

Summary:
CRISPR-based editing has revolutionized genome engineering despite the observation that many DNA sequences remain challenging to target. Unproductive interactions formed between the single guide RNA’s (sgRNA) Cas9-binding scaffold domain and DNA-binding antisense domain are often responsible for such limited editing resolution. To bypass this limitation, we develop a functional SELEX (Systematic Evolution of Ligands by Exponential Enrichment) approach, termed BLADE (Binding and Ligand Activated Directed Evolution), to identify numerous, diverse sgRNA variants that bind S. pyogenes Cas9 and support DNA cleavage. These variants demonstrate surprising malleability in sgRNA sequence. We also observe that particular variants partner more effectively with specific DNA-binding antisense domains, yielding combinations with enhanced editing efficiencies at various target sites. Using molecular evolution, CRISPR-based systems could be created to efficiently edit even challenging DNA sequences making the genome more tractable to engineering. This selection approach will be valuable for generating sgRNAs with a range of useful activities.
Keywords: CRISPR, SELEX, Cas9, guide RNA, aptamer, DNA editing, Molecular Evolution
eTOC blurb
Bush et al. present a high throughput selection and RNA evolution scheme that can identify functional sgRNA scaffold domains with distinct properties. These variants provide additional insight into the structure-activity-relationships required for effective Cas9 guided DNA editing. BLADE SELEX promises to facilitate the development of sgRNA variants with new activities.
Graphical Abstract
Introduction:
The discovery that CRISPR (clustered regularly interspaced short palindromic repeats) loci encode RNA-protein complexes that can be directed to modify a specific DNA sequence has revolutionized genome engineering 1–4. Generally, CRISPR-based DNA editing systems consist of two components: a CRISPR-associated endonuclease (Cas), which scans the genome for protospacer adjacent motifs (PAM), and a guide RNA (gRNA), which contains a 20-nt ‘spacer sequence’ complementary to a genomic site preceding a PAM sequence. Naturally occurring Cas9 associated CRISPR systems utilize the gRNA in the form of a duplex consisting of a spacer containing CRISPR RNA (crRNA) and a trans-activating crRNA (tracrRNA) which activates crRNA-guided DNA cleavage by Cas9. Alternatively, the crRNA and tracrRNA can be utilized as a single guide RNA (sgRNA) 2. In the Cas9 system, once a heteroduplex is formed between the Cas9-sgRNA complex and the complementary genomic site, Cas9 induces a double-stranded break (DSB) in the DNA. Subsequent cellular repair of the DSB by non-homologous end joining or homology directed repair (HDR) results in the knockout or alteration of the genes of interest 2,5,6.
Over the last decade, the research and therapeutic application of the CRISPR/Cas system have expanded through rigorous investigation and engineering 1–4. Modifications to the Cas9 nuclease domain have led to the development of nCas9 (Cas9 nickase) and dCas9 (nuclease deficient Cas9) and given rise to variants with higher editing fidelities or altered sequence recognition characteristics 7–10. Additionally, expanding the CRISPR/Cas system through protein fusions has generated Cas9 variants capable of transcriptional regulation and prime editing 11,12.
While much of the current focus revolves around the engineering of Cas9, the interrogation and evolution of the sgRNA scaffold represent another echelon of development and therapeutic potential. Currently, a single guide RNA (sgRNA) is composed of a spacer sequence that specifies the DNA target site and a scaffold that interacts with the Cas9 protein 13. Unfortunately, changing of the spacer sequence to edit different genomic targets while keeping the internal gRNA scaffold sequence constant can often result in sgRNAs with intramolecular interactions that give rise to alternate and sometimes undesirable secondary and tertiary structures. This sequence inflexibility makes many sites within the genome intractable to CRISPR/Cas editing either directly or through competition for Cas9 binding between nonfunctional and active sgRNAs 14–16. Furthermore, a machine learning model trained on activity data acquired from 50,000 sgRNAs revealed a correlation between editing efficiencies and sgRNA composition. sgRNA features such as secondary structure accessibility, self-folding free energy, melting temperature, spacer composition, and GC content are strongly associated with gRNA activity 17. Corsi et al. substantiated these observations and showed that low accessibility of bases unpaired in the secondary structure of the scaffold due to intramolecular interactions with the gRNA is linked to reduced editing efficiency 18,19. Thus, it stands to reason that the ability to modify the sequence composition of the sgRNA scaffold could expand the targeting range of a given CRISPR system.
Much of the current research surrounding the changeability of the sgRNA attempts to explore which bases are critical for secondary structure and Cas9 interactions and which bases are amenable to change. This knowledge in turn has enabled the appending of aptamers or enhancer elements 20–22 to specific regions of the guide RNA, which, in combination with Cas9 modifications, has expanded the system’s binding characteristics and given rise to additional capabilities such as prime editing 11. To specifically address the disparity in targeting efficiencies between different genomic sites, solutions aimed at stabilizing the secondary structure of the sgRNA and enhancing the editing efficiencies are being developed. Advances in CRISPR-target selection algorithms and high-throughput testing of potential target sites have improved the likelihood of identifying a functional target site with a reduced potential for secondary structure interactions 23,24. However, such site selection still excludes many potential genomic loci due to incorrectly folded sgRNA structures making many loci challenging to edit 14. Several groups have sought to address this problem through rational modifications to the gRNA structure itself. For example, Riesenberg et al. used a defined motif and stabilizing modifications to ‘lock’ the structure of the tracrRNA for better efficacy 14. They demonstrate that gRNAs containing a stabilized, chemically-modified secondary structure, within the tracrRNA, function more efficiently following hybridization for most of the guides tested when compared to the native gRNA. In an alternate approach, Nelson et al. stabilized the 3’ end of sgRNAs utilized in prime editing by appending additional RNA motifs 25. Similarly, the gRNA has been chemically modified to increase stability 10,14,16,26.
While these multiple structural modifications to both the sgRNA and its two-component counterpart led to enhanced editing efficiencies, they are designed to be universally applied to all guides within a given CRISPR/Cas system and often rely on chemical modifications that cannot be incorporated by intracellular processes. Furthermore, they do not explore the existing sequence space of the sgRNA scaffold for functional solutions. As the CRISPR/Cas system becomes more complex through protein and RNA engineering, it will become imperative to ensure that the spacer, scaffold and sgRNA-Cas9 retain their optimal functionality.
Here, we use a combinatorial approach utilizing SELEX (Systematic Evolution of Ligands by Exponential Enrichment) 27–29 to identify numerous, diverse sgRNA variants that bind S. pyogenes Cas9 and support DNA cleavage. These variants demonstrate surprising malleability in the sgRNA sequence and are utilized in a combinatorial approach to identify scaffolds that enhance editing efficiencies when paired with DNA-binding antisense domains. Using this molecular evolution approach, guide RNA scaffolds can be generated for specific targets and optimized to ensure that a functional sgRNA secondary structure is maintained. This approach termed BLADE (Binding and Ligand Activated Directed Evolution) illuminates the sequence space of the sgRNA and promises to significantly deepen our understanding of sgRNA structure-function relationships, expand the genomic sites amenable to specific and efficient CRISPR-based editing, and should prove valuable for evolving sgRNA variants with additional desirable properties.
Results:
A selection pool based on partially randomized sgRNA sequence:
To examine the sequence variation tolerated by the Streptococcus pyogenes gRNA scaffold while maintaining its overall secondary structure, we generated a partially randomized library based on the S. pyogenes sgRNA sequence 2. While both the sgRNA and its alternative crRNA:tracrRNA counterpart can be subject to undesirable secondary structure interactions, the sgRNA sequence was utilized as the base for our selections due to a single RNA sequence being more amenable to the SELEX process. The 5’ 20-nucleotide (nt) DNA targeting domain, directed towards a sequence in GFP, and stem-loop 3 of the sgRNA were kept constant and utilized for amplification and regeneration of the library for subsequent rounds (Figure 1). The internal 60 nucleotides encompassing the Repeat and Anti-repeat regions, Stem-loop 1, and Stem-loop 2 of the sgRNA were randomized by synthesizing each position with a phosphoramidite mixture that contained the wild type (WT) base and the three remaining nucleotides at a ratio of 58:14:14:14 such that each position contained the WT base 58% of the time and each of the other nucleotides 14% of the time (Figure 1). This level of degeneracy ensures that the WT sgRNA is statistically, but minimally, represented in the starting library and that variants that are biased toward the secondary structure of the WT sgRNA are represented. The intended base composition in the initial library was evaluated by sequencing and found to be similar to the intended ratios (see Data.S1).
Figure 1. Synthesis of biased sgRNA library template.
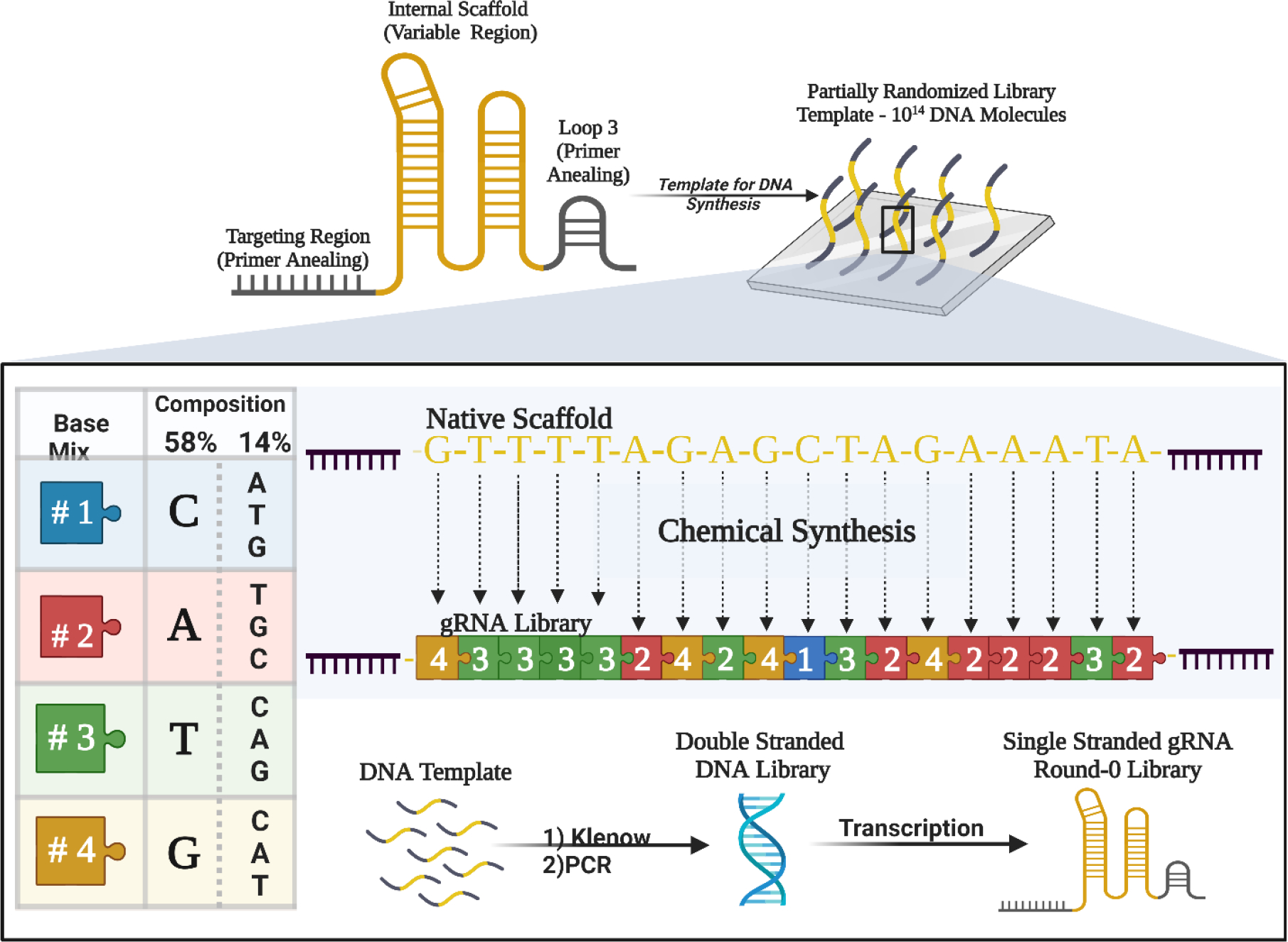
A library based on the WT SpCas9 sgRNA sequence was synthesized with 5’ and 3’ constant regions (the Targeting Region and Loop 3, respectively, shown in black) and a 60 nucleotide partially randomized region (shown in gold). The random region was synthesized using phosphoramidite mixes such that each position in the randomized region had a 58% chance of being the WT nucleotide at that position and a 14% chance of being any of the other 3 nucleotides. The synthesized library was purified, extended, and transcribed as described in the Methods.
A TdT-based approach to isolate cleavage competent sgRNAs:
To isolate sgRNA variants capable of supporting gene editing, we exploited two properties inherent to Cas9. First, Cas9 cleavage exposes a newly formed 3’ end within the non-target DNA strand without complex dissociation. Second, in vitro studies demonstrate that Cas9 has a natural dissociation time of roughly 6 hours 30. The lengthy dissociation time contributes to Cas9’s ability to serve as a targetable protein-DNA roadblock, inducing replication fork arrest 31. These properties enabled us to isolate functional sgRNA variants following Cas9 mediated DNA cleavage. The polymerase, terminal deoxynucleotidyl transferase (TdT) is capable of extending 3’ ends with free nucleotides in a template-independent manner 32. We employed TdT to add a polyA tail to the new 3’ end on substrate DNA exposed by Cas9 cleavage, and cleavage products were purified using a biotinylated oligo(dT) probe and streptavidin beads (Figure 2A). By labeling the substrate DNA with either a fluorophore or radiolabel, we were able to track the successful capture of cleavage products (Figure 2B). We were also able to capture cleaved complexes by direct incorporation of biotinylated nucleotides with TdT (data not shown). We note that for all TdT-based experiments, all ends on substrates and sgRNAs were pre-blocked with dideoxynucleotides to prevent TdT extension anywhere other than the cleavage site.
Figure 2. TdT-based capture of sgRNA cleavage.
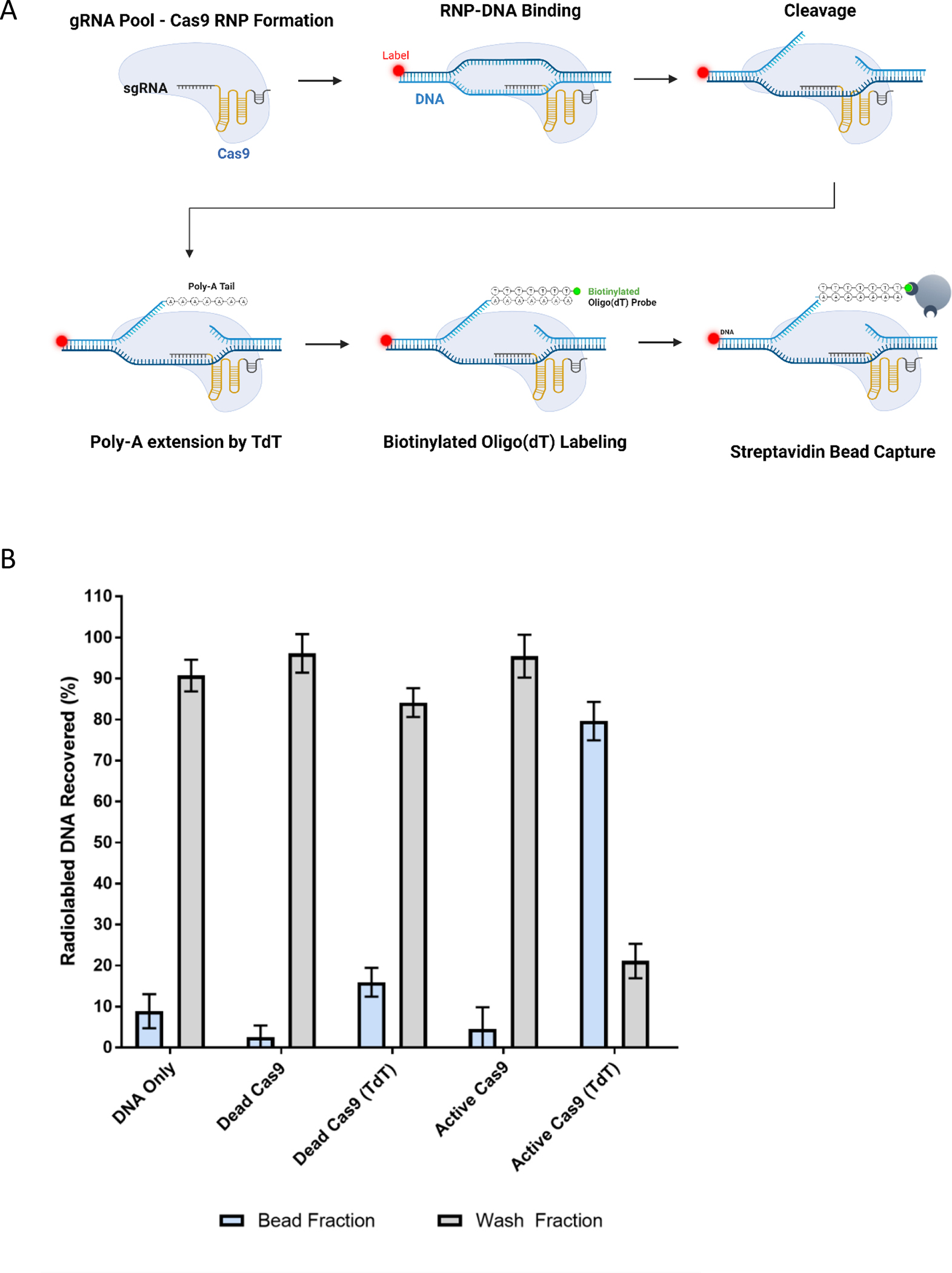
(A) Variant or WT sgRNAs are complexed to SpCas9 and a radiolabeled or fluorescently labeled DNA substrate. Upon cleavage by Cas9, TdT adds a poly(A) chain to the PAM-distal DNA’s 3’ end. A biotinylated Oligo(dT) probe binds the poly(A) tail, and the whole complex can be captured with magnetic streptavidin coated beads. Captured complexes are analyzed by scintillation counting or flow cytometry. (B) The WT sgRNA was complexed with Cas9 variants and then incubated with a radiolabeled substrate DNA and the components of an A-tailing assay as described in the Methods. As negative and positive controls, the WT sgRNA was complexed with inactive “dead” Cas9 or active Cas9 with (as indicated with ‘(TdT)’) or without TdT. Complexes containing cleaved DNA targets were bound by magnetic streptavidin beads, washed several times, and the amount of labeled DNA in the bead fraction and wash fractions was determined. The DNA only control does not contain either Cas9 or sgRNA and serves as a non-specific bead-binding background control. Error bars represent 4 independent replicates.
BLADE SELEX yielded cleavage-capable variant sgRNAs:
We recognized that traditional SELEX screening for sgRNA variants that could bind Cas9, as utilized to identify aptamers, would not necessarily ensure the isolation of sgRNAs that could complex with Cas9 and mediate cleavage. Therefore, to isolate cleavage-capable scaffolds, >1014 sgRNA variants were subjected to 5 rounds of selection using 3 different partitioning strategies: 1.) RNA pool binding to Cas9 to ensure formation of ribonucleoprotein (RNP) complexes, 2.) RNP complex binding to target DNA to ensure variant scaffold complexes could still recognize substrate DNA, and 3.) RNP-mediated cleavage of the target DNA (Figure 3A).
Figure 3. BLADE SELEX generates cleavage capable sgRNA scaffolds.
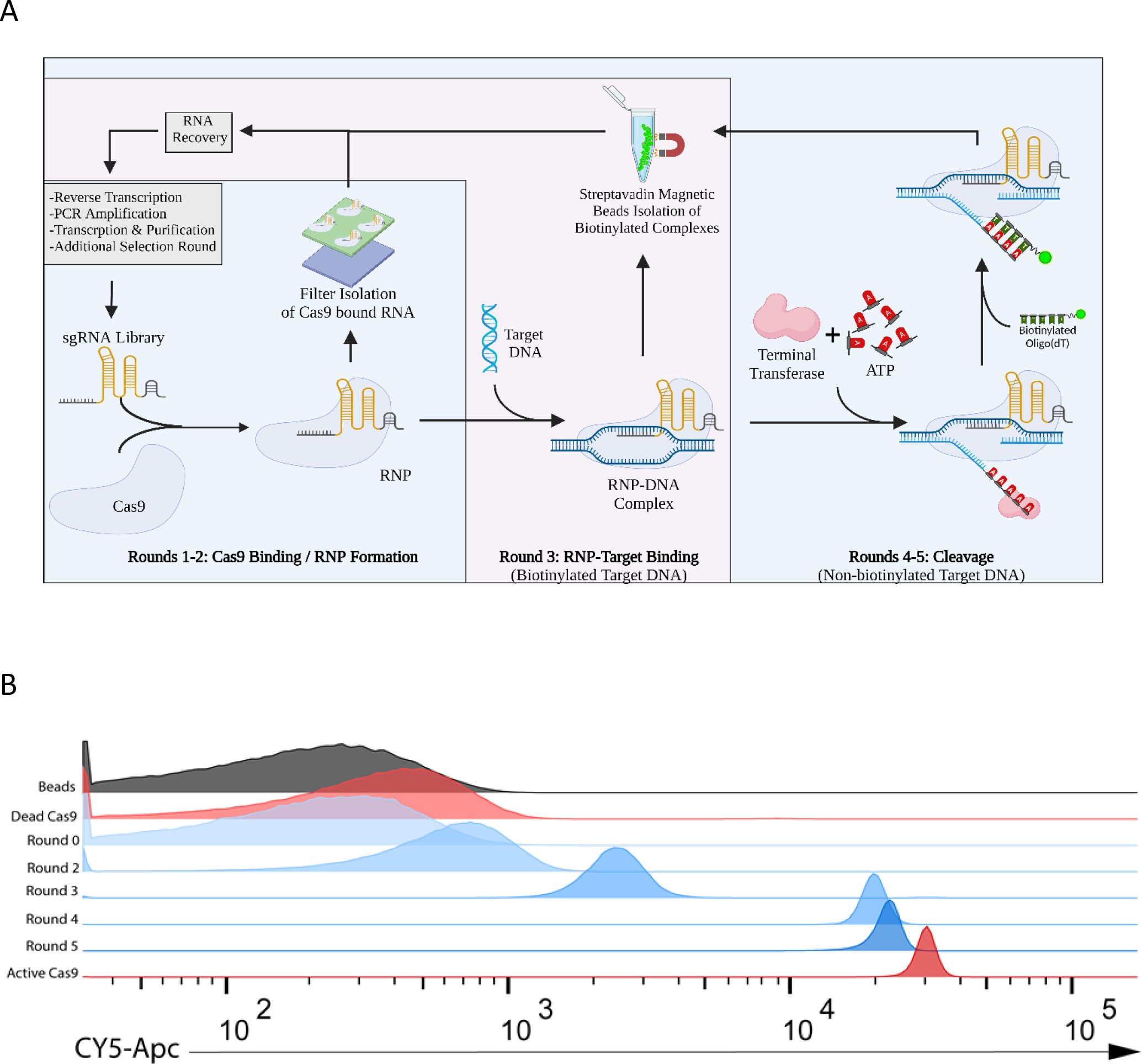
(A) A library based on the WT SpCas9 sgRNA sequence was synthesized with 5’ and 3’ constant regions and a 60 nucleotide partially randomized region. BLADE SELEX was performed with this library and consisted of 3 phases: RNA pool-RNP formation, RNP binding to DNA substrate, and a TdT-based screen for cleavage. (B) DNA cleavage by the pools of sgRNA variants isolated following various rounds of BLADE SELEX (blue) was assessed by A-tailing and flow cytometry and compared to the WT sgRNA (red) complexed with inactive “dead” Cas9 or active Cas9. In this assay, functional RNPs cleave a Cy5-labeled DNA target, TdT adds a poly(A) tail to the newly exposed 3’ end-cleavage product allowing for capture by a biotinylated oligo(dT) probe and magnetic streptavidin beads.
sgRNA variants capable of binding to Cas9 and forming RNPs were partitioned from non-binding gRNA variants via nitrocellulose filter binding during Rounds 1 and 2. In Round 3, sgRNAs amenable to RNP complex formation and DNA binding were isolated by using a biotinylated target DNA fragment allowing for partitioning of RNP-DNA complexes with streptavidin coated beads. We note that the Round 3 partitioning step does not differentiate between cleaved and non-cleaved substrates and will recover RNPs of both species. To isolate those sgRNA variants capable of supporting Cas9-mediated DNA cleavage, we used the TdT-based approach described above, wherein we utilized TdT to add a polyA tail to the new 3’ end on the DNA cleavage product exposed by Cas9 cleavage, and cleavage products were purified using a biotinylated oligo(dT) probe and streptavidin beads (Figure 3A).
Monitoring this functional selection approach using fluorescent or radiolabeled target DNA, we observed an enrichment in cleavage competent SpCas9 RNP complexes as the selection progressed. Following five rounds of sgRNA BLADE SELEX, the DNA cleavage activity of the resulting sgRNA round 5 pool approached that of the WT scaffold (Figure 3B).
BLADE SELEX yielded a highly diverse set of scaffold variants:
Rounds 1, 3 and 5 were sequenced to determine how different phases of the selection affected the sequence composition of each pool (Figure S1, Data S1). Rounds 1 and 3, which selected for sgRNA binding to Cas9 and binding of RNPs to the target DNA respectively, did not significantly alter the sequence composition of the pool when compared to the initial library. The nucleotide frequency of most positions within the sgRNA became more restricted only after the incorporation of the functional selection. This indicates that the functional SELEX developed here may be sufficient for the selection of sgRNAs capable of gene editing and complements the observation of dramatic increases in editing efficiencies between rounds 3 and 4, when the TdT assay is implemented, as observed in Figure 3B.
From the 26,419 sequences returned in the Round 5 sequencing results, 8,009 unique sgRNA scaffolds were identified with varying degrees of relatedness to the WT sgRNA. These sgRNA variants were organized based on frequency of occurrence, and the 647 most abundant sequences were clustered phylogenetically into families based on sequence deviation from the WT scaffold (Figure 4).
Figure 4. Sequence differences between selected sgRNAs and WT sgRNA.
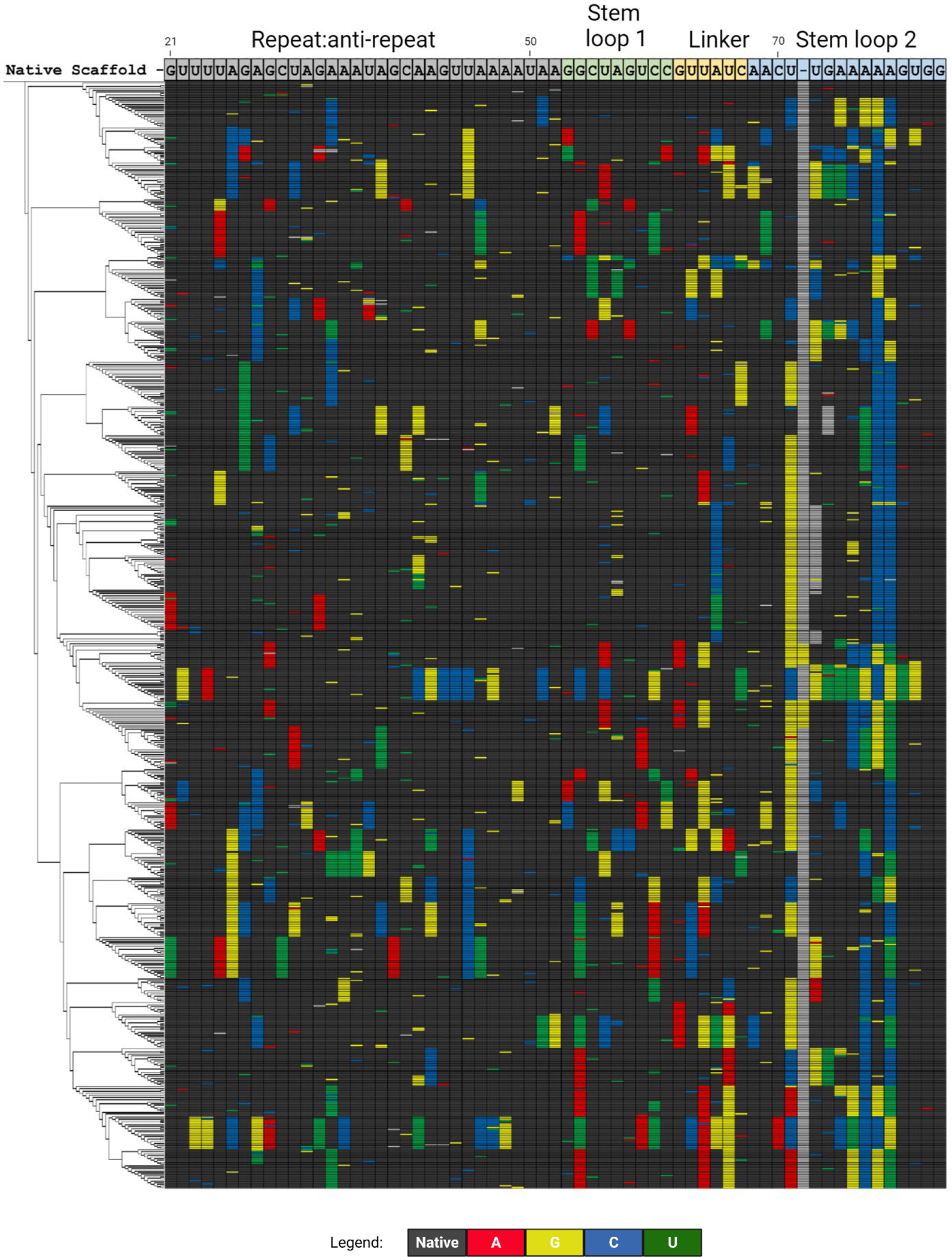
The divergence of 647 selected cleavage-capable variant sgRNAs is shown compared to the WT sgRNA sequence. Positions where the base does not change from WT (“native”) are shown in black. Changes in base identify from the WT sequence are shown in color as indicated. Certain variants have an additional nucleotide between positions 71 and 72 or 73 as marked in gray
BLADE SELEX yielded a highly diverse set of sgRNAs, and the WT sgRNA was only the fourth most abundant sequence isolated even though it was over-represented in the original library. Surprisingly only a few of the WT sgRNA nucleotides were highly conserved in the functionally selected sgRNA variants with only 14 positions out of the 60 randomized being the WT sequence more than 90% of the time respectively despite their over-representation (58%) in the starting library (Figures 1 and 4). Of note, double-stranded regions often tolerated compensatory changes that maintained base pairing, and the frequency of variation increased towards the 3’-end of the sgRNA (Figure 4). Very surprisingly, the BLADE round 5 consensus sequence even deviated from the WT scaffold sequence in Stem loop 2 indicating that the WT nucleotides are not necessarily preferred at these positions under our SELEX conditions (Figure S1). BLADE SELEX and subsequent sequencing analysis reveal that sgRNAs can accommodate multiple sequence alterations and still guide Cas9 cleavage of DNA.
BLADE selected scaffold variants are capable of mediating cleavage in vitro and in cells:
From the parsed sgRNA variant sequences, 153 representative sgRNA variants were screened for DNA cleavage in vitro, and functional ones were evaluated for GFP editing in mammalian cells. Remarkably, 89 sgRNA variants supported DNA cleavage in vitro, of which 72 were comparable to the WT scaffold in cleavage capability. Some sgRNA variants supported partial activity (<50% cleavage) despite containing up to 16 nucleotide changes (Figure 5; Figure S2; Data S2).
Figure 5. Structure-based clustering of sgRNA variants displaying a range of cleavage efficiencies in vitro and editing efficiencies in cells.
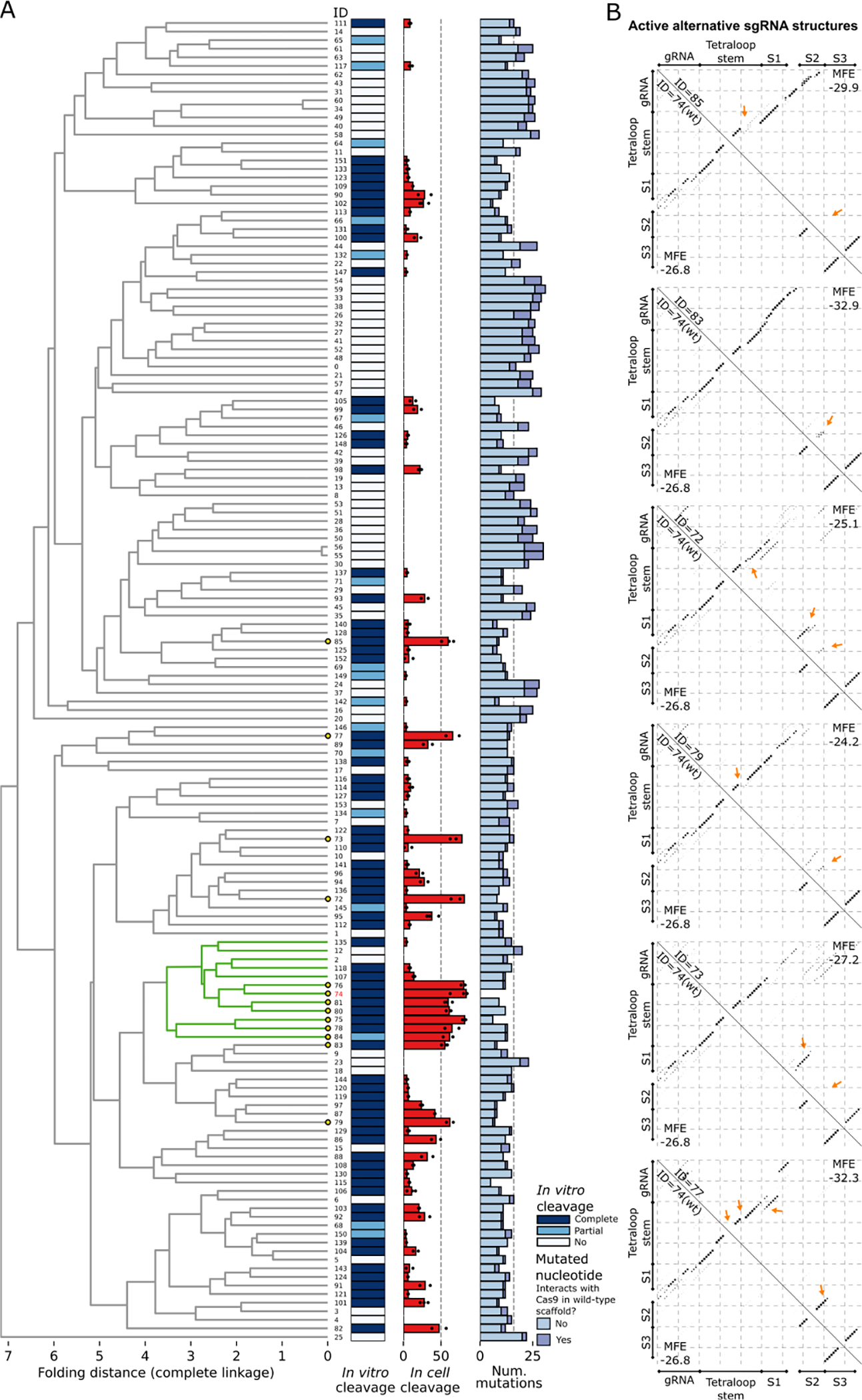
(A) Selected sgRNAs were clustered based on their structures, as described in the Methods. A subcluster with > 50% of its members having mean in cell editing efficiency ≥ 50% is colored in green (WT subcluster). Leaves representing sgRNAs with mean in cell editing efficiency ≥ 50% are highlighted by yellow circles. The in vitro cleavage efficiency of sgRNAs is shown with a colormap with Complete cleavage (> 50%) in dark blue and Partial cleavage (< 50%) in light blue. The mean in cell editing is shown as a barplot, with efficiencies obtained from each replicate scattered with dots. Absence of dots implies that the sgRNA was not tested in cell. The number of differences to the WT obtained from the aligned scaffold sequences is shown with a stacked barplot, which differentiates mutations based on their role in interacting with Cas9. (B) Base pairing probability matrices of alternative functional sgRNA structures (upper triangle) and of the WT sgRNA structure (bottom triangle) are displayed as dotplots. The sgRNA IDs, corresponding to the IDs in the clustering (A), are given along the diagonal. The Tetraloop Stem, Stem 1 (S1), Stem 2 (S2), and Stem 3 (S3) previously reported 34 are annotated for compatibility. Arrows point to the major changes between each sgRNA scaffold and the WT scaffold structure.
To evaluate their gene editing activity, these 89 functional sgRNA variants were assessed for GFP knockout efficiency in HEK293 cells. Twenty-eight of the 89 cleavage competent scaffolds yielded editing efficiencies greater than 20% in cells, of which, seven achieved levels of GFP editing comparable to the WT sgRNA (threshold: >50% cleavage; Figure 5; Figure S3; Data S2). These variant scaffolds were further tested in HeLa and PC3 cells also expressing GFP. Of note, the HeLa-GFP cells expressed a short-lived version of GFP 33. As with the HEK293 cells, certain scaffolds supported higher editing efficiencies than others, with several variants achieving editing efficiencies comparable to the WT sgRNA (Figure S4). Given that we utilized a GFP target site for sgRNA BLADE selection known to be highly receptive to WT sgRNA-based editing, the observation that seven different sgRNA variants, containing divergent 6–16 nucleotide changes, can support similarly high levels of editing as the WT sgRNA demonstrates that the SpCas9 sgRNA sequence is quite malleable; it can tolerate multiple changes and retain potent gene editing activity.
BLADE SELEX illuminates the flexibility of sequence specific Cas9 - sgRNA interactions:
The elucidation of the Cas9-sgRNA crystal structure identified a number of sequence dependent interactions vital for scaffold recognition and RNP complex stability 34. These important contacts observed between the sgRNA scaffold and the Cas9 protein are consistent with the highly conserved nucleotides identified through BLADE SELEX. Within the Repeat:Anti-repeat duplex, the nucleobases of U23, A42, G43, U44, and A49 form important protein contacts with the REC (recognition) and PI (PAM-interacting) domains within Cas9 and aid in sequence recognition. These nucleobases are conserved across 90% of the functional sequences identified through BLADE SELEX, with G43 and U44 showing essentially 100% sequence conservation (Figure S1).
Interestingly, by contrast, some scaffold variants retain editing capabilities despite having changes in nucleobases that participate in protein interactions and have high levels of sequence conservation among related Cas proteins. In the crystal structure, nucleobase A42 forms a hydrogen bond with the side chain of Arg1122 within Cas9, yet Scaffold-87 has a cytosine at position 42 and retains an editing efficiency of >80%. The nucleobases of G21 and U50 form a wobble base pair, partake in both base stacking and protein interactions, and have been shown to be highly conserved among type II-A–C Cas systems. While the majority of scaffolds that have mutations at this position have been shown to be non-functional, some variants adopted an A21:U50 pair while retaining editing efficiencies as high as 30% when compared to the WT scaffold.
A number of residues within Stem loops 1–3 reinforce the interactions between the sgRNA and Cas9. In the crystal structure, the Linker (positions 63–67) interacts with Cas9 through several phosphate backbone interactions. Positions such as U64 and A65 interact with the NUC (nuclease) lobe through hydrogen bonds on their 2’-hydroxyl group. This common nucleobase interaction is consistent with the BLADE SELEX results which reveal high variability at these positions, with only 68% conserved and 78% conserved for U64 and A65 respectively. As Cas9 interacts with the phosphate backbone or 2’-OH at these positions, the sequence identity of the bases does not appear particularly critical. On the other hand, C67, which forms 2 hydrogen bonds at the N3 and exocyclic amine of cytosine with the Cas9 PI domain, was > 90% conserved among functional scaffold variants.
Meanwhile, regions of the sgRNA that do not appear to interact with Cas9 or be involved in intramolecular stacking or stabilizing interactions tolerated significantly more changes. Our data shows that the Tetraloop connecting the Repeat and Anti-repeat regions (positions 33–36), which does not interact with Cas9 or other parts of the sgRNA in the crystal structure, tolerated changes in the GNRA tetraloop with the N position (residue A34) being the most variable at about 70% conservation. The residues adjacent to this tetraloop appear to stabilize the stem efficiently regardless of nucleobase identity. We found that many of the mutations at position at U31 were matched with compensatory mutations at A38 that would maintain base stem formation (Figure 4, Figure S1). Similarly, the tetraloop of Stem Loop 2 (positions 73–76) and its adjacent stem residues, which also do not appear to interact with the protein or have long range tertiary interactions with other parts of the sgRNA, tolerated significant changes to sequence, with position A78 of the stem only 22% conserved (Figure S1). As with the Repeat:Antirepeat stem, many mutations at this position were matched by compensatory mutations at U71 or immediately nearby to maintain stem formation. The relative flexibility of sequence at these sites has previously been noted. Indeed, Kundert et al. were able to insert a whole aptamer at the Repeat:Antirepeat tetraloop and at Stem Loop 2 to create ligand-activated sgRNAs 20.
The results of the SpCas9 sgRNA BLADE SELEX are consistent with the findings of the x-ray structure analysis and reinforce the importance of specific nucleobase interactions with Cas9 previously identified by the RNP crystal structure 34. The bases that directly interacted with the protein were highly conserved within our selection library of sgRNA variants. However, even though select positions are highly conserved, the majority of nucleobases could tolerate specific changes that enabled for gene editing, some of which functioned as efficiently as the WT scaffold indicating that sgRNA evolution may represent an important approach to optimize and engineer sgRNAs with useful and interesting functions.
Structural analysis of BLADE selected scaffold variants:
While sequence-based clustering gave some indication of the diversity of selected variants, a possibility remained that sequence divergence could include compensatory mutations or neutral mutations that would result in what is essentially the canonical scaffold structure.
To determine the structural variation of selected functional scaffolds, sgRNAs structures were analyzed using base pairing probability matrices computed in silico using RNAfold 35. The base pairing probability matrix of an sgRNA reports the probability of any two bases in the sgRNA being paired to each other in the secondary structure. The structure of the WT scaffold provided by the base pairing probability matrix well reflects the known structure of the scaffold in the Cas9-sgRNA complex 34, except for few differences. The base pairing probability matrix lacks the non-canonical base pairs A52:G62 and A68:G81, the latter of which results in the additional base pair G81:U97 in the 2D RNAfold predicted structure. Another difference is the lack of the base pair C55:G58, which creates the short loop of 2 nt in Stem 1, that becomes a loop of 4 nt in the in silico structure. Last, in the base pairing probability matrix there is an additional base pair G27:U44, which is absent in the sgRNA-Cas9 complex structure possibly due to the reported interaction between G27 and A26:U45 34 (Figure S5). Folding of the WT scaffold together with the sgRNA spacer eliminates Stem 1 in the base pairing probability matrix, supporting the concept that the effect of the sgRNA spacer pairing with the scaffold is non-negligible (Figure S5).
The mutated sgRNAs and the WT were clustered by their pairwise structural similarity. For this, the pairwise structure distances between two sgRNAs were computed by comparing the base pairing probabilities from the respective matrices position by position. In this calculation the region of the matrices representing the spacer self-folding was set to zero, as it should not be taken into account when comparing scaffold architectures. For the part of the matrix concerning the interactions between the spacer and the scaffold, we summed the base pair probabilities to obtain the probability with which each nucleotide in the spacer pairs elsewhere with the scaffold. This provides a measure of the spacer’s potential to disrupt the scaffold. Hence the resulting pairwise distance is calculated as a combination of the part of the base pairing probability matrix excluding the spacer and the spacer positional probabilities to interact with the scaffold (Figure S5).
After obtaining the pairwise Euclidian distances, the clustering was made by hierarchical complete linkage, (i.e., two clusters of sgRNAs are combined in the same cluster at a higher level in the hierarchy if the distance between the two elements in the clusters that are farthest away from each other is the minimum over all pairs of clusters). The resulting structure-based clustering provides a clear separation between active and inactive sgRNAs already at the 2nd top tree branch level (Figure 5A), with one cluster significantly enriched (Yate’s-corrected χ2= 2.19E-6, degrees of freedom =1) for sgRNAs with activity in vitro (56 active, 15 inactive) compared to sgRNAs outside of the cluster (33 active, 50 inactive). The cluster of active sgRNAs includes the WT, that is part of a subcluster with 7/12 members having mean cleavage activity in cell ≥ 50%, which we will refer to as WT subcluster. Only one highly active sgRNA (sgRNA ID = 85, mean cleavage in cell = 59.43%) lays outside of the major cluster of active sgRNAs. The scaffold folding of this sgRNA differs from the WT in that the hairpin of the Tetraloop Stem includes a wider internal loop, resulting in a shifted stem in the base pairing probability matrix, and Stem 2 is absent (Figure 5B). While this alternative structure is substantially different from the minimum free energy (MFE) structure of the WT, the shifted stem represents a possible alternative structure in the WT itself. Notably, the MFE of the sgRNA having this alternative structure (ID = 85, MFE = −29.9 kcal/mol) is lower compared to the WT (ID = 74, MFE = −26.8 kcal/mol), and hence more stable. Although the MFE is computed on the entire sgRNA molecule and includes eventual spacer self-folding interactions, in sgRNA ID = 85 there is no trace of such self-folding. Therefore, the increase in MFE is due to the different Tetraloop Stem structure, elongated by additional spacer-scaffold interactions. This alternative Tetraloop Stem structure is absent in the immediate neighbors of sgRNA ID = 85 in the dendrogram of the hierarchical clustering, which have low efficiency in cell (Figure S6).
Other sgRNAs with mean cleavage activity in cell ≥ 50% that do not belong to the WT subcluster (n = 5) differ from the WT mainly at Stem 2 (Figure 5B), which has either lower pairing probabilities (sgRNA IDs = 83, 79), is extended (sgRNA ID = 77), or shifted (sgRNA IDs = 72, 73). Changes to the hairpin of the Tetraloop Stem are also visible (sgRNA IDs = 72, 79, 77) and, notably, sgRNAs ID = 72 and 77 show two alternative Tetraloop Stems, one reflecting the WT, the other being the shifted stem described for sgRNA ID = 85. Among these sgRNAs, ID = 83 is the one with the lowest MFE (−32.9 kcal/mol), followed by sgRNA ID = 77 (−32.3 kcal/mol). In both cases the Tetraloop Stem is extended due to interactions with the spacer. Inspection of the immediate neighbors of these highly active sgRNAs in the dendrogram of the hierarchical clustering shows that either they are active (> 10% efficiency in cell) and present only minor differences compared to the highly active neighbor, or they are inefficient but hold visible difference in the configuration of the Tetraloop Stem, of Stem 2, or in terms of spacer interactions (Figure S6). These results suggest that alternative scaffold structures are not only tolerated during the formation of the Cas9-sgRNA complex, but also retain high editing efficiency in cell (mean efficiency ≥ 50%).
The base pairing probability matrices of the members of the WT subcluster point to minor changes in the organization of the internal loop within the Tetraloop Stem compared to the WT scaffold (Figure 6A). These differences are more substantial in inefficient sgRNAs (ID = 118, 12, 135) with the exception of sgRNA ID = 2, which bears one additional base pair in Stem 2 and has the lowest MFE in the whole cluster (−39.8 kcal/mol).
Figure 6. Structure differences between the members of the WT subcluster.
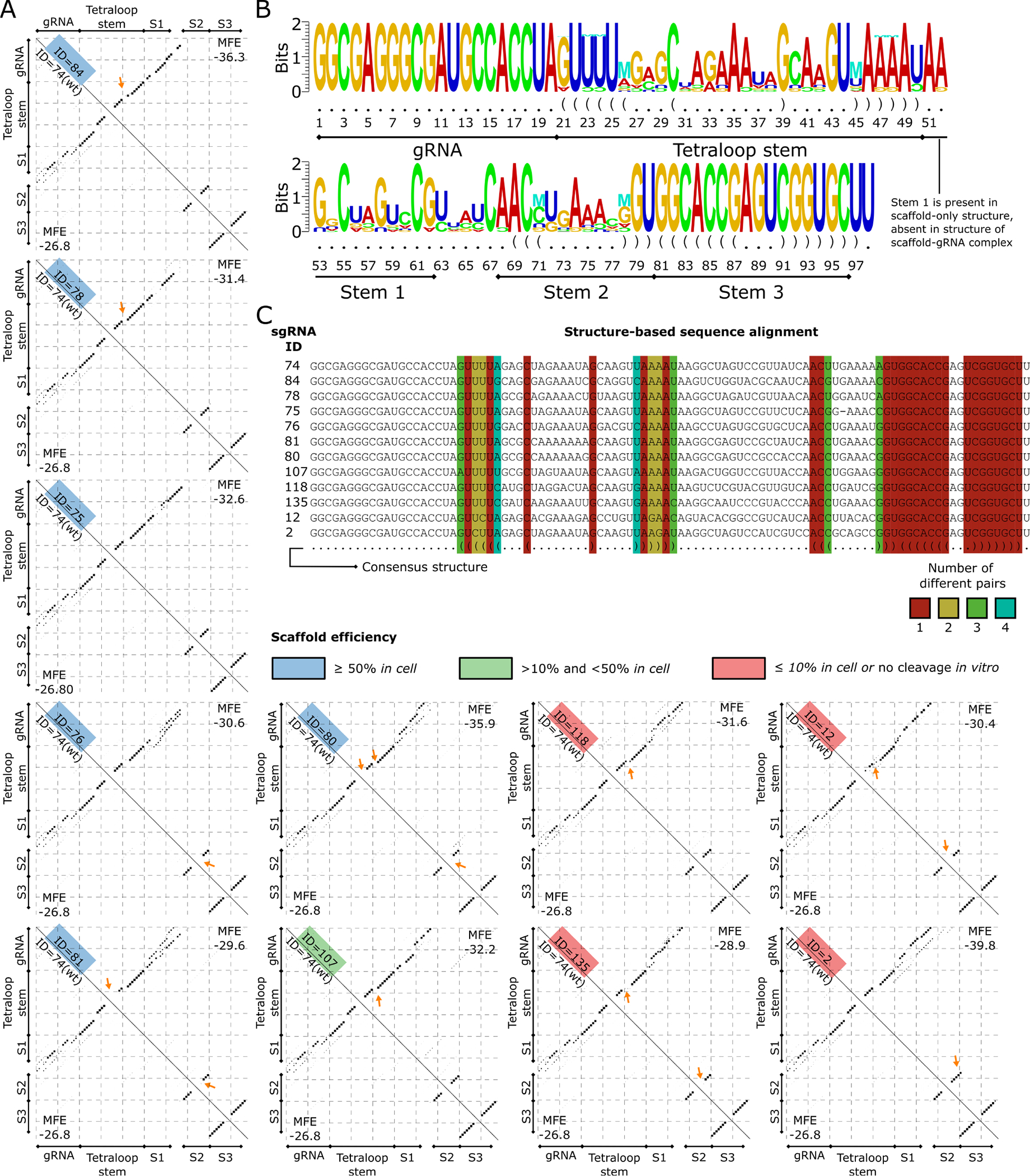
(A) Base pairing probability matrices of sgRNA structures that are part of the WT subcluster (upper triangle) and of the WT sgRNA structure (bottom triangle) are displayed as dotplots. The sgRNA IDs are given along the diagonal and colored based on the efficiency category (low ≤ 10%, medium between 10%, and 50%, high ≥ 50%). The Tetraloop Stem, Stem 1 (S1), Stem 2 (S2), and Stem 3 (S3) previously reported (34 are annotated for compatibility. Arrows point to the major changes between each sgRNA scaffold and the WT scaffold structure. (B) Structure logo of sgRNAs in the WT subcluster is shown. The structure logo annotates a sequence logo with mutual information of RNA base pairs, shown with the “M” symbol. Nucleotides that appear less than expected are shown upside down. (C) Structure-based sequence alignment of sgRNAs in the WT subcluster. The consensus structure is shown in the last row. Columns corresponding to base pairs in the consensus are colored based on the number of different base-pair types in the sequences.
The structure logo of the sgRNAs in the WT subcluster shows compensatory base changes in the hairpins of the Tetraloop Stem and in Stem 2 (Figure 6B). Inspection of these positions in the structure-based alignment of the sgRNAs in the WT subcluster shows two base pairs with compensatory base changes in highly active sgRNAs (Figure 6C). These are the variant AU>GC at the pair 26:45 and UA>GC or UA>CG at the pairs 71:78. The presence of compensatory base changes in highly active sgRNAs highlights the functional importance of the hairpins in the Tetraloop Stem and in Stem 2 for sgRNAs with folding structures similar to that of the WT.
BLADE selected sgRNA variants demonstrate altered editing abilities with different target sites:
The observation that multiple, different sgRNA scaffold domains can support SpCas9-based editing led us to explore how editing efficiencies vary when the different sgRNA scaffold domains are paired with different DNA targeting domains. The 20-nucleotide targeting domain was altered to recognize five different DNA sequences within GFP, and each was paired with ten different scaffold variants as well as the WT sgRNA scaffold (see Data S2 for sequences). These 60 GFP-targeting sgRNAs were evaluated for their ability to edit GFP in HEK293-GFP and HeLa-GFP cells, resulting in a reduction in GFP fluorescence. Different DNA target-scaffold combinations yielded functional sgRNAs with a range of editing abilities (Figure 7). Remarkably, for three out of five tested DNA target sites (GFP DNA sites 2, 5 and 6), an sgRNA scaffold variant(s) paired more effectively with the DNA targeting domain than the WT scaffold domain resulting in higher editing efficiencies than the WT sgRNA. Moreover, though the WT sgRNA was able to edit ~80% of the GFP genes when utilizing the original DNA targeting domain (Target-1), it was unable to achieve this high level of editing at 4 and 5 out of 5 new sites in HEK293 and HeLa cells respectively (Figure 7). However, various BLADE-derived scaffolds were able to support ~80% editing efficiencies at 4 out of the 6 target sites tested in both cell lines (Target sites 1, 2, 5 and 6). Surprisingly, some variant scaffolds functioned even more effectively with a different targeting domain compared to the one employed in the initial selection. For example, Scaffold-1 knocked out GFP expression in 30–40% of the cells when paired with the DNA targeting domain (Target-1) used during selection. However, when the targeting domain was changed to recognize Target-6, GFP was edited in approximately 80% of the cells. Interestingly one variant, Scaffold-3, paired particularly well with four of the six targeting sequences. Thus, analysis of these 60 sgRNAs revealed that the ten BLADE-derived scaffold domain variants and the WT scaffold domain partnered most effectively with different sets of DNA targeting domains. Some variants appear to be generalists, able to partner well with multiple DNA binding domains, while others are more selective and tend to partner best with a particular DNA binding domain. For example, variants Scaffold-1, Scaffold-16 and Scaffold-87 preferentially supported editing most effectively at different DNA target sites (Figure 7). This breadth of editing efficiencies highlights the importance of the structure-function relationship between the DNA-binding and Cas9-binding domains and reveals that changes in the scaffold sequence differentially accommodate various DNA targeting domains (Figure 7 and Figure S7). Moreover, it demonstrates that in vitro evolution can be employed to generate a variety of newfound sgRNA variants with distinct functional properties.
Figure 7. Selected variants demonstrate altered efficiencies when targeted to other sites.
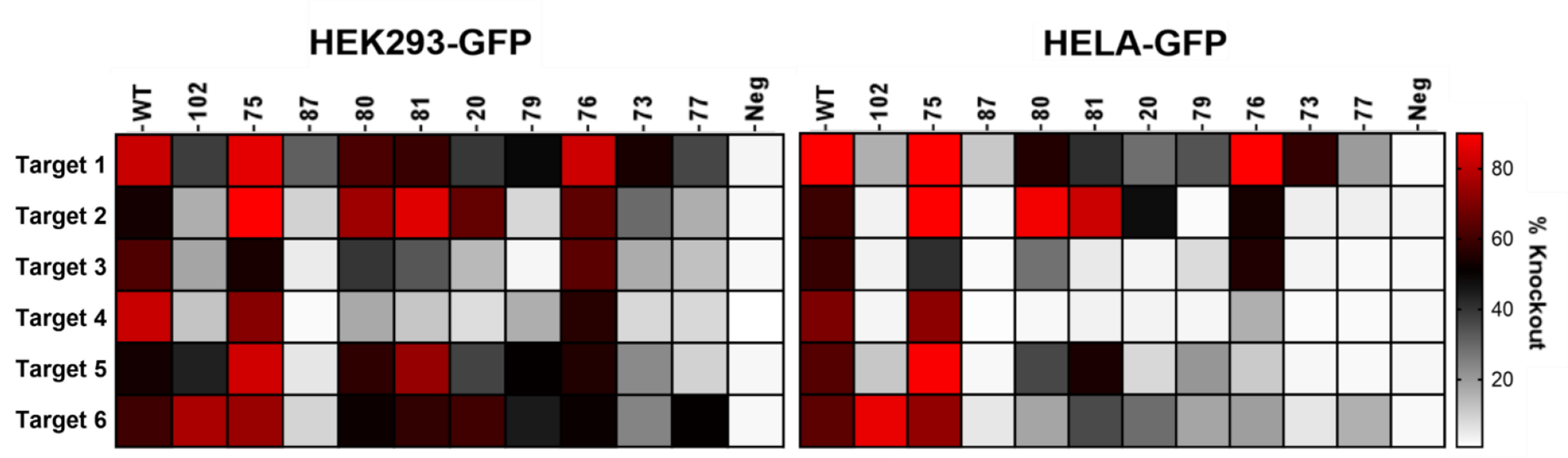
The wild type (WT) and ten selected scaffolds were retargeted to five other target sites on the GFP gene, and GFP knockout was assessed in cells. GFP Target 1, the DNA site utilized during selection, is provided for comparison. The degree of GFP knockout is shown for each combination of scaffold variant with each DNA target domain. White colored boxes represent no knockout, black indicates 50% knockout, and red represents >80% reduction in GFP 10 expressing cells (significance values are reported in Data.S2; n= 2–5 independent experiments for each of the 66 GFP targeted sgRNAs in each of the two cell types).
Discussion:
We have developed a molecular evolution method, BLADE-SELEX, to identify functional sgRNAs variants. Our results indicate that BLADE SELEX can produce numerous, functional sgRNAs from a vast sgRNA library that support SpCas9-mediated cleavage of DNA in vitro and efficient gene editing in mammalian cells. Such variants have a range of distinct activities including the ability to target certain DNA sites more effectively than the WT sgRNA. This high-throughput sgRNA selection approach can be utilized to optimize the targeting of any DNA sequence containing a SpCas9 PAM sequence which should significantly expand the repertoire of DNA sites amenable to highly efficient editing.
The selection approach described here utilized a GFP spacer sequence that was known to support a high level of editing efficiency. This strategy was taken to ensure that the combinatorial chemistry approach described would have a high probability of yielding active gRNA variants; therefore, it is not surprising that an sgRNA scaffold variant more efficient than the native scaffold was not identified when using this particularly efficient spacer. However, we believe that BLADE SELEX can be utilized to identify scaffold variants that can improve editing efficiencies when paired with less efficient spacers. The ad hoc pairings of ten scaffold variants identified here with 5 randomly chosen spacers led to improved editing efficiencies over the WT gRNA in three out of five cases (Figure 7); nevertheless, these results are limited as all ten of these variants was isolated in the context of a different spacer. Thus, additional selections utilizing gRNA libraries containing low efficiency spacers should be performed as these have an even greater potential to not only further expand the known variation tolerated but also to dramatically improve editing efficiencies against very challenging DNA target sequences. Moreover by utilizing Toggle SELEX 36 or positive-negative SELEX 37, sgRNA variants can be created that can function on more than one DNA target site or that can distinguish between highly related DNA sequences to improve editing specificity and reduce off target editing concerns. Furthermore, by incorporating an in vivo partitioning screen 38–40 during BLADE selection, optimized sgRNAs for particular cellular environments can theoretically be isolated.
Due to the limited number of different targets and scaffolds tested on multiple targets, here the structural analysis of the sgRNAs was performed using only the one target for which 154 diverse scaffolds were tested. The generation of sgRNAs with variated scaffolds for multiple targets, enabled by BLADE-SELEX, will allow us to build computational models that automatically learn the relation between structure differences and editing efficiency. Such data will also allow models to combine structure information with mutation identity, for a complete understanding of the effects of scaffold variants on Cas9 binding. This should contribute significantly to computational methods predicting optimal sgRNA variant(s) for particular cellular applications as well as to increase the probability of generating sgRNAs that work efficiently in vivo. These combinatorial approaches and the computational methods they enable should be useful for optimizing any CRISPR-based editing system that utilizes a sgRNA. Such optimization is expected to be particular impactful in situations where one wants to target a specific sequence in a gene, not just knock out or inactivate a gene, such as when using PRIME-editing, single nucleotide editing, or HDR-based editing to make specific sequence changes to correct a genetic defect or to insert a specific point mutation at a particular position in a gene for research or clinical applications. Here, targeting a particular DNA sequence in the genome at or near the site of the wanted change is vital for the efficiency of these DNA editing approaches.
Finally, the observation that the sgRNA sequence can be modified at many positions and still retain activity indicates that sgRNA evolution and selection approaches can be utilized to generate sgRNA variants with many useful properties. Moreover, the knowledge gained from the interplay of different sites critical for sgRNA structure-function could feed into in silico predictions for scaffolds to optimally target specific genes. The ability to modify the sequence of sgRNA scaffold domains yet still create highly functional CRISPR-based editing agents will facilitate the development of more efficient, higher resolution and more precise DNA or RNA editing as well as further expand the repertoire of research and medical applications amenable to CRISPR-based systems.
Limitations of the study:
Several limitations of this study stem from our library design. Spacer-scaffold interactions due to improper secondary structures can lead to potentially detrimental effects on editing efficiencies. For our experiments, the spacer was kept constant along with stem loop three for the purposes of PCR amplification. In all likelihood, many guide RNA sequences were likely lost due to improper secondary structure interactions with the constant regions. To obtain a true snapshot of the full range of sequence diversity tolerated by the guide RNA scaffold, alternative spacers would have to be utilized so that scaffolds that are lost due to inappropriate interactions with one spacer might be amplified by another. Additionally, variation was likely limited due to the utilization of stem loop 3 as a constant region for amplification purposes. Creating a library based on the entire scaffold may further enhance the level of diversity tolerated by the guide RNA scaffold. Our choice of target may have also limited our ability to produce scaffold variants with dramatically improved efficiency over the wild type. Because it was unclear how much of the scaffold sequence could be changed and remain functional when this project was started, we decided to evaluate the approach with an sgRNA library targeting a GFP sequence that was known to yield high editing efficiencies with the wild type scaffold. Thus, the library of variants created are by their nature limited as they were evolved in the context of a spacer sequence that was already known to work well for editing.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bruce Sullenger (bruce.sullenger@duke.edu)
Materials Availability
The sequences for the library, guide variants, and targets are described in the associated data files, Data S1 and Data S2, and can be purchased through oligonucleotide synthesis sources such as Integrated DNA Technologies (IDT).
Data and Code Availability
The sequences of the sgRNA variants and components for BLADE SELEX tested in this manuscript are described in the attached Data S1 and Data S2 files. Experimental data used to generate the figures in this text are also included in the file Data.S2. Additional data reported in this paper will be shared by the lead contact upon request.
Methodology and software used for structural analysis of variant guides are as described in the text. The scripts and further details on how to use them are available upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell Lines and Culture
HEK293-GFP (immortalized kidney; gender unknown) cells were a kind gift from the Charles Gersbach lab. HeLa-GFP (cervical adenocarcinoma; female) and PC3-GFP (prostate adenocarcinoma; male) cells as well as the corresponding parental lines were kind gifts from the Matthew Levy lab. HEK293 parental cells were purchased from the Duke Cell Culture Facility. HeLa and HEK293 derived lines were grown in DMEM supplemented with 4.5 g/L glucose, 0.11 g/L sodium pyruvate, 3.7 g/L sodium bicarbonate and 10% FBS. PC3 cells were grown in Ham’s F12K media supplemented with 2.5 g/L sodium bicarbonate and 10% FBS. All cell culture reagents were purchased from ThermoFisher. All cells were grown at 37°C in 5% CO2 and 95% relative humidity.
METHOD DETAILS
Guide RNA Library and Variant Clone Generation.
The starting guide SELEX library was generated by annealing 1.5 nmol of the synthesized single-stranded template library (5’-AAAAGCACCGACTCGGTGCNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTAGGTGGCATCGCCCTCGCC-3’) to 1 nmol of the Forward primer (TAATACGACTCACTATAGGCGAGGGCGATGCCACCTA) in 10 mM Tris-HCl, pH 8.0, with 10 mM MgCl2 at 95°C for 5 minutes and then snap cooled on ice. All sequences used in this paper were purchased through Integrated DNA Technologies (IDT; Coralville, IA). The underlined regions in both the library sequence and the Forward primer indicate the spacer region, a target in the GFP gene. The T7 promoter sequence is indicated in bold on the Forward primer. The annealed oligonucleotides were extended to full length with Exo(−) Klenow (New England Biolabs (NEB)) according to the manufacturer’s protocol, phenol-chloroform extracted, and subsequently concentrated and desalted with an Amicon 10 kDa Ultra-0.5 mL column (Millipore-Sigma) using 10 mM Tris pH 7.5 with 0.1 mM EDTA for washes. The DNA libraries were transcribed in vitro with T7 RNA polymerase (NEB) following manufacturer’s protocol using 250 pmol of DNA and 2 mM each NTP (ThermoFisher). Resulting RNA libraries were treated with DNAse I (NEB), phenol-chloroform extracted, concentrated, and desalted with an Amicon 10 kDa Ultra-0.5 mL column and then purified using 12% denaturing polyacrylamide gel electrophoresis (PAGE; 19:1::acrylamide:bis-acrylamide (Biorad) in 1X TBE with 7 M urea). Excised RNA was eluted overnight in TE (10 mM Tris-Cl, 1 mM EDTA, pH 8) at 4°C and desalted with an Amicon 10 kDa Ultra-0.5 mL column.
To make the variant guides, overlapping oligonucleotides comprising each variant guide was PCR amplified using Phusion HF (NEB) following manufacturer’s protocols and purified using a QIAquick PCR purification kit (Qiagen). PCR templates were transcribed with T7 RNA polymerase for 2.5 hours at 37°C. Transcription reactions (50 μL) contained 2–4 μM template DNA, 200 units T7 polymerase, 1 μg/mL pyrophosphatase (Roche), 5 mM each NTP, 30 mM Tris-CH (pH 8.1), 25 mM MgCl2, 10 mM dithithreitol (DTT), 2 mM spermidine, and 0.01% Triton X-100. Reactions were treated with DNase I (Lucigen) for 30 minutes at 37°C and loaded onto a 12% denaturing urea-polyacrylamide gel. Bands corresponding to the transcription product were excised and eluted overnight in TE at 4°C. Triphosphates were removed with 10 units of calf intestinal phosphatase (NEB) following manufacturer’s protocols.
Selection for Cas9 and Substrate Binding.
In vitro selection for binding was initially performed by isolation of bound RNA-protein complexes filtered through a 25 mm Whatman 0.45 μm nitrocellulose membrane (GE Healthcare Life Sciences; 41). Rounds 1 and 2 were performed by incubating 1 nmol of the RNA library with 0.1 nmol of EnGen (NEB) or TrueCut SpCas9 (ThermoFisher) in Binding Buffer 1 (Table S1; 20 mM HEPES pH 7.4, 100 mM NaCl, 1 mM MgCl2, and 0.01% bovine serum albumin (BSA)) at 37°C for 20 minutes to generate ribonucleoprotein complexes (RNPs). RNPs were filtered through the nitrocellulose membrane, and the RNAs were extracted via phenol:chloroform:isoamyl alcohol (25:24:1) and ethanol precipitated in 0.3 M sodium acetate and 2.5X volumes 100% ethanol. 50% of the extracted RNA was reverse transcribed (RT) with 100 pmol of the Reverse primer (5’- AAAAGCACCGACTCGGTGC-3’), 10 nmol dNTPs, and 20 units of AMV Reverse Transcriptase (Roche) according to the manufacturer’s protocol. The RT reaction was PCR amplified with 500 pmol of 5’ and 3’ primers using standard PCR conditions. Reactions were then desalted and purified with a Qiagen PCR Purification Kit according to the manufacturer’s protocol. The resulting PCR product was utilized to generate the sgRNA library necessary for the subsequent round of selection following the transcription conditions for generating the original guide RNA library, above, but using only 100 pmol selection round input.
For Round 3, 1 nmol RNA library transcribed from Round 2 was complexed with 100 pmol SpCas9 in Binding Buffer 2 (Table S1) for 30 minutes to generate RNPs. 1 pmol biotinylated Substrate 1 (5’-GAGGTTGGGCCATTCAGGCTGCGCAGGGCGAGGGCGATGCCACCTAGGGCCTGAGCACTGCACGCCGTTAGI-3’, where I is an inverted dT residue; the spacer sequence is underlined) was added and allowed to bind for 30 minutes. RNP-biotinylated DNA complexes were mixed with 2 μL of Streptavidin MyOne C1 Dynabeads (ThermoFisher) in NEB Buffer 3.1 supplemented with 0.005% Tween-20 (Sigma) and incubated at room temperature for 1 hour with rotation. Complexes bound to the magnetic beads were sequestered and washed 3X in NEB Buffer 3.1 supplemented with 0.005% Tween-20. Target DNA was then degraded with Dnase I (Lucigen), and the protein bound sgRNA library was prepped for subsequent rounds as described above.
Selection for Cleavage Capable Variant Guides.
Rounds 4 and 5 of the functional selection were performed by isolating cleavage capable RNA-protein complexes. RNPs were formed by incubating the in vitro transcribed sgRNA libraries from binding selection Round 3 with SpCas9 at an equimolar ratio of 0.1 nmol at 37°C for 30 minutes in NEB Buffer 3.1. Three pmol of 3’-end blocked, non-biotinylated Substrate 1 target DNA was then added to the RNP reaction mix for an additional 30 minute incubation. Following RNP-DNA cleavage complex formation, 100 units of recombinant E. coli terminal deoxynucleotidyl transferase (TdT; Millipore-Sigma), 5X Reaction buffer to a final concentration of 1X, 5 mM CoCl2 and 1 mM dATP was added to the reaction and incubated at 37°C for 30 minutes. Unincorporated nucleotides were removed with an Amicon 10 kDa Ultra-0.5 mL column using 1x NEB Buffer 3.1 for washes. One pmol of a biotinylated Oligo(dT) (Promega) was added to the reaction mix and incubated at 37°C for 15 minutes with rotation. Unbound probes were removed with an Amicon 30 kDa Ultra-0.5 mL column using 1x NEB buffer 3.1 for washes. The biotinylated, TdT-treated RNP-DNA complexes were mixed with 2 μL of Streptavidin MyOne C1 Dynabeads in NEB Buffer 3.1 supplemented with 0.005% Tween-20 (Sigma) and incubated at room temperature for 1 hour with rotation. Complexes bound to the magnetic beads were sequestered and washed 3X in NEB Buffer 3.1 supplemented with 0.005% Tween-20. Target DNA was then degraded with Dnase I, and the protein bound sgRNA library was prepped for subsequent rounds as described above.
Sequencing Analysis.
500 ng Qiagen PCR-cleaned DNA from rounds 0 and 5 of the selection were submitted for Amplicon EZ sequencing (GeneWiz). The returned sequences were frequency ranked through FastAptamer 42. Sequence alignments and phylogenetic trees were performed using Geneious (BioMatters Ltd.).
In Vitro Cleavage Assay.
Selected variant sgRNAs were transcribed and purified as described above. For cleavage assays, 5 pmol each variant sgRNA was mixed with equimolar amounts of SpCas9 and incubated at room temperature for 15 minutes. 0.5 pmol DNA target Substrate 1 or 2 (5’-TAGGCGCCGACCGATTAAGTTAACTTCAATTAAAGTGGATTAAACATAAAACAATTAAGTTGTCCCCAAAAACATCTCAAGAATTGTGTTGATTCTGCTCATTTTTCATTTTACACATTCTCTCTTTCGCAGATCATTAAAGAGTCCACTACGGAGAGGAAGGATCTGCCAGGATCCTCTTCTATGGTGAGCAAGGGCGAGGAGCTGTTCACCGGGGTGGTGCCCATCCTGGTCGAGCTGGACGGCGACGTAAACGGCCACAAGTTCAGCGTGTCCGGCGAGGGCGAGGGCGATGCCACCTACGGCAAGCTGACCCTGAAGTTCATCTGCACCACCGGCAAGCTGCCCGTGCCCTGGCCCACCCCG GCGTGCAGTGCTTCAGCCGCTACCCCGACCACATGAAGCAGCACGACTTCTTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTTCAAGGACGACGGCAACTACAAGACCCGCGCCGAGGTGAAGTTCGAGGGCGACACCCTGGTGAACCGCATCGAGCTGAAGGGCATCGACTTCAAGGAGGACGGCAACATCCTGGGGCACAAGCTGGAGTACAAC-3’; the spacer sequence is underlined) was added to each tube, and the reactions were incubated at 37°C for 30 minutes. Cleavage reactions were treated with 1 μL of 20 mg/ml proteinase K (Sigma) at 37°C for 30 minutes and then loaded onto 3% agarose gels stained with SYBR Safe (ThermoFisher). Gels were imaged using a BioRad Chemidoc MP Imaging System.
Radiometric Assays.
A radiolabeled A-tailing assay was used to assess TdT-based isolation of cleavage-capable variant guides. The DNA target for these assays, Substrate 1, was synthesized as forward and reverse complementary oligonucleotides with an inverted dT at the 3’ ends to block nucleotide addition by TdT (see Data S1 for sequences). 20 pmol Substrate 1 was end labeled using 20 units T4 Polynucleotide Kinase (NEB) and 20 Ci [ɣ-32P]-ATP (6000 Ci/mmol; Perkin Elmer) at 37°C for 1 hour. Radiolabeled DNA was cleaned with Micro Bio-Spin P30 columns following manufacturer’s protocols (BioRad).
20 pmol of WT scaffold RNA, Round 0 RNA, or Round 5 RNA were incubated with 20 pmol of either active SpCas9 or an inactive “dead” variant (dCas9; NEB) in a reaction that contained NEB Buffer 3.1 supplemented with 0.025% Tween-20 and incubated at 37°C for 1 hour to enable for RNP formation. Trace amounts of radiolabeled Substrate 1 DNA was added to the RNP reaction mixture and incubated at 37°C for 1. The reaction mix was then supplemented with a TdT mix that consisted of 5X TdT Buffer to a final concentration of 1X, 5 mM CoCl2, 1 mM dATP, and 100 units TdT and allowed to react at 37°C for 30 minutes. The samples were cleaned through Amicon 30 kDa Ultra-0.5 mL columns to remove excess nucleotides, and 1 pmol biotinylated Oligo(dT) probe was added to each reaction. After incubation at 37°C for 15 minutes, excess probe was removed through Amicon 30 kDa Ultra-0.5 mL columns, and the eluted complexes were added to 2 μL of Streptavidin MyOne C1 Dynabeads in NEB Buffer 3.1 supplemented with 0.005% Tween-20 (Sigma) and incubated at room temperature for 1 hour with rotation. Complexes bound to the magnetic beads were sequestered and washed 3X in NEB Buffer 3.1 supplemented with 0.005% Tween-20. Washes were collected, and both bead fractions and wash fractions were mixed with Safety-Solve scintillation fluid (Research Products International), and radiation levels were detected using a Tri-Carb 4810 TR scintillation counter (Perkin Elmer).
Flow Cytometry Assays
Rounds from the sgRNA selection, as well as Round 0 and the WT scaffold were transcribed and purified as described above. For flow cytometry assays, 5 pmol each variant sgRNA was mixed with equimolar amounts of SpCas9 and incubated at room temperature for 15 minutes. A parallel control reaction also included WT scaffold mixed with equimolar amounts of the inactive “dead” Cas9. 0.5 pmol DNA target Substrate 1 labeled with Cy5 at the 5’ end was added to each tube, and the reactions were incubated at 37°C for 30 minutes. Reactions were mixed with 1 μL Streptavidin MyOne C1 Dynabeads and incubated at room temperature for 30 minutes. The beads were washed with NEB Buffer 3.1 supplemented with 0.002% Tween-20 and analyzed on a CytoFlex flow cytometer (Beckman Coulter). An example of flow gating and analysis can be found in Figure S3.
Intracellular Cleavage Assays
On Day 0, cells were seeded at 6,000 to 8,000 cells/well (HeLa, HeLa-GFP), 12,000 to 15,000 cells/well (HEK293, HEK293-GFP), or 6,000 to 8,000 cells/well (PC3, PC3-GFP) in 96-well tissue culture plates (ThermoFisher). On Day 1, for each sample, 0.5 pmol RNA was mixed with 0.5 pmol SpCas9 in Opti-MEM (ThermoFisher) and allowed to form RNPs for 15– 20 minutes. 0.1 μL Lipofectamine CRISPRMAX Cas9 Transfection reagent (ThermoFisher) diluted in 10 μL Opti-MEM was mixed with the RNP complexes and incubated at room temperature for 15 minutes. RNP:lipofection mixes were added to cells and mixed gently with culture media.
Cells were split 1:4 on Day 3. On Day 6, cells were washed with DPBS without Mg2+ or Ca2+ and lifted from the plates by incubating in 40 μL 0.025% trypsin at 37°C. The trypsin was inactivated with flow buffer (DPBS with Mg2+ and Ca2+, 2% BSA, and 1X SYTOX Red (ThermoFisher) for live/dead contrasting, and cells were interrogated on a Beckman Coulter Cytoflex flow cytometer.
Clustering of sgRNA structures
sgRNAs sequences (20 nt gRNA + scaffold + TT transcription termination residues 43) were folded using RNAfold 2.5.1 35 with option -p to calculate the partition function and the base pairing probability matrix of each sgRNA using the McCaskill’s algorithm 44. To account for indels, the sequences of the sgRNAs were aligned using, MAFFT 7 (default parameters)45, which resulted in an 102 nt alignment (82 nt aligned scaffolds). The base pairing matrices were then modified to insert rows and columns entirely set to −1 at gap positions, to obtain matrices of the same size (102 × 102).
The pairwise distance between base pairing probability matrices was computed as follows: (i) base pairing probabilities ≤ 0.24 were set to 0 to eliminate the noise given by small probability values. This threshold was identified using the elbow method implemented in kneed v.0.7.0 46 screening values of x between 0.1 to 0.8 with a step of 0.01, and matching them with the number of base pairing probabilities in the range [x, x + 0.01) (Figure S6). Kneed was run with parameter “curve” = “convex”, “direction” = “decreasing”, and “interp_method” = “polynomial”. Variations in this threshold do not alter the general structure of the hierarchy, in which active sgRNAs cluster together at early branches, but can rearrange lower levels including the members of the wild-type subcluster. This threshold was applied solely to minimize the noise in the clustering, but alternative structures of low probability are taken into account in the scrutiny of the structures; (ii) the base pairing probabilities in the cells i.e., the probabilities of the gRNA spacer folding on itself, were set to 0; (iii) the base pairing probabilities of the first 20 rows were summed up over all columns (ignoring −1 values), obtaining 20 values representing the probability that a nucleotide in the gRNA spacer pairs anywhere with the scaffold (iv) the first 20 rows and 20 columns were removed from the base pairing probability matrices (v) the Euclidean distance was computed on the upper triangle of the base pairing probability matrices and the sum-up probabilities of the first 20 rows, ignoring comparisons with value −1 (see Figure S5). SgRNA scaffold structures were clustered via complete linkage, and a dendrogram of the linkage was generated in SciPy 1.9.0 47 (Python 3.10.6 48). The order of the branches in the WT subcluster was manually edited to group efficient elements (this has no effect on the distances represented in the cluster). The aligned sgRNA sequences obtained with MAFFT were used to count the variated nucleotides and mark variants at nucleotides reported to interact with Cas9 34 in the WT scaffold as critical (positions 1, 3, 22, 23, 24, 29, 30, 31, 32, 38, 39, 47, 48, 61, 68, 69, 70, 71 in the WT scaffold, which correspond to positions 1, 3, 24, 25, 26, 31, 32, 33, 34, 40, 41, 50, 51, 65, 72, 73, 74, 75, 24, 25, 30, 31, 32, 33, 39, 40, 48, 49, 64, 71, 72, 73, 74 in the alignment). The clustering visualization was created in matplotlib 3.5.3 49 and seaborn 0.11.0 50. The Yate’s correction is applied when comparing the number of active and inactive sgRNAs in that fall in different clusters to account for the fact that the χ2 test is applied on discrete probabilities of frequencies (from the sgRNA counts). This gives a more conservative p-value.
Alignment of sgRNA structures
A cluster of sgRNA structures with more than 50% of its members having mean efficiency in cell ≥ 50% was identified. The cluster comprises 12 sgRNA structures, including the WT. These 12 structures were structurally aligned using pm_multi 51, which computes a variant of the Sankoff’s algorithm for simultaneous structure and global sequence alignment 52. pm_multi was used with the following options: --slow_pairwise flag on, sequence score weight –seqw 0, and gap penalty −g 0, to align the sgRNAs solely based on their structures. The consensus alignment and the sgRNA sequences aligned by structure were provided as input to structure logo 53,54 (default parameters) to obtain a logo of the sgRNAs enriched with base pairs mutual information, manually annotated.
QUANTITATION AND STATISTICAL ANALYSIS
Statistical analysis was done using GraphPad Prism 9. Frequency representation of library sequences was analyzed by Geneious software and Microsoft Excel. The software and methods for further sequence alignment and probability matrix calculations are as described in the STAR Methods. Statistics values are reported in Data S1 and Data S2. Error bars and number of replicates are as indicated in the figure legends.
Supplementary Material
Data S1. Sequencing data for Rounds 0, 1, 3 and 5, related to Figure 4. Sequencing results of BLADE-SELEX rounds are reported as output from the Geneious software. At the bottom of each sheet are nucleotide composition summaries for unique sequences in each round.
Data S2. Experimental data used to generate the main figures as well as relevant replicates and variant sgRNA sequences, related to Figures 4, 5, 7, S4, and S7. The “Fig.2B - TdT Validation” data sheet provides the data from the TdT assay that was validated via radiolabeling. The “Fig.4 – Phylogenetic Tree” data sheet provides the sequences utilized to generate the phylogenetic tree based on the primary sequence analysis. The “Fig.5 - Secondary Structure” data sheet provides the information in Figure 5 organized by sequence, in vitro cleavage, in vitro cleavage, and mutation number. We note that the “gRNA Scaffold #” in this tab refers to an internal reference number we used to organize the sequences. The “Secondary Structure Table ID” number is the scaffold identifier used in the figures and throughout the text. The “Figure 7 Heat Map” data sheet provides the data utilized to make the heat map. The data used to generate statistical significance for Figure 7 can be found on the datasheet named “Sup.Fig.7 – Retargeting”. The “Sup.Fig.4 – Knockdown” data sheet displays cellular cleavage activity of selected sgRNAs across multiple cell types. “Sup.Fig.S7 – Retargeting” lists the data used to make the Figure 7 heat map as well as Figure S7 in the Supplement.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| EnGen Spy Cas9 NLS | New England Biolabs | M0646M |
| EnGen Spy dCas9 (SNAP-Tag) | New England Biolabs | M0652T |
| TrueCut Cas9 Protein v2 | ThermoFisher | A36499 |
| Terminal Transferase | Millipore Sigma | 3333574001 |
| [ɣ-32P]-ATP (6000 Ci/mmole) | Perkin Elmer | BLU502Z250UC |
| SYTOX Red Dead Cell Stain | ThermoFisher | S34859 |
| Critical commercial assays | ||
| QIAQuick PCR Purification Kit | Qiagen | 28104 |
| Lipofectamine CRISPRMAX Cas9 Transfection Reagent | ThermoFisher | CMAX00003 |
| Deposited data | ||
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Pools, primers, and other sequences | This paper | See Data.S2 |
| Biotinylated Oligo(dT) probe | Promega | Z5261 |
| Recombinant DNA | ||
| Software and algorithms | ||
| FastAptamer | FASTAptamer: A Bioinformatic Toolkit for Combinatorial Selections | Burke Lab | University of Missouri (Alam et al., 2015) | N/A |
| Geneious | BioMatters Ltd. | N/A |
| BioRender | BioRender | N/A |
| RNAfold 2.5.1 | Lorenz et al., 2011 | N/A |
| MAFFT 7 | Katoh et al., 2019 | |
| SciPy 1.9.0 | Virtanen et al., 2020 | N/A |
| Python 3.10.6 | Van Rossum and Drake, 2009 | |
| Matplotlib 3.5.3 | Hunter, 2007 | |
| pm_multi | Hofacker et al., 2004 | |
| Structure logo | Gorodkin et al., 1997 | |
| Kneed 0.7.0 | Satopaa et al., 2011 | |
| Seaborn 0.11.0 | Waskom, 2021 | |
| Other | ||
| Amicon UltraCel 10, 0.5 Centrifugal Filter Unit | Millipore-Sigma | UFC501096 |
| Amicon UltraCel 30, 0.5 Centrifugal Filter Unit | Millipore-Sigma | UFC503096 |
| Whatman Protran BA 25 mm 0.45 micron nitrocellulose | GE Healthcare Life Sciences | 10402506 |
| Dynabeads MyOne Streptavidin C1 beads | ThermoFisher | 65002 |
| Micro Bio-Spin P-30 columns | BioRad | 7326251 |
Highlights.
In vitro evolution generates multiple functional gRNA scaffolds.
sgRNAs are highly malleable.
sgRNA scaffold variants display varied efficacy with different sgRNA spacer domains.
Functional sgRNA selection can generate novel sgRNAs with useful properties.
Significance:
CRISPR-based gene editing has revolutionized biomedical research and is on the verge of changing the practice of medicine. However, many Cas-gRNA complexes do not function well making it challenging to edit many specific sequences in the genome. To attempt to understand and hopefully address this limitation, we develop a functional SELEX (Systematic Evolution of Ligands by Exponential Enrichment) approach, termed BLADE (Binding and Ligand Activated Directed Evolution) to acquire a more thorough understanding of the structure-activity relationship (SAR) of the SpCas9 gRNA scaffold domain. This high throughput approach reveals that thousands of scaffold domain variants can be isolated, and many have the ability to cleave and edit DNA as well as the wild-type gRNA despite having numerous sequence changes. Bioinformatic and functional analyses of these variants demonstrates that the scaffold domain is quite malleable with only a few nucleotides highly conserved within its folded structure. This sequence flexibility and SAR understanding allows for the generation of unique gRNAs that incorporate scaffold domain variants that can edit certain DNA sites more effectively than the wild-type gRNA. Thus, BLADE SELEX coupled with bioinformatics has the potential to greatly expand the number of genome target sites amenable to efficient editing. Thus such functional RNA selection promises to improve our SAR understanding of and accelerate the translational development of numerous gRNAs and other highly structured, non-coding RNAs.
Acknowledgments:
We thank the laboratories of Drs. Charles Gersbach and Matthew Levy for cells used in these studies. This work was supported by NIH Grant UG3 TR002852 (BAS), the Independent Research Foundation Denmark (9041-00317B; JG and GIC) and the Novo Nordisk Foundation (NNF21OC0068988; JG and GIC). Many of the figures in the text were created with the help of Biorender.com.
Inclusion and Diversity:
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of interests: Duke University has submitted a patent application on the BLADE sgRNA selection approach.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Doudna JA (2020). The promise and challenge of therapeutic genome editing. Nature 578, 229–236. 10.1038/s41586-020-1978-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sullenger BA (2020). RGEN Editing of RNA and DNA: The Long and Winding Road from Catalytic RNAs to CRISPR to the Clinic. Cell 181, 955–960. 10.1016/j.cell.2020.04.050. [DOI] [PubMed] [Google Scholar]
- 5.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, and Zhang F (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang F, Zhou K, Ma L, Gressel S, and Doudna JA (2015). STRUCTURAL BIOLOGY. A Cas9-guide RNA complex preorganized for target DNA recognition. Science 348, 1477–1481. 10.1126/science.aab1452. [DOI] [PubMed] [Google Scholar]
- 7.Bao Z, Xiao H, Liang J, Zhang L, Xiong X, Sun N, Si T, and Zhao H (2015). Homology-integrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth Biol 4, 585–594. 10.1021/sb500255k. [DOI] [PubMed] [Google Scholar]
- 8.Casini A, Olivieri M, Petris G, Montagna C, Reginato G, Maule G, Lorenzin F, Prandi D, Romanel A, Demichelis F, et al. (2018). A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat Biotechnol 36, 265–271. 10.1038/nbt.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, Sternberg SH, Joung JK, Yildiz A, and Doudna JA (2017). Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410. 10.1038/nature24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong C, Gou Y, and Lian J (2022). SgRNA engineering for improved genome editing and expanded functional assays. Curr Opin Biotechnol 75, 102697. 10.1016/j.copbio.2022.102697. [DOI] [PubMed] [Google Scholar]
- 11.Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, and Liu DR (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157. 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, and Joung JK (2013). CRISPR RNA-guided activation of endogenous human genes. Nat Methods 10, 977–979. 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Briner AE, Donohoue PD, Gomaa AA, Selle K, Slorach EM, Nye CH, Haurwitz RE, Beisel CL, May AP, and Barrangou R (2014). Guide RNA functional modules direct Cas9 activity and orthogonality. Mol Cell 56, 333–339. 10.1016/j.molcel.2014.09.019. [DOI] [PubMed] [Google Scholar]
- 14.Riesenberg S, Helmbrecht N, Kanis P, Maricic T, and Paabo S (2022). Improved gRNA secondary structures allow editing of target sites resistant to CRISPR-Cas9 cleavage. Nat Commun 13, 489. 10.1038/s41467-022-28137-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thyme SB, Akhmetova L, Montague TG, Valen E, and Schier AF (2016). Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nat Commun 7, 11750. 10.1038/ncomms11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang T, Wei JJ, Sabatini DM, and Lander ES (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84. 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang D, Zhang C, Wang B, Li B, Wang Q, Liu D, Wang H, Zhou Y, Shi L, Lan F, and Wang Y (2019). Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat Commun 10, 4284. 10.1038/s41467-019-12281-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corsi GI, Qu K, Alkan F, Pan X, Luo Y, and Gorodkin J (2022). CRISPR/Cas9 gRNA activity depends on free energy changes and on the target PAM context. Nat Commun 13, 3006. 10.1038/s41467-022-30515-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiang X, Corsi GI, Anthon C, Qu K, Pan X, Liang X, Han P, Dong Z, Liu L, Zhong J, et al. (2021). Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning. Nat Commun 12, 3238. 10.1038/s41467-021-23576-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kundert K, Lucas JE, Watters KE, Fellmann C, Ng AH, Heineike BM, Fitzsimmons CM, Oakes BL, Qu J, Prasad N, et al. (2019). Controlling CRISPR-Cas9 with ligand-activated and ligand-deactivated sgRNAs. Nat Commun 10, 2127. 10.1038/s41467-019-09985-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma H, Tu LC, Naseri A, Huisman M, Zhang S, Grunwald D, and Pederson T (2016). Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat Biotechnol 34, 528–530. 10.1038/nbt.3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roueinfar M, Templeton HN, Sheng JA, and Hong KL (2022). An Update of Nucleic Acids Aptamers Theranostic Integration with CRISPR/Cas Technology. Molecules 27. 10.3390/molecules27031114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Brien AR, Burgio G, and Bauer DC (2021). Domain-specific introduction to machine learning terminology, pitfalls and opportunities in CRISPR-based gene editing. Brief Bioinform 22, 308–314. 10.1093/bib/bbz145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, Cheng AW, Trevino AE, Konermann S, Chen S, et al. (2014). Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol 32, 670–676. 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson JW, Randolph PB, Shen SP, Everette KA, Chen PJ, Anzalone AV, An M, Newby GA, Chen JC, Hsu A, and Liu DR (2022). Engineered pegRNAs improve prime editing efficiency. Nat Biotechnol 40, 402–410. 10.1038/s41587-021-01039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mir A, Alterman JF, Hassler MR, Debacker AJ, Hudgens E, Echeverria D, Brodsky MH, Khvorova A, Watts JK, and Sontheimer EJ (2018). Heavily and fully modified RNAs guide efficient SpyCas9-mediated genome editing. Nat Commun 9, 2641. 10.1038/s41467-018-05073-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellington AD, and Szostak JW (1990). In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822. 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 28.Martell RE, Nevins JR, and Sullenger BA (2002). Optimizing aptamer activity for gene therapy applications using expression cassette SELEX. Mol Ther 6, 30–34. 10.1006/mthe.2002.0624. [DOI] [PubMed] [Google Scholar]
- 29.Tuerk C, and Gold L (1990). Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510. 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 30.Richardson CD, Ray GJ, DeWitt MA, Curie GL, and Corn JE (2016). Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol 34, 339–344. 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- 31.Whinn KS, Kaur G, Lewis JS, Schauer GD, Mueller SH, Jergic S, Maynard H, Gan ZY, Naganbabu M, Bruchez MP, et al. (2019). Nuclease dead Cas9 is a programmable roadblock for DNA replication. Sci Rep 9, 13292. 10.1038/s41598-019-49837-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramadan K, Shevelev IV, Maga G, and Hubscher U (2004). De novo DNA synthesis by human DNA polymerase lambda, DNA polymerase mu and terminal deoxyribonucleotidyl transferase. J Mol Biol 339, 395–404. 10.1016/j.jmb.2004.03.056. [DOI] [PubMed] [Google Scholar]
- 33.Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, Huang CC, and Kain SR (1998). Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem 273, 34970–34975. 10.1074/jbc.273.52.34970. [DOI] [PubMed] [Google Scholar]
- 34.Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, and Nureki O (2014). Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949. 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, and Hofacker IL (2011). ViennaRNA Package 2.0. Algorithms Mol Biol 6, 26. 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White R, Rusconi C, Scardino E, Wolberg A, Lawson J, Hoffman M, and Sullenger B (2001). Generation of species cross-reactive aptamers using “toggle” SELEX. Mol Ther 4, 567–573. 10.1006/mthe.2001.0495. [DOI] [PubMed] [Google Scholar]
- 37.Ishizaki J, Nevins JR, and Sullenger BA (1996). Inhibition of cell proliferation by an RNA ligand that selectively blocks E2F function. Nat Med 2, 1386–1389. 10.1038/nm1296-1386. [DOI] [PubMed] [Google Scholar]
- 38.Magalhaes ML, Byrom M, Yan A, Kelly L, Li N, Furtado R, Palliser D, Ellington AD, and Levy M (2012). A general RNA motif for cellular transfection. Mol Ther 20, 616–624. 10.1038/mt.2011.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mi J, Liu Y, Rabbani ZN, Yang Z, Urban JH, Sullenger BA, and Clary BM (2010). In vivo selection of tumor-targeting RNA motifs. Nat Chem Biol 6, 22–24. 10.1038/nchembio.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Powell Gray B, Kelly L, Ahrens DP, Barry AP, Kratschmer C, Levy M, and Sullenger BA (2018). Tunable cytotoxic aptamer-drug conjugates for the treatment of prostate cancer. Proc Natl Acad Sci U S A 115, 4761–4766. 10.1073/pnas.1717705115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong I, and Lohman TM (1993). A double-filter method for nitrocellulose-filter binding: application to protein-nucleic acid interactions. Proc Natl Acad Sci U S A 90, 5428–5432. 10.1073/pnas.90.12.5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alam KK, Chang JL, and Burke DH (2015). FASTAptamer: A Bioinformatic Toolkit for High-throughput Sequence Analysis of Combinatorial Selections. Mol Ther Nucleic Acids 4, e230. 10.1038/mtna.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao Z, Herrera-Carrillo E, and Berkhout B (2018). Delineation of the Exact Transcription Termination Signal for Type 3 Polymerase III. Mol Ther Nucleic Acids 10, 36–44. 10.1016/j.omtn.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCaskill JS (1990). The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers 29, 1105–1119. 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- 45.Katoh K, Rozewicki J, and Yamada KD (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20, 1160–1166. 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Satopaa V, Albrecht J, Irwin D, and Raghavan B (2011). Finding a “Kneedle” in a Haystack: Detecting Knee Points in System Behavior. held in Minneapolis, MN, (IEEE), pp. 166–171. [Google Scholar]
- 47.Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, et al. (2020). SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 17, 261–272. 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Rossum G, and Drake FL (2009). Python 3 Reference Manual (Createspace). [Google Scholar]
- 49.Hunter JD (2007). Matplotlib: A 2D Graphics Environment. Computing in Science and Engineering 9, 90–95. 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 50.Waskom ML (2021). seaborn: statistical data visualization. Journal of Open Source Software 6. 10.21105/joss.03021. [DOI] [Google Scholar]
- 51.Hofacker IL, Bernhart SH, and Stadler PF (2004). Alignment of RNA base pairing probability matrices. Bioinformatics 20, 2222–2227. 10.1093/bioinformatics/bth229. [DOI] [PubMed] [Google Scholar]
- 52.Sankoff D (1985). Simultaneous Solution of the RNA Folding, Alignment and Protosequence Problems. SIAM Journal on Applied Mathematics 45, 810–825. 10.1137/0145048. [DOI] [Google Scholar]
- 53.Gorodkin J, Heyer LJ, Brunak S, and Stormo GD (1997). Displaying the information contents of structural RNA alignments: the structure logos. Comput Appl Biosci 13, 583–586. 10.1093/bioinformatics/13.6.583. [DOI] [PubMed] [Google Scholar]
- 54.Schneider TD, and Stephens RM (1990). Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 18, 6097–6100. 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Sequencing data for Rounds 0, 1, 3 and 5, related to Figure 4. Sequencing results of BLADE-SELEX rounds are reported as output from the Geneious software. At the bottom of each sheet are nucleotide composition summaries for unique sequences in each round.
Data S2. Experimental data used to generate the main figures as well as relevant replicates and variant sgRNA sequences, related to Figures 4, 5, 7, S4, and S7. The “Fig.2B - TdT Validation” data sheet provides the data from the TdT assay that was validated via radiolabeling. The “Fig.4 – Phylogenetic Tree” data sheet provides the sequences utilized to generate the phylogenetic tree based on the primary sequence analysis. The “Fig.5 - Secondary Structure” data sheet provides the information in Figure 5 organized by sequence, in vitro cleavage, in vitro cleavage, and mutation number. We note that the “gRNA Scaffold #” in this tab refers to an internal reference number we used to organize the sequences. The “Secondary Structure Table ID” number is the scaffold identifier used in the figures and throughout the text. The “Figure 7 Heat Map” data sheet provides the data utilized to make the heat map. The data used to generate statistical significance for Figure 7 can be found on the datasheet named “Sup.Fig.7 – Retargeting”. The “Sup.Fig.4 – Knockdown” data sheet displays cellular cleavage activity of selected sgRNAs across multiple cell types. “Sup.Fig.S7 – Retargeting” lists the data used to make the Figure 7 heat map as well as Figure S7 in the Supplement.
Data Availability Statement
The sequences of the sgRNA variants and components for BLADE SELEX tested in this manuscript are described in the attached Data S1 and Data S2 files. Experimental data used to generate the figures in this text are also included in the file Data.S2. Additional data reported in this paper will be shared by the lead contact upon request.
Methodology and software used for structural analysis of variant guides are as described in the text. The scripts and further details on how to use them are available upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.