

Abstract
The hormone receptor oestrogen receptor-α (ER) orchestrates physiological mammary gland development, breast carcinogenesis and the progression of breast tumours into lethal, treatment-refractory systemic disease. Selective antagonism of ER signalling has been one of the most successful therapeutic approaches in oncology, benefiting patients as both a cancer preventative measure and a cancer treatment strategy. However, resistance to anti-oestrogen therapy is a major clinical challenge. Over the past decade, we have gained an understanding of how breast cancers evolve under the pressure of anti-oestrogen therapy. This is best depicted by the case of oestrogen-independent mutations in the gene encoding ER (ESR1), which are virtually absent in primary breast cancer but highly prevalent (20–40%) in anti-oestrogen-treated metastatic disease. These and other findings highlight the ‘evolvability’ of ER+ breast cancer and the need to understand molecular processes by which this evolution occurs. Recent development and approval of next-generation ER antagonists to target ESR1-mutant breast cancer underscores the clinical importance of this evolvability and sets a new paradigm for the treatment of ER+ breast cancers.
Introduction
Expression of oestrogen receptor-α (ER) is a defining feature of hormone-sensing luminal cells in the mammary gland. Upon activation by oestrogen, ER executes a transcriptional programme that drives proliferation and expansion of the mammary epithelia during puberty1 (Fig. 1a). Although not all cells of the normal breast gland express ER, ~70–80% of tumours that emerge from the mammary gland express ER and harbour some dependence on ER signalling for proliferation2–4. ER targeting has thus become the cornerstone of treatment in both early-stage and metastatic ER+ breast cancers5,6. Although most ER+ tumours initially appear sensitive to anti-oestrogen agents, resistance can develop over time, characterized by clinical relapse, which typically involves the emergence of genetic and epigenetic alterations that allow for reactivation of ER signalling7.
Fig. 1 |. Oestrogen receptor-α signalling and modes of inhibition.
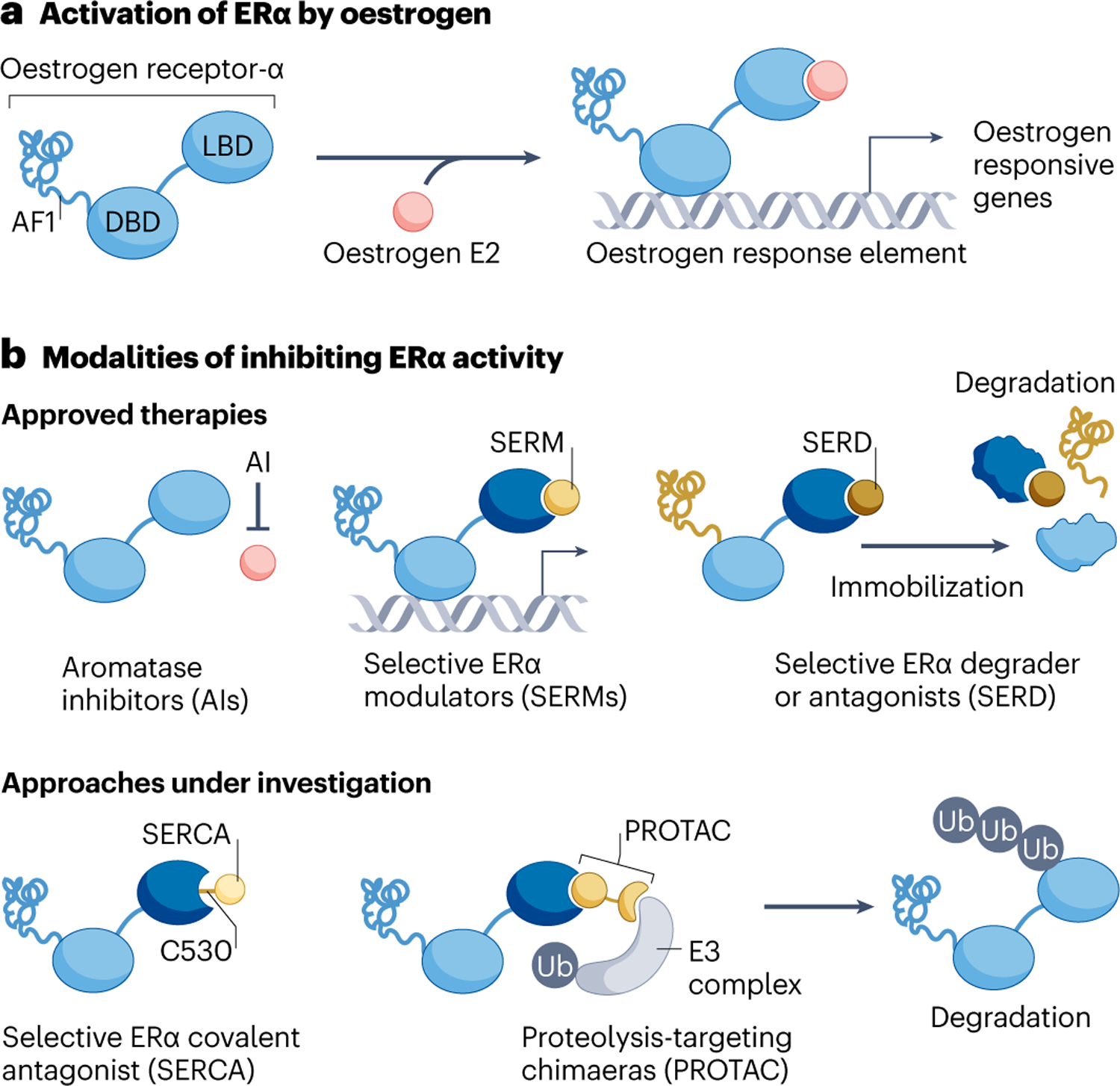
Oestrogen receptor-α (ER) signalling can be inhibited via multiple therapeutic strategies. a, ER is a multidomain ligand-inducible transcription factor that when activated drives a transcriptional programme that supports progression through the cell cycle. b, Strategies for ER targeting that are clinically approved are shown on the top. Aromatase inhibitors, which deplete oestrogen via inhibition of CYP19A1, are also known to select for oestrogen-independent ESR1 mutations. Selective ER modulators inhibit the ligand-binding domain (LBD) but not DNA-binding domain (DBD) or activation function-1 (AF1) domain of ER. They can also act as a partial ER agonist in some contexts. Selective ER degraders inhibit both LBD and AF1 domains by preventing recruitment of co-activators, immobilizing and destabilizing ER, leading to degradation. Approaches for inhibiting ER that are currently under investigation are shown on the bottom. Covalent ER antagonists react to cysteine C530 in the ER ligand-binding pocket and enforce an antagonist conformation in both wild-type and mutant ER. ER-specific PROTACs recruit E3 ligase to the ER, promoting poly-ubiquitination and proteasomal degradation. (Note: ER forms a homodimer but is depicted here as a monomer for visual clarity). Ub, ubiquitin.
Currently approved anti-oestrogen therapies use two main strategies: depletion of ER-activating ligands and/or direct modulation of ER itself (Fig. 1b). The aromatase enzyme (CYP19A1) is required for the synthesis of oestrogens in non-ovarian tissues, and aromatase inhibitors can thus be leveraged to suppress ER signalling in post-menopausal women whose ovaries no longer produce circulating hormones8. In pre-menopausal women, oestrogen is the product of both CYP19A1-independent synthesis in the ovary and CYP19A1-dependent synthesis peripherally, and thus aromatase inhibitors are given together with inhibitors of ovarian hormone production such as gonadotropin releasing hormone analogues. As an alternative approach to targeting synthesis of the activating ligand, selective oestrogen receptor modulators (SERMs) can inhibit ER transcriptional activity by directly binding to the C-terminal ligand-binding domain (LBD) of ER and preventing the recruitment of co-activators to this domain9. However, SERMs still allow for the engagement of ER with chromatin and therefore do not inhibit transcription driven by the N-terminal ER activation function-1 (AF1) domain10,11. As AF1 activity is sensitive to various cellular inputs, the impact of SERMs on ER transcriptional output depends on the tissue environment12. For instance, among the most widely utilized SERMs, tamoxifen generally attenuates oestrogen-stimulated ER signalling in the breast, but exerts agonistic or partial agonistic effects on the uterus, bone and heart tissues, which may be beneficial in some settings (for example, dampening of the osteoporosis process) but detrimental in others (for example, increasing the risk of endometrial cancer)9,13. Importantly, the agonistic potential of tamoxifen can also manifest in breast cancer cells, and this property presents a liability for therapeutic resistance. Specifically, continuous in vivo passaging of breast cancer cells in the presence of tamoxifen generates tumours in which tamoxifen acts to stimulate cell proliferation through activation of ER signalling14,15. Recognizing that further benefit might be brought to patients by avoiding the potential for ER agonism, pure oestrogen antagonists were sought through screening for oestrogen competitors that lack ER agonist potential in the rodent uterus16. Fulvestrant was the result of this campaign for pure anti-oestrogens, and its naming reflects its signalling properties as a full oestrogen antagonist17. Subsequent to its identification, fulvestrant was observed to promote ER degradation via the ubiquitination–proteasome pathway, leading to the compelling hypothesis that full ER antagonism is achieved through ER degradation and to the designation of fulvestrant as the prototypical selective ER downregulator (SERD)9,18. Subsequently, it has been proposed that ER degradation induced by SERDs may not be critical to its ER inhibitory effect19. Instead antagonism is achieved by degradation-independent suppression of the co-activator protein binding to the LBD, together with intranuclear ER immobilization, which suppresses activity from the AF1 domain of ER20–22.
The growth inhibitory effect of anti-oestrogen agents on ER+ breast cancers is due to cellular arrest in the G1 phase of the cell cycle. Given that perturbation of ER signalling results in broad transcriptional reprogramming, precisely how ER antagonists achieve G1 arrest is not fully understood. However, it is thought to involve, at least in part, suppression of cell cycle regulators such as Myc and cyclin D1. Cyclin D1 acts with cyclin-dependent kinase 4 (CDK4) and CDK6 to regulate activity of the retinoblastoma tumour suppressor protein (RB1), which in turn controls gene expression through the E2F family of transcription factors that promote G1-to-S cell cycle progression23–27.
To date, the most important predictive biomarker of response to endocrine therapy is the nuclear expression of ER, and it is unequivocal that ER-targeted therapies have brought tremendous benefit to the ER+ patient population28–31. However, individual patients with that same predictive biomarker appear to derive widely varying benefits from endocrine therapy. For instance, trials of hormonal therapy in ER+ metastatic breast cancer have identified subsets of patients with tumours that progress within 3 months, as well as those who can remain on therapy without relapse for many years32–35. The clinical vignette of this is striking: different patients may have the identical diagnosis of ER+ metastatic breast cancer, with similar prognostic and predictive biomarkers, yet the same targeted treatment may enable tumour control for decades in some patients, whereas others immediately progress. To some extent, the degree of ER expression associates with the degree of efficacy of endocrine therapy. For instance, patients with tumours that express a low level of ER (defined as 10–20 fmol ER per mg protein) derived benefit from a 5-year-course of adjuvant tamoxifen (relative risk (RR) of recurrence = 0.67, standard error (SE) = 0.08), when compared to those with tumours that express a negligible amount of ER (defined as <10 fmol ER per mg protein, which roughly corresponds to an ER scoring of 1%) (RR = 0.97, SE = 0.05). However, this benefit is far from proportional and is only slightly increased even in those with tumours expressing the highest amounts of ER (>200 fmol ER per mg protein) (RR = 0.52, SE = 0.07)28,36. From a modelling perspective, simply expressing ER is not known to drive tumour growth in the manner of other oncogenes37. As such, ER expression is more of a lineage-specific biomarker reflecting a transcriptional state poised to respond to ER activity, with various other modifiers altering the degree of dependence. This clinical heterogeneity implies that, rather than a singular phenomenon, hormonal therapy resistance in ER+ breast cancer represents a spectrum of disease states for which deeper biologic characterization is needed to understand and potentially overcome these clinical disparities.
Mechanisms of resistance to anti-oestrogen therapy
Endocrine therapy is used in various contexts in the care of patients with breast cancer. In the early-stage setting, endocrine therapy may be utilized to control tumour growth, reduce tumour size pre-operatively or prevent distant relapses post-operatively. In the advanced setting, endocrine therapy is given to prolong patient survival and palliate symptoms. Biological evidence for cooperative oncogenic signals such as from the PI3K–AKT pathway has led to the development of combinations of anti-oestrogens with other targeted therapies such as PI3K pathway inhibitors, or CDK4/CDK6 inhibitors, that have enhanced the degree and duration of therapeutic efficacy over anti-oestrogen therapy alone. Thus, the definition of what constitutes ‘endocrine resistance’ may vary on the basis of the particular clinical contexts. Moreover, as ER regulates various oncogenic effectors (for example, G1/S-specific cyclin-D1 (CCND1), Myc and so on), and as each effector can be activated by other signalling pathways, anti-oestrogen therapy can also be overcome by a wide variety of mechanisms38,39. It is therefore of little surprise that ever-increasing mechanisms of resistance to endocrine therapy have been identified (Fig. 2). Still, these may be thought of as either (1) reactivating ER signalling despite drug administration or (2) bypassing ER to drive another oncogenic programme (Table 1). Interestingly, resistance mechanisms relating to active ER signalling are more typically observed in tumours that manifest with a period of apparent sensitivity followed by resistance, that is, resistance that is acquired under the pressure of therapy, whereas ER-independent mechanisms often appear in tumours that are intrinsically resistant. This suggests that tumours that are initially dependent on oestrogen for proliferation commonly evolve to reactivate ER-directed signalling under therapeutic pressure of anti-oestrogens. The continued dependence of these tumours on ER represents a major, and highly tractable, opportunity to bring further benefit to patients through the development of next-generation ER-targeted therapeutics.
Fig. 2 |. Treatment of ER+ breast cancer with endocrine therapies creates a strong selective pressure that drives tumour evolution.
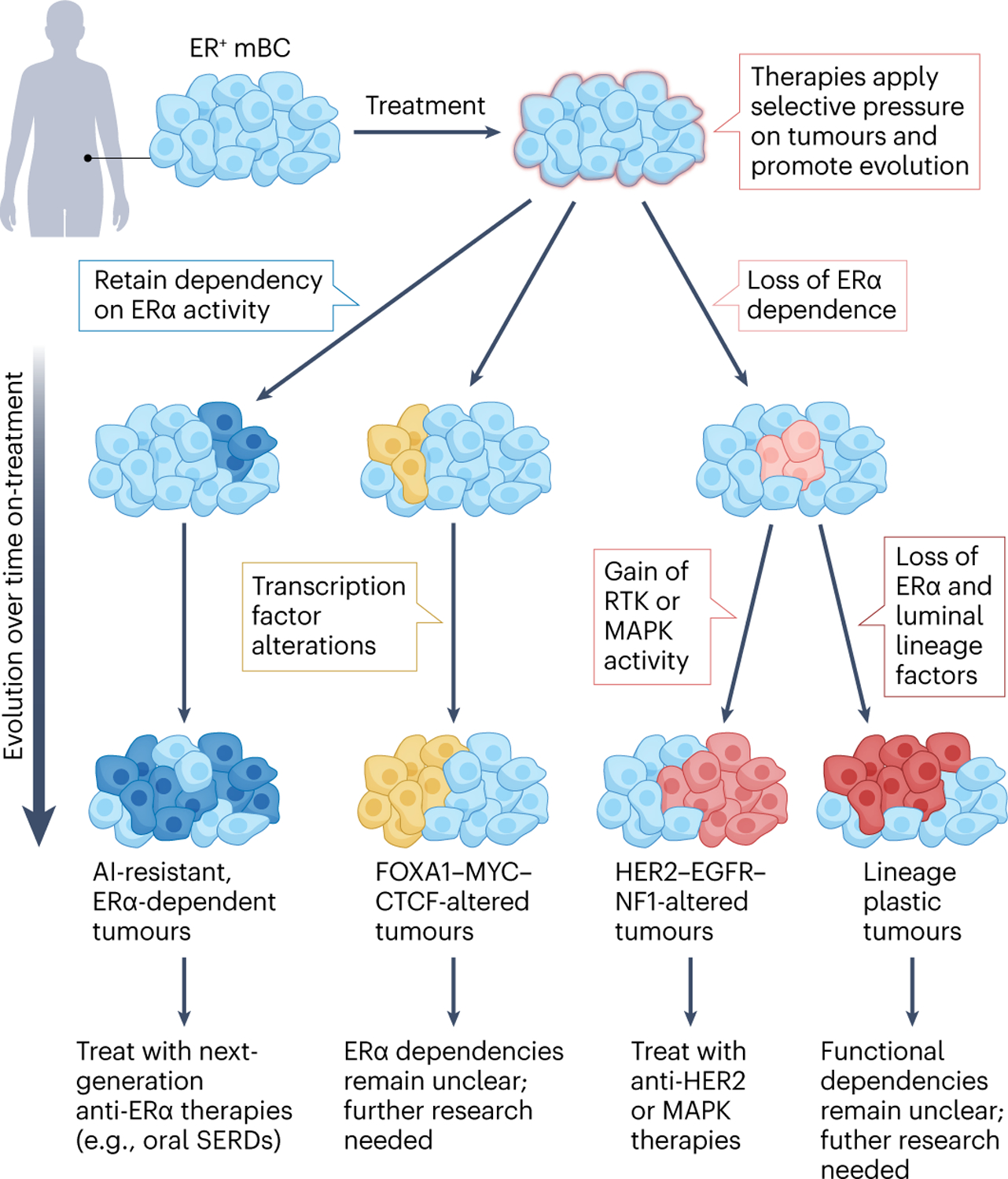
Tumours experiencing the pressure of endocrine therapies can evolve via distinct paths and can be categorized by their dependency on oestrogen receptor-α (ER) signalling (described from left to right below). Dark blue: a subset of cancers retain dependency on ER signalling but acquire features that render them resistant to first-line therapies such as aromatase inhibitors (AIs). The acquisition of ESR1 mutations exemplifies this evolution route, allowing tumours to progress in an ER-dependent but oestrogen-independent or other ligand-independent manner. These ER-dependent tumours are responsive to next-generation ER therapeutics, such as oral selective ER downregulators (SERDs). Yellow: tumours can also acquire alterations in other key transcription factors, such as FOXA1 or MYC. It remains unclear whether transcription factor-altered tumours retain ER dependency, thus further study is required to assess whether they might respond to next-generation ER-targeted therapies or require alternative treatments. Pink and red: other cancers lose dependency on ER altogether. Pink: of these, some become driven by orthogonal signalling pathways, such as HER2 or mitogen-activated protein kinases (MAPKs), and may benefit from the corresponding targeted therapies (for example, anti-HER2 therapies). Red: others lose ER expression altogether and show some evidence of undergoing lineage alterations. These tumours are reminiscent of lineage-plastic prostate cancers, which lose androgen receptor expression, although much less is known regarding lineage plasticity in breast cancer. Further investigation is required to devise treatment strategies for this subset. mBC, metastatic breast cancer; RTK, receptor tyrosine kinase.
Table 1 |.
Common molecular mechanisms of endocrine resistance in breast cancer
| Route of resistance | Mechanism | Examples | Mutation prevalence |
|---|---|---|---|
| Retain ER signalling in the presence of ER-pathway-targeted endocrine therapies | Acquisition of ligand-independent ESR1 mutations | LBD or activating ESR1 mutations | 0–5% pre-ET, 30–40% post-ET |
| ESR1 in-frame fusions with partner genes (e.g., YAP1) | 0–2% | ||
| Alterations in the ratio of ER-associated co-regulatory proteins | Elevated expression of co-activators, such as NCOA1 and NCOA3 | Not applicable | |
| Loss of co-repressors, such as N-CoR1 and N-CoR2 | |||
| Activation and acquired dependency of alternative proliferative pathways | Acquired ERBB2 activity and dependency | ERBB2 amplification; occurs before endocrine therapies and confers intrinsic resistance | ~5% |
| ERBB2 mutations; acquired following endocrine therapy | 1–2% pre-ET, 5–10% post-ET | ||
| Acquired activity and dependency on other receptor tyrosine kinases (RTKs) and downstream MAPK signalling | Amplification of EGFR | 1–5% per alteration | |
| Co-amplification of FGFR1 and associated factors | |||
| Loss-of-function mutations in negative regulators of the MAPK pathway (e.g., NF1) | |||
| Activating mutations in positive regulators of the MAPK pathway (e.g., KRAS, BRAF and MAP2K1) | |||
| Broader alterations in the landscape of transcriptional regulators | Mutations in other key transcription factors | Amplifications and hotspot mutations in MYC and CTCF | 1–10% per alteration |
| FOXA1 mutations in the Wing2-region; proposed to enhance ER-mediated transcription | |||
| FOXA1 mutations (e.g., SY242CS), which activate alternative transcriptomes via non-canonical DNA binding | |||
| Decrease or loss of ER expression, possibly owing’ to lineage plasticity (as in prostate cancer) | Abnormal DNA methylation of the ER CpG island | Not applicable | |
| Chromatin inactivation by histone deacetylation | |||
| Repression of ESR1 transcription via Twist |
Major routes of endocrine resistance, along with examples of specific mechanisms. For mechanisms driven by genomic alterations, literature-reported prevalences are shown. (Disparities in publicly available data sets — for example, assay, sampling method and so on — make it difficult to align mutational frequencies across studies; such meta-analyses are beyond the scope of this Review.) EGFR, epidermal growth factor receptor; ER, oestrogen receptor; ERBB2, erb-b2 receptor tyrosine kinase 2; ESR1, oestrogen receptor 1; ET, endocrine therapy; FGFR1, fibroblast growth factor receptor 1; LBD, ligand-binding domain; MAPK, mitogen-activated protein kinase; NCOA1, nuclear receptor co-activator 1 (also known as SRC1); N-CoR, nuclear receptor co-repressor; NF1, neurofibromin 1; YAP1, Yes1-associated transcriptional regulator.
ER-dependent resistance mechanisms
The evolving capacity of ER+ breast cancers is best illustrated by those cancers with acquired, activating mutations in ESR1, the gene encoding ER. Although first described as a case report in 1997 (ref. 40), ESR1 mutations were largely neglected in subsequent years, as initial genomic studies were focused on primary tumours before exposure to therapy and failed to capture the importance of this genetic event. It was not until 2013 that we and others sequenced tumours that had been exposed to therapy in the metastatic setting and demonstrated that acquired mutations in the LBD of ESR1 are important drivers of therapeutic resistance41–45. Across several large-scale analyses, activating ESR1 mutations are rarely found in primary breast tumours (<1%) or treatment-naive metastatic disease (<5%)45–47, but are present in a substantial number (~20–40%) of previously treated metastatic tumours, particularly those treated with aromatase inhibitors45,46,48–50. These data strongly suggest that ESR1 mutations are predominantly acquired under the selective pressure created with oestrogen deprivation. Indeed, when sequential samples were taken from the same patients with ER+ metastatic breast cancer, ESR1 mutations appear to be selected by aromatase inhibitor treatment and persist through subsequent therapies51–53.
Most ESR1 mutations are located in the LBD of ER, with mutations occurring at residues E380, L536, Y537 and D538 accounting for the vast majority of all variants detected in previously treated ER+ breast cancer. Spanning amino acids 304–554, the LBD contains 12 α-helices (named h1–h12) linked together by loop regions. Without ligand, the LBD is bound by heat shock proteins such as HSP90 and therefore kept in an inactive conformation. In the presence of oestrogen, the LBD dimerizes, ER engages with chromatin and the α-helix h12 forms a hydrophobic groove that binds to co-activator proteins to promote the expression of ER target genes. Binding of the SERM tamoxifen to the LBD prevents h12 from adopting the active conformation, thereby inhibiting recruitment of co-activators to this domain54. SERDs likewise bind the LBD and induce a conformation that prevents the recruitment of co-activators; these molecular events occur independently of ER degradation20. Recent structural and biochemical assays, as well as molecular dynamics modelling, have demonstrated that ESR1 mutations stabilize ER in an agonist conformation even in the absence of oestrogen55. Mutant ER recruits co-activators in a manner that mimics oestrogen-activated ER, enabling oestrogen-independent expression of ER target genes46,56,57. This cumulates in a growth advantage for cells harbouring ESR1 mutations, specifically in oestrogen-deprived conditions41–46. In addition, ESR1 mutations have been shown to enhance migration, invasion and metastatic capacity43,58,59.
Transcriptomic analyses of preclinical models engineered to express ESR1 mutations have revealed that in addition to oestrogen-independent activation of canonical ER target genes, cells with different ESR1 mutations (for example, those encoding ER.E380Q, ER.D538G and ER.Y537S, within the LBD region) demonstrate mutant-specific gene signatures41,60–63. The paucity of major structural differences in mutant ER compared with oestrogen-stimulated wild-type ER (as described earlier) makes it challenging to understand how mutated ER can alter cell biology beyond oestrogen independence. We speculate that these ascribed ‘neomorphic’ properties might result from the effects of constitutive mutant ER activity, when compared with periodic oestrogen stimulation of wild-type ER, but more research is necessary to fully understand the molecular mechanisms leading to mutant effects beyond oestrogen independence.
As mutant ER possesses oestrogen-independent activity, oestrogen deprivation strategies such as aromatase inhibitors have proven less effective64. Importantly, in addition to enabling oestrogen independence, the mutant-induced agonist conformation reduces the binding affinity of ER antagonists. The SERMs tamoxifen, raloxifene, bazedoxifene, as well as the SERD fulvestrant, all demonstrate decreased binding affinity to LBD-mutated ER proteins when compared with wild type (up to >60-fold)54,55,65,66. Indeed, the different ER variants (for example, ER.D538G and ER.Y537S) exhibit varying degrees of oestrogen-independent ER activity, and this phenotype roughly correlates with the reduction in ER-drug affinity for each of those variants, as both stem from the receptor pre-folding bias46,54. The predicted consequence of reduced binding affinity for direct ER antagonists on mutant versus wild-type ER (rather than completely ablated drug binding) is that inhibition of mutant ER may still be achieved by these drugs, but that higher drug concentrations would be required to do so, relative to what is required to inhibit wild-type ER. Indeed, preclinical in vitro data demonstrate that higher concentrations of tamoxifen and fulvestrant are needed to achieve the same degree of inhibition of cell proliferation in the context of ESR1 mutations relative to wild-type ESR1 (refs. 41–46). This phenomenon, whereby higher drug concentrations are required to inhibit mutant versus wild-type ER, is likely most clinically meaningful for drugs that exhibit in vivo exposure constraints, meaning that there is an upper limit on the concentration of drug that can be achieved in patients. Specifically, fulvestrant lacks oral bioavailability, which necessitates administration of high volumes of drug substance (10 ml) via intramuscular injection, most commonly into the gluteus maximus. Imaging studies have shown that this dosing regime is not sufficient to achieve saturation of ER binding in all patients67. Given that the exposure constraint of fulvestrant has been proposed as a liability even against wild-type ER, the presence of ESR1 mutations probably further exacerbates this limitation68.
The totality of data thus suggests that ESR1 mutations are a major source of resistance to aromatase inhibitors through their ability to activate ER signalling in an oestrogen-independent manner, and also presents a resistance liability for fulvestrant and tamoxifen, owing to their impact on drug binding affinity. The response of the research community to these challenges is described in a later section (entitled ‘Overcoming therapeutic resistance’).
In addition to ESR1 point mutations, structural variants have also been reported for ESR1, and these may also be relevant for therapeutic resistance. Specifically, the first six exons of ESR1 occur as in-frame fusions with various partner genes. For example, ESR1 can fuse with the Hippo pathway co-activator YAP1 or the protocadherin 11 X-linked gene (PCDH11X) to produce stable, functional protein products44,69. These fusion products lack the ER LBD but maintain the N-terminal transactivation domain and the central DNA-binding domain; they appear to function as oestrogen-independent transcription factors that drive an ER-directed proliferative and metastatic phenotype. By virtue of lacking the LBD, to which all-known ER antagonists bind, these fusion variants are insensitive to the SERM tamoxifen and the SERD fulvestrant. However, it has been suggested that inhibitors of CDK4/CDK6 retain activity against cells expressing these ESR1 fusions44,56,69. Many other fusion partners with ESR1 have been identified, including AKAP12, GYG1, SOX9, MTHFD1L, FLKHG1, TFG, NKAIN2 and CDK13. A major challenge for studies of ESR1 fusions is that most widely utilized exon-capture sequencing platforms do not reliably identify them and even when other sequencing methods (for example, whole genome sequencing) identify them, their functionality is not clear without additional information such as their level of expression. As such, their clinical prevalence as a resistance mechanism is difficult to quantify and remains a topic of active investigation70–72.
Outside alterations in the ESR1 gene, resistant tumours can augment ER signalling by changing the ratio of co-regulatory proteins. More than just bridging molecules that help ER form large protein complexes and coordinate spatiotemporal interactions, co-regulators often have intrinsic or associated enzymatic activities, such as acetylation, methylation or ubiquitination, that serve to modulate the transcription of ER target genes73. These can be exploited to confer a hormonal therapy resistant phenotype. For instance, among patients receiving tamoxifen, high expression of the co-activators nuclear receptor co-activator 1 (NCOA1, also referred to as steroid receptor co-activator 1 (SRC1)) and NCOA3 (also referred to as SRC3) have been associated with both therapy resistance and worse prognosis74–76. Conversely, loss of the ER co-repressors nuclear receptor co-repressor 1 (NCoR1) and NCoR2 (also known as SMRT) led to tamoxifen-mediated proliferation rather than inhibition77, and the low NCoR1 mRNA expression level is linked to poorer survival in patients treated with tamoxifen78.
Alterations in other signalling pathways
Mechanisms of endocrine therapy resistance in ESR1 wild-type tumours are likely highly fragmented across multiple pathways and mechanisms. This perhaps contributes to the major challenges in fully categorizing and understanding these modes of resistance. However, the activation of HER2 (encoded by ERBB2) signalling is one resistance mechanism in ESR1 wild-type tumours that has been deeply characterized and impacts current clinical treatment paradigms. ERBB2 amplification is found in ~13% of ER+ breast cancer2,3 and has a transcriptional signature that reflects, at least in part, activated HER2 signalling79. ERBB2-amplified tumours, found in both the primary and the metastatic settings, often exhibit therapeutic resistance to endocrine therapies that is apparent upon initial drug exposure. Thus, ERBB2 amplification is the classic example of intrinsic resistance to ER-directed therapeutics80,81. Tumours that are ER+ and exhibit amplification of ERBB2 are therefore recognized as a distinct breast cancer subtype (ER+ HER2+), and these patients will receive HER2-directed therapies. This is an excellent example of how understanding a particular mechanism of therapeutic resistance markedly impacts patient care.
In contrast to ERBB2 amplifications that are found in primary and metastatic tumours before endocrine therapy, ERBB2 point mutations are specifically enriched in ER+ tumours that have been exposed to endocrine therapy in the metastatic setting. This represents a mechanism of resistance that may be acquired under the selective pressure of anti-oestrogens49,82. Tumours with ERBB2 mutations show reduced sensitivity to oestrogen deprivation, the SERM tamoxifen, the SERD fulvestrant as well as inhibitors of CDK4/CDK6. Why ERBB2 amplification exists as a mechanism of intrinsic resistance before endocrine therapy exposure, and ERBB2 point mutations are more commonly an acquired mechanism, is not currently clear and will require further research. The distinction between ERBB2 amplification and point mutations is clinically relevant, as not all HER2-directed therapies that are active against ERBB2-amplified cells are relevant for ERBB2 point mutations. Understanding the sensitivity of distinct ERBB2 variants to the variety of available therapeutics is an active area of investigation82–84.
In addition to ERBB2 mutations and amplifications, alterations in other members of the receptor tyrosine kinase (RTK) family have also been associated with endocrine resistance. For instance, amplification of EGFR has been reported to confer an endocrine-resistant phenotype, which can be reversed by combining anti-oestrogen with an EGFR inhibitor49. Fibroblast growth factor receptor 1 (FGFR1) has also been implicated in preclinical and clinical studies as an important driver of endocrine therapy resistance, both intrinsically and as an acquired mechanism71,85. Interestingly, in the context of early breast cancer, co-amplification of FGFR1 with an amplicon that contains CCND1 (encoding the ER target gene cyclin D1) together with the FGFR ligands fibroblast growth factor 3 (FGF3), FGF4 and FGF19 was found to be enriched in patients whose tumours maintained a high degree of proliferation following treatment with an aromatase inhibitor71.
RTK activation promotes oncogenic signalling commonly through both the PI3K–AKT pathway and the RAS–RAF–mitogen-activated protein kinase (MAPK) pathway. Notably, activation of either pathway downstream of RTK activation can also induce breast cancer cell growth independent of ER signalling22,86. The MAPK pathway can be activated by either loss-of-function mutations in negative regulators of the pathway (for example, NF1) or activating mutations in other pathway components (for example, KRAS, BRAF and MAP2K1), and both types of alterations have been associated with resistance to endocrine therapy49,87,88. As with ERBB2 activating mutations, NF1 loss-of-function mutations were more than twice as common in tumours previously treated with endocrine therapy when compared with pre-therapy samples and are mutually exclusive with mutations in ESR1. This means that NF1 and ESR1 mutations do not tend to occur in the same tumours, suggesting that they are independent mechanisms of acquired resistance. KRAS and BRAF mutations are similarly exceedingly rare in primary ER+ breast cancers and become more frequently detected in therapy-exposed tumours, while remaining at an overall low prevalence49.
Overall, the weight of evidence from preclinical functional studies, together with clinical genomic studies, suggests that activation of RTKs and their downstream signalling effectors can support proliferation of ER+ breast cancer cells independently of ER activity and thus represents an important theme in therapeutic resistance. Conversely, inhibiting these oncogenic signals without inhibiting ER signalling limits efficacy, and so combinatorial treatments have been the mainstay of these studies. What remains a major challenge for categorizing these mechanisms, quantifying their overall abundance in different clinical scenarios, and developing strategies to overcome such resistance, is the various ways these overlapping pathways are activated.
As a distinct resistance theme, alterations in transcriptional regulators beyond ER have recently emerged as a potential mechanism for endocrine resistance. Amplifications and hotspot mutations in both the transcription factors MYC and CTCF were found more frequently in tumours that have progressed after endocrine therapy in the metastatic setting, especially in those without an ESR1 mutation or MAPK pathway alteration, suggesting that they may be independent drivers of endocrine resistance49. In addition, mutations in the transcription factor FOXA1, a protein that cooperates with ER as a pioneer factor, were associated with poorer outcomes for patients with ER+ breast cancer treated with aromatase inhibitors. FOXA1 alterations were found mutually exclusively with ESR1 mutations, meaning that they tended to occur in ESR1 wild-type tumours. In the case of mutations in the Wing2 region of FOXA1, this was thought to be related to an increase in FOXA1 chromatin occupancy at ER binding sites and thus enhanced ER-mediated transcription. Another FOXA1 mutation, SY242CS, however, appeared to activate an alternative transcriptome by binding to non-canonical DNA motifs89. Although the transcriptional consequences of different classes of FOXA1 mutations (Wing2 versus SY242CS) are distinct, functional experiments showed that both types of variants may promote oestrogen independence, consistent with resistance to aromatase inhibitors. However, they retained sensitivity to the SERM tamoxifen and the SERD fulvestrant, suggesting that overall dependency on ER signalling is likely maintained. Furthermore, in the developing mammary gland, FOXA1 directs lineage programming towards the luminal ER+ cell fate, and hormonal therapy resistance in this context may be related to an alteration in cell lineage90,91. Indeed, lineage plasticity is increasingly recognized as an important mechanism of therapy resistance in other types of cancer, including both prostate cancer under pressure of AR-directed therapies and lung cancer under the pressure of EGFR-targeted therapies92,93. The role of lineage plasticity in breast cancer hormonal therapy resistance is less well defined. However, approximately 10–20% of metastatic ER+ breast tumours appear to exhibit a loss or decrease in ER expression94–96. Several epigenetic mechanisms have been proposed to account for this change, including abnormal DNA methylation of the ER CpG island97, chromatin inactivation by histone deacetylation98 or inhibition of ER transcription by the transcription factor Twist99. Loss of ER expression as part of a mechanism of resistance likely reflects a fundamental reshaping of the transcriptome, cell identity and tumour phenotype. Sensitivity of ER+ luminal cells to endocrine therapy is underpinned by the requirement for ER signalling to drive cell cycle progression in luminal cells specifically. Thus, evolution to alternative cell identities likely comes with reduced reliance on ER signalling for proliferation and consequently resistance to endocrine therapy.
Overcoming therapeutic resistance
The frequent emergence of ESR1 mutations in endocrine therapy-exposed ER+ breast cancers suggests that a large fraction of endocrine therapy resistance is related to the particular features and limitations of today’s endocrine therapies, rather than an acquired abnegation of ER function. There has thus been significant energy and investment put towards the pursuit of fully optimized ER-targeted therapies to address refractory metastatic disease. Of the current commonly used anti-oestrogen agents, the SERD fulvestrant is capable of suppressing the activity of mutant ER in vitro, albeit with reduced potency relative to wild-type ER. This is unfortunately the case with all ER antagonists, as they engage with the LBD41,46. Efforts to develop next-generation endocrine therapies have therefore prioritized the development of molecules that have a fulvestrant-like SERD mechanism of action, but are also orally bioavailable and can be dosed to reach in-patient drug concentrations that will be capable of achieving full antagonism of mutated ER100.
There have been a plethora of ER binding ligands explored for their potential as next-generation endocrine agents. These ligands have various core structures that drive molecular interactions within the ligand-binding pocket, together with a number of different side chains which protrude from the pocket and differentially impact the positioning of helix 12 of the LBD. As described earlier, the positioning of helix 12 is critical for the recruitment of co-activator proteins to the ER LBD and therefore to the overall activity of ER as a transcription factor. These ligands can be loosely categorized as having either an acrylic acid side chain (for example, etacstil (GW5638, active metabolite GW7604), GDC-0810, AZD9496, rintodestrant (G1T48) and LSZ102) or a basic side chain (for example, elacestrant (RAD1901), GDC-0927, giredestrant (GDC-9545), amcenestrant (SAR439859) and camizestrant (AZD9833))101,102. GW5638, the first compound with an acrylic acid side chain, is a tamoxifen analogue initially synthesized as an alternative to hormonal therapy in post-menopausal women to prevent osteoporosis103. GW5638 and its active metabolite GW7604 were found, similar to tamoxifen, to displace helix 12 from the co-activator binding groove and thus inhibit ER-mediated transcription. In addition to blocking the recruitment of ER co-activators, distinct from tamoxifen, these compounds were proposed to also disrupt the ER surface charge, leading to ER ubiquitylation and proteasomal degradation104,105. Despite somewhat promising preclinical data, results from early-phase clinical trials of SERDs with an acrylic acid side chain, thought to act similar to GW5638, have been disappointing, with inconsistent response rate and a significant gastrointestinal toxicity profile9,68,100,102,106. Although the acrylic acid-containing SERDs lead to ER degradation and exhibit a fulvestrant-like full ER antagonist profile in MCF-7 cells, they exhibit partial agonist activity in other ER+ breast cancer cell lines and also in the uterus, leading to their redesignation as SERM/SERD hybrids20,107. One hypothesis is that the SERM-like, partial ER agonist properties of these molecules — as is the case with SERMs — limit their efficacy relative to what is achieved by more fully ER-inhibitory agents. Indeed, the key driver behind the discovery and development of fulvestrant was its lack of partial agonism, with the hypothesis that full ER antagonists would be more efficacious than those exhibiting context-dependent ER agonism16.
Compounds containing basic side chains display a more consistent full ER antagonist profile and have demonstrated robust on-target activity in both ESR1 wild-type and mutant models101,108–111. Several clinical trials have been conducted or are ongoing to evaluate the efficacy of these agents, both alone and in combination with CDK4/CDK6 or PI3K inhibitors, demonstrating mixed results68,101. For instance, despite strong preclinical and early clinical trial data for amcenestrant, the phase II trial AMEERA-3 (NCT04059484)112, which compared single-agent amcenestrant with physician’s choice anti-oestrogen therapy, failed on its primary end point of progression-free survival (PFS). Notably, 79% of the patients in this trial received previous CDK4/CDK6 inhibitors and 90% received fulvestrant in the physician’s choice arm, which might have complicated the interpretation of amcenestrant efficacy113,114. The phase III trial, AMEERA-5 (NCT04478266)115, aimed to evaluate the combination of amcenestrant and palbociclib (a CDK4/CDK6 inhibitor) in comparison to palbociclib and letrozole (an aromatase inhibitor), in advanced ER+ breast cancer; this trial also ended when it did not meet the prespecified efficacy boundary for continuation114,116. The clinical development of amcenestrant has subsequently been discontinued.
By contrast, elascetrant became the first oral SERD to demonstrate a statistically significant improvement in PFS in patients with ESR1-mutant tumours117 on the basis of the results of the EMERALD trial (NCT03778931)118. In January 2023, elacestrant received FDA approval for the treatment of post-menopausal women with advanced or metastatic ER+HER2−, ESR1-mutated breast cancer. The contrasting termination of amcenestrant development versus the success of elacestrant in demonstrating benefit and gaining regulatory approval illustrates both the challenge, and the importance, of optimizing multiple molecule parameters (including potency, pharmacokinetics and drug interaction properties) and clinical trial design in the drug development process. Amcenestrant and elacestrant exhibit similar potencies against ER+ breast cancer cells in vitro, and exposure differences do not obviously explain a superiority of elacestrant over amcenestrant. Different patient populations may thus have played some role in the different trial outcomes, but unique attributes of the molecules themselves cannot be ruled out as potential contributing factors. Camizestrant in the SERENA-2 trial (NCT04214288)119 and giredestrant in the acelERA (NCT04576455) trial120 both demonstrated promising clinical activity against ESR1-mutant tumours, relative to fulvestrant. These molecules also exhibit significantly higher potency than amcenestrant in preclinical studies121–123. Additionally, the drug–drug interaction between amcenestrant and palbociclib required lowering the administered dose of amcenestrant in the AMEERA-5 (NCT04478266)115 trial, whereas camizestrant and giredestrant have not required dose reductions owing to drug–drug interactions. The outcomes of the large series of clinical trials that are currently underway in both early and metastatic ER+ breast cancer will enable a more complete understanding of the molecule features most critical for driving improvements over the current standard of care and the patient populations that will gain the most benefit from these next-generation therapeutics. Interestingly, sub-group analysis in the EMERALD (NCT03778931)118, SERENA-2 (NCT04214288)119 and acelERA (NCT04576455)120 trials suggested that these agents are beneficial relative to the physician’s choice of endocrine therapy (primarily fulvestrant) mostly in the ESR1-mutant setting. The reasons behind the benefit being restricted to ESR1-mutant populations in these studies may be related to the value of ESR1 mutation as a biomarker for identifying tumours that retain a dependence on ER in the advanced setting. However, the superiority of the new endocrine agents may well manifest in ESR1-wild-type populations where endocrine resistance has not yet emerged. For instance, the coopERA trial (NCT04436744)124 showed a superiority of giredestrant versus aromatase inhibitors in treatment-naive early ER+ breast cancer124. Further transcriptional and genomic profiling of pre-treatment and on-treatment biopsies from several of these trials is currently underway to fully interrogate these hypotheses and potentially develop additional biomarkers for patient selection.
Although the development of orally bioavailable SERDs has taken centre stage in the development of next-generation ER-targeted therapies, other novel strategies to target ER have also been pursued; these include the development of a proteolysis targeting chimaera (PROTAC) and selective oestrogen receptor covalent antagonists (SERCAs). PROTACs (also known as ligand-directed degraders or chemical inducers of degradation) are heterobifunctional small molecules that combine a ligand for the target (in this case, ER), with a ligand for an E3 ligase, thereby recruiting the E3 ubiquitin ligase complex to the protein of interest and targeting it for proteasomal degradation125. It has been hypothesized that this unique catalytic-type mechanism of action will allow PROTACs to achieve a particularly high degree of selectivity and potency. This is due to their ability to induce energetically favourable protein–protein interactions between their targets and E3 ligase, to recycle each drug molecule through multiple rounds of activity and to refine binding specificity to target proteins126. The first ER-PROTAC to enter clinical trial, ARV-471, showed a favourable tolerability profile and evidence of clinical activity, including in patients previously treated with fulvestrant, CDK4/CDK6 inhibitors or oral SERDs127. The first-in-class SERCA, H3B-5942, is unique in its ability to bind to the ER LBD covalently and irreversibly via a cysteine at residue 530 of ER (versus the other ligands that display reversible binding to the same domain). This outcompetes oestrogen for binding to the LBD and prevents recruitment of co-activator proteins. SERCAs can inhibit both wild-type and mutant ER, albeit with reduced potency for mutant ER, similar to all other LBD binders. In preclinical studies, H3B-5942 demonstrates similar antiproliferative potency to fulvestrant in vitro, but greater activity versus fulvestrant in vivo128. In anticipation of the emergence of mutations at the C530 site that may render cells resistant to H3B-5942, a second SERCA, H3B-6545, was subsequently designed and shown to maintain in vitro potency even in the presence of a mutation at C530 (ref. 129). Notably, covalent antagonism of the LBD does not preclude partial ER agonism, and H3B-6545 exhibits tamoxifen-like ER stimulation in the bone and uterus. This differentiates the signalling properties of SERCAs from the more ER-inhibitory SERDs. In a phase II clinical trial (NCT03250676)130, H3B-6545 demonstrated evidence of clinical activity in heavily pretreated patients and those with ESR1 mutations131,132. Although both ARV-471 and H3B-6545 have demonstrated evidence of being clinically active, larger randomized trials with an approved endocrine therapy comparator will be required to determine whether these alternative mechanisms of ER antagonism are advantageous relative to currently approved agents.
Combining anti-oestrogen and targeted therapies
Another strategy to prevent the emergence of endocrine therapy resistance is to treat tumours with rational drug combinations that co-target the oncogenic signals that cooperate with ER to drive growth. Various approaches have been tested both preclinically and clinically but none more successfully than inhibition of CDK4 and CDK6 in combination with ER suppression. Indeed, this approach has now proven to confer PFS and overall survival benefits in both the early-stage and metastatic settings133,134.
As described earlier, ER and the CDK4/CDK6:D-cyclin complex converge on regulating the G1-S cell cycle checkpoint. The approved CDK4/CDK6 inhibitors palbociclib, abemaciclib and ribociclib suppress CDK4 and CDK6 activity and thereby attenuate the phosphorylation of RB1, which then acts as a break on the cell cycle by preventing the activity of E2F transcription factors135. The introduction of CDK4/CDK6 inhibitors into routine clinical practice was anticipated to generate resistance through loss-of-function RB1 alterations, resulting in a loss of the cell cycle ‘break’ and rendering both CDK4/CDK6 and ER inhibition irrelevant. Interestingly, however, RB1 mutations have remained relatively uncommon, with ESR1 mutations occurring more frequently. For instance, analysis of paired baseline and end-of-treatment circulating tumour DNA sequencing from patients in the PALOMA-3 trial (NCT01942135)136 reveals that, as expected, RB1 mutations emerged specifically in the treatment arm that combined the CDK4/CDK6 inhibitor palbociclib with the SERD fulvestrant; however, it was found only in a small number of patients overall at the end of treatment (4.7%). By contrast, a far greater proportion (31.3%) of patients in both the combination arm and the fulvestrant alone arm was found to have an ESR1 mutation at the end of treatment. Nevertheless, acquired resistance to combined inhibition of ER and CDK4/CDK6 can develop through genomic alterations that are more relevant for the combination than for the single-agent endocrine therapy. For instance, cells may become resistant to CDK4/CDK6 inhibitors by reactivating CDK4/CDK6 kinase activity itself, often via induction of the expression of CDK6, spawning the development of next-generation inhibitors of CDK4 and CDK6 as well as inhibitors of downstream CDK2 (refs. 137–140).
In addition to CDK4/CDK6 inhibitors, agents targeting oncogenic signals that function in parallel to ER have also been evaluated to address the challenges of endocrine resistance. In particular, the PI3K–AKT–mTOR pathway has been demonstrated to be mutationally activated in upwards of 50% of ER+ breast cancers; thus, agents targeting PI3K, AKT and mTORC1 have been combined with anti-oestrogens and shown meaningful improvements in clinical trials141–143. Agents in this class have suffered challenges with the toxicities of targeting this pathway with sufficient potency to cause complete pathway inhibition. To this end, newer approaches such as mutant-allele specific inhibitors are now under investigation. In addition, in tumours in which activation of RTK and MAPK signalling may mediate endocrine resistance, as discussed earlier, inhibitors of those pathways (for example, inhibition of EGFR, HER2, FGFR and MEK/ERK) have the potential to augment therapy response49,82–84,144. A major hurdle in bringing this concept to fruition is the fragmentation of the patient population across many distinct genetic variants, each of which requires a specific therapeutic targeting approach. These are extremely challenging trials to execute, particularly for therapeutic agents that also face significant tolerability challenges. Adding to this complexity, multiple mechanisms of resistance can also occur in the same patient. A salient example comes from a research autopsy analysis of multiple metastases from a patient with ER+ breast cancer, which revealed that different mechanisms of resistance were present in different metastases. Specifically, ESR1 and ERBB2 mutations, each sufficient to mediate endocrine resistance, were found in distinct metastatic sites. Perhaps even more telling, cell-free DNA analyses have demonstrated the presence of subclonal levels of several different ESR1 mutations in the same patient48,145. Taken together, this suggests that multiple mechanisms of resistance that may appear mutually exclusive in large sequencing cohorts may in fact be co-occurring in the same patient across different subclones.
As an additional opportunity for combination approaches, other nuclear receptors are known to cooperate with ER in both mammary phenotypes and tumour growth. For instance, progesterone receptor (PR) is highly expressed in the majority of ER+ breast cancers and is both a direct transcriptional target of ER and a modulator of ER activity146,147. Early studies of PR inhibition in ER+ PR+ tumours were limited by the availability of PR-specific compounds, but newer agents such as onapristone have shown some signal of benefit in heavily treated PR+ malignancies and may potentiate the effects of anti-oestrogen compounds148,149. Interestingly, agonism of PR has also been studied and found to exert anti-proliferative effects in combination with ER antagonists146. In fact, ample historical data spanning a few decades demonstrates that medroxy-progesterone acetate and its analogue megesterol acetate (Megace) are effective agents with comparable benefits to tamoxifen as a first-line therapy in ER+ breast cancer, as has been previously reviewed here150. The complex role of progesterone, as well as the potential for both agonists and antagonists of progesterone to function as therapeutic agents in the setting of endocrine resistant breast cancer, thus remains to be elucidated.
Similarly, the androgen receptor (AR) is expressed in the vast majority of ER+ breast cancers151–153. In the context of hormone-therapy-resistant breast cancer, AR inhibition with enzalutamide was proposed to inhibit oestrogen-mediated tumour growth in ER+ AR+ models and to act synergistically with inhibitors of oestrogen154,155. One randomized trial (NCT02007512)156 of the addition of enzalutamide to aromatase inhibition was evaluated and did not provide evidence of clinical improvement in PFS. However, a biomarker-selected group with high levels of AR mRNA showed a suggestion of benefit157. Offering an alternative hypothesis, recent work suggests that AR can act as a tumour suppressor in the context of ER+ breast cancer by opposing ER effects on selected promoters. As such, AR agonism with selective AR modulators has been proposed as a viable therapeutic opportunity that is actively being explored in the clinic158. These apparently opposing therapeutic hypotheses regarding both PR and AR highlight the complexity of hormone receptor interactions and suggest that further study is required to define patient populations within ER+ breast cancer that may benefit from therapeutic manipulation of hormone receptors beyond ER itself146.
Novel treatment modalities
There are perhaps two major challenges to the success of endocrine therapies and their combinations with signalling inhibitors. The first is that these agents frequently achieve cell cycle control without causing extensive cell death, thus allowing an opportunity for escape from therapy. Second, there is extensive heterogeneity both across and within patients with respect to the individual variants that cause therapeutic resistance. The most attractive approaches for transforming patient outcomes in ER+ breast cancer will tackle both challenges, leading to elimination of tumour cells and acting independently of specific, often relatively rare, signalling alterations. We propose that antibody–drug conjugates (ADCs) and immunotherapies have the potential to meet these criteria. ADCs have been extremely impactful in the treatment of ERBB2-amplified breast cancers, 50% of which are also ER+. In a major recent advance for patients without ERBB2 amplification, the DESTINY-Breast04 (NCT03734029)159 trial demonstrated a strong improvement in both PFS and overall survival for patients with HER2-low tumours (defined as those scoring 1 or 2 on immuno-histochemistry and a negative fluorescence in situ hybridization) and previous evidence of endocrine therapy resistance when treated with the ADC trastuzumab deruxtecan (T-DXd). This established that T-DXd, which delivers a topoisomerase I inhibitor to HER2-expressing tumour cells, was broadly active even in heavily pretreated ER+ metastatic breast cancer160. Moreover, the phase III trial TROPiCS-02 (NCT03901339)161 also recently established the benefit of the ADC sacituzumab govitecan, which delivers chemotherapy to TROP2-expressing cells, in metastatic ER+ HER2− disease162. This drug likewise received FDA approval for the treatment of advanced metastatic ER+ breast cancer in early 2023. These studies represent important advances for the utility of ADCs beyond ERBB2-amplified breast cancer, and their early success has restructured the treatment paradigm for advanced, refractory ER+ breast cancer.
Immunotherapy, such as immune checkpoint blockade, has become the cornerstone of treatment in many types of cancer. However, ER+ HER2− breast cancer has traditionally been considered ‘immune cold’ with lower numbers of tumour infiltrating lymphocytes163 and lower expression of PDL1 (ref. 164). Interestingly, ER signalling itself appears to have an immunosuppressive effect on the tumour microenvironment165. How and where we may combine endocrine therapy with immunotherapy to take advantage of their potential synergy is thus a topic of much active investigation166. One novel approach to leveraging the immune system in relatively ‘cold’ tumours might be individualized mRNA neoantigen vaccines. Although cancer vaccines have faced significant challenges in the metastatic setting, a small proof-of-concept study of personalized cancer vaccine autogene cevumeran (NCT04161755)167 was recently conducted in the adjuvant setting in pancreatic ductal adenocarcinoma168. Similar to ER+ breast cancer, pancreatic ductal adenocarcinoma is refractory to checkpoint blockade, with most tumours exhibiting an immune-excluded or immune-desert phenotype169–171. Preliminary data suggest that patients who successfully mounted an immune response gained benefit from the individualized vaccine approach. However, it remains to be seen whether this early, but highly intriguing, signal will be validated in larger studies and in other indications, including in ER+ breast cancer.
Towards a tumour-evolution informed approach
As we gain a deeper understanding of how ER+ breast tumours evolve under the pressure of hormonal therapy to cultivate mechanisms of resistance, we will also need to leverage new technologies to monitor tumour evolution in real time. For instance, liquid biopsies could provide information about response or failure to ongoing treatment and also data on tumour heterogeneity and evolution throughout the course of the disease172,173. More recently, plasma cell-free DNA was successfully utilized to assess high-resolution ER and FOXA1 DNA-binding profiles, from which ER activity, and therefore endocrine therapy sensitivity, may be inferred174. This technology would allow for a minimally invasive means to gain insights into tumour signalling and potentially ER dependence in real time, which would significantly enhance our ability to rapidly identify patients who may gain additional benefit from ER-directed therapies versus those who need treatments targeted against non-endocrine oncogenic signals. Although this approach, which measures the binding of a transcription factor to DNA, may have utility for a number of cancers, it is particularly well suited to ER+ breast cancer, given that ER is a transcription factor. These and other techniques will become more relevant in real-time clinical monitoring and adaptive treatment of patients with breast cancer in the years ahead.
Conclusions and perspective
ER signalling represents a foundational dependency in ER+ breast cancer cells, making ER-targeted therapies the foundation of treatment in this tumour type. The discovery of activating ESR1 mutations, occurring in up to 40% of the patients following exposure to aromatase inhibitors in the metastatic setting, as the major contributor to acquired resistance to commonly used endocrine therapies, suggested that ER dependency is maintained through disease progression and was thus a major spark triggering community-wide investment in further optimized endocrine therapies. These next-generation ER-targeted agents are being rigorously evaluated, with multiple pivotal trials currently playing out across both primary and metastatic breast cancers. The recent FDA approval of elacestrant for the treatment of metastatic ER+ HER2− breast cancers harbouring ESR1 mutations, has provided important validation for the hypothesis that there remains an opportunity to bring further benefit to patients through more effective targeting of ER. Although the next-generation endocrine therapies have been designed and developed to tackle the currently described oestrogen-independent ESR1 mutations, one important question is whether these endocrine agents will select for novel mechanisms of ER reactivation, or whether they will skew tumours entirely towards ER independence. The community eagerly awaits the outcomes of ongoing trials, as well as of the genomic and transcriptomic analyses of tumour biopsies and circulating tumour DNA from patients exposed to these novel therapies for prolonged periods of time. These data will represent the leading edge of ER+ tumour evolution and set the stage for future therapeutic exploits in this disease.
Competing interests
J.L. and C.M. are both Genentech/Roche employees and own shares of Roche. C.M. is a named co-inventor on patent 11081236 entitled ‘Diagnostic and therapeutic methods for the treatment of breast cancer’. S.C. reports research grants from NIH/NCI and Breast Cancer Research Foundation during the conduct of the study; personal fees from Novartis, AstraZeneca, Nuvalent, Boxer Capital, Effector and Neogenomics; equity/leadership in Totus Medicines and Odyssey Biosciences and grants from Daiichi Sankyo and AstraZeneca outside the submitted work; and a patent for CDK4/CDK6 degraders is also pending. M.W. has no competing interest.
Glossary
- ADCs
A class of drugs that combine monoclonal antibodies with cytotoxic agents to specifically target cells expressing the antigens recognized by those antibodies.
- Aromatase inhibitors
A class of drugs that block the synthesis of oestrogens by the aromatase enzyme in non-ovarian tissues.
- LBD
The C-terminal ligand-binding domain of ER, responsible for the binding of oestrogen ligand.
- Liquid biopsies
A technology that allows sampling of body fluids such as blood for molecular profiling, allowing for a less-invasive way of tumour monitoring, detection and characterization than traditional tissue-based biopsies.
- SERCA
A class of anti-oestrogen agents that bind covalently to the LBD of ER and prevent recruitment of co-activators.
References
- 1.Brisken C & O’Malley B Hormone action in the mammary gland. Cold Spring Harb. Perspect. Biol 2, a003178 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeSantis CE et al. Breast cancer statistics, 2019. CA Cancer J. Clin 69, 438–451 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Giaquinto AN et al. Breast cancer statistics, 2022. CA Cancer J. Clin 72, 524–541 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Eeckhoute J, Carroll JS, Geistlinger TR, Torres-Arzayus MI & Brown M A cell-type-specific transcriptional network required for estrogen regulation of cyclin D1 and cell cycle progression in breast cancer. Genes Dev. 20, 2513–2526 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waks AG & Winer EP Breast cancer treatment: a review. J. Am. Med. Assoc 321, 288–300 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Burstein HJ Systemic therapy for estrogen receptor-positive, HER2-negative breast cancer. N. Engl. J. Med 383, 2557–2570 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Hartkopf AD, Grischke EM & Brucker SY Endocrine-resistant breast cancer: mechanisms and treatment. Breast Care 15, 347–354 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnston SRD & Dowsett M Aromatase inhibitors for breast cancer: lessons from the laboratory. Nat. Rev. Cancer 3, 821–831 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Patel HK & Bihani T Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther 186, 1–24 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Sakamoto T et al. Estrogen receptor-mediated effects of tamoxifen on human endometrial cancer cells. Mol. Cell. Endocrinol 192, 93–104 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Liu H, Lee ES, Deb Los Reyes A, Zapf JW & Jordan VC Silencing and reactivation of the selective estrogen receptor modulator-estrogen receptor alpha complex. Cancer Res. 61, 3632–3639 (2001). [PubMed] [Google Scholar]
- 12.Shang Y & Brown M Molecular determinants for the tissue specificity of SERMs. Science 295, 2465–2468 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Jordan VC Tamoxifen: catalyst for the change to targeted therapy. Eur. J. Cancer 44, 30–38 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gottardis MM & Jordan VC Development of tamoxifen-stimulated growth of MCF-7 tumors in athymic mice after long-term antiestrogen administration. Cancer Res. 48, 5183–5187 (1988). [PubMed] [Google Scholar]
- 15.Wolf DM & Jordan VC Characterization of tamoxifen stimulated MCF-7 tumor variants grown in athymic mice. Breast Cancer Res. Treat 31, 117–127 (1994). [DOI] [PubMed] [Google Scholar]
- 16.Wakeling AE Therapeutic potential of pure antioestrogens in the treatment of breast cancer. J. Steroid Biochem. Mol. Biol 37, 771–775 (1990). [DOI] [PubMed] [Google Scholar]
- 17.Wakeling AE, Dukes M & Bowler J A potent specific pure antiestrogen with clinical potential. Cancer Res. 51, 3867–3873 (1991). [PubMed] [Google Scholar]
- 18.Long X & Nephew KP Fulvestrant (ICI 182,780)-dependent interacting proteins mediate immobilization and degradation of estrogen receptor-alpha. J. Biol. Chem 281, 9607–9615 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Wardell SE, Marks JR & McDonnell DP The turnover of estrogen receptor α by the selective estrogen receptor degrader (SERD) fulvestrant is a saturable process that is not required for antagonist efficacy. Biochem. Pharmacol 82, 122–130 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guan J et al. Therapeutic ligands antagonize estrogen receptor function by impairing its mobility. Cell 178, 949–963.e18 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Stenoien DL et al. FRAP reveals that mobility of oestrogen receptor-alpha is ligand- and proteasome-dependent. Nat. Cell Biol 3, 15–23 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Kato S et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 270, 1491–1494 (1995). [DOI] [PubMed] [Google Scholar]
- 23.Sutherland RL, Green MD, Hall RE, Reddel RR & Taylor IW Tamoxifen induces accumulation of MCF 7 human mammary carcinoma cells in the G0/G1 phase of the cell cycle. Eur. J. Cancer Clin. Oncol 19, 615–621 (1983). [DOI] [PubMed] [Google Scholar]
- 24.Butler WB & Kelsey WH Effects of tamoxifen and 4-hydroxytamoxifen on synchronized cultures of the human breast cancer cell line MCF-7. Breast Cancer Res. Treat 11, 37–43 (1988). [DOI] [PubMed] [Google Scholar]
- 25.Watts CKW, Sweeney KJE, Warlters A, Musgrove EA & Sutherland RL Antiestrogen regulation of cell cycle progression and cyclin D1 gene expression in MCF-7 human breast cancer cells. Breast Cancer Res. Treat 31, 95–105 (1994). [DOI] [PubMed] [Google Scholar]
- 26.Musgrove EA et al. Growth factor, steroid, and steroid antagonist regulation of cyclin gene expression associated with changes in T-47D human breast cancer cell cycle progression. Mol. Cell Biol 13, 3577–3587 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doisneau-Sixou SF et al. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr. Relat. Cancer 10, 179–186 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Davies C et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 378, 771–784 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elledge RM et al. Estrogen receptor (ER) and progesterone receptor (PgR), by ligand-binding assay compared with ER, PgR and pS2, by immuno-histochemistry in predicting response to tamoxifen in metastatic breast cancer: a Southwest Oncology Group Study. Int. J. Cancer 89, 111–117 (2000). [PubMed] [Google Scholar]
- 30.Yamashita H et al. Immunohistochemical evaluation of hormone receptor status for predicting response to endocrine therapy in metastatic breast cancer. Breast Cancer 13, 74–83 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Najjar S & Allison KH Updates on breast biomarkers. Virchows Arch. 480, 163–176 (2022). [DOI] [PubMed] [Google Scholar]
- 32.Robertson JF et al. Activity of fulvestrant 500 mg versus anastrozole 1 mg as first-line treatment for advanced breast cancer: results from the FIRST study. J. Clin. Oncol 27, 4530–4535 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Bonneterre J et al. Anastrozole versus tamoxifen as first-line therapy for advanced breast cancer in 668 postmenopausal women: results of the tamoxifen or arimidex randomized group efficacy and tolerability study. J. Clin. Oncol 18, 3748–3757 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Nabholtz JM, Bonneterre J, Buzdar A, Robertson JF & Thürlimann B Anastrozole (Arimidex) versus tamoxifen as first-line therapy for advanced breast cancer in postmenopausal women: survival analysis and updated safety results. Eur. J. Cancer 39, 1684–1689 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Mauri D, Pavlidis N, Polyzos NP & Ioannidis JP Survival with aromatase inhibitors and inactivators versus standard hormonal therapy in advanced breast cancer: meta-analysis. J. Natl Cancer Inst 98, 1285–1291 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Allison KH et al. Estrogen and progesterone receptor testing in breast cancer: ASCO/CAP guideline update. J. Clin. Oncol 38, 1346–1366 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Watts CK & King RJ Overexpression of estrogen receptor in HTB 96 human osteosarcoma cells results in estrogen-induced growth inhibition and receptor cross talk. J. Bone Min. Res 9, 1251–1258 (1994). [DOI] [PubMed] [Google Scholar]
- 38.Schiff R et al. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin. Cancer Res 10, 331s–336s (2004). [DOI] [PubMed] [Google Scholar]
- 39.Musheyev D & Alayev A Endocrine therapy resistance: what we know and future directions. Explor. Target. Antitumor Ther 3, 480–496 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang QX, Borg A, Wolf DM, Oesterreich S & Fuqua SA An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res. 57, 1244–1249 (1997). [PubMed] [Google Scholar]; Following the initial identification of a case in 1997, a series of publications (Toy et al., Robinson et al., Merenbakh-Lamin et al., Li et al. and Jeselsohn et al.) in the early 2010s identified a collection of activating ESR1 mutations to be recurrent, acquired mechanisms of resistance to anti-oestrogen therapy in metastatic ER+ breast cancer.
- 41.Toy W et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet 45, 1439–1445 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study characterizes the activity and drug sensitivity of various ESR1 mutations as well as the therapeutic potential of oral SERDs in antagonizing ESR1 mutants.
- 42.Robinson DR et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet 45, 1446–1451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merenbakh-Lamin K et al. D538G mutation in estrogen receptor-α: a novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 73, 6856–6864 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Li S et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 4, 1116–1130 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeselsohn R et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res 20, 1757–1767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toy W et al. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov. 7, 277–287 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koboldt DC et al. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chandarlapaty S et al. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol 2, 1310–1315 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Razavi P et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 34, 427–438.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study dissects the genomic data of more than 1,500 ER+ breast tumours and incorporates clinical treatment and outcomes to define acquired alterations that may contribute to resistance to hormonal therapy.
- 50.Fribbens C et al. Plasma ESR1 mutations and the treatment of estrogen receptor–positive advanced breast cancer. J. Clin. Oncol 34, 2961–2968 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Schiavon G et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med 7, 313ra182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study highlights the clinical acquisition of ESR1 mutants in patients with ER+ breast cancer using serial sampling of circulating tumour DNA.
- 52.Kuang Y et al. Unraveling the clinicopathological features driving the emergence of ESR1 mutations in metastatic breast cancer. npj Breast Cancer 4, 22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fribbens CV et al. Tracking evolution of aromatase inhibitor resistance with circulating tumour DNA (ctDNA) in metastatic breast cancer. J. Clin. Oncol 35, 1015–1015 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katzenellenbogen JA, Mayne CG, Katzenellenbogen BS, Greene GL & Chandarlapaty S Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat. Rev. Cancer 18, 377–388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fanning SW et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife 5, e12792 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gates LA et al. Proteomic profiling identifies key coactivators utilized by mutant ERα proteins as potential new therapeutic targets. Oncogene 37, 4581–4598 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jeselsohn R, Buchwalter G, De Angelis C, Brown M & Schiff R ESR1 mutations — a mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol 12, 573–583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gu G et al. Hormonal modulation of ESR1 mutant metastasis. Oncogene 40, 997–1011 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Z et al. Hotspot ESR1 mutations are multimodal and contextual modulators of breast cancer metastasis. Cancer Res. 82, 1321–1339 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harrod A et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 36, 2286–2296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bahreini A et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res. 19, 60 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martin LA et al. Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat. Commun 8, 1865 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liang J et al. Giredestrant reverses progesterone hypersensitivity driven by estrogen receptor mutations in breast cancer. Sci. Transl. Med 14, eabo5959 (2022). [DOI] [PubMed] [Google Scholar]
- 64.Najim O et al. The association between type of endocrine therapy and development of estrogen receptor-1 mutation(s) in patients with hormone-sensitive advanced breast cancer: a systematic review and meta-analysis of randomized and non-randomized trials. Biochim. Biophys. Acta Rev. Cancer 1872, 188315 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Carlson KE, Choi I, Gee A, Katzenellenbogen BS & Katzenellenbogen JA Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open pocket conformation is required for ligand interaction. Biochemistry 36, 14897–14905 (1997). [DOI] [PubMed] [Google Scholar]
- 66.Zhao Y et al. Structurally novel antiestrogens elicit differential responses from constitutively active mutant estrogen receptors in breast cancer cells and tumors. Cancer Res. 77, 5602–5613 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Kruchten M et al. Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer. Cancer Discov. 5, 72–81 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Ferraro E, Walsh EM, Tao JJ, Chandarlapaty S & Jhaveri K Accelerating drug development in breast cancer: new frontiers for ER inhibition. Cancer Treat. Rev 109, 102432 (2022). [DOI] [PubMed] [Google Scholar]
- 69.Lei JT et al. Functional annotation of ESR1 gene fusions in estrogen receptor-positive breast cancer. Cell Rep. 24, 1434–1444.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hartmaier RJ et al. Recurrent hyperactive ESR1 fusion proteins in endocrine therapy-resistant breast cancer. Ann. Oncol 29, 872–880 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Giltnane JM et al. Genomic profiling of ER(+) breast cancers after short-term estrogen suppression reveals alterations associated with endocrine resistance. Sci. Transl. Med 9, eaai7993 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lei JT, Gou X & Ellis MJ ESR1 fusions drive endocrine therapy resistance and metastasis in breast cancer. Mol. Cell Oncol 5, e1526005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lonard DM & O’Malley BW Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol. Cell 27, 691–700 (2007). [DOI] [PubMed] [Google Scholar]
- 74.Osborne CK et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J. Natl Cancer Inst 95, 353–361 (2003). [DOI] [PubMed] [Google Scholar]
- 75.Myers E et al. Inverse relationship between ER-beta and SRC-1 predicts outcome in endocrine-resistant breast cancer. Br. J. Cancer 91, 1687–1693 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McBryan J et al. Metastatic progression with resistance to aromatase inhibitors is driven by the steroid receptor coactivator SRC-1. Cancer Res. 72, 548–559 (2012). [DOI] [PubMed] [Google Scholar]
- 77.Keeton EK & Brown M Cell cycle progression stimulated by tamoxifen-bound estrogen receptor-α and promoter-specific effects in breast cancer cells deficient in N-CoR and SMRT. Mol. Endocrinol 19, 1543–1554 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Girault I et al. Expression analysis of estrogen receptor alpha coregulators in breast carcinoma: evidence that NCOR1 expression is predictive of the response to tamoxifen. Clin. Cancer Res 9, 1259–1266 (2003). [PubMed] [Google Scholar]
- 79.Prat A & Perou CM Deconstructing the molecular portraits of breast cancer. Mol. Oncol 5, 5–23 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Knowlden JM et al. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology 144, 1032–1044 (2003). [DOI] [PubMed] [Google Scholar]
- 81.Dowsett M et al. Relationship between quantitative estrogen and progesterone receptor expression and human epidermal growth factor receptor 2 (HER-2) status with recurrence in the arimidex, tamoxifen, alone or in combination trial. J. Clin. Oncol 26, 1059–1065 (2008). [DOI] [PubMed] [Google Scholar]
- 82.Nayar U et al. Acquired HER2 mutations in ER(+) metastatic breast cancer confer resistance to estrogen receptor-directed therapies. Nat. Genet 51, 207–216 (2019). [DOI] [PubMed] [Google Scholar]; This study identifies HER2 activating mutations in metastatic biopsies of patients with ER+ breast cancer resistant to hormonal therapy.
- 83.Croessmann S et al. Combined blockade of activating ERBB2 mutations and ER results in synthetic lethality of ER+/HER2 mutant breast cancer. Clin. Cancer Res 25, 277–289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Smyth LM et al. Efficacy and determinants of response to HER kinase inhibition in HER2-mutant metastatic. Cancer Discov. 10, 198–213 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mao P et al. Acquired FGFR and FGF alterations confer resistance to estrogen receptor (ER) targeted therapy in ER(+) metastatic breast cancer. Clin. Cancer Res 26, 5974–5989 (2020). [DOI] [PubMed] [Google Scholar]
- 86.Lupien M et al. Growth factor stimulation induces a distinct ER(alpha) cistrome underlying breast cancer endocrine resistance. Genes Dev. 24, 2219–2227 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pearson A et al. Inactivating NF1 mutations are enriched in advanced breast cancer and contribute to endocrine therapy resistance. Clin. Cancer Res 26, 608–622 (2020). [DOI] [PubMed] [Google Scholar]
- 88.Sokol ES et al. Loss of function of NF1 is a mechanism of acquired resistance to endocrine therapy in lobular breast cancer. Ann. Oncol 30, 115–123 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Arruabarrena-Aristorena A et al. FOXA1 mutations reveal distinct chromatin profiles and influence therapeutic response in breast cancer. Cancer Cell 38, 534–550.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Adams EJ et al. FOXA1 mutations alter pioneering activity, differentiation and prostate cancer phenotypes. Nature 571, 408–412 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Seachrist DD, Anstine LJ & Keri RA FOXA1: a pioneer of nuclear receptor action in breast cancer. Cancers 13, 5205 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Quintanal-Villalonga Á et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat. Rev. Clin. Oncol 17, 360–371 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chan JM et al. Lineage plasticity in prostate cancer depends on JAK/STAT inflammatory signaling. Science 377, 1180–1191 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hoefnagel LDC et al. Prognostic value of estrogen receptor α and progesterone receptor conversion in distant breast cancer metastases. Cancer 118, 4929–4935 (2012). [DOI] [PubMed] [Google Scholar]
- 95.Gutierrez MC et al. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J. Clin. Oncol 23, 2469–2476 (2005). [DOI] [PubMed] [Google Scholar]
- 96.Drury SC et al. Changes in breast cancer biomarkers in the IGF1R/PI3K pathway in recurrent breast cancer after tamoxifen treatment. Endocr. Relat. Cancer 18, 565–577 (2011). [DOI] [PubMed] [Google Scholar]
- 97.Ottaviano YL et al. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer Res. 54, 2552–2555 (1994). [PubMed] [Google Scholar]
- 98.Yang X et al. Transcriptional activation of estrogen receptor alpha in human breast cancer cells by histone deacetylase inhibition. Cancer Res. 60, 6890–6894 (2000). [PubMed] [Google Scholar]
- 99.Vesuna F et al. Twist contributes to hormone resistance in breast cancer by downregulating estrogen receptor-α. Oncogene 31, 3223–3234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang L & Sharma A The quest for orally available selective estrogen receptor degraders (SERDs). ChemMedChem 15, 2072–2097 (2020). [DOI] [PubMed] [Google Scholar]
- 101.Hernando C et al. Oral selective estrogen receptor degraders (SERDs) as a novel breast cancer therapy: present and future from a clinical perspective. Int. J. Mol. Sci 22, 7812 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ozyurt R & Ozpolat B Molecular mechanisms of anti-estrogen therapy resistance and novel targeted therapies. Cancers 14, 5206 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Willson TM et al. 3-[4-(1,2-Diphenylbut-1-enyl)phenyl]acrylic acid: a non-steroidal estrogen with functional selectivity for bone over uterus in rats. J. Med. Chem 37, 1550–1552 (1994). [DOI] [PubMed] [Google Scholar]
- 104.Wu YL et al. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol. Cell 18, 413–424 (2005). [DOI] [PubMed] [Google Scholar]
- 105.Bentrem D et al. Molecular mechanism of action at estrogen receptor alpha of a new clinically relevant antiestrogen (GW7604) related to tamoxifen. Endocrinology 142, 838–846 (2001). [DOI] [PubMed] [Google Scholar]
- 106.Lloyd MR, Wander SA, Hamilton E, Razavi P & Bardia A Next-generation selective estrogen receptor degraders and other novel endocrine therapies for management of metastatic hormone receptor-positive breast cancer: current and emerging role. Ther. Adv. Med. Oncol 14, 17588359221113694 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wardell SE, Nelson ER, Chao CA, Alley HM & McDonnell DP Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr. Relat. Cancer 22, 713–724 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Garner F, Shomali M, Paquin D, Lyttle CR & Hattersley G RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anticancer Drugs 26, 948–956 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bihani T et al. Elacestrant (RAD1901), a selective estrogen receptor degrader (SERD), has antitumor activity in multiple ER(+) breast cancer patient-derived xenograft models. Clin. Cancer Res 23, 4793–4804 (2017). [DOI] [PubMed] [Google Scholar]
- 110.Shomali M et al. SAR439859, a novel selective estrogen receptor degrader (SERD), demonstrates effective and broad antitumor activity in wild-type and mutant ER-positive breast cancer models. Mol. Cancer Ther 20, 250–262 (2021). [DOI] [PubMed] [Google Scholar]
- 111.Liang J et al. GDC-9545 (giredestrant): a potent and orally bioavailable selective estrogen receptor antagonist and degrader with an exceptional preclinical profile for ER+ breast cancer. J. Med. Chem 64, 11841–11856 (2021). [DOI] [PubMed] [Google Scholar]
- 112.US National Library of Medicine. ClinicalTrials.gov, https://classic.clinicaltrials.gov/show/NCT04059484 (2019). [DOI] [PubMed]
- 113.Tolaney SM et al. 212MO AMEERA-3, a phase II study of amcenestrant (AMC) versus endocrine treatment of physician’s choice (TPC) in patients (pts) with endocrine-resistant ER+/HER-advanced breast cancer (aBC). Ann. Oncol 33, S634–S635 (2022). [Google Scholar]
- 114.Wang Y & Tang S-C The race to develop oral SERDs and other novel estrogen receptor inhibitors: recent clinical trial results and impact on treatment options. Cancer Metastasis Rev. 41, 975–990 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.US National Library of Medicine. ClinicalTrials.gov, https://classic.clinicaltrials.gov/show/NCT04478266 (2020). [DOI] [PubMed]
- 116.Bardia A et al. AMEERA-5: a randomized, double-blind phase 3 study of amcenestrant plus palbociclib versus letrozole plus palbociclib for previously untreated ER+/HER2-advanced breast cancer. Ther. Adv. Med. Oncol 14, 17588359221083956 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bardia A et al. EMERALD: phase III trial of elacestrant (RAD1901) vs endocrine therapy for previously treated ER+ advanced breast cancer. Future Oncol. 15, 3209–3218 (2019). [DOI] [PubMed] [Google Scholar]
- 118.US National Library of Medicine. ClinicalTrials.gov, https://classic.clinicaltrials.gov/show/NCT03778931 (2019). [DOI] [PubMed]
- 119.US National Library of Medicine. ClinicalTrials.gov, https://classic.clinicaltrials.gov/show/NCT04214288 (2020). [DOI] [PubMed]
- 120.US National Library of Medicine. ClinicalTrials.gov, https://classic.clinicaltrials.gov/show/NCT04576455 (2020). [DOI] [PubMed]
- 121.Chen YC et al. Latest generation estrogen receptor degraders for the treatment of hormone receptor-positive breast cancer. Exp. Opin. Investig. Drugs 31, 515–529 (2022). [DOI] [PubMed] [Google Scholar]
- 122.Martin Jimenez M et al. 211MO giredestrant (GDC-9545) vs physician choice of endocrine monotherapy (PCET) in patients (pts) with ER+, HER2− locally advanced/metastatic breast cancer (LA/mBC): primary analysis of the phase II, randomised, open-label acelERA BC study. Ann. Oncol 33, S633–S634 (2022). [Google Scholar]
- 123.Oliveira M et al. Abstract GS3–02: GS3–02 camizestrant, a next generation oral SERD vs fulvestrant in post-menopausal women with advanced ER-positive HER2-negative breast cancer: results of the randomized, multi-dose phase 2 SERENA-2 trial. Cancer Res. 83, GS3–02 (2023). [Google Scholar]
- 124.Fasching PA et al. Neoadjuvant giredestrant (GDC-9545) plus palbociclib (P) versus anastrozole (A) plus P in postmenopausal women with estrogen receptor–positive, HER2-negative, untreated early breast cancer (ER+/HER2–eBC): final analysis of the randomized, open-label, international phase 2 coopERA BC study. J. Clin. Oncol 40, 589–589 (2022). [Google Scholar]
- 125.Békés M, Langley DR & Crews CM PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov 21, 181–200 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Flanagan JJ & Neklesa TK Targeting nuclear receptors with PROTAC degraders. Mol. Cell. Endocrinol 493, 110452 (2019). [DOI] [PubMed] [Google Scholar]
- 127.Hamilton EP et al. ARV-471, an estrogen receptor (ER) PROTAC degrader, combined with palbociclib in advanced ER+/human epidermal growth factor receptor 2-negative (HER2−) breast cancer: phase 1b cohort (part C) of a phase 1/2 study. J. Clin. Oncol 40, TPS1120 (2022). [Google Scholar]
- 128.Puyang X et al. Discovery of selective estrogen receptor covalent antagonists for the treatment of ERα(WT) and ERα(MUT). Cancer Discov. 8, 1176–1193 (2018). [DOI] [PubMed] [Google Scholar]
- 129.Furman C et al. Covalent ERα antagonist H3B-6545 demonstrates encouraging preclinical activity in therapy-resistant breast cancer. Mol. Cancer Ther 21, 890–902 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.US National Library of Medicine. ClinicalTrials.gov, https://classic.clinicaltrials.gov/show/NCT03250676 (2017). [DOI] [PubMed]
- 131.Hamilton EP et al. Abstract P1-17-10: H3B-6545, a novel selective estrogen receptor covalent antagonist (SERCA), in estrogen receptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2−) advanced breast cancer — a phase II study. Cancer Res. 82, P1-17-10–P11-17-10 (2022). [Google Scholar]
- 132.Hamilton EP et al. Phase I/II study of H3B-6545, a novel selective estrogen receptor covalent antagonist (SERCA), in estrogen receptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2−) advanced breast cancer. J. Clin. Oncol 39, 1018–1018 (2021). [Google Scholar]
- 133.Pernas S, Tolaney SM, Winer EP & Goel S CDK4/6 inhibition in breast cancer: current practice and future directions. Ther. Adv. Med. Oncol 10, 1758835918786451 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gil-Gil M et al. The role of CDK4/6 inhibitors in early breast cancer. Breast 58, 160–169 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fassl A, Geng Y & Sicinski P CDK4 and CDK6 kinases: from basic science to cancer therapy. Science 375, eabc1495 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.US National Library of Medicine. ClinicalTrials.gov, https://classic.clinicaltrials.gov/show/NCT01942135 (2013). [DOI] [PubMed]
- 137.Li Z et al. Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the hippo pathway. Cancer Cell 34, 893–905.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yang C et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 36, 2255–2264 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cornell L, Wander SA, Visal T, Wagle N & Shapiro GI MicroRNA-mediated suppression of the TGF-β pathway confers transmissible and reversible CDK4/6 inhibitor resistance. Cell Rep. 26, 2667–2680.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li Q et al. INK4 tumor suppressor proteins mediate resistance to CDK4/6 kinase inhibitors. Cancer Discov. 12, 356–371 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.André F et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med 380, 1929–1940 (2019). [DOI] [PubMed] [Google Scholar]
- 142.Turner N et al. Abstract GS3–04: GS3–04 capivasertib and fulvestrant for patients with aromatase inhibitor-resistant hormone receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer: results from the phase III CAPItello-291 trial. Cancer Res. 83, GS3–04 (2023). [Google Scholar]
- 143.Baselga J et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med 366, 520–529 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Turner N et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 70, 2085–2094 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Spoerke JM et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat. Commun 7, 11579 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mohammed H et al. Progesterone receptor modulates ERα action in breast cancer. Nature 523, 313–317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article highlights the role of PR in modulating ER function and behaviour.
- 147.Singhal H et al. Genomic agonism and phenotypic antagonism between estrogen and progesterone receptors in breast cancer. Sci. Adv 2, e1501924 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cottu PH et al. Phase I study of onapristone, a type I antiprogestin, in female patients with previously treated recurrent or metastatic progesterone receptor-expressing cancers. PLoS ONE 13, e0204973 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nishino T, Ishibashi K, Hirtreiter C & Nishino Y Potentiation of the antitumor effect of tamoxifen by combination with the antiprogestin onapristone. J. Steroid Biochem. Mol. Biol 116, 187–190 (2009). [DOI] [PubMed] [Google Scholar]
- 150.Carroll JS, Hickey TE, Tarulli GA, Williams M & Tilley WD Deciphering the divergent roles of progestogens in breast cancer. Nat. Rev. Cancer 17, 54–64 (2017). [DOI] [PubMed] [Google Scholar]
- 151.Gonzalez LO et al. Androgen receptor expresion in breast cancer: relationship with clinicopathological characteristics of the tumors, prognosis, and expression of metalloproteases and their inhibitors. BMC Cancer 8, 149 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Collins LC et al. Androgen receptor expression in breast cancer in relation to molecular phenotype: results from the Nurses’ Health Study. Mod. Pathol 24, 924–931 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Niemeier LA, Dabbs DJ, Beriwal S, Striebel JM & Bhargava R Androgen receptor in breast cancer: expression in estrogen receptor-positive tumors and in estrogen receptor-negative tumors with apocrine differentiation. Mod. Pathol 23, 205–212 (2010). [DOI] [PubMed] [Google Scholar]
- 154.D’Amato NC et al. Cooperative dynamics of AR and ER activity in breast cancer. Mol. Cancer Res 14, 1054–1067 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cochrane DR et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 16, R7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.US National Library of Medicine. ClinicalTrials.gov, https://classic.clinicaltrials.gov/show/NCT02007512 (2013). [DOI] [PubMed]
- 157.Krop I et al. A randomized placebo controlled phase II trial evaluating exemestane with or without enzalutamide in patients with hormone receptor-positive breast cancer. Clin. Cancer Res 26, 6149–6157 (2020). [DOI] [PubMed] [Google Scholar]
- 158.Hickey TE et al. The androgen receptor is a tumor suppressor in estrogen receptor-positive breast cancer. Nat. Med 27, 310–320 (2021). [DOI] [PubMed] [Google Scholar]; This study dissects the role of AR activation in modulating resistance to hormonal therapy in breast cancer.
- 159.US National Library of Medicine. ClinicalTrials.gov, https://classic.clinicaltrials.gov/show/NCT03734029 (2018). [DOI] [PubMed]
- 160.Modi S et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N. Engl. J. Med 387, 9–20 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.US National Library of Medicine. ClinicalTrials.gov, https://classic.clinicaltrials.gov/show/NCT03901339 (2019). [DOI] [PubMed]
- 162.Rugo HS et al. Primary results from TROPiCS-02: a randomized phase 3 study of sacituzumab govitecan (SG) versus treatment of physician’s choice (TPC) in patients (Pts) with hormone receptor-positive/HER2-negative (HR+/HER2−) advanced breast cancer. J. Clin. Oncol 40, LBA1001 (2022). [Google Scholar]
- 163.Stanton SE, Adams S & Disis ML Variation in the incidence and magnitude of tumor-infiltrating lymphocytes in breast cancer subtypes: a systematic review. JAMA Oncol. 2, 1354–1360 (2016). [DOI] [PubMed] [Google Scholar]
- 164.Zhang M et al. Expression of PD-L1 and prognosis in breast cancer: a meta-analysis. Oncotarget 8, 31347–31354 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Segovia-Mendoza M & Morales-Montor J Immune tumor microenvironment in breast cancer and the participation of estrogen and its receptors in cancer physiopathology. Front. Immunol 10, 348 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Vathiotis IA et al. Immune checkpoint blockade in hormone receptor-positive breast cancer: resistance mechanisms and future perspectives. Clin. Breast Cancer 22, 642–649 (2022). [DOI] [PubMed] [Google Scholar]
- 167.US National Library of Medicine. ClinicalTrials.gov, https://classic.clinicaltrials.gov/show/NCT04161755 (2019). [DOI] [PubMed]
- 168.Rojas LA et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 618, 144–150 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Muller M et al. The immune landscape of human pancreatic ductal carcinoma: key players, clinical implications, and challenges. Cancers 14, 995 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Li X et al. Immune checkpoint blockade in pancreatic cancer: trudging through the immune desert. Semin. Cancer Biol 86, 14–27 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ullman NA, Burchard PR, Dunne RF & Linehan DC Immunologic strategies in pancreatic cancer: making cold tumors hot. J. Clin. Oncol 40, 2789–2805 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Russo M & Bardelli A Lesion-directed therapies and monitoring tumor evolution using liquid biopsies. Cold Spring Harb. Perspect. Med 7, a029587 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sant M, Bernat-Peguera A, Felip E & Margelí M Role of ctDNA in breast cancer. Cancers 14, 310 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rao S et al. Transcription factor-nucleosome dynamics from plasma cfDNA identifies ER-driven states in breast cancer. Sci. Adv 8, eabm4358 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]