

Abstract
Proteasomal degradation of intrinsically disordered proteins, such as tau, is a critical component of proteostasis in both aging and neurodegenerative diseases. In this study, we investigated proteasomal activation by MK886 (MK). We previously identified MK as a lead compound capable of modulating tau oligomerization in a cellular FRET assay and rescuing P301L tau-induced cytotoxicity. We first confirmed robust proteasomal activation by MK using 20S proteasomal assays and a cellular proteasomal tau-GFP cleavage assay. We then show that MK treatment can significantly rescue tau-induced neurite pathology in differentiated SHSY5Y neurospheres. Due to this compelling result, we designed a series of seven MK analogs to determine if proteasomal activity is sensitive to structural permutations. Using the proteasome as the primary MOA, we examined tau aggregation, neurite outgrowth, inflammation, and autophagy assays to identify two essential substituents of MK that are required for compound activity: (1) removal of the N-chlorobenzyl group from MK negated both proteasomal and autophagic activity and reduced neurite outgrowth; and (2) removal of the indole-5-isopropyl group significantly improved neurite outgrowth and autophagy activity but reduced its anti-inflammatory capacity. Overall, our results suggest that the combination of proteasomal/autophagic stimulation and anti-inflammatory properties of MK and its derivatives can decrease tau-tau interactions and help rebalance dysfunctional proteostasis. Further development of MK to optimize its proteasomal, autophagic, and anti-inflammatory targets may lead to a novel therapeutic that would be beneficial in aging and neurodegenerative diseases.
Keywords: Proteasome, Tau, Neurite outgrowth, FRET, Proteostasis, Drug discovery
Introduction
Cellular protein homeostasis, or proteostasis, is a common driver of pathology in aging and neurodegeneration [1]. The proteostasis network maintains proper protein folding, assembly, and degradation through a complex interplay of chaperones, proteolytic systems, and regulatory elements. Increasing evidence suggests that deterioration in this network—either through mutations or other cellular stresses—leads to the accumulation of aggregated proteins. This buildup of misfolded proteins can reorganize into proteotoxic substrates (e.g., oligomers or fibrils), a process that is central to the pathogenesis of Alzheimer’s disease (AD) and related neurodegenerative diseases [2–4].
Misfolded oligomeric and aggregated tau proteins have been shown to disrupt cellular proteostasis [5, 6]. Inhibiting their formation or enhancing their clearance has been the target of multiple therapeutic discovery campaigns for AD [7–9]. Previously, our group identified MK886 (MK) as a lead compound capable of modulating tau misfolding and oligomerization in a cellular fluorescence-lifetime förster resonance energy transfer (FLT-FRET) high-throughput screen [10]. MK directly binds to recombinant tau and rescued SHSY5Y cytotoxicity induced by overexpression of either wildtype (WT) or P301L tau [10]. Although we observed direct tau engagement of MK to recombinant tau protein, the cellular mechanism of action (MOA) for MK could be a combination of direct tau engagement or other indirect targets; the nature of which was not thoroughly explored in that study.
MK was developed as a drug candidate to target leukotriene biosynthesis and was shown to be a potent inhibitor (IC50 of 3nM) of a key chaperone, 5-lipoxygenase-activating protein (FLAP) [11–14]. However, in a study investigating amyloidbeta (Aβ) induced toxicity in neuroblastoma cells, MK was shown to rescue Aβ induced cell death by increasing proteasomal activity and preventing Aβ accumulation [15]. Proteasomal activation by MK likely occurs through the 20S proteasome. The 20S is the proteolytic core particle of 26S proteasome and is recognized as the protease most suited for degrading intrinsically disordered proteins (IDP), like Aβ and tau [16, 17]. Due to its unstructured nature, tau is readily degraded by the 20S proteasome without the need for ubiquitination or unfolding by the 19S or 26S proteasome. Indeed, MK was identified as a proteasomal activator (with an EC50 of 32 μM) in a drug screen using a recombinant 20S proteasome cleavage assay [18]. The proteasomal stimulation by MK was confirmed using LC-MS and further validated in cellulo with GFP cleavage from a GFP-alpha synuclein fusion protein [18, 19].
In this study, we investigated the proteasomal MOA of MK and then explored the effects of proteasomal stimulation on neurite outgrowth and tau aggregation. We first validated the proteasomal activation of MK in a 20S core particle assay and then established a baseline for the proteasomal stimulation by MK in HEK293T and SHSY5Y cells. In addition to the 20S proteasome assays, we confirmed proteasomal stimulation by MK in cellulo by evaluating GFP cleavage from a tau-GFP fusion protein expressed in HEK293T cells. We then evaluated the effects of proteasomal stimulation in a more pathologically relevant neurite outgrowth model, which we developed using differentiated SHSY5Y neurospheres (SHNS) expressing wildtype (WT, 2N4R) tau. As expected, we observed robust proteasomal activity from MK and in subsequent assays show that MK was able to significantly rescue tau-induced neurite pathology and decrease tau aggregation in our FLT-FRET biosensor.
Due to these results, we expanded our scope of study to a series of seven MK analogs to understand how structural variations to the MK scaffold can alter proteasomal activity. From these seven derivatives, we identified both activators and inhibitors of the proteosome. In addition to proteasomal stimulation, MK and the analogs may also be acting through other proteostasis-related MOAs. Thus, we explored the effects of MK and a subset of analogs on rescuing tau-induced deficits in an autophagy biosensor as well as targeting the inflammatory pathway through FLAP and NF-κB inhibition. We demonstrate that analogs which were proteasomal activators exerted a neuroprotective role in promoting neurite outgrowth and reducing tau aggregation, while inhibitory compounds were more detrimental to neuronal projections and had reduced effects on tau aggregation and autophagy. Taken together, our findings suggest that further development of MK can potentially lead to the discovery of a novel therapeutic that can improve the encumbered proteostasis in aging and neurodegenerative diseases.
Results
MK Evaluation
Synthesis of MK
MK886 was synthesized in-house using the synthetic route described in Supplemental Scheme S1 [20]. The approach employed exploits the classic Fischer indole synthesis using the appropriately substituted phenylhydrazine and ketone precursors to arrive at the final product in 42% overall yield in > 95% purity.
Evaluation of Proteasomal Activity of MK
Prior studies have shown that tau can be degraded through the 20S proteasome [19, 21, 22] and that MK can stimulate proteasomal activity as a true agonist of the 20S proteasome [18, 23]. We first confirmed that MK generated a robust response in 20S activity at most concentrations tested. Measurements taken at the end of the assay show that concentrations of MK between 2 and 100 μM significantly increased the amount of AMC that was cleaved compared to 20S alone (Supplemental Figure S1). We then adapted a previously developed in cellulo GFP cleavage assay [18, 24] to monitor tau catabolism using a tau-GFP fusion protein. Due to its unstructured nature, the tau portion of the fusion protein is easily degraded by 20S proteasome whereas the GFP is left intact due to the inability of the 20S proteasome to degrade its highly stable β-barrel structure without additional modifications. Therefore, the amount of cleaved GFP can be used to approximate the amount of tau proteolysis that has occurred [25].
Using this cellular proteasomal assay, we evaluated the proteasomal-stimulating potency of MK in the cellular environment. HEK293T cells were transfected for 48 h with a tau-GFP construct then incubated with 20 μM MK for 20 h. Immunoblots probing for GFP and tau (Fig. 1a) and their quantification (Fig. 1b) confirm that MK significantly (p < 0.0001) increased the amount of cleaved GFP in the cells. In our cellular model of tau overexpression, we do not expect to see a large depreciation in the amount of tau over the duration of the in cellulo GFP cleavage assay due to the high amount of protein in constant production by the HEK293T cell line. However, MK treatment did increase the amount of shorter tau fragments that were detected by immunoblots. Since there is very little endogenous tau in HEK293T, the immunoblot bands correspond to the full-length tau-GFP fusion protein and various cleavage products due to proteasomal activity and potentially other proteostasis pathways.
Fig. 1. MK886 stimulates the proteasome and rescues tau induced ► neurite pathology.
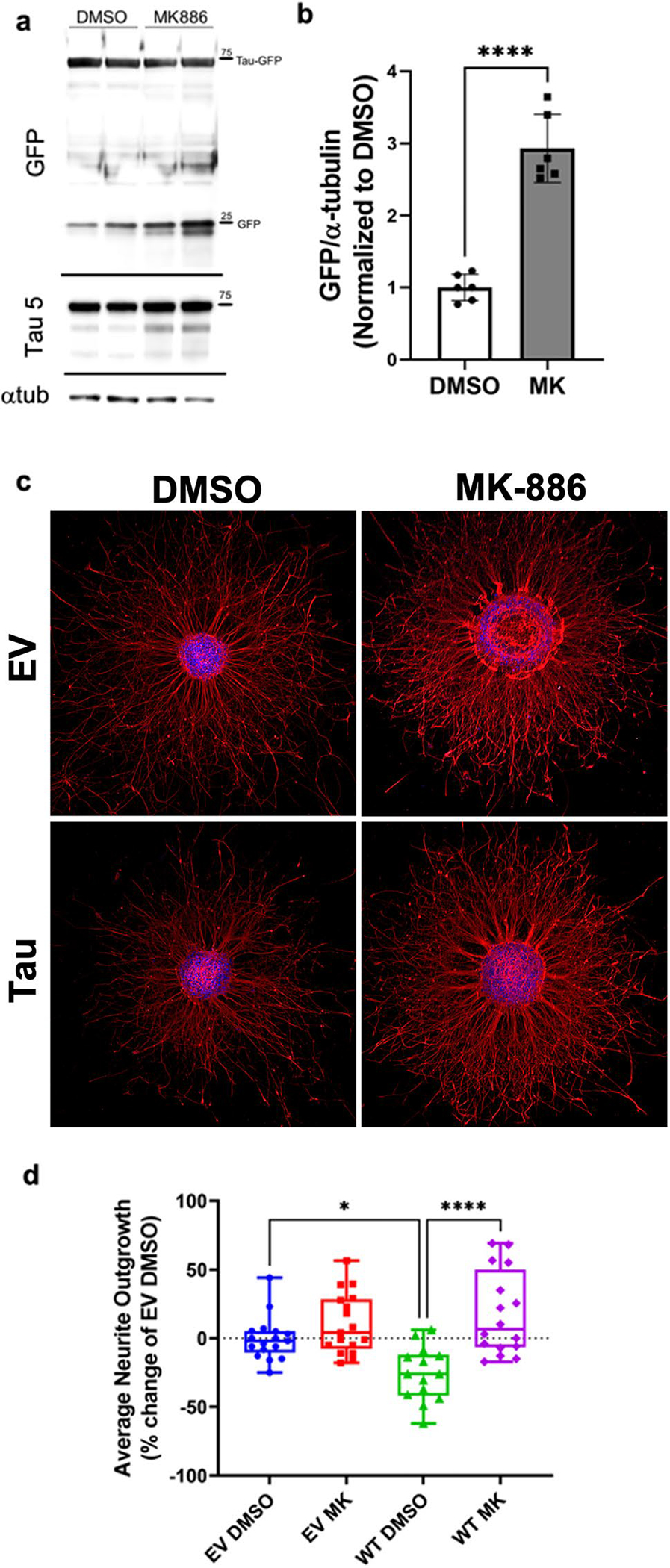
a Immunoblots from the proteasomal GFP cleavage assay conducted in HEK293T cells expressing tau-GFP. The blot probing GFP shows both tau-GFP expression and GFP cleavage in cells treated with either DMSO or 20 μM MK886 (MK) for 20 h. Blots probing tau (Tau-5) show an increase in shorter tau fragments with MK treatment. Loading control, α-tubulin, is also presented. b Quantification of GFP cleavage showing MK significantly increased the amount of free GFP (p < 0.0001) compared with DMSO controls. c Representative images of SHNS expressing empty vector (EV) or tau and treated with either DMSO or 2 μM MK for 36 h. Neurite outgrowth is labeled with β(III)-tubulin, while the neurosphere body is stained with DAPI. The bar = 300 μm for all images. d Quantification of neurite outgrowth from the SHNS using a custom module in MetaXpress image analysis program. The average neurite outgrowth from tau-expressing SHNS is significantly shorter than EV controls (p < 0.05). Treatment with MK significantly rescued this tau-induced deficit (p < 0.0001) and increased neurite density
MK Increased Neurite Outgrowth from SHSY5Y Neurospheres (SHNS)
We next evaluated the effects of proteasomal activation by MK in a more physiologically relevant model. SHSY5Y neuroblastoma cells were initially assessed for their response to MK using a cellular lysate proteasomal assay. SHSY5Y have lower proteasomal capacity and had a smaller dynamic range compared to HEK293T (Supplemental Figure S2). The HEK293T cells showed maximal activity at 20–40 μM drug concentrations, while SHSY5Y cells showed maximal activity at 0.5–2 μM concentrations. These differences in proteasomal capacity require adjustments to the treatment conditions for specific cell lines, as high concentrations of MK can increase oxidative stress and lead to cellular apoptosis [26–29].
Using differentiated SHSY5Y cells, we developed a pathologic neurite outgrowth model to evaluate the neuronal development and degeneration due to tau overexpression. Neurite outgrowth is a common assay used to assess neuronal health and to examine neurotrophic factors and environments that are conducive for neurite extension. Understanding the mechanisms that contribute to dynamic neurite outgrowth can elucidate the factors that contribute to neurodegeneration when a pathologic insult is applied. The 3D environment of the neurospheres greatly enhances differentiation capacity of the SHSY5Y and allows the formation of more mature and complex morphologies [30].
SHSY5Y were transfected with empty vector (EV) or WT 2N4R tau (unlabeled). After transfection, the SHSY5Y were cultured in suspension using a modified differentiation protocol [31–33], to encourage differentiation into dopaminergic neurons within 3D neurospheres. Neuronal-like cells are easily visualized within and on the surface of the neurospheres when the cells express Tau-GFP (Supplemental Figure S3a–b). After a 10-day differentiation period, the SHSY5Y neurospheres (SHNS) express markers for mature neurons (e.g., NeuN, β(III)-tubulin, tyrosine hydroxylase (TH), and synaptophysin; Supplemental Figure S3c–d). SHNS were then plated onto poly-L-lysine/laminin-coated plates and treated with compounds for the evaluation of neurite outgrowth.
Figure 1c presents representative SHNS expressing EV or tau that were stained for β(III)-tubulin to high-light the neurites and DAPI to label the nuclei within the SHNS bodies. Quantification of the SHNS average neurite length (Fig. 1d) indicates that tau expression significantly (p < 0.05) decreased neurite outgrowth from the SHNS. The neuronal processes from tau expressing SHNS were shorter and slightly more dystrophic. This finding is corroborated by another study in which overexpression of tau in neuroblastoma cells resulted in decreased neurite length and impaired mitochondrial and vesicle transport [34]. Tau overexpression has also been shown to significantly downregulate BDNF expression [35], a neurotrophic factor that is essential for neurite development and synaptic plasticity [36, 37] in both SHSY5Y cells and transgenic mice. We have previously shown that MK can protect SHSY5Y cells from P301L tau-induced cytotoxicity with in IC50 of 0.5 μM [10]. With 2 μM MK treatment, there is a dramatic increase in neurite outgrowth and process density from the tau SHNS compared to DMSO controls. MK significantly rescued the neuronal projections from tau-expressing SHNS (p < 0.001), recovering to levels on par with EV-expressing controls (Fig. 1d). Furthermore, immunoblots of Tau-GFP expressing SHNS treated with MK significantly reduced the amount of introduced tau (Supplemental Figure S3e–f), illustrating that treatment with MK can stimulate the degradation of tau.
Synthesis of MK Derivatives
We then designed a small set of MK analogs to determine if the proteasomal activity is sensitive to structural permutations and to ascertain if MK is a good scaffold for a future structure-activity relationship campaign. We chose the N-chlorobenzyl, S-tert-butyl, indole-5-isopropyl, and the carboxylic acid as areas of interest to modify and generated seven analogs (Fig. 2) using the aforementioned synthetic route for MK886 (Supplemental Scheme S1). Dynamic light scattering nanoparticle analysis was conducted on MK, 1H, and 5H to determine compound solubility at 20, 50, and 100 μM (Supplemental Figure S4).
Fig. 2.
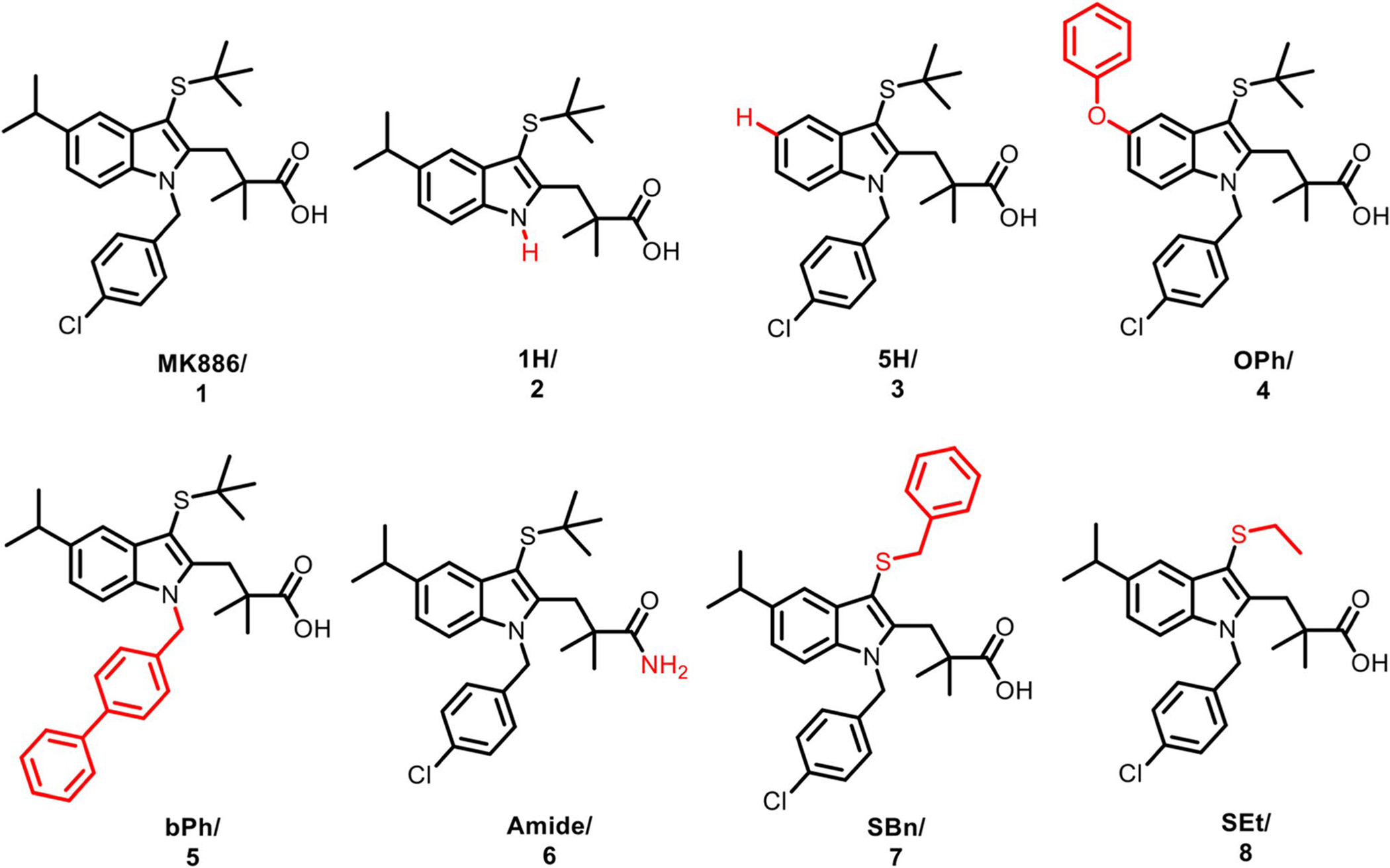
Chemical structure of MK886 and the seven analogs explored in this study with structural modifications shown in red. Amendments to the N-chlorobenzyl group resulted in compounds 1H (2) and bPh (5). The indole-5-isopropyl was replaced to generate compound 5H (3) and OPh (4). The carboxylic acid was replaced with an amino substituent to make (Amide, 6). Finally, conversion of the S-tert-butyl gave rise to compounds SBn (7) and SEt (8). Synthesis scheme is presented in Supplemental Scheme S1
Evaluation of MK Derivatives Using the in Cellulo Proteasomal GFP Cleavage Assay
Using the same in cellulo proteasome GFP cleavage assay described above, we assessed the ability of MK analogs to cleave GFP from tau-GFP fusion proteins and compared them to the baseline established by MK. HEK293T cells expressing the tau-GFP construct were incubated with the MK analogs at 20 μM concentrations for 20 h. The cumulative results from triplicate experiments are shown in Fig. 3 (and Supplemental Figure S5a with replicate immunoblots presented in Supplemental Figure S6). One derivative, 5H, significantly (p < 0.001) stimulated proteasomal activity compared with DMSO-treated controls, indicating that the removal of indole-5-isopropyl group is well tolerated and conserves proteasomal activity. Interestingly, replacing the isopropyl with a phenoxy (OPh) instead greatly diminished this activity. The indole N-substitution played a rather pivotal role to the proteasomal function as demonstrated in the contrasting activity profile of bPh and 1H. Extending the N-substituent from chlorobenzyl to phenylbenzyl in bPh, led to slightly increased GFP cleavage, albeit the results were not significantly greater than controls (p = 0.08). In contrast, cells treated with analog 1H, with the chlorobenzyl group removed, had significantly (p < 0.05) lower amounts of cleaved GFP than DMSO control. The amount of cleaved GFP from the treatment of this compound was comparable to the amount of cleaved GFP in the presence of MG132 and bortezomib (BTZ), known proteasomal inhibitors (Supplemental Figure S5). The remaining derivatives (Amide, SBn, and SEt) had significantly lower GFP cleavage compared to MK (Supplemental Figure S5), indicating that both S-tert-butyl and the carboxyl groups also contributed to MK’s proteasomal activity in this assay.
Fig. 3. Evaluation of MK and analogs using a cellular proteasomal Tau-GFP cleavage assay.
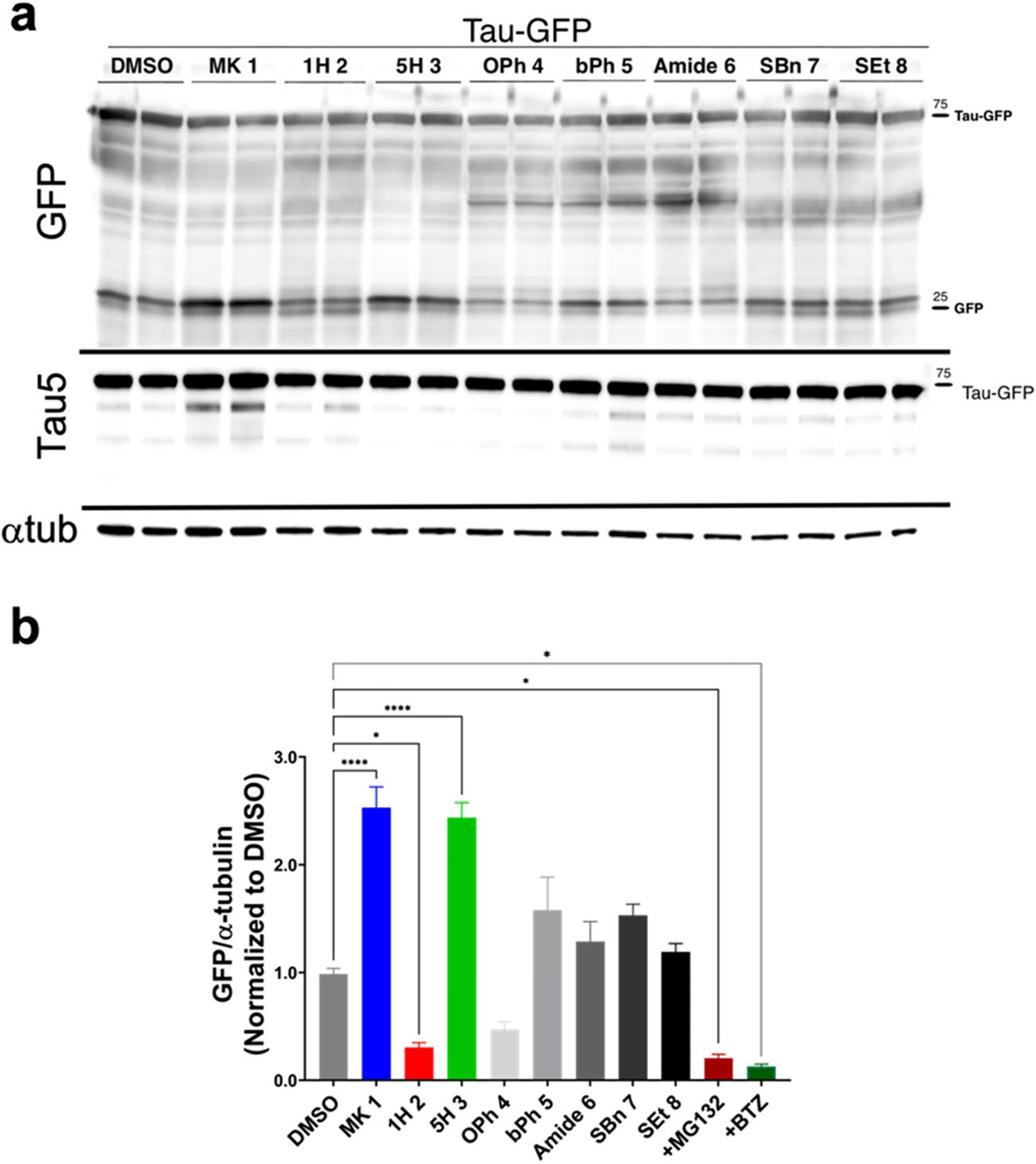
a Cellular HEK293T extracts expressing tau-GFP treated with 20 μM MK analogs (1–8) were collected after 20 h of treatment. Representative immunoblots for GFP, tau (Tau5), and α-tubulin (loading control) are presented. Treatment with MK and a few analogs increased GFP cleavage and tau fragments. b Quantification of GFP cleavage from immunoblots. One analog, 5H significantly increased GFP cleavage (p < 0.0001), while bPh also increased amount of cleaved GFP, but this was not significant (p = 0.08) compared to DMSO. In contrast, one analog, 1H, had significantly lower levels of GFP, which were comparable to the amount present when the proteasome is inhibited with MG132 and bortezomib (BTZ) (Supplemental Figure S5a, c). A replot of this GFP cleavage data with statistical comparison compared to DMSO is presented in Supplemental Figure S5b
Further evaluation of MK, 1H, and 5H was done with co-treatment of a sublethal dose of BTZ, 5 nM BTZ for 20 h [38]. Here we observed that BTZ significantly reduced the amount of 20S activity to an amount even lower than with MG132 inhibition (Supplemental Figure S5c). GFP cleavage with BTZ and MK, 1H and 5H co-treatment was also significantly reduced, indicating that GFP cleavage is primarily due to 20S proteasomal activity.
The proteasomal impact of 1H and 5H was corroborated by 20S core particle assay (Supplemental Figure S1). Analysis of 1H at all concentrations tested had an inhibitory effect on the 20S, while concentrations of 5H at 10 μM and below had significantly higher activity than the 20S core particle alone. The results for 5H were further confirmed in the cellular lysate proteasomal assay where 5H exhibited proteasomal activity between 0.5 and 20 μM in HEK293T cells and at 2 μM in SHSY5Y cells (Supplemental Figure S2a–b). Interestingly, 1H did demonstrate inhibitory activity in SHSY5Y cells, but at the lowest concentration (0.5 μM) it still maintained some activity in HEK2393T whole cell lysate (Supplemental Figure S2a) This result is in contrast with 1H’s response in the 20S core particle assay and suggests that a different MOA might be responsible for the cleavage of AMC in the HEK293T whole cell lysate, such as calpains or aminopeptidases [39].
Tau FLT-FRET Biosensor Shows MK Analogs Maintain Capacity to Reduce Tau-Tau Interactions
All analogs were evaluated in the same tau-tau FLT-FRET assay previously used to identify MK as a lead compound that attenuated FRET (increasing FLT), indicating a decrease in tau aggregation [10]. Our tau biosensor monitors tau-tau protein interactions (e.g., oligomerization) and tau conformation in cellulo via expression of WT (2N4R) tau fused with either donor (C-terminal mEGFP) or acceptor (C-terminal TagRFP) monomerized fluorescent proteins [10]. Using this tau FRET biosensor, we determined the FLT dose response (1nM–50 μM) for MK and all analogs (Supplemental Figure S7). Changes in the FLT at 50 μM compound concentration are presented in Supplemental Figure S8. Consistent with our previous study, MK significantly increased FLT (decreased FRET) relative to DMSO-treated control ((p < 0.0001); Supplemental Figure S8a). Two analogs, 5H (p < 0.05) and bPh (p < 0.0001), also resulted in significantly larger increase in FLT relative to MK (Supplemental Figure 8b), whereas the remaining five analogs (1H, OPh, Amide, SBn, and SEt) displayed a reduced increase in FLT. All but one of the analogs, SBn, were still able to induce a significant increase in FLT relative to DMSO treated control (Supplemental Figure S8a).
Two MK analogs (1H and 5H) elicited a significant change in both proteasome activity and tau-tau interactions (ΔFLT). The direction of these changes is consistent with the modulation of proteasomal activity having a direct effect on ΔFLT. Specifically, increased proteasomal activity with 5H correlates to reduced FRET (increased FLT), whereas inhibition of proteasomal activity with 1H correlates to increased FRET (reduced FLT) relative to MK. This corroboration suggests that the change in FRET may result from proteasomal degradation of tau and corresponds to the release of free donor mEGFP, which has reduced protein-protein interactions with free acceptor TagRFP (compared to the affinity of tau-mEGFP to tau-TagRFP) [10]. Interestingly, even though 1H inhibits the proteasome (Fig. 3b and Supplemental Figure S7b) it did not induce an increase in tau-tau interaction or aggregation (i.e., increased FRET), which would have resulted in a negative ΔFLT. Although this initially appears to be contradictory to observations of increased aggresome formation with disrupted proteostasis, the lack of increased FRET likely results from differences between the rates at which proteasome activators stimulate protein degradation and the time required for proteasome inhibitors to induce protein aggregation. Guthrie et al. observed protease inhibitor-induced recruitment of tau to aggresomes occurs over an 18-h window with nascent aggresomes beginning to form around 3 h [40]. In contrast, proteasome stimulation can induce a detectable response using fluorogenic probes within minutes of treatment and would have a similar response time for our tau FRET biosensors [22, 41]. These discrepancies are not captured in the different treatment protocols used for the proteasome (20-h treatment) relative to the FLT-FRET (90 min) assays.
Neurite Outgrowth with 1H and 5H Treatment
The contradistinction of 1H and 5H, which displayed opposing effects on proteasomal and tau aggregation activity, were further characterized in our SHNS assay. Neurite outgrowth from 1H- and 5H-treated SHNS also reflected the proteasomal effects of the compounds (Fig. 4). The neurite outgrowth from SHNS treated with proteasome inhibitor 1H was more truncated with altered morphologies compared with both control and MK-treated SHNS (Fig. 4a). Whereas treatment with proteasome activator 5H not only significantly increased neurite outgrowth in both EV (p < 0.0001) and WT tau (p < 0.05) SHNS compared to EV DMSO, but also increased the neurite density. Interestingly, 5H also significantly increased neurite lengths over EV SHNS treated with MK (p < 0.001), but a corresponding increase was not observed in tau expressing SHNS treated with 5H (Fig. 4b). Neuronal projections from tau SHNS treated with MK and 5H were not significantly different (Supplemental Figure S9). These results suggest that the effects of 5H (e.g., significantly increased neurite growth) are subdued when tau is overexpressed. Since the proteasomal stimulating capacity of 5H is similar to MK and 5H elicited a greater ΔFLT change, perhaps an additional non-proteasomal MOA may be involved in the SHNS tau-induced neurite deficit. To further elucidate the activity of these analogs we investigated the effects of MK, 1H, and 5H on autophagy and inflammation, alternative MOAs that may contribute to changes in tau proteostasis.
Fig. 4. MK derivatives modulate the proteasome and can also affect tau induced neurite pathology.
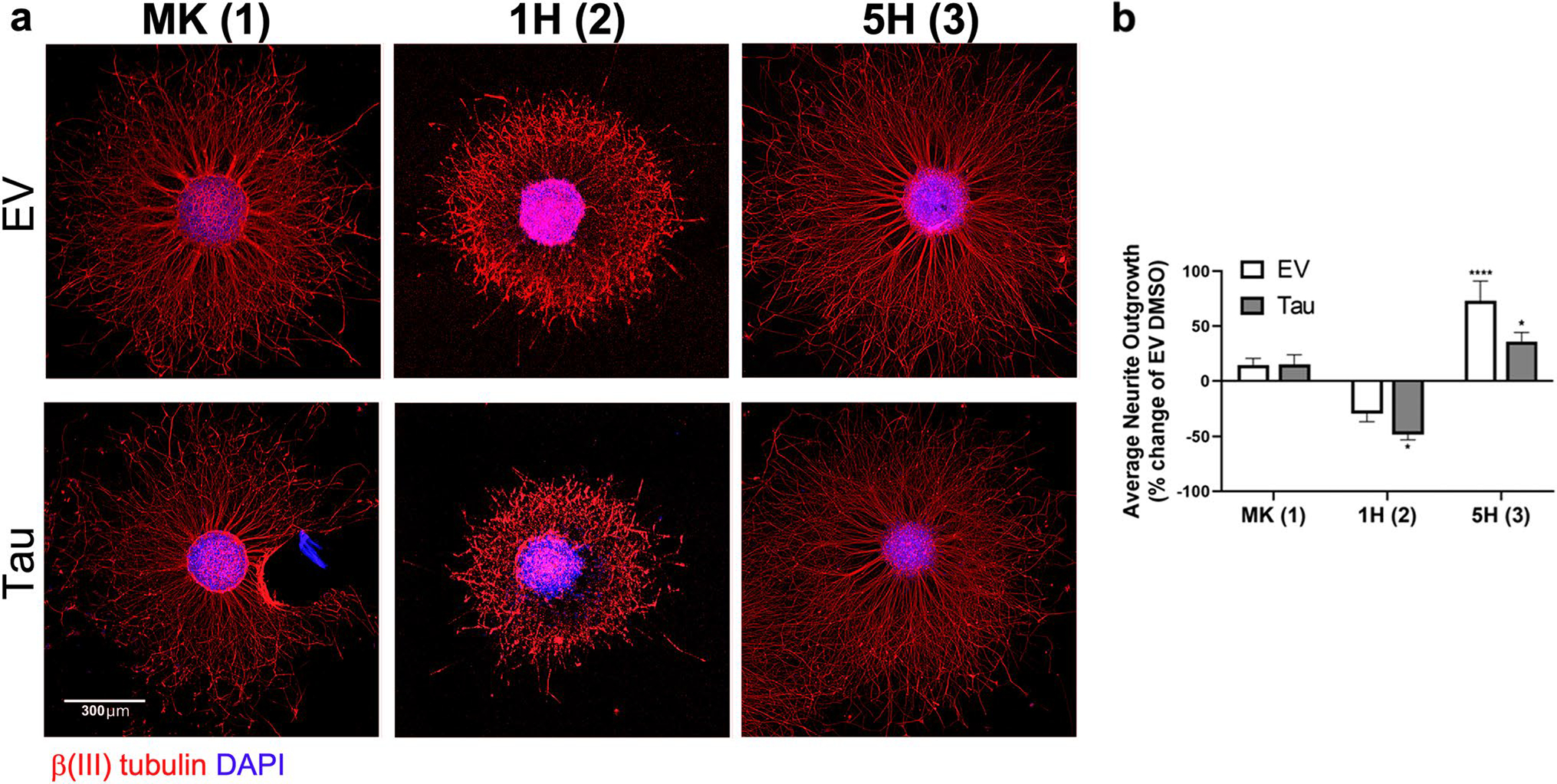
a Images of representative SHNS expressing EV or tau and treated with 2 μM 1H or 5H are shown, along with SHNS treated with MK for comparison. Neuronal projections treated with 1H were more truncated, while 5H treatment significantly enhanced the length and neurite density. Scale bar is 300 μm for all images. b Quantification of neurite outgrowth from SHNS treated with 1H or 5H compared to EV DMSO. Derivative 1H was significantly (p < 0.05) different than EV controls, but the neurite lengths were not significantly different than the Tau DMSO group. The addition of 5H to SHNS significantly increased the neurite outgrowth for both EV and tau expressing SHNS, (p < 0.0001) and (p < 0.05), respectively. Although, the length of the neurites in Tau SHNS treated with 5H was not significantly different from neurite outgrowth from Tau SHNS treated with MK (Supplemental Figure S9)
Evaluation of Autophagic Activity of MK and Analogs 1H and 5H
Interaction between the proteasome and autophagy is essential to maintaining cellular homeostasis. We investigated this association by evaluating whether proteasomal stimulation or inhibition by MK and two analogs, 1H and 5H, could also impact macroautophagy activity. The compounds were evaluated using an established autophagy LC3 biosensor probe (GFP-LC3-RFP-LC3ΔG) [42] in conjunction with tau overexpression. Endogenous Atg4 cleaves the fluorescent LC3 construct into equimolar GFP-LC3 and RFP-LC3ΔG. The GFP-LC3 is incorporated into the phagophore membrane, while the RFP-LC3ΔG remains in the cytosol as an internal control to determine autophagic flux [42]. If autophagy is functionally active, then the GFP-LC3 is degraded in the autolysosome. Therefore, the ratio of GFP/RFP provides an estimate of the autophagic flux; high ratio = low flux or inhibition of autophagy, low ratio = high flux or activation of autophagy. Characterization of the LC3 biosensor using known stimulators (starvation) and inhibitors (Bafilomycin A) is presented in Supplemental Figure S10. Tau overexpression in this LC3 biosensor resulted in an increase in the GFP/RFP ratio over empty vector (EV) controls, indicating a dysfunction in autophagy (p < 0.05, Supplemental Figure S10).
Treatment of the LC3 tau biosensor with 10 μM MK, 1H, and 5H was conducted to determine if changes in proteasomal activity could rescue tau-induced dysfunctional autophagy. The addition of MK significantly reduced the GFP/RFP ratio (p < 0.05) in tau-expressing cells, indicating stimulation of autophagy (Fig. 5). Analog 5H displayed an even greater autophagy stimulation response compared to MK, significantly decreasing the GFP/RFP ratio (p < 0.001). Conversely, treatment with 1H increased the GFP/RFP ratio above the basal WT tau levels, but the effect was not significantly different than WT controls. This effect is demonstrated by the increase in the GFP signal in the cells treated with 1H compared to those treated with MK or 5H (Fig. 5).
Fig. 5. Phenotypic autophagy LC3-biosensor suggests that MK stimulates autophagy, while 1H and 5H have contrasting effects.
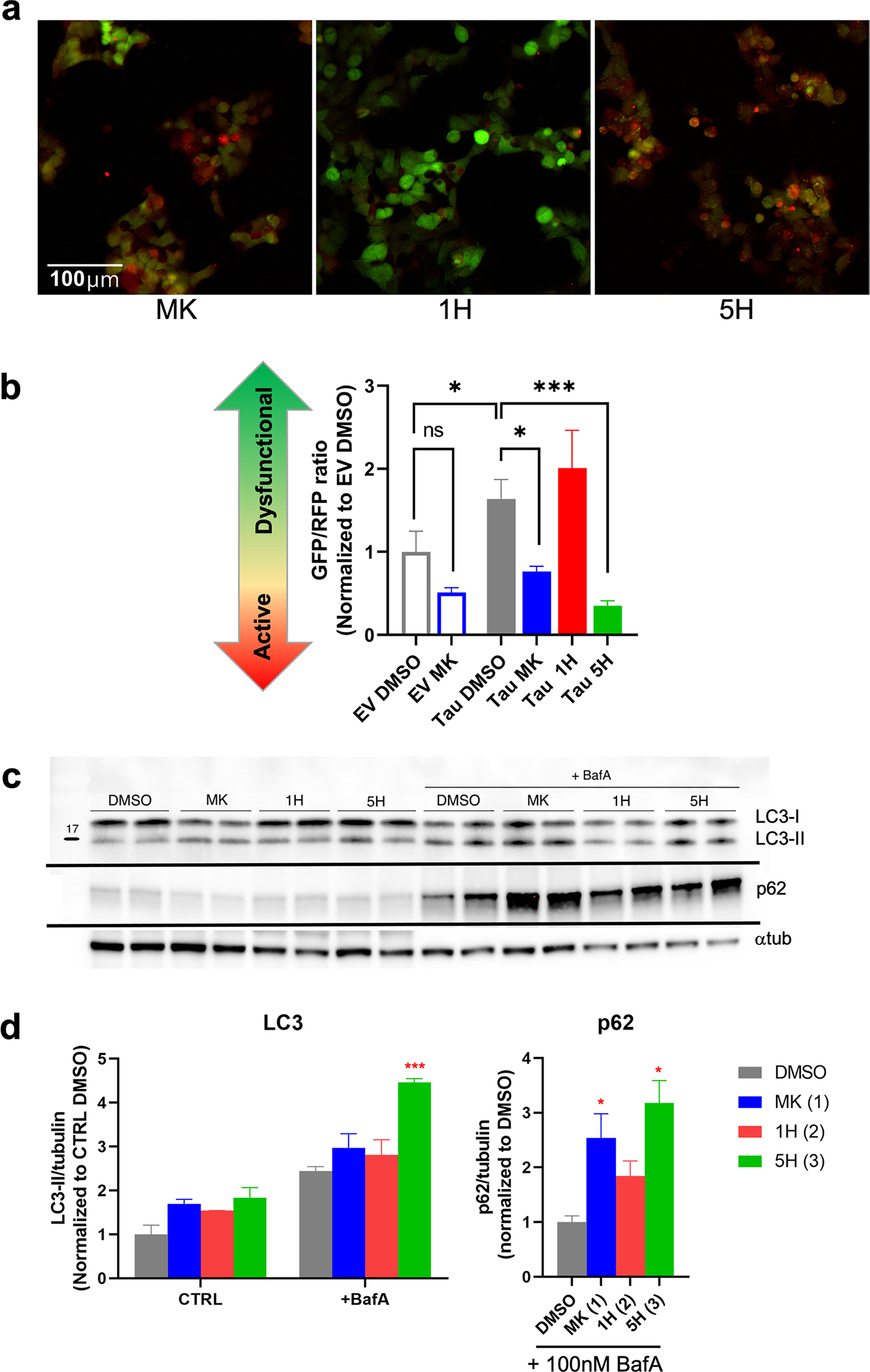
a Images of tau-expressing LC3 biosensor treated with 10 μM MK, 1H, or 5H. for 24 h. Cells treated with MK and 5H appear more red, indicating activation of autophagy. In contrast, 1H is visibly greener, which illustrates that 1H is causing greater autophagic dysfunction in these cells. Scale bar is 100 μm. b Quantification of the GFP/RFP ratio was conducted on both EV and tau expressing LC3 biosensor. Tau expression significantly increased the GFP/RFP ratio compared to EV expressing controls. Treatment with MK (p < 0.05) and 5H (p < 0.001) significantly increased the flux (lower GFP/RFP), while 1H increased the GFP/RFP ratio, but was not significant compared to Tau DMSO. c Immunoblots of LC3 and p62 from a LC3 turnover assay confirm activation of autophagy with 5H. HEK293T cells were treated with DMSO, 20 μM MK, 1H, or 5H with or without 100 nM bafilomycin A for 24 h. d Quantification of immunoblots show significant accumulation of both LC3-II and p62 with treatment of 5H, indicating that it is an autophagy activator. Chaperone, p62, is also increased with MK treatment, but LC3-II is not significantly higher than DMSO controls. The autophagic flux of 1H is also increased over controls, suggesting that autophagy mechanisms may be trying to counteract the inhibition of the proteasome
An LC3 turnover assay was conducted to further verify the results from the LC3 biosensor. LC3 and p62 expression were evaluated with and without 24-h Bafilomycin A treatment. Immunoblot quantification shows that treatment with both MK and 5H resulted in greater accumulation of both LC3-II and p62, which indicates a higher rate of autophagic flux (Fig. 5c). The significant accumulation of p62 and the increase of LC3-II with 5H treatment suggests that it is an autophagy activator. Although the flux was not significantly different than DMSO controls, treatment with 1H was higher than controls suggesting that an increase in autophagy might be compensating for inhibition of the proteasome [2, 43].
FLAP and NF-κB Inhibition by MK, 1H, and 5H
Inflammation is another component in the pathogenesis of neurodegenerative diseases. MK is a potent inhibitor of FLAP, a key chaperone in the 5-lipoxygenase (5-LO) pathway, which produces pro-inflammatory leukotrienes [11]. We explored the effects of MK, 1H, and 5H on FLAP expression in HEK293T cells. Non-immune cells, like HEK293 epithelial cells, express bioactive molecules that convey inflammatory signals to the immune cells as part of the protective immune response [44], and this response can be further modulated by inhibitors and stimulators [45, 46]. Untransfected HEK293T cells do not express FLAP under basal conditions (Supplemental Figure S11a). Seventy-two hours after transfection, Tau-GFP-expressing cells have increased FLAP expression, presumably due to the cationic lipids used to facilitate DNA delivery and overexpression of tau protein. Treatment of these cells with a range of MK doses between 5 and 30 μM reduced the FLAP expression greater than fifty percent (Supplemental Figure S11b). Although at 40 μM MK concentration, the FLAP levels increased to a level more comparable to DMSO-treated controls. FLAP expression decreased slightly with increasing 1H concentrations, but even at 30 μM concentration it was only 20% lower than DMSO controls. Additionally, treatment with 5H was not effective at reducing FLAP until the 30 μM concentration. This finding suggests that both the N-chlorobenzyl and indole-5-isopropyl groups on MK contribute to its ability to inhibit FLAP, as changing these groups decreased its anti-inflammatory capacity.
The anti-inflammatory effect of MK has been also been directly linked to decreased NF-κB signaling [46]. NF-κB is a critical mediator of pro-inflammatory responses. Activation of the IκB kinase complex (IKK) leads to phosphorylation of NF-κB p65 and its subsequent translocation into the nucleus where it initiates pro-inflammatory gene expression. MK has been shown to reduce the activation of IKK through inhibition of 5-LO. Immunoblots of phospho--F-κB p65 (S536) reflect that treatment with MK, 1H and 5H show some ability to attenuate phosphorylation of NF-κB (Supplemental Figure S11c–d). Only 5 μM MK treatment significantly reduced the phosphorylation state of NF-kB (p < 0.05). There is also a decrease in phospho-NF-κB with 15 μM 1H and 5H treatment, indicating some anti-inflammatory capacity for these compounds at certain concentrations but they are not as potent as MK.
Collectively, we observe contrasting results when comparing the overall activity profile of MK to the analogs 1H and 5H. Loss of N-chlorobenzyl in 1H was antagonistic in all the assays tested. With 5H, the removal of the indole-5-isopropyl group enhanced autophagy flux and decreased tau oligomerization (increased FLT), both of which exceeded MK’s capabilities. This resulted in significant increases in neuronal projections, but 5H was unable to surmount the tau-induced neurite deficit relative to 5H-treated EV. It is possible that this inadequacy is due to the reduced anti-inflammatory capacity of 5H.
Discussion
In our aim to further characterize MK as a proteasome activator and neuroprotective agent in cellular models of tauopathy, we found that modifying certain chemical moieties of MK resulted in distinct effects on both the proteasome and anti-inflammatory activity. Supplemental Table S1 presents a summary of the seven MK analogs synthesized and their effects on proteasome activity and other secondary assays. While 5H has comparable proteasomal activity to the original MK, our attempts to remove or modify other substituents diminished the potency of proteasomal activation. These newly introduced groups may hinder the ability of the analogs to stimulate the 20S proteasome. One compound in particular, 1H, had a large inhibitory effect on proteasomal activity. The results from our assays indicate that the loss of the N-chlorobenzyl group acted as a chemical switch in turning the proteasome activator MK into an inhibitor. This reduction in proteasomal activity also correlated to reductions in both neurite outgrowth and FLAP inhibition.
Engagement of the proteasome has been implicated in regulating tau aggregation. A recent study by Ukmar-Godec et al. found that the 20S preferentially targets the R3 repeat domain in tau and cleaves the PHF6 306VQIVYK311 motif, so that the digested peptides are unable to aggregate [47]. This finding is consistent to our ΔFLT results observed with our tau FLT-FRET biosensor. Stimulation of the 20S proteasome by MK and select analogs increases the digestion of tau into potentially non-aggregating fragments (reducing FRET, increasing FLT). Evidence of tau cleavage can be seen in the immunoblots probing for tau in Fig. 1a and Supplemental Figure S5 and S6.
In neurodegenerative diseases, the production and dysregulation of IDPs can overwhelm the 20S proteasome, creating blockage in the degradation machinery. Modifications to tau (e.g., acetylation, point mutations, and phosphorylation), diminish or inhibit 20S proteasomal degradation [47]. Under these obstructed conditions, all three branches of autophagy (macroautophagy, chaperone-mediated autophagy (CMA), and endosomal microautophagy) are recruited to remove the pathogenic tau [2, 48]. But conversely, if autophagy is inhibited, then ubiquitin-proteasome flux also becomes encumbered due to the accumulation of p62 [49]. Compound 1H elicited inhibition of the proteasome, but the slightly increased autophagic flux suggests that the autophagic mechanisms may be trying to compensate for the proteasomal inhibition.
Crosstalk between the proteasome and autophagy-lysosomal pathways help maintain proteostasis within the cell and there are multiple chaperones, such as ubiquitin and p62, that facilitate synergism between the two proteolytic systems [50–52]. Both MK and 5H elicited a strong proteasomal response as well as stimulated autophagy. The enhanced autophagic activity of 5H over MK also corresponded with significant reduction in tau-tau oligomerization (reduced FRET). Since the proteasomal stimulation of MK and 5H are similar, this difference might signify the autophagic contribution to the degradation of tau and may explain the loss of tau cleavage fragments with 5H treatment seen in tau immunoblots (Fig. 3a). Further studies are needed to delineate the contributions of each proteolytic system in our experiments.
MK was developed as a potent FLAP inhibitor as its primary MOA. In this study, we show that changing substituents on MK can affect both FLAP and NF-κB inhibition. Studies have shown that targeting the 5-LO pathway is neuroprotective in models of AD because neuroinflammation can further aggravate neurodegeneration [53–57]. Treatment of transgenic mice with FLAP inhibitors and 5-LO knockout mice significantly reduced tau pathology and rescued synaptic integrity [58–60], suggesting that inhibiting this pathway can protect neurite outgrowth and synaptic function. In our experiments, compounds with reduced FLAP inhibition (1H and 5H) limited the length of neuronal processes, possibly due to a more neuroinflammatory environment where there is higher expression of phosphorylated NF-κB p65 when tau is overexpressed. This is further substantiated by the significant neurite outgrowth seen in 5H treatment of EV SHNS without excess tau. Collectively, these studies indicate that reduction of inflammation by MK can be beneficial and is another relevant target for our compounds in future studies [61].
In conclusion, our study demonstrates that proteasomal stimulation by MK and its derivatives can be used to compensate for the unbalanced proteostasis found in aging and neurodegenerative diseases. We show that proteasomal function is highly sensitive to structural perturbations of the MK scaffold. Consequently, derivatives boosting proteasomal activity can mitigate tau oligomerization and stimulate neurite outgrowth, whereas proteasome-inhibiting derivatives exacerbate the imbalance of proteostasis and neurodegeneration. In addition to proteasomal stimulation, the MOA of MK and its derivatives also includes autophagic and inflammatory activity. Taken together, this study suggests the MK is a suitable candidate for a future in depth structure-activity investigation aimed at understanding how the chemical structure of MK is associated with these distinct biological activities. Ultimately, designing a compound that can target both the inflammatory and the proteasome/autophagy pathways would be beneficial for not only AD, but as a potential therapeutic for other neurodegenerative diseases as well.
Supplementary Material
Acknowledgements
We thank Noboru Mizushima for his generous gift (pMRX-IP-GFP-LC3-RFP-LC3dG) construct. (Addgene plasmid #84572; http://n2t.net/addgene:84572; RRID:Addgene 84572).
Funding
The authors disclose receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the U.S. National Institutes of Health (NIH) to J.N.S. (NINDS NS117968, NIA AG073734, and NIGMS GM131814).
List of Abbreviations
- Aβ
amyloid-beta
- AD
Alzheimer’s disease
- BafA
Bafilomycin A1
- BTZ
Bortezomib
- EV
empty vector
- 5-LO
5-lipoxygenase
- FLAP
5-lipoxygenase-activating protein
- FLT
fluorescence lifetime
- FLT-FRET
Fluorescence LifeTime-Förster Resonance Energy Transfer
- IDP
intrinsically disordered protein
- MK
MK-886
- MOA
mechanism of action
- SHNS
S HSY5Y neurosphere
- WT
wildtype
Footnotes
Declarations
Conflict of Interest The authors declare no competing interests.
Ethics Approval This research did not contain any studies involving human or animal participants.
Consent to Participate This research did not contain any studies involving human or animal participants.
Consent for Publication This research did not contain any studies involving human participants.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s12035-023-03417-5.
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Data Availability
The datasets generated and/or analyzed during the current study are available upon request.
References
- 1.Hipp MS, Kasturi P, Hartl FU (2019) The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol 20(7):421–435. 10.1038/s41580-019-0101-y [DOI] [PubMed] [Google Scholar]
- 2.Lee MJ, Lee JH, Rubinsztein DC (2013) Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol 105:49–59. 10.1016/j.pneurobio.2013.03.001 [DOI] [PubMed] [Google Scholar]
- 3.Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D (2016) α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci U S A 113(7):1931–1936. 10.1073/pnas.1520335113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, Heutink P, Shulman JM et al. (2017) Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140(12):3191–3203. 10.1093/brain/awx285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sweeney P, Park H, Baumann M, Dunlop J, Frydman J, Kopito R, McCampbell A, Leblanc G et al. (2017) Protein misfolding in neurodegenerative diseases: implications and strategies. Transl Neurodegener 6:6. 10.1186/s40035-017-0077-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen JJ, Nathaniel DL, Raghavan P, Nelson M, Tian R, Tse E, Hong JY, See SK et al. (2019) Compromised function of the ESCRT pathway promotes endolysosomal escape of tau seeds and propagation of tau aggregation. J Biol Chem 294(50):18952–18966. 10.1074/jbc.RA119.009432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Congdon EE, Sigurdsson EM (2018) Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol 14(7):399–415. 10.1038/s41582-018-0013-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Njomen E, Tepe JJ (2019) Proteasome activation as a new therapeutic approach to target proteotoxic disorders. J Med Chem 62(14):6469–6481. 10.1021/acs.jmedchem.9b00101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng C, Reis SA, Adams ET, Fass DM, Angus SP, Stuhlmiller TJ, Richardson J, Olafson H et al. (2021) High-content image-based analysis and proteomic profiling identifies tau phosphorylation inhibitors in a human iPSC-derived glutamatergic neuronal model of tauopathy. Sci Rep 11(1):17029. 10.1038/s41598-021-96227-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lo CH, Lim CK, Ding Z, Wickramasinghe SP, Braun AR, Ashe KH, Rhoades E, Thomas DD et al. (2019) Targeting the ensemble of heterogeneous tau oligomers in cells: a novel small molecule screening platform for tauopathies. Alzheimers Dement 15(11):1489–1502. 10.1016/j.jalz.2019.06.4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rouzer CA, Ford-Hutchinson AW, Morton HE, Gillard JW (1990) MK886, a potent and specific leukotriene biosynthesis inhibitor blocks and reverses the membrane association of 5-lipoxygenase in ionophore-challenged leukocytes. J Biol Chem 265(3):1436–1442 [PubMed] [Google Scholar]
- 12.Dixon RA, Diehl RE, Opas E, Rands E, Vickers PJ, Evans JF, Gillard JW, Miller DK (1990) Requirement of a 5-lipoxygenase-activating protein for leukotriene synthesis. Nature 343(6255):282–284. 10.1038/343282a0 [DOI] [PubMed] [Google Scholar]
- 13.Gillard J, Ford-Hutchinson AW, Chan C, Charleson S, Denis D, Foster A, Fortin R, Leger S et al. (1989) L-663,536 (MK-886) (3-[1-(4-chlorobenzyl)-3-t-butyl-thio-5-isopropylindol-2-yl]-2,2 - dimethylpropanoic acid), a novel, orally active leukotriene biosynthesis inhibitor. Can J Physiol Pharmacol 67(5):456–464. 10.1139/y89-073 [DOI] [PubMed] [Google Scholar]
- 14.Miller DK, Gillard JW, Vickers PJ, Sadowski S, Léveillé C, Mancini JA, Charleson P, Dixon RA et al. (1990) Identification and isolation of a membrane protein necessary for leukotriene production. Nature 343(6255):278–281. 10.1038/343278a0 [DOI] [PubMed] [Google Scholar]
- 15.Valera E, Dargusch R, Maher PA, Schubert D (2013) Modulation of 5-lipoxygenase in proteotoxicity and Alzheimer’s disease. J Neurosci 33(25):10512–10525. 10.1523/JNEUROSCI.5183-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ben-Nissan G, Vimer S, Tarnavsky M, Sharon M (2019) Structural mass spectrometry approaches to study the 20S proteasome. Methods Enzymol 619:179–223. 10.1016/bs.mie.2018.12.029 [DOI] [PubMed] [Google Scholar]
- 17.Zhao X, Yang J (2010) Amyloid-β peptide is a substrate of the human 20S proteasome. ACS Chem Neurosci 1(10):655–660. 10.1021/cn100067e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trader DJ, Simanski S, Dickson P (1861) Kodadek T (2017) Establishment of a suite of assays that support the discovery of proteasome stimulators. Biochim Biophys Acta Gen Subj 4:892–899. 10.1016/j.bbagen.2017.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coleman RA, Trader DJ (2019) Methods to discover and evaluate proteasome small molecule stimulators. Molecules 24(12). 10.3390/molecules24122341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt SJ, Garnes KT, Heys JR, Landvatter SW, Adams JL (1991) Synthesis of tritium labeled MK-886, a leukotriene biosynthesis inhibitor; employment of epibromohydrin as a masked electrophilic acetone synthon. J Label Compd Radiopharm 29(8):891–902. 10.1002/jlcr.2580290805 [DOI] [Google Scholar]
- 21.David DC, Layfield R, Serpell L, Narain Y, Goedert M, Spillantini MG (2002) Proteasomal degradation of tau protein. J Neurochem 83(1):176–185. 10.1046/j.1471-4159.2002.01137.x [DOI] [PubMed] [Google Scholar]
- 22.Coleman RA, Mohallem R, Aryal UK, Trader DJ (2021) Protein degradation profile reveals dynamic nature of 20S proteasome small molecule stimulation. RSC Chem Biol 2(2):636–644. 10.1039/d0cb00191k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith DM, Chang SC, Park S, Finley D, Cheng Y, Goldberg AL (2007) Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Mol Cell 27(5):731–744. 10.1016/j.molcel.2007.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu CW, Corboy MJ, DeMartino GN, Thomas PJ (2003) Endoproteolytic activity of the proteasome. Science 299(5605):408–411. 10.1126/science.1079293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nonaka T, Hasegawa M (2009) A cellular model to monitor proteasome dysfunction by alpha-synuclein. Biochemistry 48(33):8014–8022. 10.1021/bi900619j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.La E, Kern JC, Atarod EB, Kehrer JP (2003) Fatty acid release and oxidation are factors in lipoxygenase inhibitor-induced apoptosis. Toxicol Lett 138(3):193–203. 10.1016/s0378-4274(02)00407-1 [DOI] [PubMed] [Google Scholar]
- 27.Ghosh J, Myers CE (1998) Inhibition of arachidonate 5-lipoxygenase triggers massive apoptosis in human prostate cancer cells. Proc Natl Acad Sci 95(22):13182–13187. 10.1073/pnas.95.22.13182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dittmann KH, Mayer C, Rodemann HP, Petrides PE, Denzlinger C (1998) MK-886, a leukotriene biosynthesis inhibitor, induces antiproliferative effects and apoptosis in HL-60 cells. Leuk Res 22(1):49–53. 10.1016/s0145-2126(97)00132-x [DOI] [PubMed] [Google Scholar]
- 29.Klegeris A, McGeer P (2003) Toxicity of human monocytic THP-1 cells and microglia toward SH-SY5Y neuroblastoma cells is reduced by inhibitors of 5-lipoxygenase and its activating protein FLAP Abstract. J Leukoc Biol 73(3):369–378. 10.1189/jlb.1002482 [DOI] [PubMed] [Google Scholar]
- 30.Seidel D, Krinke D, Jahnke HG, Hirche A, Kloß D, Mack TG, Striggow F, Robitzki A (2012) Induced tauopathy in a novel 3D-culture model mediates neurodegenerative processes: a realtime study on biochips. PLoS One 7(11):e49150. 10.1371/journal.pone.0049150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harris G, Hogberg H, Hartung T, Smirnova L (2017) 3D differentiation of LUHMES cell line to study recovery and delayed neurotoxic effects. Curr Protoc Toxicol 73:11.23.11–11.23.28. 10.1002/cptx.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Encinas M, Iglesias M, Liu Y, Wang H, Muhaisen A, Cena V, Gallego C, Comella JX (2000) Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated neurotrophic factor-dependent human neuron-like cells. J Neurochem 75(3):991–1003. 10.1046/j.1471-4159.2000.0750991.x [DOI] [PubMed] [Google Scholar]
- 33.Smirnova L, Harris G, Delp J, Valadares M, Pamies D, Hogberg HT, Waldmann T, Leist M et al. (2016) A LUHMES 3D dopaminergic neuronal model for neurotoxicity testing allowing long-term exposure and cellular resilience analysis. Arch Toxicol 90(11):2725–2743. 10.1007/s00204-015-1637-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E (1998) Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J Cell Biol 143(3):777–794. 10.1083/jcb.143.3.777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosa E, Mahendram S, Ke YD, Ittner LM, Ginsberg SD, Fahnestock M (2016) Tau downregulates BDNF expression in animal and cellular models of Alzheimer’s disease. Neurobiol Aging 48:135–142. 10.1016/j.neurobiolaging.2016.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24:677–736. 10.1146/annurev.neuro.24.1.677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hofer MM, Barde YA (1988) Brain-derived neurotrophic factor prevents neuronal death in vivo. Nature 331(6153):261–262. 10.1038/331261a0 [DOI] [PubMed] [Google Scholar]
- 38.Dasgupta S, Castro LM, Dulman R, Yang C, Schmidt M, Ferro ES, Fricker LD (2014) Proteasome inhibitors alter levels of intracellular peptides in HEK293T and SH-SY5Y cells. PLoS One 9(7):e103604. 10.1371/journal.pone.0103604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giguere CJ, Schnellmann RG (2008) Limitations of SLLVY-AMC in calpain and proteasome measurements. Biochem Biophys Res Commun 371(3):578–581. 10.1016/j.bbrc.2008.04.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guthrie CR, Kraemer BC (2011) Proteasome inhibition drives HDAC6-dependent recruitment of tau to aggresomes. J Mol Neurosci 45(1):32–41. 10.1007/s12031-011-9502-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verplank JJS, Lokireddy S, Zhao J, Goldberg AL (2019) 26S Proteasomes are rapidly activated by diverse hormones and physiological states that raise cAMP and cause Rpn6 phosphorylation. Proc Natl Acad Sci 116(10):4228–4237. 10.1073/pnas.1809254116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaizuka T, Morishita H, Hama Y, Tsukamoto S, Matsui T, Toyota Y, Kodama A, Ishihara T et al. (2016) An autophagic flux probe that releases an internal control. Mol Cell 64(4):835–849. 10.1016/j.molcel.2016.09.037 [DOI] [PubMed] [Google Scholar]
- 43.Zhu K, Dunner K, McConkey DJ (2010) Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 29(3):451–462. 10.1038/onc.2009.343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dainichi T, Kabashima K, Ivanov II, Goto Y (2021) Editorial: regulation of Immunity by Non-Immune Cells. Front Immunol 12:770847. 10.3389/fimmu.2021.770847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poirier SJ, Boudreau LH, Flamand N, Surette ME (2020) LPS induces ALOX5 promoter activation and 5-lipoxygenase expression in human monocytic cells. Prostaglandins Leukot Essent Fatty Acids 154:102078. 10.1016/j.plefa.2020.102078 [DOI] [PubMed] [Google Scholar]
- 46.Lin HC, Lin TH, Wu MY, Chiu YC, Tang CH, Hour MJ, Liou HC, Tu HJ et al. 5-Lipoxygenase inhibitors attenuate TNF-α-induced inflammation in human synovial fibroblasts. PLoS One 9(9):e107890. 10.1371/journal.pone.0107890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ukmar-Godec T, Fang P, Ibáñez de Opakua A, Henneberg F, Godec A, Pan KT, Cima-Omori MS, Chari A et al. (2020) Proteasomal degradation of the intrinsically disordered protein tau at single-residue resolution. Sci Adv 6(30):eaba3916. 10.1126/sciadv.aba3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caballero B, Wang Y, Diaz A, Tasset I, Juste YR, Stiller B, Mandelkow EM, Mandelkow E et al. (2018) Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 17(1). 10.1111/acel.12692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC (2009) Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 33(4):517–527. 10.1016/j.molcel.2009.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korolchuk VI, Menzies FM, Rubinsztein DC (2010) Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 584(7):1393–1398. 10.1016/j.febslet.2009.12.047 [DOI] [PubMed] [Google Scholar]
- 51.Njomen E, Tepe JJ (2019) Regulation of autophagic flux by the 20S proteasome. Cell Chem Biol 26(9):1283–1294.e1285. 10.1016/j.chembiol.2019.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao J, Goldberg AL (2016) Coordinate regulation of autophagy and the ubiquitin proteasome system by MTOR. Autophagy 12(10):1967–1970. 10.1080/15548627.2016.1205770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T et al. (2015) Neuroin-flammation in Alzheimer’s disease. Lancet Neurol 14(4):388–405. 10.1016/S1474-4422(15)70016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giannopoulos PF, Joshi YB, Chu J, Praticò D (2013) The 12–15-lipoxygenase is a modulator of Alzheimer’s-related tau pathology in vivo. Aging Cell 12(6):1082–1090. 10.1111/acel.12136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chu J, Praticò D (2011) 5-lipoxygenase as an endogenous modulator of amyloid β formation in vivo. Ann Neurol 69(1):34–46. 10.1002/ana.22234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chu J, Praticò D (2011) Pharmacologic blockade of 5-lipoxygenase improves the amyloidotic phenotype of an Alzheimer’s disease transgenic mouse model involvement of γ-secretase. Am J Pathol 178(4):1762–1769. 10.1016/j.ajpath.2010.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Chu J, Praticò D (2009) The 5-lipoxygenase as a common pathway for pathological brain and vascular aging. Cardiovasc Psychiatry Neurol 2009:174657. 10.1155/2009/174657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chu J, Lauretti E, Di Meco A, Praticò D (2013) FLAP pharmacological blockade modulates metabolism of endogenous tau in vivo. Transl Psychiatry 3:e333. 10.1038/tp.2013.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vagnozzi AN, Giannopoulos PF, Praticò D (2018) Brain 5-lipoxygenase over-expression worsens memory, synaptic integrity, and tau pathology in the P301S mice. Aging Cell 17(1). 10.1111/acel.12695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Giannopoulos PF, Chu J, Joshi YB, Sperow M, Li JG, Kirby LG, Praticò D (2013) 5-lipoxygenase activating protein reduction ameliorates cognitive deficit, synaptic dysfunction, and neuropathology in a mouse model of Alzheimer’s disease. Biol Psychiatry 74(5):348–356. 10.1016/j.biopsych.2013.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Vagnozzi AN, Giannopoulos PF, Praticò D (2017) The direct role of 5-lipoxygenase on tau pathology, synaptic integrity and cognition in a mouse model of tauopathy. Transl Psychiatry 7(12):1288. 10.1038/s41398-017-0017-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the current study are available upon request.