

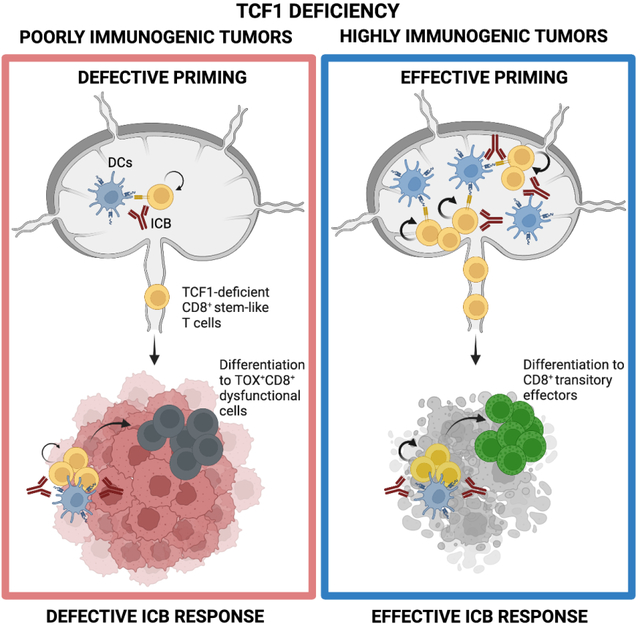
Abstract
Stem-like CD8+ T cells are regulated by T cell factor 1 (TCF1) and are considered requisite for immune checkpoint blockade (ICB) response. However, recent findings indicate that reliance on TCF1+CD8+ T cells for ICB efficacy may differ across tumor contexts. We find that TCF1 is essential for optimal priming of tumor antigen-specific CD8+ T cells and ICB response in poorly immunogenic tumors that accumulate TOX+ dysfunctional T cells, but is dispensable for T cell priming and therapy response in highly immunogenic tumors that efficiently expand transitory effectors. Importantly, improving T cell priming by vaccination or by enhancing antigen presentation on tumors rescues the defective responses of TCF1-deficient CD8+ T cells upon ICB in poorly immunogenic tumors. Our study highlights TCF1’s role during the early stages of anti-tumor CD8+ T cell responses with important implications for guiding optimal therapeutic interventions in cancers with low TCF1+CD8+ T cells and low neo-antigen expression.
eTOC Blurb
Escobar et al. uncover that TCF1 expression in CD8+ T cells is required for ICB response in poorly, but not highly, immunogenic tumors. They further show that therapeutic vaccination can rescue ICB response in low antigen-expressing tumors in TCF1-deficient settings.
Graphical Abstract
Introduction
Immune checkpoint blockade (ICB) therapy represents a breakthrough in cancer treatment but fails to elicit durable clinical benefit in many patients, underscoring the need to identify the cellular and molecular mechanisms underlying ICB response1. In this regard, a CD8+ T cell subset that retains stemness and memory potential has emerged as a key player in the response to ICB and other immune-harnessing therapies. We, and others, have shown that intra-tumoral CD8+ stem-like T cells are tumor-antigen specific, have polyfunctional effector capacity, and provide the proliferative burst that seeds the effector response upon ICB2–8. Stem-like T cells depend, at least in part, on T cell factor 1 (TCF1; encoded by Tcf7)9. Genetic ablation of Tcf7 in mature CD8+ T cells compromises, but does not completely abrogate, their maintenance and function, thereby limiting the efficacy of ICB in experimental cancer models3,4.
The clinical relevance of TCF1+CD8+ stem-like T cells is supported by studies in melanoma patients. One study showed that the frequency of TCF7-expressing CD8+ T cells correlated with positive response to ICB7. However, other studies have shown that the frequency of TCF1+CD8+ T cells associated with progression free survival (PFS) but did not stratify ICB responders versus non-responders3,8. In advanced clear cell renal carcinoma patients treated with ICB, the frequency of TCF1+CD8+ T cells failed to predict any clinical outcomes10. Such findings indicate that the reliance on TCF1+CD8+ stem-like T cells for ICB efficacy may not be equal across patients or tumor contexts. Moreover, that loss of TCF1 in mature CD8+ T cells neither completely abrogates the presence of stem-like CD8+ T cells in tumors nor the response to ICB, raises the important question of what determines reliance on TCF1 for effective anti-tumor responses. Recent studies have highlighted the tumor-draining lymph node (TDLN) as a reservoir of tumor-specific TCF1+CD8+ stem-like precursors that are the direct targets of ICB therapy11–15. How TCF1 regulates the priming of these TCF1+CD8+ precursors and shapes the quality of anti-tumor CD8+ T cell responses remains an open question.
Here, we investigated a role for TCF1 during the priming of tumor-antigen specific CD8+ T cells in the TDLN and in shaping the intra-tumoral CD8+ T cell response in different tumor contexts and upon ICB.
Results
Reliance on TCF1 for ICB response is determined by tumor immunogenicity
Our previous work and that of others indicated that loss of TCF1 in CD8+ T cells limits, but does not completely abrogate, the response to ICB and other immune-based therapies3,4. We therefore examined the reliance on TCF1 for the induction of effective anti-tumor responses upon ICB in different tumor contexts. We generated conditional knock-out mice in which Cre-mediated Tcf7 (encoding TCF1) deletion was driven by the E8i enhancer (E8i-Cre x Tcf7fl/fl; TCF1 cKO), which becomes active after thymic positive selection in the CD8+ T cell lineage only16. We observed no abnormalities in thymocyte development in TCF1 cKO mice as compared to wild-type controls (Tcf7fl/fl; WT) (Figure 1A). Indeed, we observed limited (~24%) Tcf7 deletion in CD8+ thymocytes but nearly complete (~91%) deletion in peripheral CD8+ T cells in TCF1 cKO mice (Figure 1B). Next, we implanted B16-F10 melanoma or MC38 colon carcinoma, both expressing ovalbumin (OVA) as a common ectopic tumor antigen in WT and TCF1 cKO mice. The MC38-OVA cell line we used in this study has high MHC-I expression, in contrast to the cell line we used previously4 (Figure S1A). B16-OVA had lower expression of total MHC-I (Figure S1A–B) and of MHC-I presenting SIINFEKL (Figure 1C) compared to MC38-OVA tumors in vivo. When tumors were established, we treated with isotype (ISO) or ICB (anti-TIM-3 + anti-PD-L1) (Figure 1D). Surprisingly, we found that loss of TCF1 in CD8+ T cells limited ICB response in mice bearing B16-OVA tumors but had no effect in mice bearing MC38-OVA tumors (Figure 1E). These data indicated differential reliance on TCF1+CD8+ stem-like T cells to seed the effector response in these different tumor contexts. Accordingly, we next investigated the frequency of stem-like CD8+ tumor-infiltrating lymphocytes (TILs) and their transition to effectors in the two tumor models at comparable tumor dimensions (Figure S1C). We implanted tumors into WT and Tcf7-GFP mice, in which GFP reports Tcf7 locus activity17, and used PD-1, TIM-3, SLAMF6, CX3CR1, and CD39 expression to distinguish four CD8+ TIL subsets (Figure 1F): PD-1+TIM-3−SLAMF6+CX3CR1− stem-like cells (population 1), PD-1+TIM-3−SLAMF6low/negCX3CR1+ early effectors (population 2), PD-1+TIM-3+CD39low/negSLAMF6+ pre-dysfunctional cells (population 3) and PD-1+TIM-3+CD39+SLAMF6− terminally dysfunctional cells (population 4). We observed a reduced proportion of stem-like cells (population 1) and a higher proportion and number of early effectors (population 2) in MC38-OVA compared to B16-OVA tumors (Figure 1G and S1D). Furthermore, MC38-OVA tumors contained higher numbers of PD-1+TIM-3+ CD8+ T cells (populations 3 and 4) (Figure S1D), although the proportions of these PD-1+TIM-3+ subsets were similar compared to B16-OVA tumors (Figure 1G). We observed similar differences within OVA-dextramer+ CD8+ TILs in the two tumor models (Figure 1H and S1E). Notably, OVA-dextramer+ CD8+ T cells were detected in all MC38-OVA tumors analyzed (n=15) but only in a fraction (n=7/16; 44%) of B16-OVA tumors, in line with their differential immunogenicity (Figure S1F). We next examined TCF1 expression and Tcf7-locus activity. As expected, TCF1 expression and Tcf7-locus activity was highest in stem-like T cells (population 1), reduced in both early effectors (population 2) and pre-dysfunctional cells (population 3), and mostly absent in terminally dysfunctional cells (population 4) (Figure 1G–H). Notably, TCF1 expression and/or Tcf7-locus activity were significantly diminished in populations 1 and 2 from MC38-OVA as compared to B16-OVA tumors (Figure 1G and S1G). However, when examining only OVA-dextramer+ CD8+ TILs, we observed a similar proportion of TCF1-expressing cells in population 2 between the two tumors, likely reflecting strong TCF1 downregulation by cells that have undergone successful effector transition upon TCR triggering by SIINFEKL in the two models. Altogether, these data indicated that tumors that failed to promote potent stem-like to early effector transition relied on a pool of TCF1+ CD8+ stem-like T cells to sustain ICB response.
Figure 1. TCF1 is dispensable for ICB efficacy in highly but not poorly immunogenic tumors.
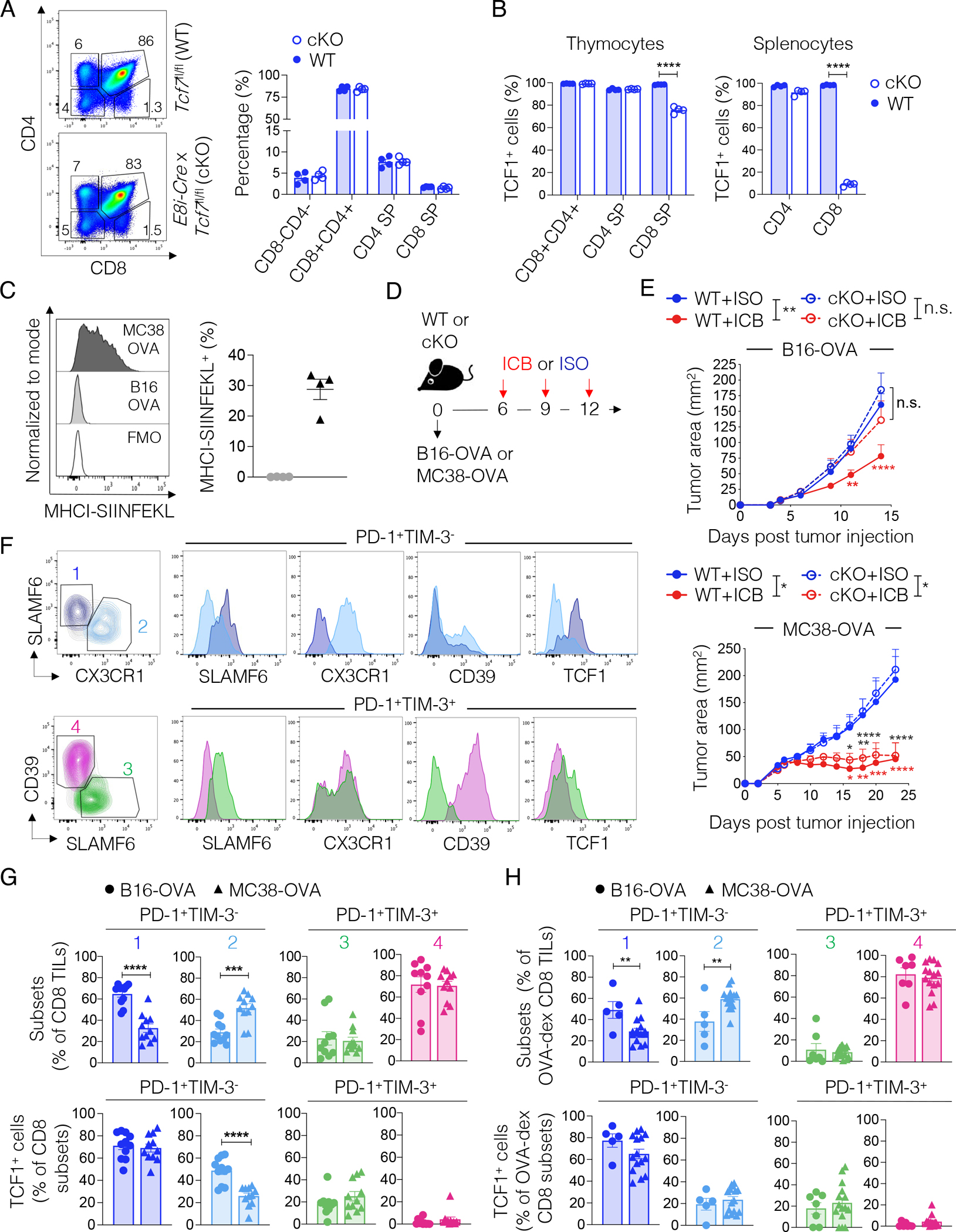
(A) Representative plots and frequency of the indicated populations in the thymus of WT or TCF1 cKO mice (n=4, one experiment). (B) Frequency of TCF1+ cells in the indicated populations in WT or TCF1 cKO mice from (A). Student’s t test, ****p<0.0001 (C) Representative histograms and frequency of MHC-I-SIINFEKL expression in B16-OVA or MC38-OVA tumors 14 days post implant (n=4, one experiment). (D) Experimental scheme. ICB: anti-PD-L1 + anti-TIM-3; ISO: IgG2a + IgG2b (E) Tumor area over time in WT and TCF1 cKO mice treated as in (D) (n=4–7 per group. B16-OVA: shown one out of 10 experiments; MC38-OVA: shown one out of 7 experiments). Two-way ANOVA and Sidak’s multiple comparisons test, *p<0.05, **p<0.01, ***p < 0.001, ****p<0.0001. (F) Gating strategy and marker expression in the indicated subsets of CD8+ TILs in MC38-OVA and B16-OVA tumors. (G-H) Frequency (top panel) of the TIL subsets defined in (F) and of TCF1-expressing cells (bottom panel) within total (G, n=11, two experiments combined,) or OVA-dextramer+ CD8+ TILs (H, n=5–15, two experiments combined) in MC38-OVA or B16-OVA tumors, 14 days post tumor implant into WT mice. Student’s t test, **p<0.01, ***p<0.001, ****p<0.0001. All values are reported as mean±SEM.
See also Figure S1.
Effective priming of tumor-antigen specific CD8+ precursors in the TDLN requires TCF1 in low tumor antigen contexts.
The observed differential requirement for TCF1 in the two tumor contexts prompted us to examine underlying mechanisms. Despite conflicting data on the contribution of peripheral tumor-antigen specific CD8+ T cells to ICB efficacy3,11,18–22, several studies have shown that the TDLN plays an important role in the priming of productive anti-tumor CD8+ T cell responses11–15. We therefore investigated whether the failure of TCF1 cKO mice to respond to ICB when bearing B16-OVA, but not MC38-OVA, tumors might reflect a differential reliance on TCF1 during the priming of tumor-antigen specific T cells in the TDLN. We transferred naïve WT or TCF1 cKO OTI T cells into B16-OVA or MC38-OVA tumor-bearing mice followed by treatment with one dose of ICB or ISO prior to assessment of T cell proliferation in the TDLN as indicated in Figure 2A. We observed equal proportions of WT and TCF1 cKO OTI T cells in the TDLN of both tumor models (Figure S2A). However, in B16-OVA-bearing mice, only WT OTI T cells underwent efficient proliferation (37.9 ± 4.5% proliferating cells) upon ICB whereas TCF1 cKO OTI T cells proliferated less irrespective of treatment (cKO ICB: 17.4 ± 4.6% proliferating cells; cKO ISO: 20 ± 2.7% proliferating cells). Accordingly, a greater proportion of WT OTI T cells achieved >5 cell divisions (7.1 ± 1.8 %) as compared to TCF1 cKO OTI T cells (3.8 ± 1.4 %) upon ICB (Figure 2B). In contrast, in MC38-OVA-bearing mice, although TCF1 cKO OTI T cells exhibited a modest defect at baseline (ISO), they proliferated equally well as WT OTI T cells upon a single dose of ICB (Figure 2C). Notably, OTI T cells proliferated to a much greater extent in MC38-OVA- (up to 8 divisions) as compared to B16-OVA-bearing (up to 5 divisions) mice. These data indicated that TCF1 was required for optimal activation of tumor antigen-specific CD8+ T cells in the TDLN of mice bearing poorly immunogenic tumors but was dispensable in the context of highly immunogenic tumors. We confirmed these results in a co-transfer setting (Figure S2B–C). We further examined WT and TCF1 cKO Pmel T cells co-transferred into B16-F10 tumor-bearing mice (Figure S2D). In line with the low affinity of the Pmel TCR for its cognate antigen, WT Pmel T cell proliferation was low even upon ICB treatment (16.2% ± 4.3 proliferating cells) compared to WT OTI T cell proliferation (24.5% ± 4.6 proliferating cells) in B16-OVA tumor-bearing mice (co-transfer setting) (Figures S2B and D). Nonetheless, we observed a proliferation defect in TCF1 cKO Pmel T cells at baseline (ISO) and upon ICB, although the latter did not reach statistical significance (Figure S2D).
Figure 2. Defective proliferation of TCF1 cKO T cells in the TDLN of poorly immunogenic tumors.
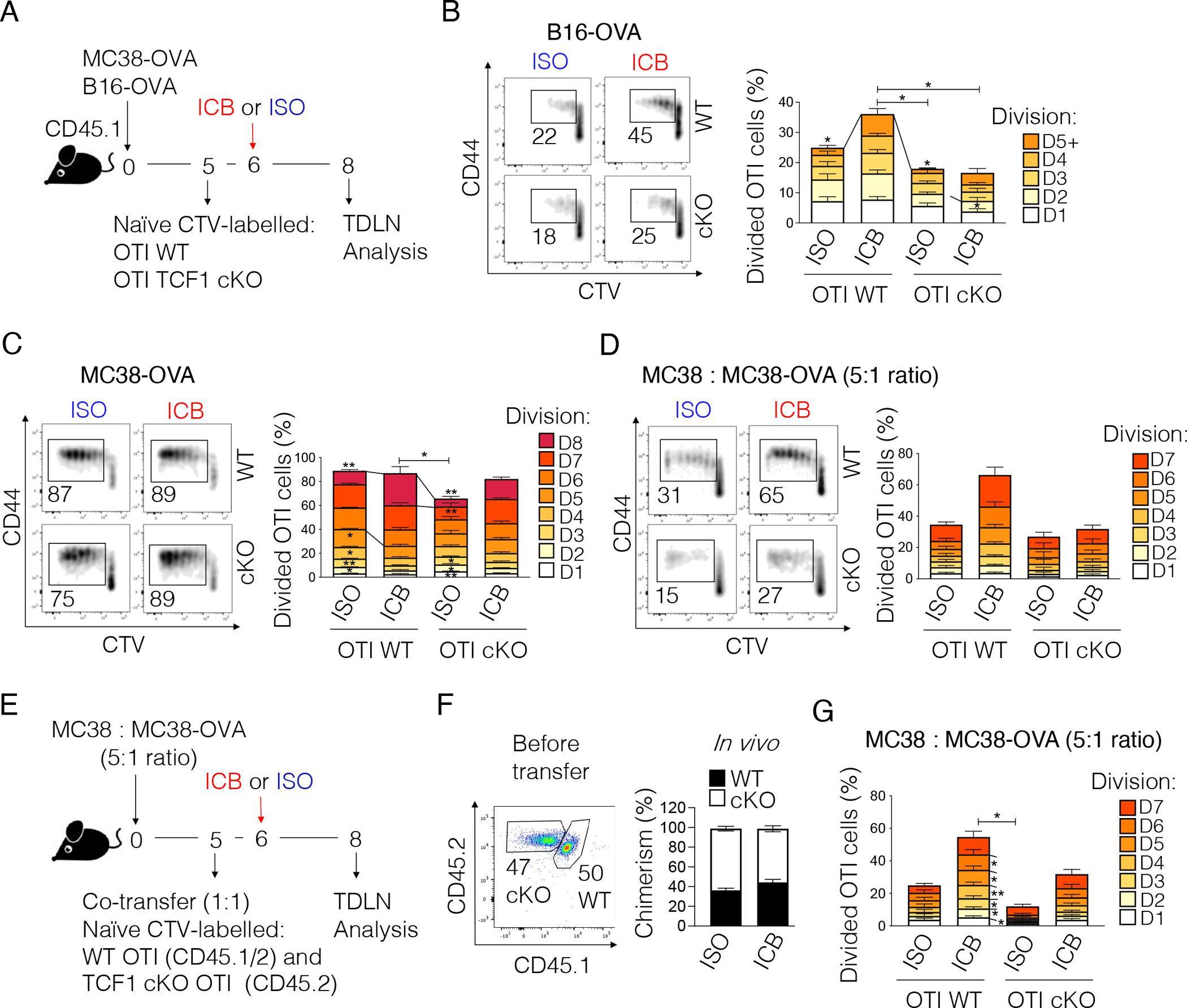
(A) Experimental scheme. ICB: anti-PD-L1 + anti-TIM-3; ISO: IgG2a + IgG2b. (B-D) Representative plots and frequency of proliferating WT or TCF1 cKO OTI T cells in the TDLN of mice implanted with (B) B16-OVA (n=6–7, two experiments combined out of 5 experiments), (C) MC38-OVA (n=3–5, one out of 3 experiments shown), or (D) a 1:5 mixture of MC38-OVA + MC38 tumor cells (n=4–5, one experiment) and treated as shown in (A). (E) Experimental scheme. ICB: anti-PD-L1 + anti-TIM-3; ISO: IgG2a + IgG2b. (F) Chimerism of WT and TCF1 cKO OTI T cells before and 3 days after adoptive transfer. (G) Frequency of proliferating WT or TCF1 cKO OTI T cells in the TDLN of mice treated as shown in (E) (n=6, one experiment). In Figure 2B–D and 2G, one-way ANOVA is used to calculate the statistics related to the total frequency of proliferating cells and within each cell division (all comparisons are done versus WT+ICB group). *p<0.05, **p<0.01. All values are reported as mean±SEM.
See also Figure S2.
The differential reliance on TCF1 for optimal T cell activation could reflect quantitative and/or qualitative differences in antigen presentation in the two models. To address this, we transferred naïve WT or TCF1 cKO OTI T cells into WT mice implanted with a 5:1 mixture of parental and OVA-expressing MC38 cells and examined proliferation upon ICB or ISO administration as in Figure 2A. Strikingly, we found that a five-fold reduction in OVA antigen levels was sufficient to reveal defective proliferation of TCF1 cKO OTI T cells upon one dose of ICB, similar to our observation in the B16-OVA model (Figure 2D). Conversely, WT OTI T cells proliferated well upon one dose of ICB. Importantly, we confirmed these data in a co-transfer setting (Figure 2E–G).
TCF1 regulates T cell responsiveness to low TCR triggering
Our observations indicated differential effects of TCF1 deficiency depending on the strength of TCR triggering. We therefore examined how WT and TCF1 cKO OTI T cells responded to different TCR signal strengths. We activated naïve WT and TCF1 cKO OTI T cells with high vs low doses of anti-CD3 + anti-CD28 antibodies in vitro. We found that WT and TCF1 cKO OTI T cells proliferated equally well, with TCF1 cKO OTI T cells showing a slightly accelerated accumulation of cells undergoing >6 divisions, when stimulated with a high dose of anti-CD3 + anti-CD28 (Figure 3A). Accordingly, both WT and TCF1 cKO OTI T cells decreased CD62L and increased CD44, PD-1, and CD28 expression with TCF1 cKO cells showing a small, but significant, increase in PD-1 and CD28 as compared to WT cells (Figure 3B and S3A). Conversely, TCF1 cKO OTI T cells showed defective proliferation commencing with the first division, reduced CD62L downregulation, and reduced acquisition of CD44, PD-1, and CD28 compared to WT OTI T cells when activated with a low dose of anti-CD3 + anti-CD28 (Figure 3A–B and S3A). Phosphoflow further revealed reduced phosphorylation of key TCR signaling molecules (LAT, ZAP70, ERK, and AKT) in TCF1 cKO OTI T cells (Figure 3C). In line with reduced ERK activation, we observed reduced phosphorylation, and thus activation, of SHP2, which sustains ERK activation23, in TCF1 cKO OTI T cells. Importantly, we further confirmed the generalizability of these results in WT and TCF1 cKO T cells expressing the Pmel TCR or a native TCR repertoire (Figure S3B–H).
Figure 3. TCF1 lowers the activation threshold and poises T cells for optimal responses to low TCR triggering.
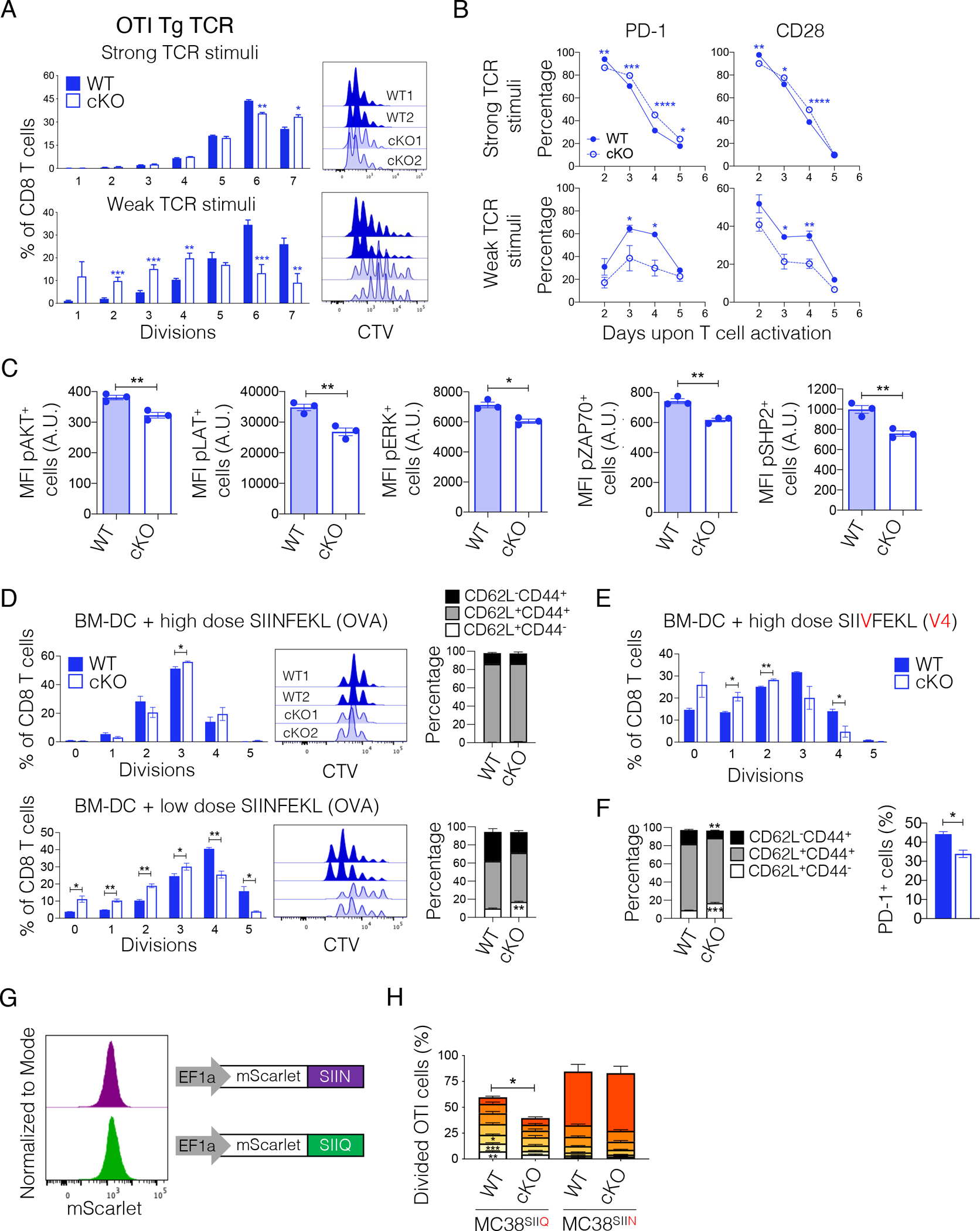
(A) WT and TCF1 cKO T cells were activated with a high (1ug/ml each, n=3, shown one out of 3 experiments) or low (0.3ug/ml each, n=5–6, two experiments combined) dose of anti-CD3 + anti-CD28 for 48 hrs. Representative histograms and frequency of WT and TCF1 cKO OTI T cells in each cell division three days post activation. Student’s t test, *p<0.05, **p<0.01, ***p<0.001. (B) Frequency of PD-1 and CD28 positive cells in WT and TCF1 cKO OTI T cells activated as in (A). Two-way ANOVA and Sidak’s multiple comparison test, *p,0.05, **p<0.01, ***p<0.001, ****p<0.0001. n=3, one experiment. (C) Level (MFI) of phospho-AKT, phospho-LAT, phospho-SHP2, phospho-ERK and phospho-ZAP70 in WT and TCF1 cKO OTI T cells, 24 hours post in vitro activation with anti-CD3 + anti-CD28 (1ug/ml each). n=3, shown one out of 2 experiments. (D) Representative histograms and frequency of WT and TCF1 cKO OTI T cells in each cell division three days post co-culture with BM-DC pulsed with a high (17.78nM) or low (3pM) dose of SIINFEKL. The phenotype of WT and TCF1 cKO T cells at day three is shown. Student’s t test, *p<0.05, **p<0.01. n=3, one experiment. (E) Frequency of WT and TCF1 cKO OTI T cells in each cell division three days post co-culture with BM-DC pulsed with a high (17.78nM) dose of SIIVFEKL (V4). n=3, one experiment. (F) Phenotype and frequency of PD-1 positive cells in WT and TCF1 cKO T cells from (E). (G) Expression of mScarlet-SIIN or mScarlet-SIIQ in MC38 tumor cells engineered with the indicated lentiviral vectors. (H) Frequency of proliferating WT or TCF1 cKO OTI T cells in the TDLN three days post transfer in mice implanted with MC38-mScarlet-SIIN or MC38-mScarlet-SIIQ tumors. n=8–10 mice, one experiment. All values are reported as mean±SEM.
See also Figures S3 and S4.
To study TCF1’s role in the context of different TCR stimulations in a more physiological setting, we co-cultured naïve WT or TCF1 cKO OTI T cells with bone marrow-derived dendritic cells (BM-DC) pulsed with different doses of SIINFEKL. In line with our results using anti-CD3 + anti-CD28 stimulation, we found defective proliferation and acquisition of an activated phenotype in TCF1 cKO OTI T cells cultured with BM-DC pulsed with a low, but not a high, dose of SIINFEKL (Figure 3D). When repeating the experiment using the SIIVFEKL variant (V4), which has low affinity for OTI (SIIV EC50 = 680 vs. SIIN EC50 = 1)24, we observed defective proliferation, reduced down-regulation of CD62L, and reduced acquisition of CD44 and PD-1 expression in TCF1 cKO T cells even when SIIVFEKL was tested at the same high (17.78nM) dose used for SIINFEKL (Figure 3E–F). Accordingly, when testing additional SIINFEKL variants (SIIQFEKL EC50=18, SIITFEKL EC50=70), we observed defective proliferation of TCF1 cKO T cells even when using high peptide doses (Figure S4A). To confirm these results in vivo, we assessed the priming of WT or TCF1 cKO OTI T cells after co-transfer into mice implanted with MC38 cells expressing comparable levels of SIINFEKL or SIIQFEKL (Figure 3G). In line with our in vitro data, we found defective proliferation of TCF1 cKO as compared to WT OTI T cells in the TDLN of mice implanted with MC38-SIIQFEKL but not MC38-SIINFEKL (Figure 3H). Of note, we did not observe WT or TCF1 cKO OTI T cell proliferation in mice bearing MC38-SIIVFEKL, indicating that the affinity of OTI for SIIVFEKL was below the threshold required for activation in vivo (Figure S4B–C). Together, these data suggested that TCF1 can poise T cells for optimal antigen responsiveness, particularly in settings with limiting doses of antigen and low TCR signals.
TCF1 is required for optimal activation of stem-like CD8+ T cells in the TDLN of mice bearing poorly immunogenic tumors
To study how TCF1 affects the activation of tumor-antigen specific T cells in the TDLN, we repeated the experiment in Figure 2A and performed single-cell RNA-seq (scRNA-seq). Unsupervised clustering of the single-cell data identified 6 cell clusters: 1) naïve cells; 2) Xcl1-expressing cells; 3) early activated cells, expressing early activation (Lck, Fyb, Slamf6) and memory-associated (Id3) transcripts but lacking effector transcripts; 4) early effectors and 5) effectors, both expressing effector-associated genes (S100a4, S100a6, Id2, Cxcr3, Cd48, S1pr4) with effectors exhibiting more proliferative capacity (Top2a, mKi67), and 6) late effectors expressing genes important for effector differentiation (Ezh2, Suv39h1, Dnmt1) and showing the highest proliferative capacity (Top2a, Miki67) (Figure 4A–B). T cell differentiation trajectory analysis using pseudotime (Methods) showed highest expression of progenitor-associated genes (Klf2, Lef1, Sell) in naïve T cells (the starting node of the trajectory) with effector-associated genes (Cxcr3, Irf8, Ezh2) progressively increasing along the trajectory and showing highest expression in late effectors (the last node of the trajectory) (Figure 4C). We then compared the distribution of WT and TCF1 cKO OTI T cells along the pseudotime trajectory. In the highly immunogenic MC38-OVA model, one dose of ICB boosted WT OTI T cell activation, as shown by the increased frequency of late effectors and concomitant decrease of naïve cells (Figure 4D–E). Interestingly, although TCF1 cKO OTI T cells showed reduced proportions of early activated and effector populations at baseline (ISO), one dose of ICB rescued the defective priming of TCF1 cKO OTI T cells (Figure 4D–E). In the poorly immunogenic B16-OVA model, one dose of ICB boosted the activation of WT OTI T cells as shown by the increased frequency of early activated cells, but could not rescue the defective activation of TCF1 cKO OTI T cells. The more robust activation of OTI T cells in MC38-OVA- compared to B16-OVA-bearing mice was consistent with the increased levels of MHC-I and MHCI-SIINFEKL in MC38-OVA tumors (Figure 1C and S1A–B). Altogether, these data indicated that TCF1 is required for effective priming of stem-like CD8+ T cells in suboptimal antigen presentation settings, providing insight into mechanisms underlying the requirement for TCF1 for ICB efficacy in B16-OVA but not MC38-OVA tumors (Figure 1E).
Figure 4. Defective activation of TCF1 cKO OTI T cells in the TDLN of poorly but not highly immunogenic tumors.
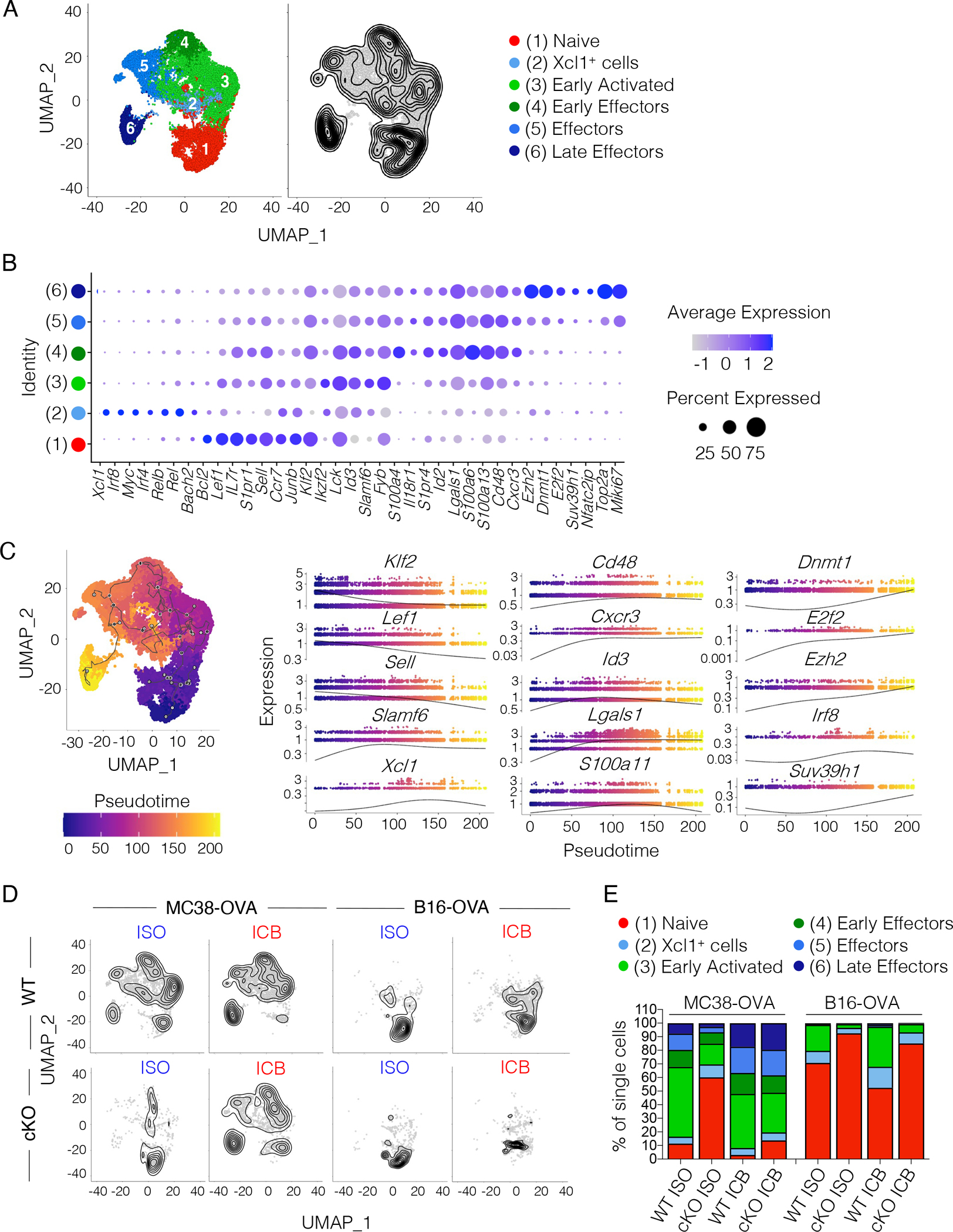
(A) Combined Uniform Manifold Approximation and Projection (UMAP) of single-cell transcriptomes from WT (n=3–5) and TCF1 cKO OTI T cells (n=3–5) sorted from the TDLN of B16-OVA- or MC38-OVA-bearing mice treated as shown in Figure 2A. Left, cluster annotation. Right, single-cell density. (B) Dot plot showing expression of cluster defining genes. Circle size indicates the percentage of cells expressing the gene in each cluster. Color scale indicates average gene expression level. (C) Pseudotime trajectory of single-cells (starting node: naïve cell cluster). Left, pseudotime projected on the UMAP. Right, expression profiles of selected genes along the pseudotime. (D) Single-cell density in WT or TCF1 cKO OTI T cells sorted from the TDLN of MC38-OVA- or B16-OVA-bearing mice treated as in Figure 2A. (E) Frequency of single-cells in each cluster and condition analyzed.
Loss of TCF1 expands naïve precursors poised to become short-lived effector cells
We next examined whether intrinsic differences in the composition or transcriptome of naïve T cells between WT and TCF1 cKO mice were responsible for the defective priming observed in the B16-OVA model. We assessed differentially expressed genes between WT and cKO OTI T cells in the naïve cluster from the TDLN of B16-OVA-bearing mice after one dose of ICB (Table S1). We found increased expression of chemokine ligands and receptors (Cxcr6, Ccr9, Ccl5) and IFN responsive genes (Irf1, Stat1, Ifi214, Ifi206, Ifi47, Ifi208, Ifi203) in naive TCF1 cKO OTI T cells. Interestingly, we also observed increased expression of both effector- (Prf1, Ly6c2, Ly6a, Runx3) and naïve-associated (Klf2, S1pr1, Sell, Il7r, Lef1, Foxp1) genes, indicating that although TCF1-deficient naïve cells are poised to become effectors, they fail to undergo efficient T cell activation in low antigenic settings. Conversely, we observed reduced naïve-associated transcripts in naïve WT OTI T cells in B16-OVA-bearing mice upon ICB, suggesting more effective initiation of activation programs. Indeed, we found increased expression of genes encoding ribosomal proteins in naïve WT as compared to TCF1 cKO OTI T cells already at baseline, indicative of their preparedness to rapidly mount an immune response25 (Table S1). Naïve TCF1 cKO OTI T cells also had decreased expression of Jun, both at baseline and upon ICB, which has been shown to enhance expansion and persistence of CAR T cells26. Thus, reduced Jun expression in TCF1 cKO T cells may contribute to their reduced ability to sustain anti-tumor responses in the B16-OVA tumor model. Finally, although we observed increased effector and chemokine ligand/receptor genes in naive TCF1 cKO OTI T cells from MC38-OVA bearing mice upon ICB, notably, we did not see increased naïve-associated genes (Table S1), indicating that TCF1-deficient cells can undergo effective T cell activation in a more antigenic setting.
To address whether these transcriptional changes reflected differences in the naïve cell composition between WT and TCF1 cKO OTI T cells, we performed unsupervised clustering of the naïve cells from our scRNA-seq data. We identified five cell clusters (Figure 5A and S5A). Cluster 5, characterized by increased expression of Ccl5, Cxcr6, and Cxcr3, stood out as the most distinct (Figure 5B). This cluster also expressed Ly6c2, which has been shown to mark a subset of naïve cells poised to give rise to short-lived effectors in acute viral infections27 (Figure 5B). Notably, cluster 5 was largely absent in WT but was expanded in naïve TCF1 cKO OTI T cells (Figure 5C).
Figure 5. TCF1 cKO T cells contain a higher proportion of CXCR3+Ly6C+ naïve cells poised to become short-lived effectors.
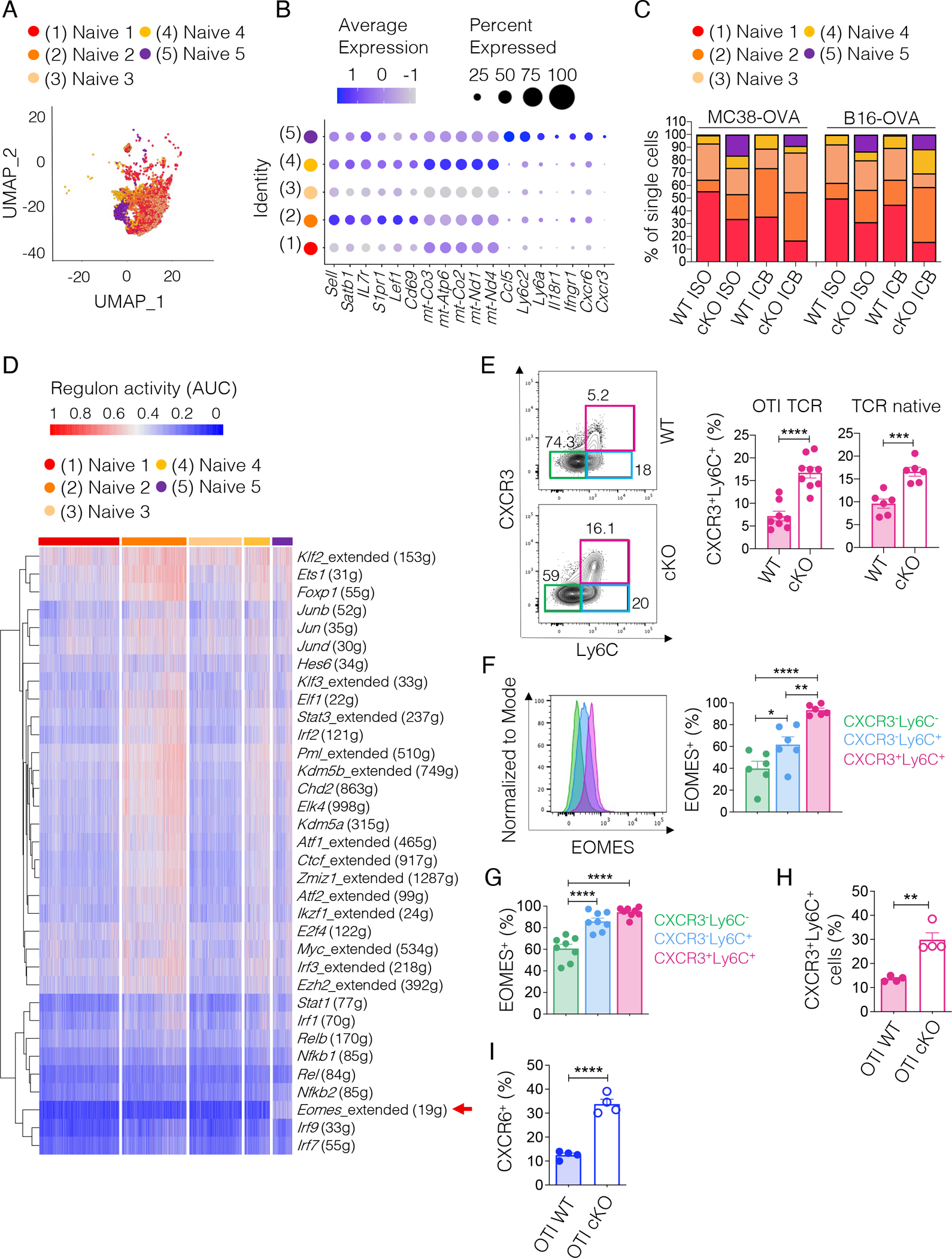
(A) UMAP of single-cell transcriptomes from naive WT and TCF1 cKO OTI T cells from Figure 4A. (B) Dot plot showing cluster defining genes. Circle size indicates the percentage of cells expressing the gene in each cluster. Color scale indicates average gene expression level. (C) Frequency of single-cells in each naïve cell cluster expressed as percentage of the overall naïve population in each condition analyzed. (D) Heatmap showing the inferred activity (red, active; blue, inactive) of 34 selected regulons (i.e. regulons relevant in stem-like CD8+ T cells) in the naïve cell clusters. Single cells are ordered in columns and grouped by cluster. Rows represent regulons named by the relative transcription factor. For each regulon, the number of the downstream target genes co-expressed with the indicated transcription factor is given. (E) Representative plots and frequency of the three naïve subsets defined using CXCR3 and Ly6C markers among naïve (CD62L+CD44−) T cells expressing either a OTI (n=8–9 mice, three experiments combined) or a native (n=6 mice, two experiments combined) TCR repertoire from tumor-free WT or TCF1 cKO mice. Student’s t test, ***p<0.001, ****p<0.0001. (F) Representative histograms and frequency of EOMES+ cells in the naïve subsets defined in (E) from tumor-free WT or TCF1 cKO mice (n=6 mice, two experiments combined). One-way ANOVA, *p<0.05, **p<0.01, ****p<0.0001 (G) Frequency of EOMES+ cells in the naïve subsets defined in (E) and present in adoptively transferred WT or TCF1 cKO OTI T cells harvested from the TDLN of MC38-OVA-bearing mice (n=8, shown one out of two experiments) three days post transfer. One-way ANOVA, ****p<0.0001. (H) Frequency of CXCR3+Ly6C+ naïve WT or TCF1 cKO OTI T cells harvested from the TDLN of MC38-OVA-bearing mice three days post adoptive transfer. Student’s t test, **p<0.01. (n=4, shown one out of two experiments). (I) Frequency of CXCR6+ cells in naive (CTVhighCD62L+CD44− ) WT or TCF1 cKO T cells harvested from the TDLN of MC38-OVA-bearing mice (n=4, shown one out of two experiments) three days post transfer. Student’s t test, **p<0.01, ****p<0.0001. All values are reported as mean±SEM.
To gain insight into the transcriptional regulation of these cells, we ran SCENIC (Single-Cell rEgulatory Network Inference and Clustering)28. We found that cluster 5 had higher activity of the Eomes regulon (comprising 19 genes) (Figure 5D). To confirm SCENIC results, we examined cluster 5 cells (CXCR3+ Ly6C+) among naïve T cells in tumor-free WT and TCF1 cKO mice. The frequency of CXCR3+Ly6C+ naïve cells was increased in TCF1 cKO mice expressing either the OTI TCR or a native TCR repertoire (Figure 5E) and this population had the highest EOMES expression among all naïve cells (Figure 5F) with no changes in EOMES levels between TCF1 cKO and WT T cells (Figure S5B). Analysis of OTI T cells in the TDLN of MC38-OVA-bearing mice confirmed highest EOMES expression in CXCR3+Ly6C+ naïve cells (Figure 5G and S5C) and increased frequency of this subset in TCF1 cKO OTI T cells (Figure 5H). Similar results were found in B16-OVA-bearing mice (not shown). Although TCF1 has been reported to positively regulate EOMES29, the observed increase in EOMES-expressing cells in naïve TCF1-deficient T cells indicates that other factors besides TCF1 regulate EOMES expression. Further, in line with our scRNA-seq data, we observed higher CXCR6 expression in naïve TCF1 cKO OTI T cells in the TDLN of MC38-OVA-bearing mice (Figure 5I). Collectively, these data indicated that TCF1 deficiency affects the composition of naïve T cells at baseline, resulting in an increased proportion of naïve cells poised to give rise to terminal effectors. In highly, but not poorly, immunogenic tumors this naïve population is readily activated to deploy potent anti-tumor responses.
Highly and poorly immunogenic tumors differentially instruct the CD8+ T cell differentiation trajectory.
To examine how the defect in early TCF1 cKO T cell activation in the TDLN affected intra-tumoral CD8+ T cell responses and ICB efficacy, we implanted WT or TCF1 cKO mice with either MC38-OVA or B16-OVA and then administered two doses of ICB or ISO prior to harvesting CD8+ TILs for scRNA-seq (Figure S6A). Unsupervised clustering identified 8 clusters of CD8+ TILs (Figure 6A–B and Table S2). These included a naïve/stem-like T cell cluster that showed highest Tcf7 expression among all clusters and a cluster characterized by Cxcr3 expression19. The CXCR3 cluster expressed intermediate Tcf7 levels and showed higher Pdcd1 (encodes PD-1) expression as compared to the naïve/stem-like cluster (Figure 6B and Table S2). Projection of a stem-like CD8+ T cell signature8 showed highest expression in the naïve/stem-like cluster (Figure S6B and Table S3). We also found a cluster characterized by high expression of interferon stimulated genes (ISG: Ifit3, Ifit1, Isg15, Ifi209), and effector transcripts (Gzma, Gzmk, Ccl5) (Figure 6B and Table S2). This cluster showed strongest enrichment for a short-lived effector T cell signature30 (Figure S6B and Table S3). We identified 5 clusters of cells co-expressing Pdcd1 and Havcr2 (encodes TIM-3). The Xcl1-expressing cluster had overall low expression of genes encoding for cytotoxic molecules and co-inhibitory receptors (Figure 6B and Table S2). The Ccl3 and Ccl4-expressing cluster had high expression of effector genes (Ifng, Prf, Tnf, Gzmb,Zeb2) and highest expression of Nuclear Receptor Subfamily 4 members (Nr4a2, Nr4a3, Nr4a1). The PD-1+TIM-3+ cells-1 cluster had low expression of effector cytokine genes but high expression of genes encoding co-inhibitory and co-stimulatory molecules (Lag3, Pdcd1, Havcr2, Icos, Cd28), effector-associated transcription factors (Id2, Runx2, Zbtb32), and chemokine receptors (Cxcr6, Cx3cr1). The PD-1+TIM-3+ cells-2 cluster was enriched for transcripts encoding granzymes (Gzmf, Gzmc, Gzmd, Gzme, Gzmb) and metallothioneins (Mt1, Mt2). Finally, we observed a cluster characterized by high expression of proliferation genes (Figure 6B and Table S2). Projection of a T cell dysfunction signature31 showed broad expression and failed to distinguish the PD-1+TIM-3+ clusters (Figure S6B and Table S3).
Figure 6. Poorly versus highly immunogenic tumors instruct distinct CD8+ T cell fates.
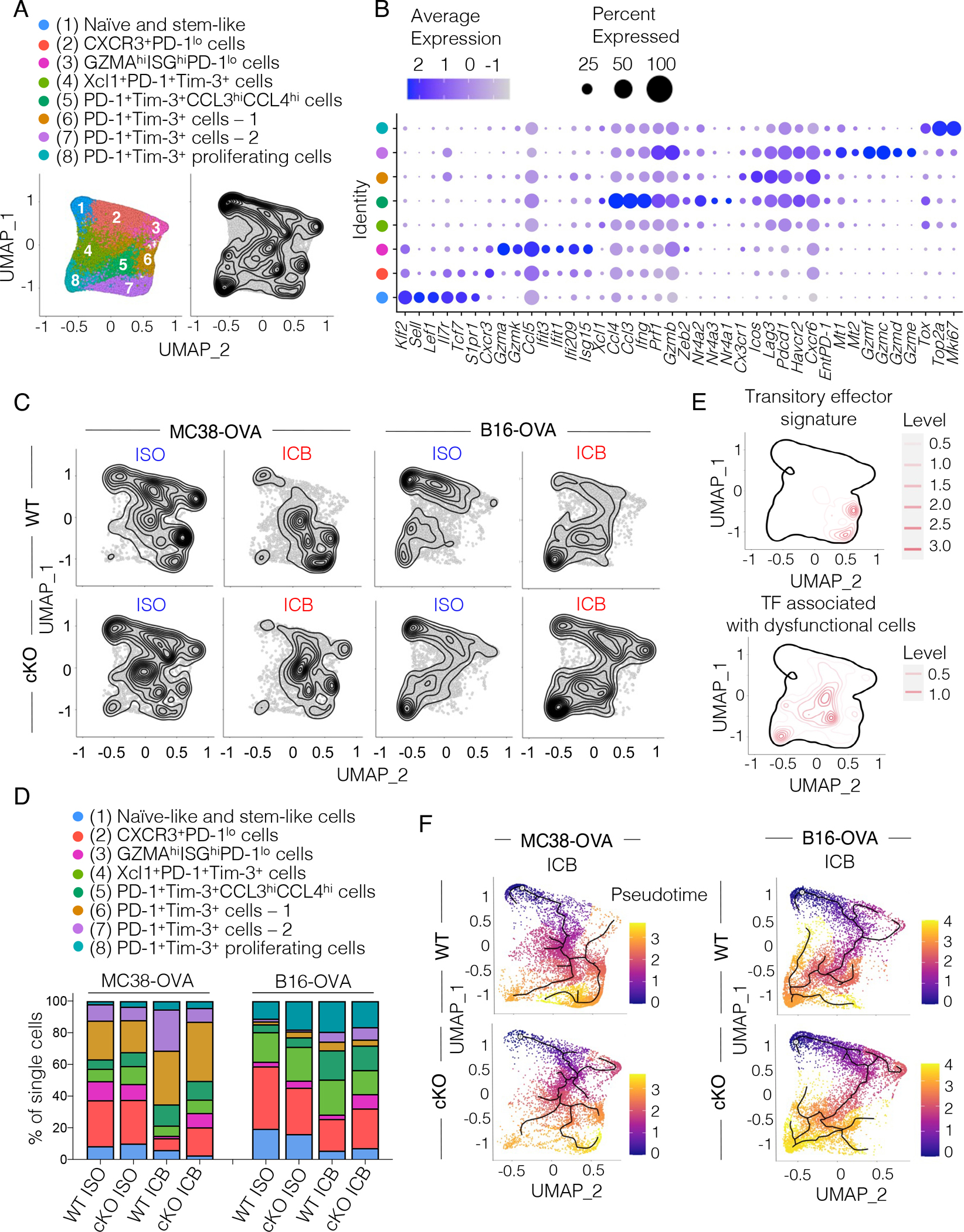
(A) UMAP of single-cell transcriptomes from CD8+ TILs harvested from WT or TCF1 cKO mice (n=3–5) inoculated with B16-OVA or MC38-OVA tumors and treated with two doses of anti-PD-L1 + anti-TIM-3 (ICB) or control antibodies (ISO) at days 6 and 9 post tumor inoculation. Left, cluster annotation. Right, single-cell density. (B) Dot plot showing cluster defining genes. Circle size indicates the percentage of cells expressing the gene within each cluster. Color scale indicates average gene expression level. (C) Density of single-cell events in WT or TCF1 cKO CD8+ TILs from MC38-OVA- or B16-OVA-bearing mice treated as indicated. (D) Frequency of single-cells in each cluster from (A) expressed as percentage of the overall population in WT or TCF1 cKO CD8 TILs from B16-OVA or MC38-OVA tumors treated as indicated. (E) Projection of a transitory effector signature32 and of a signature comprising transcription factors enriched in dysfunctional cells from melanoma patients34 on the UMAP from (A). (F) Pseudotime trajectory of single cells (starting node: naïve/stem-like cell cluster). Cells are colored along the trajectory based on a blue-to-yellow color gradient.
We compared the distribution of cells among these clusters between MC38-OVA and B16-OVA tumors and found different proportions irrespective of TCF1 expression. MC38-OVA tumors mostly accumulated cells in the PD-1+TIM-3+ cells-1 and -2 (Figure 6C–D) at baseline (ISO), which increased upon ICB. Notably, we found that the PD-1+TIM-3+ cells-1, and to a lower extent the PD-1+TIM-3+ cells-2, were enriched for the gene signature of transitory effector cells that have been shown to contribute to ICB efficacy in chronic viral infection models32,33 (Figure 6E). Conversely, B16-OVA tumors accumulated the proliferating PD-1+TIM-3+ cluster at baseline and upon ICB and expanded the PD-1+TIM-3+CCL3hiCCL4hi cluster upon ICB (Figure 6C–D). The cluster of PD-1+TIM-3+ proliferating cells that accumulated in poorly immunogenic B16-OVA tumors shared transcriptional regulators with a proliferative dysfunctional cluster described in melanoma patients34 (Figure 6E and Table S3). Importantly, pseudotime analysis identified a differentiation trajectory that stemmed from the naïve/stem-like T cell cluster and ended in the PD-1+TIM-3+ cells-1 and -2 in the MC38-OVA model or in the proliferating PD-1+TIM-3+ cells in the B16-OVA model (Figure 6F). Altogether, these data indicated that poorly and highly immunogenic tumors differentially instructed the fate of CD8+ T cells with B16-OVA tumors favoring the accumulation of dysfunctional CD8+ T cells.
Next, we compared the cluster composition between WT and TCF1 cKO mice within each tumor model upon ICB. In MC38-OVA, we found that both WT and TCF1 cKO mice similarly expanded transitory effector PD-1+TIM-3+ cells-1 upon ICB (Figure 6C–D). Moreover, upon ICB both WT, and to a lesser extent TCF1 cKO mice showed a reduction in CXCR3+ early effectors19. WT, but not TCF1 cKO, mice treated with ICB also greatly expanded PD-1+TIM-3+ cells-2 whereas TCF1 cKO mice retained a higher frequency of short-lived GZMAhiISGhi effectors (Figure 6C–D and Table S3), reflecting the increased proportion of naïve cells poised to become short-lived effectors in the TDLN of TCF1 cKO mice (Figure 5). The observed expansion of PD-1+TIM-3+ cells-1 that share features with transitory effectors in both TCF1 cKO and WT mice likely accounted for the strong anti-tumor response observed upon ICB in both groups. Conversely, in B16-OVA tumors, we found that both WT and TCF1 cKO mice accumulated proliferating PD-1+TIM-3+ cells, expanded PD-1+TIM-3+CCL3hiCCL4hi cells, and failed to increase transitory effectors (PD-1+TIM-3+ cells-1 and -2) upon ICB (Figure 6C–D). As observed in MC38-OVA, TCF1 cKO, but not WT, mice retained higher proportions of GZMAhiISGhi short-lived effectors upon ICB.
Loss of TCF1 destabilizes the dysfunctional T cell state in poorly immunogenic tumors
To understand the requirement for TCF1 for ICB response in poorly immunogenic tumors, we focused on the PD-1+TIM-3+ proliferating T cell cluster that was enriched in B16-OVA tumors. We found that TOX, which is required for the survival and persistence of dysfunctional CD8+ TILs but dispensable for the generation of memory and effector CD8+ T cells in acute viral infections35–39 was highly expressed in this cluster (Figure 7A and 6B). Indeed, we detected lower TOX protein in PD-1+TIM-3+ TILs in MC38-OVA as compared to B16-OVA tumors (Figure 7B). Accordingly, we also found that highly immunogenic Yummer 1.7 melanomas harbored low TOX+PD-1+TIM-3+ TILs, indicating that reduced accumulation of TOX+ dysfunctional cells is a feature of highly immunogenic tumors (Figure S6C). ICB further diminished TOX expression in both WT and TCF1 cKO PD-1+TIM-3+ TILs from MC38-OVA-bearing mice. Conversely, in the poorly immunogenic B16-OVA tumors TOX expression was high at baseline and post-ICB in WT but was reduced upon ICB in TCF1 cKO PD-1+TIM-3+ TILs (Figure 7B). Analysis of the differentially expressed genes in proliferating PD-1+TIM-3+ cells from mice bearing B16-OVA tumors confirmed lower Tox in TCF1 cKO as compared to WT cells upon ICB (Figure 7Cand Table S4). Reduced TOX levels in TCF1 cKO T cells correlated with reduced survival as CASPASE3+ cells were increased in PD-1+CD8+ TCF1 cKO T cells from B16-OVA-bearing mice treated with ICB (Figure 7D). Besides reduced levels of Tox, we also found increased effector-associated genes (Ly6c2, Gzmb, Gzma, Id2, Ly6a) in the PD-1+TIM-3+ proliferative cluster from TCF1 cKO mice (Figure 7C and Table S4). Conversely, PD-1+TIM-3+ proliferating WT T cells expressed more Jun, which has been shown to enhance the persistence of CAR T cells26 (Figure 7C and Table S4). Our paradoxical observation that proliferating PD-1+TIM-3+ TCF1 cKO T cells, although expressing more effector-like genes, failed to control tumor growth and survive in B16-OVA tumors was reminiscent of a destabilized dysfunctional T cell phenotype recently described for EGR2 KO CD8+ T cells in chronic viral infection40. Indeed, the signature of EGR2 KO CD8+ T cells was significantly enriched in PD-1+TIM-3+ TCF1 cKO T cells as compared to WT cells (Figure 7E). Finally, we also observed reduced Jun expression in the naïve/stem-like cluster from ICB-treated TCF1 cKO mice bearing B16-OVA, but not MC38OVA, tumors (Figure 7F and Table S5). Together, our data indicated that loss of TCF1 in poorly immunogenic tumors was associated with an altered stem-like T cell phenotype and a destabilized TOX+ dysfunctional T cell state, culminating in a reduced capacity of TCF1 cKO T cells to persist in the TME and contribute to ICB response.
Figure 7. TOX+ dysfunctional CD8+ TILs from B16-OVA-bearing TCF1 cKO mice have a destabilized phenotype resembling cells from patients that fail to respond to ICB.
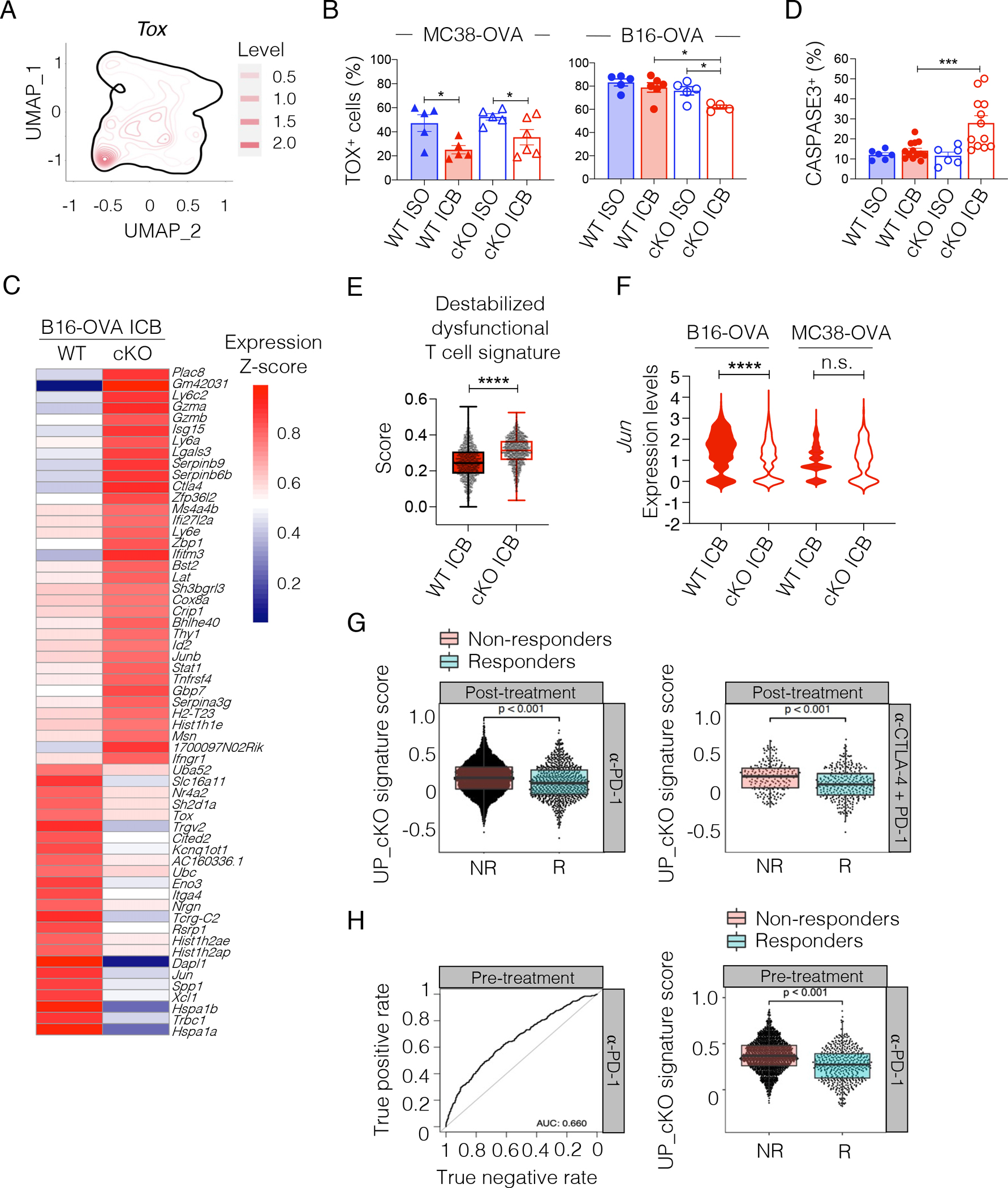
(A) Tox expression on the UMAP from Figure 6A. (B) TOX expression (frequency, mean±SEM) in PD-1+TIM-3+ CD8+ TILs in MC38-OVA- or B16-OVA-bearing WT or TCF1 cKO mice (n=4–6, one experiment) treated with anti-PD-L1 + anti-TIM-3 (ICB) or control (ISO) antibodies at day 6 and 9 post tumor injection. Analysis is done on day12. Student’s t test, *p<0.05. (C) Heatmap showing differentially expressed genes between WT and TCF1 cKO cells within the proliferating TOX+PD-1+TIM-3+CD8+ TIL (cluster 8) from Figure 6A in B16-OVA tumors treated with ICB at day 6 and 9 post tumor inoculation. (D) CASPASE-3 expression (frequency, mean±SEM) in PD-1+CD8+ TILs from B16-OVA-bearing WT or TCF1 cKO mice, three days after the second dose of ISO/ICB (n=6–13 mice, two experiments combined). ANOVA, ***p<0.001 (comparison is done vs WT ICB group). (E) Expression of the indicated signature40 on proliferating TOX+TIM-3+PD-1+CD8+ TILs (cluster 8) from figure 6A in B16-OVA tumors in WT or TCF1 cKO mice treated with two doses of ICB at day 6 and 9 post tumor inoculation. Mann-Whitney test, ****p<0.0001. (F) Violin plot of Jun expression in the naïve/stem-like T cell cluster (cluster 1) from Figure 6A from WT or TCF1 cKO TILs in B16-OVA- and MC38-OVA-bearing mice treated with ICB at day 6 and 9 post tumor inoculation. Mann-Whitney test, ****p<0.0001. Standard violin plot (G) Expression of the TCF1 cKO signature in CD8+ TILs from post-treatment melanoma patients treated as indicated and stratified as responder (R) or non-responder (NR)7. Dots represent individual cells. (H) Left: True positive (y axis) and true negative (x axis) rates when predicting the clinical response based on expression of the TCF1 cKO signature in pre-treatment CD8+ TILs from melanoma patients subsequently treated with anti-PD-17. Right: expression of the TCF1 cKO signature in CD8+ TILs from pre-treatment melanoma patients stratified as responders (R, n=4) and non-responders (NR, n=8). Dots represent individual cells. For box plot: Bottom, middle and upper lines denote lower quartile (Q1), median and upper quartile (Q3), respectively. Whiskers denote minimum (Q1 − 1.5 × interquartile range) and maximum (Q3 + 1.5 × interquartile range).
Our data showed that TCF1 was required not only for the activation/priming of stem-like precursors but also for the stability of dysfunctional T cells that contribute to anti-tumor responses. To determine the therapeutic implications of the latter, we scored a signature shown to be associated with failure to respond to ICB in patients41 (Table S3) on our scRNA-seq data. We found that this signature was enriched in the dysfuntional TOX+PD-1+TIM-3+ proliferating cluster and showed higher expression in TCF1 cKO T cells (Figure S6D–E), further supporting that dysfunctional T cells became unfit to sustain ICB responses in the absence of TCF1. To substantiate these findings, we computed a TCF1 cKO signature from the differentially expressed genes between TCF1 cKO and WT cells within the TOX+PD-1+TIM-3+ dysfunctional T cell cluster (Figure 7C) and scored this signature on the scRNA-seq data of CD8+ TILs from melanoma patients treated with ICB7. We found that the TCF1 cKO signature scored highly in the post-treatment samples from patients that failed to respond to either anti-PD-1 or anti-PD-1 + anti-CTLA-4 (Figure 7G). To further test the clinical relevance of this observation, we evaluated the predictive power of the TCF1 cKO signature. We found that expression of the TCF1 cKO signature in the pre-treatment CD8+ TIL samples could predict failure to respond to anti-PD-1 (Figure 7H). We could not assess the predictive power of the TCF1 cKO signature in the anti-CTLA-4 + anti-PD-1 cohort due to the limited numbers of pre-treatment patient samples (non-responder (NR) n=1, responder (R) n=4).
Therapeutic vaccination or enhancement of tumor antigen presentation rescues defective ICB responses in TCF1 cKO mice bearing poorly immunogenic tumors
Besides differences in the level of tumor antigen presentation, B16-OVA and MC38-OVA tumors also vary in cellular composition and soluble factors present in the TME. Such differences could contribute to the differential reliance on TCF1 for ICB efficacy in these models. We therefore tested the reliance on TCF1 for ICB response using a B16-F10 tumor cell line engineered to stably express H-2Kb MHC-I42 (B16-K1, Figure 8A). Analysis of CD8+ TILs in B16-K1 and parental B16-F10 tumors at similar tumor dimensions (Figure S7A) revealed reduced proportion and number of stem-like T cells (population 1) and increased proportions and number of early effectors (population 2) in B16-K1 tumors (Figure 8B and S7B). Furthermore, both populations 1 and 2 showed reduced TCF1 expression in B16-K1 tumors (Figure 8B). These results resembled the changes in CD8+ TIL composition and TCF1 expression observed between MC38-OVA and B16-OVA tumors (Figure 1G). Next, we implanted WT or TCF1 cKO mice with B16-F10 or B16-K1 cells and treated the mice with ISO or ICB controlling for equivalent tumor sizes at time of treatment (Figure 8C). As expected, ICB response was limited in TCF1 cKO mice bearing parental B16-F10 tumors (Figure 8D), akin to the B16-OVA model (Figure 1E), but was highly effective in TCF1 cKO mice bearing B16-K1 tumors. These data indicated that increasing the antigen presentation of poorly immunogenic melanomas rescued effector differentiation and ICB responses in TCF1 cKO mice. We additionally examined the effect of vaccination, as a clinically relevant strategy to boost CD8+ T cell priming, in combination with ICB in rescuing defective responses in TCF1 cKO mice bearing poorly immunogenic B16-OVA tumors. We generated SIINFEKL-CpG polyplex-like nanoparticles43 and confirmed their ability to activate DCs and stimulate proliferation of naïve OTI T cells in vitro (Figure S7C–D) prior to testing in vivo (Figure 8E). In line with our previous data (Figure 1E), ICB alone failed to achieve tumor control; however, the combination of ICB + vaccine induced effective tumor control (Figure 8F). Therapeutic responses were accompanied by increased frequency of total and OVA-dextramer+ CD8+ TILs (Figure 8G–H) and expansion of both stem-like (population 1) and early effector (population 2) CD8+ TILs (Figure 8I). Together, these data indicated that enhancement of either antigen presentation on tumor cells or therapeutic vaccination could overcome defective ICB responses in TCF1 cKO mice bearing poorly immunogenic tumors.
Figure 8. Enhancing antigen presentation on melanoma cells or T cell priming by therapeutic vaccination can rescue defective ICB responses in TCF1 cKO mice.
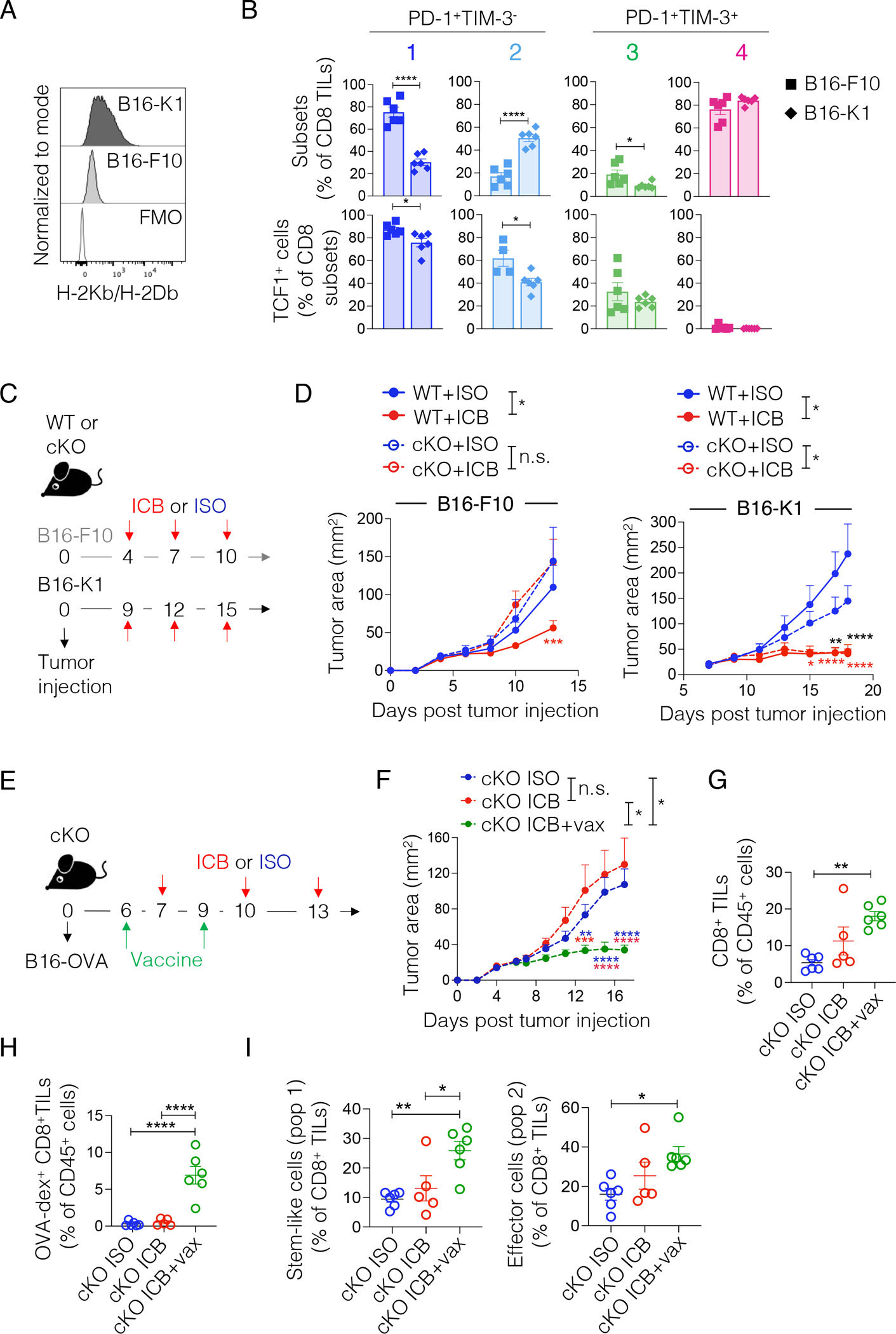
(A) Representative histograms of MHC-I (H-2Kb/H-2Db) expression on B16-F10 or B16-K1 cells. (B) Frequency (top panel) of the TIL subsets defined in Figure 1F and of TCF1 positive cells (bottom panel) in these CD8+ TIL subsets present in B16-F10 or B16-K1 tumors. Analysis is done at comparable tumor dimensions (B16-F10: day 13; B16-K1 day 17 post tumor implant into WT mice). Student’s t test, *p<0.05, ****p<0.0001, n=6, one experiment. (C) Experimental scheme. (D) Tumor area over time in WT and TCF1 cKO mice treated as shown in (C) (n = 5–6 mice per group; B16: one experiment. B16K1: shown one out of two experiments). Two-way ANOVA, Sidak’s multiple comparisons test, *p<0.05, **p<0.01, ***p < 0.001, ****p<0.0001. (E) Experimental scheme. (F) Tumor area over time in TCF1 cKO mice treated as shown in (E) (n=5–6, one experiment). Two-way ANOVA, Sidak’s multiple comparisons test, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. (G-H) Frequency of total (G) or OVA-specific (H) CD8+ TILs present in tumors from mice in (F). One-way ANOVA. **p<0.01; ****p<0.0001 (I) Frequency of stem-like cells (population 1 as in Figure 1F: PD-1+Slamf6+CX3CR1−) and effector cells (population 2 as in Figure 1F: PD-1+Slamf6low/negCX3CR1+) within total CD8+ TILs in tumors from mice in (F). ICB: anti-PD-L1 + anti-TIM-3; ISO: IgG2a + IgG2b. All values are reported as mean ± SEM.
See also Figure S7.
Discussion
Here we showed that reliance on TCF1 for ICB efficacy is dependent on tumor immunogencity. By investigating tumor-specific T cell activation in the TDLN, we found that TCF1 poises CD8+ T cells for optimal activation in suboptimal priming (i.e. low tumor antigen expression or low TCR affinity for cognate antigens) conditions commonly found in tumors with low mutational burden. Importantly, defective TCF1 expression during tumor-specific CD8+ T cell activation resulted in de-stabilized dysfunctional intra-tumoral CD8+ T cells that shared features with CD8+ T cells in ICB non-responder patients, highlighting the clinical relevance of our findings.
That TCF1 poises naïve CD8+ T cells for optimal activation is in line with the observation that TCF1 pre-programs memory CD8+ T cells for the bioenergetic and proliferative demand required during secondary challenges by enabling chromatin opening and optimal induction of genes required for rapid recall responses44. Similarly, that naive WT T cells in the TDLN retained higher expression of ribosomal genes as compared to TCF1 cKO T cells is in line with the observation that maintenance of a reservoir of idling ribosomes poises naïve CD4+ T cells to mount rapid immune responses25. Thus, TCF1 has an important role in regulating the fitness, not only of memory, but also of naïve CD8+ T cells.
We found that TCF1 deficiency led to the emergence of a CXCR3+Ly6C+CD8+ naïve T cell subset in the TDLN that resembled a subset previously described to have low memory potential and poised to give rise to short-lived effectors27. Indeed, TCF1 cKO mice had increased proportions of intra-tumoral short-lived CD8+ effectors as compared to WT mice upon ICB. In highly immunogenic tumors (i.e. MC38-OVA), the increased proportion of short-lived effectors did not impact the ability of TCF1 cKO T cells to deploy potent anti-tumor responses. However, these cells were not suited to elicit effective immune responses in poorly immunogenic tumors (i.e. B16-OVA) upon ICB. It has been suggested that TCF1 opposes the generation of short-lived effectors thereby ensuring the formation of “dysfunctional” T cells that are better equipped to persist in chronic stimulation settings45. In line with this, our data indicate that the genetic program regulated by TCF1 is pivotal for the generation of intra-tumoral TOX+ dysfunctional T cells that are fit to survive in the TME, albeit TCF1 is no longer active in these cells. These TOX+ dysfunctional T cells accumulate in poorly immunogenic tumors and, in the absence of TCF1, showed reduced TOX expression and survival. Although TOX+ dysfunctional T cells from TCF1 cKO mice shared features with destablized dysfunctional CD8+ T cells that arise in EGR2-deficient mice40, we did not observe any differences in EGR2 expression between WT and TCF1 cKO cells. This could indicate that TCF1 and EGR2 have independent, but partially overlapping functions, in shaping CD8+ T cell differentiation. We also found that JUN, which has been shown to enhance function and persistence in CAR T cells26, was reduced in both the stem-like and TOX+ dysfunctional TIL clusters in TCF1 cKO as compared to WT mice bearing B16-OVA tumors. These findings further enforce that TCF1 determines the fitness of tumor-antigen specific T cells to respond to ICB. A limitation of the study is that the extent to which TCR affinity, antigen loading and presentation, or other cues in the TME or TDLN influence the generation of TOX+ dysfunctional TILs will require further detailed study.
Our findings have multiple clinical implications. It is known that tumor environments vary widely across cancer patients, even in patients with the same tumor type. Thus, it is likely that reliance on TCF-1+ CD8+ T cells for ICB response may not be equal across patients. Indeed, retrospective studies correlating the frequency of TCF-1+ CD8+ T cell with ICB response are discordant on the ability to stratify responders and non-responders patients3,7,8,10. Although more investigations are needed, the results of previous studies combined with our data, raise questions regarding the utility of TCF1 as a sole biomarker for universally predicting response to ICB. Compounding this is the fact that single nucleotide polymorphisms in TCF7 in the human population and have been linked with susceptibility to multiple autoimmune diseases46–49. In light of our findings, it is interesting to speculate whether TCF7 polymorphisms may, in similar fashion, influence response to ICB in cancer patients. Our findings further lead us to speculate whether in highly immunogenic settings, such as in virally-induced tumors, an abundant reservoir of TCF1+ stem-like T cells may be dispensable for effective ICB responses. Conversely, in tumors expressing low antigen levels, the presence of a stable abundant reservoir of TCF1+ stem-like T cells is likely critical for effective ICB responses. It is important to note that our findings do not dispel the importance of stem-like T cells for anti-tumor responses, rather our findings indicate that some tumors may be less reliant on these cells for achieving effective anti-tumor immunity. Importantly, we believe that in tumors with low antigen expression or low frequency of tumor-specific TCF1+ precursors in the TDLN and/or intra-tumoral TCF1+ stem-like T cells, vaccination or agonistic treatments should be combined with ICB to drive effective priming of tumor-specific TCF1+ T cells.
Our work further highlights the importance of dysfunctional T cells for tumor control in low-antigen expressing, poorly immunogenic tumor settings. In this regard, it is important to remember that the “dysfunctional” T cell state is an adapted state meant to balance a certain level of tumor or viral control and protection of the host from damage caused by uncontrolled inflammation in chronic stimulation settings. Indeed, reports have shown that dysfunctional T cells are heterogenous and include cells that can contribute to therapy responses50,51. Thus, treatments aimed at promoting the stability of dysfunctional cells may represent an important therapeutic option.
RESOURCE AVAILABILITY
Lead contact
Requests for reagents should be directed to and will be fulfilled by the Lead Contact, Ana C. Anderson (acanderson@bwh.harvard.edu).
Materials availability
Requests for plasmids generated in this study should be directed to the Lead Contact, Ana C. Anderson (acanderson@bwh.harvard.edu).
Data availability
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse CD8 BUV805 Clone 53–6.7 | BD | Cat:# 612898 RRID: AB_2870186 |
| Anti-Mouse CD8 Bv421 Clone 53–6.7 | Biolegend | Cat:# 100753 RRID: AB_2562558 |
| Anti-Mouse CD45.2 Percp5.5 Clone 104 | Biolegend | Cat:# 109828 RRID: AB_893350 |
| Anti-Mouse CD45.2 Bv785 Clone 104 | Biolegend | Cat:# 109839 RRID: AB_2562604 |
| Anti-Mouse CD45.2 AlexaFluor700 Clone 104 | Biolegend | Cat:# 109822 RRID: AB_493731 |
| Anti-Mouse CD45.1 APCFire-750 Clone A20 | Biolegend | Cat:# 110752 RRID: AB_2629806 |
| Anti-Mouse CD45.1 Bv421 Clone A20 | Biolegend | Cat:# 110732 RRID: AB_2562563 |
| Anti-Mouse Ly108 APC Clone 330-AJ | Biolegend | Cat:# 134610 RRID: AB_2728155 |
| Anti-Mouse PD1 Bv605 Clone 29F.1A12 | Biolegend | Cat:# 135220 RRID: AB_2562616 |
| Anti-Mouse PD1 PE-Cy7 Clone 29F.1A12 | Biolegend | Cat:# 135216 RRID: AB_10689635 |
| Anti-Mouse Tim3 BUV395 Clone 5D12 | BD | Cat:# 747620 RRID: AB_2744186 |
| Anti-Mouse Tim3 APC Clone 5D12 | BD | Cat:# 567164 |
| Anti-Mouse Tim3 PE Clone 5D12 | BD | Cat:# 566346 RRID: AB_2739702 |
| Anti-Mouse CX3CR1 Bv785 Clone SA011F11 | Biolegend | Cat:# 149029 RRID: AB_2565938 |
| Anti-Mouse CX3CR1 Bv421 Clone SA011F11 | Biolegend | Cat:# 149023 RRID: AB_2565706 |
| Anti-Mouse CD39 PE-Dazzle Clone Duha59 | Biolegend | Cat:# 143811 RRID: AB_2750321 |
| Anti-Mouse CD39 PE-Cy7 Clone Duha59 | Biolegend | Cat:# 143806 RRID: AB_2563394 |
| Anti-Mouse/Human CD44 Bv785 Clone IM7 | Biolegend | Cat:# 103059 RRID: AB_2571953 |
| Anti-Mouse/Human CD44 APC Clone IM7 | Biolegend | Cat:# 103012 RRID: AB_312963 |
| Anti-Mouse CD62L Bv650 Clone MEL-14 | Biolegend | Cat:# 104453 RRID: AB_2800559 |
| Anti-Mouse CD62L Bv711 Clone MEL-14 | Biolegend | Cat:# 104445 RRID: AB_2564215 |
| Anti-Mouse CD62L PE-Cy7 Clone MEL-14 | Biolegend | Cat:# 104417 RRID: AB_313102 |
| Anti-Mouse CD28 PE-CY7 Clone 37.51 | Biolegend | Cat:# 102126 RRID: AB_2617011 |
| Anti-Mouse CXCR3 PE Clone CXCR3–173 | Biolegend | Cat:# 126506 RRID: AB_1027650 |
| Anti-Mouse Ly6C AF700 Clone HK1.4 | Biolegend | Cat:# 128024 RRID: AB_10643270 |
| Anti-Mouse TCRValpha2 PE Clone B20.1 | Biolegend | Cat:# 127807 RRID: AB_1134184 |
| Anti-Mouse TCRValpha2 PERCP5.5 Clone B20.1 | Biolegend | Cat:# 127814 RRID: AB_1186116 |
| Anti-Mouse TCR b-chain BUV737 Clone H57 597 | BD | Cat:# 564799 |
| Anti-Mouse H2-Kb/H2-Db APC Clone 28–8-6 | Biolegend | Cat:# 114612 RRID: AB_492931 |
| OVA-dextramer PE | Immudex | Cat:# JD2163-PE |
| Anti-Mouse/Human TCF1 PE Clone C63D9 | Cell Signaling Technology | Cat:# 14456S |
| Anti-Mouse/Human TCF1 Alexa Fluor-647 Clone C63D9 | Cell Signaling Technology | Cat:# 6709S |
| Anti-Mouse/Human TCF1 Alexa Fluor-488 Clone C63D9 | Cell Signaling Technology | Cat:# 6444S |
| Anti-Mouse TOX PE Clone TXRX10 | eBioscience | Cat:# 12–6502-82 RRID: AB_10855034 |
| Anti-Mouse Eomes Alexa Fluor-488 Clone Dan11mag | eBioscience | Cat:# 53–4875-82 RRID: AB_10854265 |
| Anti-Mouse/Human pAkt PE Clone M89 61 | BD | Cat:# 560378 RRID: AB_1645328 |
| Anti-Mouse/Human pLAT Alexa Fluor-488 | Bioss | Cat:# BS-10128R-A488 |
| Anti-Mouse/Human pSHP2 Clone L99 921 | BD | Cat:# 560390 RRID: AB_1645439 |
| Anti-Mouse/Human pZAP70 Clone 17A/P ZAP70 | BD | Cat:# 557881 RRID: AB_396919 |
| Anti-Mouse/Human pERK Unconjugated | Cell Signaling Technology | Cat:# 4370T |
| Anti-Mouse CD28 Clone PV-1 | Bio X Cell | Cat:# BE0015–5 |
| Anti-Mouse CD3 Clone 145–2C11 | Bio X Cell | Cat:# BP0001–1 |
| Anti-Mouse PDL1 Clone 10F.9G2 | Bio X Cell | Cat:# BE0101 |
| Anti-Mouse Tim3 Clone RMT3–23 | Bio X Cell | Cat:# BE0115 |
| Bacterial and virus strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Invitrogen | Cat:# C737303 |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | GIBCO | Cat#:11–965-118 |
| RPMI | Thermo Scientific | Cat:# 11875119 |
| NEEA | GIBCO | Cat:# 11140050 |
| GlutaMAX Supplement | Thermo Scientific | Cat:# 35050061 |
| NaPyruvate | GIBCO | Cat:# 11360070 |
| Fetal Bovine Serum | SIGMA | Cat:# F2442 |
| Collagenase D | GIBCO | Cat:# 17104019 |
| DMXAA | Invivogen | Cat:# tlrl-dmx |
| ODN1826 (CpG DNA) | Invivogen | Cat:# tlrl-1826–1 |
| Beta-mercaptoethanol | SIGMA | Cat:# M3148 |
| Recombinant mouse GM-CSF | Biolegend | Cat:# 576304 |
| Recombinant mouse IL2 | Miltenyi Biotec | Cat:# 130–120-662 |
| Recombinant human FLT3L-Ig | Bio X Cell | Cat:# BE0098 |
| CountBright™ Absolute Counting Beads | Thermo Fisher | Cat:# C36950 |
| Zombie UV fixable viability dye | Biolegend | Cat:# 423108 |
| CellTrace Violet Cell Proliferation Kit | Thermo Scientific | Cat:# C34557 |
| SIINFEKL peptide | Genscript | Cat:# RP10611 |
| SIIQFEKL peptide | Genscript | Custom order |
| SIIVFEKL peptide | Genscript | Custom order |
| SIITFEKL peptide | Genscript | Custom order |
| SIINFEKLRRR | Genscript | Custom order |
| BlpI | New England Bioscience | Cat:# R0585S |
| EcoRV-HF | New England Bioscience | Cat:# R3195S |
| Critical commercial assays | ||
| FoxP3 Transcription Factor Staining Buffer Set | eBioscience | Cat:# 00–5523-00 |
| Perm Buffer III | BD | Cat:# 558050 |
| Quick Blunting™ Kit | New England Bioscience | Cat:# E1201S |
| Quick Ligation™ Kit | New England Bioscience | Cat:# M2200S |
| Q5 Site-Directed Mutagenesis Kit | New England Bioscience | Cat:# E0554S |
| Deposited data | ||
| scRNA-seq data | This manuscript | GSE217038 |
| scRNA-seq data on Human CD8 TILs | Ref8 | GSE120575 |
| Experimental models: Cell lines | ||
| B16F10 melanoma | ATCC | #CRL-6475 |
| MC38 colon carcinoma | Laboratory of Dr. Mark Smyth | N/A |
| MC38-OVA colon carcinoma | Laboratory of Dr. Nick Haining | N/A |
| MC38 SIINFEKL colon carcinoma | This manuscript | N/A |
| MC38 SIIQFEKL colon carcinoma | This manuscript | N/A |
| MC38 SIIVFEKL colon carcinoma | This manuscript | N/A |
| B16-OVA melanoma | Laboratory of Dr. Kai Wucherpfennig | N/A |
| Yummer1.7 melanoma | Laboratory of Dr. Marcus Bosenberg | N/A |
| B16-K1 melanoma | Laboratory of Dr. Bruno Segui | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | Jackson laboratory | 000664 |
| C57BL/6 CD45.1 mice | Jackson laboratory | 002014 |
| OTI mice | Jackson laboratory | 003831 |
| Pmel mice | Jackson laboratory | 05023 |
| E8i-CRE+ x Tcf7fl/fl mice | Previously generated in our lab5 | N/A |
| E8i-CRE+ x Tcf7fl/fl x OTI mice | This manuscript | N/A |
| E8i-CRE+ x Tcf7fl/fl x Pmel mice | This manuscript | N/A |
| TCF7-GFP mice | Laboratory of Dr. Hai-Hui Xue | Ref17 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Med-SIINFEKL-miR-30a-shRNA-Apc | Addgene | Cat:# 185664 |
| Med-SIINFEKL | This manuscript | N/A |
| Med-SIIQFEKL | This manuscript | N/A |
| Med-SIIVFEKL | This manuscript | N/A |
| Software and algorithms | ||
| FlowJo software | Tree Star | https://flowjo.com |
| Prism 9 | GraphPad Software, Inc | https://www.graphpad.com/ |
| CellRanger software | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| SEURAT V4.0 | (https://satijalab.org/seurat/) | |
| SCENIC R package | (https://scenic.aertslab.org/) | |
| MONOCLE3 | http://cole-trapnell-lab.github.io/monocle-release/ | |
This paper analyzes existing, publicly available data. The accession number for the dataset is listed in the key resources table.
Code availability
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mouse Models
8–10-week-old male or female C57BL/6 (Stock No. 000664), OTI (Stock No. 003831), Pmel (Stock No. 005023) and CD45.1 (Stock No. 002014) mice were purchased from the Jackson Laboratory. TCF1 cKO (E8i-Cre+ x Tcf7fl/fl) were generated as previously described5 and E8i-Creneg x Tcf7fl/fl littermates were used as controls. TCF1 cKO mice were crossed to OTI or Pmel mice. Tcf7-GFP reporter mice were generously provided by Hai-Hui Xue and were generated as previously described17. All mice were housed in a vivarium under SPF conditions, in cages of up to five mice, and fed a special rodent diet. All experiments involving laboratory animals were performed under protocols approved by the Harvard Medical Area Standing Committee on Animals (Boston, MA) and followed IACUC guidelines on the ethical care and use of animals.
Mouse Tumor Cell Lines
MC38-OVA and B16-OVA cell lines were kindly provided by Dr. Kai Wucherpfennig and Dr. Nick Haining, respectively. B16-K1 were kindly provided by Dr. Bruno Segui. B16-OVA were grown in RPMI supplemented with 10% FBS, pen/strep, glutamin (2mM), sodium pyruvate (1mM), non-essential amminoacids (1X) and hepes (5mM) and injected subcutaneously into the flank of mice at 4×105 cells per mouse. Prior to in vivo injection B16-OVA cells were cultured for one passage in medium containing geneticin (0.5mg/ml) to select OVA-expressing cells followed by one passage in normal medium prior to injection into mice. MC38-OVA, MC38-mScarlet-SIINFEKL, MC38-mScarlet-SIIQFEKL and MC38-mScarlet-SIIVFEKL cells were grown in DMEM supplemented with 10% FBS, pen/strep, glutamine (2mM), sodium pyruvate (1mM), non-essential amminoacids (1X) and injected subcutaneously into the right flank of mice at 1×106 cells per mouse. Prior to in vivo injection MC38-OVA cells were cultured for one passage in medium containing puromycin (4 ug/ml) to select OVA-expressing cells followed by one passage in normal medium prior to injection into mice. B16-K1 were grown in DMEM supplemented with 10% FBS, pen/strep and GlutaMAX and injected subcutaneously into the flank of mice at 5×105 cells per mouse.
METHOD DETAILS
Generation of MC38 cells expressing SIINFEKL, SIIQFEKEL and SIIVFEKL peptides
To generate SIINFEKL expressing plasmid the Med-SIINFEKL-miR-30a-shRNA-Apc52 was purchased from Addgene (# 185664) and the miR-30a-shRNA-Apc sequence was excised by BlpI and EcoRV-HF (NEB) digestion followed by blunting (#E1201S NEB) and ligation (#M2200S NEB) according to manufacturer’s instructions. The SIIQFEKL and SIIVFEKL plasmids were generated by site-specific mutagenesis (#E0554S NEB) starting from the SIINFEKL plasmid using the following primers:
SIIQFEKL_Fw: GAGTATAATCCAGTTTGAAAAACTGTAATGACG;
SIIQFEKL_Rv: TCGAGCTGCTCCTTGTAC;
SIIVFEKL_Fw GAGTATAATCGTCTTTGAAAAACTGTAATGACGTAC;
SIIVFEKL_Rv TCGAGCTGCTCCTTGTAC;
To generate MC38 cells expressing the different constructs, VSVg-pseudotyped lentiviral vectors were generated as previously described53. Parental MC38 cells were transduced at diffent multiplicity of infections (MOIs) and the MOI resulting in one copy of integrated vector per genome was selected in order to have comparable expression of the different OVA peptide variants across the cell lines generated. Transduced cells were sorted based on mScarlet expression, expanded, and used in vivo.
Tumor experiments with immune checkpoint blockade
Tumor size was measured in two dimensions by caliper and are expressed as the product of two perpendicular diameters. For immune checkpoint blockade experiments, mice were treated intraperitoneally with a combination of anti-PD-L1 (10F.9G2, 200μg per mouse) and anti-TIM-3 (RMT3-23, 200μg per mouse) antibodies or control immunoglobulins (Rat IgG2a and Rat IgG2b) on the indicated days specified for each experiment. Mice were then monitored every two days for tumor growth.
Vaccination
SIINFEKLRRR/CpG polyplexes were assembled as previously described43. Briefly, aqueous solutions of CpG DNA (ODN 1826, Invitrogen) and SIINFEKL peptide (Genscript) modified with three arginine groups were combined at different mass ratios of SIINFEKLRRR : CpG ranging from 1:20 to 20:1. The concentration of CpG was kept fixed at 0.4mg/mL while varying the amount of SIINFEKLRRR in a fixed volume. A SYBR green exclusion assay was then used to confirm polyplex formation as previously described43. The size and polydispersity of polyplexes was measured by dynamic light scattering. Based on condensation efficiency, as measured by the SYBR green exclusion assay, and polyplex size, the mass ratio of 2:1 SIINFEKLRRR : CpG was selected for in vitro and in vivo vaccination experiments. For in vitro vaccine characterization, 105 splenic CD11c+ DCs were pulsed with increasing dilutions of SIIN-polyplexes or PBS as control for 18 hours prior to staining of DCs for surface markers of activations. 1x polyplexes contained 4ug CpG and 8ug SIINFEKLRRR. For T cell proliferation, 105 splenic CD11c+ DCs were pulsed with increasing dilutions of the SIIN-polyplexes overnight. The next day, 3 × 105 naïve CTV-labelled OTI T cells were added to the culture and three days later T cell proliferation (CTV dilution) was assessed by flow. For in vivo vaccination, mice were injected into the tail base with SIIN-polyplexes (20ug CpG + 40ug SIINFEKLRRR) on day 7 post tumor injection followed by a vaccine boost at day 9 post tumor injection.
TIL isolation
TILs were isolated by dissecting the tumor mass and mincing the tumor tissue prior to digestion with Collagenase D (2.5 mg/ml) for 20 minutes at 37°C. Tumors were then dissociated to single cell suspensions by passing tissue through a 70μM filter prior to centrifugation and analysis by flow cytometry.
Flow cytometry
Single cell suspensions were first incubated with anti-mouse FcγIII/II receptor (Cd16/Cd32) blocking antibodies for 15 min followed by staining with antibodies for 20 minutes at 4°C. Fixable viability dye Zombie UV (Biolegend) or 7AAD (Biolegend) were used to exclude dead cells. For nuclear staining, eBioscience Foxp3/Transcription Factor Staining Buffer Set was used according to manufacturer’s protocol. All data were collected on a BD Symphony (BD Biosciences) or LSR Fortessa (BD Biosciences) instruments and analyzed with FlowJo 10.7.1 software (TreeStar). For quantitative flow cytometry we used absolute counting beads (Thermo Fisher Scientific, Cat. C36950) according to manufacturer’s instruction.
Adoptive cell transfers
Tumor cells were injected into the flanks of recipient mice as outlined above. TCF1 cKO and WT OTI or Pmel T cells were purified from the spleen and lymph nodes of congenically marked donor mice using the naive CD8a+ T Cell Isolation Kit (Miltenyi, Cat. 130-096-543) or the CD8 T Cell Isolation Kit (Miltenyi, Cat. 130-104-075) following manufacturer’s instructions. Prior to in vivo transfer, T cells were stained with the cell trace violet dye (CTV) according to manufacturer’s instructions (Invitrogen, Cat. C34571). On day 5 post tumor implantation, mice were randomized by tumor size and given 1×106 CTV-labelled TCF1 cKO or WT OTI T cells resuspended in PBS via tail vein injections. For co-transfer experiments between 0.8 – 1.4 × 106 total CTV-labelled congenically marked WT and cKO OTI or Pmel T cells were co-transferred (i.e. 4 ×105 – 7×105 T cells per genotype) into mice at day 5 post tumor injection. Mice were then treated on day 6 post tumor injection with one dose of ICB (anti-PD-L1 + anti-TIM-3, 200ug each) or control antibodies (Rat IgG2a and Rat IgG2b). On day 8 post tumor injection the inguinal tumor-draining lymph node (TDLN) was dissected and dissociated to single cell suspensions by passing tissue through a 70μM filter prior to centrifugation and analysis by flow cytometry. For co-transfer experiments into mice bearing MC38 cells engineered to express SIINFEKL peptide variants, congenically marked WT and TCF1 cKO OTI T cells were co-transferred into mice (0.4 × 106 cells per genotype) at day 6 post tumor injection. The inguinal TDLN was harvested at day 9 post tumor injection for flow cytometric analysis.
scRNA-seq on CD8+ T cells from B16-OVA and MC38-OVA tumors
WT (Tcf7fl/fl) or TCF1 cKO (E8i-Cre+ x Tcf7fl/fl) mice were injected with 4×105 B16-OVA or 1×106 MC38-OVA cells subcutaneously in the right flank. On day 6 and 9, mice were randomized and treated intraperitoneally with a combination of anti-PD-L1 (10F.9G2, 200μg per mouse) and anti-TIM-3 (RMT3-23, 200μg per mouse) antibodies or control immunoglobulins (Rat IgG2a and Rat IgG2b). Two (for MC38-OVA tumors) or three (for B16-OVA tumors) days after the second ICB/ISO dose, tumors were harvested (n=3–5 per group) dissociated to single cell suspensions prior to fluorescence-activated cell sorting. An equal amount of CD45+ TCRb+ CD8+ T cells were sorted from each mouse (10,000 cells/mouse) within each experimental group. Cells from the same group were pooled (n=3–5 mice per group) and loaded for encapsulation on the Chromium system (10x Genomics). Libraries were prepared using 3’ sequencing 10X Genomics kit v2 according to the Manufacturer’s protocol. Libraries were sequenced on an Illumina HiSeq or NextSeq.
scRNA-seq on OTI T cells from the TDLN of B16-OVA and MC38-OVA-bearing mice
CD45.1 C57Bl/6 mice were injected with 4×105 B16-OVA or 1×106 MC38-OVA cells subcutaneously in the right flank. On day 5 post tumor implantation, mice were randomized by tumor size and given 1×106 TCF1 cKO or WT OTI naïve T cells resuspended in PBS via tail vein injections. Mice were then treated on day 6 post tumor injection with one dose of ICB (anti-PD-L1 + anti-TIM-3, 200ug each) or control antibodies (Rat IgG2a and Rat IgG2b). On day 8 post tumor injection the inguinal TDLN were dissected and dissociated to single cell suspensions by passing tissue through a 70μM filter prior to centrifugation and fluorescence-activated cell sorting. CD45.2+CD8+TCRVa2+ OTI T cells were purified from the TDLN of n=4–5 mice per group and loaded for encapsulation on the Chromium system (10x Genomics). Libraries were prepared using 3’ sequencing 10X Genomics kit v2 according to the Manufacturer’s protocol. Libraries were sequenced on an Illumina HiSeq or NextSeq.
Single cell RNA-seq computational analysis
The Illumina raw BCL sequencing files were processed through the CellRanger software (10x Genomics) for generating FASTQ files and count matrixes (https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome). The count matrixes were then used as input for the SEURAT V4.0 (https://satijalab.org/seurat/) and MONOCLE3 (http://cole-trapnell-lab.github.io/monocle-release/) R tools for single cell genomics analyses. Briefly, single cell barcodes were filtered for the ones containing mitochondrial gene content lower than 15%. For the single cell analysis in the tumor an additional filter was added to exclude CD4 and CD34 expressing cells to clear the CD8+ cell fraction from residual CD4+ T cells and CD34+ tumor cells contaminants, respectively. Expression data were then normalized, scaled, and searched for variable features using the SCTransform function of SEURAT V4.0 followed by UMAP dimensionality reduction and clustering using the FindClusters function. The plots shown in Figure 4A,D, Figure 5A and Figure 6A,C,E and Figure 7A as well as Figure S5A and Figure S6B,D were generated running UMAP coordinates on the R package ggplot2 (https://cran.rproject.org/web/packages/ggplot2/index.html). The analyses of Figure 4B, Figure 5B and Figure 6B were generated using the DotPlot function of SEURAT V4.0. The pseudotime analysis of Figure 4C and Figure 6F was created using MONOCLE3 using the “Naïve” and the “Naïve-like/stem-like cells” as starting clusters, respectively. To generate the barplots of Figure 4E, Figure 5C, and Figure 6D the absolute single cell counts were extracted from the meta.data table of the SEURAT objects and relative single cell representation was calculated for each cluster as percent of total single cells. The heatmap of Figure 5D was created with the ComplexHeatmap R Package (https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html) using the regulonAUC output of the SCENIC R package (https://scenic.aertslab.org/) generated starting from the SEURAT gene expression matrix and setting the parameter coexMethod=NULL. The following gene signatures (Table S3) have been used to generate the density plot of Figure 6E, and Figure S6B,D: Transitory effector signature32 (density map generated extracting single cells from the SEURAT object expressing the signature at min.cutoff=0.05); TF associated with dysfunctional cells 34 (density map generated extracting single cells from the SEURAT object expressing the signature at min.cutoff=0.01); Stem-like T cell signature8 (density map generated extracting single cells from the SEURAT object expressing the signature at min.cutoff=0.001); Short-lived effector signature30 (density map generated extracting single cells from the SEURAT object expressing the signature at min.cutoff =0.9); Dysfunctional T cell Signature31 (density map generated extracting single cells from the SEURAT object expressing the signature at min.cutoff=0.1); Signature associated with Non-Responders to ICB41 (density map generated extracting single cells from the SEURAT object expressing the signature at min.cutoff=0.001). For the analysis in Figure 7E and S6E the signature score (destabilized dysfunctional T cell signature 40 and non responder to ICB signature41, Table S3) for each single cell belonging to each sample was calculated using the UCell R package (https://github.com/carmonalab/UCell). Samples scores were then plotted with GraphPad Prism V9.3.1 and analyzed for statistical differences using a two-tailed Mann-Whitney test. For Figure 7G–H, the public scRNA-seq data of tumor infiltrating immune cells from melanoma patients before and after checkpoint blockade therapy were obtained from GSE1205757. Cell type annotations were obtained from Figure 1B and Table S1 of the original paper. To isolate CD8+ T cells from the data, we firstly filtered the cells based on the provided cell type annotations to keep only the T cells (clusters retained are lymphocytes, regulatory T-cells, exhausted/HS CD8+ T-cells, dendritic cells, memory T-cells, lymphocytes exhausted/cell-cycle, cytotoxicity lymphocytes, and exhausted CD8+ T-cells). Next, we performed a clustering analysis on the T cells to identify the CD8+ subset using Seurat v4.1.0 54. Default parameters were used in the functions unless otherwise noted. Expression data were log-normalized (NormalizeData function) and z-normalized across cells (ScaleData function). Principle Component Analysis (PCA) was then performed using the RunPCA function on the top 2000 highly variable genes (FindVariableFeatures function). Then, a Shared Neares Neighbor (SNN) graph was constructed (FindNeighbors function) and clusters were identified using a modularity maximization algorithm55 (FindCluster function; resolution set to 0.3 to avoid over-clustering). Clusters were then annotated based on their upregulated genes (identified using the FindAllMarkers function). Signature scores were computed for the CD8+ T cells (T cell clusters 0, 3, and 4) using the AddModuleScore function. Wilcoxon rank-sum test was applied to test the differences in the signature scores between responders and non-responders, stratified by therapies (anti-CTLA-4+PD-1 or anti-PD-1; anti-CTLA-4 only was excluded because data were available for only one patient). Receiver Operating Characteristic (ROC) curves and corresponding areas under the curve (AUCs) were computed for the TCF1 cKO signature at the single cell level and sample level using the pROC package in R56. The predictors are the signature scores of the pre-treatment cells, and the true class labels are the treatment responses of the patient to whom the cells belong.
In vitro T cell activation with anti-CD3 and anti-CD28 antibodies
For in vitro T cell activation experiments, naïve T cells were purified from the spleen and lymph nodes of TCF1 cKO or WT OTI mice, TCF1 cKO or WT Pmel mice, or TCF1 cKO or WT mice expressing a native TCR repertoire using the naive CD8a+ T Cell Isolation Kit (Miltenyi, Cat. 130-096-543). T cells were stained with the cell trace violet (CTV) dye (Invitrogen, Cat. C34571) and then activated in 96-well plates pre-coated with anti-CD3 and anti-CD28 antibodies. 1–4ug/ml or 0.3ug/ml of anti-CD3 and anti-CD28 antibodies were used for high and low TCR stimulation, respectively. T cells were cultured for 48 hours on anti-CD3/CD28-coated plates in DMEM supplemented with 10% FBS, pen/strep (100U/ml), glutamine (2mM), sodium pyruvate (1.5mM), non-essential amminoacids (1X, Lonza Cat. 13-114E), vitamins (1X, Thermo Fisher Scientific, Cat. 11120052), arginine, aspargine, folic acid (14uM), b-mercaptoethanol (57.2uM) and recombinant mouse IL2 (30U/ml). On day three post activation, T cells were stained for flow cytometric analysis. For phosphoflow analyses, T cells were stimulated for 24 (OTI T cells) or 48 (Pmel T cells) hours on plates pre-coated with 1ug of CD3/CD28 antibodies. Cells were then stained following the BD phosphoflow protocol II (https://www.bdbiosciences.com/content/dam/bdb/marketing-documents/Phosflow_Protocol_for_Human_PBMCs.pdf) and using the Perm Buffer III (BD, cat 558050) for staining intracellular phosphorylated proteins.
In vitro T cell activation with OVA-peptide pulsed BM-DC
Bone marrow derived DCs (BM-DC) were generated as previously described57 and activated overnight by adding 2ug/ml of DMXAA murine STING ligand (Invivogen, Cat# tlrl-dmx) prior to peptide pulsing. Pulsing of BM-DC with the indicated OVA-specific peptides (Genscripts) was done by incubating BM-DC for 2.5 hours with different concentrations of OVA peptides at 37C in RPMI supplemented with 10% FBS, pen/strep (100U/ml), glutamine (2mM), hepes (25mM), non-essential amminoacids (1X), sodium pyruvate (1 mM), b-mercaptoethanol (55uM), 100ng/mL recombinant human Flt3L-Fc (BioXcell, Cat# BE0098) and 5ng/mL recombinant mouse GM-CSF (BioLegend, Cat #576306). At the end of the incubation, BM-DC were washed three times and then plated with CTV-labelled naïve TCF1 cKO or WT OTI T cells at a 1:5 ratio (104 BM-DC : 5×104 T cells) in U bottom plates in T cell medium (described above) supplemented with low dose (30U/ml) recombinant mouse IL2. Three days later, T cells were analyzed by flow cytometry.
QUANTIFICATION AND STATISTICAL ANALYSIS
Values are expressed as mean ± standard error of the mean (SEM) as indicated in the figure legends. Statistical analyses were performed using GraphPad Prism 9 using unpaired Student’s t test or Mann-Whitney U test, One-way ANOVA followed by Dunnett’s post-test correction or two way-ANOVA followed by Sidak post-test correction, unless otherwise indicated. Differences were considered statistically significant at *p < 0.05, **p < 0.01, ***p < 0.001 and **** p<0.0001.
Supplementary Material
Table S1. Differentially expressed genes between WT and TCF1 cKO OTI cells in the naive cluster in the TDLN of MC38-OVA and B16-OVA-bearing mice. Related to Figure 5.
Table S2. Differentially expressed genes in CD8 TIL clusters from MC38-OVA and B16-OVA tumors. Related to Figure 6.
Table S3. List of gene signatures used for projection and scoring on scRNA-seq maps throughout the manuscript. Related to Figures 6 and 7 and Supplementary Figure S6.
Table S4. Differentially expressed genes between WT and TCF1 cKO PD-1+TIM-3+ proliferating cluster from B16-OVA tumors. Related to Figure 7.
Table S5. Differentially expressed genes between WT and TCF1 cKO naive/stem-like T cell cluster from B16-OVA and MC38-OVA tumors. Related to Figure 7.
Highlights.
TCF1 regulates the fitness and composition of naïve CD8+ T cells
TCF1 promotes optimal tumor-specific CD8+ T cell priming in low antigenic settings
TCF1-deficient T cells resemble dysfunctional T cells in ICB non-responders
Boosting antigen presentation can overcome dependency on TCF1 for ICB response
Acknowledgments
We thank Anita Giobbie-Hurder for help with statistics and Lionel Apetoh for advice. This work was supported by NIH grant R01CA187975 to A.C.A. A.C.A. is a recipient of the Brigham and Women’s President’s Scholar Award. G.E. is supported by the Cancer Research Institute Irvington Postdoctoral Fellowship (CRI 2934). M.B. is supported by the Cancer Research Institute Irvington Postdoctoral Fellowship to Promote Racial Diversity (CRI 4491).
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of interests
A.C.A. is a member of the SAB for Tizona Therapeutics, Trishula Therapeutics, Compass Therapeutics, Zumutor Biologics, ImmuneOncia, and Excepgen, which have interests in cancer immunotherapy. A.C.A. is a paid consultant for iTeos Therapeutics and Larkspur Biosciences. A.C.A.’s interests were reviewed and managed by the Brigham and Women’s Hospital. C.M.J. is an employee of the VA Maryland Health Care System. The views reported here do not reflect the views of the VA or United States Government. C.M.J. has an equity position with Cartesian Therapeutics. L.B. is also an employee of SANA Biotechnology, Inc. Neither SANA Biotechnology nor its subsidiaries have conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Haslam A, and Prasad V (2019). Estimation of the percentage of us patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw. Open 2, 1–9. 10.1001/jamanetworkopen.2019.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brummelman J, Mazza EMC, Alvisi G, Colombo FS, Grilli A, Mikulak J, Mavilio D, Alloisio M, Ferrari F, Lopci E, et al. (2018). High-dimensional single cell analysis identifies stem- like cytotoxic CD8 + T cells infiltrating human tumors. 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siddiqui I, Schaeuble K, Chennupati V, Marraco SAF, Calderon-copete S, Ferreira DP, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity, 1–17. 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 4.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, et al. (2019). Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1-CD8 + Tumor-Infiltrating T Cells. Immunity 50, 181–194.e6. 10.1016/j.immuni.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsui C, Kretschmer L, Rapelius S, Gabriel SS, Chisanga D, Knöpper K, Utzschneider DT, Nüssing S, Liao Y, Mason T, et al. (2022). MYB orchestrates T cell exhaustion and response to checkpoint inhibition. Nature. 10.1038/s41586-022-05105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eberhardt CS, Kissick HT, Patel MR, Cardenas MA, Prokhnevska N, Obeng RC, Nasti TH, Griffith CC, Im SJ, Wang X, et al. (2021). Functional HPV-specific PD-1+ stem-like CD8 T cells in head and neck cancer. Nature 597, 279–284. 10.1038/s41586-021-03862-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sade-feldman M, Yizhak K, Bjorgaard SL, Ray JP, Boer C.G. De, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. 998–1013. 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller BC, Sen DR, Abosy R. Al, Bi K, Virkud YV, Lafleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20. 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Escobar G, Mangani D, and Anderson AC (2020). T cell factor 1: A master regulator of the T cell response in disease. Sci. Immunol. 5. 10.1126/SCIIMMUNOL.ABB9726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ficial M, Jegede OA, Angelo MS, Hou Y, Flaifel A, Pignon JC, Braun DA, Wind-Rotolo M, Sticco-Ivins MA, Catalano PJ, et al. (2021). Expression of T-Cell exhaustion molecules and human endogenous retroviruses as predictive biomarkers for response to nivolumab in metastatic clear cell renal cell carcinoma. Clin. Cancer Res. 27, 1371–1380. 10.1158/1078-0432.CCR-20-3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, van Nimwegen M, Lau SP, Latupeirissa K, Schetters S, et al. (2020). The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell, 1–16. 10.1016/j.ccell.2020.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Fransen MF, Schoonderwoerd M, Knopf P, Camps MG, Hawinkels LJ, Kneilling M, van Hall T, and Ossendorp F (2018). Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI insight 3, 1–6. 10.1172/jci.insight.124507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Francis DM, Manspeaker MP, Schudel A, Sestito LF, O’Melia MJ, Kissick HT, Pollack BP, Waller EK, and Thomas SN (2020). Blockade of immune checkpoints in lymph nodes through locoregional delivery augments cancer immunotherapy. Sci. Transl. Med. 12, eaay3575. 10.1126/scitranslmed.aay3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Connolly KA, Kuchroo M, Venkat A, Khatun A, Wang J, William I, Hornick NI, Fitzgerald BL, Damo M, Kasmani MY, et al. (2021). A reservoir of stem-like CD8+ T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci. Immunol. 6, 1–17. 10.1126/sciimmunol.abg7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schenkel JM, Herbst RH, Canner D, Li A, Hillman M, Shanahan S-L, Gibbons G, Smith OC, Kim JY, Westcott P, et al. (2021). Conventional type I dendric cells maintain a reservoir of proliferative tumor-antigen specific TCF-1+ CD8+ T cells in tumor-draining lymph nodes. Immunity, 1–16. 10.1016/j.immuni.2021.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellmeier W, Sunshine MJ, Losos K, Hatam F, and Littman DR (1997). An enhancer that directs lineage-specific expression of CD8 in positively selected thymocytes and mature T cells. Immunity 7, 537–547. 10.1016/S1074-7613(00)80375-1. [DOI] [PubMed] [Google Scholar]
- 17.Yang Q, Li F, Harly C, Xing S, Ye L, Xia X, Wang H, Wang X, Yu S, Zhou X, et al. (2015). TCF-1 upregulation identifies early innate lymphoid progenitors in the bone marrow. Nat. Immunol. 10.1038/ni.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. (2017). Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 0, E108–6822. 10.1016/j.cell.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, Freeman GJ, Boland GM, and Luster AD (2019). Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity 50, 1498–1512.e5. 10.1016/j.immuni.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, and Gajewski TF (2014). Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+T cells directly within the tumor microenvironment. J. Immunother. Cancer 2, 1–14. 10.1186/2051-1426-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, et al. (2018). Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity, 1148–1161. 10.1016/j.immuni.2018.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Mingo Pulido Á, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, Coussens LM, and Ruffell B (2018). TIM-3 Regulates CD103 + Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 33, 60–74.e6. 10.1016/j.ccell.2017.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niogret C, Birchmeier W, and Guarda G (2019). Shp-2 in lymphocytes’ cytokine and inhibitory receptor signaling. Front. Immunol. 10, 1–11. 10.3389/fimmu.2019.02468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zehn D, Lee SY, and Bevan MJ (2009). Complete but curtailed T-cell response to very low-affinity antigen. Nature 458, 211–214. 10.1038/nature07657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolf T, Jin W, Zoppi G, Vogel IA, Akhmedov M, Bleck CKE, Beltraminelli T, Rieckmann JC, Ramirez NJ, Benevento M, et al. (2020). Dynamics in protein translation sustaining T cell preparedness. Nat. Immunol. 21. 10.1038/s41590-020-0714-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, Anbunathan H, Lattin J, Jones R, Tieu V, et al. (2019). c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300. 10.1038/s41586-019-1805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ju YJ, Lee SW, Kye YC, Lee GW, Kim HO, Yun CH, and Cho JH (2021). Self-reactivity controls functional diversity of naive CD8+ T cells by co-opting tonic type I interferon. Nat. Commun. 12, 1–15. 10.1038/s41467-021-26351-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. (2017). SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086. 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, and Xue HH (2010). Differentiation and Persistence of Memory CD8+ T Cells Depend on T Cell Factor 1. Immunity 33, 229–240. 10.1016/j.immuni.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andreatta M, Corria-Osorio J, Müller S, Cubas R, Coukos G, and Carmona SJ (2021). Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat. Commun. 12, 1–19. 10.1038/s41467-021-23324-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, Nyman J, Sakuishi K, Kurtulus S, Gennert D, et al. (2016). A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell 166, 1500–1511.e9. 10.1016/J.CELL.2016.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hudson WH, Gensheimer J, Hashimoto M, Leonard WJ, Kissick HT, Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, et al. (2019). Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1+ Stem-like CD8+ T Cells during Chronic Infection. Immunity, 1–16. 10.1016/j.immuni.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, and Cui W (2019). CD4+ T Cell Help Is Required for the Formation of a Cytolytic CD8+ T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 51, 1028–1042.e4. 10.1016/j.immuni.2019.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, van den Braber M, Rozeman EA, Haanen JBAG, Blank CU, et al. (2018). Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell, 1–15. 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, et al. (2019). TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature. 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, López-Moyado IF, Georges RO, Zhang W, Onodera A, et al. (2019). TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. U. S. A. 10.1073/pnas.1905675116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara SJ, et al. (2019). TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 10.1038/s41586-019-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, Utzschneider DT, von Hösslin M, Cullen JG, Fan Y, et al. (2019). TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 9. 10.1038/s41586-019-1326-9. [DOI] [PubMed] [Google Scholar]
- 39.Yao C, Sun H-W, Lacey NE, Ji Y, Moseman EA, Shih H-Y, Heuston EF, Kirby M, Anderson S, Cheng J, et al. (2019). Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat. Immunol. 10.1038/s41590-019-0403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wagle MV, Vervoort SJ, Kelly MJ, Van Der Byl W, Peters TJ, Martin BP, Martelotto LG, Nüssing S, Ramsbottom KM, Torpy JR, et al. (2021). Antigen-driven EGR2 expression is required for exhausted CD8+ T cell stability and maintenance. Nat. Commun. 12, 1–15. 10.1038/s41467-021-23044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaiswal A, Verma A, Dannenfelser R, Melssen M, Tirosh I, Izar B, Kim TG, Nirschl CJ, Devi KSP, Olson WC, et al. (2022). An activation to memory differentiation trajectory of tumor-infiltrating lymphocytes informs metastatic melanoma outcomes. Cancer Cell 40, 524–544.e5. 10.1016/j.ccell.2022.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bertrand F, Rochotte J, Colacios C, Montfort A, Tilkin-Mariamé AF, Touriol C, Rochaix P, Lajoie-Mazenc I, Andrieu-Abadie N, Levade T, et al. (2015). Blocking tumor necrosis factor α enhances CD8 T-cell-dependent immunity in experimental melanoma. Cancer Res. 75, 2619–2628. 10.1158/0008-5472.CAN-14-2524. [DOI] [PubMed] [Google Scholar]
- 43.Tsai SJ, Amerman A, and Jewell CM (2021). Altering Antigen Charge to Control Self-Assembly and Processing of Immune Signals During Cancer Vaccination. 11, 1–14. 10.3389/fimmu.2020.613830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shan Q, Hu SS, Zhu S, Chen X, Badovinac VP, Peng W, Zang C, and Xue HH (2022). Tcf1 preprograms the mobilization of glycolysis in central memory CD8+ T cells during recall responses. Nat. Immunol. 23, 386–398. 10.1038/s41590-022-01131-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Z, Ji Z, Ngiow SF, Vahedi G, Ji H, Wherry EJ, Chen Z, Ji Z, Ngiow SF, Manne S, et al. (2019). TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity, 1–16. 10.1016/j.immuni.2019.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leng RX, Di DS, Ni J, Wu XX, Zhang LL, Wang XF, Liu RS, Huang Q, Fan YG, Pan HF, et al. (2020). Identification of new susceptibility loci associated with rheumatoid arthritis. Ann. Rheum. Dis. 79, 1565–1571. 10.1136/annrheumdis-2020-217351. [DOI] [PubMed] [Google Scholar]
- 47.Tosti A, Lama L, Patrizi A, Annicchiarico FL, Ciavarella A, Galuppi V, and Salardi S (1987). Dermatoses associated with type 1 diabetes. G. Ital. di Dermatologia e Venereol. 122, 1579–1582. [PubMed] [Google Scholar]
- 48.Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, Cotsapas C, Shah TS, Spencer C, Booth D, Goris A, et al. (2013). Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 45, 1353–1362. 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patsopoulos NA, Baranzini SE, Santaniello A, Shoostari P, Cotsapas C, Wong G, Beecham AH, James T, Replogle J, Vlachos IS, et al. (2019). Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science (80-. ). 365. 10.1126/science.aav7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, Kiialainen A, Hanhart J, Schill C, Hess C, et al. (2018). A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 24, 994–1004. 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chow A, Perica K, Klebanoff CA, and Wolchok JD (2022). Clinical implications of T cell exhaustion for cancer immunotherapy. Nat. Rev. Clin. Oncol. 0123456789. 10.1038/s41571-022-00689-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Westcott PMK, Sacks NJ, Schenkel JM, Ely ZA, Smith O, Hauck H, Jaeger AM, Zhang D, Backlund CM, Beytagh MC, et al. (2021). Low neoantigen expression and poor T-cell priming underlie early immune escape in colorectal cancer. Nat. Cancer. 10.1038/s43018-021-00247-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Follenzi A, Ailles LE, Bakovic S, Geuna M, and Naldini L (2000). Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat. Genet. 25, 217–222. [DOI] [PubMed] [Google Scholar]
- 54.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waltman L, and van Eck NJ (2013). A smart local moving algorithm for large-scale modularity-based community detection. Eur. Phys. J. B 86, 471. 10.1140/epjb/e2013-40829-0. [DOI] [Google Scholar]
- 56.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C, and Muller M (2011). pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 8, 12–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duong E, Fessenden TB, Lutz E, Dinter T, Yim L, Blatt S, Bhutkar A, Wittrup KD, and Spranger S (2022). Type I interferon activates MHC class I-dressed CD11b+ conventional dendritic cells to promote protective anti-tumor CD8+ T cell immunity. Immunity 55, 308–323.e9. 10.1016/j.immuni.2021.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially expressed genes between WT and TCF1 cKO OTI cells in the naive cluster in the TDLN of MC38-OVA and B16-OVA-bearing mice. Related to Figure 5.
Table S2. Differentially expressed genes in CD8 TIL clusters from MC38-OVA and B16-OVA tumors. Related to Figure 6.
Table S3. List of gene signatures used for projection and scoring on scRNA-seq maps throughout the manuscript. Related to Figures 6 and 7 and Supplementary Figure S6.
Table S4. Differentially expressed genes between WT and TCF1 cKO PD-1+TIM-3+ proliferating cluster from B16-OVA tumors. Related to Figure 7.
Table S5. Differentially expressed genes between WT and TCF1 cKO naive/stem-like T cell cluster from B16-OVA and MC38-OVA tumors. Related to Figure 7.
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse CD8 BUV805 Clone 53–6.7 | BD | Cat:# 612898 RRID: AB_2870186 |
| Anti-Mouse CD8 Bv421 Clone 53–6.7 | Biolegend | Cat:# 100753 RRID: AB_2562558 |
| Anti-Mouse CD45.2 Percp5.5 Clone 104 | Biolegend | Cat:# 109828 RRID: AB_893350 |
| Anti-Mouse CD45.2 Bv785 Clone 104 | Biolegend | Cat:# 109839 RRID: AB_2562604 |
| Anti-Mouse CD45.2 AlexaFluor700 Clone 104 | Biolegend | Cat:# 109822 RRID: AB_493731 |
| Anti-Mouse CD45.1 APCFire-750 Clone A20 | Biolegend | Cat:# 110752 RRID: AB_2629806 |
| Anti-Mouse CD45.1 Bv421 Clone A20 | Biolegend | Cat:# 110732 RRID: AB_2562563 |
| Anti-Mouse Ly108 APC Clone 330-AJ | Biolegend | Cat:# 134610 RRID: AB_2728155 |
| Anti-Mouse PD1 Bv605 Clone 29F.1A12 | Biolegend | Cat:# 135220 RRID: AB_2562616 |
| Anti-Mouse PD1 PE-Cy7 Clone 29F.1A12 | Biolegend | Cat:# 135216 RRID: AB_10689635 |
| Anti-Mouse Tim3 BUV395 Clone 5D12 | BD | Cat:# 747620 RRID: AB_2744186 |
| Anti-Mouse Tim3 APC Clone 5D12 | BD | Cat:# 567164 |
| Anti-Mouse Tim3 PE Clone 5D12 | BD | Cat:# 566346 RRID: AB_2739702 |
| Anti-Mouse CX3CR1 Bv785 Clone SA011F11 | Biolegend | Cat:# 149029 RRID: AB_2565938 |
| Anti-Mouse CX3CR1 Bv421 Clone SA011F11 | Biolegend | Cat:# 149023 RRID: AB_2565706 |
| Anti-Mouse CD39 PE-Dazzle Clone Duha59 | Biolegend | Cat:# 143811 RRID: AB_2750321 |
| Anti-Mouse CD39 PE-Cy7 Clone Duha59 | Biolegend | Cat:# 143806 RRID: AB_2563394 |
| Anti-Mouse/Human CD44 Bv785 Clone IM7 | Biolegend | Cat:# 103059 RRID: AB_2571953 |
| Anti-Mouse/Human CD44 APC Clone IM7 | Biolegend | Cat:# 103012 RRID: AB_312963 |
| Anti-Mouse CD62L Bv650 Clone MEL-14 | Biolegend | Cat:# 104453 RRID: AB_2800559 |
| Anti-Mouse CD62L Bv711 Clone MEL-14 | Biolegend | Cat:# 104445 RRID: AB_2564215 |
| Anti-Mouse CD62L PE-Cy7 Clone MEL-14 | Biolegend | Cat:# 104417 RRID: AB_313102 |
| Anti-Mouse CD28 PE-CY7 Clone 37.51 | Biolegend | Cat:# 102126 RRID: AB_2617011 |
| Anti-Mouse CXCR3 PE Clone CXCR3–173 | Biolegend | Cat:# 126506 RRID: AB_1027650 |
| Anti-Mouse Ly6C AF700 Clone HK1.4 | Biolegend | Cat:# 128024 RRID: AB_10643270 |
| Anti-Mouse TCRValpha2 PE Clone B20.1 | Biolegend | Cat:# 127807 RRID: AB_1134184 |
| Anti-Mouse TCRValpha2 PERCP5.5 Clone B20.1 | Biolegend | Cat:# 127814 RRID: AB_1186116 |
| Anti-Mouse TCR b-chain BUV737 Clone H57 597 | BD | Cat:# 564799 |
| Anti-Mouse H2-Kb/H2-Db APC Clone 28–8-6 | Biolegend | Cat:# 114612 RRID: AB_492931 |
| OVA-dextramer PE | Immudex | Cat:# JD2163-PE |
| Anti-Mouse/Human TCF1 PE Clone C63D9 | Cell Signaling Technology | Cat:# 14456S |
| Anti-Mouse/Human TCF1 Alexa Fluor-647 Clone C63D9 | Cell Signaling Technology | Cat:# 6709S |
| Anti-Mouse/Human TCF1 Alexa Fluor-488 Clone C63D9 | Cell Signaling Technology | Cat:# 6444S |
| Anti-Mouse TOX PE Clone TXRX10 | eBioscience | Cat:# 12–6502-82 RRID: AB_10855034 |
| Anti-Mouse Eomes Alexa Fluor-488 Clone Dan11mag | eBioscience | Cat:# 53–4875-82 RRID: AB_10854265 |
| Anti-Mouse/Human pAkt PE Clone M89 61 | BD | Cat:# 560378 RRID: AB_1645328 |
| Anti-Mouse/Human pLAT Alexa Fluor-488 | Bioss | Cat:# BS-10128R-A488 |
| Anti-Mouse/Human pSHP2 Clone L99 921 | BD | Cat:# 560390 RRID: AB_1645439 |
| Anti-Mouse/Human pZAP70 Clone 17A/P ZAP70 | BD | Cat:# 557881 RRID: AB_396919 |
| Anti-Mouse/Human pERK Unconjugated | Cell Signaling Technology | Cat:# 4370T |
| Anti-Mouse CD28 Clone PV-1 | Bio X Cell | Cat:# BE0015–5 |
| Anti-Mouse CD3 Clone 145–2C11 | Bio X Cell | Cat:# BP0001–1 |
| Anti-Mouse PDL1 Clone 10F.9G2 | Bio X Cell | Cat:# BE0101 |
| Anti-Mouse Tim3 Clone RMT3–23 | Bio X Cell | Cat:# BE0115 |
| Bacterial and virus strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Invitrogen | Cat:# C737303 |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | GIBCO | Cat#:11–965-118 |
| RPMI | Thermo Scientific | Cat:# 11875119 |
| NEEA | GIBCO | Cat:# 11140050 |
| GlutaMAX Supplement | Thermo Scientific | Cat:# 35050061 |
| NaPyruvate | GIBCO | Cat:# 11360070 |
| Fetal Bovine Serum | SIGMA | Cat:# F2442 |
| Collagenase D | GIBCO | Cat:# 17104019 |
| DMXAA | Invivogen | Cat:# tlrl-dmx |
| ODN1826 (CpG DNA) | Invivogen | Cat:# tlrl-1826–1 |
| Beta-mercaptoethanol | SIGMA | Cat:# M3148 |
| Recombinant mouse GM-CSF | Biolegend | Cat:# 576304 |
| Recombinant mouse IL2 | Miltenyi Biotec | Cat:# 130–120-662 |
| Recombinant human FLT3L-Ig | Bio X Cell | Cat:# BE0098 |
| CountBright™ Absolute Counting Beads | Thermo Fisher | Cat:# C36950 |
| Zombie UV fixable viability dye | Biolegend | Cat:# 423108 |
| CellTrace Violet Cell Proliferation Kit | Thermo Scientific | Cat:# C34557 |
| SIINFEKL peptide | Genscript | Cat:# RP10611 |
| SIIQFEKL peptide | Genscript | Custom order |
| SIIVFEKL peptide | Genscript | Custom order |
| SIITFEKL peptide | Genscript | Custom order |
| SIINFEKLRRR | Genscript | Custom order |
| BlpI | New England Bioscience | Cat:# R0585S |
| EcoRV-HF | New England Bioscience | Cat:# R3195S |
| Critical commercial assays | ||
| FoxP3 Transcription Factor Staining Buffer Set | eBioscience | Cat:# 00–5523-00 |
| Perm Buffer III | BD | Cat:# 558050 |
| Quick Blunting™ Kit | New England Bioscience | Cat:# E1201S |
| Quick Ligation™ Kit | New England Bioscience | Cat:# M2200S |
| Q5 Site-Directed Mutagenesis Kit | New England Bioscience | Cat:# E0554S |
| Deposited data | ||
| scRNA-seq data | This manuscript | GSE217038 |
| scRNA-seq data on Human CD8 TILs | Ref8 | GSE120575 |
| Experimental models: Cell lines | ||
| B16F10 melanoma | ATCC | #CRL-6475 |
| MC38 colon carcinoma | Laboratory of Dr. Mark Smyth | N/A |
| MC38-OVA colon carcinoma | Laboratory of Dr. Nick Haining | N/A |
| MC38 SIINFEKL colon carcinoma | This manuscript | N/A |
| MC38 SIIQFEKL colon carcinoma | This manuscript | N/A |
| MC38 SIIVFEKL colon carcinoma | This manuscript | N/A |
| B16-OVA melanoma | Laboratory of Dr. Kai Wucherpfennig | N/A |
| Yummer1.7 melanoma | Laboratory of Dr. Marcus Bosenberg | N/A |
| B16-K1 melanoma | Laboratory of Dr. Bruno Segui | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | Jackson laboratory | 000664 |
| C57BL/6 CD45.1 mice | Jackson laboratory | 002014 |
| OTI mice | Jackson laboratory | 003831 |
| Pmel mice | Jackson laboratory | 05023 |
| E8i-CRE+ x Tcf7fl/fl mice | Previously generated in our lab5 | N/A |
| E8i-CRE+ x Tcf7fl/fl x OTI mice | This manuscript | N/A |
| E8i-CRE+ x Tcf7fl/fl x Pmel mice | This manuscript | N/A |
| TCF7-GFP mice | Laboratory of Dr. Hai-Hui Xue | Ref17 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Med-SIINFEKL-miR-30a-shRNA-Apc | Addgene | Cat:# 185664 |
| Med-SIINFEKL | This manuscript | N/A |
| Med-SIIQFEKL | This manuscript | N/A |
| Med-SIIVFEKL | This manuscript | N/A |
| Software and algorithms | ||
| FlowJo software | Tree Star | https://flowjo.com |
| Prism 9 | GraphPad Software, Inc | https://www.graphpad.com/ |
| CellRanger software | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| SEURAT V4.0 | (https://satijalab.org/seurat/) | |
| SCENIC R package | (https://scenic.aertslab.org/) | |
| MONOCLE3 | http://cole-trapnell-lab.github.io/monocle-release/ | |
This paper analyzes existing, publicly available data. The accession number for the dataset is listed in the key resources table.