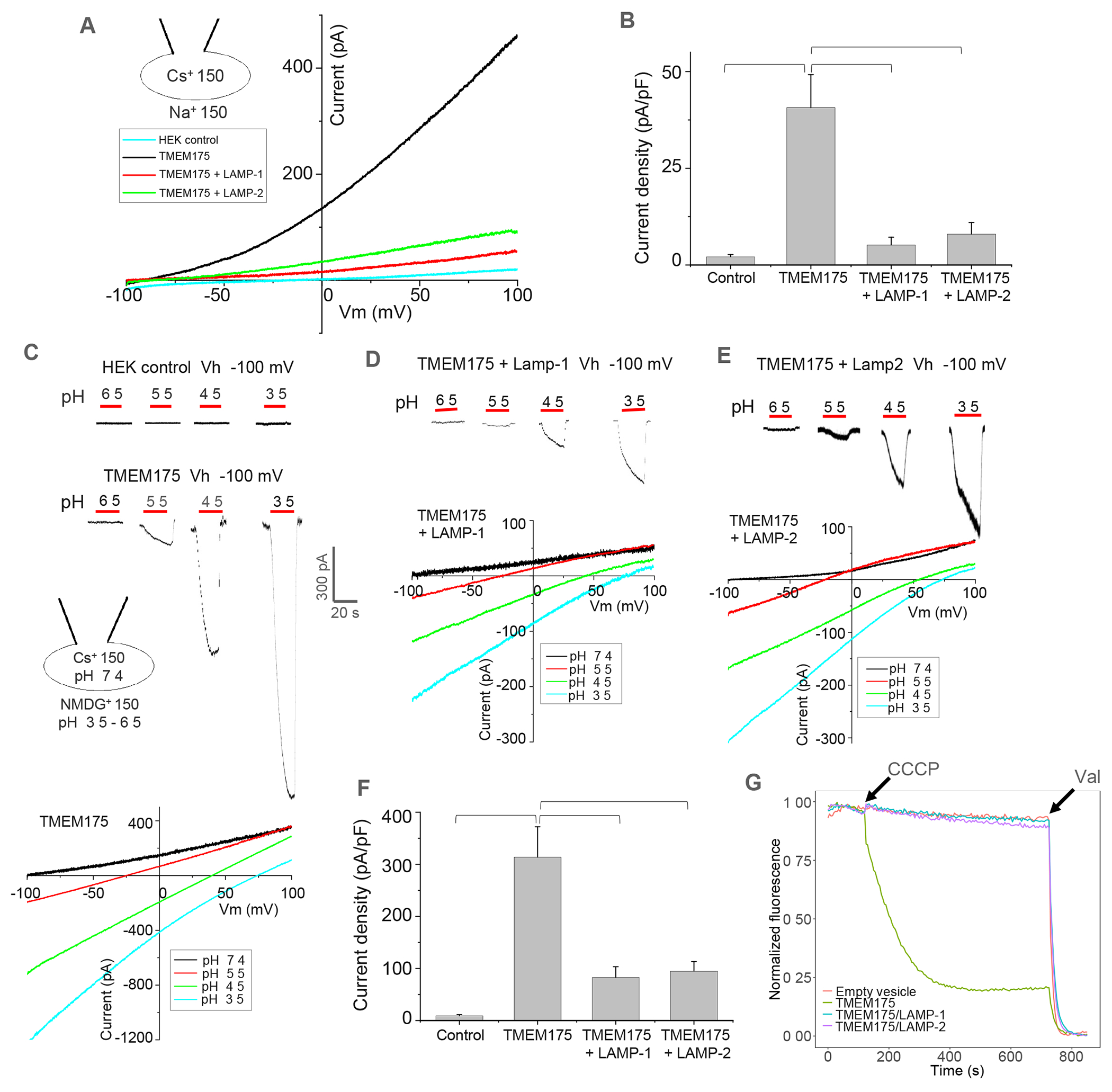
(A) Sample I-V curves of HEK293 cells expressing TMEM175 with LAMP-1 or 2. Outward Cs+ currents were recorded using whole-cell patch clamp at pH 7.4 with 150 mM Na+ in the bath (extracellular/luminal) and 150 mM Cs+ in the pipette (cytosolic).
(B) Cs+ current density of TMEM175 with or without LAMP-1 or 2 co-expression measured at 100 mV in whole-cell recordings. Data are represented as mean ± SEM (n=10, *** p <0.001, **p <0.01).
(C) Luminal acidic pH activation of TMEM175. Upper panel: sample traces of inward proton currents recorded at −100 mV with lowering bath pH. Lower panel: sample I-V curves at various bath pHs indicating the change of channel selectivity. For pH activation, Na+ was replaced by NMDG+ in low-pH bath solutions.
(D) Luminal acidic pH activation and I-V curves of TMEM175 co-expressing with LAMP-1.
(E) Luminal acidic pH activation and I-V curves of TMEM175 co-expressing with LAMP-2.
(F) Proton current density of TMEM175 with or without LAMP-1 or 2 co-expression measured at −100 mV with bath pH of 3.5 in whole-cell recordings. Data are represented as mean ± SEM (n=10, *** p <0.001, ** p <0.01).
(G) K+ flux assay using proteoliposomes containing TMEM175 or TMEM175/LAMP complexes. K+ flux was initiated by adding H+ ionophore carbonyl cyanide m-chlorophenylhydrazone (CCCP) and terminated by adding K+ ionophore valinomycin (Val). The assay was repeated 3 times with consistent results.
See also Figure S3.
We also reconstituted the purified TMEM175 or TMEM175/LAMP complexes into lipid vesicles and performed a fluorescence-based K+ flux assay as previously described50 (STAR Methods). As shown in a representative trace of liposome flux assay, the addition of CCCP to the TMEM175-containing proteoliposomes introduced a time-dependent reduction of ACMA fluorescence in the solution, indicating K+-conducting activity of reconstituted TMEM175 (Figure 2G). However, no obvious fluorescent quenching was observed when TMEM175/LAMP-1- or TMEM175/LAMP-2-containing proteoliposomes were used in the assay, indicating the inhibition of TMEM175 by LAMP-1 or 2 (Figure 2G). All proteoliposome samples used in the flux assay contained an equivalent amount of TMEM175 (Figure S3C).
Structure of the TMEM175/LAMP-1 complex
To provide structural insight into LAMP-TMEM175 interaction, we determined the structure of the TMEM175/LAMP-1 complex to 3.5 Å resolution using single particle cryo-electron microscopy (cryo-EM) (Figure 3A, Figure S4A & S4B, and Table 1). TMEM175 part of the complex is well resolved in the EM map and is similar to the recently determined EM structure of TMEM175 by itself24. LAMP-1 is predicted to contain two homologous luminal “LAMP” domains with β-prism fold followed by a single transmembrane (TM) helix39. However, only the TM helix from LAMP-1, which makes direct contact with TMEM175, is well structured in the complex. The two luminal LAMP domains, on the other hand, are poorly resolved due to their high mobility. Weak EM density from the membrane-proximal LAMP domain could be observed but is not of sufficient quality for model building (Figure 3A).
Figure 3. Structure of TMEM175 in complex with LAMP-1.
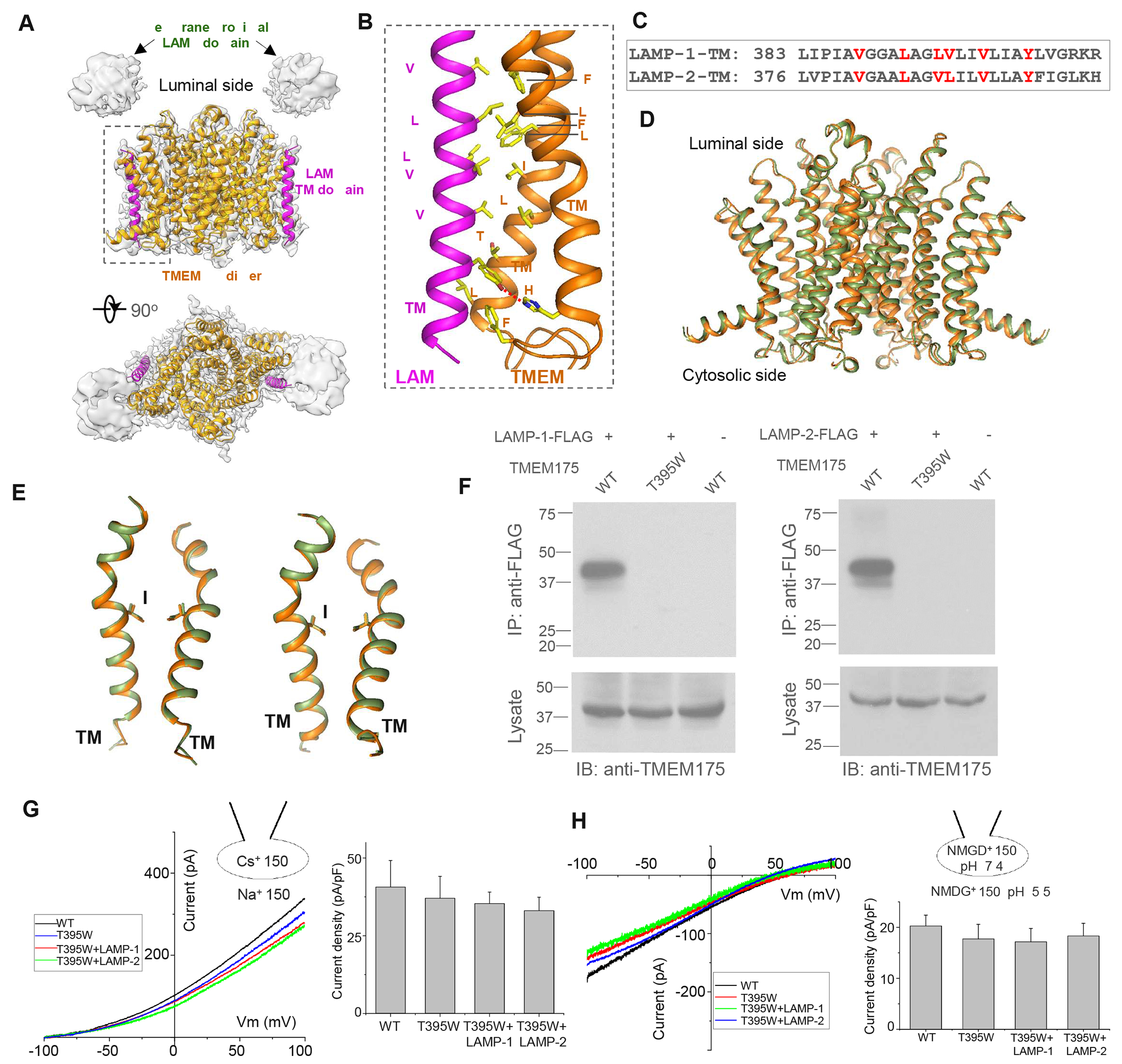
(A) Overall structure of TMEM175/LAMP-1 complex consisting of two TMEM175 and two LAMP-1 subunits. EM map (grey) for the TM region is contoured at 4σ. The weak density from the proximal LAMP domain is contoured 2σ after a local resolution filter.
(B) Inter-molecular interaction between LAMP-1 TM domain (magenta) and TMs 10 and 11 of TMEM175 (orange).
(C) Sequence comparison of the TM domains between LAMP-1 and 2.
(D) Comparison between the previously determined TMEM175 structure in a putative open conformation (green, PDB code 6WC9) and TMEM175 in complex with LAMP-1 (orange).
(E) Structural comparison of the pore-forming TMs 1 and 7 between the two structures in (D). I46 and I271 form the central constriction of the ion pathway.
(F) Co-IP of LAMP proteins and TMEM175 or its T395W mutant in HEK293 cells co-expressing un-tagged TMEM175 or T395W mutant and FLAG-tagged LAMP-1 or 2. Anti-FLAG antibodies were used for immunoprecipitation. Only WT TMEM175 but not its T395W mutant was detected in the IP samples, indicating that the mutation disrupts the interaction between TMEM175 and LAMP proteins.
(G) Sample I-V curves and Cs+ current density (at 100 mV) of HEK293 cells expressing TMEM175(T395W) mutant with or without LAMP-1 or 2. The Cs+ currents were recorded at pH 7.4 with 150 mM Na+ in the bath (extracellular) and 150 mM Cs+ in the pipette. Data are represented as mean ± SEM (n=10).
(H) Sample I-V curves and proton current density (at −100 mV) of the mutant with or without LAMP-1 or 2. The proton currents were recorded with pH 5.5 in the bath and pH 7.4 in the pipette. NMDG+ was used as monovalent in both bath and pipette solutions. Data are represented as mean ± SEM (n=10).
See also Figure S4.
Table 1.
Cryo-EM data collection, refinement and validation statistics
| TMEM175/LAMP-1 | TMEM175/LAMP-1 TM | |
|---|---|---|
| Data collection and processing | ||
| Magnification | 105,000 | 105,000 |
| Voltage (kV) | 300 | 300 |
| Electron exposure (e-/Å2) | 60 | 60 |
| Defocus range (μm) | −0.9 ~ −2.2 | −0.9 ~ −2.2 |
| Pixel size (Å) | 0.83 | 0.83 |
| Symmetry imposed | C2 | C2 |
| Initial particle images (no.) | 2,130,974 | 780,040 |
| Final particle images (no.) | 269,594 | 44,240 |
| Map resolution (Å) | 3.48 | 3.25 |
| FSC threshold | 0.143 | 0.143 |
| Refinement | ||
| Initial model used (PDB code) | 6WC9 | 6WC9 |
| Model resolution (Å) | 3.2 | 3.1 |
| FSC threshold | 0.143 | 0.143 |
| Map sharpening B factor (Å2) | −188.5 | −127.3 |
| Model composition | ||
| Non-hydrogen atoms | 6452 | 6452 |
| Protein residues | 830 | 830 |
| Ligands | 0 | 0 |
| B factors (Å2) | ||
| Protein | 88.27 | 113.78 |
| Ligand | ||
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.004 | 0.003 |
| Bond angles (°) | 0.607 | 0.581 |
| Validation | ||
| MolProbity score | 1.32 | 1.44 |
| Clashscore | 4.47 | 5.61 |
| Poor retainers (%) | 0 | 0.14 |
| Ramachandran plot | ||
| Favored (%) | 97.56 | 97.31 |
| Allowed (%) | 2.44 | 2.69 |
| Disallowed (%) | 0 | 0 |
The TMEM175/LAMP-1 complex has a 2:2 stoichiometry with two TMEM175 subunits forming a dimeric channel in the middle flanked by two LAMP-1 subunits, one on each side. The single TM of LAMP-1 directly interacts with TMs 10 and 11 of TMEM175 mostly through hydrophobic contact (Figure 3B). The key residues on LAMP-1 TM that participate in helical packing with TMEM175 are conserved in LAMP-2 (Figure 3C), suggesting similar inter-molecular interaction in TMEM175/LAMP-2 complex. The weak density of the membrane-proximal LAMP domain marks its position right above the single TM of LAMP-1, quite far from the central pore of TMEM175 (Figure 3A).
Structures of TMEM175 alone in two different conformations have been determined recently24,26. They are largely similar except for the pore-lining TM1 and TM7 helices which form a slightly wider ion pathway in one conformation proposed to represent a K+-conducting open state and a narrower ion pathway in the other conformation proposed to be in a closed state. In the complex, the TMEM175 structure is almost identical to the putative open conformation with an overall RMSD of 1.3 Å (Figure 3D & 3E). With only the TM domain from LAMP-1 being resolved and no obvious structural changes in TMEM175 upon LAMP-1 binding, the complex structure stops short of elucidating the structural mechanism of LAMP-1 inhibition of TMEM175. Nevertheless, the structure unambiguously defines the direct interaction between LAMP-1 and TMEM175. To demonstrate that the TM-mediated complex formation is essential for LAMP inhibition of TMEM175, we performed a series of single-residue mutations on interfacial residues from both TMEM175 and LAMP-1 and identify a T395W mutation on TM10 of TMEM175 that is sufficient to disrupt the complex formation (Figure 3F). The mutant exhibits the same channel activity as the wild-type TMEM175 but is no longer inhibited by LAMP-1 or LAMP-2 when co-expressed in HEK293 cells (Figure 3G & 3H). This mutant provides a useful tool to delineate the physiological role of LAMP inhibition in lysosomal pH homeostasis in our later experiments.
The proximal LAMP domain is necessary for TMEM175 inhibition
The poorly resolved structure of the LAMP-1 luminal region in the complex raises the question of whether the two luminal LAMP domains interact with TMEM175 and play any role in channel inhibition. Two pieces of evidence point to some transient interactions between the LAMP domains and TMEM175. Firstly, we generated two partial constructs of FLAG-tagged LAMP-1 containing only the luminal region or the TM domain and co-expressed them with TMEM175. As expected, the TM-only construct is sufficient to pull down TMEM175. Despite having a much weaker signal, the LAMP-1 luminal region can also pull down some TMEM175, indicating some weak interactions between them (Figure S5A). We also performed cross-linking mass spectrometry using the amine-to-amine bis(sulfosuccinimidyl)suberate (BS3) cross-linker to investigate the protein-protein interactions within the TMEM175/LAMP-1 complex (Figure S5B–S5D and STAR Methods)51. Two inter-subunit crosslinking reactions, between TMEM175 K292 and LAMP-1 K137 from the distal LAMP domain or K337 from the proximal LAMP domain, were unambiguously identified, indicating transient proximity between the LAMP domains and the luminal face of TMEM175 (Figure S5B–S5D).
To test if the luminal LAMP domains are essential for TMEM175 inhibition, we co-expressed TMEM175 with two truncation mutants of LAMP-1 (Figure 4A) and recorded its Cs+- and proton-conducting activities (Figure 4B & 4C). Both Cs+ and proton currents from TMEM175 were inhibited when the channel is co-expressed with full-length LAMP-1 or a truncation mutant without the distal LAMP domain (LAMP-1-ΔdLAMP). However, no obvious inhibition was observed when co-expressed with a LAMP-1 mutant with both distal and proximal LAMP domains deleted (LAMP-1-TM) (Figure 4B & 4C). Western-blot analysis of cell lysate and the surface pulldown confirmed that the surface expressions of TMEM175 were equivalent among all samples (Figure S6A). Thus, a LAMP protein with a single proximal LAMP domain is sufficient for TMEM175 inhibition, but the TM domain alone does not inhibit the channel. It is interesting to note that some LAMP family proteins in specific cell types contain a single LAMP domain39.
Figure 4. Effect of LAMP-1 truncation on TMEM175 inhibition.
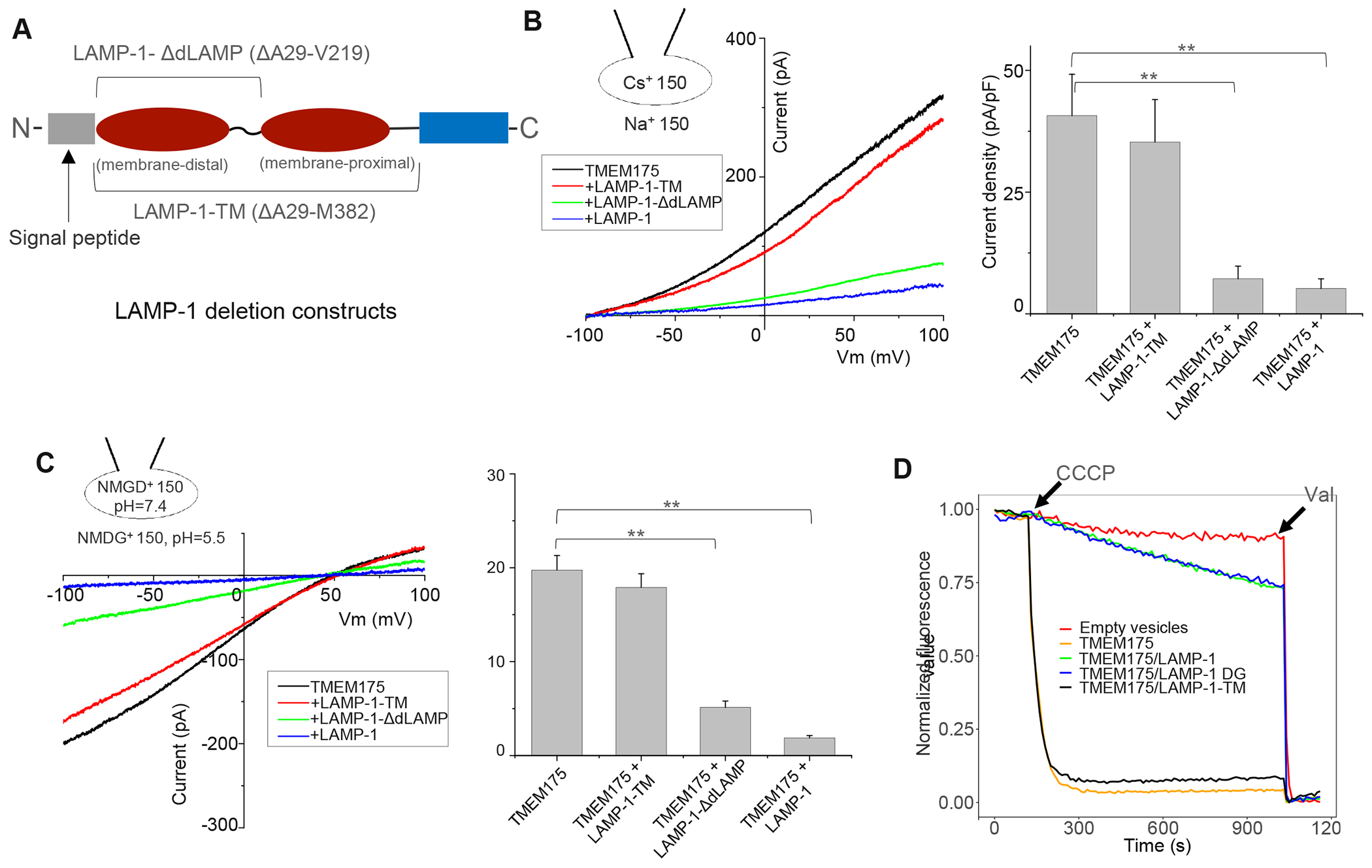
(A) Constructs of the two LAMP-1 deletion mutants. Brackets mark the deleted regions.
(B) Sample I-V curves and Cs+ current density (at 100 mV) of HEK293 cells expressing TMEM175 with LAMP-1 or its truncation mutants. The Cs+ currents were recorded in whole-cell configuration at pH 7.4. Data are represented as mean ± SEM (n=10, ** p <0.01).
(C) Sample I-V curves and proton current density (at −100 mV) of TMEM175 in complex with LAMP-1 or its truncation mutants. The proton currents were recorded with pH 5.5 in the bath and pH 7.4 in the pipette. NMDG+ was used as monovalent in both bath and pipette solutions. Data are represented as mean ± SEM (n=10, ** p <0.01).
(D) K+ flux assay using proteoliposomes containing TMEM175, TMEM175/LAMP-1 before or after de-glycosylation (DG), or TMEM175/LAMP-1-TM.
To confirm that LAMP-1-TM is expressed and forms a stable complex with TMEM175 despite the lack of inhibitory effect, we co-expressed and purified the TMEM175/LAMP-1-TM complex and determined its cryo-EM structure, which is virtually the same as the TMEM175/LAMP-1 complex (Figure S4C & S4D and Table 1). Furthermore, we also prepared proteoliposomes containing purified TMEM175, TMEM175/LAMP-1, or TMEM175/LAMP-1-TM complex and compared the channel activities using the liposome flux assay (Figure 4D). TMEM175/LAMP-1 complex showed little K+-conducting activity in the flux assay, whereas TMEM175/LAMP-1-TM complex exhibited similar activity as TMEM175 alone, confirming the lack of channel inhibition by LAMP-1-TM. The ability to bind but not inhibit TMEM175 makes LAMP-1-TM a nice molecular tool to probe the physiological function of LAMP proteins in our later experiments, as its expression in lysosome can compete against the endogenous LAMP proteins and mitigate their inhibition of TMEM175. As LAMP-1 is heavily glycosylated, we further investigated whether its glycosylation plays any role in TMEM175 inhibition. Similar inhibition of K+-conducting activity was observed in the proteoliposomes prepared using TMEM175/LAMP-1 complexes either before or after de-glycosylation (Figure 4D), suggesting that LAMP-1 glycosylation does not contribute to its inhibition of TMEM175. All proteoliposomes used in the flux assay contained an equivalent amount of TMEM175 (Figure S6B).
LAMP binding antagonizes DCPIB activation of TMEM175
DCPIB, a specific inhibitor of volume-regulated anion channel52, can potently activate the TMEM175 channel (Hu et al., 2022). We therefore also measured the effect of LAMP binding on DCPIB ligand activation of TMEM175. When only TMEM175 was expressed, DCPIB indeed elicited much larger dose-dependent Cs+ currents in our whole-cell recordings at neutral extracellular pH (luminal side of TMEM175) with EC50 of about 3.6 μM (Figure 5A & 5B). Co-expression of LAMP-1 or 2 competitively inhibited DCPIB activation by markedly reducing the DCPIB-elicited Cs+ current and also decreasing the efficacy of DCPIB activation (Figure 5A–5C). Co-expression of the LAMP-1-ΔdLAMP construct without the distal LAMP domain yielded a similar inhibition as the full-length LAMP-1 whereas no obvious inhibition was observed with LAMP-1-TM co-expression (Figure 5A–5C). Likewise, DCPIB also activates the proton conduction of TMEM175, and this activation is inhibited by the co-expression of LAMP-1, LAMP-2, or LAMP-1-ΔdLAMP, but not LAMP-1-TM (Figure 5D). Similar to the wild-type channel, the Cs+- or proton-conducting activity of TMEM175(T395W) mutant can be activated by DCPIB. However, the DCPIB activation of the mutant is no longer inhibited by LAMP-1 or 2 (Figure 5E & 5F). Thus, the DCPIB activation of the channel in both K+ (or Cs+) and proton-conducting states implies the presence of a ligand-regulated gate in TMEM175 that has not yet been defined in the structure. LAMP binding can antagonize the DCPIB activation of TMEM175. As discussed later, we suspect that the LAMP proteins inhibit TMEM175 by closing the same gate activated by DCPIB and likely function as a negative allosteric modulator to the DCPIB activation.
Figure 5. LAMP binding antagonizes DCPIB activation of TMEM175.
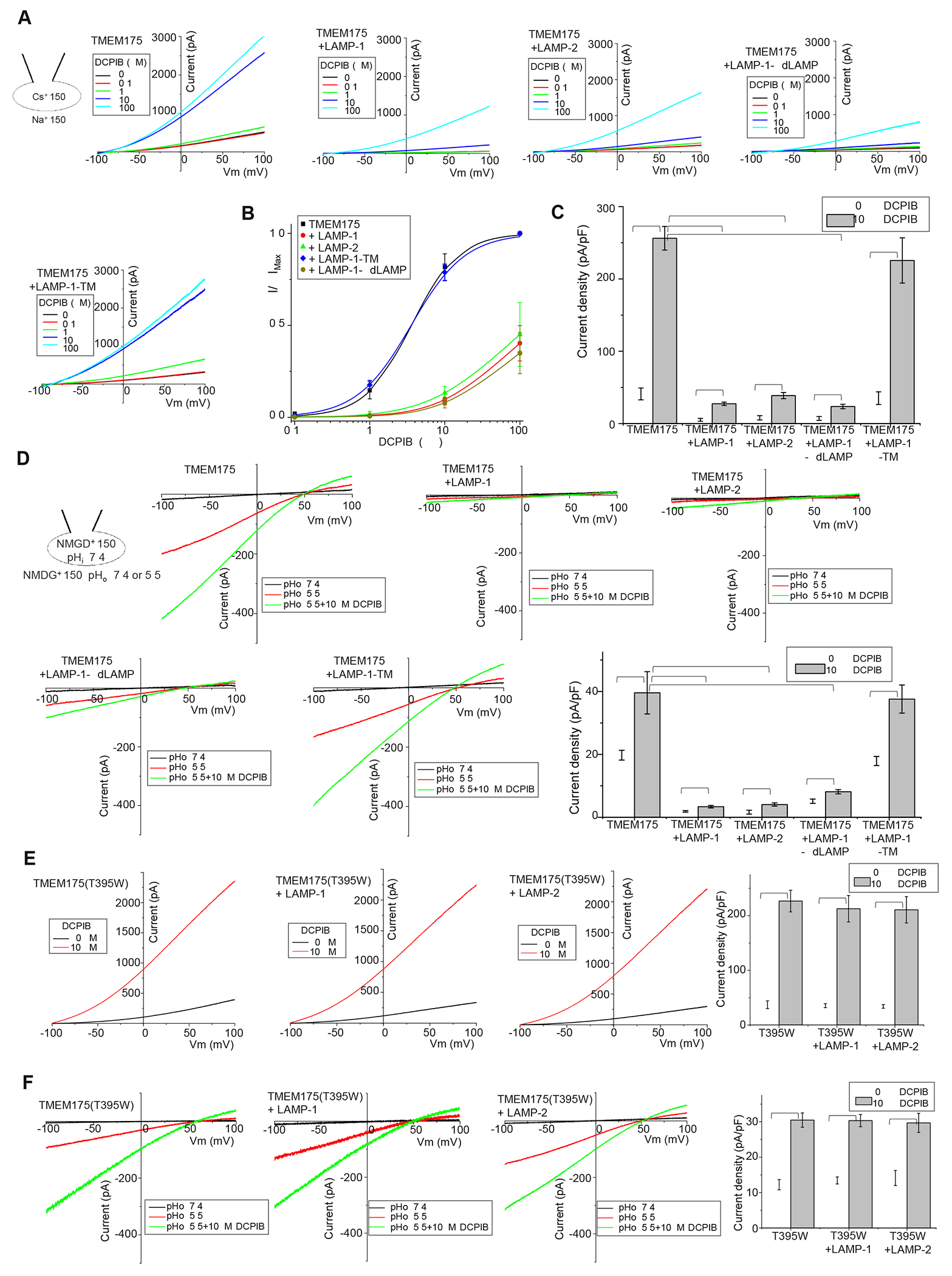
(A) Sample I-V curves of Cs+ currents from TMEM175 at various DCPIB concentrations. TMEM175 was expressed in HEK293 cells alone or with LAMP-1, LAMP-2, or LAMP-1 deletion mutant. Outward Cs+ currents were recorded using whole-cell patch clamp at pH 7.4.
(B) Concentration-dependent DCPIB activation of TMEM175 or TMEM175 in complex with various LAMP protein constructs at 100 mV. Data for DCPIB activation of TMEM175 and TMEM175/LAMP-1-TM complex were normalized against their respective currents at 100 μM DCPIB and were fitted to the Hill equation with EC50=3.6±0.3 μM, n=1.4±0.1 for TMEM175 and EC50=3.5±0.2 μM, n=1.2±0.1 for TMEM175/LAMP-1-TM complex. With markedly reduced currents and lower DCPIB efficacy of TMEM175 upon LAMP inhibition, all data from TMEM175 in complex with LAMP-1, LAMP-2, or LAMP-1-ΔdLAMP were normalized against the averaged TMEM175 currents at 100μM DCPIB. Data are represented as mean ± SEM (n=10 independent experiments).
(C) Cs+ current density from TMEM175 and TMEM175 in complex with various LAMP constructs measured at 100 mV in whole-cell recordings with or without 10 μM DCPIB. Data are represented as mean ± SEM (n=10, *** p <0.001, ** p <0.01).
(D) Sample I-V curves and proton current density (at −100 mV) from TMEM175 and TMEM175 in complex with various LAMP constructs recorded with or without 10 μM DCPIB. The proton currents were recorded with pH 7.4 (as background control) or 5.5 in the bath and pH 7.4 in the pipette. NMDG+ was used as monovalent in both bath and pipette solutions. Data are represented as mean ± SEM (n=10, *** p <0.001, ** p <0.01).
(E) Sample I-V curves and Cs+ current density (at 100 mV) from TMEM175(T395W) mutant with or without LAMP-1 or 2 co-expression. The Cs+ currents were recorded at pH 7.4 with or without 10 μM DCPIB in the bath (extracellular). Data are represented as mean ± SEM (n=10, ** p <0.01).
(F) Sample I-V curves and proton current density (at −100 mV) from TMEM175(T395W) mutant with or without LAMP-1 or 2 co-expression. The proton currents were recorded with pH 5.5 with or without 10 μM DCPIB in the bath. NMDG+ was used as monovalent in both bath and pipette solutions. Data are represented as mean ± SEM (n=10, ** p <0.01).
LAMP inhibition of TMEM175 regulates lysosomal pH homeostasis
TMEM175 was recently characterized as a low pH-activated proton channel whose proton efflux activity balances the proton import by V-ATPase and regulates lysosomal pH homeostasis18,25. Our finding of LAMP inhibition of TMEM175 implies that LAMP proteins would add extra control to the lysosomal pH by mitigating TMEM175 activity. To test that, we measured the lysosomal pH changes in response to the perturbation of complex formation between TMEM175 and LAMP proteins in HAP1 cells. We first used pH-sensitive LysoTracker staining to qualitatively monitor the lysosomal pH (STAR Methods). Consistent with the previous study18, the LysoTracker staining in TMEM175-knockout (KO) cells was more intense than that in wild-type (WT) cells indicating hyper-acidification of lysosomes in KO cells due to the loss of TMEM175 function (Figure 6A & 6B, Figure S7A). TMEM175 expression in WT or KO cells respectively decreased the staining intensity in both cells, confirming that the proton conduction of TMEM175 alkalinizes the lysosomal pH. When TMEM175(T395W) mutant was expressed in the KO cells, the staining intensity was further decreased as compared to the KO cells rescued by WT TMEM175, indicating a further increase of lysosomal pH attributable to an increase in channel activity of the mutant that is no longer inhibited by LAMP proteins. When GFP-tagged TMEM175 or its T395W mutant were expressed in the KO cells, they both co-localized with the LysoTracker staining (Figure S7B), confirming their lysosomal localization. A similar gain-of-function effect was also observed when TMEM175 was co-expressed in KO cells with LAMP-1-TM. That is because LAMP-1-TM competes against endogenous LAMP proteins in complex formation but does not inhibit TMEM175 and thereby mitigates the channel inhibition from the endogenous LAMP proteins.
Figure 6. Lysosomal pH and hydrolytic activity measurements.
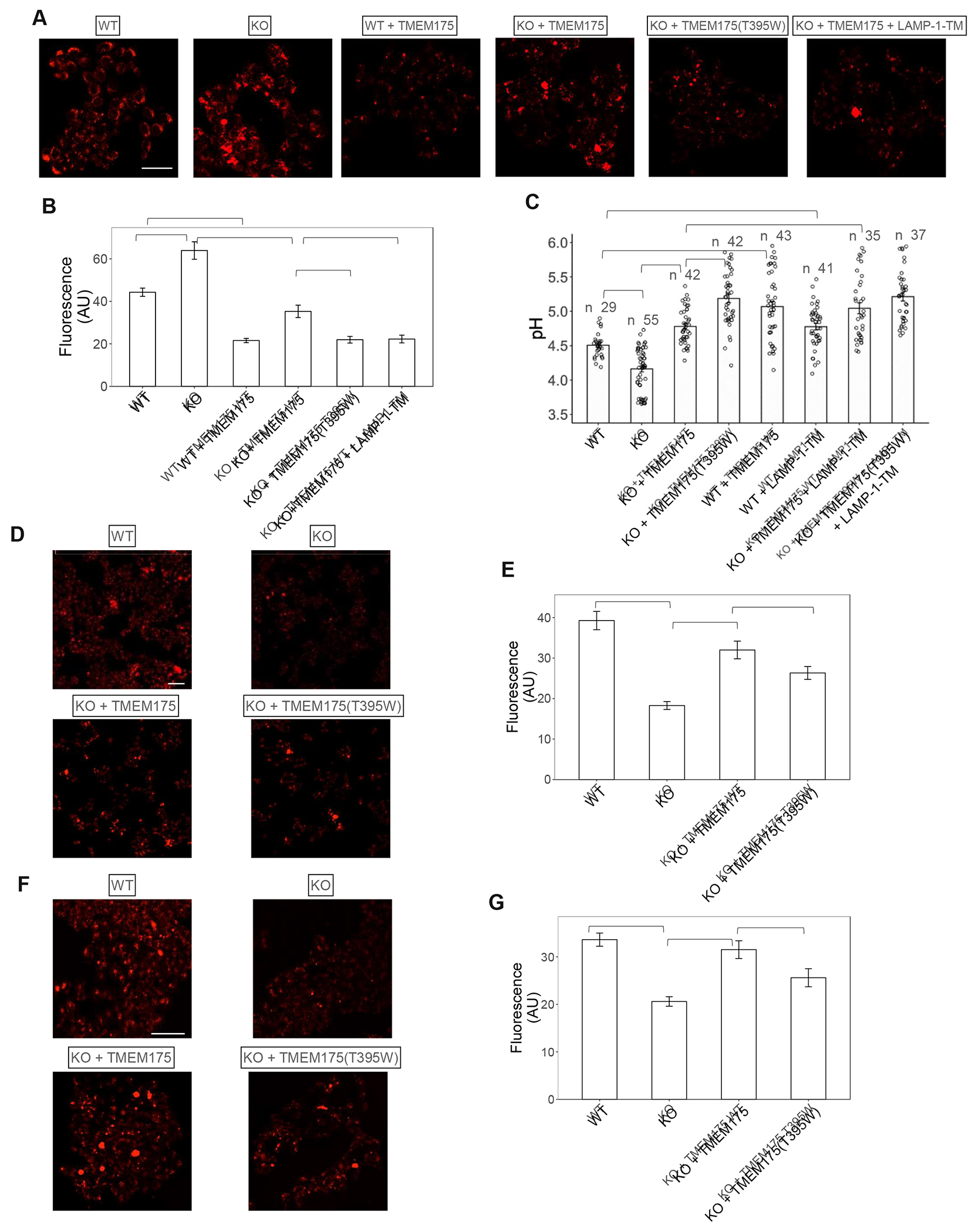
(A) Sample images of LysoTracker staining of WT and KO HAP1 cells expressing TMEM175 or its T395W mutant.
(B) Averaged LysoTracker intensity per cell. Data are represented as mean ± SEM (n= 27-30 cells, *** p <0.001).
(C) Lysosomal pH in WT and KO HAP1 cells determined using ratiometric fluorescence imaging of Oregon Green 488 Dextran. Data are represented as mean ± SEM (n = number of cells per group, *** p <0.001, ** p <0.01).
(D) & (E) Sample images of DQ™-BSA-red staining and overall intensity in WT and KO HAP1 cells expressing TMEM175 or its T395W mutant. Data are represented as mean ± SEM (n= 30 cells, *** p <0.001).
(F) & (G) Sample images Magic Red staining and overall intensity in WT and KO HAP1 cells expressing TMEM175 or its T395W mutant. Data are represented as mean ± SEM (n= 30 cells, *** p <0.001, * p <0.05).
See also Figure S7.
We also used ratiometric fluorescence imaging of the pH-sensitive dye Oregon Green™ 488 Dextran to provide a more quantitative measurement of lysosomal pH as previously described16,18 (STAR Methods). Consistent with the previous measurement18, the lysosomes of WT HAP1 cells had an average pH of about 4.5, whereas TMEM175 deficiency caused lysosome hyper-acidification, yielding an average lysosomal pH of about 4.2 in the KO cells (Figure 6C). Over-expression of TMEM175 in the WT or the KO cells increased proton efflux from lysosomes, resulting in about 0.6 pH unit increase to about 5.1 and 4.8, respectively. As expected, the expression of the non-inhibited TMEM175(T395W) mutant in KO cells resulted in a further increase of the lysosomal pH to 5.2 as compared to 4.8 when rescued by WT TMEM175. The expression of LAMP-1-TM in the wild-type HAP1 cells mitigated TMEM175 inhibition by endogenous LAMP, resulting in a higher lysosomal pH (~4.8) than that in the control cells without transfection (pH~4.5). Similarly, the lysosomal pH of KO HAP1 cells co-expressing LAMP-1-TM and TMEM175 is higher than that expressing TMEM175 alone (5.1 vs 4.8). LAMP-1-TM’s gain-of-function effect on channel activity was abolished if TMEM175(T395W) mutant was used in the rescue experiment simply because the mutant no longer interacts with LAMP or LAMP-1-TM, resulting in the same lysosome pH in KO cells with or without LAMP-1-TM co-expression. In all of our measurements, the amount of overexpressed TMEM175 and its mutant is comparable in WT and KO cells but higher than endogenous TMEM175 in the WT cells (Figure S7C). This explains the observation that the lysosomal pH of KO cells rescued by TMEM175 overexpression tends to be higher than that in the native WT cells. There were no observable changes in endogenous LAMP expression in all of our samples (Figure S7C). Thus, both pH measurements demonstrated that LAMP inhibition of TMEM175 activity facilitates lysosomal acidification to a lower pH range.
LAMP inhibition of TMEM175 is important for Lysosomal hydrolytic activity
While the proton release by TMEM175 prevents hyper-acidification of the lysosome, its inhibition by LAMP proteins appears to be necessary for the lysosome to reach an optimal pH of 4.5. As the pH range in lysosomes is tuned to be optimal for hydrolase activity, the perturbation of lysosomal pH by increasing or decreasing TMEM175 channel activity should also affect the lysosomal hydrolytic activity. When using DQ™-BSA-red staining to measure the overall lysosomal hydrolytic activity53, the TMEM175-KO HAP1 cells displayed a marked decrease of the fluorescence intensity as compared to the WT cells, indicating a reduction of the lysosomal hydrolytic activity caused by the hyper-acidification of the lysosome (Figure 6D & 6E). Overexpressing TMEM175 in KO cells can partly rescue the hydrolytic activity but not to the same level as that in the WT cells. This is likely because TMEM175 overexpression in the KO cells led to a higher lysosomal pH than that in native cells as demonstrated in pH measurement, resulting in a less optimal pH environment for lysosomal hydrolytic activity. As expected, the recovery of hydrolytic activity became even smaller when the non-inhibited TMEM175(T395W) mutant was expressed in the KO cells, attributable to a further increase in lysosomal pH. Similar results were also observed in the Cathepsin B activity assay using Magic Red, a fluorogenic substrate of Cathepsin B54 (Figure 6F & 6G): impaired Cathepsin B activity in KO cells can be largely restored by expressing wild-type TMEM175, but this recovery becomes less effective when expressing TMEM175(T395W) mutant. The expression levels of Cathepsin B remained comparable in all cell samples used for its activity measurement (Figure S7D). Thus, the effect of LAMP inhibition on lysosomal hydrolytic activity correlates with its effect on lysosome pH homeostasis, demonstrating that both hyper- and hypo-acidification can compromise lysosomal hydrolytic activity.
Discussion
As the most abundant lysosomal membrane glycoproteins, LAMP-1 and LAMP-2 were initially suggested to be structural proteins that protect lysosome integrity. However, mice deficient in both LAMP-1 and LAMP-2 are embryonic lethal and exhibit phenotypes of autophagic vacuoles accumulation and impaired cholesterol export from lysosomes, yet their lysosome integrity appears to be intact40, suggesting that LAMP proteins have some functional role in lysosomal physiology other than structural proteins. Here, using a multidisciplinary approach, we unravel a key biological function of LAMP-1 and LAMP-2 in regulating lysosomal pH homeostasis by directly interacting with and tightly controlling the function of the lysosomal cation channel TMEM175. We first used functional proteomics to identify LAMP proteins as potential interacting partners of TMEM175 and confirmed their direct interaction by Co-IP and biochemical purification. We subsequently determined the high-resolution cryo-EM structure of the TMEM175/LAMP-1 complex that reveals the molecular details underlying the complex formation mediated through their respective TM domains. Using electrophysiology, we functionally demonstrated that LAMP-1 or 2 binding inhibits TMEM175 channel activity. We performed in vivo imaging to demonstrate that disrupting the interaction between TMEM175 and LAMP proteins alkalinizes lysosomal pH and impairs lysosomal hydrolytic activity. Thus, LAMP inhibition of TMEM175 reduces proton efflux and facilitates lysosomal acidification to a lower pH necessary for their key role as cellular degradation and recycling centers.
It is worth noting that TMEM175 is localized in both early endosome and late endosome/lysosome whereas LAMP proteins are delivered to late endosome/lysosome from the trans-Golgi network (TGN)22,55–57. We suspect that the delivery of LAMP proteins and their ensued inhibition of TMEM175 facilitate the lysosomal acidification to a lower pH necessary for their degradation function and therefore play a central role in lysosome biogenesis. Thus, while the V-ATPase is responsible for organelle acidification, TMEM175 and LAMP proteins may work together to optimally offset the proton influx from V-ATPase and set different pH gradients in different organelles, from higher pH in early endosome to lower pH in the lysosome (Figure 7). It is worth noting that organelle pH regulation is a complex process and involves many protein factors. Our simplistic model only emphasizes the proton relief function of TMEM175.
Figure 7.
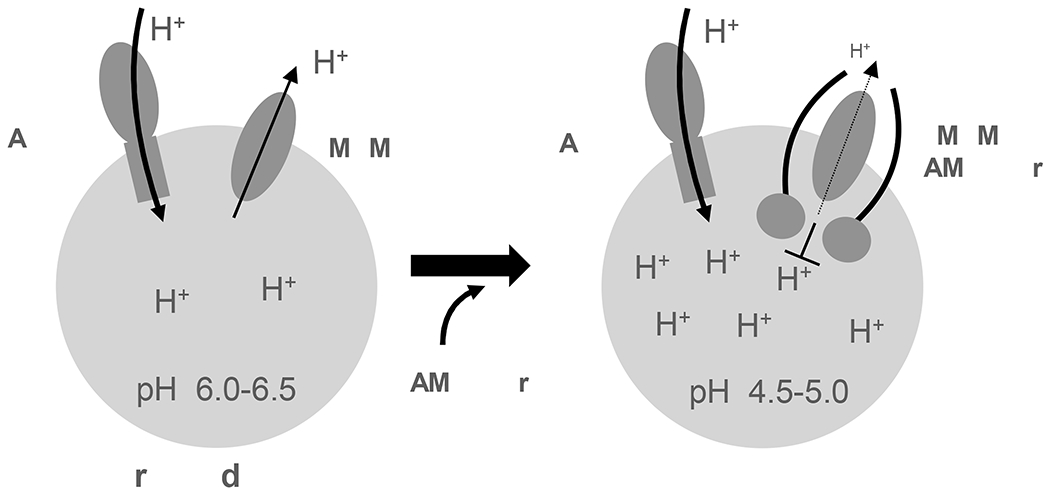
Working model of LAMP-facilitated lysosomal acidification
LAMP-1 and 2 deficient cells have impaired fusion of lysosomes with phagosomes and autophagosomes, resulting in the accumulation of autophagic vacuoles41,58. TMEM175, the target of LAMP inhibition, was suggested to regulate the fusion of the autophagosomes to lysosomes22. The defect of lysosomal acidification in V-ATPase-depleted cells also impairs autophagic flux and results in the accumulation of enlarged autophagic vesicles59. All these proteins are involved in lysosome acidification and their connections to the autophagic degradation process likely converge to their common role in regulating the lysosomal pH homeostasis.
Limitations of the Study
One outstanding question derived from our study is how LAMP proteins inhibit TMEM175 channel activity. Due to the poorly resolved luminal domain structure in the TMEM175/LAMP-1 complex as well as the lack of obvious structural changes in TMEM175 with or without LAMP-1 binding, the current complex structure is unable to elucidate the structural basis of the inhibition. Furthermore, some basic questions related to TMEM175 selectivity and gating remain unanswered despite the determination of multiple high-resolution structures, adding further complexity to resolve the structural mechanism of LAMP inhibition. For example, the TMEM175 structure in our complex is virtually identical to the putative open conformation even though the channel is inhibited by LAMP-1, raising the question of whether the two conformations observed in previous TMEM175 structures truly represent the open and closed states, respectively. In addition, the acidic luminal pH not only changes TMEM175 selectivity but also activates the channel in its proton-conducting state, yet it is unclear what structural changes occur in TMEM175 that trigger the changes of both channel selectivity and gating in response to lower luminal pH. Nevertheless, several conclusions can be drawn from our structural and functional analyses of LAMP inhibition. Firstly, the small molecule agonist DCPIB can potently activate the TMEM175 channel in both K+ and proton-conducting states suggesting that TMEM175 has an unidentified ligand gate shared in both conducting modes. Secondly, the LAMP-1 deletion mutant containing the single proximal LAMP domain is sufficient to inhibit the channel and also antagonize the DCPIB activation, yet the proximal LAMP domain is positioned at the periphery of TMEM175, far from the central pore for direct blocking, suggesting that LAMP inhibits TMEM175 activity by regulating the channel gating rather than blocking. Furthermore, if LAMP proteins inhibit TMEM175 by simple blockage, we do not expect to observe their antagonizing effect on the efficacy of DCPIB activation. Thirdly, the LAMP-1 TM domain contributes to the major inter-subunit contact between LAMP-1 and TMEM175 and it can bind but not inhibit TMEM175 or antagonize DCPIB activation, suggesting non-overlapping binding between DCPIB agonist and LAMP proteins. As the LAMP proteins inhibit TMEM175 in both K+- and proton-conducting states and also competitively antagonize DCPIB activation, we suspect they regulate TMEM175 by closing the same gate activated by DCPIB and function as a negative allosteric modulator to the DCPIB activation. However, where is this gate in TMEM175, and how this gate controls the channel opening remain open questions for further investigation.
While our findings demonstrate that LAMP-1 or LAMP-2 can individually bind and inhibit TMEM175, we cannot rule out the possibility that the dimeric TMEM175 channel can also be inhibited by binding both LAMP-1 and 2, one on each side. Considering the similarity between the two LAMP proteins in their interaction with and inhibition of TMEM175, this likely happens in lysosomes where both LAMP-1 and 2 are abundant. To test this possibility, we performed a Co-IP experiment using HEK293 cells co-expressing TMEM175 with both LAMP-1 and LAMP-2 and demonstrated that FLAG-tagged LAMP-2 can pull down both TMEM175 and LAMP-1 (Figure S2C), but LAMP-2 cannot pull down LAMP-1 in the control cells without TMEM175 co-expression. This result suggests that TMEM175 can indeed form a heterogeneous complex with LAMP-1 and LAMP-2, but LAMP-1 and 2 do not interact with each other. Further study is needed to test if this heterogeneous complex exerts a different inhibitory effect on TMEM175 than the complex formed with individual LAMP proteins.
In this study, we chose to perform the lysosomal pH and hydrolytic activity measurements using TMEM175 KO cells rather than LAMP-1 and/or LAMP-2 KO cells for two reasons. First, due to functional redundancy between LAMP-1 and 2, we might not observe obvious functional defects in LAMP-1 or LAMP-2 single-KO cells. Second, in light of the high abundance of LAMP-1 and LAMP-2 as well as their other potential functions in lysosomes, we suspect their double KO could cause significant defects to lysosome stability and properties, making their pH and hydrolytic activity measurements unreliable. An alternative approach we plan to take in the future study is to identify mutations in LAMP-1 and 2 that will disrupt their interactions with TMEM175 and use the knock-in method to test the effect of these mutations on lysosomal pH and hydrolytic activity.
STAR Methods:
Resource availability
Lead contact:
Further information and requests for resources and reagents should be directed to Youxing Jiang (youxing.jiang@utsouthwestern.edu)
Materials availability:
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
The cryo-EM density maps of the human TMEM175/LAMP-1 and TMEM175/LAMP-1-TM complexes have been deposited at the Electron Microscopy Data Bank. Atomic coordinates of TMEM175/LAMP-1 and TMEM175/LAMP-1-TM complexes have been deposited at the Protein Data Bank and are publicly available as of the date of publication. Accession numbers are listed in the key resource table. All original imaging data have been deposited at Mendeley Data and are publicly available as of the date of publication. DOIs are listed in the key resource table. The quantitative proteomics and cross linking proteomics datasets have been deposited at MassIVE database and are publicly available as of the date of publication. DOIs are listed in the key resource table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FLAG, rabbit sourced | Sigma | Cat#F7425 |
| anti-LAMP-1, rabbit sourced | Abcam | Cat#ab24170 |
| anti-LAMP-2, rabbit sourced | Abcam | Cat#ab199946 |
| Anti-FLAG, mouse sourced | Sigma | Cat#F3165 |
| Anti-TMEM175, rabbit sourced | Proteintech | Cat#19925-1-AP |
| anti-Cathepsin B, rabbit sourced | Abcam | Cat#ab125067 |
| anti-β actin, rabbit sourced | Sigma | Cat#A2066 |
| anti-LAMP1, mouse sourced | Abcam | Cat#ab25630 |
| anti-LAMP2, mouse sourced | Abcam | Cat#ab25631 |
| Bacterial and Virus Strains | ||
| DH5a competent E.coli | Thermo Fisher | Cat# 18265017 |
| E. coli DH10bac | Thermo Fisher | Cat# 10361012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| n-Dodecyl-β-D-Maltopyranoside | Anatrace | Cat#D310 |
| Dithiothreitol | Thermo | Cat#R0861 |
| Iodoacetamide | Sigma | Cat#I6125 |
| Acetonitrile | Fisher | Cat#75-05-8 |
| Trypsin Gold | Promega | Cat#V5280 |
| 6-Aminohexanoic acid | Sigma | Cat#07260 |
| Sulfo-NHS-SS-Biotin | Thermo | Cat#21331 |
| GDN | Anatrace | Cat#GDN101 |
| Endo Hf | NEB | Cat#P0703S |
| Carbonyl cyanide 3-chlorophenylhydrazone | Sigma | Cat#C2759 |
| ACMA (9-Amino-6-Chloro-2-Methoxyacridine) | Thermo | Cat#A1324 |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (POPG) | Avanti | Cat#840457C |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) | Avanti | Cat#850757P |
| n-Decyl-β-D-Maltoside | Anatrace | Cat#D322LA |
| Bio-Beads SM-2 Resin | Bio-rad | Cat#1523920 |
| Valinomycin | Invitrogen | Cat#V1644 |
| BS3 (bis(sulfosuccinimidyl)suberate) | Thermo | Cat#21580 |
| LysoTracker™ Red DND-99 | Invitrogen | Cat#L7528 |
| Oregon Green™ 488 dextran; 10,000 MW, Anionic | Invitrogen | Cat#D7170 |
| Nigericin | Invitrogen | Cat#N1495 |
| DQ™ Red BSA | Invitrogen | Cat#D12051 |
| Magic Red® Cathepsin-B Assay Kit | Immunochemistry | Cat#937 |
| FLAG peptide | Sigma | Cat#F3290 |
| Deposited Data | ||
| TMEM175-full length LAMP-1 density map | This study | EMD: EMD-29553 |
| TMEM175-full length LAMP-1 atomic model | This study | PDB: 8FY5 |
| TMEM175-TM LAMP-1 density map | This study | EMD: EMD-29572 |
| TMEM175-TM LAMP-1 atomic model | This study | PDB: 8FYF |
| The raw data of quantitative proteomics of TMEM175 affinity enrichment | This study | http://dx.doi.org/doi:10.25345/C5RN30J1P |
| The raw data of crosslinking proteomics of TMEM175-LAMP1 complex | This study | http://dx.doi.org/doi:10.25345/C5CC0V39R |
| The Mendeley dataset of raw data of gels, blots and imagings | This study | http://dx.doi.org/doi:10.17632/6d8w9vv5bv.3 |
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | ATCC#CRL-1573 |
| SH-SY5Y | ATCC | ATCC#CRL-2266 |
| Sf9 | ATCC | ATCC#CRL-1711 |
| FreeStyle™ 293-F | ThermoFisher | Cat#R79007 |
| HAP1 | HorizonDiscovery | Cat#HorizonDiscovery |
| HAP1 TMEM175 knockout | HorizonDiscovery | Cat#HZGHC005593c004 |
| Recombinant DNA | ||
| pCMV6-TMEM175-Myc-FLAG | Origene | Cat#RC201422 |
| pCMV6-LAMP1-Myc-FLAG | Origene | Cat#RC219208 |
| pCMV3-FLAG-LAMP1 | SinoBiological | Cat#HG11215-NF |
| pCMV3-FLAG-LAMP2 | SinoBiological | Cat#HG13555-NF |
| pEZT-BM | Addgene | Plasmid#74099 |
| pCMV6-LAMP1-GFP | Origene | Cat#RC100016 |
| pCMV6-LAMP2-GFP | Origene | Cat#RG200456 |
| Software and Algorithms | ||
| Maxquant | Maxquant 2.1.4 | https://www.maxquant.org |
| Mascot | Mascot 2.7.0 | http://www.matrixscience.com/ |
| RELION | Scheres, 2012 | https://www3.mrc-lmb.cam.ac.uk/relion |
| UCSF ChimeraX | Pettersen et al., 2021 | https://www.cgl.ucsf.edu/chimerax/ |
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org |
| COOT | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PyMOL | Molecular Graphics System, Version 2.5.2, Schrödinger | https://pymol.org/2/ |
| Fiji | Schindelin et al., 2012 | https://imagej.net/software/fiji/ |
| Other | ||
| R1.2/1.3 200-mesh or 300-mesh gold holey carbon grid | Quantifoil | Cat#4220G-CF/4230G-CF |
Experimental Model and Study Participant Details
Cell lines
HEK293 (ATCC# CRL-1573) and SH-SY5Y (ATCC# CRL-2266) were used in proteomics, immunoprecipitation, and immunoblotting experiments. Sf9 (ATCC# CRL-1711) and FreeStyle™ 293-F (ThermoFisher, R79007) cells were used for protein expression and purification. TMEM175 knockout HAP1 cells (HorizonDiscovery, HZGHC005593c004) and the parental control cells (HorizonDiscovery, C631) were used in the lysosomal pH and hydrolytic activity assay.
Method details
Plasmids
For proteomics, C-terminal FLAG-tagged TMEM175 was cloned into the pCMV6-Entry vector (Origene, RC201422). For biochemical and functional assays and in vivo imaging, C-terminal 8-His-tagged TMEM175 was cloned into the pEZT-BM vector, C-terminal FLAG-tagged LAMP-1 was cloned into pCMV6-Entry vector (Origene, RC219208), and N-terminal FLAG-tagged LAMP-2 was cloned into pCMV3-SP vector (SinoBiological Inc, HG13555-NF). For protein overexpression and structural studies, TMEM175 with C-terminal 8-His tag, the C-terminal FLAG-tagged human LAMP-1, and the N-terminal FLAG-tagged human LAMP-2 were all individually cloned into the pEZT-BM vector. The LAMP-1 truncation constructs used in all studies were cloned into the pEZT-BM vector. Point mutations and truncation mutations were performed with PfuUltra II Fusion High-fidelity DNA Polymerase (Agilent, 600385) using the manufacturer-recommended protocol.
Summary of proteomic screening of TMEM175-interacting proteins
FLAG-tagged TMEM175 was transiently expressed in HEK293 or SH-SY5Y cells. To ensure proper maturation and lysosomal localization of the expressed TMEM175, only a minimal amount of plasmid (about 0.3 μg per 100 mm dish) was used in cell transfection to minimize the protein expression level. The membrane fraction was isolated from the total cell lysate and the membrane proteins were extracted and solubilized in detergent (see Membrane preparation section). FLAG-tagged TMEM175 was pulled down by an anti-FLAG antibody and its putative interacting proteins were identified by high-resolution nano-LC-MS/MS (nano liquid chromatography-tandem mass spectrometry) (see Tryptic digestion of proteins and mass spectrometry section). Two control experiments were performed to differentiate TMEM175-interacting proteins from the background proteins that interact non-specifically with resins or IgGs: the use of cells transfected with the empty vector in the pull-down by anti-FLAG antibody and the use of immobilized rabbit IgGs from an un-immunized animal in the pull-down assay for cells transfected with FLAG-tagged TMEM175. The candidates of TMEM175-interacting proteins were identified by applying three criteria, namely abundance, specificity, and consistency, to the MS spectra data of all pull-down samples 60. Proteins identified from the acquired MS data were quantified using peak-volume (PV) based label-free quantitative proteomics method as previously described 60. The abundance of each putative TMEM175-interacting protein identified in our proteomic analysis was estimated by the total PVs of the protein divided by the number of MS-accessible peptides 61. See Proteome identification and quantification section for details.
Membrane preparation, immunoprecipitation, and immunoblotting
The general procedures for cell membrane preparation, immunoprecipitation, and immunoblotting were similar to what was previously described 62. After cell homogenization, the cell membranes were prepared by differential centrifugation first at 2,000 g for 10 min to collect the supernatant followed by 120,000 g for 30 min to collect the membrane pellet. Membrane proteins in the pellet were re-suspended and solubilized using the lysis buffer containing 2% dodecyl maltoside (DDM), 150 mM NaCl and 20 mM Tris-Cl (pH 7.4). The solubilized proteins in the supernatant were collected after 17,000 g centrifugation for 10 min, and then incubated with an immobilized antibody covalently linked to protein-A agarose beads. After a brief wash with the lysis buffer, the bound proteins were eluted by the lysis buffer containing either 4% SDS or 100 μg/ml FLAG peptide (for anti-FLAG pulldown only). For immunoblotting, proteins were electrically transferred to PVDF membranes after SDS-PAGE and then probed with antibodies against the specific proteins. Anti-FLAG (Sigma, F7425), anti-LAMP-1 (Abcam, 24170), and anti-LAMP-2 (Abcam, ab199946) antibodies were used for immunoprecipitation. Anti-FLAG (Sigma, F3165), Anti-TMEM175 (Proteintech, 19925-1-AP), anti-LAMP-1 (Abcam, 24170), anti-LAMP-2 (Abcam, ab199946), anti-Cathepsin B (Abcam, ab125067), and anti-β actin (Sigma, A2066) antibodies were used for immunoblotting.
Tryptic digestion of proteins and mass spectrometry
A similar protocol as previously described was used for protein digestion and mass spectrometry 63. Proteins from the pull-down experiment were briefly separated by a short run on SDS-PAGE, and the part of the protein-containing gel for each sample was excised and divided into top and bottom halves for further proteomic analysis. The gel fragments were sequentially washed with water and acetonitrile, followed by in-gel reduction with dithiothreitol (DTT) and alkylation with iodoacetamide (IAM). After in-gel tryptic digestion at 37 °C overnight, the digested peptides were extracted in 50% acetonitrile (ACN), dried in a speed vacuum concentrator, and redissolved in 2% acetonitrile and 0.1% formic acid before LC-MS/MS analysis with an Orbitrap Lumos Fusion mass spectrometer. Peptides were separated by ultra-performance liquid chromatography (UPLC) in solutions containing 0.1% formic acid and 5 to 50% linear gradient of ACN over 120 min at a flow rate of 300 nl/min and sprayed into the mass spectrometer with a nano-electrospray source. A full MS scan in the range of 300-1800 m/z was acquired. Up to 10 most intense peaks from each MS scan were selected for MS/MS fragmentation with the higher-collisional dissociation (HCD) mode.
Proteome identification and quantification
Proteome identification and quantification were performed using MaxQuant Software (v 2.1.4) 64. Raw MS spectra files for pull-down samples from either HEK193 or SH-SY5Y cells were loaded into MaxQuant. For proteome identification, a human protein database from Swissprot was used, and cysteine carbamidomethylation, Gln-to-pyroGlu cyclization, and methionine oxidation were considered as variable modifications. The False Discovery Rates (FDRs) in MaxQuant were set as follows: peptide-spectrum match (PSM) FDR as 0.01, Protein FDR as 0.05, and Site decoy fraction as 0.01. Peak-volume (PV, integral of MS signal intensity in the m/z-retention time plane) based label-free quantification (LFQ) was used for proteome quantification in MaxQuant with selected options of LFQ, match between runs, and separate LFQ in parameter groups 61. The amount of each protein was defined by the total peak volumes of multiple tryptic peptides divided by the number of identified peptides from this protein.
The quantitative protein amounts data from MS spectra were subjected to a three-staged filter to identify putative TMEM175-interacting proteins following a similar strategy as previously described 60. In the first abundance filter, proteins being identified by less than two different peptides were excluded. The remaining proteins were subject to the second target specificity filter which was defined by threshold values established in abundance ratio histograms (Figure S1B). The abundance ratio for each protein is defined by comparing its amount in the sample after anti-FLAG antibody affinity purification using TMEM175-FLAG transfected cells with that in the two negative controls: one is the sample after anti-FLAG antibody affinity purification using empty-vector transfected cells and the other is the sample after un-immunized rabbit IgG affinity purification using TMEM175-FLAG transfected cells. Based on the histogram, only proteins with abundance ratios over 16 were selected. In the third consistency filter, only proteins identified in at least 3 out of 5 independently prepared samples (3 out of 5 independent experiments) from HEK293 cells, or in 2 out of 4 samples from SH-SY5Y cells were selected.
Blue-native PAGE
A similar protocol as previously described was used for Blue-native PAGE 65. The membrane fractions were re-suspended with a solubilization buffer containing 2.5% DDM, 50 mM NaCl, 2 mM 6-aminohexanoic acid, 1 mM EDTA, and 50 mM imidazole/HCl, pH 7.0. Solubilized membrane proteins in the supernatant were collected after 170,000 g centrifugation for 30 min and supplemented with 5% glycerol and 0.25% Coomassie Blue G250 before loading onto 4-15% polyacrylamide gel. For the mobility-shift assay, 5 μg of mouse anti-LAMP-1 (Abcam, ab25630) or anti-LAMP-2 (Abcam, ab25631) antibodies were incubated with the solubilized proteins for 20 min before being supplemented with glycerol and Coomassie Blue G250. Rabbit anti-LAMP-1 (Abcam, ab24170) or anti-LAMP-2 (Abcam, ab199946) antibodies were used for the detection of LAMP-1 and LAMP-2, respectively, after mobility-shift.
Surface protein biotinylation assay
Surface protein pull-down assay was performed using Cell Surface Biotinylation and Isolation Kit (Pierce, A44390). Transfected cells were washed with PBS solution twice, and incubated with 0.5 mg/ml sulfo-NHS-SS-Biotin in darkness for 40 min. Then 100 mM Tris-Cl pH 7.4 was added to quench the unreacted biotinylation reagent. After washing with PBS, cells were scraped down and lysed with cell lysis buffer containing 2% dodecyl maltoside (DDM), 150 mM NaCl and 20 mM Tris-Cl (pH 7.4). Streptavidin beads were then incubated with the cell lysate at 4°C for 2 hrs. After washing the beads, the bound biotinylated surface proteins were eluted with cell lysis buffer supplemented with 500 mM DTT and subjected to Western blot.
Expression and purification of TMEM175/LAMP-1 and 2 complexes
The full-length human TMEM175 with C-terminal 8x His tag, the C-terminal FLAG-tagged human LAMP-1, and the N-terminal FLAG-tagged human LAMP-2 were all individually cloned into a pEZT-BM vector for heterologous expression of the TMEM175 alone and TMEM175 in complex with LAMP-1 or LAMP-2 66. E. coli DH10bac (Thermo Fisher Scientific) was used to synthesize bacmids, which were subsequently used for the production of baculovirus in Sf9 cells using Cellfectin II reagent (Thermo Fisher Scientific) following the manufacturer’s protocol.
HEK293F cells were grown to a density of 3 × 106 cells mL−1 in FreeStyle 293 Expression Media (Thermo Fisher Scientific) and then incubated with the baculoviruses for TMEM175 and LAMP-1 or LAMP-2 at a ratio of 1:40 (virus:cell) for each virus and supplemented with 10 mM sodium butyrate. The cells were grown in suspension at 37°C for 48 hours and then harvested by centrifugation at 4000 rpm for 15 minutes.
To purify TMEM175/LAMP-1 or 2 complexes, the cell pellet was re-suspended in Buffer A (50 mM Tris pH 8.0, 150 mM NaCl), supplemented with a complete protease inhibitor cocktail (Roche Inc. 11697498001), and then homogenized by sonication. The protein was extracted by adding 2% (w/v) n-dodecyl-b-D-maltopyranoside (DDM, Anatrace) supplemented with 0.2% (w/v) cholesteryl hemisuccinate (CHS, Sigma Aldrich) to the cell lysate followed by gentle agitation at 4 °C for 2 hours. Insoluble components were removed by centrifugation at 40,000 g, 4 °C for 30 minutes, and the supernatant was incubated with Ni-NTA agarose beads (ThermoFisher Scientific) pre-equilibrated with Buffer A (Buffer A + 0.1% DDM, 0.02% CHS) at 4 °C for 1 hr. After brief centrifugation at 1,000 g for 1 min, the supernatant was discarded and the beads were washed three times with 6 bead-volume of Buffer B (50 mM Tris-Cl pH 8.0, 150 mM KCl, 0.06% glyco-diosgenin (GDN), 25 mM imidazole) for 5 min each time. The proteins were eluted with 5 bead-volume of Buffer C (Buffer B with 300 mM imidazole). The protein eluate was concentrated in 100 kDa MWCO Amicon Ultra centrifugal filters (Amicon®, Millipore Inc) at 3,000 g for 30 min, and then incubated with anti-FLAG conjugated agarose beads at 4 °C for 2 hrs. The beads were washed three times with 6 bead-volume of Buffer C (buffer B without imidazole) for 5 min each time and the bound protein complex was eluted with 5 bead-volume of 100 μg/ml FLAG peptide in buffer C. For structural analysis of the TMEM175/LAMP-1 complex, a two-step de-glycosylation was performed to remove the glycan chains on LAMP-1. After the 1st affinity chromatography with Ni-NTA and concentration, Endo Hf (NEB Inc) was added and incubated with the protein sample at the ratio of 20,000 U/mg protein at 37 °C for 1 hr. After the 2nd affinity chromatography with anti-FLAG resin, the 2nd round of de-glycosylation was performed by adding Endo Hf to the protein sample at the ratio of 10,000 U/mg protein and incubated at 37 °C for 1 hr. The protein complex with or without de-glycosylation was further purified by size exclusion chromatography using a Superose 6 10/300 GL column (GE Healthcare) pre-equilibrated with Buffer C. Same protocol was used for the expression and purification of TMEM175/LAMP-1-TM complex. For cryo-EM structural determination, all protein samples were concentrated to about 1.5 mg/mL before grid preparation.
Electron microscopy data acquisition
The cryo-EM grids were prepared by applying 4 μl protein (~1.5 mg/mL) to a glow-discharged Quantifoil R1.2/1.3 200-mesh or 300-mesh gold holey carbon grid (Quantifoil, Micro Tools GmbH) and blotted for 3.0 s with blot force 5 under 100% humidity at 4 °C before being plunged into liquid ethane using a Mark IV Vitrobot (FEI). Micrographs were acquired on a Titan Krios microscope (FEI) operated at 300 kV with a K3 Summit direct electron detector (Gatan), using a slit width of 20 eV on a GIF-Quantum energy filter. Data were collected using the CDS (Correlated Double Sampling) mode of the K3 camera. The defocus range was set from −0.9 to −2.2 μm. Each movie was dose-fractionated to 60 frames with a dose rate of 1e−/Å2/frame for a total dose of 60e−/Å2. The total exposure time was between 5 to 6 s.
Image processing
Cryo-EM data were processed following the subsequent general scheme, with modifications to different datasets (see below). First, movie frames were motion corrected and binned two times and dose-weighted using MotionCor2 67. The CTF parameters of the micrographs were estimated using the GCTF program 68. The rest of the image processing steps were carried out using RELION 3.1 69–71. After CTF estimation, micrographs were manually inspected to remove images with bad defocus values, ice contamination, or carbon. Particles were selected using Gautomatch (Kai Zhang, https://www2.mrc-lmb.cam.ac.uk/download/gautomatch-056/) and extracted using a binning factor of 3. Next, particles were subjected to one round of 2D classification in Relion, followed by one round of 3D classification with alignment on selected particles from the 2D classification job. No symmetry was imposed at this step. The human TMEM175 map (EMD-21603) 24 low-pass filtered to 30 Å was used as the initial reference. Beam tilt, anisotropic magnification, and per-particle CTF estimations were performed in Relion 3.1 to improve the resolution of the final reconstruction. All resolution was reported according to the gold-standard Fourier shell correlation (FSC) using the 0.143 criterion 72. Local resolution was estimated using Relion.
For the dataset of TMEM175/LAMP-1, a total of 12,021 movies were collected and 11,556 were selected after motion correction and CTF estimation. A total number of 2,130,974 particles were extracted from the selected micrographs and were subjected to one round of 2D classification, from which 802,407 particles were selected. Next, these particles were classified into 5 classes with 50 iterations with T=4 (the default value) and an extra 50 iterations with T=8. After the initial 3D classification, 269,594 particles were selected and subjected to a 3D auto-refinement job with C2 symmetry imposed. Further CTF refinement yielded a map at 3.5Å overall resolution, with a calibrated pixel size of 0.83Åpixel (0.415Åpixel for the super-resolution mode of the K3 camera).
For the dataset of TMEM175/LAMP-1-TM complex, a total of 4,193 movies were collected and 4,109 were selected after motion correction and CTF estimation. A total number of 780,040 particles were extracted from the selected micrographs and were subjected to one round of 2D classification, from which 330,189 particles were selected. Next, these particles were classified into 8 classes with 50 iterations with T=4 (the default value) and an extra 100 iterations with T=8. After the initial 3D classification, 145,017 particles were selected and subjected to a 3D auto-refinement job. Next, a soft mask excluding the micelle density was applied and particles were sorted into 5 classes without performing the alignment. From this, 44,240 particles were selected for further refinement with C2 symmetry and CTF refinement in Relion, yielding a map at an overall resolution of 3.4Å, with a calibrated pixel size of 0.842Å/pixel (0.421/pixel for super-resolution mode of the K3 camera).
Model building, refinement, and validation
EM maps of the two complexes (TMEM175/LAMP-1 and TMEM175/LAMP-1-TM) show high-quality density for most parts of TMEM175 and the single TM of LAMP-1. The de novo model building was performed in Coot 73, facilitated by the previous cryo-EM structure of human TMEM175 (PDB code 6WC9). The model was manually adjusted in Coot and refined against the map by using the real space refinement module with secondary structure and non-crystallographic symmetry restraints in the Phenix package 74. The final structural models contain TMs 1-5 and 7-12 from TMEM175 and the single TM from LAMP-1. TM5 of TMEM175 was modeled as a poly-A helix due to poorly resolved side-chain density. The geometry statistics of the models were generated using MolProbity 75. All the structural figures were prepared in PyMol (Schrödinger, LLC.) and UCSF ChimeraX 76.
Electrophysiology
Expression vectors containing TMEM175 and LAMP-1 or 2 were co-transfected into HEK293 cells using Lipofectamine 2000 (Life Technology). To achieve comparable levels of TMEM175 expression for all recordings, 1 μg of plasmid per single well of cells (6-well plate) was used when transfecting TMEM175 alone, whereas 3 μg of TMEM175-containing plasmid and 1 μg of LAMP-1 or LAMP-2-containing plasmid were used in the co-transfection. 48 hours after transfection, cells were dissociated by trypsin treatment and kept in a complete serum-containing medium and re-plate on 35 mm tissue culture dishes in a tissue culture incubator until recording.
Patch clamps in the whole-cell configuration were used to measure TMEM175 currents in HEK293 cells. The standard extracellular solution in the bath contained (in mM): 145 sodium methanesulfonate (Na-MS), 5 NaCl, 1 MgCl2, 1 CaCl2, 10 HEPES buffered with Tris, pH 7.4. The intracellular solution in the pipette contained (in mM): 150 Cs-MS, 5 MgCl2, 10 EGTA, and 10 HEPES buffered with Tris, pH=7.4. To measure proton current, the extracellular (bath) solution contained (in mM): 150 NMDG-MS, 10 HEPES (pH=7.4, 6.5), or 10 MES (pH=5.5 and 4.5), or 10 acetic acid (pH=3.5), all buffered with Tris. The intracellular solution contained (in mM): 150 Cs-MS (for data shown in Figure 2C–2E) or 150 NMDG-MS (for data shown in Figure 3H, 4C, 5D & 5F), 5 MgCl2, 10 EGTA, 10 HEPES buffered with Tris, pH=7.4. The patch pipettes were pulled from Borosilicate glass (Harvard Apparatus) and heat polished to a resistance of 4-6 MΩ. After the patch pipette was attached to the cell membrane, the giga seal (5-10 GΩ) was formed by gentle suction. The whole cell configuration was formed by a short zap or suction to rupture the patch. Data were acquired using an AxoPatch 200B amplifier (Molecular Devices) and a low-pass analog filter set to 1 kHz. The current signal was sampled at a rate of 10 kHz using a Digidata 1550B digitizer (Molecular Devices) and further analyzed with pClamp 11 software (Molecular Devices). For continuous current recording, the membrane potential was held at −100 mV. To generate the I-V curve, the holding potential was set to 0 mV, and voltage pulses ramp from −100 to +100 mV over 800 ms duration. All data points are mean ± SEM (n ≥10).
Liposome flux assay
The flux assay was performed following the same protocol as previously described 50. In brief, the channel-containing proteoliposomes with high K+ inside (300 mM) were exposed to a low K+ solution (~6 mM). The K+ efflux was initiated by adding an H+ ionophore carbonyl cyanide m-chlorophenylhydrazone (CCCP) that allowed the influx of H+ to balance the charges caused by K+ efflux. Thus K+ efflux provides the driving force for H+ influx, resulting in the acidification of the proteoliposomes. This K+ efflux-driven H+ influx was monitored by pH-dependent quenching of 9-amino-6-chloro-2-methoxyacridine (ACMA), a fluorescent dye that is membrane-permeable at neutral pH but becomes quenched and membrane-impermeable when protonated inside the liposomes. Thus, the addition of CCCP to the proteoliposomes containing K+-conducting channels would introduce a time-dependent reduction of ACMA fluorescence in the solution.
For reconstitution, the lipids 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (POPG) in chloroform (Avanti) were mixed at a ratio of 3:1 (v:v) and dried with argon gas. Residue chloroform was vacuum-dried overnight. The dried lipids mixture was then re-suspended in a reconstitution buffer (20 mM HEPES-KOH, pH 7.4, 1 mM EDTA, and 300 mM KCl) to a concentration of 10 mg/ml. Unilamellar lipid vesicles were made by sonicating lipid resuspension until translucent and then incubated with an equal volume of solubilization buffer (reconstitution buffer with 20 mM Decylmaltoside (DM) and 20 mM DTT) at room temperature for 30 min. The purified TMEM175 alone or TMEM175 in complex with LAMP-1 or 2 was mixed with the solubilized lipid at the ratio of 1:500-1000 (w:w) and incubated at room temperature for 1 hr. The amounts of TMEM175 used in reconstitutions for TMEM175 alone and the complexes were comparable. The protein-detergent-lipid mixture was then transferred to a 20 kD MWCO dialysis bag and dialyzed against the reconstitution buffer supplemented with 3 mM DTT at 4 °C three times with buffer change every 24 hrs. During the last buffer change, 1 g of Bio-beads SM-2 resin (Biorad Inc) was added to the dialysis solution to remove residual detergent.
After dialysis, the proteoliposomes were incubated at 37 °C for 5 minutes and diluted 10 times with the vesicle buffer (20 mM HEPES-NaOH, pH 7.4, and 150 mM NaCl). The fluorescence reading was performed on a 96-well plate. Each well contains a mixture of 18 μl diluted proteoliposomes, 18 μl 6.5 μM 9-amino-6-chloro-2-methoxyacridine (ACMA) in 20 mM HEPES-NaOH, pH7.4, and 36 μl drug buffer (20 mM HEPES-NaOH, pH 7.4 and 675 mM NaCl). The baseline was recorded for 2 min using excitation/emission wavelengths of 410/480 nm with 5-second intervals, and then 18 μl H+ ionophore carbonyl cyanide m-chlorophenylhydrazone (CCCP) at 1 μM in 20 mM HEPES-NaOH, pH 7.4 and 5 mM EDTA was added to initiate the flux reaction. The reaction was monitored for 10-15 min with 5-second intervals and then terminated by adding K+ ionophore valinomycin to 50 nM.
Crosslinking between TMEM175 and LAMP-1
The purification procedure of TMEM175/LAMP-1 complex for crosslinking was the same as the sample preparation for cryo-EM, except that Tris-Cl, pH8.0 in all solutions was replaced by 20 mM HEPES-KOH, pH 7.4. The non-cleavable, bis(sulfosuccinimidyl)suberate (BS3, Thermo Scientific Pierce) was used in the crosslinking reaction. It is an amine-to-amine crosslinker with a spacer arm length of 11.4 Å. The purified protein complex at the concentration of 1.5 mg/ml was mixed with an equal volume of BS3 stock solution (100 mM in 20 mM HEPES-KOH, pH 7.4) and incubated in dark for 2 hrs. The protein sample after crosslinking was re-purified by size exclusion chromatography using a Superose 6 10/300 GL column, followed by in-gel digestion for MS spectrometry analysis as previously described. The crosslinking site identification was performed with Mascot (v 2.7.0) 77. Briefly, the MS/MS spectra were searched against the SwissProt protein database with taxonomy being specified as Human. The peptide and MS/MS tolerance were set as 20 ppm and 0.5 Da, respectively. Carbamidomethyl (C) was set as fixed modification and Oxidation (M) was set as variable modification. DSS was selected as the crosslinking method, in which crosslinking between TMEM175 and LAMP-1 was specified.
LysoTracker staining
Wild-type or TMEM175-knockout HAP1 cells were cultured on 35 mm glass bottom tissue culture dishes (Thermo Scientific) and transfected with vectors containing TMEM175, its mutant, or LAMP-1-TM using Lipofectamine 2000. 48 hours after transfection LysoTracker Red DND-99 (Invitrogen, L7528) was added into the culture medium to a final concentration of 50 nM and incubated at 37 °C for 30 min. Cells were washed three times with Tyrode’s solution (145 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES-NaOH, pH 7.4 and 10 mM glucose), and images were acquired using a fluorescence microscope (Zeiss LSM 880) at 200× magnitude with a pre-set LysoTracker Red setting in ZenPro program (Excitation/Emission wavelength of 578/590 nm). The fluorescence intensities were quantified using the Fiji image processing package 78.
Lysosomal pH measurement with Oregon Green Dextran 488
The lysosomal pH was measured by ratiometric fluorescence imaging of pH-sensitive dye Oregon Green™ 488 conjugated to dextran with 10-kDa MW (Invitrogen, D7170) as previously described18. 48 hours after transfection, cells were loaded with Oregon Green™ 488 dextran at 150μg/mL overnight. Before imaging, cells were chased without the dye in the medium for 3 hrs. Cells were washed with Ringer’s solution (155 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 2 mM NaH2PO4, 10 mM HEPES-NaOH, pH 7.4, and 10 mM glucose) three times, and then imaged using the same fluorescence microscope for the LysoTracker Red imaging at 200× magnitudes under live cell imaging conditions (37 °C and 5% CO2). The fluorescence emissions at 530 nm were acquired at the excitation wavelengths of 440 nm and 488 nm, respectively. After each imaging measurement, the in-situ pH calibration was performed by sequentially equilibrating the cells with various pH standard solutions for 5 min followed by imaging after each equilibration. The prepared pH standard solutions contain 130 mM KCl, 1 mM MgCl2, 15 mM HEPES, 15 mM MES, 10 μM nigericin, and 10 μM valinomycin with pH 3.5, 4.5, 5.5, and 6.5. The individual cell used for measurement was outlined in Fiji/ImageJ. The ratio of fluorescence emission intensity of individual cells acquired at excitation wavelengths of 488/440 nm after each pH treatment as a function of pH was fitted to a Boltzmann sigmoid equation which was then used as a standard curve to obtain the experimental measurement of lysosomal pH for each cell. Student’s t-test was used to calculate the P-values for comparison between any two given groups.
Lysosomal hydrolytic activity assay
The overall lysosomal hydrolytic activity was measured using DQ™ Red BSA (Invitrogen, D12051). Two days after cell transfection, DQ™ Red BSA was added into the culture medium to a concentration of 10 μg/ml. After overnight incubation, cells were chased in the medium without DQ-red BSA for 3 hours and washed three times with Tyrode’s solution before fluorescence imaging. Cathepsin B activities were measured using the Magic Red Cathepsin B assay kit (ImmunoChemistry Technologies, 937). Cells were incubated with Magic Red at 1:1000 dilution at 37 °C for 1 hour and washed three times with Tyrode’s solution before fluorescence imaging. The fluorescence intensities were quantified using the Fiji image processing package.
Quantification and statistical analysis
Statistical details are described in the Results and Figure Legends. All data are mean ± SEM. Exact values of n are reported where appropriate. Depending on the experiment, n represents number of independent measurements.
Supplementary Material
Highlights:
Lysosomal LAMP-1 and LAMP-2 proteins directly interact with TMEM175 channel.
Complex formation between TMEM175 and LAMPs is mediated by their TM domains.
LAMP binding inhibits the channel activity of TMEM175.
TMEM175 inhibition facilitates lysosomal acidification for optimal hydrolase activity.
Acknowledgments
Single particle cryo-EM data were collected at the University of Texas Southwestern Medical Center Cryo-EM Facility which is funded by the CPRIT Core Facility Support Award RP170644 and the Howard Hughes Medical Institute Janelia Cryo-EM Facility. We thank Rui Yan at the Janelia Cryo-EM Facility for her help in microscope operation and data collection. Cryo-EM sample grids were prepared at the Structural Biology Laboratory at UT Southwestern Medical Center which is partially supported by grant RP170644 from CPRIT. This work was supported in part by the Howard Hughes Medical Institute (to Y.J.) and by grants from the National Institute of Health (R35GM140892 to Y.J. and R01GM144479 to J.L.), and the Welch Foundation (Grant I-1578 to Y.J.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- 1.Luzio JP, Pryor PR, and Bright NA (2007). Lysosomes: fusion and function. Nat Rev Mol Cell Biol 8, 622–632. 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- 2.De Duve C, and Wattiaux R (1966). Functions of lysosomes. Annu Rev Physiol 28, 435–492. 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 3.Venkatachalam K, Wong CO, and Zhu MX (2015). The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium 58, 48–56. 10.1016/j.ceca.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong XP, Yu T, Lieberman AP, Showalter HD, and Xu H (2012). Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun 3, 731. 10.1038/ncommsl735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kilpatrick BS, Yates E, Grimm C, Schapira AH, and Patel S (2016). Endo-lysosomal TRP mucolipin-1 channels trigger global ER Ca2+ release and Ca2+ influx. J Cell Sci 129, 3859–3867. 10.1242/jcs.190322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scotto Rosato A, Montefusco S, Soldati C, Di Paola S, Capuozzo A, Monfregola J, Polishchuk E, Amabile A, Grimm C, Lombardo A, et al. (2019). TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKβ/VPS34 pathway. Nature Communications 10, 5630. 10.1038/s41467-019-13572-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu H, and Ren D (2015). Lysosomal physiology. Annu Rev Physiol 77, 57–80. 10.1146/annurev-physiol-021014-071649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ballabio A, and Bonifacino JS (2020). Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol 21, 101–118. 10.1038/s41580-019-0185-4. [DOI] [PubMed] [Google Scholar]
- 9.Butor C, Griffiths G, Aronson NN Jr., and Varki A (1995). Co-localization of hydrolytic enzymes with widely disparate pH optima: implications for the regulation of lysosomal pH. J Cell Sci 108 (Pt 6), 2213–2219. 10.1242/jcs.l08.6.2213. [DOI] [PubMed] [Google Scholar]
- 10.Ishida Y, Nayak S, Mindell JA, and Grabe M (2013). A model of lysosomal pH regulation. J Gen Physiol 141, 705–720. 10.1085/jgp.201210930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mindell JA (2012). Lysosomal acidification mechanisms. Annu Rev Physiol 74, 69–86. 10.1146/annurev-physiol-012110-142317. [DOI] [PubMed] [Google Scholar]
- 12.Johnson DE, Ostrowski P, Jaumouille V, and Grinstein S (2016). The position of lysosomes within the cell determines their luminal pH. J Cell Biol 212, 677–692. 10.1083/jcb.201507112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forgac M (2007). Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 8, 917–929. 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- 14.Graves AR, Curran PK, Smith CL, and Mindell JA (2008). The Cl−/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature 453, 788–792. 10.1038/nature06907. [DOI] [PubMed] [Google Scholar]
- 15.Steinberg BE, Huynh KK, Brodovitch A, Jabs S, Stauber T, Jentsch TJ, and Grinstein S (2010). A cation counterflux supports lysosomal acidification. J Cell Biol 189, 1171–1186. 10.1083/jcb.200911083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christensen KA, Myers JT, and Swanson JA (2002). pH-dependent regulation of lysosomal calcium in macrophages. J Cell Sci 115, 599–607. 10.1242/jcs.115.3.599. [DOI] [PubMed] [Google Scholar]
- 17.Van Dyke RW (1993). Acidification of rat liver lysosomes: quantitation and comparison with endosomes. Am J Physiol 265, C901–917. 10.1152/ajpcell.l993.265.4.C901. [DOI] [PubMed] [Google Scholar]
- 18.Hu M, Li P, Wang C, Feng X, Geng Q, Chen W, Marthi M, Zhang W, Gao C, Reid W, et al. (2022). Parkinson’s disease-risk protein TMEM175 is a proton-activated proton channel in lysosomes. Cell 185, 2292–2308 e2220. 10.1016/j.cell.2022.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jinn S, Blauwendraat C, Toolan D, Gretzula CA, Drolet RE, Smith S, Nalls MA, Marcus J, Singleton AB, and Stone DJ (2019). Functionalization of the TMEM175 p.M393T variant as a risk factor for Parkinson disease. Hum Mol Genet 28, 3244–3254. 10.1093/hmg/ddzl36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hopfner F, Mueller SH, Szymczak S, Junge O, Tittmann L, May S, Lohmann K, Grallert H, Lieb W, Strauch K, et al. (2020). Rare Variants in Specific Lysosomal Genes Are Associated With Parkinson’s Disease. Mov Disord 35, 1245–1248. 10.1002/mds.28037. [DOI] [PubMed] [Google Scholar]
- 21.Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, et al. (2014). Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46, 989–993. 10.1038/ng.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cang C, Aranda K, Seo YJ, Gasnier B, and Ren D (2015). TMEM175 Is an Organelle K(+) Channel Regulating Lysosomal Function. Cell 162, 1101–1112. 10.1016/j.cell.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 23.Lee C, Guo J, Zeng W, Kim S, She J, Cang C, Ren D, and Jiang Y (2017). The lysosomal potassium channel TMEM175 adopts a novel tetrameric architecture. Nature 547, 472–475. 10.1038/nature23269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oh S, Paknejad N, and Hite RK (2020). Gating and selectivity mechanisms for the lysosomal K(+) channel TMEM175. Elife 9. 10.7554/eLife.53430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng W, Shen C, Wang L, Rawson S, Xie WJ, Nist-Lund C, Wu J, Shen Z, Xia S, Holt JR, et al. (2022). pH regulates potassium conductance and drives a constitutive proton current in human TMEM175. Sci Adv 8, eabm1568. 10.1126/sciadv.abm1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oh S, Marinelli F, Zhou W, Lee J, Choi HJ, Kim M, Faraldo-Gomez JD, and Hite RK (2022). Differential ion dehydration energetics explains selectivity in the non-canonical lysosomal K(+) channel TMEM175. Elife 11. 10.7554/eLife.75122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jinn S, Drolet RE, Cramer PE, Wong AH, Toolan DM, Gretzula CA, Voleti B, Vassileva G, Disa J, Tadin-Strapps M, and Stone DJ (2017). TMEM175 deficiency impairs lysosomal and mitochondrial function and increases alpha-synuclein aggregation. Proc Natl Acad Sci U S A 114, 2389–2394. 10.1073/pnas.1616332114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kornfeld S, and Mellman I (1989). The biogenesis of lysosomes. Annu Rev Cell Biol 5, 483–525. 10.1146/annurev.cb.05.110189.002411. [DOI] [PubMed] [Google Scholar]
- 29.Schwake M, Schroder B, and Saftig P (2013). Lysosomal membrane proteins and their central role in physiology. Traffic 14, 739–748. 10.1111/tra.12056. [DOI] [PubMed] [Google Scholar]
- 30.Lewis V, Green SA, Marsh M, Vihko P, Helenius A, and Mellman I (1985). Glycoproteins of the lysosomal membrane. J Cell Biol 100, 1839–1847. 10.1083/jcb.100.6.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen JW, Murphy TL, Willingham MC, Pastan I, and August JT (1985). Identification of two lysosomal membrane glycoproteins. J Cell Biol 101, 85–95. 10.1083/jcb.101.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lippincott-Schwartz J, and Fambrough DM (1986). Lysosomal membrane dynamics: structure and interorganellar movement of a major lysosomal membrane glycoprotein. J Cell Biol 102, 1593–1605. 10.1083/jcb.102.5.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barriocanal JG, Bonifacino JS, Yuan L, and Sandoval IV (1986). Biosynthesis, glycosylation, movement through the Golgi system, and transport to lysosomes by an N-linked carbohydrate-independent mechanism of three lysosomal integral membrane proteins. J Biol Chem 261, 16755–16763. [PubMed] [Google Scholar]
- 34.Eskelinen EL (2006). Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med 27, 495–502. 10.1016/j.mam.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 35.Hunziker W, Simmen T, and Honing S (1996). Trafficking of lysosomal membrane proteins in polarized kidney cells. Nephrologie 17, 347–350. [PubMed] [Google Scholar]
- 36.Braulke T, and Bonifacino JS (2009). Sorting of lysosomal proteins. Biochim Biophys Acta 1793, 605–614. 10.1016/j.bbamcr.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 37.Honing S, and Hunziker W (1995). Cytoplasmic determinants involved in direct lysosomal sorting, endocytosis, and basolateral targeting of rat lgp120 (lamp-I) in MDCK cells. J Cell Biol 128, 321–332. 10.1083/jcb.128.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henell F, Ericsson JL, and Glaumann H (1983). Degradation of phagocytosed lysosomes by Kupffer cell lysosomes. Lab Invest 48, 556–564. [PubMed] [Google Scholar]
- 39.Wilke S, Krausze J, and Bussow K (2012). Crystal structure of the conserved domain of the DC lysosomal associated membrane protein: implications for the lysosomal glycocalyx. BMC Biol 10, 62. 10.1186/1741-7007-10-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eskelinen EL, Schmidt CK, Neu S, Willenborg M, Fuertes G, Salvador N, Tanaka Y, Lullmann-Rauch R, Hartmann D, Heeren J, et al. (2004). Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol Biol Cell 15, 3132–3145. 10.1091/mbc.e04-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, and Grinstein S (2007). LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J 26, 313–324. 10.1038/sj.emboj.7601511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saftig P, Beertsen W, and Eskelinen EL (2008). LAMP-2: a control step for phagosome and autophagosome maturation. Autophagy 4, 510–512. 10.4161/auto.5724. [DOI] [PubMed] [Google Scholar]
- 43.Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, et al. (2000). Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406, 906–910. 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 44.Schneede A, Schmidt CK, Holtta-Vuori M, Heeren J, Willenborg M, Blanz J, Domanskyy M, Breiden B, Brodesser S, Landgrebe J, et al. (2011). Role for LAMP-2 in endosomal cholesterol transport. J Cell Mol Med 15, 280–295. 10.1111/j.1582-4934.2009.00973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bandyopadhyay U, Kaushik S, Varticovski L, and Cuervo AM (2008). The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 28, 5747–5763. 10.1128/MCB.02070-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prentzell MT, Rehbein U, Cadena Sandoval M, De Meulemeester AS, Baumeister R, Brohee L, Berdel B, Bockwoldt M, Carroll B, Chowdhury SR, et al. (2021). G3BPs tether the TSC complex to lysosomes and suppress mTORC1 signaling. Cell 184, 655–674 e627. 10.1016/j.cell.2020.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harter C, and Mellman I (1992). Transport of the lysosomal membrane glycoprotein lgp120 (lgp-A) to lysosomes does not require appearance on the plasma membrane. J Cell Biol 117, 311–325. 10.1083/jcb.117.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kima PE, Burleigh B, and Andrews NW (2000). Surface-targeted lysosomal membrane glycoprotein-1 (Lamp-1) enhances lysosome exocytosis and cell invasion by Trypanosoma cruzi. Cell Microbiol 2, 477–486. 10.1046/j.1462-5822.2000.00071.x. [DOI] [PubMed] [Google Scholar]
- 49.Rodriguez A, Webster P, Ortego J, and Andrews NW (1997). Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol 137, 93–104. 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Su Z, Brown EC, Wang W, and MacKinnon R (2016). Novel cell-free high-throughput screening method for pharmacological tools targeting K+ channels. Proc Natl Acad Sci U S A 113, 5748–5753. 10.1073/pnas.1602815113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Reilly FJ, and Rappsilber J (2018). Cross-linking mass spectrometry: methods and applications in structural, molecular and systems biology. Nat Struct Mol Biol 25, 1000–1008. 10.1038/s41594-018-0147-0. [DOI] [PubMed] [Google Scholar]
- 52.Decher N, Lang HJ, Nilius B, Bruggemann A, Busch AE, and Steinmeyer K (2001). DCPIB is a novel selective blocker of I(Cl,swell) and prevents swelling-induced shortening of guinea-pig atrial action potential duration. Br J Pharmacol 134, 1467–1479. 10.1038/sjbjp0704413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ashcom JD, and Jacobson LA (1989). Self-quenched fluorogenic protein substrates for the detection of cathepsin D and other protease activities. Anal Biochem 176, 261–264. 10.1016/0003-2697(89)90306-0. [DOI] [PubMed] [Google Scholar]
- 54.Creasy BM, Hartmann CB, White FK, and McCoy KL (2007). New assay using fluorogenic substrates and immunofluorescence staining to measure cysteine cathepsin activity in live cell subpopulations. Cytometry A 71, 114–123. 10.1002/cyto.a.20365. [DOI] [PubMed] [Google Scholar]
- 55.Winchester BG (2001). Lysosomal membrane proteins. Eur J Paediatr Neurol 5 Suppl A, 11–19. 10.1053/ejpn.2000.0428. [DOI] [PubMed] [Google Scholar]
- 56.Karlsson K, and Carlsson SR (1998). Sorting of lysosomal membrane glycoproteins lamp-1 and lamp-2 into vesicles distinct from mannose 6-phosphate receptor/gamma-adaptin vesicles at the trans-Golgi network. J Biol Chem 273, 18966–18973. 10.1074/jbc.273.30.18966. [DOI] [PubMed] [Google Scholar]
- 57.Cook NR, Row PE, and Davidson HW (2004). Lysosome associated membrane protein 1 (Lamp1) traffics directly from the TGN to early endosomes. Traffic 5, 685–699. 10.1111/j.1600-0854.2004.00212.x. [DOI] [PubMed] [Google Scholar]
- 58.Binker MG, Cosen-Binker LI, Terebiznik MR, Mallo GV, McCaw SE, Eskelinen EL, Willenborg M, Brumell JH, Saftig P, Grinstein S, and Gray-Owen SD (2007). Arrested maturation of Neisseria-containing phagosomes in the absence of the lysosome-associated membrane proteins, LAMP-1 and LAMP-2. Cell Microbiol 9, 2153–2166. 10.1111/j.1462-5822.2007.00946.x. [DOI] [PubMed] [Google Scholar]
- 59.Mauvezin C, Nagy P, Juhasz G, and Neufeld TP (2015). Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun 6, 7007. 10.1038/ncomms8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muller CS, Haupt A, Bildl W, Schindler J, Knaus HG, Meissner M, Rammner B, Striessnig J, Flockerzi V, Fakler B, and Schulte U (2010). Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc Natl Acad Sci U S A 107, 14950–14957. 10.1073/pnas.1005940107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, and Mann M (2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13, 2513–2526. 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang J, Guan X, Li Q, Meredith AL, Pan HL, and Yan J (2018). Glutamate-activated BK channel complexes formed with NMDA receptors. Proc Natl Acad Sci U S A 115, E9006–E9014. 10.1073/pnas.1802567115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang J, Guan X, Shah K, and Yan J (2021). Lsm12 is an NAADP receptor and a two-pore channel regulatory protein required for calcium mobilization from acidic organelles. Nat Commun 12, 4739. 10.1038/s41467-021-24735-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 65.Wittig I, Braun HP, and Schagger H (2006). Blue native PAGE. Nat Protoc 1, 418–428. 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 66.Morales-Perez CL, Noviello CM, and Hibbs RE (2016). Manipulation of Subunit Stoichiometry in Heteromeric Membrane Proteins. Structure 24, 797–805. 10.1016/j.str.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang K (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12. 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nakane T, Kotecha A, Sente A, McMullan G, Masiulis S, Brown P, Grigoras IT, Malinauskaite L, Malinauskas T, Miehling J, et al. (2020). Single-particle cryo-EM at atomic resolution. Nature 587, 152–156. 10.1038/s41586-020-2829-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Henderson R, Sali A, Baker ML, Carragher B, Devkota B, Downing KH, Egelman EH, Feng Z, Frank J, Grigorieff N, et al. (2012). Outcome of the first electron microscopy validation task force meeting. Structure 20, 205–214. 10.1016/j.str.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. S0907444909052925 [pii] 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, and Ferrin TE (2021). UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci 30, 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perkins DN, Pappin DJ, Creasy DM, and Cottrell JS (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567. . [DOI] [PubMed] [Google Scholar]
- 78.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM density maps of the human TMEM175/LAMP-1 and TMEM175/LAMP-1-TM complexes have been deposited at the Electron Microscopy Data Bank. Atomic coordinates of TMEM175/LAMP-1 and TMEM175/LAMP-1-TM complexes have been deposited at the Protein Data Bank and are publicly available as of the date of publication. Accession numbers are listed in the key resource table. All original imaging data have been deposited at Mendeley Data and are publicly available as of the date of publication. DOIs are listed in the key resource table. The quantitative proteomics and cross linking proteomics datasets have been deposited at MassIVE database and are publicly available as of the date of publication. DOIs are listed in the key resource table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FLAG, rabbit sourced | Sigma | Cat#F7425 |
| anti-LAMP-1, rabbit sourced | Abcam | Cat#ab24170 |
| anti-LAMP-2, rabbit sourced | Abcam | Cat#ab199946 |
| Anti-FLAG, mouse sourced | Sigma | Cat#F3165 |
| Anti-TMEM175, rabbit sourced | Proteintech | Cat#19925-1-AP |
| anti-Cathepsin B, rabbit sourced | Abcam | Cat#ab125067 |
| anti-β actin, rabbit sourced | Sigma | Cat#A2066 |
| anti-LAMP1, mouse sourced | Abcam | Cat#ab25630 |
| anti-LAMP2, mouse sourced | Abcam | Cat#ab25631 |
| Bacterial and Virus Strains | ||
| DH5a competent E.coli | Thermo Fisher | Cat# 18265017 |
| E. coli DH10bac | Thermo Fisher | Cat# 10361012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| n-Dodecyl-β-D-Maltopyranoside | Anatrace | Cat#D310 |
| Dithiothreitol | Thermo | Cat#R0861 |
| Iodoacetamide | Sigma | Cat#I6125 |
| Acetonitrile | Fisher | Cat#75-05-8 |
| Trypsin Gold | Promega | Cat#V5280 |
| 6-Aminohexanoic acid | Sigma | Cat#07260 |
| Sulfo-NHS-SS-Biotin | Thermo | Cat#21331 |
| GDN | Anatrace | Cat#GDN101 |
| Endo Hf | NEB | Cat#P0703S |
| Carbonyl cyanide 3-chlorophenylhydrazone | Sigma | Cat#C2759 |
| ACMA (9-Amino-6-Chloro-2-Methoxyacridine) | Thermo | Cat#A1324 |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (POPG) | Avanti | Cat#840457C |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) | Avanti | Cat#850757P |
| n-Decyl-β-D-Maltoside | Anatrace | Cat#D322LA |
| Bio-Beads SM-2 Resin | Bio-rad | Cat#1523920 |
| Valinomycin | Invitrogen | Cat#V1644 |
| BS3 (bis(sulfosuccinimidyl)suberate) | Thermo | Cat#21580 |
| LysoTracker™ Red DND-99 | Invitrogen | Cat#L7528 |
| Oregon Green™ 488 dextran; 10,000 MW, Anionic | Invitrogen | Cat#D7170 |
| Nigericin | Invitrogen | Cat#N1495 |
| DQ™ Red BSA | Invitrogen | Cat#D12051 |
| Magic Red® Cathepsin-B Assay Kit | Immunochemistry | Cat#937 |
| FLAG peptide | Sigma | Cat#F3290 |
| Deposited Data | ||
| TMEM175-full length LAMP-1 density map | This study | EMD: EMD-29553 |
| TMEM175-full length LAMP-1 atomic model | This study | PDB: 8FY5 |
| TMEM175-TM LAMP-1 density map | This study | EMD: EMD-29572 |
| TMEM175-TM LAMP-1 atomic model | This study | PDB: 8FYF |
| The raw data of quantitative proteomics of TMEM175 affinity enrichment | This study | http://dx.doi.org/doi:10.25345/C5RN30J1P |
| The raw data of crosslinking proteomics of TMEM175-LAMP1 complex | This study | http://dx.doi.org/doi:10.25345/C5CC0V39R |
| The Mendeley dataset of raw data of gels, blots and imagings | This study | http://dx.doi.org/doi:10.17632/6d8w9vv5bv.3 |
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | ATCC#CRL-1573 |
| SH-SY5Y | ATCC | ATCC#CRL-2266 |
| Sf9 | ATCC | ATCC#CRL-1711 |
| FreeStyle™ 293-F | ThermoFisher | Cat#R79007 |
| HAP1 | HorizonDiscovery | Cat#HorizonDiscovery |
| HAP1 TMEM175 knockout | HorizonDiscovery | Cat#HZGHC005593c004 |
| Recombinant DNA | ||
| pCMV6-TMEM175-Myc-FLAG | Origene | Cat#RC201422 |
| pCMV6-LAMP1-Myc-FLAG | Origene | Cat#RC219208 |
| pCMV3-FLAG-LAMP1 | SinoBiological | Cat#HG11215-NF |
| pCMV3-FLAG-LAMP2 | SinoBiological | Cat#HG13555-NF |
| pEZT-BM | Addgene | Plasmid#74099 |
| pCMV6-LAMP1-GFP | Origene | Cat#RC100016 |
| pCMV6-LAMP2-GFP | Origene | Cat#RG200456 |
| Software and Algorithms | ||
| Maxquant | Maxquant 2.1.4 | https://www.maxquant.org |
| Mascot | Mascot 2.7.0 | http://www.matrixscience.com/ |
| RELION | Scheres, 2012 | https://www3.mrc-lmb.cam.ac.uk/relion |
| UCSF ChimeraX | Pettersen et al., 2021 | https://www.cgl.ucsf.edu/chimerax/ |
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org |
| COOT | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PyMOL | Molecular Graphics System, Version 2.5.2, Schrödinger | https://pymol.org/2/ |
| Fiji | Schindelin et al., 2012 | https://imagej.net/software/fiji/ |
| Other | ||
| R1.2/1.3 200-mesh or 300-mesh gold holey carbon grid | Quantifoil | Cat#4220G-CF/4230G-CF |