

Abstract
Background and Aims
Ulcerative colitis (UC) is characterized by severe inflammation and destruction of the intestinal epithelium, and associated with risk single nucleotide polymorphism in HLA class II. Given the recently discovered interactions between subsets of HLA-DP molecules and the activating natural killer (NK) cell receptor NKp44, genetic associations of UC and HLA-DP haplotypes and their functional implications were investigated.
Methods
HLA-DP haplotype and UC risk association analyses were performed (UC: N= 13,927; control: N= 26,764). Expression levels of HLA-DP on intestinal epithelial cells (IECs) in individuals with and without UC were quantified. Human intestinal 3D-organoid co-cultures with human NK cells were employed to determine functional consequences of interactions between HLA-DP and NKp44.
Results
These studies identified HLA-DPA1*01:03-DPB1*04:01 (HLA-DP401) as a risk haplotype and HLA-DPA1*01:03-DPB1*03:01 (HLA-DP301) as a protective haplotype for UC in European populations. HLA-DP expression was significantly higher on IECs of individuals with UC compared to controls. IECs in human intestinal 3D-organoids derived from HLA-DP401pos individuals showed significantly stronger binding of NKp44 compared to HLA-DP301pos IECs. HLA-DP401pos IECs in organoids triggered increased degranulation and TNF production by NKp44+ NK cells in co-cultures, resulting in enhanced epithelial cell death compared to HLA-DP301pos organoids. Blocking of HLA-DP401-NKp44 interactions (anti-NKp44) abrogated NK cell activity in co-cultures.
Conclusion
Here, we identify an UC risk HLA-DP haplotype that engages NKp44 and activates NKp44+ NK cells, mediating damage to intestinal epithelial cells in an HLA-DP haplotype-dependent manner. The molecular interaction between NKp44 and HLA-DP401 in UC can be targeted by therapeutic interventions to reduce NKp44+ NK cell-mediated destruction of the intestinal epithelium in UC.
Keywords: Ulcerative colitis, NK cells, NKp44, HLA-DP, intestinal organoids
Lay summary
We demonstrate that specific subsets of HLA-DP molecules associated with UC and expressed on intestinal epithelial cells under inflammatory conditions can activate NK cells resulting in intestinal epithelial damage.
Introduction
Ulcerative Colitis (UC) is a chronic inflammatory disease of the colon and rectum, with higher prevalence in high-income countries.1 Current therapeutic strategies of mild-to-moderate UC aim to reduce inflammation with e.g. aminosalicylates or topical corticosteroids, whereas systemic corticosteroids, biologicals (i.e. anti-tumor necrosis factor (TNF) or anti-alpha4/beta7) and small molecules (i.e. Janus kinase (JAK) inhibitors) are employed in patients with moderate-to-severe and steroid refractory UC cases.2–4 Nonetheless, even with the newest medications frequent and severe flare-ups are observed in individuals with UC and long-term remission is achieved in less than 50% of affected patients, indicating the complex mechanisms underlying UC.5
To maintain intestinal homeostasis, a balanced interplay between the intestinal microbiota, intestinal epithelial cells (IECs), and immune cells is required.6 In UC, homeostasis is disrupted and destruction of the epithelium, atrophy of crypts and immune cell infiltration is observed.7,8 Although the precise etiology of UC remains unknown, multiple factors have been suggested to contribute to the pathogenesis of UC, ranging from host to environmental factors. Specifically, genes encoding human leukocyte antigen (HLA) class I and II have been associated with UC, as well as genes regulating epithelial cell functioning.9,10 Furthermore, dysregulated responses of innate immune cells, as well as CD4+ T cells, are observed and implicated in the pathogenesis of UC.11,12 Although the role of natural killer (NK) cells in UC is less investigated, NKG2A+ NK cells have been suggested to play a role in the regulation of innate immune cell functions in UC.13 In addition, polymorphisms in killer immunoglobulin-like receptor (KIR) genes, encoding for activating and inhibitory NK cell receptors that bind to HLA I molecules, have been linked to UC.14,15 Furthermore, innate lymphoid cells (ILCs), which share features of NK cells, have been implicated in inflammatory bowel disease (IBD).16,17
Recently, our group demonstrated that the activating NK cell receptor NKp44 binds to subsets of HLA-DP molecules,18 allowing direct interactions between NK cells and HLA-DP-expressing cells. Studies by Biton et al. and other groups showed that IECs can express HLA class II molecules.19–23 Taken together, these recent observations led us to investigate the role of specific HLA-DP/NKp44 interactions in UC. An HLA-DP haplotype and association analyses of individuals with UC and controls was performed and identified HLA-DPA1*01:03-DPB1*04:01 as a risk haplotype and HLA-DPA1*01:03-DPB1*03:01 as a protective haplotype for UC. Phenotypical characterization of colon samples revealed increased HLA-DP expression levels by IECs in individuals with UC. Co-cultures of human intestinal 3D-organoids and NK cells revealed that expression of the UC risk HLA-DP401 molecule by IECs strongly activated NKp44+ NK cells and induced TNF production resulting in increased epithelial cell death of organoids, while the UC protective HLA-DP301 molecule did not. These findings reveal a molecular interaction between NKp44+ NK cells and HLA-DP401 on IECs in UC that can be targeted in therapeutic interventions to reduce NK cell-mediated epithelial tissue damage.
Materials and Methods
HLA-haplotype analysis
Analysis was performed on 13,927 UC cases and 26,764 controls with Caucasian ancestry. HLA-DPA1, -DPB1 genotypes were imputed from quality-controlled single nucleotide polymorphism (SNP) genotypes. These data as well as genotype quality control data have been previously described.9,24 To compare results from different imputation methods/imputation reference panels, we performed imputation A) with the Michigan Imputation Server (MichiganImpServer)25 and B) using HLA genotype Imputation with Attribute Bagging (HIBAG). For detailed description of both imputation methods see Supplementary Material.
Statistical analysis of HLA-haplotype analysis
Logistic regression analysis was performed for each allele and haplotype on the case-control status for UC using the first five principal components derived from whole-genome SNP data.24 Allele and haplotype frequencies were calculated and only alleles/haplotypes with frequencies > 0.5% were retained. Alleles missing in either respective dataset were assigned allele frequency “below 0.005”. The data from both analyses were combined and corrected for multiple testing using the multiple correction method of Bonferroni-Holm. All analyses were conducted in R version 3.6.2.
Recombinant human Fc construct binding to HLA class II-coated beads
Screening of HLA class II-coated beads was performed as previously described.18 In brief, recombinant human NKp44 Fc construct (R&D Systems, Minneapolis, MN, 2249-NK-050), recombinant human LAG-3 Fc construct (R&D Systems, 2319_L3–050) or recombinant human FcRL6 Fc construct26 were incubated with a mixture of HLA class II-coated beads (OneLambda, Canoga Park, CA, LS2A01). Uncoated beads were used as negative control and IgG-coated beads as positive control. Samples were washed, incubated with a F(ab’)2 goat-anti-human IgG PE secondary antibody (Thermo Fisher, Waltham, MA, 10129892) and binding of NKp44 Fc construct was quantified using Luminex xMAP technology on a Bio-Plex 200 (Bio-Rad Laboraties, Hercules, CA).
Intestinal tissue samples
Human intestinal tissues were obtained at surgery for intestinal anastomosis reconstruction or colectomy at the University Medical Center Hamburg-Eppendorf. Collection of tissue samples was approved by the ethics committee of the Medical Association of the Freie Hansestadt Hamburg (Ärztekammer Hamburg) and donors provided written informed consent. 17 male and 22 female donors were included with a median age of 57 years (Supplementary Table 1). This included 11 donors with UC. Control samples were obtained from individuals that underwent a tissue resection for carcinoma or reconstruction of anastomosis and did not have an intestinal inflammatory disease.
Immunohistochemistry staining of tissue sections
After formalin fixation and paraffin wax embedding, sections of control and UC intestinal tissue samples were deparaffinized in xylene and cooked at 121°C in presence of antigen retrieval solution (Dako Target Retrieval Solution, Agilent Technologies, Santa Clara, CA, S1699). Sections were incubated with HLA-DP (Sigma-Aldrich, St. Louis, MO, HPA017967) or isotype control antibody (rabbit poly IgG, abcam, Cambridge, UK, ab37415) followed by incubation with the secondary antibody (Goat-anti-rabbit biotinylated antibody, LS-Bio, Seattle, WA, LS-C350860). Biotinylated antibody binding sites were detected with the streptavidin-alkaline phosphatase complex (Vectastain ABC-AP Kit, Vector Laboratories, Newark, CA, AK-5000) followed by visualization using liquid permanent red (Zytomed Systems, Berlin, Germany, ZUC001–125). Sections were counterstained with hemalumn (Sigma-Aldrich, 1.09249.2500). Images were acquired using an EVOS M5000 imaging system (Thermo Fisher, Waltham, MA).
Intestinal epithelial cell and lymphocyte isolation
IECs and intraepithelial lymphocytes (IELs) were isolated from adult intestinal tissue samples as previously described and validated to isolate the superficial epithelial layer.27,28 In short, the muscular layer and fat were mechanically removed from the intestinal tissue samples. IECs and IELs from intestinal tissues were obtained after incubation of the intestinal tissue samples with IMDM (Iscove’s modified Dulbecco’s medium, Thermo Fisher Scientific, 10135083) supplemented with ethylenediaminetetraacetic acid (EDTA, 5 mM; Promega, Madison, WI, V4231), 1,4-Dithiothreitol (DTT, Carl-Roth GmbH+Co. KG, 6908.1) and 1% fetal bovine serum (FBS; Capricorn, Düsseldorf, Germany, FBS-11A). To isolate lamina propria lymphocytes (LPLs) intestinal tissue, now devoid of the epithelial layer, was minced and incubated in IMDM supplemented with 1 mg/ml Collagenase (Sigma-Aldrich, 11088882001), 1% FBS and 1000 U/ml DNaseI (StemCell Technologies, 07470). The resulting cell solution was filtered with a 70 μm cell strainer and the single cell suspension of LPL was obtained after a 60% standard isotonic Percoll (Sigma-Aldrich) density gradient centrifugation step.
For flow cytometric analyses, IECs were stained with anti-EpCAM-BV421 (Biolegend, San Diego, CA, #324220), anti-CD45-BV785 (Biolegend, #304048), anti-HLA-DP-BUV737 (BD Biosciences, San Jose, CA, #750942), anti-HLA-DQ-APC (Leinco Technologies, St.Louis, MO, H242–100), anti-HLA-DR-BV711 (Biolegend, #307644) and LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit (Thermo Fisher, L34976). Cells were fixed using BD Cellfix (BD Biosciences, 340181) and analyzed using a BD LSR Fortessa (BD Biosciences). IELs were enriched after epithelial cell layer dissociation using Biocoll density centrifugation and stained with anti-CD3-BUV395 (BD Biosciences, #563546), anti-CD45-AF700 (Biolegend, #368514), anti-CD16-BUV737 (BD Biosciences, #612786), anti-CD56-BV785 (Biolegend, #362550), anti-NKp44-PE (Biolegend, #325108), anti-CD127-PE-Dazzle (Biolegend, #351336), anti-CD14-PE-Cy7 (Biolegend, #301814), anti-CD19-PE-Cy7 (Biolegend, #363012), anti-BDCA2-PE-Cy7 (Biolegend, #354214), anti-CD1a-PE-Cy7 (Biolegend, #300122), anti-CD123-PE-Cy7 (Biolegend, #306010), anti-CD34-PE-Cy7 (Biolegend, #343516) and LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit. Fixation was performed with BD Cellfix and cells were analyzed using a BD LSR Fortessa.
RNA isolation and RT-qPCR
RNA was retrieved from cells of the intestinal epithelium using Trizol (Thermo Fisher, 15596018) according to the manufacturer’s instructions. RNA was treated with RNase Out (Thermo Fisher, 10777019) and DNase I (Thermo Fisher, AM2224) before transcribing 1 μg RNA into cDNA using the qScriber cDNA synthesis Kit (highQU, Kraichtal, Germany, RTK0104). cDNA templates were mixed with TNF, interferon-gamma (IFNγ) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primer pairs (Supplementary Table 2) and the QuantiFast SYBR Green PCR Kit (Qiagen, Hilden, Germany, 204057). qPCR reactions were performed on the Lightcycler 96 System (Roche, Basel, Switzerland). For calculating the relative gene expression, target CT values were normalized to a reference gene (GAPDH) and log2 transformed.
Human intestinal organoid culture
Human intestinal organoids from adult intestinal tissue samples were generated as previously described.28–30 In brief, isolated intestinal epithelial cells were washed with ice-cold AD+++ (Advanced Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 (Thermo Fisher, 12634–028) containing 1% GlutaMAX (Thermo Fisher, 35050061), 10 mM HEPES (Thermo Fisher, 15630056), and 1% penicillin/streptomycin (Sigma-Aldrich, P4333)) and resuspended in ice-cold growth factor-reduced Matrigel (Corning, Corning, NY, 356231). Matrigel droplets were seeded in pre-warmed 24-well plates (Greiner, Frickenhausen, Germany, 662160) and covered with expansion medium (EM; Supplementary Table 3) supplemented with 10 μM Rho kinase inhibitor Y-27632 (StemCell, Vancouver, Canada, 72308). Medium with Y-27632 was refreshed every 2–3 days until first passage. Intestinal organoids were cultured at 37°C and 5% CO2 and passaged weekly by mechanical disruption. EM was refreshed every 2–3 days.
Human intestinal organoid stimulation
Human intestinal organoids were stimulated for 12 hrs, 24 hrs, 48 hrs or 72 hrs with different concentrations of IFNγ (Pepro-Tech, Hamburg, Germany, 300–02), TNF (R&D Systems, 210-TA/CF) or both cytokines. Intestinal organoids were dissociated into single cells using TrypLE Express (Thermo Fisher, 12605028) and used for antibody staining. Cells were stained with anti-EpCAM-BV421, anti-CD45-AF700, anti-HLA-DP-BUV737 and LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit, fixed using BD Cellfix and analyzed using a BD LSR Fortessa.
Preparation of human NK cells from blood samples
All donors provided written informed consent to obtain PBMCs and studies were approved by the ethics committee of the Ärztekammer Hamburg. NK cells were isolated as previously described18 and after 5–7 day stimulation with 250 U/ml IL-2 (Pepro-Tech, 200–02) and 10 ng/ml IL-15 (Pepro-Tech, 200–15) to induce NKp44 expression used for functional analyses.
Plate-coated NK cell and ILC degranulation assay
Degranulation assays were performed as previously described.18 In brief, non-tissue culture-treated plates (Corning) were coated with HLA-DP401 or HLA-DP301 molecules (provided by OneLambda), or biotinylated anti-NKp44 (Biolegend, 325106). Isolated NK cells or sorted intestinal ILCs (viable singlets CD45+CD3−CD14−CD19−CD1a−BDCA-2−CD123−CD34−CD127+) resuspended in assay medium containing Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% FBS or IMDM supplemented with 10% FBS were distributed on coated plates. The vesicular transport protein inhibitor brefeldin A (Sigma-Aldrich, B7651) and anti-CD107a-BV785 (Biolegend, #328644) were added, and cells were incubated for 5–8 hrs. NK cells were stained with anti-CD3-BV510 (Biolegend, #344828), anti-CD56-BV605 (Biolegend, #318334), anti-CD16-FITC (Biolegend, #302006), anti-NKp44-AF647 (Biolegend, #325112), anti-TNF-PE (Biolegend, #502909) and LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit. ILCs were stained with anti-CD45-AF700, anti-NKp44-AF647, anti-TNF-BUV395, anti-IL-22-PE and LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit. Fixation was performed using the BD Cytofix/Cytoperm Kit (BD Biosciences, 554714) and cells were analyzed using a BD LSR Fortessa.
Immunofluorescence
To de-wax the samples, the samples were washed in xylol 100%, followed by incubations in a descending series of ethanol. Antigen target retrieval was performed using Agilent DAKO Target Retrieval Solution (pH9) (product number: S236884–2) in a Braun Multiquick FS20 steamer followed by washing in Agilent Wash Buffer Solution (product number: K800721–2). The staining included NCAM1/CD56 (anti-mouse, Cell Signaling #3576, 1:400), HLA-DPA (anti-rb, Sigma-Aldrich #HPA017967–100ul, 1:100), AF488 donkey anti-rabbit (Invitrogen #A-21206, 1:200), AF647 donkey anti-mouse (Invitrogen #A-31571, 1:200) diluted in Agilent Antibody diluent solution (product number: K800621–2). Primary antibodies were incubated overnight at 4°C followed by corresponding secondary antibodies for 1 hr. DAPI (Sigma-Aldrich D90542) was added within the Agilent Antibody diluent solution to counterstain the nuclei. After washing, samples were mounted with Prolong Gold (product number: P36930). Images were acquired using the LED-based widefield system THUNDER Imager 3D Live Cell and 3D Cell Culture (Leica Microsystems) in 40x (NA: 1.10) with Instant Computational Clearing (ICC) mode.
Single cell gene expression analysis
IELs from colon tissues were thawed and stained with anti-CD45-AF700, anti-CD3-BV510, anti-CD56-BV605, anti-CD16-FITC, anti-CD127-PE/Dazzle and LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit. 2000 viable CD45+CD3−CD127− and CD56+/CD16+ single or double positive NK cells were sorted from each donor, using a 5-laser FACS Aria-Fusion (BD Biosciences). Single Cell Preamp IFCs, 5–10 μm (Fluidigm) were used for the generation of cDNA. After cell capture, capture sites were visualized under a microscope and only capture sites with one single cell were included into further analysis. Generation of cDNA was performed following company protocol. Gene expression analysis was performed using Biomark HD 192.24 Dynamic Array IFCs for Gene Expression.
NKp44 Fc binding to intestinal epithelial cells
IFNγ-stimulated intestinal organoids (200 U/ml for 3–4 days) were dissociated into single cells using TrypLE Express, treated with 1 U/ml Proteinase K (Qiagen, 19131) and rested in EM + Y-27632 at 37°C and 5%CO2. Cells were washed with phosphate-buffered saline (PBS, Sigma-Aldrich, D8537) and stained with LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit, NKp44-Fc (25 μg/ml), secondary antibody F(ab) anti-human IgG-PE and anti-HLA-DP BUV737. Fixation was performed with 4% PFA (Sigma-Aldrich, P6148) and cells were analyzed using a BD LSR Fortessa.
Co-culture of NK cells and intestinal epithelial cells
In NK cell – IEC co-cultures, NK cell donors were matched for HLA class I genotypes to organoid donors to reduce allogenic reactions. All donors had heterozygous alleles encoding for the epitopes C1/C2 and had at least one allele of Bw4. NK cells were isolated as described above and cultured for 6–7 days in RPMI 1640 (Thermo Fisher, 12004997) supplemented with 10% FBS (Capricorn, FBS-11A), 250 U/ml IL-2 (Pepro-Tech, 200–02) and 10 ng/ml IL-15 (Pepro-Tech, 200–15). Unstimulated and IFNγ-stimulated intestinal organoids (3 days with 200 U/ml) were dissociated into single cells with TrypLE, treated with 1 U/ml Proteinase K and counted to enumerate the number of target cells. Enriched NK cells were co-incubated with IECs at an effector/target cell ratio of 1:2 for 5 hrs in RPMI 1640 with 10% FBS. Additionally, anti-CD107a-BV785 antibody, brefeldin A (BD Biosciences, 555029) and anti-HLA-DP (10 μg/ml; Leinco, H266) were added. For blocking experiments, NK cells were pre-incubated in the presence of an anti-NKp44 antibody (30 μg/ml, Biolegend, #325122) for 30 min at 37°C. Cells were washed with PBS, stained with anti-CD3-BV510, anti-CD16-FITC, anti-CD56-BV605, anti-NKp44-AF647, anti-TNF-PE (Biolegend, #502909), anti-HLA-DP-BUV737 and LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit, fixed using the BD Cytofix/Cytoperm Kit and analyzed using a BD LSR Fortessa. % NKp44 downregulation was calculated as follows: (% NKp44+ cells of CD56++ NK cells after co-culture with DP− IECs) – (% NKp44+ cells of CD56++ NK cells after co-culture with DP+ IECs). % CD107a upregulation was calculated by: (% CD107a+ cells of CD56++ NK cells after co-culture with DP+ IECs) – (% CD107a+ cells of CD56++ NK cells after co-culture with DP− IECs).
Co-culture of NK cells and intestinal organoids
NK cell donors were matched to organoid donors as described above, and NK cells were isolated and stimulated to induce NKp44 expression. IFNγ-stimulated intestinal organoids (3–4 days with 200 U/ml) were stained with 2 μM Calcein (Thermo Fischer, 65–0853-78) and organoids derived from one reference well were dissociated into single cells and counted to enumerate the number of target cells. Sorted viable CD3−, CD16−, CD56++ NK cells were co-incubated with intestinal organoids at an effector/target cell ratio of 1:1 for 6 hrs in RPMI 1640 with 10% FBS and anti-HLA-DP (10 μg/ml; Leinco, H266). Co-cultures were monitored using the spheroid module of the Incucyte SX5 Live Cell Imaging Analysis Instrument (Sartorius, Göttingen, Germany). The size of individual organoids were measured at 0 hrs and 6 hrs of co-incubation using Fiji. The normalized organoid size was calculated as follows: (organoid size at 6 hrs) / (organoid size at 0 hrs). To determine viability of IECs, NK cell – intestinal organoid co-cultures were harvested after 16 hrs incubation and dissociated into single cells. Cells were washed with PBS, stained with anti-CD45-BUV395 (BD Biosciences, #563792), anti-EPCAM-BV421 and LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit, fixed using the BD Cytofix/Cytoperm Kit and analyzed using a BD LSR Fortessa. To determine the effect of blocking of NKp44, NK cells were pre-incubated in the presence of an anti-NKp44 antibody (30 μg/ml, Biolegend, #325122) for 30 min at 37°C before co-cultures.
Software
BD FACS Diva (BD Biosciences) was used to acquire flow cytometry data and FlowJo (version 10.8.1; Treestar) was used to analyse flow cytometry data. GraphPad Prism (Version 9.4.0 for Windows; San Diego, CA, www.graphpad.com) was used for graphical display and statistical analysis. ImageJ (http://imagej.nih.gov/ij/) was used for organoid annotation and image display. Software used for the HLA-DP haplotype and association analyses is described above.
Statistical analysis
GraphPad Prism 9 was used to perform statistical analyses including Wilcoxon-signed match rank tests, Mann-Whitney U tests and Ordinary Two-way ANOVA and Spearman’s rank correlations. HLA-DP haplotype and association analyses are described above.
Results
HLA-DP haplotype analyses reveal HLA-DPA1*01:03-DPB1*04:01 as a risk haplotype for UC and HLA-DPA1*01:03-DPB1*03:01 and HLA-DPA1*01:03-DPB1*11:01 as protective haplotypes
To investigate whether individuals carrying certain HLA-DP haplotypes exhibit higher or lower risk to develop UC, we performed a HLA-DP haplotype and association analyses of 13,927 individuals with UC and 26,764 control individuals with Caucasian ancestry. HLA-DPA1 and -DPB1 genotypes were imputed from quality-controlled SNPs, and results of the two different imputation methods, HIBAG31 and the MichiganImpServer25, were compared and revealed similar outcomes. Out of a total of 21 imputed HLA-DPA1-DPB1 haplotypes, including 3 HLA-DPA1 alleles and 17 HLA-DPB1 alleles, two protective HLA-DPA1-DPB1 haplotypes and one risk HLA-DPA1-DPB1 haplotype with allele frequencies >1% and P < .05 were identified for UC after correction for multiple testing (Figure 1A, Supplementary Figure 1A, Supplementary Table 4). The haplotypes HLA-DPA1*02:01-DPB1*11:01 and HLA-DPA1*01:03-DPB1*03:01 were associated with a decreased risk for UC. In contrast, HLA-DPA1*01:03-DPB1*04:01, was identified as a risk haplotype for UC (Figure 1A).
Figure 1: HLA-DP risk haplotype for UC binds to NKp44.
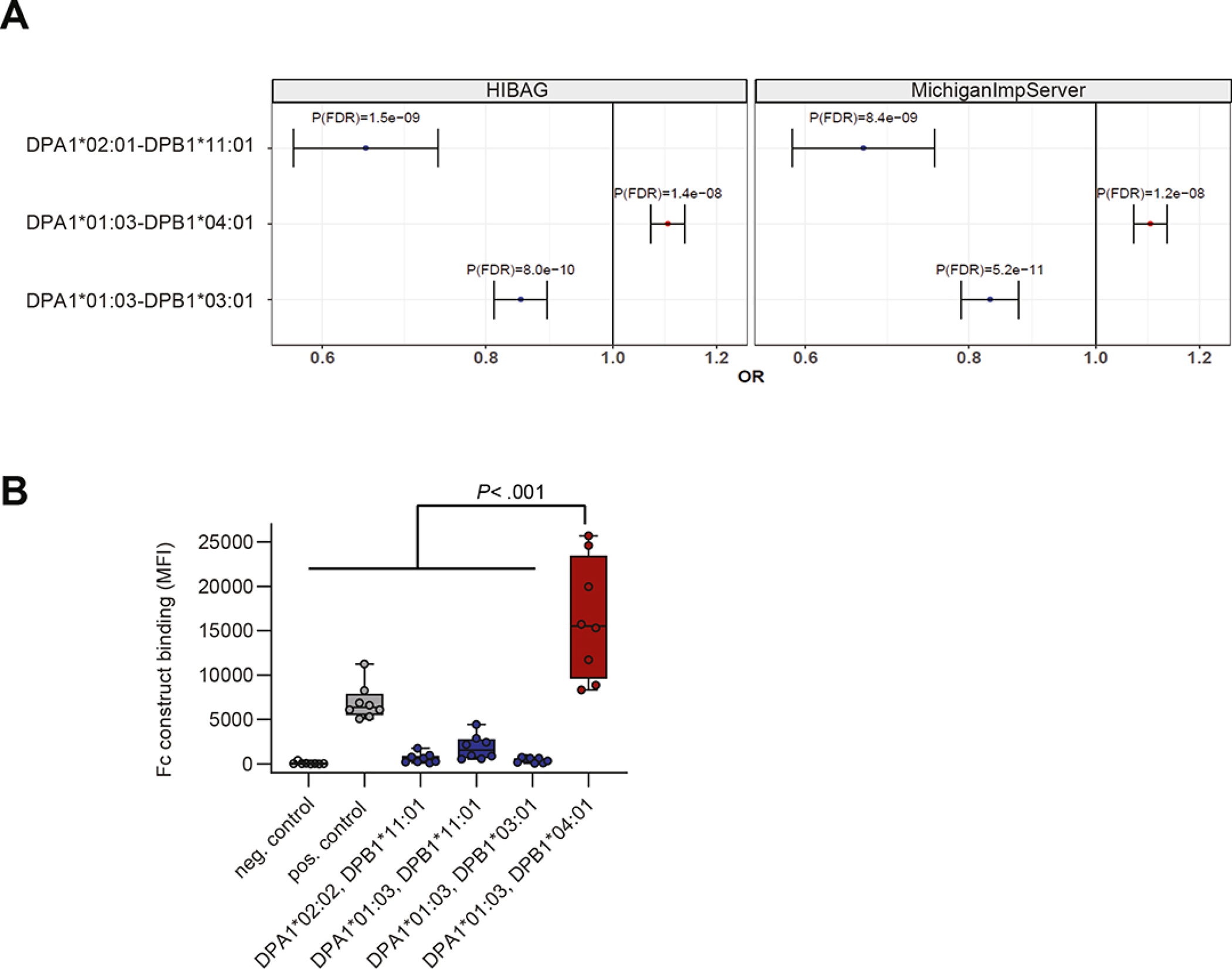
A. Imputation of HLA-DPA1-DPB1 genotypes using HIBAG (left) and MichiganImpServer (right) showing UC risk and protective HLA-DPA1-DPB1 haplotypes with a frequency of >1% and a False Discovery Rate P (P(FDR)) of < .05. Odds ratios (OR) and 95% confidence intervals are shown. B. NKp44 Fc construct binding to beads coated with different HLA-DP molecules was determined and medians of fluorescence intensity (MFIs) of all individual experiments (n= 8) are depicted. Boxes indicate medians with 25% and 75% quartile ranges, and whiskers indicate minimum and maximum MFI of each HLA-DP molecule tested. HLA-DP molecules that exhibited higher median binding to the NKp44 Fc construct than to the positive control (IgG-coated beads) are marked in red and less in blue. Statistical significance was measured using Mann-Whitney U comparisons.
Building on our recent findings that subsets of HLA-DP molecules serve ligands for the activating NK cell receptor NKp44,18 the binding of the above-identified UC-associated HLA-DP haplotypes to NKp44 was assessed using an NKp44 Fc construct and Luminex technology. Due to unavailability of HLA-DPA1*02:01-DPB1*11:01 molecules for this assay, HLA-DPB1*11:01 molecules in combination with two other alpha chains, HLA-DPA1*02:02 and -DPA1*01:03 were used. The UC protective HLA-DPB1*11:01 molecule in combination with both HLA-DP alpha chains, and the HLA-DPA1*01:03-DPB1*03:01 (HLA-DP301) molecule did not bind the NKp44 Fc construct, whereas the UC risk molecule HLA-DPA1*01:03-DPB1*04:01 (HLA-DP401) exhibited significantly strong binding to the NKp44 Fc construct across multiple experiments (Figure 1B). In contrast, we did not detect binding of NKp44 Fc construct to previously identified HLA-DR or HLA-DQ UC risk and protective molecules9,24,32,33 (Supplementary Table 5; Supplementary Figure 1B). NK cell receptors, LAG-3 and FcRL6 have also been described to bind to HLA class II molecules26,34, however no differential binding to previously described HLA-DR, DQ or DP haplotypes associated with UC were observed (Supplementary Figure 2A–B). These results confirm the specific interactions between HLA-DP401 and NKp44 molecules,18 and provided rational for further analyses into a potential role for NKp44/HLA-DP interactions in UC. Since HLA-DPA1*01:03-DPB1*04:01 (haplotype frequency: 43.29%) as a risk haplotype and HLA-DPA1*01:03-DPB1*03:01 (haplotype frequency: 10.01%) as a protective haplotype for UC belong to the most common haplotypes in the European population,35 we focused on these two HLA-DP haplotypes in further analyses.
Intestinal epithelial cells of individuals with UC express high levels of HLA-DP
To further determine whether HLA-DP is available for NKp44/HLA-DP interactions in UC, expression of HLA-DP molecules in colon-derived samples of individuals with UC was compared to controls. Immunohistochemical analyses showed HLA-DP+ cells in intestinal crypts of individuals with UC, whereas HLA-DP expression was almost absent in intestinal crypts of controls (Figure 2A; Supplementary Figure 3A). To further quantify the expression of HLA-DP on primary human IECs, flow cytometric analyses of colon-derived IECs of individuals with UC and controls were performed. Expression levels of HLA-DP on EpCAM+CD45− IECs of individuals with UC were significantly higher compared to controls, whereas HLA-DP expression levels of CD45+EPCAM− immune cells was lower and did not significantly differ between individuals with UC and controls (Figure 2B, Supplementary Figure 3B, C, D). As the expression of HLA class II molecules is regulated by the transcription factor class II transactivator (CIITA),36 we also investigated the expression levels of other HLA class II molecules. HLA-DR expression was significantly higher on IECs of individuals with UC compared to controls (Supplementary Figure 3E), whereas HLA-DQ expression levels did not significantly differ between individuals with UC and controls (Supplementary Figure 3F). These data demonstrate a significantly higher expression of HLA-DP molecules on human IECs of individuals with UC.
Figure 2: High HLA-DP expression on intestinal epithelial cells of individuals with UC.
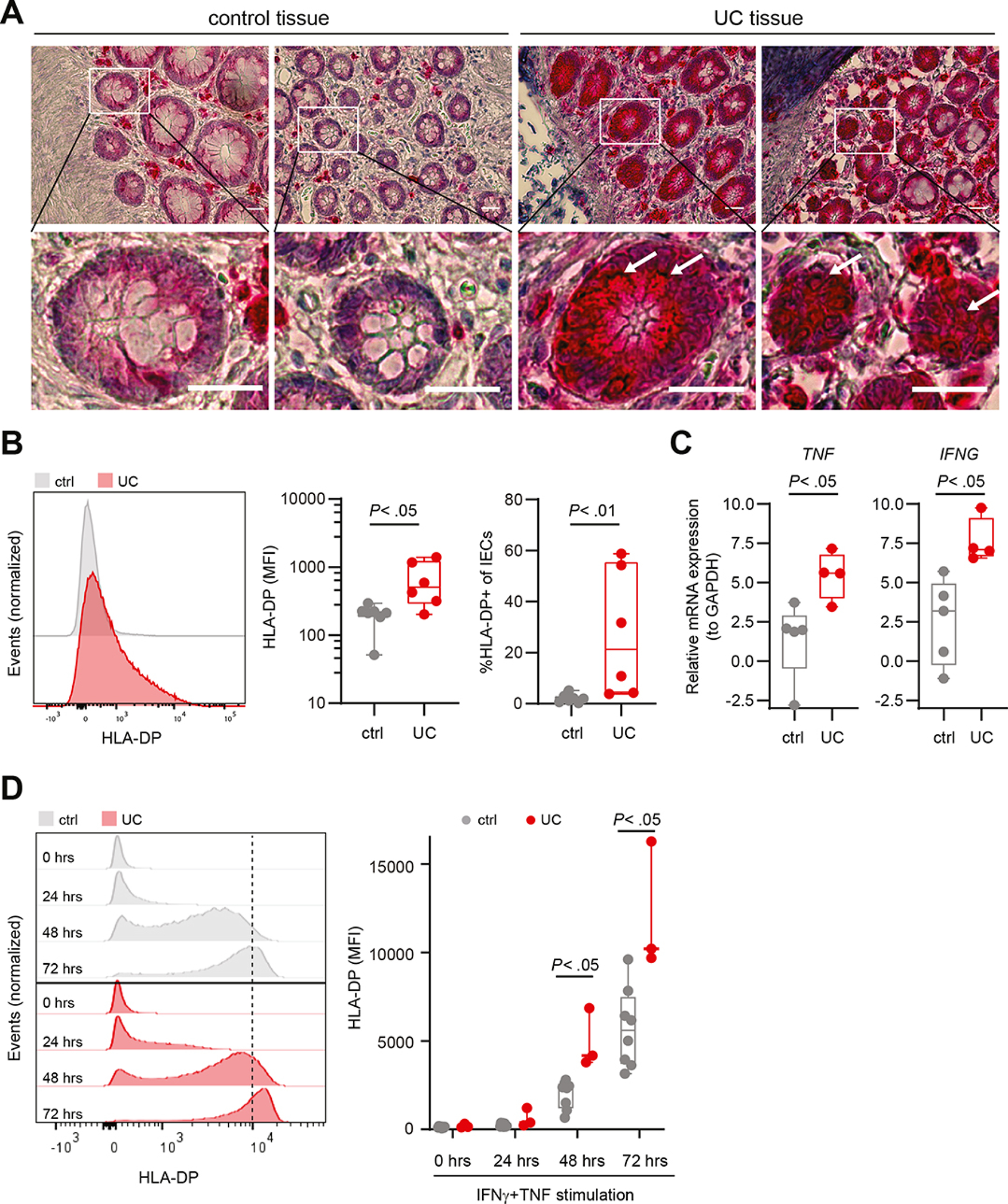
A. Images of immunohistochemical analyses of control and UC colons. Specific HLA-DP expression is labelled in red, hemalumn was used as a nuclear counterstain (blue). Lower row shows magnifications of the selected ROI (white box in images of upper row). White arrows point to HLA-DP expressing cells. Scale bars: 250 μm. B. Representative histograms of HLA-DP expression in EpCAM+ IECs of control (ctrl) and individuals with UC measured by flow cytometry (left panel). Expression of HLA-DP (MFI) on EpCAM+ IECs (middle panel). Median percentage of HLA-DP+ cells of EpCAM+ IECs of controls (n = 7) and individuals with UC (n = 6) (right panel). C. Relative mRNA expression of TNF and IFNG to reference gene GAPDH in the intestinal epithelium of individuals with UC (n = 4) compared to controls (n = 5). D. Representative histograms of HLA-DP expression in EpCAM+ epithelial cells in organoids derived from controls or individuals with UC upon stimulation with IFNγ (100 U/ml) and TNF (20 ng/ml) for the indicated time points measured by flow cytometry (left panel). Plot shows MFI of HLA-DP on IECs in intestinal organoids of controls (n = 8) and individuals with UC (n = 3) upon IFNγ and TNF stimulation (right panel).
All boxes indicate medians with 25% and 75% quartile ranges, and whiskers indicate minimum and maximum values. Statistical significance was measured using Mann-Whitney U comparisons.
Pro-inflammatory cytokines TNF and IFNγ upregulate HLA-DP expression by IECs
The pro-inflammatory cytokines IFNγ and TNF are involved in CIITA-dependent upregulation of HLA-class II molecules.37,38 TNF and IFNG were significantly upregulated in the intestinal epithelium of individuals with UC compared to controls (Figure 2C). To further assess the impact of TNF and IFNγ on HLA-DP expression of IECs, we generated adult stem cell-derived human colon organoids from intestinal tissues of individuals with UC or controls as previously described.28–30 Organoids generated from colon were stimulated with different concentrations of TNF, IFNγ or a combination of both cytokines from 12 hours up to 3 days, and expression of HLA-DP molecules was quantified over time using flow cytometric analyses. IFNγ stimulation induced increased expression of HLA-DP molecules on EpCAM+ IECs in colon organoids over time (Supplementary Figure 4A–C). This effect of IFNγ was further enhanced by addition of TNF (Supplementary Figure 4B), whereas stimulation with TNF alone did not induce HLA-DP protein expression on IECs in colon organoids (Supplementary Figure 4B, C). Remarkably, the upregulation of HLA-DP molecules upon dual stimulation with IFNγ and TNF was significantly higher on IECs in organoids generated from UC-affected colon compared to controls (Figure 2D). Previously, increased expression of HLA-DP301 compared to HLA-DP401 molecules has been described39. Although a trend towards increased expression of HLA-DP by HLA-DP301+ IECs compared to HLA-DP401+ IECs was observed, this difference decreased upon stimulation with IFNγ and TNF (Supplementary Figure 4D), in line with previous studies.39 Together, these results demonstrate that IFNγ alone and in synergy with TNF induces HLA-DP expression by IECs, including the UC risk HLA-DP401 molecule. Furthermore, IECs from UC tissues maintained an enhanced pro-inflammatory response to IFNγ and TNF over time upon culture in organoids.
HLA-DP401 engagement activates NKp44+ NK cells
Binding of activating NK cell receptors to their respective ligands leads to activation, degranulation, cytokine production and receptor internalization.40 We next investigated whether haplotype-specific HLA-DP recognition activated NK cells from individuals with UC and controls. NKp44 expression by peripheral blood-derived NK cells was induced upon culture with IL-2 and IL-15.41 Culture with HLA-DP401 molecules triggered NK cell degranulation (CD107a) and TNF production to similar levels as the positive control (anti-NKp44) and significantly higher compared to HLA-DP301 molecules for NK cells from both healthy controls and individuals with UC (Figure 3A, Supplementary Figure 5A and B). In line with NKp44-mediated activation, NKp44 expression on NK cells was significantly decreased after engagement with HLA-DP401, but not with HLA-DP301 molecules, in healthy individuals and individuals with UC (Figure 3A). These data demonstrate that binding of NKp44 to HLA-DP401, but not HLA-DP301 molecules, activates NK cells from individuals with UC and controls in a haplotype-dependent manner, and results in the downregulation of NKp44 receptor expression on NK cells.
Figure 3: Increased activation and TNF production by NK cells of individuals with UC after engagement with HLA-DP401.
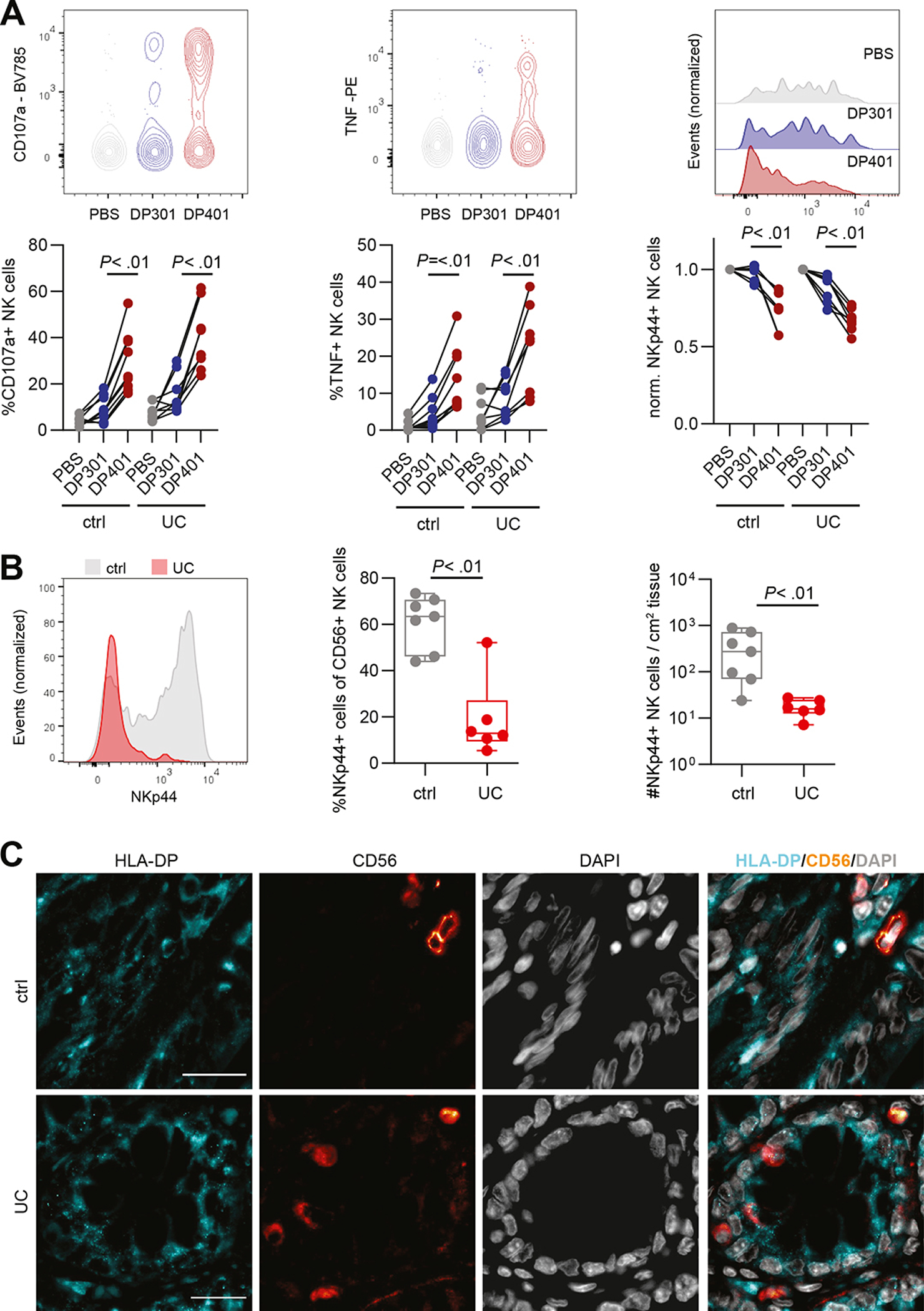
A. Representative flow plots for CD107a and TNF, representative histograms of NKp44 expression and summarized plots of peripheral blood-derived NK cells from controls and individuals with UC after incubation with PBS, HLA-DP301 and HLA-DP401 measured by flow cytometry. Each dot represents an individual donor and lines connect CD107a, TNF or NKp44 expression of one donor (ctrl: n = 9 replicates of 5 donors; UC: n = 8 replicates of 4 donors). B. Representative histograms of NKp44 expression on intestinal CD56+ NK cells in colon of non-inflammatory controls (ctrl) and individuals with UC (left panel) measured by flow cytometry. Median percentages of NKp44+ cells of CD56+ NK cells in non-inflammatory controls (n = 7) and individuals with UC (n = 6) (middle panel). Median counts of NKp44+ CD56+ NK cells per cm2 in control (n = 7) and UC-affected tissues (n = 6) (right panel). C. Representative single and merged fluorescence images of HLA-DP, CD56 and DAPI of control and UC-affected colon tissues. Scale bars: 20 μm.
All boxes indicate medians with 25% and 75% quartile ranges, and whiskers indicate minimum and maximum values. Statistical significance was measured using Wilcoxon signed rank tests (A) or Mann-Whitney U comparisons (B).
To determine whether NKp44 expression by NK cells as a reflection of activation was also altered in the intestines of individuals with UC, we compared the frequencies and total numbers per cm2 of tissue of NKp44+ CD56+ CD127− NK cells in UC affected intestinal epithelium and controls using flow cytometry. While frequencies and total numbers of CD56+ NK cells did not differ significantly between individuals with UC and controls (data not shown), frequencies of NKp44+ cells within CD56+ NK cell population and total numbers of NKp44+ CD56+ CD127− NK cells were significantly lower in intestinal samples from individuals with UC compared to controls (Figure 3B, Supplementary Figure 5C). Due to the relatively small sample size for these in-depth functional studies of intestinal samples, the analyses could not be stratified by HLA-DP haplotype. We did however observe a negative trend between NKp44+ NK cells and HLA-DP+ IECs (Spearman r = −0.38; P = .2) assessed on the same sample, suggesting that increased frequencies of HLA-DP+ IECs correlate with a decrease in frequencies of intestinal NKp44+ NK cells (Supplementary Figure 5D).
In addition to NK cells, epithelial ILCs can express NKp44 and have been shown to contribute to intestinal tissue inflammation16,17,42. To assess whether ILCs also can be activated by HLA-DP molecules in a haplotype-specific manner, sorted intestinal ILCs were incubated with HLA-DP401 or HLA-DP301 molecules. ILCs incubated with HLA-DP401 molecules exhibited a significantly higher TNF production compared to stimulation with HLA-DP301 molecules (Supplementary Figure 5E), whereas IL-22 production was absent upon co-incubation with both HLA-DP molecules (data not shown). These observations suggest that the NKp44-HLA-DP receptor-ligand interaction can be utilized by different immune cell types; however, considering the 12-fold higher numbers of epithelial NKp44+ NK cells compared to NKp44+ ILCs (Supplementary Figure 5F), NK cells are likely the principal mediators of inflammation in response to HLA-DP expressing IECs.
We next quantified mRNA expression of NKp44 (NCR2) in single NK cells isolated from the epithelium of UC-affected intestines and controls, as mRNA levels should not be affected by engagement with the ligand HLA-DP. NCR2 was higher expressed in epithelial NK cells from UC-affected epithelium compared to controls (Supplementary Figure 6A). Furthermore, significant increased expression of IFNG, LAMP1 (encoding CD107a), PRF1 (encoding perforin) was detected in UC-derived epithelial NK cells compared to controls (Supplementary Figure 6A). The percentage of TNF+ NK cells did not differ significantly between UC and control derived samples (Supplementary Figure 6B), this is in line with previous studies reporting that increased TNF protein levels depend substantially on post transcriptional regulation28,43,44. Taken together, UC-derived epithelial NK cells have an increased pro-inflammatory profile together with increased expression of mRNA of NKp44. To further visualize the anatomical location of NK cells in the intestinal epithelium, a co-staining using immunofluorescence analysis of CD56 and HLA-DP was established. In line with immunohistochemical and flow cytometric analyses, HLA-DP expression was increased in UC-affected intestines and NK cells were detected neighboring HLA-DP+ IECs (Figure 3C, Supplementary Figure 6C, D). Taken together, these data demonstrate the NKp44-expressing NK cells from individuals with UC can be activated and produce pro-inflammatory cytokines upon recognition of HLA-DP401, the HLA-DP haplotype associated with UC risk, but not by HLA-DP301, associated with UC protection.
Binding to HLA-DP-expressing IECs results in increased activation of primary NKp44+ NK cells
To determine whether HLA-DP molecules expressed on IECs can bind to NKp44 and modulate NK cell functioning in a haplotype dependent manner, we compared binding of NKp44 Fc constructs to IECs in IFNγ-stimulated intestinal organoids from HLA-DP401pos individuals to organoids from HLA-DP301pos individuals. HLA-DP401-expressing IECs displayed significantly higher binding to NKp44 Fc constructs compared to HLA-DP301-expressing IECs (Figure 4A, Supplementary Figure 7A). In contrast, IFNγ-treated HLA-DP negative or low-expressing IECs showed no detectable binding to NKp44 Fc constructs, independent of the HLA-DP allotype (Figure 4A). In order to investigate whether the differential binding efficiencies of HLA-DP401 and -DP301 to NKp44 resulted in functional consequences for NK cell activation, we quantified CD107a expression of NK cells after co-culture with IECs from unstimulated and IFNγ-stimulated intestinal organoids from homozygous HLA-DP401pos or -DP301pos individuals (Figure 4B). NK cell donors were matched to organoid donors with regard to their HLA class I genotype (HLA-B/HLA-C), which was comparable between HLA-DP401pos and HLA-DP301pos donors, and the same NK cell donors were used in co-culture experiments with IECs from HLA-DP401- and HLA-DP301-expressing intestinal organoids. As previously described,41 NKp44 expression was highest on CD56++ NK cells allowing interactions with HLA-DP in this NK cell subset. Therefore, NK cell activation was determined on CD56++ NK cells. To adjust for differential expression of NKp44 between different NK cell donors (NKp44+ cells of CD56++ NK cells ranged from 60–90% (data not shown)), degranulation of CD56++ NK cells in response to culture with HLA-DP301-expressing IECs was set to 1 for each NK cell donor and compared to NK cell degranulation after co-culture with HLA-DP401-expressing IECs. Degranulation did not significantly differ after co-culture with unstimulated organoids from either HLA-DP haplotypes, but was significantly higher in CD56++ NK cells after co-culture with IFNγ-stimulated, HLA-DP401-expressing IECs as compared to co-culture with IFNγ-stimulated, HLA-DP301-expressing IECs (Figure 4B and C, Supplementary Figure 7B). In line, a trend towards higher TNF production by CD56++ NK cells after co-culture with IFNγ-treated, HLA-DP401-expressing IECs was observed compared to cultures with HLA-DP301-expressing IECs (Figure 4D, Supplementary Figure 7C). As expected, NK cell degranulation was associated with NKp44 downregulation on CD56++ NK cells co-cultured with HLA-DP401 expressing IECs (Figure 4E, Spearman r: 0.81, P = .02). NK cell degranulation and TNF production in NK cell - IEC (HLA-DP401+) co-cultures was significantly reduced by an anti-NKp44 blocking antibody, further validating the critical interaction of NKp44 with HLA-DP (Figure 4F, G).
Figure 4: Expression of HLA-DP401 on IECs is recognized by NK cells and results in NK cell activation.
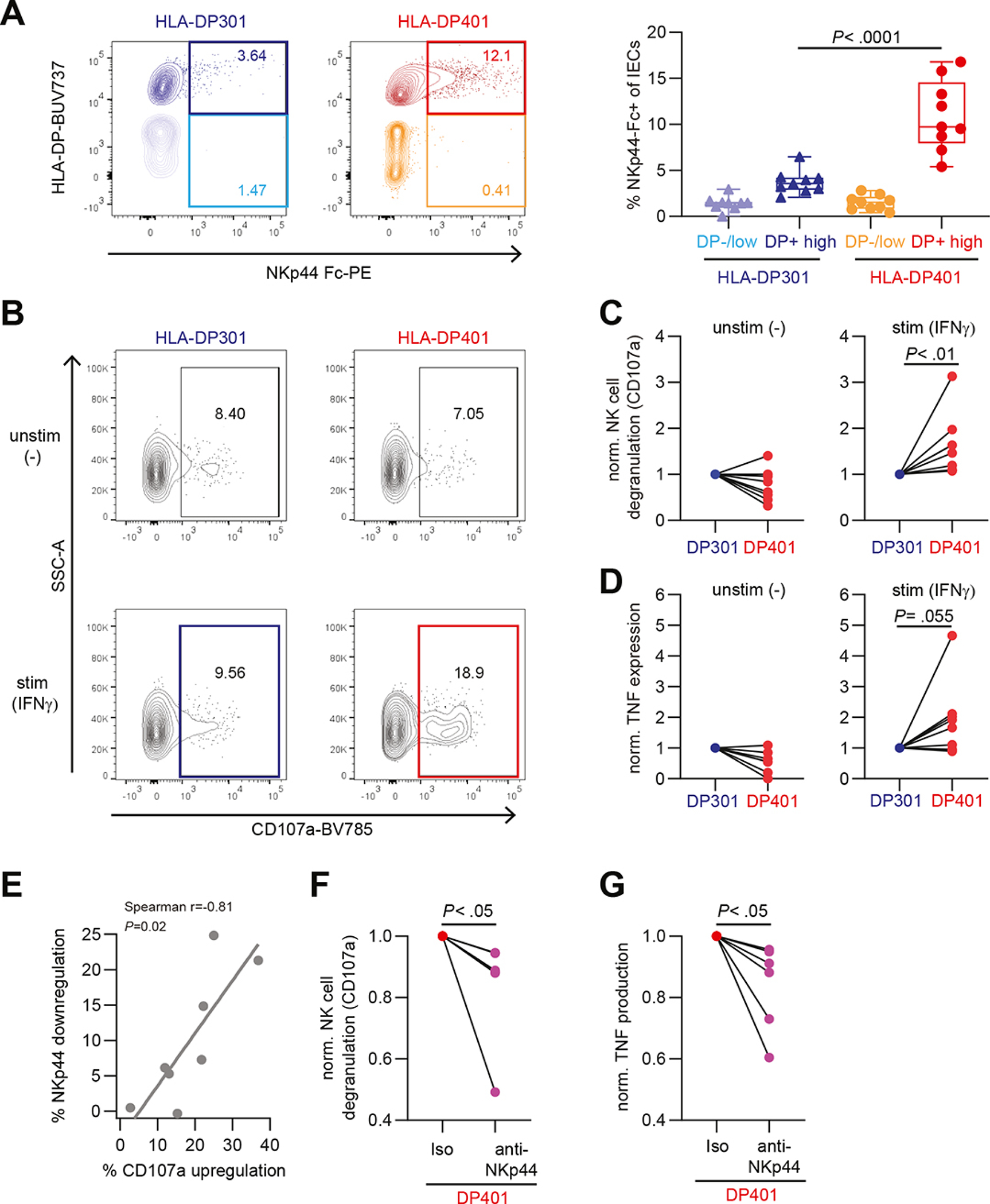
A. Representative flow cytometric plots of NKp44 Fc construct binding to HLA-DP301- or HLA-DP401-expressing IECs in organoids after IFNγ stimulation (200 U/ml for 3 days) (left panel). Median percentages of NKp44 Fc construct binding to HLA-DP−/low or HLA-DPhigh IECs (n = 9 replicates of 2 donors in 3 independent experiments for each haplotype) (right panel). B. Representative flow cytometric plots showing CD107a expression of CD56++ NK cells after co-culture with HLA-DP301- or HLA-DP401 expressing IECs. IECs were either unstimulated (−) or stimulated (IFNγ) (200 U/ml for 3 days). C. Plots show fold change degranulation of CD56++ NK cells after co-culture with unstimulated (−) (left panel) or stimulated (IFNγ) (right panel) IECs (degranulation upon culture with HLA-DP301-expressing IECs was set to 1 for each NK cell donor). Each dot represents an individual donor (n = 8 NK cell donors) and lines connect CD107a expression of one NK cell donor. D. Plots show fold change of TNF expression of CD56++ NK cells after co-culture with unstimulated (−) (left panel) or stimulated (IFNγ) (right panel) IECs (TNF expression upon culture with HLA-DP301-expressing IECs was set to 1 for each NK cell donor). Each dot represents an individual donor (n = 8 NK cell donors) and lines connect TNF expression of one NK cell donor. E. Plot showing correlation between percentage of NKp44 downregulation against percentage of CD107 upregulation on NK cells co-cultured with HLA-DP401 expressing IECs (n = 8 NK cell donors). Line indicates linear regression. F. and G. Plots show fold change degranulation (F) and TNF expression (G) of CD56++ NK cells after co-culture with IFNγ-stimulated HLA-DP401+ IECs in presence of an isotype or anti-NKp44 blocking antibody (degranulation and TNF expression of CD56++ NK cells upon co-culture with HLA-DP401-expressing IECs in presence of an isotype antibody was set to 1 for each NK cell donor). Each dot represents an individual donor (n = 6 NK cell donors) and lines connect CD107a or TNF expression of one NK cell donor.
All boxes indicate medians with 25% and 75% quartile ranges, and whiskers indicate minimum and maximum values. Statistical significance was measured using Mann-Whitney U comparisons (A) or Wilcoxon signed rank tests (C, D, F, G).
To assess whether the enhanced NK cell activation in response to HLA-DP401 expressing IECs resulted in epithelial damage, we performed co-cultures of NK cells with intestinal 3D-organoids. After 6 hours, the size of HLA-DP401-expressing intestinal 3D-organoids co-cultured with CD56++ NK cells was significantly reduced indicating destruction of the 3D-organoids by the NKp44+ NK cells (Figure 5A), whereas the size of HLA-DP301-expressing organoids in co-culture with CD56++ NK cells remained similar to cultures without NK cells (Figure 5A). In line with these morphometric analyses, the percentages of dead IECs in HLA-DP401+ organoids cultured with NK cells were significantly higher as compared to HLA-DP301+ organoids (Figure 5B, Supplementary Figure 8A). Blocking of the NKp44-HLA-DP interaction using an anti-NKp44 blocking antibody rescued viability of HLA-DP401+ IECs in organoids cultured with NK cells (Figure 5C). These findings demonstrate that NKp44+ NK cells are activated by IECs expressing UC risk HLA-DP401 molecules, resulting in intestinal epithelial tissue damage that can be blocked by anti-NKp44.
Figure 5: Expression of HLA-DP401 on IECs results in the induction of cytotoxicity by NK cells and reduces IECs viability.
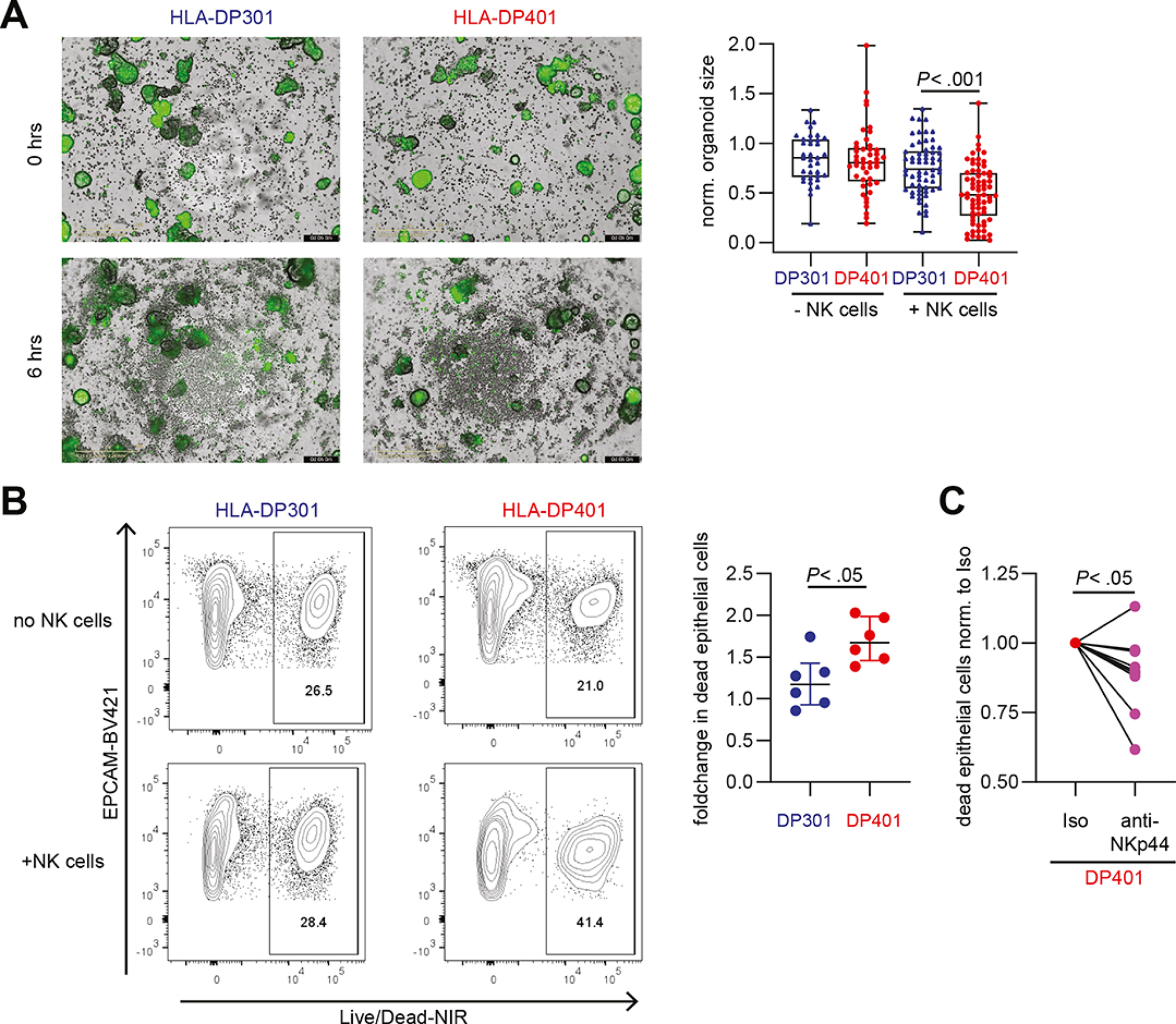
A. Representative images of co-cultures of CD56++ NK cells with IFNγ-stimulated, Calcein-labelled (green) HLA-DP301- or HLA-DP401-expressing intestinal organoids at 0 hrs and 6 hrs (left panel). Plot shows normalized organoid size of HLA-DP301- and HLA-DP401-expressing intestinal organoids in presence or absence of CD56++ NK cells (n = 36–68 organoids per condition from experiments with 3 different NK cell donors). Scale Bars: 400 μm. B. Representative flow cytometric plots showing LIVE/DEAD™ Fixable Near-IR Dead Cell Stain of HLA-DP301+ or HLA-DP401+ EpCam+ IECs after incubation without NK cells or after co-culture with CD56++ NK cells. Plot shows fold change (normalized to dead IECs (LIVE/DEAD™ Fixable Near-IR Dead Cell Stain) without NK cells) in dead HLA-DP301+ or HLA-DP401+ IECs after co-culture with CD56++ NK cells (n = 6 replicates from 2 NK cell donors). C. Plot shows fold change in dead HLA-DP401+ IECs (LIVE/DEAD™ Fixable Near-IR Dead Cell Stain) after co-culture with CD56++ NK cells in presence of an isotype or anti-NKp44 blocking antibody (percentage of dead IECs upon culture with CD56++ NK cells in presence of an isotype antibody was set to 1 for each NK cell donor). Each dot represents an individual donor (n = 10 replicates from 5 NK cell donors) and lines connect one NK cell donor.
All boxes indicate medians with 25% and 75% quartile ranges, and whiskers indicate minimum and maximum values. Statistical significance was measured using Ordinary Two-way ANOVA (A), Mann-Whitney U comparisons (B) or Wilcoxon signed rank tests (C).
Discussion
A combination of microbiota, environmental components, host genetic factors and mucosal immune dysregulation are involved in the pathogenesis of UC.5 In particular, significant genetic associations with UC have been identified in the HLA class II region.9,45 Here, we identify HLA-DPA1*01:03-DPB1*04:01 (HLA-DP401) as a risk haplotype for UC and demonstrate that HLA-DP401 molecules expressed by IECs trigger activation of primary human NK cells via NKp44 resulting in IEC cell death.
Previous studies have identified HLA-DP alleles as risk factors for UC.9,24 We extended our study to investigate the associations of UC with the HLA-DP haplotype, which includes the combination of the alpha (HLA-DPA1) and beta (HLA-DPB1) chain. These new findings are in line with previous studies reporting a decreased risk of HLA-DPB1*03:01 and *11:01 alleles and *04:01 with increased risk for UC.9 Goyette et al9 also identified the HLA-DPB*06:01 allele to be associated with a decreased risk for UC, which in our analysis only showed a trend (P = 0.09, OR = 0.9), potentially due to differential imputation accuracy score used. Of note, the UC risk allele HLA-DPB1*04:01 is in weak linkage disequilibrium (LD) with the previously reported UC risk HLA class II alleles HLA-DRB1*15:01, HLA-DQA1*01:02 and HLA-DQB1*06:01/2, whereas the UC protective alleles HLA-DP*11:01 and HLA-DP*03:01 are in weak LD with other reported UC protective alleles (HLA-DRB1*13:02, HLA-DRB1*07:01, HLA-DQA1*02:01, HLA-DQB1*02:01 and HLA-DQB1*06:04).9,24,46,47 We furthermore assessed whether binding of other NK cell receptors, such as LAG3 to HLA class II molecules34 display distinct patterns associated with UC risk or protective alleles, however these were not detected. The findings presented here highlight the need to determine the functional consequences of risk-associated haplotypes in the interactions between human immune and human tissue cells to disentangle the individual contributions of risk alleles/haplotypes in disease pathogenesis. This is particularly critical for receptors and their ligands such as NKp44 and HLA-DP, which are restricted to humans and can not be studied in mice.
Although it is well-known that HLA class II molecules are expressed on antigen-presenting cells and B cells, epithelial cells can induce HLA class II molecule expression upon stimulation with inflammatory signals, through the activation of the transcription factor CIITA.48,49 In line with previous studies investigating HLA class II molecule expression in UC-affected intestines,50,51 we observed significant upregulation of HLA-DP molecules in the intestinal epithelium of individuals with UC. Stimulation of intestinal organoids with the pro-inflammatory cytokines IFNγ and TNF, which also displayed increased expression in UC affected intestinal epithelium, induced HLA-DP upregulation on IECs, recapitulating the in vivo situation. Organoids generated from HLA-DP401pos compared to HLA-DP301pos individuals provided the opportunity to demonstrate that IFNγ-induced HLA-DP molecules on IECs are recognized by NKp44 in a haplotype-dependent manner, with UC risk HLA-DP401 molecules being recognized by NKp44, while UC protective HLA-DP301 molecules were not. Interestingly, we also observed higher HLA-DP expression upon IFNγ and TNF stimulation in intestinal organoids derived from individuals with UC than from controls, indicating that inflammatory response-signatures can be maintained in vitro in intestinal organoids. Thus, organoids recapitulate disease states and can be used to investigate the role of specific genotypes in interactions with immune cells in human diseases, especially in the context of genes restricted to humans.52
The role of NK cells in UC has been less investigated compared to other immune cells. This may be due to the molecular differences between human NK cells and those of animal models, especially critical receptors for NK cell functioning, such as KIRs but also NKp44. Nevertheless, previous studies analyzing human samples have indicated a potential role for NK cells in UC.13–15 Our studies demonstrate that human NKp44+ NK cells were significantly stronger activated by IECs expressing the UC risk molecule HLA-DP401 than by IECs expressing the UC protective molecule HLA-DP301; an observation that is consistent with the better binding of NKp44 to HLA-DP401 compared to HLA-DP301.18 NK cells have exceptional cytotoxic activity, and these new findings indicate that NKp44+ NK cells can induce epithelial tissue damage in UC in an HLA-DP haplotype-dependent manner, following the risk profile of our large genetic study. Blocking of NKp44 inhibited NK cell activation and rescued IEC viability in HLA-DP401+ organoids confirming the critical role of NKp44 in HLA-DP401-mediated NK cell activation. NKp44 can also be expressed by ILCs, and in line with previous studies NKp44-mediated activation of ILCs influenced production of TNF but not IL-22, potentially contributing to inflammation and tissue damage in UC.16 Overall, the methodological approach applied here, using organoid systems encoding for specific HLA alleles, offers a novel in vitro approach to determine interactions of immune cells in immune-mediated diseases in general.
In conclusion, our data identify a molecular and functional immune correlate for the genetic associations of HLA-DPA1*01:03-DPB1*04:01 and HLA-DPA1*01:03-DPB1*03:01 in UC by demonstrating HLA-DP haplotype-dependent activation of NKp44+ NK cells and epithelial tissue damage. Blockade of NKp44/HLA-DP interactions might therefore provide a promising future therapeutic approach to decrease epithelial damage in the over 40% of all UC patients who carry the UC risk HLA-DP haplotype, and may complement current treatment strategies.
Supplementary Material
{kind=link}
What you need to know.
Background and Context
The chronic inflammatory disease Ulcerative Colitis (UC) affects millions of individuals worldwide; however, the precise etiology of UC is unknown. HLA molecules have been associated with risk for UC and the recent finding that the activating NK cell receptor NKp44 binds subsets of HLA-DP molecules suggested that NK cells might contribute to intestinal inflammation.
New Findings
A specific genotype of the HLA class II molecule HLA-DP, HLA-DPA1*01:03-DPB1*04:01, was identified as a UC risk haplotype. Such HLA-DP molecules expressed on intestinal epithelial cells were able to bind NKp44 and activate NKp44+ NK cells leading to intestinal epithelial damage.
Limitations
Our cohort consisted only of Caucasian individuals. Whether the here identified risk haplotype also frequently occurs in other populations needs to be investigated.
Basic Research Relevance
The herein identified UC risk genotype HLA-DPA1*01:03-DPB1*04:01 is the most frequent HLA-DP haplotype in the European population with a frequency of more than 40%. Targeting the NKp44/HLA-DP interaction might therefore constitute a promising future therapeutic approach to decrease epithelial damage by NK cells in UC patients carrying this UC risk HLA-DP haplotype.
Acknowledgement
We would like to thank all donors of intestinal tissues who participated in this study. Further, we thank the colleagues from the Department of General, Visceral and Thoracic Surgery of the University Hospital Hamburg-Eppendorf for the collection of intestinal tissues. We also thank the core facility Fluorescence Cytometry at the Leibniz Institute of Virology (LIV) and the blood donors and coordinators of the Healthy Cohort at the LIV. The Hamburg Intestinal Tissue Study group includes Alaa Akar, Cornelius Flemming, Felix, Flomm, Markus Flosbach, Julia Jäger, Niklas Jeromin, Johannes Jung, Mareike Ohms, Konrad Reinshagen, Johann Rische, Adrian Sagebiel, Deborah Sandfort, Fenja Steinert, Christian Tomuschat, Jasmin Wesche. We thank the International IBD Genetics Consortium for providing access to the haplotype data.
Grant support
This work was supported by the Daisy Huet Roell Foundation, the European Research Council (#884830), the Landesforschungsförderung (LFF-75) Hamburg City of Hamburg, the DFG (CRC/1192), the BMBF (eMed Consortia Fibromap) and the Novo Nordisk Foundation (Young Investigator Award – NNF21OC0066381). This project has been funded in part with federal funds from the Frederick National Laboratory for Cancer Research, under Contract No. HHSN261200800001E, and by the Intramural Research Program of the NIH, Frederick National Lab, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The Leibniz Institute of Virology is supported by the Free and Hanseatic City of Hamburg and the Federal Ministry of Health.
Abbreviations used in this paper:
- CIITA
class II transactivator
- EM
expansion medium
- HIBAG
HLA genotype Imputation with Attribute Bagging
- HLA
human leukocyte antigen
- HLA-DP301
HLA-DPA1*01:03-DPB1*03:01
- HLA-DP401
HLA-DPA1*01:03-DPB1*04:01
- IBD
inflammatory bowel disease
- IECs
intestinal epithelial cells
- IELs
intraepithelial lymphocytes
- IFNγ
interferon-gamma
- ILCs
innate lymphoid cells
- JAK
Janus kinase
- KIRs
killer immunoglobulin-like receptors
- LD
linkage disequilibrium
- MFI
median fluorescence intensity
- MichiganImpServer
Michigan Imputation Server
- NK
natural killer
- OR
odds ratio
- SNPs
single nucleotide polymorphisms
- TNF
tumor necrosis factor
- UC
ulcerative colitis
Members of the International Inflammatory Bowel Disease Genetics Consortium (IIBDGC)
Shifteh Abedian, Clara Abraham, Jean-Paul Achkar, Tariq Ahmad, Rudi Alberts, Behrooz Alizadeh, Leila Amininejad, Ashwin N Ananthakrishnan, Vibeke Andersen, Carl A Anderson, Jane M Andrews, Vito Annese, Guy Aumais, Leonard Baidoo, Robert N Baldassano, Peter A Bampton, Murray Barclay, Jeffrey C Barrett, Johannes Bethge, Claire Bewshea, Joshua C Bis, Alain Bitton, Thelma BK, Gabrielle Boucher, Oliver Brain, Stephan Brand, Steven R Brant, Jae Hee Cheon, Angela Chew, Judy H Cho, Isabelle Cleynen, Ariella Cohain, Rachel Cooney, Anthony Croft, Mark J Daly, Mauro D’Amato, Silvio Danese, Naser Ebrahim Daryani, Lisa Wu Datta, Frauke Degenhardt, Goda Denapiene, Lee A Denson, Kathy L Devaney, Olivier Dewit, Renata D’Inca, Hazel E Drummond, Marla Dubinsky, Richard H Duerr, Cathryn Edwards, David Ellinghaus, Pierre Ellul, Motohiro Esaki, Jonah Essers, Lynnette R Ferguson, Eleonora A Festen, Philip Fleshner, Tim Florin, Denis Franchimont, Andre Franke, Yuta Fuyuno, Richard Gearry, Michel Georges, Christian Gieger, Jürgen Glas, Philippe Goyette, Todd Green, Anne M Griffiths, Stephen L Guthery, Hakon Hakonarson, Jonas Halfvarson, Katherine Hanigan, Talin Haritunians, Ailsa Hart, Chris Hawkey, Nicholas K Hayward, Matija Hedl, Paul Henderson, Georgina L Hold, Myhunghee Hong, Xinli Hu, Hailiang Huang, Jean-Pierre Hugot, Ken Y Hui, Marcin Imielinski, Omid Jazayeri, Laimas Jonaitis, Luke Jostins, Garima Juyal, Ramesh Chandra Juyal, Rahul Kalla, Tom H Karlsen, Nicholas A Kennedy, Mohammed Azam Khan, Won Ho Kim, Takanari Kitazono, Gediminas Kiudelis, Michiaki Kubo, Subra Kugathasan, Limas Kupcinskas, Christopher A Lamb, Katrina M de Lange, Anna Latiano, Debby Laukens, Ian C Lawrance, James C Lee, Charlie W Lees, Marcis Leja, Nina Lewis, Johan Van Limbergen, Paolo Lionetti, Jimmy Z Liu, Edouard Louis, Yang Luo, Gillian Mahy, Masoud Mohammad Malekzadeh, Reza Malekzadeh, John Mansfield, Suzie Marriott, Dunecan Massey, Christopher G Mathew, Toshiyuki Matsui, Dermot PB McGovern, Andrea van der Meulen, Vandana Midha, Raquel Milgrom, Samaneh Mirzaei, Mitja Mitrovic, Grant W Montgomery, Craig Mowat, Christoph Müller, William G Newman, Aylwin Ng, Siew C Ng, Sok Meng Evelyn Ng, Susanna Nikolaus, Kaida Ning, Markus Nöthen, Ioannis Oikonomou, David Okou, Timothy R Orchard, Orazio Palmieri, Miles Parkes, Anne Phillips, Cyriel Y Ponsioen, Urõs Potocnik, Hossein Poustchi, Natalie J Prescott, Deborah D Proctor, Graham Radford-Smith, Jean- Francois Rahier, Miguel Regueiro, Walter Reinisch, Florian Rieder, John D Rioux, Rebecca Roberts, Gerhard Rogler, Richard K Russell, Jeremy D Sanderson, Miquel Sans, Jack Satsangi, Eric E Schadt, Michael Scharl, John Schembri, Stefan Schreiber, L Philip Schumm, Regan Scott, Mark Seielstad, Tejas Shah, Yashoda Sharma, Mark S Silverberg, Alison Simmons, Lisa A Simms, Abhey Singh, Jurgita Skieceviciene, Suzanne van Sommeren, Kyuyoung Song, Ajit Sood, Sarah L Spain, A. Hillary Steinhart, Joanne M Stempak, Laura Stronati, Joseph JY Sung, Stephan R Targan, Kirstin M Taylor, Emilie Theatre, Leif Torkvist, Esther A Torres, Mark Tremelling, Holm H Uhlig, Junji Umeno, Homayon Vahedi, Eric Vasiliauskas, Anje ter Velde, Nicholas T Ventham, Severine Vermeire, Hein W Verspaget, Martine De Vos, Thomas Walters, Kai Wang, Ming-Hsi Wang, Rinse K Weersma, Zhi Wei, David Whiteman, Cisca Wijmenga, David C Wilson, Juliane Winkelmann, Sunny H Wong, Ramnik J Xavier, Keiko Yamazaki, Suk-Kyun Yang, Byong Duk Ye, Sebastian Zeissig, Bin Zhang, Clarence K Zhang, Hu Zhang, Wei Zhang, Hongyu Zhao, Zhen Z Zhao, Australia and New Zealand IBDGC, Belgium IBD Genetics Consortium, Italian Group for IBD Genetic Consortium, NIDDK Inflammatory Bowel Disease Genetics Consortium, Quebec IBD Genetics Consortium, United Kingdom IBDGC, Wellcome Trust Case Control Consortium.
Footnotes
Declaration of Interests
A.N. and M.A. are inventors of one provisional patent that describes the binding of NKp44 to a subset of HLA-DP molecules, and the role of these interactions in Graft versus Host Disease. All other authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability
The ImmunoChip data used in this study are proprietary to the International Inflammatory Bowel Disease Genetics Consortium and may be requested from the consortium (http://www.ibdgenetics.org/). All other data used in this study have been collected in clinical studies and are subject to the regulations of the ethics committee of the Freie und Hansestadt Hamburg (Ärztekammer Hamburg). Participant’s written informed consent has been provided for data generation and handling according to the approved protocols and scopes of the study. Data storage is performed at the Leibniz Institute of Virology. Data are available upon request and can be shared after confirming that data will be used within the scope of the originally provided informed consent. Further information and requests for data should be directed to the Lead Contact, Madeleine Bunders (ma.bunders@uke.de).
References
Author names in bold designate shared co-first authorship.
- 1.Alatab S, Sepanlou SG, Ikuta K, et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boer NKH de, Ahuja V, Almer S, et al. Thiopurine Therapy in Inflammatory Bowel Diseases: Making New Friends Should Not Mean Losing Old Ones. Gastroenterology 2019;156. [DOI] [PubMed] [Google Scholar]
- 3.Al-Horani R, Spanudakis E, Hamad B. The market for ulcerative colitis. Nat Rev Drug Discov 2022;21. [DOI] [PubMed] [Google Scholar]
- 4.Panaccione R, Ghosh S, Middleton S, et al. Combination therapy with infliximab and azathioprine is superior to monotherapy with either agent in ulcerative colitis. Gastroenterology 2014;146. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi T, Siegmund B, Berre C Le, et al. Ulcerative colitis. Nat Rev Dis Prim 2020;6. [DOI] [PubMed] [Google Scholar]
- 6.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 2009;9. [DOI] [PubMed] [Google Scholar]
- 7.Matts SG. The value of rectal biopsy in the diagnosis of ulcerative colitis. Q J Med 1961:393–407. [PubMed] [Google Scholar]
- 8.Geboes K, Riddell R, Öst A, et al. A reproducible grading scale for histological assessment of inflammation in ulcerative colitis. Gut 2000;47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goyette P, Boucher G, Mallon D, et al. High-density mapping of the MHC identifies a shared role for HLA-DRB101:03 in inflammatory bowel diseases and heterozygous advantage in ulcerative colitis. Nat Genet 2015;47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang H, Fang M, Jostins L, et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 2017;547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hegazy AN, West NR, Stubbington MJT, et al. Circulating and Tissue-Resident CD4+ T Cells With Reactivity to Intestinal Microbiota Are Abundant in Healthy Individuals and Function Is Altered During Inflammation. Gastroenterology 2017;153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leppkes M, Neurath MF. Cytokines in inflammatory bowel diseases – Update 2020. Pharmacol Res 2020;158. [DOI] [PubMed] [Google Scholar]
- 13.Hall LJ, Murphy CT, Quinlan A, et al. Natural killer cells protect mice from DSS-induced colitis by regulating neutrophil function via the NKG2A receptor. Mucosal Immunol 2013;6. [DOI] [PubMed] [Google Scholar]
- 14.Jones DC, Edgar RS, Ahmad T, et al. Killer Ig-like receptor (KIR) genotype and HLA ligand combinations in ulcerative colitis susceptibility. Genes Immun 2006;7. [DOI] [PubMed] [Google Scholar]
- 15.Fathollahi A, Aslani S, Mostafaei S, et al. The role of killer-cell immunoglobulin-like receptor (KIR) genes in susceptibility to inflammatory bowel disease: systematic review and meta-analysis. Inflamm Res 2018;67. [DOI] [PubMed] [Google Scholar]
- 16.Glatzer T, Killig M, Meisig J, et al. RORγt+ Innate Lymphoid Cells Acquire a Proinflammatory Program upon Engagement of the Activating Receptor NKp44. Immunity 2013;38. [DOI] [PubMed] [Google Scholar]
- 17.Bernink JH, Peters CP, Munneke M, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol 2013;14. [DOI] [PubMed] [Google Scholar]
- 18.Niehrs A, Garcia-Beltran WF, Norman PJ, et al. A subset of HLA-DP molecules serve as ligands for the natural cytotoxicity receptor NKp44. Nat Immunol 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biton M, Haber AL, Rogel N, et al. T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell 2018;175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiman K, Curman B, Forsum U, et al. Occurrence of Ia antigens on tissues of non-lymphoid origin [13]. Nature 1978;276. [DOI] [PubMed] [Google Scholar]
- 21.Scott H, Solheim BG, Brandtzaeg P, et al. HLA-DR-like Antigens in the Epithelium of the Human Small Intestine. Scand J Immunol 1980;12. [DOI] [PubMed] [Google Scholar]
- 22.Parr EL, McKenzie IFC. Demonstration of Ia antigens on mouse intestinal epithelial cells by immunoferritin labeling. Immunogenetics 1979;8. [Google Scholar]
- 23.Heuberger C, Pott J, Maloy KJ. Why do intestinal epithelial cells express MHC class II? Immunology 2021;162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Degenhardt F, Mayr G, Wendorff M, et al. Transethnic analysis of the human leukocyte antigen region for ulcerative colitis reveals not only shared but also ethnicity-specific disease associations. Hum Mol Genet 2021;30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das S, Forer L, Schönherr S, et al. Next-generation genotype imputation service and methods. Nat Genet 2016;48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schreeder DM, Cannon JP, Wu J, et al. Cutting Edge: FcR-Like 6 Is an MHC Class II Receptor. J Immunol 2010;185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schreurs RRCE, Drewniak A, Bakx R, et al. Quantitative comparison of human intestinal mononuclear leukocyte isolation techniques for flow cytometric analyses. J Immunol Methods 2017;445. [DOI] [PubMed] [Google Scholar]
- 28.Schreurs RRCE, Baumdick ME, Sagebiel AF, et al. Human Fetal TNF-α-Cytokine-Producing CD4 + Effector Memory T Cells Promote Intestinal Development and Mediate Inflammation Early in Life. Immunity 2019;50. [DOI] [PubMed] [Google Scholar]
- 29.Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009;459. [DOI] [PubMed] [Google Scholar]
- 30.Sato T, Stange DE, Ferrante M, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011;141. [DOI] [PubMed] [Google Scholar]
- 31.Zheng X, Shen J, Cox C, et al. HIBAG - HLA genotype imputation with attribute bagging. Pharmacogenomics J 2014;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toyoda H, Wang SJ, Yang HY, et al. Distinct associations of HLA Class II genes with inflammatory bowel disease. Gastroenterology 1993;104. [DOI] [PubMed] [Google Scholar]
- 33.Stokkers PCF, Reitsma PH, Tytgat GNJ, et al. HLA-DR and -DQ phenotypes in inflammatory bowel disease: A meta-analysis. Gut 1999;45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baixeras E, Huard B, Miossec C, et al. Characterization of the Lymphocyte Activation Gene 3-Encoded Protein. A New Ligand for Human Leukocy Antigen Class H Antigens. J Exp Med 1992;176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hollenbach JA, Madbouly A, Gragert L, et al. A combined DPA1~DPB1 amino acid epitope is the primary unit of selection on the HLA-DP heterodimer. Immunogenetics 2012;64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steimle V, Otten LA, Zufferey M, et al. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell 1993;75. [PubMed] [Google Scholar]
- 37.Thelemann C, Eren RO, Coutaz M, et al. Interferon-γ induces expression of MHC class II on intestinal epithelial cells and protects mice from colitis. PLoS One 2014;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wosen JE, Mukhopadhyay D, MacAubas C, et al. Epithelial MHC class II expression and its role in antigen presentation in the gastrointestinal and respiratory tracts. Front Immunol 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meurer T, Arrieta-Bolaños E, Metzing M, et al. Dissecting genetic control of HLA-DPB1 expression and its relation to structural mismatch models in hematopoietic stem cell transplantation. Front Immunol 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Masilamani M, Peruzzi G, Borrego F, et al. Endocytosis and intracellular trafficking of human natural killer cell receptors. Traffic 2009;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barrow AD, Martin CJ, Colonna M. The natural cytotoxicity receptors in health and disease. Front Immunol 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cella M, Gamini R, Sécca C, et al. Subsets of ILC3–ILC1-like cells generate a diversity spectrum of innate lymphoid cells in human mucosal tissues. Nat Immunol 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brook M, Sully G, Clark AR, et al. Regulation of tumour necrosis factor α mRNA stability by the mitogen-activated protein kinase p38 signalling cascade. FEBS Lett 2000;483. [DOI] [PubMed] [Google Scholar]
- 44.Kotlyarov A, Neininger A, Schubert C, et al. MAPKAP kinase 2 is essential for LPS-induced TNF-α biosynthesis. Nat Cell Biol 1999;1. [DOI] [PubMed] [Google Scholar]
- 45.Liu JZ, Sommeren S Van, Huang H, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015;47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Begovich AB, McClure GR, Suraj VC, et al. Polymorphism, recombination, and linkage disequilibrium within the HLA class II region. J Immunol 1992;148. [PubMed] [Google Scholar]
- 47.Howell WM, Evans PR, Devereux SA, et al. Absence of strong HLA-DR/DQ-DP linkage disequilibrium in the British and French Canadian Caucasoid populations. Int J Immunogenet 1993;20. [DOI] [PubMed] [Google Scholar]
- 48.Hershberg RM, Framson PE, Cho DH, et al. Intestinal epithelial cells use two distinct pathways for HLA class II antigen processing. J Clin Invest 1997;100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hershberg RM, Cho DH, Youakim A, et al. Highly polarized HLA class II antigen processing and presentation by human intestinal epithelial cells. J Clin Invest 1998;102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horie Y, Chiba M, Iizuka M, et al. Class II (HLA-DR, -DP, and -DQ) antigens on intestinal epithelia in ulcerative colitis, Crohn’s disease, colorectal cancer and normal small intestine. Gastroenterol Jpn 1990;25. [DOI] [PubMed] [Google Scholar]
- 51.Horie Y, Chiba M, Suzuki T, et al. Induction of major histocompatibility complex class II antigens on human colonic epithelium by interferon-gamma, tumor necrosis factor-alpha, and interleukin-2. J Gastroenterol 1998;33. [PubMed] [Google Scholar]
- 52.Jung JM, Ching W, Baumdick ME, et al. KIR3DS1 directs NK cell–mediated protection against human adenovirus infections. Sci Immunol 2021;6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ImmunoChip data used in this study are proprietary to the International Inflammatory Bowel Disease Genetics Consortium and may be requested from the consortium (http://www.ibdgenetics.org/). All other data used in this study have been collected in clinical studies and are subject to the regulations of the ethics committee of the Freie und Hansestadt Hamburg (Ärztekammer Hamburg). Participant’s written informed consent has been provided for data generation and handling according to the approved protocols and scopes of the study. Data storage is performed at the Leibniz Institute of Virology. Data are available upon request and can be shared after confirming that data will be used within the scope of the originally provided informed consent. Further information and requests for data should be directed to the Lead Contact, Madeleine Bunders (ma.bunders@uke.de).