

Abstract
We combined efficient sample preparation and ultra-low-flow liquid chromatography with a newly developed data acquisition and analysis scheme termed wide window acquisition (WWA) to quantify >3,000 proteins from single cells in rapid label-free analyses. WWA employs large isolation windows to intentionally co-isolate and co-fragment adjacent precursors along with the selected precursor. Optimized WWA increased the number of MS2-identified proteins by ~40% relative to standard data-dependent acquisition. For a 40-min LC gradient operated at ~15 nL/min, we identified an average of 3,524 proteins per single-cell-sized aliquot of protein digest. Reducing the active gradient to 20 min resulted in a modest 10% decrease in proteome coverage. Using this platform, we compared protein expression between single HeLa cells having an essential autophagy gene, atg9a, knocked out, with their isogenic WT parental line. Similar proteome coverage was observed, and 268 proteins were significantly up- or downregulated. Protein upregulation primarily related to innate immunity, vesicle trafficking and protein degradation.
Keywords: nanoPOTS, single-cell proteomics, nanoLC, autophagy, LFQ
Graphical Abstract
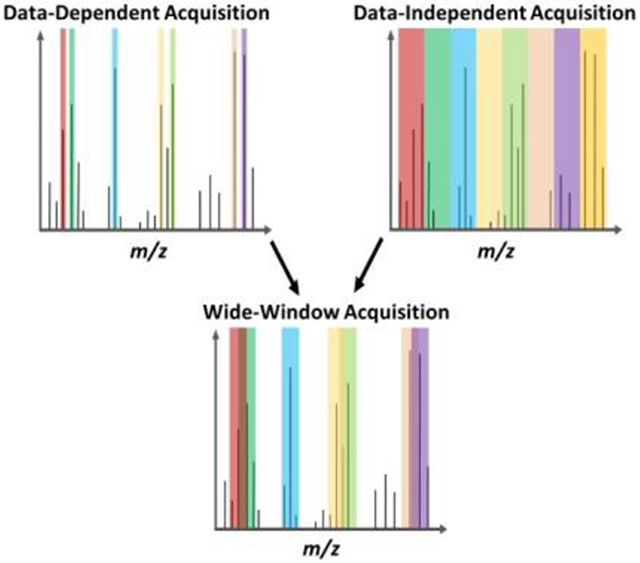
A new data acquisition strategy for biological mass spectrometry, termed wide-window acquisition, combined with efficient sample preparation and ultra-low-flow liquid chromatography, enables single-cell proteome profiling to an unprecedented depth of >3000 proteins per cell. This advance allows additional classes of proteins to be studied within single cells.
Introduction
The field of single-cell proteomics (SCP) is advancing rapidly due to improvements in experimental design, sample preparation, separations, mass spectrometry (MS) data acquisition and analysis.[1] Experimental conditions that are successful for bulk-scale analyses are often suboptimal for single cells and vice versa. For example, during bulk-scale sample preparation, cleanup steps are generally necessary to avoid clogging of liquid chromatography (LC) columns and contaminating the mass spectrometer,[2] while adsorptive analyte losses during sample cleanup may be negligible. In contrast, due to much smaller amounts of, e.g., lipids and insoluble debris in single cells, sample cleanup may not only be unnecessary, but the accompanying sample losses as a percentage of starting material are expected to be much more severe.[3] These different requirements have resulted in low-volume, one-pot methods generally being used for low-input samples, while bulk-scale sample preparation continues to employ extensive cleanup.
Similarly, mass spectrometry (MS) acquisition parameters have very different optimum settings for bulk and single-cell samples. The large ion flux from bulk samples results in rapid accumulation of a sufficient ion population to produce a productive MS2 spectrum such that tens of MS2 spectra can be collected per second on an Orbitrap instrument.[4] Since the resolution achieved by an orbitrap mass analyzer scales linearly with transient time, a low-resolution orbitrap setting such as 15,000 fwhm at m/z 200 must also be selected to achieve rapid scan rates. However, this low resolution is still sufficient to produce high-confidence peptide-spectrum matches (PSMs) for MS2 spectra that predominantly contain fragment ions from a single peptide.[4] Single-cell samples have a much lower ion flux, so longer injection times (up to hundreds of milliseconds) may be required to achieve a sufficient ion population for a productive MS2 scan.[5] This results in few collected spectra and low proteome coverage. At these slow scan speeds, a higher resolution (slower) orbitrap resolution setting (e.g., >100,000 at m/z 200) may be used with no impact on cycle time, but this higher resolution is generally excessive and of little benefit for resolving fragments from a single precursor.
The long required injection times and the accompanying high-resolution MS2 spectra that can resolve more complex ion populations point to intentional precursor coisolation for the identification of multiple peptides as a means of overcoming the slow scan speeds required for most SCP analyses. That is, multiple peptides can simultaneously accumulate for sufficient time in an ion trap, and the fragment ions from these peptides can be readily resolved using a high-resolution orbitrap scan. Indeed, data independent acquisition (DIA)-based SWATH MS[6] steps through large, overlapping precursor isolation windows to generate complex peptide spectra. In contrast, data-dependent acquisition (DDA) software has historically searched for the highest scoring single peptide from a given MS2 spectrum, although approaches have been developed to effectively identify multiple peptides from single ‘chimeric’ MS2 spectra.[7]
We supposed that the intentional coisolation of multiple precursors using much larger isolation windows (up to 48 Th vs. ~1 Th for standard DDA), termed wide-window acquisition (WWA)-DDA, would be especially beneficial for SCP performed with Orbitrap instrumentation due to the required use of long ion accumulation times and their accompanying high-resolution MS2 spectra. To test this, we utilized the recently released CHIMERYS software developed by MSAID (Munich, Germany) and included with Proteome Discoverer 3.0 (Thermo Fisher, Waltham, MA),[8] which can resolve >10 precursors per MS2 spectrum. WWA serves as a hybrid between DDA and DIA (Scheme 1), with a specific precursor being isolated for fragmentation (as with DDA), while the wide isolation windows allow for co-fragmentation and analysis of untargeted neighboring precursors (as with DIA). We evaluated WWA for rapid label-free proteome profiling of single cells that were prepared using nanoPOTS,[3a] and found that isolation windows in the range of 8 - 12 Th provide greatest peptide and protein coverage when identification is based solely on MS2 spectra (i.e., without the use of MS1-based feature matching such as the Match Between Runs (MBR) algorithm[9]).
Scheme 1.

Precursor isolation using DDA, DIA and WWA.
These optimized isolation windows agree with the findings of Mayer et al.,[10] who have concurrently explored WWA for aliquots of bulk-prepared samples. Optimized WWA provides a ~30% increase in MS2-identified peptides relative to standard DDA, although peptide and proteome coverage are similar when including MBR identifications. However, due to the increased confidence associated with false discovery rate (FDR)-controlled MS2 identification relative to MBR, it is beneficial to increase the ratio of MS2:MBR identifications. We also compared WWA to DIA analyzed using MSFragger-DIA with DIA-NN quantification,[11] and found WWA provides greater coverage, particularly for faster (20-min) LC separations. Remarkably, in combination with highly sensitive analyses afforded by low-nanoflow LC operated at ~15 nL/min, we have achieved an unprecedented proteome coverage of >3000 protein groups/cell, with ~2000 of those proteins identified by their tandem mass spectra. Using this platform, we compared protein expression between single HeLa cells having an essential autophagy gene, atg9a, knocked out, to their isogenic WT parental line. Similar proteome coverage was observed, and 268 proteins were significantly up- or downregulated. Included in the upregulated proteins in the autophagy-deficient cells were those associated with innate immunity, vesicle trafficking and protein degradation, including subunits of the proteasome. The reported workflow represents a substantial increase in both sensitivity and acquisition speed for label-free single-cell proteomics.
Results and Discussion
We anticipated that WWA would increase coverage relative to DDA by quantifying additional peptides that are coisolated with the precursor specifically selected for fragmentation. However, we also anticipated that with a sufficiently large isolation window, the complexity of the resulting spectra may impede identification. As such, we performed screening experiments using single-cell-sized (0.2 ng) aliquots of HeLa digest in which the isolation window was systematically varied from 2 to 48 Th as shown in Figure 1. These analyses were performed across four different maximum injection times/MS2 resolutions ranging from 54 ms/30,000 to 246 ms/120,000. The active chromatographic gradient was ~40 min and the AGC target was set to 1000% such that the maximum injection time rather than AGC target was generally limiting. The number of collected MS2 spectra predictably decreased with increasing maximum injection time/resolution settings, yet the number of PSMs was greatest for the intermediate MS2 resolutions of 45k and 60k (Figure 1A). For larger isolation windows, the number of identified PSMs exceeded and in some cases more than doubled the number of collected MS2 spectra, indicating effective identification of multiple precursors per MS2 spectrum by the CHIMERYS search engine.
Figure 1.

Parameter optimization experiments for 40-min gradients using 0.2 ng aliquots of HeLa digest. A) Number of collected MS2 spectra and PSMs as a function of maximum injection time/resolution and the isolation window size for MS2. (B) Number of unique peptides identified as a function of MS acquisition settings. (C) Number of high-confidence master proteins (1% FDR) for the same conitions. All identifications are based solely on MS2 identification (i.e., no MBR). Std. DDA conditions are listed in Methods. Error bars indicate ± 1 std. dev., and n=2 for all conditions.
The distribution of PSMs per MS2 spectrum using WWA is shown in Figure S1. Overall, 62% of MS2 spectra produced a high-confidence PSM. Of those productive MS2 spectra, 42% yielded two or more PSMs.
While >30,000 PSMs could remarkably result from single-cell-sized samples, far fewer unique peptides were identified (Figure 1B), indicating that many of these PSMs were redundantly sequenced due to the large and overlapping isolation windows. Still, identifying up to an average of 10,789 unique peptides with 1% FDR and without MBR is 28% greater than our DDA-based coverage (average 8,399) when using the same separation and MS instrumentation (Figure 1B). Peptide coverage was consistently greatest for isolation windows of 8 or 12 Th, which is in agreement with the findings of Mayer et al.,[10] and which points to a compromise between maximizing the number of isolated precursors and avoiding overly complex MS2 spectra. At the protein level, the number of MS2-identified high-confidence master proteins from 0.2 ng aliquots of HeLa digest increased 39% to 2,396 for WWA compared to 1,721 identified on average using standard DDA (Figure 1C).
We also employed MBR, known as Feature Mapper in PD 3.0, to evaluate proteome coverage for single-cell-sized (0.2 ng) aliquots of HeLa digest when using 10 ng aliquots of the same digest to serve as a matching library. We found that MBR substantially increased peptide coverage and that the coverage was essentially independent of MS2 acquisition parameters, as shown in Figure S2A. Indeed, 19,478 unique peptides were identified on average across all acquisition conditions including standard DDA, with just 3.9% CV. Similarly, proteome coverage including MBR was highly uniform across all screening conditions: 3,484 high-confidence master proteins were identified on average, with just 2.0% CV (Figure S2B). Only 171 proteins were identified on average from blank runs, including those identified by MBR, indicating a low degree of column carryover. Given the uniform coverage observed across screening conditions after applying the MBR algorithm, it should be determined whether there are benefits that remain for alternative acquisition strategies such as WWA. To address this, we estimated the false matching rate by generating a mixed-species library comprising human and yeast tryptic peptides in a similar manner to Woo et al.[12] Among 9,252 total peptides identified by MS2, 40 (0.43%) precursors were matched to nonhomologous yeast peptides. For MBR-based identifications, 483 of 15,070 unique peptides (3.2%) were incorrectly matched to yeast. As such, while transferred identifications from the MBR algorithm greatly increase the depth of attainable proteome coverage, it is beneficial to employ WWA in conjunction with MBR, as it maximizes the ratio of MS2: transferred identifications to reduce the overall FDR.
Shorter LC gradients having reduced peak capacities increase the number of coeluting peptides, which poses a challenge for standard DDA. Since the CHIMERYS search engine can decipher complex, highly chimeric fragmentation spectra, it should also benefit more rapid analyses. We thus performed screening experiments for faster separations having ~20-min elution windows (Figure S3) and found that 12 Th isolation windows and 60k MS2 resolution were highly effective in maximizing MS2-identified peptides. We compared proteome coverage between 40-minute and 20-minute gradients using the same acquisition parameters, as shown in Figure 2A. Impressively, MS2-based protein identifications only decreased by 9% for the faster analyses (1,953 vs. 2,150). With MBR, a 10% reduction in coverage was observed (3,160 vs 3,524). This indicates that the WWA can accommodate much faster separations with only modest impact on proteome coverage.
Figure 2.

Analysis of 0.2 ng aliquots of protein digest using WWA, standard DDA and DIA with 20- and 40-min gradients. (A) Proteome coverage using the three data acquisition modes, with MS2-based identifications shown in darker shading and additional library-matched proteins shown in lighter shading. (B) PCA plot resolving two cell types (HeLa and K562) in PC1, and cells of a given type resolved in PC2 according to gradient length. (C) Violin plots indicating %CV of protein abundance for different acquisition modes and gradient lengths, using the same color and shading scheme as (A). Median %CVs for each group are shown next to each plot, with and without MBR (WWA and DDA) or a spectral library search (DIA) shown without and with parentheses, respectively. Eight replicates were obtained in all cases except for standard DDA with 20-min gradients, which had only two replicates.
Given that WWA may be considered a hybrid between traditional DDA and DIA, we sought to also compare its performance to DIA. Using MSFragger-DIA with DIA-NN quantification.[11] We first evaluated various DIA acquisition settings (Table S1) and found no significant difference in coverage (Figure S4). As such, while the acquisition parameters for DIA were not exhaustively evaluated, we selected a fixed 50 Th isolation window scanned between m/z 400–800 as described by Gebreyesus et al.[13] for further experiments. Results were obtained both with and without employing a spectral library obtained from analyzing 10 ng of HeLa digest. Under all conditions, WWA yielded greater proteome coverage (Fig. 2A), and both DIA and WWA provided robust differentiation of cell types by PCA (Figure S5). The reduction in coverage for DIA when employing faster gradients was much greater than for WWA. As also shown in Figure 2A, average proteome coverage for DIA decreased by 45% with library-free search (1,021 vs. 1,850), and by 31% when a spectral library was employed (2,131 vs. 3,083). Clearly, shorter gradients in DIA can lead to overly complex spectra that negatively impact identification rates. The tradeoff between analysis speed and proteome coverage for DDA, WWA and DIA depends on various factors including the separation, mass spectrometer and search algorithm employed. As such, the trends observed here may be different under other experimental conditions.
The ability to differentiate distinct cell types, treatment groups, etc. is of course critical for SCP. We used principal component analysis (PCA) to determine the ability of WWA to cluster technical replicates of HeLa and K562 digest according to cell type for both 20 and 40-min gradients. As shown in Figure 2B, HeLa and K562 cell types readily cluster according to cell type along the PC1 axis, which accounts for 62% of variance. Replicates for a given cell type are also well differentiated along the PC2 axis according to gradient length, presumably due to increased coverage for the longer gradients; PC2 accounts for another 17% of explained variance.
We investigated CVs in protein intensities across 8 technical replicates for the different acquisition/analysis methods and gradient lengths (Figure 2C). Note that standard DDA experiments employing 20-min LC gradients were excluded from this comparison as only two technical replicates were obtained. The darker shades in Figure 2C indicate no MBR/spectral library, while the lighter shades show slightly increased median CVs when MBR (DDA and WWA) or spectral library matching (DIA) are included. The values of median %CVs are shown with and without matching, with values resulting from matching provided in parentheses. WWA improved the reproducibility of intensity measurements relative to DDA, while DIA provided lower median CVs than the other methods. We initially supposed that this was due to increased sampling rates across the narrow chromatographic peaks when using DIA. However, DIA-NN reports an average of 4.07 data points across the FWHM peak for 40-min separations, which is very close to the 4.09 points obtained for the corresponding WWA analyses as obtained by dividing the average FWHM peak width of 6.14 s by the 1.5 s cycle time. Similarly, DIA-NN reports an average of 3.09 points across the FWHM peak for 20 min DIA analyses, which is very similar to the 3.19 points per FWHM peak for WWA, when the average FWHM peak width decreased to 4.78 s. As such, it appears that other differences in assigning intensity values between DIA-NN and PD 3.0 account for the decreased CVs observed with DIA. We also investigated internal consistency in assigning intensity values between WWA and DIA using their respective data analysis algorithms. Figure 3A compares mean intensities observed across 8 replicates between 40-min and 20-min WWA analyses. The assigned intensities are in close agreement, with a correlation coefficient of 0.91 (Fig 3A). In contrast, DIA provided a more modest correlation of 0.74 between slow and fast analyses (Fig. 3B). Comparing intensities between 40-min WWA and DIA analyses resulted in a correlation coefficient of 0.78 (Figure 3C), with differences arising presumably from MS1-based identification of WWA vs. the use of MS2 intensities for DIA.
Figure 3.

Comparison of protein abundance values obtained using different data acquisition and analysis schemes. Additional description is in the text.
Having established the achievable proteome coverage and quantitative reproducibility of WWA with technical replicates of single-cell-sized aliquots of protein digest, nanoPOTS-prepared single HeLa and K562 cells were then analyzed. For single cells, we maintained the same maximum injection and MS2 resolution settings of 118 ms and 60,000. However, we narrowed the isolation window from 12 to 8 Th, anticipating that increased contaminant levels in single cells may limit ion accumulation times. Ten HeLa cells and 7–10 K562 cells were analyzed using each of the acquisition modes (DIA, DDA and WWA) with 40 min LC gradients. As with the aliquots, WWA provided the greatest proteome coverage, both with and without MBR/spectral library (Figure 4A). Without MBR or a spectral library, an average of 1,758 proteins were identified from HeLa cells by WWA, compared to 1,350 by DIA and 1,312 by DDA, such that WWA provided a respective increase of 30% and 34%. Including transferred identifications from MBR or a spectral library, WWA proteome coverage was 24% greater than DIA (3,042 vs. 2,463), yet only 3% greater than DDA (3042 vs. 2943). These findings were thus consistent with those resulting from the analysis of bulk-prepared digests. As expected due to biological variability, greater median CVs were observed for single-cell experiments relative to aliquots of bulk-prepared digest (Fig. 4B), and DIA continued to show less variability in observed protein intensities than the other acquisition modes. Similar proteome coverage was observed for HeLa and K562 cells, and these two cell types were readily differentiated by PCA along PC1 (Fig. 4C).
Figure 4.

Analysis of nanoPOTS-prepared single HeLa and K562 cells. (A) Proteome coverage using the three data acquisition modes, with MS2-based identification shown in darker shading and additional MBR-identified proteins shown in lighter shading. (B) Violin plots indicating CV of protein abundance for different acquisition modes and gradient lengths, using the same color and shading scheme as (A). Median CVs for each group are shown next to each plot, with and without MBR (WWA and DDA) or a spectral library search (DIA) shown without and with parentheses, respectively. (C) PCA plot resolving HeLa and K562 cells. n = 10 for each cell type/analysis method.
To contextualize the increased depth of discovery afforded by the present method, we compared the quantifiable proteins from this study to a previous study by Bekker-Jensen et al.,[14] which used bulk samples and extensive fractionation to calculate the protein copies per cell for >12,000 proteins in HeLa cells. We plotted the depth of discovery for all 3,840 detectable proteins observed in our analysis of single HeLa cells according to their copy number per cell as determined previously[14] (Figure 5A). Our single-cell data identifies more than half of the proteins at ≥105 copies per cell, while proteome coverage substantially drops off by 10,000 copies per cell. The median abundance of identified proteins is 256,626 copies per cell (426 zmol). Several low-abundance proteins are well represented in our dataset, including 111 proteins present at 6,000 copies per cell (~10 zmol) or fewer, although their presence in the single cells may be at higher levels than were observed in the bulk study.[14] 25% of quantified proteins are present at 88,945 copies per cell (148 zmol) or fewer.
Figure 5.

Depth of coverage in WWA compared to a traditional LFQ single-cell dataset. (A) The number of proteins detected based on the copy number for three datasets: a bulk proteome study,[14] the new WWA data (present study, blue), and data from our previous LFQ single-cell study (2021 study, orange).[5c] (B and C) A Gene Ontology analysis shows that the wide-window acquisition is able to achieve greater depth for proteins whose processes (B) and functions (C) are important to cancer development and cellular physiology. The scale in (B) and (C) are the portion of detected proteins relative to the bulk study.[14]
In comparison to our lab's prior efforts,[5c] we observe detection of proteins having approximately one order of magnitude fewer copies per cell (i.e., as few as 104 as shown in Figure 5A). A large portion of proteins in HeLa cells were calculated by Bekker-Jensen to be present at 1,000 to 10,000 copies per cell.[14] Detection of these ultra-low abundance proteins with LC-MS methods is very challenging, yet with additional advances in MS sensitivity and improvements in ionization efficiency afforded by low-flow separations such as open-tubular LC,[15] these species may also become amenable to profiling by single-cell MS.
With more than twice the number of quantified proteins from our previous analytical methods, we sought to understand the new biological processes that could be observed in our data. Our goal was to investigate whether these newly quantifiable proteins represent a meaningful addition to our ability to describe and characterize cellular physiology, as opposed to uncharacterized proteins or those with ambiguous function. Therefore, we classified proteins according to their Gene Ontology Biological Processes and Molecular Functions (Table S2). A total of 6,113 out of the 7,527 annotations that were found in both single cell studies had an increase in protein detection in the new study, while 326 only had a decrease in detection. An additional 3,464 annotations from the bulk study were found only in the new single cell study. 5,883 annotations were found in the bulk study but in neither single cell study. Additionally, 37 annotations were found in one or both of the single cell studies, but not in the bulk study. Select biological processes and molecular functions are shown in Figure 5. For example, several important functions relating to cancer physiology show a significant increase in proteins now detected such as cell division, chromatin remodeling, apoptosis, DNA repair, and cell cycle regulation. Moreover, proteins involved in the essentials of cellular physiology such as protein phosphorylation and de-phosphorylation, signal transduction and cellular differentiation have also substantially improved in coverage.
To evaluate the ability of our platform to differentiate closely related cells, we used our workflow to measure changes in the proteome triggered by a deficiency in autophagy, a cellular recycling process through which proteins and organelles are degraded via the lysosome. Autophagy is essential for oncogenesis and promotes the growth and metastasis of established tumors.[16] In addition, recently developed autophagy inhibitors boost anti-tumor immunity in pre-clinical mouse models.[17] However, the specific substrates of autophagy that could explain these effects, particularly the role of autophagy in tumor immunity, are still poorly understood. Single-cell proteomics offers the ability to measure autophagy-dependent changes in the proteome while also capturing cell-to-cell variability, which could be particularly important for solid tumors in which the breadth and selectivity of autophagy may differ depending on the location of the cell and its connection to nutrient supply. To generate an autophagy-deficient (AD) cell model, we used CRISPR/Cas9 in HeLa cells to knockout (KO) atg9a, an essential autophagy gene,[18] and compared these cells to their isogenic WT parental line. We validated the loss of ATG9A by immunoblot and verified that cells deficient in ATG9A also showed an increase in p62 levels, which indicates that the cells are autophagy defective as expected (Figure 6A). As with other experiments, we identified ~3,000 proteins in both KO and control (WT) cells (Figure 6B), and the WT and KO cells were well separated by PCA (Figure 6C). A string analysis of functionally related proteins that differed between WT and atg9a knockout cells is shown in Figure 6D.
Figure 6.

Comparison of protein expression between control and atg9a knockout HeLa cells. (A) Western blot showing effective knockout of atg9a in HeLa cells. (B) Depth of coverage for control (WT) and knockout cells. Proteins identified by MS2 and MBR are represented with dark and light shading, respectively. (C) PCA plot differentiating the WT and knockout cells. (D) String analysis of functionally related proteins that differed between WT and atg9a knockout cells. Red indicates proteins that were at significantly higher levels in atg9a knockout cells.
Gene ontology analysis of the enriched-in-AD protein subset contained a number of proteins associated with infectious disease/immunity and autophagy. Within the autophagy subset, a group of vesicle trafficking proteins emerged as up- or downregulated in AD cells. This aligns with our understanding of ATG9A function. ATG9A is multi-pass transmembrane protein that traffics on small vesicles between the golgi, ER, and endosomal system. The network of ATG9A interactions includes numerous vesicle trafficking proteins,[19] many of which traffic with ATG9A[20] and are likely degraded by autophagy. Notably, one of the vesicle trafficking proteins enriched in AD cells, SEC22B (Figure S6), has been implicated in autophagy through regulation of ATG9A function.[21] Spreadsheets showing all up- down- and unregulated proteins are included in Supporting Information as Tables S3–S5.
Among the enriched-in-AD infectious disease-related proteins was interferon-stimulated gene 15 (ISG15; Figure S6), a ubiquitin-like protein with roles in anti-pathogen and anti-tumor immunity.[22] ISG15 can be covalently conjugated to viral and bacterial lysine residues through a process called ISGylation, which targets these pathogens for autophagic degradation. ISGylation also occurs on core autophagy regulators, including mTORC1 and WIPI2, to boost autophagy levels even in the absence of infection.[23] The degradation of ISG15 by autophagy could be a mechanism of negative feedback to prevent autophagic cell death after an initial immune response. In addition to ISGylation, the unconjugated form of ISG15 can be secreted into the extracellular milieu where it interacts with lymphocyte function-associated antigen-1 (LFA1) receptors on NK cells and T cells to boost type I and II interferon signaling.[24] Therefore, an increase in ISG15 levels/secretion could help explain how autophagy inhibitors enhance anti-tumor immunity and interferon signaling in syngeneic mouse tumor models.[17] Together, these data demonstrate the value of the WWA approach in distinguishing closely related cell types and measuring autophagy-dependent changes in the proteome.
Conclusion
Here we report an improved workflow for label-free single proteomics that incorporates each of the unique advantages of the nanoPOTS sample preparation platform, ultra-low-flow liquid chromatography, and newly developed WWA data acquisition. Compared to our previous DDA-based workflows, the new WWA acquisition resulted in increase coverage of up to ~2150 proteins (~3524 with MBR) from 0.2 ng aliquots Hela digest, respectively. WWA also increases the overall throughput by accommodating much faster 20-min gradients with only a modest reduction (9%) in coverage. Compared to different DIA variants, WWA coverage outperformed under all conditions, especially at faster gradients. Utilizing all the advantages of the new workflow, we achieved an average of 1758 and 1642 proteins for single Hela and K562 cells without matching (3042 and 2891 with matching). Further investigation using published bulk samples as a reference revealed that the new single-cell WWA workflow significantly increased proteome coverage for proteins present at low copy numbers per cell compared to the previously reported DDA result. With the addition of 3464 annotations from the bulk study found only in the new single-cell study, new cellular functions and biological processes were also observed, as well as an increase in those already detected. Finally, by comparing wildtype and single-gene knockouts, we demonstrated that the ability of the workflow to differentiate closely related cells.
Supplementary Material
Acknowledgements
The research reported in this publication was supported by the National Institute of General Medical Sciences and the National Cancer Institute of the National Institutes of Health under award numbers R21CA272326 (RTK), R01GM138931 (RTK), R01GM147653 (SHP) and R01GM147310 (JLA). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD037527.
References
- [1].a Zhu Y, Piehowski PD, Kelly RT, Qian WJ, Expert Rev. Proteomics 2018, 15, 865–871; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kelly RT, Mol. Cell. Proteomics 2020, 19, 1739–1748; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ctortecka C, Mechtler K, Anal. Sci. Adv 2021, 2, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a Tubaon RM, Haddad PR, Quirino JP, Proteomics 2017, 17, 1700011; [DOI] [PubMed] [Google Scholar]; b Alexovič M, Sabo J, Longuespée R, Proteomics 2021, 21, 2000318. [DOI] [PubMed] [Google Scholar]
- [3].a Zhu Y, Piehowski PD, Zhao R, Chen J, Shen YF, Moore RJ, Shukla AK, Petyuk VA, Campbell-Thompson M, Mathews CE, Smith RD, Qian WJ, Kelly RT, Nat. Commun 2018, 9, 882; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhu Y, Clair G, Chrisler WB, Shen YF, Zhao R, Shukla AK, Moore RJ, Misra RS, Pryhuber GS, Smith RD, Ansong C, Kelly RT, Angew. Chem. Int. Ed 2018, 57, 12370–12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kelstrup CD, Jersie-Christensen RR, Batth TS, Arrey TN, Kuehn A, Kellmann M, Olsen JV, J. Proteome Res 2014, 13, 6187–6195. [DOI] [PubMed] [Google Scholar]
- [5].a Zhu Y, Podolak J, Zhao R, Shukla AK, Moore RJ, Thomas GV, Kelly RT, Anal. Chem 2018, 90, 11756–11759; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cong Y, Liang Y, Motamedchaboki K, Huguet R, Truong T, Zhao R, Shen Y, Lopez-Ferrer D, Zhu Y, Kelly RT, Anal. Chem 2020, 92, 2665–2671; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cong Y, Motamedchaboki K, Misal SA, Liang Y, Guise AJ, Truong T, Huguet R, Plowey ED, Zhu Y, Lopez-Ferrer D, Kelly RT, Chem. Sci 2021, 12, 1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ludwig C, Gillet L, Rosenberger G, Amon S, Collins BC, Aebersold R, Mol. Syst. Biol 2018, 14, e8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dorfer V, Maltsev S, Winkler S, Mechtler K, J. Proteome Res 2018, 17, 2581–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8]. https://www.msaid.de/chimerys .
- [9].Tyanova S, Temu T, Cox J, Nat. Protoc 2016, 11, 2301–2319. [DOI] [PubMed] [Google Scholar]
- [10].Mayer RL, Matzinger M, Schmücker A, Stejskal K, Krššáková G, Berger F, Mechtler K, bioRxiv 2022, DOI: 10.1101/2022.09.01.506203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Demichev V, Szyrwiel L, Yu F, Teo GC, Rosenberger G, Niewienda A, Ludwig D, Decker J, Kaspar-Schoenefeld S, Lilley KS, Mülleder M, Nesvizhskii AI, Ralser M, Nat. Commun 2022, 13, 3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Woo J, Clair GC, Williams SM, Feng S, Tsai C-F, Moore RJ, Chrisler WB, Smith RD, Kelly RT, Paša-Tolić L, Ansong C, Zhu Y, Cell Syst. 2022, 13, 426–434.e424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gebreyesus ST, Siyal AA, Kitata RB, Chen ES-W, Enkhbayar B, Angata T, Lin K-I, Chen Y-J, Tu H-L, Nat. Commun 2022, 13, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bekker-Jensen DB, Kelstrup CD, Batth TS, Larsen SC, Haldrup C, Bramsen JB, Sørensen KD, Høyer S, Ørntoft TF, Andersen CL, Nielsen ML, Olsen JV, Cell Syst. 2017, 4, 587–599.e584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a Li SY, Plouffe BD, Belov AM, Ray S, Wang XZ, Murthy SK, Karger BL, Ivanov AR, Mol. Cell. Proteomics 2015, 14, 1672–1683; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Xiang PL, Zhu Y, Yang Y, Zhao ZT, Williams SM, Moore RJ, Kelly RT, Smith RD, Liu SR, Anal. Chem 2020, 92, 4711–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mizushima N, Levine B, N. Engl. J. Med 2020, 383, 1564–1576. [DOI] [PubMed] [Google Scholar]
- [17].a Noman MZ, Parpal S, Van Moer K, Xiao M, Yu Y, Viklund J, De Milito A, Hasmim M, Andersson M, Amaravadi RK, Martinsson J, Berchem G, Janji B, Sci. Adv 2020, 6, eaax7881; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Deng J, Thennavan A, Dolgalev I, Chen T, Li J, Marzio A, Poirier JT, Peng DH, Bulatovic M, Mukhopadhyay S, Nat. Cancer 2021, 2, 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shoemaker CJ, Huang TQ, Weir NR, Polyakov NJ, Schultz SW, Denic V, PLoS Biol. 2019, 17, e2007044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kannangara AR, Poole DM, McEwan CM, Youngs JC, Weerasekara VK, Thornock AM, Lazaro MT, Balasooriya ER, Oh LM, Soderblom EJ, Lee JJ, Simmons DL, Andersen JL, EMBO Rep. 2021, 22, e51136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Judith D, Jefferies HBJ, Boeing S, Frith D, Snijders AP, Tooze SA, J. Cell Biol 2019, 218, 1634–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen W-L, Griffith J, Nag S, Wang K, Moss T, Baba M, James A McNew, Jiang X, Reggiori F, Melia Thomas J., Klionsky Daniel J., Cell 2011, 146, 290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Perng Y-C, Lenschow DJ, Nat. Rev. Microbiol 2018, 16, 423–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a Villarroya-Beltri C, Baixauli F, Mittelbrunn M, Fernández-Delgado I, Torralba D, Moreno-Gonzalo O, Baldanta S, Enrich C, Guerra S, Sánchez-Madrid F, Nat. Commun 2016, 7, 13588; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang Y, Thery F, Wu NC, Luhmann EK, Dussurget O, Foecke M, Bredow C, Jiménez-Fernández D, Leandro K, Beling A, Knobeloch K-P, Impens F, Cossart P, Radoshevich L, Nat. Commun 2019, 10, 5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Swaim CD, Scott AF, Canadeo LA, Huibregtse JM, Mol. Cell 2017, 68, 581–590.e585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD037527.