

Abstract
Mutations leading to colony-stimulating factor-1 receptor (CSF-1R) loss-of-function or haploinsufficiency cause CSF1R-related leukoencephalopathy (CRL), an adult-onset disease characterized by loss of myelin and neurodegeneration, for which there is no effective therapy. Symptom onset usually occurs in the fourth decade of life and the penetrance of disease in carriers is high. However, familial studies have identified a few carriers of pathogenic CSF1R mutations that remain asymptomatic even in their seventh decade of life, raising the possibility that the development and severity of disease might be influenced by environmental factors. Here we report new cases in which long-term glucocorticoid treatment is associated with asymptomatic status in elder carriers of pathogenic CSF-1R mutations. The main objective of the present study was to investigate the link between chronic immunosuppression initiated pre-symptomatically and resistance to the development of symptomatic CRL, in the Csf1r+/− mouse model. We show that chronic prednisolone administration prevents the development of memory, motor coordination and social interaction deficits, as well as the demyelination, neurodegeneration and microgliosis associated with these deficits. These findings are in agreement with the preliminary clinical observations and support the concept that pre-symptomatic immunosuppression is protective in patients carrying pathogenic CSF1R variants associated with CRL. Proteomic analysis of microglia and oligodendrocytes indicates that Prednisone suppresses processes involved in microglial activation and alleviates senescence and improves fitness of oligodendrocytes. This analysis also identifies new potential targets for therapeutic intervention.
Keywords: CSF1R, CRL, microglia, prednisone, leukodystrophy
Graphical Abstract
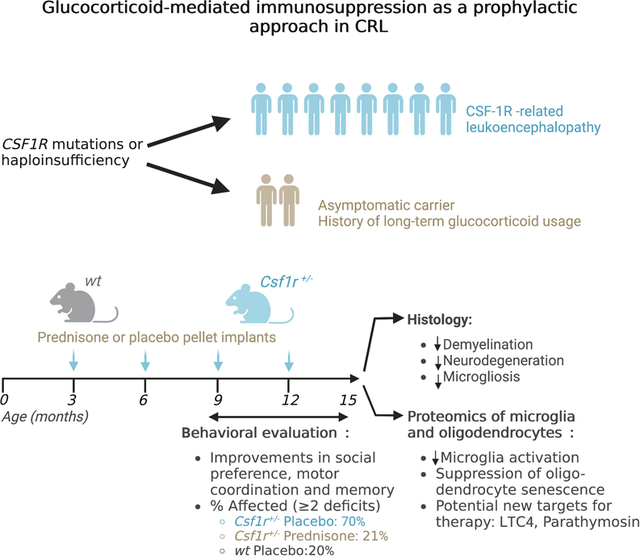
1. Introduction
Colony stimulating factor-1 (CSF-1) receptor (CSF-1R) is a receptor tyrosine kinase that transduces the signals of two cognate ligands, CSF-1 and interleukin-34 (IL-34) to regulate the differentiation, survival and proliferation of several tissue macrophage populations, including microglia and of bone-resorbing osteoclasts (reviewed in (Chitu, Gokhan, Nandi, Mehler, & Stanley, 2016; Chitu & Stanley, 2017; Stanley & Chitu, 2015)). Consistent with this, bi-allelic combinations of hypomorphic and/or amorphic mutations cause a pediatric disorder characterized by abnormalities in brain and bone development while mono-allelic inactivating mutations cause an adult-onset demyelinating disease designated CSF1R-related leukoencephalopathy (CRL) (reviewed in (Chitu, Gokhan, & Stanley, 2022)). CRL is characterized by cognitive and motor impairment, psychiatric disorders and seizures. Magnetic resonance imaging of CRL patient brains reveals initially patchy and later confluent white matter lesions, thinning of the corpus callosum and enlargement of the lateral ventricles (reviewed in (Konno, Kasanuki, Ikeuchi, Dickson, & Wszolek, 2018)). Characteristic pathological features include the loss of myelin, axonal swelling, neurodegeneration and stage-dependent microgliosis (Alturkustani, Keith, Hazrati, Rademakers, & Ang, 2015; Kinoshita et al., 2021; Konno, Tada, Tada, Nishizawa, & Ikeuchi, 2014; Oyanagi et al., 2017). The accumulation of iron and end-oxidation products in microglia leads to the formation of pigmented glia, a hallmark feature of the disease (Z. S. Ali, Van Der Voorn, & Powers, 2007).
Currently there is no effective therapy for CRL. However, studies in both CRL (Kempthorne et al., 2020) and in the Csf1r+/− mouse model of the disease (Chitu et al., 2020) revealed a loss of the homeostatic microglial phenotype and suggested that CRL is a primary microgliopathy. Indeed, in the Csf1r+/− mouse model, Csf1r heterozygosity in microglia is sufficient to produce the full CRL-like disease phenotype (Biundo et al., 2021). These findings suggested that the development of CRL might be delayed by modulating microglia function. Indeed, using mouse models, we have shown that granulocyte-macrophage colony stimulating factor (GM-CSF) and granulocyte colony stimulating factor (G-CSF) contribute to microglia dyshomeostasis in non-overalpping manner and that their genetic targeting improves several aspects of CRL pathology and behavior (Chitu 2020; Biundo 2023). Furthermore, treatment of 6-month-old Csf1r+/− mice for 6–8 weeks with a dosage of the CSF-1R inhibitor PLX5622 that eliminates <25% of microglia resulted in an improvement in cognitive function in the novel object recognition test (Arreola et al., 2021). Additional experiments are needed to investigate the consequences of longer term treatment with CSF-1R inhibitors. More recently, it has been shown that daily administration of the semi-synthetic tetracycline derivative, minocycline, to 8-month-old Csf1r+/− male mice for one month inhibits inflammatory factor production by Csf1r+/− microglia and alleviates working memory loss, increased anxiety and pathologies in the mice (X. Li et al., 2023). In addition, the activation of the microglial phagocytic receptor, Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) is being clinically evaluated as an effective treatment for neurodegenerative diseases including CRL. However, preliminary experiments with Trem2 deficiency in the Csf1r+/− mouse model (Biundo, et al., 2023b) suggest that this may not be an effective approach.
Although CRL has a typical onset in the fourth decade of life (Konno et al., 2018; Papapetropoulos et al., 2021) carriers of pathogenic CSF1R mutations, that remain healthy in their 7th decade of life, have been identified in several familial studies ((S. Ali et al., 2022; Tipton, Stanley, Chitu, & Wszolek, 2021), reviewed in (Chitu et al., 2022)). Previously we reported an association between the healthy carrier status and long-term glucocorticoid-mediated immunosuppression in two patients aged 71 and 63 years (S. Ali et al., 2022; Tipton et al., 2021). Glucocorticoids are known to limit the amplification of inflammatory responses in macrophages and microglia and to promote resolution (Ehrchen, Roth, & Barczyk-Kahlert, 2019; Ros-Bernal et al., 2011). They also curtail microglial activation in response to stimulation by GM-CSF (Ganter, Northoff, Mannel, & Gebicke-Harter, 1992; Tanaka et al., 1997), a cytokine that contributes to microglial dyshomeostasis in the mouse model of CRL (Chitu et al., 2020). Furthermore, glucocorticoids provide protection against oxidative stress in cultured oligodendrocytes and in vivo (Pitarokoili et al., 2019). These observations prompted us to investigate whether pre-symptomatic immune suppression might be protective in CRL using the Csf1r+/− mouse model. Here we show that chronic prednisone administration prevents the development of behavioral deficits and associated CRL pathology. Comparative proteomic studies in isolated cells suggest that prednisone treatment suppresses several processes related to microglia activation, including the production of reactive oxygen species and eicosanoids. They also indicate that prednisone alleviates oligodendrocyte senescence, improves energy production and promotes remyelination. Together these data suggest that prednisone treatment might be of prophylactic value in CRL
2. Material and methods
2.1. Human subjects
Clinical data were collected regarding demographics (sex, age, ethnicity), family history, symptoms of CRL, and treatment with glucocorticoid medications (indications, administration route, therapy duration). Targeted Sanger sequencing of the CSF1R gene was performed in the two newly reported cases. Information was collected under IRB no. 19–011016. Additionally, informed consent for publication was obtained from the two newly reported cases.
2.2. Mouse strains, breeding, maintenance and prednisone treatment
Csf1r+/− mice (Dai et al., 2002) backcrossed more than 10 generations to C57BL/6J mice (RRID: IMSR JAX:000664) were genotyped as described previously (Dai et al., 2002). Cohorts were developed from the progeny of matings of Csf1r+/− to wt mice, randomized with respect to the litter of origin. A total of 61 mice (32 females and 29 males) were included in this study. At 3 months of age, they were transferred from a breeder diet (PicoLab Rodent Diet 20 5058) to a lower fat maintenance diet (PicoLab Rodent Diet 20 5053). All in vivo experiments were performed in accordance with the National Institutes of Health regulations on the care and use of experimental animals and approved by the Institutional Animal Care and Use Committees of the Albert Einstein College of Medicine. Prednisone (Innovative Research of America, Inc) was administered starting at three months of age in the form of slow-release subcutaneous pellets at 1.8 mg/kg/day; four pellets were used per mouse, with the second, third and fourth pellet being implanted after 3, 6 and 9 months from the start of the initial treatment, respectively (Figure 1 A). Control mice received placebo pellets at the same time intervals. Although prednisone administration has been reported to produce significant side effects in humans, in our experimental cohort those were limited to weight loss in male mice (Figure S1). In addition, consistent with the known side effects of prednisone in the skin, ~60% of the mice exhibited impaired hair regrowth in the area where pellets were implanted. Behavioral studies involved both male and female mice. Because hallmark histopathological features of the disese (demyelination, neurodegeneration, increased densities and morphological change of microglia) were reproducibly detected in both male and female mice (Chitu 2015, Chitu 2020; Biundo 2023a) we have conducted the histopathological evaluation in female mice and proteomic analysis in male mice. The age and sex of mice used in each experiment are indicated in the figures and figure legends.
Figure 1. Presymptomatic prednisone treatment improves the neurocognitive performance of Csf1r+/− mice.
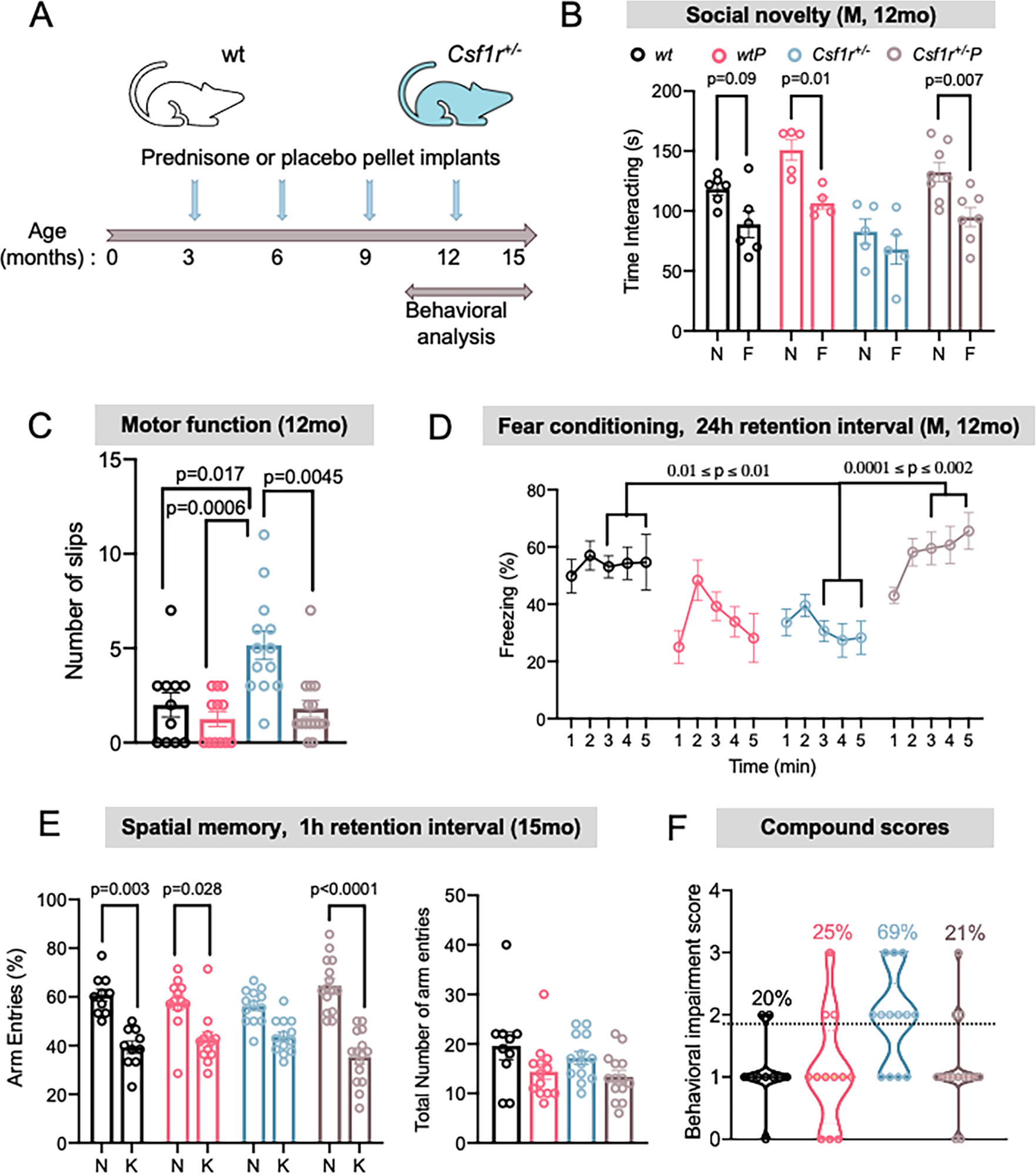
(A) Timeline of the experiment. (B) Evaluation of autistic-like behavior in the three-chamber sociability test (2-way ANOVA; group F(3,39)= 11.08, p<0.0001; preference F(1, 39) =24.24, p<0.0001)). N, novel mouse; F, familiar mouse. N= 5–8 male mice/condition. (C) Scoring of motor coordination deficits in the balance beam test (Kruskall-Wallis test p=0.0003). n= 11–15 mice/condition, both sexes. (D) Assessment of associative learning and long-term memory using the fear conditioning test (2-way ANOVA; group F(3,19)= 9.549, p=0.0005; time F(4, 76) =4.568, p=0.0023). Each data point represent average values for 5–7 male mice/condition. (E) Evidence for improvement of the short-term spatial memory deficit by prednisone. Left: Y maze test (2-way ANOVA; arm F(1,46)= 53.73, p<0.0001; group F(3, 46) =158.2, p<0.0001). N, novel arm; K, known arm. Right: Similar exploratory activity in all groups is shown by the number of total entries into the arms of the Y-maze (one-way ANOVA, p= 0.06); n= 10–15 mice/condition, both sexes. (F) Compound neurocognitive impairment scores, based on performance in the balance beam, fear conditioning and Y-maze testing. The graph includes only the mice that performed in all tests; each point represents one mouse; n= 10–14 mice/condition, both sexes. The data are presented as means ± SEM; M indicates male mice only. The p values shown for individual comparisons are based on Bonferroni’s post-hoc test following significant ANOVA or on Dunn’s post-hoc test following a significant Kruskall-Wallis test. Only significantly different changes are marked on the charts.
2.3. Behavioral studies
Behavioral studies were conducted in mice between 11 and 15 months of age as described (Biundo et al., 2023a). Social interaction deficits were assessed at 11 months of age using the three-chamber sociability test (Kana et al., 2019). Motor coordination was tested in the balance beam test (Gulinello, Chen, & Dobrenis, 2008) at 12 months of age. Spatial memory was assessed at 15 months of age using the two-stage version of the Y-maze (Biundo, Ishiwari, Del Prete, & D’Adamio, 2015). Associative learning and long-term memory were assessed using the contextual fear conditioning test (Biundo, Del Prete, Zhang, Arancio, & D’Adamio, 2018). On the first day (training), mice were given a single foot shock (0.6 mA, 2 s) coupled with a conditioned stimulus (a tone of 2.8 kHz, 85 dB). One day after (testing), mice were returned to the conditioning chamber and the percentage of time freezing recorded by tracking software (FreezeFrame 4, Actimetrics). All the experiments were conducted during the light cycle by a blinded experimenter. Data were analyzed separately for males and females. When similar trends were observed for both sexes, the data were pooled.
2.4. Ultrastructural studies
Callosal sections were prepared as described (Chitu et al., 2015) and examined by transmission electron microscopy using a JEOL 1400 transmission electron microscope. G-ratios, the ratio of the mean diameter of the axon over the mean diameter of the myelinated fiber, were determined on 200 randomly chosen fibers per mouse (4–8 mice/ genotype) using Image J software (imagej.net) in a blinded fashion. Age-related ultrastructural changes were identified according to the description provided by Peters and Sethares (the fine structure of the aging brain [http://www.bu.edu/agingbrain]) and quantified in 10 different microscopic fields/mouse.
2.5. Immunofluorescence staining and data analysis
Immunostaining was performed in brain slices prepared as described previously (Biundo et al., 2021; Chitu et al., 2015) using 30 μm thick sagittal sections. To ensure consistency and to avoid bias, tissue sections were chosen from matched anatomical regions. Brain sections were incubated with primary antibodies directed to either Ionized calcium binding adaptor molecule 1 (Iba1, 1:500) (rabbit IgG; Wako Chemicals RRID: AB_839504) or neuronal nuclei (NeuN, 1:500) (mouse IgG, Millipore RRID:AB_2149209) overnight at 4°C. Following incubation with primary antibodies, the sections were incubated with secondary antibodies conjugated to either Alexa 488 or Alexa 594 (1:1000) (Life Technologies, Grand Island, NY) for 1 hour at room temperature. Fluoromyelin staining for myelin (1:350, 30 minutes) was performed according to the manufacturer’s (Molecular Probes, Inc.) instructions. Images were captured using a Nikon Eclipse TE300 fluorescence microscope with NISElements D4.10.01 software. Quantification of cell numbers was performed manually by a blinded operator. Fluorescence intensity was quantified in ImageJ. Images were cropped and adjusted for brightness, contrast and color balance using Adobe Photoshop CS4. Morphometric analysis of microglia was carried out on maximum intensity projections of Iba-1-stained tissue sections using FIJI as described (Young & Morrison, 2018). Images were collected using a Leica SP5 Confocal microscope by a blinded operator.
2.6. Isolation of microglia and oligodendrocytes
Brains were dissociated into single cell suspensions using the Adult Brain Dissociation Kit from Miltenyi Biotech. Microglia and oligodendrocytes were isolated by immunomagnetic separation using CD11b and Anti-O4 microbeads (Miltenyi Biotech), respectively according to the manufacturer’s instructions. Cell purity was assessed in a preliminary experiment by immunostaining of each cell type preparation with antibodies to microglial (P2RY12-PE), astrocytic (ACSA-VB515) and oligodendrocyte (O4-APC) markers, followed by confocal microscopy and confirmed to be >90% (Figure S2). The number of cells isolated was determined using a hemocytometer. Cell pellets were stored at −80°C before processing for proteomic analysis.
2.7. Proteomic analysis
Cells were lysed in 5% SDS-containing sample buffer and the proteins were isolated using S-TRAP micro spin columns (Protifi) and digested to obtain peptides for mass spectrometric analysis according to the manufacturer’s instructions. Briefly, samples were homogenized in a buffer containing 5% SDS, 5 mM DTT and 50 mM ammonium bicarbonate (pH = 8), and incubated at 56 °C for 30 minutes for disulfide bond reduction. Samples were then alkylated with 20 mM iodoacetamide in the dark for 30 minutes. Phosphoric acid (12%) was then added to the sample at a final concentration of 1.2%. Samples were diluted in six volumes of binding buffer (90% methanol and 10 mM ammonium bicarbonate, pH 8.0). After gentle mixing, the protein solution was loaded to an S-trap filter (Protifi) and spun at 500 × g for 30 sec. The sample was washed twice with binding buffer. Finally, 1 μg of sequencing grade trypsin (Promega), diluted in 50 mM ammonium bicarbonate, was added into the S-trap filter and samples were digested at 37 °C for 18 h. Peptides were eluted in three steps: (i) 40 μl of 50 mM ammonium bicarbonate, (ii) 40 μl of 0.1% TFA and (iii) 40 μl of 60% acetonitrile and 0.1% TFA. The peptide solution was pooled, spun at 1,000 × g for 30 sec and dried in a vacuum centrifuge.
Prior to mass spectrometry analysis, samples were desalted using a 96-well plate filter (Orochem) packed with 1 mg of Oasis HLB C-18 resin (Waters). Briefly, the samples were resuspended in 100 μl of 0.1% TFA and loaded onto the HLB resin, which was previously equilibrated using 100 μl of the same buffer. After washing with 100 μl of 0.1% TFA, the samples were eluted with a buffer containing 70 μl of 60% acetonitrile in 0.1% TFA and then dried in a vacuum centrifuge. Samples were resuspended in 10 μl of 0.1% TFA and loaded onto a Dionex RSLC Ultimate 300 (Thermo Scientific), coupled online with an Orbitrap Fusion Lumos (Thermo Scientific). Chromatographic separation was performed with a two-column system, consisting of a C-18 trap cartridge (300 μm ID, 5 mm length) and a picofrit analytical column (75 μm ID, 25 cm length) packed in-house with reversed-phase Repro-Sil Pur C18-AQ 3 μm resin. Peptides were separated using a 120 min gradient from 4–30% buffer B (buffer A: 0.1% formic acid, buffer B: 80% acetonitrile + 0.1% formic acid) at a flow rate of 300 nl/min. The mass spectrometer was set to acquire spectra in a data-dependent acquisition (DDA) mode. Briefly, the full MS scan was set to 300–1200 m/z in the orbitrap with a resolution of 120,000 (at 200 m/z) and an AGC target of 5×10e5. MS/MS was performed in the ion trap using the top speed mode (2 secs), an AGC target of 1×10e4 and an HCD collision energy of 35.
Proteome raw files were searched using Proteome Discoverer software (v2.4, Thermo Scientific) using SEQUEST search engine and the SwissProt mouse database. The search for total proteome included variable modification of N-terminal acetylation, and fixed modification of carbamidomethyl cysteine. Trypsin was specified as the digestive enzyme with up to 2 missed cleavages allowed. Mass tolerance was set to 10 pm for precursor ions and 0.2 Da for product ions. Peptide and protein false discovery rate was set to 1%. The data were processed and analyzed as described (Aguilan, Kulej, & Sidoli, 2020). Ingenuity Pathway Analysis (IPA) was used to determine the impact of the significant changes in protein abundance detected in each sample.
2.8. Statistical analyses
Statistical analyses were computed using GraphPad Prism 8 (GraphPad, La Jolla, CA). Data were checked for outliers using the Grubbs’ method. Gaussian distribution was evaluated using the Shapiro-Wilk normality test and the Kolmogorov-Smirnov test. The screened data were analyzed using Student’s t-test, the Kruskal–Wallis test, or by analysis of variance (one- or two-way ANOVA followed by Bonferroni’s multiple comparison test). When significant effects of the independent variables were detected, single differences between or within groups were analyzed by post-hoc multiple comparisons as indicated in the figure legends. The level of significance was set at p ≤ 0.05. All comparisons in which the p value was > 0.1 are omitted from the figure panels,. Unless otherwise indicated, all data are presented as means ± SEM. Sample sizes for each experiment are plotted on the graphs.
3. Results
3.1. Case histories of CRL patients treated with glucorticoid medications (Table 1).
Table 1.
Summary of demographics, genetic and clinical information on asymptomatic CSF1R mutation carriers with a history of glucocorticoid medication intake.
| Sex, current age (years) | CSF1R mutation | Evidence of pathogenicity | Ethnicity | Positive family history of CRL | Presence of CRL symptoms | Steroid intake | Steroid indication | Steroid administration route | Steroid therapy total duration | Age during steroid intake | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Female, died at 72 | c.2625G > A (Met875Ile) |
Daughter affected | Caucasian | Yes | No | Yes | Rheumatoid arthritis | Oral | 26 years | Once a day between the ages of 46 and 72 years old | Tipton et al., 2021 Koga et al., 2022 Dulski et al, 2023 |
| Female, 64 | c.2656_2657insC (Tyr886Ser fs*56) | Multiple other individuals affected | Caucasian | Yes | No | Yes | Allergy | Oral | 10 years | Irregular intake between the ages of 20 to 30 years old | Dulski et al, 2023 |
| Arthritis | Intramuscular | 9 years | One injection a year since the age of 55 | Ali et al., 2022 | |||||||
| Male, 47 | c.1924C>T (Gln642*) | Truncating variant; close location to other CSF1R pathogenic variants; in silico models indicate pathogenicity (CADD score of 41) | Ashkenazi Jewish | Yes | No | Yes | Asthma | Oral | 8 years | Repeated 1-week courses 1–2 times a year, intake once a day, between the ages of 12 and 20 years old | Z.K. Wszolek, unpublished |
| Spondylosis | Epidural | 1 injection | 1 injection at 46 years old | ||||||||
| Female, 51 | c.2507G>A, Ser836Asn | Two affected siblings | Black American | Yes | No | Yes | Asthma | Inhaled | 5 years | Once/twice a day since the age of 45 (ongoing) | Z.K. Wszolek, unpublished |
| Intramuscular | 9 years | Repeated injections once every 3 months between the ages of 36 and 45 |
We previously reported two cases of asymptomatic CSF1R mutation carriers in their 60s and 70s treated with glucocorticoid medications (S. Ali et al., 2022; Tipton et al., 2021). We also collected information on two other asymptomatic carriers of CSF1R mutations with a history of long-term glucocorticoid therapy (Dulski, Heckman, Nowak, & Wszolek, 2023) (Table 1). As approximately 300 cases of CRL have been reported worldwide, the number of patients with detailed clinical information and comorbidities requiring concomitant glucocorticoid medication intake is limited to only a few cases. However, the four presented cases were of various ethnical backgrounds (Ashkenazi Jewish, Black American, Caucasian), carried mutations affecting the tyrosine kinase domain (amino-acids 582–910), had a positive family history and did not develop symptomatic disease, despite exceeding the average age of onset of CRL by decades. The exposure to glucocorticoid therapy was long-term (ranging from 8 to 26 years), and in 3/4 cases, more than one administration route was used. One case died of causes unrelated to CRL at the age of 72, and the autopsy showed neuropathological features of early-stage CRL (Koga, Tipton, Wierenga, Dickson, & Wszolek, 2022).
3.2. Prednisone treatment prevents development of social, cognitive and motor deficits in Csf1r+/−mice
Cognitive impairment is one of the most frequent initial manifestations of CRL, followed by gait and mood/personality disorders (reviewed in (Chitu et al., 2022; Dulski et al., 2022)). To assess the prophylactic effects of chronic, long-term immune suppression on the development of CRL in mice, a cohort of male and female mice treated either with prednisone or placebo was evaluated for a variety of behavioral deficits associated with CRL (Figure 1A). Csf1 deficiency in neural lineage cells has been reported to produce an early deficit in social memory, that is commonly found in mouse models of neurodevelopmental and neuropsychiatric disorders, such as autism spectrum disorders (Kana et al., 2019). Therefore, we tested whether the social memory deficit is an early marker of CRL in Csf1r+/− mice in the three-chamber sociability test (Kana et al., 2019). This test evaluates the preference of the test mouse to interact with a novel rather than familiar mouse. At 12 months of age Csf1r+/− male mice exhibited autistic–like behavior which was attenuated by prednisone treatment (Figure 1B). This phenotype was not observed in females of this age (data not shown), although in a previous study (Biundo et al., 2023a), it was detected in older (13–16 months) females. Similarly, there was a positive effect of prednisone treatment in preventing the development of motor coordination deficits in Csf1r+/− mice, detected by evaluating the number of slips on a balance beam (Figure 1C). Assessment of associative learning and long-term memory using the fear conditioning test revealed restoration of learning and memory in the prednisone-treated male (Figure 1D), but not female (data not shown) mice. Evaluation of short-term spatial memory at 15 months of age using the Y-maze showed that the spatial memory deficits characteristic of Csf1r+/− mice were also attenuated by prednisone treatment (Figure 1E, left panel). The total number of arm entries was comparable among the four groups tested, indicating that differences in cognitive performance detected in placebo-treated Csf1r+/− mice did not result from a decreased propensity to explore the apparatus (Fig. 1E, right panel). To determine whether prednisone treatment produced an overall behavioral improvement in Csf1r+/− mice, we calculated a compound behavioral deficit score by assigning a value of 1 to each behavioral deficit detected. Based on the observation that on average most CRL patients present with two or more deficits (reviewed in (Chitu et al., 2022)), we considered mice exhibiting two or more deficits as being affected. Using this scoring system, we show that chronic prednisone administration reduces the penetrance of CRL in mice at 15 months of age from 69% to 21% (Figure 1F). This value of 21% is comparable to that observed in the wt group, suggesting that it reflects the effects of normal aging in mice.
3.3. Chronic prednisone administration attenuates demyelination and neuronal degeneration in Csf1r+/− mice
The improvement of behavioral parameters in Csf1r+/− mice who received prednisone administration prompted us to examine the effect of treatment on myelin and neuronal pathology. We initially focused on characterizing myelination in the supraventricular region of the corpus callosum, a site that is frequently affected in CRL patients. At 15 months of age, Csf1r+/− mice that received placebo exhibited a significant decrease in callosal myelin evidenced by reduced fluoromyelin staining (Figure 2A, B). In contrast the intensity of fluoromyelin staining was normal in Csf1r+/− mice treated with prednisone (Figure 2A, B). Consistent with this, prednisone treatment also prevented the increase in G-ratio in callosal axons (Figure 2C–G) that is indicative of cycles of demyelination followed by remyelination. Axonal degeneration in the corpus callosum is another pathognomonic feature of CRL that is also encountered in the Csf1r+/− model. Prednisone treatment alleviates this phenotype (Figure 2 H, I). Together these data show that chronic administration of prednisone to young adults significantly delays the phenotypic expression of Csf1r heterozygosity.
Figure 2. Prednisone treatment improves myelination and is neuroprotective in Csf1r+/− mice.
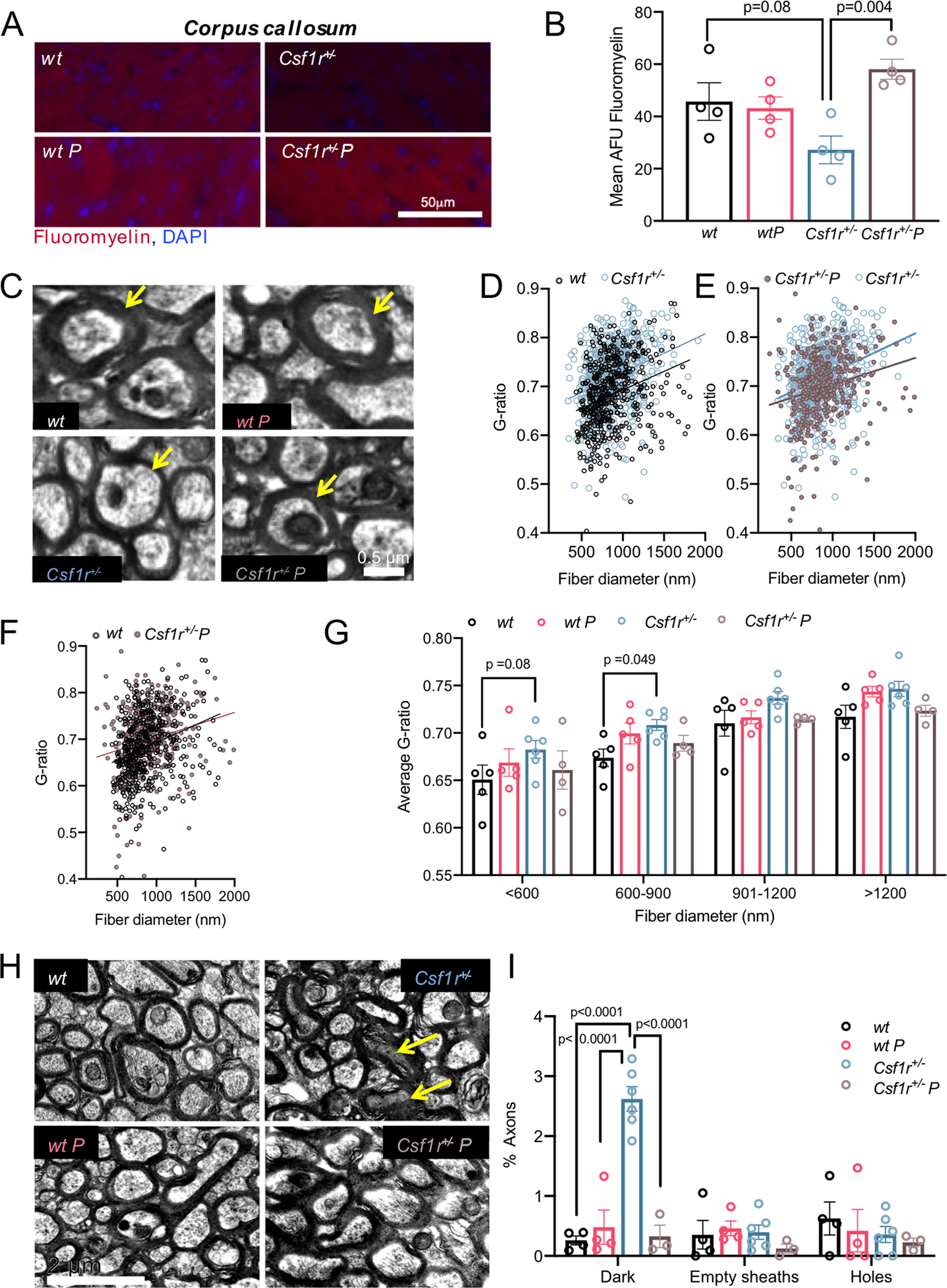
(A-G) Improvement of callosal myelination. (A) Fluoromyelin staining of the corpus callosum and (B) quantification of the fluorescence intensity (one-way ANOVA, F(3, 12) = 5.696, p = 0.012); n= 4 mice/condition, females. (C) Representative images of cross-sections through callosal axons. The arrows point to differences in myelin thickness in axons of similar diameters. Scale bar, 2.0 μm, applies to all panels. (D-F) Comparative distribution of the G-ratios for individual axons examined. The trendline in (D) indicates a reduction in myelin thickness in Csf1r+/− axons compared with wt while the trendline in (E) shows a trend to normalization that is more evident in the low and medium range of fiber diameters in axons of Csf1r+/− mice treated with prednisone. (F) Shows an almost complete superimposition of wt and Csf1r+/− prednisone scatter plots of G-ratio distribution. (G) Prednisone treatment alleviates the increase in average G-ratio values in Csf1r+/− mice. Two-way ANOVA (main effect of group F(3,64) =6.983; p =0.0004); n= 4–6 mice/condition, females. (H, I) Improvement of neurodegeneration. (H) Electron micrographs of the four experimental groups illustrating the presence of degenerating, dark axons (arrows). Scale bar, 0.5 μm, applies to all panels. (I) Quantification of various aspects of axonal pathology. Two-way ANOVA (main effect of group F(3,39) =12.95; p <0.0001). Data are presented as means ± SEM; n= 4–6 mice/condition, females. Each point on the graphs represents one mouse. The p values shown for individual comparisons are based on Bonferroni’s post-hoc test following significant ANOVA.
3.4. Prednisone prevents the expansion of microglia and partially attenuates their de-ramification
Microglia expansion occurs in the cortex and white matter of CRL patients in a stage-dependent manner and is accompanied by gradual loss of membrane processes, indicative of activation (Oyanagi et al., 2017). Csf1r+/− mice reproduce the increase in microglial densities in multiple brain regions including the motor cortex, hippocampus, corpus callosum (Fig. 3A–D) (Biundo et al., 2023a; Chitu et al., 2015). This phenotype is prevented by prednisone treatment (Fig. 3A–D). In addition, prednisone also prevented the substantial de-ramification of the microglial processes observed in the motor cortex of Csf1r+/− mice. In contrast to previous studies (Biundo et al., 2021; Chitu et al., 2020), in this cohort we did not observe significant morphological changes in callosal microglia, possibly because we have examined younger mice (15 versus 18–23 months old).
Figure 3. Prednisone treatment alleviates microglia expansion and deramification in Csf1r+/− mice.
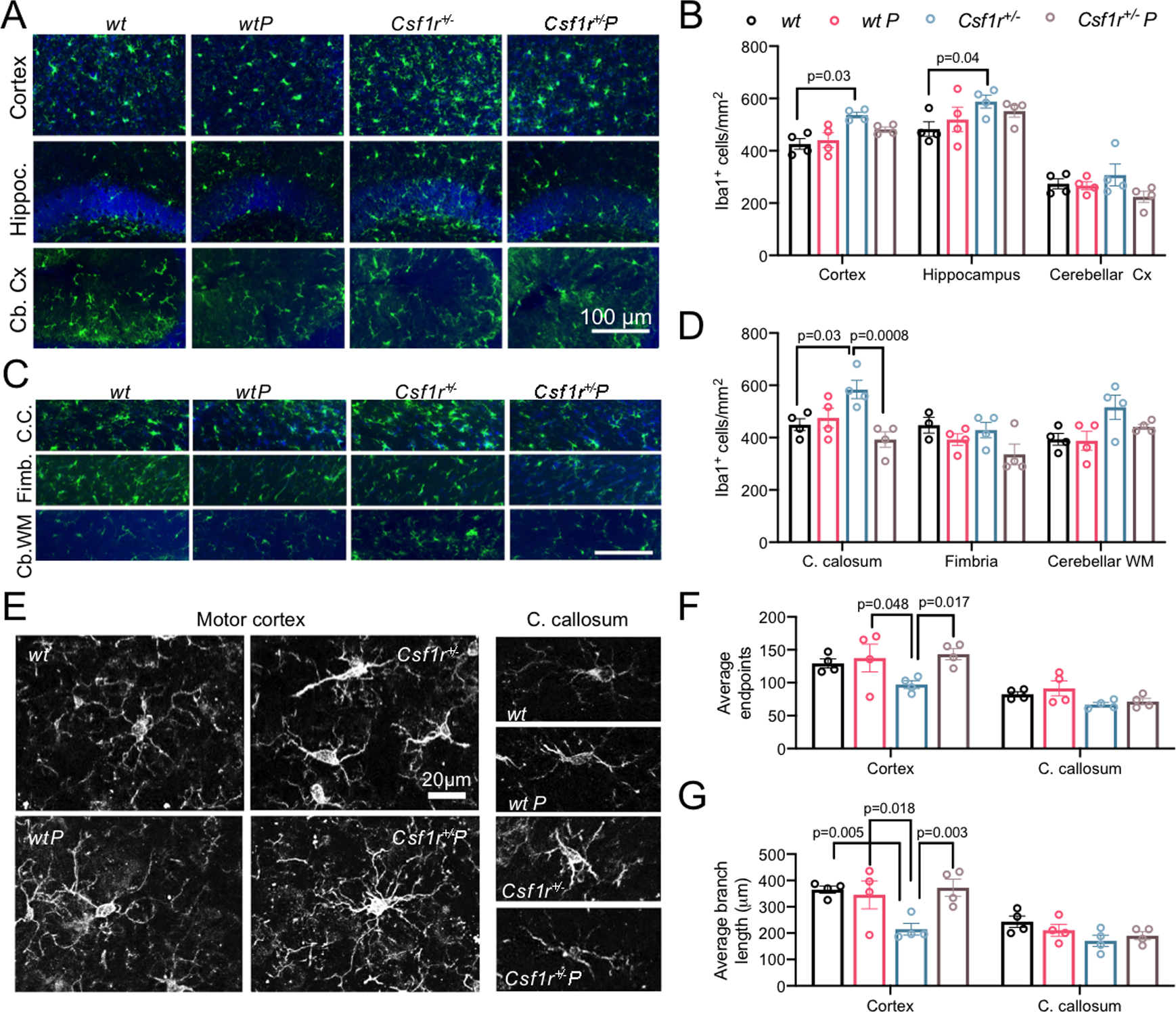
(A-D) Effect of prednisone treatment on microglial densities in various grey (A,B) and white (C,D) matter regions. (A,C) Illustrative images of Iba-1 stained microglia. Scale bar, 100 μm, applies to all panels in each composite. (B) Quantification of Iba1+ cell densities in the grey matter (two-way ANOVA, main effect of group F(3, 36) =5.79, p=0.0025); n= 4 mice/condition, females. (D) Quantification of Iba1+ cell densities in the white matter (two-way ANOVA, main effect of group F(3, 35) =7.95, p=0.0004); n= 4 mice/condition, females. (E-G) Effects of Csf1r heterozygosity and prednisone treatment on microglia morphology. (E) Illustrative images of Iba-1-stained microglia. Scale bar, 20 μm, applies to all panels. (F,G) Quantification of process branching (F, two-way ANOVA, main effect of group F(3, 24) =4.1, p=0.017) and length (G, two-way ANOVA, main effect of group F(3, 24) =6.12, p=0.003). Each point on the graphs represents average values obtained from 20±0.9 (C. callosum) or 36±1.9 (cortex) cells/mouse; n= 4 mice/condition, females. Data are presented as means ± SEM. The p values shown for individual comparisons are based on Bonferroni’s post-hoc test following significant ANOVA.
3.5. Prednisone treatment suppresses several processes relevant to microglia activation and oligodendrocyte senescence
To address the mechanism of prednisone action, we investigated the cellular states of microglia and oligodendrocytes using comparative proteomic analysis of freshly isolated cells. Because in mice, CRL is a primary microgliopathy (Biundo et al., 2021), we initially studied the effects of Csf1r heterozygosity in microglia. We found that a subset of 70 proteins were differentially expressed in Csf1r+/− microglia compared to wt (Figure 4A, B; Supplementary Table 1). Despite the small number of changes detected, these were sufficient to predict dysregulation of several biological processes including enhancement of cell fusion and autophagy and suppression of protein phosphorylation (Figure 4C), all of which were prevented by prednisone treatment. In Csf1r+/− microglia, prednisone also induced a proteomic profile consistent with suppression of a series of processes and pathways relevant to microglia activation, including the generation of reactive oxygen species, eicosanoid synthesis and metabolism, phagocytosis, migration and the inflammatory response (Figure 4C, D). Similar suppression of pathways dysregulated by Csf1r heterozygosity was observed when comparing the proteomic profile changes of Csf1r+/−P microglia versus wt control and Csf1r+/− versus Csf1r+/−P microglia (Figure 4D). In contrast, prednisone treatment had no significant effect in wt microglia (Figure 4C, D). Upstream regulator analysis indicates that dysregulation of microglial function in Csf1r+/− mice may involve the actions of IL-6 and IFN-α, both of which were alleviated by prednisone. In addition, consistent with earlier reports (Ganter et al., 1992) prednisone reduced the upstream actions of CSF-2 in Csf1r+/− microglia to levels lower than in wt controls (Figure 4 E).
Figure 4. Prednisone treatment suppresses several processes relevant to microglia activation and regulates eicosanoid synthesis.
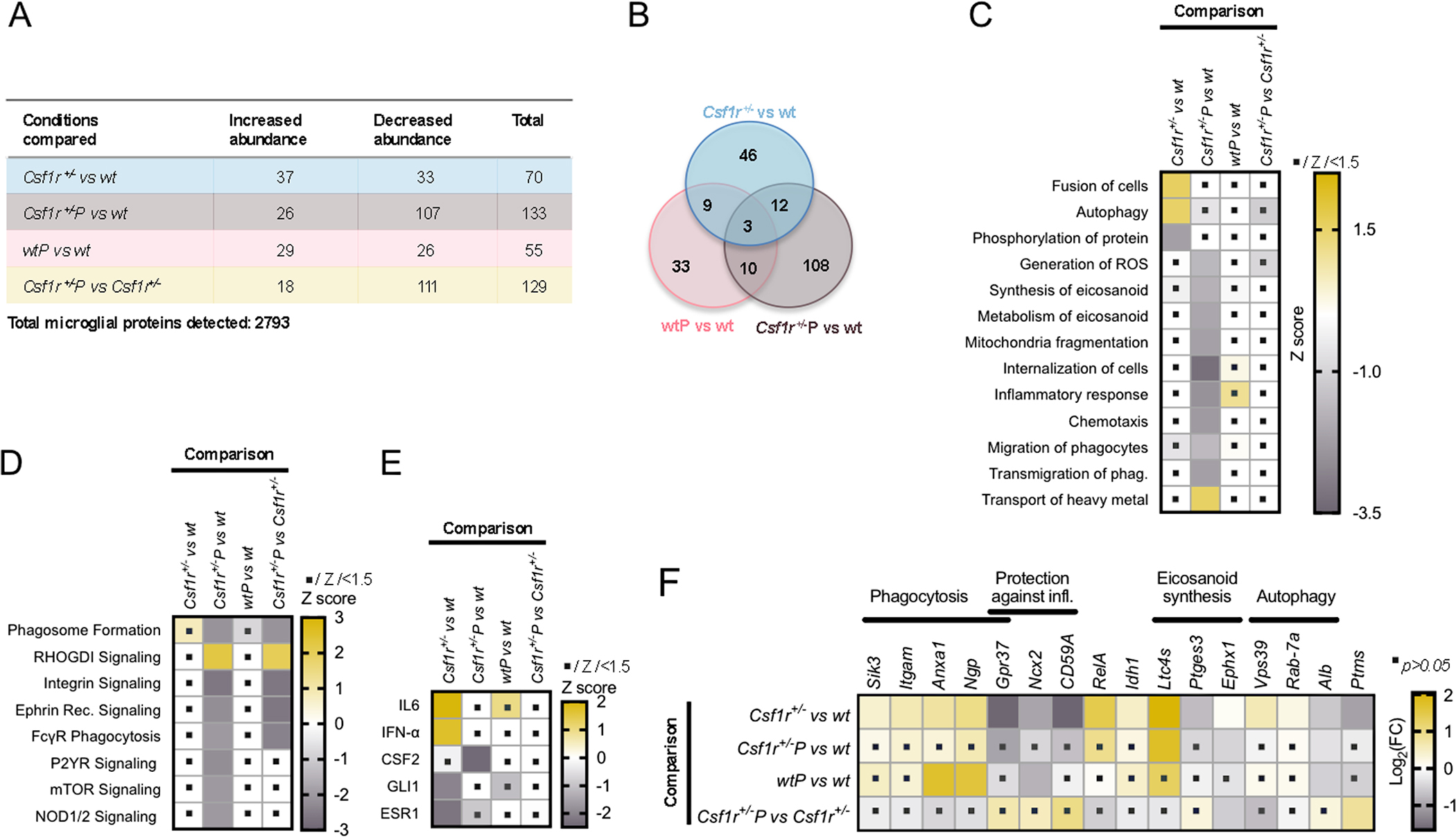
(A) The number of proteins with increased and decreased abundance compared to wt placebo. Blue, effects of Csf1r heterozygosity; Brown, combined effects of Csf1r heterozygosity and prednisone treatment; Pink, effects of prednisone treatment in wt; Light yellow, effects of prednisone treatment in Csf1r+/− microglia. Data from n= 4 mice/condition, males (B) The overlap between the effects of Csf1r heterozygosity and the effects of prednisone treatment. The Venn diagram displays the number of proteins differentially enriched compared to wt placebo in each category. (C) Biological processes predicted to be affected by Csf1r heterozygosity and prednisone treatment alone or in combination. (D) Comparative pathway analysis. (E) Predicted upstream regulators. (F) Illustration of the effects of prednisone treatment on several potential mediators of the effects of Csf1r heterozygosity. The dots within squares indicate the change is not significant (absolute Z scores<1.5 in C-E; or p>0.05 in F).
The amounts of several proteins involved in phagocytic processes, such as the salt-inducible kinase Sik3 (K. Wang et al., 2022), the CD11b (Itgam) complement receptor CR3 subunit (Norris et al., 2018), Annexin A1 (Anxa1) (McArthur et al., 2010) and the neutrophilic granule protein (NGP) (Liu et al., 2020) (Figure 4 F), were significantly increased in Csf1r+/− microglia. Potentially relevant to the pathology of CRL, Sik3 was recently reported to promote excessive myelin phagocytosis (K. Wang et al., 2022), while CR3 and Anxa1 are involved in the engulfment of neurodegenerative debris by microglia (McArthur et al., 2010; Norris et al., 2018). In addition, Csf1r+/− microglia also exhibit decreased levels of GPR37, a G protein coupled receptor that promotes phagocytosis by macrophages but also reduces post-phagocytic inflammatory activation (Bang et al., 2018). Interestingly, GPR37 also promotes macroautophagy (Marazziti et al., 2009) which in turn is essential for myelin clearance and recovery from neuroinflammation (Berglund et al., 2020). Together, these alterations in proteome composition are suggestive of an altered state associated with aberrant responses following clearance of myelin and apoptotic debris.
Csf1r+/− microglia also exhibit decreased levels of other proteins that protect against inflammation, including NCX2 (Anzilotti et al., 2021) and CD59A (Herrmann et al., 2015). (Figure 4F). Prolonged activation of the Na+/Ca2+ exchanger NCX2 was reported to reduce neuroinflammation in a mouse model of ALS (Anzilotti et al., 2021), suggesting an anti-inflammatory function and CD59A prevents the autoactivation of the complement system (Herrmann et al., 2015). In addition, upregulation of the NFkB p65 subunit (RelA) and of IDH1, a metabolic enzyme that mediates the toxic effects of oxidized LDL including foam cell formation and ferroptosis (B. Li et al., 2022) was also detected. Together these data suggest that in Csf1r+/− microglia have a propensity for abnormal activation in response to inflammatory stimuli.
Other proteomic changes in Csf1r+/− microglia are consistent with alteration of the synthesis of eicosanoids. Among these, the upregulation of leukotriene C4 synthase (LTC4s) and downregulation of prostaglandin E2 synthase (Ptges3) might be highly relevant since LTC4 has neurotoxic (Michael, Marschallinger, & Aigner, 2019; Wang, Wang, & Zou, 2022), while PGE2 has anti-inflammatory (Fadok et al., 1998), properties. With the exception of Ltc4s, prednisone atenuates the dysregulated expression of all the proteins discussed above (Fig. 4F). In addition, prednisone treatment reduced the abundance of epoxide hydrolase Ephx1, that inactivates epoxyeicosatrienoic acids, a class of arahidonic acid metabolites with anti-inflammatory and vasoactive properties (Gautheron & Jeru, 2020).
Prednisone also prevented the decrease in parathymosin (Ptms), a hormone with anti-senescent effects (Yu, Tang, & Cai, 2020) and a nuclear suppressor of NFκB (Okamoto et al., 2016). Together, these data suggest that Csf1r+/− microglia exhibit an aberrant, tissue damaging phenotype that is attenuated by prednisone-mediated immunosuppression.
3.6. Prednisone treatment suppresses oligodendrocyte senescence
Next, we sought to examine the consequences of microglial dyshomeostasis on oligodendrocytes. Ex vivo proteomic analysis identified 122 proteins differentially expressed in Csf1r+/− oligodendrocytes compared to wt counterparts (Figure 5A, B; Supplementary Table 2). In silico analysis indicates that these alterations perturb cellular organization and membrane trafficking (Figure 5C). Pathway analysis revealed the activation of the senescence pathway in Csf1r+/− oligodendrocytes (Figure 5D). These phenotypes were attenuated by prednisone treatment (Fig 5 C,D). Of interest, prednisone also promoted gap junction formation, metal ion homeostasis and energy production in oligodendrocytes as well as the activation of the PDGF and CXCR4 signaling pathways that promote oligodendrocyte differentiation and remyelination (Patel, McCandless, Dorsey, & Klein, 2010; Vana et al., 2007). Notably, many of the proteins involved in the transport and homeostasis of metal regulated by prednisone control iron and calcium homeostasis (Figure 5C,F) and their changes in abundance are consistent with increased cytosolic levels of Fe and decreased cytosolic Ca2+. Oligodendrocytes require iron for their proliferation, differentiation and for the production lipid myelin components (Cheli, Correale, Paez, & Pasquini, 2020). While the prednisone induced increase in Fe ions might also contribute to oxidative stress via the Fenton reaction, this is probably compensated by the concomitant upregulation of Metallothionein 3 (Mt3), a scavenger of reactive oxygen species (Koh & Lee, 2020) (Figure 5F). Upstream regulator analysis identifies RICTOR, a component of the mTORC2 complex and LARP1, a substrate of mTORC1 (Hong 2017) as activated upstream regulators of the proteomic alterations in Csf1r+/− oligodendrocytes suggesting that dysregulation of the mTOR pathway may play an important role (Figure 5E), possibly by regulating translation. The involvement of GABA was also detected which is important for the myelination of interneurons (Benamer, Vidal, Balia, & Angulo, 2020) and proper neuronal cortical circuits. Together, these data demonstrate that prednisone treatment is beneficial for oligodendrocyte fitness in CRL.
Figure 5. Prednisone treatment alleviates oligodendrocyte senescence and improves energetic and metal ion homeostasis.
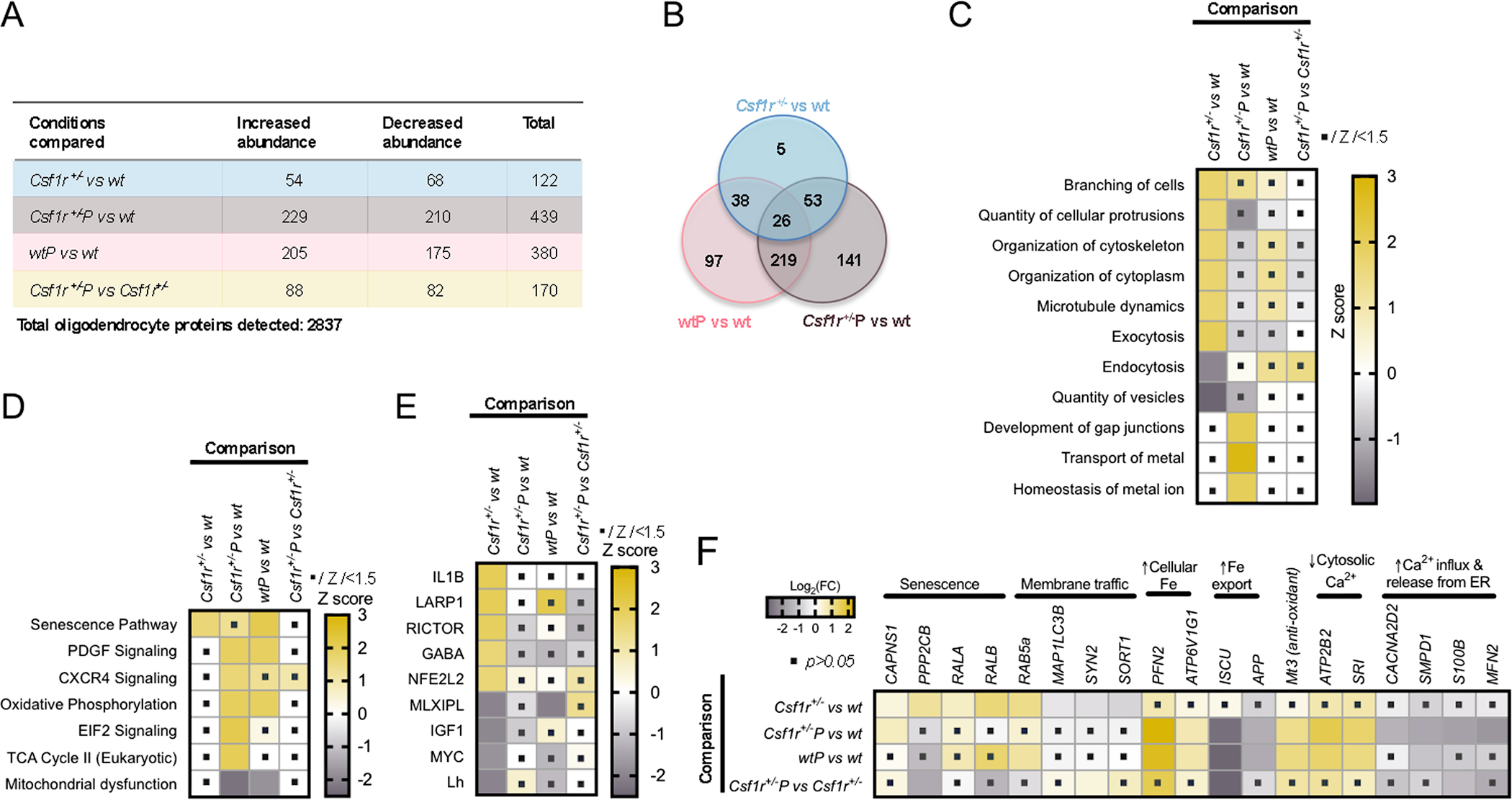
(A) The number of proteins with increased and decreased abundance compared to wt placebo. Blue, effects of Csf1r heterozygosity; Brown, combined effects of Csf1r heterozygosity and prednisone treatment; Pink, effects of prednisone treatment in wt. Light; Light yellow, effects of prednisone treatment in Csf1r+/− microglia. (B) The overlap between the effects of Csf1r heterozygosity and the effects of prednisone treatment. The Venn diagram displays the number of proteins enriched compared to wt placebo in each category. (C) Biological processes predicted to be affected by Csf1r heterozygosity and prednisone treatment alone or in combination. (D) Comparative pathway analysis. (E) Predicted upstream regulators. (F) Illustration of the effects of prednisone treatment on several potential mediators of the effects of Csf1r heterozygosity. The dots within squares indicate the change is not significant (absolute Z scores<1.5 in C-E; or p>0.05 in F).
4. Discussion
The involvement of dominantly inherited, monoallelic, CSF1R mutations in an adult-onset hereditary leukoencephalopathy was first recognized more than a decade ago (Rademakers et al., 2011). To date more than 100 different mutations have been identified in CRL patients, most of which reside in exons encoding the kinase domain (Papapetropoulos et al., 2021). Many of these mutations have been shown to lead to loss or decrease of the tyrosine kinase activity and a small proportion of these cause loss of expression mediated by nonsense-mediated RNA decay (NMD) (reviewed in (Chitu et al., 2022)). Although no clear genotype–phenotype correlation is apparent, mutations that trigger NMD or lead to protein truncation are associated with an earlier time of onset (Chitu et al., 2022). However, there is also evidence of marked intra-familial variability of the phenotypic expression of CSF1R mutations which raises the possibility that the development and severity of clinical CRL might be influenced by other genetic or environmental factors. We have previously described two carriers of pathogenic CSF1R mutations that remained healthy despite their advanced age (71 and 63 years old) (S. Ali et al., 2022; Tipton et al., 2021) and have recently identified more cases (Dulski 2023 in press; Z. Wszolek, unpublished data in Table 1). All of these patients have been subjected to long-term steroid-mediated immunosuppression for unrelated causes.
Glucocorticoids can readily cross the blood-brain barrier and the glucocorticoid receptor is expressed on neurons and glial cells, including microglia (Witt & Sandoval, 2014). They have been shown to curtail microglial activation in response to neuronal injury (Ros-Bernal et al., 2011) and to provide protection against oxidative stress in cultured oligodendrocytes and in vivo (Pitarokoili et al., 2019). Relevant to CRL, previous studies indicate that oxidative stress caused by microglial dyshomeostasis might play a role in the pathology of disease (Chitu et al., 2020). Together, these data suggested that glucocorticoids might provide protection in CRL, a demyelinating disease triggered by microglial dyshomeostasis. However, due to the very limited number of patients in which an association between chronic immunosuppression and CRL resilience was observed, more evidence was needed to support a link between glucocorticoid-mediated immunosuppression and CRL protection. We therefore investigated the role of presymptomatic prednisone administration in the development of CRL using the Csf1r+/− mouse model. We show that, consistent with the clinical observations, mice treated with prednisone are protected against the development of neurocognitive deficits (Figure 1) and pathognomonic histopathology (Figures 2–3) and provide experimental evidence for successful pharmacological intervention in an adult organism.
We used proteomic profiling of isolated cells to investigate the mechanisms contributing to disease and the effect of prednisone. We show that prednisone administration attenuates a series of biological processes relevant to microglia activation (Figure 4), while suppressing the senescence and improving the fitness of oligodendrocytes (Figure 5). It is unclear whether the protective actions of prednisone result from suppression of microglia activation, direct actions in oligodendrocytes (e.g., protection against oxidative stress), or a combination of both. However, since we have previously demonstrated that in the mouse, CRL is a primary microgliopathy (Biundo et al., 2021), we focused on identifying candidate factors produced by microglia that could trigger demyelination and neuronal cell loss. Previous investigations using transcriptomic profiling of brain tissue or isolated microglia indicated that microglial dyshomeostasis was related to the increase in the bioavailability of GM-CSF (also known as CSF-2), a proinflammatory cytokine that is also a microglial mitogen and of G-CSF (Biundo et al., 2023a; Chitu et al., 2020). Notably, glucocorticoids are known to suppress microglial activation by GM-CSF (Ganter et al., 1992; Tanaka et al., 1997), and, in our proteomic study, upstream mediator analysis suggests that the effects of prednisone on microglia were mediated in part by suppression of CSF-2 signaling (Fig. 4E). While the previous transcriptomic studies (Chitu et al., 2020) showed that Csf1r+/− microglia were dyshomeostatic, they did not produce evidence of classical neuroinflammation or identify candidate mediators of microglia toxicity to oligodendrocytes and neurons. In contrast, in the present work, proteomic profiling of isolated microglia raises the possibility that altered eicosanoid production (Figure 4 C, F) might contribute to the functional alteration of both microglia (Fadok et al., 1998; Michael et al., 2019) and oligodendrocytes (S. Wang et al., 2022), an aspect that deserves further investigation. In addition, Csf1r heterozygosity leads to decreased levels of parathymosin in microglia. Since parathymosin is both an anti-inflammatory protein (Okamoto et al., 2016) and an anti-senescence hormone (Yu et al., 2020), it is tempting to speculate that decreased levels of Ptms in microglia might alter their responses to stimuli and their ability to provide trophic support to the neighboring cells, e.g. oligodendrocytes and neurons. A hypothetical model of how changes in the microglial secretome could affect oligodendrocyte and neuronal cells is presented in Figure 6.
Figure 6. Potential impact of Csf1r heterozygosity on microglial secretome and effect of these changes on neural lineage cells.
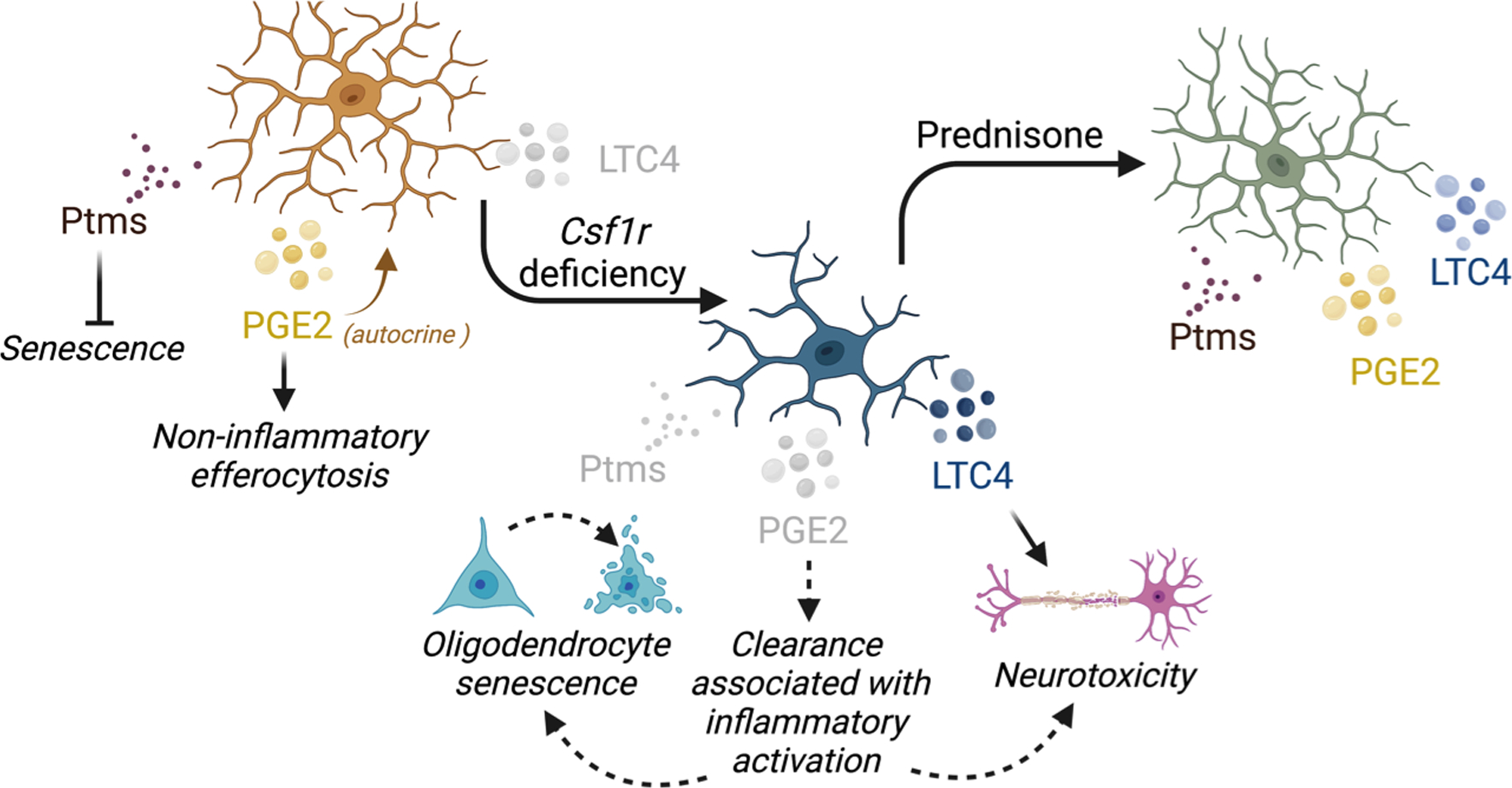
Compared to wild-type microglia, Csf1r+/− microglia produce less Ptms, a hormone with anti-senescence actions. Its decreased availability in the extracellular milieu might contribute to oligodendrocyte senescence. The decreased levels of prostaglandin E2 synthase (Ptges3) in Csf1r+/− microglia might cause reduced production of PGE2. Since PGE2 normally acts in an autocrine manner to suppress the inflammatory activation of macrophages following the uptake of apoptotic cells, this might contribute to aberrant activation of microglia after efferocytosis. The increased levels of leukotriene C4 synthase (Ltc4s) in Csf1r+/− microglia might result in increased production of LTC4 which is neurotoxic. Application of prednisone treatment restores the levels of Ptms and Ptges3 and slightly reduces those of Ltc4s, driving Csf1r+/− microglia towards a more homeostatic phenotype. Light gray coloring denotes decreased levels; dotted lines mark hypothetical events.
Importantly, the present study demonstrates that CRL can be managed pharmaceutically. However, chronic glucocorticoid administration can have significant side effects both peripherally (e.g., infection, diabetes, osteoporosis) and centrally (e.g., psychosis, depression, memory decline, seizures) (Witt & Sandoval, 2014). Clearly more research is needed to determine the optimal timing of glucocorticoid therapy, type of glucocorticoid medication, dosing regimen, and route of administration. Also, the lack of improvement following steroid treatment in patients after the onset of demyelination (Kim et al., 2015; Konno et al., 2017), suggests that prednisone treatment should be prophylactic. Fortunately, as most CRL cases are familial and the identification of CSF1R mutation carriers is straight-forward, it is likely that prophylactic treatment could significantly reduce the incidence of disease.
In addition, this study also prompts the exploration of novel, more specific, therapeutic approaches. In this regard, although parathymosin was originally described as a co-activator of the glucocorticoid receptor (Okamoto & Isohashi, 2005), more recent studies show that parathymosin can independently inhibit NFκB and that parathymosin-based drugs inhibit acute and chronic inflammation in vivo without producing significant side effects (Okamoto et al., 2016). Furthermore, the reduction in parathymosin levels with concomitant activation of NFκB target genes was shown in microglia stimulated with aggregated amyloid beta, suggesting a contribution to microglial dyshomeostasis in Alzheimer’s disease (Walker, Lue, & Beach, 2001). The development of brain-permeant parathymosin-based drugs and the evaluation of their effects in the mouse model of CRL might be an important step in identifying novel strategies to control microglia activation and suppress cellular senescence in the central nervous system. Therefore, observations from the present study may be translated into clinical applications and pave the way toward effective treatment for asymptomatic carriers of CSF1R mutations.
Supplementary Material
Acknowledgements
The authors thank Hillary Guzik, Leslie Cummings and Dr. Vera Des-Marais of the Einstein Analytical Imaging Facility for help with electron microscopy, imaging and histomorphometry and Dr. Deyou Zheng of the Saul R. Corey Department of Neurology, Dominick P. Purpura Department of Neuroscience and the Department of Genetics of the Albert Einstein College of Medicine for discussions of the proteomic data processing. This work was supported by grants from the National Institutes of Health: Grant R01NS091519 (to E. R. S.) and the P30CA013330 NCI Cancer Center Grant, the Renee and Robert A. Belfer Chair in Developmental Biology (to E. R. S.) and a gift from David and Ruth Levine.
Footnotes
Declaration of competing interests
None
Data availability
All data are available on request from the corresponding author.
References
- Aguilan JT, Kulej K, & Sidoli S (2020). Guide for protein fold change and p-value calculation for non-experts in proteomics. Mol Omics, 16(6), 573–582. doi: 10.1039/d0mo00087f [DOI] [PubMed] [Google Scholar]
- Ali S, Tipton PW, Koga S, Middlebrooks EH, Josephs KA, Strongosky A, . . . Wszolek ZK. (2022). A novel CSF1R variant in a South Dakota family with CSF1R-related leukoencephalopathy. Parkinsonism Relat Disord, 102, 51–53. doi: 10.1016/j.parkreldis.2022.07.016 [DOI] [PubMed] [Google Scholar]
- Ali ZS, Van Der Voorn JP, & Powers JM (2007). A comparative morphologic analysis of adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia--a role for oxidative damage. J Neuropathol Exp Neurol, 66(7), 660–672. doi: 10.1097/nen.0b013e3180986247 [DOI] [PubMed] [Google Scholar]
- Alturkustani M, Keith J, Hazrati LN, Rademakers R, & Ang LC (2015). Pathologic staging of white matter lesions in adult-onset leukoencephalopathy/leukodystrophy with axonal spheroids. J Neuropathol Exp Neurol, 74(3), 233–240. doi: 10.1097/NEN.0000000000000168 [DOI] [PubMed] [Google Scholar]
- Anzilotti S, Valsecchi V, Brancaccio P, Guida N, Laudati G, Tedeschi V, . . . Pignataro G. (2021). Prolonged NCX activation prevents SOD1 accumulation, reduces neuroinflammation, ameliorates motor behavior and prolongs survival in a ALS mouse model. Neurobiol Dis, 159, 105480. doi: 10.1016/j.nbd.2021.105480 [DOI] [PubMed] [Google Scholar]
- Arreola MA, Soni N, Crapser JD, Hohsfield LA, Elmore MRP, Matheos DP, . . . Green KN. (2021). Microglial dyshomeostasis drives perineuronal net and synaptic loss in a CSF1R(+/−) mouse model of ALSP, which can be rescued via CSF1R inhibitors. Sci Adv, 7(35). doi: 10.1126/sciadv.abg1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang S, Xie YK, Zhang ZJ, Wang Z, Xu ZZ, & Ji RR (2018). GPR37 regulates macrophage phagocytosis and resolution of inflammatory pain. J Clin Invest, 128(8), 3568–3582. doi: 10.1172/JCI99888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benamer N, Vidal M, Balia M, & Angulo MC (2020). Myelination of parvalbumin interneurons shapes the function of cortical sensory inhibitory circuits. Nat Commun, 11(1), 5151. doi: 10.1038/s41467-020-18984-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund R, Guerreiro-Cacais AO, Adzemovic MZ, Zeitelhofer M, Lund H, Ewing E, . . . Jagodic M. (2020). Microglial autophagy-associated phagocytosis is essential for recovery from neuroinflammation. Sci Immunol, 5(52). doi: 10.1126/sciimmunol.abb5077 [DOI] [PubMed] [Google Scholar]
- Biundo F, Chitu V, Shlager GGL, Park ES, Gulinello ME, Saha K, . . . Stanley ER. (2021). Microglial reduction of colony stimulating factor-1 receptor expression is sufficient to confer adult onset leukodystrophy. Glia, 69, 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biundo F, Chitu V, Tindi J, Burghardt NS, Shlager GGL, Ketchum HC, . . . Stanley ER. (2023a). Elevated granulocyte colony stimulating factor (CSF) causes cerebellar deficits and anxiety in a model of CSF-1 receptor related leukodystrophy. Glia, 71:775–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biundo F, Chitu V, Gökhan Ş, Chen E, Oppong-Asare J and Stanley ER (2023b). Trem2 enhances demyelination in the Csf1r+/− mouse model of leukoencephalopathy. Biomedicines, 11(8):2094. doi: 10.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biundo F, Del Prete D, Zhang H, Arancio O, & D’Adamio L (2018). A role for tau in learning, memory and synaptic plasticity. Sci Rep, 8(1), 3184. doi: 10.1038/s41598-018-21596-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biundo F, Ishiwari K, Del Prete D, & D’Adamio L (2015). Interaction of ApoE3 and ApoE4 isoforms with an ITM2b/BRI2 mutation linked to the Alzheimer disease-like Danish dementia: Effects on learning and memory. Neurobiol Learn Mem, 126, 18–30. doi: 10.1016/j.nlm.2015.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheli VT, Correale J, Paez PM, & Pasquini JM (2020). Iron Metabolism in Oligodendrocytes and Astrocytes, Implications for Myelination and Remyelination. ASN Neuro, 12, 1759091420962681. doi: 10.1177/1759091420962681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, Biundo F, Shlager GGL, Park ES, Wang P, Gulinello ME, . . . Stanley ER. (2020). Microglial Homeostasis Requires Balanced CSF-1/CSF-2 Receptor Signaling. Cell Rep, 30(9), 3004–3019 e3005. doi: 10.1016/j.celrep.2020.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, Gokhan S, Gulinello M, Branch CA, Patil M, Basu R, . . . Stanley ER. (2015). Phenotypic characterization of a Csf1r haploinsufficient mouse model of adult-onset leukodystrophy with axonal spheroids and pigmented glia (ALSP). Neurobiol Dis, 74, 219–228. doi:S0969-9961(14)00372-6 [pii] 10.1016/j.nbd.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, Gokhan S, Nandi S, Mehler MF, & Stanley ER (2016). Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends Neurosci, 39(6), 378–393. doi: 10.1016/j.tins.2016.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, Gokhan S, & Stanley ER (2022). Modeling CSF-1 receptor deficiency diseases - how close are we? The FEBS journal, 289(17), 5049–5073. doi: 10.1111/febs.16085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, & Stanley ER (2017). Regulation of Embryonic and Postnatal Development by the CSF-1 Receptor. Curr Top Dev Biol, 123, 229–275. doi: 10.1016/bs.ctdb.2016.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, . . . Stanley ER. (2002). Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood, 99(1), 111–120. [DOI] [PubMed] [Google Scholar]
- Dulski J, Heckman MG, Nowak J, & Wszolek Z (2023). Protective effect of glucocorticoids against symptomatic disease in CSF1R variant carriers. Movement Disorders, In Press. [DOI] [PubMed] [Google Scholar]
- Dulski J, Heckman MG, White LJ, Zur-Wyrozumska K, Lund TC, & Wszolek ZK (2022). Hematopoietic Stem Cell Transplantation in CSF1R-Related Leukoencephalopathy: Retrospective Study on Predictors of Outcomes. Pharmaceutics, 14(12). doi: 10.3390/pharmaceutics14122778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrchen JM, Roth J, & Barczyk-Kahlert K (2019). More Than Suppression: Glucocorticoid Action on Monocytes and Macrophages. Front Immunol, 10, 2028. doi: 10.3389/fimmu.2019.02028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, & Henson PM (1998). Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest, 101(4), 890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganter S, Northoff H, Mannel D, & Gebicke-Harter PJ (1992). Growth control of cultured microglia. J Neurosci Res, 33(2), 218–230. doi: 10.1002/jnr.490330205 [DOI] [PubMed] [Google Scholar]
- Gautheron J, & Jeru I (2020). The Multifaceted Role of Epoxide Hydrolases in Human Health and Disease. Int J Mol Sci, 22(1). doi: 10.3390/ijms22010013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulinello M, Chen F, & Dobrenis K (2008). Early deficits in motor coordination and cognitive dysfunction in a mouse model of the neurodegenerative lysosomal storage disorder, Sandhoff disease. Behavioural brain research, 193(2), 315–319. doi:S0166-4328(08)00311-2 [pii] 10.1016/j.bbr.2008.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann P, Cowing JA, Cristante E, Liyanage SE, Ribeiro J, Duran Y, . . . Ali RR. (2015). Cd59a deficiency in mice leads to preferential innate immune activation in the retinal pigment epithelium-choroid with age. Neurobiol Aging, 36(9), 2637–2648. doi: 10.1016/j.neurobiolaging.2015.05.019 [DOI] [PubMed] [Google Scholar]
- Kana V, Desland FA, Casanova-Acebes M, Ayata P, Badimon A, Nabel E, . . . Merad M. (2019). CSF-1 controls cerebellar microglia and is required for motor function and social interaction. J Exp Med, 216(10), 2265–2281. doi: 10.1084/jem.20182037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempthorne L, Yoon H, Madore C, Smith S, Wszolek ZK, Rademakers R, . . . Dickson DW. (2020). Loss of homeostatic microglial phenotype in CSF1R-related Leukoencephalopathy. Acta Neuropathol Commun, 8(1), 72. doi: 10.1186/s40478-020-00947-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Shin JH, Lee JH, Kim JH, Na DL, Suh YL, . . . Huh GY. (2015). Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia linked CSF1R mutation: Report of four Korean cases. J Neurol Sci, 349(1–2), 232–238. doi:S0022-510X(14)00786-2 [pii] 10.1016/j.jns.2014.12.021 [doi] [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Oyanagi K, Kondo Y, Ishizawa K, Ishihara K, Yoshida M, . . . Ikeda SI. (2021). Pathologic basis of the preferential thinning of thecorpus callosum in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP). eNeurologicalSci, 22, 100310. doi: 10.1016/j.ensci.2021.100310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga S, Tipton PW, Wierenga KJ, Dickson DW, & Wszolek ZK (2022). Neuropathological Findings of CSF1R-Related Leukoencephalopathy After Long-Term Immunosuppressive Therapy. Mov Disord, 37(2), 439–440. doi: 10.1002/mds.28919 [DOI] [PubMed] [Google Scholar]
- Koh JY, & Lee SJ (2020). Metallothionein-3 as a multifunctional player in the control of cellular processes and diseases. Mol Brain, 13(1), 116. doi: 10.1186/s13041-020-00654-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno T, Broderick DF, Mezaki N, Isami A, Kaneda D, Tashiro Y, . . . Ikeuchi T. (2017). Diagnostic Value of Brain Calcifications in Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia. AJNR Am J Neuroradiol, 38(1), 77–83. doi: 10.3174/ajnr.A4938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno T, Kasanuki K, Ikeuchi T, Dickson DW, & Wszolek ZK (2018). CSF1R-related leukoencephalopathy: A major player in primary microgliopathies. Neurology, 91(24), 1092–1104. doi: 10.1212/WNL.0000000000006642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno T, Tada M, Tada M, Nishizawa M, & Ikeuchi T (2014). [Hereditary Diffuse Leukoencephalopathy with Spheroids (HDLS): A Review of the Literature on its Clinical Characteristics and Mutations in the Colony-Stimulating Factor-1 Receptor Gene]. Brain Nerve, 66(5), 581–590. doi:1416101796 [pii] [PubMed] [Google Scholar]
- Li B, Wang C, Lu P, Ji Y, Wang X, Liu C, . . . Wang X. (2022). IDH1 Promotes Foam Cell Formation by Aggravating Macrophage Ferroptosis. Biology (Basel), 11(10). doi: 10.3390/biology11101392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Hu B, Guan X, Wang Z, Zhou Y, Sun H, . . . Zheng H. (2023). Minocycline protects against microgliopathy in a Csf1r haplo-insufficient mouse model of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP). J Neuroinflammation, 20(1), 134. doi: 10.1186/s12974-023-02774-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Tian LX, Tang X, Wang J, Tang WQ, Ma ZF, . . . Liang HP. (2020). Neutrophilic granule protein (NGP) attenuates lipopolysaccharide-induced inflammatory responses and enhances phagocytosis of bacteria by macrophages. Cytokine, 128, 155001. doi: 10.1016/j.cyto.2020.155001 [DOI] [PubMed] [Google Scholar]
- Marazziti D, Di Pietro C, Golini E, Mandillo S, Matteoni R, & Tocchini-Valentini GP (2009). Induction of macroautophagy by overexpression of the Parkinson’s disease-associated GPR37 receptor. Faseb J, 23(6), 1978–1987. doi: 10.1096/fj.08-121210 [DOI] [PubMed] [Google Scholar]
- McArthur S, Cristante E, Paterno M, Christian H, Roncaroli F, Gillies GE, & Solito E (2010). Annexin A1: a central player in the anti-inflammatory and neuroprotective role of microglia. J Immunol, 185(10), 6317–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael J, Marschallinger J, & Aigner L (2019). The leukotriene signaling pathway: a druggable target in Alzheimer’s disease. Drug Discov Today, 24(2), 505–516. doi: 10.1016/j.drudis.2018.09.008 [DOI] [PubMed] [Google Scholar]
- Norris GT, Smirnov I, Filiano AJ, Shadowen HM, Cody KR, Thompson JA, . . . Kipnis J. (2018). Neuronal integrity and complement control synaptic material clearance by microglia after CNS injury. J Exp Med, 215(7), 1789–1801. doi: 10.1084/jem.20172244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Hirata-Tsuchiya S, Kitamura C, Omoteyama K, Sato T, Arito M, . . . Kato T. (2016). A Small Nuclear Acidic Protein (MTI-II, Zn(2+) Binding Protein, Parathymosin) That Inhibits Transcriptional Activity of NF-kappaB and Its Potential Application to Antiinflammatory Drugs. Endocrinology, 157(12), 4973–4986. doi: 10.1210/en.2016-1746 [DOI] [PubMed] [Google Scholar]
- Okamoto K, & Isohashi F (2005). Macromolecular translocation inhibitor II (Zn(2+)-binding protein, parathymosin) interacts with the glucocorticoid receptor and enhances transcription in vivo. J Biol Chem, 280(44), 36986–36993. doi: 10.1074/jbc.M506056200 [DOI] [PubMed] [Google Scholar]
- Oyanagi K, Kinoshita M, Suzuki-Kouyama E, Inoue T, Nakahara A, Tokiwai M, . . . Ikeda SI. (2017). Adult onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) and Nasu-Hakola disease: lesion staging and dynamic changes of axons and microglial subsets. Brain Pathol, 27(6), 748–769. doi: 10.1111/bpa.12443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos S, Pontius A, Finger E, Karrenbauer V, Lynch DS, Brennan M, . . . Wszolek ZK. (2021). Adult-Onset Leukoencephalopathy With Axonal Spheroids and Pigmented Glia: Review of Clinical Manifestations as Foundations for Therapeutic Development. Front Neurol, 12, 788168. doi: 10.3389/fneur.2021.788168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JR, McCandless EE, Dorsey D, & Klein RS (2010). CXCR4 promotes differentiation of oligodendrocyte progenitors and remyelination. Proc Natl Acad Sci U S A, 107(24), 11062–11067. doi: 10.1073/pnas.1006301107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitarokoili K, Sgodzai M, Gruter T, Bachir H, Motte J, Ambrosius B, . . . Gold R. (2019). Intrathecal triamcinolone acetonide exerts anti-inflammatory effects on Lewis rat experimental autoimmune neuritis and direct anti-oxidative effects on Schwann cells. J Neuroinflammation, 16(1), 58. doi: 10.1186/s12974-019-1445-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, Soto-Ortolaza A, . . . Wszolek ZK. (2011). Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet, 44(2), 200–205. doi:ng.1027 [pii] 10.1038/ng.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ros-Bernal F, Hunot S, Herrero MT, Parnadeau S, Corvol JC, Lu L, . . . Vyas S. (2011). Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proc Natl Acad Sci U S A, 108(16), 6632–6637. doi: 10.1073/pnas.1017820108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley ER, & Chitu V (2014). CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol, 6(6), 1–21. doi:6/6/a021857 [pii] 10.1101/cshperspect.a021857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J, Fujita H, Matsuda S, Toku K, Sakanaka M, & Maeda N (1997). Glucocorticoid- and mineralocorticoid receptors in microglial cells: The two receptors mediate differential effects of corticosteroids. Glia, 20(1), 23–37. doi:Doi [DOI] [PubMed] [Google Scholar]
- Tipton PW, Stanley ER, Chitu V, & Wszolek ZK (2021). Is Pre-Symptomatic Immunosuppression Protective in CSF1R-Related Leukoencephalopathy? Mov Disord. doi: 10.1002/mds.28515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vana AC, Flint NC, Harwood NE, Le TQ, Fruttiger M, & Armstrong RC (2007). Platelet-derived growth factor promotes repair of chronically demyelinated white matter. J Neuropathol Exp Neurol, 66(11), 975–988. doi: 10.1097/NEN.0b013e3181587d46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DG, Lue LF, & Beach TG (2001). Gene expression profiling of amyloid beta peptide-stimulated human post-mortem brain microglia. Neurobiol Aging, 22(6), 957–966. doi: 10.1016/s0197-4580(01)00306-2 [DOI] [PubMed] [Google Scholar]
- Wang K, Wang C, Chen D, Huang Y, Li J, Wei P, . . . Gao Y. (2022). The role of microglial/macrophagic salt-inducible kinase 3 on normal and excessive phagocytosis after transient focal cerebral ischemia. Cell Mol Life Sci, 79(8), 439. doi: 10.1007/s00018-022-04465-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wang Y, & Zou S (2022). A Glance at the Molecules That Regulate Oligodendrocyte Myelination. Curr Issues Mol Biol, 44(5), 2194–2216. doi: 10.3390/cimb44050149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt KA, & Sandoval KE (2014). Steroids and the blood-brain barrier: therapeutic implications. Adv Pharmacol, 71, 361–390. doi: 10.1016/bs.apha.2014.06.018 [DOI] [PubMed] [Google Scholar]
- Young K, & Morrison H (2018). Quantifying Microglia Morphology from Photomicrographs of Immunohistochemistry Prepared Tissue Using ImageJ. J Vis Exp(136). doi: 10.3791/57648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Tang Y, & Cai D (2020). Brain is an endocrine organ through secretion and nuclear transfer of parathymosin. Life Sci Alliance, 3(12). doi: 10.26508/lsa.202000917 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available on request from the corresponding author.