

Abstract
Type 1 diabetes (T1D) is widely considered to result from the autoimmune destruction of insulin-producing β-cells. This concept has been a central tenet for decades of attempts seeking to decipher the disorder’s pathogenesis and prevent/reverse the disease. Recently, this and many other disease related notions have come under increasing question, particularly given knowledge gains from analyses of human T1D pancreas. Perhaps most crucial are findings suggesting that a collective of cellular constituents — immune, endocrine, and exocrine in origin — mechanistically coalesce to facilitate T1D. This Review considers these emerging concepts, from basic science to clinical research, and identifies several key remaining knowledge voids.
Keywords: Type 1 diabetes, islet of Langerhans, immune cells, exocrine pancreas, insulin, autoimmunity, insulitis, inflammation, endocrinology
The Pathological Basis of Type 1 Diabetes
From a historical perspective, the autoimmune basis for type 1 diabetes (T1D) has largely been centered, in terms of its pathogenic nature, on three seminal observations including an inflammatory infiltrate of pancreatic islets (i.e., insulitis), genetic susceptibility associated with the Major Histocompatibility Complex (MHC), and autoantibodies reactive against β cell antigens1. These concepts formed key components for a natural history model of T1D, developed in the mid-1980s2, that has served as a multi-generational roadmap for efforts seeking to understand the disorder’s pathogenesis, determine disease risk, and uncover methods for preventing β cell loss and preserving endocrine function. While subject to modification over the years, the benefits of this model have been numerous, including the ability to stage pre-diabetes (i.e., stage 1 and 2 disease) for therapeutic interventions based on risk for stage 3 T1D diagnosis (Figure 1), developing predictive biomarkers, guiding studies examining environmental constituents influencing the disorder’s development, and more3–8. However, despite such knowledge advances, a major therapeutic shortcoming remains: pragmatic challenges allowing for preventing T1D in those at increased disease risk or providing for stable, long-term preservation of β cell function in those with recently diagnosed T1D. Indeed, to date only one agent, an anti-CD3 antibody (i.e., teplizumab), has received regulatory approval in the United States for attenuating loss of β cell function and delaying progression to stage 3 T1D by approximately two years in those at the highest risk for the disorder9,10. Further advances towards this end are needed and must be based, optimally, on an improved understanding of the disorder’s pathogenesis.
Figure 1. A re-depiction of the current “staging” model for type 1 diabetes.
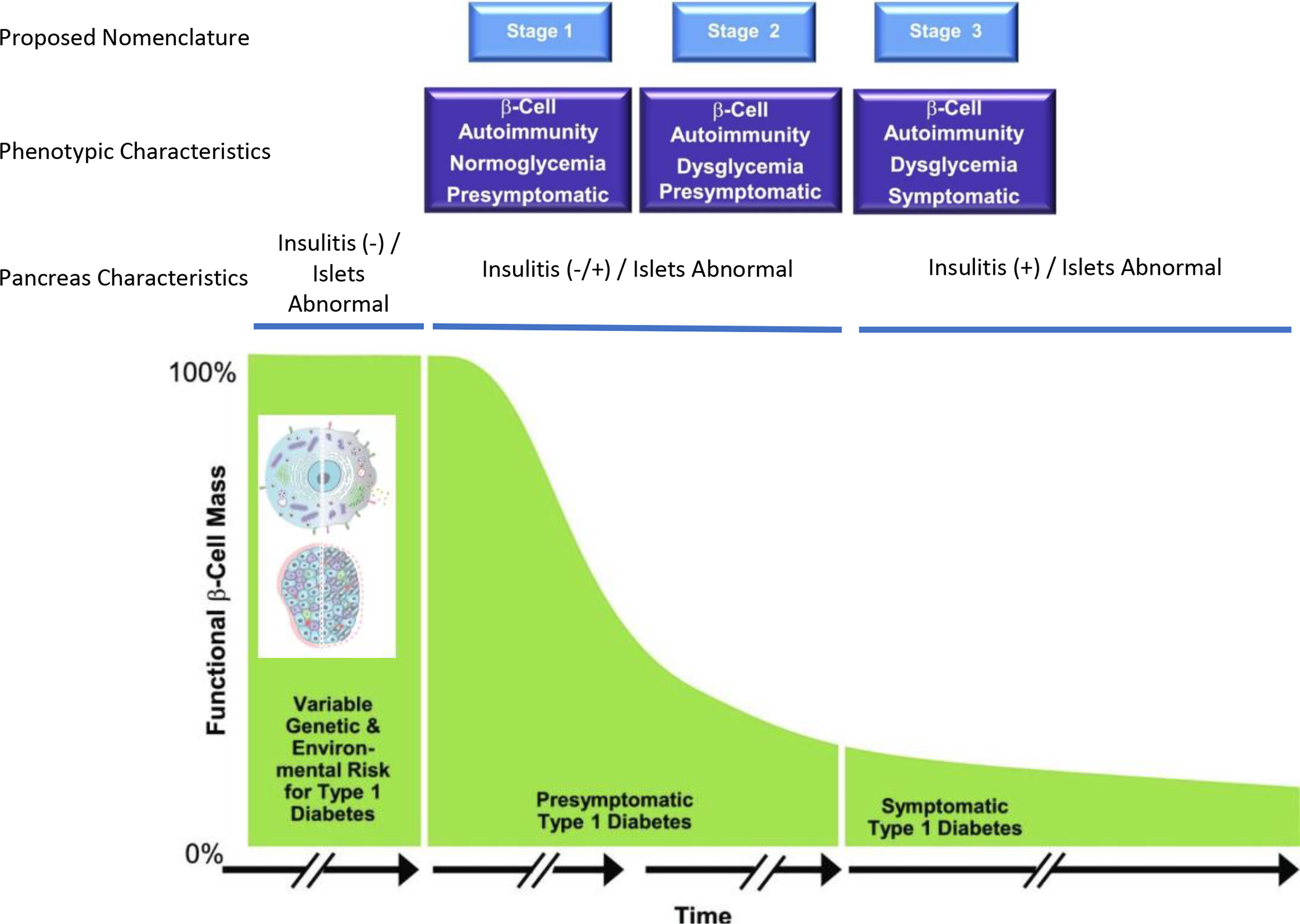
Following decades of natural history studies of type 1 diabetes (i.e., monitoring the period prior to and after traditional disease diagnosis), a consensus group within the community of type 1 diabetes developed a model whose intention was to better define risk for disease development. This model was largely developed based on analysis of both immunological (autoantibody type, number) and metabolic markers (response to glucose stimulation). Other considerations not strictly considered in this published model are those of having a relative with the disease, relative relationship, age, and more. Here we depict emerging concepts related to recent studies of pancreatic pathology to improve the staging of type 1 diabetes, as described herein. Adapted from Insel, et al.3 with insets from Figures 2–3.
Effectively addressing this therapeutic challenge for T1D has not been for lack of effort in performing clinical trials attempting to meet these goals. Indeed, since the inception of the cyclosporin trials in the early 1980s, hundreds of trials, both small and large, have attempted to avert autoimmunity and preserve existing β cell function11–13. Multiple factors contributed to limited therapeutic successes, including limited knowledge regarding mechanisms of β cell death, an inability to safely biopsy the human pancreas, a failure to understand the role for environment in T1D, the absence of a means to image pancreatic islets in vivo, issues of therapeutic equipoise, defining the relationship between the disorder’s polygenic nature with specific mechanistic outcomes, and the relative rarity of the disease in the general population, among others14–17. An additional and likely major contributor includes the “Streetlight Effect” for T1D18, a concept that posits advances in T1D research have, to a certain degree, been restrained by the failure to address truly novel if not controversial hypotheses and perhaps, more importantly, accept pedagogical dogma with limited experimental questioning.
Thankfully, this situation appears to be changing as many long-standing dogmas in T1D have recently come under question and are becoming subject to intellectual change (Table 1), creating a situation where multiple aspects of research involving this disease, including clinical research and trials, will benefit. Examples include, but are not limited to, improvements in diagnostic testing, seeing an appreciation for disease heterogeneity (albeit still somewhat an undefined notion in T1D)19–21, and movement toward precision-based therapies (e.g., based on age, HLA-DR diplotype, etc.)22–25, which ultimately will require definitive data linking disease phenotypes with treatment response. Such knowledge gains have emanated from a series of sources including large-scale efforts involving longitudinal analysis of blood samples from living subjects with T1D and those at varying levels of risk for the disease, as well as genome wide association studies (GWAS), implementation of emerging technologies (e.g., continuous glucose monitoring, single cell analyses), and identification of improved disease biomarkers7. However, among the most transformative findings challenging conventional thought regarding the pathogenesis of T1D have emerged from studies of the human pancreas from persons with this disease, non-diabetic islet autoantibody positive subjects at increased risk for the disorder (including those with two or more autoantibodies indicative of early-stage disease), and those without diabetes26–28.
Table 1.
Dogmas or long-standing beliefs that have come under change, or seen correction in recent years, largely if not significantly due to studies of the human pancreas.
| Long-Standing Dogma | Reality | References |
|---|---|---|
| T1D occurs when 85–95% of β-cells are destroyed | The degree of β-cell loss resulting in symptomatic disease is quite variable (i.e., 40–95% loss). | 33,104,215 |
| Individuals with longstanding T1D are devoid of insulin positive β-cells | Proinsulin and insulin expression can be detected within scattered single cells as well as small clusters of islets in pancreas from donors with longstanding T1D. | 33,104,121,138,215–217 |
| T1D represents a singular disease | T1D likely represents a collective of distinct disorders (i.e., endotypes) with commonalities of insulinopenia and loss of β-cells. Heterogeneity exists in the degree of β-cell cell loss at onset, rate of insulitis, age and onset, and more. | 22,77,218,219 |
| β-cells are subject to replication | In those without T1D, β-cell replication rates are highest in the neonatal period and decline exponentially throughout childhood with little to no replication observed in human adults. Limited evidence exists that such replication occurs in settings of T1D. | 144,220–222 |
| T1D is an autoimmune disease, with limited pathogenic contributions of β-cells | An extensive number of β-cell alterations preceed T1D onset and may contribute toward autoimmunity (e.g., ER stress, oxidative stress, protein misfolding, neoepitope formation, impaired autophagy, aberrant prohormone processing, senescence, and increased expression of MHC-I and PD-L1). | 15,35,53,74,87,99,104,111,113,115,116,120,216,223–237 |
| The exocrine pancreas is unaffected by T1D | Reduced pancreas organ mass and volume, as well as reduced exocrine pancreatic enzyme levels in serum, are characteristics of recent-onset and pre- T1D. T1D donors also exhibit reduced acinar tissue area and increase acinar cell density. | 40,104,175,178,180,183,187,188,238,239 |
| Insulitis is a chronic and predominant feature in the natural history of T1D | Insulitis is restricted to insulin containing islets and has been primarily reported in individuals with recent-onset T1D, with rare cases of insulitis observed in pancreata from persons with single autoantibody positivity, with slightly more in those with multiple islet autoantibodies. | 33,34,40,43,44,89,104,106,215,240,241 |
| T1D and T2D are distinct in terms of pancreatic features | Reductions in islet number, pancreas size, β-cell mass, and β-cell function have been observed in both T1D and T2D. Genetic analyses have provided new insight into the contribution of specific polymorphisms, including how the presence of variants associated with T2D can influence the phenotype of donors with T1D. | 27,192–194 |
| Insulitis, as reflected in NOD mice, is representative of human disease | Compared to NOD mice, human insulitis is far less severe (fewer immune cells per inflammed islet), less pervasive (fewer islets affected) and occurs in a patchy “mosaic” pattern | 37,40,89,111,242–244 |
This Review article considers how studies of the human pancreas, largely conducted over the past 15 years, have resulted in dramatic changes with respect to our understanding of mechanisms underlying T1D pathogenesis, the disorder’s heterogeneity, and identification of cellular contributors to disease formation. Importantly, revelations garnered from these studies have also led to a major rethinking of cause versus effect in terms of the damage observed throughout the T1D pancreas and apparent pathogenic contributions of cells beyond those purely autoimmune in nature, providing for a more expansive view in terms of the roles that β cells, islets, and the exocrine pancreas provide toward defining the development of this disorder. It seems evident that an improved understanding of such notions will have major implications for efforts to improve disease prediction and prevention, alongside of those seeking to preserve metabolic function in individuals with T1D29–32.
T1D is an Autoimmune Disorder
Insulitis
From a perspective of pathology, T1D is characterized by a selective loss of β cells within the pancreatic islets33–36. While exceptions exist, the majority of literature suggests that insulitis is more predominant in individuals with youth onset T1D (≤14 years of age) versus those with an older age at onset37. This finding is consistent with the notion that a more vigorous autoimmune response occurs when disease develops in young children compared to adults38,39. However, in contrast to the NOD mouse model of the disease, the presence of insulitis in human T1D is more limited quantitatively33, with an expert panel establishing a standardized histological definition for this lesion as three islets, each containing more than 15 CD45+ cells, within a pancreas34. This standard has, however, recently come under question alongside a call for its replacement using a “30–30 rule,” whereby the presence of 30 white blood cells within a 30mm region defines insulitis40. Hence, while acknowledging that islet inflammation in T1D remains a central tenant, its definition continues to be a subject of debate.
Beyond simply the definition of insulitis is the finding that its occurrence (relative to the NOD mouse model of T1D) prior to disease diagnosis in humans is very limited. Prior to symptomatic onset, insulitis is rare if not totally absent in single glutamic acid decarboxylase autoantibody (GADA)-positive subjects and, surprisingly, has only been observed in a subset of multiple autoantibody-positive subjects — persons at a high 10-year risk of T1D33,41–44. This observation, often overlooked or underappreciated, should be given consideration for clinical interventions that are based on targeting insulitis in the early (i.e., pre-symptomatic) stages of the disease versus therapies whose presumed mechanism involves immunological activities acting in the periphery.
The immunophenotype of insulitic lesions shows a predominance of CD8+ T lymphocytes and macrophages (CD68+), but cells of other phenotypes are also observed (e.g., CD4+, CD20+)45,46. Recent studies indicate that younger age of onset is associated with greater frequencies of CD20+ B cells, CD45+ cells, and CD8+ T cells in insulitis lesions, alongside fewer insulin-positive islets46,47. With respect to B-lymphocytes, these efforts reported a CD20Hi profile in the insulitis lesion of donors diagnosed before 7 years of age versus a CD20Lo profile in the those diagnosed after 13 years of age. As a result, it has been suggested that the cellular composition of insulitis or, more specifically, the quantitative presence of insulitis, serves as one form of an “endotype” of T1D, a concept to explain potential diversity in T1D disease pathogenesis22. This said, not all studies have supported the notion of a predominance of B-lymphocytes in individuals with an early onset of disease33, and limited progress has been made in forming a consensus for defining T1D endotypes in the research and care community20. This information void regarding age-associated insulitis could be tested for association with circulating biomarkers (e.g., TCR and BCR repertoire, comprehensive immunophenotyping, serological markers) measurable in clinical cohorts and organ donors, allowing for specific hypothesis testing based on the observed differences in islets.
An abundance of studies suggest that islet-invading T cells are largely directed against known β cell antigens (e.g., insulin, proinsulin, GAD), post-translationally modified peptides, viral antigens, and so-called hybrid peptides48–58. CD8+ T cells expressing T cell receptors (TCRs) that bind MHC class I tetramers loaded with the β cell autoantigens resident to insulitis lesions are also detectable in the peripheral blood of individuals with T1D59,60. Toward this latter notion, a series of investigations have undertaken the task of evaluating TCR utilization within the insulitis lesion and of addressing clone publicity (i.e., shared amongst those with T1D versus unique to an individual), abundancy, and disease-specificity61. Notably, most such efforts to date have suggested a bias in terms of receptor utilization55. Hope exists that these TCR datasets will eventually form novel biomarkers of T1D development as well as provide an approach for optimizing therapeutic intervention61,62. For example, such discoveries have facilitated exciting functional studies using primary human T cells engineered to express an islet antigen-specific TCR or chimeric antigen receptor (CAR) to model T1D63–68. Finally, the frequency of CD8 T cells directed against β cell antigens in the pancreas uniquely discriminates subjects with T1D versus those without the disease, unlike what is often observed in analysis of peripheral blood69. Taken as a collective, this body of information, regarding both peripheral and islet invading T cells, is highly supportive for the notion that analysis of the human pancreas in T1D is of importance.
Potential pathogenic “drivers” of T cell infiltration into pancreatic islets are quite numerous (e.g., chemokines, β cell abnormalities, extracellular matrix [ECM] expression)70,71. However, among the most often discussed, insulin-containing islets of T1D subjects as well as single and double autoantibody-positive non-diabetic subjects overexpress class I HLA, purportedly unmasking β cells to immune-mediated killing35,72. To be clear, this MHC class I hyperexpression is not specific for islet β cells and, as discussed later, the exocrine pancreas of those with T1D also shows higher levels of exocrine immune infiltration73. Finally, while its pathogenic importance remains unclear, class II HLA upregulation was also recently reported on β cells within insulin containing islets from T1D donors74.
The Natural History of Insulitis in T1D
The non–insulin-producing cells of the islets (i.e., α-, δ-, ε-, and pancreatic polypeptide cells) persist in patients with long-standing T1D, with these remaining islets, lacking insulitis and β cells, termed “pseudoatrophic”34. This information sets the stage for another remarkable feature of the T1D pancreas, namely, heterogeneity of islet lesions (Figure 2). Histopathologic examination of the pancreas in settings of multi-autoantibody positivity without T1D, recent-onset disease, or established (long duration) T1D note that within the same tissue section, a “normal” islet with no immune infiltrate can coexist with an islet containing β cells with intense infiltration as well as a pseudoatrophic islet that has no infiltrate33,75,76. Such heterogeneity of lesions may underlie the variable natural history of T1D, whereby studies suggest the time period between initial β cell autoimmunity and overt symptoms can be one of months, years, or even decades41,77. Interestingly and as mentioned previously, contrary to studies of the NOD mouse model of T1D, analysis of pancreata from non-diabetic subjects positive for one or multiple islet autoantibodies often show limited to no insulitis43,78,79. Specifically, the presence of insulitis using the aforementioned criteria33 is extremely rare in single GADA-positive patients whereas this lesion is observed in approximately one-half of those with multiple autoantibodies. Such information is consistent with data from longitudinally followed cohorts and the staging of T1D80. Detection of insulitis in multiple autoantibody positive pancreata may be further impacted by the cross-sectional nature of these studies. This finding has been considered by some to support the notion that T1D represents a relapsing/remitting disease, with waves of immunologic destruction81–83. However, this concept remains speculative, in part, due to the inability to perform pancreatic biopsy, lack of in vivo imaging, and absence of a circulating biomarker of insulitis.
Figure 2. Histopathologic progression of type 1 diabetes within the human pancreatic islet.
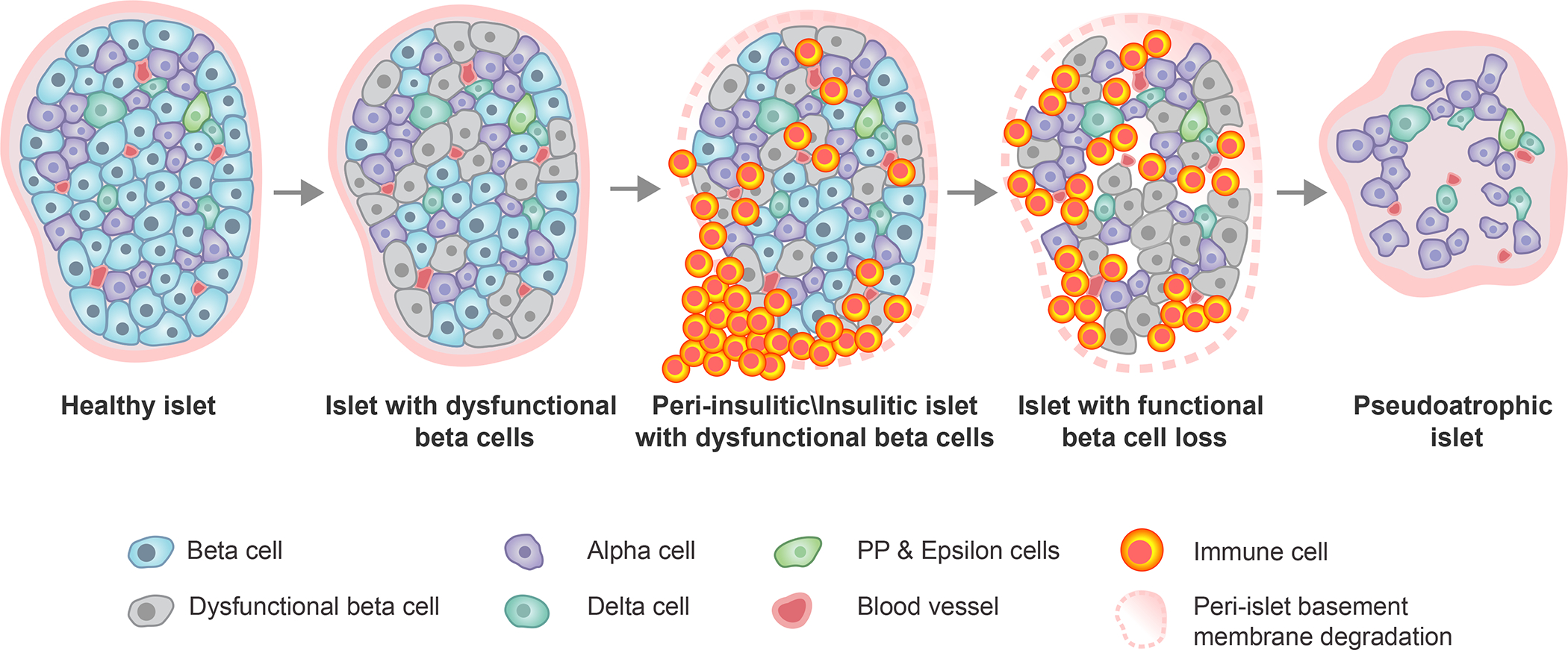
Within an initially healthy islet, a portion of β-cells develop phenotypic and functional impairments. This precipitates immune cell infiltration around (peri-insulitic) and within (insulitic) the islet, coupled with degradation of the peri-islet basement membrane. Following the immune-mediated destruction of functional β-cells, the immune cells exit leaving behind an irregularly shaped, insulin-negative, pseudoatrophic islet characterized by presence of the remaining endocrine cell types (alpha, delta, PP, epsilon), reduced vessel diameter, increased microvascular density, and restored peri-islet basement membrane integrity. PP, pancreatic polypeptide.
Mechanisms of Immune Mediated β Cell Death
Whereas the exact mechanisms of β cell death remain subject to debate, considerable enthusiasm exists for the notion that certain cytokines (e.g., interleukin 1 beta [IL-1β], tumor necrosis factor [TNF], type 1 interferons (T1-IFN)) and CD8+ cytotoxic T lymphocytes are likely to be major contributors to β cell destruction84. Indeed, that T1-IFN potentiates CD8+ T cell cytotoxicity as well as HLA class I hyperexpression on β cells representing a “catastrophic feature” of the T1D pancreas is a popular notion67,85. However, to be clear, HLA class I hyperexpression is not limited to β cells and has been observed in other islet endocrine cells as well as additional pancreatic constituents. With this, the question becomes one of why only β cells would be subject to destruction. One can speculate that specificity of autoimmunity for β cell-specific antigens, increased sensitivity of β cells to inflammatory based insults or metabolic stress, and/or an ill-defined contribution of environmental agent(s) may underlie such differences. The demonstration that human gene-edited pseudoislets lacking HLA class I/II can escape rejection and autoimmunity in a humanized mouse emphasizes the potential catastrophic role of HLA expression on β cells86. Additionally, analysis of pancreata from multi-autoantibody-positive and recent-onset T1D donors note that β cells express TET2, which has been suggested to augment inflammation and autoimmune killing87. T follicular regulatory cells (Tfr) are reduced in frequency within the pancreas draining lymph nodes and spleen, but not blood, of T1D donors88, and tertiary lymphoid organs (TLOs) were recently detected in the T1D pancreas89 — TLO presence was most commonly associated with residual insulin containing islets and younger onset age89. CD8+ T cells specific for the islet antigen ZnT8 are also reportedly enriched in the pancreas but not blood of T1D donors, altogether supporting local defects in immunoregulation as potentiating cytotoxic β cell killing69.
In contrast, several pathways active within the human T1D pancreas are suggested to represent attempts to constrain the autoimmune destruction of islets during T1D. For example and perhaps counterintuitively to the aforementioned notion, exposure to the proinflammatory cytokine γ-interferon (IFN-γ) during activation reduces cytotoxic β cell killing by CD8+ T cells engineered to express an islet antigen-specific TCR66. In situ, IFN-γ and IFN-α drive programmed death ligand-1 (PD-L1) expression on β cells in order to limit T cell function during T1D progression90,91. Indeed, the PD-1/PD-L1 pathway has been demonstrated as a key checkpoint for T1D development in murine models of the disease and in cancer patients with T1D-associated HLA genotypes. Specifically, treatment involving a PD-1 checkpoint inhibitor has been shown to precipitate the development of islet autoantibodies and T1D onset92. Studies of residual β cells in T1D donors demonstrate the expression of PD-L1 on the surface of these β cells, suggesting that these residual cells may have escaped T cell-mediated killing91. The signal regulatory protein gamma (SIRPg)/CD47 co-inhibitory pathway has raised similar interest where signaling is speculated to potentiate protection of β cells93. Additionally, murine studies have demonstrated MERTK expressing mononuclear phagocytes to regulate T cell activation in both T1D islets and melanoma tumors94. With this, increased numbers of MERTK+ cells observed within insulin containing islets from pancreata with longstanding T1D may represent an intrinsic mechanism to preserve residual β cells94.
T1D is a Disease of Islets and β Cells
Despite clear evidence for an autoimmune etiology in T1D, a growing minority in the T1D research and care community have increasingly questioned the sole or primary role for this concept95, resulting in an expanding debate regarding key contributors to the disorder’s pathogenesis96. This controversy has largely occurred based on a mounting appreciation for an active participation of β cells and islets in human T1D beyond processes of autoimmunity.
The concept that β cells themselves influence disease pathogenesis and participate in their own demise is not new, having been proposed in the mid-1980s by a seminal publication questioning whether β cell loss in T1D occurs via a “homicide” or “suicide” like mechanism97. The rationale underlying this reemerging notion is largely tied to recent morphologic and functional studies of rodent and human islets, both within and outside settings of inflammation. These efforts suggest a host of metabolic and cellular aberrations occur in the natural history of T1D involving β cells98 (Figure 3A), as well as other islet cell types (Figure 3B). Among the many cellular aberrations, we would highlight stress and dysfunction of various organelles within β-cells (as detailed below), β-cell senescence and its release of senescence-related secretory cytokines that attract monocytes99, aberrant accumulation of immunogenic GAD65 in β-cells100, the loss of regulated glucagon secretion in α cells101, as well as alterations in cells that maintain the integrity of islet ECM and its isolation from invading immune cells71,102. It is important to note that many of these abnormalities are observed in single GADA-positive patients who do not have detectable insulitis. As we will discuss below, research efforts need to be directed at further understanding the dynamic, interactive, and complex dance between β cells, islets, exocrine tissue and the immune system in T1D development. Also, while the potential impact of such abnormalities on disease formation remains unclear, it is conceivable that these islet-immune cell interactions contribute toward the heterogeneous progression of human T1D23,103. Indeed, as will be discussed throughout this article, we believe that a contemporaneous view of this question would suggest both mechanisms (i.e., β cell homicide and suicide) are crucial to T1D development.
Figure 3. Alterations associated with type 1 diabetes at the β-cell and whole islet level.
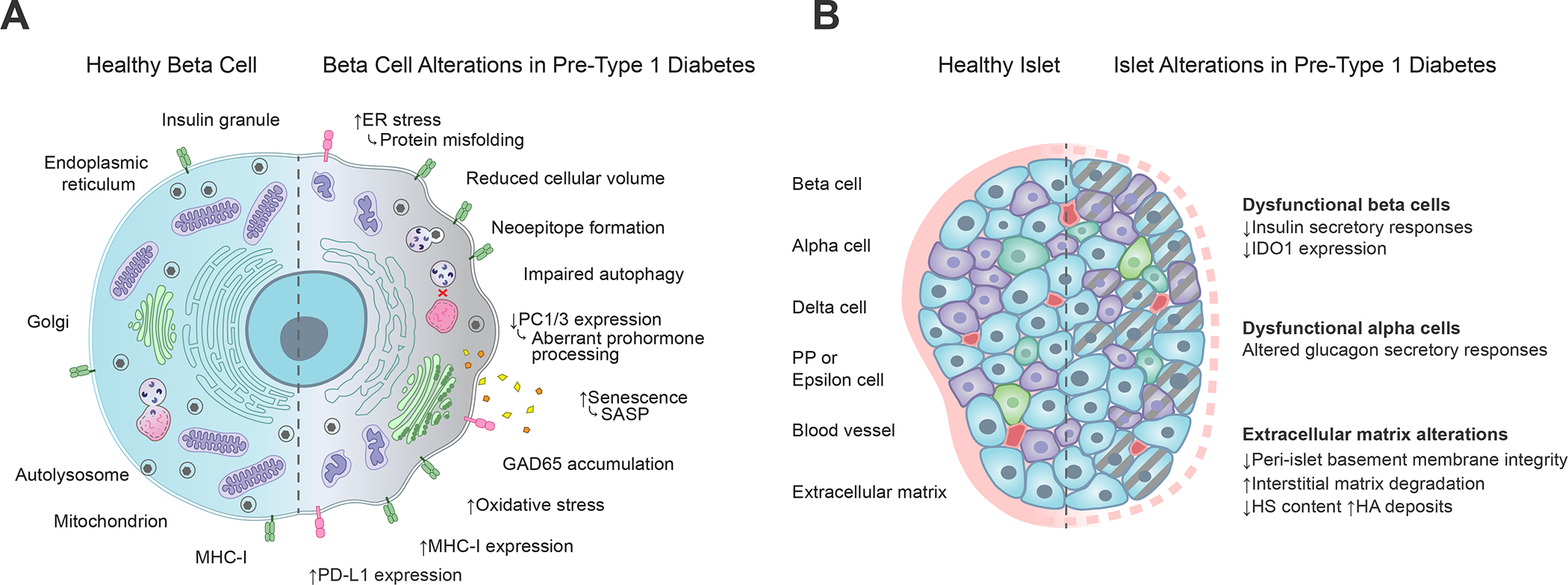
(A) Functional and phenotypic changes within the pancreatic β-cell during type 1 diabetes development include protein misfolding reflective of ER stress, neoepitope formation, impairments in autophagy and prohormone processing, secretion of SASP molecules, accumulation of GAD65 in the Golgi apparatus, mitochondrial oxidative stress, and increased expression of MHC class I and PD-L1 molecules, which respectively serve opposing roles related to T cell activation through antigen presentation and co-inhibitory receptor stimulation (Figure 2). Straight arrows depict increases (up) or decreases (down) in type 1 diabetes as compared to healthy β-cells. Curved arrows depict the functional consequences of those changes. (B) Pancreatic islet alterations during type 1 diabetes development include loss of insulin secretion and IDO1 expression within dysfunctional β-cells, dysfunctional glucagon secretion from alpha cells, and degradation of extracellular matrix comprising the peri-islet basement membrane. Gray dashed lines depict cellular dysfunction. Pink dashed lines depict basement membrane degradation. ER, endoplasmic reticulum; PC1/3, proprotein convertase 1/3; SASP, senescence-associated secretory phenotype; GAD65, glutamic acid decarboxylase 65; MHC, major histocompatibility complex; PD-L1, programmed death ligand 1; IDO1, indoleamine 2, 3-dioxygenase 1; HS, heparan sulphate; HA, hyaluronic acid.
β Cell Stress
The β cell is a professional secretory unit that is predominantly tasked with the production of insulin to maintain glucose homeostasis. As part of this function, the biosynthetic machinery, from gene transcription and mRNA translation to protein folding and secretion, is under tenuous homeostasis. In the pre-diabetic state, stress signals (e.g., viral infection, exposure to proinflammatory cytokines, increased metabolic demand) may pose a threat to these homeostatic processes104–110. The resultant effects on RNA processing, protein production and folding, as well as protein clearance and release would provide for disruption of cellular function and survival independent of direct immune-mediated damage111–116.
Studies utilizing NOD mice, human islets, and human pancreas from T1D donors have identified several molecular responses that likely contribute to β cell impairment in T1D and the pre-diabetic state (Figure 3). These include altered mRNA splicing115,117, endoplasmic reticulum (ER) and oxidative stress118,119, reductions in proinsulin to insulin processing (resulting from reductions in PC1/3 processing enzyme)120,121, impaired autophagy114, and cellular senescence99. These processes may not only impair the function of β cells but, in addition, promote their enhanced visibility to the immune system through formation of neoepitopes, augmented expression of MHC class I molecules, and other processes capable of immune stimulation59,122–124.
Beyond offering new insights into T1D pathophysiology, the identification of early β cell stress in the natural history of disease also offers an opportunity for developing a novel means of therapeutic intervention, especially if reliable serological biomarkers of these processes can be identified125. Among biomarkers having such potential, increasing attention has been directed at the proinsulin-to-C-peptide ratio (PI:C); an indicator reflecting defects in proinsulin processing resulting from ER stress and loss of PC1/3 production. Elevated PI:C ratios have been observed in multiple autoantibody-positive youth who are more likely to progress to T1D126. A second biomarker example identifies epigenetically modified DNA fragments uniquely emanating from dying, stressed β cells127,128. These biomarkers appear to identify an increase in β cell death in recently diagnosed T1D individuals, those receiving islet transplants, and in those with high risk for T1D (i.e., first-degree relatives)127–130,131. Importantly, unlike T1D associated autoantibodies, biomarkers of β cell stress remain in their infancy and uncertainty exists regarding their sensitivity and utility to reliably identify early-stage T1D132.
β Cell Function Versus β-Cell Mass
Loss of insulin secretion has historically been viewed as a hallmark of T1D. Yet, the pathophysiology of this loss may not be as simple as the loss of β cells themselves, as it occurs many months to years before the onset of clinically apparent diabetes, and may therefore emanate from some combination of a functional deficit and a deficit in β cell mass7. In early studies of autoantibody-positive individuals at increased-risk for T1D, intravenous glucose infusion studies indicated a loss of early phase insulin secretion that preceded alterations in glucose homeostasis133. Similarly, in later studies of high-risk co-twins134, those progressing to T1D exhibited similar defects in early phase insulin secretion compared to those who did not progress; moreover, these individuals had impaired insulin responses to different levels of glucose infusion, suggesting a defect in β cell glucose sensitivity. Whereas this early, prediabetic defect in insulin secretion might justifiably be attributed to a loss of β-cell mass, analysis of pancreas pathology suggests a greater complexity. Studies of human pancreas have demonstrated the presence of normal β cell mass135, but impaired proinsulin processing in single and multi-islet autoantibody positive donors without diabetes111, implying that glycemic dysregulation in the early stages of T1D may reflect impairments in β cell function prior to a catastrophic decline in β cell mass near the time of disease onset. Interestingly, a small number of studies of islets isolated from individuals with T1D have suggested that insulin secretion can be restored ex vivo with prolonged culture under non-diabetogenic conditions; however, these findings remain controversial given the observations from the Diabetes Virus Detection (DiViD) study showing only partial recovery of islet function following 3–6 days of culture136,137.
The original Eisenbarth model depicting the loss in β cell mass or number being associated with T1D has also come under recent question2, with the notion that β cell function, with or without concurrent loss in mass, may be just as relevant. Whether or not β cell dysfunction in T1D arises from a loss in mass or a loss in β cell function (or both) has potential therapeutic implications, since approaches to avert or reverse either concept would differ substantially138. Seemingly, the aforementioned studies documenting the loss in early-phase insulin secretion, reduced glucose sensitivity, and preservation of β cell mass in the pre-T1D period support the loss in function model, wherein underlying cellular dysregulation precludes glucose sensing and/or prohormone processing resulting in dysfunctional early docking and release of insulin-containing granules96,111. However, analysis of subjects undergoing partial (~50%) pancreatectomy revealed that the decline in glucose sensitivity and early-phase insulin release can accompany this procedure and thereby predict the likelihood of developing diabetes139. Thus, loss of β cell mass can also manifest as defects in glucose sensing and insulin release kinetics.
Estimating residual β cell mass during disease progression represents a challenge, since histological examination of pancreatic tissue from living persons with T1D is not considered feasible or ethical140. Nevertheless, several key observations from living subjects note that C-peptide and proinsulin (measures of β cell metabolic function) remain detectable in many subjects, even after decades of T1D, although these levels decline with disease duration141–143. Furthermore, histological studies of β cell numbers in whole-organ cross-sections (i.e., a measure of mass) are highly variable (60–95%loss) at the time of T1D diagnosis. β cell number is also variable in those with longstanding disease (i.e., decades), albeit with significant reductions (80–98%) relative to non-diabetic controls144–146. Finally, when measures of β cell function (intravenous glucose tolerance test) and mass (glucose-potentiated arginine stimulation) are simultaneously measured in living subjects at low or high risk of T1D, there is significant variability in the two measures suggesting heterogeneity in the relative loss of mass versus function147, and more importantly, that the two variables may coexist in any given individual. Collectively, these studies suggest that although the loss of β cell mass predominates in T1D, much less is known regarding the pre-diabetic period when disease pathogenesis is likely most active and when therapies may be most impactful.
Contributions of Islet Microvasculature, Extracellular Matrix, and Sympathetic Innervation
The islets receive a disproportionately large fraction of the total pancreatic blood flow supplied by a dense network of fenestrated capillaries148, which are subject to neurovascular control through activation of pericytes that densely cover the capillary endothelial cells149. Indeed, nearly every β cell is supplied by an intra-islet or peri-islet capillary facilitating systemic metabolic control through rapid nutrient sensing and responsive secretion of endocrine hormones150. In shorter duration T1D (<10 years), islet microvascular density is increased, and vessel diameter is reduced151–153. Interestingly, these alterations are most pronounced in pseudoatrophic islets (i.e., insulin-negative) while vessel densities and diameters within residual insulin containing islets from T1D donors are comparable to those of non-diabetic controls151. Moreover, islet capillary density appears to return to normal levels in longstanding T1D152,153.
Recent efforts have demonstrated the structural and functional polarization of human β cells in relation to islet capillaries, but the implications of this observed enrichment of scaffold proteins along the β cell-vascular interface remain subject to investigation, both as it relates to diabetes pathogenesis and the development of β cell-targeted therapeutics154. Efferent blood is generally thought to travel through venules from the islet to the surrounding acini, forming what is known as the insulo-acinar portal system148. However, recent work supports the existence of open circulation characterized by bidirectional blood flow between the endocrine and exocrine pancreas compartments155, providing a potential mechanistic link to exocrine pancreas alterations associated with T1D, as discussed below.
The islet microvasculature is largely responsible for the production of ECM linking capillaries with β cells150, and the passage of immune cells through the islet ECM represents a key step in forming the insulitis lesion (Figure 2)71. ECM is comprised of a complex network of molecules (e.g., heparan sulfate/heparan sulfate proteoglycans, hyaluronan, laminins, collagens) both within and surrounding the islets, constituting the interstitial matrix and basement membrane (BM). The importance of islet ECM in T1D found early evidence from in vitro analyses suggesting hyaluronan and the hyaluronan binding protein (HBP) are capable of modulating T-cell adhesion and migration156. These concepts gained further support by histological examination of human T1D pancreata where hyaluronan and HBP, including inter-α-inhibitor (IαI) and versican, not only accumulated in T1D islets but were also associated with the degree of inflammatory cell accumulation (i.e., insulitis)71,157. Specifically, hyaluronan was dramatically increased both within the islet and outside the islet endocrine cells, juxtaposed against islet microvessels in T1D pancreata. Moreover, when extending such models to additional vascular constituents, the loss of the peri-islet basement membrane was noted to only occur at sites of leukocyte infiltration into an islet, suggesting that leukocyte penetration of the peri-islet BM is a critical step in the process of insulitis development102. Importantly, hyaluronan accumulation in islets has been observed in the pancreas of islet autoantibody-positive individuals prior to immune cell infiltration and T1D onset with the degree of hyaluronan staining positively associating with the number of autoantibodies158, and treatment with a hyaluronan synthesis inhibitor (4-methylumbelliferone, 4-MU) prevented diabetes onset in NOD mice and rendered insulitis non-destructive in the TCR transgenic DORmO mouse model of autoimmune diabetes159, altogether supporting a potential role for such ECM alterations early in the disease process.
Another ECM constituent, the glycoaminoglycan heparan sulfate (HS), was demonstrated in studies of NOD mice to protect β cells from oxidative damage160. Examination of insulin-positive islets in T1D pancreases noted significant loss of HS, whereas heparanase expression was pronounced in islet-infiltrating leukocytes161, further supporting the concept that loss of HS could contribute to β cell death in T1D. Taken together, it would appear that in settings of T1D, islet ECM composition represents a significant factor in maintaining immunological tolerance, activation of immune cell populations, islet invasion, and eventual destruction of β cells.
The sympathetic nervous system, a key constituent of the autonomic nervous system, plays a major role in organ homeostasis. With respect to the pancreas, it is intensively innervated with sympathetic efferent nerves originating from the superior mesenteric and celiac ganglia162. This innervation is considered a key contributor to establishing islet architecture163 and with respect to function, sympathetic neural activity inhibits insulin secretion while promoting glucagon release, together increasing systemic glucose levels. Emerging technologies combined with the availability of pancreatic and islet tissues from persons at various stages of T1D have allowed for recent studies that have amplified a potential contributory role for disordered innervation in metabolic disorders164. While sometimes conflicting, analysis of humans with T1D do suggest that islets undergo a series of key modifications to their sympathetic innervation as part of the pathogenic processes underlying this disease and in particular, the formation of insulitis164.
At one extreme, a sustained and pronounced loss of sympathetic innervation in islets from pancreas autopsies has been noted for those with T1D, both at disease diagnosis and in those with established disease165. A second and more recent effort suggested both recent-onset and longstanding T1D donors had greater islet nerve fiber density compared to non-diabetic donors, whereas no changes in sympathetic axons were observed with diabetes of any duration153. Sympathetic tyrosine hydroxylase (TH) axon numbers and density were decreased in islet autoantibody-positive non-T1D subjects in comparison to those with T1D166. Through a series of molecular investigations, this latter study also identified variances in noradrenalin degradation, α-adrenergic signaling, cardiac β-adrenergic signaling, catecholamine biosynthesis, and additional neuropathology pathways.
T1D is a Disorder of the Exocrine Pancreas
Pancreas Size/Weight/Volume
In the absence of T1D, the human pancreas increases in size with age, plateauing in the mid- to late-20s, followed by a decrease after 60 years of age (in most), with its size also reflective of overall BMI167. Over a century ago, pancreas autopsy studies of individuals with diabetes (albeit undefined by today’s standards) showed reduced pancreas size168. Later, a series of efforts published between 1975 and 1985 demonstrated low serum levels of exocrine pancreatic enzymes in patients with established T1D, an observation that was initially presumed a consequence of long-term insulinopenia169–172. Unfortunately, the potential pathogenic significance of these observations remained underappreciated173 for decades until recent studies, discussed below, confirmed these observations in individuals definitively diagnosed with stage 1–2 and recent-onset stage 3 T1D, revealing a variety of exocrine changes across the natural history of T1D (Figure 4).
Figure 4. Alterations to pancreas size and organization during type 1 diabetes progression.
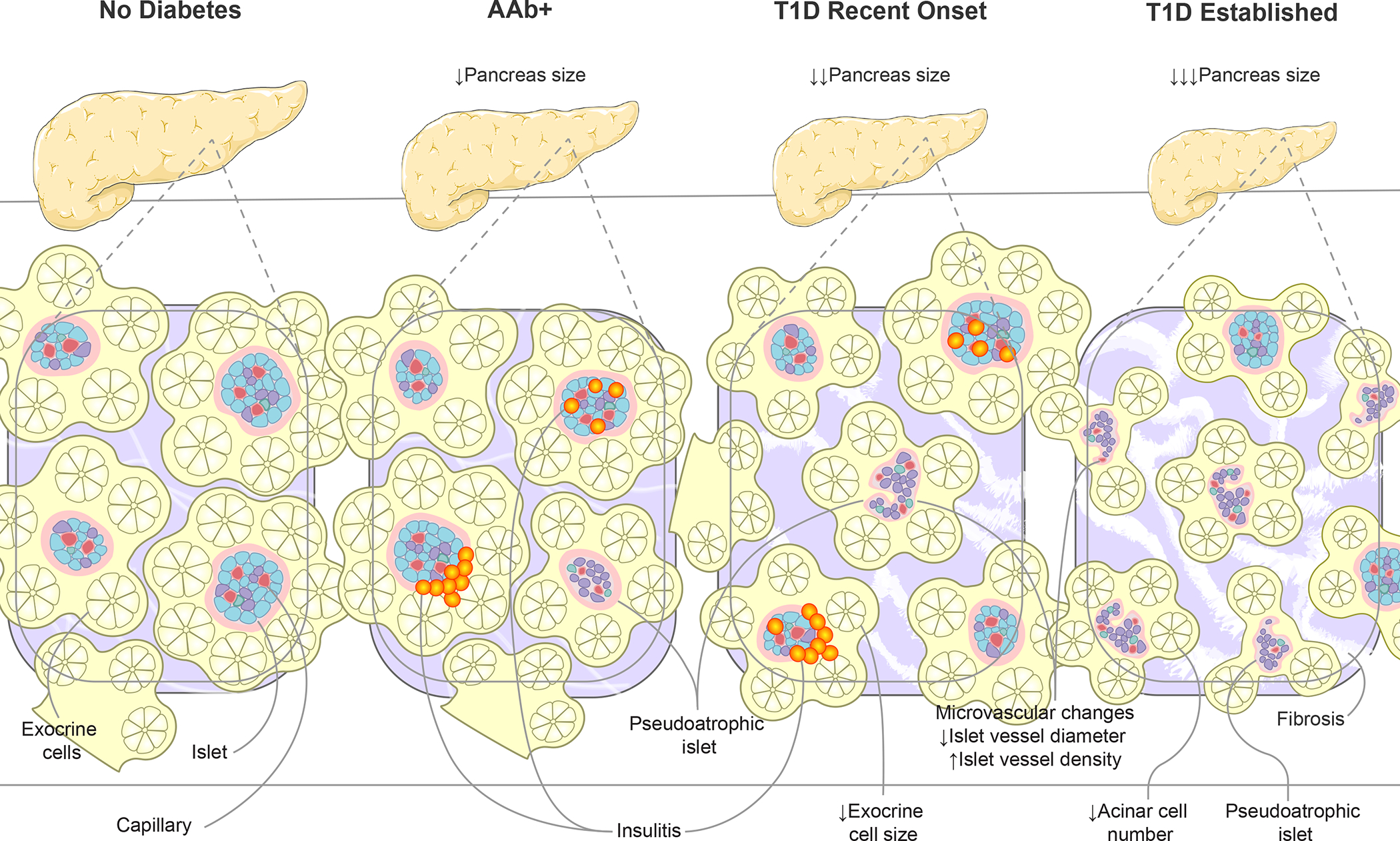
Disease progression, from AAb seroconversion (stage 1–2) to recent-onset and established type 1 diabetes (stage 3), is associated with increasingly smaller pancreas organ size. Insets show highly vascularized islets surrounded by exocrine cell bundles arranged in acini. Asynchronous islet infiltration and β-cell destruction result in a heterogeneous mixture of normal, insulitic, and pseudoatrophic islets in AAb+ and recent onset type 1 diabetes pancreas while the majority of islets are pseudoatrophic in longstanding type 1 diabetes. Insulin negative pseudoatrophic islets have distinct microvascular alterations including reduced vessel diameter and increase vessel density. Within the exocrine compartment, recent onset type 1 diabetes is associated with smaller exocrine cells, eventually leading to reduced acinar cell number in established disease. The exocrine pancreas is commonly characterized by periductal and interlobular fibrosis after type 1 diabetes onset. Arrows depict direction of change. AAb+, islet autoantibody positive; T1D, type 1 diabetes.
Although patient entry criteria vary amongst reported studies, reductions in pancreatic volume of approximately 25–30% have been noted in recent-onset T1D cases and reductions of 30–50% in long-standing disease in comparison to control subjects, usually matched for age and/or normalized for BMI174–177. Importantly, the notion of reduced pancreatic volume in T1D has found support irrespective of the methodology employed and has been confirmed in living subjects, as assessed by magnetic resonance imaging (MRI)176,178,179. However, from a mechanistic perspective, perhaps the strongest support for a pathogenic link to T1D arises from findings by MRI that pancreas volume is reduced by approximately 25% in those with one or multiple T1D-associated autoantibodies (in the absence of overt disease) with reduced pancreas volume also observed in autoantibody-negative first degree relatives of a T1D proband (compared to unrelated control subjects)178,180. These observations are especially salient, as they suggest that pancreatic size likely reflects a genetic contribution toward disease and potentially parallels the progressive loss of functional β cell mass.
Considering that islets comprise only 1–2% of pancreas volume, the mechanistic relationship between loss in pancreatic size with T1D also suggests that any reduction in volume or weight should be attributed to a loss in exocrine tissue. Histological evidence suggests that acinar cells are reduced in size and number, with loss of characteristic peri-islet amylase-negative cell clusters in T1D pancreata181–183,184. The mechanisms underlying loss of pancreatic size in T1D are not well understood but have largely been hypothesized to result from loss of insulinotrophic effects on the exocrine pancreas185.
Given the reductions in exocrine volume, a feature that has gone largely underappreciated in T1D is that of diminished exocrine function. Decreases in exocrine function in T1D patients were identified years ago through analysis of sera7,186. More recent efforts suggest that subjects with T1D are characterized by diminished levels of serum trypsinogen and lipase as well as stool chymotrypsinogen and elastase178,187,188. Moreover, serological levels of the exocrine pancreas enzymes lipase and trypsinogen are directly associated with pancreas size in subjects with T1D as well as single and multiple autoantibody positive relatives188.
Pancreas Morphology
Both the endocrine and exocrine compartments of the normal human pancreas are defined by notable morphological heterogeneity. This heterogeneity includes variations in islet cell composition, size, and density. Similarly, acinar tissues display regionally distinct physiological and morphological variations. Such findings raise the question of their potential impact on health and disease, including T1D18. Specifically, in T1D, loss of β-cell mass is less severe in the pancreatic head and more dominant in body and tail174,175,189. In concert, the aforementioned reductions in pancreas weight and volume are largely related to reductions of the ventral portion of the organ, especially in the tail region174,175,189. Beyond these variations, pancreatic islet infiltration is often “patchy,” akin to the loss of melanocytes as seen in vitiligo and often segregated to neighboring lobules75. β-cell loss is accordingly patchy and it remains unclear why β-cell destruction proceeds asynchronously, even within the same lobule. Pancreatic duct gland proliferation is observed in T1D190 and exhibits a unique exocrine proteomic pattern as determined by LC/MS191. Despite the lack of morphological heterogeneity in the healthy exocrine and endocrine pancreas when compared to the heterogeneous pancreas pathology in T1D, it remains unclear how the three-dimensionally organized cellular environment and morphology might direct pancreatic function and contribute toward T1D pathogenies. Finally, it is also worth noting that while many of these features distinguish the pancreas of T1D from type 2 diabetes (T2D), there is a growing appreciation for how specific polymorphisms, including certain loci classically associated with T2D (e.g., TCF7L2), can influence the phenotype of donors with T1D27,192–194 (Table 1).
Exocrine:Immune and Exocrine:Islet Interactions within the Pancreas
Beyond size and function, many additional pancreatic abnormalities in T1D are associated with innate and adaptive immunity. Indeed, T1D is often noted to be a disorder where inflammation is limited to the pancreatic islets (i.e., insulitis). Nevertheless, a curious increase in immune cell types has been observed in the exocrine compartment of the pancreas of donors with T1D, including neutrophils39, CD8+ and CD4+ T cells, and CD11c+ cells73. Additionally, enhanced levels of complement deposition, specifically C4d, has been observed195. The potential exists that these features are the result of what has long been termed “gut leakiness” in T1D196. Curiously, however, preproinsulin-reactive CD8+ T cells are present within the exocrine pancreas at comparable frequencies from T1D, single GADA+ positive and control donors197 raising questions surrounding the mechanisms that allow for their migration into the islets and β-cell destruction during T1D. The T1D in Acute Pancreatitis Consortium (T1DAPC)198,199 is poised to address many of the questions, potentially linking acute pancreatitis and exocrine pancreatic inflammation with T1D.
The Chicken and Egg Scenario for T1D Pathogenesis
With the aforementioned evidence in place (i.e., aberrations in the immune response, β-cells, islet cells, and exocrine pancreas in T1D), the classic “Chicken or the Egg” scenario arises for the disorder’s pathogenesis. Specifically, does the initiation of T1D pathogenesis begin via the conventional autoimmune model (i.e., a failure to recognize self-versus non-self in terms of β-cell antigens)? Multiple contributors are key to this concept including genetic susceptibility, largely related to the ability for a given HLA type (class I and class II) to present β-cell self-antigens; failure of central tolerance (i.e., inability to delete autoreactive T-cells in the thymus); together with inefficient peripheral tolerance (i.e., dysregulated immune regulation)96. Each of these features has been well established in various research settings and/or experimental models of T1D. More speculative and long unproven is the concept of “molecular mimicry”200 and the identification of environmental factors capable of initiating anti-β-cell autoimmunity200,201. Toward this latter notion, a number of studies have implicated the gut microbiome202,203 and prolonged infection with enterovirus B204 as potential modulators of T1D development.
Or is it possible that an alternative model represents actuality — one where β-cells, through specific features (e.g., stress, number), or perhaps the pancreas itself, drive the immune response to function as would be expected? In this setting, the role of the β-cell may be to augment versus initiate the pathogenic processes (including but perhaps not limited to autoimmunity), with pancreatic alterations being subject to the influences of β-cell number and function. Indeed, despite providing an unparalleled opportunity to visualize the site of disease pathogenesis, organ donor tissue studies are inherently limited in that longitudinal analyses are not possible, and unlike studies of mouse models or cell lines, they are not readily poised to make focused/mechanistic manipulations that would address key features of disease development.
Evidence in support for each notion exists, especially when one considers information beyond studies highlighted in this Review96. Perhaps both scenarios occur in T1D pathogenesis, with a sliding scale between autoimmune first versus β-cell first forms of T1D (Figure 1); a concept that might shed light onto the notion of disease heterogeneity. It is now becoming clear that the seminal notion of β-cell “homicide vs. suicide”97 likely represents a false choice as a growing body of literature supports the notion that islet-specific, whole-pancreas, and autoimmune mechanisms contribute altogether toward the multifactorial pathogenesis of T1D. And, while a detailed discussion of genetics is beyond the scope of this Review, the balance between autoimmune, exocrine, and β-cell drivers of this disease may be subject to polygenic control205–208, including the HLA class II, insulin, and 77 additional regions identified by genome-wide association studies (GWAS), which are associated with genes thought to impact both leukocyte and pancreatic β-cell function209,210.
Questions and Future Directions
Recent revelations derived from studies of the human T1D pancreas have clearly improved our understanding of the disorder’s pathogenesis, yet knowledge voids in the autoimmune and pancreatic pathogeneses of the disorder remain. With respect to autoimmunity, questions remain regarding its contributions to disease heterogeneity (including endotypes211) and the role for environmental “triggers” vs. environmental factors that exacerbate autoimmunity. One important consideration for studies on organ donor tissues is that of age. The reality is, only a limited number of young people (thankfully) become organ donors, resulting in the greatest percentage of organ donors being between 50–64 years of age in the United States212. That said, nPOD is amazingly efficient at procuring cases at younger ages when they are available. Beyond this, while insulin autoantibodies (IAA) are measured for accepted cases, nPOD does not perform rapid screens for IAA due to technical limitations of the available validated assays. Hence, studies of organ donor pancreas are biased to older ages and likely miss an important subgroup of IAA+ donors, representing key areas for improvement and advance of our current knowledge. As noted above, studies of pancreata from at risk individuals suggest that T1D is preceded by a period of “waxing and waning” insulitis and β-cell loss213. If true, what factors stimulate or reduce this autoimmune response and might it be chronic in certain individuals?
As for pancreatic contributions, the rapidly gaining notion of “functional β-cell mass” still remains conceptual, at least until the ability to image the endocrine pancreas in vivo becomes a reality214. Such a technology would also address other key endocrine issues, including the contribution of islet cell number in the first years of life to T1D risk, the number of islets present at various stages of T1D, and the issue of whether loss of β cells occurs in a linear2 versus waxing and waning fashion213. Furthermore, while a role for β-cell stressors has become increasingly appreciated, their exact identification and mechanisms of action remains elusive and largely relies on in vitro models to mimic human disease. Finally, we need see clearer information regarding the question of why some β-cells are destroyed while others are spared. Future efforts investigating the role of the exocrine pancreas in T1D should include whether the many features described herein contribute to the disorder’s pathogenesis. In addition, studies should evaluate how these exocrine features might serve as a biomarker of disease stage as a function of its natural history3. Finally, exocrine features may serve in the pre-diabetic period, as well as post-onset, as a therapeutic sentinel of a positive therapeutic intervention, through preservation of exocrine volume or function.
For each of these areas, hope exists that advances in multi-omics, imaging technologies, novel biomarkers, screening programs defining those at risk for T1D (including in the general population), artificial intelligence systems, investigations across historically underrepresented/minoritized groups in biomedical research, and more will lead to improvements in our understanding of the symphony underscoring the pathogenesis of T1D.
Acknowledgements
The authors would like to thank Dr. MacKenzie Williams (University of Florida) for creating the figures and Drs. Amanda Posgai and Sara Williams (University of Florida) for editorial assistance.
Funding
This Review was prepared with support from the National Institutes of Health (NIH) R01 DK123292, R01 DK131059, and Human Pancreas Analysis Program-Type 1 Diabetes (HPAP-T1D) U01 DK112217; along with the Human Atlas of Neonatal Development and Early Life Pancreas/Immunity (HANDEL-P/I) supported by The Leona M. & Harry B. Helmsley Charitable Trust (2004-03813); and the Network for Pancreatic Organ donors with Diabetes (nPOD; RRID:SCR_014641), a collaborative type 1 diabetes research project supported by JDRF (nPOD: 5-SRA-2018-557-Q-R) and The Leona M. & Harry B. Helmsley Charitable Trust (Grant#2018PG-T1D053, G-2108-04793). The content and views expressed are the responsibility of the authors and do not necessarily reflect the official view of nPOD or HPAP.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Atkinson MA, Eisenbarth GS, and Michels AW (2014). Type 1 diabetes. Lancet 383, 69–82. 10.1016/S0140-6736(13)60591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisenbarth GS (1986). Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med 314, 1360–1368. 10.1056/NEJM198605223142106. [DOI] [PubMed] [Google Scholar]
- 3.Insel RA, Dunne JL, Atkinson MA, Chiang JL, Dabelea D, Gottlieb PA, Greenbaum CJ, Herold KC, Krischer JP, Lernmark A, et al. (2015). Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 38, 1964–1974. 10.2337/dc15-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenbaum C, Lord S, and VanBuecken D (2017). Emerging Concepts on Disease-Modifying Therapies in Type 1 Diabetes. Curr Diab Rep 17, 119. 10.1007/s11892-017-0932-x. [DOI] [PubMed] [Google Scholar]
- 5.DiMeglio L, Evans-Molina C, and Oram R (2018). Type 1 diabetes. Lancet 391, 2449–2462. 10.1016/S0140-6736(18)31320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krischer JP, Lynch KF, Lernmark Å, Hagopian WA, Rewers MJ, She JX, Toppari J, Ziegler AG, Akolkar B, and Group TS (2017). Genetic and Environmental Interactions Modify the Risk of Diabetes-Related Autoimmunity by 6 Years of Age: The TEDDY Study. Diabetes Care 40, 1194–1202. 10.2337/dc17-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lernmark Å (2021). Etiology of Autoimmune Islet Disease: Timing Is Everything. Diabetes 70, 1431–1439. 10.2337/dbi18-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michels AW, Redondo MJ, and Atkinson MA (2022). The pathogenesis, natural history, and treatment of type 1 diabetes: time (thankfully) does not stand still. Lancet Diabetes Endocrinol 10, 90–92. 10.1016/S2213-8587(21)00344-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, Gitelman SE, Gottlieb PA, Krischer JP, Linsley PS, et al. (2019). An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N Engl J Med 381, 603–613. 10.1056/NEJMoa1902226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans-Molina C, and Oram RA (2023). Teplizumab approval for type 1 diabetes in the USA. Lancet Diabetes Endocrinol 11, 76–77. 10.1016/S2213-8587(22)00390-4. [DOI] [PubMed] [Google Scholar]
- 11.Atkinson MA, Roep BO, Posgai A, Wheeler DCS, and Peakman M (2019). The challenge of modulating beta-cell autoimmunity in type 1 diabetes. Lancet Diabetes Endocrinol 7, 52–64. 10.1016/s2213-8587(18)30112-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roep BO, Wheeler DCS, and Peakman M (2019). Antigen-based immune modulation therapy for type 1 diabetes: the era of precision medicine. Lancet Diabetes Endocrinol 7, 65–74. 10.1016/s2213-8587(18)30109-8. [DOI] [PubMed] [Google Scholar]
- 13.Bingley PJ, Wherrett DK, Shultz A, Rafkin LE, Atkinson MA, and Greenbaum CJ (2018). Type 1 Diabetes TrialNet: A Multifaceted Approach to Bringing Disease-Modifying Therapy to Clinical Use in Type 1 Diabetes. Diabetes Care 41, 653–661. 10.2337/dc17-0806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skyler JS (2015). Prevention and reversal of type 1 diabetes--past challenges and future opportunities. Diabetes Care 38, 997–1007. 10.2337/dc15-0349. [DOI] [PubMed] [Google Scholar]
- 15.Sims E, Mirmira R, and Evans-Molina C (2020). The role of beta-cell dysfunction in early type 1 diabetes. Curr Opin Endocrinol Diabetes Obes 27, 215–224. 10.1097/MED.0000000000000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eizirik D, Szymczak F, Alvelos M, and Martin F (2021). From Pancreatic β-Cell Gene Networks to Novel Therapies for Type 1 Diabetes. Diabetes 70, 1915–1925. 10.2337/dbi20-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Powers A (2021). Type 1 diabetes mellitus: much progress, many opportunities. The Journal of Clinical Investigation 131, e142242. 10.1172/JCI142242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Battaglia M, and Atkinson MA (2015). The streetlight effect in type 1 diabetes. Diabetes 64, 1081–1090. 10.2337/db14-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taka AM, Härkönen T, Vähäsalo P, Lempainen J, Veijola R, Ilonen J, Knip M, and Register FPD (2022). Heterogeneity in the presentation of clinical type 1 diabetes defined by the level of risk conferred by human leukocyte antigen class II genotypes. Pediatr Diabetes 23, 219–227. 10.1111/pedi.13300. [DOI] [PubMed] [Google Scholar]
- 20.Parviainen A, Härkönen T, Ilonen J, But A, Knip M, and Register FPD (2022). Heterogeneity of Type 1 Diabetes at Diagnosis Supports Existence of Age-Related Endotypes. Diabetes Care 45, 871–879. 10.2337/dc21-1251. [DOI] [PubMed] [Google Scholar]
- 21.Redondo MJ, Evans-Molina C, Steck AK, Atkinson MA, and Sosenko J (2019). The Influence of Type 2 Diabetes-Associated Factors on Type 1 Diabetes. Diabetes Care 42, 1357–1364. 10.2337/dc19-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Battaglia M, Ahmed S, Anderson M, Atkinson M, Becker D, Bingley P, Bosi E, Brusko T, DiMeglio L, Evans-Molina C, et al. (2020). Introducing the Endotype Concept to Address the Challenge of Disease Heterogeneity in Type 1 Diabetes. Diabetes care 43. 10.2337/dc19-0880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roep B, Thomaidou S, van Tienhoven R, and Zaldumbide A (2021). Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?). Nature Reviews Endocrinology 17, 150–161. 10.1038/s41574-020-00443-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roep B, Montero E, van Tienhoven R, Atkinson M, Schatz D, and Mathieu C (2021). Defining a cure for type 1 diabetes: a call to action. The Lancet Diabetes & Endocrinology 9, 553–555. 10.1016/S2213-8587(21)00181-9. [DOI] [PubMed] [Google Scholar]
- 25.Charles M, and Leslie R (2021). Diabetes: Concepts of β-Cell Organ Dysfunction and Failure Would Lead to Earlier Diagnoses and Prevention. Diabetes 70, 2444–2456. 10.2337/dbi21-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pugliese A, Yang M, Kusmarteva I, Heiple T, Vendrame F, Wasserfall C, Rowe P, Moraski JM, Ball S, Jebson L, et al. (2014). The Juvenile Diabetes Research Foundation Network for Pancreatic Organ Donors with Diabetes (nPOD) Program: goals, operational model and emerging findings. Pediatr Diabetes 15, 1–9. 10.1111/pedi.12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atkinson M, Campbell-Thompson M, Kusmartseva I, and Kaestner K (2020). Organisation of the human pancreas in health and in diabetes. Diabetologia 63, 1966–1973. 10.1007/s00125-020-05203-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richardson S, and Pugliese A (2021). 100 YEARS OF INSULIN: Pancreas pathology in type 1 diabetes: an evolving story. The Journal of Endocrinology 252, R41–R57. 10.1530/JOE-21-0358. [DOI] [PubMed] [Google Scholar]
- 29.Roep BO, Montero E, van Tienhoven R, Atkinson MA, Schatz DA, and Mathieu C (2021). Defining a cure for type 1 diabetes: a call to action. Lancet Diabetes Endocrinol 9, 553–555. 10.1016/S2213-8587(21)00181-9. [DOI] [PubMed] [Google Scholar]
- 30.Mathieu C, Martens PJ, and Vangoitsenhoven R (2021). One hundred years of insulin therapy. Nat Rev Endocrinol 17, 715–725. 10.1038/s41574-021-00542-w. [DOI] [PubMed] [Google Scholar]
- 31.Ziegler AG, and Bonifacio E (2021). Shortening the paths to type 1 diabetes mellitus prevention. Nat Rev Endocrinol 17, 73–74. 10.1038/s41574-020-00450-5. [DOI] [PubMed] [Google Scholar]
- 32.Bluestone JA, Buckner JH, and Herold KC (2021). Immunotherapy: Building a bridge to a cure for type 1 diabetes. Science 373, 510–516. 10.1126/science.abh1654. [DOI] [PubMed] [Google Scholar]
- 33.Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, and Atkinson MA (2016). Insulitis and beta-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes 65, 719–731. 10.2337/db15-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campbell-Thompson ML, Atkinson MA, Butler AE, Chapman NM, Frisk G, Gianani R, Giepmans BN, von Herrath MG, Hyöty H, Kay TW, et al. (2013). The diagnosis of insulitis in human type 1 diabetes. Diabetologia 56, 2541–2543. 10.1007/s00125-013-3043-5. [DOI] [PubMed] [Google Scholar]
- 35.Richardson SJ, Rodriguez-Calvo T, Gerling IC, Mathews CE, Kaddis JS, Russell MA, Zeissler M, Leete P, Krogvold L, Dahl-Jorgensen K, et al. (2016). Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia 59, 2448–2458. 10.1007/s00125-016-4067-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flatt A, Greenbaum C, Shaw J, and Rickels M (2021). Pancreatic islet reserve in type 1 diabetes. Annals of the New York Academy of Sciences 1495, 40–54. 10.1111/nyas.14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.In’t Veld P (2014). Insulitis in human type 1 diabetes: a comparison between patients and animal models. Semin Immunopathol 36, 569–579. 10.1007/s00281-014-0438-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vecchio F, Lo Buono N, Stabilini A, Nigi L, Dufort MJ, Geyer S, Rancoita PM, Cugnata F, Mandelli A, Valle A, et al. (2018). Abnormal neutrophil signature in the blood and pancreas of presymptomatic and symptomatic type 1 diabetes. JCI Insight 3. 10.1172/jci.insight.122146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valle A, Giamporcaro GM, Scavini M, Stabilini A, Grogan P, Bianconi E, Sebastiani G, Masini M, Maugeri N, Porretti L, et al. (2013). Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes 62, 2072–2077. 10.2337/db12-1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Apaolaza PS, Balcacean D, Zapardiel-Gonzalo J, and Rodriguez-Calvo T (2023). The extent and magnitude of islet T cell infiltration as powerful tools to define the progression to type 1 diabetes. Diabetologia. 10.1007/s00125-023-05888-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, Winkler C, Ilonen J, Veijola R, Knip M, et al. (2013). Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. Jama 309, 2473–2479. 10.1001/jama.2013.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jackson J, Posgai A, Campbell-Thompson M, and Kusmartseva I (2017). Insulitis in Autoantibody-Positive Pancreatic Donor With History of Gestational Diabetes Mellitus. Diabetes Care 40, 723–725. 10.2337/dc16-2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smeets S, De Paep D, Stangé G, Verhaeghen K, Van der Auwera B, Keymeulen B, Weets I, Ling Z, In’t Veld P, and Gorus F (2021). Insulitis in the pancreas of non-diabetic organ donors under age 25 years with multiple circulating autoantibodies against islet cell antigens. Virchows Archiv, Online ahead of print. 10.1007/s00428-021-03055-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.In’t Veld P (2011). Insulitis in human type 1 diabetes: The quest for an elusive lesion. Islets 3, 131–138. 15728 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leete P, Willcox A, Krogvold L, Dahl-Jørgensen K, Foulis AK, Richardson SJ, and Morgan NG (2016). Differential Insulitic Profiles Determine the Extent of β-Cell Destruction and the Age at Onset of Type 1 Diabetes. Diabetes 65, 1362–1369. 10.2337/db15-1615. [DOI] [PubMed] [Google Scholar]
- 46.Arif S, Leete P, Nguyen V, Marks K, Nor NM, Estorninho M, Kronenberg-Versteeg D, Bingley PJ, Todd JA, Guy C, et al. (2014). Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes 63, 3835–3845. 10.2337/db14-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leete P, Willcox A, Krogvold L, Dahl-Jorgensen K, Foulis AK, Richardson SJ, and Morgan NG (2016). Differential insulitic profiles determine the extent of beta cell destruction and the age at onset of type 1 diabetes. Diabetes. 10.2337/db15-1615. [DOI] [PubMed] [Google Scholar]
- 48.Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, Roep BO, and von Herrath MG (2012). Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med 209, 51–60. 10.1084/jem.20111187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, Bottino R, Naji A, Kaddis J, Elyaman W, et al. (2016). Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med 22, 1482–1487. 10.1038/nm.4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kent SC, and Babon JA (2017). Narrowing in on the anti-β cell-specific T cells: looking ‘where the action is’. Curr Opin Endocrinol Diabetes Obes 24, 98–102. 10.1097/MED.0000000000000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kent SC, Mannering SI, Michels AW, and Babon JAB (2017). Deciphering the Pathogenesis of Human Type 1 Diabetes (T1D) by Interrogating T Cells from the “Scene of the Crime”. Curr Diab Rep 17, 95. 10.1007/s11892-017-0915-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laban S, Suwandi J, van Unen V, Pool J, Wesselius J, Höllt T, Pezzotti N, Vilanova A, Lelieveldt B, and Roep B (2018). Heterogeneity of circulating CD8 T-cells specific to islet, neo-antigen and virus in patients with type 1 diabetes mellitus. PLoS One 13, e0200818. 10.1371/journal.pone.0200818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, Armstrong M, Powell RL, Reisdorph N, Kumar N, et al. (2016). Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 351, 711–714. 10.1126/science.aad2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Michels AW, Landry LG, McDaniel KA, Yu L, Campbell-Thompson M, Kwok WW, Jones KL, Gottlieb PA, Kappler JW, Tang Q, et al. (2017). Islet-Derived CD4 T Cells Targeting Proinsulin in Human Autoimmune Diabetes. Diabetes 66, 722–734. 10.2337/db16-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seay HR, Yusko E, Rothweiler SJ, Zhang L, Posgai AL, Campbell-Thompson M, Vignali M, Emerson RO, Kaddis JS, Ko D, et al. (2016). Tissue distribution and clonal diversity of the T and B cell repertoire in type 1 diabetes. JCI Insight 1, e88242. 10.1172/jci.insight.88242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anderson A, Landry L, Alkanani A, Pyle L, Powers A, Atkinson M, Mathews C, Roep B, Michels A, and Nakayama M (2021). Human islet T cells are highly reactive to preproinsulin in type 1 diabetes. Proc Natl Acad Sci U S A 118, e2107208118. 10.1073/pnas.2107208118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Landry L, Anderson A, Russ H, Yu L, Kent S, Atkinson M, Mathews C, Michels A, and Nakayama M (2021). Proinsulin-Reactive CD4 T Cells in the Islets of Type 1 Diabetes Organ Donors. Frontiers in Endocrinology 12, 622647. 10.3389/fendo.2021.622647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rodriguez-Calvo T, Johnson J, Overbergh L, and Dunne J (2021). Neoepitopes in Type 1 Diabetes: Etiological Insights, Biomarkers and Therapeutic Targets. Frontiers in Immunology 12, 667989. 10.3389/fimmu.2021.667989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gonzalez-Duque S, Azoury ME, Colli ML, Afonso G, Turatsinze JV, Nigi L, Lalanne AI, Sebastiani G, Carré A, Pinto S, et al. (2018). Conventional and Neo-antigenic Peptides Presented by β Cells Are Targeted by Circulating Naïve CD8+ T Cells in Type 1 Diabetic and Healthy Donors. Cell Metab 28, 946–960.e946. 10.1016/j.cmet.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 60.James E, Abreu J, McGinty J, Odegard J, Fillié Y, Hocter C, Culina S, Ladell K, Price D, Alkanani A, et al. (2018). Combinatorial detection of autoreactive CD8+ T cells with HLA-A2 multimers: a multi-centre study by the Immunology of Diabetes Society T Cell Workshop. Diabetologia 61, 658–670. doi: 10.1007/s00125-017-4508-8. [DOI] [PubMed] [Google Scholar]
- 61.Nakayama M, and Michels AW (2021). Using the T Cell Receptor as a Biomarker in Type 1 Diabetes. Front Immunol 12, 777788. 10.3389/fimmu.2021.777788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ahmed S, Cerosaletti K, James E, Long SA, Mannering S, Speake C, Nakayama M, Tree T, Roep BO, Herold KC, and Brusko TM (2019). Standardizing T-Cell Biomarkers in Type 1 Diabetes: Challenges and Recent Advances. Diabetes 68, 1366–1379. 10.2337/db19-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rana J, Perry D, Kumar S, Muñoz-Melero M, Saboungi R, Brusko T, and Biswas M (2021). CAR- and TRuC-redirected regulatory T cells differ in capacity to control adaptive immunity to FVIII. Molecular Therapy 29, 2660–2676. 10.1016/j.ymthe.2021.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dean J, Peters L, Fuhrman C, Seay H, Posgai A, Stimpson S, Brusko M, Perry D, Yeh W, Newby B, et al. (2020). Innate Inflammation Drives NK Cell Activation to Impair Treg Activity. Journal of autoimmunity 108. 10.1016/j.jaut.2020.102417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brusko M, Stewart J, Posgai A, Wasserfall C, Atkinson M, Brusko T, and Keselowsky B (2020). Immunomodulatory Dual-Sized Microparticle System Conditions Human Antigen Presenting Cells Into a Tolerogenic Phenotype In Vitro and Inhibits Type 1 Diabetes-Specific Autoreactive T Cell Responses. Frontiers in Immunology 11, 574447. 10.3389/fimmu.2020.574447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Driver JP, Racine JJ, Ye C, Lamont DJ, Newby BN, McPhee-Leeth CG, Chapman HD, Brusko TM, Chen YG, Mathews CE, and Serreze DV (2017). Interferon gamma Limits Diabetogenic CD8 T-Cell Effector Responses in Type-1 Diabetes. Diabetes 66, 710–721. 10.2337/db16-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Newby BN, Brusko TM, Zou B, Atkinson MA, Clare-Salzler M, and Mathews CE (2017). Type 1 Interferons Potentiate Human CD8+ T Cell Cytotoxicity Through a STAT4 and Granzyme B Dependent Pathway. Diabetes 66, 3061–3071. 10.2337/db17-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yeh WI, Seay HR, Newby B, Posgai AL, Moniz FB, Michels A, Mathews CE, Bluestone JA, and Brusko TM (2017). Avidity and Bystander Suppressive Capacity of Human Regulatory T Cells Expressing De Novo Autoreactive T-Cell Receptors in Type 1 Diabetes. Front Immunol 8, 1313. 10.3389/fimmu.2017.01313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Culina S, Lalanne A, Afonso G, Cerosaletti K, Pinto S, Sebastiani G, Kuranda K, Nigi L, Eugster A, Østerbye T, et al. (2018). Islet-reactive CD8+ T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci Immunol 3, pii: eaao4013. doi: 10.1126/sciimmunol.aao4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Christen U, and Kimmel R (2020). Chemokines as Drivers of the Autoimmune Destruction in Type 1 Diabetes: Opportunity for Therapeutic Intervention in Consideration of an Optimal Treatment Schedule. Front Endocrinol (Lausanne) 11, 591083. 10.3389/fendo.2020.591083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bogdani M, Korpos E, Simeonovic CJ, Parish CR, Sorokin L, and Wight TN (2014). Extracellular matrix components in the pathogenesis of type 1 diabetes. Curr Diab Rep 14, 552. 10.1007/s11892-014-0552-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Benkahla M, Sabouri S, Kiosses W, Rajendran S, Quesada-Masachs E, and von Herrath M (2021). HLA class I hyper-expression unmasks beta cells but not alpha cells to the immune system in pre-diabetes. Journal of Autoimmunity 119, 102628. 10.1016/j.jaut.2021.102628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rodriguez-Calvo T, Ekwall O, Amirian N, Zapardiel-Gonzalo J, and von Herrath MG (2014). Increased immune cell infiltration of the exocrine pancreas: a possible contribution to the pathogenesis of type 1 diabetes. Diabetes 63, 3880–3890. 10.2337/db14-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Quesada-Masachs E, Zilberman S, Rajendran S, Chu T, McArdle S, Kiosses W, Lee J, Yesildag B, Benkahla M, Pawlowska A, et al. (2022). Upregulation of HLA class II in pancreatic beta cells from organ donors with type 1 diabetes. Diabetologia, 387–401. 10.1007/s00125-021-05619-9. [DOI] [PubMed] [Google Scholar]
- 75.Eisenbarth GS (2010). Banting Lecture 2009: An unfinished journey: molecular pathogenesis to prevention of type 1A diabetes. Diabetes 59, 759–774. 59/4/759 [pii] 10.2337/db09-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krogvold L, Wiberg A, Edwin B, Buanes T, Jahnsen FL, Hanssen KF, Larsson E, Korsgren O, Skog O, and Dahl-Jørgensen K (2016). Insulitis and characterisation of infiltrating T cells in surgical pancreatic tail resections from patients at onset of type 1 diabetes. Diabetologia 59, 492–501. 10.1007/s00125-015-3820-4. [DOI] [PubMed] [Google Scholar]
- 77.Jacobsen L, Bocchino L, Evans-Molina C, DiMeglio L, Goland R, Wilson D, Atkinson M, Aye T, Russell W, Wentworth J, et al. (2020). The risk of progression to type 1 diabetes is highly variable in individuals with multiple autoantibodies following screening. Diabetologia 63, 588–596. 10.1007/s00125-019-05047-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wiberg A, Granstam A, Ingvast S, Harkonen T, Knip M, Korsgren O, and Skog O (2015). Characterization of human organ donors testing positive for type 1 diabetes-associated autoantibodies. Clin Exp Immunol 182, 278–288. 10.1111/cei.12698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rodriguez-Calvo T, Zapardiel-Gonzalo J, Amirian N, Castillo E, Lajevardi Y, Krogvold L, Dahl-Jorgensen K, and von Herrath MG (2017). Increase in Pancreatic Proinsulin and Preservation of beta-Cell Mass in Autoantibody-Positive Donors Prior to Type 1 Diabetes Onset. Diabetes 66, 1334–1345. 10.2337/db16-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jacobsen LM, Bocchino L, Evans-Molina C, DiMeglio L, Goland R, Wilson DM, Atkinson MA, Aye T, Russell WE, Wentworth JM, et al. (2020). The risk of progression to type 1 diabetes is highly variable in individuals with multiple autoantibodies following screening. Diabetologia 63, 588–596. 10.1007/s00125-019-05047-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.von Herrath M, Sanda S, and Herold K (2007). Type 1 diabetes as a relapsing-remitting disease? Nat Rev Immunol 7, 988–994. nri2192 [pii] 10.1038/nri2192. [DOI] [PubMed] [Google Scholar]
- 82.van Belle TL, Coppieters KT, and von Herrath MG (2011). Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev 91, 79–118. 10.1152/physrev.00003.2010. [DOI] [PubMed] [Google Scholar]
- 83.Herold KC, Vignali DA, Cooke A, and Bluestone JA (2013). Type 1 diabetes: translating mechanistic observations into effective clinical outcomes. Nat Rev Immunol 13, 243–256. 10.1038/nri3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bender C, Rajendran S, and von Herrath M (2021). New Insights Into the Role of Autoreactive CD8 T Cells and Cytokines in Human Type 1 Diabetes. Frontiers in Endocrinology (Lausanne) 11, 606434. 10.3389/fendo.2020.606434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Newby BN, and Mathews CE (2017). Type I Interferon Is a Catastrophic Feature of the Diabetic Islet Microenvironment. Front Endocrinol (Lausanne) 8, 232. 10.3389/fendo.2017.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hu X, Gattis C, Olroyd AG, Friera AM, White K, Young C, Basco R, Lamba M, Wells F, Ankala R, et al. (2023). Human hypoimmune primary pancreatic islets avoid rejection and autoimmunity and alleviate diabetes in allogeneic humanized mice. Sci Transl Med 15, eadg5794. 10.1126/scitranslmed.adg5794. [DOI] [PubMed] [Google Scholar]
- 87.Rui J, Deng S, Perdigoto A, Ponath G, Kursawe R, Lawlor N, Sumida T, Levine-Ritterman M, Stitzel M, Pitt D, et al. (2021). Tet2 Controls the Responses of β cells to Inflammation in Autoimmune Diabetes. Nature Communications 12, 5074. 10.1038/s41467-021-25367-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vecchione A, Jofra T, Gerosa J, Shankwitz K, Di Fonte R, Galvani G, Ippolito E, Cicalese MP, Schultz AR, Seay HR, et al. (2021). Reduced Follicular Regulatory T Cells in Spleen and Pancreatic Lymph Nodes of Patients With Type 1 Diabetes. Diabetes 70, 2892–2902. 10.2337/db21-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Korpos É, Kadri N, Loismann S, Findeisen CR, Arfuso F, Burke GW, Richardson SJ, Morgan NG, Bogdani M, Pugliese A, and Sorokin L (2021). Identification and characterisation of tertiary lymphoid organs in human type 1 diabetes. Diabetologia 64, 1626–1641. 10.1007/s00125-021-05453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Osum KC, Burrack AL, Martinov T, Sahli NL, Mitchell JS, Tucker CG, Pauken KE, Papas K, Appakalai B, Spanier JA, and Fife BT (2018). Interferon-gamma drives programmed death-ligand 1 expression on islet β cells to limit T cell function during autoimmune diabetes. Sci Rep 8, 8295. 10.1038/s41598-018-26471-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Colli ML, Hill JLE, Marroquí L, Chaffey J, Dos Santos RS, Leete P, Coomans de Brachène A, Paula FMM, Op de Beeck A, Castela A, et al. (2018). PDL1 is expressed in the islets of people with type 1 diabetes and is up-regulated by interferons-α and-γ via IRF1 induction. EBioMedicine 36, 367–375. 10.1016/j.ebiom.2018.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tucker C, Dwyer A, Fife B, and Martinov T (2021). The Role of Programmed Death-1 in Type 1 Diabetes. Current Diabetes Reports 21, 20. 10.1007/s11892-021-01384-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sharp R, Brown M, Shapiro M, Posgai A, and Brusko T (2021). The Immunoregulatory Role of the Signal Regulatory Protein Family and CD47 Signaling Pathway in Type 1 Diabetes. Frontiers in Immunology 12, 739048. 10.3389/fimmu.2021.739048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lindsay R, Whitesell J, Dew K, Rodriguez E, Sandor A, Tracy D, Yannacone S, Basta B, Jacobelli J, and Friedman R (2021). MERTK on mononuclear phagocytes regulates T cell antigen recognition at autoimmune and tumor sites. The Journal of Experimental Medicine 218, e20200464. 10.1084/jem.20200464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Skog O, Korsgren S, Melhus A, and Korsgren O (2013). Revisiting the notion of type 1 diabetes being a T-cell-mediated autoimmune disease. Curr Opin Endocrinol Diabetes Obes 20, 118–123. 10.1097/MED.0b013e32835edb89. [DOI] [PubMed] [Google Scholar]
- 96.Roep BO, Thomaidou S, van Tienhoven R, and Zaldumbide A (2021). Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?). Nat Rev Endocrinol 17, 150–161. 10.1038/s41574-020-00443-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bottazzo GF (1986). Lawrence lecture. Death of a beta cell: homicide or suicide? Diabet Med 3, 119–130. 10.1111/j.1464-5491.1986.tb00722.x. [DOI] [PubMed] [Google Scholar]
- 98.Kulkarni A, Muralidharan C, May SC, Tersey SA, and Mirmira RG (2022). Inside the β Cell: Molecular Stress Response Pathways in Diabetes Pathogenesis. Endocrinology 164. 10.1210/endocr/bqac184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, and Bhushan A (2019). Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab. 10.1016/j.cmet.2019.01.021. [DOI] [PubMed] [Google Scholar]
- 100.Phelps EA, Cianciaruso C, Michael IP, Pasquier M, Kanaani J, Nano R, Lavallard V, Billestrup N, Hubbell JA, and Baekkeskov S (2016). Aberrant Accumulation of the Diabetes Autoantigen GAD65 in Golgi Membranes in Conditions of ER Stress and Autoimmunity. Diabetes 65, 2686–2699. 10.2337/db16-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Doliba NM, Rozo AV, Roman J, Qin W, Traum D, Gao L, Liu J, Manduchi E, Liu C, Golson ML, et al. (2022). α Cell dysfunction in islets from nondiabetic, glutamic acid decarboxylase autoantibody-positive individuals. J Clin Invest 132. 10.1172/JCI156243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Korpos É, Kadri N, Kappelhoff R, Wegner J, Overall CM, Weber E, Holmberg D, Cardell S, and Sorokin L (2013). The peri-islet basement membrane, a barrier to infiltrating leukocytes in type 1 diabetes in mouse and human. Diabetes 62, 531–542. 10.2337/db12-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Peters L, Posgai A, and Brusko T (2019). Islet-immune interactions in type 1 diabetes: the nexus of beta cell destruction. Clinical and Experimental Immunology 198, 326–340. 10.1111/cei.13349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Damond N, Engler S, Zanotelli VRT, Schapiro D, Wasserfall CH, Kusmartseva I, Nick HS, Thorel F, Herrera PL, Atkinson MA, and Bodenmiller B (2019). A Map of Human Type 1 Diabetes Progression by Imaging Mass Cytometry. Cell Metab 29, 755–768.e755. 10.1016/j.cmet.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Geravandi S, Richardson S, Pugliese A, and Maedler K (2021). Localization of enteroviral RNA within the pancreas in donors with T1D and T1D-associated autoantibodies. Cell Rep Med 2, 100371. 10.1016/j.xcrm.2021.100371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Apaolaza PS, Balcacean D, Zapardiel-Gonzalo J, Nelson G, Lenchik N, Akhbari P, Gerling I, Richardson SJ, Rodriguez-Calvo T, and Group n.-V. (2021). Islet expression of type I interferon response sensors is associated with immune infiltration and viral infection in type 1 diabetes. Sci Adv 7. 10.1126/sciadv.abd6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jeffery N, Richardson S, Chambers D, Morgan NG, and Harries LW (2019). Cellular stressors may alter islet hormone cell proportions by moderation of alternative splicing patterns. Hum Mol Genet 28, 2763–2774. 10.1093/hmg/ddz094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Krogvold L, Genoni A, Puggioni A, Campani D, Richardson SJ, Flaxman CS, Edwin B, Buanes T, Dahl-Jørgensen K, and Toniolo A (2022). Live enteroviruses, but not other viruses, detected in human pancreas at the onset of type 1 diabetes in the DiViD study. Diabetologia 65, 2108–2120. 10.1007/s00125-022-05779-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nigi L, Brusco N, Grieco GE, Fignani D, Licata G, Formichi C, Aiello E, Marselli L, Marchetti P, Krogvold L, et al. (2022). Increased Expression of Viral Sensor MDA5 in Pancreatic Islets and in Hormone-Negative Endocrine Cells in Recent Onset Type 1 Diabetic Donors. Front Immunol 13, 833141. 10.3389/fimmu.2022.833141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ifie E, Russell MA, Dhayal S, Leete P, Sebastiani G, Nigi L, Dotta F, Marjomäki V, Eizirik DL, Morgan NG, and Richardson SJ (2018). Unexpected subcellular distribution of a specific isoform of the Coxsackie and adenovirus receptor, CAR-SIV, in human pancreatic beta cells. Diabetologia 61, 2344–2355. 10.1007/s00125-018-4704-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rodriguez-Calvo T, Zapardiel-Gonzalo J, Amirian N, Castillo E, Lajevardi Y, Krogvold L, Dahl-Jørgensen K, and von Herrath MG (2017). Increase in Pancreatic Proinsulin and Preservation of β-Cell Mass in Autoantibody-Positive Donors Prior to Type 1 Diabetes Onset. Diabetes 66, 1334–1345. 10.2337/db16-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hiller H, Beachy DE, Lebowitz JJ, Engler S, Mason JR, Miller DR, Kusmarteva I, Jacobsen LM, Posgai AL, Khoshbouei H, et al. (2021). Monogenic Diabetes and Integrated Stress Response Genes Display Altered Gene Expression in Type 1 Diabetes. Diabetes 70, 1885–1897. 10.2337/db21-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hiller H, Yang C, Beachy D, Kusmartseva I, Candelario-Jalil E, Posgai A, Nick H, Schatz D, Atkinson M, and Wasserfall C (2021). Altered cellular localisation and expression, together with unconventional protein trafficking, of prion protein, PrP C, in type 1 diabetes. Diabetologia 64, 2279–2291. 10.1007/s00125-021-05501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Muralidharan C, Conteh AM, Marasco MR, Crowder JJ, Kuipers J, de Boer P, and Linnemann AK (2021). Pancreatic beta cell autophagy is impaired in type 1 diabetes. Diabetologia 64, 865–877. 10.1007/s00125-021-05387-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wu W, Syed F, Simpson E, Lee CC, Liu J, Chang G, Dong C, Seitz C, Eizirik DL, Mirmira RG, et al. (2021). The Impact of Pro-Inflammatory Cytokines on Alternative Splicing Patterns in Human Islets. Diabetes. 10.2337/db20-0847. [DOI] [PubMed] [Google Scholar]
- 116.Nomoto H, Pei L, Montemurro C, Rosenberger M, Furterer A, Coppola G, Nadel B, Pellegrini M, Gurlo T, Butler PC, and Tudzarova S (2020). Activation of the HIF1α/PFKFB3 stress response pathway in beta cells in type 1 diabetes. Diabetologia 63, 149–161. 10.1007/s00125-019-05030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Eizirik DL, Sammeth M, Bouckenooghe T, Bottu G, Sisino G, Igoillo-Esteve M, Ortis F, Santin I, Colli ML, Barthson J, et al. (2012). The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet 8, e1002552. 10.1371/journal.pgen.1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, Evans-Molina C, Rickus JL, Maier B, and Mirmira RG (2012). Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 61, 818–827. 10.2337/db11-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marhfour I, Lopez XM, Lefkaditis D, Salmon I, Allagnat F, Richardson SJ, Morgan NG, and Eizirik DL (2012). Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 55, 2417–2420. 10.1007/s00125-012-2604-3. [DOI] [PubMed] [Google Scholar]
- 120.Teitelman G (2022). Abnormal Expression of an Insulin Synthesizing Enzyme in Islets of Adult Autoantibody Positive Donors. J Histochem Cytochem 70, 695–706. 10.1369/00221554221138368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sims EK, Syed F, Nyalwidhe J, Bahnson HT, Haataja L, Speake C, Morris MA, Balamurugan AN, Mirmira RG, Nadler J, et al. (2019). Abnormalities in proinsulin processing in islets from individuals with longstanding T1D. Transl Res 213, 90–99. 10.1016/j.trsl.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Buitinga M, Callebaut A, Marques Câmara Sodré F, Crèvecoeur I, Blahnik-Fagan G, Yang ML, Bugliani M, Arribas-Layton D, Marré M, Cook DP, et al. (2018). Inflammation-Induced Citrullinated Glucose-Regulated Protein 78 Elicits Immune Responses in Human Type 1 Diabetes. Diabetes 67, 2337–2348. 10.2337/db18-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Marre ML, McGinty JW, Chow IT, DeNicola ME, Beck NW, Kent SC, Powers AC, Bottino R, Harlan DM, Greenbaum CJ, et al. (2018). Modifying Enzymes Are Elicited by ER Stress, Generating Epitopes That Are Selectively Recognized by CD4. Diabetes 67, 1356–1368. 10.2337/db17-1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Coomans de Brachène A, Dos Santos RS, Marroqui L, Colli ML, Marselli L, Mirmira RG, Marchetti P, and Eizirik DL (2018). IFN-α induces a preferential long-lasting expression of MHC class I in human pancreatic beta cells. Diabetologia 61, 636–640. 10.1007/s00125-017-4536-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sims EK, Evans-Molina C, Tersey SA, Eizirik DL, and Mirmira RG (2018). Biomarkers of islet beta cell stress and death in type 1 diabetes. Diabetologia 61, 2259–2265. 10.1007/s00125-018-4712-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sims EK, Chaudhry Z, Watkins R, Syed F, Blum J, Ouyang F, Perkins SM, Mirmira RG, Sosenko J, DiMeglio LA, and Evans-Molina C (2016). Elevations in the Fasting Serum Proinsulin-to-C-Peptide Ratio Precede the Onset of Type 1 Diabetes. Diabetes Care 39, 1519–1526. 10.2337/dc15-2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fisher MM, Watkins RA, Blum J, Evans-Molina C, Chalasani N, DiMeglio LA, Mather KJ, Tersey SA, and Mirmira RG (2015). Elevations in Circulating Methylated and Unmethylated Preproinsulin DNA in New-Onset Type 1 Diabetes. Diabetes 64, 3867–3872. 10.2337/db15-0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Akirav EM, Lebastchi J, Galvan EM, Henegariu O, Akirav M, Ablamunits V, Lizardi PM, and Herold KC (2011). Detection of β cell death in diabetes using differentially methylated circulating DNA. Proc Natl Acad Sci U S A 108, 19018–19023. 10.1073/pnas.1111008108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Speake C, Ylescupidez A, Neiman D, Shemer R, Glaser B, Tersey SA, Usmani-Brown S, Clark P, Wilhelm JJ, Bellin MD, et al. (2020). Circulating Unmethylated Insulin DNA As a Biomarker of Human Beta Cell Death: A Multi-laboratory Assay Comparison. J Clin Endocrinol Metab 105, 781–791. 10.1210/clinem/dgaa008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Syed F, Tersey SA, Turatsinze JV, Felton JL, Kang NJ, Nelson JB, Sims EK, Defrance M, Bizet M, Fuks F, et al. (2020). Circulating unmethylated CHTOP and INS DNA fragments provide evidence of possible islet cell death in youth with obesity and diabetes. Clin Epigenetics 12, 116. 10.1186/s13148-020-00906-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lehmann-Werman R, Neiman D, Zemmour H, Moss J, Magenheim J, Vaknin-Dembinsky A, Rubertsson S, Nellgård B, Blennow K, Zetterberg H, et al. (2016). Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci U S A 113, E1826–1834. 10.1073/pnas.1519286113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Neiman D, Gillis D, Piyanzin S, Cohen D, Fridlich O, Moss J, Zick A, Oron T, Sundberg F, Forsander G, et al. (2020). Multiplexing DNA methylation markers to detect circulating cell-free DNA derived from human pancreatic β cells. JCI Insight 5. 10.1172/jci.insight.136579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Srikanta S, Ganda OP, Gleason RE, Jackson RA, Soeldner JS, and Eisenbarth GS (1984). Pre-type I diabetes. Linear loss of beta cell response to intravenous glucose. Diabetes 33, 717–720. 10.2337/diab.33.8.717. [DOI] [PubMed] [Google Scholar]
- 134.Lo SS, Hawa M, Beer SF, Pyke DA, and Leslie RD (1992). Altered islet beta-cell function before the onset of type 1 (insulin-dependent) diabetes mellitus. Diabetologia 35, 277–282. 10.1007/BF00400930. [DOI] [PubMed] [Google Scholar]
- 135.Diedisheim M, Mallone R, Boitard C, and Larger E (2016). β-cell Mass in Nondiabetic Autoantibody-Positive Subjects: An Analysis Based on the Network for Pancreatic Organ Donors Database. J Clin Endocrinol Metab 101, 1390–1397. 10.1210/jc.2015-3756. [DOI] [PubMed] [Google Scholar]
- 136.Krogvold L, Skog O, Sundström G, Edwin B, Buanes T, Hanssen KF, Ludvigsson J, Grabherr M, Korsgren O, and Dahl-Jørgensen K (2015). Function of Isolated Pancreatic Islets From Patients at Onset of Type 1 Diabetes: Insulin Secretion Can Be Restored After Some Days in a Nondiabetogenic Environment In Vitro: Results From the DiViD Study. Diabetes 64, 2506–2512. 10.2337/db14-1911. [DOI] [PubMed] [Google Scholar]
- 137.Lupi R, Marselli L, Dionisi S, Del Guerra S, Boggi U, Del Chiaro M, Lencioni C, Bugliani M, Mosca F, Di Mario U, et al. (2004). Improved insulin secretory function and reduced chemotactic properties after tissue culture of islets from type 1 diabetic patients. Diabetes Metab Res Rev 20, 246–251. 10.1002/dmrr.460. [DOI] [PubMed] [Google Scholar]
- 138.Oram RA, Sims EK, and Evans-Molina C (2019). Beta cells in type 1 diabetes: mass and function; sleeping or dead? Diabetologia 62, 567–577. 10.1007/s00125-019-4822-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mezza T, Ferraro PM, Di Giuseppe G, Moffa S, Cefalo CM, Cinti F, Impronta F, Capece U, Quero G, Pontecorvi A, et al. (2021). Pancreaticoduodenectomy model demonstrates a fundamental role of dysfunctional β cells in predicting diabetes. J Clin Invest 131. 10.1172/JCI146788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Atkinson MA (2014). Pancreatic biopsies in type 1 diabetes: revisiting the myth of Pandora’s box. Diabetologia 57, 656–659. 10.1007/s00125-013-3159-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sims EK, Bahnson HT, Nyalwidhe J, Haataja L, Davis AK, Speake C, DiMeglio LA, Blum J, Morris MA, Mirmira RG, et al. (2019). Proinsulin Secretion Is a Persistent Feature of Type 1 Diabetes. Diabetes Care 42, 258–264. 10.2337/dc17-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang L, Lovejoy NF, and Faustman DL (2012). Persistence of prolonged C-peptide production in type 1 diabetes as measured with an ultrasensitive C-peptide assay. Diabetes Care 35, 465–470. 10.2337/dc11-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Oram RA, Jones AG, Besser RE, Knight BA, Shields BM, Brown RJ, Hattersley AT, and McDonald TJ (2014). The majority of patients with long-duration type 1 diabetes are insulin microsecretors and have functioning beta cells. Diabetologia 57, 187–191. 10.1007/s00125-013-3067-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lam CJ, Jacobson DR, Rankin MM, Cox AR, and Kushner JA (2017). β Cells Persist in T1D Pancreata Without Evidence of Ongoing β-Cell Turnover or Neogenesis. J Clin Endocrinol Metab 102, 2647–2659. 10.1210/jc.2016-3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, and Atkinson MA (2016). Insulitis and β-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes 65, 719–731. 10.2337/db15-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Butler AE, Galasso R, Meier JJ, Basu R, Rizza RA, and Butler PC (2007). Modestly increased beta cell apoptosis but no increased beta cell replication in recent-onset type 1 diabetic patients who died of diabetic ketoacidosis. Diabetologia 50, 2323–2331. 10.1007/s00125-007-0794-x. [DOI] [PubMed] [Google Scholar]
- 147.Hao W, Woodwyk A, Beam C, Bahnson HT, Palmer JP, and Greenbaum CJ (2017). Assessment of β Cell Mass and Function by AIRmax and Intravenous Glucose in High-Risk Subjects for Type 1 Diabetes. J Clin Endocrinol Metab 102, 4428–4434. 10.1210/jc.2017-01713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Muratore M, Santos C, and Rorsman P (2021). The vascular architecture of the pancreatic islets: A homage to August Krogh. Comp Biochem Physiol A Mol Integr Physiol 252, 110846. 10.1016/j.cbpa.2020.110846. [DOI] [PubMed] [Google Scholar]
- 149.Almaça J, Weitz J, Rodriguez-Diaz R, Pereira E, and Caicedo A (2018). The Pericyte of the Pancreatic Islet Regulates Capillary Diameter and Local Blood Flow. Cell Metab 27, 630–644.e634. 10.1016/j.cmet.2018.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Burganova G, Bridges C, Thorn P, and Landsman L (2021). The Role of Vascular Cells in Pancreatic Beta-Cell Function. Front Endocrinol (Lausanne) 12, 667170. 10.3389/fendo.2021.667170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Canzano JS, Nasif LH, Butterworth EA, Fu DA, Atkinson MA, and Campbell-Thompson M (2019). Islet Microvasculature Alterations With Loss of Beta-cells in Patients With Type 1 Diabetes. J Histochem Cytochem 67, 41–52. 10.1369/0022155418778546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Granlund L, Hedin A, Korsgren O, Skog O, and Lundberg M (2022). Altered microvasculature in pancreatic islets from subjects with type 1 diabetes. PLoS One 17, e0276942. 10.1371/journal.pone.0276942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Richardson TM, Saunders DC, Haliyur R, Shrestha S, Cartailler JP, Reinert RB, Petronglo J, Bottino R, Aramandla R, Bradley AM, et al. (2023). Human pancreatic capillaries and nerve fibers persist in type 1 diabetes despite beta cell loss. Am J Physiol Endocrinol Metab 324, E251–E267. 10.1152/ajpendo.00246.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Cottle L, Gan WJ, Gilroy I, Samra JS, Gill AJ, Loudovaris T, Thomas HE, Hawthorne WJ, Kebede MA, and Thorn P (2021). Structural and functional polarisation of human pancreatic beta cells in islets from organ donors with and without type 2 diabetes. Diabetologia 64, 618–629. 10.1007/s00125-020-05345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dybala MP, Kuznetsov A, Motobu M, Hendren-Santiago BK, Philipson LH, Chervonsky AV, and Hara M (2020). Integrated Pancreatic Blood Flow: Bidirectional Microcirculation Between Endocrine and Exocrine Pancreas. Diabetes 69, 1439–1450. 10.2337/db19-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Evanko SP, Potter-Perigo S, Bollyky PL, Nepom GT, and Wight TN (2012). Hyaluronan and versican in the control of human T-lymphocyte adhesion and migration. Matrix Biol 31, 90–100. 10.1016/j.matbio.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bogdani M, Johnson PY, Potter-Perigo S, Nagy N, Day AJ, Bollyky PL, and Wight TN (2014). Hyaluronan and Hyaluronan-Binding Proteins Accumulate in Both Human Type 1 Diabetic Islets and Lymphoid Tissues and Associate With Inflammatory Cells in Insulitis. Diabetes 63, 2727–2743. 10.2337/db13-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bogdani M, Speake C, Dufort MJ, Johnson PY, Larmore MJ, Day AJ, Wight TN, Lernmark Å, and Greenbaum CJ (2020). Hyaluronan deposition in islets may precede and direct the location of islet immune-cell infiltrates. Diabetologia 63, 549–560. 10.1007/s00125-019-05066-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nagy N, Kaber G, Johnson PY, Gebe JA, Preisinger A, Falk BA, Sunkari VG, Gooden MD, Vernon RB, Bogdani M, et al. (2015). Inhibition of hyaluronan synthesis restores immune tolerance during autoimmune insulitis. J Clin Invest 125, 3928–3940. 10.1172/JCI79271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ziolkowski A, Popp S, Freeman C, Parish C, and Simeonovic C (2012). Heparan sulfate and heparanase play key roles in mouse β cell survival and autoimmune diabetes. The Journal of clinical investigation 122, 132–141. 10.1172/JCI46177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Simeonovic CJ, Popp SK, Starrs LM, Brown DJ, Ziolkowski AF, Ludwig B, Bornstein SR, Wilson JD, Pugliese A, Kay TWH, et al. (2018). Loss of intra-islet heparan sulfate is a highly sensitive marker of type 1 diabetes progression in humans. PLoS One 13, e0191360. 10.1371/journal.pone.0191360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Scott-Solomon E, Boehm E, and Kuruvilla R (2021). The sympathetic nervous system in development and disease. Nat Rev Neurosci 22, 685–702. 10.1038/s41583-021-00523-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Borden P, Houtz J, Leach SD, and Kuruvilla R (2013). Sympathetic innervation during development is necessary for pancreatic islet architecture and functional maturation. Cell Rep 4, 287–301. 10.1016/j.celrep.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hampton RF, Jimenez-Gonzalez M, and Stanley SA (2022). Unravelling innervation of pancreatic islets. Diabetologia 65, 1069–1084. 10.1007/s00125-022-05691-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Mundinger TO, Mei Q, Foulis AK, Fligner CL, Hull RL, and Taborsky GJ (2016). Human Type 1 Diabetes Is Characterized by an Early, Marked, Sustained, and Islet-Selective Loss of Sympathetic Nerves. Diabetes 65, 2322–2330. 10.2337/db16-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Campbell-Thompson M, Butterworth E, Boatwright J, Nair M, Nasif L, Nasif K, Revell A, Riva A, Mathews C, Gerling I, et al. (2021). Islet sympathetic innervation and islet neuropathology in patients with type 1 diabetes. Scientific Reports 11, 6562. 10.1038/s41598-021-85659-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Saisho Y, Butler AE, Meier JJ, Monchamp T, Allen-Auerbach M, Rizza RA, and Butler PC (2007). Pancreas volumes in humans from birth to age one hundred taking into account sex, obesity, and presence of type-2 diabetes. Clin Anat 20, 933–942. 10.1002/ca.20543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Cecil RL (1909). A STUDY OF THE PATHOLOGICAL ANATOMY OF THE PANCREAS IN NINETY CASES OF DIABETES MELLITUS. Journal of Experimental Medicine 11, 266–290. 10.1084/jem.11.2.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Domschke W, Tympner F, Domschke S, and Demling L (1975). Exocrine pancreatic function in juvenile diabetics. Am J Dig Dis 20, 309–312. 10.1007/BF01237787. [DOI] [PubMed] [Google Scholar]
- 170.Frier BM, Saunders JH, Wormsley KG, and Bouchier IA (1976). Exocrine pancreatic function in juvenile-onset diabetes mellitus. Gut 17, 685–691. 10.1136/gut.17.9.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Foo Y, Rosalki SB, Ramdial L, Mikhailidis D, and Dandona P (1980). Serum isoamylase activities in diabetes mellitus. J Clin Pathol 33, 1102–1105. 10.1136/jcp.33.11.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Junglee D, De Albarran R, Katrak A, Freedman DB, Beckett AG, and Dandona P (1983). Low pancreatic lipase in insulin-dependent diabetics. J Clin Pathol 36, 200–202. 10.1136/jcp.36.2.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Creutzfeldt W, Gleichmann D, Otto J, Stöckmann F, Maisonneuve P, and Lankisch PG (2005). Follow-up of exocrine pancreatic function in type-1 diabetes mellitus. Digestion 72, 71–75. 10.1159/000087660. [DOI] [PubMed] [Google Scholar]
- 174.Williams AJ, Thrower SL, Sequeiros IM, Ward A, Bickerton AS, Triay JM, Callaway MP, and Dayan CM (2012). Pancreatic volume is reduced in adult patients with recently diagnosed type 1 diabetes. J Clin Endocrinol Metab 97, E2109–2113. 10.1210/jc.2012-1815. [DOI] [PubMed] [Google Scholar]
- 175.Campbell-Thompson ML, Kaddis JS, Wasserfall C, Haller MJ, Pugliese A, Schatz DA, Shuster JJ, and Atkinson MA (2016). The influence of type 1 diabetes on pancreatic weight. Diabetologia 59, 217–221. 10.1007/s00125-015-3752-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Virostko J, Williams J, Hilmes M, Bowman C, Wright JJ, Du L, Kang H, Russell WE, Powers AC, and Moore DJ (2019). Pancreas Volume Declines During the First Year After Diagnosis of Type 1 Diabetes and Exhibits Altered Diffusion at Disease Onset. Diabetes Care 42, 248–257. 10.2337/dc18-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gaglia J, Harisinghani M, Aganj I, Wojtkiewicz G, Hedgire S, Benoist C, Mathis D, and Weissleder R (2015). Noninvasive mapping of pancreatic inflammation in recent-onset type-1 diabetes patients. Proceedings of the National Academy of Sciences 112, 2139–2144. 10.1073/pnas.1424993112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Campbell-Thompson ML, Filipp SL, Grajo JR, Nambam B, Beegle R, Middlebrooks EH, Gurka MJ, Atkinson MA, Schatz DA, and Haller MJ (2019). Relative Pancreas Volume Is Reduced in First-Degree Relatives of Patients With Type 1 Diabetes. Diabetes Care 42, 281–287. 10.2337/dc18-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Williams J, Hilmes M, Archer B, Dulaney A, Du L, Kang H, Russell W, Powers A, Moore D, and Virostko J (2020). Repeatability and Reproducibility of Pancreas Volume Measurements Using MRI. Scientific Reports 10, 4767. 10.1038/s41598-020-61759-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Campbell-Thompson M, Wasserfall C, Montgomery EL, Atkinson MA, and Kaddis JS (2012). Pancreas organ weight in individuals with disease-associated autoantibodies at risk for type 1 diabetes. JAMA 308, 2337–2339. 10.1001/jama.2012.15008. [DOI] [PubMed] [Google Scholar]
- 181.Kusmartseva I, Beery M, Hiller H, Padilla M, Selman S, Posgai A, Nick HS, Campbell-Thompson M, Schatz DA, Haller MJ, et al. (2020). Temporal Analysis of Amylase Expression in Control, Autoantibody-Positive, and Type 1 Diabetes Pancreatic Tissues. Diabetes 69, 60–66. 10.2337/db19-0554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wright J, Saunders D, Dai C, Poffenberger G, Cairns B, Serreze D, Harlan D, Bottino R, Brissova M, and Powers A (2020). Decreased Pancreatic Acinar Cell Number in Type 1 Diabetes. Diabetologia 63, 1418–1423. 10.1007/s00125-020-05155-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tang X, Kusmartseva I, Kulkarni S, Posgai A, Speier S, Schatz D, Haller M, Campbell-Thompson M, Wasserfall C, Roep B, et al. (2020). Image-Based Machine Learning Algorithms for Disease Characterization in the Human Type 1 Diabetes Pancreas. American Journal of Pathology S0002–9440, 30551–30554. 10.1016/j.ajpath.2020.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Malaisse-Lagae F, Ravazzola M, Robberecht P, Vandermeers A, Malaisse WJ, and Orci L (1975). Exocrine pancreas: evidence for topographic partition of secretory function. Science 190, 795–797. 10.1126/science.1105788. [DOI] [PubMed] [Google Scholar]
- 185.Atkinson MA, Campbell-Thompson M, Kusmartseva I, and Kaestner KH (2020). Organisation of the human pancreas in health and in diabetes. Diabetologia 63, 1966–1973. 10.1007/s00125-020-05203-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Saisho Y (2016). Pancreas Volume and Fat Deposition in Diabetes and Normal Physiology: Consideration of the Interplay Between Endocrine and Exocrine Pancreas. Rev Diabet Stud 13, 132–147. 10.1900/RDS.2016.13.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Li X, Campbell-Thompson M, Wasserfall CH, McGrail K, Posgai A, Schultz AR, Brusko TM, Shuster J, Liang F, Muir A, et al. (2017). Serum Trypsinogen Levels in Type 1 Diabetes. Diabetes Care 40, 577–582. 10.2337/dc16-1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ross J, Wasserfall C, Bacher R, Perry D, McGrail K, Posgai A, Dong X, Muir A, Li X, Campbell-Thompson M, et al. (2021). Exocrine Pancreatic Enzymes Are a Serological Biomarker for Type 1 Diabetes Staging and Pancreas Size. Diabetes, db200995. 10.2337/db20-0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Campbell-Thompson ML, Schatz DA, Kaddis JS, and Atkinson MA (2016). Pancreatic duct hyperplasia/dysplasia in type 1 diabetes and pancreatic weight in individuals with and without diabetes. Reply to Kobayashi T, Aida K, Fukui T et al [letter] and Saisho Y [letter]. Diabetologia 59, 870–872. 10.1007/s00125-016-3889-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Moin AS, Butler PC, and Butler AE (2017). Increased Proliferation of the Pancreatic Duct Gland Compartment in Type 1 Diabetes. J Clin Endocrinol Metab 102, 200–209. 10.1210/jc.2016-3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Liu CW, Atkinson MA, and Zhang Q (2016). Type 1 diabetes cadaveric human pancreata exhibit a unique exocrine tissue proteomic profile. Proteomics 16, 1432–1446. 10.1002/pmic.201500333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Redondo M, Evans-Molina C, Steck A, Atkinson M, and Sosenko J (2019). The Influence of Type 2 Diabetes-Associated Factors on Type 1 Diabetes. Diabetes Care 42, 1357–1364. 10.2337/dc19-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Dooley J, Tian L, Schonefeldt S, Delghingaro-Augusto V, Garcia-Perez J, Pasciuto E, Di Marino D, Carr E, Oskolkov N, Lyssenko V, et al. (2016). Genetic predisposition for beta cell fragility underlies type 1 and type 2 diabetes. Nature Genetics 48, 519–527. 10.1038/ng.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Josefsen K, Krogvold L, Gerling IC, Pociot F, Dahl-Jørgensen K, and Buschard K (2022). Development of Type 1 Diabetes may occur through a Type 2 Diabetes mechanism. Front Endocrinol (Lausanne) 13, 1032822. 10.3389/fendo.2022.1032822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Rowe P, Wasserfall C, Croker B, Campbell-Thompson M, Pugliese A, Atkinson M, and Schatz D (2013). Increased complement activation in human type 1 diabetes pancreata. Diabetes Care 36, 3815–3817. 10.2337/dc13-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Li X, and Atkinson MA (2015). The role for gut permeability in the pathogenesis of type 1 diabetes--a solid or leaky concept? Pediatr Diabetes 16, 485–492. 10.1111/pedi.12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bender C, Rodriguez-Calvo T, Amirian N, Coppieters KT, and von Herrath MG (2020). The healthy exocrine pancreas contains preproinsulin-specific CD8 T cells that attack islets in type 1 diabetes. Sci Adv 6. 10.1126/sciadv.abc5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Serrano J, Laughlin MR, Bellin MD, Yadav D, Chinchilli VM, Andersen DK, and (T1DAPC) T.D.i.A.P.C. (2022). Type 1 Diabetes in Acute Pancreatitis Consortium: From Concept to Reality. Pancreas 51, 563–567. 10.1097/MPA.0000000000002073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Casu A, Grippo PJ, Wasserfall C, Sun Z, Linsley PS, Hamerman JA, Fife BT, Lacy-Hulbert A, Toledo FGS, Hart PA, et al. (2022). Evaluating the Immunopathogenesis of Diabetes After Acute Pancreatitis in the Diabetes RElated to Acute Pancreatitis and Its Mechanisms Study: From the Type 1 Diabetes in Acute Pancreatitis Consortium. Pancreas 51, 580–585. 10.1097/MPA.0000000000002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Girdhar K, Huang Q, Chow IT, Vatanen T, Brady C, Raisingani A, Autissier P, Atkinson MA, Kwok WW, Kahn CR, and Altindis E (2022). A gut microbial peptide and molecular mimicry in the pathogenesis of type 1 diabetes. Proc Natl Acad Sci U S A 119, e2120028119. 10.1073/pnas.2120028119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Rewers M, and Ludvigsson J (2016). Environmental risk factors for type 1 diabetes. Lancet 387, 2340–2348. 10.1016/S0140-6736(16)30507-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, Lernmark Å, Hagopian WA, Rewers MJ, She JX, et al. (2018). The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature 562, 589–594. 10.1038/s41586-018-0620-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.de Groot P, Nikolic T, Pellegrini S, Sordi V, Imangaliyev S, Rampanelli E, Hanssen N, Attaye I, Bakker G, Duinkerken G, et al. (2021). Faecal microbiota transplantation halts progression of human new-onset type 1 diabetes in a randomised controlled trial. Gut 70, 92–105. 10.1136/gutjnl-2020-322630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Vehik K, Lynch KF, Wong MC, Tian X, Ross MC, Gibbs RA, Ajami NJ, Petrosino JF, Rewers M, Toppari J, et al. (2019). Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat Med 25, 1865–1872. 10.1038/s41591-019-0667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ramos-Rodríguez M, Pérez-González B, and Pasquali L (2021). The β-Cell Genomic Landscape in T1D: Implications for Disease Pathogenesis. Curr Diab Rep 21, 1. 10.1007/s11892-020-01370-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Chiou J, Geusz RJ, Okino ML, Han JY, Miller M, Melton R, Beebe E, Benaglio P, Huang S, Korgaonkar K, et al. (2021). Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 594, 398–402. 10.1038/s41586-021-03552-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Alvelos MI, Juan-Mateu J, Colli ML, Turatsinze JV, and Eizirik DL (2018). When one becomes many-Alternative splicing in β-cell function and failure. Diabetes Obes Metab 20 Suppl 2, 77–87. 10.1111/dom.13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Eizirik DL, Szymczak F, Alvelos MI, and Martin F (2021). From Pancreatic β-Cell Gene Networks to Novel Therapies for Type 1 Diabetes. Diabetes 70, 1915–1925. 10.2337/dbi20-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Robertson CC, Inshaw JRJ, Onengut-Gumuscu S, Chen WM, Santa Cruz DF, Yang H, Cutler AJ, Crouch DJM, Farber E, Bridges SL, et al. (2021). Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat Genet 53, 962–971. 10.1038/s41588-021-00880-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Onengut-Gumuscu S, Chen WM, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, Farber E, Bonnie JK, Szpak M, Schofield E, et al. (2015). Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet 47, 381–386. 10.1038/ng.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Battaglia M, Ahmed S, Anderson MS, Atkinson MA, Becker D, Bingley PJ, Bosi E, Brusko TM, DiMeglio LA, Evans-Molina C, et al. (2020). Introducing the Endotype Concept to Address the Challenge of Disease Heterogeneity in Type 1 Diabetes. Diabetes Care 43, 5–12. 10.2337/dc19-0880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Elflein J (2022). Number of deceased organ donors in the United States in 2021, by age group. https://www.statista.com/statistics/398442/total-number-of-us-organ-donors-by-age-group/.
- 213.Warshauer JT, Bluestone JA, and Anderson MS (2020). New Frontiers in the Treatment of Type 1 Diabetes. Cell Metab 31, 46–61. 10.1016/j.cmet.2019.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Virostko J (2020). Quantitative Magnetic Resonance Imaging of the Pancreas of Individuals With Diabetes. Front Endocrinol (Lausanne) 11, 592349. 10.3389/fendo.2020.592349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wang YJ, Traum D, Schug J, Gao L, Liu C, Atkinson MA, Powers AC, Feldman MD, Naji A, Chang KM, and Kaestner KH (2019). Multiplexed In Situ Imaging Mass Cytometry Analysis of the Human Endocrine Pancreas and Immune System in Type 1 Diabetes. Cell Metab 29, 769–783.e764. 10.1016/j.cmet.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wasserfall C, Nick HS, Campbell-Thompson M, Beachy D, Haataja L, Kusmartseva I, Posgai A, Beery M, Rhodes C, Bonifacio E, et al. (2017). Persistence of Pancreatic Insulin mRNA Expression and Proinsulin Protein in Type 1 Diabetes Pancreata. Cell Metabolism 26, 568–575.e563. 10.1016/j.cmet.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lam CJ, Chatterjee A, Shen E, Cox AR, and Kushner JA (2019). Low-Level Insulin Content Within Abundant Non-β Islet Endocrine Cells in Long-standing Type 1 Diabetes. Diabetes 68, 598–608. 10.2337/db18-0305. [DOI] [PubMed] [Google Scholar]
- 218.Cefalu W, Andersen D, Arreaza-Rubín G, Pin C, Sato S, Verchere C, Woo M, Rosenblum N, and Symposium Planning Committee Moderators and Speakers (2021). Heterogeneity of Diabetes: β-Cells, Phenotypes, and Precision Medicine: Proceedings of an International Symposium of the Canadian Institutes of Health Research’s Institute of Nutrition, Metabolism and Diabetes and the U.S. National Institutes of Health’s National Institute of Diabetes and Digestive and Kidney Diseases. Diabetes, db210777. 10.2337/db21-0777. [DOI] [Google Scholar]
- 219.Redondo M, Hagopian W, Oram R, Steck A, Vehik K, Weedon M, Balasubramanyam A, and Dabelea D (2020). The clinical consequences of heterogeneity within and between different diabetes types. Diabetologia 63, 2040–2048. 10.1007/s00125-020-05211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.van der Meulen T, Mawla AM, DiGruccio MR, Adams MW, Nies V, Dolleman S, Liu S, Ackermann AM, Caceres E, Hunter AE, et al. (2017). Virgin Beta Cells Persist throughout Life at a Neogenic Niche within Pancreatic Islets. Cell Metab 25, 911–926.e916. 10.1016/j.cmet.2017.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, Wright AJ, Atkinson MA, and Rhodes CJ (2012). Formation of a human beta-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab 97, 3197–3206. 10.1210/jc.2012-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wang Y, Golson M, Schug J, Traum D, Liu C, Vivek K, Dorrell C, Naji A, Powers A, Chang K, et al. (2016). Single-Cell Mass Cytometry Analysis of the Human Endocrine Pancreas. Cell Metabolism 24, 616–626. 10.1016/j.cmet.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Atkinson MA, Bluestone JA, Eisenbarth GS, Hebrok M, Herold KC, Accili D, Pietropaolo M, Arvan PR, Von Herrath M, Markel DS, and Rhodes CJ (2011). How does type 1 diabetes develop?: the notion of homicide or beta-cell suicide revisited. Diabetes 60, 1370–1379. 60/5/1370 [pii] 10.2337/db10-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Toren E, Burnette K, Banerjee R, Hunter C, and Tse H (2021). Partners in Crime: Beta-Cells and Autoimmune Responses Complicit in Type 1 Diabetes Pathogenesis. Frontiers in Immunology 12, 756548. 10.3389/fimmu.2021.756548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kracht MJ, van Lummel M, Nikolic T, Joosten AM, Laban S, van der Slik AR, van Veelen PA, Carlotti F, de Koning EJ, Hoeben RC, et al. (2017). Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat Med 23, 501–507. 10.1038/nm.4289. [DOI] [PubMed] [Google Scholar]
- 226.Hiller H, Beachy D, Lebowitz J, Engler S, Mason J, Miller D, Kusmarteva I, Jacobsen L, Posgai A, Khoshbouei H, et al. (2021). Monogenic Diabetes and Integrated Stress Response Genes Display Altered Gene Expression in Type 1 Diabetes. Diabetes 70, 1885–1897. 10.2337/db21-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Muralidharan C, Conteh A, Marasco M, Crowder J, Kuipers J, de Boer P, and Linnemann A (2021). Pancreatic beta cell autophagy is impaired in type 1 diabetes. Diabetologia. 10.1007/s00125-021-05387-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Rodriguez-Calvo T, Chen Y, Verchere C, Haataja L, Arvan P, Leete P, Richardson S, Morgan N, Qian W, Pugliese A, et al. (2021). Altered β-Cell Prohormone Processing and Secretion in Type 1 Diabetes. Diabetes 70, 1038–1050. 10.2337/dbi20-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sims E, Syed F, Nyalwidhe J, Bahnson H, Haataja L, Speake C, Morris M, Balamurugan A, Mirmira R, Nadler J, et al. (2019). Abnormalities in proinsulin processing in islets from individuals with longstanding T1D. Translational Research 213, 90–99. 10.1016/j.trsl.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Colli M, Hill J, Marroquí L, Chaffey J, Dos Santos R, Leete P, Coomans de Brachène A, Paula F, Op de Beeck A, Castela A, et al. (2018). PDL1 is expressed in the islets of people with type 1 diabetes and is up-regulated by interferons-α and-γ via IRF1 induction. EBioMedicine 36, 367–375. 10.1016/j.ebiom.2018.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Parveen N, Wang JK, Bhattacharya S, Cuala J, Singh Rajkumar M, Butler AE, Wu X, Shih HP, Georgia SK, and Dhawan S (2023). DNA methylation Dependent Restriction of Tyrosine Hydroxylase Contributes to Pancreatic β-cell Heterogeneity. Diabetes. 10.2337/db22-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Syed F, Singhal D, Raedschelders K, Krishnan P, Bone RN, McLaughlin MR, Van Eyk JE, Mirmira RG, Yang ML, Mamula MJ, et al. (2023). A discovery-based proteomics approach identifies protein disulphide isomerase (PDIA1) as a biomarker of β cell stress in type 1 diabetes. EBioMedicine 87, 104379. 10.1016/j.ebiom.2022.104379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Chen CW, Guan BJ, Alzahrani MR, Gao Z, Gao L, Bracey S, Wu J, Mbow CA, Jobava R, Haataja L, et al. (2022). Adaptation to chronic ER stress enforces pancreatic β-cell plasticity. Nat Commun 13, 4621. 10.1038/s41467-022-32425-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Russell MA, Redick SD, Blodgett DM, Richardson SJ, Leete P, Krogvold L, Dahl-Jørgensen K, Bottino R, Brissova M, Spaeth JM, et al. (2019). HLA Class II Antigen Processing and Presentation Pathway Components Demonstrated by Transcriptome and Protein Analyses of islet β-Cells from Donors with Type 1 Diabetes. Diabetes. 10.2337/db18-0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Anquetil F, Mondanelli G, Gonzalez N, Rodriguez Calvo T, Zapardiel Gonzalo J, Krogvold L, Dahl-Jørgensen K, Van den Eynde B, Orabona C, Grohmann U, and von Herrath MG (2018). Loss of IDO1 Expression From Human Pancreatic β-Cells Precedes Their Destruction During the Development of Type 1 Diabetes. Diabetes 67, 1858–1866. 10.2337/db17-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Horwitz E, Krogvold L, Zhitomirsky S, Swisa A, Fischman M, Lax T, Dahan T, Hurvitz N, Weinberg-Corem N, Klochendler A, et al. (2018). β-Cell DNA Damage Response Promotes Islet Inflammation in Type 1 Diabetes. Diabetes 67, 2305–2318. 10.2337/db17-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Nakayasu ES, Syed F, Tersey SA, Gritsenko MA, Mitchell HD, Chan CY, Dirice E, Turatsinze JV, Cui Y, Kulkarni RN, et al. (2020). Comprehensive Proteomics Analysis of Stressed Human Islets Identifies GDF15 as a Target for Type 1 Diabetes Intervention. Cell Metab 31, 363–374.e366. 10.1016/j.cmet.2019.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Campbell-Thompson M, Rodriguez-Calvo T, and Battaglia M (2015). Abnormalities of the Exocrine Pancreas in Type 1 Diabetes. Curr Diab Rep 15, 79. 10.1007/s11892-015-0653-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Foster T, Bruggeman B, Campbell-Thompson M, Atkinson M, Haller M, and Schatz D (2020). Exocrine Pancreas Dysfunction in Type 1 Diabetes. Endocrine Practice 26, 1505–1513. 10.4158/EP-2020-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Benkahla MA, Sabouri S, Kiosses WB, Rajendran S, Quesada-Masachs E, and von Herrath MG (2021). HLA class I hyper-expression unmasks beta cells but not alpha cells to the immune system in pre-diabetes. J Autoimmun 119, 102628. 10.1016/j.jaut.2021.102628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Culina S, Lalanne AI, Afonso G, Cerosaletti K, Pinto S, Sebastiani G, Kuranda K, Nigi L, Eugster A, Østerbye T, et al. (2018). Islet-reactive CD8. Sci Immunol 3. 10.1126/sciimmunol.aao4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.O’Kell A, Wasserfall C, Catchpole B, Davison L, Hess R, Kushner J, and Atkinson M (2017). Comparative Pathogenesis of Autoimmune Diabetes in Humans, NOD Mice, and Canines: Has a Valuable Animal Model of Type 1 Diabetes Been Overlooked? Diabetes 66, 1443–1452. 10.2337/db16-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Anderson AM, Landry LG, Alkanani AA, Pyle L, Powers AC, Atkinson MA, Mathews CE, Roep BO, Michels AW, and Nakayama M (2021). Human islet T cells are highly reactive to preproinsulin in type 1 diabetes. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2107208118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Huber MK, Drotar DM, Hiller H, Beery ML, Joseph P, Kusmartseva I, Speier S, Atkinson MA, Mathews CE, and Phelps EA (2021). Observing Islet Function and Islet-Immune Cell Interactions in Live Pancreatic Tissue Slices. J Vis Exp. 10.3791/62207. [DOI] [PMC free article] [PubMed] [Google Scholar]