

Abstract
Parkinson's disease (PD) is a neurodegenerative disease that is common in middle‐aged and elderly people, and its onset is related to multiple factors, such as heredity, environment, and age. The vesicle protein sorting 35 (VPS35) gene was found to be a late‐onset autosomal dominant familial PD (PARK17) causative gene. The protein encoded by this gene is located in the endosome and aggregates with other membrane proteins to form a retromer complex, which participates in the membrane protein cycle between the endosome and the Golgi network. Increasing evidence shows that VPS35 may participate in the pathogenesis of PD by affecting autophagy, mitochondria, neurosynaptic transmission, dopamine signaling pathways, and so forth, and it can interact with other disease‐causing genes of familial PD. This article aimed to review the functions of VPS35 and the mechanism of its mutations in PD that have been discovered in recent years.
Keywords: mechanism, Parkinson's disease (PD), retromer complex, vesicle protein sorting 35 (VPS35)
VPS35 is the core component of the retromer complex; it may play a role in the pathogenesis of Parkinson's disease (PD) by affecting autophagy, mitochondria, neurosynaptic transmission, dopamine signaling pathways, and so forth, and it can interact with other disease‐causing genes (PARKIN, LRRK2) of familial PD.
1. INTRODUCTION
Parkinson's disease (PD) is a common degenerative disease of the nervous system. It has been reported that the prevalence rate of PD in people older than 60 years of age is approximately 1%, and the prevalence rate in people older than 85 years of age is approximately 4%–5%; in our country, among individuals older than 65 years of age, the prevalence is 1700/100,000, and it increases with age. 1 However, the pathogenesis of PD is not yet clear and may be related to multiple factors, such as genetic factors, environmental factors, and aging. 2 , 3 With the development of molecular genetics, more than 20 disease‐causing genes have been cloned thus far. 4 , 5 In 2011, Vilariñno‐Güell et al. used an exome sequencing method to find the c.1858G>A (p.D620N) mutation of vesicle sorting protein 35 (VPS35) in a family with late‐onset autosomal dominant PD in Switzerland for the first time and named the disease subtype PARK17. 6 In the same year, Zimprich et al. also used this method to discover the c.1858G>A (p.D620N) mutation of VPS35 in an Austrian PD family with an average age of 53 years. 7 Subsequently, other research groups also obtained similar findings in some individuals and families of PD patients worldwide, suggesting that the VPS35 gene mutation is obviously related to the progression of PD. 8 It has been found that c. A1858G>(p.D620N) is the most common type of VPS35 mutation. The VPS35 D620N variant in the general population has not yet been determined exactly. At present, the clinical symptoms and neuroimaging of VPS35‐related PD patients suggest that their typical disease spectrum is similar to that of idiopathic PD. 7 PD patients with VPS35 gene mutations have at least 3/4 of the main motor symptoms of PD clinically, mainly tremor. Subjects occasionally show mild cognitive impairment, and all reported subjects respond to levodopa treatment. 9 , 10 , 11 Studying the function of VPS35 and revealing the pathogenesis of its development in PD could enhance the understanding of DA neuron dysfunction and degeneration in PD and contribute to the genetic diagnosis and treatment of the disease.
1.1. Expression distribution, structure, and function of VPS35
The retromer complex can promote the transport and circulation of transmembrane proteins in the endosome–GTN and endosome–plasma membrane. It is usually composed of two major subunits: the cargo recognition trimer (CSC) and the sorting nexin dimer (SNX). The CSC is composed of VPS26, VPS29, and VPS35, and is responsible for identifying and combining goods to be classified. 12 VPS35 is the core component of the retromer complex, which can regulate the sorting and assembly of transmembrane cargo and direct the transport of specific proteins to the Golgi apparatus or cell surface to play its role. 11 VPS35 is widely expressed in human tissues, and it is highly expressed in organs and tissues, such as the brain, heart, testis, ovary, small intestine, spleen, skeletal muscle, and placenta. 13 , 14 The VPS35 gene is located on chromosome 16q11.2, with a full length of 33 kb, including 17 exons (NM_018206.6), and the encoded protein contains 796 amino acid residues, with a molecular weight of approximately 92 kDa.
2. VPS35 PROTEIN AND PD
2.1. VPS35 mutation phenotype profile
VPS35 PD has significant clinical and genetic heterogeneity. Different mutation sites, mutation types, and races have different phenotypes. The currently known mutation sites in the VPS35 gene in PD are summarized in Table 1. Also, the cellular processes affected by the VPS35 mutation are detailed in Figure 1.
Table 1.
Known mutation sites of the VPS35 gene in Parkinson's disease
| Type of mutation | Base change | Amino acid change | Phenotype | References |
|---|---|---|---|---|
| Missense/nonsense | c.96A>C | R32S | PD? | [15] |
| c.171G>A | M57I | ADLP? | [7] | |
| c.723T>G | I241M | ADLP? | [7] | |
| c.946C>T | P316S | ADLP? | [6] | |
| c.1463A>G | Q488R | PD17 | [16] | |
| c.1520A>T | Y507F | PD? | [17] | |
| c.1570C>T | R524W | ADLP | [7] | |
| c.1576C>T | R526C | PD? | [18] | |
| c.1679T>C | I560T | LBD? | [19] | |
| c.1796A>G | H599R | LBD? | [19] | |
| c.1819A>G | M607V | LBD? | [19] | |
| c.1858G>A | D620N | ADLP | [6] | |
| c.1874T>C | L625P | EOAD | [20] | |
| c.2210C>T | A737V | ADLP? | [21] | |
| c.2320C>A | L774M | ADLP? | [7] | |
| c.2359G>A | E787K | PD? | [17] | |
| Splicing | IVS2+33G>A | – | PD? | [22] |
| IVS5+79G>A | – | LBD? | [19] | |
| IVS10−70G>A | – | LBD? | [19] | |
| Small deletion mutation | c.2141 2142delCT | – | – | [23] |
| Complex rearrangements | Translocation t(15;16) (p11.2;q12.1) | – | – | [24] |
Abbreviations: ADLP, autosomal dominant late‐onset Parkinson's disease; EOAD, early‐onset dementia; LBD, Lewy body disease; PD, Parkinson's disease.
Figure 1.
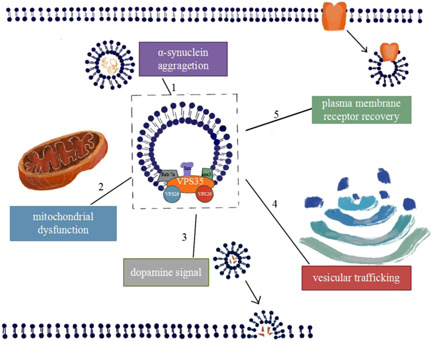
Cellular processes affected by the VPS35 mutation. VPS35, which is the core component of the retromer, along with VPS26 and VPS29, sits at the endosomal membrane and recognizes cargo to be sorted. The retromer, along with retromer‐associated proteins that aid in membrane binding (Snx3 and Rab7a), is responsible for the retrograde transport of several cargo proteins from the endosomal network to either the trans‐Golgi network or the plasma membrane. (1) VPS35 can control α‐synuclein degradation through macrophage autophagy, and endosomal pathways, etc. The VPS35‐d620n mutation and the VPS35 deletion cause α‐synuclein degradation dysfunction; (2) VPS35 indirectly regulates Mfn2 in the mitochondrial membrane. The VPS35 mutation increases the interaction with DLP1, making mitochondrial dynamics prone to excessive division; (2) the VPS35 mutation loses its function of regulating DRD1 transport and signal transduction, and the mutation or defect of VPS35 may interfere with the normal dopamine signaling pathway; and (4 and 5) VPS35 is the core component of the retromer complex, which can regulate the sorting and assembly of transmembrane cargo and direct the transport of specific proteins to the Golgi apparatus or cell surface to play its role [Color figure can be viewed at wileyonlinelibrary.com]
3. VPS35 AND THE PATHOGENESIS OF PD
3.1. VPS35 affects the degradation and aggregation of α‐synuclein
α‐synuclein plays an important role in maintaining synapses and neurotransmitter transport, but the aggregation of α‐synuclein after mutation is a pathological sign of PD. Decreased VP35 levels or VPS35 mutations in the hippocampus of PD mice led to defects in α‐synuclein clearance, leading to the extensive accumulation of aggregates. Additionally, in mouse models, a lack of VPS35 or pathogenic VPS35 mutations can lead to the accumulation and aggregation of α‐synuclein, accompanied by DA neuronal degeneration, decreased DA levels, motor behavior disorders, and lysosomal morphological changes. 25 In contrast, excessive wild‐type VPS35 expression can inhibit the accumulation of α‐synuclein aggregates and lead to decreased neuron loss and astrocyte proliferation in PD mouse models overexpressing α‐synuclein. 26 These data indicate that endosomal dysfunction caused by the loss of VPS35 impairs the ability of neurons to cope with the accumulation and removal of α‐synuclein, thereby promoting the progression of PD pathology. In addition, studies have suggested that the lack of retromer complex activity caused by the mutation of the VPS35 gene can cause DWT1 to regulate iron‐ion transmembrane transport pathway obstacles, cause a large amount of iron‐ion deposition, and induce α‐Synuclein aggregation. 27
Under normal circumstances, α‐synuclein can enter the lysosome through macrophage autophagy, CMA, and endosomal pathways and be degraded by lysosomal hydrolase. 28 VPS35 indirectly regulates lysosomal activity by sorting receptors for lysosomal hydrolases (such as Sortilin and CI‐MPR). The impaired lysosomal function caused by defects in endosome‐to‐Golgi transport and the incorrect sorting of lysosomal hydrolases (such as CTSD) may promote the accumulation of α‐synuclein in the brain of Drosophila. 29 , 30 In addition, the loss of VPS35 in heterozygous KO mice resulted in a decrease in the CMA marker (LAMP2A) because dopamine neurons lacking VPS35 impaired the endosome–Golgi transport of LAMP2A and accelerated the degradation of LAMP2A. Impaired recycling leads to increased lysosomal degradation. 25 The level of LAMP2A was also significantly reduced in mouse dopaminergic neurons overexpressing the VPS35 D620N mutant, which induced the accumulation of α‐synuclein. 25 Moreover, the retromer binding to the WASH complex is necessary for autophagy. The VPS35 mutation in a retromer component impairs WASH complex recruitment and the ability of ATG9A to traffic to autophagic compartments. 31 Therefore, the deletion of VPS35 or the expression of mutant VPS35 D620N had a major impact on lysosomal function and weakened autophagy. These mechanisms are likely to be the cause of neuronal death.
3.2. VPS35 and mitochondrial dynamic balance
Mitochondrial dysfunction has always been considered to play an important role in the pathogenesis of familial and idiopathic PD. Mitochondrial fusion protein (MFN2) is a GTPase embedded in the outer mitochondrial membrane that is essential for mitochondrial fusion. Tang et al. found that the absence of VPS35 in neuronal cell lines, DA neurons, and brain tissues reduces the level of MFN2. 32 The mitochondria of dopaminergic neurons lacking VPS35 are shorter and rounder, suggesting that the loss of VPS35 leads to mitochondrial fragmentation. The expression of wild‐type VPS35 can rescue mitochondrial fusion, while expression of the D620N mutant cannot. 32 In addition, the interaction of VPS35 and dynein‐like protein 1 (DLP1) can regulate mitochondrial division. The VPS35 mutation increases the interaction with DLP1, enhances the operation of mitochondrial DLP1 through the MDV‐mediated retromer complex, and makes mitochondrial dynamics prone to excessive division, leading to mitochondrial dysfunction and neuron loss. 33 In short, increasing evidence shows that VPS35 deletion and mutation can affect the dynamic balance of mitochondria, leading to mitochondrial dysfunction.
3.3. VPS35 and plasma membrane receptor recovery
Neurotransmitters play an important role in maintaining normal neuronal function. Studies have confirmed that VPS35 deficiency can cause damage to the maturation of mouse dendritic spines and reduce glutamatergic transmission. In VPS35‐deficient neurons, the level of the glutamatergic receptor AMPAR on the plasma membrane surface is reduced. 34 Another study found that VPS35 is located in dendritic spines and participates in the transport of excitatory AMPAR. The overexpression of VPS35 changes the basic physiological processes of neurons, including excitatory synaptic transmission, AMPAR expression, and synaptic cycling. 35 In addition, the D620N mutation causes a defect in GluR1 transport, which leads to a defect in downstream synaptic transmission, and neurons lacking VPS35 also show abnormal synaptic transmission and abnormal GluR1 and GluR2 transport. 34 , 35 The D620N mutation affects the maturation of dendritic spines and disrupts synaptic transmission, but it is unclear whether these synaptic effects cause neurodegeneration. The glucose transport receptor (GLUT1) is located in the plasma membrane, and the absence of VPS35 in HeLa cells has been shown to cause GLUT1 to be missorted. 36 However, some studies have shown that the VPS35 D620N mutation does not impair the transport of GLUT1 from the endosome to the plasma membrane. 37
3.3.1. VPS35 and the dopamine signaling pathway
Dopamine plays a key role in regulating various brain physiological functions by binding to receptors and triggering their endocytosis and signal transduction pathways. 38 , 39 The pathological feature of PD is the loss of dopamine neurons in the substantia nigra of the brain. Cataldi et al. reported an independent VPS35 D620N knock‐in mouse model that showed increased dopamine release from the striatum by rapid scanning cyclic voltammetry. They found that VPS35 interacts with dopamine receptor D1 (DRD1), and the overexpression and downregulation of VPS35 increase and decrease the cell surface steady‐state level of DRD1, respectively, and it is accompanied by changes in the phosphorylation levels of the effector molecules of the dopamine signaling pathway, CREB and ERK. 40 Vanan et al. used a Rosa26‐based transgene expression platform to generate a VPS35 D620N mouse model. The study found that the dopamine level of VPS35 D620N mice was significantly higher than that of nontransgenic control mice. 41
4. INTERACTION BETWEEN VPS35 AND OTHER PD PATHOGENIC PROTEINS
PARKIN is an E3 ubiquitin ligase. Mutations in the PARK2 gene encoding PARKIN can lead to autosomal recessive early‐onset PD. Drosophila lacking PARKIN have a shortened lifespan, decreased climbing ability, and increased sensitivity to paraquat. 42 , 43 Malik et al. 43 crossed VPS35‐deficient fruit flies with VPS35‐mutant transgenic fruit flies, proved that VPS35 overexpression can rescue the defects formed by PARKIN mutant phenotype fruit flies, and speculated that VPS35 and PARKIN genetically interact. Williams et al. 44 proved that PARKIN interacts with VPS35 and stabilizes ubiquitination in human nerve cells, and the PARKIN mutation weakens its ability to ubiquitinate VPS35. In addition, they used adenovirus vectors to express human VPS35 D620N in the substantia nigra of PARKIN knockout mice or wild‐type mice, and the overexpression of VPS35 D620N mutants in wild‐type and PARKIN‐deficient mice can induce the loss of large numbers of DA neurons. 44 These results indicate that VPS35 may be downstream of PARKIN, 14 but they did not identify the role of PARKIN in mediating the pathogenic effect of the VPS35 mutation, thereby accelerating neurodegeneration.
Leucine‐rich repeat protein kinase‐2 (LRRK2) is a kinase that regulates vesicle trafficking via the phosphorylation of Rab protein subgroups. Its dysfunction may affect the aggregation of α‐synuclein and its pathological changes. 45 Mutations in the LRRK2 gene can lead to autosomal dominant delayed‐type PD, and G2019S is its most common pathogenic mutation. 46 , 47 MacLeod et al. provided evidence to support retromer complex dysfunction in the context of LRRK2‐RAB7L1 pathway defects. In mouse N2A neuroblastoma cells, the expression of LRRK2 G2019S or the knockout of RAB7L1 will cause a significant decrease in the levels of VPS35 and VPS29. 48 To further study the interaction between VPS35 and LRRK2, Mir et al. discussed the effect of VPS35 on LRRK2‐mediated Rab protein phosphorylation. The knockout of VPS35 resulted in a significant increase in the LRRK2‐mediated phosphorylation of many other Rab proteins. 49 These data indicate that VPS35 may be located upstream of LRRK2 and play a role in regulating the catalytic activity of LRRK2 kinase. VPS35 controls the activity of LRRK2, and the VPS35 D620N mutation leads to an increase in its function, which may lead to PD through the excessive activation of LRRK2 kinase. 49
5. OUTLOOK
Previous studies on the correlation between VPS35 and PD mainly focused on the clinical manifestations and some possible pathogenesis caused by VPS35 mutations. However, in recent years, research on VPS35 in the field of PD has undergone a qualitative change. As a key protein of the retromer complex, VPS35 participates in a variety of possible mechanisms in the pathogenesis of PD, including affecting autophagy, the dynamic balance of mitochondria, neurosynaptic transmission, dopamine signaling pathway conduction, vesicle transport, other PD pathogenic protein interactions, etc. These findings suggest that increasing the expression level of VPS35 protein in neurons or enhancing its function may be a potential therapeutic target for the treatment of PD. With the rapid development of molecular biology technology, it may be possible to accurately target VPS35 in the future to regulate the clinical symptoms of PD patients.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
ETHICS STATEMENT
The ethics statement is not available.
AUTHOR CONTRIBUTIONS
Ai‐Di Luo was involved in the main conception of this study and drafted the manuscript; Zu‐Cai Xu was involved in reviewing and editing this paper; and Shu‐Sheng Liao was in charge of acquiring funding, supervising, reviewing, and editing this paper.
TRANSPARENCY STATEMENT
The authors affirm that this manuscript is an honest, accurate, and transparent account of the study being reported; that no important aspects of the study have been omitted; and that any discrepancies from the study as planned (and, if relevant, registered) have been explained.
ACKNOWLEDGMENTS
The acknowledgment is not available. This study was funded by the National Natural Science Foundation of China (No: 82160316), the National Natural Science Foundation of China (No: 81260177), and the Guizhou Provincial Science and Technology Foundation [No: (2019)1351].
Luo A‐D, Xu Z‐C, Liao S‐S. VPS35, the core component of the retromer complex, and Parkinson's disease. ibrain. 2021;7:318‐324. 10.1002/ibra.12004
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Tysnes OB, Storstein A. Epidemiology of Parkinson's disease. J Neural Transm. 2017;124(8):901‐905. [DOI] [PubMed] [Google Scholar]
- 2. Simon DK, Tanner CM, Brundin P. Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin Geriatr Med. 2020;36(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhao Y, Qin L, Pan H, et al. The role of genetics in Parkinson's disease: a large cohort study in Chinese mainland population. Brain. 2020;143(7):2220‐2234. [DOI] [PubMed] [Google Scholar]
- 4. Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson's disease. Lancet Neurol. 2020;19(2):170‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Billingsley KJ, Bandres‐Ciga S, Saez‐Atienzar S, Singleton AB. Genetic risk factors in Parkinson's disease. Cell Tissue Res. 2018;373(1):9‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vilariño‐Güell C, Wider C, Ross OA, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89(1):162‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zimprich A, Benet‐Pagès A, Struhal W, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late‐onset Parkinson disease. Am J Hum Genet. 2011;89(1):168‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klein C, Hattori N, Marras C. MDSGene: closing data gaps in genotype‐phenotype correlations of monogenic Parkinson's disease. J Parkinsons Dis. 2018;8(s1):S25‐25S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deng H, Gao K, Jankovic J. The VPS35 gene and Parkinson's disease. Mov Disord. 2013;28(5):569‐575. [DOI] [PubMed] [Google Scholar]
- 10. Struhal W, Presslauer S, Spielberger S, et al. VPS35 Parkinson's disease phenotype resembles the sporadic disease. J Neural Transm. 2014;121(7):755‐759. [DOI] [PubMed] [Google Scholar]
- 11. Williams ET, Chen X, Moore DJ. VPS35, the retromer complex and Parkinson's disease. J Parkinsons Dis. 2017;7(2):219‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cui Y, Yang Z, Teasdale RD. The functional roles of retromer in Parkinson's disease. FEBS Lett. 2018;592(7):1096‐1112. [DOI] [PubMed] [Google Scholar]
- 13. Zhang P, Yu L, Gao J, et al. Cloning and characterization of human VPS35 and mouse Vps35 and mapping of VPS35 to human chromosome 16q13‐q21. Genomics. 2000;70(2):253‐257. 10.1006/geno.2000.6380 [DOI] [PubMed] [Google Scholar]
- 14. Sassone J, Reale C, Dati G, Regoni M, Pellecchia MT, Garavaglia B. The role of VPS35 in the pathobiology of Parkinson's disease. Cell Mol Neurobiol. 2021;41(2):199‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bandrés‐Ciga S, Mencacci NE, Durán R, et al. Analysis of the genetic variability in Parkinson's disease from Southern Spain. Neurobiol Aging. 2016;37(210):210.e1‐210.e5. [DOI] [PubMed] [Google Scholar]
- 16. Farwell KD, Shahmirzadi L, El‐Khechen D, et al. Enhanced utility of family‐centered diagnostic exome sequencing with inheritance model‐based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2015;17(7):578‐586. [DOI] [PubMed] [Google Scholar]
- 17. Nuytemans K, Bademci G, Inchausti V, et al. Whole exome sequencing of rare variants in EIF4G1 and VPS35 in Parkinson disease. Neurology. 2013;80(11):982‐989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pihlstrøm L, Rengmark A, Bjørnarå KA, Toft M. Effective variant detection by targeted deep sequencing of DNA pools: an example from Parkinson's disease. Ann Hum Genet. 2014;78(3):243‐252. [DOI] [PubMed] [Google Scholar]
- 19. Verstraeten A, Wauters E, Crosiers D, et al. Contribution of VPS35 genetic variability to LBD in the Flanders‐Belgian population. Neurobiol Aging. 2012;33(8):1844.e11‐13. [DOI] [PubMed] [Google Scholar]
- 20. Rovelet‐Lecrux A, Charbonnier C, Wallon D, et al. De novo deleterious genetic variations target a biological network centered on Aβ peptide in early‐onset Alzheimer disease. Mol Psychiatry. 2015;20(9):1046‐1056. 10.1038/mp.2015.100 [DOI] [PubMed] [Google Scholar]
- 21. Ando M, Funayama M, Li Y, et al. VPS35 mutation in Japanese patients with typical Parkinson's disease. Mov Disord. 2012;27(11):1413‐1417. [DOI] [PubMed] [Google Scholar]
- 22. Bartonikova T, Mensikova K, Mikulicova L, et al. Familial atypical parkinsonism with rare variant in VPS35 and FBXO7 genes: a case report. Medicine. 2016;95(46):e5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Foo JN, Tan LC, Liany H, et al. Analysis of non‐synonymous‐coding variants of Parkinson's disease‐related pathogenic and susceptibility genes in East Asian populations. Hum Mol Genet. 2014;23(14):3891‐3897. [DOI] [PubMed] [Google Scholar]
- 24. Finelli P, Sirchia SM, Masciadri M, et al. Juxtaposition of heterochromatic and euchromatic regions by chromosomal translocation mediates a heterochromatic long‐range position effect associated with a severe neurological phenotype. Mol Cytogenet. 2012;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang FL, Erion JR, Tian Y, et al. VPS35 in dopamine neurons is required for endosome‐to‐golgi retrieval of Lamp2a, a receptor of chaperone‐mediated autophagy that is critical for α‐synuclein degradation and prevention of pathogenesis of Parkinson's disease. J Neurosci. 2015;35(29):10613‐10628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dhungel N, Eleuteri S, Li LB, et al. Parkinson's disease genes VPS35 and EIF4G1 interact genetically and converge on α‐synuclein. Neuron. 2015;85(1):76‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hirayama T, Inden M, Tsuboi H, et al. A Golgi‐targeting fluorescent probe for labile Fe(ii) to reveal an abnormal cellular iron distribution induced by dysfunction of VPS35. Chem Sci. 2019;10(5):1514‐1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. 2015;47(3):e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cullen V, Lindfors M, Ng J, et al. Cathepsin D expression level affects alpha‐synuclein processing, aggregation, and toxicity in vivo. Mol Brain. 2009;2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Macías‐Calvio V, Fuentealba LM, Marzolo MP. An update on cellular and molecular determinants of Parkinson's disease with emphasis on the role of the retromer complex. J Neurosci Res. 2021;99(1):163‐179. [DOI] [PubMed] [Google Scholar]
- 31. Zavodszky E, Seaman MN, Moreau K, et al. Mutation in VPS35 associated with Parkinson's disease impairs WASH complex association and inhibits autophagy. Nat Commun. 2014;5:3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tang FL, Liu W, Hu JX, et al. VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. 2015;12(10):1631‐1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang W, Wang X, Fujioka H, et al. Parkinson's disease‐associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat Med. 2016;22(1):54‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tian Y, Tang FL, Sun X, et al. VPS35‐deficiency results in an impaired AMPA receptor trafficking and decreased dendritic spine maturation. Mol Brain. 2015;8(1):70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Munsie LN, Milnerwood AJ, Seibler P, et al. Retromer‐dependent neurotransmitter receptor trafficking to synapses is altered by the Parkinson's disease VPS35 mutation p.D620N. Hum Mol Genet. 2015;24(6):1691‐1703. [DOI] [PubMed] [Google Scholar]
- 36. Steinberg F, Gallon M, Winfield M, et al. A global analysis of SNX27‐retromer assembly and cargo specificity reveals a function in glucose and metal ion transport. Nat Cell Biol. 2013;15(5):461‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McGough IJ, Steinberg F, Jia D, et al. Retromer binding to FAM21 and the WASH complex is perturbed by the Parkinson disease‐linked VPS35(D620N) mutation. Curr Biol. 2014;24(14):1670‐1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hasbi A, O'Dowd BF, George SR. Dopamine D1‐D2 receptor heteromer signaling pathway in the brain: emerging physiological relevance. Mol Brain. 2011;4:26. 10.1186/1756-6606-4-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang C, Niu M, Zhou Z, et al. VPS35 regulates cell surface recycling and signaling of dopamine receptor D1. Neurobiol Aging. 2016;46:22‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cataldi S, Follett J, Fox JD, et al. Altered dopamine release and monoamine transporters in Vps35 p.D620N knock‐in mice. NPJ Parkinsons Dis. 2018;4(27):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vanan S, Zeng X, Chia SY, et al. Altered striatal dopamine levels in Parkinson's disease VPS35 D620N mutant transgenic aged mice. Mol Brain. 2020;13(1):164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cha GH, Kim S, Park J, et al. Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc Natl Acad Sci U S A. 2005;102(29):10345‐10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Malik BR, Godena VK, Whitworth AJ. VPS35 pathogenic mutations confer no dominant toxicity but partial loss of function in Drosophila and genetically interact with Parkin. Hum Mol Genet. 2015;24(21):6106‐6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Williams ET, Glauser L, Tsika E, Jiang H, Islam S, Moore DJ. Parkin mediates the ubiquitination of VPS35 and modulates retromer‐dependent endosomal sorting. Hum Mol Genet. 2018;27(18):3189‐3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Daher JP. Interaction of LRRK2 and α‐synuclein in Parkinson's disease. Adv Neurobiol. 2017;14:209‐226. [DOI] [PubMed] [Google Scholar]
- 46. Paisán‐Ruiz C, Lewis PA, Singleton AB. LRRK2: cause, risk, and mechanism. J Parkinsons Dis. 2013;3(2):85‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ren C, Ding Y, Wei S, et al. G2019S variation in LRRK2: an ideal model for the study of Parkinson's disease? Front Hum Neurosci. 2019;13:306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. MacLeod DA, Rhinn H, Kuwahara T, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron. 2013;77(3):425‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mir R, Tonelli F, Lis P, et al. The Parkinson's disease VPS35[D620N] mutation enhances LRRK2‐mediated Rab protein phosphorylation in mouse and human. Biochem J. 2018;475(11):1861‐1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.