

SUMMARY
The bone marrow microenvironment (BME) drives drug resistance in acute lymphoblastic leukemia (ALL) through leukemic cell interactions with bone marrow (BM) niches, but the underlying mechanisms remain unclear. Here, we show that the interaction between ALL and mesenchymal stem cells (MSCs) through integrin β1 induces an epithelial-mesenchymal transition (EMT)-like program in MSC-adherent ALL cells, resulting in drug resistance and enhanced survival. Moreover, single-cell RNA sequencing analysis of ALL-MSC co-culture identifies a hybrid cluster of MSC-adherent ALL cells expressing both B-ALL and MSC signature genes, orchestrated by a WNT/β-catenin-mediated EMT-like program. Blockade of interaction between β-catenin and CREB binding protein impairs the survival and drug resistance of MSC-adherent ALL cells in vitro and results in a reduction in leukemic burden in vivo. Targeting of this WNT/β-catenin-mediated EMT-like program is a potential therapeutic approach to overcome cell extrinsically acquired drug resistance in ALL.
Graphical Abstract
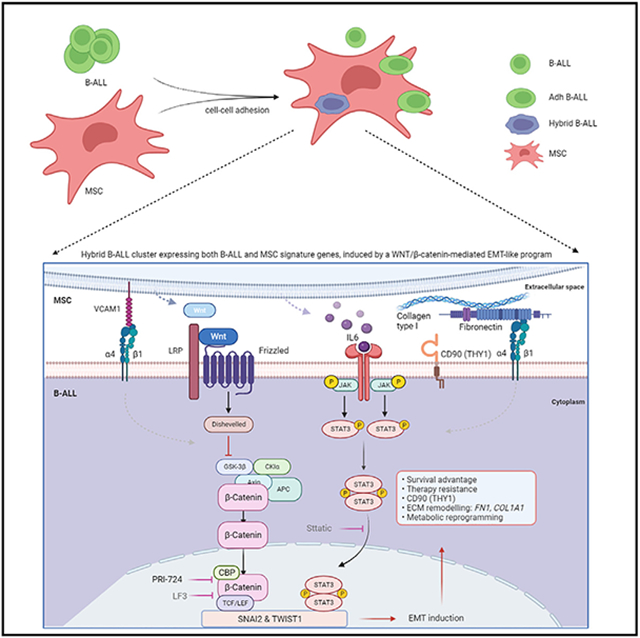
In brief
Acute lymphoblastic leukemia (ALL) cell interaction with bone marrow stroma results in drug resistance. Park et al. show that ALL interaction with mesenchymal stromal cells is mediated by integrin beta 1 and drives an EMT phenotype that activates WNT/β-catenin signaling that may be targeted to ameliorate drug resistance.
INTRODUCTION
Acute lymphoblastic leukemia (ALL) comprises multiple subtypes in which a variety of genetic variations have biological and clinical relevance.1 B-ALL comprises 23 subtypes defined by genomic alterations that perturb various cellular processes, several of which drive not only leukemogenesis but also cause therapy failure and relapse.1-3 Despite advances in ALL treatment, patients with relapsed or refractory ALL still have poor outcomes partly due to cell-intrinsic genetic changes. In addition, prior data suggest that cell-extrinsic cues from the bone marrow microenvironment (BME) contribute to ALL cell survival and chemoresistance.4,5 Single-cell RNA sequencing (scRNA-seq) of matched diagnosis and relapsed B-ALL bone marrow (BM) samples has identified remodeling of the myeloid compartment upon disease initiation and subsequent relapse,6 but it has not directly interrogated crosstalk between B-ALL cells and non-leukemic cells.
The BME is a complex milieu composed of stromal cells, immune cells, adipocytes, and neural cells7,8 that contains niches that modulate leukemia initiation and progression.9-13 The BME mesenchymal stem cells (MSCs) and vascular niche cells promote ALL cell survival, quiescence, and therapeutic resistance via soluble factors and adhesion-dependent signaling.8,12,14-22 Conversely, leukemic cells reprogram the BME through crosstalk with stromal cells, creating a favorable environment for leukemic cell survival.23
We previously showed that cell-intrinsic and -extrinsic pathways intersect to promote resistance in B-ALL. IKZF1 alterations in high-risk B-ALL result in derepression of stemness and adhesion programs, resulting in BM niche mislocalization, aberrant MSC adherence, and drug resistance.19 Limited RNA-seq studies of B-ALL cells revealed chemotherapy-refractory minimal residual disease-like transcriptional signatures in cells adherent to human osteoblast or BM stromal cells.15 Additionally, human induced pluripotent stem cell-derived MSCs can protect leukemic cells, both dormant and cycling, against dexamethasone.24 Thus, ALL-MSC interactions may promote the survival of ALL cells regardless of genetic subtype, but the molecular mechanisms underlying MSC-induced drug resistance have yet to be elucidated.
Here, we used an ex vivo co-culture model to culture primary B-ALL patient samples and cell lines with primary MSCs. Through CRISPR-Cas9 genomic perturbation screens and RNA-seq, we discovered that ALL-MSC interaction induces EMT in ALL cells by binding to MSCs through integrin β1. Additionally, scRNA-seq analyses identified a hybrid cluster of MSC-adherent ALL cells displaying both ALL and MSC gene signatures, induced by EMT activation, with increased β-catenin-mediated transcriptional activity. Inhibiting β-catenin-CBP interaction reduced the survival of patient-derived xenograft (PDX) cells co-cultured with MSCs ex vivo and decreased the leukemic burden in vivo. These findings highlight the role of ALL-MSC interaction in promoting EMT in ALL cells through WNT/β-catenin signaling, leading to targetable drug resistance.
RESULTS
Dynamic change of cell adhesion and extracellular matrix genes in adherent ALL cells
MSCs are known to contribute to therapy resistance in ALL through direct interactions, the secretion of soluble factors, and formation of tunneling nanotubes.7,14,25,26 To investigate MSC-mediated mechanisms of enhanced survival and drug resistance in ALL, we used an ex vivo co-culture model of B-ALL cells with BM-derived stromal/stem cells. First, we utilized human telomerase reverse transcriptase-immortalized BM MSCs (hTERT-MSCs)27 co-cultured with five B-ALL cell lines of differing genotypes (Figure 1A). ALL cells could be divided into three groups: (1) ALL cells cultured with MSCs that remained nonadherent (Nonadh ALL), (2) ALL cells adherent to MSCs (Adh ALL), and (3) ALL cells cultured without MSCs (ALL only). Nonadh cells in suspension were collected from the co-culture media of ALL with stroma, followed by collection of Adh ALL cells by digestion and cell sorting to >99% purity to avoid hTERT-MSC contamination. Importantly, all ALL lines tested displayed substantial adhesion ability regardless of the type of culture medium, although the REH (ETV6::RUNX1) cell line showed less adherence in RPMI than in α-MEM (Figure S1A). Principal component analysis (PCA) of RNA-seq data showed that Adh ALL cells were distinct from both Nonadh ALL and ALL-only cells, which were closely grouped irrespective of leukemia subtype (Figure 1B). There were 2,138 differentially expressed genes (DEGs) between Adh vs. Nonadh ALL cells (2,107 upregulated and 31 downregulated; log2(Adh/Nonadh) > 1; FDR < 0.05; Table S1), indicating that ALL-MSC crosstalk primarily augmented ALL gene expression in a subtype-independent manner.
Figure 1. Enrichment of genes encoding cell adhesion molecules and extracellular matrix proteins in adherent ALL cells.

(A) Schema of ex vivo co-culture of ALL with MSCs. Five B-ALL cell lines and 11 B-ALL patient samples were co-cultured with hTERT-MSCs and primary MSCs, respectively. The RNA isolated from ALL-only, Adh ALL, and Nonadh ALL cells was subjected to RNA-seq.
(B) PCA plot for five ALL cell lines.
(C) PCA plot for 11 primary B-ALLs.
(D) Common DEGs in both cell lines and primary patient samples.
(E) Pathway analysis of the upregulated DEGs in Adh ALL was performed by using the Reactome database. NES, normalized enrichment score. Red color, ECM-associated pathways. Blue color, integrin-associated pathway.
(F) Enrichment plots of integrin cell surface and reactome extracellular matrix organization pathways. FDR, false discovery rate.
(G) Enriched integrin family genes in 11 patient Adh ALL samples compared to Nonadh ALL samples (false discovery rate [FDR] < 0.05). Dots, individual value. Bar, mean with SD.
(H) Enriched ECM genes in 11 patient Adh ALL samples compared to Nonadh ALL samples (FDR < 0.05). Dots, individual value. Bar, mean with SD. Statistical testing was performed using two-tailed Student’s t test, ****p < 0.0001, ***p < 0.0005, **p < 0.005, *p < 0.05. See also Figure S1.
hTERT-MSCs have limitations, including lack of CD90 (THY1)28-31 expression, which may bias ALL-MSC interactions. Accordingly, we generated primary MSCs from BM aspirates,32 which fulfilled criteria for MSCs,33 including adherence, expression of MSC cell surface markers, and the ability to differentiate into osteoblasts, adipocytes, and chondroblasts in vitro (Figures S1B-S1D); hTERT-MSCs lacked expression of CD90 (Figure S1E).
These cells were co-cultured with fresh, flow-sorted primary samples obtained at diagnosis from 11 patients with various subtypes of B-ALL (Table S2). In contrast to leukemic cell lines, primary leukemic cells did not grow ex vivo without MSCs (Figure S1F) but proliferated over a 4-day period in the presence of MSCs. On gene expression analysis, the 11 Adh ALL samples were grouped together distinct from Nonadh ALL samples (Figure 1C), supporting the notion of subtype-independent deregulation of gene expression driven by ALL-MSC interaction in leukemic cells regardless of the presence of subtype-defining genomic drivers. Differential gene expression analysis identified 1,329 upregulated and 10 downregulated genes in Adh versus Nonadh samples (Table S2), with 879 genes being upregulated in both patient sample and cell lines (Figure 1D). Integrin cell surface interaction and extracellular matrix (ECM) organization pathways were significantly enriched34 in Adh cells (Figures 1E and 1F), with upregulation of genes encoding collagen, fibronectin, and laminin and their corresponding ligands including integrin α11, αv, α3, β5, β3, and β1 (Figures 1G and 1H).
Multiple mechanisms of interaction between ALL and MSCs
Cytokine array analysis using cytokine-directed antibodies (Table S3) and the NALM6 (IGH::DUX4) cell line, which is derived from ALL in relapse,35 revealed that 24 cytokines including IGFBP2, VEGF, IL6, and CXCL12 were enriched in MSC-ALL co-culture media compared to NALM6 mono-culture; conversely, no cytokines were enriched in co-culture media compared to MSC mono-culture, indicating that enriched cytokines in co-culture were secreted by MSCs (Figures 2A-2C).
Figure 2. Multiple interactions between B-ALL cells and primary MSCs.

(A) Identification of enriched cytokines in the co-culture media.
(B) All enriched cytokines in the co-culture media compared to the NALM6 media (fold change [FC] > 2.0).
(C) Top 10 increased cytokines in the co-culture media. Representative data are from one of two experiments.
(D) Scheme of TMT-MS analysis of proteins in the media of NALM6, MSCs, and co-culture of NALM6 and MSCs.
(E) Enriched proteins in the co-culture media compared to NALM6 media (FC > 1.5).
(F) Dot plots displaying the gating strategy for flow cytometry experiments on TNT-mediated transfer of cellular vesicles. The transfer of the lipophilic membrane stain Dil from labeled donor cells to unlabeled recipient cells was analyzed by flow cytometry.
(G) Cumulative efficiency of cellular vesicle trafficking between B-ALL and MSCs (n = 3). Data, mean ± SD. Statistical testing was performed using two-tailed Student’s t test; ****p < 0.0001.
Proteome profiling of media from both mono-culture and co-culture conditions using tandem mass tag mass spectrometry (TMT-MS) (Figure 2D) identified that 31 proteins, including DCN, CASP8, and POSTN (periostin), were enriched in the co-culture media compared to NALM6 mono-culture media (Figure 2E, Table S4). Notably, POSTN derived from MSCs is known to promote B-ALL proliferation by activating the integrin-ILK-NF-κB-CCL2 pathway.17 No proteins were enriched by more than 1.5-fold in co-culture media compared to MSC mono-culture (Figure 2E, Table S4) showing that soluble mediators driving ALL-MSC interactions are MSC derived.
Tunneling nanotubes (TNTs), long membrane nanotubes that connect distant cells, have been reported to mediate B-ALL-stroma interaction and drug resistance.26 Thus, we evaluated TNT-mediated communication between NALM6 and MSCs and observed bidirectional TNT-mediated B-ALL-MSC interaction (Figures 2F and 2G), in contrast to prior reports of predominantly ALL to hTERT-MSC transfer. Previous studies indicated that TNT signaling between B-ALL and hTERT-MSCs induced prednisolone resistance.26 Therefore, we did not test the impact of TNT-mediated B-ALL-MSC interaction on chemoresistance.
Integrin beta 1 mediates ALL cell adhesion to MSCs
The adhesome is a network comprised of interactions between cellular receptors and components, and TGFβ is known to regulate cell-matrix and cell-cell adhesions.36 Thus, to systematically identify cell adhesion genes implicated in adhesion of ALL to MSCs, we performed a CRISPR-Cas9 screen targeting adhesome37,38 and TGFβ pathway genes39 (Table S5) (Figures 3A, S2A, and S2B). Integrin β1 was the only gene enriched in Nonadh leukemia cell lines and depleted in Adh leukemia cell lines (Figures 3B, 3C, and S2A-S2C). Next, we performed cell adhesion assays in the presence of the integrin β1-blocking antibodies AIIB2 and P5D2 and found that only AIIB2 diminished NALM6, 697, and REH cell adhesion to MSCs in a dose-dependent manner (Figures 3D and 3E).
Figure 3. Identification of integrin beta 1 in ALL cells mediating the adhesion to MSCs.

(A) Functional CRISPR-Cas9 library screening of cell adhesion genes implicated in adhesion of ALL cells to MSCs. NALM6 cells transduced with lentivirus expressing Cas9 and gRNAs that target 285 genes, adhesome (232 genes) and TGFβ pathway (53 genes), were co-cultured with MSCs, and then Nonadh and Adh NALM6 cells were collected for sequencing and MAGeCK analysis.
(B) Volcano plots displaying the log fold change on the X axis and −log10 p value in Y axis for all sgRNAs identified in Nonadh NALM6 (left panel) and Adh NALM6 (right panel).
(C) Venn diagram showing number of genes that are either depleted or enriched genes in both Nonadh and Adh NALM6.
(D) Cell adhesion assay of NALM6 cells to MSCs in the presence of P5D2 or AIIB2 (n = 3). Data, mean ± SD.
(E) Cell adhesion assay of 697 and REH to MSCs in the presence of AIIB2 (n = 3). Data, mean ± SD.
(F) Cell adhesion assay of ITGB1 KO NALM6 clones to MSCs (n = 3). Data, mean ± SD.
(G) Cell adhesion assay of NALM6 cells to MSCs in the presence of blocker of either 25 μM ATN-161 or BIO-1211 (n = 3). Data, mean ± SD.
(H) TEM of integrin β1 in NALM6 co-cultured with MSCs. White arrows, integrin β1 localized at the synapse between NALM6 and MSCs. Scale bars represent 2 μm. Student’s t test compared to WT or no treatment of Adh leukemic cells, ****p < 0.0001, ***p < 0.0005, **p < 0.005, *p < 0.05. See also Figure S2.
Moreover, AIIB2 efficiently abrogated NALM6 adhesion onto fibronectin but not P5D2 (Figure S2D). Consistent with these data, AIIB2 reduced adhesion of PDX B-ALL cells to MSCs (Figure S2E). Moreover, integrin β1 knockout (ITGB1 KO) cells (Figure S2F) exhibited strikingly reduced adhesion to MSCs (Figure 3F). We also found that fibronectin blocking peptide (FN1 CS1) and VCAM-1 blocking antibody reduced NALM6 cell adhesion to MSCs (Figures S2G and S2H), indicating that leukemic cells bind to both FN1 and VCAM-1 on MSCs. Integrin α4β1 and α5β1 complexes have been reported to mediate interaction of B-ALL cells to BM stromal cells or chemoresistance.40-44 Thus, we performed cell adhesion assays in the presence of the α5β1 blocker (ATN-161) or the α4β1 blocker (BIO-1211).45 We found that integrin α4β1 (VLA-4) participated in leukemic cell adhesion to MSCs (Figure 3G). To verify whether integrin α4 (ITGA4) is implicated in the adhesion of leukemia cells to MSCs, we knocked down ITGA4 and ITGA5 and examined cell adhesion. Despite downregulation of protein level (Figure S2I), knockdown had only a modest effect on cell adhesion (Figure S2J). In contrast, ITGA4 knockdown augmented ITGA5 expression, indicating that ITGA4 knockdown results in compensatory deregulated expression of other integrins (Figure S2K). Using an ECM array slide, NALM6 exhibited strong affinity to fibronectin and fibronectin-included ECM combinations and weak adhesion to collagen I-included ECM combinations (Figure S2L). Using immunofluorescence, we observed that integrin β1 protein was redistributed from diffuse cytoplasmic location to contacts between NALM6 and MSCs upon engagement with MSCs (Figure S2M). Further, transmission electron microscopy (TEM) analysis showed that integrin β1 was located in the synapse between NALM6 and MSCs (Figure 3H). Collectively, these data demonstrate that ALL cells adhered to MSCs via integrin β1 localized at the synapse between ALL cells and MSCs by binding to fibronectin and VCAM-1 on MSCs.
Next, we tested whether integrin β1-mediated cell adhesion is implicated in the chemoresistance of NALM6 cells co-cultured with MSCs using ITGB1 KO NALM6 cells expressing yellow fluorescent protein (YFP) and firefly luciferase (ffLuc). ITGB1 KO did not sensitize NALM6 cells co-cultured with MSCs to dexamethasone (DEX) or cytarabine (Figures S3A and S3B). However, integrin β1 is also critical for multiple various cell functions, including survival, homeostasis, and cell adhesion,46 and we observed poor cell viability of ITGB1 KO NALM6 cells even in the absence of drugs and MSCs (Figures S3A and 3B). Therefore, we also performed cell viability assays in the presence of blockers of integrin β1-mediated adhesion. Both the blocking antibody of integrin β1 (AIIB2) and the inhibitor of integrin α4β1 (BIO-1211) sensitized YFP-positive NALM6 cells co-cultured with MSCs (Figures S3C and S3D), indicating that the cell adhesion of ALL cells to MSCs via integrin β1 as part of the α4β1 complex contributes to the chemoresistance of B-ALL cells in contact with MSCs.
ALL-MSC interactions drive an EMT-like program in ALL
Pathway analysis of genes deregulated upon ALL-MSC engagement of five ALL cell lines and 11 patient samples showed EMT to be the top enriched gene set (NES = 1.46; FDR < 0.0001) in Adh ALL samples (Figure 4A and 4B). Enriched genes in Adh ALL patient samples included the EMT transcription factors (TFs) SNAI2 and TWIST1, the cadherins CDH2 and CDH11, collagens COL1A1 and COL3A1, FN1 (fibronectin), ACTA2 (actin), and MMP2 genes (Figure 4C). Additional TFs known to activate EMT47 including SOX9, ZNF703, and GATA6 were also overexpressed in Adh patient samples (Figure 4D). This striking upregulation of EMT genes in Adh ALL cells is shown in a gene expression heatmap of five ALL cell lines (Figure 4E).
Figure 4. Induction of an EMT-like program in Adh ALL cells.

(A) Gene set enrichment analysis against hallmark gene sets, comparing Adh ALL with Nonadh ALL, for each cell line, combined all cell lines as well as combined 11 patients. Only gene sets with FDR < 0.05 in at least one comparison and nominal p value < 0.01 in at least four comparisons were selected.
(B) Enrichment plot of hallmark EMT dataset enriched in Adh ALL cell lines.
(C) The upregulation of EMT markers, including SNAI2, TWIST1, CDH2, FN1, and MMP2, was determined by mRNA-seq in 11 patients with Adh ALL (FDR < 0.05). Data, mean ± SD.
(D) Expression level of conventional and novel EMT TFs in 11 patient Adh ALL samples. n.d., not detectable. n.s., not significant. Data, mean ± SD.
(E) Heatmap of top 70 gene set enrichment analysis EMT genes in five Adh ALL cell lines.
(F) Immunoblotting analysis of EMT markers in Adh cells.
(G) Analysis of the EMT state in mono- or co-cultured YFP+ NALM6 cells with MSCs for 48 h by immunoblotting.
(H) Cell aggregation assay of Adh YFP+ NALM6 cells (n = 3). Y axis shows the number of cell clusters consisting of more than four cells. Data, mean with individual value.
(I and J) Cell migration and invasion assay of Adh YFP+ NALM6 cells, respectively (n = 3). Y axis shows the number of YFP+ NALM6 cells that migrated from the upper chamber and subsequently bound to MSCs. Data, mean with individual value. Statistical testing was performed using two-tailed Student’s t test, ****p < 0.0001, ***p < 0.0005, **p < 0.005. See also Figures S4A-S4D.
Immunoblotting showed the EMT makers fibronectin (FN1), TWIST (TWIST1), and SLUG (SNAI2) were highly expressed in the five Adh ALL cell lines (Figure 4F). Although SNAI1, ZEB1, and ZEB2 were not upregulated in RNA-seq data of Adh ALL patient samples (Figure 4D), they have putative roles in leukemia development/progression,48-50 and we observed SNAIL (SNAI1) and ZEB1 but not ZEB2 to be highly expressed in Adh leukemic cell lines (Figure 4F). Interestingly, SNAIL and ZEB1 proteins were not highly expressed in MSCs, in contrast to TWIST and SLUG. We also tested if the EMT property of Adh ALL cells was reversible by measuring the EMT markers in Adh leukemic cells after additional culture with or without MSCs. The protein levels of the EMT markers in Adh ALL cells were diminished after mono-culture for 48 h, while they were maintained in Adh ALL cells after co-culture with MSCs, indicating that MSC-derived EMT in Adh leukemic cells is dynamic and reversible (Figure 4G). Next, we found that Adh ALL cells form larger clusters compared to mono-cultured ALL cells (Figures 4H and S4A). Consistent with this, a substantial EMT phenotype of Adh ALL cells was retained after mono-culture for 24 h, with a decline after 48 h (Figures 4G and S4B). Surprisingly, the migration and invasion ability were suppressed in Adh ALL cells (Figures 4I, 4J, S4C, and S4D). Collectively, this indicates that elevated adherence in Adh ALL cells promotes aggregation, which in turn impedes migration and invasion. As ALL-MSC interactions drive an EMT-like state, which is strongly associated with the maintenance of stemness, modulation of metabolism, and drug resistance,51 we hypothesized that MSC-induced EMT in B-ALL might be implicated in the survival and drug resistance of B-ALL.
Hybrid ALL-MSC-adherent cells driven by an EMT-like program
To characterize the heterogeneity of ALL cells induced by interaction with MSC, we performed scRNA-seq of NALM6 cells (1) and MSCs (2), each cultured in isolation, and from co-culture of NALM6 cells with MSCs, Nonadh NALM6 (3), and Adh NALM6 plus MSCs (4) (Figure 5A). We generated a heatmap showing normalized expression of NALM6 and MSC signature genes across cells from all samples (Figure 5B). Consistent with bulk RNA-seq, the Nonadh NALM6 sample displayed a similar expression signature to NALM6 alone. In the NALM6-MSC co-culture, we observed a population of cells expressing both NALM6 and MSC signature genes (cluster 2, C2), in addition to the expected NALM6 and MSC populations, which was also evident in a t-distributed stochastic neighbor embedding (tSNE) plot analysis of scRNA-seq data (Figures 5B and 5C). Cells in this hybrid cluster expressed hallmark B-ALL genes such as IGLC7, IGLL1, VPREB1, TCL1A, and IGHM, albeit at lower levels than NALM6 (C1), and expressed MSC genes such as FN1, TAGLN, COL1A1, COL1A2, and LGALS1 (Figure 5D), indicating that this hybrid cluster (C2) is derived from B-ALL cells but had acquired MSC features.
Figure 5. Identification of novel hybrid cluster retaining both ALL and MSC features in Adh ALL cell-driven EMT-like program by scRNA-seq analysis.

(A) Scheme of droplet-based scRNA-seq analysis of NALM6, MSCs, Nonadh NALM6, and Adh NALM6 plus MSCs.
(B) Heatmap showing normalized expression of the NALM6 and MSC signature genes across cells from all four samples. Cell cluster for Adh NAML6 plus MSC sample and aggregated signature scores are shown on the top.
(C) tSNE plot showing three clusters of cells in Adh NALM6 plus MSC sample.
(D) Violin plot showing expression pattern of top five genes of NALM6 and MSC signature across all the samples/clusters.
(E) Enrichment analysis for MSigDB Hallmark 2020 analysis in clusters C1, C2, and C3 of Adh NALM6 plus MSC samples by analyzing top 100 genes of each cluster using EnrichR. Log2 (enrichment combined score).
(F) Dot plots showing the expression of EMT, MSCs, and B-ALL markers, respectively (Y axis) in each cluster (X axis). The size of the dot represents the percentage of cells expressing the gene, and saturation of color represents average normalized expression level.
(G) Immunoblotting analysis of the levels of FN1, TWIST, SLUG, and actin in YFP-positive NALM6 cells with (+) or without (−) MSCs. Asterisk(*), nonspecific bands.
(H) Volcano plots displaying the expression level of EMT genes, which were enriched hybrid clusters, is enriched in patient sample compared to normal BM-derived CD19+ cells. See also Figures S4E and S4F.
Pathway analysis using the top 100 expressed genes in each cluster showed that the hybrid C2 cluster exhibited evidence of EMT signaling that was not present in the remaining NALM6 cells (C1 cluster; Figure 5E). Thus, the EMT observed in Adh ALL elicited by crosstalk with MSCs is heterogeneous and likely mediates acquisition of hybrid phenotype of the C2 cells that exhibit expression of B-ALL and MSC genes. In addition, hypoxia and glycolysis pathways, which contribute to the emergence of stem cell-like leukemia-initiating cells in B-ALL,52 were also found to be enriched in the cluster C2 compared to C1 (Figure 5E).
In contrast, cell proliferation-related pathways (E2F targets, MYC targets V1, and G2-M checkpoint genes) were enriched in C1 (Figure 5E). Consistent with induction of EMT, we observed upregulation of the EMT genes SNAI2, TWIST1, CDH2, CDH11, COL1A1, COL3A1, FN1, ACTA2, and MMP2, in hybrid C2 cells, which were also upregulated in MSC-adherent patient ALL cells in bulk RNA-seq analyses (Figure 4C) but not in C1, NALM6, and Nonadh NALM6 samples (Figure 5F). Collectively, these data indicate EMT activation contributes to the formation of the hybrid C2 cell cluster.
To examine whether the novel hybrid C2 cluster is ALL derived, and to exclude the possibility of contamination by MSC, we measured the protein expression level of fibronectin, TWIST, and SLUG, which were upregulated EMT markers in the hybrid cluster, in YFP-positive NALM6 cells. Indeed, the levels of these proteins were elevated in Adh YFP-positive NALM6 cells (Figure 5G), indicating that the hybrid cluster was derived from ALL via activation of EMT and previous findings were not due to MSC contamination. Consistent with this, most of the MSC-derived genes expressed in the hybrid cluster (Figure 5B) were also deregulated in bulk RNA-seq data from both cell line and patient samples and were components of the EMT, coagulation, and hypoxia hallmark pathways enriched in bulk RNA-seq data (Figure S4E).
The majority of EMT genes (50/70) were also upregulated in patient samples compared to normal BM, emphasizing the clinical relevance of these findings (Figure 5H). To examine whether this hybrid population could be identified by immunophenotyping, we examined ITGAV, a cell surface protein enriched in hybrid cluster (Figure S4E), and found ITGAV to be highly expressed in Adh NALM6 compared to NALM6 alone (Figure S4F).
Targeting of EMT-associated signaling pathways impairs leukemic cell viability
Several signaling pathways collaborate to induce EMT in response to signaling cues from the BME, including WNT, NOTCH1, TGFβ, receptor tyrosine kinase (RTK), cytokine signaling, and BMP53,54 (Figure 6A). We investigated activation of these pathways in Adh ALL cells by measuring expression of the main executor protein of each pathway: β-catenin (WNT signaling), cleaved NOTCH1 (NOTCH1 signaling), phosphorylated SMAD3 (pSMAD3) (TGFβ signaling), pNF-κB p65 (RTK signaling), pSTAT3 (cytokine signaling), and pSMAD1/5/8 (BMP signaling). Unexpectedly, several signaling pathways known to drive EMT, including WNT, NOTCH1, and TGFβ, were activated in Adh ALL cell lines, based on increase of protein level of β-catenin, cleaved NOTCH1, and pSMAD3, respectively (Figure 6B). We also examined the level of phosphorylation of AKT, NF-κB p65, STAT3, SMAD2/3, and SMAD1/5/8 at earlier time points to detect transient activation. We observed activation of RTK (pAKT and pNF-κB p65), cytokine signaling (pSTAT3), and TGFβ (pSMAD2/3) at earlier time points (Figure 6C). Various cells in the BME secrete IL6, leading to activation of STAT3 signaling in tumor cells55; moreover, we observed enrichment of IL6 in co-culture media due to secretion from MSCs (Figures 2B and 2C) that demonstrated that IL6 secreted from MSCs led to transient activation of STAT3 signaling in Adh ALL cells.
Figure 6. Targeting WNT/β-catenin and STAT3 signaling pathways impairs cell viability of Adh cells.

(A) Signaling pathways leading to EMT activation based on two review journals.53,54 Inhibitors of each signaling pathways are shown.
(B) Immunoblotting analysis of the levels of fibronectin, TWIST, SLUG, β-catenin, cleaved NOTCH1, pSMAD3 (pS423/S425), pSMAD1/5/8 (pS463/S465 on SMAD1, pS463/S465 on SMAD5, pS466/S467 on SMAD8), pNF-κB p65 (pS536), pSTAT3 (pY705), and actin in Adh ALL cells after 48 h of co-culture with MSCs. "p," an abbreviation for "phosphorylated."
(C) Phospho-flow cytometry in Adh NALM6 at indicated time. All samples were analyzed for the mean fluorescence intensity (MFI) of pAKT (pS473), pNF-κB p65, pSTAT3, pSMAD2/3 (pS465/S467 on SMAD2, pS423/S425 on SMAD3), and pSMAD1/5/8 in Adh NALM6. (n = 3). Data, mean ± SD. Statistical testing was performed using two-tailed Student’s t test; ****p < 0.0001, ***p < 0.0005, and **p < 0.005.
(D) Cell viability assay in the presence of inhibitors of activated signaling pathways in Adh cells with or without DEX. DEX = 1 μM and other drugs = 40 μM
(E) Effects of LF3 and Stattic in ALL cells with or without DEX (n = 3). Data, mean ± SD. Significance was assessed by two-way ANOVA with a Sidak adjustment for multiple comparison between control and drug-treated samples, ****p < 0.0001.
(F) Generation of NALM6-TCF/LEF reporter cell line expressing TCF/LEF transcriptional response elements.
(G) Upregulated β-catenin-mediated transcriptional activity in Adh NALM6 compared to NALM6. For analysis, the signal from NALM6, CD19+ cells is gated, and then GFP signals were measured.
(H) Analysis of β-catenin-mediated transcription activity in Adh NALM6 by measuring percentage of relatively high level of GFP signal (GFPhigh) (n = 3). Data, individual value and mean.
(I) Flow cytometry of CD90 in NALM6 and Adh NALM6 after gating CD19+.
(J) Flow cytometric measurement of β-catenin-mediated transcriptional activity in either CD19+CD90− or CD19+CD90+ subset of Adh NALM6 by assessing MFI of GFP (n = 3). Data, individual value and mean. Statistical testing was performed using two-tailed Student’s t test, **p < 0.005. See also Figures S5 and S6.
As expected, NALM6 co-cultured with MSCs displayed strong resistance to DEX compared to NALM6 (Figure S5A). Next, to determine which of the WNT, NOTCH1, TGFβ, and cytokine signaling pathways activated in Adh ALL mediated leukemic cell survival and drug resistance, we performed cell viability assays in the presence of inhibitors (Figure 6A). To prioritize pathways for detailed investigation, we first used inhibitors of activated signaling pathways at high concentration (40 μM; Figure 6D) and 1 μM DEX. Notably, only inhibitors of WNT (the β-catenin-TCF inhibitor LF3 and the stimulator of β-catenin degradation by stabilization of Axin, XAV-939) and cytokine signaling (the inhibitor of nuclear translocation pSTAT3, Stattic) reduced viability of NALM6 cells co-cultured with MSCs. The inhibitor of nuclear translocation of NF-κB, BAY 11–7082, was only effective in NALM6 mono-culture (Figure 6D), indicating that activated WNT and cytokine signaling pathways represent potential targets to eradicate B-ALL cells adherent to MSCs. Thus, we tested the WNT inhibitor LF3 and cytokine signaling inhibitor Stattic at 15 μM concentration alone or with 1 μM DEX. Both drugs reduced cell viability as single agents and with enhanced toxicity in combination with DEX, with greatest efficacy for the WNT pathway inhibitor LF3 in NALM6-MSC co-culture (Figure 6E).
Cell viability assay using YFP-positive NALM6 cells following TWIST1 or SNAI2 siRNA-mediated knockdown, which sensitized NALM6 cells co-cultured with MSCs to DEX (Figures S5B and S5C), demonstrates that the upregulation of TWIST1 and SNAI2 in Adh ALL cells is implicated in MSC-mediated drug resistance. We investigated whether DEX treatment could potentiate EMT in NALM6 cells that were co-cultured with MSCs by enriching Adh ALL cells with EMT phenotype by treating the cells with either vehicle or 50 nM DEX (the IC75 of DEX in NALM6 mono-culture) and found that DEX treatment slightly increased EMT, as evidenced by the increased fibronectin expression in DEX-treated NALM6 with MSCs (Figure S5D).
MSCs confer dormancy, proliferation, and senescent characteristics to adherent ALL cells
We investigated the cell-cycle status of NALM6 and Adh NALM6 cells, showing that Adh ALL cells had increased dormant (G0) and proliferative (S-G2/M) populations (Figure S6A), both of which were also increased in MSC-co-cultured ALL PDX cells (Figure S6B). Consistent with these results, there was upregulation of both cell-cycle promoting (CCND1, CDK15, and CDK20) and inhibitory genes (CDKN2B and CDKN2A) in Adh leukemic cell lines (Figure S6C). Thus, Adh leukemic cells were heterogeneously composed of chemoresistant and proliferative subsets. We also found that MSCs protect Adh PDX cells from apoptosis (Figure S6D). The senescence-associated β-galactosidase activity assay showed that senescence program was augmented in Adh PDX cells (Figure S6E). Consistent with these data, many senescence-associated secretory phenotype genes were enriched in Adh cell lines (Figure S6F).
β-Catenin-mediated transcription activity is enhanced in adherent and hybrid ALL cells
The canonical WNT/β-catenin signaling pathway involves the nuclear translocation of β-catenin and activation of target genes via TCF/LEF TFs in response to WNT ligands.53 Accumulation of β-catenin in Adh ALL cells and inhibition of its interaction with TCF reduced cell viability, indicating that increased β-catenin forms a complex with TCF/LEF, leading to transcription activation in Adh ALL cells. Thus, we established a NALM6 reporter cell line (NALM6-TCF/LEF) expressing TCF/LEF transcriptional response elements under control of the minimal CMV (mCMV) promoter and dscGFP-T2A-luciferase, which was used to assess β-catenin-mediated transcriptional activity by measuring GFP or luciferase activity (Figure 6F). We observed increased GFP-positive cells in NALM6-TCF/LEF co-cultured with MSCs (Figures 6G and 6H).
Prior data showed that THY1 (CD90) is transcriptionally repressed by IKZF1 (Ikaros) and is overexpressed in drug-resistant, IKZF1-altered BCR::ABL1 ALL.19 As we here observed that THY1 is enriched in the hybrid cluster C2 (Figure 5F), we tested if THY1 could be used as a cell surface marker for this hybrid cluster. THY1 protein expression was increased in Adh ALL cells (Figure 6I), and the CD90-positive subpopulation of Adh NALM6-TCF/LEF cells displayed elevated β-catenin-mediated transcriptional activity (Figures 6I and 6J) demonstrating that the hybrid cluster is dependent upon on WNT/β-catenin-mediated transcriptional activity.
Inhibition of β-catenin-CBP interaction reduces the viability of B-ALL cells co-cultured with MSCs
Targeting of WNT signaling by inhibition of β-catenin-TCF interaction was effective in reducing viability of MSC-adherent ALL cells but at relatively high inhibitor concentration, so we sought alternative approaches to target this signaling pathway. For example, blocking of β-catenin-CBP interaction reverses pulmonary fibrosis, which is mediated by EMT56; and the β-catenin-CBP complex promotes self-renewal of stem cells, while β-catenin-p300 complex induces differentiation.57 Moreover, targeting of β-catenin-CBP interaction reduces drug resistance of chronic myeloid leukemia and the survival of leukemic stem cells in acute myeloid leukemia.58,59 Therefore, we explored if WNT/β-catenin target genes associated with stemness and self-renewal were enriched in Adh ALL cells.60 Expression of FN1, SNAI2, HAS2, DCLK1, DDND1, CD44, CLDN1, DKK1, and TERT was enriched in all five Adh cell lines (Figure S7A), and expression of genes with promoter TCF4 binding sites was also enriched in Adh ALL cell lines (Figure S7B). PRI-724, a selective inhibitor of β-catenin-CBP interaction, reduced the β-catenin-mediated transcriptional activity of NALM6 co-cultured with MSCs at nanomolar range (IC50 = 172 nM; Figure 7A), and in contrast to DEX, it inhibited the viability of NALM6 cells co-cultured with MSCs as a single agent (Figure 7B). In addition, PRI-724 was synergistic with DEX to inhibit the cell viability of Adh NALM6 cells (Figures 7B and 7C).
Figure 7. Targeting of β-catenin-CBP interaction reduces leukemic burden of B-ALL mouse model and inhibits the viability of B-ALL PDX cells co-cultured with MSCs.

(A) Inhibition of β-catenin-mediated transcriptional activity by PRI-724 (n = 3). Data, mean ± SD. Statistical testing was performed using two-tailed Student’s t test; ****p < 0.0001 and ***p < 0.0005.
(B) Dose-response matrix (inhibition) for PRI-724 and DEX in YFP-positive NALM6 cells co-cultured with MSCs. The left two graphs show dose-response curves for PRI-724 (upper) and DEX (lower). Y axis shows the inhibition rate. The right graph shows drug response of combination of both drugs. Inhibition scores are analyzed by SynergyFinder 2.0.
(C) ZIP synergy score used for synergy score. Less than −10, antagonistic; from −10 to 10, additive; larger than 10, synergistic.
(D) Schematic of the NALM6 xenograft model. NSG mice were treated with vehicle, DEX (4 mg/L), PRI-723 (40 mg/kg), or combination of both drugs on day 7 and stopped at day 24. Leukemic burdens were analyzed by BLI.
(E) BLI images of day 6, 11, 17, and 25. (n = 7 per group).
(F) Quantification of the total flux (p/s) of BLI. Data, mean ± SD. Statistical testing was performed using two-way ANOVA with Tukey’s multiple comparisons test, **p < 0.005, *p < 0.05. See also Figure S7.
PRI-724 reduces the leukemic burden of cell-line-derived xenograft mice
Pharmacokinetic (PK) studies showed that the concentration of C-82, a dephosphorylated form of PRI-724, in the plasma treated with 40 mg/kg of PRI-724 reached 185 nM (Figures S7C and S7D), which might be enough to target Adh NALM6 cells (IC50 = 172 nM; Figure 7B). However, it rapidly degraded within 6 h. Despite low stability of PRI-724/C-82 in vivo, we tested the efficacy of PRI-724 as a single drug or in combination with DEX using NALM6 xenograft model by measuring leukemia burden with bioluminescence imaging (BLI) (Figure 7D). The data showed that PRI-724 was able to reduce leukemia burden compared to the vehicle control (Figures 7E and 7F). When combined with DEX, there was a trend toward further reduction in tumor burden over 4 weeks of therapy, compared to the single-drug regimen groups. These results suggest that targeting the β-catenin-CBP interaction is effective in vivo, but stable drug is required to fully target β-catenin-CBP-mediated drug resistance.
Finally, we assessed the ability of PRI-724, either as a single drug or in combination with DEX, to inhibit the viability of B-ALL PDX leukemic cells co-cultured with MSCs ex vivo to faithfully model human leukemic cell-BME interactions. PRI-724 as a single drug displayed antileukemic activity in all B-ALL PDX models (Figures S7E and S7F), with two types of responses in combination with DEX: either more potent activity (Figure 7E) or no greater activity compared to PRI-724 alone (Figure 7F). These results suggest that targeting the β-catenin-CBP interaction is effective in alleviating DEX-resistant Adh ALL cells, either as a single agent or in combination with DEX. The varying efficacy of PRI-724 in combination with DEX among PDX samples may be attributed to specific ALL subtype-asociated genomic alterations.
DISCUSSION
Prior studies have implicated cellular components of the BME as influencing ALL cell survival and drug responsiveness and have nominated specific mediators of interaction (e.g., integrin-mediated adherence or intercellular tubule formation) and leukemic cell tropism (e.g., provision of amino acids such as asparagine).5,13,16,26,42,43,61 However, most prior studies have focused on individual pathways of interaction and have commonly been limited by analysis of cell lines,6,15 which may not fully reflect leukemia biology in vivo as such lines have lost the requirement for stromal cell interaction to proliferate.
To address these issues, we detailed analyses characterizing the mechanisms of interaction and the alterations in leukemia cell phenotype and resistance using complementary experimental approaches: comparison of leukemia and stromal cell line interactions with those of primary leukemia cell lines and stroma, and genomic analyses at the bulk and single-cell levels, coupled with interrogation and perturbation of implicated downstream signaling pathways. Collectively, the results were striking in the diversity of pathways and mechanisms perturbed at leukemia cell engagement with stroma. These include induction of a gene expression signature primarily in leukemic cells characterized by EMT, coagulation, and hypoxia, several of which, including EMT, are known to mediate tumor resistance and metastasis in other cancer contexts.51 scRNA-seq of isolated ALL cell line, MSC, and ALL-MSC co-culture demonstrated that a subset of MSC-adherent leukemia cells adopt hybrid ALL-MSC characteristics, and that EMT is a driver of acquisition of this phenotype. Although only a minority of MSC-adherent leukemia cells in the adherent cell population exhibited this hybrid phenotype on scRNA-seq, these analyses likely underestimate the magnitude of this phenomenon, as we showed it to be dependent on integrin-mediated cell adhesion (which is disrupted on harvesting cells for scRNA-seq analysis), and that the gene expression signature of this hybrid cell cluster was recapitulated in the bulk RNA-seq analysis of adherent cells, suggesting that this phenotype is present in the majority of MSC-adherent ALL cells.
Parallel functional genomic and biochemical assays provided additional insights into the causes and consequences of this adherence-induced EMT transition. First, the focused CRISPR-Cas9 genome editing screen identified ITGB1 (as part of the VLA-4 heterodimer) as a mediator of this adhesion. Second, blockade of ITGB1 and VLA-4 resensitized ALL cells adhered to MSCs to DEX. Lastly, the knockdown of EMT TFs TWIST1 and SNAI2 alleviated the chemoresistance in MSC-adherent ALL cells, indicating that EMT drives the acquisition of this chemoresistant phenotype in adherent ALL cells.
Biochemical assays were notable for showing activation of WNT, NOTCH1, TGFβ, RTK, and cytokine signaling pathways known to drive EMT in MSC-adherent leukemia cells. However, only the inhibition of WNT/β-catenin and cytokine signaling reduced the viability of leukemic cells interacting with MSCs, indicating that the NOTCH1, TGFβ, and RTK signaling pathways may have other functions (e.g., ECM remodeling) or may be dispensable during EMT. Our findings are consistent with prior data showing that the inhibition of the WNT/β-catenin pathway sensitized leukemic cell lines co-cultured with stromal cell lines and improved overall survival in a mouse model of leukemia cell line, but high concentrations of XAV-939, a stimulator of β-catenin degradation by stabilization of Axin, were required.62
These assays also revealed that only a subset of these EMT-associated activated pathways were suitable for targeting to achieve therapeutic benefits. Indeed, we discovered that β-catenin-mediated transcriptional activity is increased in hybrid ALL cells exhibiting THY1 expression, and specific inhibition of the β-catenin-CBP complex, which is known to promote self-renewal, diminished the survival and drug resistance of PDX ALL cells adhered to MSCs ex vivo and reduced leukemic burden of NALM6 xenograft model in vivo. This indicates that hybrid, MSC-adherent ALL cells utilize β-catenin-CBP complex-mediated transcription and are vulnerable to inhibition of β-catenin-CBP interaction.
Limitations of the study
The ex vivo and in vivo studies described here have several limitations. Our studies solely focused on the interaction of ALL with MSCs within BME, although other components of the BME, including vascular niche cells, are also important for chemoresistance. Additionally, we used NALM6 cells in many assays due to their facility for experimental manipulation such as transduction, but importantly, results were corroborated in studies with primary or PDX cells and co-culture with primary MSCs. While it is possible that culture conditions (lower cell density and reduced serum) in co-culture might affect leukemic adherence, we demonstrated that the culture media type did not play a major role in leukemic adhesion to MSCs. Further, while in vivo PDX models are extensively used to test antileukemic drug efficacy in ALL, such models are of limited utility in studying the interaction between human leukemic cells and human BME because NSG mice lack human BME. Furthermore, although PRI-724 is very effective as a single drug and in combination with DEX in vitro, due to its short half-life in vivo, it should be viewed as a tool compound demonstrating potential utility of inhibiting this pathway. However, our data demonstrate that deregulation of leukemic cell biology by physical interaction with the BME is an important determinant of resistance to therapy. These results extend our findings of genotype (IKZF1 alteration)-determined perturbation of adhesion and stemness as a driver of drug resistance in BCR::ABL1 leukemia to show that ALL-stromal interactions are important across the spectrum of ALL genetic subtypes. Moreover, these data identify therapeutic approaches that may ameliorate this stromal-mediated resistance.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Charles Mullighan (charles.mullighan@stjude.org). An MTA will be required for all requested materials.
Materials availability
This study did not generate new unique reagents. Information on reagents used in this study in available in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Integrin Beta1, clone AIIB2 (Azide Free) Antibody | Sigma-Aldrich | Cat# MABT409, RRID:AB_2893323 |
| Integrin beta 1 antibody [P5D2] | Abcam | Cat# ab24693, RRID:AB_448230 |
| Human VCAM-1/CD106 MAb (Clone BBIG-V1) | R and D Systems | Cat# BBA5, RRID:AB_356958 |
| Fibronectin antibody | Abcam | Cat# ab2413, RRID:AB_2262874 |
| Slug (C19G7) Rabbit mAb | Cell Signaling Technology | Cat# 9585, RRID:AB_2239535 |
| Twist antibody [Twist2C1a] - ChIP Grade | Abcam | Cat# ab50887, RRID:AB_883294 |
| Snail (C15D3) Rabbit mAb | Cell Signaling Technology | Cat# 3879, RRID:AB_2255011 |
| TCF8/ZEB1 (D80D3) Rabbit mAb | Cell Signaling Technology | Cat# 3396, RRID:AB_1904164 |
| ZEB2 (E6U7Z) Rabbit mAb | Cell Signaling Technology | Cat# 97885, RRID:AB_2934315 |
| β-Catenin (D10A8) XP® Rabbit mAb | Cell Signaling Technology | Cat# 8480, RRID:AB_11127855 |
| Cleaved Notch1 (Val1744) (D3B8) Rabbit mAb | Cell Signaling Technology | Cat# 4147, RRID:AB_2153348 |
| Recombinant Anti-Smad3 (phospho S423 + S425) antibody [EP823Y] | Abcam | Cat# ab52903, RRID:AB_882596 |
| Phospho-Smad1 (Ser463/465)/Smad5 (Ser463/465)/Smad9 (Ser465/467) (D5B10) Rabbit mAb | Cell Signaling Technology | Cat# 13820, RRID:AB_2493181 |
| Ultrasmall gold conjugated Goat Mouse Ab (Aurion) | Electron Microscopy Sciences | Cat# 25121 |
| Phospho-NF-κB p65 (Ser536) (93H1) Rabbit mAb | Cell Signaling Technology | Cat# 3033, RRID:AB_331284 |
| Phospho-Stat3 (Tyr705) (D3A7) XP Rabbit mAb | Cell Signaling Technology | Cat# 9145, RRID:AB_2491009 |
| Mouse Anti-beta-Actin Monoclonal Antibody, Unconjugated, Clone AC-74 | Sigma-Aldrich | Cat# A5316, RRID:AB_476743 |
| Alexa Fluor® 647 Anti-Smad2 (pS465/ pS467)/Smad3 (pS423/pS425) antibody | BD Biosciences | Cat# 562696, RRID:AB_2716578 |
| Mouse Anti-Human Akt, phospho (Ser473) Monoclonal Antibody, Alexa Fluor??647 Conjugated | BD Biosciences | Cat# 560343, RRID:AB_1645397 |
| Goat Anti-Rabbit IgG H&L (Alexa Fluor® 647) | Abcam | Cat# ab150079, RRID:AB_2722623 |
| APC-H7 Anti-Human CD45 Antibody | BD Biosciences | Cat# 560178, RRID:AB_1645479 |
| PE Anti-Human CD14 Antibody | BD Biosciences | Cat# 561707, RRID:AB_10924593 |
| APC Anti-Human HLA-DR Antibody | BD Biosciences | Cat# 560896, RRID:AB_10563218 |
| FITC Anti-Human CD34 Antibody | BD Biosciences | Cat# 560942, RRID:AB_10562559 |
| FITC Anti-Human CD90 Antibody | BD Biosciences | Cat# 555595, RRID:AB_395969 |
| FITC Anti-Human CD19 Antibody | BioLegend | Cat# 363008, RRID:AB_2564171 |
| PE Anti-Human CD90 Antibody | BioLegend | Cat# 328110, RRID:AB_893433 |
| APC Anti-Human CD90 Antibody | BD Biosciences | Cat# 559869, RRID:AB_398677 |
| BV605 Anti-Human CD73 Antibody | BD Biosciences | Cat# 563199, RRID:AB_2738063 |
| FITC Anti-Human CD105 Antibody | BD Biosciences | Cat# 561443, RRID:AB_10714629 |
| APC Anti-Human CD105 Antibody | BD Biosciences | Cat# 562408, RRID:AB_11154045 |
| PerCP-Cy™5.5 Anti-Human CD19 Antibody | BD Biosciences | Cat# 340950, RRID:AB_400191 |
| PerCP-Cy™5.5 Anti-Human CD90 Antibody | BD Biosciences | Cat# 561557, RRID:AB_10712762 |
| PE Anti-Human CD49d (Integrin alpha 4) Antibody | Thermo Fisher Scientific | Cat# 12-0499-42, RRID:AB_10717245 |
| BV421 Anti-Human CD49E (Integrin α5 chain) Antibody | BD Biosciences | Cat# 740077, RRID:AB_2739840 |
| APC Anti-Human CD51 (Integrin alpha V) Antibody | BioLegend | Cat# 327913, RRID:AB_2876633 |
| Bacterial and virus strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Thermo Fisher Scientific | Cat# C737303 |
| Biological samples | ||
| Human Bone Marrow Aspirates | SJCRH | N/A |
| Human B cell Acute Lymphoblastic Leukemia Patient Samples | ECOG-ACRIN Cancer Research Group (ECOG-ACRIN) | SJALL054548, SJALL054550, SJALL054551, SJALL054554, SJALL054563, SJALL055623, SJALL055625, SJALL055626, SJALL054535, SJALL057947, SJALL057948 |
| Patient-derived xenograft (PDX) cells derived from B-ALL patients | This paper | SJBALL022611_R4, SJBALL047370_R1, SJBALL055854_X1, SJBALL021896_D1, SJBALL265_D1, SJBALL020589_D1 |
| Chemicals, peptides, and recombinant proteins | ||
| Dexamethasone | Sigma-Aldrich | Cat# D4902 |
| ATN-161 | Selleckchem | Cat# S8454 |
| XAV-939 | Selleckchem | Cat# S1180 |
| LF3 | Selleckchem | Cat# S8474 |
| LY2157299 | Selleckchem | Cat# S2230 |
| BAY 11-7082 | Selleckchem | Cat# S2913 |
| Stattic | Selleckchem | Cat# S7024 |
| PRI-724 | Selleckchem | Cat# S8968 |
| LY411575 | Selleckchem | Cat# S2714 |
| APC Annexin V | BioLegend | Cat# 640941 |
| Fibronectin | Advanced BioMatrix | Cat# 5050 |
| 7-AAD | BioLegend | Cat# 420403 |
| Hydrocortisone | Sigma-Aldrich | Cat# H0888 |
| Lymphoprep™ | STEMCELL Technologies | Cat# 07851 |
| Minimum Essential Medium Eagle (MEME) | Sigma-Aldrich | Cat# M4526 |
| DMEM 4.5 g/L Glucose w/o L-Gln w/Phenol Red | Lonza | Cat# BE12-614F |
| RPMI 1640 Medium, no glutamine | Thermo Fisher Scientific | Cat# 21870092 |
| RetroNectin® | TaKaRa | Cat# T100B |
| L-Proline | Sigma-Aldrich | Cat# P5607 |
| HyClone Fetal Bovine Serum (FBS) | Cytiva | Cat# SH30071.03HI |
| L-Ascorbic Acid 2-Phosphate Sesquimagnesium Salt Hydrate | Sigma-Aldrich | Cat# A8960 |
| ITS+1 Liquid Media Supplement (100×) | Sigma-Aldrich | Cat# I2521 |
| Recombinant Human TGF-β3 | Peprotech | Cat# 100-36E |
| Ascorbic Acid | STEMCELL Technologies | Cat# 72132 |
| β-Glycerophosphate Disodium Salt Hydrate | Sigma-Aldrich | Cat# G9422 |
| Toluidine blue O | Sigma-Aldrich | Cat# 1.15930 |
| Alcian blue Solution | Sigma-Aldrich | Cat# 1.01647 |
| Alizarin Red Staining Solution | Sigma-Aldrich | Cat# TMS-008 |
| Safranin O Solution | Sigma-Aldrich | Cat# HT90432 |
| Oil Red O Staining Solution | Sigma-Aldrich | Cat# 1.02419 |
| Silver enhancement (Aurion R-Gent SE-EM) | Electron Microscopy Sciences | Cat# 25521 |
| EmBed-812 resin | Electron Microscopy Sciences | Cat# 14120 |
| Sodium Pyruvate (100 mM) | Thermo Fisher Scientific | Cat# 11360070 |
| Penicillin-Streptomycin-Glutamine (100X) | Thermo Fisher Scientific | Cat# 10378016 |
| TrypLE™ Express Enzyme (1X), No Phenol Red | Thermo Fisher Scientific | Cat# 12604013 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat# 34096 |
| ProLong™ Diamond Antifade Mountant with DAPI | Thermo Fisher Scientific | Cat# P36962 |
| Critical commercial assays | ||
| MesenCult™ Osteogenic Differentiation Kit (Human) | STEMCELL Technologies | Cat# 05465 |
| MesenCult™ Adipogenic Differentiation Kit (Human) | STEMCELL Technologies | Cat# 05412 |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# G7571 |
| Gene Knockout Kit v2 - human - ITGB1 - 1.5 nmol | Synthego | N/A |
| BD Cytofix/Cytoperm™ | BD Biosciences | Cat# 554714 |
| Vybrant™ DiD Cell-Labeling Solution | Thermo Fisher Scientific | Cat# V22887 |
| Cell Meter™ Cellular Senescence Activity Assay Kit *Red Fluorescence* | AAT Bioquest | Cat# 23007 |
| AllPrep DNA/RNA Mini Kit (50) | Qiagen | Cat# 80204 |
| QCM™ 24-Well Fluorimetric Cell Migration Assay | Sigma-Aldrich | Cat# ECM 509 |
| Neon™ Transfection System 100 μL Kit | Thermo Fisher Scientific | Cat# MPK10025 |
| Neon™ Transfection System 10 μL Kit | Thermo Fisher Scientific | Cat# MPK1025 |
| Deposited data | ||
| RNA-seq data from human primary patient and normal samples | This paper | EGA: EGAS00001006120 |
| RNA-seq data from cell lines | This paper | GEO: GSE200039 |
| Experimental models: Cell lines | ||
| NALM6 | ATCC | Cat# CRL-3273 |
| REH | ATCC | Cat# CRL-8286 |
| SUP-B15 | ATCC | Cat# CRL-1929 |
| HEK293T | ATCC | Cat# CRL-3216 |
| NALM6 marked with YFP-Luc2 (YFP-positive NALM6) | This paper | N/A |
| hTERT-MSC | Mihara et al.27 | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) | The Jackson Lab | Cat# 005557 |
| Oligonucleotides | ||
| ITGB1 PCR forward primer: AAACTGGTCA GAGTTTGCATAAAGC | This paper | N/A |
| ITGB1 PCR reverse primer: AAGGACTT TTCAAAGGGAAATTTTG | This paper | N/A |
| ITGB1 sequencing primer: GTAAACACG CAGGTATTCACAGAG | This paper | N/A |
| ITGB1 guide_#1: U*G*C*UGUUCCUUU GCUACGGU *Synthego modified EZ Scaffold |
Synthego | N/A |
| ITGB1 guide_#2: G*A*U*GACAUAGA AAAUCCCAG *Synthego modified EZ Scaffold |
Synthego | N/A |
| ITGB1 guide_#3: A*U*U*UAGACAUUUUUACAGGA *Synthego modified EZ Scaffold |
Synthego | N/A |
| Non-targeting control Silencer®Select siRNA | Thermo Fisher Scientific | Cat# 4390843 |
| ITGA4 Silencer®Select siRNA (ITGA4-1) | Thermo Fisher Scientific | Cat# s7544 |
| ITGA4 Silencer®Select siRNA (ITGA4-2) | Thermo Fisher Scientific | Cat# s7545 |
| ITGA5 Silencer®Select siRNA (ITGA5-1) | Thermo Fisher Scientific | Cat# s7548 |
| ITGA5 Silencer®Select siRNA (ITGA5-2) | Thermo Fisher Scientific | Cat# s7549 |
| TWIST1 Silencer®Select siRNA (TWIST1-1) | Thermo Fisher Scientific | Cat# s14524 |
| TWIST1 Silencer®Select siRNA (TWIST1-2) | Thermo Fisher Scientific | Cat# s529181 |
| SNAI2 Silencer®Select siRNA (SNAI2-1) | Thermo Fisher Scientific | Cat# s13128 |
| SNAI2 Silencer®Select siRNA (SNAI2-2) | Thermo Fisher Scientific | Cat# s13129 |
| Recombinant DNA | ||
| pGreenFire1-TCF/LEF (EF1 α-neo) Lentivector | System Biosciences | Cat# TR013PA-N |
| vCL20SF2-Luc2a-YFP | Iacobucci et al.63 | N/A |
| pLentiCRISPRv2 | Addgene | Cat# 82416 |
| Software and algorithms | ||
| Morpheus | https://software.broadinstitute.org/morpheus | RRID:SCR_017386 |
| FlowJo 10.7.2 | https://flowjo.com/ | RRID:SCR_008520 |
| EnrichR | https://maayanlab.cloud/Enrichr/ | RRID:SCR_001575 |
| SynergyFinder 2.0 | Ianevski et al.64 | https://synergyfinder.fimm.fi |
| ImageJ 1.50i | https://imagej.nih.gov/ij/download.html | RRID:SCR_003070 |
| GraphPad Prism | https://www.graphpad.com:443/ | RRID:SCR_002798 |
| Other | ||
| Adhesome | Winograd-Katz et al.37; Zaidel-Bar et al.38 | http://www.adhesome.org/components.html |
| Amersham Hyperfilm MP | Cytiva | Cat# 28906846 |
Data and code availability
Bulk and single-cell RNA-seq data from cell lines have been deposited at the Gene Expression Omnibus (GEO) under accession number GEO: GSE200039 and are publicly available as of the date of publication. Bulk RNA-seq data from human primary patient and normal samples have been deposited at the European Genome-phenome Archive (EGA) under accession number EGA: EGAS00001006120.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANTS DETAILS
Human subjects
Leukemia samples were obtained from subjects were enrolled on St. Jude Children’s Research Hospital (SJCRH) and ECOG-ACRIN Cancer Research Group studies. This study was approved by institutional review boards at SJCRH and ECOG-ACRIN and informed consent was obtained from parents, guardians, or patients, and assent from the patient, as appropriate.
Mouse studies
All mice work was carried out according to Office of Laboratory Animal Welfare guidelines and was approved by the Institutional Animal Care and Use Committee of St Jude Children’s Research Hospital.
Cell lines
All the cell lines were confirmed as Mycoplasma spp. free using the Universal Mycoplasma Detection Kit (American Type Culture Collection, Manassas, VA). Cell lines were maintained in RPMI 1640 medium supplemented with 10% FBS (Hyclone), Antibiotic (1X, Gibco), and GlutaMAX (1X, Gibco) and incubated in a 37°C humidity-controlled incubator with 5% CO2. Each cell line identity was confirmed by STR profiling using PowerPlex Fusion System (Promega) and confirmed using ATCC or DSMZ STR database.
HEK293T cells (ATCC, Manassas, VA) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Lonza) supplemented with 10% fetal bovine serum (FBS) (Cytiva), and 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM glutamine (P/S/G) (Thermo Fisher Scientific). 697, NALM6, and REH were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% FBS and P/S/G. SUP-B15 and MHH-CALL-2 were cultured in RPMI 1640 medium supplemented with 20% FBS and P/S/G. For either coculture with human BM-MSC or mono-culture for experiments, all leukemic cell lines were cultured in α-modified Minimum Essential Medium (α-MEM) (Sigma-Aldrich) supplemented with 10% FBS, and P/S/G. All cells were incubated in a humidified incubator at 37°C with 5% CO2.
Generation of primary human BM-MSC
Human BM-MSC were prepared by available protocol32 with some modification of procedure. BM aspirates from special hematology laboratory of SJRCH were mixed with Lymphoprep (STEMCELL Technologies) followed by centrifugation at 800 x g for 20 min. Middle layer which contains mononuclear cells were collected and plated into tissue culture plate with α-MEM supplemented with 10% FBS, and P/S/G. After the initial 48 h of incubation, the medium containing nonadherent cells were removed and adherent cells were washed twice with phosphate-buffered saline (PBS) to get rid of the remaining floating cells. The adherent cells were further expanded until the outgrowth of colonies (colony unit forming fibroblasts, CFU-F) was observed. These cells are considered passage 0 (p0).
Cell culture of primary BM-MSC and hTERT-MSC
Primary BM-MSC were cultured in α-MEM supplemented with 10% FBS, and P/S/G. hTERT-MSC27 were maintained in RPMI 1640 medium supplemented with 10% FBS, P/S/G and 1.0 μM Hydrocortisone (Sigma-Aldrich).
In vitro differentiation of primary BM-MSC
For osteogenic differentiation, BM-MSC were pre-cultured in 6 well plates using α-MEM supplemented with 10% FBS, and P/S/G until reaching confluence and were replaced with 2mL of complete MesenCult Osteogenic Stimulatory Medium which was made of MesenCult Osteogenic Differentiation Kit (Human) (STEMCELL Technologies) per well. Once multilayering had been observed, aspirated medium and replaced with 2mL of complete Medium with β-glycerophosphate (Sigma-Aldrich). Medium was changed every 3 days for 2–3 weeks. Osteogenic differentiation is visualized by staining with Alizarin Red S (Sigma).
For chondrogenic differentiation, we followed methods reported in Reinisch et al.32 For optimized 3-dimensional (3-D) chondrogenic differentiation, BM-MSCs were centrifuged onto 0.4 μm pore size membranes of transwell inserts (24 well plates, Corning), and cultivated in DMEM medium containing 40 μg/mL L-proline, 25 μg/mL L-ascorbic acid 2-phosphate, insulin-transferrin-sodium selenite plus linoleic-bovine serum albumin (BSA) (ITS+1 Liquid Media Supplement) (Sigma-Alrich), 10−7 M dexamethasone (Selleckchem), sodium pyruvate (Thermo Fisher Scientific) and 10 ng/mL TGF-β3 (PeproTech). After 28 days of differentiation, formalin-fixed and paraffin-embedded tissue was stained with Alcian blue, Toluidine Blue and Safranin O (Sigma).
Adipogenic differentiation, BM-MSC were pre-cultured in 6 well plates using α-MEM supplemented with 10% FBS, and P/S/G until reaching confluence and were replaced with 2mL of complete MesenCult Adipogenic Differentiation Medium made of MesenCult Adipogenic Differentiation Kit (Human) (STEMCELL Technologies) per well. Media was changed every 3 days for 10–20 days. Adipogenic differentiation was visualized by staining with Oil Red O staining (Sigma).
RNA-seq
hTERT-MSC were cultured until confluent. 5 ALL cell lines including 697, MHH-CALL-2, NALM6, REH, and SUP-B15 were co-cultured with hTERT-MSC for two days. After two days, the cells exist in supernatant from the dish were collected as Nonadh ALL cells, whereas Adh ALL cells bound to MSC were dissociated by TrypLE treatment after washing with PBS. Cells were stained with fluorescent antibody for CD19 and CD90, then sorted by flow cytometry (minimum 99%). The total RNAs from the Nonadh and Adh cells were isolated using Qiagen kit. The RNA-seq libraries were prepared using the Illumina TruSeq Stranded Total RNA library preparation kit and sequenced on HiSeq 4000 (Illumina) with 100-bp paired-end setting. For B-ALL patient samples (Table S3), primary MSC were used for co-culture. Since there is limited numbers of sorted cell from patient samples, a low-input RNA library preparation kit (NuGen Ovation V2) was used to generate the RNA-seq libraries, followed by sequencing on HiSeq 4000 (Illumina) with 100-bp paired-end setting.
The sequencing libraries from cell lines were mapped to the GRCh37 human genome using StrongArm mapping pipeline65 and gene read counts were generated using HTSeq (version 0.6.1) and differential express analysis was done using R/Limma-Voom.
For sequencing libraries from patient samples, the reads were mapped to the GRCh37 human genome reference by STAR (version 2.5.3a).66 Gene expression was quantified using RSEM67 (v1.3.0) against Ensembl gene annotation (v75) (http://www.ensembl.org/). Differential expression between Adh and Nonadh patient cells was done using paired moderated t test (R/limma-Voom).68 In addition, gene fusions and B-ALL subtype analyses was performed as described in Gu et al.2 for the RNA-seq data from patient samples.
Gene set enrichment analysis (GSEA) was done using Pre-ranked GSEA34 with ratios from individual cell line comparisons, combined cell line comparison or paired patient sample comparison. Multiple gene sets collected in MSigDB were explored.
Single cell RNA-seq
To unravel cell heterogeneity in co-cultured cells, 1.5 × 106 NALM6 cells were co-cultured with BM-MSC in T175 flask for two days. The cells exist in supernatant from the dish were collected as Nonadh NALM6 and Adh NALM6 with BM-MSC were collected after dissociation with TrypLE. In comparison, NALM6 and MSC were also collected and subjected to single cell RNAseq using 10x Chromium Next GEM Single Cell 3′ v3. The Cell Ranger Single-Cell Software Suite (version 1.1.0) was used to perform sample demultiplexing, barcode processing, single-cell gene counting. Raw base call (BCL) files were demultiplexed using the Cell Ranger mkfastq pipeline into sample-specific FASTQ files that were then processed individually using the Cell Ranger count pipeline with default setting. Each sample was analyzed using Seurat69 package (version 4.0.6) in R. The tSNE plots were generated using res = 0.4. Differential expression analysis between NALM6 and MSC was performed using FindMarkers function in Seurat with default parameters (logfc.threshold = 0.25, test.use = "wilcox", min.pct = 0.1). Only the significant genes expressed in >50% of cells in one group (pct.1 > 0.5 from FindMarkers output) and <10% in the other (pct.2 < 0.1) were retained as marker genes, resulting in 178 for NALM6 and 244 for MSC. These markers were further aggregated to calculate enrichment scores for NALM6 and MSC using AddModuleScore function in Seurat and plot the heatmap in Figure 5B.
METHOD DETAILS
Cell viability assay
To measure selectively leukemic cells co-cultured with BM-MSC, ALL cells were modified to express a previously described yellow fluorescent protein (YFP) and firefly luciferase (ffLuc) by transduction with a vector (vCL20SF2-Luc2a-YFP).63 Marked ALL with vCL20SF2-Luc2a-YFP were plated into the 96 well plate with or without MSC and co-cultured for 24 h. Subsequently, drugs were treated for 72 h and cell viability was determined by measuring relative luminescence units (RLU) using CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI) with GloMax Discover (Promega). Three technical replicates were used for each of three biological replicate experiments. Cell Cultures and maintained in RPMI 1640 with 10% FBS and 2 mM GlutaMAX. To assess drug synergisms of between two drugs, SynergyFinder 2.0 was used.64
CRISPR screening
Two leukemia cell lines were used to study genes responsible for adhesion by targeted CRISPR KO library: B-progenitor ALL cell lines NALM-6 (IGH::DUX4) and 697 (TCF3::PBX1. Guide RNAs (gRNAs) targeting the adhesome (http://www.adhesome.org/components.html), TGFβ pathway, and non-template control were prepared and cloned into LentiCRISPRv2 (Cas9+eGFP+gRNAs) plasmid backbone which expresses Cas9 and gRNAs by the Center for Advanced Genome Engineering (CAGE) at SJRCH.
The sgRNA sequences for 284 coding genes were compiled from GeCKO70 and Brunello71 CRISPR libraries (Table S5). Duplicate gRNAs were removed, and off-target analysis was performed. Ten gRNA sequences per gene were selected with unique target sites; these sequences were supplemented with gRNA sequences designed using the Broad GPP sgRNA design tool CRISPick (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design. Non-targeting control gRNAs were added and comprise 10% of the total library. The final library design contained 3105 sgRNAs targeting 284 genes and 283 non-targeting control gRNAs. The resulting oligo library was designed according to the GeCKO protocol72 and synthesized (Twist Bioscience). Library amplification and Gibson Assembly into the pLentiCRISPRv2 backbone (Addgene #82416) was performed according to the prior method.73 The python scripts count_spacers.py72 and calc_auc_v1.1.py74 were used to validate library representation and quality control. The validated library was subsequently used to produce viral particles. Production and titration of lentiviral vectors were performed as described previously.75
Optimal infection conditions were determined for each cell line in order to achieve 20–60% infection efficiency, corresponding to a multiplicity of infection (MOI) of ~0.5–1. RetroNectin (TaKaRa)-coated plates were preloaded with virus supernatant by centrifugation for 2 h, 1500xg, at 4°C and then 2x106 leukemic cells were added and cultured for 2 days at CO2 incubator. Infections were performed in three biological replicates per cell line using cells and viral amount that yielded around 40% infection efficiency to achieve target representation of ~500 cells per sgRNA. At 48 h post transduction, GFP positive cells were isolated using FACSAria III cell sorter (BD Biosciences). Thereafter, 0.7-1.1x106 cells were harvested as the baseline counts for the control, and 2x106 cells were co-cultured with MSC in T175 flask considering that each sgRNA, on average, was represented ~500 times. Adherent and non-adherent cells were collected after 48 h of co-culture. Cells harvested at each time point were subjected to genomic DNA/RNA extraction using AllPrep DNA/RNA Mini Kit. Genomic DNA was quantitated by Nanodrop 2000 (Thermo Fisher scientific). PCR amplification was performed using protocols described in the Broad Genome Perturbation Portal (https://portals.broadinstitute.org/gpp/public/resources/protocols). Single-end, 100 cycle sequencing was performed on a NovaSeq 6000 (Illumina) by the Hartwell Center for Genome Sequencing Facility at SJRCH. CRISPR KO screens were analyzed using Mageck-Vispr/0.5.6.76
Cell adhesion assay
NALM6 were preincubated with isotype antibody or integrin β1 antibodies (P5D2 and AIIB2) for 30 min on ice and these mixtures were plated into confluent MSC plate. Four hours after coculture, Nonadh and Adh NALM6 were counted. In addition, cell adhesion assays were performed in the presence of integrin α4β1 inhibitor (BIO-1211), α5β1 inhibitor (ATN-161), or combination of both with same procedure above. NALM6 WT or ITGB1 knockout (KO) were co-cultured with confluent MSC for 4 h and then Nonadh and Adh NALM6 were counted. When using YFP-positive NALM6 cells, Nonadh cells were washed out with PBS three time, the level of Adh cells bound either MSC or fibronectin (50 μg/mL) was determined by measuring RLU using CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI) with GloMax Discover (Promega).
Cell aggregation assay
Cells were seeded at a density of 2x105 cells/2mL into 6 well non-tissue plate (triplicate) and incubated for 24 h before being imaged at 10x using an Eclipse TS100 light microscope. The number of cell clusters consisting of more than four cells was counted and area (pixel per square) of cell cluster was measured by ImageJ.
Cell migration assay
Migration assays were performed using the QCM 24-Well Fluorimetric Cell Migration Assay kit (Cat No: ECM 509). To facilitate cell migration, MSC were plated at lower chamber and cultured for 4 days. YFP+ NALM6 and Adh YFP+ NALM6 cell suspensions were prepared, containing 1 × 106 cells/mL in serum-free α-MEM. Then, 300 μL of the prepared suspension was added to each insert. Next, 500 μL of α-MEM supplemented with 10% FBS and P/S/G was added to the lower chamber, and the plates were incubated for 24 h at 37°C in a CO2 incubator. Afterward, the cells/media from the top side of the insert were carefully removed by pipetting out the remaining cell suspension. Then, the invasion chamber insert was removed and the image of the YFP+ NALM6 cells that migrated from top chamber and subsequently bound to MSC was captured by fluorescence microscopy and the number of migrated YFP+ NALM6 was analyzed ImageJ.
Cell invasion assay
Invasion assays were performed using the QCM 24-Well Fluorimetric Cell Migration Assay kit (Cat No: ECM 554). To facilitate cell invasion, MSC were plated at lower chamber and cultured for 4 days. YFP+ NALM6 and Adh YFP+ NALM6 cell suspensions were prepared, containing 1 × 106 cells/mL in serum-free α-MEM. Then, 300 μL of the prepared suspension was added to each insert. Next, 500 μL of α-MEM supplemented with 10% FBS and P/S/G was added to the lower chamber, and the plates were incubated for 24 h at 37°C in a CO2 incubator. Afterward, the cells/media from the top side of the insert were carefully removed by pipetting out the remaining cell suspension. Then, the invasion chamber insert was removed and the image of the YFP+ NALM6 cells that migrated from upper chamber through the ECM layer and subsequently bound to MSCs was captured by fluorescence microscopy and the number of migrated YFP+ NALM6 was analyzed ImageJ.
ECM array
NALM6 cell line was plated at cell density of 1 × 106 cells/mL onto the ECM Select Array Kit (Advanced Biomatrix) containing 36 different single ECM protein or combinations, each in 9 micro-spots. Twenty-four hours incubation, the ECM Select Array Kit was washed with PBS to remove non-specifically bound cells and was imaged at 4x magnification using an Eclipse TS100 light microscope (Nikon, Melville, NY).
Electron microscopy
For electron microscopy, MSC were plated in 8 well Lab-Tek Permanox slide chambers and cultured for 48 h and then NALM6 cells were added into MSC-cultured chamber and cultured for 48 h. Nonadh NALM6 cells were washed out by PBS. Adherent NALM6 with MSC were fixed by EM fixative composed of 4% paraformaldehyde and 0.5% glutaraldehyde in 0.1M phosphate buffer (PB) for 10 min at room temperature. After removal of fixative and immediate washing with PB, co-cultured sample was quenched with 0.1% sodium borohydride in PB. Integrin beta 1 is visualized by labeling with anti-Integrin beta 1 antibody (P5D2) from Abcam, an ultrasmall gold conjugated goat anti-mouse antibody (Aurion) and silver enhancement (Aurion R-Gent SE-EM). Following labeling and silver enhancement, samples were osmicated, contrasted en bloc with uranyl acetate, dehydrated and infiltrated in EmBed-812 resin (Electron Microscopy Sciences). Fully infiltrated samples were polymerized at 60°C for 48 h. Samples were sectioned at 70nm on a Leica UC-7 ultramicrotome and imaged in a ThermoFisher Scientific TF-20 operating at 80kV equipped with an AMT NanoSprint15 MkII imaging system.
Generation of stable NALM6 cell line expressing lentivector co-expressing dscGFP and luciferase in response to TCF/LEF
pGreenFire1-TCF/LEF (EF1α-neo) Lentivector (System Biosciences) were packaged into replication-incompetent, amphotropic lentiviral particles by transient transfection of HEK293T cells with pHDM-G, CAG4-RTR2, and CAG-KGP1-1R. NALM6 was transduced with lentivirus in the presence of RetroNectin (Takara) for 48 h. For the selection of a stable cell line expressing pGreenFire1-TCF/LEF (EF1α-neo) NALM6 (NALM6-TCF/LEF) cells were grown in RPMI 1640 medium supplemented with 10% FBS and P/S/G plus 400 μg/mL G418. Selection media was refreshed every 3 days until 14 days.
Immunoblotting
Cells are lysed in RIPA buffer (Sigma-Aldrich) containing 150 mM NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0 supplemented with proteosome and phosphatase inhibitors for 1hr on ice. Protein samples (15 μg of lysate) electrophoresed through 4–12% NuPAGE Bis-Tris gels (Life Technologies) at 200 V for 60 min. Blots were probed with anti-fibronectin, anti-TWIST, anti-SLUG, anti-β-catenin, anti-Cleaved NOTCH1, anti-pSMAD3, anti-pSMAD1/5/8, anti-pNFκB p65, anti-pSTAT3 and anti-Actin.
Immunofluorescence
Cytospins of NALM6 alone and co-cuture samples of NALM6 with BM-MSC were fixed with 4% paraformaldehyde and washed with PBS, followed by 1 h incubation in a 3% BSA blocking buffer. After removal of blocking buffer, Cells were permeabilized by incubation with 0.1% Triton X-100 in PBS for 10 min at room temperature and then incubated for 1 h with primary anti-ITGB1 antibody (P5D2, Abcam) diluted in 3% NGS/0.1% Triton X-100/PBS. Slides were washed three times in PBS, and then incubated for 1 h in 3% NGS/0.1% Triton X-100/PBS containing goat anti-rabbit Alexa Fluor 568 (Invitrogen, Carlsbad, CA). Slides were washed three times in PBS and mounted with ProLong Diamond Antifade Mountant with 4′-6-diamidino-2-phenylindol (DAPI) (ThermoFisher Scientific). All steps were carried out at room temperature. Images were captured using a Nikon C2 confocal fluorescence microscope and analyzed using NIS Elements software.
Cytokine array assay
Cytokine profiling was performed using Proteome Profiler Array Human (XL) Cytokine Array kit (R&D Systems), which detects 105 human cytokines simultaneously. We used cell culture supernatants collected from BM-MSC only, NALM6 only, and co-culture of NALM6 and BM-MSC, which were cultured for two days. Cytokine arrays were incubated overnight at 4°C with 500 μL of cell culture supernatant on a rocking platform. Following incubation with antibody detection cocktail, antibody conjugation, and recommended washes, membranes were washed, incubated with the antibody cocktail diluted for 1 h, washed, and incubated with streptavidin-HRP for 30 min, and finally treated with chemiluminescent reagent mix; membranes were exposed to film and imaged. Arbitrary values of cytokine abundance were calculated as integrated densities of each dot plot normalized by the reference spots. Integrated densities were measured using the ImageJ software (v1.50i).
Tunneling nanotube-mediated vesicle transfer
Donor cells were labeled with Vybrant DiD Cell-Labeling Solution (Thermo Fisher Scientific), 5 μL per ml of cell culture medium for 15 min at 37°C, washed three times. Afterward, cells were seeded in co-culture with acceptor cells in 6-well cell culture plates (1 × 105 MSC cells plus 1 × 105 YFP-positive NALM6 cells) and co-cultured for 24 h. To physical separate the interaction between NALM6-YFP and MSC, MSC and YFP-positive NALM6 cells were plated in the lower and upper chambers of a transwell with 0.4 μm pore polyester membrane insert (Corning). After 24 h, acceptor cells received DiD-labeled components were analyzed by flow cytometry analysis.
Flow cytometric analysis
Flow antibodies listed in REAGENT or RESOURCE were used for flow cytometric analysis. Cellular fluorescence data were collected for three technical replicates on BD LSRFortessa cytometer (Becton Dickinson Poland) using DIVA software (BD Biosciences, Franklin Lakes, NJ), and analyzed with FlowJo vX.0.6 (Tree Star, Inc., Ashland, OR).
For intracellular phosphorylation analysis, NALM6 cells were co-cultured with BM-MSC for indicated times. Cells were dissociated by TrypLE and neutralized by adding of culture medium followed by centrifugation. Cells were resuspended in PBS and fixed with same volume of BD Fixation/Permeabilization solution at 37°C for 10 min. Fixation was stopped by addition of medium followed by centrifugation. Cells were resuspended in 2% FBS-PBS. After Fc block, cells were stained with FITC Mouse Anti-Human CD19 (SJ25C1) and PE Mouse Anti-Human CD90 (5E10) antibodies. After permeabilization with 1x BD Perm/Wash Buffer for 30 min on ice and cells were stained with Fluorochrome conjugated antibodies or Phospho-primary antibodies followed by staining with Alexa Fluor 647 Goat Anti-Rabbit IgG H&L.
siRNA knock-down
For knockdown experiments with small interfering RNA (siRNA), NALM6 cells mixed with 300 nM SilencerSelect siRNAs (Thermo Fisher Scientific) against ITGA4 (1: s7544, 2: s7545), ITGA5 (1: s7548, 2:s7549), TWIST1 (1: s14524, 2: s529181), SNAI2 (1: s13128, 2: s13129) or non-targeting control siRNA (Cat.# 4390843) were electroporated in Resuspension Buffer R with the Neon transfection system using 3 pulse at 1410V for 10 ms each. 48h after nucleofection, target protein expression was determined by flow cytometry or immunoblotting.
Cell cycle analysis
Cells were fixed and permeabilized in 250 μL of fixation/permeabilization solution from BD Cytofix/Cytoperm kit for 20 min at 4°C and washed in 1 mL of BD Perm/Wash buffer. Then, cells were stained with APC anti-human Ki-67 antibody for 20 min at 4° C and washed 2 mL of BD Perm/Wash buffer. Cells were incubated in staining buffer in the presence of 7-AAD at 1:40 dilution for 20 min at room temperature (BD). The cells were analyzed with BD LSRFortessa.
CRISPR/Cas9 genome editing
To achieve gene knockout (KO) of ITGB1, NALM6 cells were mixed with ribonucleoprotein (RNP) complexes that consist of 10pmole of purified Cas9 nuclease duplexed with 90 pmol of multi-guide chemically modified ITGB1 synthetic single guide RNA (sgRNA) (predesigned Gene Knockout Kit v2, Synthego). The cells were then resuspended in 12μL of R Buffer (Thermo Fisher Scientific) for electroporation in the Neon Transfection System (Thermo Fisher Scientific). Cells were loaded in 10μL tips and electroporated according to the manufacturer’s instructions using the setting of 1410 V, 10ms width, and 3 pulse. Following electroporation, cells were recovered in RPMI (Thermo Fisher Scientific) containing 20% FBS (Thermo Fisher Scientific) for 24 h before undergoing another round of electroporation to increase the knockout efficiency. Cells were then harvested for single cell sorting 24 h later. Knockout efficiencies of all clones were analyzed by flow cytometry.
Proteome profiling by tandem mass tag mass spectrometry (TMT-MS)
Proteome profiling was performed using a previously published protocol77 with slight modifications. Cell pellets and media were resuspended in lysis buffer (50 mM HEPES, pH 8.5, 8.0 M urea and 0.5% sodium deoxycholate) and protein concentrations were determined by a Coomassie stained short gel with BSA as a standard.78 Approximately 100 μg of protein per sample were proteolyzed using Lys-C (Wako) at an enzyme-to-substrate ratio of 1:100 (w/w) for 3 h at 21°C. Following this the samples were diluted with 50 mM HEPES (pH 8.5) to a final 2.0 M urea. Disulfide bonds were reduced with DTT (5 mM and 45 min incubation at 21°C) and alkylated with iodoacetamide (20 mM and 45 min incubation at 21 °C in the dark). Proteins were further digested with trypsin (Promega) at an enzyme-to-substrate ratio of 1:50 (w/w) overnight at 21°C. Finally, the enzyme digestions were terminated by adding formic acid (FA) to 1% and samples were clarified at 21,000g, desalted on tC18 SepPak solid-phase extraction cartridges (Waters), and dried by SpeedVac.
The purified TMT-labeled peptides were resuspended in 50 μL 100 mM HEPES (pH 8.5) and labeled with 10-plex TMT reagents (Thermo Fisher Scientific, 10 μg/μL in 100% acetonitrile (ACN), 1:2 (w/w) peptide-TMT ratio, 1 h at 21°C). The labeling reactions were quenched by adding hydroxylamine to 0.3% (v/v), and labeled peptides were combined equally, acidified by adding FA to 1% (v/v), desalted and dried.
The pooled peptides were resuspended in 10 mM ammonium formate (pH 8.5) buffer and fractionated on an offline HPLC (Agilent 1220) using basic pH reverse phase liquid chromatography (pH 8.5, XBridge C18 column, 4.6 mm × 25 cm, 3.5 μm particle size, Waters) with a 180 min gradient of 15%–50% Buffer B [90% ACN, 10 mM ammonium formate (pH8.5)] and collected into 90 concatenated fractions. Fractions were dried in SpeedVac and resuspended in 5 μL 5% FA.
A QE-HF (Thermo Fisher Scientific) mass spectrometer connected in-line to a Dionex Ultimate 3000 ultra-high pressure liquid chromatography (UHPLC) system was used for LC-MS/MS analysis. Peptides were separated on a CoAnn column (75 μm × 15 cm, 1.9 μm C18, column at 50°C) using a 60 min gradient of 12%–56% Buffer B [67% ACN/2.5% DMSO/0.1% FA] at 0.25 μL/min flow rate. The mass spectrometer was operated in data-dependent mode with a survey scan in Orbitrap (60,000 resolution, 410–1600 m/z, 1 × 106 AGC target, 50 ms maximal ion time), followed by 20 data-dependent MS2 scans in Orbitrap (60,000 resolution, scan range starting from 120 m/z, 1 × 105 AGC target, ~105 ms maximal ion time, 32 HCD normalized collision energy, 1.0 m/z isolation window with 0.2 m/z offset,15 s dynamic exclusion, charge state screening enabled to reject precursor charge states that were unassigned, +1, or >+4).
MS raw files were searched against Uniprot HUMAN protein database using our in-house developed JUMP pipeline.79 Database search parameters included precursor and fragment ion mass tolerance 20 ppm, fully tryptic with maximal 2 missed cleavages, and maximal 3 modification sites per peptide. TMT10 tags on Lys residues and N-termini (+229.162932 Da) and carbamidomethylation of Cys (+57.02146 Da) were set as static modifications while Met oxidation (+15.99492 Da) were set as dynamic modifications. Peptide-spectrum match (PSM) were filtered to reduce the protein false discovery rate (FDR) to below 1% based on the target-decoy strategy using reversed database.80 TMT-based quantification and statistical analysis were performed based on our previous method.81 TMT quantification data quality was evaluated by comparing positive control protein intensities with Western blot results and by utilizing statistical methods (PCA or unsupervised clustering) to determine outliers or sample mislabeling. Protein fold change and p values of different comparisons were calculated based on protein intensities. Differential expression analysis was performed based on our previous method,82 by applying different combinations of cutoff threshold [i.e., FDR (Benjamini-Hochberg corrected p) < 0.05] based on the positive protein list (proteins known to be dysregulated in the comparisons).
In vivo pharmacokinetic (PK) studies
Female NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice aged 8 to 12 weeks were dosed by intraperitoneal injections with single dose daily for 3 days of PRI-724 at 20 mg/kg and 40 mg/kg, respectively. PRI-724 was suspended in 4.2% DMSO+95.8% PBS. After last injections of PRI-724, blood samples (3 mice per group) were collected at 2, 6 and 24 h by retro-orbital eye bleeding. Plasmas were prepared by centrifugation for 10 min at 1,500xg using a refrigerated centrifuge. Concentration of PRI-724 and its active form C-82 in plasma was determined by LC-MS.
Cellular senescence activity assay
We followed instruction of Cell Meter Cellular Senescence Activity Assay Kit (AAT Bioquest). Briefly, Xite Red beta-D-galactopyranoside working solution were prepared by adding 10 μL Xite Red beta-D-galactopyranoside stock solution dissolved in DMSO to 1 mL of assay buffer. Cells were washed with PBS, incubated with 100 μL Xite Red beta-D-galactopyranoside working solution for 30 min at 37°C, 5% CO2 incubator. After removing the working solution, cells were washed with PBS and resuspended in the assay buffer (Component B) and monitored the fluorescence intensity (MFI) with flow cytometer using 575/26 nm filter set.
In vivo preclinical studies
Female NSG mice aged 8 to 12 weeks were inoculated by tail vein injection with 1 × 106 YFP+ NALM6 cells, which were modified to express YFP and ffLuc by transduction with a vector (vCL20SF2-Luc2a-YFP). Leukemia burden was determined by bioluminescence imaging using a Xenogen IVIS-200 system and Living Image software (Caliper Life Sciences). Treatment was started 7 days post injection (luminescence averaged ~4x107 photons per second) and continued until 24 days post injection. Mice were randomized following injection 6 days post injection and were treated with either vehicle, dexamethasone (4 mg/L) alone, PRI-723 alone (40 mg/kg per day) via intraperitoneal injection, or combination of dexamethasone and PRI-724. Dexamethasone was given daily in drinking water with tetracycline (1 g/L; Sigma-Aldrich, St. Louis, MO), and half of each week the water contained Sulfamethoxazole (600 mg/L) and trimethoprim (120 mg/L; from Aurobindo).83 Mice were sacrificed when they became moribund.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analyses of functional data were performed using GraphPad Prism Version 8.0 (GraphPad, La Jolla, CA). All data are presented as mean ± SD. Differential expression between Adh and Nonadh patient cells was done using paired moderated t test (R/limma-Voom). Enriched genes in 11 patient Adh ALL samples compared to Nonadh ALL samples (Log2(Adh/Nonadh) > 1, FDR<0.05) are shown. The significance of the drug effect on cell viability was assessed by two-way ANOVA with a Sidak adjustment for multiple comparisons between control and drug-treated samples. Analysis of drug synergisms between two drugs was performed using SynergyFinder 2.0. ZIP synergy score is used for the synergy score (Less than −10, antagonistic; from −10 to 10, additive; larger than 10, synergistic). Statistical testing of BLI was performed using two-way ANOVA with Tukey’s multiple comparisons test. For comparison between two groups, data were analyzed with a two-tailed Student’s t-test. A p value of less than 0.05 was considered significant. *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.0005. All the statistical details of the experiments can be found in the figure legends.
Supplementary Material
Highlights.
Acute lymphoblastic leukemia interaction with mesenchymal stromal cells drives drug resistance
Interaction is mediated by integrin beta 1 and drives an EMT phenotype
Activated WNT/β-catenin signaling may be targeted to reverse drug resistance
ACKNOWLEDGMENTS
We thank the St. Jude Children’s Research Hospital (SJCRH) Center for In Vivo Imaging and Therapeutics, Animal Resource Center, the Genome Sequencing Facility, Hartwell Center for Bioinformatics and Biotechnology, and flow cytometry, cell sorting core facility, and Vector Core of SJCRH. Images were acquired at the Cell & Tissue Imaging Center, which are supported by SJCRH and NCI P30 CA021765. This study was supported in part by an Alex’s Lemonade Stand Foundation Innovative Research Grant and the National Cancer Institute of the National Institutes of Health under award numbers R35 CA197695, U10 CA180820, UG1 CA189859, and UG1 CA232760. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112804.
DECLARATION OF INTERESTS
I.I. received honoraria from Mission Bio. C.G.M. received research funding from AbbVie and Pfizer, honoraria from Amgen and Illumina, and royalty payments from Cyrus. C.G.M. is on an advisory board for Illumina. N.H. and M.P.V. have patent applications in the field of immunotherapy.
REFERENCES
- 1.Iacobucci I, Kimura S, and Mullighan CG (2021). Biologic and Therapeutic Implications of Genomic Alterations in Acute Lymphoblastic Leukemia. J. Clin. Med 10, 3792. 10.3390/jcm10173792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, Hagiwara K, Pelletier S, Gingras S, Berns H, et al. (2019). PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet 51, 296–307. 10.1038/s41588-018-0315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iacobucci I, and Mullighan CG (2017). Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol 35, 975–983. 10.1200/JCO.2016.70.7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lane SW, Scadden DT, and Gilliland DG (2009). The leukemic stem cell niche: current concepts and therapeutic opportunities. Blood 114, 1150–1157. 10.1182/blood-2009-01-202606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwamoto S, Mihara K, Downing JR, Pui CH, and Campana D (2007). Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J. Clin. Invest 117, 1049–1057. 10.1172/JCI30235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Witkowski MT, Dolgalev I, Evensen NA, Ma C, Chambers T, Roberts KG, Sreeram S, Dai Y, Tikhonova AN, Lasry A, et al. (2020). Extensive Remodeling of the Immune Microenvironment in B Cell Acute Lymphoblastic Leukemia. Cancer Cell 37, 867–882.e12. 10.1016/j.ccell.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Witkowski MT, Kousteni S, and Aifantis I (2020). Mapping and targeting of the leukemic microenvironment. J. Exp. Med 217, e20190589. 10.1084/jem.20190589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding L, Saunders TL, Enikolopov G, and Morrison SJ (2012). Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462. 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morrison SJ, and Scadden DT (2014). The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334. 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reagan MR, and Rosen CJ (2016). Navigating the bone marrow niche: translational insights and cancer-driven dysfunction. Nat. Rev. Rheumatol 12, 154–168. 10.1038/nrrheum.2015.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schepers K, Campbell TB, and Passegué E (2015). Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell 16, 254–267. 10.1016/j.stem.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moses BS, Slone WL, Thomas P, Evans R, Piktel D, Angel PM, Walsh CM, Cantrell PS, Rellick SL, Martin KH, et al. (2016). Bone marrow microenvironment modulation of acute lymphoblastic leukemia phenotype. Exp. Hematol 44, 50-9.e1–2. 10.1016/j.exphem.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Domínguez Á, Pinho S, Akhmetzyanova I, Gao J, Witkowski M, et al. (2019). The bone marrow microenvironment at single-cell resolution. Nature 569, 222–228. 10.1038/s41586-019-1104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiarini F, Lonetti A, Evangelisti C, Buontempo F, Orsini E, Evangelisti C, Cappellini A, Neri LM, McCubrey JA, and Martelli AM (2016). Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: From biology to therapeutic targeting. Biochim. Biophys. Acta 1863, 449–63. 10.1016/j.bbamcr.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 15.Rellick SL, Hu G, Piktel D, Martin KH, Geldenhuys WJ, Nair RR, and Gibson LF (2021). Co-culture model of B-cell acute lymphoblastic leukemia recapitulates a transcription signature of chemotherapy-refractory minimal residual disease. Sci. Rep 11, 15840. 10.1038/s41598-021-95039-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manabe A, Coustan-Smith E, Behm FG, Raimondi SC, and Campana D (1992). Bone marrow-derived stromal cells prevent apoptotic cell death in B-lineage acute lymphoblastic leukemia. Blood 79, 2370–2377. [PubMed] [Google Scholar]
- 17.Ma Z, Zhao X, Deng M, Huang Z, Wang J, Wu Y, Cui D, Liu Y, Liu R, and Ouyang G (2019). Bone Marrow Mesenchymal Stromal Cell-Derived Periostin Promotes B-ALL Progression by Modulating CCL2 in Leukemia Cells. Cell Rep. 26, 1533–1543.e4. 10.1016/j.celrep.2019.01.034. [DOI] [PubMed] [Google Scholar]
- 18.Fei F, Joo EJ, Tarighat SS, Schiffer I, Paz H, Fabbri M, Abdel-Azim H, Groffen J, and Heisterkamp N (2015). B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through Galectin-3. Oncotarget 6, 11378–11394. 10.18632/oncotarget.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Churchman ML, Low J, Qu C, Paietta EM, Kasper LH, Chang Y, Payne-Turner D, Althoff MJ, Song G, Chen SC, et al. (2015). Efficacy of Retinoids in IKZF1-Mutated BCR-ABL1 Acute Lymphoblastic Leukemia. Cancer Cell 28, 343–356. 10.1016/j.ccell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joshi I, Yoshida T, Jena N, Qi X, Zhang J, Van Etten RA, and Georgopoulos K (2014). Loss of Ikaros DNA-binding function confers integrin-dependent survival on pre-B cells and progression to acute lymphoblastic leukemia. Nat. Immunol 15, 294–304. 10.1038/ni.2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vitanza NA, Zaky W, Blum R, Meyer JA, Wang J, Bhatla T, Morrison DJ, Raetz EA, and Carroll WL (2014). Ikaros deletions in BCR-ABL-negative childhood acute lymphoblastic leukemia are associated with a distinct gene expression signature but do not result in intrinsic chemoresistance. Pediatr. Blood Cancer 61, 1779–1785. 10.1002/pbc.25119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nwabo Kamdje AH, Mosna F, Bifari F, Lisi V, Bassi G, Malpeli G, Ricciardi M, Perbellini O, Scupoli MT, Pizzolo G, and Krampera M (2011). Notch-3 and Notch-4 signaling rescue from apoptosis human B-ALL cells in contact with human bone marrow-derived mesenchymal stromal cells. Blood 118, 380–389. 10.1182/blood-2010-12-326694. [DOI] [PubMed] [Google Scholar]
- 23.Duan CW, Shi J, Chen J, Wang B, Yu YH, Qin X, Zhou XC, Cai YJ, Li ZQ, Zhang F, et al. (2014). Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell 25, 778–793. 10.1016/j.ccr.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 24.Pal D, Blair H, Parker J, Hockney S, Beckett M, Singh M, Tirtakusuma R, Nelson R, McNeill H, Angel SH, et al. (2022). hiPSC-derived bone marrow milieu identifies a clinically actionable driver of niche-mediated treatment resistance in leukemia. Cell Rep. Med 3, 100717. 10.1016/j.xcrm.2022.100717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dander E, Palmi C, D’Amico G, and Cazzaniga G (2021). The Bone Marrow Niche in B-Cell Acute Lymphoblastic Leukemia: The Role of Microenvironment from Pre-Leukemia to Overt Leukemia. Int. J. Mol. Sci 22, 4426. 10.3390/ijms22094426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polak R, de Rooij B, Pieters R, and den Boer ML (2015). B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 126, 2404–2414. 10.1182/blood-2015-03-634238. [DOI] [PubMed] [Google Scholar]
- 27.Mihara K, Imai C, Coustan-Smith E, Dome JS, Dominici M, Vanin E, and Campana D (2003). Development and functional characterization of human bone marrow mesenchymal cells immortalized by enforced expression of telomerase. Br. J. Haematol 120, 846–849. 10.1046/j.1365-2141.2003.04217.x. [DOI] [PubMed] [Google Scholar]
- 28.Skårn M, Noordhuis P, Wang MY, Veuger M, Kresse SH, Egeland EV, Micci F, Namløs HM, Håkelien AM, Olafsrud SM, et al. (2014). Generation and characterization of an immortalized human mesenchymal stromal cell line. Stem Cells Dev. 23, 2377–2389. 10.1089/scd.2013.0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simonsen JL, Rosada C, Serakinci N, Justesen J, Stenderup K, Rattan SIS, Jensen TG, and Kassem M (2002). Telomerase expression extends the proliferative life-span and maintains the osteogenic potential of human bone marrow stromal cells. Nat. Biotechnol 20, 592–596. 10.1038/nbt0602-592. [DOI] [PubMed] [Google Scholar]
- 30.James S, Fox J, Afsari F, Lee J, Clough S, Knight C, Ashmore J, Ashton P, Preham O, Hoogduijn M, et al. (2015). Multi parameter Analysis of Human Bone Marrow Stromal Cells Identifies Distinct Immunomodulatory and Differentiation-Competent Subtypes. Stem Cell Rep. 4, 1004–1015. 10.1016/j.stemcr.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elsafadi M, Manikandan M, Atteya M, Hashmi JA, Iqbal Z, Aldahmash A, Alfayez M, Kassem M, and Mahmood A (2016). Characterization of Cellular and Molecular Heterogeneity of Bone Marrow Stromal Cells. Stem Cells Int. 2016, 9378081. 10.1155/2016/9378081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reinisch A, Hernandez DC, Schallmoser K, and Majeti R (2017). Generation and use of a humanized bone-marrow-ossicle niche for hematopoietic xenotransplantation into mice. Nat. Protoc 12, 2169–2188. 10.1038/nprot.2017.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, and Horwitz E (2006). Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317. 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 34.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hurwitz R, Hozier J, LeBien T, Minowada J, Gajl-Peczalska K, Kubonishi I, and Kersey J (1979). Characterization of a leukemic cell line of the pre-B phenotype. Int. J. Cancer 23, 174–180. 10.1002/ijc.2910230206. [DOI] [PubMed] [Google Scholar]
- 36.Wang H, Radjendirane V, Wary KK, and Chakrabarty S (2004). Transforming growth factor beta regulates cell-cell adhesion through extracellular matrix remodeling and activation of focal adhesion kinase in human colon carcinoma Moser cells. Oncogene 23, 5558–5561. 10.1038/sj.onc.1207701. [DOI] [PubMed] [Google Scholar]
- 37.Winograd-Katz SE, Fässler R, Geiger B, and Legate KR (2014). The integrin adhesome: from genes and proteins to human disease. Nat. Rev. Mol. Cell Biol 15, 273–288. 10.1038/nrm3769. [DOI] [PubMed] [Google Scholar]
- 38.Zaidel-Bar R, Itzkovitz S, Ma’ayan A, Iyengar R, and Geiger B (2007). Functional atlas of the integrin adhesome. Nat. Cell Biol 9, 858–867. 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Caestecker M (2004). The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 15, 1–11. 10.1016/j.cytogfr.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 40.Messinger Y, Chelstrom L, Gunther R, and Uckun FM (1996). Selective homing of human leukemic B-cell precursors to specific lymphohematopoietic microenvironments in SCID mice: a role for the beta 1 integrin family surface adhesion molecules VLA-4 and VLA-5. Leuk. Lymphoma 23, 61–69. 10.3109/10428199609054803. [DOI] [PubMed] [Google Scholar]
- 41.Shalapour S, Hof J, Kirschner-Schwabe R, Bastian L, Eckert C, Prada J, Henze G, von Stackelberg A, and Seeger K (2011). High VLA-4 expression is associated with adverse outcome and distinct gene expression changes in childhood B-cell precursor acute lymphoblastic leukemia at first relapse. Haematologica 96, 1627–1635. 10.3324/haematol.2011.047993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu Z, and Slayton WB (2014). Integrin VLA-5 and FAK are Good Targets to Improve Treatment Response in the Philadelphia Chromosome Positive Acute Lymphoblastic Leukemia. Front. Oncol 4, 112. 10.3389/fonc.2014.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsieh YT, Gang EJ, Geng H, Park E, Huantes S, Chudziak D, Dauber K, Schaefer P, Scharman C, Shimada H, et al. (2013). Integrin alpha4 blockade sensitizes drug resistant pre-B acute lymphoblastic leukemia to chemotherapy. Blood 121, 1814–1818. 10.1182/blood-2012-01-406272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Toni-Costes F, Despeaux M, Bertrand J, Bourogaa E, Ysebaert L, Payrastre B, and Racaud-Sultan C (2010). A New alpha5beta1 integrin-dependent survival pathway through GSK3beta activation in leukemic cells. PLoS One 5, e9807. 10.1371/journal.pone.0009807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang W, Wang F, Lu F, Xu S, Hu W, Huang J, Gu Q, and Sun X (2011). The antiangiogenic effects of integrin alpha5beta1 inhibitor (ATN-161) in vitro and in vivo. Invest. Ophthalmol. Vis. Sci 52, 7213–7220. 10.1167/iovs.10-7097. [DOI] [PubMed] [Google Scholar]
- 46.Mezu-Ndubuisi OJ, and Maheshwari A (2021). The role of integrins in inflammation and angiogenesis. Pediatr. Res 89, 1619–1626. 10.1038/s41390-020-01177-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lamouille S, Xu J, and Derynck R (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol 15, 178–196. 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carmichael CL, Wang J, Nguyen T, Kolawole O, Benyoucef A, De Mazière C, Milne AR, Samuel S, Gillinder K, Hediyeh-Zadeh S, et al. (2020). The EMT modulator SNAI1 contributes to AML pathogenesis via its interaction with LSD1. Blood 136, 957–973. 10.1182/blood.2019002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goossens S, Radaelli E, Blanchet O, Durinck K, Van der Meulen J, Peirs S, Taghon T, Tremblay CS, Costa M, Farhang Ghahremani M, et al. (2015). ZEB2 drives immature T-cell lymphoblastic leukaemia development via enhanced tumour-initiating potential and IL-7 receptor signalling. Nat. Commun 6, 5794. 10.1038/ncomms6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stavropoulou V, Kaspar S, Brault L, Sanders MA, Juge S, Morettini S, Tzankov A, Iacovino M, Lau IJ, Milne TA, et al. (2016). MLL-AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell 30, 43–58. 10.1016/j.ccell.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 51.Brabletz S, Schuhwerk H, Brabletz T, and Stemmler MP (2021). Dynamic EMT: a multi-tool for tumor progression. EMBO J. 40, e108647. 10.15252/embj.2021108647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morris V, Wang D, Li Z, Marion W, Hughes T, Sousa P, Harada T, Sui SH, Naumenko S, Kalfon J, et al. (2022). Hypoxic, glycolytic metabolism is a vulnerability of B-acute lymphoblastic leukemia-initiating cells. Cell Rep. 39, 110752. 10.1016/j.celrep.2022.110752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dongre A, and Weinberg RA (2019). New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol 20, 69–84. 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- 54.Wu M, Chen G, and Li YP (2016). TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 4, 16009. 10.1038/boneres.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson DE, O’Keefe RA, and Grandis JR (2018). Targeting the IL-6/ JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol 15, 234–248. 10.1038/nrclinonc.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henderson WR Jr., Chi EY, Ye X, Nguyen C, Tien YT, Zhou B, Borok Z, Knight DA, and Kahn M (2010). Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 107, 14309–14314. 10.1073/pnas.1001520107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Teo JL, Ma H, Nguyen C, Lam C, and Kahn M (2005). Specific inhibition of CBP/beta-catenin interaction rescues defects in neuronal differentiation caused by a presenilin-1 mutation. Proc. Natl. Acad. Sci. USA 102, 12171–12176. 10.1073/pnas.0504600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou H, Mak PY, Mu H, Mak DH, Zeng Z, Cortes J, Liu Q, Andreeff M, and Carter BZ (2017). Combined inhibition of beta-catenin and Bcr-Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia 31, 2065–2074. 10.1038/leu.2017.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou HS, Carter BZ, and Andreeff M (2016). Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol. Med 13, 248–259. 10.20892/j.issn.2095-3941.2016.0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bild A, Teo JL, and Kahn M (2019). Enhanced Kat3A/Catenin transcription: a common mechanism of therapeutic resistance. Cancer Drug Resist. 2, 917–932. 10.20517/cdr.2019.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, Kokkaliaris KD, Mercier F, Tabaka M, Hofree M, et al. (2019). A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 177, 1915–1932.e16. 10.1016/j.cell.2019.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang Y, Mallampati S, Sun B, Zhang J, Kim SB, Lee JS, Gong Y, Cai Z, and Sun X (2013). Wnt pathway contributes to the protection by bone marrow stromal cells of acute lymphoblastic leukemia cells and is a potential therapeutic target. Cancer Lett. 333, 9–17. 10.1016/j.canlet.2012.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iacobucci I, Li Y, Roberts KG, Dobson SM, Kim JC, Payne-Turner D, Harvey RC, Valentine M, McCastlain K, Easton J, et al. (2016). Truncating Erythropoietin Receptor Rearrangements in Acute Lymphoblastic Leukemia. Cancer Cell 29, 186–200. 10.1016/j.ccell.2015.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ianevski A, Giri AK, and Aittokallio T (2020). SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res. 48, W488–W493. 10.1093/nar/gkaa216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wen J, Rusch M, Brady SW, Shao Y, Edmonson MN, Shaw TI, Powers BB, Tian L, Easton J, Mullighan CG, et al. (2021). The landscape of coding RNA editing events in pediatric cancer. BMC Cancer 21, 1233. 10.1186/s12885-021-08956-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dobin A, and Gingeras TR (2015). Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinformatics 51, 11.14.1 14 11.14.19–11. 10.1002/0471250953.bi1114s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 12, 323. 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME, Vaimberg EW, Goodale A, Root DE, Piccioni F, and Doench JG (2018). Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun 9, 5416. 10.1038/s41467-018-07901-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, and Zhang F (2017). Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc 12, 828–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dickerson KM, Qu C, Gao Q, Iacobucci I, Gu Z, Yoshihara H, Backhaus EA, Chang Y, Janke LJ, Xu B, et al. (2022). ZNF384 Fusion Oncoproteins Drive Lineage Aberrancy in Acute Leukemia. Blood Cancer Discov. 3, 240–263. 10.1158/2643-3230.BCD-21-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li W, Köster J, Xu H, Chen C-H, Xiao T, Liu JS, Brown M, and Liu XS (2015). Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome biology 16, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pagala VR, High AA, Wang X, Tan H, Kodali K, Mishra A, Kavdia K, Xu Y, Wu Z, and Peng J (2015). Quantitative protein analysis by mass spectrometry. Methods Mol. Biol 1278, 281–305. 10.1007/978-1-4939-2425-7_17. [DOI] [PubMed] [Google Scholar]
- 78.Xu P, Duong DM, and Peng J (2009). Systematical optimization of reverse-phase chromatography for shotgun proteomics. J. Proteome Res 8, 3944–3950. 10.1021/pr900251d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang X, Li Y, Wu Z, Wang H, Tan H, and Peng J (2014). JUMP: a tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol. Cell. Proteomics 13, 3663–3673. 10.1074/mcp.O114.039586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Peng J, Elias JE, Thoreen CC, Licklider LJ, and Gygi SP (2003). Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res 2, 43–50. 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 81.Niu M, Cho JH, Kodali K, Pagala V, High AA, Wang H, Wu Z, Li Y, Bi W, Zhang H, et al. (2017). Extensive Peptide Fractionation and y(1) Ion-Based Interference Detection Method for Enabling Accurate Quantification by Isobaric Labeling and Mass Spectrometry. Anal. Chem 89, 2956–2963. 10.1021/acs.analchem.6b04415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bai B, Wang X, Li Y, Chen PC, Yu K, Dey KK, Yarbro JM, Han X, Lutz BM, Rao S, et al. (2020). Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 105, 975–991.e7. 10.1016/j.neuron.2019.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Finch ER, Janke LJ, Smith CA, Karol SE, Pei D, Cheng C, Kaste SC, Inaba H, Pui CH, Wolf J, and Relling MV (2019). Bloodstream infections exacerbate incidence and severity of symptomatic glucocorticoid-induced osteonecrosis. Pediatr. Blood Cancer 66, e27669. 10.1002/pbc.27669. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk and single-cell RNA-seq data from cell lines have been deposited at the Gene Expression Omnibus (GEO) under accession number GEO: GSE200039 and are publicly available as of the date of publication. Bulk RNA-seq data from human primary patient and normal samples have been deposited at the European Genome-phenome Archive (EGA) under accession number EGA: EGAS00001006120.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.