

Abstract
Transcriptional coregulators have been widely pursued as targets for disrupting oncogenic gene regulatory programs. However, many proteins in this target class are universally essential for cell survival, which limits their therapeutic window. Here, we unveil a genetic interaction between histone deacetylase 1 (HDAC1) and HDAC2, wherein each paralog is synthetically lethal with hemizygous deletion of the other. This collateral synthetic lethality is caused by recurrent chromosomal translocations that occur in diverse solid and hematological malignancies, including neuroblastoma and multiple myeloma. Using genetic disruption or dTAG-mediated degradation, we show that targeting HDAC2 suppresses the growth of HDAC1-deficient neuroblastoma in vitro and in vivo. Mechanistically, we find that targeted degradation of HDAC2 in these cells prompts the degradation of several members of the nucleosome remodeling and deacetylase (NuRD) complex, leading to diminished chromatin accessibility at HDAC2/NuRD-bound sites of the genome and impaired control of enhancer-associated transcription. Furthermore, we reveal that several of the degraded NuRD complex subunits are dependencies in neuroblastoma and multiple myeloma, providing motivation to develop paralog-selective HDAC1 or HDAC2 degraders that could leverage HDAC1/2 synthetic lethality to target NuRD vulnerabilities. Altogether, we identify HDAC1/2 collateral synthetic lethality as a potential therapeutic target and reveal an unexplored mechanism for targeting NuRD-associated cancer dependencies.
Editor summary:
Here, the authors show that HDAC1 and HDAC2 genetically interact, with each paralog being synthetically lethal with hemizygous deletion of the other. Mechanistically, HDAC1/2 co-deficiency leads to degradation of the NuRD complex, decreased chromatin accessibility and aberrant enhancer-based interactions.
Introduction:
Currently, 4 HDAC inhibitors are approved for use as anti-cancer agents in humans1, but the vast majority of clinical development has failed due to low response rates and poor tolerability2. Notably, all but one of the currently approved HDAC inhibitors are broadly active against most or all HDAC enzymes, which likely contributes to their toxicities2. The other, romidepsin (FK228), inhibits Class I HDACs, including HDAC1, HDAC2, HDAC3, and HDAC8. These enzymes are predominantly localized to the nucleus and catalyze zinc-dependent removal of acyl groups from the ε-amino lysine of various histone and non-histone substrates3. While romidepsin is approved for the treatment of cutaneous T cell lymphoma, it was recently withdrawn for peripheral T cell lymphoma after failing to improve progression-free survival in a confirmatory Phase III trial4. Other class I HDAC inhibitors, like the ortho-aminoanilides that selectively inhibit HDAC1/2/3, are available as preclinical tools, but the pan-essentiality of these targets remains a persistent challenge for drug development2,5,6.
HDAC1 and HDAC2 are present in multiple protein complexes, including NuRD, Sin3A, CoREST, and MiDAC3,7. These paralogs, which were originally discovered through target identification studies for the HDAC inhibitor, trapoxin8, play key roles as transcriptional co-regulators in normal development as well as tumorigenesis3,9. While a limited number of non-redundant roles have been identified for both paralogs6,10, simultaneous deletion of the two is detrimental to the growth and survival of most, if not, all cell types11–13. The widespread importance of HDAC1/2 has been shown extensively in normal cells and tissues by mouse genetics13–17 and validated in cancer cell lines using chemical and genetic approaches18–20. Moreover, simultaneous deletion of HDAC1/2 has reliably scored among the top hits in pooled CRISPR/Cas9-based paralog knockout screens that measure cell fitness21,22. The pan-essential nature of these HDACs explains, at least in part, the difficulty of achieving a therapeutic window in patients, even with Class-I-selective HDAC inhibitors. However, the redundant effects of HDAC1 and HDAC2 may represent an opportunity for identifying a cancer-specific synthetic lethality.
In cancer biology, synthetic lethality is used as a framework to identify genes that are selectively essential for cell survival in the presence of particular tumor-associated alterations23. As drug targets, these gene products promise a favorable therapeutic window since many or most normal cell types do not require them for survival. The study of synthetic lethality has historically depended on hypothesis-driven research that is well demonstrated by the classic example of PARP inhibition in BRCA1/2-mutated cancers24. However, modern tools for genetic loss-of-function screens in mammalian cells have enabled the discovery of synthetic lethal relationships at scale. Pooled genetic screens have, for example, revealed therapeutically compelling synthetic lethalities between SMARCA2 and SMARCA425–27, CBP and p30028, STAG1 and STAG229, and many other paralogs with redundant or semi-redundant function23. Surveying paralogous proteins by combinatorial genetic screens, or examining existing dependency maps for genetic interactions between paralogs, could expedite the identification of actionable synthetic lethalities in cancer21,30,31.
In this study, we systematically explored the Cancer Dependency Map32,33 for Class I HDAC dependencies across dozens of cancer lineages. Through this effort, we identified a requirement for HDAC1 or HDAC2 in cancers harboring hemizygous deletions of the other paralog. HDAC1 is located in proximity to a region of chromosome 1p that is deleted in approximately 20% of neuroblastoma34–37. Likewise, HDAC2, is located within chromosome 6q, which experiences deletions in nearly 40% of lymphoid malignancies38–41. Using complementary genetic and pharmacological approaches, we investigated the effects of targeting this synthetic lethality and identified a link between HDAC1/2 abundance and NuRD complex structure and function.
Results:
Lineage selective HDAC1 and HDAC2 cancer dependencies
To systematically evaluate the requirement for individual class I HDACs in cancer, we extracted scaled gene-level dependency scores (Chronos) from the 21Q4 release of the Cancer Dependency Map (DepMap) (Fig. 1a). In contrast to HDAC3, which is indiscriminately required for cell survival, most cancer lineages are not affected by loss of HDAC1 or HDAC2 (Fig. 1a). This is consistent with the known redundancy between HDAC1 and HDAC2, whereby loss of one paralog is compensated by increased abundance of the other13,14,17. However, we noticed that multiple myeloma cell lines (plasma cell lineage) are highly dependent on HDAC1, whereas neuroblastoma cell lines (peripheral nervous system) are highly dependent on HDAC2 (Fig. 1a and Extended Data Fig. 1a). In neuroblastoma, HDAC2 registers as one of the most selective dependencies, comparable to the core regulatory circuitry (CRC) transcription factors (TF) that are known to be selectively required in this disease42,43 (Fig. 1b and Extended Data Fig. 1b) and consistent with the nomination of HDAC2 as a neuroblastoma dependency by the Pediatric Cancer DepMap44,45. Likewise, a significantly higher dependency on HDAC1 is observed in multiple myeloma compared to other cancer types (Fig. 1c).
Figure 1: HDAC1 and HDAC2 are selective dependencies in multiple myeloma and neuroblastoma respectively.
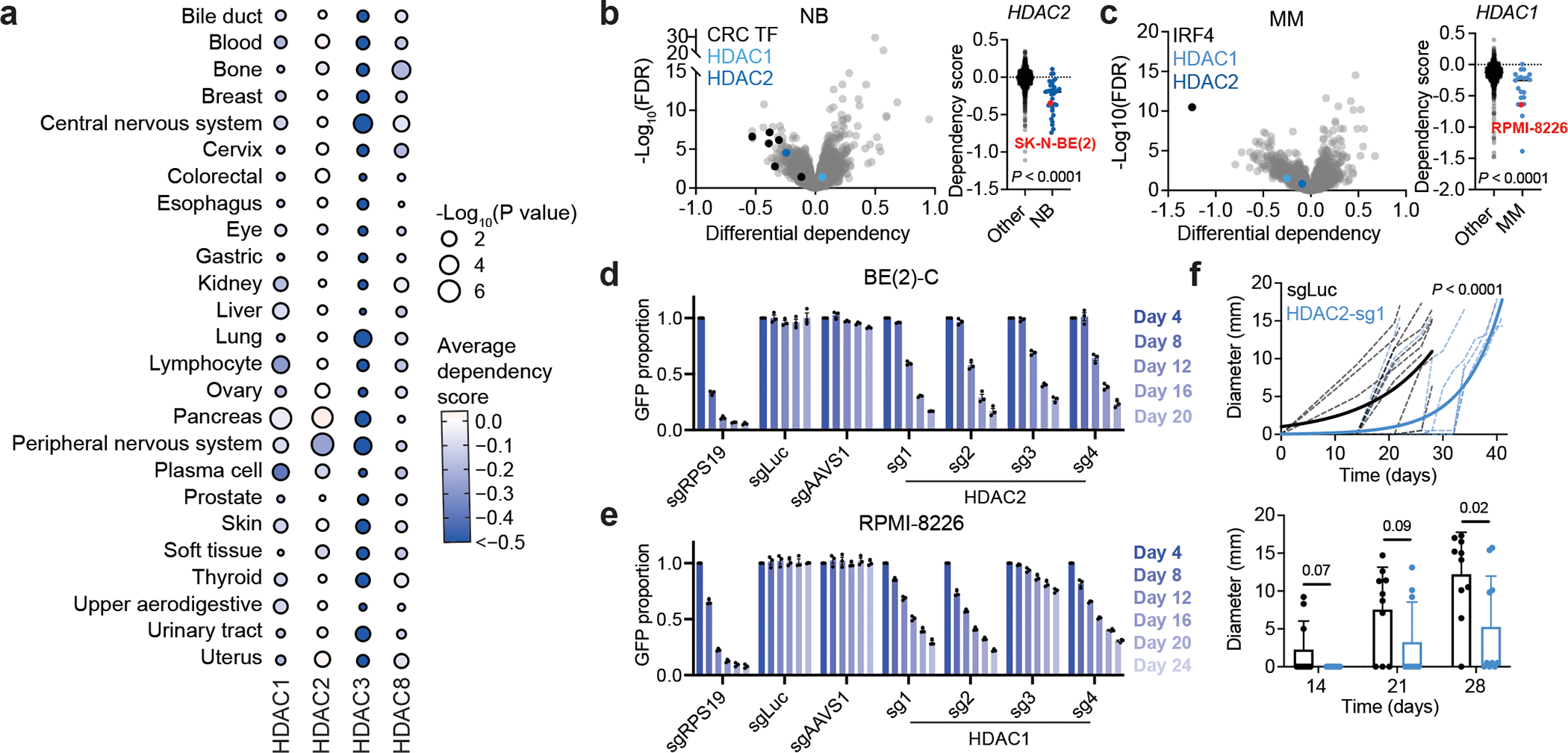
a, Average Chronos gene effect scores (dependency scores) shown for each cancer lineage (circle color). P value (circle size) given by two-tailed Student’s t-test comparing cell lines within each lineage to all other cell lines. b, Left, volcano plot of differential dependencies (average dependency score in neuroblastoma compared to average of all other cell lines). Core regulatory circuitry (CRC) transcription factor genes in neuroblastoma include MYCN, GATA3, HAND2, ISL1, PHOX2B, and TBX2. False discovery rate (FDR) was determined by P values given by two-tailed Student’s t-test. Right, dependency scores of HDAC2 in neuroblastoma lines (blue, n = 34) and all other lines (black, n = 1,020). P value was determined by two-tailed Student’s t-test. c, Left, volcano plot of differential dependencies in multiple myeloma. False discovery rate (FDR) was determined by P values given by two-tailed Student’s t-test. Right, dependency scores of HDAC1 in multiple myeloma cell lines (blue, n = 21) and all other lines (black, n = 1,033). P value was determined by two-tailed Student’s t-test. d, CRISPR-Cas9-based competitive growth assays in BE(2)-C cells. Negative controls include nontargeting (sgLuc) and cutting (sgAAVS1). All measurements of GFP-positive proportion are normalized to the day 4 measurements in the same group. Mean ± s.e.m., n = 3. e, As in (d) for RPMI-8226 cells. f, Top, growth of individual subcutaneous BE(2)-C-Cas9 xenografts expressing sgLuc or HDAC2-sg1 (n = 10 per group). Non-linear fit with exponential (Malthusian) model shown for each group (solid lines). P value (two-tailed) determined by the extra-sum-of-squares F test. Bottom, tumor diameters over time. Mean ± s.d., n = 10.
To validate HDAC1 and HDAC2 dependencies, we performed CRISPR-Cas9-based competitive growth assays (Extended Data Fig. 1c–e). We found that HDAC2 disruption impairs the competitive proliferation of BE(2)-C neuroblastoma cells, whose parental line, SK-N-BE(2), shows a requirement for HDAC2 on DepMap (Fig. 1b,d and Extended Data Fig. 2a). Meanwhile, HDAC1 disruption impairs the competitive proliferation of RPMI-8226 cells, which is among the more sensitive multiple myeloma cell lines on DepMap (Fig. 1c,e and Extended Data Fig. 2a). We extended these findings in vivo using a subcutaneous xenograft model of neuroblastoma wherein we observed delayed tumor growth as a result of HDAC2 disruption by CRISPR/Cas9 (Fig. 1f). Tumors with HDAC2 knockdown collected at the end of this experiment showed overexpression of HDAC1 protein and/or a rebound in the levels of HDAC2 (Extended Data Fig. 2b). These data, together with the known redundancy between HDAC1 and HDAC2, prompted the hypothesis that these dependencies reflected a paralog-based synthetic lethality.
Collateral synthetic lethality between HDAC1 and HDAC2
Integrating data from the Cancer Cell Line Encyclopedia (CCLE)46 and DepMap, we discovered that sensitivity to HDAC2 disruption correlates with chromosome 1p deletions encompassing the HDAC1 locus (Fig. 2a). This is consistent with our demonstration that BE(2)-C neuroblastoma cells, whose parental line, SK-N-BE(2), harbor a monoallelic loss of HDAC1 (Extended Data Fig. 2c), are sensitive to loss of HDAC2 (Fig. 1d). We therefore hypothesized that deletion of HDAC1 creates a synthetic vulnerability to HDAC2 disruption by reducing HDAC1 expression. Indeed, HDAC1 expression is both significantly lower in neuroblastoma cells with an HDAC1 deletion and is correlated with HDAC2 dependency (Fig. 2b,c and Extended Data Fig. 2c,e). The HDAC1 locus (1p35) is proximal to 1p36, a region that is deleted in nearly a quarter of neuroblastoma cases. These deletions are tightly associated with MYCN amplifications (a key feature of aggressive disease34,37) and lead to the inactivation of the tumor suppressor, ARID1A47,48. We observe HDAC1 deletions in 12 of 31 neuroblastoma cell lines in CCLE (39%, Extended Data Fig. 2c) and reduced HDAC1 expression in MYCN-amplified tumors on cBioPortal49 (Fig. 2d). This confirms that deletion of HDAC1 sensitizes cells to HDAC2 disruption and is a common feature of high-risk, MYCN-amplified neuroblastoma. Furthermore, we observed no change in proliferation as a result of HDAC1 overexpression in BE(2)-C cells, suggesting HDAC1 loss is a collateral effect of recurrent 1p36 deletions in neuroblastoma (Extended Data Fig. 2f).
Figure 2: Collateral synthetic lethality between HDAC1 and HDAC2.
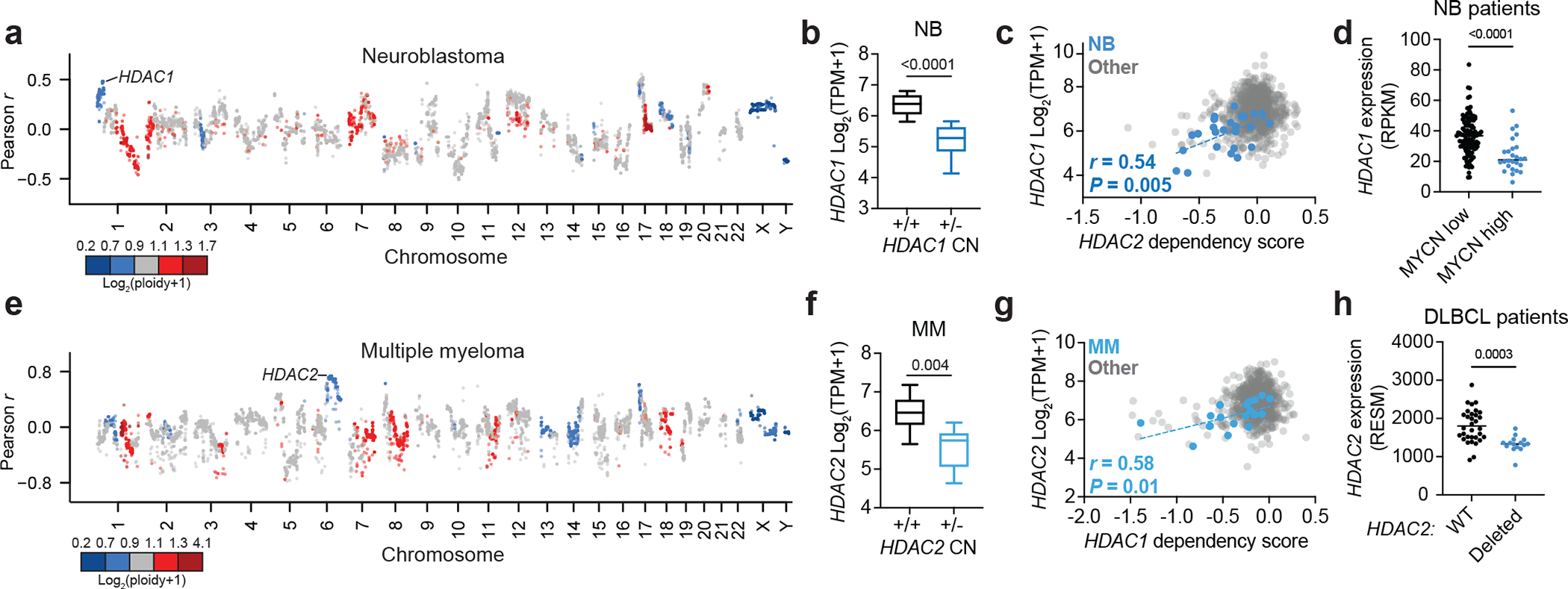
a, Correlations between gene copy number and HDAC2 dependency in neuroblastoma cell lines on DepMap (Pearson, n = 34 neuroblastoma cell lines). Points colored by average copy number in neuroblastoma cell lines. b, Boxplot of HDAC1 expression in neuroblastoma cell lines with (n = 12) and without (n = 19) HDAC1 deletion (see Methods for cutoff). c, Correlation of HDAC2 dependency versus HDAC1 expression in neuroblastoma cell lines and other lineages (n = 973). P value (two-tailed) was determined by Pearson correlation coefficient (r). d, HDAC1 expression in neuroblastoma patient sample with low MYCN expression (black, n = 115) and high MYCN expression (black, n = 26) (cutoff: RPKM < 90) (see Methods for data accessibility). e, Correlation between gene copy number and HDAC1 dependency in multiple myeloma (n = 21). f, Boxplot of HDAC2 expression in multiple myeloma lines with (n = 10) and without (n = 20) HDAC2 deletion (see Methods for cutoff). g, HDAC1 dependency versus HDAC2 expression in multiple myeloma and other lineages (n = 973). P value (two-tailed) was determined by Pearson correlation coefficient (r). h, HDAC2 expression in DLBCL patients with wild-type HDAC2 (black, n = 32) and HDAC2 loss (blue, n = 15) (see Methods for data accessibility). Unless specified, P values were determined by two-tailed Student’s t-test. Boxplots represent 25–75 percentiles with whiskers extending to 10–90 percentiles and the center line represents the median of the data.
Repeating these analyses in multiple myeloma, we discovered that 6q deletions, which cause hemizygous HDAC2 deletions, correlate with sensitivity to loss of HDAC1 (Fig. 2e). Moreover, HDAC2 deletion is associated with reduced HDAC2 expression (Fig. 2f and Extended Data Fig. 2d,e), which is in turn correlated with sensitivity to HDAC1 disruption by CRISPR/Cas9 (Fig. 2g). This is consistent with our demonstration that RPMI-8226 multiple myeloma cells, which harbor an HDAC2 deletion, are sensitive to loss of HDAC1 in competitive proliferation assays (Fig. 1e and Extended Data Fig. 2d). Since 6q deletions are known to be common in multiple myeloma as well as several other lymphoid malignancies, HDAC1 is likely to represent a broad dependency in multiple cancer subtypes38–40 (Extended Data Fig. 1a and 2d,e). Indeed, we find that diffuse large B-cell lymphoma (DLBCL) are significantly more dependent on HDAC1 than other lineages (Extended Data Fig. 2g) and have significantly reduced HDAC2 expression in both cell lines and human patient samples with HDAC2 deletions (Fig. 2h and Extended Data Fig. 2h).
Multiple myeloma cell lines also show a weakly selective dependency on HDAC2 in DepMap (Fig. 1a and Extended Data Fig. 1a), which we validated in RPMI-8226 cells using competitive growth assays (Extended Data Fig. 3a). The requirement for both HDAC1 and HDAC2 in RPMI-8226 cells is different from BE(2)-C cells, which require only HDAC2 (Fig. 1d and Extended Data Fig. 3b). Here, HDAC2 dependency is correlated with HDAC1 mRNA expression but not with HDAC1 copy number alterations (Extended Data Fig. 3c,d). The same was observed for HDAC2 dependencies and HDAC1 expression in soft tissue (Fig. 1a, Extended Data Fig. 1a, and Extended Data Fig. 3c,d), which includes rhabdomyosarcoma, a lineage previously shown to be HDAC2-dependent by the Pediatric Cancer DepMap44. These data suggest that HDAC1/2 synthetic lethality can be caused by expression changes independent of copy number alterations.
To functionally validate that HDAC1 deletion sensitizes cells to loss of HDAC2 by decreasing HDAC1 expression, we found that exogenously expressing HDAC1 in BE(2)-C neuroblastoma cells fully rescued the anti-proliferative effects of HDAC2 disruption by CRISPR/Cas9 (Fig. 3a). As further validation, we tested the effects of HDAC1 and HDAC2 disruption in three other neuroblastoma cell lines: CHP-212, SK-N-AS, and KELLY. Only the cell line harboring a deletion of HDAC1 and reduced HDAC1 protein levels (CHP-212; Extended Data Fig. 2c,e) was sensitive to loss of HDAC2 and none were sensitive to HDAC1 (Fig. 3b–d and Extended Data Fig. 3e,f). In contrast, combined deletion of HDAC1 and HDAC2 dramatically impaired the fitness of multiple cell lines, including BE(2)-C, SK-N-AS, and OCI/AML-2 (Fig. 3e and Extended Data Fig. 4a–c). Critically, GI-ME-N cells, which harbor a 1p36 deletion that does not affect the HDAC1 locus, do not require HDAC2 for survival (Extended Data Fig. 4d–f). This strongly suggests that the hemizygous deletion of HDAC1 is responsible for the HDAC2 requirement in del1p neuroblastoma cells. In multiple myeloma, we find that HDAC1 is dispensable for the growth of MM.1S cells, as these do not harbor an HDAC2 deletion (Fig. 3f and Extended Data Fig. 2d, 4g). Altogether, these data demonstrate that HDAC1 or HDAC2 deletions are sufficient to sensitize cancer cells to loss of the remaining paralog.
Figure 3: Hemizygous HDAC1 deletion is necessary and sufficient to sensitize neuroblastoma to loss of HDAC2.
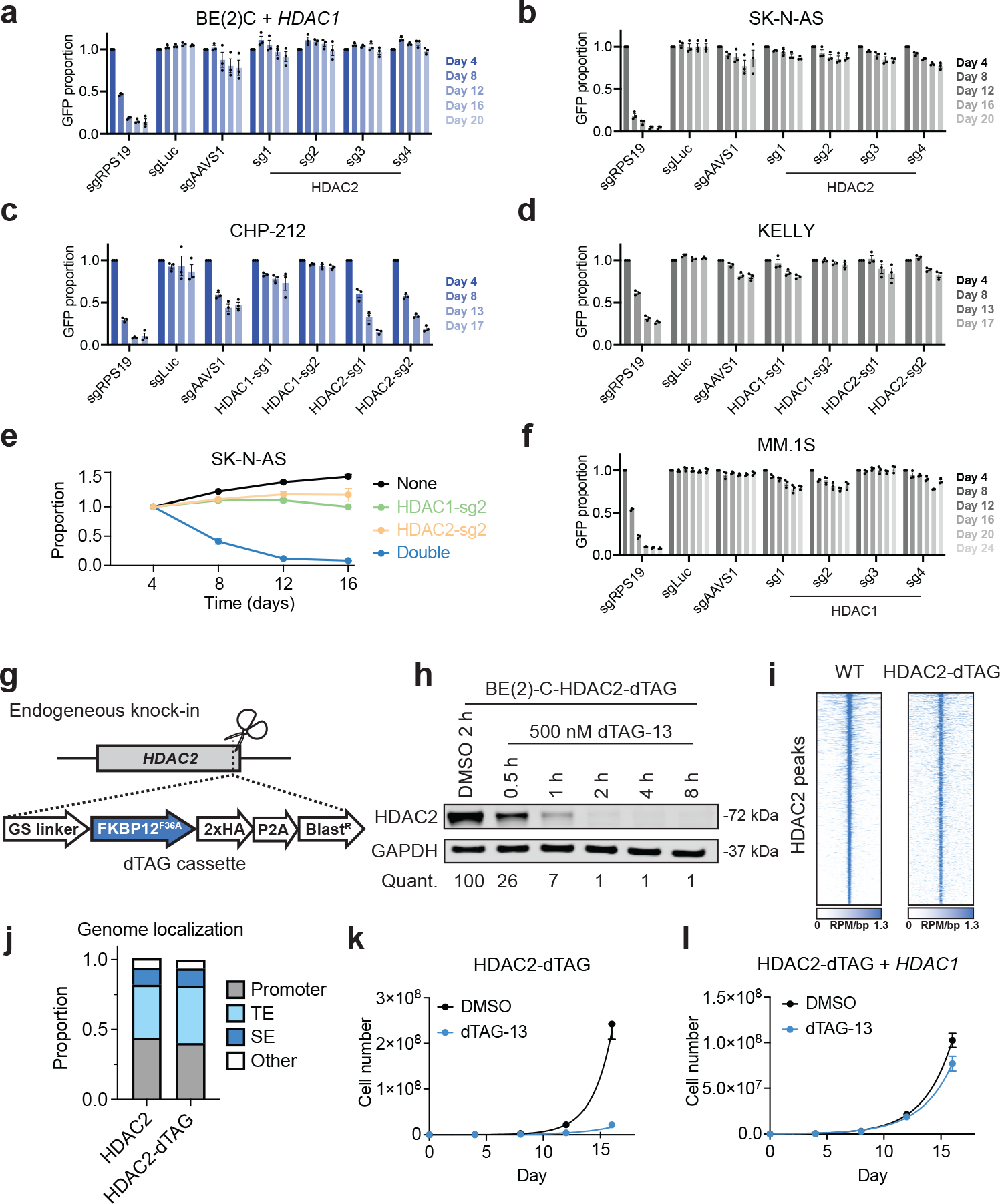
a, Competitive growth assay in BE(2)-C cells overexpressing HDAC1. Mean ± s.e.m., n = 3. b-d, Competitive growth assays in SK-N-AS cells (b), CHP-212 cells (c), and KELLY cells (d). Mean ± s.e.m., n = 3. e, Two-color competitive growth assay in SK-N-AS cells. Mean ± s.d., n = 3. Proportion of each sub-population normalized to day 4. f, Competitive growth assay in MM.1S cells. Mean ± s.e.m., n = 3. g, Schematic depiction of dTAG knock-in. h, Kinetics of HDAC2-dTAG degradation in BE(2)-C-HDAC2-dTAG cells following dTAG-13 treatment (500 nM). DMSO-normalized quantification shown below. i, Rank-ordered heatmaps of CUT&RUN signal for HDAC2 in wild-type BE(2)-C cells and HA in BE(2)-C-HDAC2-dTAG cells (ranked based on HDAC2 signal at HDAC2 binding sites in wild-type BE(2)-C cells). j, Genomic feature distribution of HDAC2- and HDAC2-dTAG-bound sites. Promoters are defined as transcription start site (TSS) ± 1kb. Enhancers are defined by TSS-distal H3K27ac signal by ROSE2. TE, typical enhancer. SE, super enhancer. k,l Proliferation of BE(2)-C-HDAC2-dTAG cells (k) and BE(2)-C-HDAC2-dTAG with HDAC1 overexpression (l) in response to dTAG-13 treatment (500 nM). Mean ± s.e.m., n = 3.
A dTAG-based approach to degrade and study HDAC2
To enable an orthogonal validation of these data, we employed the dTAG system for chemically induced protein degradation. The dTAG system relies on proteolysis targeting chimeras (PROTACs) to elicit rapid and selective degradation of proteins that have been tagged with an engineered “bump-and-hole” variant of FKBP1250,51 (Extended Data Fig. 5a). We reasoned that the rapid kinetics of this approach would support subsequent mechanistic investigations while also modeling the effects of therapeutic disruption in a manner that could inform future undertakings in drug discovery52,53. Using CRISPR/Cas9, we engineered BE(2)-C cells to express HDAC2 with a carboxy-terminal fusion of FKBP12F36V (Fig. 3g and Extended Data Fig. 5b,c). This “HDAC2-dTAG” chimera can be degraded by the PROTAC, dTAG-13, in a dose-responsive and time-dependent fashion, with virtually complete degradation achieved within 2 hours (Fig. 3h and Extended Data Fig. 5d). By Cleavage Under Targets and Release Using Nuclease (CUT&RUN)54 there was a strong correlation between the genomic localization of HDAC2-dTAG in BE(2)-C-HDAC2-dTAG cells and HDAC2 in wild-type BE(2)-C cells (Fig. 3i,j and Extended Data Fig. 5e–g) and HDAC2-dTAG also retains protein-protein interactions with members of the NuRD complex (Extended Data Fig. 5h). The tagged allele was expressed at lower levels than wild-type cells (Extended Data Fig. 5c), which increased BE(2)-C-HDAC2-dTAG dependency on HDAC1, but did not affect their HDAC2 dependency (Extended Data Fig. 5i). These results confirmed that the HDAC2-dTAG allele is functional despite its lower expression and further reinforced the co-essentiality of HDAC1 and HDAC2. Exposure to dTAG-13 potently inhibited the growth BE(2)-C-HDAC2-dTAG without impacting the growth of wild-type BE(2)-C cells, confirming the growth effect was due to HDAC2 degradation (Fig.3k and Extended Data Fig. 5j). Importantly, overexpression of HDAC1 was able to rescue this effect (Fig. 3l and Extended Data Fig. 5c), providing an orthogonal validation of HDAC1/2 synthetic lethality in neuroblastoma.
HDAC2 degradation disrupts enhancer-mediated transcription
To identify the direct transcriptional consequences of HDAC2 degradation, we performed thiol(SH)-linked alkylation for the metabolic sequencing of RNA (SLAM)-seq55, which quantifies changes in the abundance of newly synthesized mRNAs. After 2 hours, both HDAC2 degradation and HDAC1/2 inhibition (ACY-95756) predominantly upregulated transcriptional activity, producing tightly correlated SLAM-seq responses (Fig. 4a,b). Interestingly, genes with HDAC2 bound at their promoter were not preferentially upregulated by HDAC2 degradation compared to those without HDAC2 (Extended Data Fig. 6a). This prompted us to inspect promoter-distal sites of HDAC2 enrichment using ROSE2 (ranking of super enhancer 2), which was previously developed to identify typical enhancers (TE), super enhancers (SE), and their putative target genes57. From this analysis, we were able to classify genes as being proximal to sites of typical HDAC2 enrichment (94% of promoter-distal HDAC2-bound sites) or disproportionately high HDAC2 enrichment (6% of sites), which we termed asymmetrically bound sites (Fig. 4c). After integrating these data with the 2-h SLAM-seq, we found that genes associated with typical or asymmetric sites were upregulated less than other genes in response to dTAG-13 treatment (Fig. 4d). This effect was most pronounced for genes associated with asymmetric sites, but did not occur following HDAC1/2 inhibition (Fig. 4d). Given the otherwise similar transcriptional response to HDAC1/2 inhibition and HDAC2 degradation, we were intrigued by this apparent difference.
Figure 4: HDAC2 degradation dysregulates transcription.
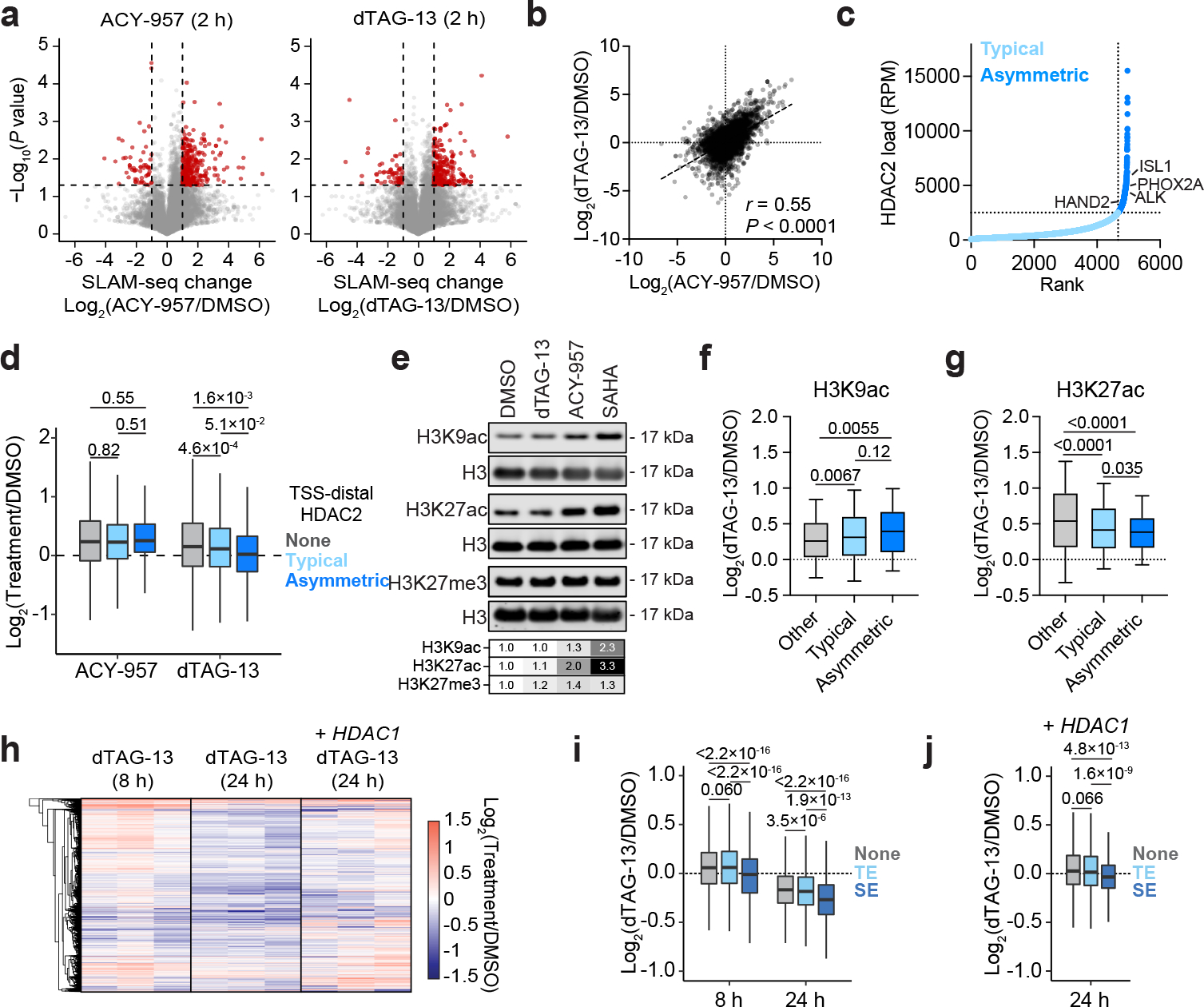
a, Volcano plots depicting changes in newly synthesized mRNAs (SLAM-seq) following 2-h treatments of 5 μM ACY-957 (left) or 500 nM dTAG-13 (right). n = 3. b, Correlation of SLAM-seq changes induced by ACY-957 and dTAG-13 treatments. P value (two-tailed) was determined by Pearson correlation coefficient (r). n = 13,199. c, Asymmetric genomic localization of HDAC2 is depicted by plotting the rank-ordered CUT&RUN signal at each HDAC2-occupied site. d, Boxplot of SLAM-seq changes. Genes are classified as not being associated with TSS-distal HDAC2 sites (n = 11,210), being associated with typical TSS-distal HDAC2 sites (n = 1,826), or being associated with asymmetric TSS-distal HDAC2 sites (n = 163). e, Immunoblot detection of H3K9ac, H3K27ac, and H3K27me3 following 2-h treatments of DMSO, dTAG-13 (500 nM), ACY-957 (5 μM), or SAHA (5 μM). f, Boxplots of changes in H3K9ac CUT&RUN signal following 2-h dTAG-13 treatment (500 nM) at H3K9ac-enriched sites that overlap with TSS-distal HDAC2-typical sites (n = 882), with TSS-distal HDAC2-aysmmetric sites (n = 133), and other sites (n = 13,984). g, same as (f) but for H3K27ac (Other, n = 9,863; typical, n = 2,149; asymmetric, n = 280). Boxes represent 25–75 percentiles with whiskers extending to 10–90 percentiles and the center line represents the median of the data. h, Heatmap of changes in total mRNA abundance (3’-end mRNA-seq) following dTAG-13 treatment (500 nM) treatments in BE(2)-C-HDAC2-dTAG cells with or without HDAC1 overexpression, n = 3. i, Boxplots of changes in total mRNA abundance for genes not associated with an enhancer (none, n = 7,588), associated with typical enhancers (TE, n = 5,026), or associated with super enhancers (SE, n = 585). j, As in (i) but with 24-h dTAG-13 treatment (500 nM) in BE(2)C-HDAC2-dTAG cells overexpressing HDAC1. Analyses of gene expression changes were restricted to active genes (see Methods). For volcano plots, P values were determined by two-tailed Student’s t-test. For boxplots, P values were determined by two-tailed Welch’s t-test. Unless specified, boxplots represent 25–75 percentile with whiskers extending 1.5 interquartile range (IQR) and the center line represents the median.
We considered that residual HDAC1 activity might be able to contribute sufficient deacetylase function at asymmetric sites to compensate for HDAC2 degradation, whereas ACY-957 would inhibit both enzymes. This would potentially explain why genes associated with asymmetric sites were not coordinately upregulated by dTAG-13. To investigate this possibility, we measured the effect of dTAG-13 treatment on H3K9ac and H3K27ac levels, two well-established HDAC1/2 substrates3,58. While we observed minimal changes by immunoblot analysis (Fig. 4e), we detected a slight increase of H3K9ac and H3K27ac abundance by CUT&RUN (Extended Data Fig. 6b). The increase in H3K27ac was indeed muted at typical and asymmetric HDAC2 sites, but this was not the case for H3K9ac (Fig. 4f,g). Since both modifications are known to be regulated by HDAC1/2, it is unclear if the difference in H3K27ac reflects residual HDAC1 activity or an alternative mechanism. Another possibility, not mutually exclusive with the first, is that the differences we observe at asymmetric sites might be the result of disrupting a non-enzymatic function of HDAC2 that is not addressed by HDAC1/2 inhibition. HDAC1 and HDAC2 are members of several multiprotein complexes, including the NuRD complex, which regulates chromatin accessibility through the nucleosome remodeler, CHD47. CHD4/NuRD-mediated chromatin remodeling positively regulates the transcription of genes associated with super enhancers59, which we found to share considerable overlap with sites of asymmetric HDAC2 localization (Extended Data Fig. 6c). Therefore, HDAC2 degradation might negatively impact NuRD function at super enhancers, which would, in principle, result in suppression of super enhancer target genes. This effect, overlayed with the more widespread transcriptional de-repression caused by loss of HDAC2 catalytic activity, might explain why most genes are upregulated by HDAC2 degradation, except those associated with asymmetric HDAC2 sites (Fig. 4d).
To determine how super-enhancer-associated transcripts are regulated in response to HDAC2 degradation over time, we performed kinetic gene expression profiling experiments using 3’-end mRNA-seq to detect changes in total transcript abundance. After 8 hours of dTAG-13 treatment, we observed a general increase of mRNAs (Fig. 4h and Extended Data Fig. 6d), which is consistent with the general upregulation we observed by SLAM-seq after 2 hours (Fig. 4a). Indeed, the genes found to be significantly upregulated after 2 hours by SLAM-seq were found to be preferentially upregulated after 8 hours by 3’-end mRNA-seq (Extended Data Fig. 6e). However, after 24 and 72 hours of dTAG-13 treatment, transcriptional activity was generally suppressed (Fig. 4h and Extended Data Fig. 6f,g), consistent with previous reports that class I HDAC inhibition causes only transient transcriptional upregulation60. Again, we saw no obvious difference between genes with and without promoter-bound HDAC2 (Extended Data Fig. 6h), but super-enhancer-associated transcripts were distinctly affected at every time point (Fig. 4i and Extended Data Fig. 6i). That is, despite otherwise widespread increases in transcriptional activity after 8 hours, the transcription of SE-linked genes was relatively unchanged. Then, after 24 and 72 hours, when transcriptional downregulation becomes predominant, SE-linked genes were more severely suppressed than others (Fig. 4i and Extended Data Fig. 6i). Altogether, these data suggest that SE-associated transcripts, which are enriched with lineage-specific dependencies (Extended Data Fig. 6j), are potently inhibited by HDAC2 degradation (Fig. 4i and Extended Data Fig. 6i). For instance, the enhancer-controlled neuroblastoma-selective dependencies, IRS261, ISL142, and CDK662 were downregulated by HDAC2 degradation (Extended Data Fig. 6l). Importantly, exogenous expression of HDAC1 rescued the transcriptional effects caused by HDAC2 degradation, substantially muting the preferential repression of SE-associated transcripts (Fig. 4j and Extended Data Fig. 6k).
In the 3’-end mRNA-seq experiments described above, metabolic labeling with 4-thiouridine was included for the final hour of the dTAG-13 treatment. This allowed us to not only interpret changes in total mRNAs over the duration of the dTAG-13 treatment (3’-end mRNA-seq), but also to detect changes in newly synthesized mRNAs over the course of metabolic labeling (SLAM-seq). Comparing to SLAM-seq following a 2-h dTAG-13 treatment, the 8-h dTAG-13 treatment elicited a similar increase of transcriptional activity by SLAM-seq, yet to a lesser degree (Extended Data Fig. 7a). Similar to the results from the 8-hour 3’-end mRNA seq (Fig. 4i), the 8-hour SLAM-seq showed that super-enhancer-associated genes were not upregulated, confirming that the effect was not biased by half-life (Extended Data Fig. 7c). After 24 hours, the pronounced suppression of super-enhancer-associated genes that we observed by 3’-end mRNA-seq (Fig. 4i) was not observed by SLAM-seq (Extended Data Fig. 7b,c), potentially reflecting the onset of more indirect effects.
HDAC2 degradation destabilizes the NuRD complex
To explain the class-selective effects on SE-linked transcription, we wondered whether loss of HDAC2 may destabilize the NuRD complex and lead to its degradation. This phenomenon, whereby chemically induced degradation results in destabilization of other subunits within a large multiprotein complex, has been reported for PRC2, BAF (by both direct-acting PROTACs and dTAG-based degradation), and some HDAC-associated complexes63–66. Conditional knockout of HDAC1 and/or HDAC2 has been shown to decrease the abundance of MTA2 protein, a NuRD subunit that interacts with HDACs, in mouse embryonic stem cells and mouse T cells12,17. Although it is unclear if this downregulation of MTA2 protein levels was caused through transcriptional or post-transcriptional effects, these results likely imply a link between HDAC1/2 levels and the integrity of NuRD complex12,17. Indeed, a saturating CRISPR/Cas9-based genetic screen of NuRD components has previously identified MTA2 mutations that disrupt NuRD structure and function67. Moreover, a recent study reported an electrophilic small molecule that causes the selective and coordinated degradation of several NuRD subunits in T cells, although the mechanism by which this occurs has yet to be reported68.
Using unbiased quantitative expression proteomics, we confirmed that dTAG-13 selectively degrades HDAC2 in BE(2)-C-HDAC2-dTAG cells but also discovered that it decreases the abundance of HDAC-associated proteins (Fig. 5a–d and Extended Data Fig. 8a). Specifically, several members of the HDAC1/2-containing complexes, mitotic deacetylase complex (MiDAC) and NuRD, were depleted by dTAG-13 treatment without any change in mRNA expression (Fig. 5a,b and Extended Data Fig. 8b,c). We were able to reproduce the depletion of NuRD subunits by immunoblot but failed to validate the effects on MiDAC (Extended Data Fig. 8d–f). We therefore focused our efforts on studying NuRD structure and function in response to HDAC2 degradation. The NuRD complex consists of subunits that catalyze ATP-dependent chromatin remodeling (CHD3/4) and histone deacetylation (HDAC1/2), accompanied by several scaffolding and chromatin-binding proteins, including MTA1/2/3, GATAD2A/B, MBD2/3 and RBBP4/769. MTA paralogs bind directly to HDAC1 and HDAC2, forming a subcomplex together with RBBP4/7, while CHD3/4 and GATAD2A/B form the other half of the complex70,71. MBD2/3 are thought to bridge the two halves together to constitute a complete structure and are thus critical for NuRD complex integrity70,71. Moreover, as several paralogous members of the complex are mutually exclusive, distinct complex compositions can preferentially affect certain cell types or cellular processes72. Mechanistically, NuRD degradation might be caused by direct PROTAC-facilitated ubiquitination of non-HDAC subunits or occur as a secondary consequence of protein complex destabilization following HDAC2 degradation. We strongly favor the latter explanation, as we detect decreased abundance of NuRD subunits following CRISPR/Cas9-mediated disruption of HDAC2 (Fig. 5e), and because more subunits were affected after 24 hours of dTAG-13 treatment than after 2 hours (Fig. 5b).
Figure 5: HDAC2 degradation destabilizes the NuRD complex in HDAC1-deficient neuroblastoma cells.
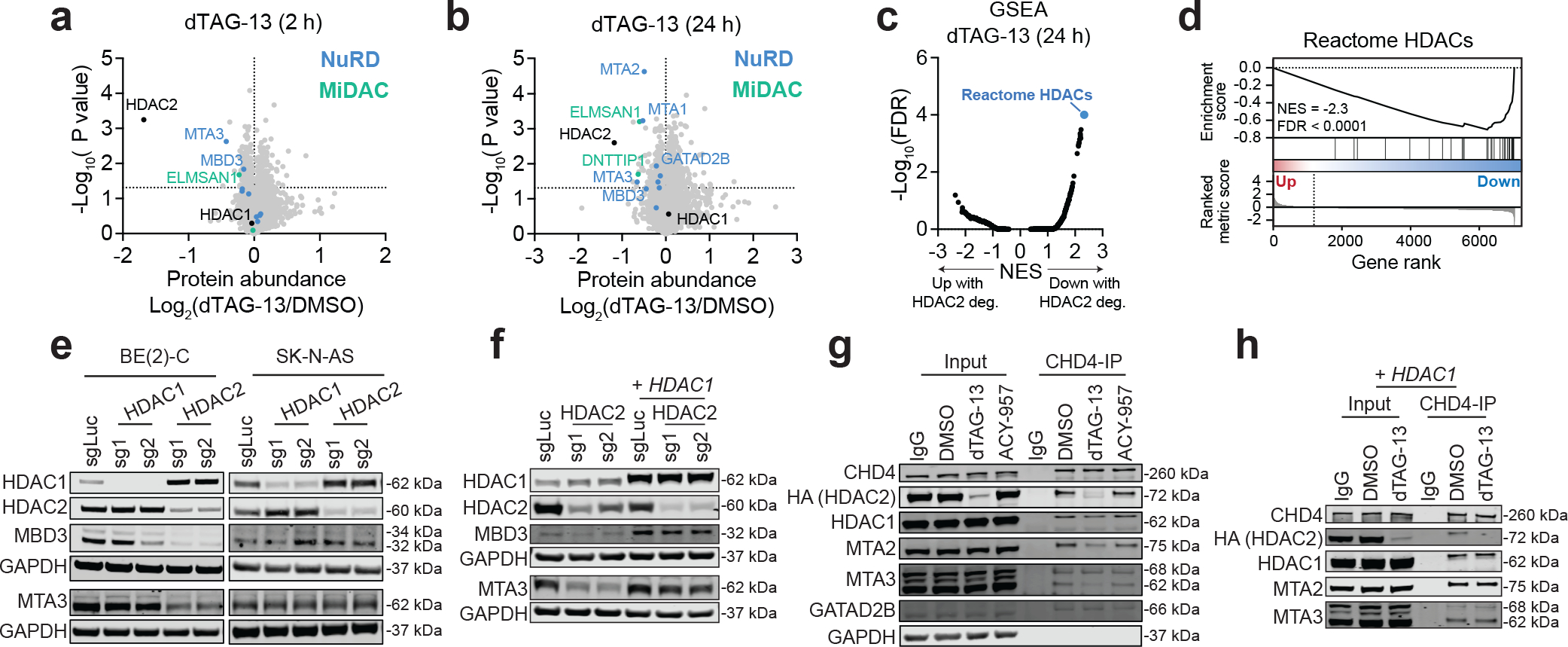
a,b Volcano plots of changes in protein abundance determined by unbiased quantitative proteomics following dTAG-13 (500 nM) treatment for 2 h (a) or 24 h (b). DMSO n = 2, dTAG-13 n = 3. P values were determined by two-tailed Student’s t-test. c, Gene set enrichment analysis (GSEA) for protein abundance changes after 24-h dTAG-13 treatment. NES, normalized enrichment score. d, GSEA of Reactome HDACs gene set for protein abundance changes after 24-h dTAG-13 treatment. e, Protein levels of NuRD subunits following genetic disruption of HDAC1 and HDAC2 by CRISPR/Cas9 in BE(2)-C (left) and SK-N-AS (right) determined by immunoblot analysis. f, As in (d) but in BE(2)-C cells with or without HDAC1 overexpression. g, Co-immunoprecipitation of CHD4 in BE(2)-C-HDAC2-dTAG cells with 2-h dTAG-13 (500 nM) and ACY-957 (5 μM) treatments. h, As in (f) but with 2-h dTAG-13 treatment in BE(2)-C-HDAC2-dTAG cells with HDAC1 overexpression.
We next asked whether NuRD degradation was specific to cells with compromised levels of HDAC1. Indeed, CRISPR/Cas9-mediated disruption of HDAC2 in the HDAC1-diploid SK-N-AS cells did not affect the abundance of other NuRD subunits (Fig. 5e) and the effect on BE(2)-C cells could be rescued by overexpression of HDAC1 (Fig. 5f). We therefore hypothesized that HDAC2 degradation might result in compromised intra-complex interactions in cells without sufficient levels of HDAC1 to stabilize the complex. This would be consistent with experiments in mouse thymocytes that demonstrated a single allelic loss of HDAC1 can cause a decrease in MTA2 protein levels17. To probe NuRD interactions, we performed CHD4 co-immunoprecipitations following a 2-h dTAG-13 treatment and noted weakened interactions between CHD4 and HDAC1, MTA2 and MTA3, but not between CHD4 and GATAD2B (Fig. 5g). These data indicate that the HDAC/MTA subcomplex is immediately dissociated from the CHD4 subcomplex upon HDAC2 degradation, which results in degradation of MTA and MBD subunits. Moreover, the impact of HDAC2 degradation on NuRD interactions was rescued by overexpression of HDAC1 (Fig. 5h), confirming that the effect is dependent on diminished HDAC1 protein abundance.
HDAC2 degradation disrupts NuRD remodeling activity
To determine if the effect of HDAC2 degradation on NuRD stability impacts its chromatin remodeling activity, we first mapped the genomic localization of MBD3 and CHD4 in BE(2)-C cells by CUT&RUN (Fig. 6a). In comparison to HDAC2, which occupies both promoters and enhancers, MBD3 and CHD4 show a predominant localization to enhancers (Fig. 6a and Extended Data Fig. 9a). About 60% of HDAC2-bound sites overlap with either MBD3 or CHD4 (Fig. 6b) and these sites (HDAC2-NuRD) are preferentially associated with enhancers (Fig. 6c). Approximately 70% of super enhancers and 16% of typical enhancers are associated with HDAC2-NuRD (Fig. 6d), whereas HDAC2-bound sites that do not co-localize with MDB3/CHD4 consist mostly of promoter regions (Fig. 6c). We interpret this to indicate that HDAC2 localization to enhancers is commonly associated with the NuRD complex, whereas its localization to promoters is associated with other complexes. For example, we observe all three members of the NuRD complex highly enriched at a super enhancer proximal to ISL1, which is a highly selective neuroblastoma dependency that is downregulated by HDAC2 degradation (Extended Data Fig. 1b and Extended Data Fig. 6l), but only HDAC2 bound to its promoter (Fig. 6e).
Figure 6: HDAC2 degradation compromises NuRD function and exploits cancer-specific NuRD dependencies.
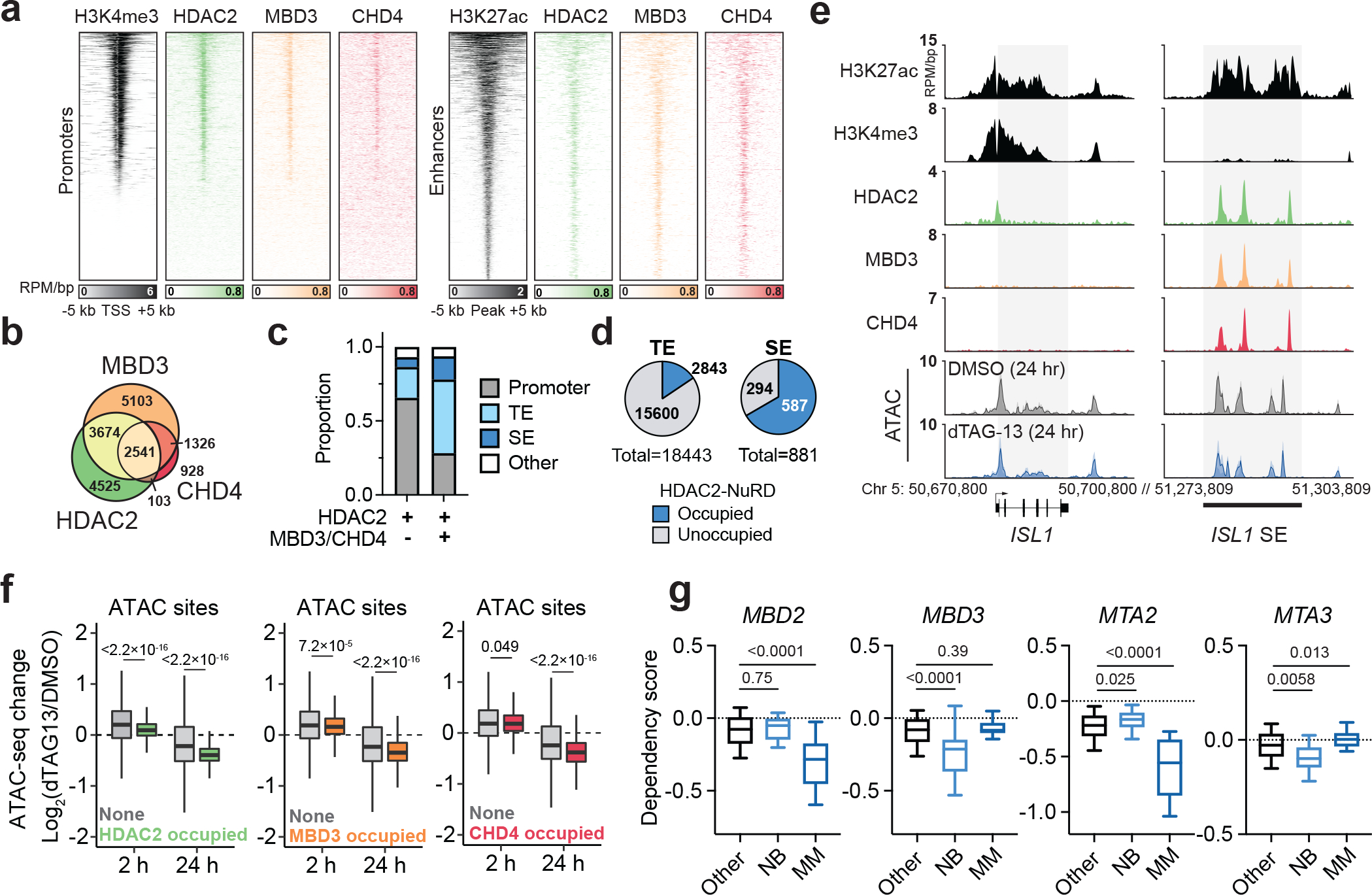
a, Heatmaps of CUT&RUN signals for HDAC2, MBD3, and CHD4 mapped to promoters (left, ranked by H3K4me3 ChIP signal from GSM2113519) and enhancers (right, ranked by H3K27ac ChIP signal from GSM2113518). b, Overlap of HDAC2-, MBD3-, and CHD4-bound sites. HDAC2 bound-sites shared with MBD3 and/or CHD4 are defined as HDAC2-NuRD sites. c, HDAC2-only sites (HDAC2 sites that do not overlap MBD3 or CHD4) mainly localize to promoters while HDAC2-NuRD predominantly occupy enhancers. d, Proportion of enhancers and super enhancers that are occupied by HDAC2-NuRD. e, Gene track representation of ChIP-seq, CUT&RUN, or ATAC-seq signal at the ISL1 locus (left) and its proximal enhancer (right). ATAC-seq signal is shown as meta-track representation of triplicates following 24-h DMSO control (grey) or 500 nM dTAG-13 treatment (blue). f, Boxplots of chromatin accessibility changes grouped by HDAC2 occupancy (left) (2 h, unoccupied n = 97,849, occupied n = 9,581; 24 h, unoccupied n = 94,967, occupied n = 9,573), MBD3 occupancy (middle) (2 h, unoccupied n = 96,391, occupied n = 11,039; 24 h, unoccupied n = 93,608, occupied n = 10,932), or CHD4 occupancy (right) (2 h, unoccupied n = 103,049, occupied n = 4,381; 24 h, unoccupied n = 100,223, occupied n = 4,317) at ATAC-seq sites. Boxes represent 25–75 percentiles with whiskers extending 1.5 IQR and the center line represents the median of the data. P values were determined by two-tailed Welch’s t-test. g, Boxplots of MBD2, MBD3, MTA2, and MTA3 dependency scores in neuroblastoma (n = 34), multiple myeloma (n = 21), or other cell lines (n = 999). Boxes represent 25–75 percentiles with whiskers extending to 10–90 percentiles and the center line represents the median of the data. P values were determined by two-tailed Student’s t-test.
Next, we performed assay for transposase-accessible chromatin (ATAC)-seq73 following 2-h and 24-h exposures to dTAG-13 to determine if NuRD chromatin remodeler activity is impacted by HDAC2 degradation. Similar to the transcriptional effects we observed after 2 hours, acute HDAC2 degradation causes widespread increases of chromatin accessibility (Extended Data Fig. 9b–d). However, HDAC2-bound sites of the genome are significantly less affected (Fig. 6f), similar to our observation that genes associated with asymmetric HDAC2 sites were less upregulated at 2 hours (Fig. 4d). After 24 hours, chromatin accessibility was widely diminished, with pronounced effects evident at HDAC2-bound sites (Fig. 6f and Extended Data Fig. 9e). This decrease in chromatin accessibility following extended HDAC2 degradation is likewise consistent with the transcriptional suppression we observe at 24 hours (Extended Data Fig. 7f), but contrary to past reports that link HDAC inhibition (both Class I and pan-HDAC) with increased chromatin accessibility74,75. Therefore, we wondered whether the differential regulation of accessibility at HDAC2-bound sites might reflect a competition between HDAC2 catalytic activity, which represses chromatin accessibility, and NuRD remodeler function, which maintains open chromatin structures at super enhancers59. Indeed, we find that accessibility at MBD3- and CHD4-bound sites becomes preferentially repressed following HDAC2 degradation for 24 hours (Fig. 6f and Extended Data Fig. 9e). These data indicate that HDAC2 degradation not only disrupts NuRD integrity, but also disrupts its chromatin remodeler function, which is consistent with the pronounced suppression of SE-associated transcripts (Fig. 4i and Extended Data Fig. 7f). We probed the subcellular localization of CHD4 by immunoblot analysis following subcellular fractionation and did not observe an obvious change of CHD4 localization (Extended Data Fig. 9f). This might suggest that dissociation of the NuRD complex affects CHD4 activity rather than its localization, but we cannot rule out the possibility that CHD4 binding to the genome is impacted in a locus-specific manner.
NuRD harbors lineage-specific cancer dependencies
Interested in how NuRD degradation might impact cancer cell survival, we used DepMap to discover that specific paralogs within the NuRD complex, but not other HDAC1/2-associated complexes, register as highly selective dependencies in neuroblastoma and multiple myeloma (Fig. 6g and Extended Data Fig. 10a). Interestingly, discrete sets of paralogs are selectively required in each lineage – MBD2 and MTA2 in multiple myeloma and MBD3 and MTA3 in neuroblastoma (Fig. 6g). Using DepMap to identify co-dependencies of these NuRD subunits, we find that MBD2 and MTA2 are most highly correlated with each other, followed by several top multiple myeloma dependencies, like IRF4 (Extended Data Fig. 10b). On the other hand, MBD3 and MTA3 dependency scores are highly correlated with HDAC2 and other selective neuroblastoma dependencies, such as MYCN and ISL1 (Extended Data Fig. 10b). Consistent with the known capacity of the NuRD complex to form functionally non-redundant subcomplexes through differential assembly of paralogous subunits72, this potentially suggests that distinct NuRD subcomplexes are relied on in neuroblastoma and multiple myeloma. We used unsupervised clustering of all NuRD dependency scores across all cancer cell lines on DepMap to identify functional trends among NuRD paralogs, as this type of co-dependency analysis has previously been used to identify functionally and physically distinct protein complex assemblies76,77. This revealed a cluster containing MBD2, MTA2, and HDAC1, and another containing MBD3, MTA3, and HDAC2 (Extended Data Fig. 10c), which tracks with the former being required by multiple myeloma and the latter being required by neuroblastoma. It is therefore attractive to speculate that these cancer types rely on distinct NuRD subcomplex compositions for tumor maintenance. Since these NuRD dependencies are degraded following loss of HDAC2, we suggest that exploiting HDAC1/2 synthetic lethality represents an effective strategy to target lineage-specific NuRD vulnerabilities.
Discussion:
This study applied a generalizable framework for identifying synthetic lethal genetic interactions. We identified HDAC1/2 as a reciprocal synthetic lethality caused by common chromosomal deletions observed in multiple human cancers. Using genetic perturbations and chemically responsive degradation, we demonstrate that hemizygous deletions of HDAC1 or HDAC2 are sufficient to sensitize cells to loss of the other.
The dTAG system for chemically induced degradation was particularly useful for determining the mechanisms underlying this synthetic lethality, ultimately revealing an effect on NuRD complex stability, chromatin accessibility, and cell-type-specific transcriptional regulation. Its fast kinetics allowed us to dissect HDAC2 function with exceptional temporal resolution. Immediately following the loss of HDAC2, we observed transient transcriptional upregulation and increased chromatin accessibility (Fig. 7). This gave way to transcriptional repression, coinciding with NuRD destabilization and chromatin compaction at NuRD-binding sites (Fig. 7). Transcriptionally, the preferential downregulation of SE-linked genes, which are regulated by CHD4/NuRD59, results in the repression of many genes that are selectively required for neuroblastoma survival (Fig. 7). These findings join an increasing body of evidence that links HDAC/NuRD to transcriptional activation, contrasting with the paradigmatic role of HDACs as transcriptional repressors78,79.
Figure 7: HDAC2 degradation leads to NuRD complex collapse and dysregulations of chromatin accessibility and transcriptome in HDAC1-deficient neuroblastoma.
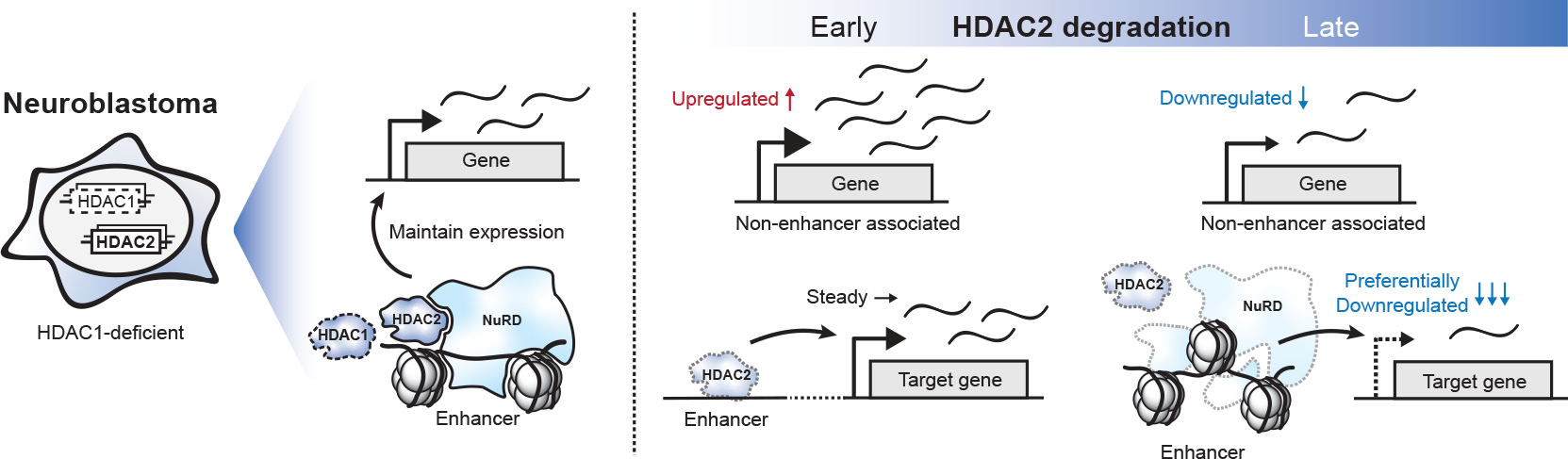
We demonstrate that targeting HDAC1/2 synthetic lethality compromises the structure and function of the NuRD complex, leading to the degradation of NuRD subunits that are selectively essential in neuroblastoma and compromising the function of CHD4, which is pan-essential (Fig. 7). Structurally, MTA proteins are bound directly to HDAC1/2 through the ELM2-SANT domain71. Exposing the intrinsically disordered regions of the ELM2-SANT domain upon HDAC2 degradation likely subjects MTAs for degradation and triggers the collapse of the NuRD complex71,80. As a result, CHD4, which is pan-essential, can no longer interact with several NuRD subunits, likely indicating that it is functionally compromised in these cells, consistent with the loss of chromatin accessibility at CHD4-bound sites. Our study follows the discovery of a conceptually similar synthetic lethality between NXT1 and NXT2, which causes degradation of a pan-essential binding partner, NXF131. Therefore, we conclude that perturbing the stability and/or function of essential proteins (either lineage-specific, like the MTA/MBD subunits, or pan-essential, like CHD4) is likely to be a generalizable consequence of many paralog synthetic lethalities.
These data provide further motivation to develop direct-acting HDAC1/2 PROTACs63,81–84. To effectively capitalize on this synthetic lethality therapeutically, it will be necessary to develop compounds that selectively degrade one paralog or the other. While orthosteric ligands are unlikely to bind one paralog selectively, it is well demonstrated that selectivity can improved with degradation-based pharmacologies. The additional constraints that are unique to ligand-induced degradation (e.g. ternary complex formation) can allow for unselective ligands to be developed into selective degraders. There are now many examples of degraders that can differentiate between two highly similar paralogs85–89, joined by the recent report of an agent that degrades HDAC1/3 but not HDAC290. Therefore, we and others may now pursue paralog-selective degraders, with dTAG-inspired mechanistic guidance, to address HDAC1/2-NuRD vulnerabilities.
Methods:
Cell culture and lentivirus production.
BE(2)-C (DMEM supplemented with 10% fetal bovine serum (FBS) and Gibco Antibiotic-Antimycotic), SK-N-AS (DMEM supplemented with 10% FBS and Gibco Antibiotic-Antimycotic), and OCI-AML2 (RPMI supplemented with 10% FBS and Gibco Antibiotic-Antimycotic) cell lines were provided by the laboratory of Prof. James E. Bradner. MM.1S (RPMI supplemented with 10% FBS and Gibco Antibiotic-Antimycotic) cells were provided by the laboratory of Prof. Christopher Ott. KELLY (RPMI supplemented with 10% FBS and Gibco Antibiotic-Antimycotic), CHP-212 (RPMI supplemented with 10% FBS and Gibco Antibiotic-Antimycotic), and GI-M-EN (RPMI supplemented with 10% FBS and Gibco Antibiotic-Antimycotic) cells were provided by the laboratory of Prof. Adam D. Durbin. RPMI-8226 (RPMI supplemented with 15% FBS and Gibco Antibiotic-Antimycotic) cells were purchased from DSMZ. Lenti-X 293T (DMEM supplemented with 10% FBS and Gibco Antibiotic-Antimycotic) cells were purchased from Takara for lentivirus production. All cell lines were tested negative for mycoplasma infections regularly. Lentiviral packaging plasmids pMD2.G (a gift from Didier Trono, Addgene plasmid #12259; http://n2t.net/addgene:12259; RRID:Addgene_12259), psPAX2 (a gift from Didier Trono, Addgene plasmid #12260; http://n2t.net/addgene:12260; RRID:Addgene_12260), and the lentiviral expression plasmid were co-transfected to Lenti-X 293T cells to produce corresponding lentivirus. Supernatants with viral particles were harvested at 48 and 72 hours after transfection, filtered with 0.22 μm membrane and concentrated by 50-fold with Lenti-X Concentrator (Takara, #631232). All cells were transduced by spinoculation at 800 g for 1 hour at room temperature supplemented with 8 μg/mL polybrene. For MM.1S and RPMI-8226 cells polybrene was removed 8 hours after the spinoculation to prevent toxicity.
Analyses of datasets from DepMap.
For HDAC1/2 copy number analyses in Fig. 2b&f we used Log2(relative to ploidy + 1) of 0.7 as cutoff for hemizygous deletion to include all cell lines potentially harboring deleted HDAC1/2, based on the calculation that 1 copy of the gene equals Log2(relative to ploidy + 1) of 0.58.
Analyses of datasets from cBioportal49.
The analysis of neuroblastoma patient data is based upon data generated by the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) initiative, phs000218, managed by the NCI. Specifically, data used in this study were obtained from Pediatric Neuroblastoma (TARGET, 2018) (cancer study identifier: nbl_target_2018_pub). Information about TARGET can be found at http://ocg.cancer.gov/programs/target. DLBCL patient data were obtained from Lymphoid Neoplasm Diffuse Large B-cell Lymphoma (TCGA, Firehose Legacy) (cancer study identifier: dlbc_tcga). Source data is from http://gdac.broadinstitute.org/runs/stddata__2016_01_28/data/DLBC/20160128/.
Immunoblotting.
Cells were resuspended with CelLytic M buffer (Sigma-Aldrich, #C2978) supplemented with 1X Halt Protease Inhibitor Cocktail (Thermo Scientific, #78430) and 0.1% benzonase endonuclease (Millipore, #70746) for whole cell lysates. Lysates were incubated on ice for 30 mins with occasional vortexing and cleared by 13,000 g centrifugation at 4° C for 10 mins. The total protein concentration was then measured with Pierce BCA protein assay kit (Thermo Scientific, #23225). Samples on one blot were normalized to the same total protein content. To extract histones cells were resuspended with 0.5% Triton X 100 in PBS supplemented with 1X Halt Protease Inhibitor Cocktail (Thermo Scientific, #78430) and incubated on ice for 10 mins. The pellets were collected after 500 g centrifugation at 4° C for 10 mins. Histones were extracted with 0.2 N HCl at 4° C overnight. The protein content was then determined by Bradford assay (Thermo Scientific, #23200) and equal amount of protein across samples was run on the same blot. Protein bands were detected by fluorescently labeled infrared secondary antibodies (LI-COR IRDye 800CW Donkey anti-Rabbit IgG, #926–32213, 1:7,000; IRDye 800CW Goat anti-Mouse IgG, #926–32210, 1:7,000; IRDye 680RD Goat anti-Mouse IgG, #926–68070,1:7,000; IRDye 680RD Donkey anti-Rabbit IgG, #926–68073, 1:7,000) on the Odyssey CLx Imager (LI-COR). The antibodies used are all commercially available: anti-CHD4 (Cell Signaling Technology, #12011S, 1:1,000), anti-HDAC1 (Cell Signaling Technology, #34589S, 1:1,000), anti-HDAC2 (Cell Signaling Technology, #5113S, 1:1,000), anti-HA (Cell Signaling Technology, #3724S, 1:1,000), anti-MBD3 (Cell Signaling Technology, #99169S, 1:500), anti-MTA2 (Bethyl, #A300–395A, 1:1,000), anti-MTA3 (Proteintech, #14682–1-AP, 1:1,000), anti-H3 (Cell Signaling Technology, #14269S, 1:2,000), anti-H3K9ac (Cell Signaling Technology, #9649S, 1:1,000), anti-H3K27ac (Abcam, #ab4729, 1:1,000), anti-H3K27me3 (Cell Signaling Technology, #9733S, 1:1,000), anti-GAPDH (Santa-Cruz, #sc-32233, 1:3,000).
Subcellular fractionation.
Lysates of different subcellular fractions from 1 million cells per sample were prepared with Subcellular Protein Fractionation Kit for Cultured Cells (Thermo Scientific, #78840) following manufacturer’s instructions and subjected to immunoblot. GAPDH (Santa-Cruz, #sc-32233, 1:3,000) was used as marker for cytoplasmic fraction; snRNP70 (Santa-Cruz, #sc-390899, 1:1,000) was used as the marker for nuclear fraction. H3 (Cell Signaling Technology, #14269S, 1:2,000) was used as the marker for chromatin fraction.
Engineering of dTAG cell line.
The HDAC2-dTAG cell line was engineered using the PITCh system91, as previously described51, using microhomology-mediate end joining (sgRNA: 5’-GTTGCTGAGCTGTTCTGATT-3’) to insert a linker-FKBP12F36V-2xHA-P2A-BSDR cassette into the C-terminus of HDAC2. BE(2)-C cells were transfected with both CRIS-PITCh plasmids, selected by 10 μg/mL Blasticidin S HCl, and clonally expanded to confirm biallelic knock-in by PCR and immunoblot.
Cellular proliferation.
BE(2)-C cells were plated into 12-well plates with 12,000 cells in 1 ml medium per well. DMSO and compounds were added 1:1000 in triplicates. Every 3 to 4 days cells were trypsinized, counted by Countess automated cell counter (Invitrogen), and re-plated at the concentration of 12,000 cells/ml with fresh DMSO or compounds.
CRISPR-Cas9 competitive growth assays.
Cas9 expressing cells were created by transduction of lentiCas9-Blast (a gift from Feng Zhang, Addgene plasmid #52962; http://n2t.net/addgene:52962; RRID:Addgene_52962) virus and selected by 10 μg/mL Blasticidin S HCl. sgRNA sequences were cloned into the LRG backbone92 (a gift from Christopher Vakoc, Addgene plasmid #65656; http://n2t.net/addgene:65656; RRID:Addgene_65656) for co-expression of an sgRNA and GFP. A modified version of the LRG vector with mCherry replacing GFP was also developed for this study. Cas9-expressing cells were transduced in triplicate with the corresponding sgRNA-LRG virus at 30–60% efficiency in 96-well plates. For two-color experiments, cells were transduced with GFP-sgRNA1 and mCherry-sgRNA2 simultaneously to achieve ~25% of each population (non-fluorescent, GFP or mCherry positive, GFP and mCherry positive). Transduced cells were passaged and subjected to flow cytometry to measure GFP- and/or mCherry-positive percentage every 3 to 4 days. sgRNA sequences, which were taken from the Brunello sgRNA library93, are as follows: AAVS1 5’-GGGGCCACTAGGGACAGGAT-3’; RPS19 5’-GTAGAACCAGTTCTCATCGT-3’; Luciferase 5’-CCCGGCGCCATTCTATCCGC-3’; HDAC1-sg1 5’-CATCCGTCCAGATAACATGT-3’; HDAC1-sg2 5’-TGAGTCATGCGGATTCGGTG-3’; HDAC1-sg3 5’-GCACCGGGCAACGTTACGAA-3’; HDAC1-sg4 5’-GGAGATGTTCCAGCCTAGTG-3’; HDAC2-sg1 5’-GATGTATCAACCTAGTGCTG-3’; HDAC2-sg2 5’-TACAACAGATCGTGTAATGA-3’; HDAC2-sg3 5’-CCTCCTCCAAGCATCAGTAA-3’; HDAC2-sg4 5’-TCAAAGAGTCCATCAAACAC-3’. For sgAAVS1, cutting efficiency was determined by quantitative sequence trace decomposition (tracking of indels by decomposition software; TIDE)94 following PCR amplification and Sanger sequencing of the cut site (5’-AGGTGGGGGTTAGACCCAAT-3’, 5’-CTTCTCCGACGGATGTCTCC-3’). The efficiency of all other sgRNAs was confirmed by immunoblot.
Generation of knockout populations.
To create loss-of-function knockout populations, Cas9-expressing cells were transduced with the corresponding sgRNA-LRG vector to achieve 90–100% efficiency, as confirmed by flow cytometry. Population-level knockouts were validated by immunoblot of the corresponding protein.
Immunoprecipitation.
50 million cells were collected after drug treatment and washed twice with 5 mL cold PBS. Cells were then lysed in 1 ml lysis buffer (50 mM HEPES, 150 mM NaCl, 1.5 mM MgCl2, 1% NP40, 125 U benzonase, and 1X Halt Protease Inhibitor Cocktail) with mild sonication and incubated on a rotating platform at 4° C for 3 hours. The supernatant was collected following 18,000 g centrifugation at 4° C for 20 mins. The protein content for each sample was quantified by BCA assay and normalized to the same level. For CHD4 immunoprecipitations, 50 μL of Dynabeads protein G (Invitrogen, #10003D) were combined with 10 mg antibody (anti-CHD4, Abcam, #ab70469–10) and rotated at room temperature for 30 mins to make antibody-beads conjugates, which were then washed three times supplemented with 1 mL PBS with 0.05% Tween20. For HA immunoprecipitations, 30 μL Pierce anti-HA Magnetic Beads (Thermo Fisher Scientific, #88836) were used for each sample. The antibody-bead conjugates were added to the protein lysate and rotated at 4° C overnight for immunoprecipitation. The beads were washed with 1 mL cold wash buffer (50 mM HEPES, 150 mM NaCl, and 0.2% NP40) for three times and eluted with 1X Bolt LDS Sample Buffer (Invitrogen, #B0007) supplemented with 2.5% 2-Mercaptoethanol by incubation at 95° C for 10 mins. Eluted samples were subjected to immunoblot.
Animal experiments.
Mouse experiments were performed in accordance with the Institutional Animal Care and Use Committee and conform to the guidelines for the care and use of laboratory animals approved at The Scripps Research Institute (protocol #18–031-01). Xenografts were established in six-week-old female NOG (NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac) mice (Taconic) by flank injection of 0.8 million cells in 50% Matrigel (BD Biosciences) in DMEM (Gibco, ThermoFisher Scientific), total volume 50 μL. Experimental groups consisted of 5 mice, with two flank tumors each. Tumor burden was assessed by measuring tumor length and width using digital calipers once per week for the first two weeks then three times per week for up to six weeks. Caliper measurements of the longest axis were used to calculate the diameter. Serial caliper measurements of perpendicular axes were used to calculate tumor volume by the following formula: (short diameter × 2) × (long diameter) × 0.5. Mice were sacrificed once the longest diameter of at least one flank tumor reached 15 mm. At the endpoint, tumors were collected for molecular studies. All animals were maintained at sterile conditions with an ambient temperature of 68 F (20 c) to 79 F (26 c), humidity of 30–70% (on average 50%), and a light/dark cycle of 12 hours.
Cleavage Under Targets & Release Using Nuclease (CUT&RUN).
All CUT&RUN experiments were performed as previously described54 with minor modifications. Briefly, 50,000 cells were collected and washed twice with 1 mL wash buffer (20 mM HEPES-KOH, 150 mM NaCl, 0.5 mM spermidine, 1X HALT protease inhibitor cocktail) at room temperature for each sample. Concanavalin A coated magnetic beads (BioMag, #BP531, 40 μL per sample) were activated and used to capture cells by incubating at room temperature on a rotating platform for 15 mins. Cells were permeabilized on beads with 250 μL antibody buffer (20 mM HEPES-KOH, 150 mM NaCl, 0.5 mM spermidine, 0.02% digitonin, 2 mM EDTA, 1 mM EGTA, 1X HALT protease inhibitor cocktail) with 1:100 corresponding antibody (anti-CHD4, Cell Signaling Technology, #12011S; anti-HDAC2, Active motif, #39533; anti-HA, Cell Signaling Technology, #3724S; anti-MBD3, Cell Signaling Technology, #99169S; anti-H3K9ac, Cell Signaling Technology, #9649S; anti-H3K27ac, Abcam, #ab4729; anti-IgG, CiteAb, ABIN101961). Samples were incubated at 4° C for 2 hours on a rotating platform, washed with 1 mL cold digitonin buffer (20 mM HEPES-KOH, 150 mM NaCl, 0.5 mM spermidine, 0.02% digitonin) twice, and then incubated with 350 ng of pA-MNase (a gift from the Henikoff lab) in 250 μL of digitonin buffer at 4° C for 1 hour. Samples were then washed three times with 1 mL cold digitonin buffer, resuspended in 150 μl digitonin buffer, and placed on a metal block equilibrated to 0° C. The MNase digestion was activated with 2 mM CaCl2 and performed for 30 mins at 0° C with occasional mixing of the beads. The reaction was stopped by addition of 150 μL 2X stop buffer (340 mM NaCl, 20 mM EDTA, 4 mM EGTA, 0.02% digitonin, 0.05 mg/mL RNase A, 0.05 mg/mL glycogen). The CUT&RUN fragments were released from cells by incubation at 37° C for 30 mins and collected from the supernatant. Additional 2 μL of RNase A (10 mg/mL) was added to each sample and incubated at 37° C for 30 mins. 0.1% SDS and 0.2 mg/mL proteinase K were added for protein digestion performed at 70° C for 30 mins. The DNA fragments were collected by phenol-chloroform extraction and resuspended in 25 μL of DNase-free water. The fragment distribution was then checked by TapeStation (Agilent). Sequencing libraries were prepared using Thruplex DNA-seq kit (Takara, # R400675) and 10 μL of input material. Libraries were amplified with modified conditions to allow preferential amplification of the short target fragments from CUT&RUN (16 cycles of 98° C for 15 s and 60° C for 10 s). Libraries were then purified with AMPure XP beads (Beckman Coulter, #A63880) and sequenced on Illumina NextSeq 2000 (paired-end, 35-bp reads). The sequencing reads were aligned to human genome build hg19 and RefSeq genes by Bowtie2 with the following parameters: --local --very-sensitive-local --no-unal --no-mixed --no-discordant --phred33 -I 10 -X 700 --threads 12 -x. Using the IgG sample as background, the signal-enriched regions were identified by Model-based Analysis of ChIP-seq (MACS) peak-finding algorithm (v1.4.1) with the P value threshold of 1e-9. For differential analyses across samples, the read density calculator Bamliquidator (v1.0) (http://github.com/BradnerLab/pipeline/wiki/bamliquidator) was used to map sequencing reads to designated loci with a 200 bp extension window in either direction. The read density per base pair was calculated with the normalization to the total number of million mapped read (RPM/bp). ROSE2 was used with default parameters to identify asymmetric binding by HDAC2 (https://github.com/linlabbcm/rose2). H3K27ac ChIP-seq in wild-type BE(2)-C (GEO ID: GSM2113518) and its input sample (GEO ID: GSM2113520) were obtained from a previous study43 and used to predict enhancers and super enhancers with ROSE2. H3K4me3 ChIP-seq in wild-type BE(2)-C (GEO ID: GSM2113519) from a previous study43 was used as a mark for active promoters.
Thiol(SH)-linked alkylation for the metabolic sequencing of RNA (SLAM-seq).
SLAM-seq was performed as previously described55 using the SLAMseq Kinetics Kit (Lexogen, #061.24). Briefly, 1 million cells were pretreated with 0.1% DMSO, 500 nM dTAG-13, or 5 μM ACY-957 in triplicate. 100 μM 4-thiouracil (4sU) was added to the cells to label newly synthesized mRNA for 1 hour before the end of compound treatment. Total RNA for each sample was extracted with RNeasy miniprep kit (Qiagen) and quantified by Qubit RNA XR Assay kit (Invitrogen, # Q33223). 5 μg RNA was subjected to alkylation by 100 mM iodoacetamide. The alkylated RNA was purified by ethanol precipitation. 1 μg of RNA for each sample was used to prepare the library for sequencing with QuantSeq 3’ mRNA-Seq Library Prep Kit FWD for Illumina (Lexogen, #015) and sequenced on Illumina NovaSeq 6000 (single-end, 101-bp reads). The sequencing data was analyzed by SlamDunk94 with default settings to align to human genome build hg19, and to calculate counts per million (CPM) and T-to-C conversion rate at 3’ UTRs (built from ncbiRefSeqCurated table for hg19 accessible on UCSC table browser, including 29,870 3’ UTRs for 22,514 genes).
3’ mRNA-seq with ERCC RNA Spike-In Mix normalization.
Cells were plated in 6-well plates with equal number and treated in triplicates. Prior to collection of the cells, equal cell numbers and viability were confirmed across samples. Total RNA was extracted with RNeasy miniprep kit (Qiagen). 350 μL of lysis buffer (RLT) supplemented with 2.1 ng/mL of ERCC RNA Spike-In Control SIRV Set 3 (Lexogen, #051.0) for spike-in normalization was used to lyse cells for each sample. 1 μg of RNA for each sample was used to prepare sequencing library with QuantSeq 3’ mRNA-Seq Library Prep Kit FWD for Illumina (Lexogen, #015). The libraries of 8-h and 24-h DMSO and 500 nM dTAG-13 treatments were sequenced on Illumina NovaSeq (single-end, 101-bp reads); The libraries of 24-h DMSO and 5 μM ACY-957 treatments were sequenced on Illumina Nextseq 500 (single-end, 100-bp reads); The libraries of 72-h DMSO and 500 nM dTAG-13 treatments were sequenced on Illumina Nextseq 2000 (single-end, 75-bp reads). SlamDunk was used to align the sequencing reads to human genome build hg19 and ERCC SIRV Set3 reference with -q flag to ignore the conversion rate calculation for SLAM-seq. To normalize the CPM value for each transcript in each sample based on spike-in RNA, the normalize.loess function of the affy v1.50.0 R package76 was used. Locally estimated scatterplot smoothing (LOESS) normalization was applied to all CPM values of transcripts with the model specified by the distribution of the SIRV-Set-3 spike-ins. Unexpressed genes were filtered out with the cutoff of CPM < 3 in all 8-h and 24-h DMSO and 500 nM dTAG-13 treated samples.
Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq).
50,000 treated BE(2)-C cells for each sample were washed with 50 μL cold PBS once. Pellets were resuspended in 50 μL cold Omni-ATAC lysis buffer (10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40, 0.1% Tween20, 0.01% digitonin) and incubated on ice for 3 mins, then mixed with 1 mL Omni-ATAC wash buffer (10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween20). Nuclei were pelleted by centrifugation and resuspended in 50 μL Omni-ATAC transposition reaction mixture (25 μL Nextera TD buffer, 16.5 μL PBS, 0.1% Tween20, 0.01% digitonin, 2.5 μL Tn5 transposase) and incubated at 37 °C for 30 mins. DNA was purified using Quick-DNA MicroPrep column purification kit (Zymo Research, #D3020). The libraries were generated by PCR amplification with the cycle numbers determined by qPCR side-reactions as previously described95. DNA libraries were purified with Quick-DNA MicroPrep column purification kit (Zymo Research, #D3020). The quality of the libraries was assessed with TapeStation and the DNA contents were quantified by Qubit. The libraries were sequenced on Illumina NextSeq 500 (paired-end, 36-bp reads). Reads were processed with the ENCODE ATAC-seq pipeline (v1.8.0) and were aligned to the human genome build hg19. Adapters were trimmed with Cutadapt (v3.0) and reads were aligned with Bowtie2 (v2.4.2). Blacklist regions excluded using data file wgEncodeDacMapabilityConsensusExcludable.bed.gz. Unmapped/low quality reads were filtered with Samtools (v1.11) and duplicated reads were removed with Picard Tools (v2.23.9). Peaks were called by MACS2 (v2.2.7.1) with call-peak P value threshold of 0.01, maximum peaks of 5×105, size of smoothing window of 150, and IDR (irreproducible discovery rate) threshold of 0.05. GC bias computation was performed and TSS (transcription start site) enrichment was calculated on filtered reads. For the differential analysis, the bed files from the same time point were merged with Bedtools (v2.27.1) to generate a peak list including peaks from all treatments. In the merged bed files the adjacent peaks with distances less than 1 kb were merged. The read density calculator Bamliquidator (v1.0) (http://github.com/BradnerLab/pipeline/wiki/bamliquidator) was used to quantify the read density of each treatment at each peak in the corresponding merged bed file with RPM normalization. Peaks with low density (< 0.1 RPM/bp) from all treatments were filtered out. The Log2-transformed fold change and the P values given by Student’s t test were calculated for each peak.
Quantitative proteomics.
BE(2)C-HDAC2-dTAG cells were treated with 500 nM dTAG-13 for 2 h or 24 h, collected by trypsinization, and washed twice with PBS before flash freezing in liquid nitrogen. Cell pellets were lysed by probe sonication in resuspension buffer (10 mM NaHPO4 pH 7.5, 125 mM NaCl, 25 mM KCl, 1.5 mM MgCl2, 10% glycerol, 1X HALT Halt Protease Inhibitor Cocktail). Protein concentration was adjusted to 2 mg/mL and 200 μg protein per sample was combined with 48 mg urea to give 8 M final concentration. Samples were reduced by addition of DTT (10 mM final concentration) at 65 °C for 20 mins, before alkylation with iodoacetamide (20 mM) for 30 mins on a 37°C shaking incubator protected from light. Ice-cold H2O (500 μL), MeOH (600 μL), and CHCl3 (100 μL), were added and the mixture was vortexed and centrifuged (10,000 g, 10 mins, 4°C) to afford a protein precipitate at the interface of CHCl3 and aqueous layers. Solvent was removed and protein washed with additional MeOH (600 μL) before being allowed to dry briefly at room temperature. The resulting protein pellets were resuspended in EPPS buffer (160 μL, 200 mM, pH 8) by probe sonication. LysC solution (0.5 ug/sample in diH2O) was added and the samples were incubated at 37 °C with shaking for 2 h. Trypsin (1 ug in trypsin buffer) and CaCl2 (1 μL, 100 mM in H2O) were then added and the samples were incubated at 37 °C with shaking overnight. Peptide concentration was determined using the microBCA assay (Thermo Scientific) according to manufacturer’s instructions and samples normalized. To a volume corresponding to 25 μg per sample, CH3CN was added to a final concentration of 30% before incubation with the corresponding TMT tags (60 μg/sample) at room temperature for 30 mins. Additional TMT tag (60 μg/sample) was added and the samples were incubated for another 30 mins. Labeling was quenched by the addition of hydroxylamine (6 μL, 5% in H2O). Following a 15 mins incubation at room temperature, formic acid was added to a concentration of 5% and the samples were stored at −80°C until further analysis. Sep Pak desalting, HPLC high-pH fractionation, Orbitrap Fusion (Thermo) Nano-LCMS data collection, and Integrated Proteomics Pipeline (IP2) data analysis were performed as described previously68. Data was filtered for 10,000 reporter ion control sum intensity, maximum control channel coefficient of variation < 0.5, and 2 unique peptides identified per protein.
Gene set enrichment analysis (GSEA).
The averages of the replicates from the control group or the treatment group were calculated respectively for each gene and used as the input for GSEA. The GSEA software developed by UC San Diego and Broad Institute98,97 was used with the following parameters: gene sets database c2.all, number of permutations 1000, collapse no_collapse, permutation type gene_set.
qRT-PCR.
RNA was extracted with the RNeasy miniprep kit (Qiagen). 2.5 μg of RNA from each sample was used to synthesize cDNA with the SuperScript VILO cDNA Synthesis Kit (Life Technologies, #11755050). Quantification of each transcript of interest was performed in triplicates with SYBR Select Master Mix (Life Technologies, #4472908) on BioRad CFX 384 with primer pairs listed below. Cycle threshold (Ct) values were determined by the default setting on BioRab CFX Manager (v3.0) and the relative expression level of each gene was calculated with normalization to B2M transcript levels and DMSO control using the ddCt method.
Primer pairs for qRT-PCR.
B2M: forward 5’-TCTCTGCTGGATGACGTGAG-3’, reverse 5’-TAGCTGTGCTCGCGCTACT-3’. GAPDH: forward 5’-CATCATCCCTGCCTCTACTG-3’, reverse 5’-GCCTGCTTCACCACCTTC-3’. ACTB: forward 5’-TGGCACCCAGCACAATGAA-3’, reverse 5’-CTAAGTCATAGTCCGCCTAGAAGCA-3’. CHD4: forward 5’-GCTGCAACCATCCATACCTC-3’, reverse 5’-ACCATCGATGCGTTCGTATT-3’. HDAC1: forward 5’-GGAAATCTATCGCCCTCACA-3’, reverse 5’-AACAGGCCATCGAATACTGG-3’. GATAD2A: forward 5’-ACGAGTTCATCTACCTGGTCGG-3’, reverse 5’-ACGTGAAGTCCGTCTTGCACTG-3’. GATAD2B: forward 5’-CAAAAGCTGTGCCTCACTTC-3’, reverse 5’-TTCCAGTGAGGGGTGAAATC-3’. MBD2: forward 5’-AAGTGCTGGCAAGAGCGATGTCTA-3’, reverse 5’-TTTCCCAGGTACCTTGCCAACTGA-3’. MBD3: forward 5’-GGCCACAGGGATGTCTTTTACT-3’, reverse 5’-CGGCTTGCTGCGGAACT-3’. MTA1: forward 5’-TGCTCAACGGGAAGTCCTACC-3’, reverse 5’-GGGCATGTAGAACACGTCACC-3’.
MTA2: forward 5’-TATCACTCTGTTTCACGCCA-3’, reverse 5’-ACCATTCCTCCATCTCATCC-3’. MTA3: forward 5’-AAGCCTGGTGCTGTGAAT-3’, reverse 5’-AGGGTCCTCTGTAGTTGG-3’. IRS2: forward 5’-GAGTGCACCCGTACCTATGGAA-3’, reverse 5’-GAAATCCGGCTTTACCTTGAACT-3’.
Statistics and Reproducibility.
Experiments shown in Figures 3h, 4e, 5e–h and Extended Data Figures 1d, 2b, 2e, 5c–d, 5h, 8d–e were repeated independently for at least two times with consistent results ovserved. The exact P values not shown in the figures are as following: Fig. 1b: P < 1E-10; Fig. 1c: P < 1E-10; Fig. 1f: P = 3E-10; Fig. 2b: P = 3.62E-07; Fig. 2d: P = 3.57E-06; Fig. 4g: Other vs Typical P = 1.66E-08, Other vs Asymmetric P = 5.30E-04; Fig. 6g: MBD2 Other vs MM P < 1E-10, MBD3 Other vs NB P = 1E-10; MTA2 Other vs MM P < 1E-10; Extended Data Fig. 2g: P = 1.60E-05; Extended Data Fig. 2h: P < 1E-10; Extended Data Fig. 6e: P < 1E-10; Extended Data Fig. 6j: No enhancer vs SE P = 3E-09, TE vs SE P = 7.00E-06; Extended Data Fig. 8f: ELMSAN1 P = 2.90E-05, DNTTIP1 P = 7.6E-08; Extended Data Fig. 10a: SAP30 Other vs NB P = 2.00E-06.
Extended Data
Extended Data Fig. 1. HDAC1 and HDAC2 are selective dependencies in neuroblastoma and lymphoid malignancies.
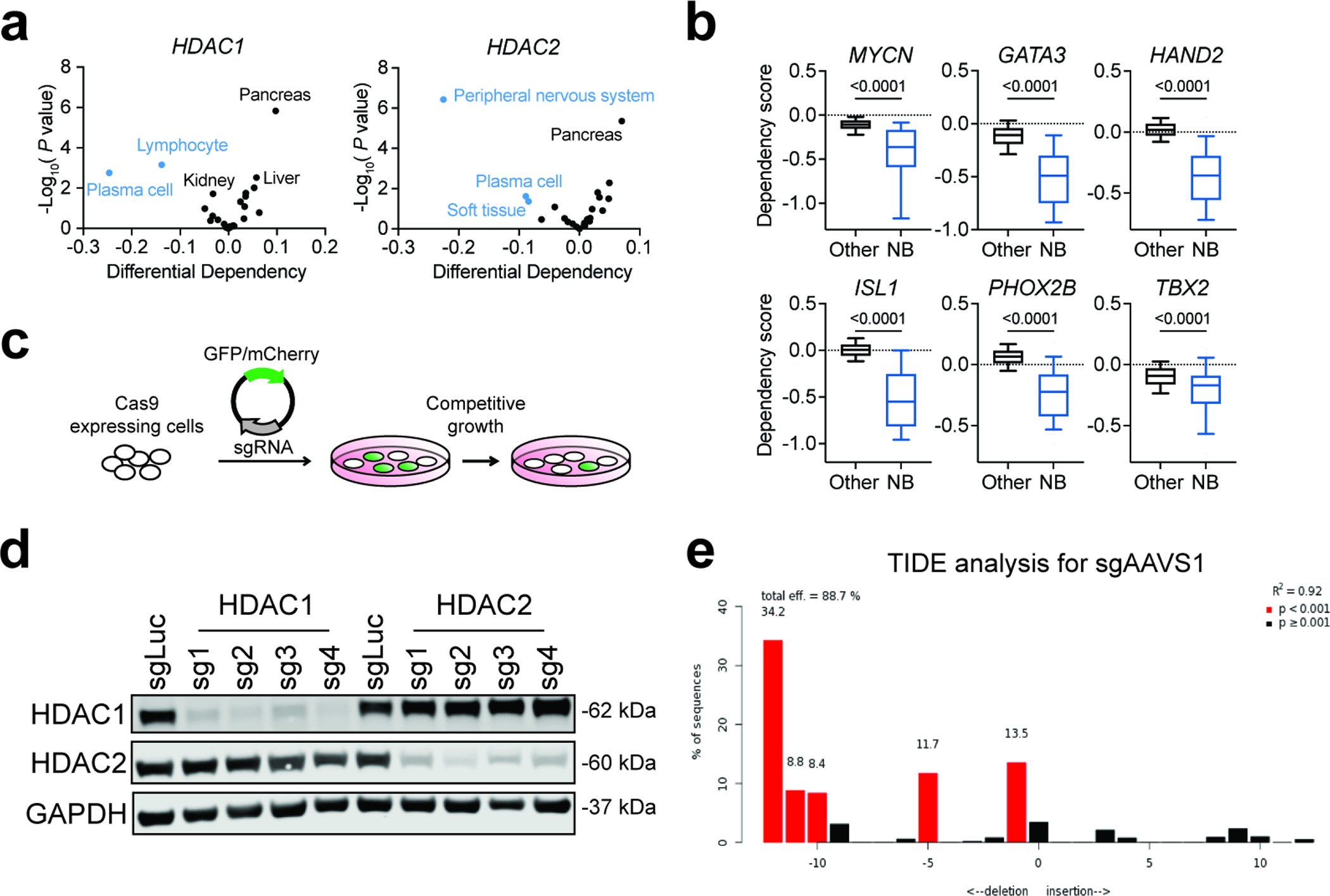
a, Volcano plots of HDAC1 (left) and HDAC2 (right) differential dependencies in cancer lineages. P value were determined by two-tailed Student’s t-test. b, Boxplots of dependency scores of core regulatory circuitry (CRC) transcription factor genes in neuroblastoma (blue, n = 34) and other cancer cell lines (black, n = 1,020). Boxes represent 25–75 percentiles with whiskers extending to 10–90 percentiles and the center line represents the median of the data. P values were determined by two-tailed Student’s t-test. c, Schematic illustration of CRISPR/Cas9-based competitive growth assay. d, Validation of on-target effects of HDAC1 and HDAC2 guides by immunoblot. e, TIDE (tracking of indels by decomposition) analysis shows a cutting efficiency of 88.7% for sgAAVS1. P values were determined by two-tailed t-test of the variance.
Extended Data Fig. 2. Selective dependencies of HDAC1 and HDAC2 are validated in human cell lines and in vivo.
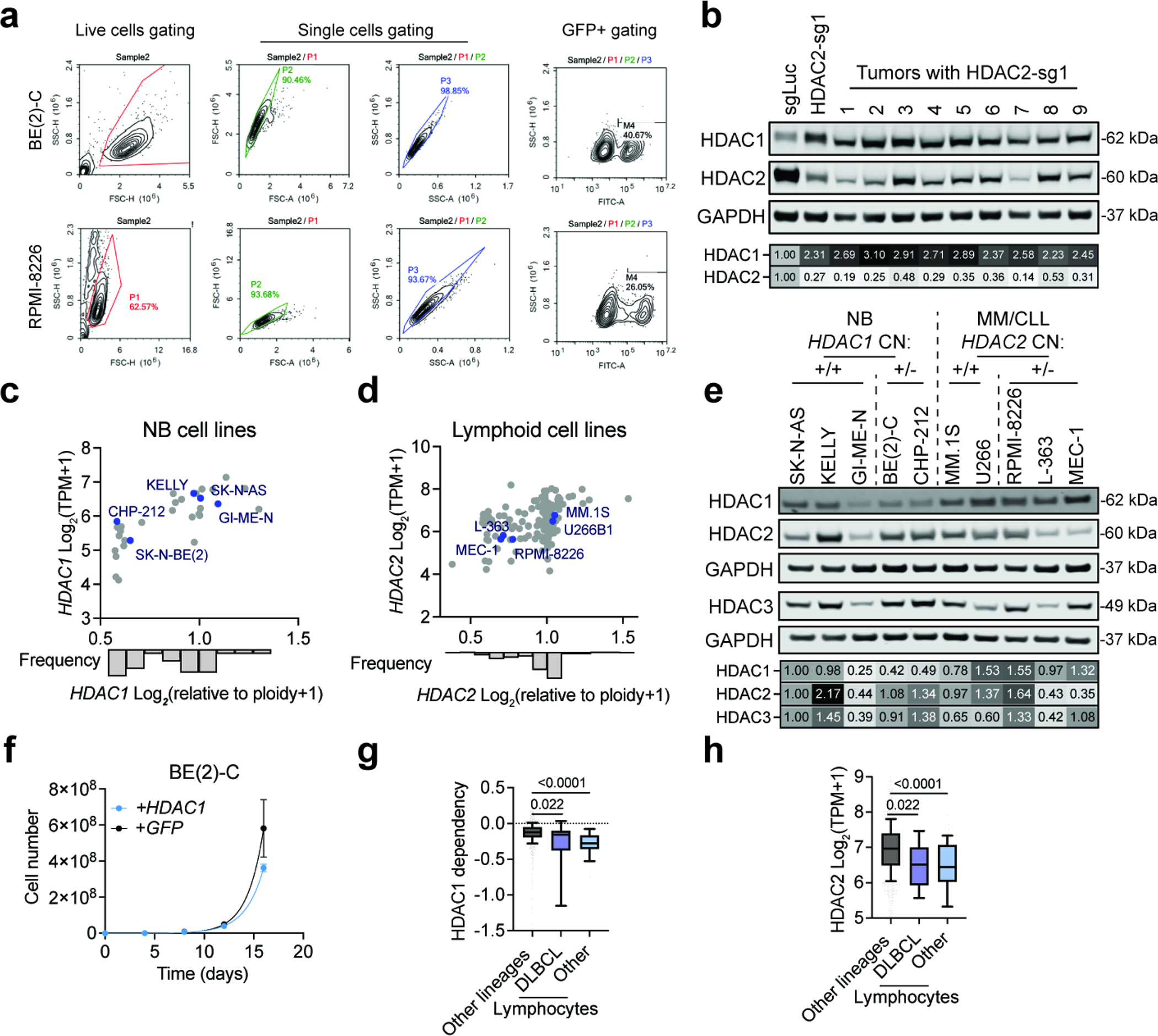
a, Representative plots of flow cytometry gating for CRISPR/Cas9-based competitive growth assay in BE(2)-C and RPMI-8226 cells. b, Protein levels of HDAC1 and HDAC2 in pre-xenografting BE(2)-C cells with sgLuc or HDAC2-targeting guide and end-point tumors with HDAC2-targeting guide. c. HDAC1 expression versus HDAC1 copy number in all neuroblastoma cell lines. d. HDAC2 expression versus HDAC2 copy number in all lymphoid cell lines. Data from DepMap, CCLE expression and gene copy number 22Q4. e. Protein levels of HDAC1, HDAC2, HDAC3 in the neuroblastoma and lymphoid cell line, including multiple myeloma (MM) and chronic lymphocytic lymphoma (CLL) with/without HDAC1/2 hemizygous deletions highlighted in (c) and (d). f. Proliferation of BE(2)-C cells overexpressing GFP or HDAC1. Mean ± s.e.m., n = 3. g, Boxplots of HDAC1 dependency scores in DLBCL lines (purple, n = 8), non-DLBCL lymphocyte lines (blue, n = 19), and other lineages (grey, n = 962). h, Boxplot of HDAC2 transcript levels in DLBCL lines (purple, n = 20), non-DLBCL lymphocyte lines (blue, n = 63), and other lineages (grey, n = 1,292). Data from DepMap, CRISPR_genetic_effect 22Q1 and CCLE_expression 22Q1. Boxes represent 25–75 percentiles with whiskers extending to 10–90 percentiles and the center line represents the median of the data. P values were determined by two-tailed Student’s t-test.
Extended Data Fig. 3. Hemizygous deletion of HDAC1 leads to high dependency on HDAC2 neuroblastoma.
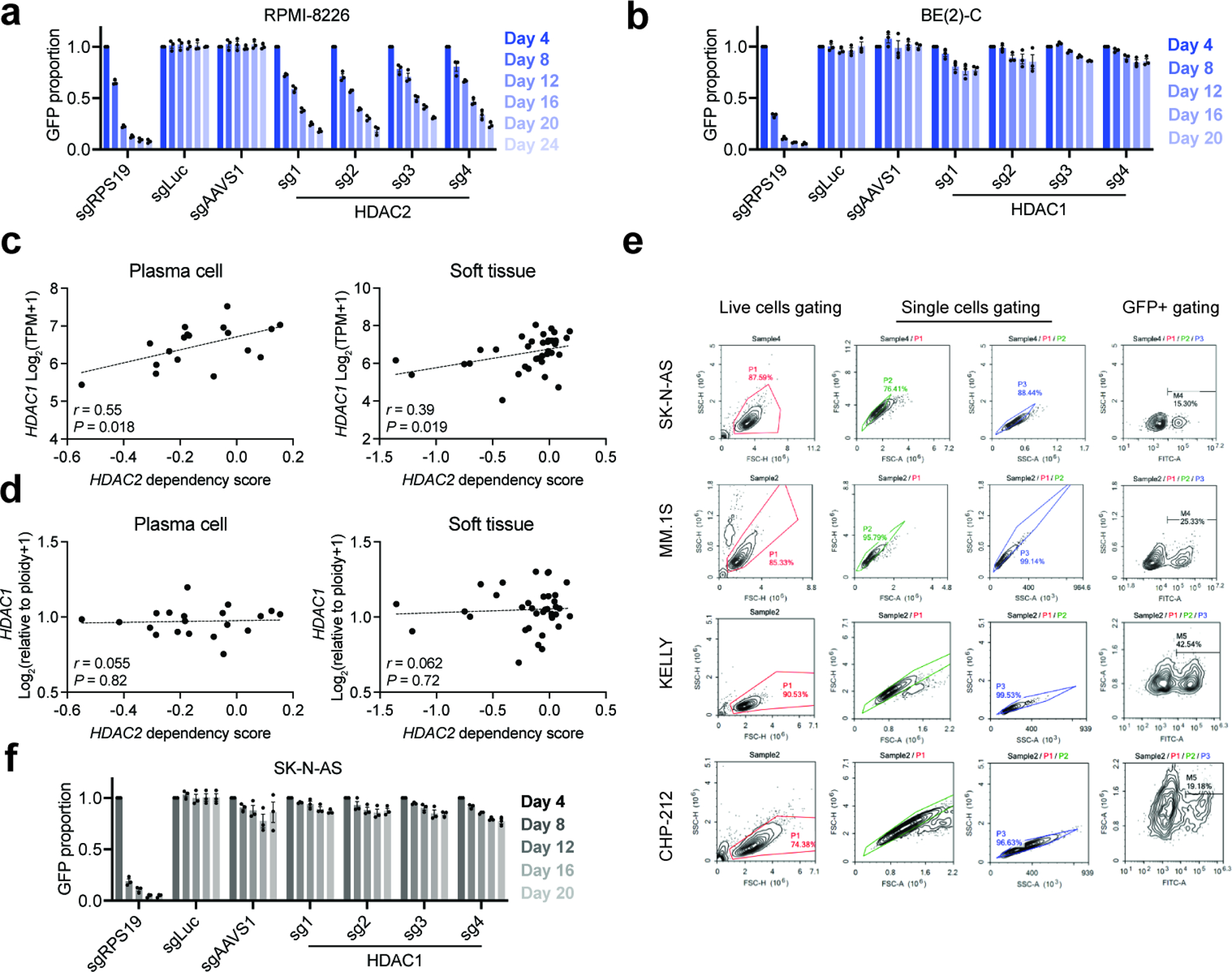
a, Competitive growth assays with HDAC2-targeting guides in and RPMI-8226. Mean ± s.e.m., n = 3. Experiments were performed at the same time with Fig. 1e, hence shared the same control groups. b, Competitive growth assays with HDAC1-targeting guides in and BE(2)-C cells. Mean ± s.e.m., n = 3. Experiments were performed at the same time with Fig. 1d, hence shared the same control groups. c,d HDAC1 RNA expression (c) or HDAC1 copy number (d) versus HDAC2 dependency in plasma cell lines (left) and soft tissue lines (right). P values (two-tailed) were determined by Pearson correlation coefficient (r). Data from DepMap, CCLE expression and gene copy number 22Q4. e, Representative plots of flow cytometry gating for CRISPR/Cas9-based competitive growth assay in SK-N-AS, MM.1S, KELLY, and CHP-212 cells. f, Competitive growth assays with HDAC1-targeting guides in and SK-N-AS cells. Mean ± s.e.m., n = 3. Experiments were performed at the same time with Fig. 3b, hence shared the same control groups.
Extended Data Fig. 4. HDAC1 and HDAC2 form synthetic lethality in multiple myeloma and neuroblastoma.
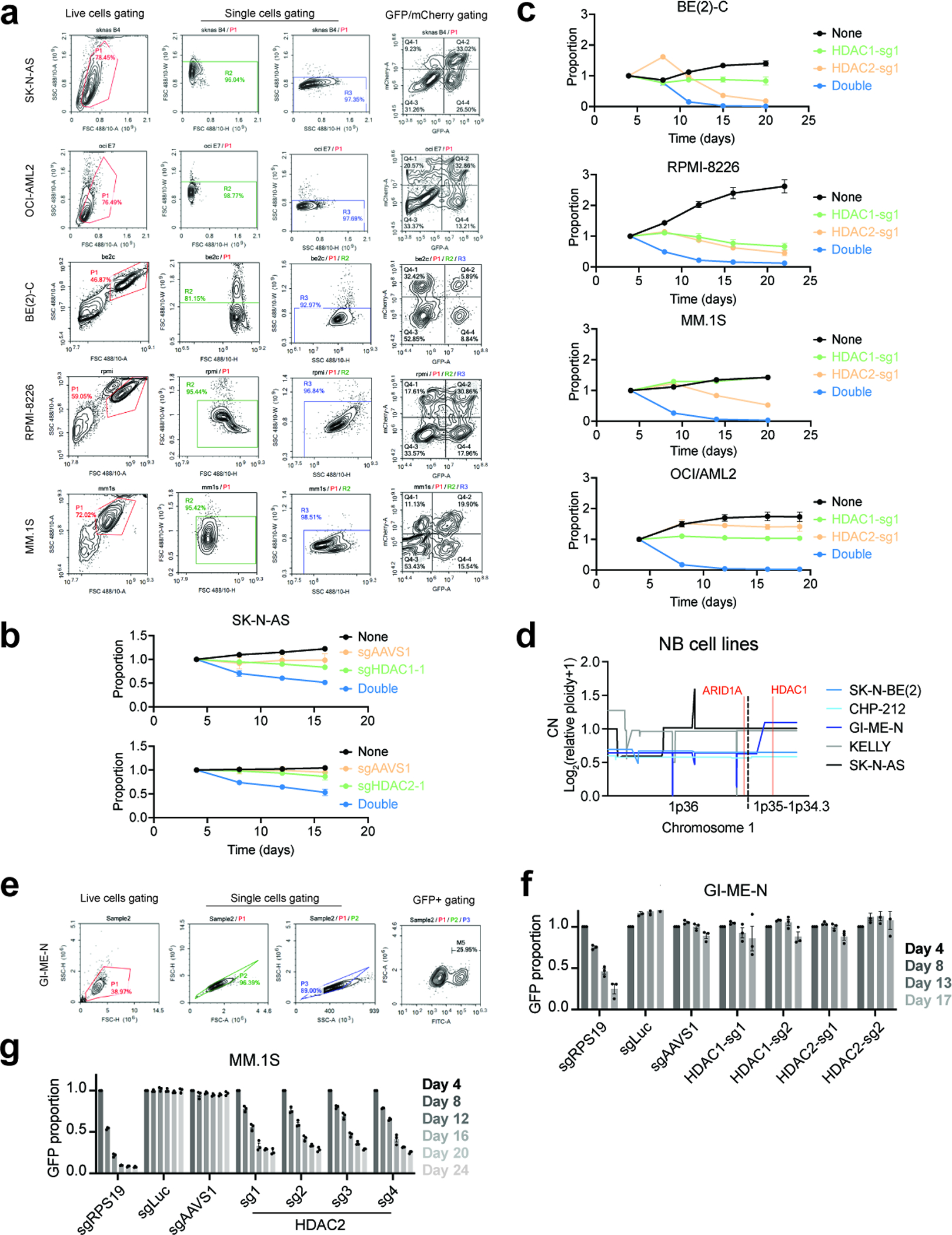
a. Representative plots of flow cytometry gating for CRISPR/Cas9-based two-color competitive growth assay in SK-N-AS, OCI-AML2, BE(2)-C, RPMI-8226, and MM.1S cells. b, Control groups related to the two-color competitive growth assay in Fig. 3e. c, Two-color competitive growth assay with HDAC1-sg1 and HDAC2-sg1 in BE(2)-C, RPMI-8226, MM.1S, and OCI-AML2 cells. Mean ± s.d., n = 3. Proportion of each sub-population normalized to day 4. d, Copy number of genes located at 1p36–1p34.3 in neuroblastoma cell lines highlighted in Extended Data Fig. 3a. Data from CCLE copy number, 22Q4. e. Representative plots of flow cytometry gating for CRISPR/Cas9-based competitive growth assay in GI-ME-N cells. f. Competitive growth assay with HDAC1 or HDAC2 targeting guides in GI-ME-N cells which harbor 1p36 deletion but not HDAC1 deletion. Mean ± s.e.m., n = 3. g, Competitive growth assay with HDAC2 targeting guides MM.1S cells. Mean ± s.e.m., n = 3. Experiments were performed at the same time with Fig. 3f, hence shared the same control groups.
Extended Data Fig. 5. dTAG system allows efficient degradation of HDAC2 without affecting its normal functions.
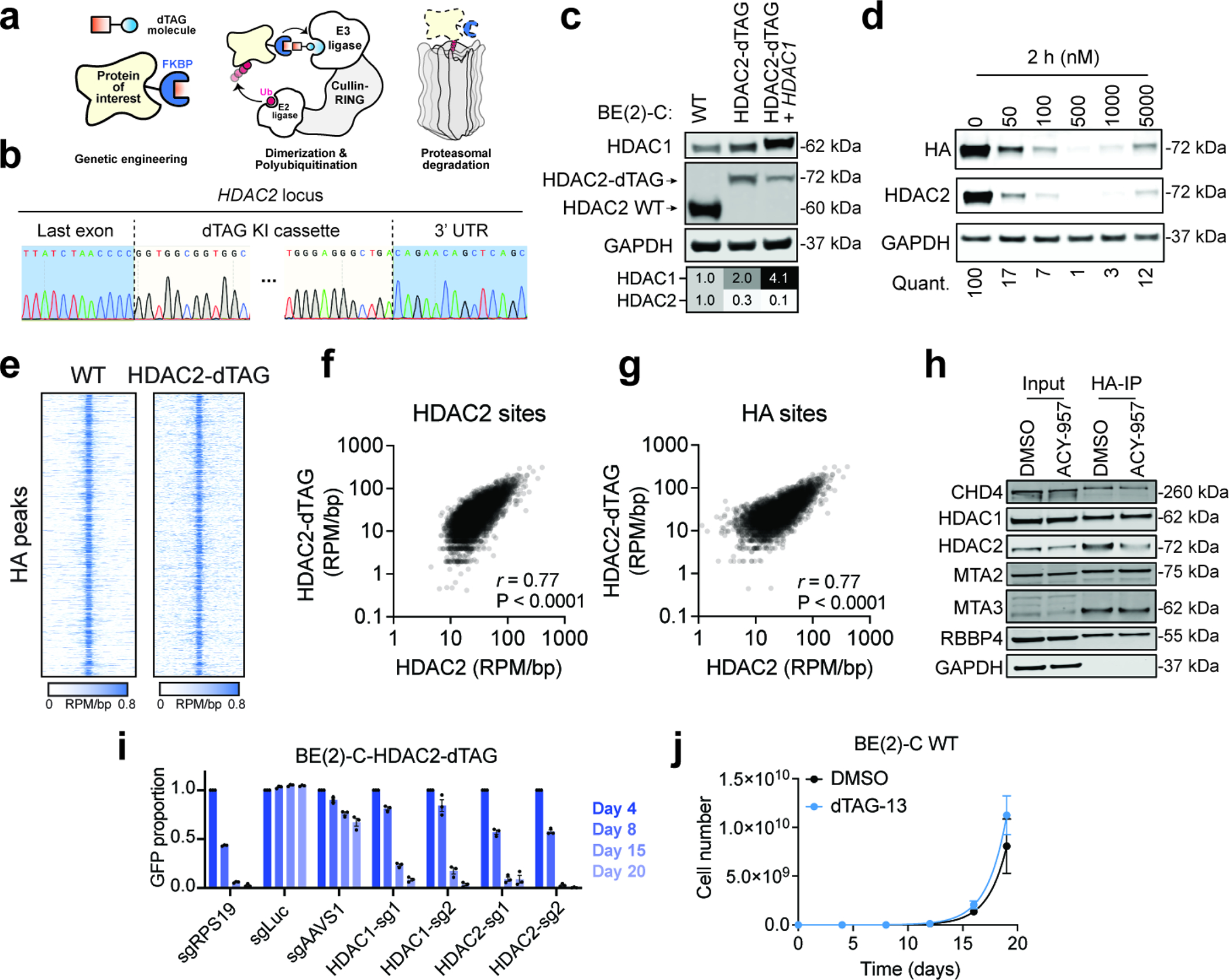
a, Schematic illustration of the dTAG system. dTAG PROTACs mediate dimerization of the FKBP12F36A-tagged protein of interest and an E3 ubiquitin ligase, which results in ubiquitination and proteasomal degradation of the target protein. b, Representative Sanger sequencing chromatograms of HDAC2 locus of a clone with successful dTAG knock-in. c, Immunoblot validation of HDAC2-dTAG cell lines with and without HDAC1 overexpression. d, Dose response of dTAG-13 treatment in BE(2)-C-HDAC2-dTAG cells (2 h). e, Rank-ordered heatmaps of CUT&RUN signal for HDAC2 in wild-type BE(2)-C cells and HA in BE(2)-C-HDAC2-dTAG cells (ranked based on HA signal at HDAC2-HA binding sites in BE(2)-C-HDAC2-dTAG cells). f,g Correlations of HDAC2 CUT&RUN in BE(2)-C cells versus HA CUT&RUN in BE(2)-C-HDAC2-dTAG cells at HDAC2 binding sites in wild-type BE(2)-C cells (f) (n = 10,832) and at HDAC2-HA binding sites in BE(2)-C-HDAC2-dTAG cells (g) (n = 8,661). P values (two-tailed) were determined by Pearson correlation coefficient (r). h, Co-immunoprecipitation of HDAC2-dTAG (IP: HA) and NuRD subunits. i, Competitive growth assay with HDAC1- or HDAC2-targeting guides in BE(2)-C-HDAC2-dTAG cells. Mean ± s.e.m., n = 3. j, Proliferation of BE(2)-C wild-type cells are not affected by dTAG-13 (500 nM) (blue) compared to the DMSO group (black). Mean ± s.e.m., n = 3.
Extended Data Fig. 6. HDAC2 degradation disrupts transcriptional regulation.
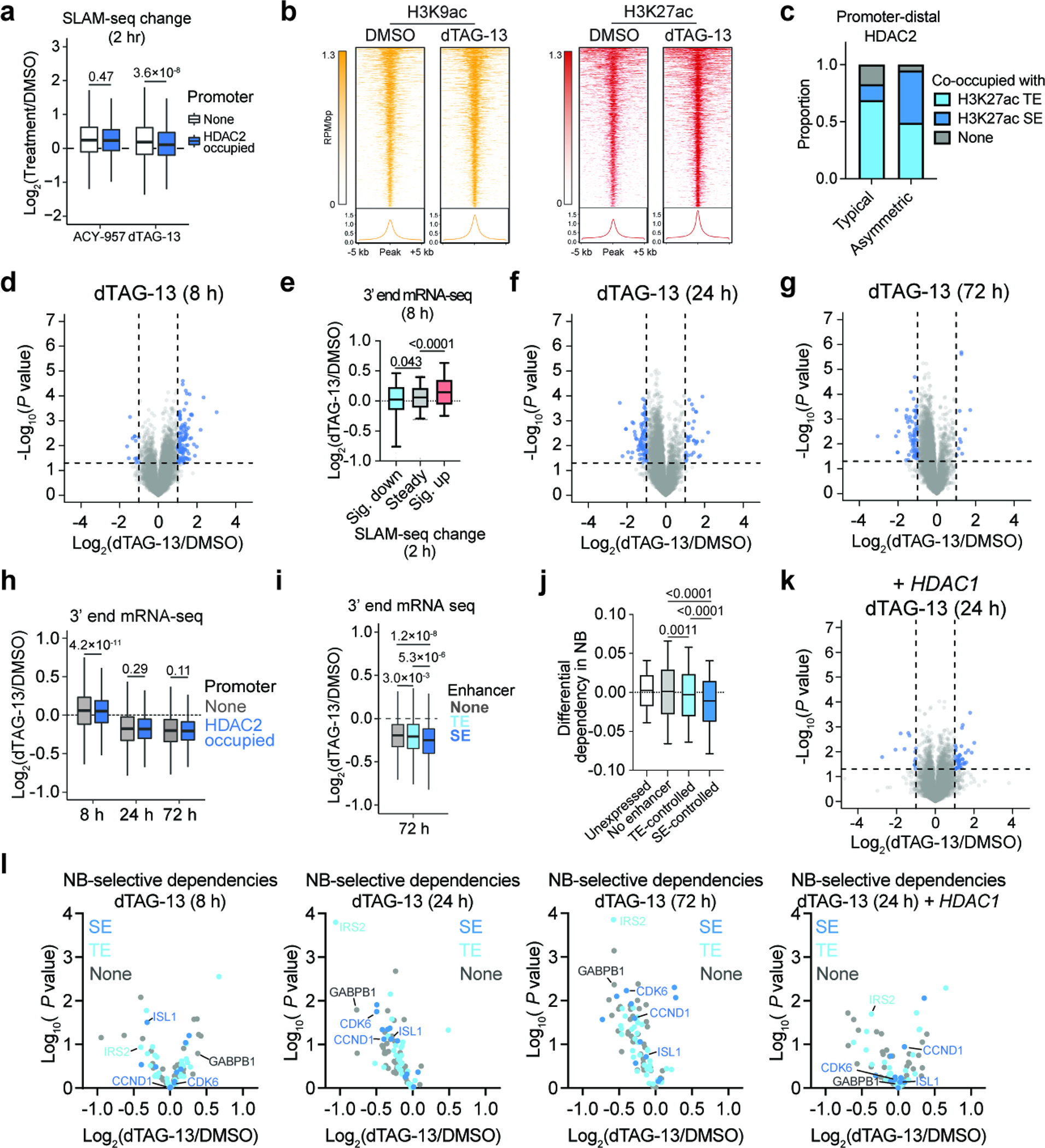
a, Boxplot of SLAM-seq changes at genes with (n = 5,082) or without (n = 8,117) HDAC2 bound at the promoter (TSS ± 1kb) with ACY-957 (5 μM) or dTAG-13 (500 nM) for 2 h. b, CUT&RUN signals with DMSO control and 2-h dTAG-13 (500 nM) treatment. c, Genomic feature distribution of typical and asymmetric HDAC2 sites. d, Volcano plots of changes in total mRNA abundance (3’-end mRNA-seq) following dTAG-13 treatment (500 nM) for 8 h. e, Boxplots of 3’-end mRNA-seq changes following 8-h dTAG-13 (500 nM) treatment. Genes are grouped by 2-h SLAM-seq data (significantly downregulated, n = 59; significantly upregulated, n = 224; steady, n = 12,916). f,g Volcano plots of 3’-end mRNA-seq changes following dTAG-13 treatment (500 nM) for 24 h (f), and 72 h (g). n = 3. h, Boxplots of 3’-end mRNA-seq change of genes with (n = 5,082) or without (n = 8,117) HDAC2-bound promoter. i, Boxplot of 3’-end mRNA-seq changes following 72-h dTAG-13 treatment (500 nM) in BE(2)C-HDAC2-dTAG cells (No enhancer, n = 7,588; TE, n = 5,026; SE, n = 585). j, Boxplot of neuroblastoma-specific differential dependencies for genes in SK-N-BE(2) cells (unexpressed, CPM < 3 by 3’-end mRNA-seq data, n = 7,973; expressed-and-no-enhancer, n = 7,588; TE-controlled, n = 5,026; SE-controlled, n = 585). k, Volcano plot of total transcript changes following 24-h dTAG-13 (500 nM) treatment in BE(2)-C-HDAC2-dTAG cells overexpressing HDAC1. l, Volcano plots of changes of genes with neuroblastoma-selective dependencies (differential dependency < −0.2, n = 91) by 3’-end mRNA-seq following 8-h, 24-h, and 72-h dTAG-13 treatments (500 nM) in BE(2)C-HDAC2-dTAG cells and 24-h treatment in HDAC1-overexpressing BE(2)C-HDAC2-dTAG cells. For volcano plots, P values were determined by two-tailed Student’s t-test. For boxplots in e and i boxes represent 25–75 percentiles with whiskers extending to 10–90 percentiles and the center line represents the median. P values were determined by two-tailed Student’s t-test. For other boxplots, boxes represent 25–75 percentiles with whiskers extending 1.5 IQR and the center line represents the median. P values were determined by two-tailed Welch’s t-test.
Extended Data Fig. 7. HDAC2 degradation transiently upregulated transcriptome and suppressed mRNA synthesis in long term.
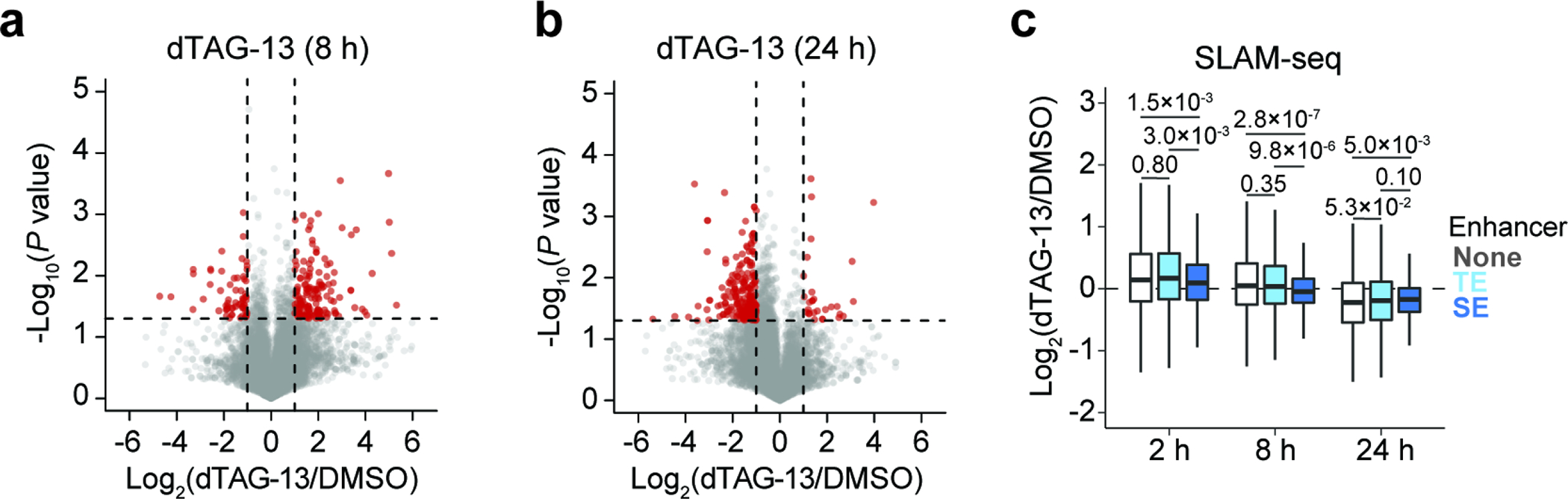
a,b Volcano plots depicting in SLAM-seq changes following 500 nM dTAG-13 treatments for 8 h (a) and 24 h (b). n = 3. P values were determined by two-tailed Student’s t-test. c. Boxplot of SLAM-seq changes at genes not associated with an enhancer (n = 7,588), associated with typical enhancers (n = 5,026), or associated with super enhancers (n = 585) with dTAG-13 (500 nM) for 2 h, 8 h, or 24 h. Boxes represent 25–75 percentiles with whiskers extending 1.5 IQR and the center line represents the median of the data. P values were determined by two-tailed Welch’s t-test.
Extended Data Fig. 8. HDAC2 loss destabilizes the NuRD complex.
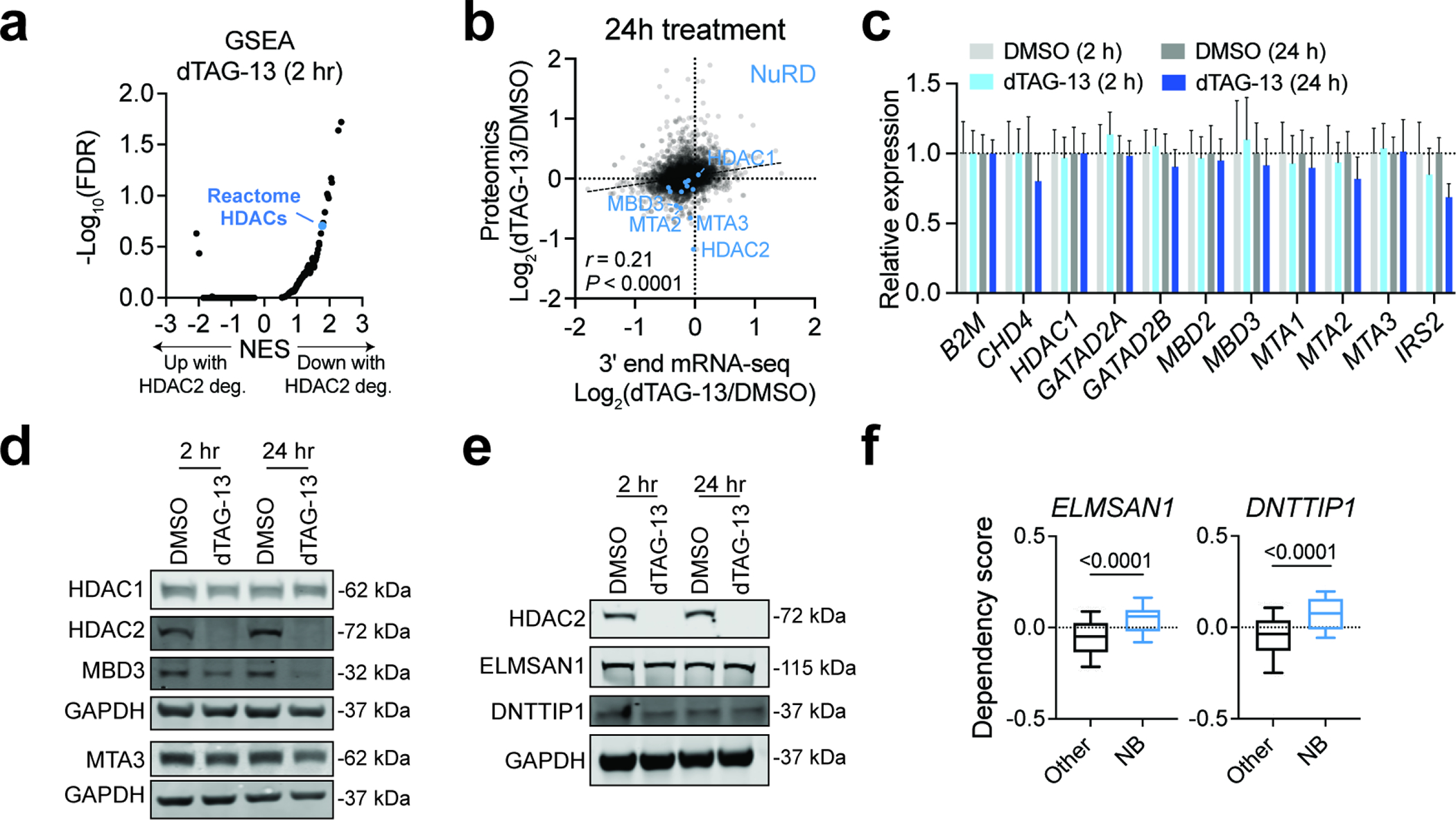
a, GSEA of proteomic changes following 2-h dTAG-13 (500 nM) treatment. b, Changes of mRNA abundance given by 3’ end mRNA-seq from Extended Data Fig. 6e versus changes of protein abundance given by quantitative proteomics from Fig. 5b. P value (two-tailed) was determined by Pearson correlation coefficient (r). n = 8,109. c, DMSO-normalized changes in gene expression (qRT-PCR) following dTAG-13 treatments (500 nM). Gene expression levels are normalized to B2M transcript level. See Source Data File for raw data. Mean ± propagated error (ΔΔCt), n = 3. IRS2 was used as a positive control as it was significantly downregulated by 24-h dTAG-13 treatment shown by 3’-end mRNA-seq (Extended Data Fig. 6l). d,e Immunoblots of NuRD subunits (d) and MiDAC subunits (e) with 2-h and 24-h dTAG-13 treatments in BE(2)-C-HDAC2-dTAG cells. f. Dependency scores of MiDAC subunits in neuroblastoma (n = 34) and other cell lines (n = 1,020). Boxes represent 25–75 percentiles with whiskers extending to 10–90 percentiles and the center line represents the median of the data. P values were determined by two-tailed Student’s t-test.
Extended Data Fig. 9. Destabilized NuRD complex leads to dysregulated chromatin accessibility.
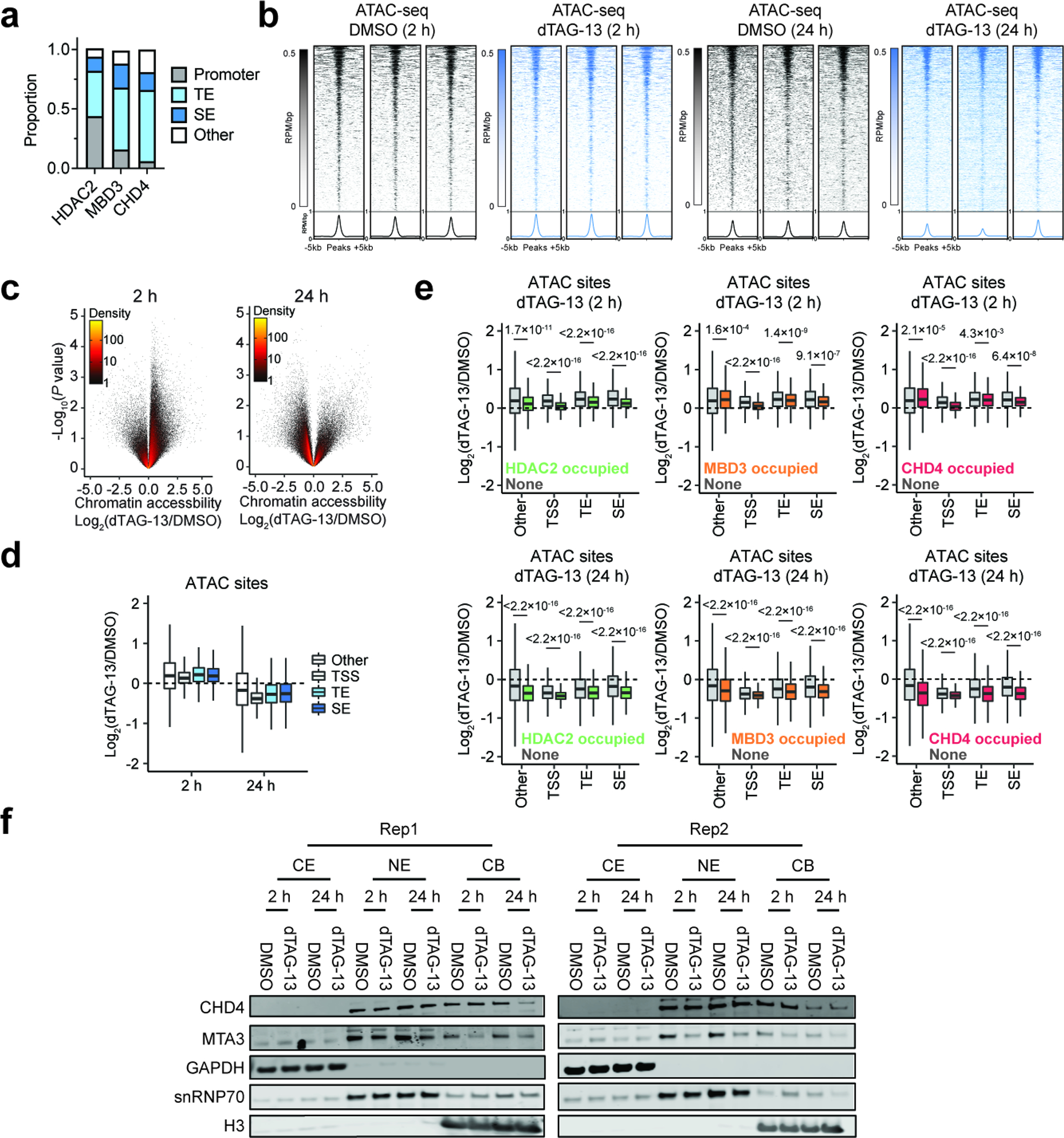
a, Genomic feature distribution of HDAC2, MBD3, and CHD4 sites. b. Heatmaps (top) and metaplots (bottom) of ATAC-seq signals with DMSO and dTAG-13 (500 nM) treatments for 2 h (left) and 24 h (right) in triplicates. c, Volcano plots of chromatin accessibility changes measured by ATAC-seq following dTAG-13 (500 nM) treatment for 2 h (left) or 24 h (right). n = 3. d, Boxplots of chromatin accessibility changes grouped by genomic localizations. For 2-h treatment, TSS (n = 14,142), TE (n = 19,687), SE (n = 2,557), and other sites (n = 71,044). For 24-h treatment, TSS (n = 14,167), TE (n = 19,369), SE (n = 2,546), and other sites (n = 68,458). e. Boxplots of chromatin accessibility changes grouped by genomic localizations and HDAC1, MBD3, and CHD4 co-occupancy. For 2-h treatment, at TSS HDAC2-occupied sites n = 4,370, MBD3-occupied sites n = 2,041, and CHD4-occupied sites n = 324; at TE HDAC2-occupied sites n = 3,565, MBD3-occupied sites n = 5,871, and CHD4-occupied sites n = 2,697; at SE HDAC2-occupied sites n = 880, MBD3-occupied sites n = 1,102, and CHD4-occupied sites n = 612; at other sites HDAC2-occupied sites n = 766, MBD3-occupied sites n = 2,045, and CHD4-occupied sites n = 748. For 24-h treatment, at TSS HDAC2-occupied sites n = 4,361, MBD3-occupied sites n = 2,028, and CHD4-occupied sites n = 320; at TE HDAC2-occupied sites n = 3,572, MBD3-occupied sites n = 5,837, and CHD4-occupied sites n = 2,655; at SE HDAC2-occupied sites n = 874, MBD3-occupied sites n = 1,098, and CHD4-occupied sites n = 604; at other sites HDAC2-occupied sites n = 766, MBD3-occupied sites n = 1,969, and CHD4-occupied sites n = 738. f, Immunoblots with subcellular fractionation in BE2C-HDAC2-dTAG cells with dTAG-13 treatments (500 nM). Experiments were performed in two biological replicates. For volcano plots, P values were determined by two-tailed Student’s t-test. Boxplots represent 25–75 percentiles with whiskers extending 1.5 IQR and the center line represents the median. P values were determined by two-tailed Welch’s t-test.
Extended Data Fig. 10. HDAC2 degradation exploits lineage-specific NuRD dependencies.
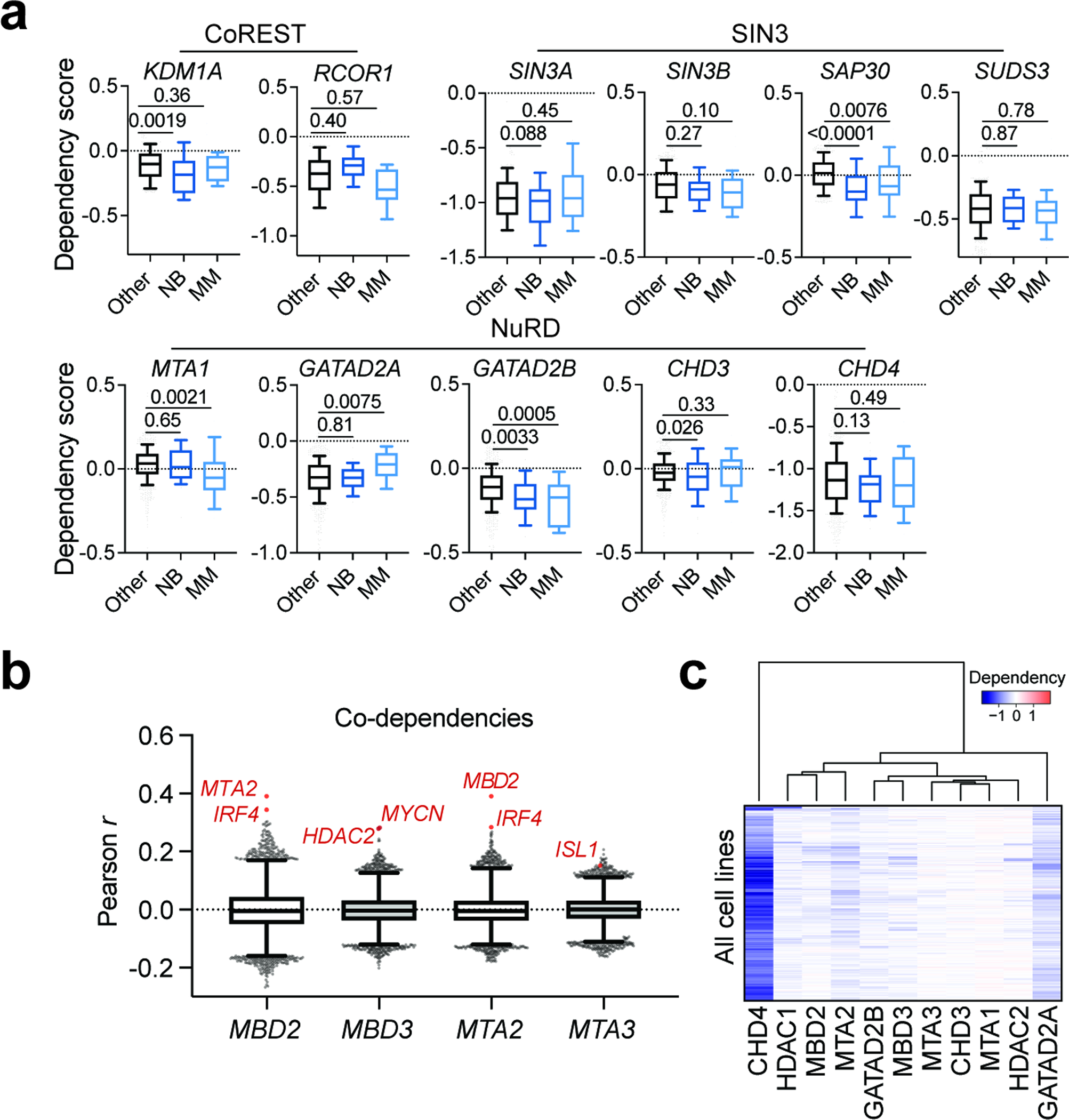
a, Dependency scores of HDAC1/2-containing complexes in neuroblastoma (n = 34), multiple myeloma (n = 21), and other cell lines (n = 999). b, Boxplots of Pearson correlation coefficients between MBD2, MBD3, MTA2, and MTA3 dependency scores compared to all other genes (n = 17,386). c, Unsupervised clustering of dependency scores of NuRD subunits in all cell lines. For boxplots, boxes represent 25–75 percentiles with whiskers extending to 10–90 percentiles and the center line represents the median of the data. P values were determined by two-tailed Student’s t-test.
Supplementary Material
Acknowledgements:
This work was supported by the National Institutes of Health (NIH) through a NIH Director’s Early Independence Award (DP-5-OD26380, M.A.E.) and by the Ono Pharma Foundation. We thank Dr. Martin G. Jaeger for his critical feedback on the manuscript prior to submission. We also thank the flow cytometry core, the NGS core at The Scripps Research Institute, and the NGS at La Jolla Institute for Immunology for supporting this work.
Footnotes
Competing Interests Statement:
The authors declare no competing interests.
Code Availability:
The sources of the codes used in this study can be found in corresponding method sections and are available online.
Peer review Information:
Nature Structural & Molecular Biology thanks Shaun Cowley and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Editor recognition statement (if applicable to your journal):
Carolina Perdigoto and Dimitris Typas were the primary editors on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Data Availability:
For availability of the data from DepMap and cBioportal see Analyses of datasets from DepMap and Analyses of datasets from cBioportal. All raw next-generation sequencing data (SLAM-seq, 3’ end mRNA-seq, CUT&RUN, and ATAC-seq) and the related processed data were deposited in the NCBI Gene Expression Omnibus (GEO) database under accession number GSE202706. Quantitative proteomics data were deposited in the PRIDE database by EMBL’s European Bioinformatics Institute (EMBL-EBI) under accession number PXD034444. Previously published datasets (H3K27ac ChIP and H3K4me3 ChIP in BE(2)-C cells) used in this study can be found on GEO under accession number GSE80154. No restrictions on data availability apply.
References:
- 1.Shortt J, Ott CJ, Johnstone RW, Bradner JE. A chemical probe toolbox for dissecting the cancer epigenome. Nat Rev Cancer. 2017;17(3):160–183. doi: 10.1038/nrc.2016.148 [DOI] [PubMed] [Google Scholar]
- 2.Chang L, Ruiz P, Ito T, Sellers WR. Targeting pan-essential genes in cancer: Challenges and opportunities. Cancer Cell. 2021;39(4):466–479. doi: 10.1016/j.ccell.2020.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seto E, Yoshida M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb Perspect Biol. 2014;6(4):a018713–a018713. doi: 10.1101/cshperspect.a018713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachy E, Camus V, Thieblemont C, et al. Final Analysis of the Ro-CHOP Phase III Study (Conducted by LYSA): Romidepsin Plus CHOP in Patients with Peripheral T-Cell Lymphoma. Blood. 2020;136(Supplement 1):32–33. doi: 10.1182/blood-2020-134440 [DOI] [Google Scholar]
- 5.Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13(9):673–691. doi: 10.1038/nrd4360 [DOI] [PubMed] [Google Scholar]
- 6.Kelly RDW, Cowley SM. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem Soc Trans. 2013;41(3):741–749. doi: 10.1042/BST20130010 [DOI] [PubMed] [Google Scholar]
- 7.Millard CJ, Watson PJ, Fairall L, Schwabe JWR. Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharmacol Sci. 2017;38(4):363–377. doi: 10.1016/j.tips.2016.12.006 [DOI] [PubMed] [Google Scholar]
- 8.Taunton J, Hassig CA, Schreiber SL. A Mammalian Histone Deacetylase Related to the Yeast Transciptional Regulator Rpd3p Published by : American Association for the Advancement of Science Stable URL : https://www.jstor.org/stable/2890320 American Association for the Advancement of Science is. Science (80- ). 1996;272(5260):408–411. [DOI] [PubMed] [Google Scholar]
- 9.Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6(10):1–34. doi: 10.1101/cshperspect.a026831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dovey OM, Foster CT, Cowley SM. Histone deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell differentiation. Proc Natl Acad Sci U S A. 2010;107(18):8242–8247. doi: 10.1073/pnas.1000478107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lagger G, O’Carroll D, Rembold M, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21(11):2672–2681. doi: 10.1093/emboj/21.11.2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jamaladdin S, Kelly RDW, O’Regan L, et al. Histone deacetylase (HDAC) 1 and 2 are essential for accurate cell division and the pluripotency of embryonic stem cells. Proc Natl Acad Sci. 2014;111(27):9840–9845. doi: 10.1073/pnas.1321330111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamaguchi T, Cubizolles F, Zhang Y, et al. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010;24(5):455–469. doi: 10.1101/gad.552310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilting RH, Yanover E, Heideman MR, et al. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 2010;29(15):2586–2597. doi: 10.1038/emboj.2010.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LeBoeuf M, Terrell A, Trivedi S, et al. Hdac1 and Hdac2 Act Redundantly to Control p63 and p53 Functions in Epidermal Progenitor Cells. Dev Cell. 2010;19(6):807–818. doi: 10.1016/j.devcel.2010.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heideman MR, Wilting RH, Yanover E, et al. Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood. 2013;121(11):2038–2050. doi: 10.1182/blood-2012-08-450916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dovey OM, Foster CT, Conte N, et al. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood. 2013;121(8):1335–1344. doi: 10.1182/blood-2012-07-441949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matthews GM, Mehdipour P, Cluse LA, et al. Functional-genetic dissection of HDAC dependencies in mouse lymphoid and myeloid malignancies. Blood. 2015;126(21):2392–2403. doi: 10.1182/blood-2015-03-632984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stubbs MC, Kim W, Bariteau M, et al. Selective inhibition of HDAC1 and HDAC2 as a potential therapeutic option for B-ALL. Clin Cancer Res. 2015;21(10):2348–2358. doi: 10.1158/1078-0432.CCR-14-1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frumm SM, Fan ZP, Ross KN, et al. Selective HDAC1/HDAC2 inhibitors induce neuroblastoma differentiation. Chem Biol. 2013;20(5):713–725. doi: 10.1016/j.chembiol.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ito T, Young MJ, Li R, et al. Paralog knockout profiling identifies DUSP4 and DUSP6 as a digenic dependence in MAPK pathway-driven cancers. Nat Genet. 2021;53(12):1664–1672. doi: 10.1038/s41588-021-00967-z [DOI] [PubMed] [Google Scholar]
- 22.DeWeirdt PC, Sanson KR, Sangree AK, et al. Optimization of AsCas12a for combinatorial genetic screens in human cells. Nat Biotechnol. 2021;39(1):94–104. doi: 10.1038/s41587-020-0600-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang A, Garraway LA, Ashworth A, Weber B. Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discov. 2020;19(1):23–38. doi: 10.1038/s41573-019-0046-z [DOI] [PubMed] [Google Scholar]
- 24.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase.[erratum appears in Nature. 2007 May 17;447(7142):346]. Nature. 2005;434(7035):913–917. [DOI] [PubMed] [Google Scholar]
- 25.Hoffman GR, Rahal R, Buxton F, et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc Natl Acad Sci U S A. 2014;111(8):3128–3133. doi: 10.1073/pnas.1316793111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson BG, Helming KC, Wang X, et al. Residual Complexes Containing SMARCA2 (BRM) Underlie the Oncogenic Drive of SMARCA4 ( BRG1 ) Mutation. Mol Cell Biol. 2014;34(6):1136–1144. doi: 10.1128/mcb.01372-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oike T, Ogiwara H, Tominaga Y, et al. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res. 2013;73(17):5508–5518. doi: 10.1158/0008-5472.CAN-12-4593 [DOI] [PubMed] [Google Scholar]
- 28.Ogiwara H, Sasaki M, Mitachi T, et al. Targeting p300 addiction in CBP-deficient cancers causes synthetic lethality by apoptotic cell death due to abrogation of MYC expression. Cancer Discov. 2016;6(4):430–445. doi: 10.1158/2159-8290.CD-15-0754 [DOI] [PubMed] [Google Scholar]
- 29.Lelij P van der, Lieb S, Jude J, et al. Synthetic lethality between the cohesin subunits STAG1 and STAG2 in diverse cancer contexts. Elife. 2017;6:e26980. doi: 10.7554/elife.26980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parrish PCR, Thomas JD, Gabel AM, Kamlapurkar S, Bradley RK, Berger AH. Discovery of synthetic lethal and tumor suppressor paralog pairs in the human genome. Cell Rep. 2021;36(9):109597. doi: 10.1016/j.celrep.2021.109597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malone CF, Dharia NV, Kugener G, et al. Selective modulation of a pan-essential protein as a therapeutic strategy in cancer. Cancer Discov. 2021;11(9):2282–2299. doi: 10.1158/2159-8290.CD-20-1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsherniak A, Vazquez F, Montgomery PG, et al. Defining a Cancer Dependency Map. Cell. 2017;170(3):564–576.e16. doi: 10.1016/j.cell.2017.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyers RM, Bryan JG, McFarland JM, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49(12):1779–1784. doi: 10.1038/ng.3984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caron H, van Sluis P, van Hoeve M, et al. Allelic loss of chromosome 1p36 in neuroblastoma is of preferential maternal origin and correlates with N–myc amplification. Nat Genet. 1993;4(2):187–190. doi: 10.1038/ng0693-187 [DOI] [PubMed] [Google Scholar]
- 35.Maris JM, White PS, Beltinger CP, et al. Significance of Chromosome 1p Loss of Heterozygosity in Neuroblastoma. Cancer Res. 1995;55(20):4664–4669. [PubMed] [Google Scholar]
- 36.Janoueix-Lerosey I, Novikov E, Monteiro M, et al. Gene expression profiling of 1p35–36 genes in neuroblastoma. Oncogene. 2004;23(35):5912–5922. doi: 10.1038/sj.onc.1207784 [DOI] [PubMed] [Google Scholar]
- 37.Komotar RJ, Otten ML, Starke RM, Anderson RCE. Chromosome 1p and 11q deletions and outcome in neuroblastoma—A critical review. Clin Med Oncol. 2008;2:419–420. doi: 10.4137/cmo.s391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merup M, Moreno TC, Heyman M, et al. 6q deletions in acute lymphoblastic leukemia and non-Hodgkin’s lymphomas. Blood. 1998;91(9):3397–3400. doi: 10.1182/blood.v91.9.3397 [DOI] [PubMed] [Google Scholar]
- 39.Thelander EF, Ichimura K, Corcoran M, et al. Characterization of 6q deletions in mature B cell lymphomas and childhood acute lymphoblastic leukemia. Leuk Lymphoma. 2008;49(3):477–487. doi: 10.1080/10428190701817282 [DOI] [PubMed] [Google Scholar]
- 40.Taborelli M, Tibiletti MG, Martin V, Pozzi B, Bertoni F, Capella C. Chromosome band 6q deletion pattern in malignant lymphomas. Cancer Genet Cytogenet. 2006;165(2):106–113. doi: 10.1016/j.cancergencyto.2005.06.025 [DOI] [PubMed] [Google Scholar]
- 41.Aktas Samur A, Minvielle S, Shammas M, et al. Deciphering the chronology of copy number alterations in Multiple Myeloma. Blood Cancer J. 2019;9(4). doi: 10.1038/s41408-019-0199-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Durbin AD, Zimmerman MW, Dharia NV., et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat Genet. 2018;50(9):1240–1246. doi: 10.1038/s41588-018-0191-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeid R, Lawlor MA, Poon E, et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat Genet. 2018;50(4):515–523. doi: 10.1038/s41588-018-0044-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dharia NV, Kugener G, Guenther LM, et al. A first-generation pediatric cancer dependency map. Nat Genet. 2021;53(4):529–538. doi: 10.1038/s41588-021-00819-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen L, Alexe G, Dharia NV, et al. CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J Clin Invest. 2018;128(1):446–462. doi: 10.1172/JCI90793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghandi M, Huang FW, Jané-Valbuena J, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569(7757):503–508. doi: 10.1038/s41586-019-1186-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.García-López J, Wallace K, Otero JH, et al. Large 1p36 Deletions Affecting Arid1a Locus Facilitate Mycn-Driven Oncogenesis in Neuroblastoma. Cell Rep. 2020;30(2):454–464.e5. doi: 10.1016/j.celrep.2019.12.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi H, Tao T, Abraham BJ, et al. ARID1A loss in neuroblastoma promotes the adrenergic-to-mesenchymal transition by regulating enhancer-mediated gene expression. Sci Adv. 2020;6(29). doi: 10.1126/sciadv.aaz3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cerami E, Gao J, Dogrusoz U, et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Erb MA, Scott TG, Li BE, et al. Transcription control by the ENL YEATS domain in acute leukaemia. Nature. 2017;543(7644):270–274. doi: 10.1038/nature21688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nabet B, Roberts JM, Buckley DL, et al. The dTAG system for immediate and target-specific protein degradation. Nat Chem Biol. 2018;14(5):431–441. doi: 10.1038/s41589-018-0021-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jaeger MG, Winter GE. Fast-acting chemical tools to delineate causality in transcriptional control. Mol Cell. 2021;81(8):1617–1630. doi: 10.1016/j.molcel.2021.02.015 [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, Erb MA. Enabling cancer target validation with genetically encoded systems for ligand-induced protein degradation. Curr Res Chem Biol. 2021;1:100011. doi: 10.1016/j.crchbi.2021.100011 [DOI] [Google Scholar]
- 54.Skene PJ, Henikoff JG, Henikoff S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc. 2018;13(5):1006–1019. doi: 10.1038/nprot.2018.015 [DOI] [PubMed] [Google Scholar]
- 55.Herzog VA, Reichholf B, Neumann T, et al. Thiol-linked alkylation of RNA to assess expression dynamics. Nat Methods. 2017;14(12):1198–1204. doi: 10.1038/nmeth.4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shearstone JR, Golonzhka O, Chonkar A, et al. Chemical inhibition of histone deacetylases 1 and 2 induces fetal hemoglobin through activation of GATA2. PLoS One. 2016;11(4):1–27. doi: 10.1371/journal.pone.0153767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. doi: 10.1016/j.cell.2013.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schölz C, Weinert BT, Wagner SA, et al. Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat Biotechnol. 2015;33(4):415–425. doi: 10.1038/nbt.3130 [DOI] [PubMed] [Google Scholar]
- 59.Marques JG, Gryder BE, Pavlovic B, et al. NURD subunit CHD4 regulates super-enhancer accessibility in rhabdomyosarcoma and represents a general tumor dependency. Elife. 2020;9:1–30. doi: 10.7554/ELIFE.54993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gryder BE, Pomella S, Sayers C, et al. Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nat Genet. 2019;51(12):1714–1722. doi: 10.1038/s41588-019-0534-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Emdal KB, Pedersen AK, Bekker-Jensen DB, et al. Integrated proximal proteomics reveals IRS2 as a determinant of cell survival in ALK-driven neuroblastoma. Sci Signal. 2018;11(557):1–18. doi: 10.1126/scisignal.aap9752 [DOI] [PubMed] [Google Scholar]
- 62.Rihani A, Vandesompele J, Speleman F, Van Maerken T. Inhibition of CDK4/6 as a novel therapeutic option for neuroblastoma. Cancer Cell Int. 2015;15(1):4–11. doi: 10.1186/s12935-015-0224-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiong Y, Donovan KA, Eleuteri NA, et al. Chemo-proteomics exploration of HDAC degradability by small molecule degraders. Cell Chem Biol. 2021;28(10):1514–1527.e4. doi: 10.1016/j.chembiol.2021.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hsu JHR, Rasmusson T, Robinson J, et al. EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex. Cell Chem Biol. 2020;27(1):41–46.e17. doi: 10.1016/j.chembiol.2019.11.004 [DOI] [PubMed] [Google Scholar]
- 65.Farnaby W, Koegl M, Roy MJ, et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat Chem Biol. 2019;15(7):672–680. doi: 10.1038/s41589-019-0294-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schick S, Grosche S, Kohl KE, et al. Acute BAF perturbation causes immediate changes in chromatin accessibility. Nat Genet. 2021;53(3):269–278. doi: 10.1038/s41588-021-00777-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sher F, Hossain M, Seruggia D, et al. Rational targeting of a NuRD subcomplex guided by comprehensive in situ mutagenesis. Nat Genet. 2019;51(7):1149–1159. doi: 10.1038/s41588-019-0453-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vinogradova EV, Zhang X, Remillard D, et al. An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell. 2020;182(4):1009–1026.e29. doi: 10.1016/j.cell.2020.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Denslow SA, Wade PA. The human Mi-2/NuRD complex and gene regulation. Oncogene. 2007;26(37):5433–5438. doi: 10.1038/sj.onc.1210611 [DOI] [PubMed] [Google Scholar]
- 70.Low JKK, Silva APG, Sharifi Tabar M, et al. The Nucleosome Remodeling and Deacetylase Complex Has an Asymmetric, Dynamic, and Modular Architecture. Cell Rep. 2020;33(9):108450. doi: 10.1016/j.celrep.2020.108450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Millard CJ, Varma N, Saleh A, et al. The structure of the core NuRD repression complex provides insights into its interaction with chromatin. Elife. 2016;5:1–21. doi: 10.7554/elife.13941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reid XJ, Low JKK, Mackay JP. A NuRD for all seasons. Trends Biochem Sci. 2023;48(1):11–25. doi: 10.1016/j.tibs.2022.06.002 [DOI] [PubMed] [Google Scholar]
- 73.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 2015;2015(January):21.29.1–21.29.9. doi: 10.1002/0471142727.mb2129s109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gryder BE, Wu L, Woldemichael GM, et al. Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat Commun. 2019;10(1):1–12. doi: 10.1038/s41467-019-11046-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qu K, Zaba LC, Satpathy AT, et al. Chromatin Accessibility Landscape of Cutaneous T Cell Lymphoma and Dynamic Response to HDAC Inhibitors. Cancer Cell. 2017;32(1):27–41.e4. doi: 10.1016/j.ccell.2017.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pan J, Meyers RM, Michel BC, et al. Interrogation of Mammalian Protein Complex Structure, Function, and Membership Using Genome-Scale Fitness Screens. Cell Syst. 2018;6(5):555–568.e7. doi: 10.1016/j.cels.2018.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Michel BC, D’Avino AR, Cassel SH, et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat Cell Biol. 2018;20(12):1410–1420. doi: 10.1038/s41556-018-0221-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lai AY, Wade PA. Cancer biology and NuRD: A multifaceted chromatin remodelling complex. Nat Rev Cancer. 2011;11(8):588–596. doi: 10.1038/nrc3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bornelöv S, Reynolds N, Xenophontos M, et al. The Nucleosome Remodeling and Deacetylation Complex Modulates Chromatin Structure at Sites of Active Transcription to Fine-Tune Gene Expression. Mol Cell. 2018;71(1):56–72.e4. doi: 10.1016/j.molcel.2018.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jamaladdin S, Kelly RDW, O’Regan L, et al. Histone deacetylase (HDAC) 1 and 2 are essential for accurate cell division and the pluripotency of embryonic stem cells. Proc Natl Acad Sci U S A. 2014;111(27):9840–9845. doi: 10.1073/pnas.1321330111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smalley JP, Baker IM, Pytel WA, et al. Optimization of Class I Histone Deacetylase PROTACs Reveals that HDAC1/2 Degradation is Critical to Induce Apoptosis and Cell Arrest in Cancer Cells. J Med Chem. Published online 2022. doi: 10.1021/acs.jmedchem.1c02179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Smalley JP, Adams GE, Millard CJ, et al. PROTAC-mediated degradation of class i histone deacetylase enzymes in corepressor complexes. Chem Commun. 2020;56(32):4476–4479. doi: 10.1039/d0cc01485k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cross JM, Coulson ME, Smalley JP, et al. A ‘click’ chemistry approach to novel entinostat (MS-275) based class I histone deacetylase proteolysis targeting chimeras. RSC Med Chem. 2022;13(12):1634–1639. doi: 10.1039/d2md00199c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Baker IM, Smalley JP, Sabat KA, Hodgkinson JT, Cowley SM. Comprehensive Transcriptomic Analysis of Novel Class I HDAC Proteolysis Targeting Chimeras (PROTACs). Biochemistry. 2022;62(3):645–656. doi: 10.1021/acs.biochem.2c00288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Scholes NS, Mayor-Ruiz C, Winter GE. Identification and selectivity profiling of small-molecule degraders via multi-omics approaches. Cell Chem Biol. 2021;28(7):1048–1060. doi: 10.1016/j.chembiol.2021.03.007 [DOI] [PubMed] [Google Scholar]
- 86.Remillard D, Buckley DL, Paulk J, et al. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew Chemie - Int Ed. 2017;56(21):5738–5743. doi: 10.1002/anie.201611281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Olson CM, Jiang B, Erb MA, et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol. 2018;14(2):163–170. doi: 10.1038/nchembio.2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cantley J, Ye X, Rousseau E, et al. Selective PROTAC-mediated degradation of SMARCA2 is efficacious in SMARCA4 mutant cancers. Nat Commun. 2022;13(1). doi: 10.1038/s41467-022-34562-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gopalsamy A. Selectivity through Targeted Protein Degradation (TPD). J Med Chem. 2022;65(12):8113–8126. doi: 10.1021/acs.jmedchem.2c00397 [DOI] [PubMed] [Google Scholar]
- 90.Toriki ES, Papatzimas JW, Nishikawa K, et al. Rational Chemical Design of Molecular Glue Degraders. bioRxiv. Published online 2022:2022.11.04.512693. doi: 10.1021/acscentsci.2c01317 [DOI] [Google Scholar]
Methods-only references:
- 91.Sakuma T, Nakade S, Sakane Y, Suzuki KIT, Yamamoto T. MMEJ-Assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat Protoc. 2016;11(1):118–133. doi: 10.1038/nprot.2015.140 [DOI] [PubMed] [Google Scholar]
- 92.Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33(6):661–667. doi: 10.1038/nbt.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Doench JG, Fusi N, Sullender M, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34(2):184–191. doi: 10.1038/nbt.3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brinkman EK, Chen T, Amendola M, Van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42(22):1–8. doi: 10.1093/nar/gku936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Neumann T, Herzog VA, Muhar M, et al. Quantification of experimentally induced nucleotide conversions in high-throughput sequencing datasets. BMC Bioinformatics. 2019;20(1):1–16. doi: 10.1186/s12859-019-2849-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Corces MR, Trevino AE, Hamilton EG, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14(10):959–962. doi: 10.1038/nmeth.4396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mootha VK, Lindgren CM, Eriksson K-F, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For availability of the data from DepMap and cBioportal see Analyses of datasets from DepMap and Analyses of datasets from cBioportal. All raw next-generation sequencing data (SLAM-seq, 3’ end mRNA-seq, CUT&RUN, and ATAC-seq) and the related processed data were deposited in the NCBI Gene Expression Omnibus (GEO) database under accession number GSE202706. Quantitative proteomics data were deposited in the PRIDE database by EMBL’s European Bioinformatics Institute (EMBL-EBI) under accession number PXD034444. Previously published datasets (H3K27ac ChIP and H3K4me3 ChIP in BE(2)-C cells) used in this study can be found on GEO under accession number GSE80154. No restrictions on data availability apply.