

SUMMARY
Ionotropic glutamate receptors (GluRs) are targets for modulation in Hebbian and homeostatic synaptic plasticity and are remodeled by development, experience, and disease. We have probed the impact of synaptic glutamate levels on the two postsynaptic GluR subtypes at the Drosophila neuromuscular junction, GluRA and GluRB. We first demonstrate that GluRA and GluRB compete to establish postsynaptic receptive fields, and that proper GluR abundance and composition can be orchestrated in the absence of any synaptic glutamate release. However, excess glutamate adaptively tunes postsynaptic GluR abundance, echoing GluR scaling observed in mammalian systems. Furthermore, when GluRA vs. GluRB competition is eliminated, GluRB becomes insensitive to glutamate modulation. In contrast, GluRA is now homeostatically regulated by excess glutamate to maintain stable miniature activity, where Ca2+ permeability through GluRA receptors is required. Thus, excess glutamate, GluR competition, and Ca2+ signaling collaborate to selectively target GluR subtypes for homeostatic regulation at postsynaptic compartments.
Graphical Abstract
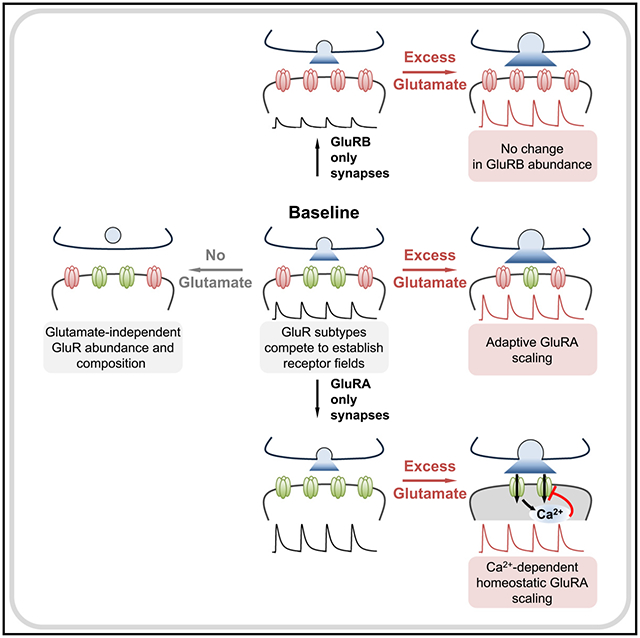
In brief
Differences in glutamate receptor abundance and composition underlie numerous forms of synaptic plasticity, but how receptors respond to changes in glutamate itself is unclear. Han et al. demonstrate that excess glutamate release provokes subtype-specific, homeostatic receptor downscaling. Calcium permeability through the glutamate receptor is necessary for homeostatic regulation.
INTRODUCTION
Ionotropic glutamate receptors (GluRs) are dynamically regulated at postsynaptic densities during development, plasticity, aging, and disease. A variety of intracellular signaling systems in the postsynaptic compartment coordinate the establishment, maintenance, and remodeling of GluR abundance and composition during development, plasticity, and disease.1,2 Another layer of regulation involves competition between GluR subtypes, where, for example, Ca2+ permeable and impermeable AMPA receptor subtypes are selectively modulated during plasticity,1,3,4 and alterations in synaptic activity can drive both Hebbian and homeostatic remodeling of GluRs.2,5–8 However, the role of the neurotransmitter glutamate itself in regulating postsynaptic GluRs is somewhat counterintuitive: while glutamate is dispensable for the development of GluR fields,9–11 extracellular glutamate alone is capable of provoking GluR assembly de novo.12 To what extent levels of synaptic glutamate itself influences GluR composition and abundance has not been clearly defined.
The glutamatergic Drosophila neuromuscular junction (NMJ) is an attractive system to interrogate the role of synaptic glutamate in establishing and adaptively modulating postsynaptic GluR fields. At this synapse, the postsynaptic GluRs are classified as kainate-type GluRs (KARs13), tetramers composed of three essential subunits (GluRIIC, GluRIID, and GluRIIE) and one of two alternative subunits, GluRIIA or GluRIIB14,15 (Figure 1A). Here, we abbreviate these distinct KAR subtypes as “GluRA” and “GluRB” to define receptors containing either the GluRIIA or GluRIIB subunit. Studies in vivo and in heterologous systems have shown that the majority of postsynaptic Ca2+ influx and depolarizing currents are driven by GluRA receptors, while GluRB passes much less current due to rapid desensitization.16,17 In addition, the recent development of a botulinum neurotoxin that blocks neurotransmitter release in Drosophila now enables the silencing of synaptic glutamate release,11 while excess glutamate can be released from individual synaptic vesicles by overexpression of the vesicular glutamate transporter in Drosophila motor neurons.18–21 However, whether changes in synaptic glutamate levels are capable of adaptively modulating GluRs at the fly NMJ is not known.
Figure 1. GluRIIA- and GluRIIB-containing receptor subtypes compete to establish postsynaptic glutamate receptor fields.
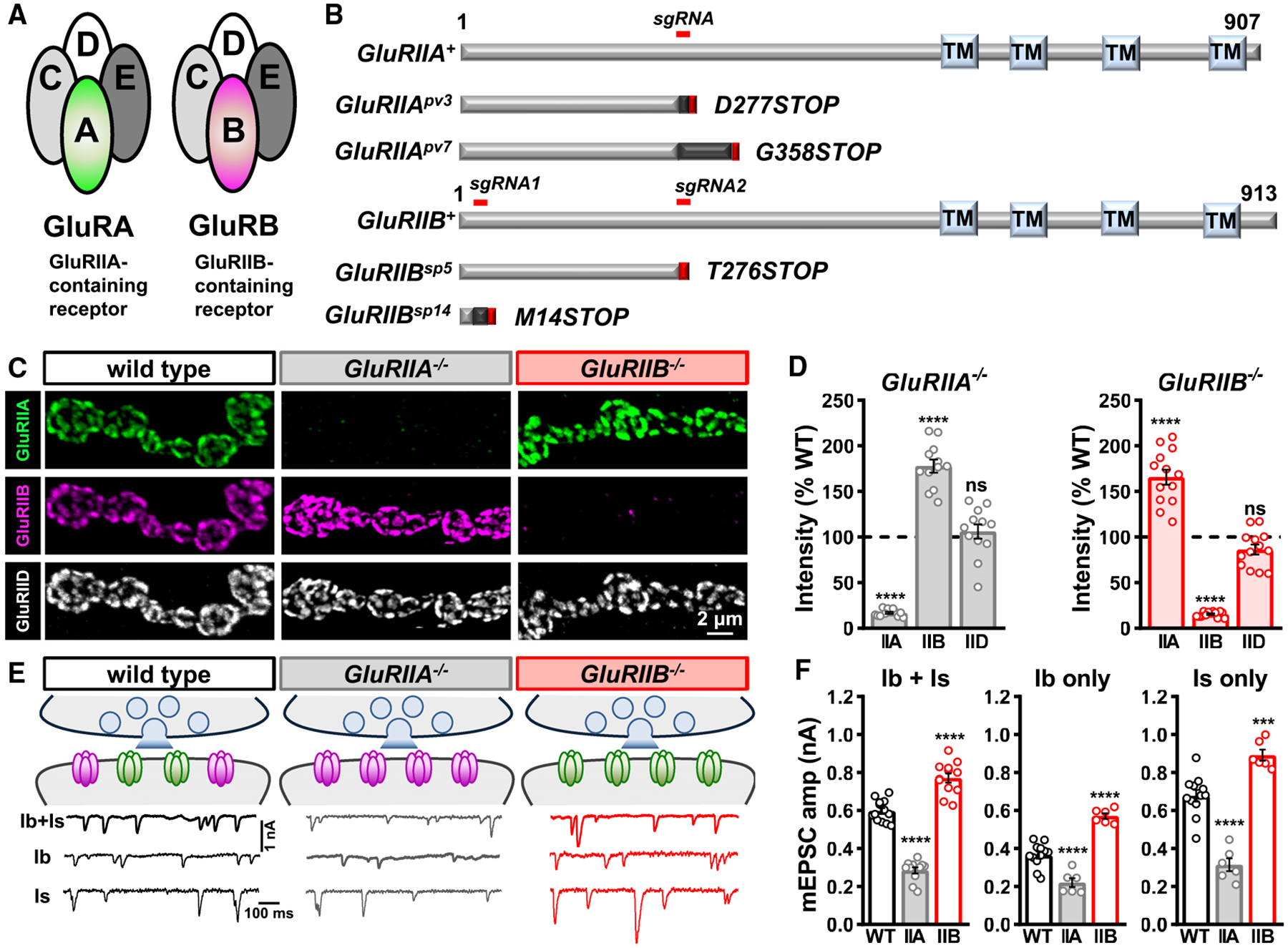
(A) Schematic illustrating the subunit composition of GluRIIA- and GluRIIB-containing postsynaptic GluR subtypes at the Drosophila NMJ, referred to here as GluRA and GluRB receptors.
(B) Domain structure of GluRIIA and GluRIIB subunits with the region targeted by the single- guide (sg)RNA used to generate the CRISPR mutant alleles and their predicted protein products indicated below.
(C) Representative images of muscle 6 MN-Ib NMJs in wild type (w1118), GluRIIA mutants (w;GluRIIApv3), and GluRIIB mutants (w;GluRIIBsp5) immunostained with antibodies against three postsynaptic GluR subunits (GluRIIA, GluRIIB, and GluRIID).
(D) Quantification of mean GluR fluorescence intensity normalized to wild-type values confirms GluRIIA subunits are not detected at NMJs of GluRIIApv3 mutants, while GluRIIB levels are significantly increased. An inverse change is found in GluRIIBSP5 mutants. Similar results are observed in GluRIIApv7 and GluRIIBsp14 alleles. Overall levels of the common GluRIID subunit do not significantly change in either mutant.
(E) Schematic summarizing the data in (C) and (D). Representative electrophysiological traces of blended mEPSC events from Is+Ib motor inputs, and isolated MN-Ib only (WT: w;+;Is-GAL4/UAS-BoNT-C; GluRIIA−/−: w;GluRIIApv3;Is-GAL4/UAS-BoNT-C; GluRIIB−/−: w;GluRIIBsp5;Is-GAL4/UAS-BoNT-C) or MN-Is only (same genotypes as Ib only except Ib-GAL4 used instead of Is-GAL4) in the indicated genotypes are shown below.
(F) Quantification of mEPSC frequency and amplitude in the indicated genotypes. Note that while baseline mEPSC amplitudes are different in Ib- vs. Is-only NMJs, as expected, mEPSC amplitudes from each input are significantly reduced in GluRIIApv3 and increased in GluRIIBsp5 mutants compared to their baseline, consistent with the staining results. Error bars indicate ±SEM, with the following statistical significance: ***p < 0.001, ****p < 0.0001; ns, not significant. Additional statistics and sample number values (n) for all experiments are summarized in Table S1.
We have generated null mutations that specifically ablate GluRIIA and GluRIIB receptor subunits using CRISPR-Cas9 gene editing. These mutants have provided an opportunity to investigate how GluR fields are established during development, how they respond to synaptically released glutamate, and to define how competition between GluRA and GluRB influences GluR plasticity. These studies reveal a hierarchy of control through competition between GluR subtypes and selective, homeostatic regulation by synaptic glutamate that requires Ca2+ permeability through GluRA receptors.
RESULTS
GluRIIA and GluRIIB mutants reveal competition between receptor subtypes
To understand how postsynaptic receptive fields are established at the Drosophila NMJ, we generated targeted genetic mutations in the two distinctive GluR subunits, GluRIIA and GluRIIB (Figures 1A and 1B). Although a mutant allele of GluRIIA was generated over 20 years ago using imprecise transposon excision22 (GluRIIASP16), this lesion did not specifically mutate GluRIIA; expression of a neighboring gene, oscillin, was also disrupted.23 Specific mutations in GluRIIB have not been reported. We used single-guide RNAs (sgRNAs) targeting early exons of the GluRIIA or GluRIIB coding regions combined with Cas9-mediated mutagenesis to generate a series of specific mutations in either subunit; two independent null mutations in GluRIIA and GluRIIB were chosen for further analysis (see STAR Methods; Figure 1B). To validate these alleles, we co-immunostained the larval NMJ with antibodies against GluRIIA, GluRIIB, and the common essential subunit GluRIID. As expected, the GluRIIA and GluRIIB signals were similar to background in the GluRIIA or GluRIIB mutants, while the common GluRIID signal was maintained in both (Figure 1C). We confirmed the GluRIIA and GluRIIB subunits are exclusively expressed in muscle (Figure S1A), that muscle-specific expression of either subunit in mutant backgrounds can largely restore GluR levels (Figures S1B–S1G), and that mRNA levels of the receptor subunits are substantially reduced or eliminated in the mutant backgrounds (Figures S1D and S2H). Thus, this approach has generated clean null alleles in the GluRIIA and GluRIIB receptor subunits.
Next, we used immunostaining and electrophysiology to determine the composition and functionality of receptive fields exclusively composed of GluRA or GluRB. In GluRIIA mutants, a compensatory ~170% enhancement in GluRIIB intensity was found, with no significant change in the common GluRIID subunit (Figures 1C and 1D). However, mEPSC amplitude, which reflects the postsynaptic current induced by the spontaneous release of single synaptic vesicles, was reduced by over 50% without changing mEPSC frequency (Figures 1E and 1F), as expected by exclusive expression of the rapidly desensitizing GluRB receptors and as observed in previous GluRIIA mutant studies.16,22 Conversely, GluRIIA levels were similarly increased in GluRIIB mutants, with no overall change in GluRIID (Figures 1C and 1D). This increased GluRA expression was reflected by a large increase in mEPSC amplitude (Figures 1E and 1F). Hence, loss of one GluR subtype leads to a concomitant increase in the other receptor subtype at postsynaptic receptive fields.
We performed several important controls. First, most Drosophila muscle fibers are co-innervated by two distinct motor neuron subtypes, MN-Is and MN-Ib, that differ in morphology and function.11,24,25 mEPSCs are therefore blended events of spontaneous activity emanating from both inputs. Because our electrophysiological recordings are a blend of events deriving from both MN-Ib and MN-Is inputs, it is possible that the average mEPSC amplitudes recorded do not accurately reflect GluR changes at both NMJs. To isolate transmission from Is vs. Ib, we used selective expression of BoNT-C, which silences all neurotransmitter release without inducing altered innervation or heterosynaptic plasticity.11 mEPSC events are larger at MN-Is compared to MN-Ib NMJs due to Is terminals having larger synaptic vesicles, which enhances glutamate emitted from individual synaptic vesicles.11,26 As expected, mEPSCs were larger at MN-Is NMJs compared to -Ib, and mEPSCs were reduced or increased in GluRIIA or GluRIIB mutants relative to these different baseline states, as expected (Figures 1E and 1F). To control for possible differences in the decay kinetics of mEPSCs, we found the total charge transfer was similarly reduced in GluRIIA mutants and enhanced in GluRIIB mutants (Table S1). Additional analyses of GluRs revealed that GluRA and GluRB receptors are intermixed at individual postsynaptic clusters, with GluRA typically localized in the center and GluRB more peripherally (Figure S2A), as previously observed.11,14,27 In addition, total GluR cluster number per NMJ did not significantly change in GluRIIA or GluRIIB mutants (Figures S2B and S2C), although GluR cluster volume was moderately reduced in the mutants compared to wild type (Figure S2D). As expected, GluRIIA and GluRIIB subunits exhibit a high degree of co-localization with the common subunit GluRIID at wild-type NMJs (Figures S2E and S2F), which is enhanced with loss of the other subunit (Figures S2E and S2F).
Finally, we probed the relationship between GluRIIA and GluRIIB competition further by asking whether overexpression of one subunit outcompetes the other. When we transgenically overexpressed the GluRIIA or GluRIIB subunits in muscle (GluRIIA-OE or GluRIIB-OE), we observed high levels of GluRIIA or GluRIIB, as expected (Figures 2A and 2B). Interestingly, transcriptional overexpression of either subunit reduced the other subunit to levels undetectable above background, while total GluR levels, as assessed by the common subunit GluRIID, remained unchanged (Figures 2A and 2B). Correspondingly, mEPSC amplitudes were enhanced in GluRIIA-OE or reduced in GluRIIB-OE (Figures 2C and 2D). Hence, GluRIIA-OE essentially phenocopies GluRIIB mutants, while GluRIIB-OE phenocopies GluRIIA mutants. Taken together, these experiments suggest that GluRA and GluRB receptors compete to establish postsynaptic receptive fields without changing total GluR abundance at postsynaptic compartments.
Figure 2. GluR subunit overexpression outcompetes the opposing subunit.
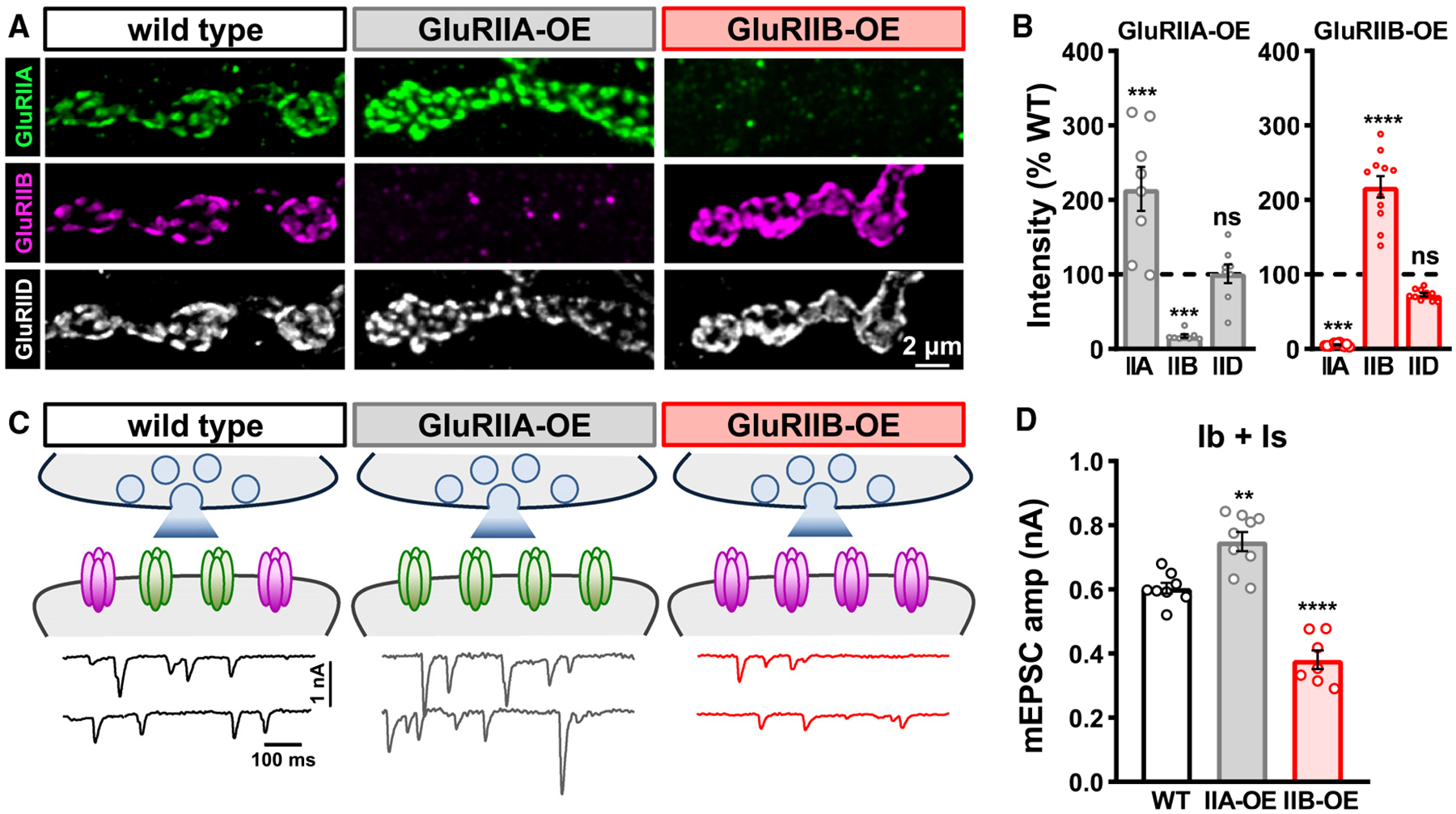
(A) Representative images of MN-Ib NMJs at muscle 6 in wild-type, muscle-specific GluRIIA overexpression (GluRIIA-OE: w,MHC-GluRIIA;+;+), and muscle-specific GluRIIB overexpression (GluRIIB-OE: w;G14-GAL4/+;UAS-GluRIIB/+) immunostained with anti-GluRIIA, -GluRIIB, and -GluRIID.
(B) Quantification of mean GluR intensity levels normalized to wild-type values in the indicated genotypes. Note that GluRIIB levels are essentially eliminated in GluRIIA-OE, while GluRIIA levels are largely absent in GluRIIB-OE.
(C) Schematic and mEPSC traces from blended Is+Ib inputs in the indicated genotypes.
(D) Quantification of mEPSC frequency and amplitude in the indicated genotypes. While mEPSC frequency does not significantly change, mEPSC amplitude is enhanced in GluRIIA-OE and reduced in GluRIIB-OE, as expected. Error bars indicate ±SEM, with the following statistical significance: **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant. Additional statistics and sample number values (n) for all experiments are summarized in Table S1.
Excess glutamate release adaptively reduces GluR abundance
GluRA and GluRB receptors compete to establish postsynaptic receptive fields, but it is not clear what physiologic signals regulate this competition. We hypothesized that the levels of synaptically released glutamate may modulate postsynaptic GluR abundance and/or composition. We first eliminated all synaptic glutamate release to determine whether GluR abundance or organization requires synaptic activity or glutamate itself. To accomplish this, we used a botulinum neurotoxin (BoNT) transgene that targets the SNARE protein syntaxin for cleavage at release sites to block all neurotransmitter release.11 Specifically, selective expression of BoNT-C in both MN-Is and -Ib that innervate muscle 6, using the driver OK319-GAL4, eliminates all miniature and evoked glutamate release and does not impact NMJ growth or morphology11 (Figures 3A and 3B). We then quantified GluRIIA, GluRIIB, and GluRIID immuno-intensity levels and found no significant difference in their levels or organization compared to wild-type controls (Figures 3C and 3D). Thus, glutamate release from presynaptic release sites is not required to establish or maintain GluRA or GluRB abundance at the Drosophila NMJ.
Figure 3. Excess presynaptic glutamate release induces a compensatory reduction in postsynaptic GluR abundance.
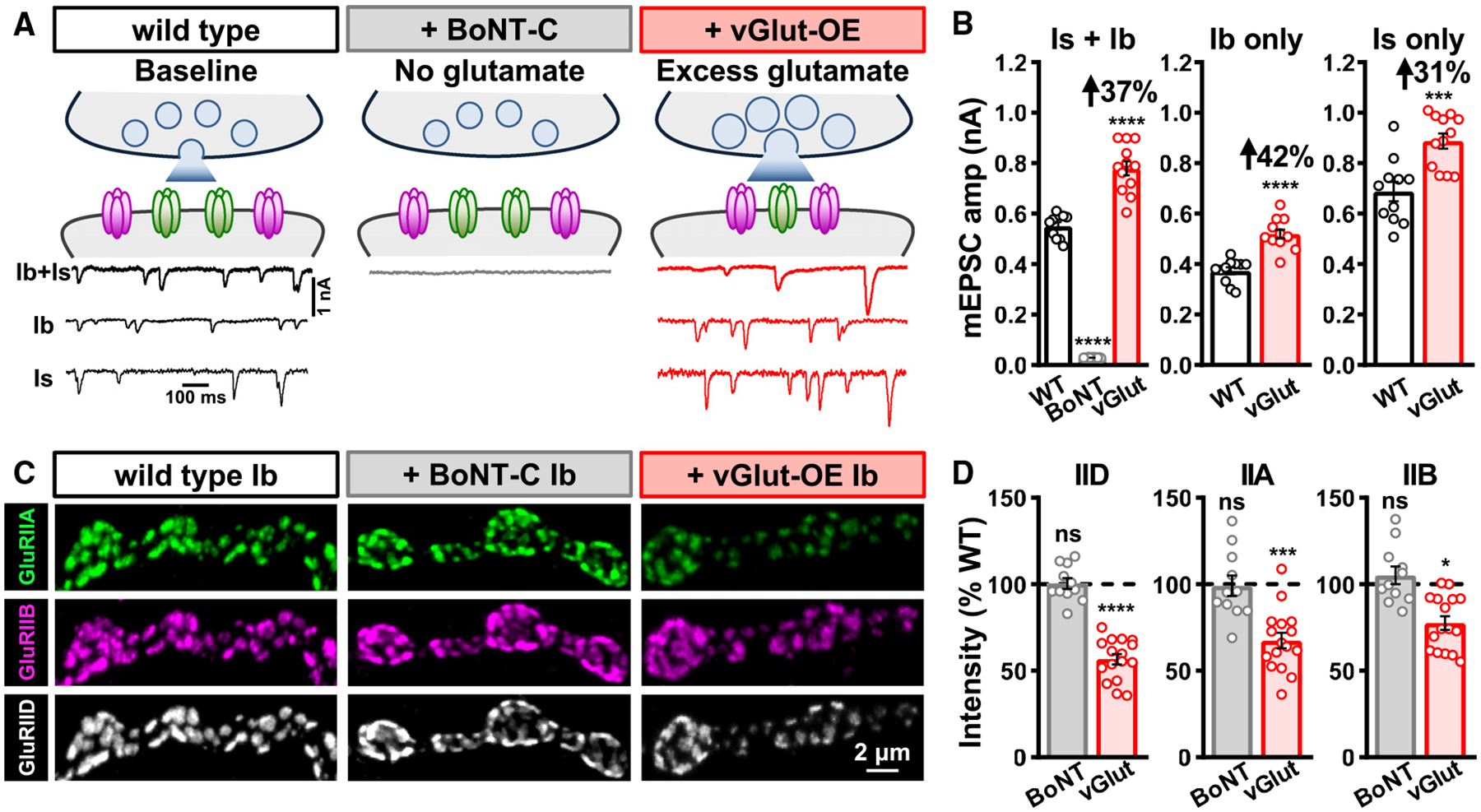
(A) Schematics and representative mEPSC traces of wild type, NMJs with no synaptic glutamate release due to BoNT-C expression (w;OK319-GAL4/+;UAS-BoNT-C/+), and NMJs releasing excess glutamate due to vGlut overexpression in motor neurons (vGlut-OE: w;OK371-Gal4/UAS-vGlut). mEPSC events from MN-Ib or -Is were isolated from vGlut-OE using LexA-Op-vGlut (w;OK6-LexA/LexAop-vGlut;dHb9-GAL4/UAS-BoNT-C and w;OK6-LexA/LexAop-vGlut;R27E09-GAL4/UAS-BoNT-C).
(B) Quantification of mEPSC amplitude in the indicated genotypes and inputs. Note that while BoNT-C expression eliminates all synaptic vesicle release and miniature activity, vGlut-OE leads to enhanced quantal size from both MN-Ib and -Is inputs, as expected.
(C) Representative images of MN-Ib NMJs from wild type, BoNT-C, and vGlut-OE immunostained with anti-GluRIIA, -GluRIIB, and -GluRIID. While GluR abundance is unchanged in the absence of glutamate release, excess glutamate induces a compensatory reduction in GluR abundance.
(D) Quantification of mean fluorescence intensity of individual GluRIID, GluRIIA, and GluRIIB puncta in the indicated genotypes normalized to wild-type values, indicating a reduction in total GluR abundance in vGlut-OE. Similar results are observed at MN-Is NMJs (Figures S3A and S3B). Error bars indicate ±SEM, with the following statistical significance: *p < 0.05, ***p < 0.001, ****p < 0.0001; ns, not significant. Additional statistics and sample number values (n) for all experiments are summarized in Table S1.
Next, we tested whether enhanced presynaptic glutamate release from individual synaptic vesicles impacts postsynaptic GluR fields. Neuronal overexpression of the vesicular glutamate transporter (vGlut) increases the size of synaptic vesicles and leads to a concomitant increase in the abundance of glutamate emitted from single synaptic vesicles.18–20 We use the term “excess” glutamate release in vGlut-OE simply to refer to the greater glutamate abundance released from individual synaptic vesicles following vGlut-OE compared to wild-type values. Transgenic overexpression of vGlut in motor neurons using OK371-GAL4 (vGlut-OE) enhanced mEPSC amplitude at blended Is+Ib NMJs, as well as at isolated MN-Is or -Ib NMJs, as expected (Figures 3A and 3B). Interestingly, total GluR abundance, as assessed by GluRIID immunofluorescence intensity, was reduced by ~50% in vGlut-OE (Figures 3C and 3D), with a substantial diminishment of GluRIIA and a small but significant reduction in GluRIIB at MN-Ib postsynaptic compartments (Figures 3C and 3D). Similar changes were observed at MN-Is NMJs, except that no significant change in GluRIIB levels following vGlut-OE was found (Figures S3A and S3B). We determined that surface GluRA receptors were adaptively downregulated by performing GluR staining in intact vs. perforated muscle (Figures S3A and S3B). To control for potential artifacts of vGlut overexpression, we characterized GluR levels in minibrain mutants (mnb1), an endocytotic gene that when lost leads to enhanced synaptic vesicle size and glutamate release.28 Similar to vGlut-OE, we observed increased mEPSC amplitude and reduced GluR levels in mnb1 mutants compared to wild type (Figures S5A–S5D). Therefore, excess presynaptic glutamate release from both Is and Ib motor neurons induces an adaptive reduction in postsynaptic GluRA abundance at both NMJs.
Interestingly, these data likely explain the distinction in GluR subtypes at MN-Is vs. MN-Ib NMJs. Synaptic vesicle size is enlarged at MN-Is terminals compared to MN-Ib,26 which leads to enhanced miniature amplitude at MN-Is compared to MN-Ib.11 In addition, GluR composition is also different at MN-Is vs. MN-Ib NMJs, with a higher ratio of GluRA::GluRB receptors at MN-Ib postsynaptic compartments.11,14 Thus, enhanced glutamate is released from individual synaptic vesicles at MN-Is relative to MN-Ib, indicating that adaptive GluR scaling happens naturally at wild-type synapses.
GluRA receptors are homeostatically regulated in the absence of GluRB competition
Although excess glutamate appears to adaptively reduce postsynaptic GluRA levels, this apparent change in GluRA abundance is compensatory but not homeostatic. In particular, mEPSC amplitudes are still increased in vGlut-OE, indicating that the reduction in GluRA is not sufficient to maintain baseline miniature activity. We hypothesized that GluRA vs. GluRB competition may obscure the individual responses of GluRA vs. GluRB to excess glutamate. Therefore, we next sought to isolate the behavior of GluRA receptors to excess glutamate in the absence of GluRB receptors and vice versa.
To characterize the behavior of GluRA or GluRB receptors in the absence of subtype competition, we manipulated glutamate in GluRIIA or GluRIIB null mutants, where NMJs were composed exclusively of GluRB or GluRA. First, we found that GluRIIA or GluRIIB mutant synapses devoid of glutamate release by BoNT-C expression had no significant impact on GluR levels in GluRB- or GluRA-only NMJs (Table S1). Next, we examined the impact of vGlut-OE on GluRIIA mutant NMJs, which exclusively express GluRB receptors (Figures 4A and 4B). mEPSCs were reduced by ~50% in GluRIIA mutants alone compared to wild type, as expected, but they were increased by 64% in GluRIIA+vGlut-OE compared to baseline values (GluRIIA mutants; Figure 4A). Correspondingly, immunostaining revealed that GluRIIB and GluRIID levels did not change in GluRIIA+vGlut-OE compared to GluRIIA mutants alone (Figure 4B). Similar results were observed at GluRIIA mutant MN-Is NMJs (Figure S3C). These data indicate that in the absence of GluRA receptors, GluRB receptor abundance is not adaptively modulated by excess glutamate release.
Figure 4. Excess glutamate does not adaptively downregulate GluRB in the absence of GluRA.
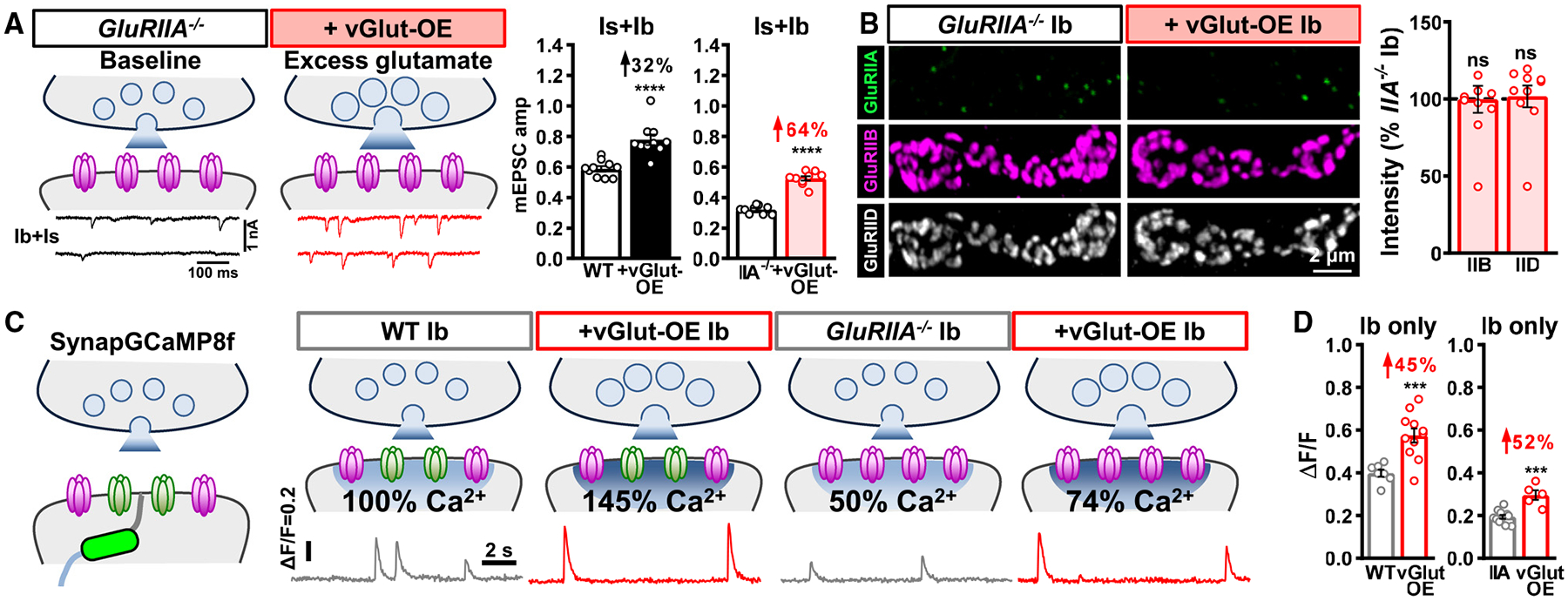
(A) Schematics and representative mEPSC traces of blended Is+Ib NMJs containing only GluRB receptors (GluRIIA−/−mutants: w;GluRIIApv3) at baseline and following vGlut-OE (GluRIIA+vGlut-OE: w;GluRIIApv3,UAS-vGlut/GluRIIApv3,OK371-Gal4). Right: quantification of mEPSC amplitude in the indicated genotypes; the percentage above indicates the increase observed compared to baseline (wild type or GluRIIA mutants). Note that mEPSC amplitude is enhanced by 64% in GluRIIA+vGlut-OE over GluRIIA mutants alone.
(B) Representative images of GluRs from MN-Ib NMJs immunostained in the indicated genotypes. Right: quantification of GluR mean fluorescence intensity in GluRIIA+vGlut-OE normalized to GluRIIA mutants alone. In the absence of GluRA receptors, excess glutamate release does not significantly change GluRB abundance.
(C) Schematics and representative quantal events using SynapGCaMP8f Ca2+ imaging at MN-Ib NMJs from the indicated genotypes. Note that quantal events are enhanced by 50% in vGlut-OE or GluRIIA−/−+vGlut-OE and reduced by ~50% in GluRIIA−/−, compared to baseline values (wild type or GluRIIA−/−), as expected.
(D) Quantification of average fluorescence intensity of quantal events in the indicated genotypes. Similar results are observed at MN-Is NMJs from the same genotypes (Figures S3C and S3D). Error bars indicate ±SEM, with the following statistical significance: ***p < 0.001, ****p < 0.0001; ns, not significant. Additional statistics and sample number values (n) for all experiments are summarized in Table S1.
To determine mEPSC amplitude at isolated MN-Is or -Ib NMJs, we attempted to selectively express BoNT-C in GluRIIA mutant backgrounds, but unfortunately these larvae did not survive. Therefore, we performed quantal Ca2+ imaging at postsynaptic MN-Ib and -Is NMJ compartments using GCaMP8f targeted to postsynaptic densities11 (Figure 4C, SynapGCaMP8f) in wild-type, vGlut-OE, GluRIIA, and GluRIIA+vGlut-OE mutants. Quantal Ca2+ events were reduced by ~50% in GluRIIA mutants compared to wild type at both MN-Ib and -Is NMJs and enhanced by ~50% in vGlut-OE at both MN-Ib and -Is NMJs, as expected (Figures 4C, 4D, and S3D). Quantal Ca2+ events were also enhanced in GluRIIA+vGlut-OE compared to GluRIIA mutants alone at both MN-Ib and -Is NMJs (Figures 4C, 4D, and S3D). Thus, in the absence of competition with GluRA receptors, GluRB receptors are not adaptively modulated in either MN-Ib or -Is synapses.
Finally, we characterized the impact of excess glutamate in GluRIIB mutants. In these mutants, baseline mEPSC amplitudes at blended Ib+Is NMJs were elevated by ~50% compared to wild type due to the exclusive and enhanced expression of GluRA receptors (Figure 5A). Remarkably, however, no significant difference in mEPSC amplitude was observed in GluRIIB mutants with excess glutamate release compared to GluRIIB mutants alone (Figure 5A). This suggests that in the absence of GluRB, GluRA receptors are homeostatically downregulated by excess glutamate to maintain baseline miniature amplitude. Indeed, vGlut-OE led to a >60% reduction in GluRIIA levels compared to baseline (GluRIIB mutants) at both MN-Ib and Is NMJs (Figures 5B and S3E). Quantal Ca2+ imaging of miniature events revealed excess glutamate release did not change quantal events relative to baseline in GluRIIB mutants at either MN-Ib or -Is NMJs (Figures 5C, 5D, and S3F). This quantitative reduction in GluRA abundance was sufficient in amplitude to explain the stable mEPSC values despite excess glutamate driven by vGlut-OE. Thus, the elimination of competition between GluRA and GluRB receptors reveals two distinct, subtype-specific responses to excess synaptic glutamate release at fly NMJ receptive fields: (1) GluRB receptors are immutable, remaining fixed and unresponsive to glutamate, while (2) GluRA receptors are plastic, precisely downregulated in response to excess synaptic glutamate to homeostatically maintain stable miniature activity.
Figure 5. Excess glutamate homeostatically downregulates GluRA in the absence of GluRB.
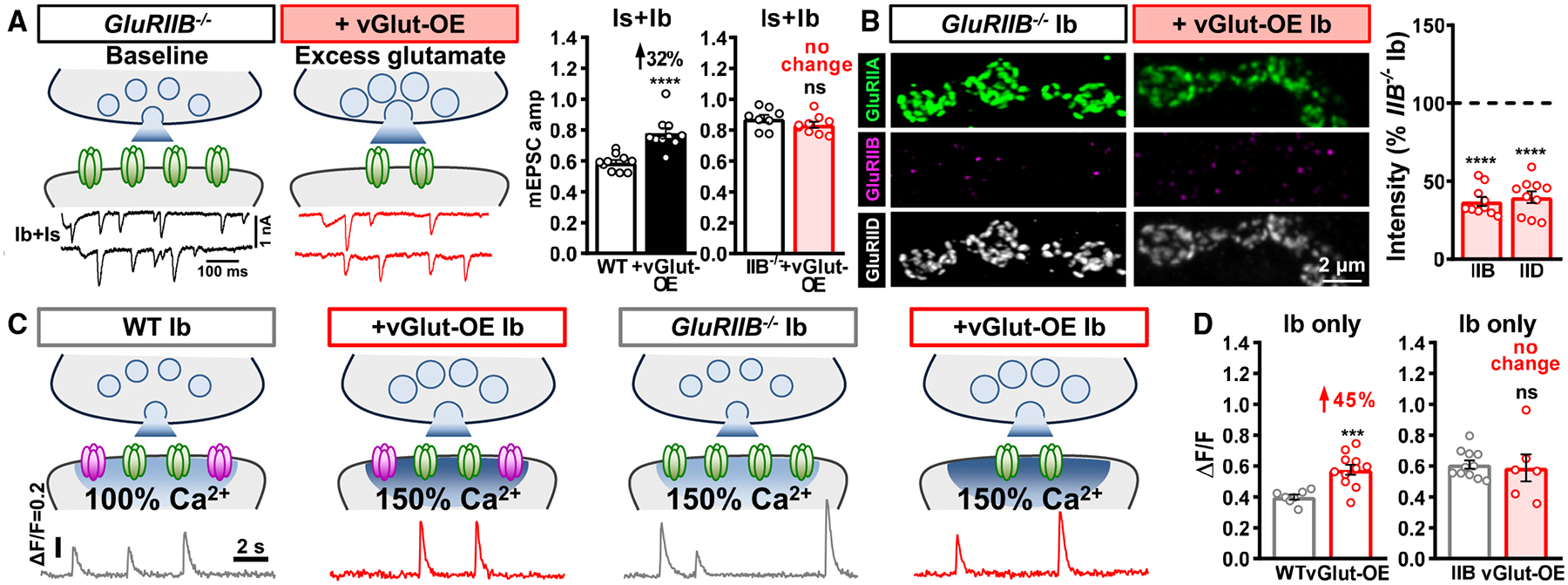
(A) Schematics and representative mEPSC traces of blended Is+Ib NMJs containing only GluRA receptors (GluRIIB−/−mutants: w;GluRIIBsp5) at baseline and following vGlut-OE (GluRIIB+vGlut-OE: w;GluRIIBsp5,UAS-vGlut/GluRIIBsp5,OK371-Gal4). Right: quantification of mEPSC amplitude in the indicated genotypes; the percentage above indicates the change observed compared to baseline (wild type or GluRIIB mutants). Note that mEPSC amplitude is not significantly changed in GluRIIB+vGlut-OE over GluRIIB mutants alone.
(B) Representative images of GluRs from MN-Ib NMJs immunostained in the indicated genotypes. Right: quantification of GluR mean fluorescence intensity in GluRIIB+vGlut-OE normalized to GluRIIB mutants alone. In the absence of GluRB receptors, excess glutamate substantially diminishes GluRA abundance.
(C) Schematics and representative quantal events using SynapGCaMP8f Ca2+ imaging at MN-Ib NMJs from the indicated genotypes. Note that quantal events are enhanced by ~50% in vGlut-OE or GluRIIB−/−compared to wild type, as expected, but no significant change is observed in GluRIIB+vGlut-OE compared to GluRIIB mutants alone.
(D) Quantification of average fluorescence intensity of quantal events in the indicated genotypes. Similar results are observed at MN-Is NMJs from the same genotypes (Figures S3E and S3F). Error bars indicate ±SEM, with the following statistical significance: ***p < 0.001, ****p < 0.0001; ns, not significant. Additional statistics and sample number values (n) for all experiments are summarized in Table S1.
Ca2+ permeability through GluRA is necessary for homeostatic receptor scaling
Postsynaptic GluRA receptor abundance is homeostatically diminished by enhanced glutamate release. Ca2+ influx through GluRs and related signaling in postsynaptic compartments orchestrate a number of forms of plasticity that ultimately modulate GluR abundance and composition at postsynaptic receptive fields.1,29 GluRA receptors are Ca2+ permeable and pass the majority of glutamate-elicited currents at postsynaptic compartments of the Drosophila NMJ.16,17 We therefore hypothesized that Ca2+ influx through GluRA receptors may be necessary for homeostatic receptor scaling induced by excess glutamate release.
To test this hypothesis, we generated a GluRIIA allele designed to specifically eliminate Ca2+ influx through GluRA using CRISPR-Cas9 gene editing. Like most Ca2+-permeable GluRs, the GluRIIA subunit encodes a neutral glutamine amino acid (Q) in the pore-forming M2 loop (Q615; Figure 6A). To render GluRA Ca2+ impermeable while remaining permeable to other ions, we used CRISPR-Cas9 genome editing to mutate this glutamine to the positively charged amino acid arginine (R) at the endogenous GluRIIA locus to make GluRIIAQ615R alleles. In heterologous systems and in vivo, this Q to R transition renders both AMPA- and Kainate-GluRs impermeable to Ca2+.13,30–33 GluRIIAQ615R mutants are homozygous viable and healthy, exhibit normal synaptic transmission (miniature and evoked release), and express GluRA and GluRB receptors that do not significantly change in abundance compared to wild type,34 as expected. Thus, GluRIIAQ615R mutants render GluRA receptors Ca2+ impermeable without impacting basal synaptic transmission or relative GluR composition or abundance.
Figure 6. Ca2+ permeability is required for the homeostatic control of GluRA abundance.
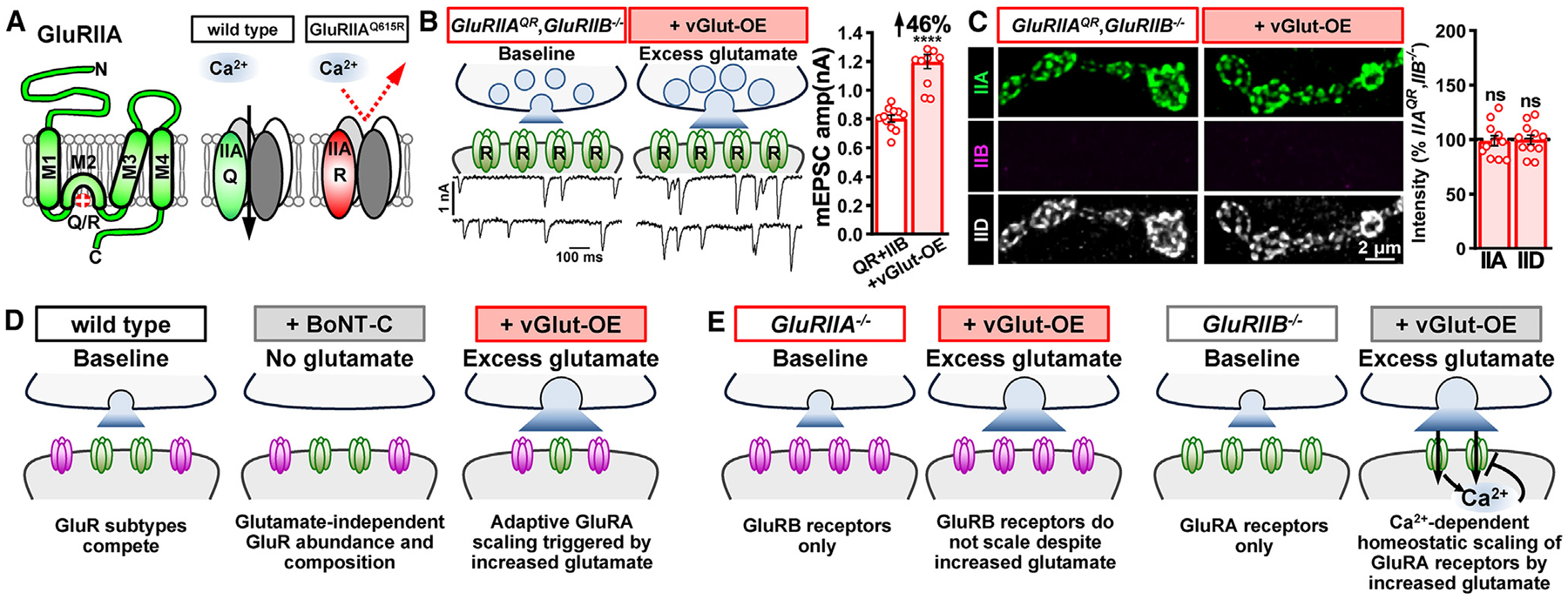
(A) Schematics illustrating GluRIIA subunit topology with the Q615R mutation engineered in the pore-forming M2 domain, rendering GluRA receptors Ca2+ impermeable.
(B) Schematics and representative mEPSC traces from Is and Ib NMJs containing only Ca2+-impermeable GluRA receptors (GluRIIAQ615R,GluRIIB: w;GluRIIAQ615R,GluRIIBsp6/GluRIIAQ615R,GluRIIBsp6;+) at baseline and following vGlut-OE (GluRIIAQR, GluRIIB+vGlut-OE: w;OK371-GAL4,GluRIIAQ615R,GluRIIBsp6/UAS-vGlut,GluRIIAQ615R,GluRIIBsp6;+). Right: quantification of mEPSC amplitude in both genotypes. Note that a significant increase in mEPSC amplitude is observed following excess glutamate released by vGlut-OE, which was not observed with Ca2+-permeable GluRA (Figure 5A).
(C) Representative images of GluRs immunostained in the indicated genotypes. Right: quantification of GluR mean fluorescence intensity in GluRIIAQR, GluRIIB+vGlut-OE normalized to GluRIIAQR, GluRIIB mutants alone. Without Ca2+ permeability, excess glutamate no longer downregulates GluRA abundance in the absence of competition with GluRB receptors.
(D) Summary of glutamate loss or enhancement on GluR receptive fields when GluRA and GluRB are in competition.
(E) Summary of the impact of excess glutamate on GluR receptive fields when GluR subtype competition is lost. Error bars indicate ±SEM, with the following statistical significance: ****p < 0.0001; ns, not significant. Additional statistics and sample number values (n) for all experiments are summarized in Table S1.
We next determined whether homeostatic GluRA receptor scaling is affected when GluRA receptors are rendered Ca2+ impermeable. To eliminate competition with GluRB receptors, we generated CRISPR-induced GluRIIB null mutations in GluRIIAQ615R mutant backgrounds, leading to synapses composed entirely of Ca2+-impermeable GluRA receptors (Figure 6B). In GluRIIAQ615R,GluRIIB double mutants, baseline mEPSC amplitudes were enhanced, as expected, but following vGlut overexpression, they were now further enhanced by ~50% (Figure 6B). Consistently, we observed no change in GluRIIA or GluRIID levels in GluRIIAQ615R,GluRIIB double mutants with vGlut-OE compared to GluRIIAQ615R mutants alone (Figure 6C). These data indicate that when GluRA is rendered impermeable to Ca2+ and competition with GluRB is eliminated, excess glutamate is no longer capable of inducing homeostatic reductions in GluRA levels at postsynaptic compartments. We summarize these results in Figures 6D and 6E.
DISCUSSION
By generating null mutations in GluRIIA and GluRIIB subunits, we have shown a competition exists between GluR subtypes that establishes stable postsynaptic fields. While synaptically released glutamate is not required to organize this process, excess glutamate triggers an adaptive downscaling of both GluR subtypes in the postsynaptic compartment. However, when this GluR subtype competition is eliminated, a clear and distinctive relationship is revealed between excess glutamate and GluR plasticity: GluRB receptors become completely insensitive to excess glutamate, with stable GluRB levels maintained. In contrast, GluRA receptors constitute the “plastic” receptor subtype, homeostatically tuned to excess synaptic glutamate release to maintain stable miniature activity. Further, Ca2+ influx through GluRA receptors is a key transducer of this signaling system, rendering GluRA non-plastic when Ca2+ permeability is lost. Together, these results highlight the interplay between GluR subtype competition, synaptic glutamate, and Ca2+ signaling at postsynaptic compartments and reveal the existence of homeostatic receptor scaling at the Drosophila NMJ.
Several layers of regulation operate at postsynaptic compartments to establish GluR fields at the fly NMJ. First, the relative level of GluR transcription and translation between subtypes can ultimately set GluRs at synapses. This is demonstrated by overexpression of either GluRIIA or GluRIIB subunits, which can saturate the entire GluR field at postsynaptic compartments and lead to the concomitant loss of the other GluR subtype.14,16,20 Second, post-translational processes, mediated by such factors as enzymatic cleavage, phosphorylation, and degradation modulate GluR activity and abundance. For example, Ca2+-dependent protein cleavage by calpain, phosphorylation control by p21-activated kinase, and proteosomal degradation by the E3 ubiquitin ligase adapter Diablo have all been shown to modulate GluRs at the fly NMJ,35,36 which parallel findings in vertebrates.37–40 Third, while glutamate released from synaptic vesicles is not necessary to establish or maintain GluR fields in Drosophila (Figure 3) or in mammals,9,10 there is evidence that ambient glutamate modulation from non-vesicular glutamate release and glial transporters might regulate GluR clustering and receptor field size,41,42 while excess vesicular glutamate release triggers adaptive reductions in GluRs (Figure 3). There is also evidence that correlated or diminished activity selectively regulates GluRA abundance.43 In C. elegans, parallel processes regulate GluR trafficking and plasticity.44,45 It will be of interest to determine how these many layers of control intersect and are coordinated to establish GluR fields during development and remodeling in plasticity.
There appears to be a hierarchy of regulatory steps controlling GluR plasticity in response to excess glutamate. While excess glutamate downregulates both GluRA and GluRB abundance when both receptor subtypes are present, this plasticity is adaptive but not homeostatic—miniature amplitude is still enhanced. However, when GluRA vs. GluRB competition is eliminated, a complete distinction in GluR behavior is revealed: GluRB is not responsive to excess glutamate, while GluRA receptors are sensitively tuned to glutamate to now enable the homeostatic control of miniature activity. Ca2+ influx through GluRA is crucial to this plasticity, where loss of this secondary messenger converts GluRA to behave like static GluRB receptors. It is possible that excess glutamate drives Ca2+-related signaling through GluRA in postsynaptic compartments that ultimately acts on both GluRA and GluRB when both receptor subtypes are present, at least at MN-Ib NMJs, and this signaling may be lost in GluRB-only NMJs. Although the downstream effectors that respond to excess glutamate and Ca2+ to modulate postsynaptic GluRs are not known, an attractive candidate is the auxiliary KAR subunit Neto, which regulates GluR abundance at the fly NMJ46 and functionally modulates AMPARs in worms.47 Interestingly, KARs in mammals are also under homeostatic control,48 where the auxiliary subunit Neto controls key properties of these receptors.49
What purpose might two GluR subtypes, differing in their current amplitudes, biophysics, and plasticity, subserve? One idea is that GluRB receptors provide a basal signal at postsynaptic compartments to maintain synaptic dialogue, while GluRA receptors are the potent subtype that sets synaptic strength, drives muscle contraction, and is targeted for plasticity. These differential functions may be reflected in their distinct subsynaptic localizations, with GluRA enriched opposite active zone centers where glutamate is released, while GluRB is enriched in the outside periphery of these areas.11,14,50 Another possibility, not mutually exclusive, is that GluRBs serve as “back-up” receptors to maintain NMJ transmission and locomotion when GluRAs are blocked, a phenomenon that occurs naturally at larval NMJs due to toxins injected by parasitoid wasps and other organisms.51–53 Indeed, presynaptic homeostatic potentiation, a conserved form of retrograde plasticity modeled at the fly NMJ, is induced when GluRA is lost or pharmacologically inhibited to maintain stable NMJ excitation.54 Hence, stable GluRB receptors provide robustness to buffer NMJ function from perturbations while also allowing flexibility for GluRA receptors to dynamically change with plasticity.
Although the Drosophila NMJ has long been used as a model to study presynaptic forms of adaptive plasticity such as homeostatic potentiation and depression,54,55 recent work has found parallel modes of adaptive plasticity that target postsynaptic GluR abundance at this model glutamatergic synapse. In addition to excess glutamate targeting GluRs, at least three additional examples of adaptive GluR plasticity have been observed at the fly NMJ. First, in synaptic undergrowth mutants, where presynaptic innervation is reduced, overall synaptic strength can be maintained at least in some cases through an adaptive enhancement of postsynaptic GluR abundance.56 Second, when innervation by a single motor neuron is biased at adjacent muscles, stable synaptic strength is maintained through both pre- and postsynaptic mechanisms,57 with a homeostatic increase in postsynaptic GluR abundance necessary at hypo-innervated NMJs.58 Third, activation of injury-related signaling in motor neurons induces a downregulation in postsynaptic GluR abundance to adaptively reduce the set point of synaptic strength.59 These examples suggest an underappreciated level of postsynaptic plasticity exists at the Drosophila NMJ, which when combined with the sophisticated genetic and functional tools available, highlights the great potential for this system to illuminate how GluR subtype competition and presynaptic function target GluRs for adaptive modulation.
Limitations of the study
There are several limitations regarding our understanding of the timing and dynamics of the GluR plasticity described in our study. Because we assessed GluR composition, abundance, and function through imaging and electrophysiology at fixed times in later stages of development (third-instar larvae), the time course of the changes in GluR levels (minutes, hours, days) is uncertain. It is also not clear whether the reduction in GluRA levels, in response to excess glutamate release, is due to reduced transcription, translation, trafficking, and/or enhanced degradation. Finally, it is not clear to what extent action-potential-driven patterns of activity are altered during development in vGlut-OE NMJs and whether these potential differences contribute to GluR plasticity in addition to the enhanced glutamate emitted from individual synaptic vesicles. Future studies, including intravital live imaging of GluRs, will help to address these limitations.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Dion Dickman (dickman@usc.edu).
Materials availability
Fly stocks and constructs generated in this study are available from the lead contact upon request.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Fly husbandry, stocks, and handling
Drosophila melanogaster stocks were raised at 25°C on standard molasses food. Both male and female third-instar larvae were used. The w1118 strain is used as the wild-type control unless otherwise noted as this is the genetic background in which all genotypes are bred. To selectively express BoNT-C using the GAL4/UAS system while also overexpressing vGlut in both MN-Is and -Ib, we engineered a LexAOp-vGlut transgene that can be overexpressed in motor neurons using OK6-LexA. Details of all fly stocks used, including their sources, are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-GluRIIA (8B4D2) (1:50) | Developmental Studies Hybridoma Bank | RRID: AB_528269 |
| Rabbit anti-GluRIIB (1:1000) | Reference # Perry et al.60 | N/A |
| Guinea pig anti-GluRIID (1:1000) | Reference # Perry et al.60 | N/A |
| Alexa Fluor 488 conjugated secondary antibodies (1:400) | Jackson ImmunoResearch Laboratories (Jackson) | Cat#715-545-150; RRID: AB_2340846 |
| Cy3-conjugated secondary antibodies (1:400) | Jackson | Cat#706-165-148; RRID: AB_2340460, Cat#711-165-152; RRID: AB_2307443 |
| DyLight 405-conjugated secondary antibodies (1:400) | Jackson | Cat#706-475-148; RRID: AB_2340470, Cat#711-475-152; RRID: AB_2340616 |
| Experimental models: Organisms/strains | ||
| w 1118 | Bloomington Drosophila Stock Center (BDSC) | Cat#5905 |
| nos.Cas9-DsRed | BDSC | Cat#79004 |
| P{TKO.GS00444} (gRNA targeting GluRIIA) | BDSC | Cat#68059 |
| dHb9-Gal4 (Ib-Gal4) | BDSC | Cat#83004 |
| GMR2 7E09-Gal4 (Is-Gal4) | BDSC | Cat#49227 |
| UAS-RedStinger | BDSC | Cat#8547 |
| GluRIIA PV3 | This study | N/A |
| GluRIIA PV7 | This study | N/A |
| GluRIIB SP5 | This study | N/A |
| GluRIIB SP14 | This study | N/A |
| pU6-BbsI-{GluRIIB.gRNA} | This study | N/A |
| GluRIIA Q615R , GluRIIB SP6 | This study | N/A |
| LexAOp-vGlut | This study | N/A |
| GluRIIA SP22 | Reference # Diantonio et al.16 | N/A |
| GluRIIA Q615R | Reference # Perry et al.34 | N/A |
| OK371-GAL4 | Reference # Mahr et al.61 | N/A |
| UAS-vGlut | Reference # Daniels et al.18 | N/A |
| OK319-GAL4 | Reference # Sweeney et al.62 | N/A |
| G14-Gal4 | Reference # Aberle et al.63 | N/A |
| UAS-BoNT-C | Reference # Han et al.11 | N/A |
| SynapGCaMP8f | Reference # Han et al.11 | N/A |
| mnb 1 | Reference # Chen et al.28 | N/A |
| vGlut-LexA | Reference # Sherer et al.64 | N/A |
| GluRIIA mRFP | Reference # Qin et al.15 | N/A |
| UAS-GluRIIB | Reference # Perry et al.34 | N/A |
| GluRIIA-T2A-Gal4 | Reference # Perry et al.65 | N/A |
| GluRIIB-T2A-Gal4 | Reference # Kondo et al.65 | N/A |
| Recombinant DNA | ||
| pU6-BbsI-chiRNA | Addgene | Cat#45946 |
| pDEST-APLO | Addgene | Cat#112805 |
| pACU2 | Addgene | Cat#31223 |
| Software and algorithms | ||
| NIS Elements software | Nikon | 4.51.01 |
| Huygens Object Analysis | Scientific Volume Imaging | 22.04 |
| Axon pCLAMP Clampfit | Molecular Devices | 10.7 |
| MiniAnalysis | Synaptosoft | 6.0.3 |
| GraphPad Prism | GraphPad | 8.0.1 |
| ImageJ (Fiji) | Reference # Rueden et al.66 | N/A |
METHOD DETAILS
Molecular biology
GluRIIAPV3, GluRIIAPV7, GluRIIBSP5, and GluRIIBSP14 mutants were generated using a CRISPR/Cas9 genome editing strategy as described.67 For GluRIIA mutants, a TKO stock was obtained from BDSC (#68059) that ubiquitously expressed a sgRNA (5′ CAATCGCACCGACGTAATGTTGG 3′) targeting the sixth exon of the GluRIIA locus (Figure 1B). To generate GluRIIB mutants, we generated two independent sgRNA lines that targeted the first and sixth exons (sgRNA1: 5′ GGTGTCTTCATTGGCGCCGCTGG 3’; sgRNA2: 5′CATTGATGGATTCTACTCCCGGG 3′) and cloned each into the pU63 vector (#49410; Addgene). Constructs were sent to BestGene Inc. (Chino Hill, CA) for targeted insertion into the VK18 attP site on the second chromosome. sgRNA flies were crossed to a nos-Cas9 line (#54591; BDSC) on the second chromosome to induce active germline CRISPR mutagenesis, and 20 independent lines generated from each sgRNA were screened by PCR for mutations. This identified at least 8 independent indel mutations for each sgRNA that shifted the open reading frame, with GluRIIAPV3, GluRIIAPV7, GluRIIBSP5, and GluRIIBSP14 alleles kept for additional analysis (Figure 1B). This strategy was also used to generate the GluRIIBSP6 mutation in the GluRIIAQ615R background, which resulted in a similar allele as GluRIIBSP14.
quantitative PCR (qPCR) was performed using the Luna Universal One-Step RT-qPCR Kit (NEB, E3005S) according to the manufacturer’s instructions. RNA was isolated and prepared from body wall tissue as previously described.23 20 ng of total RNA was used as the template for each reaction. Three biological replicates were performed for each sample and the comparative Ct method was used for qPCR data analysis. The following primers were used (fwd; rev: 5’−3′): GluRIIA: TCCTCAACTTGGAACTGGAAAG; CGTACTTTTCCCTGCCTCTG. GluRIIB: GCGAATACAGATGAATGGGATG; TGCATGAAGGGTACAGTGAAG.
Electrophysiology
All dissections and two-electrode voltage clamp (TEVC) recordings were performed as described68 using modified hemolymph-like saline (HL-3) containing: 70mM NaCl, 5mM KCl, 10mM MgCl2, 10mM NaHCO3, 115mM Sucrose, 5mM Trehelose, 5mM HEPES, and 0.5mM CaCl2, pH 7.2, from cells with an initial resting potential between −60 and −75 mV, and input resistances >6 MΩ. Recordings were performed on an Olympus BX61 WI microscope using a 40x/0.80 NA water-dipping objective and acquired using an Axoclamp 900A amplifier, Digidata 1440A acquisition system and pClamp 10.5 software (Molecular Devices). Miniature excitatory postsynaptic currents (mEPSCs) were recorded in the absence of any stimulation with a voltage clamp of −80 mV, and low pass filtered at 1 kHz. All recordings were made on abdominal muscle 6, segment A3 of third-instar larvae with the leak current never exceeding 5 nA mEPSCs were recorded for 60 s and analyzed using MiniAnalysis (Synaptosoft) and Excel (Microsoft) software. The average mEPSC amplitude and total charge transfer values for each NMJ were obtained from approximately 100 events in each recording.
Immunocytochemistry
Third-instar larvae were dissected in ice-cold 0 Ca2+ HL-3 and immunostained as described.19,23,69 In brief, larvae were either fixed in Bouin’s fixative for 5 min (Sigma, HT10132-1L), 100% ice-cold ethanol for 5 min, or 4% paraformaldehyde (PFA) for 10 min. Larvae were then washed with PBS containing 0.1% Triton X-100 (PBST) for 30 min, blocked with 5% Normal Donkey Serum followed by overnight incubation in primary antibodies at 4°C. Preparations were then washed 3x in PBST, incubated in secondary antibodies for 2 h, washed 3x in PBST, and equilibrated in 70% glycerol. Prior to imaging, samples were mounted in VectaShield (Vector Laboratories). To assess surface vs. total receptors (Figure S4), samples were stained as stated above but without Triton X-100 added so that the cell membranes remained intact. Some muscles were selectively damaged through perforation by sharp forceps to allow antibodies to enter these cells. Successful perforation was confirmed by clear GluRIID intracellular staining. Details of all antibodies, their source, dilution used, and references are listed in key resource table.
Imaging and analysis
Samples were imaged using a Nikon A1R Resonant Scanning Confocal microscope equipped with NIS Elements software and a 100x APO 1.4NA oil immersion objective using separate channels with four laser lines (405 nm, 488 nm, 561 nm, and 647 nm) as described.60 For fluorescence intensity quantifications of GluRIIA, GluRIIB and GluRIID, z-stacks from MN-Is or MN-Ib NMJs at muscle 6, segment A3 were obtained on the same day using identical gain and laser power settings with z axis spacing between 0.15 and 0.20 μm for all genotypes within an individual experiment. Maximum intensity projections were utilized for quantitative image analysis using the general analysis toolkit of NIS Elements software. Immunofluorescence intensity levels were quantified by applying intensity thresholds and filters to binary layers in the 405 nm, 488 nm, and 561 nm channels. The mean intensity for each channel was quantified by obtaining the average total fluorescence signal for each individual punctum and dividing this value by the puncta area. A mask was created around the HRP channel, used to define the neuronal membrane, and only puncta within this mask were analyzed to eliminate background signals. For additional image analyses (Figures S1, S2 and S4), Scientific Volume Imaging Huygen’s software was used to perform 3D object analysis of individual receptor clusters to determine cluster volume and sum intensity values. All measurements based on confocal images were taken from NMJs acquired from at least six different animals.
Ca2+ imaging and analysis
Third-instar larvae were dissected in ice-cold saline. Imaging was performed in modified HL-3 saline with 1.5 mM Ca2+ added using a Nikon A1R Resonant Scanning Confocal microscope equipped with NIS Elements software and a 60x APO 1.0NA water immersion objective as detailed.34 NMJs on muscle 6/7 were imaged at a resonant frequency of 60 fps (256 × 256 pixels). Spontaneous Ca2+ events were recorded at 4–8 individual NMJs during 120 s imaging sessions from at least three different larvae. Horizontal drifting was corrected using ImageJ plugins70 and imaging data with severe muscle movements were rejected as described.71 Three ROIs were manually selected using the outer edge of terminal Ib or Is boutons observed by baseline GCaMP signals with ImageJ.66,72 Ib and Is boutons were defined by baseline GCaMP8f fluorescence levels, which are 2–3 fold higher at Ib NMJs compared to their Is counterparts at a particular muscle. Fluorescence intensities were measured as the mean intensity of all pixels in each individual ROI. ΔF for a spontaneous event was calculated by subtracting the baseline GCaMP fluorescence level F from the peak intensity of the GCaMP signal during each spontaneous event at a particular bouton as previously detailed. ΔF/F was calculated for each spontaneous Ca2+ transient event as detailed.11,34
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed using GraphPad Prism (version 7.0), MiniAnalysis (Synaptosoft), or Microsoft Excel software (version 16.22). Sample values were tested for normality using the D’Agostino & Pearson omnibus normality test which determined that the assumption of normality of the sample distribution was not violated. Data were then compared using either a one-way ANOVA and tested for significance using a Tukey’s multiple comparison test or using an unpaired 2-tailed Student’s t-test with Welch’s correction.
Supplementary Material
Highlights.
Glutamate receptor subtypes compete to establish postsynaptic receptive fields
Excess glutamate release triggers adaptive receptor scaling
GluRA receptors are selectively targeted for homeostatic scaling in Drosophila
Homeostatic receptor scaling requires calcium permeability through GluRA
ACKNOWLEDGMENTS
We thank Aaron DiAntonio (Washington University, MO, USA) and Karen Chang (USC, CA, USA) for sharing Drosophila stocks. We acknowledge the Developmental Studies Hybridoma Bank (Iowa, USA) for antibodies used in this study and the Bloomington Drosophila Stock Center for fly stocks (NIH P40OD018537). We thank Giwoo Kim, Veronica Haro-Acosta, and Surbhi Trivedi for assistance in validating reagents. This work was supported by grants from the National Institutes of Health (NS111414 and NS126654) to D.D.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research. We support inclusive, diverse, and equitable conduct of research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112775.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Herring BE, and Nicoll RA (2016). Long-Term Potentiation: From CaMKII to AMPA Receptor Trafficking. Annu. Rev. Physiol 78, 351–365. 10.1146/annurev-physiol-021014-071753. [DOI] [PubMed] [Google Scholar]
- 2.Diering GH, and Huganir RL (2018). The AMPA Receptor Code of Synaptic Plasticity. Neuron 100, 314–329. 10.1016/J.NEURON.2018.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park P, Kang H, Sanderson TM, Bortolotto ZA, Georgiou J, Zhuo M, Kaang BK, and Collingridge GL (2018). The Role of Calcium-Permeable AMPARs in Long-Term Potentiation at Principal Neurons in the Rodent Hippocampus. Front. Synaptic Neurosci 10.3389/FNSYN.2018.00042/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Purkey AM, and Dell’Acqua ML (2020). Phosphorylation-Dependent Regulation of Ca2+-Permeable AMPA Receptors During Hippocampal Synaptic Plasticity. Front. Synaptic Neurosci 12, 8. 10.3389/Fnsyn.2020.00008/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chowdhury D, and Hell JW (2018). Homeostatic synaptic scaling: Molecular regulators of synaptic AMPA-type glutamate receptors. F1000Res. 7, 234. 10.12688/f1000research.13561.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turrigiano GG (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435. 10.1016/J.CELL.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pozo K, and Goda Y (2010). Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 66, 337–351. 10.1016/j.neuron.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Park E, Zhong LR, and Chen L (2019). Homeostatic synaptic plasticity as a metaplasticity mechanism — a molecular and cellular perspective. Curr. Opin. Neurobiol 54, 44–53. 10.1016/j.conb.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sando R, Bushong E, Zhu Y, Huang M, Considine C, Phan S, Ju S, Uytiepo M, Ellisman M, and Maximov A (2017). Assembly of Excitatory Synapses in the Absence of Glutamatergic Neurotransmission. Neuron 94, 312–321.e3. 10.1016/J.NEURON.2017.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sigler A, Oh WC, Imig C, Altas B, Kawabe H, Cooper BH, Kwon HB, Rhee JS, and Brose N (2017). Formation and Maintenance of Functional Spines in the Absence of Presynaptic Glutamate Release. Neuron 94, 304–311.e4. 10.1016/J.NEURON.2017.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han Y, Chien C, Goel P, He K, Pinales C, Buser C, and Dickman D (2022). Botulinum neurotoxin accurately separates tonic vs. phasic transmission and reveals heterosynaptic plasticity rules in Drosophila. Elife 11, e77924. 10.7554/elife.77924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwon H-B, and Sabatini BL (2011). Glutamate induces de novo growth of functional spines in developing cortex. Nature 474, 100–104. 10.1038/NATURE09986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Dharkar P, Han TH, Serpe M, Lee CH, and Mayer ML (2016). Novel Functional Properties of Drosophila CNS Glutamate Receptors. Neuron 92, 1036–1048. 10.1016/j.neuron.2016.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marrus SB, Portman SL, Allen MJ, Moffat KG, and DiAntonio A (2004). Differential Localization of Glutamate Receptor Subunits at the Drosophila Neuromuscular Junction. J. Neurosci 24, 1406–1415. 10.1523/JNEUROSCI.1575-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin G, Schwarz T, Kittel RJ, Schmid A, Rasse TM, Kappei D, Ponimaskin E, Heckmann M, and Sigrist SJ (2005). Four different subunits are essential for expressing the synaptic glutamate receptor at neuromuscular junctions of Drosophila. J. Neurosci 25, 3209–3218. 10.1523/JNEUROSCI.4194-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diantonio A, Petersen SA, Heckmann M, and Goodman CS (1999). Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J. Neurosci 19, 3023–3032. 10.1523/jneurosci.19-08-03023.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han TH, Dharkar P, Mayer ML, and Serpe M (2015). Functional reconstitution of Drosophila melanogaster NMJ glutamate receptors. Proc. Natl. Acad. Sci. USA 112, 6182–6187. 10.1073/pnas.1500458112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, and DiAntonio A (2004). Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J. Neurosci 24, 10466–10474. 10.1523/JNEUROSCI.3001-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Chien C, Han Y, Sun Z, Chen X, and Dickman D (2021). Auto-crine inhibition by a glutamate-gated chloride channel mediates presynaptic homeostatic depression. Sci. Adv 7, eabj1215. 10.1126/sciadv.abj1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Goel P, Wondolowski J, Paluch J, and Dickman D (2018). A Glutamate Homeostat Controls the Presynaptic Inhibition of Neurotransmitter Release. Cell Rep. 23, 1716–1727. 10.1016/J.CELREP.2018.03.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaviño MA, Ford KJ, Archila S, and Davis GW (2015). Homeostatic synaptic depression is achieved through a regulated decrease in presynaptic calcium channel abundance. Elife 2015. 10.107554/ELIFE.05473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, and DiAntonio A (1997). Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19, 1237–1248. 10.1016/S0896-6273(00)80415-8. [DOI] [PubMed] [Google Scholar]
- 23.Chen X, and Dickman D (2017). Development of a tissue-specific ribo-some profiling approach in Drosophila enables genome-wide evaluation of translational adaptations. PLoS Genet. 13, e1007117. 10.1371/JOURNAL.PGEN.1007117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aponte-Santiago NA, and Littleton JT (2020). Synaptic Properties and Plasticity Mechanisms of Invertebrate Tonic and Phasic Neurons. Front. Physiol 11, 611982. 10.3389/FPHYS.2020.611982/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He K, Han Y, Li X, Hernandez RX, Riboul DV, Feghhi T, Justs KA, Mahneva O, Perry S, Macleod GT, et al. (2023). Physiologic and nanoscale distinctions define glutamatergic synapses in tonic vs phasic neurons. Journal of Neuroscience, JN-RM-0046-123. 10.1523/JNEUROSCI.0046-23.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karunanithi S, Marin L, Wong K, and Atwood HL (2002). Quantal Size and Variation Determined by Vesicle Size in Normal and Mutant Drosophila Glutamatergic Synapses. J. Neurosci 22, 10267–10276. 10.1523/JNEUROSCI.22-23-10267.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmid A, Hallermann S, Kittel RJ, Khorramshahi O, Frölich AMJ, Quentin C, Rasse TM, Mertel S, Heckmann M, and Sigrist SJ (2008). Activity-dependent site-specific changes of glutamate receptor composition in vivo. Nat. Neurosci 11, 659–666. 10.1038/nn.2122. [DOI] [PubMed] [Google Scholar]
- 28.Chen CK, Bregere C, Paluch J, Lu JF, Dickman DK, and Chang KT (2014). Activity-dependent facilitation of Synaptojanin and synaptic vesicle recycling by the Minibrain kinase. Nat. Commun 5, 4246–4314. 10.1038/ncomms5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bayer KU, and Schulman H (2019). CaM Kinase: Still Inspiring at 40. Neuron 103, 380–394. 10.1016/j.neuron.2019.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ni L (2020). The Structure and Function of Ionotropic Receptors in Drosophila. Front. Mol. Neurosci 13, 638839. 10.3389/fnmol.2020.638839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Köhler M, Burnashev N, Sakmann B, and Seeburg PH (1993). Determinants of ca2+ permeability in both TM1 and TM2 of high affinity kainate receptor channels: Diversity by RNA editing. Neuron 10, 491–500. 10.1016/0896-6273(93)90336-P. [DOI] [PubMed] [Google Scholar]
- 32.Hume RI, Dingledine R, and Heinemann SF (1991). Identification of a site in glutamate receptor subunits that controls calcium permeability. Science 253, 1028–1031. 10.1126/SCIENCE.1653450. [DOI] [PubMed] [Google Scholar]
- 33.Kiragasi B, Wondolowski J, Li Y, and Dickman DK (2017). A Presynaptic Glutamate Receptor Subunit Confers Robustness to Neurotransmission and Homeostatic Potentiation. Cell Rep. 19, 2694–2706. 10.1016/J.CELREP.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perry S, Han Y, Qiu C, Chien C, Goel P, Nishimura S, Sajnani M, Schmid A, Sigrist SJ, and Dickman D (2022). A glutamate receptor C-tail recruits CaMKII to suppress retrograde homeostatic signaling. Nat. Commun 13, 7656–7716. 10.1038/s41467-022-35417-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metwally E, Zhao G, Li W, Wang Q, and Zhang YQ (2019). Calcium-Activated Calpain Specifically Cleaves Glutamate Receptor IIA But Not IIB at the Drosophila Neuromuscular Junction. J. Neurosci 39, 2776–2791. 10.1523/JNEUROSCI.2213-17.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang M, Chen PY, Wang CH, Lai TT, Tsai PI, Cheng YJ, Kao HH, and Chien CT (2016). Dbo/Henji Modulates Synaptic dPAK to Gate Glutamate Receptor Abundance and Postsynaptic Response. PLoS Genet. 12, e1006362. 10.1371/JOURNAL.PGEN.1006362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simpkins KL, Guttmann RP, Dong Y, Chen Z, Sokol S, Neumar RW, and Lynch DR (2003). Selective Activation Induced Cleavage of the NR2B Subunit by Calpain. J. Neurosci 23, 11322–11331. 10.1523/JNEUROSCI.23-36-11322.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hawasli AH, Benavides DR, Nguyen C, Kansy JW, Hayashi K, Chambon P, Greengard P, Powell CM, Cooper DC, and Bibb JA (2007). Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat. Neurosci 10, 880–886. 10.1038/NN1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, and Vaux DL (2000). Identification of DIABLO, a Mammalian Protein that Promotes Apoptosis by Binding to and Antagonizing IAP. Cell 102, 43–53. 10.1016/S0092-8674(00)00009-X. [DOI] [PubMed] [Google Scholar]
- 40.Murata Y, and Constantine-Paton M (2013). Postsynaptic Density Scaffold SAP102 Regulates Cortical Synapse Development through EphB and PAK Signaling Pathway. J. Neurosci 33, 5040–5052. 10.1523/JNEUROSCI.2896-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Augustin H, Grosjean Y, Chen K, Sheng Q, and Featherstone DE (2007). Nonvesicular release of glutamate by glial xCT transporters suppresses glutamate receptor clustering in vivo. J. Neurosci 27, 111–123. 10.1523/JNEUROSCI.4770-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Featherstone DE, Rushton E, and Broadie K (2002). Developmental regulation of glutamate receptor field size by nonvesicular glutamate release. Nat. Neurosci 5, 141–146. 10.1038/NN789. [DOI] [PubMed] [Google Scholar]
- 43.Ljaschenko D, Ehmann N, and Kittel RJ (2013). Hebbian Plasticity Guides Maturation of Glutamate Receptor Fields In. Cell Rep. 3, 1407–1413. 10.1016/J.CELREP.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 44.Aronoff R, Mellem JE, Maricq AV, Sprengel R, and Seeburg PH (2004). Neuronal Toxicity in Caenorhabditis elegans from an Editing Site Mutant in Glutamate Receptor Channels. J. Neurosci 24, 8135–8140. 10.1523/JNEUROSCI.2587-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grunwald ME, Mellem JE, Strutz N, Maricq AV, and Kaplan JM (2004). Clathrin-mediated endocytosis is required for compensatory regulation of GLR-1 glutamate receptors after activity blockade. Proc. Natl. Acad. Sci. USA 101, 3190–3195. 10.1073/PNAS.0306156101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim YJ, Bao H, Bonanno L, Zhang B, and Serpe M (2012). Drosophila neto is essential for clustering glutamate receptors at the neuromuscular junction. Genes Dev. 26, 974–987. 10.1101/gad.185165.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang R, Mellem JE, Jensen M, Brockie PJ, Walker CS, Hoerndli FJ, Hauth L, Madsen DM, and Maricq AV (2012). The SOL-2/Neto Auxiliary Protein Modulates the Function of AMPA-Subtype Ionotropic Glutamate Receptors. Neuron 75, 838–850. 10.1016/J.NEURON.2012.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan D, Yamasaki M, Straub C, Watanabe M, and Tomita S (2013). Homeostatic Control of Synaptic Transmission by Distinct Glutamate Receptors. Neuron 78, 687–699. 10.1016/J.NEURON.2013.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tomita S, Castillo PE, Castillo PE, and Purpura DP (2012). Neto1 and Neto2: auxiliary subunits that determine key properties of native kainate receptors. J. Physiol 590, 2217–2223. 10.1113/JPHYSIOL.2011.221101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muttathukunnel P, Frei P, Perry S, Dickman D, and Müller M (2022). Rapid homeostatic modulation of transsynaptic nanocolumn rings. PNAS 119, e2119044119. 10.1073/pnas.2119044119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karst H, and Piek T (1991). Structure-activity relationship of philanthotoxins–II. Effects on the glutamate gated ion channels of the locust muscle fibre membrane. Comp. Biochem. Physiol. C Comp. Pharmacol. Toxicol 98, 479–489. 10.1016/0742-8413(91)90237-N. [DOI] [PubMed] [Google Scholar]
- 52.Eldefrawi AT, Eldefrawi ME, Konno K, Mansour NA, Nakanishi K, Oltz E, and Usherwood PN (1988). Structure and synthesis of a potent glutamate receptor antagonist in wasp venom. Proc. Natl. Acad. Sci. USA 85, 4910–4913. 10.1073/PNAS.85.13.4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hwang RY, Zhong L, Xu Y, Johnson T, Zhang F, Deisseroth K, and Tracey WD (2007). Nociceptive Neurons Protect Drosophila Larvae from Parasitoid Wasps. Curr. Biol 17, 2105–2116. 10.1016/J.CUB.2007.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goel P, and Dickman D (2021). Synaptic homeostats: latent plasticity revealed at the Drosophila neuromuscular junction. Cell. Mol. Life Sci 78, 3159–3179. 10.1007/s00018-020-03732-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frank CA, James TD, and Müller M (2020). Homeostatic control of Drosophila neuromuscular junction function. Synapse 74. 10.1002/syn.22133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goel P, Dufour Bergeron D, Böhme MA, Nunnelly L, Lehmann M, Buser C, Walter AM, Sigrist SJ, and Dickman D (2019). Homeostatic scaling of active zone scaffolds maintains global synaptic strength. J. Cell Biol 218, 1706–1724. 10.1083/jcb.201807165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davis GW, and Goodman CS (1998). Synapse-specific control of synaptic efficacy at the terminals of a single neuron. Nature 392, 82–86. 10.1038/32176. [DOI] [PubMed] [Google Scholar]
- 58.Goel P, Nishimura S, Chetlapalli K, Li X, Chen C, and Dickman D (2020). Distinct Target-Specific Mechanisms Homeostatically Stabilize Transmission at Pre- and Post-synaptic Compartments. Front. Cell. Neurosci 14, 196. 10.3389/fncel.2020.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goel P, and Dickman D (2018). Distinct homeostatic modulations stabilize reduced postsynaptic receptivity in response to presynaptic DLK signaling. Nat. Commun 9, 1856–1914. 10.1038/s41467-018-04270-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perry S, Han Y, Das A, and Dickman D (2017). Homeostatic plasticity can be induced and expressed to restore synaptic strength at neuromuscular junctions undergoing ALS-related degeneration. Hum. Mol. Genet 26, 4153–4167. 10.1093/hmg/ddx304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mahr A, and Aberle H (2006). The expression pattern of the Drosophila vesicular glutamate transporter: A marker protein for motoneurons and glutamatergic centers in the brain. Gene Expr. Patterns 6, 299–309. 10.1016/J.MODGEP.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 62.Sweeney ST, Broadie K, Keane J, Niemann H, and O’Kane CJ (1995). Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron 14, 341–351. 10.1016/0896-6273(95)90290-2. [DOI] [PubMed] [Google Scholar]
- 63.Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhães TR, and Goodman CS (2002). Wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron 33, 545–558. 10.1016/S0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- 64.Sherer LM, Catudio Garrett E, Morgan HR, Brewer ED, Sirrs LA, Shearin HK, Williams JL, McCabe BD, Stowers RS, and Certel SJ (2020). Octopamine neuron dependent aggression requires dVGLUT from dual-transmitting neurons. PLoS Genet. 16, e1008609. 10.1371/JOURNAL.PGEN.1008609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kondo S, Takahashi T, Yamagata N, Imanishi Y, Katow H, Hiramatsu S, Lynn K, Abe A, Kumaraswamy A, and Tanimoto H (2020). Neurochemical Organization of the Drosophila Brain Visualized by Endogenously Tagged Neurotransmitter Receptors. Cell Rep. 30, 284–297.e5. 10.1016/J.CELREP.2019.12.018. [DOI] [PubMed] [Google Scholar]
- 66.Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, and Eliceiri KW (2017). ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinf. 18, 529–626. 10.1186/s12859-017-1934-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kikuma K, Li X, Kim D, Sutter D, and Dickman DK (2017). Extended Synaptotagmin Localizes to Presynaptic ER and Promotes Neurotransmission and Synaptic Growth in Drosophila. Genetics 207, 993–1006. 10.1534/GENETICS.117.300261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kikuma K, Li X, Perry S, Li Q, Goel P, Chen C, Kim D, Stavropoulos N, and Dickman D (2019). Cul3 and insomniac are required for rapid ubiquitination of postsynaptic targets and retrograde homeostatic signaling. Nat. Commun 10, 2998–3013. 10.1038/s41467-019-10992-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goel P, Li X, and Dickman D (2017). Disparate Postsynaptic Induction Mechanisms Ultimately Converge to Drive the Retrograde Enhancement of Presynaptic Efficacy. Cell Rep. 21, 2339–2347. 10.1016/j.celrep.2017.10.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li K (2008). Kang Li @ CMU - Image Stabilizer Plugin for ImageJ. https://www.cs.cmu.edu/~kangli/code/Image_Stabilizer.html.
- 71.Ding K, Han Y, Seid TW, Buser C, Karigo T, Zhang S, Dickman DK, and Anderson DJ (2019). Imaging neuropeptide release at synapses with a genetically engineered reporter. Elife 8, e46421. 10.7554/elife.46421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.