

Abstract
The Hippo signaling pathway is a master regulator of organ size and tissue homeostasis. Hippo integrates a broad range of cellular signals to regulate numerous processes, such as cell proliferation, differentiation, migration and mechanosensation. Ca2+ is a fundamental second messenger that modulates signaling cascades involved in diverse cellular functions, some of which are also regulated by the Hippo pathway. Studies published over the last five years indicate that Ca2+ can influence core Hippo pathway components. Nevertheless, comprehensive understanding of the crosstalk between Ca2+ signaling and the Hippo pathway, and possible mechanisms through which Ca2+ regulates Hippo, remain to be elucidated. In this review, we summarize the multiple intersections between Ca2+ and the Hippo pathway and address the biological consequences.
Keywords: Calcium, Calmodulin, S100, Hippo, Signaling, YAP
1. Introduction
The Hippo pathway and the Ca2+ signaling network have crucial roles in regulating diverse physiological processes, including cell proliferation, differentiation, and apoptosis. The highly conserved Hippo pathway [1] controls organ size and tissue homeostasis by regulating cell growth and proliferation [2]. Ca2+ influences a wide range of signaling cascades via a complex network to control diverse cellular processes, ranging from muscle contraction and neurotransmitter release to gene expression [3]. The recent observations that Ca2+ regulates Hippo signaling has stimulated investigation into the interactions between Ca2+ and the Hippo pathway. In this review, we summarize the evidence and discuss potential mechanisms underlying the regulation of Hippo by Ca2+.
2. Hippo signaling
Hippo regulates expression of genes that modulate numerous processes, including cell polarity, adhesion, nutrient sensing, proliferation, migration, anti-apoptosis, response to stress and self-renewal [4]. Unlike many other signaling pathways, Hippo is not controlled by a dedicated receptor; rather it functions as a cellular hub, constantly receiving signals from diverse receptors and stimuli [5].
The intracellular core components of the mammalian Hippo pathway comprise i) adaptor protein MOB1 and Salvador (SAV1); ii) a kinase module consisting of sterile 20-like protein kinase 1 and 2 (MST1, MST2) and large tumor suppressor kinase 1 and 2 (LATS1, LATS2); and iii) transcriptional co-activators Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) [5] (Figure 1).
Figure 1. Core components of the Hippo pathway.
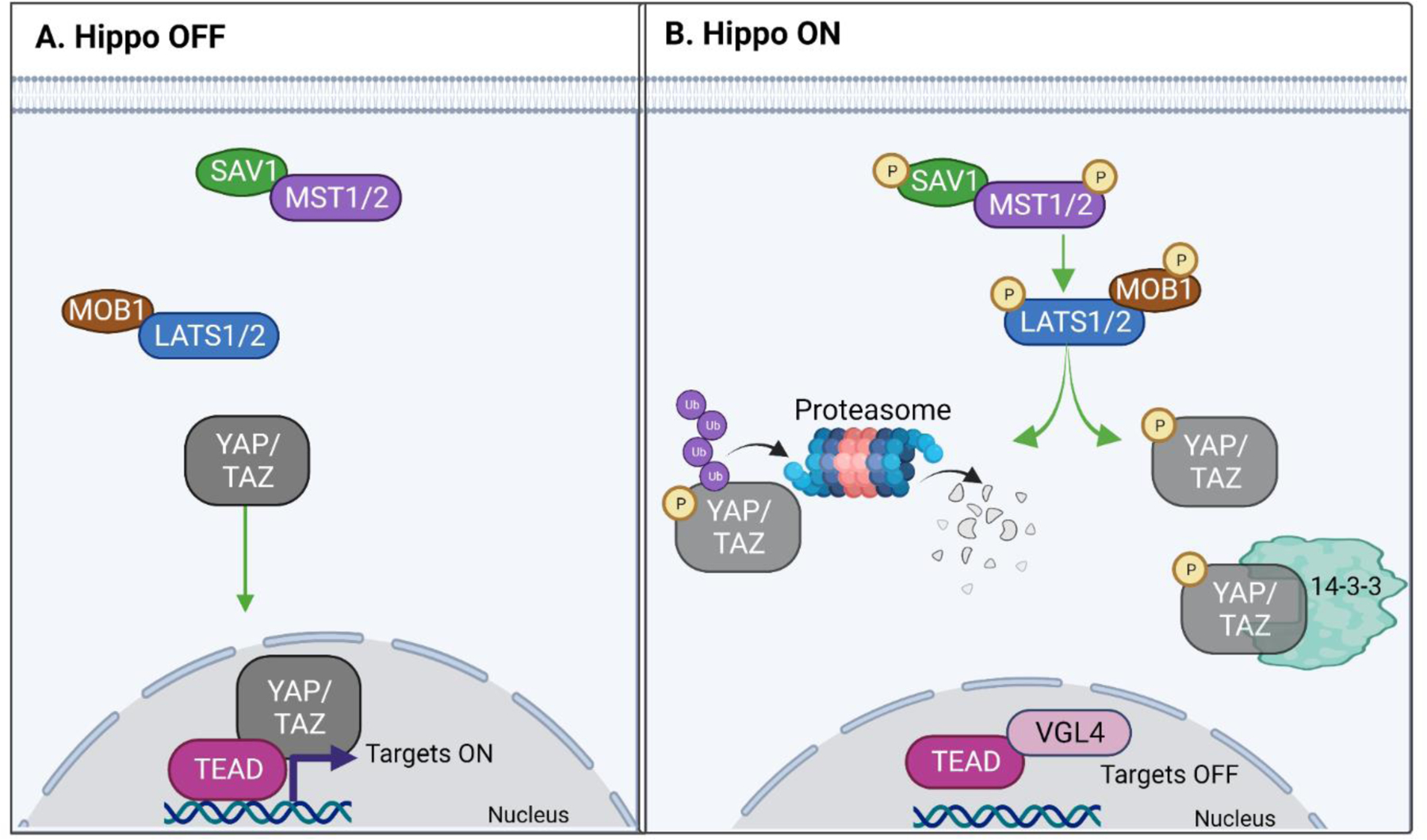
A. Schematic diagram illustrating the interactions among the core components of the Hippo pathway. When Hippo is OFF, YAP and TAZ accumulate in the nucleus, where they bind to and activate TEAD to promote the expression of target genes. B. When Hippo is ON, SAV1 allows MST to phosphorylate LATS, and MOB1 binds to LATS to enhance its catalytic activity. LATS then phosphorylates YAP/TAZ, causing their sequestration in the cytoplasm, either through binding to adaptor proteins, such as 14–3-3, or by inducing YAP/TAZ degradation in the proteasome. In the absence of nuclear YAP/TAZ, TEAD, which is bound to the transcription cofactor VGL4, suppresses the expression of YAP/TAZ target genes. Green arrows represent activation. Figure generated in BioRender.
Posttranslational modifications, predominantly phosphorylation, control the abundance and subcellular localization of YAP/TAZ, which ultimately determine their activity [6]. When the Hippo pathway is ON, SAV1 enables MST to catalyze phosphorylation of LATS, and MOB1 binds to LATS to enhance its catalytic activity [7] (Figure 1B). Active LATS directly phosphorylates multiple sites on YAP/TAZ [2]. When phosphorylated, YAP/TAZ are either sequestered in the cytoplasm or are ubiquitinated and undergo degradation in the proteasome [2]. Thus, phosphorylated YAP/TAZ do not enter the nucleus and are unable to stimulate gene transcription. When the Hippo pathway is OFF, hypophosphorylated YAP/TAZ translocate into the nucleus, where they induce gene expression by binding to the TEA domain (TEAD) family of transcription factors (Figure 1A) [8]. It is important to note that both in vitro and in vivo YAP/TAZ undergo rapid and dynamic phosphorylation-dephosphorylation states, which determine the spatial and temporal regulation of the Hippo pathway [9, 10]. The mechanisms mentioned above describe the canonical core Hippo pathway; however recent evidence has revealed additional regulatory mechanisms for YAP/TAZ that depend on both the cell type and microenvironment [11].
3. Ca2+ signaling
Ca2+ is a highly versatile messenger that modulates diverse signaling cascades and impacts nearly every aspect of cellular life [12]. The Ca2+ signaling network controls processes, such as embryonic development, cell differentiation, migration, apoptosis, and tumorigenesis [3].
The specific components of Ca2+ signaling vary among cell types, depending on the function of the cell [13]. In excitable cells, such as striated muscle and neurons, short-term Ca2+ spikes trigger rapid responses, while slow Ca2+ signals in non-excitable cells control functions, such as transcription and cell division [3]. The dynamics of Ca2+ signaling can be attributed to different types of receptors, sources of Ca2+, Ca2+ transporters, Ca2+ buffers, as well as Ca2+-binding proteins (CaBPs) [13]. Thus, cells have evolved molecular machinery to decode the changes in intracellular free Ca2+ concentrations ([Ca2+]i) into distinct signaling cascades using Ca2+-sensitive signaling molecules.
The large gradient of Ca2+ concentrations across the plasma membrane (1 mM in the extracellular space and 100 nM inside the cell) necessitates that [Ca2+]i be tightly regulated by a delicate balance between “ON” mechanisms that promote Ca2+ entry into the cytosol and “OFF” mechanisms that remove Ca2+ from the cytosol (Figure 2). The Ca2+ signaling toolkit includes CaBPs (e.g., calmodulin), Ca2+-buffering proteins (e.g., parvalbumin), pumps (e.g, Ca2+-ATPase), channels (e.g., transient receptor potential (TRP) channels and Piezo1) and exchangers (e.g., Na+/Ca2+ exchanger) [14] (Figure 2). The regulation of Ca2+ is highly intricate and has been comprehensively addressed in several excellent reviews [12–15]. Here, we provide a brief overview of Ca2+ signaling that focuses on the main components which may influence Hippo signaling.
Figure 2. Ca2+ homeostasis in cells.
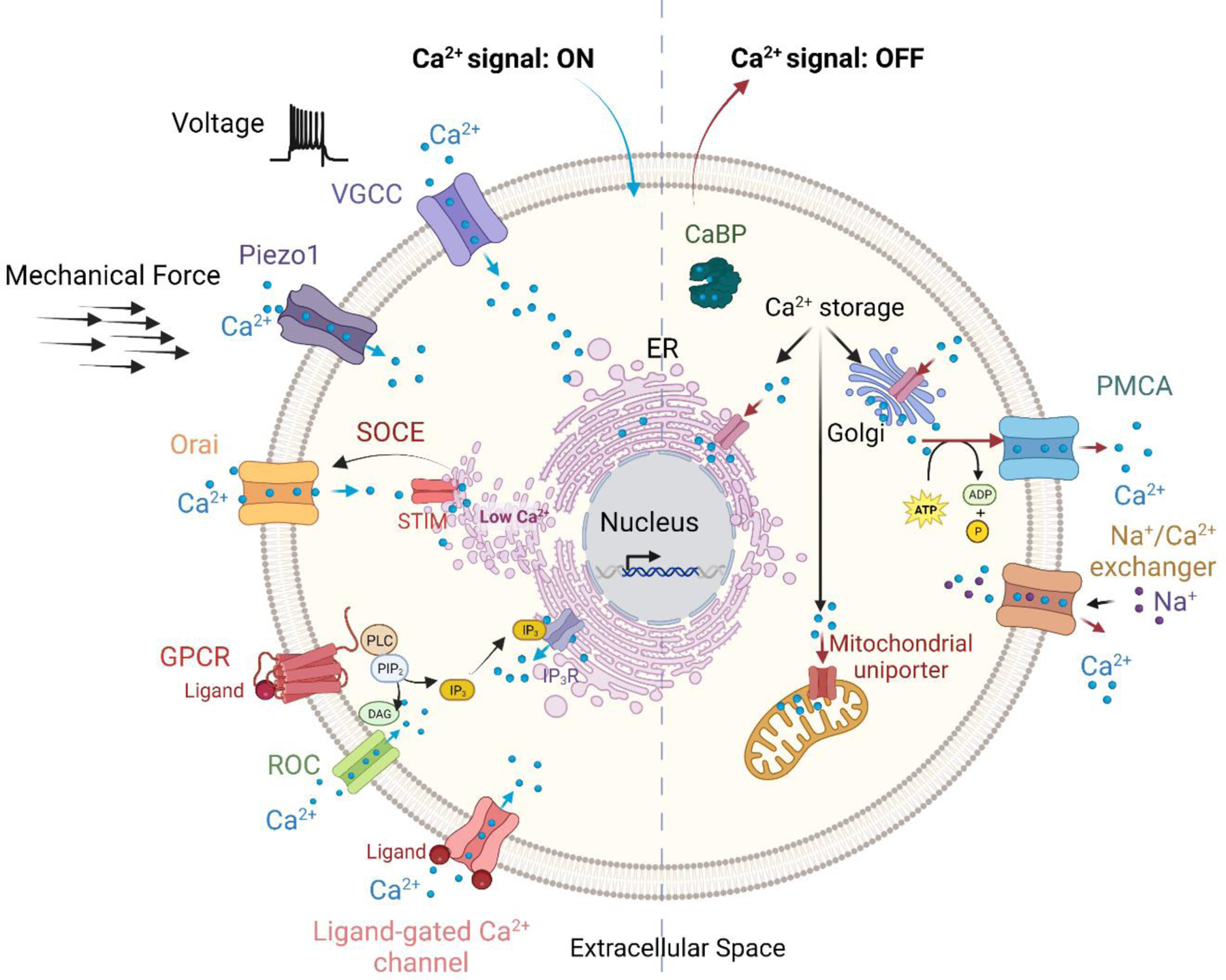
Intracellular free Ca2+ concentrations ([Ca2+]i) are tightly regulated by a network of proteins and channels, such as Ca2+-binding proteins, Ca2+pumps (e.g, Ca2+-ATPase), Ca2+ channels (e.g., Piezo1) and exchangers (e.g., Na+/Ca2+ exchanger). Diverse stimuli, including membrane depolarization, mechanical forces, receptor/ligand interactions, and intracellular messengers, induce “ON” mechanisms. These reactions increase [Ca2+]i by promoting the entry of Ca2+ from the extracellular environment or by the release of Ca2+ from intracellular stores, e.g., the ER, Golgi and mitochondria. The mobilized Ca2+ acts as a messenger to modulate numerous Ca2+-sensitive processes. When the ER Ca2+ reservoir is depleted (Low Ca2+), Ca2+ influx is triggered by store-operated Ca2+ entry (SOCE), which results from coupling between Orai channels and STIM. Once Ca2+ has carried out its signaling function, it is rapidly removed from the cytosol through “OFF” mechanisms, which either extrude Ca2+ to the extracellular space or sequester Ca2+ into internal stores. Blue arrows indicate “ON” mechanisms, red arrows depict “OFF” mechanisms and black arrows indicate induction. Ca2+ is depicted by blue circles. ER, endoplasmic reticulum; CaBP, Ca2+-binding proteins; TRP, transient receptor potential; PMCA, plasma membrane Ca2+ pumps; GPCR, G-protein coupled receptor; ROC, receptor-operated channel; VGCC, voltage-gated Ca2+ channel. The schematic depicts only the main transporters that regulate Ca2+signaling. The figure was generated in BioRender.
Intracellular Ca2+ acts as a messenger to modulate numerous Ca2+-sensitive processes. An increase in [Ca2+]i is induced by a plethora of mechano-electrochemical and biological stimuli, including, but not limited to, membrane depolarization, mechanical stretch, extracellular agonists, and intracellular messengers. These stimuli generate Ca2+-mobilizing signals (ON mechanisms), which increase [Ca2+]i by promoting the entry of Ca2+ either from the extracellular environment or by the release of Ca2+ from intracellular stores, primarily the endoplasmic reticulum (ER), but also the Golgi, mitochondria, and lysosomes (Figure 2). Moreover, agonists such as growth factors, bind to transmembrane G protein-coupled receptors (GPCRs) or receptor tyrosine kinases (not shown), to generate intracellular second messengers, including inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). The latter can activate receptor-operated channels, enabling the influx of Ca2+ across the plasma membrane, while the binding of IP3 to IP3 receptors (IP3R) located on the ER stimulates the release of Ca2+ from ER stores (Figure 2). As a consequence of depletion of Ca2+ from the ER, Ca2+ influx is triggered to replenish Ca2+ stores or to maintain [Ca2+]i. This feedback mechanism, known as store-operated Ca2+ entry (SOCE), is mediated by the coupling of Orai at the plasma membrane and STIM on the ER [16] (Figure 2). SOCE is a major mechanism for maintaining Ca2+ homeostasis in non-excitable cells [17]. The increased [Ca2+]i influences a wide array of cellular processes by binding to and altering localization, interaction, and function of CaBPs [14].
Ca2+ signaling is terminated by removal of [Ca2+]i from the cytosol (Figure 2, OFF). This occurs through channels, pumps and exchangers, which either extrude Ca2+ to the extracellular space or sequester Ca2+ in internal stores, as well as by Ca2+-buffering proteins, which bind to and sequester free Ca2+. As a result, only a small fraction of the Ca2+ that enters the cytosol remains as [Ca2+]i.
4. Ca2+-binding proteins modulate Hippo signaling
Ca2+ ions exert their secondary messenger functions in part by binding to CaBPs. CaBPs are characterized by Ca2+-binding motifs that can reversibly bind Ca2+, allowing transitions between Ca2+-free (apo) and Ca2+-bound states [18]. CaBPs are conveniently divided into (i) buffering proteins that regulate [Ca2+]i by sequestering Ca2+; (ii) transport proteins that shuttle Ca2+ across the membranes of the organelles; and (iii) signaling proteins that interact in a Ca2+-regulated manner with selected targets to influence their activity, thereby converting changes in [Ca2+]i into specific signaling outcomes [18]. The S100 proteins and calmodulin (CaM) are signaling CaBPs that contain two and four helix-loop-helix EF-hand Ca2+-binding motifs, respectively. Binding of Ca2+ to these motifs induces a conformational change, leading to exposure of hydrophobic surfaces that mediate regulatory interactions with selected targets [18]. Although most published literature reports signaling functions for the Ca2+-bound forms of S100 and CaM, their apo counterparts also regulate the activity of selected proteins [19, 20]. Via these Ca2+-modulated interactions, S100 and CaM coordinate various essential cellular processes, ranging from cell growth and differentiation to cell cycle regulation and DNA transcription [18]. The CaBPs S100A1, S100B, and CaM have been documented to modulate Hippo signaling through direct binding to Hippo core proteins [21–23]. Moreover, studies identified that the S100 proteins S100A7, S100A8, S100A9, and S100A14 modulate Hippo signaling by influencing the activity of Hippo regulators [24–26]. Both inhibitory (S100A1, S100A8, S100A9, CaM) [22, 26, 27] and activating (S100B, S100A7, S100A14, CaM) [21, 24, 25, 28] effects on Hippo signaling have been reported, which illustrates the complex, tissue-specific crosstalk between Ca2+ and Hippo. It is important to note that, except for Ca2+/CaM and Ca2+/S100B, these studies did not distinguish whether the influence on Hippo activation was produced by the apo- and/or Ca2+-bound proteins.
4.1. S100
4.1.1. Hippo inhibitory S100 proteins
S100A1 binds to LATS1 kinase in hepatocellular carcinoma cells [22]. Silencing S100A1 increases YAP phosphorylation, which accelerates YAP degradation and reduces its nuclear co-transcriptional activity. Interestingly, LATS1 knockdown reduces S100A1-mediated proliferation of liver carcinoma cells [22]. Consistent with this study, S100A1 knockdown in thyroid carcinoma cells increases YAP phosphorylation, which correlates with reduced cell proliferation and increased apoptosis [29]. In breast carcinoma cells, the long non-coding RNA FOXD2-AS1 upregulates S100A1 expression, likely by sponging S100A1 micro-RNAs, leading to S100A1-mediated reduction of LATS1 kinase activity and increased cell proliferation, migration, and invasion [30]. Together, these findings indicate that S100A1 binding to LATS1 reduces its kinase activity towards YAP, which increases oncogenic YAP nuclear activity (Figure 3A).
Figure 3. Ca2+-binding proteins modulate Hippo activation.
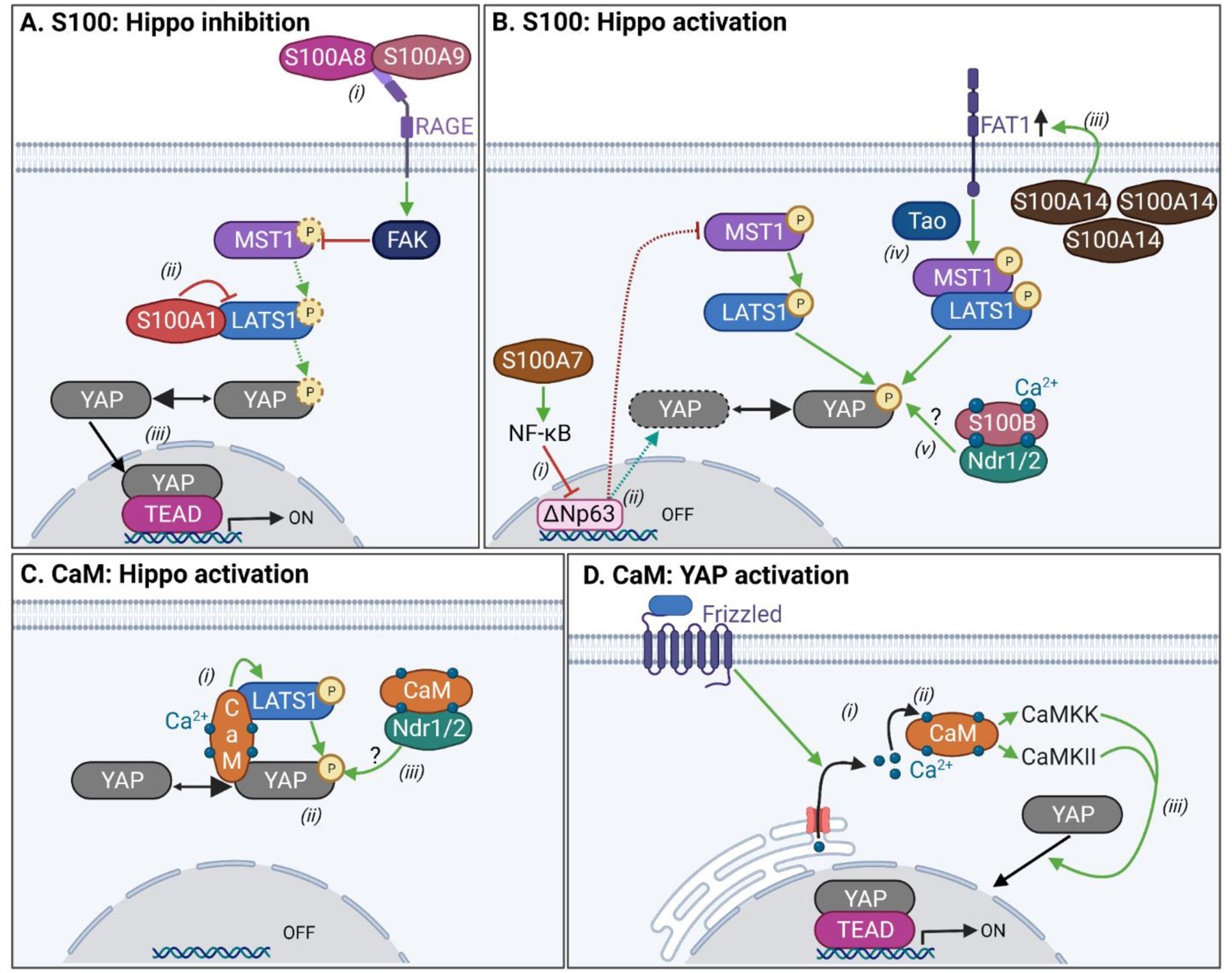
A. Hippo inhibition by S100 proteins: (i) Activation of RAGE by extracellular S100A8/S100A9 activates FAK, which inhibits MST1. (ii) Binding of S100A1 to LATS1 inhibits its kinase activity towards YAP. (iii) In both cases, this increases active, non-phosphorylated YAP, which stimulates the expression of YAP/TEAD target genes. B. Hippo activation by S100 proteins. (i) S100A7 activates NF-κB signaling, which inhibits ΔNp63. (ii) This mechanism prevents ΔNp63-mediated reduction and stimulation of MST1 and YAP expression, respectively. (iii) Separately, S100A14 overexpression increases FAT1 cellular abundance. (iv) In turn, FAT1 stimulates Tao-mediated Hippo kinase activation. (v) Binding of Ca2+/S100B to Ndr1/2 stimulates their kinase activities, which may also increase YAP phosphorylation. C. Hippo activation by calmodulin (CaM). (i) Ca2+/CaM binds to both LATS1 and YAP in a ternary complex in which Ca2+/CaM directly stimulates LATS1 kinase activity. (ii) This increases phosphorylated YAP, which impairs its nuclear translocation. (iii) Ca2+/CaM also interacts with Ndr1/2 kinases, which could possibly stimulate Ndr1/2-catalyzed phosphorylation of YAP. D. YAP activation by CaM. (i) Activation of the Frizzled receptor initiates non-canonical Wnt signaling, which increases cytosolic Ca2+ concentration via Ca2+ release from intracellular stores. (ii) This increases Ca2+/CaM which, via activation of CaMKK and CaMKII, (iii) stimulates YAP nuclear activity. Green, red, and dotted arrows represent activation, inhibition, and decrease, respectively. ? indicates speculative mechanisms not experimentally demonstrated. Figure generated in BioRender.
The S100A8/S100A9 heterocomplex also suppresses Hippo activation [26]. Secreted extracellular S100A8/S100A9 can bind to the receptor for advanced glycation end-products (RAGE) [31]. Exposure of triple-negative breast carcinoma cells to S100A8/S100A9 activates RAGE, which stimulates focal adhesion kinase (FAK). By initiating formation of focal adhesions, FAK inhibits MST1, which ultimately increases nuclear YAP (Figure 3A). S100A8/S100A9 stimulation promotes triple-negative breast carcinoma cell growth and migration, suggesting that it may have carcinogenic properties by inactivating Hippo through RAGE-FAK [26].
4.1.2. Hippo activating S100 proteins
S100A7 is an activator of Hippo [24]. Overexpression of S100A7 in epidermoid squamous carcinoma cells increases phosphorylation of LATS1 and YAP, and decreases YAP cellular abundance. S100A7 knockdown produces the opposite effect, which confirms that S100A7 inhibits YAP expression and activity. The proposed mechanism is that S100A7 activates the NF-κB pathway, thereby repressing expression of the transcription factor ΔNp63. ΔNp63 decreases MST1 and stimulates YAP expression [24]. Hence, by downregulating ΔNp63, S100A7 increases Hippo activation by MST1 and decreases YAP abundance [24] (Figure 3B). YAP phosphorylation is enhanced in squamous cell carcinoma tissues with increased expression of S100A7. Moreover, overexpression of S100A7 prevents YAP-induced apoptosis during chemotherapy [24]. Although the Hippo pathway is most commonly perceived as a tumor suppressor, these findings intimate that Hippo activation by S100A7 through NF-κB/ΔNp63 may promote carcinogenesis.
S100A14 also stimulates Hippo activation. Overexpression of S100A14 in prostate cancer cells increases phosphorylation of MST1, LATS1, and YAP, which correlates with upregulated expression of fat atypical cadherin-1 (FAT1) [25]. The intracellular region of FAT1 assembles a Hippo signalosome comprising MST1 and LATS1, and coordinates MST1 activation by the Tao kinases [32]. Together, these findings suggest that S100A14 activates Hippo by upregulating FAT1 (Figure 3B). In mice, S100A14 overexpression increases both FAT1 expression and YAP phosphorylation, which correlates with reduced prostate carcinoma growth. These observations led the authors to speculate that S100A14 is a tumor suppressor in prostate carcinoma through FAT1-mediated Hippo activation [25].
The nuclear Dbf2-related kinases 1 and 2 (Ndr1 and Ndr2), members of the same kinase family as LATS1, are additional Hippo proteins that can inhibit YAP activity by catalyzing its phosphorylation [33]. The Ca2+-bound form of S100B binds to both Ndr1 and Ndr2, which stimulates their activating autophosphorylation [23, 28]. Although not experimentally demonstrated, the authors speculate that Ca2+/S100B could inactivate YAP via the Ndr kinases (Figure 3B). S100B and S100P interact with the scaffold protein IQGAP1 [34, 35], which downregulates YAP co-transcriptional activity [36]. It is therefore feasible that YAP inhibition by S100 proteins could be coordinated by IQGAP1 scaffolding.
4.1.3. Hippo-mediated expression of S100 proteins
Reciprocally, the expression of several S100 proteins is regulated by Hippo in carcinoma cell lines, implying that Hippo signaling influences Ca2+-regulated, S100-mediated signaling. Hippo activation stimulates expression of both S100A8 and S100A9 in squamous carcinoma cells via the TEAD1 transcription factor [37]. Similarly, Hippo activation induces TEAD1-mediated expression of S100A7 in lung carcinoma, epidermoid carcinoma, and squamous carcinoma cells [38–40]. These observations suggest that nuclear YAP and TEAD1, most commonly perceived as transcriptional co-activators, also repress transcription of selected target genes, as previously reported [41]. In contrast, YAP stimulates S100A4 expression [42]. Because S100A4, S100A7, S100A8, and S100A9 promote cell proliferation, invasion, and/or carcinoma phenotypic transition, these studies imply that Hippo-regulated expression of S100 proteins drives carcinogenesis [37–40].
4.2. Calmodulin
Unlike the S100 proteins that are expressed in a cell- and tissue-specific manner, CaM is ubiquitous [18]. Ca2+/CaM was recently documented to bind LATS1 and YAP [21]. Ca2+ initiates the formation of a ternary complex of pure CaM:LATS1:YAP in vitro. The interaction of CaM with LATS1 and YAP was confirmed in HeLa cells. Functionally, CaM antagonism decreases YAP phosphorylation, increases YAP:TEAD1 interactions in the nucleus, and promotes YAP co-transcriptional activity. Moreover, Ca2+/CaM directly stimulates LATS1 kinase activity [21]. Together, these observations demonstrate that Ca2+/CaM stimulates LATS1-catalyzed phosphorylation of YAP, which increases YAP retention in the cytoplasm to inhibit its nuclear activity (Figure 3C). Ca2+/CaM also binds Ndr1 and Ndr2 kinases [28] (Figure 3C), implying that it may regulate YAP phosphorylation by acting on several upstream kinases.
In contrast to the results summarized above, Ca2+/CaM signaling was recently observed to increase the nuclear co-transcriptional activity of YAP in mouse intestinal organoids [27]. In this system, activation of non-canonical Wnt signaling triggers the release of Ca2+ from intracellular stores into the cytosol, leading to increased binding of Ca2+ to CaM and subsequent activation of the Ca2+/CaM-dependent kinases CaMKK and CaMKII. Chemical inhibition of Ca2+/CaM, CaMKK, or CAMKII suppresses YAP nuclear translocation induced by non-canonical Wnt. Moreover, inhibition of Ca2+/CaM signaling reduces the cystic morphology of mouse intestinal organoids that is observed upon activation of non-canonical Wnt (Figure 3D). These results led the authors to postulate that Ca2+/CaM signaling in intestinal cells with active non-canonical Wnt upregulates YAP nuclear activity, with consequences on tissue morphology [27].
There is evidence for bidirectional crosstalk between CaM and Hippo. CaM is encoded by three genes, CALM1, CALM2, and CALM3 [43], one of which (CALM2) was identified as a YAP target gene in colon carcinoma cells [44]. YAP and PRDI-BF- & RIZ-homology-domain-containing transcriptional regulator, PRDM14, are enriched at the CALM2 promoter. YAP suppression decreases CaM mRNA and protein levels [44]. Analysis in a mouse xenograft model indicates that PRDM14 rescues YAP suppression to promote colon carcinoma progression and maintain YAP-mediated growth in a process which is dependent on CALM2 expression. Taken together, these findings emphasize the intricate interplay and feedback between Ca2+/CaM and the Hippo pathway.
5. Increased intracellular Ca2 + enhances YAP nuclear translocation
Activation of mechanosensitive Ca2+ channels, including stretch activated channels (e.g., Piezo1), transient receptor potential (TRP) channels (e.g., TRPV), and gap junction channels (e.g., connexins), rapidly increases [Ca2+]i and triggers Ca2+-dependent signaling [45] in response to intrinsic and extrinsic mechanical cues, such as stretching, shear forces, cell-cell contact, substrate stiffness, increased extracellular viscosity, and contractile forces. Activation of the channels by mechanical cues enhances YAP nuclear localization and /or activity through increased [Ca2+]i [46–49] (Figure 4A).
Figure 4. Ca2+ and Hippo signaling crosstalk.
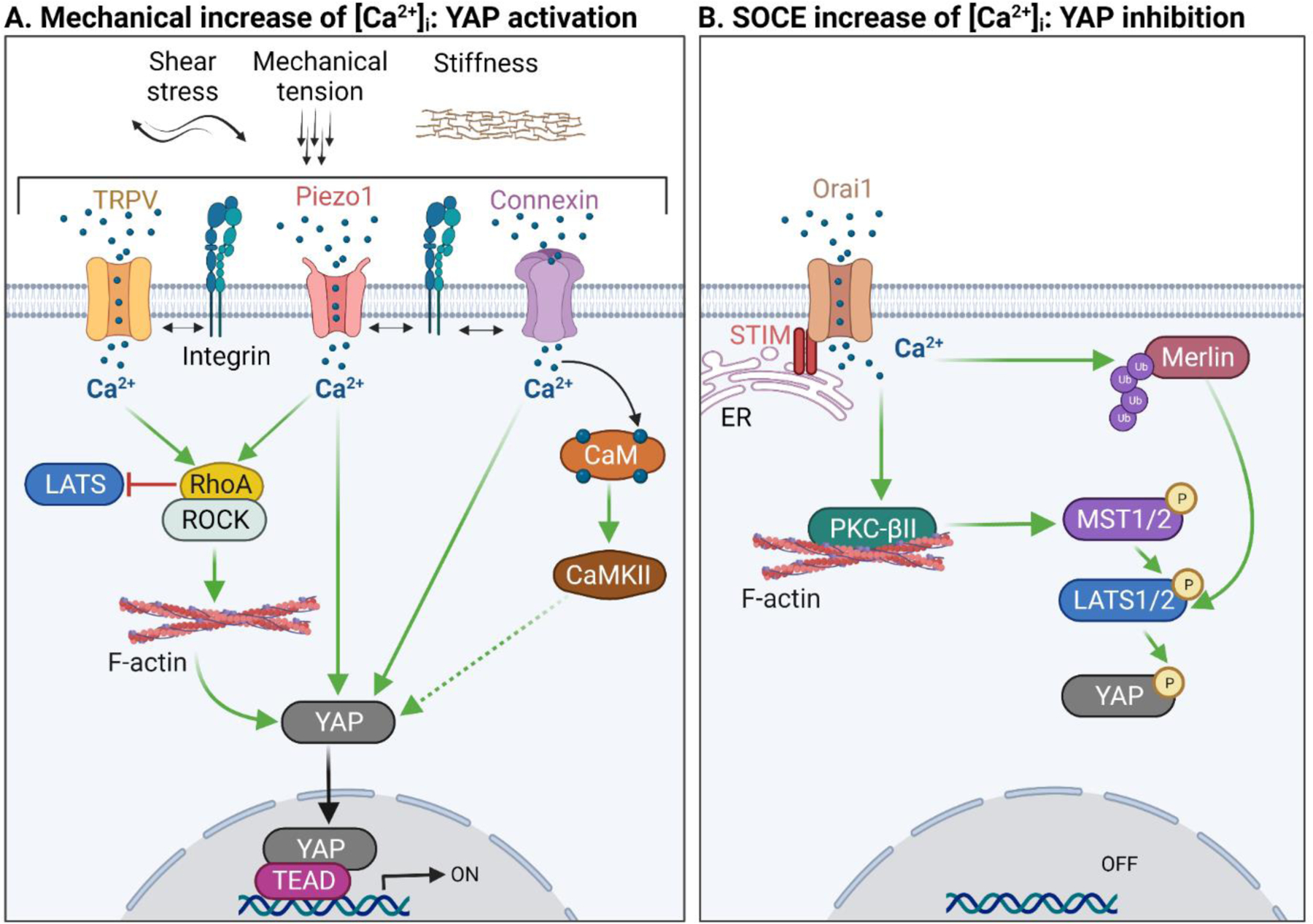
A. Mechanical stimuli, such as shear stress, mechanical tension and substratum stiffness, elicit a cascade of signaling events by inducing an influx of Ca2+ through the activation of Ca2+-permeable mechanosensitive channels, including the TRPV family of proteins, Piezo1, and connexins. The increased [Ca2+]i activates downstream effectors, RhoA/ROCK and subsequent actin remodeling. Active RhoA inhibits LATS1/2 phosphorylation, leading to nuclear translocation and transcriptional activation of YAP. Increased [Ca2+]i also activates CaMKII via CaM. CaMKII may promote nuclear translocation of YAP. B. Store-operated calcium entry (SOCE) triggers an increase in [Ca2+]i, leading to the activation of PKC-βII and ubiquitination of Merlin. Active PKC-βII stimulates the Hippo kinase cascade, while ubiquitinated Merlin promotes LATS1 activity, all resulting in increased YAP phosphorylation and inhibition of its nuclear activity. Green arrows represent activation. Dashed line depicts speculative mechanism. CaM, calmodulin; CaMKII, Ca2+/calmodulin-dependent kinase II; ER, endoplasmic reticulum; ROCK, Rho kinase; and TRPV, Transient Receptor Potential Vanilloid. Figure generated in BioRender.
5.1. Piezo1
Piezo1 is activated by traction forces and mechanical tension [50]. Ca2+ influx through Piezo1 occurs early after mechanical stimulation of cells [51]. This Ca2+ influx results in RhoA-induced stress fiber formation [52, 53]. In a separate study of stress fiber formation, constitutively active RhoA enhanced YAP nuclear localization and increased the expression of YAP target genes [54]. Activation of Piezo1 in response to mechanical forces promotes YAP nuclear localization (Figure 4A), while knocking down Piezo1 prevents this translocation [49]. Moreover, pharmacological stimulation of Piezo1 by Yoda1 increases [Ca2+]i and induces YAP-mediated transcription of genes that are related to mechanical stress [50]. Collectively, these data suggest that Piezo1 enhances YAP nuclear localization via Ca2+-stimulated activation of RhoA.
Reciprocally, the expression of an active YAP mutant construct in oral squamous carcinoma cells significantly increases Piezo1 mRNA levels, implying that Piezo1 is a transcriptional target of YAP [55]. These observations raise the possibility of a positive feedback mechanism between Ca2+ signaling by Piezo1 and the Hippo pathway, whereby active Piezo1 induces YAP nuclear translocation, which potentially enhances both expression of and signaling by Piezo1.
5.2. Transient Receptor Potential (TRP) channels
TRPV4 is a Ca2+-permeable channel and a member of a widely expressed subfamily of TRPs that crosstalks with integrins and adhesion molecules to transmit mechanical cues [56]. In response to mechanical forces, TRPV4 causes adhesion, spreading, and morphological changes of fibroblasts [57]. Knockdown or chemical inhibition of TRPV4 decreases [Ca2+]i, which suppresses actin re-organization resulting from RhoA/ROCK [48, 58]. Consistent with the role of the RhoA/ROCK/actin network in inhibiting the Hippo kinase LATS1/2 [59, 60], TRPV4-mediated Ca2+ influx initiates YAP/TAZ nuclear translocation [61, 62] (Figure 4A). Functionally, knockout or chemical antagonism of TRPV4 suppresses cytoskeletal remodeling and prevents mechanically-induced YAP/TAZ nuclear translocation [61, 62].
Actin polymerization is a priming signal for YAP/TAZ nuclear localization [63]. It is tempting to speculate that TRPV4 could also modulate YAP/TAZ through direct regulation of actin dynamics in response to Ca2+ signaling. The C-terminal domain of TRPV4 contains multiple actin and tubulin binding sites [64, 65]. TRPV4 activation disassembles microtubules, while promoting actin polymerization [64]. Moreover, the C-terminal region of TRPV4 binds to CaM [66, 67]. Ca2+/CaM regulates actin remodeling by activating CaMKII and dissociating it from actin bundles to allow actin re-organization [68]. Overall, the Ca2+ influx induced by TRPV4 stimulates YAP/TAZ subcellular localization via RhoA activation and likely via direct action on actin.
5.3. Connexins
Connexons, the structural components of gap junctions, are composed of two sets of hexameric connexin hemichannels, one set contributed by each adjacent cell. Connexin hemichannels align on the plasma membrane, creating a pore between the cytoplasm of adjacent cells, which enables the direct transfer of cations (Na+, K+, and Ca2+), small molecules and metabolites [69]. At the plasma membrane, connexins associate with integrins and focal adhesions to transmit mechanical information to cells and tissues [70]. Connexin43 (Cx43), a widely expressed and well-studied member of the connexin family, participates in Ca2+ signaling [71]. siRNA depletion of Cx43 significantly reduces [Ca2+]i, while overexpression of Cx43 enhances Ca2+ influx and increases [Ca2+]i [71, 72].
Cx43 modulates the Hippo pathway [46]. In mammary epithelial tissue Cx43 hemichannels are opened in cells located in regions of high mechanical stress. Channel opening promotes membrane depolarization, leading to nuclear translocation of YAP/TAZ and increased cell proliferation (Figure 4A) [46]. Selective inhibition of Cx43 reduces both nuclear translocation of YAP/TAZ and cell proliferation. Taken together, these findings suggest that nuclear localization of YAP/TAZ in response to high mechanical stress is contingent on Cx43 hemichannel function.
[Ca2+]i modulates Cx43 gating. Increased [Ca2+]i enhances Ca2+/CaM binding to the C-terminal region of Cx43. This closes the channel and attenuates its permeability [73–75], and also inhibits phosphorylation of Cx43 by Src and Erk1/2 [74]. Cx43 phosphorylation influences its interaction with binding partners, such as the actin binding protein, drebrin [76]. Interestingly, both Cx43 [77] and Ca2+/CaM [21] bind directly to YAP. Moreover, Ca2+/CaM binds to LATS1 and stimulates LATS1 kinase activity [21]. Thus, Ca2+/CaM could participate in the response to mechanical cues by integrating signals from Cx43 to the Hippo pathway, providing a potential mechanism for coordinating mechanical and molecular signaling.
6. Increased intracellular Ca2+ suppresses YAP nuclear localization
In contrast to the studies mentioned in the previous section, increased [Ca2+]i has also been documented to suppress YAP and TAZ nuclear co-transcriptional activity. For example, elevation of [Ca2+]i through chemical stimulation of SOCE in glioblastoma cells increases YAP/TAZ phosphorylation and cytoplasmic retention [78]. The increased [Ca2+]i promotes the association of PKC-βII with F-actin, and activates PKC-βII. In turn, PKC-βII activates MST1/2 and LATS1/2 Hippo kinases, resulting in YAP/TAZ inhibition [78] (Figure 4B). Another study reports that increasing [Ca2+]i by incubating cells with the Ca2+ ionophore A23187 leads to phosphorylation and cytoplasmic retention of YAP through activation of PKC-α [79]. Whether the PKC kinases directly phosphorylate the Hippo proteins or require intermediate effectors remains to be determined.
Chemically-induced SOCE also activates Hippo via Merlin, a membrane-associated protein that can activate MST1/2 and recruit LATS1/2 [80, 81]. Induction of SOCE by thapsigargin triggers both dephosphorylation and NEDD4L-mediated ubiquitination of Merlin. Merlin ubiquitination promotes its interaction with LATS1, which increases LATS1 activity [81] (Figure 4B). Additionally, there is a potential role for endolysosomal Ca2+ release in regulating YAP/TAZ activity brought about by SOCE. Knockdown of two-pore channel2 (TPC2), which causes Ca2+ release from endolysosomal compartments, significantly reduces the mRNA and protein levels of Orai1, a plasma membrane SOCE channel, which correlates with increased YAP/TAZ co-transcriptional activity [82]. Together, these observations further suggest that SOCE-mediated increase of [Ca2+]i inhibits YAP/TAZ.
7. Complexity of Ca2+-Hippo crosstalk
A recent study investigated YAP cytoplasmic-nuclear shuttling in response to Ca2+ in breast carcinoma cells [83]. Wounding a cell monolayer transiently increased [Ca2+]i and induced YAP nuclear accumulation within 1 min. Nuclear depletion of YAP then occurred over 20 min, followed by slow nuclear re-accumulation ~3 h after the initial stimulation. In contrast, increasing [Ca2+]i by pharmacological induction of SOCE causes YAP accumulation in the cytoplasm for 25 min, followed by slow nuclear enrichment over 50 min [83]. These data indicate that YAP spatial dynamics are influenced by the nature of the Ca2+ stimulus and emphasize the complexity of the effect of Ca2+ on the Hippo pathway.
In human adipose-derived stem cells, Ca2+ influx induced by mechanical stretching at 37°C significantly enhances YAP nuclear localization, which promotes cell proliferation [84]. In contrast, stretching while simultaneously reducing temperature from 37°C to 10°C suppresses YAP nuclear translocation and cell proliferation, despite a significant increase in [Ca2+]i. Chemical inhibition of SOCE restores YAP nuclear activity at the low temperature [84]. Together, these studies emphasize that Ca2+-Hippo crosstalk is influenced by several parameters, including temperature and the stimulus eliciting Ca2+ influx.
Actin remodeling impacts Hippo activation, thereby affecting YAP subcellular localization. Ca2+ influences actin both by binding directly to actin and via CaBP. A potential mechanism for regulation of YAP by Ca2+ is through Ca2+-mediated actin reset (CaAR) [85, 86]. In this process, Ca2+ triggers rapid disassembly of cortical actin, which is followed by the translocation of actin filaments to the endoplasmic reticulum and perinuclear rim [85, 86]. Depletion of formin-2 impairs CaAR and decreases phosphorylation of LATS and YAP [87]. Therefore, CaAR activates Hippo. However, actin remodeling via a Ca2+-mediated increase in active RhoA induces nuclear translocation of YAP [88]. This illustrates that the effects of cytoskeleton components on YAP/TAZ are complex and likely are regulated by both the dynamics and subcellular site of actin remodeling.
8. Ca2+ and Hippo crosstalk in health and disease
All the studies described above were carried out in cultured cell lines. The potential implications of the crosstalk between Ca2+ and Hippo in physiology and pathology are illustrated by in vivo studies using tissues from mouse models and human patients.
8.1. Skeletal remodeling
The skeleton is remodeled by removal of damaged bone by osteoclasts and synthesis of collagen by osteoblasts to form new bone tissue. Disruption of skeletal remodeling can lead to pathological conditions, such as osteoporosis. Patients with osteoporosis have decreased amounts of Piezo1, which is expressed in both osteoclasts and osteoblasts, in their bones [89]. Similarly, as described above, deletion of Piezo1 reduces bone mass in mice, while the Piezo1 agonist, Yoda1, increases mouse bone mass by promoting YAP transcriptional activity [50]. In a conditional knockout mouse, osteoblast-specific loss of Piezo1 results in marked bone resorption and osteoporosis [90]. Loss of Piezo1 from the mouse osteoblasts significantly decreases YAP nuclear localization, thus suppressing YAP-mediated transcription of collagen. Moreover, knockdown of YAP by shRNA decreases collagen expression, while pharmacological inhibition of MST1/2 increases collagen abundance in stem cells [90]. Based on these findings, the authors propose that collagen expression induced by YAP provides protection against mechanical stress in bone tissue.
8.2. Atherosclerosis
High shear stress at the apical surface of arterial endothelial cells increases [Ca2+]i [91], which can initiate inflammation and formation of atherosclerotic lesions [92]. These lesions can be effectively suppressed by treating patients with Ca2+-channel blocking agents [93].
Cx43 was recently identified as a mediator of inflammation and atherogenesis [94, 95]. Pharmacological inhibition of Cx43 inhibits the release of inflammatory mediators and prevents the formation of atherosclerotic lesions in mice [94]. Under mechanical stress, phosphorylation of Cx43 by MST1 suppresses its signaling by inhibiting the opening of Cx43 channels in vitro and preventing translocation of Cx43 to lipid rafts in vivo, thus protecting against atherosclerosis (Figure 5A). Plaque formation in mice with knockout of both MST1 and YAP was comparable to that seen in mice with knockout of MST1 alone [94]. While the authors concluded that the role of MST1 in atherosclerosis is not mediated through YAP and is independent of the Hippo pathway, several other studies showed that activation of the Hippo cascade and consequent inhibition of YAP/TAZ attenuates endothelial cell inflammation and suppresses formation of atherosclerotic lesions [96–98]. It is likely that MST1 knockout alone is sufficient to promote plaque formation, and TAZ could potentially compensate for the lack of YAP in the MST1/YAP double knockout mouse model used in the study by Quan et al [94]. Activation of Cx43 substantially increases [Ca2+]i in endothelial cells [99]. Therefore, it seems reasonable to postulate that MST1-mediated inhibition of Cx43 attenuates Ca2+ permeability, further contributing to the protective effect of MST1 against atherosclerosis.
Figure 5. Ca2+ and Hippo crosstalk in health and disease.
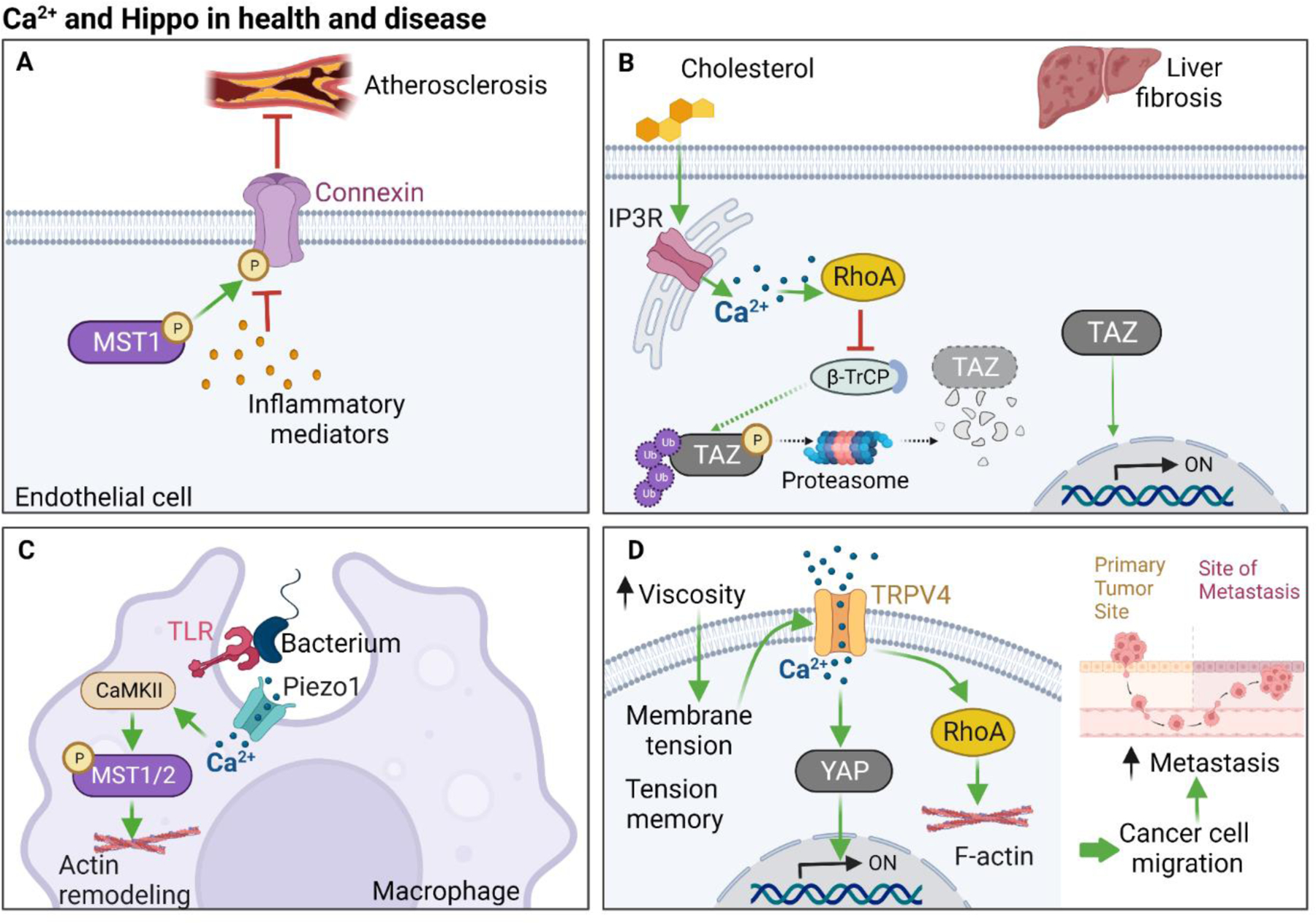
A. MST1 phosphorylates Cx43 and attenuates its ability to release inflammatory molecules, thus protecting endothelial cells from atherosclerosis. B. Cholesterol loading in hepatocytes suppresses degradation of TAZ in the proteasome by β-TrCP, an E3 ubiquitin ligase, via an IP3R-Ca2+-RhoA pathway, thereby promoting TAZ-mediated liver fibrosis. C. The interaction of bacterial cell wall components with toll-like receptors (TLRs) on macrophages initiates a cascade of events. Piezo1 couples with TLRs, mediating the influx of Ca2+, which activates MST1. This leads to actin remodeling and culminates in phagocytosis of the bacteria by the macrophages. D. Carcinoma cells secrete macromolecules, which increase the viscosity of the extracellular environment. This induces membrane tension and increases [Ca2+]i via TRPV channels. Ca2+ activates RhoA and promotes actin remodeling, leading to cancer cell migration and metastasis. Green, red and dashed arrows represent activation, inhibition, and decrease, respectively. CaMKII, Ca2+/calmodulin-dependent kinase II; IP3R, inositol 1,4,5-triphosphate (IP3) receptor; TLR, Toll-like receptor; and TRPV4, Transient Receptor Potential Vanilloid 4. Figure generated in BioRender.
8.3. Bullous skin diseases
Pemphigus vulgaris is an autoimmune blistering disorder of the skin or mucous membranes caused by autoantibodies to the Ca2+-dependent adhesion proteins desmoglein1 and/or 3 (Dsg1, Dsg3). The desmogleins are present in desmosomes and maintain the structural integrity of the epidermis [100]. In cultured keratinocytes, pemphigus autoantibodies increase [Ca2+]i via phospholipase C (PLC)-mediated activation of IP3R [101, 102]. PLC-induced release of Ca2+ from the ER is recognized as an early event in the pathogenesis of pemphigus [100]. Increased [Ca2+]I leads to Dsg3 depletion, which results in loss of intercellular adhesion [103]. Findings reported in a preprint suggest that Dsg3 forms a complex with YAP, sequestering YAP at cell-cell junctions and preventing its nuclear translocation, while depletion of Dsg3 by autoantibodies dissociates YAP from these junctions [104]. In agreement with the inhibitory effect of Dsg3 on YAP, incubating cultured keratinocytes with serum antibodies from patients with pemphigus increases both the total abundance of YAP and its translocation to the nucleus in a dose- and time-dependent manner [105]. Consistent with the in vitro observations, immunohistological analysis revealed overexpression of YAP in pemphigus lesions [105]. Collectively, these findings suggest that crosstalk between Ca2+ signaling and YAP has a role in the pathogenesis of pemphigus.
8.4. Nonalcoholic steatohepatitis (NASH)
The incidence of nonalcoholic fatty liver disease (NAFLD) and the stage known as nonalcoholic steatohepatitis (NASH) are increasing worldwide and pose a substantial healthcare concern [106]. NASH is characterized by inflammation and hepatocellular damage, increased cholesterol in the blood and accumulation of fat in the liver, which can eventually lead to fibrosis, cirrhosis, and liver failure. The connection between cholesterol and NASH progression is not fully understood. However, a link between cholesterol, Ca2+and TAZ was recently identified (Figure 5B). High levels of cholesterol increase [Ca2+]i through IP3R-mediated release of Ca2+ from the ER, which increases the amount of active RhoA in hepatocytes [107]. Active RhoA stabilizes TAZ by preventing the E3 ubiquitin ligase β-TrCP from inducing its proteasomal degradation, thereby enhancing TAZ activity. The transcriptional activation resulting from increased nuclear TAZ induces fibrosis and NASH [107]. The activation of TAZ by Ca2+ in hepatocytes may provide an opportunity for therapeutic intervention to prevent the progression of liver fibrosis in NASH.
8.5. Autosomal dominant polycystic kidney disease
The proteins PC1 and PC2 form a heterodimer that allows Ca2+ influx at the plasma membrane and cilia. Mutations within their corresponding genes (PKD1 and PKD2, respectively) causes autosomal dominant polycystic kidney disease (ADPKD), which is the most common genetic disorder of the kidney [108]. The Ca2+ imbalance caused by mutations in PC1/PC2 contributes to the development of cysts in the kidney, liver and pancreas, ultimately leading to failure of all these organs [108]. YAP activity is increased in the cysts with low Ca2+ concentration [109, 110], which suggests that influx of Ca2+ through PC1/PC2 regulates Hippo. In a mouse model of ADPKD, knockout of YAP and TAZ reduces cystogenesis, implying that their activity contributes to the pathogenesis of ADPKD [110]. Knockout of TAZ from mouse kidneys also decreases ubiquitin-dependent degradation of PC2 [111], illustrating bidirectional crosstalk between the two proteins.
8.6. Immune responses
In macrophages, Piezo1 has an important role in coordinating toll-like receptor (TLR)-mediated innate immunity [112]. Binding of the cell wall of bacteria to macrophages activates Piezo1 and induces the assembly of Piezo1 and TLR4 complexes (Figure 5C). Active Piezo1 enables influx of Ca2+, which activates CaMKII. In turn, CaMKII stimulates an increase in active Rac1 through MST1/2, leading to actin re-organization, which is required for efficient phagocytosis by the macrophages [112]. Elimination of extracellular or intracellular Ca2+ effectively inhibits MST1/2 activation and suppresses the bactericidal function of macrophages. Therefore, activation of the core Hippo kinase MST1/2 during macrophage phagocytosis is dependent on Ca2+.
Several pathological conditions, including lower respiratory tract disease, coronary artery disease, hematologic malignancies and thrombocytosis, result from altered and/or attenuated tension-induced signaling of dendritic cells [113]. General inhibition of Ca2+ signaling by ruthenium red or pharmacological antagonism of YAP/TAZ transcriptional activity suppresses the proinflammatory response of dendritic cells to mechanical stimulation [114]. Thus, the crosstalk between Ca2+ signaling and Hippo core components integrates mechanosensation with both nonspecific cellular innate immunity and humoral adaptive immunity.
8.7. Carcinoma
The role of dysregulated Ca2+ homeostasis in cancer initiation, progression, metastasis and multidrug resistance is an area of active investigation [115]. Dysregulation of the Hippo pathway is associated with the hallmarks of malignancy, including hyperproliferation, invasion, metastasis and chemotherapeutic resistance [116]. In this section we present examples of Ca2+ crosstalk with the Hippo pathway during tumorigenesis.
The Ca2+-sensing receptor (CaSR), a member of the GPCR family, maintains systemic Ca2+ homeostasis by binding to and sensing free ionized Ca2+ in the blood. Upon activation, the CaSR induces intracellular Ca2+ release through signaling molecules, such as PLC and PKC [117]. The CaSR regulates cell-cell adhesion, differentiation, gene transcription, tumorigenesis and acts as a tumor suppressor or oncogene, depending on the tissue origin of the tumor [118, 119]. In normal human parathyroid cells, activation of the CaSR reduces expression of LATS1/2, which results in YAP nuclear accumulation and increased expression of YAP target genes [120]. This effect is abrogated by RhoA inhibitors, suggesting that it is mediated by RhoA/ROCK signaling. By contrast, in parathyroid carcinoma CaSR expression is downregulated [121]. YAP nuclear localization is reduced in parathyroid cells with CaSR downregulation [120], suggesting that the CaSR modulates the Hippo pathway and influences cellular processes involved in tumorigenesis.
The Ca2+ transporter, SPCA2 (secretory pathway Ca2+/Mn2+-ATPase 2), which conveys Ca2+ from the cytosol into the Golgi apparatus [84], is primarily expressed in secretory tissues, such as mammary glands. There is a negative correlation between the expression of SPCA2 and that of YAP/TAZ in patient samples from all subtypes of breast carcinoma, suggesting that Ca2+ transport by SPCA2 reduces YAP/TAZ expression [122]. Studies in cultured breast epithelial cells revealed that Ca2+ signaling brought about by SPCA2 suppresses the oncogenic effects of nuclear YAP/TAZ, likely by regulating the expression of E-cadherin [122]. E-cadherin mediates contact inhibition and suppresses epithelial mesenchymal transition (EMT), in part by activating Hippo and attenuating YAP/TAZ nuclear accumulation [122, 123].
Mechanosensitive Ca2+ channels, such as Piezo1 and TRPV4, participate in tumorigenesis by regulating Hippo signaling. Piezo1 activation promotes EMT and metastasis of ovarian carcinoma [124] and cholangiocarcinoma [125] cells by inhibiting Hippo and activating YAP nuclear function. Furthermore, TRPV4 and YAP promote cancer cell migration regulated by viscosity. TRPV4 activation increases RhoA-mediated contractility of cancer cells in vitro [47]. In vivo, enhanced contractility facilitates the escape of cancer cells with metastatic advantage from the primary tumor site in a mouse model of metastasis.
Tumor cells establish a mechanical memory, characterized by decreased expression of MST1 and LATS2, and increased expression of YAP and TEAD2, which enables the malignant cells to retain migratory capacity while exposed to different degrees of tissue stiffness during metastasis [47]. The YAP inhibitor, verteporfin, abrogates the mechanical memory, and reduces the migration and metastasis of the neoplastic cells. Taken together, these findings suggest a role for Ca2+-mediated YAP regulation in hypermotility and metastasis of the malignant cells (Figure 5D).
In order to investigate a potential link between CaBPs and Hippo in cancer, we used the Cancer Genome Atlas (TCGA). The data available on the cBio Cancer Genomics Portal (http://cbioportal.org) allowed us to examine mRNA expression, gene alterations (e.g., mutations, copy number variations) to query a panel of 10, 967 tumor biopsies from 32 studies of all cancer types for the core Hippo components (MST1/2, LATS1/3, SAV1, MOB1/2, YAP, and TAZ) and CaM (CALM1, CALM2, CALM3) or S100 proteins known to modulate Hippo (S100A1, S100A7, S100A8, S100A9, S100A14, S100B). In examining post-disease survival, the only significant link between genetic alteration and decreased patient survival was observed between each CaM gene and TAZ across all cancer types (Table 1). Additionally, we examined the correlation between the expression of mRNA from multiple genes and identified a very weak, but significant, correlation between CaM expression and TAZ, but not YAP, despite previous reports that CALM2 is a YAP target gene [44]. These findings imply a potential functional relationship between CaM and TAZ in the development and/or progression of cancer. However, the molecular mechanism by which genetic changes in CaM or TAZ may regulate one another to affect patient survival remains to be determined. Further investigations are required to elucidate the nature and extent of the possible interactions between CaM and TAZ in tumorigenesis.
Table 1.
mRNA correlation and gene alteration of CaM and TAZ across all types of cancer.
| Gene (Protein) | mRNA correlation coefficient †Spearman (p value) |
Cases with genetic alteration (%) | Decreased survival of patients with altered genes Logrank Test p value |
|---|---|---|---|
|
CALM1 (Calmodulin) WWTR1 (TAZ) |
0.04 (p = 2.354e-5) | 97/10950 (0.9%) 383/10950 (3%) |
0.0302* |
|
CALM2 (Calmodulin) WWTR1 (TAZ) |
−0.11 (p = 4.27e-26) | 108/10950 (1%) 383/10950 (3%) |
0.0334* |
|
CALM3 (Calmodulin) WWTR1 (TAZ) |
−0.06 (p = 1.96e-9) | 87/10950 (0.8%) 383/10950 (3%) |
2.240e-3* |
P <0.05
Spearman: 0–0.19 is regarded as very weak, 0.2–0.39 as weak, 0.40–0.59 as moderate, 0.6–0.79 as strong and 0.8–1 as very strong correlation.
Concluding remarks
Signaling pathways have historically been perceived as linear, independent networks. Instead, numerous studies over the last decade have demonstrated that different signaling cascades can intersect, with one pathway influencing the activation of another. As illustrated in this review, this concept applies to Ca2+ and Hippo signaling. Importantly, both activating and inhibitory effects of Ca2+ on Hippo have been reported, illustrating the complex nature of the crosstalk. The influence of Ca2+ on Hippo is most likely determined by the cellular context, since Ca2+ and Hippo signaling proteins, as well as their regulators, exhibit differential expression, localization, and/or activity in different cell types or tissues. In addition, Ca2+-Hippo crosstalk appears to be influenced by the nature of the stimulus applied to the cells. Most studies that investigated mechanically-induced influx of Ca2+ through plasma membrane channels report that increased [Ca2+]i activates YAP via RhoA and actin. By contrast, SOCE-induced elevation of [Ca2+]i generally inhibits YAP, for example via PKC or Merlin. The apparent discrepancies in the role of Ca2+ as a Hippo regulator emphasize the importance of accurately reporting the experimental conditions under which signaling was studied in order to enable readers to interpret and integrate different studies.
Most results mentioned in this review were obtained using carcinoma cells with artificially-induced increase of [Ca2+]i, for example through application of mechanical stress or chemical treatment. The use of such cellular systems clearly demonstrates crosstalk and provides molecular insights into how the pathways intersect, both via CaBPs and Ca2+-regulated proteins. Future research should focus on validating the identified crosstalk in systems with physiologically-induced activation of Ca2+ and Hippo. Due to the localized and transient nature of signaling, this requires tools with high spatial and temporal resolution, such as imaging protein interactions and phosphorylation in live cells, neither of which is a trivial undertaking. Importantly, the influence of Ca2+ on Hippo has also been observed in more complex in vivo systems, where it influences both physiology and pathology. Further elucidation of the molecular mechanisms underlying the intersection of Ca2+ and Hippo may lead to the development of new therapeutic agents for a wide array of diseases.
Highlights.
The Hippo signaling pathway is a master regulator of organ size and tissue homeostasis.
Ca2+ is a fundamental second messenger that modulates diverse signaling cascades.
Ca2+ can influence Hippo pathway components.
This review summarises the biological consequences of the intersections between Ca2+ and Hippo.
Acknowledgements
The authors thank the many wonderful colleagues in the Sacks laboratory who have contributed to studies investigating Ca2+ and Hippo crosstalk. We thank Andrew Hedman for his valuable insight and helpful discussion. We apologize to authors whose primary work was omitted because of space restrictions.
Funding
Funding was provided by the Intramural Research Program of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
Authors declare no competing interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
The results reported in Table1 are in whole based upon data generated by the TCGA Pan Cancer Atlas Studies: https://www.cbioportal.org
References
- [1].Hilman D, Gat U, The evolutionary history of YAP and the hippo/YAP pathway, Mol Biol Evol 28(8) (2011) 2403–17. [DOI] [PubMed] [Google Scholar]
- [2].Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Halder G, Lai ZC, Guan KL, Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control, Genes Dev 21(21) (2007) 2747–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Berridge MJ, Lipp P, Bootman MD, The versatility and universality of calcium signalling, Nat Rev Mol Cell Biol 1(1) (2000) 11–21. [DOI] [PubMed] [Google Scholar]
- [4].Ma S, Meng Z, Chen R, Guan KL, The Hippo Pathway: Biology and Pathophysiology, Annu Rev Biochem 88 (2019) 577–604. [DOI] [PubMed] [Google Scholar]
- [5].Yu FX, Zhao B, Guan KL, Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer, Cell 163(4) (2015) 811–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yan F, Qian M, He Q, Zhu H, Yang B, The posttranslational modifications of Hippo-YAP pathway in cancer, Biochim Biophys Acta Gen Subj 1864(1) (2020) 129397. [DOI] [PubMed] [Google Scholar]
- [7].Chan EH, Nousiainen M, Chalamalasetty RB, Schafer A, Nigg EA, Sillje HH, The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1, Oncogene 24(12) (2005) 2076–86. [DOI] [PubMed] [Google Scholar]
- [8].Wu S, Liu Y, Zheng Y, Dong J, Pan D, The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway, Dev Cell 14(3) (2008) 388–98. [DOI] [PubMed] [Google Scholar]
- [9].Ege N, Dowbaj AM, Jiang M, Howell M, Hooper S, Foster C, Jenkins RP, Sahai E, Quantitative Analysis Reveals that Actin and Src-Family Kinases Regulate Nuclear YAP1 and Its Export, Cell Syst 6(6) (2018) 692–708 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Manning SA, Dent LG, Kondo S, Zhao ZW, Plachta N, Harvey KF, Dynamic Fluctuations in Subcellular Localization of the Hippo Pathway Effector Yorkie In Vivo, Curr Biol 28(10) (2018) 1651–1660 e4. [DOI] [PubMed] [Google Scholar]
- [11].Li FL, Guan KL, The two sides of Hippo pathway in cancer, Semin Cancer Biol 85 (2022) 33–42. [DOI] [PubMed] [Google Scholar]
- [12].Clapham DE, Calcium signaling, Cell 131(6) (2007) 1047–58. [DOI] [PubMed] [Google Scholar]
- [13].Leybaert L, Sanderson MJ, Intercellular Ca(2+) waves: mechanisms and function, Physiol Rev 92(3) (2012) 1359–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Berridge MJ, Bootman MD, Roderick HL, Calcium signalling: dynamics, homeostasis and remodelling, Nat Rev Mol Cell Biol 4(7) (2003) 517–29. [DOI] [PubMed] [Google Scholar]
- [15].Luan S, Wang C, Calcium Signaling Mechanisms Across Kingdoms, Annu Rev Cell Dev Biol 37 (2021) 311–340. [DOI] [PubMed] [Google Scholar]
- [16].Venkatachalam K, van Rossum DB, Patterson RL, Ma HT, Gill DL, The cellular and molecular basis of store-operated calcium entry, Nat Cell Biol 4(11) (2002) E263–72. [DOI] [PubMed] [Google Scholar]
- [17].Hogan PG, Rao A, Store-operated calcium entry: Mechanisms and modulation, Biochem Biophys Res Commun 460(1) (2015) 40–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Elíes J, Yáñez M, Pereira TMC, Gil-Longo J, MacDougall DA, Campos-Toimil M, An Update to Calcium Binding Proteins, in: Islam MS (Ed.), Calcium Signaling, Springer International Publishing, Cham, 2020, pp. 183–213. [DOI] [PubMed] [Google Scholar]
- [19].Zimmer DB, Dubuisson JG, Identification of an S100 target protein: glycogen phosphorylase, Cell Calcium 14(4) (1993) 323–332. [DOI] [PubMed] [Google Scholar]
- [20].Tsvetkov PO, Protasevich II, Gilli R, Lafitte D, Lobachov VM, Haiech J, Briand C, Makarov AA, Apocalmodulin Binds to the Myosin Light Chain Kinase Calmodulin Target Site *, Journal of Biological Chemistry 274(26) (1999) 18161–18164. [DOI] [PubMed] [Google Scholar]
- [21].Thines L, Gorisse L, Li Z, Sayedyahossein S, Sacks DB, Calmodulin activates the Hippo signaling pathway by promoting LATS1 kinase-mediated inhibitory phosphorylation of the transcriptional coactivator YAP, J Biol Chem 298(5) (2022) 101839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Guo Q, Wang J, Cao Z, Tang Y, Feng C, Huang F, Interaction of S100A1 with LATS1 promotes cell growth through regulation of the Hippo pathway in hepatocellular carcinoma, International journal of oncology 53(2) (2018) 592–602. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [23].Stegert MR, Tamaskovic R, Bichsel SJ, Hergovich A, Hemmings BA, Regulation of NDR2 Protein Kinase by Multi-site Phosphorylation and the S100B Calcium-binding Protein *, Journal of Biological Chemistry 279(22) (2004) 23806–23812. [DOI] [PubMed] [Google Scholar]
- [24].Li Y, Kong F, Shao Q, Wang R, Hu E, Liu J, Jin C, He D, Xiao X, YAP Expression and Activity Are Suppressed by S100A7 via p65/NFκB-mediated Repression of ΔNp63, Molecular Cancer Research 15(12) (2017) 1752–1763. [DOI] [PubMed] [Google Scholar]
- [25].Jiang S, Zhu Y, Chen Z, Huang Z, Liu B, Xu Y, Li Z, Lin Z, Li M, S100A14 inhibits cell growth and epithelial-mesenchymal transition (EMT) in prostate cancer through FAT1-mediated Hippo signaling pathway, Hum Cell 34(4) (2021) 1215–1226. [DOI] [PubMed] [Google Scholar]
- [26].Rigiracciolo DC, Nohata N, Lappano R, Cirillo F, Talia M, Adame-Garcia SR, Arang N, Lubrano S, De Francesco EM, Belfiore A, Gutkind JS, Maggiolini M, Focal Adhesion Kinase (FAK)-Hippo/YAP transduction signaling mediates the stimulatory effects exerted by S100A8/A9-RAGE system in triple-negative breast cancer (TNBC), Journal of Experimental & Clinical Cancer Research 41(1) (2022) 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yun J, Hansen S, Morris O, Madden DT, Libeu CP, Kumar AJ, Wehrfritz C, Nile AH, Zhang Y, Zhou L, Liang Y, Modrusan Z, Chen MB, Overall CC, Garfield D, Campisi J, Schilling B, Hannoush RN, Jasper H, Senescent cells perturb intestinal stem cell differentiation through Ptk7 induced noncanonical Wnt and YAP signaling, Nat Commun 14(1) (2023) 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Millward TA, Heizmann CW, Schäfer BW, Hemmings BA, Calcium regulation of Ndr protein kinase mediated by S100 calcium-binding proteins, The EMBO Journal 17(20) (1998) 5913–5922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang G, Li HN, Cui XQ, Xu T, Dong ML, Li SY, Li XR, S100A1 is a Potential Biomarker for Papillary Thyroid Carcinoma Diagnosis and Prognosis, J Cancer 12(19) (2021) 5760–5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Huang P, Xue J, Long non‑coding RNA FOXD2‑AS1 regulates the tumorigenesis and progression of breast cancer via the S100 calcium binding protein A1/Hippo signaling pathway, Int J Mol Med 46(4) (2020) 1477–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Narumi K, Miyakawa R, Ueda R, Hashimoto H, Yamamoto Y, Yoshida T, Aoki K, Proinflammatory Proteins S100A8/S100A9 Activate NK Cells via Interaction with RAGE, J Immunol 194(11) (2015) 5539–48. [DOI] [PubMed] [Google Scholar]
- [32].Martin D, Degese MS, Vitale-Cross L, Iglesias-Bartolome R, Valera JLC, Wang Z, Feng X, Yeerna H, Vadmal V, Moroishi T, Thorne RF, Zaida M, Siegele B, Cheong SC, Molinolo AA, Samuels Y, Tamayo P, Guan KL, Lippman SM, Lyons JG, Gutkind JS, Assembly and activation of the Hippo signalome by FAT1 tumor suppressor, Nature Communications 9(1) (2018) 2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang L, Tang F, Terracciano L, Hynx D, Kohler R, Bichet S, Hess D, Cron P, Hemmings BA, Hergovich A, Schmitz-Rohmer D, NDR functions as a physiological YAP1 kinase in the intestinal epithelium, Curr Biol 25(3) (2015) 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mbele GO, Deloulme JC, Gentil B.t.J., Delphin C, Ferro M, Garin J, Takahashi M, Baudier J, The Zinc- and Calcium-binding S100B Interacts and Co-localizes with IQGAP1 during Dynamic Rearrangement of Cell Membranes*, Journal of Biological Chemistry 277(51) (2002) 49998–50007. [DOI] [PubMed] [Google Scholar]
- [35].Heil A, Nazmi AR, Koltzscher M, Poeter M, Austermann J, Assard N, Baudier J, Kaibuchi K, Gerke V, S100P is a novel interaction partner and regulator of IQGAP1, J Biol Chem 286(9) (2011) 7227–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sayedyahossein S, Li Z, Hedman AC, Morgan CJ, Sacks DB, IQGAP1 Binds to Yes-associated Protein (YAP) and Modulates Its Transcriptional Activity, The Journal of biological chemistry 291(37) (2016) 19261–19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Li Y, Kong F, Jin C, Hu E, Shao Q, Liu J, He D, Xiao X, The expression of S100A8/S100A9 is inducible and regulated by the Hippo/YAP pathway in squamous cell carcinomas, BMC Cancer 19(1) (2019) 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wang R, Li Y, Hu E, Kong F, Wang J, Liu J, Shao Q, Hao Y, He D, Xiao X, S100A7 promotes lung adenocarcinoma to squamous carcinoma transdifferentiation, and its expression is differentially regulated by the Hippo-YAP pathway in lung cancer cells, Oncotarget 8(15) (2017) 24804–24814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li Y, Kong F, Wang J, Hu E, Wang R, Liu J, Xiao Q, Zhang W, He D, Xiao X, S100A7 induction is repressed by YAP via the Hippo pathway in A431 cells, Oncotarget 7(25) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kong F, Li Y, Hu E, Wang R, Wang J, Liu J, Zhang J, He D, Xiao X, The Characteristic of S100A7 Induction by the Hippo-YAP Pathway in Cervical and Glossopharyngeal Squamous Cell Carcinoma, PLoS One 11(12) (2016) e0167080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kim M, Kim T, Johnson RL, Lim DS, Transcriptional co-repressor function of the hippo pathway transducers YAP and TAZ, Cell Rep 11(2) (2015) 270–82. [DOI] [PubMed] [Google Scholar]
- [42].Hellinger JW, Hüchel S, Goetz L, Bauerschmitz G, Emons G, Gründker C, Inhibition of CYR61-S100A4 Axis Limits Breast Cancer Invasion, Front Oncol 9 (2019) 1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ikeshima H, Yuasa S, Matsuo K, Kawamura K, Hata J, Takano T, Expression of three nonallelic genes coding calmodulin exhibits similar localization on the central nervous system of adult rats, Journal of Neuroscience Research 36(1) (1993) 111–119. [DOI] [PubMed] [Google Scholar]
- [44].Kim M, Ly SH, Xie Y, Duronio GN, Ford-Roshon D, Hwang JH, Sulahian R, Rennhack JP, So J, Gjoerup O, Talamas JA, Grandclaudon M, Long HW, Doench JG, Sethi NS, Giannakis M, Hahn WC, YAP1 and PRDM14 converge to promote cell survival and tumorigenesis, Dev Cell 57(2) (2022) 212–227 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gu Y, Gu C, Physiological and pathological functions of mechanosensitive ion channels, Mol Neurobiol 50(2) (2014) 339–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Silver BB, Wolf AE, Lee J, Pang MF, Nelson CM, Epithelial tissue geometry directs emergence of bioelectric field and pattern of proliferation, Mol Biol Cell 31(16) (2020) 1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bera K, Kiepas A, Godet I, Li Y, Mehta P, Ifemembi B, Paul CD, Sen A, Serra SA, Stoletov K, Tao J, Shatkin G, Lee SJ, Zhang Y, Boen A, Mistriotis P, Gilkes DM, Lewis JD, Fan CM, Feinberg AP, Valverde MA, Sun SX, Konstantopoulos K, Extracellular fluid viscosity enhances cell migration and cancer dissemination, Nature 611(7935) (2022) 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].He L, Tao J, Maity D, Si F, Wu Y, Wu T, Prasath V, Wirtz D, Sun SX, Role of membrane-tension gated Ca(2+) flux in cell mechanosensation, J Cell Sci 131(4) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pathak MM, Nourse JL, Tran T, Hwe J, Arulmoli J, Le DT, Bernardis E, Flanagan LA, Tombola F, Stretch-activated ion channel Piezo1 directs lineage choice in human neural stem cells, Proc Natl Acad Sci U S A 111(45) (2014) 16148–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Li X, Han L, Nookaew I, Mannen E, Silva MJ, Almeida M, Xiong J, Stimulation of Piezo1 by mechanical signals promotes bone anabolism, Elife 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lu XL, Huo B, Park M, Guo XE, Calcium response in osteocytic networks under steady and oscillatory fluid flow, Bone 51(3) (2012) 466–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tsuchiya M, Hara Y, Okuda M, Itoh K, Nishioka R, Shiomi A, Nagao K, Mori M, Mori Y, Ikenouchi J, Suzuki R, Tanaka M, Ohwada T, Aoki J, Kanagawa M, Toda T, Nagata Y, Matsuda R, Takayama Y, Tominaga M, Umeda M, Cell surface flip-flop of phosphatidylserine is critical for PIEZO1-mediated myotube formation, Nat Commun 9(1) (2018) 2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jetta D, Shireen T, Hua SZ, Epithelial cells sense local stiffness via Piezo1 mediated cytoskeletal reorganization, Front Cell Dev Biol 11 (2023) 1198109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Liu Z, Li S, Qian X, Li L, Zhang H, Liu Z, RhoA/ROCK-YAP/TAZ Axis Regulates the Fibrotic Activity in Dexamethasone-Treated Human Trabecular Meshwork Cells, Front Mol Biosci 8 (2021) 728932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hasegawa K, Fujii S, Matsumoto S, Tajiri Y, Kikuchi A, Kiyoshima T, YAP signaling induces PIEZO1 to promote oral squamous cell carcinoma cell proliferation, J Pathol 253(1) (2021) 80–93. [DOI] [PubMed] [Google Scholar]
- [56].Morini M, Bergqvist CA, Asturiano JF, Larhammar D, Dufour S, Dynamic evolution of transient receptor potential vanilloid (TRPV) ion channel family with numerous gene duplications and losses, Front Endocrinol (Lausanne) 13 (2022) 1013868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sharma S, Goswami R, Merth M, Cohen J, Lei KY, Zhang DX, Rahaman SO, TRPV4 ion channel is a novel regulator of dermal myofibroblast differentiation, Am J Physiol Cell Physiol 312(5) (2017) C562–C572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Li X, Cheng Y, Wang Z, Zhou J, Jia Y, He X, Zhao L, Dong Y, Fan Y, Yang X, Shen B, Wu X, Wang J, Xiong C, Wei L, Li X, Wang J, Calcium and TRPV4 promote metastasis by regulating cytoskeleton through the RhoA/ROCK1 pathway in endometrial cancer, Cell Death Dis 11(11) (2020) 1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL, Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis, Genes Dev 26(1) (2012) 54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wada K, Itoga K, Okano T, Yonemura S, Sasaki H, Hippo pathway regulation by cell morphology and stress fibers, Development 138(18) (2011) 3907–14. [DOI] [PubMed] [Google Scholar]
- [61].Sharma S, Goswami R, Rahaman SO, The TRPV4-TAZ mechanotransduction signaling axis in matrix stiffness- and TGFbeta1-induced epithelial-mesenchymal transition, Cell Mol Bioeng 12(2) (2019) 139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sharma S, Goswami R, Zhang DX, Rahaman SO, TRPV4 regulates matrix stiffness and TGFbeta1-induced epithelial-mesenchymal transition, J Cell Mol Med 23(2) (2019) 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kim MH, Kim J, Hong H, Lee SH, Lee JK, Jung E, Kim J, Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation, EMBO J 35(5) (2016) 462–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Goswami C, Kuhn J, Heppenstall PA, Hucho T, Importance of non-selective cation channel TRPV4 interaction with cytoskeleton and their reciprocal regulations in cultured cells, PLoS One 5(7) (2010) e11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Shin SH, Lee EJ, Hyun S, Chun J, Kim Y, Kang SS, Phosphorylation on the Ser 824 residue of TRPV4 prefers to bind with F-actin than with microtubules to expand the cell surface area, Cell Signal 24(3) (2012) 641–51. [DOI] [PubMed] [Google Scholar]
- [66].Garcia-Elias A, Lorenzo IM, Vicente R, Valverde MA, IP3 receptor binds to and sensitizes TRPV4 channel to osmotic stimuli via a calmodulin-binding site, J Biol Chem 283(46) (2008) 31284–8. [DOI] [PubMed] [Google Scholar]
- [67].Masuyama R, Mizuno A, Komori H, Kajiya H, Uekawa A, Kitaura H, Okabe K, Ohyama K, Komori T, Calcium/calmodulin-signaling supports TRPV4 activation in osteoclasts and regulates bone mass, J Bone Miner Res 27(8) (2012) 1708–21. [DOI] [PubMed] [Google Scholar]
- [68].Wang Q, Chen M, Schafer NP, Bueno C, Song SS, Hudmon A, Wolynes PG, Waxham MN, Cheung MS, Assemblies of calcium/calmodulin-dependent kinase II with actin and their dynamic regulation by calmodulin in dendritic spines, Proc Natl Acad Sci U S A 116(38) (2019) 18937–18942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lampe PD, Laird DW, Recent advances in connexin gap junction biology, Fac Rev 11 (2022) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Riquelme MA, Gu S, Hua R, Jiang JX, Mechanotransduction via the coordinated actions of integrins, PI3K signaling and Connexin hemichannels, Bone Res 9(1) (2021) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Li C, Meng Q, Yu X, Jing X, Xu P, Luo D, Regulatory effect of connexin 43 on basal Ca2+ signaling in rat ventricular myocytes, PLoS One 7(4) (2012) e36165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chen J, Li L, Li Y, Liang X, Sun Q, Yu H, Zhong J, Ni Y, Chen J, Zhao Z, Gao P, Wang B, Liu D, Zhu Z, Yan Z, Activation of TRPV1 channel by dietary capsaicin improves visceral fat remodeling through connexin43-mediated Ca2+ influx, Cardiovasc Diabetol 14 (2015) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Arellano RO, Ramon F, Rivera A, Zampighi GA, Calmodulin acts as an intermediary for the effects of calcium on gap junctions from crayfish lateral axons, J Membr Biol 101(2) (1988) 119–31. [DOI] [PubMed] [Google Scholar]
- [74].Peracchia C, Leverone Peracchia LM, Calmodulin-Connexin Partnership in Gap Junction Channel Regulation-Calmodulin-Cork Gating Model, Int J Mol Sci 22(23) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hu Z, Riquelme MA, Gu S, Jiang JX, Regulation of Connexin Gap Junctions and Hemichannels by Calcium and Calcium Binding Protein Calmodulin, Int J Mol Sci 21(21) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Zheng L, Li H, Cannon A, Trease AJ, Spagnol G, Zheng H, Radio S, Patel K, Batra S, Sorgen PL, Phosphorylation of Cx43 residue Y313 by Src contributes to blocking the interaction with Drebrin and disassembling gap junctions, J Mol Cell Cardiol 126 (2019) 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Yang Y, Ren J, Sun Y, Xue Y, Zhang Z, Gong A, Wang B, Zhong Z, Cui Z, Xi Z, Yang GY, Sun Q, Bian L, A connexin43/YAP axis regulates astroglial-mesenchymal transition in hemoglobin induced astrocyte activation, Cell Death Differ 25(10) (2018) 1870–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Liu Z, Wei Y, Zhang L, Yee PP, Johnson M, Zhang X, Gulley M, Atkinson JM, Trebak M, Wang H-G, Li W, Induction of store-operated calcium entry (SOCE) suppresses glioblastoma growth by inhibiting the Hippo pathway transcriptional coactivators YAP/TAZ, Oncogene 38(1) (2019) 120–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Sinclear CK, Maruyama J, Nagashima S, Arimoto-Matsuzaki K, Kuleape JA, Iwasa H, Nishina H, Hata Y, Protein kinase Cα activation switches YAP1 from TEAD-mediated signaling to p73-mediated signaling, Cancer Sci 113(4) (2022) 1305–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Su T, Ludwig MZ, Xu J, Fehon RG, Kibra and Merlin Activate the Hippo Pathway Spatially Distinct from and Independent of Expanded, Dev Cell 40(5) (2017) 478–490.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Wei Y, Yee PP, Liu Z, Zhang L, Guo H, Zheng H, Anderson B, Gulley M, Li W, NEDD4L-mediated Merlin ubiquitination facilitates Hippo pathway activation, EMBO Rep 21(12) (2020) e50642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].D’Amore A, Hanbashi AA, Di Agostino S, Palombi F, Sacconi A, Voruganti A, Taggi M, Canipari R, Blandino G, Parrington J, Filippini A, Loss of Two-Pore Channel 2 (TPC2) Expression Increases the Metastatic Traits of Melanoma Cells by a Mechanism Involving the Hippo Signalling Pathway and Store-Operated Calcium Entry, Cancers (Basel) 12(9) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Franklin JM, Ghosh RP, Shi Q, Reddick MP, Liphardt JT, Concerted localization-resets precede YAP-dependent transcription, Nat Commun 11(1) (2020) 4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Deng Z, Wang W, Xu X, Gould OEC, Kratz K, Ma N, Lendlein A, Polymeric sheet actuators with programmable bioinstructivity, Proc Natl Acad Sci U S A 117(4) (2020) 1895–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Shao X, Li Q, Mogilner A, Bershadsky AD, Shivashankar GV, Mechanical stimulation induces formin-dependent assembly of a perinuclear actin rim, Proc Natl Acad Sci U S A 112(20) (2015) E2595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wales P, Schuberth CE, Aufschnaiter R, Fels J, Garcia-Aguilar I, Janning A, Dlugos CP, Schafer-Herte M, Klingner C, Walte M, Kuhlmann J, Menis E, Kang LH, Maier KC, Hou WY, Russo A, Higgs HN, Pavenstadt H, Vogl T, Roth J, Qualmann B, Kessels MM, Martin DE, Mulder B, Wedlich-Soldner R, Calcium-mediated actin reset (CaAR) mediates acute cell adaptations, Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Liu Z, Wei Y, Zhang L, Yee PP, Johnson M, Zhang X, Gulley M, Atkinson JM, Trebak M, Wang HG, Li W, Induction of store-operated calcium entry (SOCE) suppresses glioblastoma growth by inhibiting the Hippo pathway transcriptional coactivators YAP/TAZ, Oncogene 38(1) (2019) 120–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Pardo-Pastor C, Rubio-Moscardo F, Vogel-Gonzalez M, Serra SA, Afthinos A, Mrkonjic S, Destaing O, Abenza JF, Fernandez-Fernandez JM, Trepat X, Albiges-Rizo C, Konstantopoulos K, Valverde MA, Piezo2 channel regulates RhoA and actin cytoskeleton to promote cell mechanobiological responses, Proc Natl Acad Sci U S A 115(8) (2018) 1925–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Sun W, Chi S, Li Y, Ling S, Tan Y, Xu Y, Jiang F, Li J, Liu C, Zhong G, Cao D, Jin X, Zhao D, Gao X, Liu Z, Xiao B, Li Y, The mechanosensitive Piezo1 channel is required for bone formation, Elife 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wang L, You X, Lotinun S, Zhang L, Wu N, Zou W, Mechanical sensing protein PIEZO1 regulates bone homeostasis via osteoblast-osteoclast crosstalk, Nat Commun 11(1) (2020) 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Falcone JC, Kuo L, Meininger GA, Endothelial cell calcium increases during flow-induced dilation in isolated arterioles, Am J Physiol 264(2 Pt 2) (1993) H653–9. [DOI] [PubMed] [Google Scholar]
- [92].Toba H, Nakagawa Y, Miki S, Shimizu T, Yoshimura A, Inoue R, Asayama J, Kobara M, Nakata T, Calcium channel blockades exhibit anti-inflammatory and antioxidative effects by augmentation of endothelial nitric oxide synthase and the inhibition of angiotensin converting enzyme in the N(G)-nitro-L-arginine methyl ester-induced hypertensive rat aorta: vasoprotective effects beyond the blood pressure-lowering effects of amlodipine and manidipine, Hypertens Res 28(8) (2005) 689–700. [DOI] [PubMed] [Google Scholar]
- [93].Shetty S, Malik AH, Feringa H, El Accaoui R, Girotra S, Meta-Analysis Evaluating Calcium Channel Blockers and the Risk of Peripheral Arterial Disease in Patients With Hypertension, Am J Cardiol 125(6) (2020) 907–915. [DOI] [PubMed] [Google Scholar]
- [94].Quan M, Lv H, Liu Z, Li K, Zhang C, Shi L, Yang X, Lei P, Zhu Y, Ai D, MST1 Suppresses Disturbed Flow Induced Atherosclerosis, Circ Res 131(9) (2022) 748–764. [DOI] [PubMed] [Google Scholar]
- [95].Howe KL, Fish JE, Transforming endothelial cells in atherosclerosis, Nat Metab 1(9) (2019) 856–857. [DOI] [PubMed] [Google Scholar]
- [96].Xu S, Koroleva M, Yin M, Jin ZG, Atheroprotective laminar flow inhibits Hippo pathway effector YAP in endothelial cells, Transl Res 176 (2016) 18–28 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, Liu J, Deng D, Lau CW, Wan S, Ai D, Mak KK, Tong KK, Kwan KM, Wang N, Chiu JJ, Zhu Y, Huang Y, Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow, Nature 540(7634) (2016) 579–582. [DOI] [PubMed] [Google Scholar]
- [98].Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, Guan KL, Li YJ, Chien S, Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis, Proc Natl Acad Sci U S A 113(41) (2016) 11525–11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Amador HE, Cordova A, Figueroa X, Control of the Ca2+-Mediated Endothelial Cell Migration by Connexin 43-Formed Hemichannels, Faseb Journal 33 (2019). [Google Scholar]
- [100].Schmitt T, Waschke J, Autoantibody-Specific Signalling in Pemphigus, Front Med (Lausanne) 8 (2021) 701809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Walter E, Vielmuth F, Wanuske MT, Seifert M, Pollmann R, Eming R, Waschke J, Role of Dsg1- and Dsg3-Mediated Signaling in Pemphigus Autoantibody-Induced Loss of Keratinocyte Cohesion, Front Immunol 10 (2019) 1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Esaki C, Seishima M, Yamada T, Osada K, Kitajima Y, Pharmacologic evidence for involvement of phospholipase C in pemphigus IgG-induced inositol 1,4,5-trisphosphate generation, intracellular calcium increase, and plasminogen activator secretion in DJM-1 cells, a squamous cell carcinoma line, J Invest Dermatol 105(3) (1995) 329–33. [DOI] [PubMed] [Google Scholar]
- [103].Spindler V, Endlich A, Hartlieb E, Vielmuth F, Schmidt E, Waschke J, The extent of desmoglein 3 depletion in pemphigus vulgaris is dependent on Ca(2+)-induced differentiation: a role in suprabasal epidermal skin splitting?, Am J Pathol 179(4) (2011) 1905–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Rehman A, Cai Y, Hünefeld C, Jedličková H, Huang Y, Teh MT, Uttagomol J, Kang A, Warnes G, Ahmad U, Harwood C, Bergamaschi D, Parkinson EK, Röcken M, Hart I, Wan H, The Pemphigus Vulgaris antigen desmoglein-3 suppresses p53 function via the YAP-Hippo pathway, bioRxiv (2018) 399980.
- [105].Huang Y, Jedlickova H, Cai Y, Rehman A, Gammon L, Ahmad US, Uttagomol J, Parkinson EK, Fortune F, Wan H, Oxidative Stress-Mediated YAP Dysregulation Contributes to the Pathogenesis of Pemphigus Vulgaris, Front Immunol 12 (2021) 649502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Le MH, Yeo YH, Zou B, Barnet S, Henry L, Cheung R, Nguyen MH, Forecasted 2040 global prevalence of nonalcoholic fatty liver disease using hierarchical bayesian approach, Clin Mol Hepatol 28(4) (2022) 841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Wang X, Cai B, Yang X, Sonubi OO, Zheng Z, Ramakrishnan R, Shi H, Valenti L, Pajvani UB, Sandhu J, Infante RE, Radhakrishnan A, Covey DF, Guan KL, Buck J, Levin LR, Tontonoz P, Schwabe RF, Tabas I, Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis, Cell Metab 31(5) (2020) 969–986 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Cornec-Le Gall E, Alam A, Perrone RD, Autosomal dominant polycystic kidney disease, The Lancet 393(10174) (2019) 919–935. [DOI] [PubMed] [Google Scholar]
- [109].Happé H, van der Wal AM, Leonhard WN, Kunnen SJ, Breuning MH, de Heer E, Peters DJM, Altered Hippo signalling in polycystic kidney disease, The Journal of Pathology 224(1) (2011) 133–142. [DOI] [PubMed] [Google Scholar]
- [110].Cai J, Song X, Wang W, Watnick T, Pei Y, Qian F, Pan D, A RhoA-YAP-c-Myc signaling axis promotes the development of polycystic kidney disease, Genes Dev 32(11–12) (2018) 781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Tian Y, Kolb R, Hong JH, Carroll J, Li D, You J, Bronson R, Yaffe MB, Zhou J, Benjamin T, TAZ promotes PC2 degradation through a SCFbeta-Trcp E3 ligase complex, Mol Cell Biol 27(18) (2007) 6383–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Geng J, Shi Y, Zhang J, Yang B, Wang P, Yuan W, Zhao H, Li J, Qin F, Hong L, Xie C, Deng X, Sun Y, Wu C, Chen L, Zhou D, TLR4 signalling via Piezo1 engages and enhances the macrophage mediated host response during bacterial infection, Nat Commun 12(1) (2021) 3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Lee M, Du H, Winer DA, Clemente-Casares X, Tsai S, Mechanosensing in macrophages and dendritic cells in steady-state and disease, Front Cell Dev Biol 10 (2022) 1044729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Chakraborty M, Chu K, Shrestha A, Revelo XS, Zhang X, Gold MJ, Khan S, Lee M, Huang C, Akbari M, Barrow F, Chan YT, Lei H, Kotoulas NK, Jovel J, Pastrello C, Kotlyar M, Goh C, Michelakis E, Clemente-Casares X, Ohashi PS, Engleman EG, Winer S, Jurisica I, Tsai S, Winer DA, Mechanical Stiffness Controls Dendritic Cell Metabolism and Function, Cell Rep 34(2) (2021) 108609. [DOI] [PubMed] [Google Scholar]
- [115].Zheng S, Wang X, Zhao D, Liu H, Hu Y, Calcium homeostasis and cancer: insights from endoplasmic reticulum-centered organelle communications, Trends Cell Biol 33(4) (2023) 312–323. [DOI] [PubMed] [Google Scholar]
- [116].Mohajan S, Jaiswal PK, Vatanmakarian M, Yousefi H, Sankaralingam S, Alahari SK, Koul S, Koul HK, Hippo pathway: Regulation, deregulation and potential therapeutic targets in cancer, Cancer Lett 507 (2021) 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Godwin SL, Soltoff SP, Calcium-sensing receptor-mediated activation of phospholipase C-gamma1 is downstream of phospholipase C-beta and protein kinase C in MC3T3-E1 osteoblasts, Bone 30(4) (2002) 559–66. [DOI] [PubMed] [Google Scholar]
- [118].Tu CL, Chang W, Bikle DD, The calcium-sensing receptor-dependent regulation of cell-cell adhesion and keratinocyte differentiation requires Rho and filamin A, J Invest Dermatol 131(5) (2011) 1119–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Thiel G, Lesch A, Keim A, Transcriptional response to calcium-sensing receptor stimulation, Endocrinology 153(10) (2012) 4716–28. [DOI] [PubMed] [Google Scholar]
- [120].Tavanti GS, Verdelli C, Morotti A, Maroni P, Guarnieri V, Scillitani A, Silipigni R, Guerneri S, Maggiore R, Mari G, Vicentini L, Dalino Ciaramella P, Vaira V, Corbetta S, Yes-Associated Protein 1 Is a Novel Calcium Sensing Receptor Target in Human Parathyroid Tumors, Int J Mol Sci 22(4) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Witteveen JE, Hamdy NA, Dekkers OM, Kievit J, van Wezel T, Teh BT, Romijn JA, Morreau H, Downregulation of CASR expression and global loss of parafibromin staining are strong negative determinants of prognosis in parathyroid carcinoma, Mod Pathol 24(5) (2011) 688–97. [DOI] [PubMed] [Google Scholar]
- [122].Dang DK, Makena MR, Llongueras JP, Prasad H, Ko M, Bandral M, Rao R, A Ca(2+)-ATPase Regulates E-cadherin Biogenesis and Epithelial-Mesenchymal Transition in Breast Cancer Cells, Mol Cancer Res 17(8) (2019) 1735–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Kim NG, Koh E, Chen X, Gumbiner BM, E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components, Proc Natl Acad Sci U S A 108(29) (2011) 11930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Xiong Y, Dong L, Bai Y, Tang H, Li S, Luo D, Liu F, Bai J, Yang S, Song X, Piezo1 activation facilitates ovarian cancer metastasis via Hippo/YAP signaling axis, Channels (Austin) 16(1) (2022) 159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Zhu B, Qian W, Han C, Bai T, Hou X, Piezo 1 activation facilitates cholangiocarcinoma metastasis via Hippo/YAP signaling axis, Mol Ther Nucleic Acids 24 (2021) 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The results reported in Table1 are in whole based upon data generated by the TCGA Pan Cancer Atlas Studies: https://www.cbioportal.org