

SUMMARY
Endothelial-to-mesenchymal transition (EndMT), a process initiated by activation of endothelial TGFβ signaling, underlies numerous chronic vascular diseases and fibrotic states. Once induced, EndMT leads to a further increase in TGFβ signaling thus establishing a positive feedback loop with EndMT leading to more EndMT. While EndMT is understood at the cellular level, the molecular basis of TGFβ-driven EndMT induction and persistence remains largely unknown. Here we show that metabolic modulation of endothelium, triggered by atypical production of acetate from glucose, underlies TGFβ-driven EndMT. Induction of EndMT suppresses expression of the enzyme PDK4 that leads to an increase in ACSS2-dependent Ac-CoA synthesis from pyruvate-derived acetate. This increased Ac-CoA production results in acetylation of the TGFβ receptor ALK5 and SMADs 2 and 4 leading to activation and long-term stabilization of TGFβ signaling. Our results establish the metabolic basis of EndMT persistence and unveil novel targets, such as ACSS2, for the potential treatment of chronic vascular diseases.
Graphical Abstract
eTOC
Endothelial-to-mesenchymal transition (EndMT) is initiated by activation of endothelial TGFβ signaling. Induction of EndMT results in increased ACSS2-dependent Ac-CoA synthesis from pyruvate-derived acetate. Increased Ac-CoA production leads to acetylation of the TGFβ receptor ALK5 and SMADs 2 and 4 resulting in activation and long-term stabilization of TGFβ signaling.
INTRODUCTION
Endothelial-to-mesenchymal transition (EndMT) has emerged as an important pathophysiological process responsible for maintenance of chronic vascular inflammation and pathogenesis of several important diseases, including atherosclerosis, pulmonary hypertension, and graft arteriopathy as well as aging.1–6 EndMT is largely driven by TGFβ signaling7 that is normally suppressed in the endothelium due to very low level of expression the TGFβ receptor ALK5 (TGFβR1). An increase in ALK5 level, induced, for example, by inflammatory cytokines, initiates TGFβ signaling and leads to EndMT. One key consequence of EndMT is increased endothelial expression of leukocyte adhesion molecules. This facilitates recruitment of white blood cells, thereby promoting further vascular inflammation and further EndMT, thus establishing a vicious cycle with EndMT leading to more EndMT.8 Since recent studies have demonstrated the importance of endothelial metabolism in regulation of its biological functions9–11, to better understand the molecular basis of EndMT, we investigated metabolic effects of prolonged TGFβ stimulation on endothelial cells.
Broadly speaking, endothelial cells (ECs) display a metabolic wiring with similarities to that of many cancer cells, including a high glycolytic rate without concomitant glucose oxidation, a phenomenon known as the Warburg effect.12 Most of ATP in ECs is generated by glycolysis, and most of glucose-derived carbons are secreted as lactate.13 Glucose enters ECs largely via the glucose transporter GLUT1 and proceeds to enter various pathways including the pentose phosphate pathway (PPP), the hexosamine pathway, and glycolysis, all of which are active in ECs.14 Glycolysis ultimately yields the 3-carbon molecule pyruvate. In many cells, pyruvate enters mitochondria where it is metabolized by the pyruvate dehydrogenase (PDH) complex to Acetyl-CoA (Ac-CoA), which then enters the TCA cycle. In ECs, however, glutamine is the preferred carbon contributor to the TCA cycle13, and mitochondrial catabolism of pyruvate is in part inhibited by PDH-kinases (PDKs)-mediated phosphorylation of PDH. Pyruvate therefore is largely hydrogenated to lactate by LDH and secreted.
The large PDH complex is located in the mitochondrial matrix and is comprised of three subunits. The rate-limiting E1 subunit catalyzes the decarboxylation and dehydrogenation of pyruvate. The E2 subunit catalyzes the ligation of the resulting acetyl group to Coenzyme A. The E3 subunit transfers to harvested electrons to NAD+, yielding NADH. The PDKs inhibit PDH by phosphorylation of one or more of three residues in the BCKDHA subunit of E1. Interestingly, recent biochemical work has indicated that under some circumstances, atypical activity of the PDH complex can produce free acetate instead of Ac-CoA.15 Unlike Ac-CoA, acetate that is produced in mitochondria by PDH can exit the mitochondria. Moreover, cytosolic acetate can generate cytosolic acetyl-CoA via action of acyl-CoA synthase short chain 2 (ACSS2).16 This hypothetical PDH/acetate/ACSS2 pathway thus provides a potential alternative to canonical transport of acetyl groups out of the mitochondria by the citrate shuttle and the cytosolic generation of Ac-CoA by ATP citrate lyase (ACLY). A physiological context for this phenomenon is largely unknown.
Our results now point to an important role played by TGFβ-dependent regulation of endothelial Ac-CoA levels in the development of EndMT. We observed increased acetylation of TGFβ signaling complex proteins including SMAD 2,3 and 4 as well as Alk5, thereby augmenting and sustaining TGFβ signaling and EndMT. Unexpectedly, ACSS2 but not ACLY was required for Ac-CoA synthesis, and acetate production by a disinhibited PDH served as the major source of Ac-CoA. We thus find that the atypical production of acetate plays a central role in EndMT.
RESULTS
Metabolic effects of endothelial TGFβ signaling
To study the effect of TGFβ-driven induction of EndMT on endothelial cell metabolism, we subjected human umbilical artery endothelial cells (HUAECs) to TGFβ stimulation in vitro. Bulk RNA-seq assessment of gene expression after 7 days of treatment demonstrated the expected activation of TGFβ-related gene expression and downregulation of endothelial fate markers genes, a pattern consistent with the development of EndMT 7 (Fig 1A). Analysis of metabolism-related genes showed a prominent increase in expression of the glucose transporter gene SLC2A1 (GLUT1), a reduction in expression of glycolytic and most TCA cycle genes, and a nearly 90% decline in expression of PDK4 (Fig 1B, C). In agreement with increased GLUT1 protein levels (Fig 1D), there was a significant increase in the glycolytic flux (Fig 1E, F) and glucose uptake (Fig 1G) as well as increased lactate secretion (Fig 1H). To further analyze the effect of these changes on endothelial metabolism, HUAECs were treated with 13C-glucose after 7 days of TGFβ stimulation. This revealed a significant increase in the level of key intermediaries, as well as 13C-glucose contribution to their biosynthesis (Fig 1I).
Figure 1. Metabolic effects of endothelial TGFβ signaling.
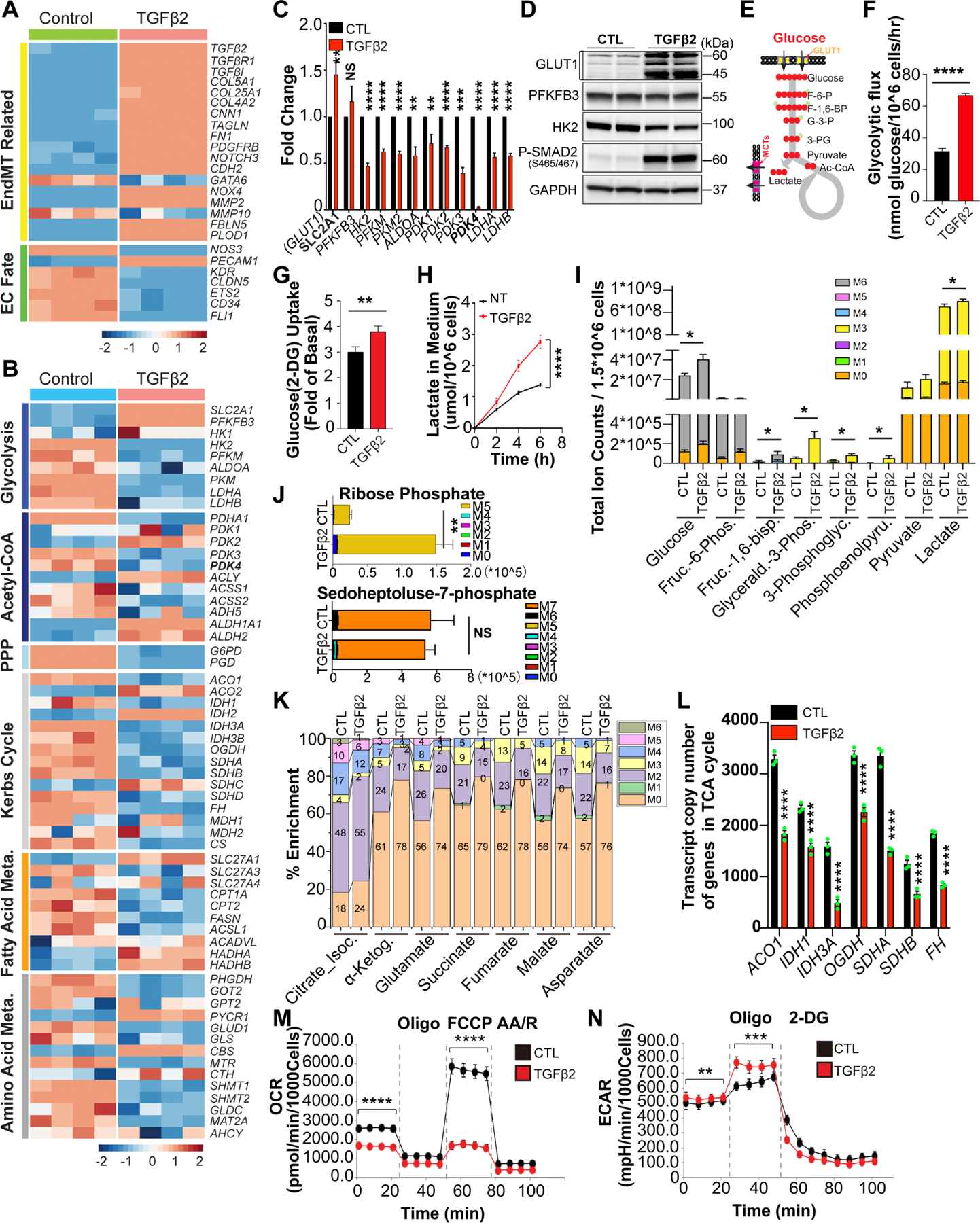
A-B, Bulk RNA-seq analysis of EndMT markers and endothelial cell (EC)-specific gene expression (A), and metabolic genes expression (B) in Human Umbilical Artery Endothelial Cells (HUAECs) with or without TGFβ2 stimulation (10ng/ml) in complete EGM-2 medium for 7 days. C, Quantitative Polymerase Chain Reactions (QPCR) analysis of glycolytic genes expression in HUAECs before and after TGFβ2 stimulation (10ng/ml) in complete EGM-2 medium for 7 days. D, Representative blots for key glycolytic proteins in HUAECs before and after TGFβ2 stimulation for 7 days. E, Diagram of carbon flux in glycolysis. F-H, Glycolytic activity was measured in HUAECs with or without TGFβ2 stimulation (10ng/ml) for 7 days. Glycolytic flux was then determined by testing the conversion of glucose, D-[5–3H(N)] to 3H2O (F). 2-Deoxyglucose (2-DG) and conversion to 2-DG-6-phosphate (2-DG6P) was measured to show the glucose uptake changes (G). Lactate concentration in the media collected from HUAECs culture under indicated conditions at specified time points normalized by the cell number (H). 5% dialyzed fetal bovine serum (D-FBS) with lower basal lactate was to the the medium instead of FBS. I-K, Liquid chromatography-mass spectrometry (LC-MS) metabolomics analysis of key metabolites in HUAECs treated with 13C-glucose (U-13C6-Glucose, 10mM) for 24 hours after 7 days of TGFβ2 stimulation. Total ion counts of key intermediaries of glycolysis can be found in (I), the change in the ribose phosphate which indicates the level of pentose phosphate pathway is shown in (J), 13C-glucose contribution to biosynthesis of TCA intermediates is shown (K). Changes in the transcript copy number of TCA related genes from RNAseq (L). Changes in the oxygen consumption rate (OCR) (M) and extracellular acidification rate (ECAR) (N) in HUAECs during TGFβ driven EndoMT were analyzed. The data in C, F, G, H, I, J, L, M and N were normalized to those of the non-TGFβ treated control or scramble siRNA–treated cells and are presented as the mean ± SEM from at least three independent experiments. ****P ≤ 0.0001, ***P ≤ 0.001, **P ≤ 0.01, *P ≤ 0.05, NS, not significant.
We further observed a reduction in expression of pentose phosphate pathway (PPP) enzymes, with no significant changes in glucose-derived sedoheptulose-7-phosphate levels but a large increase in glucose-derived ribose-5-phosphate (Fig 1J), likely driven by increased glucose uptake. Tracing of 13C -glucose contribution to the TCA cycle showed that while its incorporation into citrate/isocitrate was only mildly reduced, its contribution to α-ketoglutarate and subsequent TCA metabolites production decreased significantly (Fig 1K), implying either a block at the level of isocitrate dehydrogenase (IDH) (Fig 1L), or increased production of α-ketoglutarate from glutamine. In agreement with a block in TCA metabolism and a rise in glycolysis and lactate secretion, oxygen consumption rate (OCR) was profoundly reduced (Fig 1M) and extracellular acidification rate (ECAR) was increased (Fig 1N) in TGFβ-treated cells.
TGFβ regulates PDK4 expression and Ac-CoA synthesis.
One surprising effect of TGFβ stimulation was a near complete reduction in expression of PDK4, a key inhibitor of PDH-dependent Acetyl-CoA (Ac-CoA) biosynthesis (Fig 1C and 2A)17 In agreement with the PDH-inhibiting role of PDK4, the TGFβ-induced reduction in PDK4 expression was associated with a near-complete loss of PDH phosphorylation (Fig 2A) and a significant increase in cellular Ac-CoA levels (Fig 2B). Treatment of HUAECs with siRNAs to PDK4 led to a similar loss of PDH phosphorylation and increase in Ac-CoA levels (Fig 2C, D), confirming that the loss of PDK4 is sufficient to link TGFβ stimulation with increased Ac-CoA production. Furthermore, inhibition of PDK4 activity with the pan-PDK inhibitor dichloroacetate (DCA) similarly resulted in decreased PDH-E1α phosphorylation (Fig 2E) and increased Ac-CoA production (Fig 2F).
Figure 2. TGFβ regulates PDK4 expression and Ac-CoA synthesis, and increase in endothelial Ac-CoA levels induces EndMT in ACSS2-dependent manner.
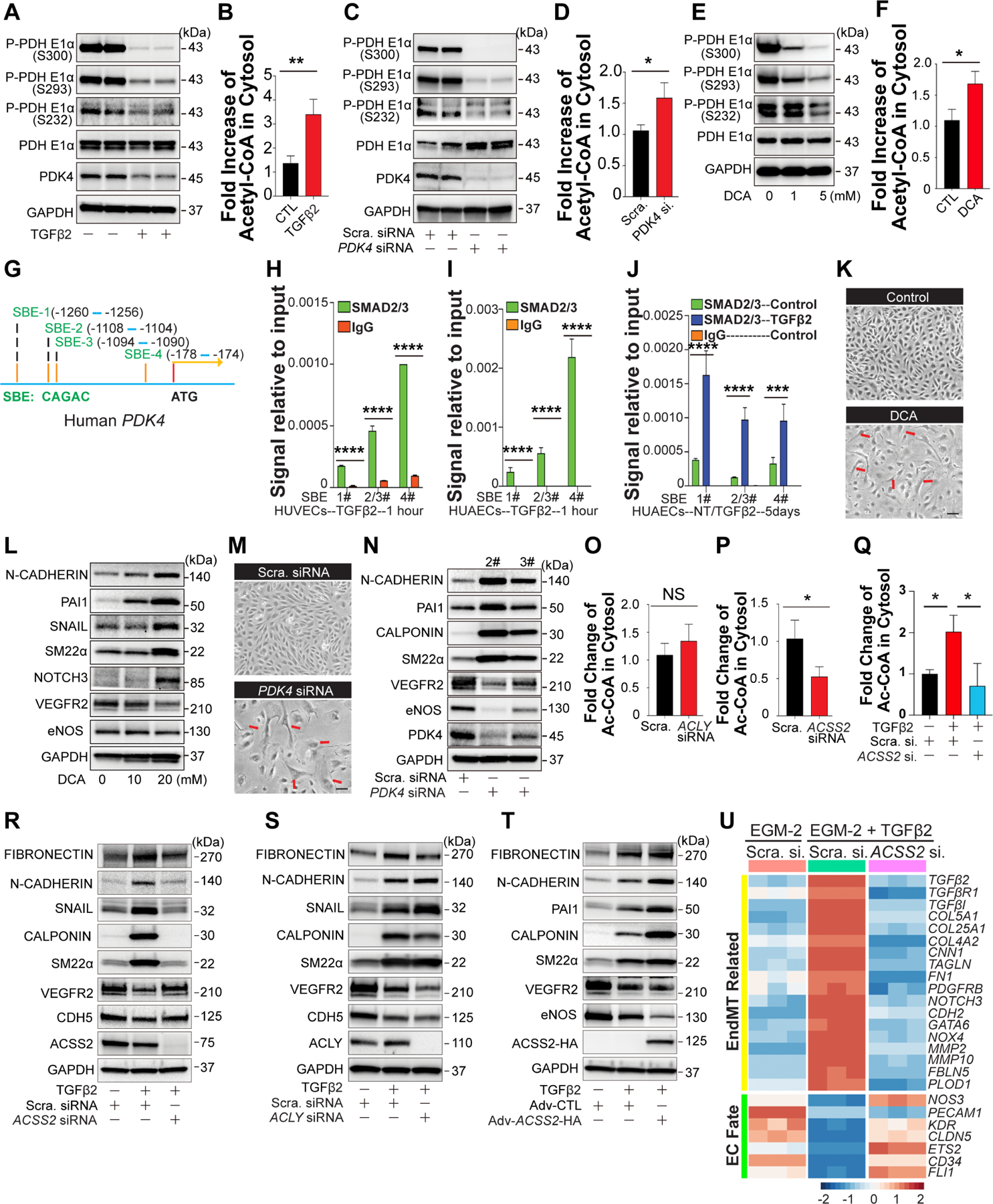
A-B, Following 7 days of TGFβ2 stimulation (10ng/ml), the levels of phosphorylated PDHE1α and total PDK4 in HUAECs cell lysates (A) and Ac-CoA level in the cytosol of HUAECs (B) were determined. C-D, Scramble siRNA or PDK4 siRNA were transduced into HUAECs for 7 days, blocking efficiency of PDK4 siRNA and the levels of phosphorylated PDHE1α were analyzed using 20 μg lysate of HUAECs (C). Cytosolic Ac-CoA levels in 1.5 million HUAECs (D). E, Representative blots showing the effect of the PDKs inhibitor, dichloroacetate sodium (DCA) on PDHE1α activity in HUAECs treated with DCA for 3 days. F, Following DCA (10mM) treatment for 7 days, the cytosolic Ac-CoA level in 1.5 million HUAECs were determined. G, Four potential SMAD-binding elements (SBEs, Motif: CAGAC) were identified in the Human PDK4 promoter. H-J, Chromatin immunoprecipitation(CHIP)-QPCR testing of SMAD-2/3 binding to SBEs in Human PDK4 promoter in human umbilical vein endothelial cells (HUVECSs) (H) and HUAECs(I). After 5 days of TGFβ2 stimulation, the direct regulation of PDK4 expression by SMADs was analyzed (J). K-N, Pharmacological inhibition of PDKs and PDK4 knockdown induce EndMT. Cell shape transition of HUAECs after DCA treatment(K) or siRNA mediated PDK4 deficiency (M) for 7 days. Scale Bar: 15 μm. Representative blots showing EndMT markers and EC-specific gene expression in HUAECs treated with DCA(L) or PDK4 siRNA(N) for 7 days. O-P, In HUAECs, the contributions of ACLY(O) and ACSS2(P) to Ac-CoA production. Q, Cytosolic Ac-CoA level were measured in HUAECs transduced with Scramble siRNA, Scramble siRNA + TGFβ2, or ACSS2 siRNA + TGFβ2 respectively. R-S, Blots showing EndMT markers and EC-specific markers in HUAECs treated with Scramble siRNA, Scramble siRNA + TGFβ2, or ACSS2 siRNA + TGFβ2(R) / ACLY siRNA + TGFβ2(S) separately for 7 days. T, Changes of EndMT markers and EC-specific markers were determined in HUAECs treated with Adv-CTL, Adv-CTL + TGFβ2, or Adv-ACSS2-HA + TGFβ2 separately for 3 days. U, Bulk RNA-seq analysis of EndMT markers and EC-specific gene expression in HUAECs under different conditions: Scramble siRNA, Scramble siRNA + TGFβ2, or ACSS2 siRNA + TGFβ2. The data in B, D, F, O, P, and Q were normalized to those of the non-TGFβ treated control or scramble siRNA–treated cells and are presented as the mean ± SEM from at least three independent experiments. **P ≤ 0.01, *P ≤ 0.05, NS, not significant.
To confirm that PDK4 is the primary PDK affected by TGFβ treatment, we measured protein levels of all PDK isoforms after 5 days of TGFβ stimulation. In agreement with RNAseq data, Western blotting showed a profound reduction in PDK3 and PDK4 expression, while PDK1 and 2 levels were not affected (Fig S1A). Similar changes were observed after PDK4 siRNA-mediated knockdown (Fig S1B). To further assess functional role of PDK isoforms, each isoform was knocked down using siRNA. While knockdowns of either PDK1 or PDK2 had no effect on expression of endothelial fate and ENDMT markers (Fig S1C), a knockdown of PDK4 induced a profound reduction in expression of endothelial fate genes and a strong EndMT marker induction while PDK3 knockdown had only mild effects (Fig S1D).
To understand how TGFβ stimulation leads to a reduction in PDK4 expression, we performed in silico analysis of the human PDK4 promoter. This identified four potential SMAD-binding elements (SBEs, Fig 2G). ChiP-PCR testing of TGFβ-treated HUAECs showed a rapid increase in Smad-2/3 binding to three of the four SBEs starting at 1hr. and to all four SBEs after 5 days (Fig 2H–J) suggesting a direct regulation of PDK4 expression by SMADs. Furthermore, adenoviral overexpression of a constitutively active form of ALK5 (ALK5-CA) that leads to increased TGFβ signaling as measured by increased SMAD-2 phosphorylation, resulted in decreased PDK4 expression (Fig S1E). Finally, inhibition of TGFβ signaling by SMAD4 siRNA interference restored PDK4 protein expression even in the presence of TGFβ (Fig S1F). These results demonstrate that canonical TGFβ/ALK5/R-SMADs signaling plays a critical role in regulating PDK4 expression.
Increase in endothelial Ac-CoA induces EndMT in ACSS2-dependent manner.
Since inhibition of PDK4 activity or expression even in the absence of TGFβ leads to an increase in Ac-CoA levels, we asked if that increase is sufficient for induction of EndMT. Seven days after initiation of DCA treatment, HUAECs acquired a distinct spindleform morphology, losing their normal cobblestone appearance (Fig 2K). Analysis of gene expression documented increased expression of a range of EndMT markers including N-Cadherin, SM22α and PAI1 (Fig 2L). Similarly to DCA-induced inhibition of PDK activity, siRNA-mediated PDK4 knockdown also resulted in EndMT induction, verified both morphologically and by Western blotting (Fig 2M, N)
These results link the increase in endothelial Ac-CoA levels to EndMT induction, in a manner dependent on PDH activity. Ac-CoA generated by PDH, which is located in the mitochondrial matrix, generally has one of two fates: incorporation into, and oxidation by, the TCA cycle, or export from the mitochondria via the citrate shuttle, a process dependent on regeneration of Ac-CoA from citrate in the cytoplasm by the enzyme ACLY18. To test if ACLY is indeed involved in Ac-CoA generation in the endothelium, we knocked down expression of ACLY in HUAECs. Surprisingly, ACLY knockdown had no effect on baseline endothelial Ac-CoA level (Fig 2O), indicating that the bulk of Ac-CoA is produced by other means. Ac-CoA can be alternatively generated by conjugation of CoA directly with acetate, a process requiring the enzyme ACCS219. Consistent with the use of this alternative means of CoA generation, knockdown of ACSS2 significantly reduced baseline endothelial Ac-CoA (Fig 2P). In agreement with the observed reduction in baseline Ac-CoA, a knockdown of ACSS2 also markedly reduced the extent of TGFβ-induced increase in Ac-CoA (Fig 2Q) and EndMT (Fig 2R) while ACLY KD had no effect (Fig 2S). Conversely, increased expression of ACSS2, achieved by adenoviral-mediated transduction of HUAECs, potentiated the extent of TGFβ-induced EndMT (Fig 2T). Importantly, in addition to reducing EndMT, ACSS2 knockdown also restored expression of endothelial fate genes (Fig 2U). Similar results were obtained in HAECs (Fig S2A–C)
Finally, ACSS2 knockdown in HUAECs reversed TGFβ-induced increased expression of GLUT1 and restored normal expression of various metabolic enzymes among others (Fig S2D). In particular, PDK4 expression returned to normal levels (Fig S2E). In agreement with these changes in PDK4 expression, we observed decreased Smad-2/3 binding to PDK4 SBEs following ACSS2 knockdown (Fig S2F). To test whether EndMT induction by TGFβ stimulation could be rescued by restoring PDK4 levels, we employed adenoviral PDK4 overexpression after 3 days of TGFβ stimulation. This resulted in a PDK4 overexpression level-dependent decrease in EndMT markers (Fig S2G). Of note, a decrease of ALK1 induced by TGFβ stimulation was reversed by ACSS2 knockdown in the presence of TGFβ stimulation (Fig S2H).
Endothelial Ac-CoA is largely derived from acetate, and acetate drives EndMT.
Since ACSS2 mediates Ac-CoA synthesis from acetate, these data implicated acetate as a central driver of EC metabolism and of EndMT and prompted us to probe more deeply into endothelial acetate metabolism. We used 13C-labeled substrate studies to investigate if endothelial cells can in fact generate endogenous acetate, and if so, from what substrates and to what end. We found that in the presence of physiological levels of ambient acetate (100μM), ECs both consume and secrete substantial amounts of acetate, i.e., labeled acetate in the media was consumed while simultaneously unlabeled acetate was secreted into the media (Fig 3A, B).
Figure 3. Endothelial Ac-CoA is largely derived from acetate in an ACLY-independent manner, and acetate drives EndMT.
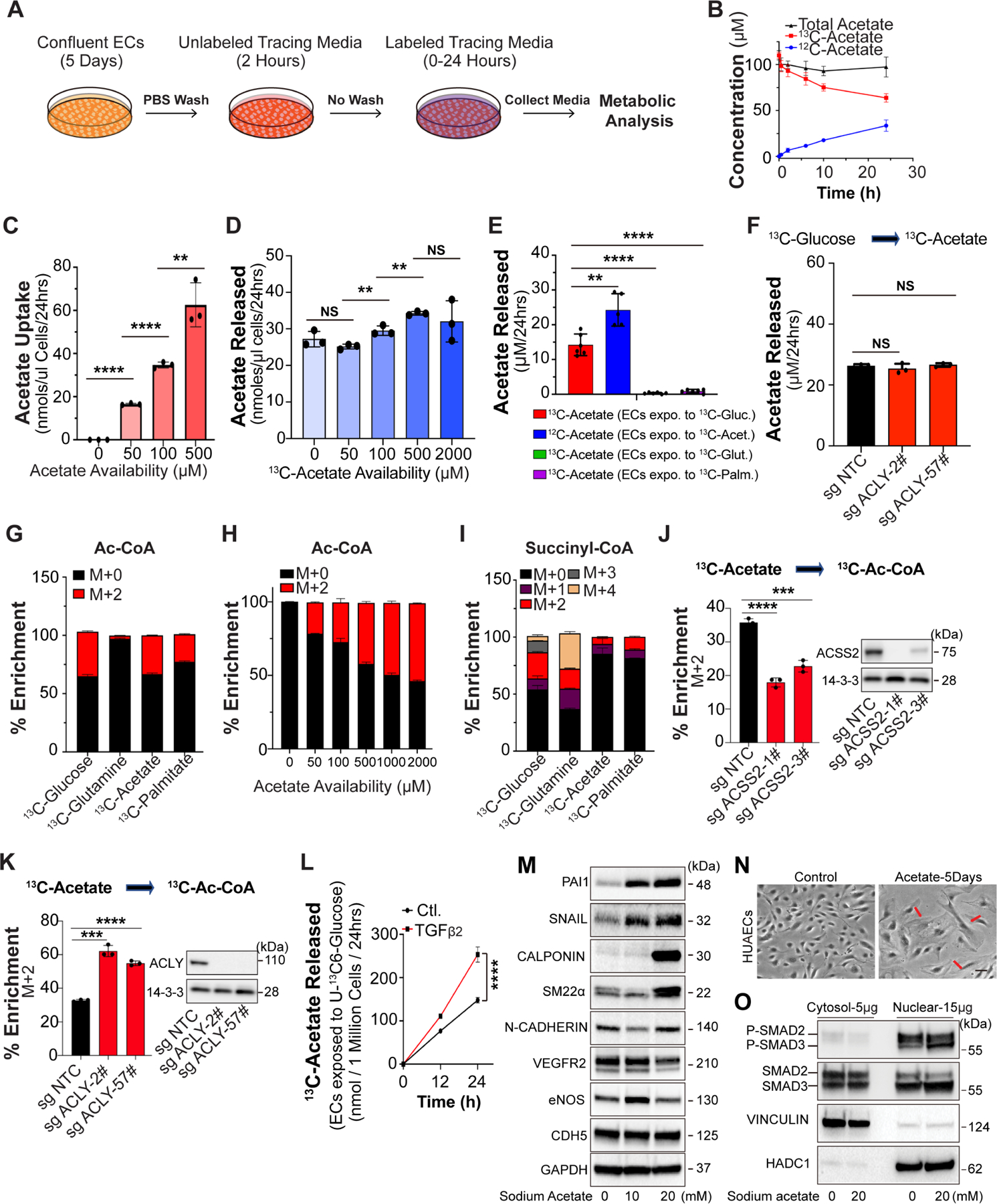
A, Schematic representation of the stable isotope tracing experimental design. Confluent endothelial colony forming cells (ECFCs) were pre-treated in tracing medium with unlabeled substrates for two hours before being switched to labeled tracing medium. B, Concentration of extracellular 12C-acetate, 13C2-acetate and total acetate (sum of 12C-acetate and 13C2-acetate) when ECFCs were exposed to media containing 100 μM of 13C2-acetate. N=3. C, Acetate uptake over 24 hrs at varying concentrations of 13C2-acetate. Acetate concentration in medium was normalized to packed cell volume in μL. N=3. D, Acetate released (i.e. extracellular 12C-acetate) over 24 hrs at varying concentrations of starting 13C2-acetate. Acetate concentration was normalized to packed cell volume in μL. N=3. E, Measurement of indicated acetate stable isotope in the cell culture medium after incubation with either U-13C-glucose, U-13C-glutamine, 13C2-acetate or U-13C-palmitate for 24 hrs. Only incubation with U-13C-glucose led to appearance of extracellular 13C2-acetate. N=5–6. F, Measurement of glucose-derived acetate release (13C2-acetate from U-13C-glucose) upon ACLY KO with two different sgRNAs over 24 hrs. N=3. Single pair-wise comparison between two groups (C, D, E and F) performed by unpaired, two-tailed Student’s t-test using Prism 9. NS, no significance. **P values < 0.01. ***P values < 0.001. ****P values < 0.0001. G, Percent isotopologue enrichment of acetyl-CoA when ECFCs are exposed to 5 mM U-13C-glucose, 500 μM U-13C-glutamine, 100 μM 13C2-acetate and 100 μM U-13C-palmitate for 24 hours. The enrichment from 13C2-acetate is corrected for label dilution by intracellular acetate release. N=3. H, Percent isotopologue enrichment of acetyl-CoA when ECFCs are exposed to varying concentrations of 13C-acetate over 24 hours. Enrichment from 13C2-acetate is corrected for media label dilution. N=3. I, Same as in (G) but for succinyl-CoA. J, Percent isotopologue enrichment of M+2 acetyl-CoA from 100 μM 13C2-acetate upon ACSS2 KO with two different sgRNAs. Enrichment from 13C2-acetate is corrected for media label dilution. N=3. K, Percent isotopologue enrichment of M+2 acetyl-CoA from 100 μM 13C2-acetate upon ACLY KO with two different sgRNAs. Enrichment from 13C2-acetate is corrected for media label dilution. N=3. L, 13C2-acetate released from U-13C-glucose upon TGFβ2 treatment at time points indicated. Acetate concentration is media was normalized to cell number. N=3. M, Representative blots showing the changes of EndMT markers and EC-specific gene expression in HUAECs treated with acetate for 7 days in a dose-dependent manner. N, Cell shape transition of HUAECs after 5 days treatment of acetate (20mM). Scale Bar: 6 μm. O, Blots showing the subcellular localization and phosphorylation of SAMD2/3 in HUAECs treated with acetate for 7 days. The data in J, K, and L were normalized to those of the non-TGFβ treated control or sg control–treated cells and are presented as the mean ± SEM from at least three independent experiments. ****P ≤ 0.0001, ***P ≤ 0.001.
Interestingly, changing the amount of available acetate in the media changed the rate of uptake, but the rate of production remained constant, indicating an endogenous regulated process (Fig 3C, D). Using 13C-Glucose, 13C-glutamine, and 13C-palmitate as tracers, we found that 60% of the acetate that is released into the media is derived from glucose, and almost none from fatty acids or glutamine, an otherwise major contributor to TCA intermediates in ECs13 (Fig 3E), suggesting that labeled acetate does not originate indiscriminately from mitochondrial acetyl-CoA pools, but specifically from glucose. Equally surprisingly, CRISPR deletion of ACLY in endothelial cells in vitro had no impact on acetate secretion, indicating that acetate production by ECs was entirely independent of ACLY (Fig 3F), despite being generated by glucose, a pathway that typically requires ACLY. These findings mirror the lack of ACLY contribution to Ac-CoA levels (Fig 2O) or EndMT (Fig 2S).
To further probe the link between acetate and Ac-CoA, we used the same 13C tracers to measure acetate incorporation into the Ac-CoA pool. We found that acetate indeed is a prominent contributor to the Ac-CoA pool in ECs, accounting for a striking 30–50% of Ac-CoA carbons in the acyl group, depending on ambient acetate concentrations (plasma acetate concentrations range from 100–500 μM) (Fig 3G, H). The vast majority of this acetate-derived Ac-CoA pool is cytoplasmic because we see nearly no contribution by 13C-acetate to succinyl-CoA (Fig 3I) (glucose and acetate provide carbons to succinyl-CoA via mitochondrial Ac-CoA, while glutamine bypasses Ac-CoA)20. Correspondingly, 13C-palmitate labeling accounted for only a small percent of Ac-CoA labeling suggesting that in ECs the mitochondrial Ac-CoA pool is smaller than the cytoplasmic pool. Furthermore, we found that the incorporation of acetate into Ac-CoA was largely dependent on ACSS2 (Fig 3J) but not ACLY (Fig 3K). In agreement with these data, treatment of HUAECs with TGFβ resulted in increased accumulation of glucose-derived acetate in the cell culture media (Fig 3L).
These results are consistent with a pathway whereby TGFβ stimulates glucose uptake and glucose-derived production of acetate, in an atypical ACLY-independent fashion, which is then converted to Ac-CoA in the cytoplasm by ACSS2 to drive EndMT. To directly test this implication of acetate as a central modulator of EndMT, we examined whether increasing acetate concentration in the media would, by itself, induce EndMT. Five days of supplementation of cultured HUAECs with acetate induced a dose-dependent increase in expression of EndMT markers (Fig 3M), a change in morphology (Fig 3N) and an increased shift of P-SMAD2/3 to the nucleus (Fig 3O), all consistent with induction of TGFβ signaling and EndMT. Edu labeling was used to test whether proliferation of endothelial cells was suppressed by decreased PDK4 expression or sodium acetate treatment. We also observed a significant reduction in endothelial proliferation, measured by Edu labeling, following PDK4 knockdown (Fig S3A), and a dose-dependent decrease following treatment with sodium acetate (Fig S3B). Of note, sodium acetate treatment in concentrations below 20 mM had no effect on tissue culture medium pH (Fig S3C). Furthermore, ACCS2 knockdown in cells subjected to PDK4 siRNA treatment restored normal EC morphology (Fig S3D). Expression of mesenchymal markers and the inhibition of cell proliferative activity induced by PDK4 KD were also rescued by knocking down ACSS2 expression (Fig S3E, F).
These data place acetate squarely at the heart of Ac-CoA biology in endothelial cells and reveal a picture whereby acetate is robustly produced by ECs, in an atypical fashion independent of ACLY, and substantially contributes to the Ac-CoA pool, in an-ACCS2-dependent fashion.
ACSS2-generated Ac-CoA regulates TGFβ signaling.
We next examined how an increase in the cytoplasmic Ac-CoA level induces EndMT. Since TGFβ signaling is the main driver of EndMT, we first evaluated changes in the cytosolic and nuclear SMADs content. As expected, TGFβ stimulation resulted in a shift of R-SMADs 2 and 3 and the common SMAD4 from the cytoplasm to the nucleus (Fig 4A, B). Importantly, the nuclear shift of SMADs 2 and 4 (but not that of SMAD3) was strongly reduced by ACSS2 knockdown while ACLY knockdown had no effect (Fig 4A–C). Furthermore, phosphorylation of R-SMADs induced by TGFβ treatment was almost completely attenuated in the presence of ACSS2 knockdown (Fig S4A, B). In addition, while TGFβ treatment of HUAECs led to a robust 3.4-fold increase in expression of the inhibitory SMAD7, there was only a minor (~1.4–1.6 fold) increase in SMAD7 acetylation (Fig S4C). Finally, even in the absence of TGFβ treatment, acetylation of SMAD2 and 4 (but not SMAD3) was diminished following ACSS2 knockdown (Fig 4D). TGFβ treatment of HUAECs induced a further increase in acetylation of R-SMADs that was reduced by ACSS2 knockdown (Fig S4D). Consistent with these TGFβ treatment results, both SMAD2 acetylation and the nuclear shift of SMAD2/3 were enhanced following adenoviral expression of ACSS2 in HUAECs (Fig S4E, F).
Figure 4. ACSS2-generated Ac-CoA regulates TGFβ signaling.
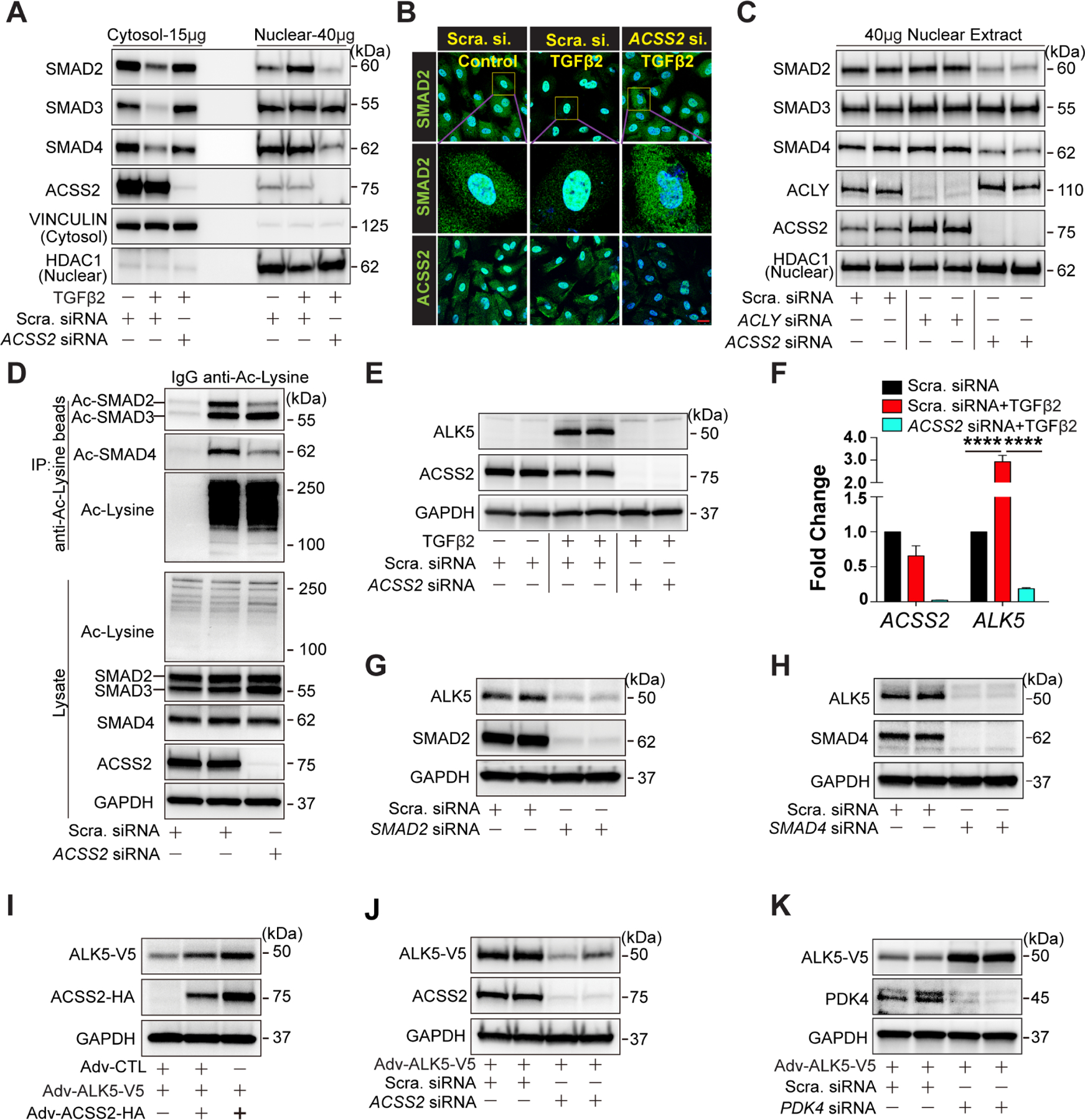
A-B, Cytosolic and nucleic R-SMADs examined by western blotting (A) and immunostaining (B) in HUAECs treated with Scramble siRNA, Scramble siRNA + TGFβ2, or ACSS2 siRNA + TGFβ2 respectively. Scale Bar: 25 μm. C, Representative blots of nucleic R-SMADs in HUAECs treated with Scramble siRNA, ACLY siRNA, or ACSS2 siRNA separately. D, Representative blots of acetylated R-SMADs in HUAECs transduced with Scramble siRNA or ACSS2 siRNA. Acetylated proteins were captured using agarose beads conjugated with anti-Acetylated lysine antibody, and acetylated R-SMADs (Ac-SMAD2/3/4) were detected using anti-SMAD2/3 antibody and anti-SMAD4 antibody. E, Blots showing the expression of ALK5 in HUAECs transduced with Scramble siRNA or ACSS2 siRNA in the presence of TGFβ2 for 7 days. F, mRNA level of ALK5 in HUAECs under different conditions: Scramble siRNA, Scramble siRNA + TGFβ2, or ACSS2 siRNA + TGFβ2 respectively. G, Blots of ALK5 level in HUAECs following with SMAD2 deficiency for 4 days. H, Blot showing ALK5 level in HUAECs transduced with Scramble siRNA or SMAD4 siRNA for 4 days. I, Representative blots of V5-tagged ALK5 (ALK5-V5) protein level following gradient overexpression of ACSS2-HA in HUAECs using adenoviral strategy(i). J-K, Representative blots of ALK5-V5 in HUAECs transfected with Scramble siRNA or ACSS2 siRNA(J) / PDK4 siRNA(K) separately for 4 days. ALK5-V5 was adenoviral overexpressed in HUAECs on the next day after siRNA transfection.
In addition to affecting SMAD2 and SMAD4 acetylation and nuclear shift, TGFβ stimulation also led to a significant increase in ALK5 expression that was reversed by ACSS2 KD (Fig 4E, F). Since ALK5 expression is known to be TGFβ-dependent, we measured ALK5 levels after SMAD2 or SMAD4 knockdowns and observed a significant decrease in its expression (Fig 4G, H).
These results show that TGFβ-induced increase in Ac-CoA levels is leading to increased acetylation of SMADs and ALK5 and increased ALK5 expression. To further link these changes in ALK5 expression to acetylation, we expressed a tagged ALK5 construct in HUAECs. Increasing ACSS2 levels, using an adenoviral-mediated ACSS2 transduction, resulted in increased ALK5 expression (Fig 4I), while ACSS2 KD led to a decrease in ALK5 expression (Fig 4J). Finally, a PDK4 knockdown that mimics the effect of TGFβ treatment also resulted in increased ALK5 expression and increased phosphorylation of R-SMADs (Fig 4K; Fig S4G–I). Treatment with the PDK inhibitor DCA had a similar effect (Fig S4J)
Taken together, these data show that ACSS2-driven Ac-CoA production leads to ALK5 acetylation, thereby increasing its expression. To test this directly, we carried out blotting with an anti-Ac-lysine antibody of TGFβ-treated HUAECs transduced with a tagged ALK5 construct. In agreement with above data, TGFβ treatment resulted in increased ALK5 acetylation (Fig 5A) as did a PDK4 knockdown (Fig 5B), while ACSS2 knockdown strongly reduced it (Fig 5C).
Figure 5. ACSS2-generated Ac-CoA promotes protein stability of ALK5.
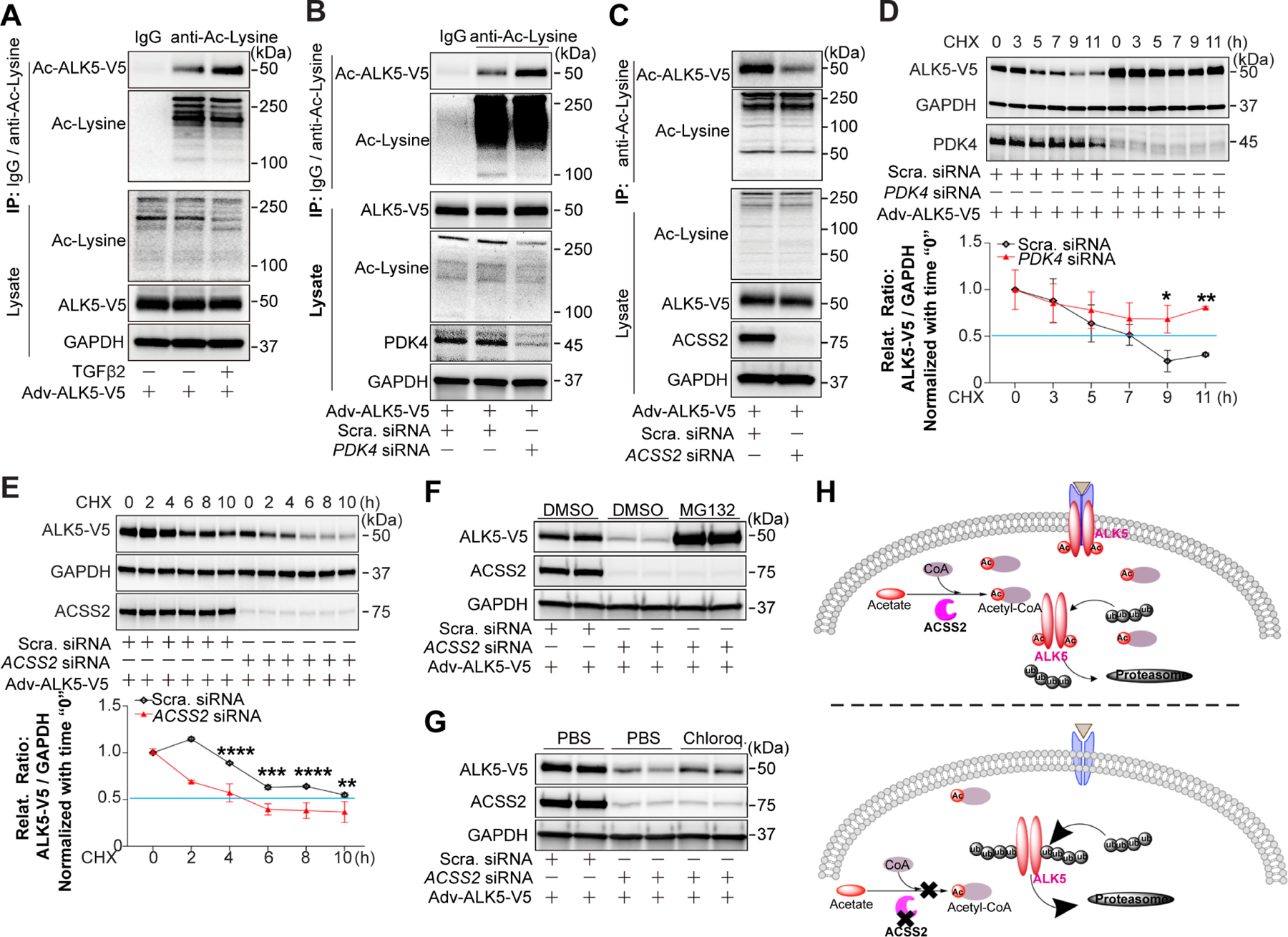
ALK5-V5 was adenovirally overexpressed in HUAECs three days before assays. A, Representative blot of acetylated ALK5-V5 in HUAECs treated or non-treated with TGFβ2 (10 ng/ml in complete EGM-2 medium) for 7 days. Acetylated proteins were captured using agarose beads conjugated with anti-Acetylated lysine antibody, and acetylated ALK5-V5 (Ac-ALK5-V5) was detected using anti-V5 tag antibody. B-C, Blots of Ac-ALK5-V5 in HUAECs transduced with Scramble siRNA or PDK4 siRNA(B) / ACSS2 siRNA(C) separately for 7 days. Acetylated proteins were captured and detected as described above. D-E, Half-life of ALK5-V5 in HUAECs transduced with Scramble siRNA or PDK4 siRNA(D) / ACSS2 siRNA (E). ALK5-V5 was adenovirally overexpressed in HUAECs cycloheximide (CHX, 10 μg/ml) treatment at indicated time points. ALK5-V5 protein level was determined by anti-V5 antibody. F-G, Representative blots showing the regulation of ALK5 degradation in HUAECs. HUAECs were first transduced with Scramble siRNA or ACSS2 siRNA, then ALK5-V5 was adenovirally overexpressed in HUAECs followed by treatment with a proteasome inhibitor MG132(F) or a lysosome inhibitor chloroquine (G). H, Diagram for Ac-CoA-regulated ALK5 acetylation results in increased protein half-life due to decreased proteasomal degradation.
To determine how acetylation affects ALK5 expression, we measured ALK5 protein half-life in HUAECs and HUVECs. There was a significant increase in ALK5 protein stability following acetylation-inducing PDK4 knockdown (Fig 5D) while ACSS2 knockdown had the opposite effect (Fig 5E, Fig S4K). Next, HUAECs and HUVECs expressing a tagged ALK5 were treated with a proteasome inhibitor MG132 and lysosome inhibitor chloroquine. Treatment with the former, but not the latter, increased ALK5 levels after ACSS2 knockdown suggesting that deacetylated ALK5 undergoes rapid proteasome-dependent degradation (Fig 5F, G; Fig S4L, M). Together, these data show that ALK5 acetylation results in increased protein half-life due to decreased proteasomal degradation (Fig 5H).
We next set out to identify the ALK5 acetylation sites. Expression of a human ALK5 protein fused with the Flag tag in 293T cells led to detection of strongly acetylated ALK5 following immunoprecipitation with an anti-Flag antibody (Fig S5A). LC-MS/MS analysis of the fusion construct pointed to Lys490 as the likely acetylation residue in the ALK5 protein (Fig S5B, C). However, transfection of 293T cells with either wild type ALK5 or an ALK5 carrying a K490R mutation (lysine (K) to arginine (R) substitutions at the Lys490 site) did not show any decrease in ALK5 acetylation implying that other site(s) were responsible (Fig S5D). We therefore separately mutated to arginine each one of the 25 lysine residues in the human ALK5 protein creating 25 different ALK5 mutant-Flag constructs. Every mutant construct was then transfected into 293T cells, and the extent of acetylation assessed after immunoprecipitation with an anti-Flag antibody. We observed a strong reduction in acetylation of ALK5 proteins carrying K223R, K343R, K391R, and K449R mutations (Fig S5E). These five lysine residues (K223, K343, K391, K449, and K490) therefore are the principal acetylation sites in ALK5.
ACSS2 regulates EndMT in vivo.
To assess if endothelial ACSS2 plays a similar role in endothelial metabolism in vivo, we investigated its role in the development and progression of atherosclerosis, an End-MT driven disease 7,21. Immunocytochemical analysis of endothelial ACSS2 expression in ascending aortas of 13 normal organ donors with either minimal (N=7) or mild/moderate (N=6) extent of atherosclerosis revealed a significant increase in ACSS2 expression (Fig 6A, B). A similar analysis of endothelial ACSS2 expression in the severely atherosclerotic brachiocephalic trunk and aortic root from Apoe−/− mice after 3 months of high fat diet revealed substantial ACSS2 expression in the endothelium (Fig 6C, D)
Figure 6. Immunostaining of ACSS2 in human aorta and mice brachiocephalic trunk and aortic root.
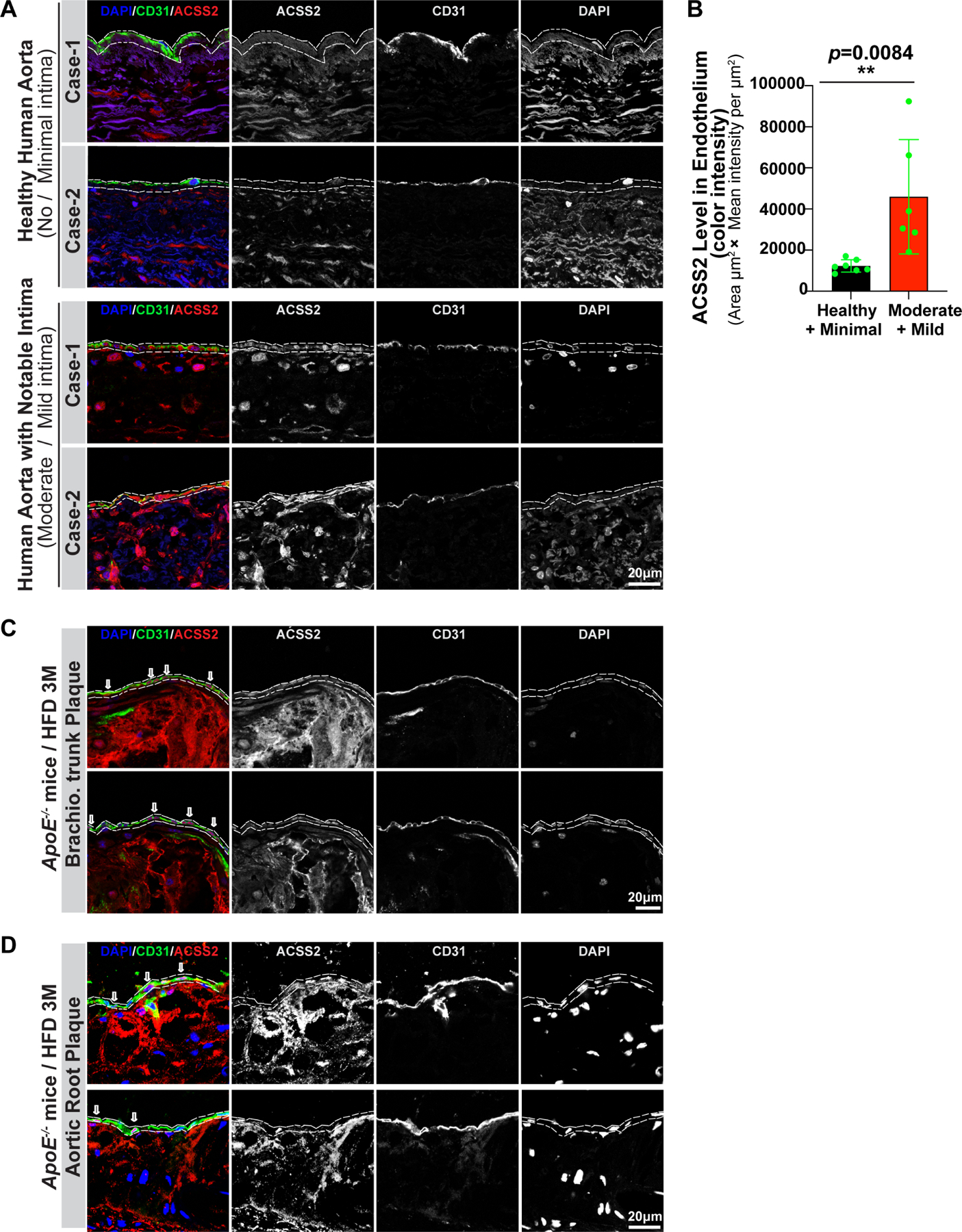
A, ACSS2 expression was studied in human aortas (n=13) from normal organ donors with minimal or moderate atherosclerosis extent. In all cases, ACSS2 expression was examined in relatively normal aortic segments (no/minimal disease) and in segments demonstrating mild/moderate atherosclerosis as judged by the extent of neointima development. ACSS2 expression (red signal), endothelial cells identified with anti-CD31 (green staining), nuclei identified with DAPI (blue). B, Quantification of ACSS2 expression in endothelial cells areas (as indicated with dashed line) from normal/minimal disease and mild/moderate disease specimens. Statistical analysis was performed by unpaired, two-tailed Student’s t-test using Prism 9. **P values < 0.01. Representative frozen sections of the brachiocephalic trunk (C) and aortic root (D) dissected from control ApoE−/− mice after three months of high fat diet were immunostained with anti-CD31 (green), DAPI (blue) and anti-Acss2 (red) antibodies. Merged channels, single channel of Acss22/CD31/DAPI were displayed in grey color. And the area of endothelium was indicated with dashed lines.
Given in vitro and in vivo data pointing to likely ACSS2 involvement in the development and progression of atherosclerosis, we evaluated whether suppression of endothelial ACSS2 expression would influence the development of atherosclerosis. To this end, we generated mice with an inducible EC-specific deletion of Acss2 on the ApoE null background (Cdh5CreERT2;Accs2fl/fl;Apoe−/− thereafter ACSS2iECKO;ApoE) (Fig S6A, B). Activation of the Cdh5Cre by tamoxifen in mice of 5–6 weeks of age resulted in a high efficiency deletion of the endothelial Acss2 gene (Fig S6D). Two weeks later, ~8 week old ApoE−/− control mice and ACSS2iECKO;ApoE mice were placed on a high fat diet (n=10 each, Fig S6C) and then sacrificed 3 months later. The whole aorta Oil-Red-O staining was used to assess the total atherosclerotic burden. Compared to Apoe−/− mice, ACSS2iECKO;ApoE mice showed a highly significant reduction in atherosclerosis extent uniformly along the length of the entire aorta (Fig 7A,B; Fig S6E, F). Histologic analysis of aortic root sections demonstrated a significant reduction of plaque area in ACSS2iECKO;ApoE mice (Fig 7C). Analysis of the total cholesterol and triglycerides levels in the plasma showed no differences between ACSS2iECKO;ApoE and ApoE−/− mice demonstrating that the plasma lipids are not affected by the EC-specific deletion of Acss2 (Fig 7D, E; Fig S6G, H). Finally, in agreement with the overall reduction of the atherosclerotic burden and atherosclerotic plaque sizes, immunostaining of the aortic root sections demonstrated a decrease in fibronectin and collagen-1 deposition, reduced VCAM1 expression and reduces number of blood-derived monocytes/macrophages (CD68) in ACSS2iECKO;ApoE mice (Fig 7F–I).
Figure 7. Reduced development of atherosclerosis following endothelial-specific deletion of Acss2 in ApoE−/− mice.
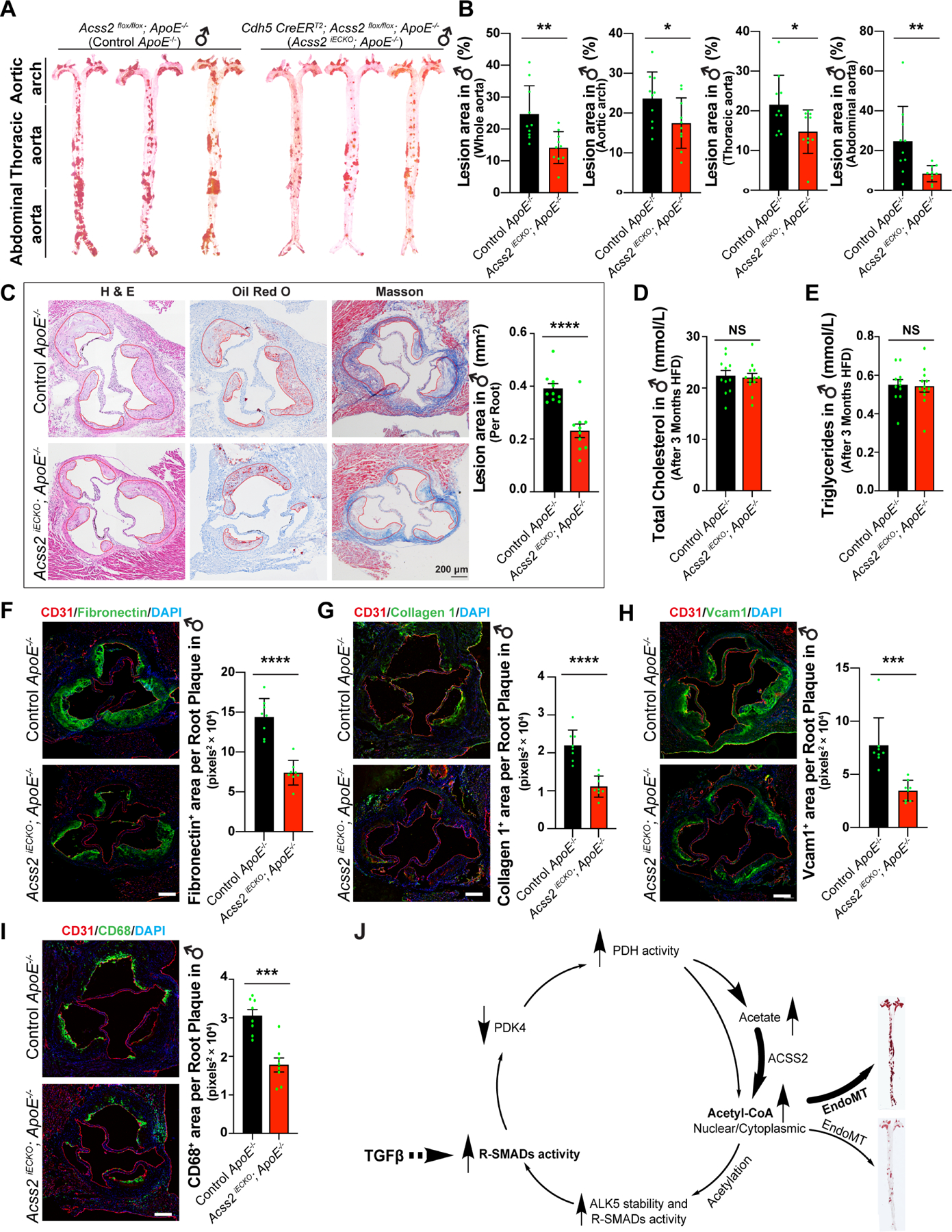
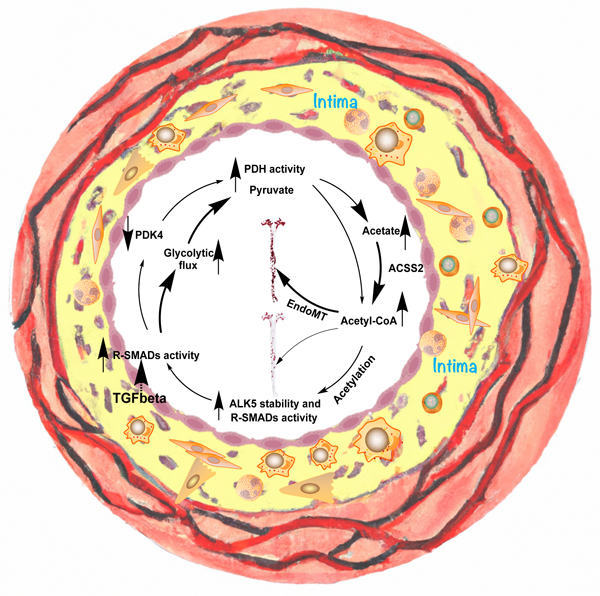
A, Plaques in aortas of control ApoE−/− mice (♂) and Acss2 iECKO; ApoE−/− mice (♂) were stained with Oil-Red-O. B, Oil-Red-O analysis of whole aortas, aortic arch, thoracic aorta, and abdominal aorta from control (n=10) and Acss2 iECKO; ApoE−/− mice (♂) (n=10). The plaque lesion area and total surface area of aortas were quantified using ImageJ software. A single pair-wise comparison between two groups performed by unpaired, two-tailed Student’s t-test using Prism 9. *P values < 0.05, **P values < 0.01. C, Plaques in aortic roots of control ApoE−/− mice and Acss2 iECKO; ApoE−/− mice were analyzed with H&E staining, Oil-Red-O staining and Masson staining. The area of the plaque per root were quantified. A single pair-wise comparison between two groups performed by unpaired, two-tailed Student’s t-test using Prism 9. ****P values < 0.0001. D-E, total cholesterol level (D) and triglycerides (E) in plasma collected from control ApoE−/− mice and Acss2 iECKO; ApoE−/−mice after 3 months of high fat diet. NS, no significance. F-I, immunostaining of Fibronectin 1 (F), Collagen 1 (G), Vcam1 (H), and CD68 (I) on aortic root sections from both control ApoE−/− mice and Acss2 iECKO; ApoE−/−mice. A single pair-wise comparison between two groups performed by unpaired, two-tailed Student’s t-test using Prism 9. ***P values < 0.001, ****P values < 0.0001. J, Diagram of metabolic reprogramming regulated by TGFβ signaling in EndMT. TGFβ signaling promotes glucose uptake and glycolysis in endothelial cells and increases the activity of pyruvate dehydrogenase complex (PDC) by suppressing PDK4 expression. TGFβ signaling induces glucose conversion to acetate which is then followed by the conversion of acetate to cytosolic acetyl-CoA mediated by ACSS2, ultimately leading to increased activity of R-SMADs and ALK5 subsequently that in turn further promotes TGFβ signaling there by establishing a positive feedback loop. Blocking the acetate conversion to Ac-CoA by ACSS2 knockdown decreased the acetylation of R-SMADs and ALK5 and disrupts TGFβ signaling thereby interrupting in this positive feedback loop.
To evaluate ACSS2 as a potential therapeutic target in atherosclerosis, we used nanoparticle-delivered ACSS2 siRNA to knockdown endothelial ACSS2 expression. To this end, 2 months old ApoE−/− mice were placed on high fat diet and assigned to every 10-day injections of 7C1 nanoparticles loaded with either unmodified or modified ACSS2 siRNA (n=9 each) or no treatment controls (n=12, Fig S7A, B). The mice were sacrificed 3 months later and whole aorta Oil-Red-O staining was used to assess total atherosclerotic burden. Both ACSS2 siRNA-treated groups showed a highly significant reduction in atherosclerosis extent in agreement with ACSS2 knockout data (Fig S7C, D).
ACSS2 and inhibition of PDK4 drive positive feedback loop
Together, our data unveil a linear pathway whereby TGFβ induces glucose conversion to acetate, followed by conversion of acetate to cytosolic acetyl-CoA by ACSS2, ultimately leading to increased expression of SMADs and ALK5 and persistent activation of the TGFβ signaling pathway. However, we also observed that TGFβ signaling suppresses PDK4 (Fig 2), thus setting up a positive feedback loop (Fig 7J). Further substantiating the existence of this positive feedback loop, we found that ACSS2 knockdown partially re-induced PDK4 (i.e. antagonized the siRNA effect), thereby reversing PDH complex activation (i.e. dephosphorylation) induced by PDK4 knockdown (Fig S8A). Consequently, ACSS2 knockdown also reduced EndMT markers and restoration of EC-specific gene expression induced by PDK4 siRNA (Fig S8B, C). Conversely, in the context of pro-EndMT TGFβ signaling, knockdown of PDK4 partially reversed the siACSS2-mediated block of EndMT induction by TGFβ (Fig S8D–G). Thus, ACSS2 acts both upstream of PDK4, via generation of Ac-CoA and promotion of SMAD2,3 binding to PDK4 promoter that leads to downregulation of PDK4 expression, and downstream of PDK4, via induced production of Ac-CoA, establishing a positive feedback loop that drives EndMT as manifested in vivo by the development of atherosclerosis (Fig 7J).
DISCUSSION
Endothelial cells metabolism relies on glycolysis for most (85%+) of its ATP generation 10,12,14,22. The results of our study show that in an unusual twist, ECs also use glucose to generate acetate that is then converted to cytoplasmic Ac-CoA by ACSS2. TGFβ stimulation increases endothelial glucose uptake while decreasing expression of PDK4 thereby activating PDH. This augments cytoplasmic acetate production leading, ultimately, to an increase in ACSS2-driven Ac-CoA synthesis. Increased availability of cytoplasmic Ac-CoA results, in turn, in increased acetylation of TGFβ secondary messengers SMAD2 and 4 as well as its receptor ALK5. Acetylation increases half-life of these proteins as well as enhances binding of acetylated SMADs to the ALK5 promoter. The overall effect is a marked increase in ALK5 levels leading to a large increase in endothelial TGFβ signaling. This further increases cellular Ac-CoA levels establishing a positive feedback loop. Once established, the TGFβ/PDK4/ACSS2/ALK5 feed-forward loop stabilizes EC TGFβ signaling at a high level, inducing and perpetuating EndMT (Fig 7J). Importantly, a knockdown of EC ACSS2 expression completely disrupted this positive feedback loop, fully eliminating expression of all EndMT molecular markers. This series of events, driven by TGFβ modulation of endothelial acetate metabolism fully aligns with and explains the previously described positive feedback TGFβ/EndMT loop.
These results place acetate metabolism at the center of endothelial biology and point to a critical role that acetate-dependent Ac-CoA synthesis plays in the development of EndMT and EndMT-related disorders. Prior work suggested that fatty-acid-derived acetyl-CoA decreased early (i.e. 24 hours) after induction of EndMT with a combination of TGFβ and IL-1β treatment, leading to lower SMAD7 protein levels and promotion of EndMT through ACLY 11. In contrast, we find here that after 7 days of TGFβ treatment alone, EndMT is robustly promoted, in ACLY-independent manner. A number of the differences between the two studies can be attributed to the use of IL-1β and a short time course by Xiong et al11.
There appear to be two critical controllers of EndMT developments under our conditions: acetate levels themselves and the activity/expression of PDK4. The fact that excessive acetate alone is enough to induce and accelerate TGFβ signaling is illustrated by the observation that simply increasing acetate concentration in the endothelial tissue culture media is enough to induce EndMT in a TGFβ-dependent manner. At the same time, the importance of PDK4 activity is demonstrated by the observation that a decrease in its expression (PDK4 RNAi knockdown) or inhibition of PDK activity (DCA) also results in induction of EndMT in a TGFβ-dependent manner. The final proof of the role of this cascade in EndMT is the fact that ACSS2 knockdown prevents its induction by TGFβ and reverses the already established transformation.
Our finding that EC-specific ablation of ACSS2 markedly reduced atherosclerosis disease burden underscores the cell-autonomous role of EC acetate metabolism and ACSS2 in driving EndMT. The work also highlights inhibition of EC ACSS2 as a potential therapeutic avenue for atherosclerosis and other EndMT-related pathologies. ACSS2 whole-body knockout mice are viable, fertile and have no overt phenotypes under basal conditions23,24, de-risking the notion of ACSS2 inhibition. Furthermore, acetate-dependency has emerged as a hallmark of nutrient-poor cancers, and there is thus already substantial interest and developing pipelines in inhibiting ACSS2 across multiple cancer types.25–27 Our work now highlights EndMT as another pathological context driven by acetate metabolism, in which ACSS2 inhibition may have therapeutic potential.
Atypical production of acetate by PDH has been described15, but whether the process occurs in any physiologically relevant contexts has been unclear. Our study provides such a context. Our data show that this atypical production of acetate by PDH, in conjunction with cytosolic ACSS2 activity, allows for shuttling of glucose-derived 2-carbon units to the cytosol without relying on the ACLY-dependent citrate shuttle, and that this process is critical to TGFβ-dependent induction of EndMT. It will be interesting to evaluate this pathway in other cell types. For example, inhibition of PDKs in vascular smooth muscle cells (VSMCs) with DCA and other agents promotes apoptosis and protects from various forms of vasculopathy including pulmonary hypertension and arterial restenosis.28,29 To what extent acetate production by PDH plays a role in these effects is not known. The central role of acetate in EndMT may also in part explain how hyperglycemia promotes EndMT and atherosclerosis30, i.e. high glycolysis in ECs may yield high pyruvate concentrations that increase flux through PDH, leading to higher acetate production and, hence, EndMT. The potentially therapeutically opposite consequence of modulating this pathway in different cells also warrants consideration. For example, PDK inhibition appears on balance to benefit pulmonary hypertension likely via beneficial effects on SMCs, outweighing negative effects on EndMT. Nevertheless, the complexity of these effects should incite caution. For example, DCA is actively being tested in humans to treat pulmonary hypertension31, but our current work suggests such studies should include careful evaluation of impact on atherosclerosis and other EndMT processes.
In addition to atypical production by PDH, acetate can be generated by deacetylation reactions (e.g. histone deacetylation), acetyl-CoA transferases, alcohol consumption32, and by the gut microbiome as a by-product of dietary fiber fermentation33. The latter likely generates most of circulating acetate. To what extent these other sources of acetate modulate EndMT and atherosclerosis will also be interesting to study. In general, circulating levels of acetate and other short chain fatty acids (SCFAs) have been inversely correlated to atherosclerosis, but these observations have been ascribed to the impact of SCFAs on immune cells rather than vascular cells31,34, again underscoring the complexity of organismal acetate metabolism.
In summary, our work reveals the surprisingly high contribution of acetate to endothelial metabolism; the key role played by acetate-derived acetyl-CoA in sustaining a powerful positive-feedback loop that drives EndMT; and endothelial ACSS2 as a potential novel target for the treatment of atherosclerosis. Taken together, our results illustrate how specific metabolic events regulate cellular signaling and drive pathophysiologic processes.
Limitations of Study
We observed that both TGFβ2 or DCA treatments had similar effect the extent of reduction of PDH phosphorylation and induction of EndMT markers in vitro. However, given that DCA is a pan-PDK inhibitor in cells, it is possible that other PDK isoforms, especially PDK3, could be also involved. This is mitigated by the observation that a PDK4 knockdown had very similar effects suggesting that this is the key regulator of EndMT. While EndMT plays a key role in maintenance of the chronic vascular inflammation and pathogenesis of several common diseases, including atherosclerosis, pulmonary hypertension and graft arteriopathy, here we studied the role of TGFβ-driven acetate derived Ac-CoA production only in an atherosclerosis model.
Whether other EndMT-dependent processes are also PDK4/Ac-CoA-dependent requires further study. We should also note that only human aortas with minimal and mild/moderate atherosclerotic intima were used for immunostaining analysis. We could not use severely atherosclerotic aortas due to extensive calcification and the loss of endothelial coverage in these specimens.
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Dr. Michael Simons (michael.simons@yale.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Bulk RNA-seq sequencing data associated with this study have been deposited Gene Expression Omnibus and the approved GEO accession numbers for these datasets are GSE224907 and GSE224939 (Data will be available on May 1, 2023). Original western blot images and analyzed data are available as Data S1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal experimental protocols used in this study were approved by the Institutional Animal Care and Use Committee of Zhejiang University (permission number: ZJU20190036) and Yale University Institutional Animal Care and Use committee (protocol 202–11231). Mice were housed in the animal facilities at Zhejiang University or Yale University and exposed to light on 12hrs cycles in a humidity- and temperature-controlled environment with no pathogenic microorganisms.
Exclusion criteria: Mice were euthanized and excluded from the defined experimental conditions if they exhibited rapid body weight reduction, loss of appetite for over 3 days, serious alopecia, weakness and hard to take food or drink water, unexpectable organ infection and death.
Generation of mice.
Cdh5-CreERT2 mice were obtained from Ralf Adams (Max Planck Institute for Molecular Biomedicine, Münster, Germany). Acss2 fl/fl mice were obtained from GemPharmatech, Nanjing, China. To generate Cdh5-CreERT2; Acss2 fl/fl mice, we mated Acss2 fl/fl mice with Cdh5-CreERT2 mice. To generate Cdh5-CreERT2; Acss2 fl/fl; Apoe−/− mice and Acss2 fl/fl; Apoe−/− mice, we mated Cdh5-CreERT2; Acss2 fl/fl mice with Apoe−/− mice (JAX Lab, B6.129P2-Apoetm1Unc/J, Stock No. 002052). Both Cdh5-CreERT2; Acss2 fl/fl; Apoe−/− mice and Acss2 fl/fl; Apoe−/− mice are C57BL/6 background. Both male mice (♂ Cdh5-CreERT2; Acss2 fl/fl; Apoe−/− mice and ♂Acss2 fl/fl; Apoe−/− mice) and female mice (♀ Cdh5-CreERT2; Acss2 fl/fl; Apoe−/− mice and ♀ Acss2 fl/fl; Apoe−/− mice) were used in atherosclerosis study, and both male and female mice showed the similar trend.
Mouse genotyping.
The genotyping was performed by PCR using mouse ear DNA. To this end, mouse ear tissues were boiling in 150ul 25mM NaOH at 95°C for 1hr, samples were vortexed for 15 sec, then neutralized by adding 150ul 40mM Tris-HCl pH 5.0. The samples were centrifuged at 10,000 g for 10 min at room temperature. Primers for genotyping used in this study can be found in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | ||||||
|---|---|---|---|---|---|---|---|---|
| Antibodies | ||||||||
| Rabbit monoclonal Anti-ACSS2 (D19C6) | Cell Signaling Technology | Cat#: 3658S; RRID:AB_2222710 | ||||||
| Rabbit polyclonal Anti-Acss2 | LSBio | Cat#: LS–C334743–100; RRID:AB_2877651 | ||||||
| Rabbit monoclonal Anti-ACLY (D1X6P) | Cell Signaling Technology | Cat#: 13390S; RRID:AB_2798203 | ||||||
| Rabbit monoclonal anti-ALK1 | Abcam | Cat#: ab108207; RRID:AB_10858289 | ||||||
| Rabbit polyclonal anti-ALK5 | Abcam | Cat#: ab31013; RRID:AB_778352 | ||||||
| Rabbit monoclonal anti-phospho-AKT1 (D9E) | Cell Signaling Technology | Cat#: 4060s; RRID:AB_2315049 | ||||||
| Rabbit monoclonal anti-AKT1 (C67E7) | Cell Signaling Technology | Cat#: 4691s; RRID:AB_915783 | ||||||
| Mouse monoclonal anti-N-CADHERIN (H-4) | Santa Cruz | Cat#: sc–271386; RRID:AB_10610922 | ||||||
| Rabbit monoclonal anti-CALPONIN | Abcam | Cat#: ab46794; RRID:AB_2291941 | ||||||
| Mouse monoclonal anti-CDH5 (F-8) | Santa Cruz | Cat#: sc–9989; RRID:AB_2077957 | ||||||
| Rabbit monoclonal anti-ENOS (D9A5L) | Cell Signaling Technology | Cat#: 32027s; RRID:AB_2728756 | ||||||
| Rabbit monoclonal anti-phospho-ERK1/2 (197G2) | Cell Signaling Technology | Cat#: 4377S; RRID:AB_331775 | ||||||
| Rabbit polyclonal anti-ERK1/2 | Cell Signaling Technology | Cat#: 9102s; RRID:AB_330744 | ||||||
| Rabbit polyclonal anti-Fibronectin 1 | Sigma-Aldrich | Cat#: F3648; RRID:AB_476976 | ||||||
| Mouse monoclonal anti-Flag-HRP | Sigma-Aldrich | Cat#: A8592; RRID:AB_439702 | ||||||
| Rabbit monoclonal anti-GAPDH (D16H11) | Cell Signaling Technology | Cat#: 5174S; RRID:AB_10622025 | ||||||
| Rabbit polyclonal anti-GLUT1 | Abcam | Cat#: ab652; RRID:AB_305540 | ||||||
| Rabbit monoclonal anti-HA antibody (C29F4) | Cell Signaling Technology | Cat#: 3724S; RRID:AB_1549585 | ||||||
| Mosue monoclonal anti-HDAC1 (10E2) | Cell Signaling Technology | Cat#: 5356S; RRID:AB_10612242 | ||||||
| Rabbit monoclonal anti-HK2 (C64G5) | Cell Signaling Technology | Cat#: 2867S; RRID:AB_2232946 | ||||||
| Rabbit monoclonal anti-Ac-Lysine-HRP | Cell Signaling Technology | Cat#: 6952S; RRID:AB_10891806 | ||||||
| Rabbit monoclonal anti-phospho-mTORC1 (S2448) (D9C2) | Cell Signaling Technology | Cat#: 5536T; RRID:AB_10691552 | ||||||
| Rabbit monoclonal anti-mTORC1 (7C10) | Cell Signaling Technology | Cat#: 2983T; RRID:AB_2105622 | ||||||
| Rabbit polyclonal anti-NOTCH3 | Cell Signaling Technology | Cat#: 2889S; RRID:AB_2298413 | ||||||
| Rabbit monoclonal anti-PAI1 (D9C4) | Cell Signaling Technology | Cat#: 11907S; RRID:AB_2797763 | ||||||
| Rabbit monoclonal anti-PDGFRβ (28E1) | Cell Signaling Technology | Cat#: 3169S; RRID:AB_2162497 | ||||||
| Rabbit polyclonal anti-phospho-PDHE1α(S232) | Sigma-Aldrich | Cat#: AP1063; RRID:AB_2934115 | ||||||
| Rabbit polyclonal anti-phospho-PDHE1α (S293) | Abcam | Cat#: ab92696; RRID:AB_10711672 | ||||||
| Rabbit polyclonal anti-phospho-PDHE1α (S300) | Sigma-Aldrich | Cat#: ABS194; RRID:AB_2934117 | ||||||
| Rabbit monoclonal anti-PDHE1α (C54G1) | Cell Signaling Technology | Cat#: 3205S; RRID:AB_2162926 | ||||||
| Rabbit polyclonal anti-PDK1 | Cell Signaling Technology | Cat#: 3062S; RRID:AB_2236832 | ||||||
| Rabbit monoclonal anti-PDK2 | Abcam | Cat#: ab68164; RRID:AB_11156499 | ||||||
| Rabbit polyclonal anti-PDK3 | Abcam | Cat#: ab154549; RRID:AB_2934113 | ||||||
| Rabbit polyclonal anti-PDK4 | Proteintech | Cat#: 12949–1–AP; RRID:AB_2161499 | ||||||
| Rabbit monoclonal anti-PDP1 (D8Y6L) | Cell Signaling Technology | Cat#: 65575S; RRID:AB_2799686 | ||||||
| Rabbit polyclonal anti-PFKFB3 | Proteintech | Cat#: 13763–1–AP; RRID:AB_2162854 | ||||||
| Rabbit monoclonal anti-phospho-P38 (D3F9) | Cell Signaling Technology | Cat#: 4511T; RRID:AB_2139682 | ||||||
| Rabbit monoclonal anti-P38 (D13E1) | Cell Signaling Technology | Cat#: 8690T; RRID:AB_10999090 | ||||||
| Rabbit polyclonal anti-SM22α | Abcam | Cat#: ab14106; RRID:AB_443021 | ||||||
| Rabbit polyclonal anti-Phospho-SMAD2 (S465/467) | Sigma-Aldrich | Cat#: AB3849–1; RRID:AB_2934118 | ||||||
| Rabbit monoclonal anti-SMAD2 (D43B4) | Cell Signaling Technology | Cat#: 5339S; RRID:AB_10626777 | ||||||
| Rabbit recombinant monoclonal anti-Phospho-SMAD3 (S423/425) | Abcam | Cat#: ab52903; RRID:AB_882596 | ||||||
| Rabbit monoclonal anti-SMAD3 | Abcam | Cat#: ab40854; RRID:AB_777979 | ||||||
| Rabbit polyclonal anti-Phospho-SMAD4 (T277) | ABCEPTA | Cat#: AP3251a; RRID:AB_2193620 | ||||||
| Rabbit monoclonal anti-SMAD4 (D3R4N) | Cell Signaling Technology | Cat#: 46535S; RRID:AB_2736998 | ||||||
| Rabbit monoclonal anti-SMAD2/3 (D7G7) | Cell Signaling Technology | Cat#: 8685S; RRID:AB_10889933 | ||||||
| Rabbit monoclonal anti-SMAD2/3-Biotinylated (D7G7) | Cell Signaling Technology | Cat#: 12470S; RRID:AB_2797930 | ||||||
| anti-phospho-SMAD1/5 (S463/465) (41D10) | Cell Signaling Technology | Cat#: 9516S; RRID:AB_491015 | ||||||
| Rabbit monoclonal anti-SMAD5 (D4G2) | Cell Signaling Technology | Cat#: 12534S; RRID:AB_2797946 | ||||||
| Mouse monoclonal anti-SMAD7 (Clone 293039) | R&D Systems | Cat#: MAB2029; RRID:AB_2193479 | ||||||
| Rabbit monoclonal anti-SNAIL1 (C15D3) | Cell Signaling Technology | Cat#: 3879S; RRID:AB_2255011 | ||||||
| Streptavidin-HRP | Cell Signaling Technology | Cat#: 3999S; RRID:AB_10830897 | ||||||
| Rabbit monoclonal anti-VEGFR2 (55B11) | Cell Signaling Technology | Cat#: 2479S; RRID:AB_2212507 | ||||||
| Rabbit monoclonal anti-VINCULIN (E1E9V) | Cell Signaling Technology | Cat#: 13901S; RRID:AB_2728768 | ||||||
| Rabbit monoclonal anti-V5 tag (D3H8Q) | Cell Signaling Technology | Cat#: 13202S; RRID:AB_2687461 | ||||||
| Goat polyclonal anti-Cd31 | R&D Systems | Cat#: AF3628; RRID:AB_2161028 | ||||||
| Rabbit monoclonal anti-CD68 (E3O7V) | Cell Signaling Technology | Cat#: 97778S; RRID:AB_2928056 | ||||||
| Rabbit polyclonal anti-Collagen 1 | Novus Biologicals | Cat#: NB600–408; RRID:AB_10000511 | ||||||
| Rat monoclonal anti-Vcam 1 | BD Biosciences | Cat#: 553330; RRID:AB_394786 | ||||||
| Bacterial and virus strains | ||||||||
| Adenovirus Adv-ALK5 WT-V5 tag | Lab made | CDS plasmid from addgene ID: #80876 | ||||||
| Adenovirus Adv-ALK5 CA-V5 tag | Lab made | CDS plasmid from addgene ID : #80877 | ||||||
| Adenovirus Adv-PDK4 | Applied Biological Materials | Cat#: 363230510100 | ||||||
| Adenovirus Adv-ACSS2-HA tag | Applied Biological Materials | Cat#: 112270510200 | ||||||
| lentiCRISPRv2-ACLY 2# | Lab made | LentiCRISPRv2 backbone: addgene ID: 52961 | ||||||
| lentiCRISPRv2-ACLY 57# | Lab made | LentiCRISPRv2 backbone: addgene ID: 52961 | ||||||
| lentiCRISPRv2-ACSS2 1# | Lab made | LentiCRISPRv2 backbone: addgene ID: 52961 | ||||||
| lentiCRISPRv2-ACSS2 3# | Lab made | LentiCRISPRv2 backbone: addgene ID: 52961 | ||||||
| Biological samples | ||||||||
| Ascending aortas of normal organ donors with either minimal or mild/moderate extent of atherosclerosis | This paper | |||||||
| Chemicals, peptides, and recombinant proteins | ||||||||
| Tamoxifen | Sigma-Aldrich | Cat.#: T5648 | ||||||
| Cycloheximide | Cell Signaling Technology | Cat.#: 2112s | ||||||
| DAPI | Cell Signaling Technology | Cat.#: 4083 | ||||||
| Sodium dichloroacetate | Sigma-Aldrich | Cat.#: 347795 | ||||||
| Trichostatin A | Sigma-Aldrich | Cat.#: T8552 | ||||||
| MG132 | Sigma-Aldrich | Cat.#: 474790 | ||||||
| Chloroquine phosphate | Sigma-Aldrich | Cat.#: PHR1258 | ||||||
| Nicotinamide | Sigma-Aldrich | Cat.#: N0636 | ||||||
| Oil Red O Solution | Sigma-Aldrich | Cat.#: O1391 | ||||||
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat.#: 11836170001 | ||||||
| Glucose, D-[5–3H(N)] | Perkin Elmer | Cat.#: NET531001MC | ||||||
| D-GLUCOSE (U-13C6, 99%) | Cambridge Isotope Laboratories | Cat #: CLM–1396–PK | ||||||
| SODIUM ACETATE (1,2–13C2, 99%; D3, 98%) | Cambridge Isotope Laboratories | Cat #: CDLM–3457–PK | ||||||
| L-GLUTAMINE (13C5, 99%) | Cambridge Isotope Laboratories | Cat #: CLM–1822–H–0.1MG | ||||||
| SODIUM PALMITATE (U-13C16, 98%+) | Cambridge Isotope Laboratories | Cat #: CLM–6059–PK | ||||||
| Critical commercial assays | ||||||||
| Click-iT® Plus EdU Imaging Kit | Thermo Fisher Scientific | Cat #: C10639 | ||||||
| agarose beads conjugated with anti-Acetylated lysine antibody | Cytoskeleton | Cat #: AAC04–Beads | ||||||
| Nuclear/Cytosol Fractionation Kit | BioVision | Cat #: K266 | ||||||
| Deproteinizing Sample Preparation Kit | BioVision | Cat #: K808 | ||||||
| PicoProbe™Acetyl-CoA Fluorometric Assay Kit | BioVision | Cat #: K317 | ||||||
| PicoProbe™Acetyl-CoA Fluorometric Assay Kit | Sigma-Aldrich | Cat #: MAK039 | ||||||
| Glucose uptake colorimetric assay kit | BioVision | Cat #: K676–100 | ||||||
| SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads) | Cell Signaling Technology | Cat #: 9003S | ||||||
| iScript cDNA synthesis lit | Bio-RAD | Cat #: 1708890 | ||||||
| iQ™ SYBR® Green Supermix | Bio-RAD | Cat #: 1708880 | ||||||
| RNeasy Plus Mini Kit | Qiagen | Cat #: 74134 | ||||||
| Lactate Colorimetric Assay Kit | BioVision | Cat #: K627–100 | ||||||
| Deposited data | ||||||||
| RNA-seq raw data for HUAECs: Control VS TGFβ2 | This paper | GSE224907 | ||||||
| RNA-seq raw data for HUAECs: Scra. siRNA VS TGFβ2 + Scra. SiRNA VS TGFβ2 + ACSS2 SiRNA | This paper | GSE224939 | ||||||
| Experimental models: Cell lines | ||||||||
| Human Umbilical Artery Endothelial Cells (HUAEC) | Promocell | Cat #: C–12202 | ||||||
| Human Aortic Endothelial Cells (HAEC) | Lonza | Cat #: CC–2535 | ||||||
| Human Umbilical Venous Endothelial Cells (HUVEC) | Yale VBT Program | Cat #: T75SC–1 | ||||||
| Human: HEK 293T/17 | ATCC | ATCC Cat# CRL–11268, RRID:CVCL_1926 | ||||||
| Experimental models: Organisms/strains | ||||||||
| Mouse: B6.129P2-Apoetm1Unc/J | The Jackson Laboratory | JAX: 002052 | ||||||
| Mouse: Apoe−/−; Cdh5 CreERT2; Acss2 flox/flox | This paper | N/A | ||||||
| Mouse: Cdh5 CreERT2; | Wang, Y. et al. 2010; Mara E P., et al. 2010. | N/A | ||||||
| Oligonucleotides | ||||||||
| siRNAs and qRT-PCR primers | This paper | Table S1 | ||||||
| ACLY sg2 oligo1: 5’ - CACCGTGGTGCTGTCATAGGCAG AG - 3’ | This paper | N/A | ||||||
| ACLY sg2 oligo2: 5’ - AAACCTCTGCCTATGACAGCACCA C - 3’ | This paper | N/A | ||||||
| ACLY sg57 oligo1: 5’ - CACCGCGTGGAAAACTTGGTCTC GT - 3’ | This paper | N/A | ||||||
| ACLY sg57 oligo2: 5’ - AAACACGAGACCAAGTTTTCCACG C - 3’ | This paper | N/A | ||||||
| ACSS2 sg1 oligo1: 5’ - CACCGCGAGCTGCACCGGCGCTCCG - 3’ | This paper | N/A | ||||||
| ACSS2 sg1 oligo2: 5’ - AAACCGGAGCGCCGGTGCAGCTCGC - 3’ | This paper | N/A | ||||||
| ACSS2 sg3 oligo1: 5’ - CACCGGCTGGCATGTGCCCGCA TTG - 3’ | This paper | N/A | ||||||
| ACSS2 sg3 oligo2: 5’ - AAACCAATGCGGGCACATGCC AGCC - 3’ | This paper | N/A | ||||||
| Mouse Acss2 primers for genotyping: Acss2–3wt-tF1: ATTCAAAGCTTAGAAGCCTTGGTA GG | This paper | N/A | ||||||
| Mouse Acss2 primers for genotyping: Acss2–3wt-tR1: GGAAGCAGAATGAGCTGTTAGTG AAAC | This paper | N/A | ||||||
| Mouse ApoE primers for genotyping: oIMR0180: GCC TAG CCG AGG GAG AGC CG; | The Jackson Laboratory; Pei-yu Chen, et al. 2019. | N/A | ||||||
| Mouse ApoE primers for genotyping: oIMR0181: TGT GAC TTG GGA GCT CTG CAG C; | The Jackson Laboratory; Pei-yu Chen, et al. 2019. | N/A | ||||||
| Mouse ApoE primers for genotyping: oIMR0182 mCdh5 F-IN1 GCC GCC CCG ACT GCA TCT | The Jackson Laboratory; Pei-yu Chen, et al. 2019. | N/A | ||||||
| Mouse Cdh5-Cre primers for genotyping: mCdh5 F-IN1: cagatcagctcctccacgaa | Wang, Y. et al. 2010; Mara E P., et al. 2010. Pei-yu Chen, et al. 2019. | N/A | ||||||
| Mouse Cdh5-Cre primers for genotyping: mCdh5 R-EX2: tggtgggcaggtagcatgtt | Wang, Y. et al. 2010; Mara E P., et al. 2010. Pei-yu Chen, et al. 2019. | N/A | ||||||
| Mouse Cdh5-Cre primers for genotyping: CRE-Rev: CATTGCTGTCACTTGGTCGT | Wang, Y. et al. 2010; Mara E P., et al. 2010. Pei-yu Chen, et al. 2019. | N/A | ||||||
| Recombinant DNA | ||||||||
| Human ALK5 wild type | Addgene | addgene ID: #80876 | ||||||
| Human constitutively active ALK5 mutant | Addgene | addgene ID: #80877 | ||||||
| Human ALK5 K-R mutants | This paper | N/A | ||||||
| Software and algorithms | ||||||||
| ImageJ | NIH | https://imagej.nih.gov/ij/ | ||||||
| GraphPad Prism 9 | GraphPad Software | https://www.graphpad.com | ||||||
| Other | ||||||||
| Table S1 | This paper | siRNAs in this study and qRT-PCR primers | ||||||
| Table S2 | This paper | information of normal donors with different extent of atherosclerosis | ||||||
ACSS2 siRNA therapy.
Thirty two-months old ApoE−/− mice were placed on a high fat diet (HFD) and randomized to two different siRNA treatment groups (n=9 each) and or no intervention (n=12). ACSS2 siRNAs, stabilized with two different chemical modifications, were loaded into 7C1 nanoparticles and injected intravenously every 10 days for 12 weeks (total 8 injections per mouse in each group). After 12 weeks of HFD, the animals were euthanized and perfusion-fixed with 4% paraformaldehyde (16% PFA from Polysciences, Inc.) via the left ventricle for 5 min. To measure the effect on atherosclerotic burden, the whole aorta was dissected and fixed in 4%PFA and gently cleaned of adventitia tissues. The aortas were then carefully opened with scissors, flat-mounted and stained with Oil-Red-O (Sigma no. O0625). The extent of atherosclerotic burden was then quantified using ImageJ software.
In vivo ACSS2 knockdown.
Mouse Acss2 siRNA was synthesized by AXOLABS, and then modified to increase in vivo stability. Sequences of original and modified mouse Acss2 siRNA are:
mouse Acss2 siRNA - original
Sense strand: 5`-CAGCAAUGUUCUCCGUAAAdTdT-3`
Antisense strand: 5`-UUUACGGAGAACAUUGCUGdGdT-3`
mouse Acss2 siRNA - modified
Sense strand: 5’-caGcaaUGUucUccguAasa-3’
Antisense strand: 5’-dTUUACGGAGAACaUUGCUGusu-3’
N: RNA residues; n: 2-O-Methyl residues; s: phosphorothioate backbone modification; dN: DNA residues Both modified and unmodified ACSS2 siRNAs were packaged into 7C1 nanoparticles by VasoRx, Inc. and used as previously described7. Two ACSS2 siRNA with different chemical modifications were encapsulated in LNPs formulated with the lipid 7C1. Briefly, lipid 7C1 was synthesized, purified and combined with C14PEG2000 in a glass syringe (Hamilton Company), then diluted with 100% ethanol. Two ACSS2 siRNA were diluted in 10 mM citrate buffer separately and loaded into a separate syringe. The two syringes containing ACSS2 siRNA different were connected to a microfluidic mixing device, before the 7C1 and RNA solutions were mixed together at a flow rate of 300 and 900 μl min−1, respectively. The resulting nanoparticles were dialysed into 1X PBS, before being sterile filtered using a 0.22 μm filter. The amount of 7C1 used in in vivo formulations was kept constant.
Animal treatment.
Cre-Lox recombination was induced by tamoxifen (Sigma T5648) at 1.5 mg/day i.p. for 8 days versus vehicle (corn oil, Sigma C8267) alone. For high cholesterol high fat diet treatment, 8 week old mice were placed on a Western diet (40 kcal% Fat, 1.25% Cholesterol, 0% Cholic Acid) for 3 months (Research Diets, product #D12108).
METHOD DETAILS
Cell culture and treatment of TGFβ-driven EndMT
Primary Human Aortic Endothelial Cells (HAECs) and Human Umbilical Artery Endothelial Cells (HUAECs) were purchased from Promocell and Lonza respectively, and Primary Human Umbilical Vein Endothelial Cells (HUVECs) were supplied by primary cells facility of Yale Vascular Biology & Therapeutics Program. All the primary endothelial cells were maintained in EGM-2 complete growth media with 5% FBS and cultured in a humidified 37 °C incubator with 5% CO2. EndMT was induced in vitro by treatment HAECs or HUAECs with TGFβ2 at 10 ng/ml in the complete EGM-2 growth media (EndMT induction media) for 7 days with daily media changes. The cells were maintained at 100% confluence and were sub-cultured using 0.05% trypsin-EDTA (Gibco 25300120) for dissociation.
Total RNA isolation, Bulk RNA-SEQ analysis and qPCR analysis
In brief, the quality of total RNA was analyzed and cDNA libraries were then constructed. Novaseq sequencing with 25 million reads per sample was performed to detect the whole transcriptome. RNA-seq data was aligned to the human reference genome (human genome build 38GRCh38) using the short reads aligner STAR. Quantification of gene expression was performed using RSEM with GENCODE annotation (human release 27, http://www.gencodegenes.org). Read counts were normalized using the trimmed mean of M-values (TMM) method and differentially expressed genes were identified (edgeR R package). Differentially expressed genes with a false discovery corrected P ≤ 0.05 were used for further analysis (heatmaps and functional enrichment). We have uploaded the Bulk RNA-seq datasets into NCBI database, and the approved GEO accession numbers for these datasets are GSE224907 and GSE224939 (Data will be available on May 1, 2023). To confirm the different expression of genes involved in EndMT and Glucose/Acetyl-CoA metabolism, first-strand cDNA was synthesized using iScript cDNA synthesis lit (Bio-RAD, Cat#: 1708890), and gene’s expression was analyzed using iQ™ SYBR® Green Supermix (Bio-RAD, Cat#: 1708880). Primers are listed in Supplemental Table 1.
Plasmids and adenoviral constructs
To generate the adenovirus, AdEasy™ Adenoviral Vector System (Agilent Technologies) was used. Expression constructs for ALK5WT (addgene ID: #80876) and constitutively active ALK5T204D (addgene ID: #80877) were obtained from Addgene and they were inserted into pShuttle-CMV vector with C-terminal V5 tag. After DNA purification, those vectors were digested and linearized by Pme-I enzyme and electronically transformed into BJ5183 competent cells with pAd-Easy vector for homologous recombination. After colony screening, the final adenoviral vectors were linearized with Pac-I and transfected into HEK293A cells for viral packaging. Virus was titered using an Adeno-X Rapid Titer Kit (Clontech Laboratories). The constructs were verified by sequencing analysis. Protein expression of ALK5-WT-V5 and ALK5-CA-V5 was confirmed by immunoblot analysis and infection efficiency was tested using immunostaining. Adv-PDK4 for overexpressing PDK4 was ordered from Applied Biological Materials (abm). And the adenoviral expression of PDK4 was confirmed by immunoblot analysis.
Chromatin immunoprecipitation and qPCR tests
Chromatin immunoprecipitation (CHIP) was performed using SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads) (Cell signaling technology, #9003S). Briefly, cells were fixed with 1% formaldehyde (Polysciences Cat #: 18814–10; methanol free) at room temperature for 10min and neutralized with 100mM Glycine. Cells were collected and nuclei were isolated, the genomic DNA was digested with Micrococcal nuclease at 37°C for 20 min. The digested DNA was completely lysed using sonication after 2 sets of 30-seconds pulses with 30% power using Sonic Dismembrator Model 500. After immunoprecipitation with Anti-SMAD2/3 antibody (Cell Signaling Technology, 8685s) or Rabbit IgG (Cell Signaling Technology, 2729s), ChIP-grade magnetic beads were used to pull down the SMAD2/3-DNA complexes. DNA was amplified by qPCR with primers to Human PDK4 promotor. Primers were designed to SMADs binding element–containing core promoter regions for Human PDK4. All samples were performed in triplicate, and data were normalized to percentage of input. Primers are listed in Supplemental Table 1.
Liquid chromatography-mass spectrometry (LC-MS) metabolomics analysis
HUAECs were incubated with the complete EGM-2 media (Control group) or EndMT-induction media (TGFβ2 group) for 7 days. Switch to DMEM media (No Glucose/no pyruvate, Gibco, Cat#: 11966025) with growth factors and 5% dialyzed serum, and incubate cells for 1 day at 37 °C in a CO2 incubator. Then switch to DMEM media with 1% dialyzed serum to starve cells for 12 hrs. Aspirate of all media, and replace with new, respective DMEM media containing growth factors, 5% dialyzed serum and isotope tracers (10 mM U-13C6-Glucose, Cambridge Isotope, Cat#: CLM-1396–1). Then incubate cells for 24 hrs in a CO2 incubator. For TGFβ2 group, TGFβ2 was contained in all medias.
Metabolites were extracted from 1.5*10^6 cells per sample. 3 ml of dry ice-cold 80% MeOH (extraction solvent) were added to each plate, and incubated plates at −80°C for 15min. Cells were scraped and the entire content of the well were transferred to 1.5ml Eppendorf tubes on dry ice. The plates were rinsed with 1ml of dry ice-cold 80% MeOH and the remaining cells were added to another 1.5 ml Eppendorf tubes. Samples were vortexed for 10 seconds, and centrifuged at 16,000 x g for 15 min at 4°C. The supernatant extract from one sample was mixed in one 15ml conical tube on dry ice, and extract was dried down with nitrogen blow to concentrate metabolites 10 times. The samples were centrifuged at 16,000 x g for 10 min at 4°C and 40 μl of supernatant was used for LC-MS run.
Cell extracts were loaded to individual vials and analyzed by quadrupole-orbitrap mass spectrometer (Q-Exactive Plus Hybrid Quadrupole-Orbitrap, Thermo Fisher) coupled to hydrophilic interaction chromatography (HILIC) via electrospray ionization. LC separation was on an Xbridge BEH amide column (2.1 mm x 150 mm, 2.5 µm particle size, 130 Å pore size; Waters) at 25°C using a gradient of solvent A (5% acetonitrile in water with 20 mM ammonium acetate and 20 mM ammonium hydroxide) and solvent B (100% acetonitrile). Flow rate was 150 µL/min. The LC gradient was: 0 min, 90% B; 2 min, 90% B; 3 min, 75% B; 7 min, 75% B; 8 min, 70% B; 9 min, 70% B; 10 min, 50% B; 12 min, 50% B; 13 min, 25% B; 14 min, 20% B; 15 min, 20% B; 16 min, 0% B; 20.5 min, 0% B; 21 min, 90% B; 25 min, 90% B. Autosampler temperature was set at 4°C and the injection volume of the sample was 3 μL. MS data were acquired in negative ion mode with a full-scan mode from m/z 70 to 830 and 140,000 resolution. Peaks were identified with MAVEN software and Microsoft Excel was used for data interpretation.
Oxygen consumption rate and extracellular acidification rate analysis using seahorse analyzer
To measure the changes of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) in HUAECs during EndMT, OCR and ECAR analysis were performed with a Seahorse XF96 analyzer according to the manufacturer’s recommendations (Agilent Technologies). In brief, a day before the experiment, a 96-well sensor cartilage was hydrated with Agilent Seahorse XF Calibrant solution and incubated at 37 °C without CO2. Cells were seeded at 30,000 cells per well in a fibronectin (Sigma-Aldrich) coated Seahorse 96-well plate (Agilent Technologies) and incubated overnight at 37 °C in a CO2 incubator. On the day of the experiment, the Seahorse XF96 analyzer was calibrated using the hydrated cartilage and cells were maintained in FBS-free Agilent Seahorse XF DMEM Media (pH7.4) in a non-CO2 incubator for 1hr before the assay. After measurement of OCR in basal level, Oligomycin (3 μM), FCCP (1 μM) and AA/R (1.5 μM / 3 μM) were sequentially added to the cells and the OCR was monitored over time according to the manufacturer’s recommendation. For ECAR measurements, oligomycin (3 μM) and 2-DG (100 mM) were sequentially added to the cells and the ECAR was monitored over time according to the manufacturer’s protocols.
2-Deoxyglucose (DG) uptake, Lactate, Acetate, and Acetyl-CoA analysis
2-DG uptake assay was analyzed using Glucose uptake colorimetric assay kit (BioVision; Catalog #: K676–100). As with glucose, 2-DG can be taken up by glucose transporters and metabolized to 2-DG-6-phosphate (2-DG6P). However, 2-DG6P cannot be further metabolized, and thus accumulates in the cells. The accumulated 2-DG6P is directly proportional to 2-DG (or glucose) uptake by cells. During this assay, 2-DG6P is oxidized to generate NADPH, which is determined by an enzymatic recycling amplification reaction. For the sample preparation in this study, briefly, HUAECs were maintained in complete EGM-2 media or EndMT induction media for 5 days with daily media changes. Cells were seeded at 30,000 cells per well in a fibronectin coated 96-well plate (Corning) and incubated 12 hr. at 37 °C in a CO2 incubator. Cells were washed with PBS twice with PBS and starved in 100ul serum free basic EBM-2 media with or without TGFβ2 overnight (to increase glucose uptake), then rewashed three times with PBS. The cells were starved for glucose by preincubating with 100 μl Krebs-Ringer-Phosphate-HEPES (KRPH) buffer containing 2 % BSA for 40min, then 10 μl of 10 mM 2-DG was added and the cells incubated for 20 min. Subsequently, proceeded to the reaction and measurement steps following instruction manual.
Lactate or acetate level in the culture media
HUAECs were incubated with the complete EGM-2 media (Control group) or EndMT induction media (TGFβ2 group) for 7 days. To detect the secreted lactate or acetate in media, HUAECs were incubated with the complete EGM-2 media (Control group) or EndMT induction media (TGFβ2 group) containing 5% dialyzed FBS. Medias from different conditions was collected at indicated time points, and cell numbers for each plate were counted. The lactate or acetate level in collected samples were measured using Lactate Colorimetric Assay Kit (BioVision, Catalog #: K627–100) or Acetate Colorimetric Assay Kit (MilliporSigma, Catalog #: MAK086) according to the manufacturer’s instruction and adjusted for cell counts.
Sample preparation and Acetyl-CoA measurement
Cells were dissociated with 0.05% trypsin-EDTA (Gibco 25300120) and collected using a centrifuge. Cell pellets were resuspended in EGM-2 media and cell number were counted for sample preparation. Cytosol were extracted from 1.5–2*10^6 cells per sample using a Nuclear/Cytosol Fractionation Kit (BioVision, Cat. #: K266) according to manufacturer’s instruction. And cytosolic extraction was subsequently deproteinized using Deproteinizing Sample Preparation Kit (BioVision, Cat. #: K808). Proteins in cytosolic extraction were precipitated with perchloric acid (PCA), then PCA was neutralized using neutralization solution and pH-indicator strips (MilliporSigma, Catalog #: 1.09533.0001). The neutralized cytosolic extraction (6.0<pH<7.5) was used for the analysis of Acetyl-CoA based on enzymatic reactions using PicoProbe™Acetyl-CoA Fluorometric Assay Kit (BioVision, Cat. #: K317) or Acetyl-Coenzyme A Assay Kit (MilliporSigma, Catalog #: MAK039).
Radioactive measurement for Glycolic capacity
Radiolabeled glucose was used to measure glycolysis in 12-well dishes. Briefly, cells were washed twice with PBS prior to a 1-hour incubation with 5 mCi of glucose, D-[5–3H(N)] (Perkin Elmer) in KREBS-Ringer HEPES buffer supplemented with 5 mM cold glucose. Following incubation, the entirety of reaction mixture (500 uL) was transferred to 5 mL cell culture tube that contained a secondary trap tube (0.25 mL microdialysis tube) pre-filled with 200 uL of water. The culture tube was sealed with a rubber gasket and 100 uL of 1M of perchloric acid was carefully injected through the rubber gasket into the reaction mixture. The culture tubes were then placed upright in a shaking water bath at 37°C for 72 hours. Following incubation, the entirety of the water in the trap tube was transferred to a scintillation vial. 4 mL of liquid scintillation fluid was added to the vials and counted on a liquid scintillation counter (Perkin Elmer) for 3 minutes. The reaction mixture was also counted to derive specific activity (DPM of input/ total nanomoles of glucose). The conversion of glucose, D-[5–3H(N)] to 3H2O over the reaction time was then determined. Finally, the data was normalized by cell number per well and expressed as nmol/hr/1 million cells.
Acetylation test for ALK5/RSMADS proteins
Following HUAECs treatment with TGFβ or PDK4 siRNA, cells were treated with 5 uM Trichostatin A (TSA) for 5 hours and lysed in RIPA buffer containing protease inhibitors and 5 uM TSA, and cell lysate were precleared with agarose beads conjugated with protein G, and precleared cell lysate were incubated with agarose beads conjugated with anti-Acetylated lysine antibody (Cytoskeleton, Cat.#: AAC04-Beads) to capture acetylated proteins at 4 °C with gentle rotation. The beads were collected by gentle spin for 2min at 3, 000rpm using a cold tabletop centrifuge, and beads compound with acetylated proteins were washed seven times with ice cold RIPA buffer to remove nonspecifically bound proteins. The bound acetylated proteins were eluted by 2*Laemmli Sample buffer at room temperature according to manufacturer’s instruction, the eluted proteins in 2*Laemmli Sample were transferred into a sterile 1.5 ml EP tube containing 2 μl β-mercaptoethanol. After heating for 5 min at 100°C, samples were placed in ice and cooled for use.
Characterization of acetylation sites of the human ALK5 protein.
Gel pieces were de-stained in 50 mM NH4HCO3 in 50% acetonitrile (v/v) until clear and then dehydrated with 100 μl of 100% acetonitrile for 5 min followed by the liquid removed and the gel pieces rehydration in 10 mM dithiothreitol, at 56 °C for 60 min. Gel pieces were again dehydrated in 100% acetonitrile, liquid was removed. and gel pieces were rehydrated with 55 mM iodoacetamide. Samples were incubated at room temperature, in the dark, for 45 min. Gel pieces were washed with 50 mM NH4HCO3 and dehydrated with 100% acetonitrile. Gel pieces were rehydrated with 10 ng/μl trypsin resuspended in 50 mM NH4HCO3 on ice for 1 h. Excess liquid was removed and gel pieces were digested with trypsin at 37 °C overnight. Peptides were extracted with 50% acetonitrile/5% formic acid, followed by 100% acetonitrile. Peptides were dried to completion and resuspended in 2% acetonitrile/0.1% formic acid.
LC-MS/MS Analysis.
The tryptic peptides were dissolved in 0.1% formic acid (solvent A), directly loaded onto a home-made reversed-phase analytical column. The gradient was comprised of an increase from 6% to 35% of solvent B (0.1% formic acid in 98% acetonitrile) over 22 min, 35% to 80% in 4 min then holding at 80% for the last 4 min, all at a constant flow rate of 450 nl/min on an EASY-nLC 1000 UPLC system. The peptides were subjected to NSI source followed by tandem mass spectrometry (MS/MS) in Q ExactiveTM Plus (Thermo) coupled online to the UPLC. The applied electrospray voltage was 2.1 kV. The m/z scan range was 350 to 1800 for a full scan, and intact peptides were detected in the Orbitrap at a resolution of 70,000. Peptides were then selected for MS/MS using NCE setting as 28 and the fragments were detected in the Orbitrap at a resolution of 17,500. A data-dependent procedure that alternated between one MS scan followed by 20 MS/MS scans with 15.0s dynamic exclusion. Automatic gain control (AGC) was set at 5E4. The resulting MS/MS data were processed using Proteome Discoverer 2.1. Trypsin/P was specified as cleavage enzyme allowing up to 4 missing cleavages. Mass error was set to 10 ppm for precursor ions and 0.02 Da for fragments ions. Peptide confidence was set at high, and peptide ion score was set > 20.
EdU staining for Cell Proliferation
Endothelial cells were plated on 4-chamber culture slides (Thermo Scientific) at ~70% confluence. A 2X working solution of EdU in the complete EGM-2 medium was prepared from the 20 mM EdU stock solution. The 2X EdU solution was pre-warmed and then 500 μl of the 2X EdU solution was added to slide chambers. 3hr. later the cells were collected and immediately fixed and permeabilized. EdU incorporation was detected using manufacture’s Click-iT® Plus EdU Imaging Kits and quantified microscopically.
Acetate Uptake/Release and Stable Isotope Tracing
Endothelial colony-forming cells (ECFCs) were freshly isolated as described previously from primary human cord blood 35,36. Briefly, cord blood was drawn from the human umbilical vein. Mononuclear cells were separated by using Ficoll-Paque solution. Carefully collect the Mononuclear cell layer and transfer it to a 50-ml conical tube on ice, and Mononuclear cells were washed twice with isolation buffer (12 mM D-glucose, 8.5 mM sodium citrate, 4 mM citric acid and 0.5% BSA in PBS). Mononuclear cells were collected and transferred to a 10-cm dish coated with 1% gelatin. When cells grew up to 80% confluence, the CD31-positive cells were purified by using Dynabead-conjugated with anti-CD31 antibody. The purified cells were cultured on a 10-cm dish coated with 0.1% gelatin. ECFCs were maintained in endothelial cell basal media (EBM2, Lonza CC-3156) supplemented with endothelial growth supplement (EGM2; Lonza CC-4176), 10% fetal bovine serum (Hyclone #SH30071.03) and 1% penicillin/streptomycin antibody solution (Gibco #15140122). ECFCs were used between passage 4 and 8. For acetate uptake/release and stable isotope tracing experiments, ECFCs were seeded in six-well plates with 2mL of culture media replenished every 2–3 days until confluence. ECFCs were maintained in contact-inhibited, confluent state for at least three days prior to beginning experiment. Tracing media was made in glucose- and glutamine-free DMEM (Gibco #A1443001) and 5mM glucose, 500uM glutamine, 100uM acetate, 100uM palmitate and 10% dialyzed FBS (dFBS, Gemini #100–108) were added. For relative substrate contribution experiments, only one substrate was isotopically labeled at one time, with the rest unlabeled. On the day of the experiment, cells were pre-treated with tracing media containing all unlabeled substrates for two hours prior to switching to tracing media with one isotope tracer. 13C6-glucose (Cambridge Isotope #CLM-1396), 13C5-glutamine (Cambridge Isotope #CLM-1822), 13C2 sodium acetate (Cambridge Isotope #CLM-440) were used. To calculate relative contribution of palmitate to intracellular acyl-CoAs, U-13C sodium palmitate (Cambridge Isotope #CLM-6059) was conjugated to fatty acid free bovine serum albumin (Sigma #A7030) at a concentration of 4mM palmitate to 1mM albumin, as described previously 37,38.
Lentiviral Generation and Transduction
ACLY and ACSS2 KO ECFCs were generated by CRISPR/Cas9. Target gRNAs were inserted into lentiCRISPRv2 (Addgene #52961) backbone using the “Target Guide Sequence Cloning Protocol” 39,40. Lentivirus was generated by transfecting HEK 293T/17 cells (CRL-11268) with the lentiCRISPRv2 plasmid containing gRNA sequence, pMD2.G transfer vector (Addgene #12259) and psPAX2 (AddGene #12260) using Fugene HD (Promega #E2311) according to manufacturer’s instruction. Lentivirus-containing media was collected 48 and 72 hours after transfection and filtered through a 0.45um low protein-binding filter. Confluent ECFCs were transfected with lentivirus by spinfection: polybrene (final conc. 8ug/mL; Sigma #TR-1003) was added to virus-containing media in six well plates, and the plates were then spun at 800 x g for 40 minutes. No-infection control with polybrene and virus-free media was included in each plate. Viral media was then removed, and fresh media was added. Antibiotic selection was started 24 hours later and maintained until all cells in the no-infection control were dead. Experiments were initiated 7 days after infection. ACLY sgRNA was cloned in puromycin resistant backbone, ACSS2 in hygromycin resistant backbone. Oligonucleotide sequences for lentivirus generation can be found in the Key Resources Table.
Extracellular Acetate Measurement by Gas-Chromatography Mass Spectrometry
To harvest media for extracellular acetate measurement, 1mL of media was collected, spun down at 2500 x g for 5 minutes and then frozen at −80C until further processing. For time 0 a no-cell control aliquot of media was used. A standard curve was made by spiking in known concentrations of acetate into no acetate media (i.e. DMEM + glucose + glutamine + 10% dFBS) for each experiment in order to calculate absolute concentrations. Acetate uptake or release was normalized to packed cell volume (measured in uL) by using packed cell volume tubes (TPP #87005) or to cell number.
Acetate quantification in cell culture media was done by adapting a method previously described 41. 200uL of media was added to a 2mL tube followed by 40uL of 1mM 13C2; D3 sodium acetate (Cambridge Isotope #CDLM-3457) as internal standard. 50uL of 1-propanol and 50uL of pyridine were added, and samples were incubated on ice for 5 minutes. 100uL of 1M sodium hydroxide was then added and immediately followed by 30uL methyl chloroformate. Samples were vigorously vortexed for 20 seconds, pausing to vent halfway through. Finally, 300uL of methyl tert-butyl ether was added, samples were vortexed for 20 seconds without venting and centrifuged at 10,000 x g for 5 minutes at 4C. 100–200uL of the upper layer was then transferred to gas-chromatography vials for analysis.
An agilent 7890B gas chromatograph (GC) coupled with an Agilent 5977B mass selective detector (MSD) (Agilent Technologies) was used for analysis. The GC column was a 30 m x 250 μm x 0.25 μm HP-5ms Ultra Inert column (Agilent 19091S-433UI). 2uL of sample was injected at 25mL/min split flow, with inlet temperature at 280C. The oven was held at 45C for 0.8 minutes, then ramped from 45C to 60C at 25C/min and held for 0 minutes and ramped from 60C to 190C at 50C/min and held for 0 min. The mass spectrometer was operated in SIM mode for m/z 61 (12C-acetate), 63 (13C-acetate) and 66 (13C2; D3-acetate) with a dwell time of 50ms for each ion. Agilent MassHunter Qualitative Analysis software (B.07.00) was used for visualization of chromatograms and Agilent MassHunter Quantitative Analysis software was used for peak area integration. Peak area of m/z 61 and 63 for each sample was divided by m/z 66 to normalize to the internal standard. Absolute quantity of acetate in the media was calculated from a standard curve of known acetate concentrations spiked into acetate-free media. Standard curve samples also included 13C2; D3-acetate internal standard. In each experiment, background 12C-acetate was subtracted out of all samples by using media blank, i.e. the 0uM standard.
Short-Chain Acyl CoA Extraction and Liquid-Chromatography Mass Spectrometry
Isotope tracing of acyl-CoAs was measured by liquid chromatography-high resolution mass spectrometry as previously described in detail 42. Briefly, media was aspirated, and cells were quenched in 1 mL 10% (w/v) trichloroacetic acid (Sigma #T6399) in water, scraped, and extracted. Samples were homogenized by using a probe tip sonicator in 0.5 second pulses 30 times then centrifuged at 17,000 x g for 10 min at 4°C. Samples were purified by solid phase extraction (SPE) cartridges (Oasis HLB 10 mg, Waters) that were conditioned with 1 mL of methanol and 1 mL of water. Acid-extracted supernatants were loaded onto the cartridges and washed with 1 mL of water. Acyl-CoAs were eluted with 1 mL of 25 mM ammonium acetate in methanol and evaporated to dryness under nitrogen. Samples were resuspended in 50 µL of 5% (w/v) 5-Sulfosalicyilic acid and 10 µL injections were analyzed on an Ultimate 3000 UHPLC using a Waters HSS T3 2.1×100mm 3.5 µm column coupled to a Q Exactive Plus. The analysts were blinded to sample identity during processing and quantification. Data was analyzed using Tracefinder 5.1 (Thermo) and isotopic normalization via FluxFix 42. To calculate acetate contribution to acyl-CoAs, the intracellular fractional labeling was corrected for the label dilution in the media as a result of unlabeled intracellular acetate release by normalizing to the fractional labeling of acetate in the media.
Immunostaining of ACSS2 in human and mouse tissues
Tissue blocks were sectioned at 5 μm intervals using a Leica cryostat (frozen blocks) or a Paraffin Microtome. For frozen tissue sections, slides were fixed in acetone for 10 min at −20 °C. For paraffin sections, slides were dewaxed in xylene, boiled for 30 min in a citrate buffer (10 mM, pH 6.0) for antigen retrieval and rehydrated. After washing three times with tris-buffered saline (TBS), tissue sections were incubated with primary antibodies (anti-CD31, R&D; anti-ACSS2, CST for Human samples; anti-Acss2, LSBio for mice samples) diluted in blocking solution (10% BSA and horse serum in PBS) overnight at 4 °C in a humidified chamber. Sections were washed three times with TBS, incubated with appropriate Alexa Fluor 488 or Alexa Fluor 594 conjugated secondary antibodies diluted 1:200 in blocking solution for 1 hr at room temperature, washed again three times and mounted on slides with ProLong Gold mounting reagent with DAPI (Life Technologies no. P36935). All immunofluorescence micrographs were acquired using Leica SP8 microscopes.
Analysis of atherosclerotic lesions
The animals were euthanized and then immediately perfused with 30mL 0.9% saline buffer for 5 min. Whole aortas, including the aortic arch and the thoracic and abdominal segments, were dissected, gently cleaned of adventitial tissues, and fixed with freshly prepared 4% paraformaldehyde overnight at 4°C. The entire aorta was then carefully opened with scissors, flat-mounted and rinsed three times with 60% isopropyl alcohol and stained with 0.6% Oil Red O(Sigma no. O0625)at room temperature for 70 min, then washed with dH2O five times before stripping the remaining tissues. Whole aorta images were captured with Nikon Digital Sight DS-Fi1c camera and the surface lesion area was quantified with ImageJ software.
To measure lesions in the aortic root, the heart and the proximal aorta were excised, the apex and lower half of the ventricles removed, and the root-containing tissues then fixed in 4% paraformaldehyde for 4hr. followed by dehydration in 15% sucrose saline buffer at 4°C overnight, and OCT embedding. OCT blocks were sectioned at 5 μm intervals using a Microm cryostat. For ORO staining, frozen tissue sections were stained with ORO for 70 min and then stained with hematoxylin for 1.5 min. Masson staining was performed with the Modified Masson’s Trichrome Stain Kit (Solarbio G1346). Images of aortic roots were captured with Nikon Digital Sight DS-Fi1c camera and the lesion area was quantified with ImageJ software.
Plasma lipid analysis
After 12 weeks of HFD diet, blood samples were collected and mixed with EDTA (50ul 0.5M EDTA/ML plasma) in 1.5ML EP tube. The samples were then centrifuged at 10,000 g at 4°C for 10 min and serum was removed to a sterile tube and stored at −80°C. Measurements of total cholesterol and triglycerides levels in serum were performed by biochemical analyzer (Roche Cobas c311) in the Research Center for Drug Safety Evaluation of Zhejiang University.
QUANTIFICATION AND STATISTICAL ANALYSIS.
Statistical analysis was performed using GraphPad Prism 9 (GraphPad Software, Inc.). The data are presented as the mean ± SEM from at least three independent experiments. Single comparisons between two groups were performed by unpaired, two-tailed Student’s t-test. Multiple comparisons were performed by two-way ANOVA followed by Šídák’s multiple comparisons test. P < 0.05 (*), P < 0.01 (**), P < 0.001 (***) and P < 0.0001(****) were considered statistically significant. More than three independent experiments were performed to guarantee reproducibility of findings.
Supplementary Material
Highlights.
Acetate is the main source of acetyl-CoA in endothelial cells
ACSS2 is the key enzyme responsible for endothelial acetyl-CoA production.
Increased acetyl-CoA levels promote TGFβ signaling that inhibits PDK4 expression
Inhibition or knockout of endothelial ACSS2 markedly reduced atherosclerosis
ACKNOWLEDGMENTS
This work was supported, in part, by NIH grant (R01 HL135582 for M.S., DK135958 for ZA, T32GM007229 and F31HL158152 for I.S.), Open Philanthropy Foundation (MS), the Biotechnology and Biological Sciences Research Council (BBSRC BB/P013406/1 for S.T.), the National Natural Science Foundation of China (Grants 91839104 and 81770444 for L.Y.) and the National Key R&D Program of China (Grants 2018YFA0800504 for L.Y.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare that there are no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Chen PY, Schwartz MA, and Simons M (2020). Endothelial-to-Mesenchymal Transition, Vascular Inflammation, and Atherosclerosis. Front Cardiovasc Med 7, 53. 10.3389/fcvm.2020.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dejana E, Hirschi KK, and Simons M (2017). The molecular basis of endothelial cell plasticity. Nature communications 8, 14361. 10.1038/ncomms14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G, and Baker AH (2019). Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. Journal of the American College of Cardiology 73, 190–209. 10.1016/j.jacc.2018.09.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piera-Velazquez S, and Jimenez SA (2019). Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiological reviews 99, 1281–1324. 10.1152/physrev.00021.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Benedetto P, Ruscitti P, Berardicurti O, Vomero M, Navarini L, Dolo V, Cipriani P, and Giacomelli R (2021). Endothelial-to-mesenchymal transition in systemic sclerosis. Clinical and experimental immunology 205, 12–27. 10.1111/cei.13599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ungvari Z, Tarantini S, Donato AJ, Galvan V, and Csiszar A (2018). Mechanisms of Vascular Aging. Circulation research 123, 849–867. 10.1161/CIRCRESAHA.118.311378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen PY, Qin L, Li G, Wang Z, Dahlman JE, Malagon-Lopez J, Gujja S, Kauffman KJ, Sun L, Sun H, et al. (2019). Endothelial TGF-beta signalling drives vascular inflammation and atherosclerosis. Nat Metab 1, 912–926. 10.1038/s42255-019-0102-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartz MA, Vestweber D, and Simons M (2018). A unifying concept in vascular health and disease. Science 360, 270–271. 10.1126/science.aat3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Facchinello N, Astone M, Audano M, Oberkersch RE, Spizzotin M, Calura E, Marques M, Crisan M, Mitro N, and Santoro MM (2022). Oxidative pentose phosphate pathway controls vascular mural cell coverage by regulating extracellular matrix composition. Nat Metab 4, 123–140. 10.1038/s42255-021-00514-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rohlenova K, Veys K, Miranda-Santos I, De Bock K, and Carmeliet P (2018). Endothelial Cell Metabolism in Health and Disease. Trends in cell biology 28, 224–236. 10.1016/j.tcb.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Xiong J, Kawagishi H, Yan Y, Liu J, Wells QS, Edmunds LR, Fergusson MM, Yu ZX, Rovira II, Brittain EL, et al. (2018). A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Molecular cell 69, 689–698 e687. 10.1016/j.molcel.2018.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eelen G, de Zeeuw P, Simons M, and Carmeliet P (2015). Endothelial Cell Metabolism in Normal and Diseased Vasculature. Circulation research 116, 1231–1244. 10.1161/CIRCRESAHA.116.302855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim B, Li J, Jang C, and Arany Z (2017). Glutamine fuels proliferation but not migration of endothelial cells. The EMBO journal 36, 2321–2333. 10.15252/embj.201796436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eelen G, de Zeeuw P, Treps L, Harjes U, Wong BW, and Carmeliet P (2018). Endothelial Cell Metabolism. Physiological reviews 98, 3–58. 10.1152/physrev.00001.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X, Cooper DE, Cluntun AA, Warmoes MO, Zhao S, Reid MA, Liu J, Lund PJ, Lopes M, Garcia BA, et al. (2018). Acetate Production from Glucose and Coupling to Mitochondrial Metabolism in Mammals. Cell 175, 502–513 e513. 10.1016/j.cell.2018.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vysochan A, Sengupta A, Weljie AM, Alwine JC, and Yu Y (2017). ACSS2-mediated acetyl-CoA synthesis from acetate is necessary for human cytomegalovirus infection. Proceedings of the National Academy of Sciences of the United States of America 114, E1528–E1535. 10.1073/pnas.1614268114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wynn RM, Kato M, Chuang JL, Tso SC, Li J, and Chuang DT (2008). Pyruvate dehydrogenase kinase-4 structures reveal a metastable open conformation fostering robust core-free basal activity. The Journal of biological chemistry 283, 25305–25315. 10.1074/jbc.M802249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi L, and Tu BP (2015). Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Current opinion in cell biology 33, 125–131. 10.1016/j.ceb.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bose S, Ramesh V, and Locasale JW (2019). Acetate Metabolism in Physiology, Cancer, and Beyond. Trends in cell biology 29, 695–703. 10.1016/j.tcb.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang L, Venneti S, and Nagrath D (2017). Glutaminolysis: A Hallmark of Cancer Metabolism. Annu Rev Biomed Eng 19, 163–194. 10.1146/annurev-bioeng-071516-044546. [DOI] [PubMed] [Google Scholar]
- 21.Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, Tellides G, Schwartz MA, and Simons M (2015). Endothelial-to-mesenchymal transition drives atherosclerosis progression. The Journal of clinical investigation 125, 4514–4528. 10.1172/JCI82719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim B, Jang C, Dharaneeswaran H, Li J, Bhide M, Yang S, Li K, and Arany Z (2018). Endothelial pyruvate kinase M2 maintains vascular integrity. The Journal of clinical investigation 128, 4543–4556. 10.1172/JCI120912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su X, Wellen KE, and Rabinowitz JD (2016). Metabolic control of methylation and acetylation. Curr Opin Chem Biol 30, 52–60. 10.1016/j.cbpa.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Z, Zhang M, Plec AA, Estill SJ, Cai L, Repa JJ, McKnight SL, and Tu BP (2018). ACSS2 promotes systemic fat storage and utilization through selective regulation of genes involved in lipid metabolism. Proc Natl Acad Sci U S A 115, E9499–E9506. 10.1073/pnas.1806635115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamphorst JJ, Chung MK, Fan J, and Rabinowitz JD (2014). Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab 2, 23. 10.1186/2049-3002-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schug ZT, Vande Voorde J, and Gottlieb E (2016). The metabolic fate of acetate in cancer. Nature reviews. Cancer 16, 708–717. 10.1038/nrc.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, et al. (2014). Acetate dependence of tumors. Cell 159, 1591–1602. 10.1016/j.cell.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deuse T, Hua X, Wang D, Maegdefessel L, Heeren J, Scheja L, Bolanos JP, Rakovic A, Spin JM, Stubbendorff M, et al. (2014). Dichloroacetate prevents restenosis in preclinical animal models of vessel injury. Nature 509, 641–644. 10.1038/nature13232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, and Michelakis ED (2004). Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circulation research 95, 830–840. 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- 30.Caolo V, Peacock HM, Kasaai B, Swennen G, Gordon E, Claesson-Welsh L, Post MJ, Verhamme P, and Jones EAV (2018). Shear Stress and VE-Cadherin. Arterioscler Thromb Vasc Biol 38, 2174–2183. 10.1161/ATVBAHA.118.310823. [DOI] [PubMed] [Google Scholar]
- 31.Michelakis ED, Gurtu V, Webster L, Barnes G, Watson G, Howard L, Cupitt J, Paterson I, Thompson RB, Chow K, et al. (2017). Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Science translational medicine 9. 10.1126/scitranslmed.aao4583. [DOI] [PubMed] [Google Scholar]
- 32.Mews P, Egervari G, Nativio R, Sidoli S, Donahue G, Lombroso SI, Alexander DC, Riesche SL, Heller EA, Nestler EJ, et al. (2019). Alcohol metabolism contributes to brain histone acetylation. Nature 574, 717–721. 10.1038/s41586-019-1700-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL, Petersen KF, Kibbey RG, Goodman AL, and Shulman GI (2016). Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature 534, 213–217. 10.1038/nature18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gawad C, Koh W, and Quake SR (2016). Single-cell genome sequencing: current state of the science. Nature reviews. Genetics 17, 175–188. 10.1038/nrg.2015.16. [DOI] [PubMed] [Google Scholar]
- 35.Prasain N, Meador JL, and Yoder MC (2012). Phenotypic and functional characterization of endothelial colony forming cells derived from human umbilical cord blood. J Vis Exp 10.3791/3872. [DOI] [PMC free article] [PubMed]
- 36.Yoder MC (2007). Developing reagents and conditions to induce mesoderm subsets from ES cells. Cell Stem Cell 1, 603–604. 10.1016/j.stem.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 37.Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Z, Yanxiang Guo J, et al. (2017). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. 10.1038/nature24057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shetty S, Ramos-Roman MA, Cho YR, Brown J, Plutzky J, Muise ES, Horton JD, Scherer PE, and Parks EJ (2012). Enhanced fatty acid flux triggered by adiponectin overexpression. Endocrinology 153, 113–122. 10.1210/en.2011-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nature methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tumanov S, Bulusu V, Gottlieb E, and Kamphorst JJ (2016). A rapid method for quantifying free and bound acetate based on alkylation and GC-MS analysis. Cancer Metab 4, 17. 10.1186/s40170-016-0157-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frey AJ, Feldman DR, Trefely S, Worth AJ, Basu SS, and Snyder NW (2016). LC-quadrupole/Orbitrap high-resolution mass spectrometry enables stable isotope-resolved simultaneous quantification and (1)(3)C-isotopic labeling of acyl-coenzyme A thioesters. Anal Bioanal Chem 408, 3651–3658. 10.1007/s00216-016-9448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNA-seq sequencing data associated with this study have been deposited Gene Expression Omnibus and the approved GEO accession numbers for these datasets are GSE224907 and GSE224939 (Data will be available on May 1, 2023). Original western blot images and analyzed data are available as Data S1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.