

Abstract
Introduction:
Non-angiotensin converting enzyme mechanisms of angiotensin II production remain underappreciated in part due to the success of current therapies to ameliorate the impact of primary hypertension and atherosclerotic diseases of the heart and the blood vessels. This review scrutinize the current literature to highlight chymase role as a critical participant in the pathogenesis of cardiovascular disease and heart failure.
Areas covered:
We review the contemporaneous understanding of circulating and tissue biotransformation mechanisms of the angiotensins focusing on the role of chymase as an alternate tissue generating pathway for angiotensin II pathological mechanisms of action.
Expert opinion:
While robust literature documents the singularity of chymase as an angiotensin II-forming enzyme, particularly when angiotensin converting enzyme is inhibited, this knowledge has not been fully recognized to clinical medicine. This review discusses limitations of clinical trials’ that explored the benefits of chymase inhibition in accounting for the failure to duplicate in humans what has been demonstrated in experimental animals.
Keywords: Angiotensin Converting Enzyme, Angiotensin II, Angiotensin peptides, Angiotensin-(1-12), Blood Pressure, Chronic Kidney Disease, Chymase, Diabetes Mellitus, Heart Failure, Myocardial Infarction, Primary Hypertension, Vascular Disease
1. Introduction
More people die each year from cardiovascular diseases than any other cause [1]. High blood pressure remains a significant risk factor for premature cardiovascular mortality. The magnitude of this problem is reflected in epidemiological surveys revealing that hypertension is present in 1.4 billion people worldwide, with less than one in five people with a hypertension diagnosis having their blood pressure under control [1]. The global antihypertensive drug market is forecast to increase from USD 20 billion in 2021 to USD 40 billion by 2031 [2]. The expansion in market size reflects a staggering rise in the global incidence of hypertension, in part attributable to a rise in cardiovascular disease, increased salt consumption, expansion of the geriatric population, and obesity. While the adoption of combination therapies or single-pill combinations (polypill) may afford greater therapeutic efficacy and increased adherence [3,4], hypertension treatment management remains suboptimal. Angiotensin converting enzyme (ACE) inhibitors and angiotensin II (Ang II) receptor blockers (ARBs) are the most common antihypertensive prescription drug classes in the United States [5]. Both types of medicines are recommended as first-line agents for initiating antihypertensive therapy based on the highest level of evidence [6]. The importance of these drugs in managing hypertension, type 2 diabetes, and heart failure are reflected by a projected market increase in the use of ARBs from 7.85 billion in 2020 to 9.95 billion in 2028. While ACE inhibitors and ARBs demonstrate comparable effects on blood pressure control, randomized clinical trials (RCT) and large meta-analyses indicate that the magnitude of the relative benefit in terms of reducing all-cause and cardiovascular mortality, stroke, heart failure, myocardial infarction, composite cardiovascular events, kidney disease, or diabetes remains uncertain [7-9]. While comparative assessment of the two drugs classes in terms of clinical outcome yields similar effectiveness [10], those analyses do not take into consideration the magnitude of the absolute risk reduction achieved with either of the two drug classes compared to placebo or other antihypertensive medications [9,11,12]. Evidence that blood pressure-independent mechanisms contribute to the adverse cardiovascular remodeling associated with high blood pressure is often not appreciated. Retrospective analysis of clinical endpoints with drugs that block the generation or activity of Ang II by us [9,11] and others [13-15] reveals non-superior efficacy of ACE inhibitors and ARBs over other antihypertensive agents for reducing the risk of myocardial infarction, heart failure (HF) or cardiovascular death. The quantification of the lifetime residual risk of cardiovascular events in hypertensive patients is several orders of magnitude greater than the risk reduction [9]. This is a disconcerting finding, given the strength of the research implicating the renin angiotensin system (RAS) in the pathogenesis of experimental and human hypertension.
Both extrinsic and intrinsic factors explain the limitations of RAS inhibitors to attain greater reductions in adverse clinical outcomes beyond what is attributed to blood pressure control [9]. A systemic chronic inflammatory response is present in non-communicable diseases such as cardiovascular disease, cancer, diabetes mellitus, chronic kidney disease, non-alcoholic fatty liver disease, autoimmune, and neurodegenerative disorders [16]. The non-superiority of RAS inhibitors over other agents may reflect the failure of these drugs to reach the intracellular sites where Ang II can be generated [17]. Angiotensin II (Ang II) formation occurs in multiple tissues because RAS genes in these target tissues generate functional proteins [18-20]. Therefore, the production of Ang II intracellularly (intracrine pathway) or in the environmental milieu of the interstitial spaces of tissues (paracrine pathway) appears critical in mediating the pathophysiology of cardiovascular diseases in specific tissues.
Furthermore, there is a general ignorance of the interplay among the RAS, the kallikrein-kinin system (KKS), and chymase [21]. These peptidergic systems exert proinflammatory and procoagulant effects via the Ang II type 1 receptor (AT1-R) and the bradykinin type 1 (BKB-1R) receptor, respectively. In addition, bradykinin vasodilator, natriuretic, and antithrombotic effects induce chymase upregulation via activation of BKB-2R [21]. Interwoven interactions among the RAS, K_KS, and chymase play a critical role in regulating cardiovascular function. This review article summarizes the potential criticality of alternate mechanisms of Ang II production via a chymase/Ang II pathway in human heart disease. Furthermore, the material presented here updates a previously published article that recapitulated the patent literature on chymase inhibitors for treating cardiovascular disease between 2010 and 2018 [22].
2. Current understanding of angiotensins forming mechanisms
Today’s generally accepted view of the biochemical cascade that accounts for the generation of angiotensin peptides possessing biological activity did not become apparent until the 1970s. The classic literature of that time had concluded that Ang II biological activity was the product of sequential reactions initiated by the generation of the decapeptide angiotensin I (Ang I) through the hydrolysis of hepatic angiotensinogen (AGT) and the subsequent Ang I cleavage by ACE into the active octapeptide pressor hormone Ang II [23,24]. However, it was not until the 1980s that seminal publications from Ferrario and colleagues uncovered the presence of the heptapeptide -angiotensin-(1-7) [Ang-(1-7)] in rat cerebrospinal fluid (CSF) [25] and the concurrent demonstration of its biological activity in the release of neurohypophysial vasopressin [26]. The further demonstration that Ang-(1-7) possessed vasodilator activity in areflexic rats [27] strengthened the potential for Ang-(1-7) to exert a role in the control of arterial pressure. While these discoveries did not immediately alter conceptualizations of the RAS, persistence in the study of Ang-(1-7) biological activity and its potential role in the pathogenesis of hypertension culminated with the discovery that the heptapeptide functions as an endogenous “inhibitor” of Ang II pressor and trophic mechanisms of action [28]. Increased recognition of Ang-(1-7) as an integral component of the RAS was accelerated by the identification of an ACE homolog -ACE 2 (ACE2) [29,30]- functioning as a mono peptidase with high catalytic efficiency for conversion of Ang II into Ang-(1-7) [31,32].
The complexity of enzymatic mechanisms with the ability to generate the angiotensins is influenced by the composition of the tissue sites at which the AGT substrate can be processed to generate Ang I, Ang II, Ang-(1-7), Ang III, the pentapeptide Ang-(1-5), and the alanine substituted angiotensins [Angiotensin A [33]and Alamandine [34]] (Figure 1). These discoveries have enriched our understanding of both the homeostatic control and RAS’s dysregulation in human diseases by revealing that the vasoconstrictive, pro-proliferative, and proinflammatory actions of the ACE/Ang II/AT1-R are counterbalanced by the vasodilator, antiproliferative, and anti-inflammatory actions of ACE2/Ang-(1-7)/Mas-R axis [35]. A potential third axis is constituted by Angiotensin-A/Alamandine/MrgD-receptor [36].
Figure 1.
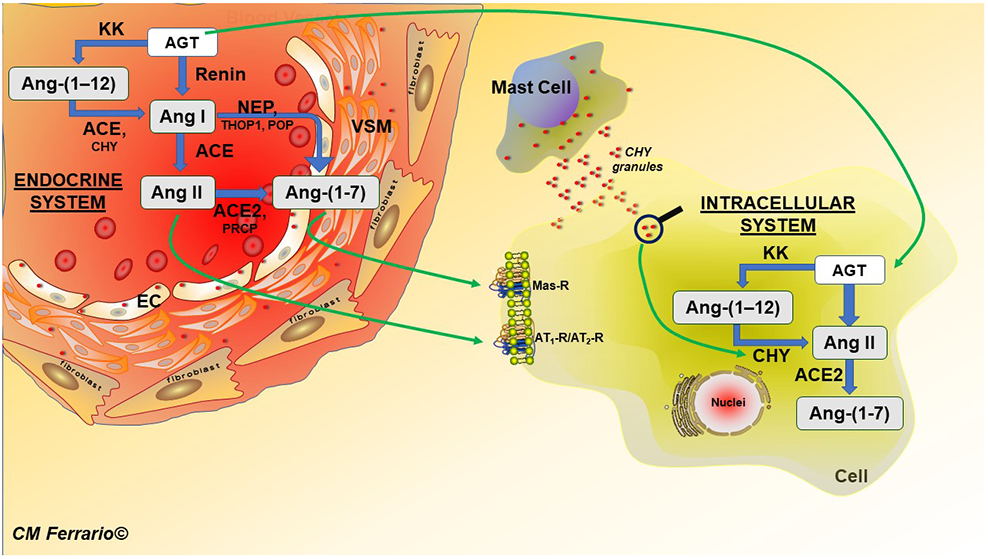
Schematic diagram of the main biochemical pathways of the angiotensin peptide production via the circulation (endocrine system depicted in the insert as a blood vessel segment) and intracellularly. The illustration depicts a role of mast cell degranulation of chymase for intracellular incorporation and direct metabolism of Ang-(1–12) into Ang II. Abbreviations, EC, endothelial cell; VSM, vascular smooth muscle. POP, prolyl oligopeptidase; THOP1, thimet oligopeptidase. Other abbreviations as in text.
Figure 2 documents the principal cleavage sites of the angiotensins. The diversity of the branching mechanisms within the RAS that can be activated to generate the different angiotensins subserving endocrine, paracrine, autocrine, and intracrine mechanisms is being further expanded with the more recent identification of a catalytic pathway upstream of Ang I and Ang-(1-9) [9,35]. In 2006, Nagata and colleagues [37] at the University of Miyazaki, Japan, reported the existence of a peptide composed of the first 12 amino acids of the AGT protein in the blood and tissues of a Japanese strain of Wistar rats. Proangiotensin-12 [Asp1-Arg2-Val3-Tyr4-Ile5-His6-Pro7-Phe8-His9-Leu10-Leu11-Tyr12], as named by the Japanese investigators, was reported to be present in highest concentrations in the gastrointestinal tract. Nevertheless, a substantial concentration of this alternate substrate was detected in the brain, heart, kidney, adrenal gland, and blood [37]. The biological activity of Proangiotensin-12 was evidenced by the abolition of the pressor response induced by intravenous injection of the dodecapeptide by either the ACE inhibitor captopril or the ARB candesartan [37]. The existence of an alternate source for Ang II generation from a molecule composed of 12 amino acids rather than the 425 amino acids contained in AGT was of interest to Ferrario’s laboratory because, in past collaboration with one of the authors of this review (RS), we had reported the existence of a family of endogenous Ang II precursors in canine cerebrospinal fluid (CSF) [38]. Since the Nagata et al. [37] report of proangiotensin-12, we and others have provided extensive evidence for the functional role of this alternate substrate as an endogenous source for the production of Ang II., Proangiotensin-12 was renamed as angiotensin-(1-12) [Ang-(1–12)] to maintain consistency with the recommendations of the Nomenclature for Angiotensin Receptors Committee of the American Heart Association [39].
Figure 2.
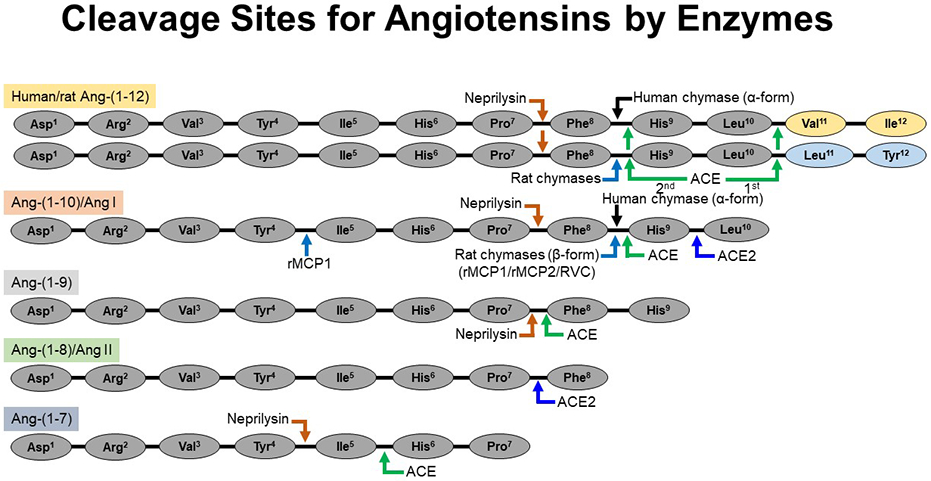
Cleavage sites for the processing of angiotensin peptides by chymase, ACE, ACE2, and neprilysin. Humans (α-chymase) and rat (β-chymases) directly cleave the Phe8-His9 bond of the Ang-(1-12) to generate Ang II directly. Similarly, rat β-chymases (rMCP-1, rMCP-2 and RVC) cleave the Phe8-His9 bond of the Ang I to generate Ang II directly. ACE cleaves Ang-(1-12) [sequentially, 1st Leu10-Val11 (human)/Leu10-Leu11 (rat) and then Phe8-His9 bonds] and Ang I [Phe8-His9 bond] to generate Ang II. ACE2 also cleaves the Ang I [Phe8-His9 bond] to generate Ang-(1-9). Ang-(1-12), Ang I and Ang-(1-9) are cleaved by neprilysin (an endopeptidase) at Pro7-Phe8 bond to generate Ang-(1-7). Rat β-chymase (rMCP-1) may hydrolyze Ang II at the Tyr4-Ile5 bond. Ang II is not further cleaved by human α-chymase but, ACE2 cleaves the Pro7-Phe8 bond of Ang II to generate Ang-(1-7). Ang-(1-7) is cleaved by ACE and neprilysin to generate Ang-(1-5) and Ang-(1-4), respectively. Abbreviations: rat mast cell protease-1, rMCP-1; rat mast cell protease-2, rMCP-2 and rat vascular chymase, RVC.
Ang-(1-12) is a precursor for renin-independent Ang II formation in the circulation[40], the human and rat heart [41-46], kidney[47], and bone marrow [48]. Ang-(1-12), cleaved from AGT by a kallikrein-like enzyme [49,50], is preferentially metabolized by endothelial ACE into Ang I in the circulation [43] and cleaved directly into Ang II by chymase in the human heart, rat kidney, and rat bone marrow [41,44,48] (Figure 1). The rate of Ang II formation from the Ang-(1-12) substrate is several-fold higher than from Ang I in plasma membranes isolated from the heart of adult spontaneously hypertensive rats (SHR) [51]. Ang-(1-12) augments rat cardiac myocyte contractility via increasing intracellular K+ currents in WKY[52] and intracellular Ca2+ mobilization in SD rats[53] and TGR(hAGT)L1623 hypertensive rats[53]. In a humanized model of hypertension expressing the human AGT gene (Tg+-hAGT), cardiac hypertrophy and systolic dysfunction were accompanied by a 4-fold increase in left ventricular (LV) Ang II content that failed to be reversed following a fortnight treatment with valsartan[54]. These data implicate a compensatory activation of intracellular Ang II generation from internalized AGT, Ang-(1–12), or both. This interpretation is in keeping with: (a)- internalization of AGT and Ang-(1–12) in human pigment retina cells by a non-AT1-R mechanism [45,55]; (b)- augmented incorporation of Ang-(1–12) in neonatal cardiac myocytes in culture[51]; (c)- mobilization of intracellular Ca2+[53] and increase in K+ currents[52] in primary cardiac myocyte cultures during superfusion with Ang-(1–12); (d)- the demonstration that inotropic stimulation of cardiomyocytes by Ang-(1–12) is inhibited by superfusion of a chymase inhibitor [56] or intracellular application of valsartan[52].
3. Chymase functions and inhibition
The original demonstration that chymase is the major Ang II forming enzyme in the human heart dates to 1990 [57,58] based upon the inhibition of 75% of Ang I conversion to Ang II by serine protease inhibitors. In 2010, Park et al. [59] noted that while healthy mouse kidneys depended primarily upon ACE to form Ang II from Ang I, diabetic mouse (db/db) kidneys relied primarily upon a serine protease to form Ang II based upon the ability of the non-selective serine protease inhibitor phenylmethylsulfonylfluoride (PMSF) to inhibit a renal arteriolar vasoconstrictor response to Ang I. In an accompanying editorial, Lorenz [60] cited work indicating chymase involvement in human diabetic nephropathy and in the formation of Ang II in human arteries.
Kaltenecker et al. [61], also reported that chymase played a major role in the elevation of renal Ang II levels in chronic kidney disease compared to those of healthy kidney donors.
Chymase is the predominant Ang II forming enzyme in failing hearts as well as in the right ventricles of healthy transplanted hearts [62]. Indeed, no ACE activity was observed in the failing hearts. Of note, Pavo et al. [62] inability to detect Ang I in the failing human hearts suggest that plasma renin did not directly influence myocardial RAS regulation while supporting the conclusion that chymase is the main enzyme responsible for tissue Ang II formation. We have proposed [63] that Ang-(1–12) might be the Ang II precursor in the heart, an interpretation that is keeping with high cardiac levels of Ang-(1–12) in the heart of SHR [64] and transgenic hypertensive rats expressing the human Agt gene in their genome [54,65].
Chymase, a member of serine proteases primarily secreted by mast cells, exhibits a twenty-fold higher catalytic efficiency for conversion of Ang I into Ang II [66,67] by its preference for phenylalanine or leucine being in the P1 position (proximate to the scissile bond) according to the Schechter and Berger nomenclature system [68]. Of note, while the action of chymase to form Ang II from Ang I mimics that of ACE, it does not mimic the proficiency of ACE to metabolize Ang-(1-7) to Ang-(1-5) since isoleucine in the P1 position is less optimal for binding to chymase. Additional chymase proteolytic properties include the lysis of extracellular proteins (laminin and fibronectin), activation of transforming growth factor-β1 (TGF-β1), interleukin-1β (IL-1β), generation of endothelin (ET)-1(1-31), and degradation of high-density lipoprotein-3 (HDL3) [67,69,70]. The newly identified potency of chymase in direct tissue generation of Ang II from Ang-(1–12) strengthens the importance of chymase as a critical enzymatic pathway for the intracellular actions of the hormone [17,41-43].
Mammalian chymases are classified into two subfamilies, α-chymase and β-chymase [71-73]. A single α-form of mast cell (MC) chymase is expressed in humans while both α- and β-isoforms of chymases (also known as mast cell proteases, rMCP in rat and mMCP in mice) are found in rodents. The α-chymases in rodents (rats and mice) are elastase-like proteases, and the β-chymases are chymotrypsin-like proteases [74]. While both α- and β-chymases generate Ang II, only a β-form (preferentially rMCP-1) degrades Ang II by cleaving the Tyr4-Ile5 bond of the peptide [75]. More recently, a rat vascular chymase (RVC, a β-form) expressed primarily in vascular smooth muscle cells of SHR has been reported to convert Ang I into Ang II [76]. Upregulation of this novel RCV chymase in hypertensive rats may significantly affect vascular proliferation and blood pressure regulation [70,76]. The two β-chymases (rMCP-1 and rMCP-2) expressed in rats MCs have been widely studied; both enzyme isoforms weakly convert Ang I into Ang II compared to human α-form chymase [77,78]. Except for rMCP-1, rMCP-2, and RVC, the substrate specificity of rMCP-3 and rMCP-4 for generating Ang II from Ang I remains unknown [22].
Because chymase has many substrates other than angiotensins, a dysregulated processing of chymase substrates is predominantly detrimental [79]. For example, chymase activates matrix metalloproteases and releases transforming growth factor (TGF)-β1 from its latent state, both of which are implicated in albuminuria, mesangial cell expansion, and interstitial fibrosis, hallmarks of diabetic kidney disease (DKD) [80]. So there are multiple reasons why chymase inhibitors have therapeutic potential.
Chymase can be inhibited by a large number of serine protease inhibitors such as chymostatin, 4-(2-Aminoethyl)benzene sulfonyl fluoride hydrochloride (AEBSF), soybean trypsin inhibitor, SoybeanBowman–Birk protease inhibitor (BBI), Eglin c, secretory leukocyte protease inhibitor [(SLPI), also known as mucus proteinase inhibitor [81]], and several endogenous serpins (see below) that are described in various protein databases [81].
The endogenous serpins (SERineProteaseINhibitors) that inhibit the activity of chymase include α-1-antitrypsin (AAT, α-1-proteinase inhibitor) [68]; α-1 antichymotrypsin (ACT, Serpin A3) [68]; seminal human inhibitor I (HUSI-I), anti leukoprotease (ALP) [82]; and squamous cell carcinoma antigen 2 (SCCA2, serpin B4) [83]. Other serpins can inhibit chymase, but their low potency makes them physiologically inconsequential.
Fulacimstat (BAY1142524) is the only specific chymase-inhibiting drug to be developed for potential clinical application. It is reported to have an IC50 of 4 nM for human chymase [84]. In hamsters with cardiac fibrosis, Fulacimstat dose-dependently reduced the extent of the fibrosis. Additionally, hamsters with experimentally induced myocardial infarctions treated with Fulacimstat had a significantly lower end-diastolic pressure than the placebo group [84].
3.1. Chymase inhibitors and cardiovascular disease
Chymase has been called the “chameleon of host defense and tissue remodeling due to its many effects on tissue remodeling [67,85]. Numerous chymase inhibitors have been tested in preclinical animal models of acute and chronic injury targeting various organs with largely beneficial effects (Table 1). However, despite this great success in preclinical studies, only one clinical trial of a chymase inhibitor is reported in the literature. Recently, a Phase II clinical trial in patients with ST-segment elevation myocardial infarction reported no improvement in adverse LV remodeling and LV systolic function when Fulacimstat was added to conventional post-myocardial infarction medical therapy [86,87].
Table 1.
Studies of Chymase Inhibition in Mice, Rats, Hamsters and Humans
| Condition | Model | Chymase inhibition effects | Reference |
|---|---|---|---|
| Intermittent Hypoxia | mouse | Decreased perivascular fibrosis and cardiomyocyte hypertrophy, inflammatory cytokines, oxidative stress, Ang II, and superoxide production in LV myocardium | [127] |
| Dextran sodium sulfate-induced colitis | mouse | Decreased neutrophil infiltration and MMP-9 activity resulting in improved disease activity index and histological scores | [128] |
| Ang II induced aneurysm | ApoE-deficient mouse | Decreased MMP-9 activity and macrophage infiltration. Prevented Ang II-induced aortic aneurysm formation in ApoE-deficient mice | [129] |
| Increased salt diet | mouse | Suppressed hypertension and decreased plasma Ang II and aldosterone | [110] |
| Myosin-immunized myocarditis | rat | Decreased MMP-9 activation, TGF-β expression, and macrophage infiltration; Improved survival and LV function 4 weeks after immunization | [130] |
| Stroke-Prone SHR | rat | Decreased aortic MMP-9 activity, TNF-α, MCP-1 levels and macrophage infiltration; Improved vascular function in vitro and survival | [113] |
| Indomethacin-induced colitis | rat | Decreased intestinal wall MMP-9 activation and myeloperoxidase activity; Decreased intestinal lesions and damage | [131] |
| Lipopolysaccharide induced liver injury | hamster | Improved liver function and reduced liver necrosis and fibrosis; Decreased liver MMP-9 activation and myeloperoxidase and TNF-α levels | [132] |
| Carbon tetrachloride-induced chronic liver failure | hamster | Decreased liver myofibroblasts and fibrosis and decreased liver Ang II levels | [133] |
| Elastase-induced aneurysm formation | hamster | Decreased abdominal aortic aneurysm size and mast cell infiltration | [134] |
| Streptozotocin-induced diabetes | hamster | Decreased LV NOX4-induced oxidative stress, malonaldehyde levels, and interstitial fibrosis; Attenuated kidney oxidative stress, decreased renal fibrosis and TGF-β, and improved renal function | [135-137] |
| Cigarette smoking and bleomycin-induced lung injury | hamster | Decreased lung TGF-β signaling, ET-1 levels, and pulmonary hypertension and fibrosis | [138, 139] |
| Coronary artery ligation | hamster | Improved LV systolic function and hemodynamics, hypertrophy and fibrosis; Improved survival | [90,91,140] |
| Obstructed kidney | hamster | Attenuated tubulointerstitial fibrosis and TGF-β and α-smooth muscle actin expression | [141] |
| Prolonged high-fat diet | hamster | Prevented lipid deposition in the aortas | [142] |
| Cardiac Ischemia Reperfusion | mouse | Decrease infarction size and improved LV function | [94] |
| Cardiac Ischemia Reperfusion | pig | Decreased myocardial infarction size and tissue MMP-9 activity | [89] |
| Cardiac Ischemia Reperfusion | dog | Decreased troponin, MMP-9 activity, and cardiac interstitial chymase activity and attenuated mitochondrial damage | [88] |
| Post-Transmural Myocardial Infarction | human | Did not improve LVEF or LV volumes six months after drug initiation 5-10 days after myocardial infarction | [86,87] |
| Hypertension | human | Polygonum inhibition of chymase caused a depressor effect especially in hypertensive subjects with excessive salt intake | [143] |
| Carotid artery bypass | dog | Inhibited vascular proliferation in grafted vein | [144] |
| Orally active chymase inhibitor | hamster | Suppressed heart chymase activity | [145] |
Abbreviations: LV - Left ventricular, LVEF - Left ventricular ejection fraction, TNF – Tumor necrosis factor, MMP-9 Matrix metallopeptidase 9, MCP-1 - Mast cell protease 1, NOX4 - NADPH oxidase 4, ET-1 – Endothelin 1.
In the (CHIARA MIA) 2 trial, patients were randomized to the chymase inhibitor Fulacimstat (n=54) or placebo (n=53) five to nine days after an ST Elevation Myocardial Infarction [86,87]. Left ventricular ejection fraction (LVEF) obtained by cardiac magnetic resonance imaging was < 45% in all patients, with the change in LVEF at 6 months being equivalent between treated and placebo groups. There were no differences in cardiovascular demographics or the use of beta-blockers, ACE inhibitors, or Ang II type 1 receptor antagonists. In addition, no significant differences were reported between the treatment groups in LVEF, LV end-diastolic or end-systolic volume index, or infarct size at baseline or after 6 months of treatment in this patient cohort with a high risk of LV remodeling. These findings contradict the beneficial effects of various chymase inhibitors in preclinical in vivo animal models of ischemia-reperfusion injury or coronary ligation. Comparing the timing of chymase inhibitor administration in the preclinical animal studies and this human study provides insight into this disappointing result.
We have shown a significant influx of chymase into cardiomyocytes and an increase in interstitial fluid chymase-mediated Ang II formation within two hours of coronary ligation in the dog [88]. Pretreatment with the chymase inhibitor significantly decreases interstitial fluid Ang II formation and troponin release [88]. Studies in the pig [89], hamster [90-92], rat [93], and dog [88] demonstrate that early treatment with a chymase inhibitor within 24 hours after coronary artery ligation decreases infarct size resulting in improved LV remodeling and LV function. Other studies of ischemia-reperfusion injury show a decrease in infarct size when a chymase inhibitor is administered shortly after induction of ischemia/reperfusion injury [94]. Treatment with a chymase-specific inhibitory RNA aptamer during coronary occlusion in the hamster also improves LV remodeling and function in four weeks [95]. Finally, the absence of mast cell protease in the mouse following coronary artery ligation results in decreased infarct size and improved LV size and function compared to wild-type mice [96-98].
Taken together, blockade or absence of chymase from the outset of coronary ligation or ischemia/reperfusion confers acute tissue protection that reduces infarct size, improves LV remodeling, and LVEF at one month. In addition to a decrease in Ang II, a significant effect of chymase inhibition in these studies is the significant decrease in MMP-9 [88,89,99], a major component of the polymorphonuclear cells that are essential mediators of acute injury of ischemia or infarction. In support of acute chymase activation in the human with ischemia/reperfusion, we have shown a 4-fold increase in pericardial fluid chymase activity within 4 hours after cardiopulmonary bypass [100]. This increase in chymase activity relates to intraoperative cross-clamp and total operative time. In another study of patients with myocardial infarction, early peak values of chymase- and Cathepsin G-dependent angiotensin II formation in circulating mononuclear leukocytes correlate with elevations of creatine kinase [101]. These studies may explain the failure of chymase inhibition to improve LV remodeling and function in the recent CHIARA MIA 2 clinical trial, where the chymase inhibitor was started six to 12 days post-myocardial infarction [86,87].
Preclinical studies in chronic heart failure report beneficial effects of chymase inhibition in the dog with pacing tachycardia [102] and with experimentally induced mitral regurgitation [103]. The nature of these two models of heart failure is a chronic state of inflammation due to the extreme nature of the stress that is unabated throughout its course until animal euthanasia. The dog model is especially clinically relevant because instead of multiple α- and β- isoforms of mast cell chymase in the rodent, the dog possessed only the alpha isoform that is very similar to the human. The silica and bleomycin models of acute pulmonary injury and subsequent pulmonary fibrosis are other models where the acute stress is treated within onset or 24 hours of inception. Chymase inhibition started within 24 hours results in reduced pulmonary fibrosis at 14 and 30 days after administration of these toxic agents [104,105].
One area of potential use of a chymase inhibitor in a chronic setting is preventing atrial fibrillation [106]. Chymase activity is four-fold higher in the left atrium compared to the right atrium and LV [99,107]. In patients with Primary Mitral Regurgitation (PMR) chymase activity was associated with extensive fibrosis, increased left atrial volume, and depressed left atrial function. Chymase-mediated Ang II formation also has an important potential intracellular electrophysiological effect. Rapid field electrical stimulation of HL-1 atrial cells induced a sustained augmentation of intracellular Ca2+ associated with increases in ACE and chymase activities and AGT expression [107]. Furthermore, transmission electron microscopy reveals chymase presence within rat cardiomyocytes [108] and human left atrial myocytes [99]. Intracellular administration of Ang (1-12), the preferred chymase substrate [41,43], into adult rat cardiomyocytes caused depolarization of the cell surface membrane with an increase of duration of the action potential followed by the generation of early afterdepolarizations due to a decrease in the potassium current [52]. Administration of the chymase inhibitor -chymostatin- (10−9 M), abolished the effect of intracellular Ang-(1-12) on the potassium current[52]. Thus, extracellular effects of chymase on inflammation and fibrosis and its intracellular effects of potassium current combine for important mechanisms underlying the genesis of atrial fibrillation.
The large presence of circulating serine protease inhibitors has clouded a potential chymase-mediated mechanism of ACE escape in the intravascular space and for blood pressure control. Urata and coworkers demonstrated a positive correlation between circulating mononuclear cell chymase-mediated Ang II formation and atrial fibrillation [109] and blood pressure in large cohorts of patients undergoing routine cardiovascular examination. Furthermore, chymase presence in endothelial cells can also serve as a means of chymase-mediated intracellular Ang II formation and secretion into the circulation. Urata and coworkers [110] have reported a major role of chymase in salt-sensitive elevation of blood pressure. A chymase inhibitor (TPC-806) prevented the elevation of blood pressure in mice with excessive salt intake [111]. In a follow-up study in humans, TSP-806 decreased blood pressure with mild hypertension after one dose [111]. Responders had a higher estimated salt intake than the non-responders, suggesting that chymase inhibition is more effective for salt-dependent hypertension. In addition, chymase inhibition prevented vascular inflammation and dysfunction in angiotensin I-infused and stroke-prone hypertensive rats, there were important anti-inflammatory vascular effects of chymase inhibition, improving survival without blood pressure reduction [112,113].
3.2. Chymase inhibition, diabetes, and cardiometabolic disease
Experimental observations suggest renoprotective and anti-diabetic actions of chronic chymase inhibition [114]. In a phase II trial (NCT03412006), Fulacimstat was administered for six months to type II diabetes mellitus (T2DM) patients diagnosed with diabetic kidney disease. The outcome measure was a reduction in the urinary albumin to creatine ratio (UACR). In addition, the trial assessed the safety and tolerability of Fulacimstat. While Fulacimstat was well tolerated and safe, the 19.6% reduction in UACR relative to the placebo group fell short of the anticipated 30% reduction for statistical significance. One of the conclusions made by the authors was that the animal models of diabetic kidney disease (DKD) that showed marked elevation of chymase activity and improvements with chymase inhibition might not be representative of DKD in humans [115]. However, some preliminary data was obtained from humans, including a demonstration of 10 - 15-fold increases in chymase activity in diabetic kidneys [116], and upregulation of chymase activity in human mesangial cells cultured in a high-glucose medium [117]. It may also be of note that chymase activity is also increased in polycystic kidney disease patients [118]. Another chymase inhibitor under development was JNJ-10311795/RWJ-355871. This drug inhibited inflammatory responses to glycogen-induced peritonitis and lipopolysaccharide-induced bronchial inflammation by reversing increases in the proinflammatory cytokines IL-1α, IL-1ß and TNFα- [119]. RWJ-355871 also showed anti-inflammatory effects in several additional rat models of inflammation, with therapeutic promise for treating airway inflammatory diseases [87,120]. The potential translation of these experimental findings to the clinical management of type-2 diabetes mellitus awaits resolution, as no clinical trials are registered for any chymase-inhibiting drugs on ClinicalTrials.gov as of June 2023.
Mast cell degranulation inhibitors may be an alternate, albeit indirect, way to suppress chymase contribution to diseases. These drugs, acting to prevent mast cell degranulation, impede the liberation of chymase to act upon its extracellular substrates. The first mast cell degranulation inhibitor, sodium cromoglycate disodium 5,5’-[(2-hydroxytrimethylene)dioxy]bis[4-oxo-4H-1-benzopyran-2-carboxylate], also known as cromolyn sodium (Intal®) was marketed as an inhalant for the treatment of asthma in the early 1970’s. Subsequently, it was marketed as a nasal spray (Nasalcrom®) for the treatment of allergic rhinitis and eye drops (Opticrom®) for allergic irritation of the eyes. A subsequent preparation was developed as an enema to treat inflammatory bowel disease with limited efficacy. An orally administered concentrated solution form of cromolyn sodium (Gastrocrom®) is currently marketed for the treatment of mastocytosis. The “membrane stabilizing” mechanism of action of cromolyn sodium remains uncertain but has many proposed mechanisms based upon a relatively recent review [121]. Additional approaches to inhibiting mast cell degranulation block the post-receptor signaling pathways leading to mast cell degranulation [122]; inhibition of Bruton’s tyrosine kinase with ibrutinib (Imbruvica®) [123] to antagonize the substance P receptor [124]; coumarins possessing high affinity agonism for GPR35 [125]; and flavonoids [126]. No information exists on the potential effect of these drugs on heart and blood vessel diseases or type 2 diabetes mellitus.
4. Conclusion
An abundance of evidence indicates that mast cell chymase plays a crucial role in tissue Ang II formation from Ang I and upstream precursors. Mast cell-derived chymase activity contributes to tissue damage, cardiac remodeling, and cardiovascular disease. However, clinical trials of chymase inhibitors have not yet led to their implementation as cardiovascular therapeutics despite promising preclinical results. The importance of chymase in contributing to Ang II pathological actions may be most critical in conditions in which ACE activity is suppressed, so investigation of the therapeutic benefits of chymase inhibitors should continue.
5. Expert opinion
Chymase inhibition remains largely an obscure therapeutic concept in 21st-century medicine. However, its relative novelty and therapeutic potential have made this a promising area of research. Interestingly, with all the chymase inhibitors developed to date, no chronic human study has demonstrated their efficacy in treating hypertension, heart failure, and diabetes. However, it is tempting to speculate that given the many proinflammatory effects of chymase, in addition to Ang II forming capacity, the plan for any clinical trial should be highly contextual concerning the timing of administration and type of remodeling process. In that respect, there may be a role for a chymase inhibitor in patients with resistant hypertension who cannot reduce a high salt intake and have a high risk for atrial fibrillation. In addition, preclinical studies suggest that a trial of chymase inhibitor in the acute phase of myocardial infarction may attenuate acute myocardial injury and result in improved LV remodeling and function over time.
Article highlights.
The therapeutic potential of chymase in the treatment of cardiovascular disease remains to be appreciated.
Chymase represents an important enzymatic mechanism for the formation of Ang II from either Ang-(1–12) or Ang I.
Chymase has multiple substrates other than angiotensins, the metabolism of which is predominantly detrimental.
As a member of the serine protease family, chymase can be inhibited by many serine protease inhibitors.
Funding
This manuscript was funded by grant 1 R21 AG070371-01 (CM Ferrario) from the National Institutes of Health, Bethesda, MD.
Footnotes
Declaration of interests
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants, or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Zhou B, Carrillo-Larco RM, Danaei G, et al. Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: a pooled analysis of 1201 population-representative studies with 104 million participants. The Lancet. 2021. 2021/September/11/;398(10304):957–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Global Antihypertensive Drugs Market Scope: Pub-ID-54; 2022. [Google Scholar]
- 3.Coca A, Kreutz R, Manolis AJ, et al. A practical approach to switch from a multiple pill therapeutic strategy to a polypill-based strategy for cardiovascular prevention in patients with hypertension. J Hypertens. 2020. Oct;38(10):1890–1898. [DOI] [PubMed] [Google Scholar]
- 4.Joseph P, Roshandel G, Gao P, et al. Fixed-dose combination therapies with and without aspirin for primary prevention of cardiovascular disease: an individual participant data meta-analysis. Lancet. 2021. Sep 25;398(10306):1133–1146. [DOI] [PubMed] [Google Scholar]
- 5.Aitken M, Kleinrock M, Pritchett J. The Use of Medicines in the U.S.: Usage and Spending Trends and Outlook to 2026. IQVIA Institute for Human Data Science; 2022. p. 64. [Google Scholar]
- 6.Williams B, Mancia G. Ten Commandments of the 2018 ESC/ESH HTN Guidelines on Hypertension in Adults. Eur Heart J. 2018. Sep 1;39(33):3007–3008. [DOI] [PubMed] [Google Scholar]
- 7.Matchar DB, McCrory DC, Orlando LA, et al. Systematic review: comparative effectiveness of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers for treating essential hypertension. Ann Intern Med. 2008. Jan 1;148(1):16–29. [DOI] [PubMed] [Google Scholar]
- 8.Sanders GD, Powers B, Crowley M, et al. Future Research Needs for Angiotensin Converting Enzyme Inhibitors or Angiotensin II Receptor Blockers Added to Standard Medical Therapy for Treating Stable Ischemic Heart Disease. Identification of Future Research Need. Agency for Healthcare Research and Quality, U.S. Department of Health and Human Services; 2010. [PubMed] [Google Scholar]
- 9. Ferrario CM, Saha A, VonCannon JL, et al. Does the Naked Emperor Parable Apply to Current Perceptions of the Contribution of Renin Angiotensin System Inhibition in Hypertension? Curr Hypertens Rep. 2022. Dec;24(12):709–721. **Of Considerable Interest: a Review article documenting the limitations of current renin angiotensin system inhibitors in reversing the residual risk for cardiovascular morbidity and mortality.
- 10.Chen R, Suchard MA, Krumholz HM, et al. Comparative First-Line Effectiveness and Safety of ACE (Angiotensin-Converting Enzyme) Inhibitors and Angiotensin Receptor Blockers: A Multinational Cohort Study. Hypertension. 2021. Sep;78(3):591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reyes S, Varagic J, Ahmad S, et al. Novel Cardiac Intracrine Mechanisms Based on Ang-(1-12)/Chymase Axis Require a Revision of Therapeutic Approaches in Human Heart Disease. Curr Hypertens Rep. 2017. Feb;19(2):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vanuzzo D The epidemiological concept of residual risk. Intern Emerg Med. 2011. Oct;6 Suppl 1:45–51. [DOI] [PubMed] [Google Scholar]
- 13.Blood Pressure Lowering Treatment Trialists C, Turnbull F, Neal B, et al. Effects of different regimens to lower blood pressure on major cardiovascular events in older and younger adults: meta-analysis of randomised trials. BMJ. 2008. May 17;336(7653):1121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blood Pressure Lowering Treatment Trialists C, Turnbull F, Neal B, et al. Blood pressure-dependent and independent effects of agents that inhibit the renin-angiotensin system. J Hypertens. 2007. May;25(5):951–8. [DOI] [PubMed] [Google Scholar]
- 15.Dusing R, Sellers F. ACE inhibitors, angiotensin receptor blockers and direct renin inhibitors in combination: a review of their role after the ONTARGET trial. Curr Med Res Opin. 2009. Sep;25(9):2287–301. [DOI] [PubMed] [Google Scholar]
- 16.Furman D, Campisi J, Verdin E, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019. Dec;25(12):1822–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferrario CM, Ahmad S, Varagic J, et al. Intracrine angiotensin II functions originate from noncanonical pathways in the human heart. Am J Physiol Heart Circ Physiol. 2016. Aug 1;311(2):H404–14. *Of interest: a Review article summarizes current understanding of the biochemical physiology of the renin angiotensin system.
- 18.Abadir PM, Walston JD, Carey RM. Subcellular characteristics of functional intracellular renin-angiotensin systems. Peptides. 2012. Dec;38(2):437–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar R, Singh VP, Baker KM. The intracellular renin-angiotensin system: implications in cardiovascular remodeling. Curr Opin Nephrol Hypertens. 2008. Mar;17(2):168–73. [DOI] [PubMed] [Google Scholar]
- 20.Re RN. Mechanisms of disease: local renin-angiotensin-aldosterone systems and the pathogenesis and treatment of cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2004. Nov;1(1):42–7. [DOI] [PubMed] [Google Scholar]
- 21.Abassi Z, Skorecki K, Hamo-Giladi DB, et al. Kinins and chymase: the forgotten components of the renin-angiotensin system and their implications in COVID-19 disease. Am J Physiol Lung Cell Mol Physiol. 2021. Mar 1;320(3):L422–L429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmad S, Ferrario CM. Chymase inhibitors for the treatment of cardiac diseases: a patent review (2010-2018). Expert Opin Ther Pat. 2018. Nov;28(11):755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ondetti MA, Cushman DW. Enzymes of the renin-angiotensin system and their inhibitors. Annu Rev Biochem. 1982;51:283–308. [DOI] [PubMed] [Google Scholar]
- 24.Skeggs LT, Dorer FE, Kahn JR, et al. The Biological Production of Angiotensin. In: Page IH, Bumpus FM, editors. Angiotensin Handbook of Experimental Pharmacology. Vol. XXXVII. XXXVII ed: Springer-Verlag; Berlin -Heidelberg - New York; 1974. p. 1–7. [Google Scholar]
- 25.Santos RA, Brosnihan KB, Chappell MC, et al. Converting enzyme activity and angiotensin metabolism in the dog brainstem. Hypertension. 1988. Feb;11(2 Pt 2):I153–7. [DOI] [PubMed] [Google Scholar]
- 26.Schiavone MT, Santos RA, Brosnihan KB, et al. Release of vasopressin from the rat hypothalamo-neurohypophysial system by angiotensin-(1-7) heptapeptide. Proc Natl Acad Sci U S A. 1988. Jun;85(11):4095–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benter IF, Diz DI, Ferrario CM. Cardiovascular actions of angiotensin(1-7). Peptides. 1993. Jul-Aug;14(4):679–84. [DOI] [PubMed] [Google Scholar]
- 28.Ferrario CM. Contribution of angiotensin-(1-7) to cardiovascular physiology and pathology. Curr Hypertens Rep. 2003. Apr;5(2):129–34. [DOI] [PubMed] [Google Scholar]
- 29.Tikellis C, Thomas MC. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int J Pept. 2012;2012:256294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turner AJ, Tipnis SR, Guy JL, et al. ACEH/ACE2 is a novel mammalian metallocarboxypeptidase and a homologue of angiotensin-converting enzyme insensitive to ACE inhibitors. Can J Physiol Pharmacol. 2002. Apr;80(4):346–53. [DOI] [PubMed] [Google Scholar]
- 31.Rice GI, Thomas DA, Grant PJ, et al. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J. 2004. Oct 1;383(Pt 1):45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vickers C, Hales P, Kaushik V, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002. Apr 26;277(17):14838–43. [DOI] [PubMed] [Google Scholar]
- 33.Jankowski V, Vanholder R, van der Giet M, et al. Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler Thromb Vasc Biol. 2007. Feb;27(2):297–302. [DOI] [PubMed] [Google Scholar]
- 34.Lautner RQ, Villela DC, Fraga-Silva RA, et al. Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res. 2013. Apr 12;112(8):1104–11. [DOI] [PubMed] [Google Scholar]
- 35.Ferrario CM, Groban L, Wang H, et al. The renin-angiotensin system biomolecular cascade: a 2022 update of newer insights and concepts. Kidney Int Suppl (2011). 2022. Apr;12(1):36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hrenak J, Paulis L, Simko F. Angiotensin A/Alamandine/MrgD Axis: Another Clue to Understanding Cardiovascular Pathophysiology. Int J Mol Sci. 2016. Jul 20;17(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nagata S, Kato J, Sasaki K, et al. Isolation and identification of proangiotensin-12, a possible component of the renin-angiotensin system. Biochem Biophys Res Commun. 2006. Dec 1;350(4):1026–31. *Of Interest: First demonstration of an extended form of angiotensin I as an alternate source of angiotensin II formation.
- 38.Husain A, Bumpus FM, Smeby RR, et al. Evidence for the existence of a family of biologically active angiotensin I-like peptides in the dog central nervous system. Circ Res. 1983. Apr;52(4):460–4. [DOI] [PubMed] [Google Scholar]
- 39.Bumpus FM, Catt KJ, Chiu AT, et al. Nomenclature for angiotensin receptors. A report of the Nomenclature Committee of the Council for High Blood Pressure Research. Hypertension. 1991;17(5):720–721. [DOI] [PubMed] [Google Scholar]
- 40.Moniwa N, Varagic J, Simington SW, et al. Primacy of angiotensin converting enzyme in angiotensin-(1-12) metabolism. Am J Physiol Heart Circ Physiol. 2013. Sep 1;305(5):H644–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ahmad S, Simmons T, Varagic J, et al. Chymase-dependent generation of angiotensin II from angiotensin-(1-12) in human atrial tissue. PLoS One. 2011;6(12):e28501. *Of interest: the study highlights the expression of angiotensin-(1-12) in human atrial tissue and its metabolism by chymase.
- 42.Ahmad S, Varagic J, Groban L, et al. Angiotensin-(1-12): a chymase-mediated cellular angiotensin II substrate. Curr Hypertens Rep. 2014. May;16(5):429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmad S, Varagic J, VonCannon JL, et al. Primacy of cardiac chymase over angiotensin converting enzyme as an angiotensin-(1-12) metabolizing enzyme. Biochem Biophys Res Commun. 2016. Sep 16;478(2):559–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahmad S, Wei CC, Tallaj J, et al. Chymase mediates angiotensin-(1-12) metabolism in normal human hearts. J Am Soc Hypertens. 2013. Mar-Apr;7(2):128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahmad S, Wright KN, Ferrario CM, et al. Internalization of Angiotensin-(1-12) in Adult Retinal Pigment Epithelial-19 Cells. The FASEB Journal. 2022;36(S1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trask AJ, Jessup JA, Chappell MC, et al. Angiotensin-(1-12) is an alternate substrate for angiotensin peptide production in the heart. Am J Physiol Heart Circ Physiol. 2008. May;294(5):H2242–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Westwood BM, Chappell MC. Divergent pathways for the angiotensin-(1-12) metabolism in the rat circulation and kidney. Peptides. 2012. Jun;35(2):190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamashita T, Ahmad S, Wright KN, et al. Noncanonical Mechanisms for Direct Bone Marrow Generating Ang II (Angiotensin II) Predominate in CD68 Positive Myeloid Lineage Cells. Hypertension. 2020. Feb;75(2):500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simington SW, Moniwa N, Ahmad S, et al. Renin does not participate in the production of plasma Ang-(1–12) from angiotensinogen. Hypertension. 2013;60:A628. [Google Scholar]
- 50.Simington SW, Moniwa N, Ahmad S, et al. Abstract 628: Renin Does not Participate in the Production of Plasma Ang-(1-12) from Angiotensinogen. Hypertension. 2012;60(suppl_1):A628–A628. [Google Scholar]
- 51.Ahmad S, Varagic J, Westwood BM, et al. Uptake and metabolism of the novel peptide angiotensin-(1-12) by neonatal cardiac myocytes. PLoS One. 2011. Jan 10;6(1):e15759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Mello WC, Dell'Itallia LJ, Varagic J, et al. Intracellular angiotensin-(1-12) changes the electrical properties of intact cardiac muscle. Mol Cell Biochem. 2016. Nov;422(1-2):31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reyes S, Cheng CP, Roberts DJ, et al. Angiotensin-(1-12)/chymase axis modulates cardiomyocyte L-type calcium currents in rats expressing human angiotensinogen. Int J Cardiol. 2019. Dec 15;297:104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferrario CM, VonCannon J, Ahmad S, et al. Activation of the Human Angiotensin-(1-12)-Chymase Pathway in Rats With Human Angiotensinogen Gene Transcripts. Front Cardiovasc Med. 2019;6:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pulgar VM, Cruz-Diaz N, Westwood BM, et al. Angiotensinogen uptake and stimulation of oxidative stress in human pigment retinal epithelial cells. Peptides. 2022. Feb 18;152:170770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li T, Zhang X, Cheng HJ, et al. Critical role of the chymase/angiotensin-(1-12) axis in modulating cardiomyocyte contractility. Int J Cardiol. 2018. Aug 1;264:137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Urata H, Healy B, Stewart RW, et al. Angiotensin II-forming pathways in normal and failing human hearts. Circ Res. 1990. Apr;66(4):883–90. [DOI] [PubMed] [Google Scholar]
- 58. Urata H, Kinoshita A, Misono KS, et al. Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J Biol Chem. 1990. Dec 25;265(36):22348–57. **Of considerable interest: the study highlights the identification of chymase as the main enzyme converting angiotensin I into angiotensin II in the human left ventricle.
- 59.Park S, Bivona BJ, Kobori H, et al. Major role for ACE-independent intrarenal ANG II formation in type II diabetes. Am J Physiol Renal Physiol. 2010. Jan;298(1):F37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lorenz JN. Chymase: the other ACE? Am J Physiol Renal Physiol. 2010. Jan;298(1):F35–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaltenecker CC, Domenig O, Kopecky C, et al. Critical Role of Neprilysin in Kidney Angiotensin Metabolism. Circ Res. 2020. Aug 14;127(5):593–606. [DOI] [PubMed] [Google Scholar]
- 62. Pavo N, Prausmuller S, Spinka G, et al. Myocardial Angiotensin Metabolism in End-Stage Heart Failure. J Am Coll Cardiol. 2021. Apr 13;77(14):1731–1743. *Of interest: this study highlights the critical importance of tissue chymase, but not ACE for cardiac Ang II generation, and demonstrates that Ang II is metabolized to Ang-(1-7) by prolyl carboxypeptidase but not to ACE2.
- 63.Ferrario CM, Santos RA, Brosnihan KB, et al. A hypothesis regarding the function of angiotensin peptides in the brain. Clin Exp Hypertens A. 1988;10 Suppl 1:107–21. [DOI] [PubMed] [Google Scholar]
- 64.Jessup JA, Trask AJ, Chappell MC, et al. Localization of the novel angiotensin peptide, angiotensin-(1-12), in heart and kidney of hypertensive and normotensive rats. Am J Physiol Heart Circ Physiol. 2008. Jun;294(6):H2614–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferrario CM, VonCannon J, Jiao Y, et al. Cardiac angiotensin-(1-12) expression and systemic hypertension in rats expressing the human angiotensinogen gene. Am J Physiol Heart Circ Physiol. 2016. Apr 15;310(8):H995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Balcells E, Meng QC, Johnson WH Jr., et al. Angiotensin II formation from ACE and chymase in human and animal hearts: methods and species considerations. Am J Physiol. 1997. Oct;273(4):H1769–74. [DOI] [PubMed] [Google Scholar]
- 67.Dell'Italia LJ, Collawn JF, Ferrario CM. Multifunctional Role of Chymase in Acute and Chronic Tissue Injury and Remodeling. Circ Res. 2018. Jan 19;122(2):319–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967. Apr 20;27(2):157–62. [DOI] [PubMed] [Google Scholar]
- 69. Dell’Italia LJ, Husain A. Dissecting the role of chymase in angiotensin II formation and heart and blood vessel diseases. Curr Opin Cardiol. 2002. Jul;17(4):374–9. *Of interest: the study highlights the differential role of chymase and ACE in the cardiac compartments.
- 70.Doggrell SA, Wanstall JC. Vascular chymase: pathophysiological role and therapeutic potential of inhibition. Cardiovascular research. 2004;61(4):653–662. [DOI] [PubMed] [Google Scholar]
- 71.Berglund P, Akula S, Fu Z, et al. Extended Cleavage Specificity of the Rat Vascular Chymase, a Potential Blood Pressure Regulating Enzyme Expressed by Rat Vascular Smooth Muscle Cells. Int J Mol Sci. 2020. Nov 12;21(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chandrasekharan UM, Sanker S, Glynias MJ, et al. Angiotensin II-forming activity in a reconstructed ancestral chymase. Science. 1996. Jan 26;271(5248):502–5. [DOI] [PubMed] [Google Scholar]
- 73.Lutzelschwab C, Pejler G, Aveskogh M, et al. Secretory granule proteases in rat mast cells. Cloning of 10 different serine proteases and a carboxypeptidase A from various rat mast cell populations. J Exp Med. 1997. Jan 6;185(1):13–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kunori Y, Koizumi M, Masegi T, et al. Rodent alpha-chymases are elastase-like proteases. Eur J Biochem. 2002. Dec;269(23):5921–30. [DOI] [PubMed] [Google Scholar]
- 75. Caughey GH. Mast cell proteases as pharmacological targets. Eur J Pharmacol. 2016. May 5;778:44–55. *Of interest: an excellent review of specific protease targets for therapeutic inhibition and choice of topical versus systemic routes of inhibitor administration.
- 76.Guo C, Ju H, Leung D, et al. A novel vascular smooth muscle chymase is upregulated in hypertensive rats. J Clin Invest. 2001. Mar;107(6):703–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kishi K, Jin D, Takai S, et al. Role of chymase-dependent angiotensin II formation in monocrotaline-induced pulmonary hypertensive rats. Pediatr Res. 2006. Jul;60(1):77–82. [DOI] [PubMed] [Google Scholar]
- 78.Wintroub BU, Schechter NB, Lazarus GS, et al. Angiotensin I conversion by human and rat chymotryptic proteinases. J Invest Dermatol. 1984. Nov;83(5):336–9. [DOI] [PubMed] [Google Scholar]
- 79.Pejler G Novel Insight into the in vivo function of mast cell chymase: lessons from knockouts and inhibitors. Journal of Innate Immunity. 2020;12(5):357–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wasse H, Naqvi N, Husain A. Impact of Mast Cell Chymase on Renal Disease Progression. Curr Hypertens Rev. 2012. Feb 1;8(1):15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rawlings ND, Barrett AJ, Thomas PD, et al. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018. Jan 4;46(D1):D624–d632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fritz H Human mucus proteinase inhibitor (human MPI). Human seminal inhibitor I (HUSI-I), antileukoprotease (ALP), secretory leukocyte protease inhibitor (SLPI). Biol Chem Hoppe Seyler. 1988. May;369 Suppl:79–82. [PubMed] [Google Scholar]
- 83.Schick C, Kamachi Y, Bartuski AJ, et al. Squamous cell carcinoma antigen 2 is a novel serpin that inhibits the chymotrypsin-like proteinases cathepsin G and mast cell chymase. J Biol Chem. 1997. Jan 17;272(3):1849–55. [DOI] [PubMed] [Google Scholar]
- 84.Tinel H, Zubov D, Zimmermann K, et al. Abstract 13624: A Novel Chymase Inhibitor BAY 1142524 Reduces Fibrosis and Improves Cardiac Function After Myocardial Infarction in Hamster. Circulation. 2017;136(suppl_1):A13624–A13624. [Google Scholar]
- 85.Trivedi NN, Caughey GH. Mast cell peptidases: chameleons of innate immunity and host defense. Am J Respir Cell Mol Biol. 2010. Mar;42(3):257–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Duengen HD, Kim RJ, Zahger D, et al. 87 Effects of the chymase inhibitor fulacimstat on adverse cardiac remodelling after acute myocardial infarction - Results of the CHIARA MIA 2 trial. European Heart Journal. 2019;40(Supplement_1). **Of considerable interest: the chymase inhibitor -Fulacimstat- was safe and well tolerated in patients with left-ventricular dysfunction after first STEMI.
- 87.Duengen HD, Kim RJ, Zahger D, et al. Effects of the chymase inhibitor fulacimstat on adverse cardiac remodeling after acute myocardial infarction-Results of the Chymase Inhibitor in Adverse Remodeling after Myocardial Infarction (CHIARA MIA) 2 trial. Am Heart J. 2020. Jun;224:129–137. [DOI] [PubMed] [Google Scholar]
- 88.Zheng J, Wei CC, Hase N, et al. Chymase mediates injury and mitochondrial damage in cardiomyocytes during acute ischemia/reperfusion in the dog. PLoS One. 2014;9(4):e94732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Oyamada S, Bianchi C, Takai S, et al. Chymase inhibition reduces infarction and matrix metalloproteinase-9 activation and attenuates inflammation and fibrosis after acute myocardial ischemia/reperfusion. J Pharmacol Exp Ther. 2011. Oct;339(1):143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jin D, Takai S, Yamada M, et al. Impact of chymase inhibitor on cardiac function and survival after myocardial infarction. Cardiovasc Res. 2003. Nov 1;60(2):413–20. [DOI] [PubMed] [Google Scholar]
- 91.Jin D, Takai S, Yamada M, et al. Beneficial effects of cardiac chymase inhibition during the acute phase of myocardial infarction. Life Sci. 2002. Jun 14;71(4):437–46. [DOI] [PubMed] [Google Scholar]
- 92. Wei CC, Hase N, Inoue Y, et al. Mast cell chymase limits the cardiac efficacy of Ang I-converting enzyme inhibitor therapy in rodents. J Clin Invest. 2010. Apr;120(4):1229–39. *Of interest: superior effect of combined chymase and ACE inhibitor in cardiac remodeling.
- 93.Kanemitsu H, Takai S, Tsuneyoshi H, et al. Chymase inhibition prevents cardiac fibrosis and dysfunction after myocardial infarction in rats. Hypertens Res. 2006. Jan;29(1):57–64. [DOI] [PubMed] [Google Scholar]
- 94.Hooshdaran B, Kolpakov MA, Guo X, et al. Dual inhibition of cathepsin G and chymase reduces myocyte death and improves cardiac remodeling after myocardial ischemia reperfusion injury. Basic Res Cardiol. 2017. Sep 14;112(6):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jin D, Takai S, Nonaka Y, et al. A Chymase Inhibitory RNA Aptamer Improves Cardiac Function and Survival after Myocardial Infarction. Mol Ther Nucleic Acids. 2019. Mar 1;14:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Houde M, Schwertani A, Touil H, et al. Mouse Mast Cell Protease 4 Deletion Protects Heart Function and Survival After Permanent Myocardial Infarction. Front Pharmacol. 2018;9:868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang Y, Liu CL, Fang W, et al. Deficiency of mouse mast cell protease 4 mitigates cardiac dysfunctions in mice after myocardium infarction. Biochim Biophys Acta Mol Basis Dis. 2019. Jun 1;1865(6):1170–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tejada T, Tan L, Torres RA, et al. IGF-1 degradation by mouse mast cell protease 4 promotes cell death and adverse cardiac remodeling days after a myocardial infarction. Proc Natl Acad Sci U S A. 2016. Jun 21;113(25):6949–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Butts B, Ahmed MI, Bajaj NS, et al. Reduced Left Atrial Emptying Fraction and Chymase Activation in Pathophysiology of Primary Mitral Regurgitation. JACC Basic Transl Sci. 2020. Feb;5(2):109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Butts B, Goeddel LA, George DJ, et al. Increased Inflammation in Pericardial Fluid Persists 48 Hours After Cardiac Surgery. Circulation. 2017. Dec 5;136(23):2284–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Okamura K, Okuda T, Shirai K, et al. Positive correlation between blood pressure or heart rate and chymase-dependent angiotensin II-forming activity in circulating mononuclear leukocytes measured by new ELISA. Clin Exp Hypertens. 2018;40(2):112–117. [DOI] [PubMed] [Google Scholar]
- 102.Matsumoto T, Wada A, Tsutamoto T, et al. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation. 2003. May 27;107(20):2555–8. [DOI] [PubMed] [Google Scholar]
- 103.Pat B, Chen Y, Killingsworth C, et al. Chymase inhibition prevents fibronectin and myofibrillar loss and improves cardiomyocyte function and LV torsion angle in dogs with isolated mitral regurgitation. Circulation. 2010. Oct 12;122(15):1488–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Takato H, Yasui M, Ichikawa Y, et al. The specific chymase inhibitor TY-51469 suppresses the accumulation of neutrophils in the lung and reduces silica-induced pulmonary fibrosis in mice. Exp Lung Res. 2011. Mar;37(2):101–8. [DOI] [PubMed] [Google Scholar]
- 105.Tomimori Y, Muto T, Saito K, et al. Involvement of mast cell chymase in bleomycin-induced pulmonary fibrosis in mice. Eur J Pharmacol. 2003. Oct 8;478(2-3):179–85. [DOI] [PubMed] [Google Scholar]
- 106.Coppini R, Santini L, Palandri C, et al. Pharmacological Inhibition of Serine Proteases to Reduce Cardiac Inflammation and Fibrosis in Atrial Fibrillation. Front Pharmacol. 2019;10:1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang H, Varagic J, Nagata S, et al. Atrial angiotensin-(1-12)/chymase expression data in patient of heart diseases. Data Brief. 2020. Aug;31:105744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Powell PC, Wei CC, Fu L, et al. Chymase uptake by cardiomyocytes results in myosin degradation in cardiac volume overload. Heliyon. 2019. Apr;5(4):e01397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang H, Varagic J, Nagata S, et al. Differential Expression of the Angiotensin-(1-12)/Chymase Axis in Human Atrial Tissue. J Surg Res. 2020. Sep;253:173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Devarajan S, Yahiro E, Uehara Y, et al. Depressor effect of chymase inhibitor in mice with high salt-induced moderate hypertension. Am J Physiol Heart Circ Physiol. 2015. Dec 1;309(11):H1987–96. [DOI] [PubMed] [Google Scholar]
- 111.Okamura K, Kuroda R, Nagata K, et al. Prospective single-arm observational study of human chymase inhibitor Polygonum hydropiper L in subjects with hypertension. Clinical and Experimental Hypertension. 2019. 2019/November/17;41(8):717–725. [DOI] [PubMed] [Google Scholar]
- 112.Ansary TM, Urushihara M, Fujisawa Y, et al. Effects of the selective chymase inhibitor TEI-F00806 on the intrarenal renin-angiotensin system in salt-treated angiotensin I-infused hypertensive mice. Exp Physiol. 2018. Nov;103(11):1524–1531. [DOI] [PubMed] [Google Scholar]
- 113.Takai S, Jin D, Chen H, et al. Chymase inhibition improves vascular dysfunction and survival in stroke-prone spontaneously hypertensive rats. J Hypertens. 2014. Aug;32(8):1637–48; discussion 1649. [DOI] [PubMed] [Google Scholar]
- 114. Bivona BJ, Takai S, Seth DM, et al. Chymase inhibition retards albuminuria in type 2 diabetes. Physiol Rep. 2019. Dec;7(24):e14302. *Of interest: the study shows the antiproteinuric effect of chymase inhibition in diabetic mice.
- 115.Rossing P, Strand J, Avogaro A, et al. Effects of the chymase inhibitor fulacimstat in diabetic kidney disease—results from the CADA DIA trial. Nephrology Dialysis Transplantation. 2020. [DOI] [PubMed] [Google Scholar]
- 116.Huang XR, Chen WY, Truong LD, et al. Chymase is upregulated in diabetic nephropathy: implications for an alternative pathway of angiotensin II-mediated diabetic renal and vascular disease. J Am Soc Nephrol. 2003. Jul;14(7):1738–47. [DOI] [PubMed] [Google Scholar]
- 117.Cristovam PC, Arnoni CP, de Andrade MC, et al. ACE-dependent and chymase-dependent angiotensin II generation in normal and glucose-stimulated human mesangial cells. Exp Biol Med (Maywood). 2008. Aug;233(8):1035–43. [DOI] [PubMed] [Google Scholar]
- 118.McPherson EA, Luo Z, Brown RA, et al. Chymase-like angiotensin II-generating activity in end-stage human autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2004. Feb;15(2):493–500. [DOI] [PubMed] [Google Scholar]
- 119.de Garavilla L, Greco MN, Sukumar N, et al. A novel, potent dual inhibitor of the leukocyte proteases cathepsin G and chymase: molecular mechanisms and anti-inflammatory activity in vivo. J Biol Chem. 2005. May 6;280(18):18001–7. [DOI] [PubMed] [Google Scholar]
- 120.Dungen HD, Kober L, Nodari S, et al. Safety and Tolerability of the Chymase Inhibitor Fulacimstat in Patients With Left Ventricular Dysfunction After Myocardial Infarction-Results of the CHIARA MIA 1 Trial. Clin Pharmacol Drug Dev. 2019. Oct;8(7):942–951. [DOI] [PubMed] [Google Scholar]
- 121.Zhang M, Huang W, Bai J, et al. Chymase inhibition protects diabetic rats from renal lesions. Mol Med Rep. 2016. Jul;14(1):121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lee JH, Kim JW, Ko NY, et al. Mast cell-mediated allergic response is suppressed by Sophorae flos: inhibition of SRC-family kinase. Exp Biol Med (Maywood). 2008. Oct;233(10):1271–9. [DOI] [PubMed] [Google Scholar]
- 123.Dispenza MC. The Use of Bruton's Tyrosine Kinase Inhibitors to Treat Allergic Disorders. Curr Treat Options Allergy. 2021;8(3):261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Louis RE, Radermecker MF. Substance P-induced histamine release from human basophils, skin and lung fragments: effect of nedocromil sodium and theophylline. Int Arch Allergy Appl Immunol. 1990;92(4):329–33. [DOI] [PubMed] [Google Scholar]
- 125.Wei L, Wang J, Zhang X, et al. Discovery of 2H-Chromen-2-one Derivatives as G Protein-Coupled Receptor-35 Agonists. J Med Chem. 2017. Jan 12;60(1):362–372. [DOI] [PubMed] [Google Scholar]
- 126.Theoharides TC. Potential association of mast cells with coronavirus disease 2019. Ann Allergy Asthma Immunol. 2021. Mar;126(3):217–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Matsumoto C, Hayashi T, Kitada K, et al. Chymase plays an important role in left ventricular remodeling induced by intermittent hypoxia in mice. Hypertension. 2009. Jul;54(1):164–71. [DOI] [PubMed] [Google Scholar]
- 128.Ishida K, Takai S, Murano M, et al. Role of chymase-dependent matrix metalloproteinase-9 activation in mice with dextran sodium sulfate-induced colitis. J Pharmacol Exp Ther. 2008. Feb;324(2):422–6. [DOI] [PubMed] [Google Scholar]
- 129.Inoue N, Muramatsu M, Jin D, et al. Effects of chymase inhibitor on angiotensin II-induced abdominal aortic aneurysm development in apolipoprotein E-deficient mice. Atherosclerosis. 2009. Jun;204(2):359–64. [DOI] [PubMed] [Google Scholar]
- 130.Palaniyandi SS, Nagai Y, Watanabe K, et al. Chymase inhibition reduces the progression to heart failure after autoimmune myocarditis in rats. Exp Biol Med (Maywood). 2007. Oct;232(9):1213–21. [DOI] [PubMed] [Google Scholar]
- 131.Kakimoto K, Takai S, Murano M, et al. Significance of chymase-dependent matrix metalloproteinase-9 activation on indomethacin-induced small intestinal damages in rats. J Pharmacol Exp Ther. 2010. Feb;332(2):684–9. [DOI] [PubMed] [Google Scholar]
- 132. Imai Y, Takai S, Jin D, et al. Chymase inhibition attenuates lipopolysaccharide/ d-galactosamine-induced acute liver failure in hamsters. Pharmacology. 2014;93(1-2):47–56. *Of interest: study suggests immuno-protective actions of chymase inhibitors in rodents.
- 133.Komeda K, Takai S, Jin D, et al. Chymase inhibition attenuates tetrachloride-induced liver fibrosis in hamsters. Hepatol Res. 2010. Aug;40(8):832–40. [DOI] [PubMed] [Google Scholar]
- 134.Tsunemi K, Takai S, Nishimoto M, et al. A specific chymase inhibitor, 2-(5-formylamino-6-oxo-2-phenyl-1,6-dihydropyrimidine-1-yl)-N-[[3,4-dioxo-1-phenyl-7-(2-pyridyloxy)]-2-heptyl]acetamide (NK3201), suppresses development of abdominal aortic aneurysm in hamsters. J Pharmacol Exp Ther. 2004. Jun;309(3):879–83. [DOI] [PubMed] [Google Scholar]
- 135.Takai S, Jin D, Ohzu M, et al. Chymase inhibition provides pancreatic islet protection in hamsters with streptozotocin-induced diabetes. J Pharmacol Sci. 2009. Aug;110(4):459–65. [DOI] [PubMed] [Google Scholar]
- 136.Maeda Y, Inoguchi T, Takei R, et al. Chymase inhibition prevents myocardial fibrosis through the attenuation of NOX4-associated oxidative stress in diabetic hamsters. J Diabetes Investig. 2012. Aug 20;3(4):354–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Maeda Y, Inoguchi T, Takei R, et al. Inhibition of chymase protects against diabetes-induced oxidative stress and renal dysfunction in hamsters. Am J Physiol Renal Physiol. 2010. Dec;299(6):F1328–38. [DOI] [PubMed] [Google Scholar]
- 138.Kosanovic D, Luitel H, Dahal BK, et al. Chymase: a multifunctional player in pulmonary hypertension associated with lung fibrosis. Eur Respir J. 2015. Oct;46(4):1084–94. [DOI] [PubMed] [Google Scholar]
- 139.Wang T, Han SX, Zhang SF, et al. Role of chymase in cigarette smoke-induced pulmonary artery remodeling and pulmonary hypertension in hamsters. Respir Res. 2010. Mar 31;11(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hoshino F, Urata H, Inoue Y, et al. Chymase inhibitor improves survival in hamsters with myocardial infarction. J Cardiovasc Pharmacol. 2003. Jan;41 Suppl 1:S11–8. [PubMed] [Google Scholar]
- 141.Fan YY, Nishiyama A, Fujisawa Y, et al. Contribution of chymase-dependent angiotensin II formation to the progression of tubulointerstitial fibrosis in obstructed kidneys in hamsters. J Pharmacol Sci. 2009. Sep;111(1):82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Uehara Y, Urata H, Ideishi M, et al. Chymase inhibition suppresses high-cholesterol diet-induced lipid accumulation in the hamster aorta. Cardiovasc Res. 2002. Sep;55(4):870–6. [DOI] [PubMed] [Google Scholar]
- 143. Okamura K, Kuroda R, Nagata K, et al. Prospective single-arm observational study of human chymase inhibitor Polygonum hydropiper L in subjects with hypertension. Clin Exp Hypertens. 2019;41(8):717–725. *Of interest: Polygonum hydropiper L (Polygonum), a chymase inhibitor, shows beneficial antihypertensive effects in salt-dependent hypertensive subjects.
- 144.Takai S, Jin D, Nishimoto M, et al. Oral administration of a specific chymase inhibitor, NK3201, inhibits vascular proliferation in grafted vein. Life Sci. 2001;69(15):1725–1732. [DOI] [PubMed] [Google Scholar]
- 145.Takai S, Jin D, Sakaguchi M, et al. An orally active chymase inhibitor, BCEAB, suppresses heart chymase activity in the hamster. Jpn J Pharmacol. 2001;86(1):124–6. [DOI] [PubMed] [Google Scholar]