

SUMMARY
The ATR kinase safeguards genomic integrity during S phase, but how ATR protects DNA replication forks remains incompletely understood. Here, we combine four distinct assays to analyze ATR functions at ongoing and newly assembled replication forks upon replication inhibition by hydroxyurea. At ongoing forks, ATR inhibitor (ATRi) increases MRE11- and EXO1-mediated nascent DNA degradation from PrimPol-generated, single-stranded DNA (ssDNA) gaps. ATRi also exposes template ssDNA through fork uncoupling and nascent DNA degradation. Electron microscopy reveals that ATRi reduces reversed forks by increasing gap-dependent nascent DNA degradation. At new forks, ATRi triggers MRE11- and CtIP-initiated template DNA degradation by EXO1, exposing nascent ssDNA. Upon PARP inhibition, ATRi preferentially exacerbates gap-dependent nascent DNA degradation at ongoing forks in BRCA1/2-deficient cells and disrupts the restored gap protection in BRCA1-deficient, PARP-inhibitor-resistant cells. Thus, ATR protects ongoing and new forks through distinct mechanisms, providing an extended view of ATR’s functions in stabilizing replication forks.
In brief
Leung et al. systematically investigated how ATR protects stressed replication forks using four distinct assays. At ongoing forks, ATR antagonizes fork uncoupling, prevents nascent DNA degradation from gaps, and promotes fork reversal, whereas at new forks, ATR prevents MRN-CtIP-initiated template DNA degradation.
Graphical Abstract
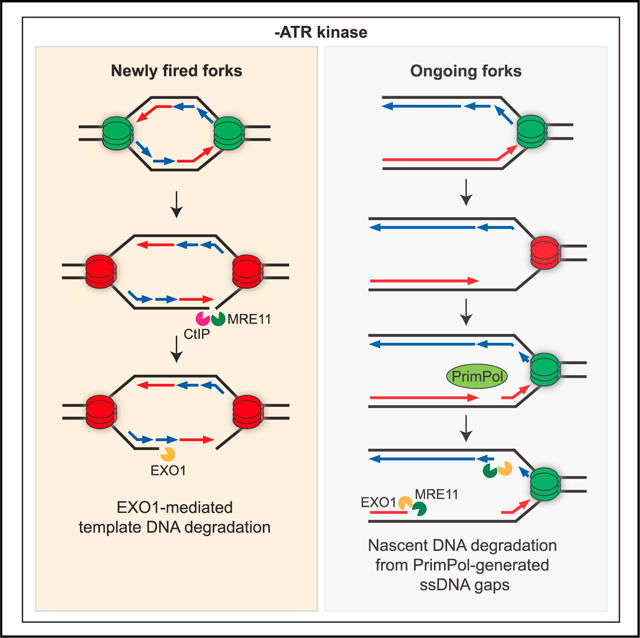
INTRODUCTION
The integrity of the genome is constantly challenged by both intrinsic and extrinsic stresses that perturb the progression of replication forks. Intrinsic stresses arise from insufficient or imbalanced deoxynucleotide (dNTP) supplies, R-loops and transcription-replication conflicts, DNA secondary structures, protein-DNA crosslinks, and reactive oxygen species.1,2 Extrinsic stresses include DNA lesions and/or adducts generated by chemical mutagens, UV light, and ionizing radiation.3 The ataxia telangiectasia and Rad3-related (ATR) kinase is the master regulator of the replication stress response.4–6 In response to genotoxic stresses that compromise DNA replication, ATR is recruited to stressed or stalled forks by replication protein A (RPA)-coated single-stranded DNA (ssDNA) and activated by TOPBP17–9 or ETAA1.10–12 Once activated, ATR and its downstream effectors play crucial roles in limiting origin firing, stabilizing stressed forks, and promoting fork restart. While the requirement of ATR for protecting stressed replication forks has been long appreciated, how exactly ATR acts at stressed forks is still unclear.
Several mechanisms have been proposed to explain the functions of ATR in protecting stressed forks. One of these mechanisms is to restrict the formation of ssDNA at replication forks. When cells are exposed to the replication inhibitor hydroxyurea (HU) and ATR inhibitor (ATRi), very high levels of ssDNA are generated, leading to exhaustion of RPA and replication catastrophe.13 Even in the absence of HU, ATRi increases the exposure of ssDNA in S phase, inducing replication catastrophe in a population of early S phase cells undergoing robust replication.14 Although ATR clearly suppresses ssDNA accumulation during DNA replication, how ssDNA is generated at stressed forks upon ATR inhibition remains unclear. A second mechanism by which ATR protects stressed forks is to prevent excessive cleavage of forks by nucleases. In human cells, inhibition of the ATR effector kinase Chk1 results in cleavage of replication forks by the MUS81 nuclease.15 In cells treated with HU and ATRi, DNA double-stranded breaks (DSBs) are generated by SLX4-associated nuclease activities.16,17 In both human and budding yeast, loss of the ATR/Mec1 pathway leads to unrestricted activity of the EXO1/Exo1 nuclease at replication forks.18,19 In Xenopus extracts, ATR prevents DSB accumulation at stressed forks by enabling the MRE11 nuclease-mediated fork restart,20 suggesting that ATR not only prevents improper nuclease activities at stressed forks but also promotes proper nucleolytic processing. The third protective function of ATR at stressed forks is related to fork remodeling. When replication forks encounter impediments to fork progression, they undergo regression through a process called fork reversal, generating four-way DNA junctions known as reversed forks or “chicken feet.”21,22 Fork reversal stabilizes stressed forks and promotes their recovery under mild replication stress, but it can also lead to excessive nuclease cleavage and an increase of DSBs under high replication stress. ATR was shown to inhibit the fork reversal activity of SMARCAL1, reducing the formation of DSBs by SLX4-associated nuclease activities.16 However, in response to DNA interstrand crosslinks (ICLs), ATR promotes reversed forks and global fork slowing.23 The mechanism by which ATR affects the levels of reversed forks still remains elusive.
Although ATR has been implicated in the regulation of ssDNA, nucleases, and fork remodeling at stressed forks, it is still largely unknown whether these functions of ATR are distinct or interconnected. A comprehensive understanding of the functions of ATR in fork protection is still lacking. One obstacle to assessing the relationships among ATR functions is the reliance of previous studies on different assays. While several assays have been successfully used to study the impact of ATR on stressed forks, it is not clear whether these assays reflect the same or different functions of ATR. In addition, it is not known whether ATR executes the same functions at all stressed forks or acts differently at distinct subsets of stressed forks. Of note, several studies using human and yeast cells suggest that the stability of ongoing replication forks and forks assembled under stress at newly fired replication origins may be differently regulated (see Discussion). For example, in cells treated with HU and ATRi, DSBs are rapidly induced at forks from newly fired origins, suggesting that ATR preferentially prevents the collapse of new forks.24 To obtain a more comprehensive understanding of the functions of ATR in protecting stressed forks, we seek to use multiple assays to systematically analyze how ATR regulates ssDNA, nucleases, and fork remodeling at replication forks upon stress, and determine whether ATR carries out these functions differently at ongoing and new forks.
In this study, we used four distinct assays to mechanistically examine the functions of ATR at ongoing and new replication forks. We found that ATR prevents degradation of nascent DNA from PrimPol-generated ssDNA gaps at ongoing forks, suppresses uncoupling of ongoing forks, antagonizes template DNA nicking and degradation at new forks, and promotes fork reversal. Notably, in response to PARP inhibitor (PARPi), ATRi preferentially exacerbates nascent DNA degradation in BRCA1/2-deficient cells, and disrupts restored gap protection in BRCA1-deficient, PARPi-resistant cells, explaining the ability of ATRi to overcome PARPi resistance. Altogether, these results present a comprehensive view of the functions of ATR in protecting stressed forks, providing insights into the distinct roles of ATR at ongoing and new forks and opportunities to target ATR in cancer therapy.
RESULTS
ATR suppresses distinct HU-induced alterations at ongoing and new replication forks
To investigate how ATR suppresses ssDNA formation at stressed forks, we first used three distinct DNA labeling-based assays to analyze the effects of ATRi on forks upon HU treatment. Furthermore, to test whether ATR functions differently at ongoing and new forks, we carried out the assays in the presence or absence of CDC7 inhibitors (CDC7i; XL-413 and PHA-767491), which reduce the firing of replication origins even in the presence of ATRi.
First, we tested the effects of ATRi on pulse-labeled nascent DNA. Nascent DNA was sequentially labeled with 5-chloro-2-deoxyuridine (CldU, red) and 5-iodo-2′-deoxyuridine (IdU, green) in U2OS cells, and then cells were treated with HU for 5 h in the presence or absence of three distinct ATRis (VE-821, AZ20, AZD6738) and processed for DNA fiber analysis (Figure 1A). In this assay, degradation of IdU-labeled nascent DNA reduces the IdU/CldU ratio in replication tracts.25 All three ATRis significantly reduced the IdU/CldU ratio (Figure 1B), suggesting that the nascent DNA at HU-stalled forks is increasingly degraded upon ATR inhibition. The Chk1 inhibitor, MK-8776, also reduced the IdU/CldU ratio but to a slightly lesser extent compared with ATRi, whereas the combination of ATRi and Chk1i exerted the same effects as ATRi alone (Figure S1A). Thus, ATR prevents degradation of nascent DNA at stressed forks primarily through Chk1, but other ATR substrates may also contribute to this function.
Figure 1. ATR suppresses distinct HU-induced alterations at ongoing and new replication forks.

(A) Cells are sequentially labeled with thymidine analogs CldU (50 μM) and IdU (100 μM) and incubated in 4 mM hydroxyurea (HU) for 5 h. Samples are then processed for fiber assay analysis.
(B) Cells were treated as in (A) with three ATR inhibitors: VE-821 (10 μM), AZ20 (1 μM), and AZD6738 (5 μM). Number (n) of fibers quantified >450 across three biological replicates. Significance was calculated using the Mann-Whitney Ranked Sum Test with ****p < 0.0001.
(C) Cells were analyzed as in (A) in the presence or absence of CDC7i (XL-413, 5 μM) and ATRi (VE-821, 10 μM). Number (n) of fibers quantified >300 across two biological replicates. Significance was calculated using the Mann-Whitney Ranked Sum Test with *p < 0.05, ****p < 0.0001.
(D) Cells are first labeled for 48 h with 20 μM BrdU, and then incubated in media without BrdU for 2 h before treatment with 4 mM HU for 4 h. Samples are then processed for immunofluorescence detection of PCNA and BrdU in non-denaturing conditions. Only PCNA-positive, S phase cells are selected for the analysis.
(E) Cells were analyzed as depicted in (D) in the presence or absence of ATRi (VE-821, 10 mM) and CDC7i (XL-413, 5 μM). Number (n) of nuclei quantified >500 across three biological replicates. Significance was calculated using Mann-Whitney Ranked Sum Test with ****p < 0.0001.
(F) Nascent DNA is labeled with 20 μM BrdU for 15 min prior to exposure to 4 mM HU for 4 h in the presence of BrdU. Samples are then processed for immunofluorescence detection of PCNA and BrdU in non-denaturing conditions. Only PCNA-positive S phase cells are selected for the analysis.
(G) Cells were analyzed as depicted in (F) in the presence or absence of ATRi (VE-821, 10 μM) and CDC7i (5 μM XL-413). Number (n) of nuclei quantified >500 across three biological replicates. Significance was calculated using Mann-Whitney Ranked Sum Test with ****p < 0.0001.
Given that ATR limits the firing of replication origins, we asked whether ATRi affects the stability of nascent DNA by increasing origin firing. As expected, ATRi increased the percentage of DNA fibers containing newly fired origins, and this effect of ATRi was reversed by CDC7is in a dose-dependent manner (Figure S1B). The ATRi-induced nascent DNA degradation was observed even in the presence of CDC7i (Figures 1C and S1C), indicating that this effect of ATRi is largely independent of increased origin firing. We noted that the ATRi-induced reduction in the IdU/CldU ratio was slightly decreased by CDC7i, which may reflect a contribution of CDC7 to nascent DNA degradation.26 These results suggest that nascent DNA degradation assay primarily analyzes ongoing forks, and that ATR plays an important role in protecting the nascent DNA at ongoing forks in response to HU.
Next, we tested whether ATRi affects the exposure of template DNA in response to HU. Increased exposure of template ssDNA at forks may reflect several changes in the fork structure, such as (1) uncoupling of the replicative helicase and DNA polymerases, (2) nascent DNA degradation, (3) ssDNA gap formation, and (4) resection of nascent DNA from DNA ends (Figure 1D). We labeled genomic DNA with BrdU for two consecutive cell cycles, and then briefly cultured cells in BrdU-free media to ensure that nascent DNA is not labeled. Subsequently, we treated cells with HU for 4 h in the presence or absence of ATRi and performed anti-BrdU staining under a non-denaturing condition. This assay specifically measures the exposure of non-nascent DNA as ssDNA at or behind replication forks. Because replication forks progress slowly in HU, the HU-induced BrdU staining mainly reflects the exposure of template ssDNA at stalled/collapsed forks. Both ATRi and Chk1i significantly increased the exposure of template ssDNA in HU-treated cells (Figures 1E and S1D), suggesting that the fork structure is altered in the absence of the ATR-Chk1 pathway.13,14 The induction of template ssDNA by ATRi still occurred robustly in CDC7i (Figures 1E and S1E), suggesting that increased new origin firing is not required. Thus, template ssDNA exposure assay primarily analyzes ongoing forks, and ATR suppresses one or more fork-remodeling/processing activities that expose template ssDNA at ongoing forks.
Last, we tested how ATRi affects the exposure of nascent ssDNA in response to HU. Increased exposure of nascent ssDNA may reflect the presence of ssDNA in the reversed arm of replication forks or degradation of template DNA from DNA ends (Figure 1F). We pulse-labeled nascent DNA with BrdU, treated cells with HU for 4 h in the presence or absence of ATRi, and then performed native anti-BrdU staining. Both ATRi and Chk1i drastically increased the exposure of nascent ssDNA (Figures 1G and S1F).16 Notably, the ATRi-induced increase of nascent ssDNA exposure was largely suppressed by CDC7i (Figures 1G and S1G), showing that this effect of ATRi is dependent on new origin firing. Thus, in contrast to nascent DNA degradation and template ssDNA exposure assays, the nascent ssDNA exposure assay primarily detects the effect of ATRi on new forks. Furthermore, ATR is critical for suppressing one or more fork-remodeling/processing activities that expose nascent ssDNA at new forks.
ATR prevents resection from PrimPol-generated gaps at ongoing replication forks
To understand how ATR prevents nascent DNA degradation at ongoing forks in response to HU, we sought to use nascent DNA degradation assay (Figure 2A) to identify the nuclease(s) and DNA structures involved. Treatment with Mirin, an inhibitor of the MRE11 nuclease, and knockdown of the EXO1 nuclease partially reversed the effect of ATRi on nascent DNA degradation (Figures 2B, 2C, and S2A), In contrast, knockdown of DNA2, another nuclease implicated in DNA end resection, did not affect ATRi-induced nascent DNA degradation (Figure S2B). Thus, in response to HU, ATR protects the nascent DNA at ongoing forks from degradation by MRE11 and EXO1.
Figure 2. ATR prevents resection from PrimPol-generated gaps at ongoing forks.

(A) Schematic representation of the nascent DNA degradation fiber assay.
(B and C) Cells were treated as in (A) with or without ATRi (VE-821, 10 μM) and MRE11i (Mirin, 50 μM) in (B), and with or without ATRi (VE-821, 10 μM) following 48 h siRNA knockdown of EXO1 in (C). Number (n) of fibers quantified >250 across two biological replicates. Significance was calculated using the Mann-Whitney Ranked Sum Test with ***p < 0.001, ****p < 0.0001.
(D and E) siRNAs against HLTF, SMARCAL1, and ZRANB3 (siTriple) (D) or PrimPol (E) were transfected 48 h prior to fiber analysis. Number (n) of fibers quantified >500 across three biological replicates. Significance was calculated using the Mann-Whitney Ranked Sum Test with ****p < 0.0001.
(F) U2OS cells were analyzed as in (E) except cells were treated for only 1 h with or without ATRi (VE-821, 10 μM) and HU (4 mM) followed by S1 nuclease digestion. Number (n) of fibers quantified >135 in each sample across two biological replicates. Significance was calculated using the Mann-Whitney Ranked Sum Test with *p < 0.05, ****p < 0.0001.
(G) Model for how nascent DNA is degraded at ongoing forks.
Nascent DNA is degraded in BRCA1/2-deficient cells in response to HU.25,27 The degradation of nascent DNA in BRCA1/2-deficient cells is dependent on replication fork reversal,28–31 indicating that degradation is initiated from the reversed arm. Three translocases, HLTF, SMARCAL1, and ZRANB3, are all required for the nascent DNA degradation in BRCA1/2-deficient cells.28–30 To understand whether fork reversal is required for ATRi-induced nascent DNA degradation, we simultaneously knocked down HLTF, SMARCAL1, and ZRANB3, generating “triple-knockdown” cells (Figure S2C). Surprisingly, the IdU/CldU ratio was still significantly reduced by ATRi in triple-knockdown cells (Figure 2D), suggesting that ATRi-induced nascent DNA degradation is largely, if not completely, independent of fork reversal.
In addition to reversed forks, ssDNA gaps could also serve as an entry point for nucleases to degrade nascent DNA. The primase and DNA-directed polymerase PrimPol is capable of repriming for DNA synthesis ahead of stalled DNA polymerases, leading to ssDNA gaps at and behind forks.32–37 To test whether PrimPol is required for ATRi-induced nascent DNA degradation, we depleted PrimPol using siRNA (Figure S2D). The nascent DNA degradation induced by ATRi was completely suppressed by the loss of PrimPol (Figure 2E), showing that PrimPol is required for this process. To test whether PrimPol promotes nascent DNA degradation by generating ssDNA gaps, we treated DNA fibers with or without the S1 nuclease, which specifically cleaves ssDNA.22 In the absence of S1, IdU tracts were shortened after ATRi treatment (Figure 2F, lanes 1–2), reflecting the degradation of nascent DNA. In the presence of S1, IdU tracts were shortened further (Figure 2F, lanes 2 and 6), confirming the presence of ssDNA gaps in nascent DNA. Importantly, in PrimPol knockdown cells, nascent DNA was no longer shortened after ATRi treatment even in the presence of S1 (Figure 2F, lanes 7–8), suggesting that the ssDNA gaps in nascent DNA are generated in a PrimPol-dependent manner and subjected to degradation. Given that the shortening of replication tracts by S1 requires cleavage of both daughter strands, ssDNA gaps are likely generated on both daughter strands by PrimPol and possibly DNA polymerase α-primase, and these gaps are protected by ATR (Figure 2G).
To confirm the findings of nascent DNA degradation, we used isolation of proteins on nascent DNA (iPOND) to capture the ssDNA-binding protein RPA at replication forks. Treatment with HU and ATRi increased the levels of RPA captured by iPOND (Figure S2E), indicating ssDNA accumulation at replication forks.24 Knockdown of PrimPol reduced the RPA captured by iPOND (Figure S2E), suggesting that PrimPol contributes to ssDNA accumulation. Furthermore, knockdown of EXO1 but not DNA2 reduced the RPA captured by iPOND (Figure S2F), supporting the idea that EXO1 but not DNA2 contributes to the nascent DNA degradation from ssDNA gaps. Thus, in cells treated with HU and ATRi, ssDNA gaps are formed and expanded at ongoing replication forks (Figure 2G).
In BRCA1/2-deficient cells, loss of stable RAD51 filaments results in nucleolytic degradation of reversed forks.25,27 Even in BRCA1/2-proficient cells, compromised RAD51 function leads to fork degradation.24,29,31 In Xenopus egg extracts, RAD51 protects ssDNA gaps resulting from fork uncoupling.38 Consistent with previous studies,39,40 we found that HU-induced RAD51 foci were significantly reduced by ATRi (Figure S2G), supporting the possibility that ATR protects ssDNA gaps by recruiting RAD51.
ATR promotes the accumulation of reversed replication forks
Given that fork reversal is not required for the effect of ATRi on nascent DNA degradation, we asked whether fork reversal occurs efficiently when ATR is inhibited. We treated cells with or without HU and used electron microscopy (EM) to quantify the fraction of replication forks carrying reversed arms (Figures 3A and 3B). As shown previously, HU treatment significantly increased fork reversal (Figures 3B and 3C).41 ATRi did not affect the levels of reversed forks in the absence of HU, but it decreased fork reversal by 50% in the presence of HU (Figures 3B, 3C, and S3A). Thus, ATR promotes fork reversal in response to HU.
Figure 3. ATR promotes accumulation of reversed forks.

(A) Schematic representation of the ATRi-induced fork structures analyzed by EM.
(B) Representative electron microscopy images of normal (left) and reversed (right) replication forks in cells treated with HU, as well as replication forks with ssDNA gaps in cells treated with HU and ATRi (bottom). Insets show the magnified fork junctions. Red arrows show the location of the ssDNA gaps. P, parental strand; D, daughter strand; R, regressed arm; red arrow, ssDNA. Note that DNA breaks distal or very close to fork junctions are not detected by EM.
(C) Frequency of EM-detectable reversed forks in U2OS cells mock-treated or treated with HU (4 mM), HU + ATRi (VE-821, 10 μM), or HU + ATRi + siControl/siPrimPol. The total numbers of replication forks (n) analyzed by EM in each sample are indicated across two biological replicates (n = 2).
(D and E) Frequency and length of EM-detectable ssDNA gaps at fork junctions and internal gaps in U2OS cells treated with HU (4 mM) or HU + ATRi (VE-821, 10 μM) + siControl/siPrimPol. Gap threshold: +20 nm. Significance of ssDNA gap length was calculated using Welch’s t test with **p < 0.01. ATRi treatment was 2 h long and siControl or siPrimPol was transfected 48 h before analysis in (C)–(E).
(F) Frequency of EM-detectable reversed forks in U2OS cells mock-treated or treated with HU (4 mM), HU + ATRi (VE-821, 10 μM), or HU + ATRi + MRE11i (Mirin, 50 μM). ATRi and MRE11i (Mirin) treatments were 2 h long. The total numbers of replication forks (n) analyzed by EM in each sample are indicated across one biological replicate (n = 1).
(G) Model for how ATR inhibitors impact the observed proportion of reversed forks.
We next investigated why fork reversal is inhibited by ATRi. We reasoned that degradation of nascent DNA at fork junctions may reduce the probability for the two nascent strands to form a reversed arm. This possibility predicts that inhibition of gap-initiated nascent DNA degradation should restore fork reversal. Indeed, knockdown of PrimPol almost fully rescued fork reversal in HU and ATRi (Figure 3C, lanes 4 and 8, S3A). In the presence of HU, ATRi did not significantly alter the fraction of forks displaying ssDNA gaps at junctions or internal gaps in daughter strands but increased the length of ssDNA in gaps (Figures 3A, 3D, and 3E), suggesting that gaps are extended in ATRi. Importantly, PrimPol depletion in cells treated with HU and ATRi significantly reduced the fraction of forks displaying ssDNA gaps and the length of ssDNA in gaps (Figures 3D and 3E), supporting the idea that ssDNA is generated by extension of PrimPol-generated gaps. In addition, the levels of reversed forks in HU and ATRi were partially rescued by Mirin (Figure 3F, lanes 2 and 4), suggesting that MRE11-mediated nascent DNA degradation contributes to the reduction in fork reversal. The partial effect of Mirin is likely attributed to the involvement of both MRE11 and EXO1 in nascent DNA degradation (Figures 2B and 2C). Thus, in cells treated with HU and ATRi, nascent DNA degradation from ssDNA gaps reduces the frequency of fork reversal (Figure 3G).
ATR prevents helicase-polymerase uncoupling at ongoing replication forks
To understand how ATRi increases the exposure of template ssDNA at ongoing forks (Figure 1E), we used template ssDNA exposure assay to investigate which factors are involved (Figure 4A). One possible mechanism to expose template ssDNA is through degradation of nascent DNA. Mirin and PFM-01, which inhibit the exonuclease and endonuclease activity of MRE11, respectively, both reduced ATRi-induced template ssDNA (Figure S4A). Knockdown of EXO1 but not DNA2 also modestly reduced template ssDNA exposure (Figure S4B). The combination of Mirin and EXO1 knockdown significantly reduced template ssDNA exposure (Figure 4B), suggesting that ATRi-induced exposure of template ssDNA is partially driven by MRE11 and EXO1.
Figure 4. ATR prevents fork uncoupling at ongoing forks.

(A) Schematic representation of the template ssDNA exposure assay.
(B) Cells were treated as in (A) with MRE11i (Mirin, 50 μM) with or without ATRi (VE-821, 10 μM) following 48 h siRNA knockdown of EXO1 and processed for the template strand exposure assay. Number (n) of nuclei quantified >300 in each sample across two biological replicates. Significance was calculated using the Mann-Whitney Ranked Sum Test with ****p < 0.0001.
(C and D) Cells were treated as in (A) with or without ATRi (VE-821, 10 μM) following 48 h siRNA knockdown of PrimPol (C) or the combination of HLTF, SMARCAL1, and ZRANB3 (siTriple, D). Number (n) of nuclei quantified >300 across three biological replicates. Significance was calculated using the Mann-Whitney Ranked Sum Test with ****p < 0.0001.
(E) Cells were treated as in (A) with or without ATRi (VE-821, 10 μM) following 48 h siRNA knockdown of Tipin. Number (n) of nuclei quantified >130 across two biological replicates. Significance was calculated using the Mann-Whitney Ranked Sum Test with ****p < 0.0001.
(F) Model for how ATR prevents template DNA exposure at ongoing forks.
Because nascent DNA degradation from ssDNA gaps may expose template ssDNA (Figures 2E and 2F), we tested whether PrimPol is involved. Surprisingly, ATRi still significantly increased template ssDNA exposure after PrimPol knockdown, although the levels of template ssDNA might be slightly reduced in PrimPol-depleted cells (Figure 4C). Furthermore, triple knockdown of HLTF, SMARCAL1, and ZRANB3 only slightly reduced template ssDNA (Figure 4D). These results show that ATRi-induced template ssDNA exposure still occurs efficiently in the absence of PrimPol and fork reversal. In contrast to nascent DNA degradation assay, which relies on shortening of both nascent strands to detect changes in replication tracts, template ssDNA exposure assay can detect ssDNA even when it accumulates on one daughter strand. Indeed, inhibition of POLα modestly increased template ssDNA (Figure S4C), which is likely a result of defective lagging strand synthesis. Notably, in cells treated with POLα inhibitor (POLαi), ATRi induced higher levels of template ssDNA, suggesting that gaps on the lagging strand promote ATRi-induced template ssDNA exposure. These results raise the possibility that gaps on the lagging strand are sufficient to provide an entry point for MRE11 and EXO1 to degrade nascent DNA and expose template ssDNA.
The degradation of nascent DNA at collapsed forks may also expose template ssDNA. To test this possibility, we knocked down MUS81 (Figure S4D), which is implicated in Chk1i-induced fork collapse.15 In neutral comet assay, ATRi increased DSBs in HU-treated cells, and this increase was not observed in MUS81 knockdown cells (Figure S4E). However, knockdown of MUS81 did not suppress ATRi-induced template ssDNA exposure (Figure S4F). Consistently, in cells treated with HU and ATRi, knockdown of MUS81 did not reduce the levels of RPA captured by iPOND (Figure S4G). Furthermore, inhibition of the endonuclease activity of MRE11, which is implicated in fork collapse,42 did not reduce ATRi-induced DSBs in HU (Figure S4H), suggesting that the effects of MRE11 on template ssDNA exposure are not attributed to DSB formation. Thus, nascent DNA degradation from collapsed forks is unlikely a major mechanism driving ATRi-induced template ssDNA exposure.
Functional uncoupling of the replicative helicase and DNA polymerases is a mechanism to expose template ssDNA and activate the ATR checkpoint.43 The human replication fork proteins Timeless and Tipin, as well as their yeast homologs, interact with both the replicative helicase and DNA polymerases, coupling the helicase to DNA synthesis.44–47 Indeed, knockdown of Tipin increased HU-induced template ssDNA even in the absence of ATRi (Figures 4E and S4I), supporting the idea that fork uncoupling is a mechanism to expose template ssDNA. Importantly, while ATRi and Tipin knockdown each increased template ssDNA similarly, the combination of ATRi and Tipin knockdown did not increase template ssDNA further (Figure 4E), suggesting that they act through the same mechanism. Similarly, knockdown of Timeless also increased template ssDNA exposure, and this effect was largely unchanged by ATRi (Figures S4J and S4K). Thus, in cells exposed to HU and ATRi, replication fork uncoupling is likely the main driver of template ssDNA exposure (Figure 4F). Notably, ATRi-induced replication tract shortening was still observed in Tipin knockdown cells (Figure S4L), showing that nascent DNA degradation still occurs at uncoupled forks.
Together, the results above suggest that ATRi has two distinct effects at ongoing forks. ATRi induces fork uncoupling, driving most of the template ssDNA exposure. In addition, ATRi also induces MRE11-and EXO1-mediated nascent DNA degradation from ssDNA gaps, increasing the overall exposure of template ssDNA.
ATR prevents degradation of template DNA at new replication forks
To understand how ATR suppresses nascent ssDNA exposure at replication forks generated at newly fired origins (Figure 1G), we used the nascent ssDNA exposure assay to identify the factors involved. The presence of ssDNA in the reversed arm or degradation of template DNA from DNA nicks or ends could lead to nascent ssDNA exposure (Figure 5A). In cells treated with HU and ATRi, the exposure of nascent ssDNA was not reduced by triple knockdown of HLTF, SMARCAL1, and ZRANB3 (Figure 5B), nor knockdown of PrimPol (Figure 5C). These results suggest that neither fork reversal nor ssDNA gaps in nascent DNA contribute significantly to ATRi-induced nascent ssDNA exposure. To test whether the collapse of new forks is involved, we carried out neutral comet assay. Consistent with previous studies, ATRi increased DSBs in HU-treated cells,13,24 and this increase was suppressed by CDC7i (Figure 5D), confirming that ATRi induces collapse of new forks. SLX4 associates with MUS81 and other structure-specific nucleases and promotes fork collapse in cells treated with HU and ATRi.16,17 Depletion of SLX4 or MUS81 did not reduce nascent ssDNA exposure (Figures 5E, S4D, and S5A), suggesting that SLX4-associated nucleases are not involved. Although fork reversal and the cleavage of reversed forks by SLX4-associated nucleases48 contribute to fork collapse, they are not critical for ATRi-induced nascent ssDNA exposure.
Figure 5. ATR prevents degradation of template DNA at new forks.

(A) Schematic representation of the nascent DNA exposure assay.
(B and C) Cells were treated as in (A) with or without ATRi (VE-821, 10 μM) following 48 h siRNA knockdown of HLTF, SMARCAL1, and ZRANB3 (siTriple, B) or PrimPol (C). Number (n) of nuclei quantified >1,000 across four biological replicates in (B) and >300 across two biological replicates in (C). Significance was calculated using the Mann-Whitney Ranked Sum Test with ****p < 0.0001.
(D) Cells were exposed to HU (4 mM) and ATRi (VE-821, 10 μM) for 5 h and processed for neutral comet assay. Box plots represent the tail moment of comets. Number (n) of cells quantified >100 across two biological replicates. Significance was calculated using Mann-Whitney ranked sum test with ****p < 0.0001.
(E) Following 48 h siRNA knockdown of SLX4 or MUS81, cells were treated as in (A) with or without ATRi (VE-821, 10 μM). Number (n) of nuclei quantified >250 across two biological replicates. Significance was calculated using the Mann-Whitney Ranked Sum Test.
(F–H) Cells were treated as in (A) with or without ATRi (VE-821, 10 μM) following 48 h siRNA knockdown of MRE11, EXO1, and DNA2 in (F); treated with MRE11i (Mirin, 50 μM or PFM-01, 100 μM) in (G); or with CDC7i (XL-413, 5 μM) following CtIP knockdown in (H). Number (n) of nuclei quantified >250 across two biological replicates. Significance was calculated using Mann-Whitney Ranked Sum Test with *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
(I) Model for how nascent ssDNA is exposed in the absence of ATR.
In contrast to the depletion of SLX4 and MUS81, knockdown of MRE11 reduced ATRi-induced nascent ssDNA exposure (Figures 5F and S5B). Interestingly, the exposure of nascent ssDNA was significantly reduced by PFM-01 but not by Mirin (Figure 5G), suggesting that the endonuclease activity of MRE11 is involved. To exclude the possibility that the effect of PFM-01 is attributed to altered replication, we knocked down CtIP, which stimulates the endonuclease activity of the MRN complex.49 CtIP knockdown also reduced nascent ssDNA exposure (Figures 5H and S5C). Notably, the effect of CtIP knockdown on nascent ssDNA exposure was no longer observed when origin firing was inhibited by CDC7i (Figure 5H), suggesting that CtIP specifically acts on new forks. Knockdown of EXO1 but not DNA2 also decreased the exposure of nascent ssDNA (Figure 5F). Thus, in cells treated with HU and ATRi, the endonuclease activity of the MRN-CtIP complex processes template DNA at new forks, which allows degradation of template DNA by EXO1 and exposure of nascent ssDNA (Figure 5I). Of note, the endonuclease activity of MRE11 did not contribute to the formation of ATRi-induced DSB in HU (Figure S4H), suggesting that MRE11 nicks template DNA without causing DSBs. Furthermore, nascent ssDNA exposure was increased by Tipin knockdown independently of ATRi (Figure S5D), suggesting that the nicking and degradation of template DNA may be triggered by uncoupling of new forks. These results raise the possibility that uncoupling of new forks generates a structure in which template DNA can be nicked by MRN-CtIP, providing an entry point for EXO1 to degrade template DNA and expose nascent ssDNA.
ATR prevents resection from PARP inhibitor-induced ssDNA gaps at ongoing forks
We and others recently showed that PARPi induces ssDNA gaps in nascent DNA during replication,35,50–52 raising the possibility that ATRi stimulates nascent DNA degradation from ssDNA gaps in PARPi-treated cells. To test this possibility, we used nascent DNA degradation assay to analyze the effects of ATRi in BRCA1-proficient U2OS cells treated with Olaparib (PARPi, Figure 6A). Cells were pulse-labeled with CldU and IdU and then exposed to PARPi in the presence or absence of ATRi. ATRi reduced the IdU/CldU ratio in PARPi-treated cells and this effect was largely unchanged by CDC7i (Figure 6A), suggesting that nascent DNA is increasingly degraded at ongoing forks upon ATR inhibition. Importantly, ATRi-induced nascent DNA degradation was dependent on PrimPol (Figure 6B), but not HLTF, SMARCAL1, and ZRANB3 (Figure S6A). Thus, ATRi increases nascent DNA degradation at ongoing forks in PARPi-treated cells, and this effect is dependent on ssDNA gaps but not reversed forks.
Figure 6. ATR inhibition enhances nascent DNA degradation from PARPi-induced ssDNA gaps.

(A) U2OS cells were treated with DMSO or CDC7i (XL-413, 5 μM) with or without ATRi (VE-821, 10 μM) in the presence of PARPi (Olaparib, 10 μM) for 5 h and processed for fiber assay analysis.
(B and C) Following 48 h knockdown of PrimPol, U2OS (B) or UWB1 and UWB1+B1 (C) cells were treated with or without ATRi (VE-821, 10 μM) in the presence of PARPi (Olaparib, 10 μM) for 5 h and processed for fiber assay analysis.
(D) UWB1 and UWB1+B1 cells were sequentially labeled in CldU (50 μM) followed by IdU (100 μM) in the presence of PARPi (Olaparib, 10 μM). Cells were then incubated in PARPi (Olaparib, 10 μM) with or without ATRi (VE-821, 10 μM) for 5 h followed by S1 nuclease digestion.
(E) UWB1, UWB1+B1, SYr12, and SYr13 cells were sequentially labeled in CldU (50 μM) followed by IdU (100 μM) in the presence of PARPi (Olaparib, 10 μM). Cells were then incubated in media containing DMSO or PARPi (Olaparib, 10 μM) with or without ATRi (VE821, 10 μM) for 5 h and processed for fiber assay analysis.
(F) Following 48 h knockdown of PrimPol, SYr12 and SYr13 cells were sequentially labeled in CldU (50 μM) followed by IdU (100 μM) in the presence of PARPi (Olaparib, 10 μM). Cells were then incubated in PARPi (Olaparib, 10 μM) with or without ATRi (VE-821, 10 μM) for 5 h. Number (n) of fibers quantified >250 across two biological replicates. Significance was calculated using the Mann-Whitney Ranked Sum Test with **p < 0.01, ***p < 0.001, ****p < 0.0001 in (A)–(F).
(G) Viability assay of UWB1, UWB1+B1, SYr12, and SYr13 cells after 6 days of treatment with increasing doses of PARPi (Olaparib, 10 μM), ATRi (VE-821, 10 μM), and PARPi + ATRi following 24 h siRNA knockdown of PrimPol.
ATRi disrupts the restored gap protection in BRCA1-deficient and PARPi-resistant cells
To investigate whether ATRi affects nascent DNA degradation in BRCA1-deficient cells after PARPi treatment, we analyzed the BRCA1-deficient ovarian cancer cell line UWB1 and UWB1+B1, a UWB1 derivative line complemented with wild-type BRCA1. ATRi reduced the IdU/CldU ratio in both UWB1 and UWB1+B1, but the effect in UWB1 was significantly more pronounced (Figure 6C, lanes 1–2 and 5–6), showing that ATRi preferentially exacerbates nascent DNA degradation in BRCA1-deficient cells. Importantly, knockdown of PrimPol suppressed ATRi-induced nascent DNA degradation in both UWB1 and UWB1+B1 (Figure 6C, lanes 3–4 and 7–8), showing that degradation initiates from ssDNA gaps in both contexts. In contrast, triple knockdown of HLTF, SMARCAL1, and ZRANB3 did not suppress ATRi-induced nascent DNA degradation in UWB1 and UWB1+B1 (Figure S6B). Thus, fork reversal, which is required for HU-induced nascent DNA degradation in BRCA1-deficient cells, is dispensable for ATRi and PARPi-induced degradation. In the presence of PARPi, IdU tracts in both UWB1 and UWB1+B1 cells were significantly shortened by S1 even in the absence of ATRi (Figure 6D, lanes 1, 3, 5, 7), consistent with our previous finding that PARPi induces ssDNA gaps regardless of the BRCA1 status.35 ATRi pronouncedly increased the shortening of IdU tracts by S1 in UWB1 than in UWB1+B1 (Figure 6D, lanes 3–4 and 7–8), suggesting that ATRi preferentially exacerbates ssDNA gaps in BRCA1-deficient cells. Similarly, in the presence of PARPi, ATRi preferentially stimulated nascent DNA degradation and increased S1 cleavage of IdU tracts in BRCA2 knockdown cells compared with control cells (Figures S6C–S6E). Furthermore, in BRCA2 knockdown cells, PrimPol depletion reduced the ATRi-induced shortening of nascent DNA in the presence of S1 (Figure S6E), suggesting that ATRi also exacerbates PARPi-induced and PrimPol-generated ssDNA gaps in BRCA2-deficient cells.
Finally, we asked whether the degradation of nascent DNA from ssDNA gaps in BRCA1-deficient cells contributes to PARPi sensitivity. To address this question, we tested SYr12 and SYr13, two UWB1 derivative lines that acquired PARPi resistance.39 After PARPi treatment, the IdU/CldU ratio was decreased in UWB1 and UWB1+B1 (Figure 6E, lanes 1–2, 4–5), suggesting that PARPi alone is sufficient to induce nascent DNA degradation. This effect of PARPi was more pronounced in UWB1, which is consistent with the defective gap protection in BRCA1-deficient cells.36 ATRi further reduced the IdU/CldU ratio in UWB1 and UWB1+B1, and the effect was also more pronounced in UWB1 (Figure 6E, lanes 2–3, 5–6). Similar observations were made in BRCA2 knockdown cells (Figure S6D). These results suggest that PARPi preferentially induces nascent DNA degradation in BRCA1/2-deficient cells, and this process is exacerbated by ATRi. Compared with UWB1 cells, SYr12 and SYr13 cells displayed higher IdU/CldU ratios after PARPi treatment (Figure 6E, lanes 1–2, 7–8, 10–11), showing that gap protection is partially restored. Notably, ATRi significantly reduced the IdU/CldU ratios in SYr12 and SYr13 (Figure 6E, lanes 8–9, 11–12), showing that ATR inhibition disrupts the restored gap protection. The ATRi-induced nascent DNA degradation in SYr12 and SYr13 cells was dependent on PrimPol (Figure 6F), but not HLTF, SMARCAL1, and ZRANB3 (Figure S6F), suggesting that the degradation in resistant cells also initiates from ssDNA gaps but not reversed forks. Importantly, loss of PrimPol reduced the sensitivity of UWB1, UWB1+B1, SYr12, and SYr13 to the combination of PARPi and ATRi (Figure 6G), showing that ssDNA gaps are a key determinant of ATRi sensitivity and the ability of ATRi to overcome PARPi resistance is dependent on ssDNA gaps. Together, these results suggest that restored gap protection in BRCA1-deficient cells is associated with PARPi resistance, and that ATRi disrupts the restored gap protection to overcome PARPi resistance.
DISCUSSION
While it is well appreciated that the stability of replication forks is critical for genomic integrity, whether all replication forks respond to stress in the same way remains unclear. In fission yeast, increased recombination intermediates and homologous integration hotspots are detected at origins.53 In budding yeast, loss of the replication checkpoint leads to fork arrest and collapse in close proximity to active origins.54 In mammalian cells, DNA breaks are detected near origins when cells progress through S phase in HU.55 When human cells are exposed to HU and ATRi, non-homologous end-joining proteins are recruited to replication forks in a CDC7-dependent manner, suggesting that DSBs are preferentially formed at new forks.24 Although these studies implied that the stress response at ongoing and new forks may be different, molecular details are lacking. In this study, we used four different assays, including (1) nascent DNA degradation, (2) template ssDNA exposure, (3) nascent ssDNA exposure, and (4) EM, to analyze the effects of ATRi on HU-stalled replication forks. We found that some of these assays have distinct preferences for ongoing or new forks. By combining these assays, we provide evidence that ATR plays distinct roles at ongoing and new forks. Our results not only support the concept that ATR functions differently at distinct subpopulations of replication forks, but also provide insights into the unique stress responses at ongoing and new forks.
Our data suggest that ATR suppresses ssDNA formation at forks through multiple mechanisms. Using nascent DNA degradation assay, we found that ATR protects nascent DNA against MRE11 and EXO1-mediated degradation from ssDNA gaps at ongoing forks (Figure 7A). Consistently, in both human and budding yeast, the ATR/Mec1 pathway restricts EXO1/Exo1 activity at stalled forks.18,19 Using template ssDNA exposure assay, we found that ATR suppresses helicase-polymerase uncoupling at ongoing forks (Figure 7A). Given that helicase-polymerase uncoupling is a stress-induced mechanism to activate ATR,43 ATR acts in a feedback loop to prevent further fork uncoupling, thereby avoiding fork collapse. Our finding is consistent with a recent study using fission yeast,56 and it establishes an anti-uncoupling function of ATR in human cells. ATRi-induced ssDNA formation at ongoing forks may occur in two phases. In the first phase, fork uncoupling drives the formation of ssDNA on one or two daughter strands. In the second phase, priming and repriming by POLα-primase and PrimPol leave ssDNA gaps on lagging and leading strands, respectively, providing entry points for MRE11 and EXO1 to degrade nascent DNA. Notably, while nascent DNA degradation assay relies on degradation of both nascent strands to detect shortening of replication tracts, template ssDNA exposure assay can detect ssDNA even when it is exposed on one daughter strand. This difference between the two assays may explain why PrimPol loss prevents shortening of replication tracts in nascent DNA degradation assay but does not affect template ssDNA exposure substantially. Using the nascent ssDNA exposure assay, we found that ATR prevents the exposure of nascent ssDNA by restricting the activities of MRN-CtIP and EXO1 at new forks (Figure 7A). While the endonuclease activity of MRE11 is required for nascent ssDNA exposure, it does not drive DSB formation. These results suggest that a structure generated by uncoupling of new forks may allow MRN-CtIP to nick template DNA without forming DSBs, providing an entry point for EXO1 to degrade template DNA and expose nascent ssDNA. In contrast to MRN-CtIP, MUS81 contributes to ATRi-induced DSB formation but not nascent ssDNA exposure in HU. It is possible that template DNA degradation cannot be initiated efficiently at MUS81-generated DSBs.
Figure 7. Model for distinct functions of ATR in the protection of replication forks.

(A) Models for distinct functions of ATR in protecting ongoing and new forks.
(B) Model for how ATRi exacerbates PARPi-induced ssDNA gaps in BRCA1/2-deficient cells.
(C) Model for how ATRi overcomes the restored gap protection in BRCA1-deficient, PARPi-resistant cells.
Our EM analysis clarifies the role of ATR in fork reversal. Upon ATR inhibition, the levels of reversed forks detected by EM are reduced, which is consistent with a previous study.23 However, our EM data suggest that ATR functions indirectly in the accumulation of reversed forks. ATR inhibition leads to degradation of nascent DNA from PrimPol-generated gaps, which reduces the nascent DNA that can form the reversed arm (Figure 7A). Consistent with this possibility, the budding yeast Exo1 processes stalled forks to counteract fork reversal in checkpoint defective cells.57 Interestingly, an increase of fork reversal was observed in yeast rad53 mutant cells, which are compromised for the Mec1/ATR pathway,58 suggesting that the regulation of fork reversal may be different between yeast and human cells in some ways. It should be noted that our data do not exclude the possibility that ATR directly regulates the activities of fork reversal factors.16 The impact of ATR on nascent DNA may outweigh its other effects and increase the overall levels of reversed forks.
Our results also reveal an intricate relationship between ATR and PrimPol-mediated repriming. On the one hand, ATR promotes the accumulation of reversed forks and may indirectly reduce the use of PrimPol at stressed forks. On the other hand, when PrimPol is active at stressed forks, ATR protects nascent DNA against degradation from ssDNA gaps. Through these functions, ATR suppresses the genomic instability arising from ssDNA gaps. A recent study reported that Chk1 promotes PrimPol activity by directly phosphorylating PrimPol,59 raising the possibility that ATR may promote PrimPol-mediated repriming and protect ssDNA gaps at the same time. Our results show that PrimPol still generates ssDNA gaps when cells were acutely treated with ATRi. It is possible that the function of PrimPol is only partially dependent on ATR and Chk1. It is also possible that the dephosphorylation of PrimPol occurs slowly, which limits the effects of acute ATR inhibition on PrimPol. Finally, ATR may affect repriming through factors other than PrimPol independently of Chk1, making the effects of ATR and Chk1 on repriming not identical. Although the role of ATR in repriming still requires further investigations, ATR clearly acts to protect the genome against the instability arising from ssDNA gaps.
BRCA1/2-deficient cells are unable to protect nascent DNA in HU, reflecting a defect in the protection of stalled forks.25,27 In HU, the degradation of nascent DNA in BRCA1/2-deficient cells is dependent on fork reversal.28–30 Based on the HU-induced nascent DNA degradation in BRCA1/2-deficient cells, defective fork protection has been linked to PARPi sensitivity.29,39,60,61–63 However, in contrast to HU, PARPi prevents the accumulation of reversed forks,64 raising a question as to whether the fork protection defects of BRCA1/2-deficient cells in HU are relevant in PARPi. We and others showed that the ssDNA gaps in BRCA1/2-deficient cells are critical for their PARPi sensitivity.29,35–37,52,65 In this study, we show that PARPi alone is sufficient to induce nascent DNA degradation and this effect is more pronounced in BRCA1/2-deficient cells (Figure 6E and S6D), consistent with the role of BRCA1/2 in protecting ssDNA gaps.36 Notably, ATRi enhances nascent DNA degradation in a ssDNA gap-dependent manner, and this effect is also more pronounced in BRCA1/2-deficient cells (Figures 6C and S6E). These results suggest that the function of ATR in protecting ssDNA gaps at ongoing forks is not only relevant in HU but also in PARPi, particularly in BRCA-deficient cells (Figure 7B).
When BRCA1-deficient cells acquire PARPi resistance, they regain the ability to protect forks in HU39 and protect ssDNA gaps in PARPi (Figure 6D), suggesting that the two protective activities are linked. It is possible that the loading of RAD51 to reversed forks in HU and to ssDNA gaps in PARPi protect both structures from nucleolytic degradation. Indeed, we detected the loading of RAD51 to HU-stalled forks and PARPi-induced ssDNA gaps by iPOND in previous studies,35,39 and HU-induced RAD51 focus formation requires ATR activity (Figure S2G). While loss of the protective function of RAD51 at ssDNA gaps does not induce DSBs right away, it promotes gap expansion and generates persistent gaps that are converted to DSBs in a cell cycle-dependent manner.35 An RNF168- and PALB2-mediated pathway was shown to promote RAD51 loading independently of BRCA1,66 providing a possible mechanism to restore fork/gap protection in BRCA1-deficient cells. Importantly, in the presence of PARPi, ATRi induces robust nascent DNA degradation in BRCA1-deficient, PARPi-resistant cells, showing an effective strategy to overcome PARPi resistance (Figure 7C). Thus, compared with HU-induced nascent DNA degradation from reversed forks, PARPi-induced nascent DNA degradation from ssDNA gaps is a more direct measurement of the functional status of fork/gap protection in PARPi-treated tumor cells. When applied to patient-derived tumor cells and organoids,61 this assay is likely useful for monitoring the fork/gap protecting activities against PARPi, allowing us to predict the PARPi response of tumors and assess therapeutic strategies to overcome PARPi resistance.
Limitations of the study
It is important to note that the assays used in this study analyze replication forks at different levels. While the nascent DNA degradation assay and EM analyze individual replication forks, template and nascent ssDNA exposure assays analyze fork populations in individual cells. The various DNA structures protected by ATR may be present at different positions of the same forks, at different subsets of forks, or even in distinct subpopulations of replicating cells. It should also be noted that each of these assays has limitations. For example, EM cannot detect DNA gaps that are too small or too far from fork junctions. DNA breaks too distal or too close to fork junctions are also undetectable by EM. The S1 nuclease does not distinguish ssDNA at fork junctions and internal gaps in daughter strands. It is important to consider these limitations when the assays are used in future studies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Inquiries about methods, reagents, or data should be directed to the lead contact, Lee Zou (lee.zou@duke.edu).
Materials availability
This study did not generate unique materials and reagents.
Data and code availability
All data reported in this paper will be available from the lead contact upon request.
This study does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
U2OS and HEK293T cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin (P/S), and 2 mM L-glutamine. The ovarian cancer cell line UWB1.249 and its derivatives, UWB1+B1, SYr12, and SYr13 were cultured in 1:1 Roswell Park Memorial Institute (RPMI) 1640-Mammary Epithelial Cell Growth Medium (MEGM) supplemented with 3% FBS and 1% P/S. UWB1+B1 cells were maintained with 200 μg/mL G-418 and SYr12 and SYr13 cells were cultured with 1 μM Olaparib. All cell lines were incubated at 37◦C with 5% CO2.
METHOD DETAILS
RNA interference
Cell transfection was carried out by reverse transfection with RNAiMAX and 4 nM silencer select siRNAs, unless stated otherwise. Experiments were initiated 48 h after transfection.
DNA fiber assay
DNA fiber assays were performed as described previously.35 In brief, cells were labeled for the indicated time with CldU (50 μM), washed twice with prewarmed media, and incubated in fresh warm media containing IdU (100 μM) for the indicated time. Cells were then incubated with HU (4 mM), HU and ATRi (10 μM VE-821, 1 μM AZ20, 5 μM AZD6738), HU, ATRi, and Mirin (50 μM), HU, ATRi, and PFM-01 (100 μM), or HU, ATRi, and CDC7i (5 μM XL-413, 0.1 μM PHA-767491) for 5 h. In conditions where cells were further incubated with S1 nuclease, cells were washed twice with prewarmed media and S1 nuclease treatment was carried out according to previously published protocols.22 Cells were washed with phosphate-buffered saline (PBS) and permeabilized with CSK100 buffer (100 mM MOPS at pH 7, 100 mM NaCl, 3 mM MgCl2, 300 mM sucrose, 0.5% Triton X-100) for 10 min at room temperature (RT). Cells were washed once with PBS and once with S1 nuclease buffer (30 mM sodium acetate at pH 4.6, 10 mM zinc acetate, 5% glycerol, 50 mM NaCl) before incubation for 30 min at 37◦C in 1.5 mL of S1 nuclease buffer + 20 U of S1 nuclease. S1 nuclease buffer was then replaced with PBS + 0.1% bovine serum albumin (BSA) and cells were collected using a cell lifter. Cell suspensions containing ~3000 cells (in 3 μL) were then dropped onto clean microscope slides for 2 min. Cells were then lysed with DNA fiber lysis buffer (7 μL; 200 mM Tris-HCl at pH 7.4, 0.5% SDS, 50 mM EDTA) and slides were incubated for 8 min at RT before tilting at a 15◦ angle to stretch the fibers. Slides were then fixed in 3:1 methanol/acetic acid solution for 5 min at −20◦C and allowed to dry overnight. DNA fibers were denatured in 2.5 M HCl for 1 h, washed five times with 1X PBS for 1 min and blocked for 30 min at 37◦C in PBS + 0.05% Tween 20 (PBS-T) containing 2% BSA in a humid chamber. Slides were then incubated with mouse anti-BrdU (BD Biosciences 347580; clone B44, 1:50) and rat anti-BrdU (BU1/75 (ICR1), Abcam ab6326, 1:100) antibodies in PBS-T + 2% BSA for 1 h at 37◦C in a humid chamber. Slides were washed three times for 5 min in PBS-T before incubation with 1:100 anti-mouse and anti-rat secondary antibodies conjugated to Alexa Fluor 488 and Alexa Fluor 594, respectively for 1 h at 37◦C in a humid chamber. Slides were further washed three times for 5 min in PBS-T and mounted with Prolong Gold. Images were captured using NIS element software with a Nikon i90 microscope and analyzed using Fiji software. Statistical analyses were performed in GraphPad Prism v9.
Immunofluorescence
Anti-BrdU
U2OS cells were cultured on #1.5 22x22-mm coverslips (Fisher Scientific 12–542-B) in 6-well plates. Cells were pre-extracted for 10 min at 4◦C in PBS + 0.5% Triton X-100 followed by fixation for 10 min at RT in 3% paraformaldehyde (PFA) and 2% sucrose. Cells were further fixed with 100% methanol for 10 min at −20◦C, allowed to dry for 1 min at RT, and then washed twice with PBS. Cells were then incubated with blocking solution consisting of PBS-T and 2% BSA for 30 min at RT. After, coverslips were incubated with mouse anti-BrdU (BD Biosciences 347580; clone B44, 1:250) and rabbit anti-PCNA (Abcam ab18197, 1:500) primary antibodies at 37◦C for 1 h. Coverslips were washed three times for 5 min in PBS-T and incubated for 1 h at RT with secondary antibodies (1:500) conjugated with Alexa Fluor 488 (BrdU) or Alexa Fluor 594 (PCNA). Coverslips were then washed three times with PBS-T and stained with DAPI (1 μg/mL) and mounted on microscope slides with ProLong Gold. 10 fields per coverslip were acquired using NIS elements software with a Nikon i90 microscope. Images were scored using MATLAB and statistical analyses were performed in GraphPad Prism v9.
Rad51
U2OS cells were seeded on 12 mm coverslips 24 h prior to treatment. Cells were treated with DMSO, HU (4 mM), or HU (4 mM) and ATRi AZD6738 (10 μM) (Selleck, S7693) for 5 h. Coverslips were incubated in 0.5% NP-40 in PBS for 2.5 min on ice and fixed with 2% PFA for 10 min at RT. Coverslips were then washed twice for 10 min in PBS, permeabilized with 0.5% Triton X-100 in PBS for 10 min, and blocked in 2% BSA, 2% horse serum, 0.25% for Triton X-100 in PBS overnight. Primary incubation was performed overnight in blocking solution using the anti-RAD51 antibody (Abcam; ab133534; 1:100). Coverslips were then washed three times using PBS containing 0.25% Tween 20. Secondary incubation was performed in PBS using Alexa 488 conjugated goat anti-rabbit antibody (ThermoFisher A11008; 1:500) for 1 h at room temperature. Coverslips were then washed three times in PBS containing 0.25% Tween 20, stained with DAPI, and mounted using Mowiol (Sigma-Aldrich 81381). Cells were imaged using the Nikon i90 microscope, quantified using FIJI, and statistical analyses were performed in GraphPad Prism v9.
Isolation of proteins on nascent DNA (iPOND)
iPOND was performed as previously described.35,67 Briefly, HEK293T cells were pulse-labeled with 10 μM EdU for 15 min. Fork samples were immediately fixed with 1% formaldehyde in PBS. To look at proteins behind the replication fork, cells were washed twice with prewarmed media and incubated in fresh media containing HU (4 mM) and VE-821 (10 μM) or an equal volume of DMSO prior to fixation. Formaldehyde was then quenched with 0.125 M glycine and cells were collected by scraping with a cell lifter. Cells were washed with PBS and permeabilized in PBS + 0.25% Triton X-100 for 30 min at RT. EdU was then labeled with biotin by click chemistry by resuspending cells in reaction buffer (1X PBS, 2 mM CuSO4, 10 mM sodium ascorbate, 1 μM biotin azide) and incubating for 2 h at RT. Cells were washed with PBS, resuspended in 1 mL lysis buffer (100 mM HEPES pH 8, 1% SDS), and sonicated with a 4710 series ultrasonic homogenizer (Cole-Parmer) at setting 3 three times for 30 s at 4◦C interspersed with 1 min incubations on ice. Lysates were spun at 13,000 rpm for 10 min at RT, and supernatant was collected and quantified using a Pierce BCA protein assay kit (Thermo Fisher Scientific 23227). Protein concentrations were normalized and diluted 1:1 in 100 mM HEPES (pH 8). To each sample, a prewashed streptavidin agarose bead slurry (100 μL; Millipore Sigma 69203–3) was added, and the bead-lysate mixtures were incubated with rotation overnight at 4◦C. Beads were then successively washed for 5 min on a rotating platform with lysis buffer, low-salt wash buffer (20 mM Tris-HCl at pH 8, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100), high-salt wash buffer (20 mM Tris-HCl at pH 8, 500 mM NaCl, 2 mM EDTA, 1% Triton X-100), 50 mM Tris-HCl pH 8, and 1% SDS. Beads were then resuspended in 2X sample buffer (100 mM Tris-HCl pH 6.8, 12% glycerol, 3.5% SDS, 0.2 M DTT), boiled for 30 min and processed for immunoblotting.
Immunoblots
Cells were resuspended and lysed in lysis buffer (100 mM Tris-HCl, pH 6.8, 1% SDS), sonicated for 10 s with a 4710 series ultrasonic homogenizer (Cole-Parmer), and boiled for 5 min. Protein concentrations were normalized using a Pierce BCA protein assay kit (Thermo Fisher Scientific 23227) and mixed 1:1 with 2X SDS-PAGE loading buffer (sample buffer, 100 mM Tris-HCl, pH 6.8, 12% glycerol, 3.5% SDS, 0.2 M DTT). Samples were boiled for 5 min, loaded on Bolt Bis-Tris Plus 4–12% gels, and run at 120 V for 70–90 min. Proteins were transferred onto PVDF membranes using a CBS Scientific electrophoretic blotting liquid transfer system (EBX-700) for 90 min at 110 V. Membranes were blocked in 5% milk (Boston Bioproducts, P-1400) in Tris-buffered saline with 0.05% Tween 20 (TBS-T) for 1 h at RT and then incubated overnight with primary antibodies at 4◦C with mild shaking. Primary antibodies were incubated in 5% milk in TBS-T at a concentration of 1:1000, except for EXO1 (1:500), GAPDH (1:500), H3 (1:20,000), RPA70 (1:2000) and PrimPol (1:50). Antibodies are listed in the reagent table. Membranes were washed three times with TBS-T for 10 min and incubated for 1 h at RT with secondary antibodies conjugated to horseradish peroxidase. Membranes were then washed three times with TBS-T for 10 min and developed using enhanced chemiluminescence (ECL Bio-Rad 1705061) substrate. Signals were detected using a Chemidoc imaging system (Bio-Rad) with ImageLab v6.0.1. software.
Cellular fractionation
Cells were trypsinized, washed with PBS, and incubated for 5 min on ice in hypotonic buffer (10 mM HEPES pH 7.9, 50 mM NaCl, 0.1 mM EDTA, 0.5 M sucrose, 1 mM DTT, 20 mM N-Ethylmaleimaide (NEM), 0.5% Triton X-100, 1X protease inhibitor cocktail (PIC, Millipore Sigma P8340)), followed by centrifugation at 1300 g at 4◦C. Supernatant (S1) was transferred into a new tube, centrifuged at 15,700 g for 15 min at 4◦C and transferred to a new tube with an equivalent volume of 2X sample buffer (100 mM Tris-HCl at pH 6.8, 12% glycerol, 3.5% SDS, 0.2 M DTT). The pellet was further washed with HS-ii-A hypotonic buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 20 mM NEM, 1X PIC) and resuspended in HS-ii-C hypertonic buffer (10 mM HEPES pH 7.9, 500 mM NaCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 20 mM NEM, 0.1% NP-40, 1X PIC) and incubated for 15 min at 4◦C. The pellet was centrifuged at 15,700 g for 5 min at 4◦C and the supernatant (S2) was transferred it a new tube with an equivalent volume of 2X sample buffer prior to combining with S1 (soluble fraction). The remaining pellet was resuspended in 2X sample buffer (nuclear fraction).
Cell viability assay
Cells were seeded in 96-well flat-bottom plates at a density of 2000 cells/well following 24 h of PrimPol siRNA knockdown. Stock solutions of each drug were prepared in sterile water or dimethyl sulfoxide (DMSO) as appropriate and further diluted in growth medium. Cells were allowed to grow for 6 days in drug containing medium (VE-821, SelleckChem S8007, Olaparib, SelleckChem S1060) and cell viability was measured with the CellTiter-Glo Luminescent Cell Viability Assay (Promega G7572) following manufacturer’s instructions. Luminescence was then measured with a PerkinElmer Envision 2103 multilabel plate reader. Viability was calculated as the luminescence signal ratio of treated versus untreated samples. Analysis and statistical test were performed using Microsoft Excel and GraphPad Prism v9.
Neutral comet assay
To visualize double-strand DNA breaks, U2OS cells were either treated with ATRi (VE-821, 10 μM) and HU (4 mM) for 5 h following siRNA knockdown of SLX4 or MUS81 or co-treated with CDC7i (XL-413, 5 μM) or PFM-01 (100 μM). Breaks were measured with CometAssay Single Cell Gel Electrophoresis Assay (R&D Systems 4250–050-K) following manufacturer’s instructions. At least 100 comet images from each condition were scored using OpenComet software.68
Electron microscopy (EM)
For EM analysis of replication intermediates, approximately 5 x 106 siControl or siPrimPol cells were collected immediately after treatment with either HU (4 mM), ATRi (VE-821, 10 μM) or Mirin (50 μM) for 2 h. DNA was cross-linked by incubating with 10 μg/mL 4,5′,8-trimethylpsoralen followed by a 3-min exposure to 366 nm UV light on a precooled metal block, for a total of three rounds. Cells were lysed and genomic DNA was isolated from the nuclei by proteinase K digestion and chloroform-isoamyl alcohol extraction. Genomic DNA was purified by isopropanol precipitation and digested with PvuII HF with the appropriate buffer for 4 h at 37◦C. Replication intermediates were enriched on a benzoylated naphthoylated DEAE-cellulose (Sigma-Aldrich) column. Samples were prepared for visualization by EM by spreading the purified, concentrated DNA on a carbon-coated grid in the presence of benzyl-dimethyl-alkylammonium chloride, followed by platinum rotary shadowing. Images were obtained on a JEOL JEM-1400 electron microscope using a bottom mounted AMT XR401 camera. Analysis was performed using ImageJ software (National Institute of Health). EM analysis allows distinguishing duplex DNA—which is expected to appear as a 10 nm thick fiber after the platinum/carbon coating step necessary for EM visualization—from ssDNA, which has a reduced thickness of 5–7 nm. Criteria used for the assignment of a three-way junction, indicative of a replication fork, include the joining of three DNA fibers into a single junction, with two symmetrical daughter strands and single parental strand. Reversed replication forks consist of four DNA fibers joined at a single junction, consisting of two symmetrical daughter strands, one parental strand and the addition of a typically shorter fourth strand, representative of the reversed arm. The length of the two daughter strands corresponding to the newly replicated duplex should be equal (b = c), whereas the length of the parental arm and the regressed arm can vary (a ≠ b= c ≠ d). Conversely, canonical Holliday junction structures will be characterized by arms of equal length (a = b, c = d). Particular attention is paid to the junction of the reversed replication fork to observe the presence of a bubble structure, indicating that the junction is opened and that it is simply not the result of the occasional crossover of two DNA molecules. These four-way junctions of reversed replication forks may also be collapsed and other indicators such as daughter strand symmetry, presence of single-stranded DNA at the junction or the entire structure itself, all are considered during analysis. The frequency of reversed forks in a sample is computed using the GraphPad Prism v9 software.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism v9 was used for data analysis and statistical significance was calculated using Mann-Whitney Ranked Sum Test, one-way ANOVA with Tukey’s multiple comparisons test, or Welch’s t test. Statistically significant differences are indicated in figures. In all cases, ns: not significant (p > 0.05), *: p < 0.05, **: p < 0.01; ***: p < 0.001 and ****: p < 0.0001. Error bars in figures indicate the standard error of the mean (SEM) for the number of replicates (n) across 2–3 biological replicates.ll
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| α-Tubulin | Sigma-Aldrich | Cat#:T5168; RRID:AB_477579 |
| anti-Mouse IgG Alexa Fluor 488 | Jackson ImmunoResearch | Cat#:715–545-151; RRID:AB_2341099 |
| anti-Rabbit IgG Alexa Fluor 488 | Thermo Fisher Scientific | Cat#:A11008; RRID:AB_143165 |
| anti-Rabbit IgG Alexa Fluor 594 | Thermo Fisher Scientific | Cat#:A21207; RRID:AB_141637 |
| anti-Rat IgG Alexa Fluor 594 | Thermo Fisher Scientific | Cat#:A11007; RRID:AB_10561522 |
| BRCA2 | Millipore | Cat#:OP95; RRID:AB_2067762 |
| BrdU [BU1/75 (ICR1)] | Abcam | Cat#:ab6326; RRID:AB_305426 |
| BrdU (Clone B44) | BD Biosciences | Cat#:BD347580 |
| CtIP | Cell Signaling Technologies | Cat#: 9201; RRID:AB_10828593 |
| DNA2 | Abcam | Cat#: ab96488; RRID:AB_10677769 |
| EXO1 | Abcam | Cat#: ab95068; RRID:AB_10675762 |
| GAPDH | Santa Cruz Biotechnology | Cat#:sc-32233; RRID:AB_627679 |
| H3 | Abcam | Cat#:ab1791; RRID:AB_302613 |
| HLTF | Santa Cruz Biotechnology | Cat#:sc-398357 |
| Ku80 | NeoMarker/Thermo Fisher Scientific | Cat#:MS-285-P1 |
| MRE11 (12D7) | Genetex | Cat#:GTX70212; RRID:AB_372398 |
| MUS81 | Abcam | Cat#:ab14387; RRID:AB_301167 |
| PCNA | Abcam | Cat#:ab18197; RRID:AB_444313 |
| PrimPol | Kindly provided by the Mendez laboratory | N/A |
| RAD51 | Abcam | Cat#:ab133534; RRID:AB_2722613 |
| RPA70 | Bethyl | Cat#:A300–241A; RRID:AB_2180681 |
| SLX4 (BTBD12) | Bethyl | Cat#:A302–270A; RRID:AB_1850156 |
| SMARCAL1 (A2) | Santa Cruz Biotechnology | Cat#:sc-376377; RRID:AB_10987841 |
| TIMELESS | Bethyl | Cat#:A300–961A; RRID:AB_805855 |
| TIPIN | Bethyl | Cat#:A301–474A; RRID:AB_999573 |
| ZRANB3 | Bethyl | Cat#:A303–033A; RRID:AB_10773114 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| AZ20 | SelleckChem | Cat#:S7050 |
| AZD6738 | SelleckChem | Cat#:S7693 |
| Benzoylated Naphthoylated DEAE-Cellulose | Sigma-Aldrich | Cat#:B6385 |
| Biotin Azide | Click Chemistry Tools | Cat#:1265 |
| BrdU | Sigma-Aldrich | Cat#:B5002 |
| CD437 | Sigma-Aldrich | Cat#:C5865 |
| CldU | Sigma-Aldrich | Cat#:I7125 |
| DAPI | Invitrogen | Cat#:D1306 |
| EdU | Click Chemistry Tools | Cat#:1149–25 |
| Hydroxyurea | Sigma-Aldrich | Cat#:H8627 |
| IdU | Sigma-Aldrich | Cat#:C6891 |
| Lipofectamine RNAiMAX | Invitrogen | Cat#:13778150 |
| Mirin | SelleckChem | Cat#:S8096 |
| Mowiol | Sigma-Aldrich | Cat#81381 |
| MK-8776 | SelleckChem | Cat#:S2735 |
| Olaparib | SelleckChem | Cat#:S1060 |
| PFM-01 | SelleckChem | Cat#:S3549 |
| PHA-767491 | SelleckChem | Cat#:S2742 |
| ProLong Gold Antifade Mountant | Invitrogen | Cat#:P36930 |
| PvuII HF | New England Biolabs | Cat#: R3151L |
| S1 nuclease | Invitrogen | Cat#:18001016 |
| Streptavidin Agarose | Millipore Sigma | Cat#:69203–3 |
| SYBR Gold Nucleic Acid Gel Stain | Invitrogen | Cat#:S11494 |
| VE-821 | SelleckChem | Cat#:S8007 |
| XL-413 | SelleckChem | Cat#:S7547 |
|
| ||
| Critical commercial assays | ||
|
| ||
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat#:G7571 |
| CometAssay Single Cell Gel Electrophoresis | R&D Systems | Cat#:4250–050K |
|
| ||
| Deposited data | ||
|
| ||
| Raw data files | This paper | Mendeley doi: https://doi.org/10.17632/6zfz2k2mcr.1 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| U2OS | ATCC | N/A |
| UWB1.249 | ATCC | N/A |
| UWB1+B1 | ATCC | N/A |
| SYr12 | Yazinski et al.39 | N/A |
| SYr13 | Yazinski et al.39 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S1 for siRNAs | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism 9 | GraphPad Software, Inc. | https://www.graphpad.com/scientific-software/prism/ |
| Image Lab | Bio-Rad | https://www.bio-rad.com/en-us/category/chemidoc-imaging-systems |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| MATLAB | Mathworks | https://www.mathworks.com/products/matlab.html |
| OpenComet plugin for ImageJ | Graduate School for Integrative Sciences and Engineering, National University of Singapore and Laboratory of Systems | https://cometbio.org/index.html |
| NIS element viewer | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements-advance-research |
Highlights.
ATR prevents DNA degradation from gaps; promotes reversal at stressed ongoing forks
ATR prevents uncoupling of ongoing forks under stress
ATR prevents MRN-CtIP-initiated template DNA degradation at stressed new forks
ATRi exacerbates nascent DNA degradation from PARPi-induced gaps in BRCA-deficient cells
ACKNOWLEDGMENTS
We thank members of the Vindigni and Zou labs for discussions. A.S. was the recipient of postdoctoral fellowships from the Canadian Institutes of Health Research and Fonds de Recherche du Québec - Santé. L.Z. was the James & Patricia Poitras Endowed Chair in Cancer Research. This work is supported by grants from the NIH (CA248526 to A.V. and L.Z., CA263934 and CA240243 to L.Z., CA237263 to A.V.), the Gray Foundation (to L.Z.), the US Department of Defense (DOD) Breast Cancer Research Program (BRCP) Expansion Award (BC191374 to A.V.), the Alvin J. Siteman Cancer Center Siteman Investment Program (supported by The Foundation for Barnes-Jewish Hospital, Cancer Frontier Fund to A.V.), and the Barnard Foundation (to A.V.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112792.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Tubbs A, and Nussenzweig A. (2017). Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 168, 644–656. 10.1016/j.cell.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saxena S, and Zou L. (2022). Hallmarks of DNA replication stress. Mol. Cell 82, 2298–2314. 10.1016/j.molcel.2022.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeman MK, and Cimprich KA (2014). Causes and consequences of replication stress. Nat. Cell Biol 16, 2–9. 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saldivar JC, Cortez D, and Cimprich KA (2017). The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol 18, 622–636. 10.1038/nrm.2017.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blackford AN, and Jackson SP (2017). ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 66, 801–817. 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 6.Simoneau A, and Zou L. (2021). An extending ATR–CHK1 circuitry: the replication stress response and beyond. Curr. Opin. Genet. Dev 71, 92–98. 10.1016/j.gde.2021.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Zou L, Liu D, and Elledge SJ (2003). Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc. Natl. Acad. Sci. USA 100, 13827–13832. 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumagai A, Lee J, Yoo HY, and Dunphy WG (2006). TopBP1 activates the ATR-ATRIP complex. Cell 124, 943–955. 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 9.Lee J, Kumagai A, and Dunphy WG (2007). The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J. Biol. Chem 282, 28036–28044. 10.1074/jbc.M704635200. [DOI] [PubMed] [Google Scholar]
- 10.Bass TE, Luzwick JW, Kavanaugh G, Carroll C, Dungrawala H, Glick GG, Feldkamp MD, Putney R, Chazin WJ, and Cortez D. (2016). ETAA1 acts at stalled replication forks to maintain genome integrity. Nat. Cell Biol 18, 1185–1195. 10.1038/ncb3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haahr P, Hoffmann S, Tollenaere MAX, Ho T, Toledo LI, Mann M, Bekker-Jensen S, Rä schle M, and Mailand N. (2016). Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol 18, 1196–1207. 10.1038/ncb3422. [DOI] [PubMed] [Google Scholar]
- 12.Lee YC, Zhou Q, Chen J, and Yuan J. (2016). RPA-Binding Protein ETAA1 Is an ATR Activator Involved in DNA Replication Stress Response. Curr. Biol 26, 3257–3268. 10.1016/j.cub.2016.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, and Lukas J. (2013). ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103. 10.1016/j.cell.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 14.Buisson R, Boisvert JL, Benes CH, and Zou L. (2015). Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol. Cell 59, 1011–1024. 10.1016/j.molcel.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forment JV, Blasius M, Guerini I, and Jackson SP (2011). Structure-specific DNA endonuclease mus81/eme1 generates DNA damage caused by chk1 inactivation. PLoS One 6, e23517. 10.1371/journal.pone.0023517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Bétous R, Carroll CM, Jung SY, Qin J, Cimprich KA, and Cortez D. (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 27, 1610–1623. 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ragland RL, Patel S, Rivard RS, Smith K, Peters AA, Bielinsky AK, and Brown EJ (2013). RNF4 and PLK1 are required for replication fork collapse in ATR-deficient cells. Genes Dev. 27, 2259–2273. 10.1101/gad.223180.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Segurado M, and Diffley JFX (2008). Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev. 22, 1816–1827. 10.1101/gad.477208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li S, Lavagnino Z, Lemacon D, Kong L, Ustione A, Ng X, Zhang Y, Wang Y, Zheng B, Piwnica-Worms H, et al. (2019). Ca2+-Stimulated AMPK-Dependent Phosphorylation of Exo1 Protects Stressed Replication Forks from Aberrant Resection. Mol. Cell 74, 1123–1137.e6. 10.1016/j.molcel.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trenz K, Smith E, Smith S, and Costanzo V. (2006). ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 25, 1764–1774. 10.1038/sj.emboj.7601045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neelsen KJ, and Lopes M. (2015). Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol 16, 207–220. NIHMS1920513-supplement-1.pdf. [DOI] [PubMed] [Google Scholar]
- 22.Quinet A, Carvajal-Maldonado D, Lemacon D, and Vindigni A. (2017). DNA Fiber Analysis: Mind the Gap, 1st ed (Elsevier Inc.). 10.1016/bs.mie.2017.03.019. [DOI] [PubMed] [Google Scholar]
- 23.Mutreja K, Krietsch J, Hess J, Ursich S, Berti M, Roessler FK, Zellweger R, Patra M, Gasser G, and Lopes M. (2018). ATR-Mediated Global Fork Slowing and Reversal Assist Fork Traverse and Prevent Chromosomal Breakage at DNA Interstrand Cross-Links. Cell Rep. 24, 2629–2642.e5. 10.1016/j.celrep.2018.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, and Cortez D. (2015). The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 59, 998–1010. 10.1016/j.molcel.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M. (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rainey MD, Bennett D, O’Dea R, Zanchetta ME, Voisin M, Seoighe C, and Santocanale C. (2020). ATR Restrains DNA Synthesis and Mitotic Catastrophe in Response to CDC7 Inhibition. Cell Rep. 32, 108096. 10.1016/j.celrep.2020.108096. [DOI] [PubMed] [Google Scholar]
- 27.Schlacher K, Wu H, and Jasin M. (2012). A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116. 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemaçon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, and Vindigni A. (2017). MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun 8, 860. 10.1038/s41467-017-01180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R, et al. (2017). Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 68, 414–430.e8. 10.1016/j.molcel.2017.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, and Lopes M. (2017). Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun 8, 859–911. 10.1038/s41467-017-01164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Técher H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017). Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 67, 867–881.e7. 10.1016/j.molcel.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quinet A, Tirman S, Jackson J, Šviković S, Lemaçon D, Carvajal-Maldonado D, González-Acosta D, Vessoni AT, Cybulla E, Wood M, et al. (2020). PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol. Cell 77, 461–474.e9. 10.1016/j.molcel.2019.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piberger AL, Bowry A, Kelly RDW, Walker AK, González-Acosta D, Bailey LJ, Doherty AJ, Méndez J, Morris JR, Bryant HE, and Petermann E. (2020). PrimPol-dependent single-stranded gap formation mediates homologous recombination at bulky DNA adducts. Nat. Commun 11, 5863–5914. 10.1038/s41467-020-19570-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Genois MM, Gagné JP, Yasuhara T, Jackson J, Saxena S, Langelier MF, Ahel I, Bedford MT, Pascal JM, Vindigni A, et al. (2021). CARM1 regulates replication fork speed and stress response by stimulating PARP1. Mol. Cell 81, 784–800.e8. 10.1016/j.molcel.2020.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simoneau A, Xiong R, and Zou L. (2021). The trans cell cycle effects of PARP inhibitors underlie their selectivity toward BRCA1/2-deficient cells. Genes Dev. 35, 1271–1289. 10.1101/GAD.348479.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tirman S, Quinet A, Wood M, Meroni A, Cybulla E, Jackson J, Pegoraro S, Simoneau A, Zou L, and Vindigni A. (2021). Temporally distinct post-replicative repair mechanisms fill PRIMPOL-dependent ssDNA gaps in human cells. Mol. Cell 81, 4026–4040.e8. 10.1016/j.molcel.2021.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang Z, Fu P, Alcivar AL, Fu H, Redon C, Foo TK, Zuo Y, Ye C, Baxley R, Madireddy A, et al. (2021). BRCA2 associates with MCM10 to suppress PRIMPOL-mediated repriming and single-stranded gap formation after DNA damage. Nat. Commun 12, 5966. 10.1038/s41467-021-26227-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hashimoto Y, Ray Chaudhuri A, Lopes M, and Costanzo V. (2010). Rad51 protects nascent DNA from Mre11 dependent degradation and promotes continuous DNA synthesis Yoshitami. Nat. Struct. Mol. Biol 17, 1305–1311. 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, Todorova Kwan T, Morris R, Lauffer S, Nussenzweig A, et al. (2017). ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 31, 318–332. 10.1101/gad.290957.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sørensen CS, Hansen LT, Dziegielewski J, Syljuåsen RG, Lundin C, Bartek J, and Helleday T. (2005). The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol 7, 195–201. 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 41.Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M. (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol 208, 563–579. 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mann A, Ramirez-Otero MA, De Antoni A, Hanthi YW, Sannino V, Baldi G, Falbo L, Schrempf A, Bernardo S, Loizou J, and Costanzo V. (2022). POLq prevents MRE11-NBS1-CtIP-dependent fork breakage in the absence of BRCA2/RAD51 by filling lagging-strand gaps. Mol. Cell 82, 4218–4231.e8. 10.1016/j.molcel.2022.09.013. [DOI] [PubMed] [Google Scholar]
- 43.Byun TS, Pacek M, Yee MC, Walter JC, and Cimprich KA (2005). Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 19, 1040–1052. 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chou DM, and Elledge SJ (2006). Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc. Natl. Acad. Sci. USA 103, 18143–18147. 10.1073/pnas.0609251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nedelcheva MN, Roguev A, Dolapchiev LB, Shevchenko A, Taskov HB, Shevchenko A, Stewart AF, and Stoynov SS (2005). Uncoupling of unwinding from DNA synthesis implies regulation of MCM helicase by Tof1/Mrc1/Csm3 checkpoint complex. J. Mol. Biol 347, 509–521. 10.1016/j.jmb.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 46.Katou Y, Kanoh Y, Bando M, and Noguchi H. (2003). S-phase checkpoint proteins replication pausing complex424, pp. 1–6. [DOI] [PubMed] [Google Scholar]
- 47.Numata Y, Ishihara S, Hasegawa N, Nozaki N, and Ishimi Y. (2010). Interaction of human MCM2–7 proteins with TIM, TIPIN and Rb. J. Biochem 147, 917–927. 10.1093/jb/mvq028. [DOI] [PubMed] [Google Scholar]
- 48.Wyatt HDM, Sarbajna S, Matos J, and West SC (2013). Coordinated actions of SLX1-SLX4 and MUS81-EME1 for holliday junction resolution in human cells. Mol. Cell 52, 234–247. 10.1016/j.molcel.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 49.Anand R, Ranjha L, Cannavo E, and Cejka P. (2016). Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 64, 940–950. 10.1016/j.molcel.2016.10.017. [DOI] [PubMed] [Google Scholar]
- 50.Hanzlikova H, Kalasova I, Demin AA, Pennicott LE, Cihlarova Z, and Caldecott KW (2018). The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 71, 319–331.e3. 10.1016/j.molcel.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vaitsiankova A, Burdova K, Sobol M, Gautam A, Benada O, Hanzlikova H, and Caldecott KW (2022). PARP inhibition impedes the maturation of nascent DNA strands during DNA replication. Nat. Struct. Mol. Biol 29, 329–338. 10.1038/s41594-022-00747-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cong K, Peng M, Kousholt AN, Lee WTC, Lee S, Nayak S, Krais J, VanderVere-Carozza PS, Pawelczak KS, Calvo J, et al. (2021). Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol. Cell 81, 3128–3144.e7. 10.1016/j.molcel.2021.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Segurado M, Gómez M, and Antequera F. (2002). Increased Recombination Intermediates and Homologous Integration Hot Spots at DNA Replication Origins. Mol. Cell 10, 907–916. [DOI] [PubMed] [Google Scholar]
- 54.Raveendranathan M, Chattopadhyay S, Bolon YT, Haworth J, Clarke DJ, and Bielinsky AK (2006). Genome-wide replication profiles of S-phase checkpoint mutants reveal fragile sites in yeast. EMBO J. 25, 3627–3639. 10.1038/sj.emboj.7601251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tubbs A, Sridharan S, van Wietmarschen N, Maman Y, Callen E, Stanlie A, Wu W, Wu X, Day A, Wong N, et al. (2018). Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse. Cell 174, 1127–1142.e19. 10.1016/j.cell.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, Wang L, Xu X, Yuan Y, Zhang B, Li Z, Xie Y, Yan R, Zheng Z, Ji J, et al. (2021). The intra-S phase checkpoint directly regulates replication elongation to preserve the integrity of stalled replisomes. Proc. Natl. Acad. Sci. USA 118, 20191831188–e2019183211. 10.1073/pnas.2019183118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cotta-Ramusino C, Fachinetti D, Lucca C, Doksani Y, Lopes M, Sogo J, and Foiani M. (2005). Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol. Cell 17, 153–159. 10.1016/j.molcel.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 58.Sogo JM, Lopes M, and Foiani M. (2002). Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297, 599–602. 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- 59.Mehta KPM, Thada V, Zhao R, Krishnamoorthy A, Leser M, Lindsey Rose K, and Cortez D. (2022). CHK1 phosphorylates PRIMPOL to promote replication stress tolerance. Sci. Adv 8, eabm0314. 10.1126/sciadv.abm0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387. 10.1038/nature18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hill SJ, Decker B, Roberts EA, Horowitz NS, Muto MG, Worley MJ, Feltmate CM, Nucci MR, Swisher EM, Nguyen H, et al. (2018). Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discov. 8, 1404–1421. 10.1158/2159-8290.CD-18-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lim KS, Li H, Roberts EA, Gaudiano EF, Clairmont C, Sambel LA, Ponnienselvan K, Liu JC, Yang C, Kozono D, et al. (2018). USP1 Is Required for Replication Fork Protection in BRCA1-Deficient Tumors. Mol. Cell 72, 925–941.e4. 10.1016/j.molcel.2018.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parmar K, Kochupurakkal BS, Lazaro JB, Wang ZC, Palakurthi S, Kirschmeier PT, Yang C, Sambel LA, Färkkilä A, Reznichenko E, et al. (2019). The CHK1 inhibitor prexasertib exhibits monotherapy activity in high-grade serous ovarian cancer models and sensitizes to PARP inhibition. Clin. Cancer Res 25, 6127–6140. 10.1158/1078-0432.CCR-19-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, et al. (2013). Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol 20, 347–354. 10.1038/nsmb.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paes Dias M, Tripathi V, van der Heijden I, Cong K, Manolika EM, Bhin J, Gogola E, Galanos P, Annunziato S, Lieftink C, et al. (2021). Loss of nuclear DNA ligase III reverts PARP inhibitor resistance in BRCA1/53BP1 double-deficient cells by exposing ssDNA gaps. Mol. Cell 81, 4692–4708.e9. 10.1016/j.molcel.2021.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zong D, Adam S, Wang Y, Sasanuma H, Callén E, Murga M, Day A, Kruhlak MJ, Wong N, Munro M, et al. (2019). BRCA1 Haploinsufficiency Is Masked by RNF168-Mediated Chromatin Ubiquitylation. Mol. Cell 73, 1267–1281.e7. 10.1016/j.molcel.2018.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, and Cortez D. (2011). Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 25, 1320–1327. 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, and Clement MV (2014). OpenComet: An automated tool for comet assay image analysis. Redox Biol. 2, 457–465. 10.1016/j.redox.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be available from the lead contact upon request.
This study does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.