

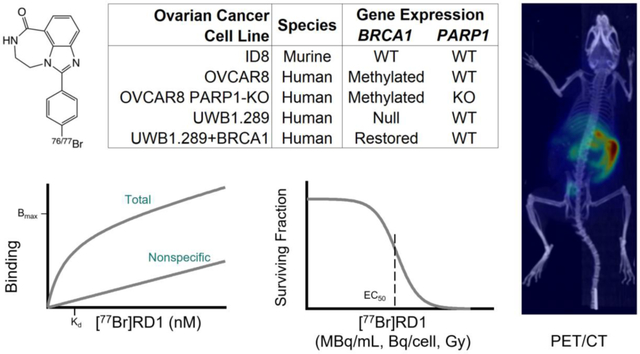
Abstract
Advanced ovarian cancer currently has few therapeutic options. Poly(ADP-ribose) polymerase (PARP) inhibitors bind to nuclear PARP and trap the protein-inhibitor complex to DNA. This work investigates a theranostic PARP inhibitor for targeted radiopharmaceutical therapy of ovarian cancer in vitro and PET imaging of healthy mice in vivo.
Methods:
[77Br]RD1 was synthesized and assessed for pharmacokinetics and cytotoxicity in human and murine ovarian cancer cell lines. [76Br]RD1 biodistribution and organ uptake in healthy mice were quantified through longitudinal PET/CT imaging and ex vivo radioactivity measurements. Organ-level dosimetry following [76/77Br]RD1 administration was calculated using RAPID, an in-house platform for absorbed dose in mice, and OLINDA for equivalent and effective dose in human.
Results:
The maximum specific binding (), equilibrium dissociation constant (), and nonspecific binding slope (NS) were calculated for each cell line. These values were used to calculate the cell specific activity uptake for cell viability studies. The half maximal effective concentration (EC50) was measured as 0.17 (95% CI: 0.13–0.24) nM and 0.46 (0.13–0.24) nM for PARP(+) and PARP(−) expressing cell lines, respectively. The EC50 was 0.27 (0.21–0.36) nM and 0.30 (0.22–0.41) nM for BRCA1(−) and BRCA1(+) expressing cell lines, respectively. When measuring the EC50 as a function of cellular activity uptake and nuclear dose, the EC50 ranges from 0.020–0.039 Bq/cell and 3.3–9.2 Gy, respectively. Excretion through the hepatobiliary and renal pathways were observed in mice, with liver uptake of 2.3±0.4 %ID/g after 48 h, contributing to estimated absorbed dose values in mice of 19.3±0.3 mGy/MBq and 290±10 mGy/MBq for [77Br]RD1 and [76Br]RD1, respectively.
Conclusion:
[77Br]RD1 cytotoxicity was dependent on PARP expression and independent of BRCA1 status. The in vitro results suggest that [77Br]RD1 cytotoxicity is driven by the targeted Meitner-Auger electron (MAe) radiotherapeutic effect of the agent. Further studies investigating the theranostic potential, organ dose, and tumor uptake of [76/77Br]RD1 are warranted.
Keywords: bromine-77, bromine-76, Meitner-Auger radionuclide therapy, positron emission tomography, PARP-1 inhibitor, theranostic, ovarian cancer therapy
Graphical Abstract
1. Introduction
An estimated 20,000 new cases of ovarian cancer will be diagnosed in 2023 with approximately 50% of patients presenting with distant-staged disease [1]. Upon metastasis of ovarian cancer, patients have a 5-year relative survival rate of 31%, only a 1–3% increase over the last 20 years [1], [2]. The current standard of care for advanced ovarian cancer includes cytoreductive surgery and platinum-based chemotherapy with curative intent [3]. Platinum-based chemotherapy drugs are known for their lack of selectivity, high systemic toxicity, and drug resistance [4]. Patients may have good initial response to this treatment, but approximately 75% of patients with advanced stage ovarian cancer will relapse, requiring further chemotherapy treatments. Furthermore, 30% of these patients will develop chemotherapy drug resistance, at which point, treatment shifts from curative intent to maximizing quality of life [5].
Comprehensive molecular characterization of epithelial ovarian cancer (EOC) and especially high-grade serous ovarian cancer demonstrated an over-representation of BRCA1/2 pathogenic variants [6]. This demonstrates the importance of double strand DNA repair pathways, such as homologous recombination (HR), with further studies confirming that dysfunction in this pathway leads to a higher risk of development of EOC [7], [8]. Drugs targeting the DNA damage response systems provides an opportunity to target this advanced disease for patients who otherwise have poor prognosis and few treatment options. Poly(ADP-ribose) polymerase (PARP) enzymes are nuclear proteins with a critical role in DNA damage response. PARP inhibitors exploit synthetic lethality through single-strand break and double-strand break repair pathways, specifically the HR impaired pathway [9]–[12]. FDA approved PARP inhibitors olaparib, rucaparib, and talazoparib trap the PARP-drug complex nanometers from target DNA [13] making them viable diagnostic and therapeutic radiopharmaceutical candidates in cancers with PARP expression [14].
The use of positron emission tomography (PET) imaging to assess physiologic PARP expression in patients has been established as a noninvasive imaging tool [14]. Two 18F-labeled PARP inhibitors are in the process of being translated from preclinical research to clinical PET application [15]–[17]. The first agent, [18F]FTT, is a rucaparib analog which has shown potential for imaging PARP [18]. Multiple studies have demonstrated [18F]FTT’s efficacy in preclinical models, and human trials are currently underway [16], [19]. The second agent is an olaparib analog, [18F]PARPi, that targets PARP and has shown promise in imaging breast, ovarian and brain cancers [17], [20]. [18F]PARPi has also been shown to have potential in imaging other DNA damage response pathways, including DNA-PK, ATR, and ATM [21] and is currently undergoing clinical trials investigating its potential as a diagnostic tool for cancer. Additional 18F-labeled isotopologues that have been preclinically investigated include [18F]rucaparib [22], [23], [18F]talazoparib [24], [25], and [18F]olaparib [26], [27].
PARP has been targeted in radiopharmaceutical therapy using olaparib- and rucaparib-derived agents labeled with various radionuclides, including emitter 131I [28], alpha-emitter 211At [29]–[31], and Meitner-Auger electron (MAe) emitters 125I [32]–[35], 123I [28], [35] and 77Br [36]. These preclinical studies have investigated responses in ovarian cancer [35] as well as glioblastoma [28], [37], [38], neuroblastoma [32], colon [33], breast [34], prostate cancers [36]. The biological mechanism of PARP that brings its inhibitors within nanometers of DNA is well suited for use with the low energy (eV–keV), short range (0.001–100 μm), and high linear energy transfer (LET, 1–23 keV/μm) of MAe [39]. In vitro cell cytotoxicity studies show MAe PARP inhibitors to be highly cytotoxic across several cancer cell lines [32], [34], [35], [37], [40], and tumor-bearing mice treated with MAe PARP inhibitors were observed to have prolonged survival in vivo [28], [33], [36]. Higher cytotoxicity was observed compared to radioinert PARP inhibitors with no dependence on BRCA1 expression [35]. These results show promising radiotherapeutic potential that warrants further preclinical and clinical investigation across a multitude of cancers.
Rucaparib is FDA approved for both treatment and maintenance therapy of ovarian cancer [41] and a derivative of this molecule was investigated as a theranostic radiopharmaceutical labeled with MAe-emitting 77Br (h, 6–7 MAe per decay) [42] and positron emitting 76Br (h, 55%). The short-range MAe from 77Br deposits their energy locally [43], providing low dose to off-target cells. Bromine forms a more stable bond with carbon compared with iodine, resulting in less dehalogenation of radiopharmaceuticals in vivo. Additionally, unlike radioiodine, free radiobromine does not accumulate in the thyroid, diffusing evenly throughout the body with only the stomach reaching higher uptake than blood [44], [45]. The PARP inhibitor mechanism provides a biological approach to localize the radioactive drug to the DNA. The imaging analog, labeled with 76Br, allows for noninvasive in vivo PET imaging [46], [47] of radiopharmaceutical biodistribution. This study examines the in vitro radiopharmacology and toxicology of rucaparib derived [77Br]RD1. Additionally, it investigated the potential for in vivo PET imaging of its diagnostic analog, [76Br]RD1, in healthy mice.
2. Materials and Methods
2.1. Radiosynthesis of [76/77Br]RD1
Production of 76/77Br and [76/77Br]RD1 synthesis and quality control were performed according to published procedures as detailed in the supplementary methods [48], [49]. Proton irradiation of Co76/77Se yielded 76/77Br which was isolated via dry distillation. [76/77Br]RD1 was synthesized by Cu-mediated bromo deborylation chemistry of a boronic acid pinacol ester precursor (Figure 1). [76/77Br]RD1 was isolated by preparative high performance liquid chromatography (HPLC), and subsequently formulated for in vitro and in vivo studies in sterile phosphate buffered saline (PBS). Molar activity and radiochemical purity of [76/77Br]RD1 were evaluated by analytical HPLC and autoradiography-visualized thin layer chromatography (radioTLC, Fig. S1–2) with methods further described in the supplementary materials.
Figure 1:
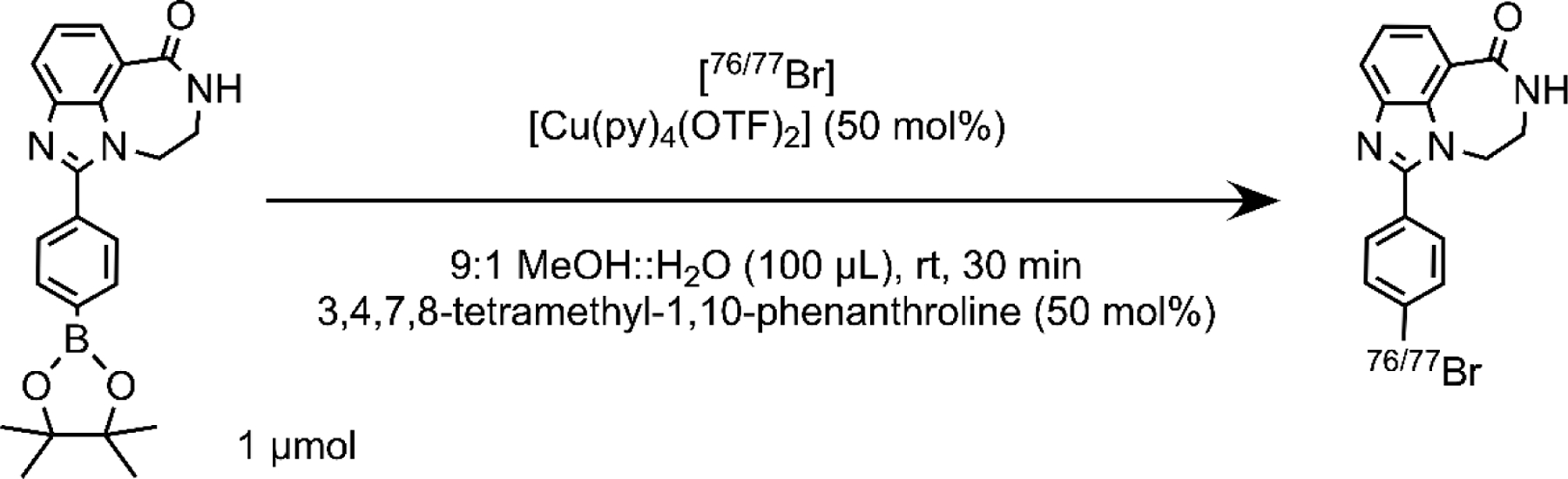
Radiosynthesis of 76/77Br-labeled PARP1 inhibitor [76/77Br]RD1.
2.2. Cell Culture
Murine ID8 ovarian cancer cells were gifted from Dr. Katherine Roby (University of Kansas, KS) and maintained in Dulbecco’s modified Eagle’s medium with 4.5 g/L glucose, L-glutamine, and sodium pyruvate (Corning) supplemented with 5% fetal bovine serum (FBS, Corning), 1% penicillin-streptomycin (Corning) and 0.2% 500X insulin-transferrin-selenium supplement (BioWhittiker). OVCAR8 and OVCAR8 PARP1-KO G1 (isogenic pair to OVCAR8 with PARP1 knocked out, [50]) human ovarian cancer cells were maintained in RPMI-1640 medium (Gibco) supplemented with 10% FBS and 1% penicillin-streptomycin. OVCAR8 PARP1-KO G1 had an additional 2 μg/mL puromycin (Gibco) added to the medium. UWB1.289 and UWB1.289+BRCA1 (isogenic pair to UWB1.289 with BRCA1 restored) human ovarian cancer cells were purchased from American Type Culture Collection and maintained in 1:1 RPMI-1640 (Gibco) and mammary epithelial growth medium (Lonza) supplemented with 3% FBS. UWB1.289+BRCA1 media also had 200 μg/mL Geneticin selective antibiotic (G418 Sulfate, Gibco). All cells were cultured in a humidified environment at 37°C and 5% CO2.
2.3. [77Br]RD1 in vitro binding assays
All cell lines were grown in clear, sterile, tissue-culture (TC)-treated 96-well plates (Corning) at a seeding density between 5,000–50,000 cells/well in a volume of 180 μL cell medium for 15–24 hours. The cells were treated in triplicate with 0.0086–128 nM of [77Br]RD1 (122–500 MBq/μmol) with and without a blocking dose of 1 μM non-radioactive BrRD1. [77Br]RD1 in PBS (BioWhittaker) was added to the wells in a volume of 20 μL for a total volume in wells of 200 μL for 2 hours.
A hemocytometer was used to verify the cell density per well at the time of contact in triplicate using wells with zero activity. The medium containing [77Br]RD1 was removed and placed in corresponding polypropylene cluster tubes (Corning). The wells were rinsed with 100 μL of PBS and added to the same corresponding cluster tubes. The cells were then dispersed with 50 μL of 0.25% trypsin-EDTA (Corning) for 20 minutes at 5% CO2 and 37°C and this volume collected. The wells were rinsed with 100 μL of PBS and collected in the same corresponding vial. The radioactivity in the cells+rinse and the medium+rinse was measured using a Wizard2 Automatic Gamma Counter (PerkinElmer) calibrated by high purity germanium (HPGe) gamma spectroscopy (HPGe, Canberra C1519). Background and dead time were manually measured. For dead times higher than 10%, measurements were retaken after a period of decay. Cellular molar uptake (in attomol/cell) was calculated by dividing the cell-associated radioactivity (MBq) by the measured number of cells per well and the measured molar activity (MBq/nmol).
The total binding curve measured from the cells contacted with [77Br]RD1 and the nonspecific binding curve measured from the cells contacted with [77Br]RD1 + 1 μM BrRD1 were fitted using GraphPad Prism v9 (GraphPad Software, San Diego, CA, USA). The maximum number of binding sites per cell (), equilibrium dissociation constant (), and nonspecific binding slope (NS) were calculated (Eq. S1–S3).
2.4. Cytotoxicity following exposure to [77Br]RD1
Cells were plated at a seeding density of 200 cells/well in 96-well white, opaque TC-treated plates (Falcon) for 2 hours before contact with varying concentrations of [77Br]RD1 from 0.001–60 MBq/mL (0.0005–450 nM) for 7 days. Cell viability was assayed using Cell-Titer Glo 2.0 (Promega) by measuring adenosine triphosphate (ATP) with luminescence. Dose response curves were fitted using GraphPad Prism v9 and the half maximal effective concentrations (EC50) were calculated as a function of molar concentration, activity concentration, and specific uptake using Equation S4. Radiation dose to the cell nucleus was calculated using Monte Carlo simulation with Medical Internal Radiation Dosimetry Cell (MIRDcell) v3.12, as previously described [51]. Further protocol details are provided in the supplementary methods.
2.5. PET/CT imaging and biodistribution of [76Br]RD1
Imaging and biodistribution studies were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin—Madison (IACUC M006459). Four female C57BL/6 mice (Jackson Laboratory, ME) aged 12 weeks were used for in vivo studies. Mice were imaged prone two at a time on an Inveon microPET/CT scanner (Siemens Medical Solutions) under anesthesia, with 1.5–3.5% isoflurane at a rate of 1–2 L/min of oxygen via nose cone after an induction with 3–5% isoflurane. X-ray computed tomography (CT) imaging lasted 10 minutes, followed by PET image acquisition. Mice received 5.1±0.1 MBq of 23 MBq/nmol [76Br]RD1 via tail vein (IV) injections at the beginning of a 1-h dynamic PET scan. Images were histogrammed into 1-minute bins between 1–5 minutes post injection (p.i.), 5-minute bins between 5–30 minutes p.i., and 10-minute bins between 30–60 minutes p.i. Static PET scans were performed at 4 h, 24 h, and 48 h p.i. and histogrammed into one frame (50 million counts/scan for all the time points except 20 million counts/scan at 48 h). PET images for static and dynamic scans were reconstructed using ordered-subset expectation maximization of 3 dimensions followed by the maximum a posteriori algorithm (18 iterations and 16 subsets). The corresponding CT was used for attenuation correction. PET/CT images were automatically coregistered and analyzed using Inveon Research Workplace 4.2 (Siemens Medical Solutions).
Regions of interest (ROIs) were drawn in the heart, lungs, brain, kidneys, bladder, liver, stomach, intestines, femur, and quadricep muscle. The mean percentage injected dose per volume (%ID/g) was measured after decay-correcting to time of injection. After the final PET/CT image at 48 h, mice were euthanized via CO2 asphyxiation followed by exsanguination. The blood, heart, lungs, pancreas, spleen, kidneys, liver, gallbladder, large intestine, small intestine, stomach, femur bone, quadriceps muscle, and enteric (combined stomach and small and large intestine) contents were harvested and weighed. The radioactivity was measured using an automated HPGe-calibrated gamma counter (PerkinElmer). Measurements were background- and decay-corrected to time of injection to calculate the %ID/g. A partial volume (PV) correction phantom (Phantech, WI) with sphere volumes from 8–900 mm3 filled with 142±6 kBq/mL of [76Br]RD1 was PET imaged as described above, and the recovery coefficient (RC) curves were calculated.
2.6. [76/77Br]RD1 three-dimensional dosimetry
PET/CT images were used to define the activity distribution and geometry for use in an in-house Monte Carlo (MC) dosimetry platform, RAPID [52]. PET/CT were coregistered and resampled in Amira (v5.3.3, Mercury Computer Systems, Berlin, Germany) and used to calculate the mean absorbed dose rate for each voxel after decay correction. This was done for each organ region at each timepoint. This was imported into the MC framework (Geant4, v9.6.p2) and the Auger electron function (ARMflag) was utilized for calculations using data from the Evaluated Atomic Data Library [53]. CT data were transformed from Hounsfield units into mass density using a CT scanner specific calibration curve. Evaluated Nuclear Structure Data Files (ENSDF, Brookhaven National Laboratory) data, which includes all MAe (>1 keV), , and radiation emitted per decay, were used to calculate the source decay sampled uniformly in a voxel. The energy deposition was tracked to create a 3D cumulative dose distribution with the PET contours defining organ regions to calculate organ specific dose. To calculate the dose distribution for [77Br]RD1, the PET images acquired with [76 Br]RD1 were corrected for physical decay differences between the two radionuclides.
The PET quantified [76Br]RD1 organ %ID/g uptake in mice was used to estimate the human organ dose using OLINDA/EXM v1.1 [54] as described previously [55]. Briefly, the mouse biodistribution was assumed to be equal to human biodistribution. The mouse organ [76Br]RD1 uptakes (%ID/g) were multiplied by the ratio of the average mouse mass in the study to the average human woman mass to get the estimated human organ %ID/g uptakes, which were multiplied by average human organ masses [56] to obtain organ-specific %ID in human. These values were used in OLINDA/EXM to calculate organ-level equivalent dose.
3. Statistical analysis
All data were obtained in technical and biological triplicate unless otherwise stated. Statistical analyses and nonlinear regression were performed using GraphPad Prism v9. Results are reported as mean standard deviation (SD) or with stated 95% confidence interval (CI).
4. Results
4.1. Radiosynthesis
The average cyclotron physical yield was 16±2 MBq·μA−1·h−1 (n = 7) for 77Br, 59±4 MBq·μA−1·h−1 (n = 4) for 76Br and the distillation yield was 67±13% (n=11). The non-decay corrected radiochemical yield (RCY) and molar activity (MA) for [77Br]RD1 was 58±25% and 300±120 MBq/nmol (n = 7). The RCY and MA for [76Br]RD1 was 5±3% and 23 MBq/nmol (n=2), respectively, when GBq-quantity [76Br]bromide was stored in solution overnight prior to reaction. When stored dry, it was 73% and 740 MBq/nmol (n=1), respectively.
4.2. In vitro analysis
The total, nonspecific (Figure 2A–B) and specific (Figure 2C) binding were used to calculate the , and NS, which are summarized in Table 1. For the OVCAR8 PARP1-KO cell line, there was no statistical difference between the nonspecific and total binding measurements for molar concentrations above 25 nM (n = 4). This indicated a low number of specific binding sites that were small compared with the nonspecific binding above 25 nM. Thus, values measured above 25 nM were excluded from the , and NS fitting analysis for this cell line.
Figure 2:
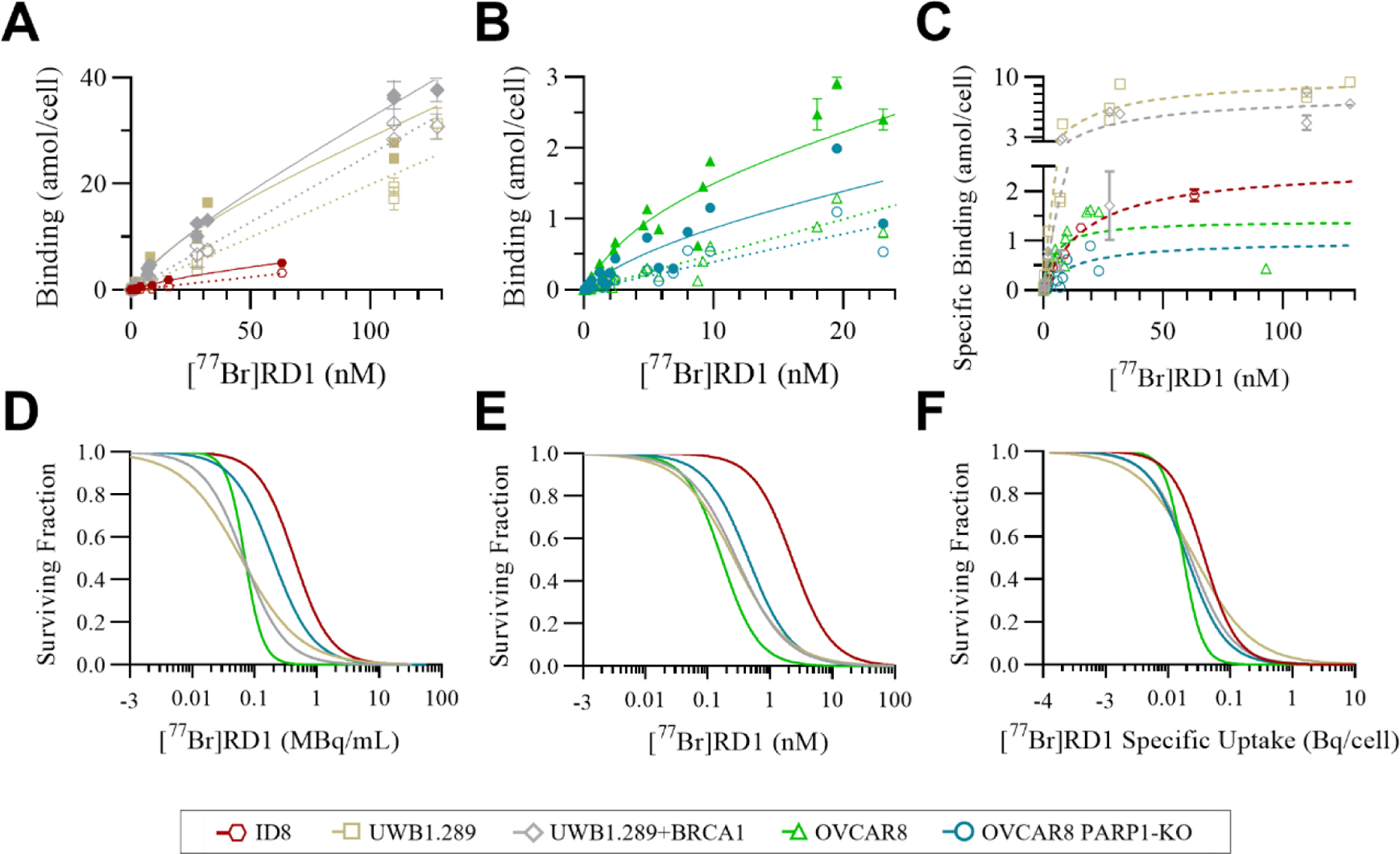
Total (closed symbols, solid lines) and nonspecific (open symbols, dotted lines) binding of (A) UWB1.289 (gold, n=3), UWB1.289 +BRCA1 (gray, n=3), ID8 (red, n=2) (B) OVCAR8 (green, n=4), and OVCAR8 PARP1-KO (blue, n=4) and (C) the specific binding (dashed lines) of [77Br]RD1 as a function of radiopharmaceutical molar concentration. (D) The cell survival of the same cell lines as a function of radiopharmaceutical activity concentration, (E) molar concentration, and (F) specific uptake (Bq/cell) inherent to the cell lines.
Table 1:
The [77Br]RD1 pharmacological parameters calculated from total and nonspecific binding studies in the various ovarian cancer cell lines and their corresponding 95% CI.
| Ovarian Cancer Cell Line |
(95% CI) (attomol/cell) |
(95% CI) (nM) |
NS (95% CI) (attomol/cell/nM) |
|---|---|---|---|
| ID8 | 3.0 (2.4–3.9) | 31 (20–49) | 0.049 (0.047–0.052) |
| OVCAR8 | 1.6 (1.4–1.8) | 6.0 (4.2–8.2) | 0.050 (0.048–0.052) |
| OVCAR8 PARP1-KO | 0.83 (0.50–1.5) | 7.5 (2.7–20) | 0.039 (0.033–0.046) |
| UWB1.289 | 11 (7.9–16) | 24 (10–57) | 0.20 (0.19–0.21) |
| UWB1.289+BRCA1 | 8.6 (6.6–11) | 25 (13–48) | 0.26 (0.25–0.26) |
4.3. In vitro cytotoxicity
The cell survival curves of the various ovarian cancer cell lines are summarized in Figure 2D–F as a function of activity concentration (Figure 2D), molar concentration (Figure 2E and S3), and specific uptake (Figure 2F). The EC50 and the corresponding 95% confidence intervals (CI) are summarized in Table 2.
Table 2:
The [77Br]RD1 toxicological EC50 values of the various ovarian cancer cell lines and their corresponding 95% CI.
| Ovarian | Molar Concentration (nM) | Activity Concentration (10−2 MBq/mL) | Cellular Activity Uptake (10−2 Bq/cell) | Nuclear Dose (Gy) |
|---|---|---|---|---|
| Cancer Cell Line | EC50 95% CI | EC50 95% CI | EC50 95% CI | D 50 |
| ID8 | 2.3 (1.8–2.9) | 44 (36–53) | 3.9 (3.3–4.6) | 9.2±1.0 |
| OVCAR8 | 0.17 (0.13–0.24) | 7.0 (6.2–8.0) | 1.8 (1.6–2.0) | 4.3±0.3 |
| OVCAR8 PARP1-KO | 0.46 (0.34–0.62) | 20 (15–28) | 2.0 (1.5–2.7) | 3.4±0.6 |
| UWB1.289 | 0.27 (0.21–0.36) | 6.1 (4.4–8.6) | 2.7 (2.0–3.7) | 3.9±0.8 |
| UWB1.289+BRCA1 | 0.30 (0.22–0.41) | 6.8 (5.5–8.3) | 2.3 (1.9–2.8) | 3.3±0.4 |
4.4. PET imaging, biodistribution, and dosimetry
Partial volume correction phantom results and recovery coefficient calibration curves are summarized in Figure S4. Significant [76Br]RD1 uptake in the liver (brown arrow) occurred within 5 minutes p.i. and the gallbladder (blue arrow) between 1–4 hours p.i. (Figure 3A). The 48 h p.i. in vivo PET quantification was compared to the ex vivo biodistribution measurements (Figure 3B, Table S2). Time activity curves showed biphasic blood clearance with a calculated α-phase half-life of 1.5 min (95% CI: 1.2–1.9 min) and -phase half-life of 85 min (95% CI: 67–107 min, Figure 3C). Based on the organ-averaged PET results from 1–48 h, 85±10% of [77Br]RD1 was excreted from 1–24 hours and 69±15% of that remaining excreted from 24–48 hours. Dosimetry analyses of [76/77Br]RD1 following injection in mice and humans are summarized in Figure 4 and Table S3.
Figure 3:
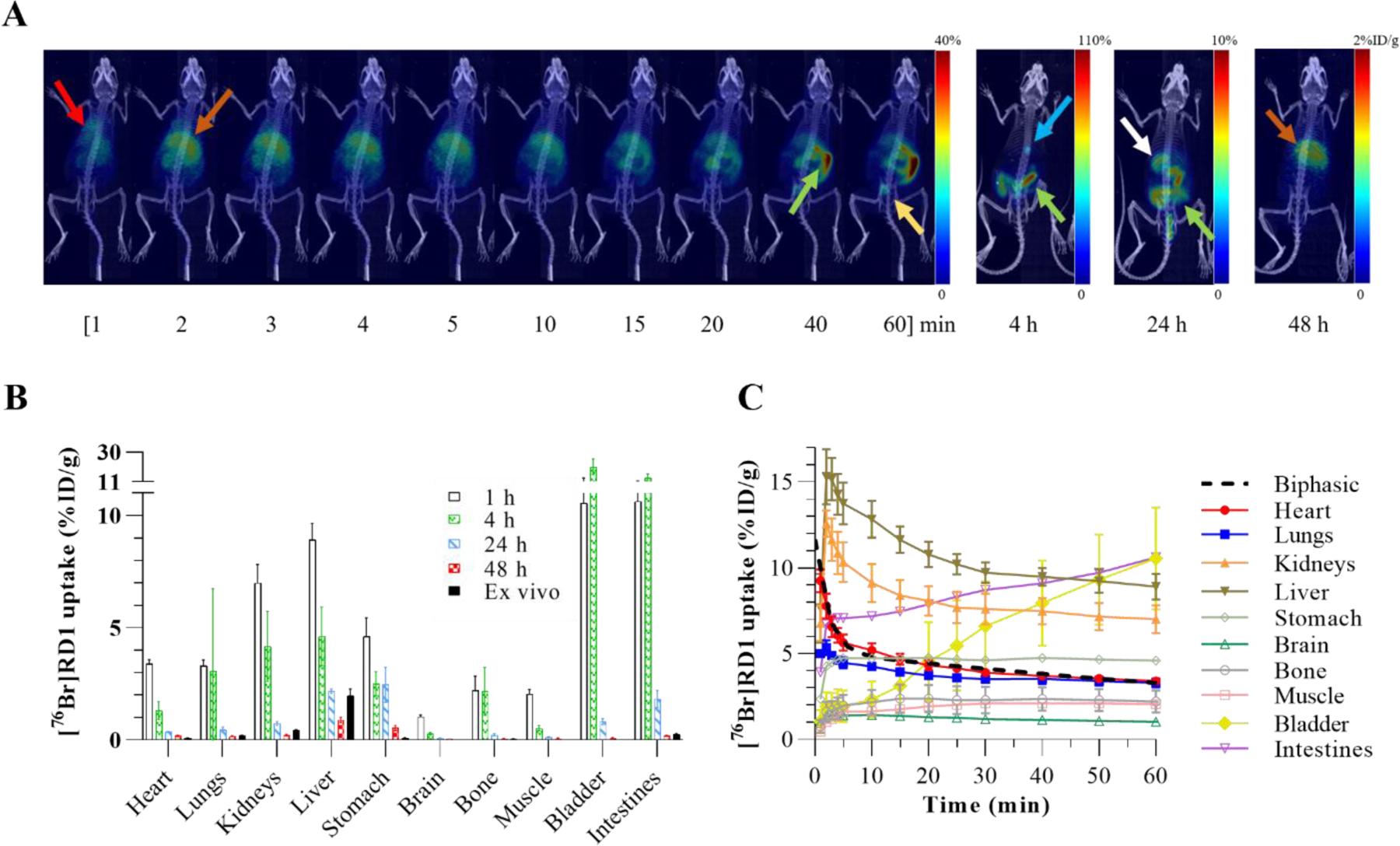
(A) PET maximum intensity projection (MIP) images of a representative mouse injected intravenously with [76Br]RD1. Red arrow: heart; brown arrow: liver; green arrow: intestines; yellow arrow: bladder; blue arrow: gallbladder; white arrow: stomach contents. (B) A comparison between the in vivo measurements at four time points p.i. and the ex vivo measurements (n=4). Ex vivo intestinal uptake was the average measurement of the small intestine, large intestine, and enteric contents. (C) Time activity curves based on ROI analysis of dynamic PET images.
Figure 4:
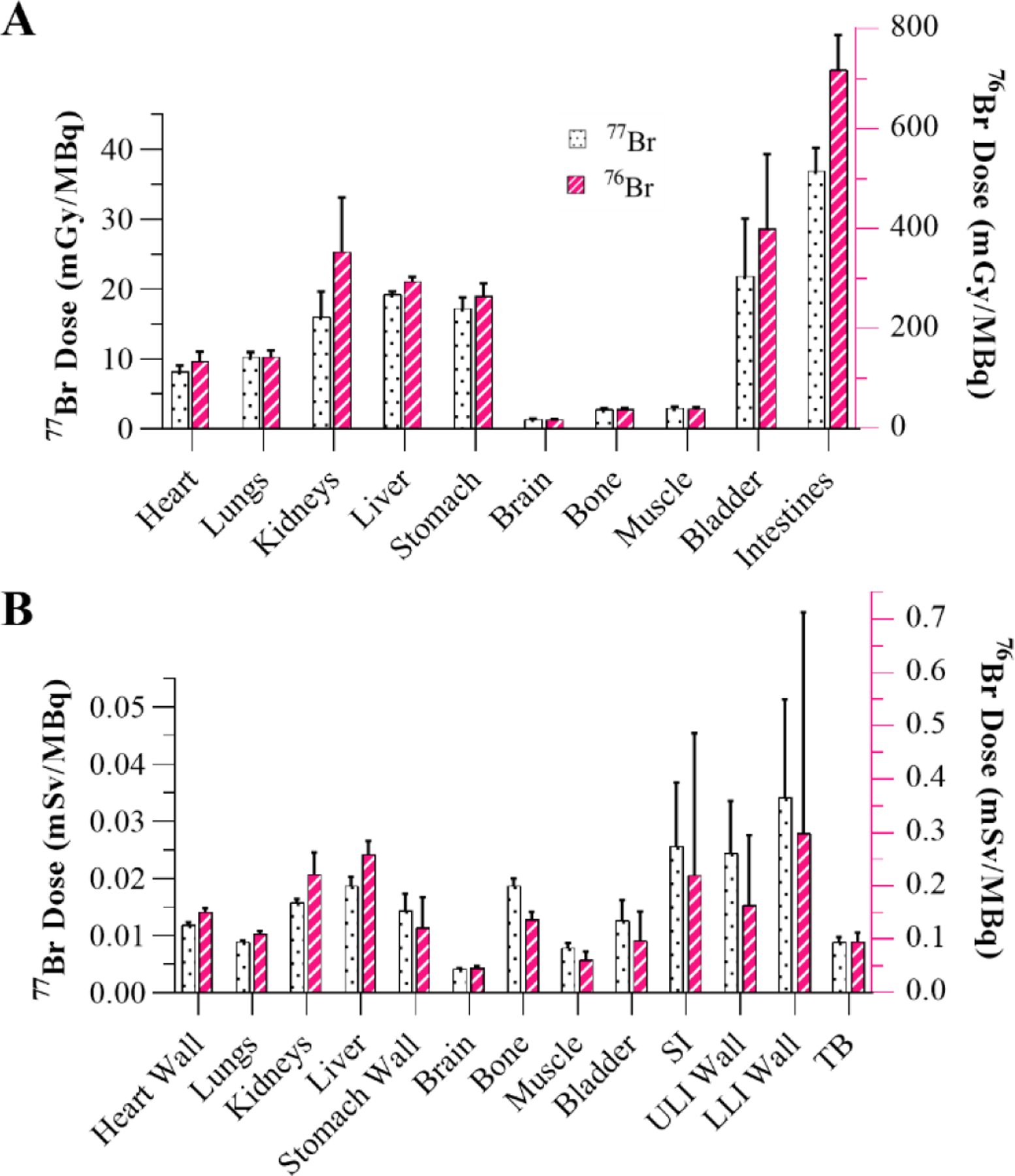
Dosimetry analysis showing (A) organ-level absorbed dose following [76/77Br]RD1 administration in mice using RAPID and (B) equivalent dose in female humans using OLINDA. SI: small intestine; ULI: upper large intestine; LLI: lower large intestine; TB: total body.
5. Discussion
Radiopharmacological and toxicological properties of a PARP-targeted MAe emitting radiopharmaceutical [77Br]RD1 were measured in ovarian cancer cell lines. [77Br]RD1 demonstrated specific, blockable uptake with nanomolar binding affinity and attomoles of binding sites per cell. The binding affinity, represented by the equilibrium dissociation constant , ranged from 6–30 nM with cell line variability potentially due to differences in relative expression levels of the various PARP isoforms. The number of specific binding sites per cell, represented by , ranged from 0.8 – 11 amol/cell (0.5–6.6 million sites per cell). Radiotracer studies utilizing structurally analogous [125I]KX1 have shown to be strongly positively correlated to PARP1 expression, as determined by Western blot analysis [57]. Supporting this, the of the OVCAR8 cell line (1.6 amol/cell) is a factor of two higher than the analogous PARP1 knock out cell line (0.83 amol/cell).
The EC50 (in nM) of [77Br]RD1 was thousands of times lower compared to rucaparib for OVCAR8 (2,400x), OVCAR8 PARP1-KO (6,700x) and UWB1.289+BRCA1 (3,000x), and 70x lower for UWB1.289 [50]. These results show the radiotoxicity of [77Br]RD1 is significantly greater than the chemotoxicity of structurally-related rucaparib in ovarian cancer. Compared to the radioiodinated MAe-emitting analog, [125I]KX1, the EC50 (in MBq/mL) of [77Br]RD1 is 5x and 4x higher for OVCAR8 and OVCAR8 PARP1-KO, respectively, and the D50 (Gy) is approximately 3x higher for OVCAR8 and OVCAR8 PARP1-KO, 1.7x higher for UWB1.289, and approximately the same for UWB1.289+BRCA1 [35]. This difference may be due to the smaller average number of MAes emitted with 77Br (6–7 MAe/decay) compared with 125I (~23 MAe/decay) [42]. When measured in terms of activity concentration, the OVCAR8 PARP1-KO cell line was significantly more [77Br]RD1-tolerant (P=0.0140) compared to OVCAR8 (Figure 2D), with EC50 of 0.20 (95% CI: 0.15–0.28) MBq/mL and 0.070 (0.062–0.080) MBq/mL (Table 2), respectively. This agrees with Makvandi, et al. [57], which demonstrates that PARP expression is essential to the efficacy of MAe-emitting PARP inhibitors, where lower values were associated with higher EC50 values. However, after accounting for the cell line’s lower number of specific binding sites, this differential sensitivity was no longer observed (P=0.6228), with EC50 of 0.020 (0.015–0.027) Bq/cell and 0.018 (0.016–0.020) Bq/cell for OVCAR8 PARP1-KO and OVCAR8 cell lines, respectively (Figure 2F, Table 2). These results indicate that the PARP-expression dependence of [77Br]RD1 radiotoxicity is driven by differences in specific binding site expression, in which the loss of PARP1 did not change the radiosensitivity of the cancer cell line.
[77Br]RD1 had similar cytotoxic effects regardless of BRCA1 gene expression (Figure 2D–E). The chemotherapeutic effect of rucaparib and other PARP inhibitors results in increased cytotoxicity in homologous recombination deficient BRCA1 mutated cells [50], [58]. When adding the radiotherapeutic effect of the MAe emitting isotope 77Br, this biomarker-dependency was no longer apparent—the [77Br]RD1 showed similar cytotoxicity in UWB1.289 cells even with upregulated BRCA1 expression (Figure 2D–E) with EC50 values of 0.061 (0.044–0.086) MBq/mL and 0.068 (0.055–0.083) MBq/mL for UWB1.289 and UWB1.289+BRCA cell lines, respectively.
Hepatobiliary and renal excretion pathways were observed for [76Br]RD1 (Figure 3A) in agreement with other radiolabeled PARP inhibitors [23], [36], [59], [60]. The excretion rate of [76Br]RD1 (70–85% per day) was significantly faster than that of [82Br]bromide (5% per day) [45], supporting the hypothesis that minimal debromination occurred in vivo. Future metabolite analyses are necessary to confirm this. Terminal in vivo and ex vivo biodistributions differed for organs with contents that were removed for ex vivo measurement, e.g., blood and enteric contents, and in organs with inhomogeneous radiopharmaceutical distribution, e.g., liver. At 48 h, [76Br]RD1 resides mainly in the liver region. This prolonged retention of [77Br]RD1 may cause toxicity in the liver, with dose estimates of mGy/MBq and mSv/GBq in mice and humans, respectively (Fig. 4, Table S3). The liver dose limit for external-beam radiotherapy is 30 Gy, which would limit the [77Br]RD1 activity to approximately 1,600 GBq (Table S4). However, liver uptake of an olaparib-based fluorescent PARP inhibitor was observed to be cytoplasmic, significantly decreasing the equivalent dose to liver cell DNA compared to the nuclear uptake observed in tumor cells [27]. In mice, stomach and intestinal absorbed doses were and mGy/MBq, respectively, for [76Br]RD1. However, as mentioned above, the ex vivo and in vivo differences indicate that most of the activity and thus dose is in the contents of the organ. Human dose estimates were compared to organ dose reported for 18F-FTT [16]. All [76Br]RD1 human equivalent organ dose estimates were times larger compared to PARP-targeted imaging agent, 18F-FTT, except for lower large intestine (20 times larger). Dose limits for the reported organs [61] show that the dose limiting organ is the bone marrow, which limits [77Br]RD1 injected activity to approximately 110 GBq.
6. Conclusion
The cytotoxicity of [77Br]RD1 was found to be PARP expression dependent and BRCA1 status independent. These results together indicate the radiotherapeutic effect of the MAe emitting radionuclide is driving the cytotoxicity of [77Br]RD1 beyond the PARP inhibitor chemotherapeutic effect. This study offers insights into the fundamental radiation biology of low energy electron emitting radiopharmaceuticals targeting the nuclear DNA damage response system. Future imaging and therapeutic studies should be conducted with further refined in vivo tumor models to show the efficacy of [76Br]RD1 as a theranostic agent for ovarian cancer.
Supplementary Material
Acknowledgements
We thank the staff of the University of Wisconsin Carbone Cancer Center (UWCCC) Biostatistics Shared Resource for their valuable contributions to this research. We also thank the staff of the UWCCC Small Animal Imaging & Radiotherapy Facility for their technical support. Shared research services at the UWCCC are supported by Cancer Center Support Grant P30 CA014520.
Funding
This research is supported by the U.S. Department of Energy Isotope Program, managed by the Office of Science for Isotope R&D and Production, grant number DE-SC0020960 and the U.S. Department of Defense Ovarian Cancer Research Program Pilot Award, grant number W81XWH2110351. J.C.M. is supported by the National Cancer Institute of the National Institutes of Health under Award Number T32CA009206. The project was supported by the Clinical and Translational Science Award (CTSA) program, through the NIH National Center for Advancing Translational Sciences (NCATS), grant UL1TR002373. B.P.B. is supported by NCI P01 CA250972-01 and NCI U01 CA233102-01. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors have no competing interests to report.
Declaration of interests
Preclinical studies of a PARP targeted, Meitner-Auger emitting, theranostic radiopharmaceutical for metastatic ovarian cancer.
SLV Hoffman, JC Mixdorf, O Kwon, TR Johnson, M Makvandi, H Lee, E Aluicio-Sarduy, TE Barnhart, JJ Jeffery, MS Patankar, JW Engle, BP Bednarz, and PA Ellison
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
10. References
- [1].Siegel RL, Miller KD, Wagle NS, and Jemal A, ‘Cancer statistics, 2023.’, CA Cancer J Clin, vol. 73, no. 1, pp. 17–48, Jan. 2023, doi: 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- [2].Jemal A, Thomas A, Murray T, and Thun M, ‘Cancer Statistics, 2002’, CA Cancer J Clin, vol. 52, no. 1, pp. 23–47, Jan. 2002, doi: 10.3322/canjclin.52.1.23. [DOI] [PubMed] [Google Scholar]
- [3].Marth C, Reimer D, and Zeimet AG, ‘Front-line therapy of advanced epithelial ovarian cancer: standard treatment’, Annals of Oncology, vol. 28, pp. viii36–viii39, Nov. 2017, doi: 10.1093/annonc/mdx450. [DOI] [PubMed] [Google Scholar]
- [4].Zhang C, Xu C, Gao X, and Yao Q, ‘Platinum-based drugs for cancer therapy and anti-tumor strategies’, Theranostics, vol. 12, no. 5, pp. 2115–2132, 2022, doi: 10.7150/thno.69424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Colombo PE, Fabbro M, Theillet C, Bibeau F, Rouanet P, and Ray-Coquard I, ‘Sensitivity and resistance to treatment in the primary management of epithelial ovarian cancer’, Crit Rev Oncol Hematol, vol. 89, no. 2, pp. 207–216, Feb. 2014, doi: 10.1016/j.critrevonc.2013.08.017. [DOI] [PubMed] [Google Scholar]
- [6].The Cancer Genome Atlas Research Network, ‘Integrated genomic analyses of ovarian carcinoma’, Nature, vol. 474, no. 7353, pp. 609–615, Jun. 2011, doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ledermann JA, Drew Y, and Kristeleit RS, ‘Homologous recombination deficiency and ovarian cancer’, Eur J Cancer, vol. 60, pp. 49–58, Jun. 2016, doi: 10.1016/j.ejca.2016.03.005. [DOI] [PubMed] [Google Scholar]
- [8].Prakash R, Zhang Y, Feng W, and Jasin M, ‘Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins’, Cold Spring Harb Perspect Biol, vol. 7, no. 4, 2015, doi: 10.1101/cshperspect.a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lord CJ and Ashworth A, ‘PARP inhibitors: Synthetic lethality in the clinic’, Science (1979), vol. 355, pp. 1152–1158, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yi M, Dong B, Qin S, Chu Q, Wu K, and Luo S, ‘Advances and perspectives of PARP inhibitors’, Exp Hematol Oncol, vol. 8, no. 29, pp. 1–12, Nov. 2019, doi: 10.1186/s40164-019-0154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sonnenblick A, de Azambuja E, Azim HA Jr, and Piccart M, ‘An update on PARP inhibitors—moving to the adjuvant setting’, Nat Rev Clin Oncol, vol. 12, pp. 27–41, 2015. [DOI] [PubMed] [Google Scholar]
- [12].Lin Q et al. , ‘PARP inhibitors as maintenance therapy in newly diagnosed advanced ovarian cancer: a meta-analysis’, BJOG, vol. 128, no. 3, pp. 485–493, Feb. 2020, doi: 10.1111/1471-0528.16450. [DOI] [PubMed] [Google Scholar]
- [13].Zandarashvili L et al. , ‘Structural basis for allosteric PARP-1 retention on DNA breaks’, Science (1979), vol. 368, no. 6486, Apr. 2020, doi: 10.1126/SCIENCE.AAX6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ambur Sankaranarayanan R, Kossatz S, Weber W, Beheshti M, Morgenroth A, and Mottaghy FM, ‘Advancements in PARP1 targeted nuclear imaging and theranostic probes’, J Clin Med, vol. 9, no. 7, 2020, doi: 10.3390/jcm9072130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].McDonald ES et al. , ‘Positron emission tomography imaging of poly-(adenosine diphosphate-ribose) polymerase 1 expression in breast cancer: A nonrandomized clinical trial’, JAMA Oncol, vol. 6, no. 6, pp. 921–923, Jun. 2020, doi: 10.1001/jamaoncol.2020.0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Michel LS et al. , ‘PET of poly (ADP-ribose) polymerase activity in cancer: Preclinical assessment and first in-human studies’, Radiology, vol. 282, no. 2, pp. 453–463, Mar. 2017, doi: 10.1148/radiol.2016161929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Young RJ et al. , ‘Preclinical and first-in-human-brain-cancer applications of [18F]poly (ADP-ribose) polymerase inhibitor PET/MR’, Neurooncol Adv, vol. 2, no. 1, Jan. 2020, doi: 10.1093/noajnl/vdaa119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Puentes LN, Makvandi M, and Mach RH, ‘Molecular imaging: PARP-1 and beyond’, Journal of Nuclear Medicine, vol. 62, no. 6, pp. 765–770, Jun. 2021, doi: 10.2967/jnumed.120.243287. [DOI] [PubMed] [Google Scholar]
- [19].McDonald ES et al. , ‘In vivo visualization of PARP inhibitor pharmacodynamics’, JCI Insight, vol. 6, no. 8, Apr. 2021, doi: 10.1172/jci.insight.146592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Carney B et al. , ‘Non-invasive PET imaging of PARP1 expression in glioblastoma models’, Mol Imaging Biol, vol. 18, no. 3, pp. 386–392, Jun. 2016, doi: 10.1007/s11307-015-0904-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chan CY, Tan KV, and Cornelissen B, ‘PARP inhibitors in cancer diagnosis and therapy’, Clinical Cancer Research, vol. 27, no. 6, pp. 1585–1594, Mar. 2021, doi: 10.1158/1078-0432.CCR-20-2766. [DOI] [PubMed] [Google Scholar]
- [22].Chen Z, Destro G, Guibbal F, Chan CY, Cornelissen B, and Gouverneur V, ‘Copper-Mediated Radiosynthesis of [18F]Rucaparib’, Org Lett, vol. 23, no. 18, pp. 7290–7294, Sep. 2021, doi: 10.1021/acs.orglett.1c02770. [DOI] [PubMed] [Google Scholar]
- [23].Chan CY et al. , ‘Imaging PARP with [18F]rucaparib in pancreatic cancer models’, Eur J Nucl Med Mol Imaging, vol. 49, no. 11, pp. 3668–3678, 2022, doi: 10.1007/s00259-022-05835-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bowden GD et al. , ‘DoE Optimization Empowers the Automated Preparation of Enantiomerically Pure [18F]Talazoparib and its In Vivo Evaluation as a PARP Radiotracer’, J Med Chem, vol. 64, no. 21, pp. 15690–15701, Nov. 2021, doi: 10.1021/acs.jmedchem.1c00903. [DOI] [PubMed] [Google Scholar]
- [25].Zhou D et al. , ‘Radiosynthesis and Evaluation of Talazoparib and Its Derivatives as PARP-1-Targeting Agents’, Biomedicines, vol. 9, no. 5, 2021, doi: 10.3390/biomedicines9050565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wilson TC et al. , ‘PET Imaging of PARP Expression Using 18F-Olaparib’, Journal of Nuclear Medicine, vol. 60, no. 4, p. 504, Apr. 2019, doi: 10.2967/jnumed.118.213223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Guibbal F et al. , ‘Manual and automated Cu-mediated radiosynthesis of the PARP inhibitor [18F]olaparib’, Nat Protoc, vol. 15, no. 4, pp. 1525–1541, 2020, doi: 10.1038/s41596-020-0295-7. [DOI] [PubMed] [Google Scholar]
- [28].Jannetti SA et al. , ‘PARP-1-targeted radiotherapy in mouse models of glioblastoma’, Journal of Nuclear Medicine, vol. 59, no. 8, pp. 1225–1233, Aug. 2018, doi: 10.2967/jnumed.117.205054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Makvandi M et al. , ‘Targeting PARP-1 with Alpha-Particles Is Potently Cytotoxic to Human Neuroblastoma in Preclinical Models’, Mol Cancer Ther, vol. 18, no. 7, pp. 1195–1204, Jul. 2019, doi: 10.1158/1535-7163.MCT-18-0837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dabagian H et al. , ‘PARP Targeted Alpha-Particle Therapy Enhances Response to PD-1 Immune-Checkpoint Blockade in a Syngeneic Mouse Model of Glioblastoma’, ACS Pharmacol Transl Sci, vol. 4, no. 1, pp. 344–351, Feb. 2021, doi: 10.1021/acsptsci.0c00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Makvandi M et al. , ‘Pre-clinical investigation of astatine-211-parthanatine for high-risk neuroblastoma’, Commun Biol, vol. 5, no. 1, p. 1260, 2022, doi: 10.1038/s42003-022-04209-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee H et al. , ‘PARP-1-Targeted auger emitters display high-LET cytotoxic properties in vitro but show limited therapeutic utility in solid tumor models of human neuroblastoma’, Journal of Nuclear Medicine, vol. 61, no. 6, pp. 850–856, Jun. 2020, doi: 10.2967/jnumed.119.233965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wilson T et al. , ‘PARP-Targeted Auger Therapy in p53 Mutant Colon Cancer Xenograft Mouse Models’, Mol Pharm, vol. 18, no. 9, pp. 3418–3428, Sep. 2021, doi: 10.1021/acs.molpharmaceut.1c00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sankaranarayanan RA et al. , ‘Auger Emitter Conjugated PARP Inhibitor for Therapy in Triple Negative Breast Cancers: A Comparative In-Vitro Study’, Cancers (Basel), vol. 14, no. 1, Jan. 2022, doi: 10.3390/cancers14010230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Riad A et al. , ‘PARP Theranostic Auger Emitters Are Cytotoxic in BRCA Mutant Ovarian Cancer and Viable Tumors from Ovarian Cancer Patients Enable Ex-Vivo Screening of Tumor Response’, Molecules, vol. 25, no. 24, Dec. 2020, doi: 10.3390/MOLECULES25246029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sreekumar S et al. , ‘Preclinical efficacy of a PARP-1 targeted Auger-emitting radionuclide in prostate cancer’, Int J Mol Sci, vol. 24, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pirovano G et al. , ‘Targeted brain tumor radiotherapy using an auger emitter’, Clinical Cancer Research, vol. 26, no. 12, pp. 2871–2881, Jun. 2020, doi: 10.1158/1078-0432.CCR-19-2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Salinas B et al. , ‘Radioiodinated PARP1 tracers for glioblastoma imaging’, EJNMMI Res, vol. 5, no. 1, p. 46, 2015, doi: 10.1186/s13550-015-0123-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ku A, Facca VJ, Cai Z, and Reilly RM, ‘Auger electrons for cancer therapy – a review’, EJNMMI Radiopharm Chem, vol. 4, no. 1, Dec. 2019, doi: 10.1186/s41181-019-0075-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Destro G et al. , ‘A radioiodinated rucaparib analogue as an Auger electron emitter for cancer therapy’, Nucl Med Biol, vol. 116–117, p. 108312, Jan. 2023, doi: 10.1016/j.nucmedbio.2022.108312. [DOI] [PubMed] [Google Scholar]
- [41].Syed YY, ‘Rucaparib: First Global Approval’, Drugs, vol. 77, no. 5, pp. 585–592, Apr. 2017, doi: 10.1007/s40265-017-0716-2. [DOI] [PubMed] [Google Scholar]
- [42].Eckerman K and Endo A, ‘Nuclear decay data for dosimetric calculations. ICRP Publication 107’, Annals ICRP, vol. 38, no. 3, pp. 1–123, 2008. [DOI] [PubMed] [Google Scholar]
- [43].Cornelissen B and Vallis KA, ‘Targeting the nucleus: An overview of Auger-electron radionuclide therapy’, Curr Drug Discov Technol, vol. 7, p. 263, 2010. [DOI] [PubMed] [Google Scholar]
- [44].Rowland DJ, McCarthy TJ, and Welch MJ, ‘Radiobromine for Imaging and Therapy’, in Handbook of Radiopharmaceuticals, John Wiley & Sons, Ltd, 2005, pp. 441–465. doi: 10.1002/0470846380.ch14. [DOI] [Google Scholar]
- [45].Pavelka S, Babický A, Vobecký M, Lener J, and Švandová E, ‘Bromide kinetics and distribution in the rat. I.’, Biol Trace Elem Res, vol. 76, pp. 57–66, 2000, doi: 10.1385/BTER:76:1:57. [DOI] [PubMed] [Google Scholar]
- [46].Ribeiro MJ et al. , ‘Comparison of fluorine-18 and bromine-76 imaging in positron emission tomography’, Eur J Nucl Med, vol. 26, no. 7, 1999. [DOI] [PubMed] [Google Scholar]
- [47].Welch MJ and Mcelvany KD, ‘Radionuclides of bromine for use in biomedical studies’, Radiochim Acta, vol. 34, pp. 41–46, 1983. [Google Scholar]
- [48].Ellison PA et al. , ‘Improved production of 76Br, 77Br and 80mBr via CoSe cyclotron targets and vertical dry distillation’, Nucl Med Biol, vol. 80–81, pp. 32–36, 2020, doi: 10.1016/j.nucmedbio.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mixdorf JC, Hoffman SLV, Aluicio-Sarduy E, Barnhart TE, Engle JW, and Ellison PA, ‘Copper-mediated radiobromination of (hetero)aryl boronic pinacol esters’, Journal of Organic Chemistry, no. 88, pp. 2089–2094, Feb. 2023, doi: 10.1021/acs.joc.2c02420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Makvandi M et al. , ‘A PET imaging agent for evaluating PARP-1 expression in ovarian cancer’, Journal of Clinical Investigation, vol. 128, no. 5, pp. 2116–2126, May 2018, doi: 10.1172/JCI97992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Vaziri B et al. , ‘MIRD pamphlet No. 25: MIRDcell V2.0 software tool for dosimetric analysis of biologic response of multicellular populations’, Journal of Nuclear Medicine, vol. 55, no. 9, pp. 1557–1564, Sep. 2014, doi: 10.2967/jnumed.113.131037. [DOI] [PubMed] [Google Scholar]
- [52].Besemer AE, Yang YM, Grudzinski JJ, Hall LT, and Bednarz BP, ‘Development and validation of RAPID: A patient-specific Monte Carlo three-dimensional internal dosimetry platform’, Cancer Biother Radiopharm, vol. 33, no. 4, pp. 155–165, May 2018, doi: 10.1089/cbr.2018.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Perkins ST, Cullen DE, Chen MH, Rathkopf J, Scofield J, and Hubbell JH, ‘Tables and graphs of atomic subshell and relaxation data derived from the LLNL Evaluated Atomic Data Library (EADL), Z = 1−-100’, United States, 1991. doi: 10.2172/10121422. [DOI] [Google Scholar]
- [54].Stabin MG, Sparks RB, and Crowe E, ‘OLINDA/EXM: The Second-Generation Personal Computer Software for Internal Dose Assessment in Nuclear Medicine’, Journal of Nuclear Medicine, vol. 46, no. 6, p. 1023, Jun. 2005, [Online]. Available: http://jnm.snmjournals.org/content/46/6/1023.abstract [PubMed] [Google Scholar]
- [55].Marsh IR et al. , ‘Preclinical pharmacokinetics and dosimetry studies of 124I/131I-CLR1404 for treatment of pediatric solid tumors in murine xenograft models’, Journal of Nuclear Medicine, vol. 60, no. 10, pp. 1414–1420, Oct. 2019, doi: 10.2967/jnumed.118.225409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Valentin J, ‘Basic anatomical and physiological data for use in radiological protection: reference values: ICRP Publication 89’, Ann ICRP, vol. 32, no. 3, pp. 1–277, 2002, doi: 10.1016/S0146-6453(03)00002-2. [DOI] [PubMed] [Google Scholar]
- [57].Makvandi M et al. , ‘A radiotracer strategy to quantify PARP-1 expression in vivo provides a biomarker that can enable patient selection for PARP inhibitor therapy’, Cancer Res, vol. 76, no. 15, pp. 4516–4524, Aug. 2016, doi: 10.1158/0008-5472.CAN-16-0416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Makvandi M et al. , ‘A radiotracer strategy to quantify PARP-1 expression in vivo provides a biomarker that can enable patient selection for PARP inhibitor therapy’, Cancer Res, vol. 76, no. 15, pp. 4516–4524, Aug. 2016, doi: 10.1158/0008-5472.CAN-16-0416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhou D et al. , ‘Synthesis, [18F] radiolabeling, and evaluation of poly (ADP-ribose) polymerase-1 (PARP-1) inhibitors for in vivo imaging of PARP-1 using positron emission tomography’, Bioorg Med Chem, vol. 22, no. 5, pp. 1700–1707, Mar. 2014, doi: 10.1016/j.bmc.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhou D et al. , ‘Preliminary evaluation of a novel 18F-labeled PARP-1 ligand for PET imaging of PARP-1 expression in prostate cancer’, Nucl Med Biol, vol. 66, pp. 26–31, 2018, doi: 10.1016/j.nucmedbio.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wahl RL et al. , ‘Normal-Tissue Tolerance to Radiopharmaceutical Therapies, the Knowns and the Unknowns’, Journal of Nuclear Medicine, vol. 62, no. Supplement 3, p. 23S, Dec. 2021, doi: 10.2967/jnumed.121.262751. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.