

Abstract
Systemic Lupus Erythematosus (SLE) is a chronic, multisystem, inflammatory autoimmune disease that disproportionately affects women. Trends in SLE prevalence and clinical course differ by ancestry, with those of African American ancestry presenting with more active, severe and rapidly progressive disease than European Americans. Previous research established altered epigenetic signatures in SLE patients compared to controls. However, the contribution of aberrant DNA methylation (DNAm) to the risk of SLE by ancestry and differences among patients with SLE-associated Lupus Nephritis (LN) has not been well described. We evaluated the DNA methylomes of 87 individuals including 41 SLE patients, with and without LN, and 46 controls enrolled in an ancestry diverse, well-characterized cohort study of established SLE (41 SLE patients [20 SLE-LN+, 21 SLE-LN-] and 46 sex-, race- and age-matched controls; 55% African American, 45% European American). Participants were genotyped using the Infinium Global Diversity Array (GDA), and genetic ancestry was estimated using principal components. Genome-wide DNA methylation was initially measured using the Illumina MethylationEPIC 850K Beadchip array followed by methylation-specific qPCR to validate the methylation status at putative loci. Differentially Methylated Positions (DMP) were identified using a case-control approach adjusted for ancestry. We identified a total of 51 DMPs in CpGs among SLE patients compared to controls. Genes proximal to these CpGs were highly enriched for involvement in type I interferon signaling. DMPs among European American SLE patients with LN were similar to African American SLE patients with and without LN. Our findings were validated using an orthogonal, methyl-specific PCR for three SLE-associated DMPs near or proximal to MX1, USP18, and IFITM1. Our study confirms previous reports that DMPs in CpGs associated with SLE are enriched in type I interferon genes. However, we show that European American SLE patients with LN have similar DNAm patterns to African American SLE patients irrespective of LN, suggesting that aberrant DNAm alters activity of type I interferon pathway leading to more severe disease independent of ancestry.
Keywords: Systemic Lupus Erythematosus, Lupus Nephritis, DNA methylation, genotyping, ancestry, disparities
1. Introduction
Systemic Lupus Erythematosus (SLE) is a chronic, multisystem autoimmune disease with complex pathogenesis dependent on environmental, genetic, and epigenetic factors. SLE is characterized by a loss of self-tolerance in immune cells, the production of autoantibodies that target self-antigens, and a formation of immune complexes that results in chronic inflammation associated with end organ damage [1–4]. Standardized incidence rates vary worldwide, but are consistently higher among ethnic minorities and women [5]. Among patients with SLE, persons of African ancestry typically present with severe disease features including lupus nephritis (LN) and a more rapid accumulation of organ damage compared to SLE patients of European ancestry [6–10]. African American (AA) SLE patients also have a higher risk for developing SLE at a younger age [7, 11], and notably higher prevalence of renal damage [12] compared to European American (EA) SLE patients. The relative risk of death from SLE in AAs is also significantly higher than in other populations [13]. Although social determinants of health [14] can play a role in the etiology and natural history of SLE [15, 16], the basis of the more aggressive clinical phenotype observed among patients of non-European ancestries remains unclear despite genetic studies having identified population-specific risk loci that also contribute to differences in the prevalence and severity of SLE between ancestral populations [17, 18].
Evidence from studies of monozygotic twins demonstrate discordance rates for SLE [19], suggesting that environmental factors play an important role in this common complex disease. Genome-wide DNA methylation (DNAm) analysis of discordant monozygotic twins revealed differentially methylated positions (DMP) in genes involved in immune regulation among SLE affected twins compared to unaffected siblings [20]. In murine models, inducing global DNA hypomethylation in T cells resulted in a lupus-like phenotype and MHC-autoreactivity [21]. In a previous report, we found hypomethylation of CpGs near interferon genes in CD4+ T cells, CD14+ monocytes, and CD19+ B cells [22] in a cohort of SLE patients. Additional exploration of these CpGs in sorted B cells from AA and EA SLE patients revealed that most of the interferon CpGs associated with SLE were hypomethylated specifically among AA SLE patients. However, neither of these investigations included SLE patients with LN nor controlled for proportions of ancestry or other potential confounders.
In this study, we conducted a genome-wide DNAm analysis in SLE patients of AA and EA ancestry, both with and without LN and age-, race-, and sex- matched controls. Using DNAm microarray data spanning the genome, we identified differences and similarities between the methylation profiles of EA and AA SLE patients. To confirm these findings, we used methylation-specific qPCR assays specific to CpGs near three SLE-associated genes (MX1, IFITM1, and USP18). To determine whether ancestry plays a role in the observed epigenetic phenotype in SLE, we genotyped cases and controls and performed local ancestry estimation. Our study highlights the importance of incorporating ancestry analyses in DNAm studies and increasing sample size across multiple populations, which is imperative to better understand the differences in methylation that may contribute to the severity of SLE in different populations.
2. Methods
2.1. Study population
Using a total of 96 female cases and controls enrolled at the University of Alabma at Birmingham in the Genetic Profile Predicting the Phenotype (PROFILE) SLE cohort [23] and Integrative Molecular And Genetic Epidemiology (IMAGE) study [24], respectively, we evaluated the contribution of DMPs across the genome relative to the risk of SLE, stratified by AA and EA ancestry. Approvals by the appropriate Institutional Review Boards were obtained prior to study initiation. All participants provided informed consent.
Eligible cases were unrelated AA and EA SLE female patients aged ≥21 years who met the revised and updated ACR classification criteria for SLE (1997), or those cases who fulfilled the Systemic Lupus International Collaborating Clinics (SLICC) classification criteria [25, 26], and with disease duration ≤10 years at enrollment. SLE patients with reported drug-induced lupus, or other systemic autoimmune diseases were excluded. Controls were randomly selected from the IMAGE study, which includes population-based controls enrolled from US Census and Centers for Disease Control population databases established from list-assisted random digit dialing methods. Eligible controls were residents of the Southeast US, at least 21 years of age, without a self-reported history of SLE, other autoimmune disease or HIV-1 infection. Controls were individually matched to cases based on self-reported race (Black, White), female sex and age (±5 years).
2.2. Genotyping, QC, and Local Ancestry Analysis
Genotyping for ancestry assessment was conducted on 96 samples using the Illumina Infinium Global Diversity array with enhanced PGx (GDA; Illumina, San Diego, CA, USA) with 50ng genomic DNA (gDNA) input extracted from buffy coat using Qiagen Puregene DNA Extraction kits (Puregene, Qiagen 158026) according to the manufacturer’s instructions and quantitated using a Qubit™ dsDNA BR assay kit (Thermo Q32850). Genotypes were called using Illumina Array Analysis Platform GenCall software v1.1.0 and converted to variant call format files (VCF) using bcftools v1.9 [27]. Standard quality control measures were applied in PLINK v2.00 [28]. Samples with poor call rates, discordant sex, and abnormal heterzygosity were excluded and SNPs with low call rates or Hardy-Weinberg equilibrium p-value < 1 × 10−5 were excluded. Principal component analysis (PCA), as implemented in PLINK [28], was used in to evaluate population stratification with sample outliers removed (Supplemental Fig. S1). Imputation was conducted using the 1000 Genomes Project (Phase 3v5) [29] as the reference panel. Missing genotypes were imputed using Minimac4 [30]. SNPs with poor imputation scores and with minor allele frequency < 1 percent were excluded, yielding 1.07 million SNPs in 92 samples for analysis. The resulting quality-controlled PLINK files were converted to VCF format and phased with Beagle v5.4 [31]. Local Ancestry was then estimated for each samples using RFmix (v2.03) [32]. Global ancestry was estimated for each sample using ADMIXTURE (v1.3.0) [33].
2.3. MethylationEPIC Assays, QC, and Batch Normalization
Genome-wide DNAm was measured using Illumina Infinium MethylationEPIC BeadChip in more than 856,187 methylation sites that include CpG sites inside and outside of CpG islands, non-CpG and differentially methylated sites, RefSeq genes, ENCODE open chromatin and transcription factor binding sites, FANTOM5 enhancers and miRNA promoter regions. Genomic DNA (1 μg) was bisulfite-treated using the Zymo EZ DNA Methylation™ Kit (Zymo Research, Irvine, CA, USA) and 500ng of bisulfite-converted DNA (500ng) was amplified using the Illumina Infinium HD Methylation assay. Raw intensity files in the form of idat files were generated for each sample and processed using R statistical suite (version 4.2.1) [34]. The methylation data were imported and pre-processed using the R package, Minfi (version 1.44.0) [35]. A sample containing more than 78% of failed probes (detection p-value > 0.05) was removed prior to normalization. Background correction was conducted using Minfi preprocessNoob and normalized using the preprocessQuantile function from Minfi. Probes with detection p-values > 0.01 in more than 98% of samples, those in close proximity to single nucleotide polymorphisms (SNP) (27,069 CpGs), those previously associated with smoking status (816 CpGs) [36], and probes that mapped to multiple loci (36,442 CpGs) were removed. Non-autosomal probes were included. Methylation samples that did not have genotyping data due to quality control or were considered genetic outliers were removed before downstream analysis. The resulting quality control measures yielded 704,312 CpGs in 87 samples.
2.4. Statistical analysis
We examined epigenome-wide DMPs associated with the presence of SLE compared to controls, stratified by ancestry. For this disease association model, we incorporated a qualitative (presence/absence) and quantitative (level) multivariable approach. After normalization, we considered log ratios of the methylation percentage (M-value), a homoscedastic measure of methylation [37], as a continuous classifier. We controlled for cellular hetergeneity using an algorithm Houseman et al [38] created as implemented in refbase from R package, ChAMP [39] to estimate the proportion of CD8+ T, CD4+ T, natural killer (NK), B, monocytes, and granulocytes cells using reference methylation data. Principal components were calculated to account for cellular heterogeneity across samples using the R package, PCAmixdata (v3.1) [40]. The first three principal components accounted for 81.5% of the variance and were included as covariates in the regression models. We performed linear regressions at each CpG using limma (v3.54.0) [41] to test for associations between methylation and disease status while accounting for age, cellular proportion principal components 1–3, and ancestry principal components 1–10 as indicated in the following model.
The resulting p-values from those models were FDR-corrected using the Benjamini-Hochberg method. CpGs with an FDR ≤ 0.05 were considered significantly associated with disease status.
2.5. Interferon Methylation Score
Twenty-one CpGs passed multiple testing (FDR<0.05) and were differentially methylated by at least 10% between cases and controls, and were used as input to calculate a summary score of DNAm at these sites. All significantly associated CpGs isolated were found to be near interferon (IFN) pathway genes. The IFN methylation score was calculated for each sample by finding the median z-score of the beta-values for twenty differentially methylated interferon-associated CpGs.
2.6. Validation of MX1, IFITM1, and USP18 DNAm Status using Methylation-Specific qPCR Assays
Standard curve-based real-time absolute quantitative PCR was performed using PrimeTime qPCR Probe Assays (Integrated DNA Technologies, Coralville, IA, USA) in the 384-well QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). Each ten-microliter reaction had an annealing temp of 58°C. Ten nanograms of bisulfite-treated DNA from each sample were used in each reaction. Assays were designed to be specific to the methylated and unmethylated CpGs near MX1, USP18, and IFITM1, which were cg21549285, cg14293575, and cg23570810 respectively. Probe sequences for each assay can be found in (Supplemental Table S1). All target probes were normalized to a stable reference gene, MYOD1. qPCR methylation scores were determined by calculating z-scores for each probe per sample. For each sample, the median z-score was used for downstream analysis. The z-scores resulting from the methylated probes were adjusted by taking the additive inverse to ensure similar directionality as the unmethylated probes.
3. Results
3.1. Participant Characteristics
Demographic characteristics of SLE cases and controls are shown in Table 1. Although we included an equal number of controls by ancestry, notably more AA SLE patients with LN (16.7%) were included in this investigation than EA SLE patients with LN (6.3%) and the median age at diagnosis was significantly younger among SLE patients with LN (38 years, range 21 to 63) compared to SLE patients without LN (50.5 years, range 26 to 70) and among controls (55 years, range 24 to 79; P=0.001), irrespective of ancestry. No notable differences were observed by smoking history. On average, there was no significant difference in SLEDAI scores between EA and AA SLE cases with and without LN, with scores ranging from 2.17±2.04 to 4.38±4.75 (P=0.14) and 2.42±2.61 to 2.93±3.38 (P=0.67), respectively. However, the average SDI scores were not significantly different for AA SLE cases with and without LN (1.88±2.06 vs 0.857±1.23, P=0.1085), but were significantly different between EA and AA cases with LN (0.5±0.548 vs 1.88±2.06, P=0.02), consistent with the presence of LN.
Table 1.
Demographic and treatment characteristics of SLE patients included in the whole genome wide DNAm study
| AA | EA | |||
|---|---|---|---|---|
| Demographic and treatment characteristic | LN− (n=14) | LN+ (n=16) | LN− (n=12) | LN+ (n=6) |
| Age, median (IQR) years | 46.5 (35, 56.5) | 36.5 (31.5, 47.2) | 54 (46, 59) | 40 (38, 55) |
| Smoking History, n (%) | ||||
| Ever | 3 (21.4%) | 0 (0%) | 1 (8.3%) | 1 (16.7%) |
| Never | 11 (78.6%) | 16 (100%) | 11 (91.7%) | 5 (83%) |
| Disease Duration, median (IQR) years | 6.1 (3.2, 8.3) | 8.6 (3.9, 13.7) | 6.0 (3.1, 13.0) | 2.4 (1.7, 4.9) |
| Histologic Subtype for LN, n (%) | ||||
| II | 0 (0%) | 2 (12.5%) | 0 (0%) | 0 (0%) |
| III | 0 (0%) | 4 (25%) | 0 (0%) | 2 (33.3%) |
| IV | 0 (0%) | 1 (6.25%) | 0 (0%) | 2 (33.3%) |
| V | 0 (0%) | 9 (56.2%) | 0 (0%) | 2 (33.3%) |
| SLEDAI, median (IQR) | 2.0 (0.0, 6.3) | 2.5 (0.0, 6.3) | 2.0 (0.0, 4.0) | 2.0 (0.5, 3.5) |
| SDI, median (IQR) | 0.5 (0.0, 1.0) | 1.5 (0.00, 3.3) | 0.0 (0.0, 0.0) | 0.50 (0.0, 1.0) |
| Hydroxychloroquine, n (%) | 12 (85.7%) | 15 (93.8%) | 9 (75%) | 2 (33%) |
| Systemic Glucocorticoids, n (%) | 7 (50%) | 6 (37.5%) | 4 (33%) | 3 (50%) |
| Immunosuppressants, n (%) | 7 (50%) | 9 (56.2%) | 4 (33%) | 4 (67%) |
| Methotrexate | 4 (28.6%) | 3 (18.8%) | 4 (33%) | 0 (0%) |
| Mycophenolate | 3 (21.4%) | 5 (31.3%) | 0 (0%) | 3 (50%) |
| Belimumab | 1 (7.1%) | 1 (6.3%) | 3 (25%) | 2 (33%) |
AA: African American; EA: European American; LN: lupus nephritis; SLEDAI: systemic lupus erythematosus disease activity index; SLICC-DI: Systemic Lupus International Collaborating Clinics Damage Index; IQR: interquartile range; LN: Lupus Nephritis
3.2. Genome Wide Methylation Suggests SLE-specific CpGs in EA LN and AA SLE Patients
The methylation profiles of buffy coat samples isolated from both SLE cases and controls were analyzed using the Illumina Infinium MethylationEPIC BeadChip. We identified 51 CpGs (Table 2) that were significantly associated with SLE compared to controls (FDR<0.05) (Figure 1A). As we and others previously reported, many of these CpGs are near genes associated with IFN signaling. Hierarchical clustering of 21 CpGs with a change in methylation ≥ 10% revealed EA LN cases clustering with AA SLE cases with and without LN (Figure 1B). Multidimensional scaling further indicated that methylation status is similar at these loci between EA LN and AA SLE without LN (Figure 1C).
Table 2.
Top 20 differentially methylated CpGs between SLE cases and controls
| CpG | Gene | Chr | Position | CpG Position in Gene | AA Control β | AA LN− β | AA LN+ β | EA Control β | EA LN− β | EA LN+ β | p-value | FDR-adjusted p-value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cg13452062 | IFI44L | 1 | 79088559 | 5’UTR | 0.703 | 0.177 | 0.134 | 0.798 | 0.843 | 0.08 | 1.439E-09 | 0.001 |
| cg10549986 | RSAD2 | 2 | 7018153 | 1stExon | 0.443 | 0.132 | 0.153 | 0.418 | 0.333 | 0.099 | 4.194E-09 | 0.001 |
| cg21549285 | MX1 | 21 | 42799141 | 5’UTR | 0.714 | 0.232 | 0.195 | 0.744 | 0.788 | 0.156 | 7.607E-09 | 0.002 |
| cg00959259 | PARP9 | 3 | 122281975 | 5’UTR | 0.501 | 0.198 | 0.226 | 0.537 | 0.523 | 0.161 | 1.714E-08 | 0.003 |
| cg16400320 | NA | 8 | 144105210 | NA | 0.919 | 0.807 | 0.808 | 0.907 | 0.887 | 0.781 | 3.876E-08 | 0.005 |
| cg18693345 | C5orf38 | 5 | 2754148 | Body | 0.149 | 0.184 | 0.172 | 0.136 | 0.269 | 0.238 | 4.986E-08 | 0.006 |
| cg11702942 | LY6E | 8 | 144102584 | Body | 0.799 | 0.636 | 0.633 | 0.778 | 0.757 | 0.628 | 7.439E-08 | 0.007 |
| cg07815522 | PARP9 | 3 | 122282157 | 5’UTR | 0.623 | 0.277 | 0.284 | 0.649 | 0.704 | 0.234 | 9.795E-08 | 0.009 |
| cg22862003 | MX1 | 21 | 42797588 | TSS1500 | 0.535 | 0.265 | 0.281 | 0.581 | 0.635 | 0.242 | 1.351E-07 | 0.011 |
| cg24678928 | DDX60 | 4 | 169240829 | TSS1500 | 0.787 | 0.478 | 0.461 | 0.83 | 0.808 | 0.371 | 1.708E-07 | 0.012 |
| cg10959651 | RSAD2 | 2 | 7018020 | 1stExon | 0.424 | 0.099 | 0.113 | 0.341 | 0.26 | 0.058 | 1.843E-07 | 0.012 |
| cg08924203 | MX1 | 21 | 42798747 | 5’UTR | 0.148 | 0.073 | 0.061 | 0.125 | 0.123 | 0.051 | 2.461E-07 | 0.014 |
| cg22016995 | IRF7 | 11 | 614787 | Body | 0.969 | 0.912 | 0.877 | 0.973 | 0.971 | 0.893 | 3.064E-07 | 0.014 |
| cg01079652 | IFI44 | 1 | 79118191 | Body | 0.915 | 0.577 | 0.507 | 0.91 | 0.889 | 0.47 | 3.213E-07 | 0.014 |
| cg14943355 | PARP11 | 12 | 3980704 | 5’UTR | 0.484 | 0.235 | 0.235 | 0.532 | 0.568 | 0.209 | 3.254E-07 | 0.014 |
| cg17593958 | PRIC285 | 20 | 62199034 | 5’UTR | 0.127 | 0.215 | 0.244 | 0.086 | 0.093 | 0.192 | 3.363E-07 | 0.014 |
| cg20791089 | FBRS | 16 | 30669590 | TSS1500 | 0.084 | 0.112 | 0.102 | 0.079 | 0.14 | 0.118 | 3.377E-07 | 0.014 |
| cg25998594 | IRF9 | 14 | 24632095 | Body | 0.862 | 0.716 | 0.730 | 0.852 | 0.861 | 0.689 | 3.967E-07 | 0.016 |
| cg06899530 | ILDR2 | 1 | 166916866 | Body | 0.068 | 0.047 | 0.051 | 0.062 | 0.047 | 0.048 | 4.726E-07 | 0.018 |
| cg06561932 | LOC389641 | 8 | 23081981 | TSS1500 | 0.049 | 0.041 | 0.039 | 0.059 | 0.047 | 0.041 | 5.145E-07 | 0.018 |
Fig 1.
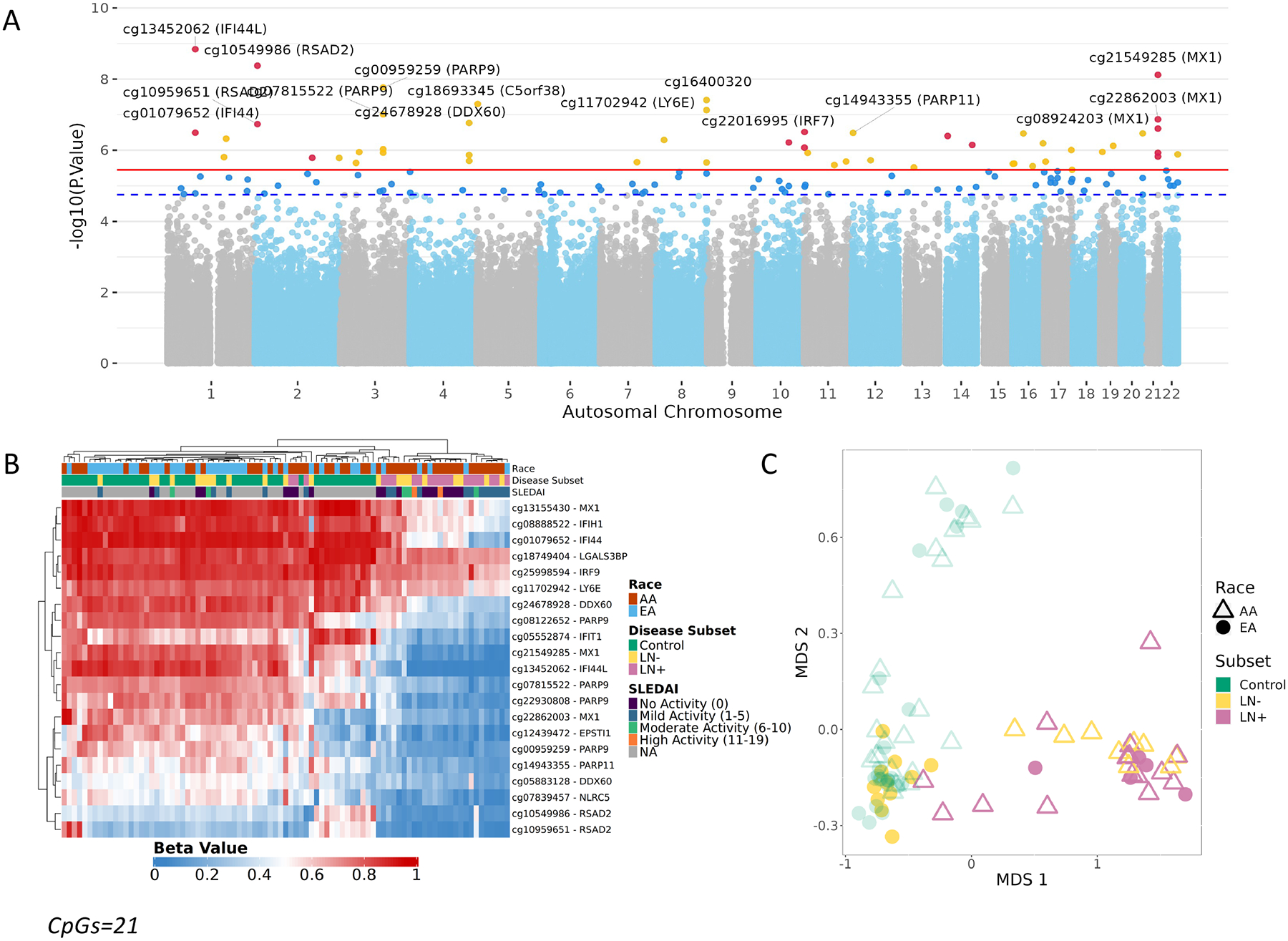
DNA Methylation was assessed using the MethylationEPIC microarray on 87 buffy coat samples (46 controls and 41 SLE patients [20 SLE-LN+, 21 SLE-LN-]) A. Manhattan plot from the DNA Methylation Analysis. CpGs above the suggestive line (dotted blue) passed FDR<0.1. CpGs above the significance line (solid red) passed FDR<0.05. CpGs near interferon genes are highlighted in red. B. Unsupervised hierarchical cluster heatmap of beta values of CpGs (CpGs=21, false discovery rate < 0.05 & difference of 10% between cases and controls) significantly associated with Systemic Lupus Erythematosus. Each column represents an individual sample which is annotated by race (AA – Red, EA – Blue) and disease subset (Control, green; lupus without nephritis (LN-), yellow; lupus nephritis (LN+), pink) whereas each row represents a CpG with a color gradient based on their methylation status (Red – Methylated, Blue – Unmethylated). C. MDS Plot separates control and SLE samples, using the top SLE-associated CpGs. EA LN+ clusters with AA cases.
3.3. Methylation Module Score using Top Associated IFN CpGs
Further exploring the enrichment of differentially methylated CpGs near IFN pathway genes, we found that EA SLE patients with LN have an increased proportion of hypomethylated CpGs nearby IFN genes compared to the EA SLE patients without LN. To focus on the differences in CpG methylation impacting IFN signaling, we established a methylation module score using the median Z-score beta values from the CpGs with a differential methylation proportion of at least 10% between cases and controls. Notably, all of these CpGs were situated near IFN pathway genes, with many of them positioned within 1500 bases of the respective gene’s transcription start site (Table 2). Comparison of this score based on disease status and ancestry shows that the IFN methylation score distinguishes AA SLE patients, with and without LN, and EA SLE patients with LN from AA controls, EA SLE cases without LN, and EA controls (Figure 2) suggesting that EA patients with LN are more similar to the average AA patient with hypomethylation observed at IFN-associated CpGs. We did not find any significant differences in the SLE methylation score after stratifying by SLE-related treatments (Supplemental Figures S2), disease activity (Supplemental Figures S3A), organ damage (Supplemental Figures S3B) or LN histologial subtype 1 thorugh VI (Supplemental Figures S3C); however, we did note that longer disease duration correlated with less severe DNAm among AA SLE cases (Supplemental Figure S3D).
Fig 2.
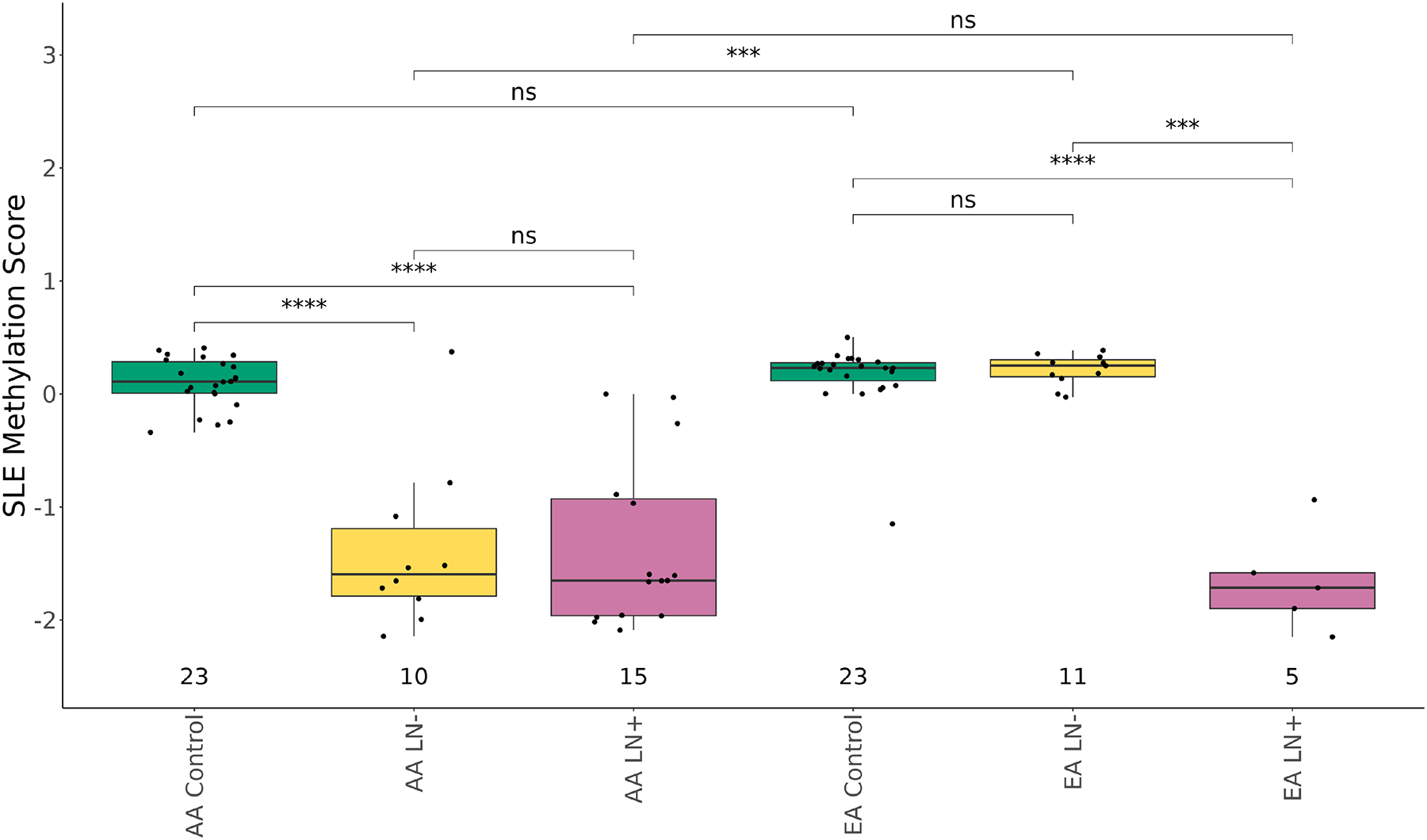
Box plot of methylation score (Median z-score across the 21 IFN CpGs) using the MethylationEPIC microarray data. Y-axis is the methylation score, and the x-axis is the disease status (Control, green; lupus without nephritis (LN-), yellow; lupus nephritis (LN+), pink)
3.4. Altered methylation of MX1, IFITM1, and USP18 loci is validated by methylation-specific PCR
Using methylation specific PCR, we validated the methylation status of three CpGs, located in MX1, IFITM1, and USP18, which were found to be associated with SLE status in our study and previously, in other studies of SLE. Methylation-specific qPCR probes were designed for cg21549285, cg14293575, and cg23570810, which are CpGs located in the 5’UTR of MX1, the 5’UTR of USP18, and the gene body of IFITM1, respectively (Figure 3A). We calculated a normalized methylation score for each sample from the resulting probe data. Using these values, we observed the same pattern we saw previously, that methylation of interferon-associated CpGs differentiated AA SLE patients with LN and AA SLE patients without LN in addition to EA SLE patients with LN from controls and EA SLE patients without LN (Figure 3B). The methylation module scores calculated from the methylation-specific qPCR probes were highly correlated with the methylation beta score derived from the microarray data (R=0.84, p < 2.2 × 10−16 Figure 3C). We observed a higher correlation if we considered only samples from AA patients in microarray methylation score vs methylation-specific qPCR methylation score (Figure 3D, AA – R=0.89, EA R=0.59). The greater correlation among AA participants may be due to the broader variation of methylation score values and larger sample size when compared to the small sample size of the EA SLE patients with LN. The majority of the EA participants cluster together with a higher methylation score.
Fig 3.
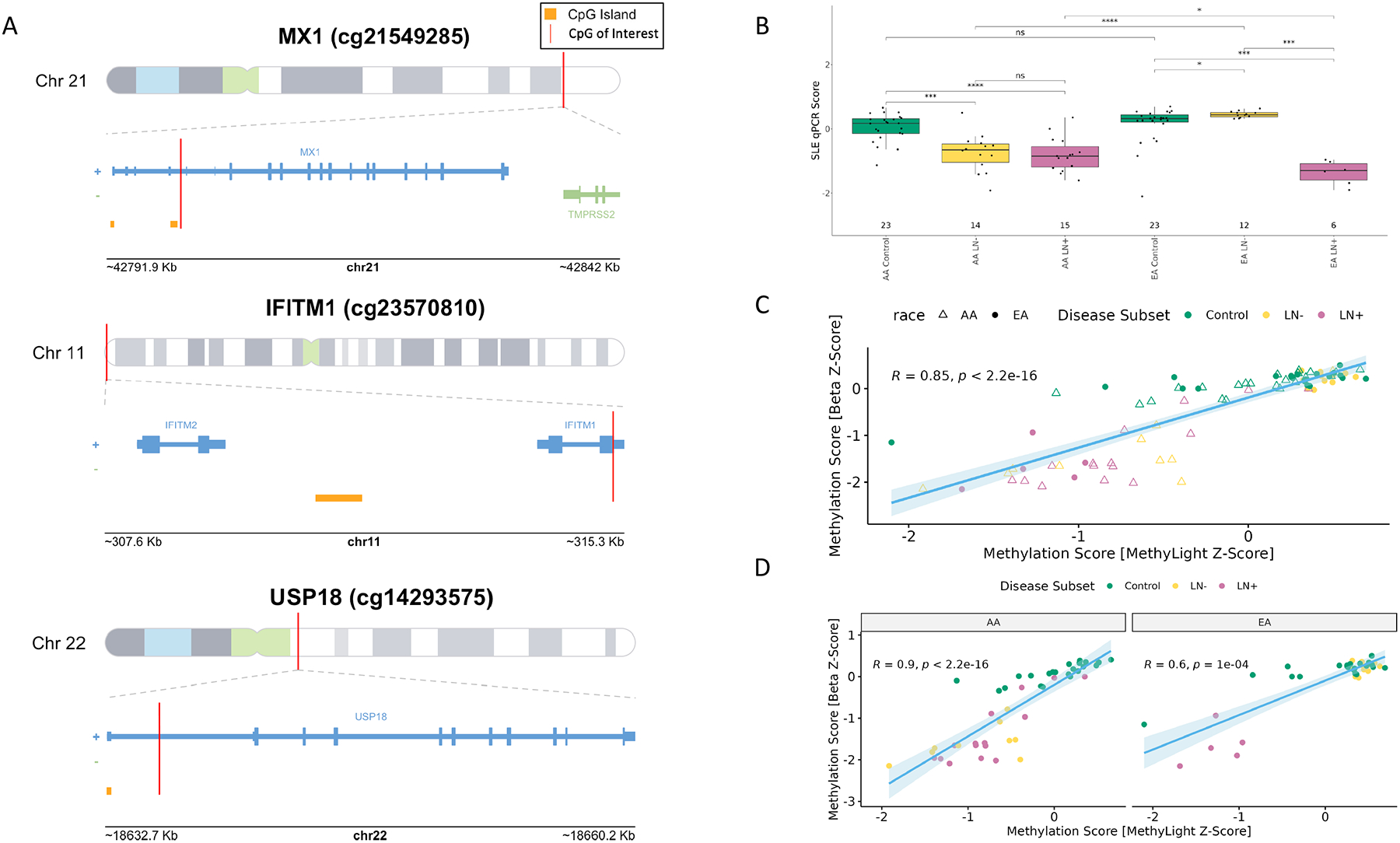
A. CpGs targeted for methylation-specific PCR in MX1, IFITM1, and USP18. B. Methylation Score from the MethyLight Assays. X-axis: Sample categories (AA/EA Control, AA/EA without lupus nephritis (AA/EA LN-), AA/EA with lupus nephritis (AA/EA LN+), Y-axis: SLE qPCR Score as calculated from median z-scores. C. Correlation of Methylation Scores between the microarray and qPCR assays. D. Correlation between methylation scores between microarray and qPCR assays stratified by race.
3.5. Local Ancestry Analysis reveals IFN hypomethylation signature in EA SLE patients with LN
Given the association of LN with the methylation status of IFN-associated CpGs and the observation that LN is more common among individuals of African ancestry, we hypothesized that methylation status at these loci could be explained by common ancestry between AA SLE patients and EA SLE patients with LN. To test this hypothesis, we genotyped individuals in our study and estimated local ancestry (African, European, or East Asian) for windows of the chromosome using RFmix [32] and compared it with 1000 Genomes populations (Figure 4A). After calculating the local ancestry for each sample, we tested whether the windows containing the CpGs found to be hypomethylated in AA SLE patients and EA patients with LN had a common ancestry.
Fig 4.
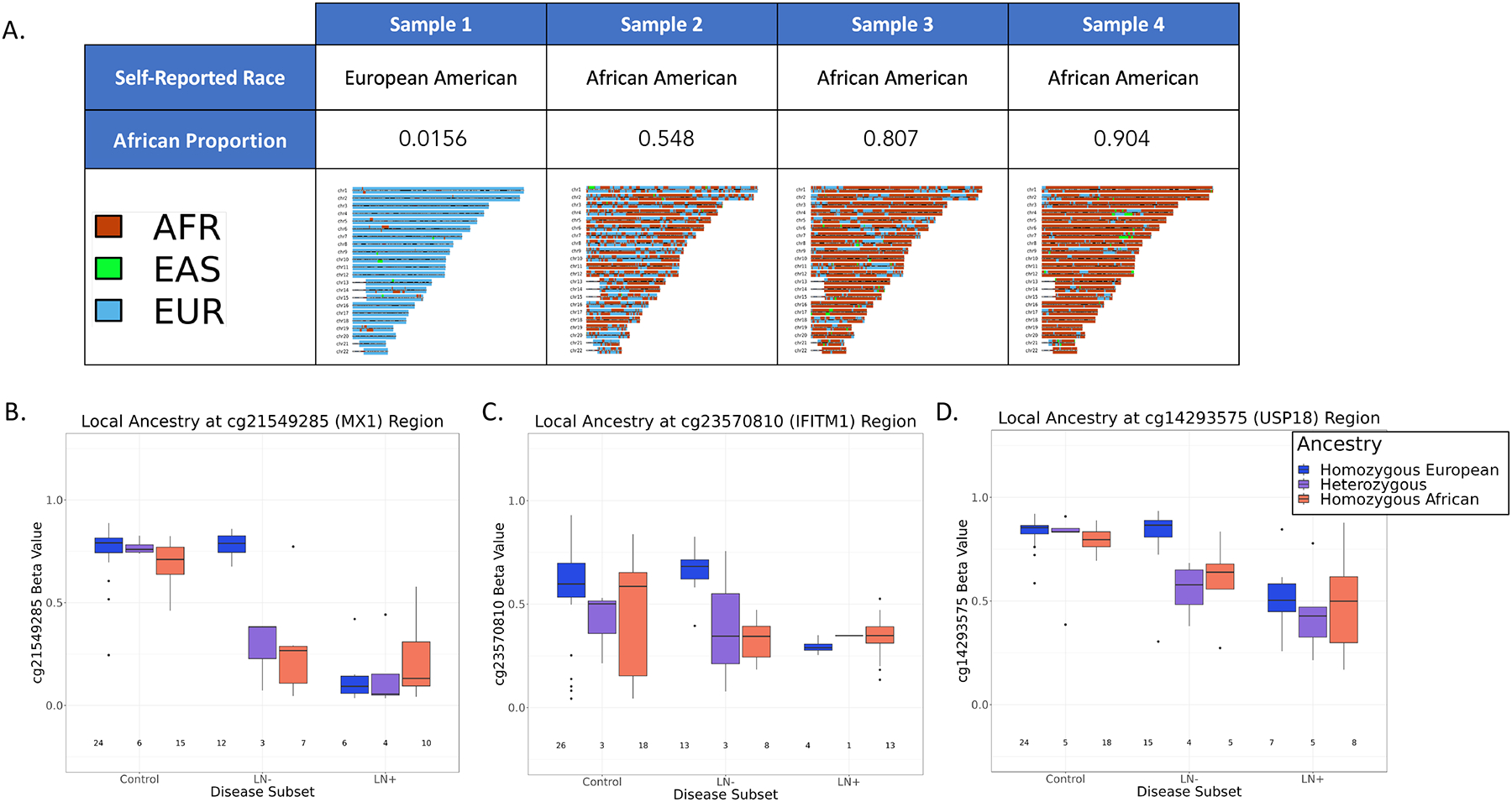
A. Table of self-reported race, the proportion of genome calculated to be of African ancestry, and karyograms where windows of the genome were assigned ancestry (African, red; Eastern Asian, green; European, blue) for a selection of four patients in our cohort. B-D. Local Ancestry at windows containing CpG of interest (Methylation Beta Value, y-axis; disease subset, x-axis). Genotype was colored by ancestry (Homozygous European, blue; Heterozygous, purple; Homozygous African, red)
We found a significant difference in methylation beta value based on genotype in the SLE patient group without LN. SLE patients without LN who were homozygous for EA alleles displayed methylation values comparable to controls who were also homozygous for EA alleles. However, SLE patients with and without LN containing at least one AA allele exhibited lower methylation proportion compared to those with both EA alleles (Figure 4B–D). Although we hypothesized that common genetic ancestry in the IFN CpG regions contributed to the reduction in methylation proportion among the EA SLE patients with LN, no correlation between ancestry at these CpGs and methylation was observed. (Figure 4 B–D).
To evaluate whether genotypes more frequent in the African population were associated with methylation at our SLE-associated CpG sites, we calculated global genetic ancestry and tested whether the methylation score and proportion of the genome mapping to African ancestry were correlated. All self-reported EAs had less than 20% African ancestry (Supplemental Figure S4). Upon stratifying by African proportion across all samples, we observed a trend in SLE patients without LN. Those with 0% to 20% African ancestry across their genome aligned with controls having 0% to 100% African proportions. However, with increasing African ancestry proportion, we found a reduction in the methylation score, suggesting a decrease in methylation proportion at many of the interferon CpGs constituting the score. SLE patients with LN had consistent SLE methylation scores across African proportion ranges, indicating that the decrease in methylation at the IFN CpGs was independent of ancestry (Supplemental Figure S5). In addition, we evaluated differences in disease activity (SLEDAI) and damage accrual (SDI) with the increasing proportion of African ancestry. Although there were no significant differences SLEDAI score among SLE patients with and without LN, among SLE patients with LN, SLEDAI score exhibited a broader distribution of SLEDAI scores when compared to lower proportions of African ancestry (Supplemental Figure S6A). The SDI in SLE patients with and without LN increases with the increased proportion of African ancestry, suggesting that with a positive association of African ancestry and increasing extent of end organ damage (Supplemental Figure S6B).
4. Discussion
In this study, we investigated the association between DMPs from across the genome in SLE in patients with and without LN, and differences by AA and EA ancestry. In EA patients with LN, we discovered an IFN-associated methylation signature that was independent of SLEDAI. By utilizing these CpGs, we established a methylation module score, which revealed similar methylation scores between AA SLE patients with and without LN and EA SLE patients with LN. We then utilized local ancestry analysis to determine if African ancestry could account for the decreased methylation in EA SLE patients with LN, which it was found not to. To our knowledge, this study is the first to incorporate local ancestry analysis with methylation in a diverse cohort of SLE patients.
In previous studies involving genome-wide DNAm in SLE, CpGs near genes in the IFN pathway were found to have a lower methylation proportion in B cells [22, 42, 43], T cells [22, 44], and monocytes [22]. The IFN-associated CpGs were utilized in predictive modeling to classify the SLE status in AA patient B cells with a specificity of 98%. However, when the model was tested on EA patient B cells, it exhibited poor performance in predicting SLE status, suggesting that the IFN methylation signature was specific to ancestry [43]. To confirm whether the signature was exclusively ancestry-specific, we analyzed DNA methylomes from a larger population that included additional AA and EA SLE patients with LN to compare with the methylomes of AA SLE patients in addition to healthy controls. Our previous studies did not include EA SLE patients with LN .
In our preliminary DNAm analysis, after correcting for age, ancestry principal components, and cellular proportions, 51 CpGs were significantly associated with SLE. Among the CpGs significantly associated with SLE were CpGs located in or near MX1, IFI44, PARP9, DDX60, and IFIT1 genes. Our DNAm analysis results largely replicate other SLE DNAm studies; these CpGs are located in IFN type I pathway genes and exhibit a lower methylation proportion in immune cell types than in healthy controls [22, 42, 43, 45, 46].
By establishing a methylation module score (median z-score across significant CpGs that showed a 10% decrease in DNAm between cases and controls), we reduced the complexity of the data. We showed that, on average, AA SLE patients and EA SLE patients with LN were at least one standard deviation below the population mean. Our study is significant because it challenges previous findings that reported a correlation between methylation and disease activity in EA SLE patients [45, 47]. Specifically, we found that although EA SLE patients with LN had lower SDI scores compared to both AA SLE patients with LN, they displayed a similar epigenetic phenotype, indicating that disease activity was not correlated with methylation status in EA SLE patients with LN. This suggests that the relationship between methylation and disease activity may be different in EA SLE patients with LN compared to EA SLE patients without LN. Further studies comparing AA SLE patients with and without LN and EA SLE patients with LN may elucidate disease pathogenesis and explain why clinical manifestations in AA SLE patients, on average, are more severe than in EA SLE patients. One possible explanation could be due to an upstream effect where progenitor hematopoietic cells are slated to be epigenetically primed to dysregulation at IFN signaling CpGs propagating the effect to downstream lymphoid and myeloid cell lineages. Another potential mechanism could be a genetic polymorphism affecting levels of IFN signaling proteins such as those in the STAT family. A SNP in STAT4 in the context of SLE has been reported to be associated with the earlier onset of the disease in addition to LN [48]. In addition, Stat4 in murine models has been reported to alter methylation of IL-18Rα, an interleukin receptor, during T helper cell (Th1) development [49]. This receptor has been associated with IFN-γ production and dysregulation of the receptor could lead to increased IFN secretion, which could lead to amplified immune system activation and heightened disease severity.
We validated our significant findings from the genome-wide methylation array using methylation-specific qPCR for CpGs in MX1, USP18, and IFITM1. The methylation score generated from the methylation-specific qPCR data positively correlated with the methylation score generated from the microarray data, showing strong agreement between the two platforms. Because these data yielded similar results as the methylation array by stratifying patients with the IFN hypomethylation signature, the methylation-specific qPCR assays could serve as an initial screening tool to identify samples with the epigenetic signature for downstream studies and could eventually prove relevant in a clinical setting with an improved understanding of the implications of these CpGs for risk of developing LN.
A previous study found that expression of IFN-stimulated genes (ISG) was more tightly correlated with African ancestry than with SLE [50], so we investigated the association between ancestry and DNAm at CpGs, found to be significantly associated with SLE. In several of the CpGs, the genotype of the samples that were found to have the hypomethylation signature largely reflected the same findings as the self-reported race. Ancestry estimates indicated that all EA SLE patients with LN were homozygous European at the windows that contained the disease-associated CpGs. This finding showed that ancestry at, or near, the CpGs associated with SLE does not fully explain the hypomethylation of IFN genes. Our global ancestry analysis also failed to identify a correlation between the global percentage of African ancestry and hypomethylation at these interferon associated CpGs.
One limitation of this study is that DNAm data were generated from buffy coat samples. DNAm is cell type-specific and by measuring signals from multiple cell types, potentially meaningful biology may be lost. In our study we corrected for this using the Houseman et al. method [38] to estimate cell type proportions using methylation data and adjusting our data to account for cellular heterogeneity, but analyzing DNAm in individual cell types could yield additional insights that cannot be observed from deconvoluted bulk interrogations. In addition, our findings may be limited by small sample size. Our study consisted of only 6 EA SLE patients with LN. Although every EA SLE patient with LN showed DNAm signatures similar to AA SLE patients, a larger, independent, well-characterized population would strengthen future investigations. Local ancestry analysis with a larger population will provide greater precision ot define cis- and trans-effects of ancestry on DNAm in the context of SLE. Finally, we cannot exclude the possibility that medication use among our SLE patients confounded DNAm-disease relationships [51] [52] [53],[54]. Immunosuppressants primarily work by restricting the activation and growth of immune cells [55]. As a result, the medication can alter the proportion of cells in a patient’s body, leading to changes in methylation proportion. To mitigate the impact of medication on methylation proportion, we accounted for cellular heterogeneity in our model. Although our findings are consistent with prior research, larger and more diverse studies with well-characterized SLE patients are necessary to validate this signature independent of immune-modulating medications. To this end, we also tested for the potential correlation between the duration of disease and the DNAm module score. Our analysis revealed that in African American (AA) SLE cases, a longer duration of the disease was associated with a less severe DNAm score, possibly indicating a relationship with the duration of their treatment.
Despite these limitations, our study was the largest investigation of DNAm in SLE and SLE-associated LN, with a focus on differences in ancestry. Our findings highlight the possibility that a common mechanism involving IFN signaling impacts LN risk among EAs, which underscores the importance of our study. Additionally, the observation of a similar methylation signature in AA SLE patients with and without LN suggests that this mechanism could potentially explain why, on average, their disease is more severe irrespective of LN status.
5. CONCLUSIONS
Our genome-wide DNAm study yielded important findings that were validated through an orthogonal method. Our results indicate that the IFN hypomethylation signature, previously reported by us to be population-specific [19, 34], is also common in EA SLE patients with LN. This leads us to conclude that IFN hypomethylation is associated with an increased risk of developing LN, rather than ancestry-specific. We found that local and global ancestry do not explain the observed differences in methylation signatures between ancestries. Additional analyses in larger, well-charcterized, diverse populations as well as mechanistic studies aimed at revealing the link between IFN signaling and LN risk are warranted to improve our understanding of SLE pathogenesis.
Supplementary Material
Highlights.
Buffy coats from African American SLE patients and European Americans with Lupus Nephritis exhibit similar methylation profiles with an enrichment of CpGs near interferon genes.
Findings were validated using an orthogonal method, methylation-specific qPCR designed for three CpGs significant in our study and previous studies, those that lie near MX1, IFITM1, and USP18.
Local ancestry at windows that contain disease-associated CpGs do not explain the observed differences in methylation signatures between ancestries.
Acknowledgements
The authors thank the participants for their contribution to the study, and members of the Myers and G. Cooper Labs at HudsonAlpha Institute for Biotechnology for helpful discussions on the study and the manuscript.
Funding
This research was supported, in part, by the National Institute of Arthritis, Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Numbers: R01 AR073850 (EEB) and R01 AR064820 (EEB) and from funds from The HudsonAlpha Institute for Biotechnology. SJC is supported by the State of Alabama. SJC and RMM are supported by CCTS grant UL1TR003096 (PI, Robert Kimberly).
List of abbreviations
- AA
African American
- DMP
Differentially Methylated Positions
- DNAm
DNA methylation
- EA
European American
- GDA
Global Diversity Array
- IFN
Interferon
- ISG
Interferon-stimulated Gene
- LN
Lupus Nephritis
- PCA
Principal Component Analysis
- SDI
Systemic Lupus International Collaborating Clinics - Damage Index
- SLE
Systemic Lupus Erythematosus
- SLEDAI
Systemic Lupus Erythematosus Disease Activity Index
- SLICC
Systemic Lupus International Collaborating Clinics
- SNP
Single Nucleotide Polymorphism
- VCF
Variant Call Format
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization: EEB, DMA; Formal analysis: PCA; Software: PCA; Investigation: PCA; Data curation: PCA, KR; Visualization: PCA; Resources: EEB, DMA, SJC, RMM; Supervision: EEB, DMA, SJC, RMM; Writing (original draft): PCA; Writing (review and editing): PCA, EEB, HKT, SJC, RMM. Project administration: EEB, DMA, SJC, RMM; Funding acquisition: EEB, DMA, SJC, RMM. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board at the University of Alabama at Birmingham.
Competing Interests
The authors declare that they have no competing interests.
Availability of data and materials
All data are available from the authors on request. All code to reproduce these analyses is available at https://github.com/PeterCAllen/sle_ancestry_manuscript2023.
References
- 1.Lisnevskaia L, Murphy G, Isenberg D (2014) Systemic lupus erythematosus. Lancet 384:1878–1888 [DOI] [PubMed] [Google Scholar]
- 2.Liu Z, Davidson A (2012) Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med 18:871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsokos GC (2011) Systemic lupus erythematosus. The New England journal of medicine 365 22:2110–21 [DOI] [PubMed] [Google Scholar]
- 4.Tan EM (1989) Antinuclear Antibodies: Diagnostic Markers for Autoimmune Diseases and Probes for Cell Biology. In: Advances in Immunology. Elsevier, pp 93–151 [DOI] [PubMed] [Google Scholar]
- 5.Izmirly PM, Parton H, Wang L, et al. (2021) Prevalence of Systemic Lupus Erythematosus in the United States: Estimates from a Meta-Analysis of the Centers for Disease Control and Prevention National Lupus Registries. Arthritis Rheumatol 73:991–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lau CS, Yin G, Mok MY (2006) Ethnic and geographical differences in systemic lupus erythematosus: an overview. Lupus 15:715–719 [DOI] [PubMed] [Google Scholar]
- 7.Fernández M, Alarcón GS, Calvo-alén J, Andrade R, McGwin G, Vilá LM, Reveille JD, LUMINA Study Group (2007) A multiethnic, multicenter cohort of patients with systemic lupus erythematosus (SLE) as a model for the study of ethnic disparities in SLE. Arthritis Rheum 57:576–584 [DOI] [PubMed] [Google Scholar]
- 8.Kumar K, Chambers S, Gordon C (2009) Challenges of ethnicity in SLE. Best Practice & Research Clinical Rheumatology 23:549–561 [DOI] [PubMed] [Google Scholar]
- 9.Lewis MJ, Jawad AS (2016) The effect of ethnicity and genetic ancestry on the epidemiology, clinical features and outcome of systemic lupus erythematosus. Rheumatology kew 399 [DOI] [PubMed] [Google Scholar]
- 10.Bastian HM, Roseman JM, Mcgwin G, Alarcón GS, Friedman AW, Fessler BJ, Baethge BA, Reveille JD, Lumina Study Group (2002) Systemic lupus erythematosus in three ethnic groups. XII. Risk factors for lupus nephritis after diagnosis. Lupus 11:152–160 [DOI] [PubMed] [Google Scholar]
- 11.Mccarty DJ, Manzi S, Medsger TA, Ramsey-Goldman R, Laporte RE, Kwoh CK (1995) Incidence of systemic lupus erythematosus race and gender differences. Arthritis & Rheumatism 38:1260–1270 [DOI] [PubMed] [Google Scholar]
- 12.Plantinga LC, Lim SS, Patzer RE, McClellan WW, Kramer MR, Klein M, Pastan SO, Gordon C, Helmick CG, Drenkard C (2016) Incidence of End-Stage renal disease among newly diagnosed systemic lupus erythematosus patients: The georgia lupus registry. Arthritis Care & Research 68: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh RR, Yen EY (2018) SLE mortality remains disproportionately high, despite improvements over the last decade. Lupus 27:1577–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams JN, Drenkard C, Lim SS (2023) The impact of social determinants of health on the presentation, management and outcomes of systemic lupus erythematosus. Rheumatology (Oxford) 62:i10–i14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barr RG, Seliger SL, Appel GB, Zuniga R, D’Agati VD, Salmon J, Radhakrishnan J (2003) Prognosis in proliferative lupus nephritis: the role of socio-economic status and race/ethnicity. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association 18 10:2039–46 [DOI] [PubMed] [Google Scholar]
- 16.Petri M, Perez-Gutthann S, Longenecker JC, Hochberg M (1991) Morbidity of systemic lupus erythematosus: Role of race and socioeconomic status. The American Journal of Medicine 91:345–353 [DOI] [PubMed] [Google Scholar]
- 17.Larsen CP, Beggs ML, Saeed M, Walker PD (2013) Apolipoprotein L1 Risk Variants Associate with Systemic Lupus Erythematosus-Associated Collapsing Glomerulopathy. JASN 24:722–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramos PS, Oates JC, Kamen DL, et al. (2013) Variable Association of Reactive Intermediate Genes with Systemic Lupus Erythematosus in Populations with Different African Ancestry. J Rheumatol 40:842–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deapen D, Escalante A, Weinrib L, Horwitz D, Bachman B, Roy-Burman P, Walker A, Mack TM (1992) A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum 35:311–318 [DOI] [PubMed] [Google Scholar]
- 20.Javierre BM, Fernandez AF, Richter J, et al. (2010) Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res 20:170–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richardson BC, Ray D, Yung R (2004) Murine models of lupus induced by hypomethylated T cells. Methods in molecular medicine 102:285–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Absher DM, Li X, Waite LL, Gibson A, Roberts K, Edberg J, Chatham WW, Kimberly RP (2013) Genome-Wide DNA Methylation Analysis of Systemic Lupus Erythematosus Reveals Persistent Hypomethylation of Interferon Genes and Compositional Changes to CD4+ T-cell Populations. PLoS Genet 9:e1003678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alarcón GS, Mcgwin GJ, Petri MA, Reveille JD, Ramsey-Goldman R, Kimberly RP (2002) Baseline characteristics of a multiethnic lupus cohort: PROFILE. Lupus 11:101–95 [DOI] [PubMed] [Google Scholar]
- 24.VanValkenburg ME, Pruitt GI, Brill IK, et al. (2015) Family history of hematologic malignancies and risk of multiple myeloma: differences by race and clinical features. Cancer Causes & Control 27:81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hochberg MC (1997) Updating the American college of rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis & Rheumatism 40:1725–1725 [DOI] [PubMed] [Google Scholar]
- 26.Petri M, Orbai A-M, Alarcón GS, et al. (2012) Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis & Rheumatism 64:2677–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Danecek P, Bonfield JK, Liddle J, et al. (2021) Twelve years of SAMtools and BCFtools. GigaScience 10:giab008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weeks JP (2010) plink: An R package for linking mixed-format tests using IRT-Based methods. Journal of Statistical Software 35:1–3321603108 [Google Scholar]
- 29.Fairley S, Lowy-Gallego E, Perry E, Flicek P (2020) The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Research 48:D941–D947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das S, Forer L, Schönherr S, et al. (2016) Next-generation genotype imputation service and methods. Nature Genetics 48:1284–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Browning SR, Browning BL (2007) Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies By Use of Localized Haplotype Clustering. The American Journal of Human Genetics 81:1084–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maples BK, Gravel S, Kenny EE, Bustamante CD (2013) RFMix: A Discriminative Modeling Approach for Rapid and Robust Local-Ancestry Inference. The American Journal of Human Genetics 93:278–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.R Core Team (2022) R: A language and environment for statistical computing. [Google Scholar]
- 35.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA (2014) Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30:1363–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Christiansen C, Castillo-Fernandez JE, Domingo-Relloso A, et al. (2021) Novel DNA methylation signatures of tobacco smoking with trans-ethnic effects. Clin Epigenet 13:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du P, Zhang X, Huang C-C, Jafari N, Kibbe WA, Hou L, Lin SM (2010) Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 11:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Houseman EA, Kile ML, Christiani DC, Ince TA, Kelsey KT, Marsit CJ (2016) Reference-free deconvolution of DNA methylation data and mediation by cell composition effects. BMC Bioinformatics 17:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, Beck S (2014) ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 30:428–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chavent M, Kuentz-Simonet V, Labenne A, Saracco J (2014) Multivariate analysis of mixed data: The R package PCAmixdata. arXiv: Computation [Google Scholar]
- 41.Ritchie ME, Phipson B, Wu D-L, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research 43:e47–e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scharer CD, Blalock EL, Mi T, et al. (2019) Epigenetic programming underpins B cell dysfunction in human SLE. Nat Immunol 20:1071–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Breitbach ME, Ramaker RC, Roberts K, Kimberly RP, Absher D (2020) Population-Specific Patterns of Epigenetic Defects in the B Cell Lineage in Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol 72:282–291 [DOI] [PubMed] [Google Scholar]
- 44.Coit P, Jeffries MA, Altorok N, Dozmorov MG, Koelsch KA, Wren JD, Merrill JT, Mccune WJ, Sawalha AH (2013) Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naïve CD4+ T cells from lupus patients. Journal of autoimmunity 43:78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Joseph S, George NI, Green-knox B, Treadwell EL, Word B, Yim S, Lyn-cook B (2019) Epigenome-wide association study of peripheral blood mononuclear cells in systemic lupus erythematosus: Identifying DNA methylation signatures associated with interferon-related genes based on ethnicity and SLEDAI. Journal of Autoimmunity 96:147–157 [DOI] [PubMed] [Google Scholar]
- 46.Coit P, Ortiz-Fernandez L, Lewis EE, McCune WJ, Maksimowicz-McKinnon K, Sawalha AH (2020) A longitudinal and transancestral analysis of DNA methylation patterns and disease activity in lupus patients. JCI Insight 5:e143654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ulff-Møller CJ, Asmar F, Liu Y, Svendsen AJ, Busato F, Grønbaek K, Tost J, Jacobsen S (2018) Twin DNA Methylation Profiling Reveals Flare-Dependent Interferon Signature and B Cell Promoter Hypermethylation in Systemic Lupus Erythematosus. Arthritis Rheumatol 70:878–890 [DOI] [PubMed] [Google Scholar]
- 48.Taylor KE, Remmers EF, Lee AT, et al. (2008) Specificity of the STAT4 Genetic Association for Severe Disease Manifestations of Systemic Lupus Erythematosus. PLoS Genet 4:e1000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu Q, Thieu VT, Kaplan MH (2007) Stat4 limits DNA methyltransferase recruitment and DNA methylation of the IL-18Rα gene during Th1 differentiation. EMBO J 26:2052–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Siddiqi KZ, Wilhelm TR, Ulff-Møller CJ, Jacobsen S (2021) Cluster of highly expressed interferon-stimulated genes associate more with African ancestry than disease activity in patients with systemic lupus erythematosus. A systematic review of cross-sectional studies.: Expression of interferon stimulated genes in systemic lupus. Translational research : the journal of laboratory and clinical medicine [DOI] [PubMed] [Google Scholar]
- 51.Wiechmann T, Röh S, Sauer S, et al. (2019) Identification of dynamic glucocorticoid-induced methylation changes at the FKBP5 locus. Clinical Epigenetics 11: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oosterom N, Griffioen PH, den Hoed MAH, Pieters R, de Jonge R, Tissing WJE, n den Heuvel-Eibrink MMM, Heil SG (2018) Global methylation in relation to methotrexate-induced oral mucositis in children with acute lymphoblastic leukemia. PLoS ONE 13: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang Y, Tang Q, Zhao M, et al. (2015) The effect of mycophenolic acid on epigenetic modifications in lupus CD4+T cells. Clinical immunology 158 1:67–76 [DOI] [PubMed] [Google Scholar]
- 54.Jones PA, Taylor SM (1980) Cellular differentiation, cytidine analogs and DNA methylation. Cell 20:85–93 [DOI] [PubMed] [Google Scholar]
- 55.Suthanthiran M, Morris RE, Strom TB (1996) Immunosuppressants: Cellular and molecular mechanisms of action. American Journal of Kidney Diseases 28:159–172 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the authors on request. All code to reproduce these analyses is available at https://github.com/PeterCAllen/sle_ancestry_manuscript2023.