

Abstract
In eukaryotes and many aerobic prokaryotes, the final step of aerobic respiration is catalyzed by an aa3-type cytochrome c oxidase, which requires a modified heme cofactor, heme a. The conversion of heme b, the prototypical cellular heme, to heme o and ultimately to heme a requires two modifications, the latter of which is conversion of a methyl group to an aldehyde, catalyzed by heme a synthase (HAS). The N- and C-terminal halves of HAS share homology, and each half contains a heme-binding site. Previous reports indicate that the C-terminal site is occupied by a heme b cofactor. The N-terminal site may function as the substrate (heme o) binding site, although this has not been confirmed experimentally. Here, we assess the role of conserved residues from the N- and C-terminal heme-binding sites in HAS from prokaryotic (Shewanella oneidensis) and eukaryotic (Saccharomyces cerevisiae) species – SoHAS/CtaA and ScHAS/Cox15, respectively. A glutamate within the N-terminal site is found to be critical for activity in both types of HAS, consistent with the hypothesis that a carbocation forms transiently during catalysis. In contrast, the residue occupying the analogous C-terminal position is dispensable for enzyme activity. In SoHAS, the C-terminal heme ligands are critical for stability, while in ScHAS, substitutions in either heme-binding site have little effect on global structure. In both species, in vivo accumulation of heme o requires the presence of an inactive HAS variant, highlighting a potential regulatory role for HAS in heme o biosynthesis.
Keywords: Cox15, CtaA, cytochrome c oxidase (Complex IV), heme a synthase, heme o, oxygen activation
Graphical Abstract
Introduction
Aerobic respiration is a key metabolic process in eukaryotes and many prokaryotes and is used to generate the chemiosmotic gradient that ultimately powers ATP synthesis. During aerobic respiration, electrons are transferred through the electron transport chain to oxygen, the terminal electron acceptor, reducing molecular oxygen to water [1]. This reduction is most commonly performed by a dedicated oxidase from the heme-copper oxidase superfamily [2–4].
As their name suggests, all heme-copper oxidases possess metal centers harboring heme and/or copper ions, including a heterobimetallic heme a3-CuB active site [1, 2]. All eukaryotic (mitochondrial) heme-copper oxidases and the majority of known prokaryotic heme-copper oxidases are aa3-type cytochrome c oxidases (CcO), meaning that the heme cofactor is a uniquely modified heme, commonly known as heme a [2–8]. Failure to synthesize heme a typically halts assembly of CcO, a multi-subunit protein complex, and abolishes aerobic respiration in eukaryotes and many prokaryotes [9–19]. Despite the critical role of heme a in aerobic respiration, many aspects of the catalytic mechanism of the final enzyme in its biosynthetic pathway – heme a synthase (HAS) – have not yet been elucidated.
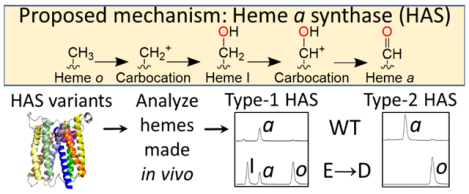
Heme a is synthesized from heme b by two sequential enzymatic reactions. In the first reaction, the vinyl group from pyrrole ring A is converted to a hydroxyethylfarnesyl group by heme o synthase (HOS), producing heme o, a stable intermediate [20]. In the second reaction, HAS oxidizes the C8 methyl group of pyrrole ring D, converting heme o to heme a (Figure 1) [19, 21–28]. HOS and HAS are both multi-span integral membrane proteins located either in the eukaryotic inner mitochondrial membrane or in the prokaryotic cell membrane. HOS is a member of the intramembrane aromatic prenyltransferase superfamily, which is distantly related to soluble isoprenyl pyrophosphate synthases [19, 24, 25, 29–34]. HOS is proposed to transfer a farnesyl group from farnesyl diphosphate to heme b via an ionization-condensation-hydroxylation mechanism [25, 35–38]. HAS, on the other hand, is an oxygen-activating enzyme without close phylogenetic relationships to any other enzyme family.
Figure 1. Overview of the proposed reaction mechanism for the conversion of heme o to heme a by HAS.
In this proposed mechanism, the C8 methyl group (pyrrole ring D substituent, shown in red) first loses a proton and two electrons, generating a carbocation intermediate. An oxygen atom from water (shown in red) then traps this carbocation, yielding heme I, an alcohol intermediate that does not significantly accumulate in vivo. A second oxidation step ultimately converts the alcohol to an aldehyde, generating heme a. The substituent of pyrrole ring A shown in pink is the hydroxyethylfarnesyl group formed by the HOS-catalyzed reaction. Pyrrole rings are labeled A-D on the first heme depicted. This scheme is adapted from Brown et al., 2004 [28].
HAS catalyzes the stepwise oxidation of the C8 methyl group of heme o to an alcohol (heme I) and then to an aldehyde (heme a) in successive oxygen-dependent reactions (Figure 1) [27]. Although HAS was initially proposed to be a monooxygenase, like cytochrome P450 or chlorophyll a oxygenase [22, 26, 27, 39–42], isotope labeling studies indicate that HAS probably oxidizes heme o via electron transfer using a peroxidase-like mechanism [28]. Thus, while HAS uses molecular oxygen as an electron acceptor, water is the source of the oxygen atom that is ultimately incorporated into the C8 alcohol of heme I and the aldehyde group of heme a.
In addition to its primary role in cofactor synthesis, it has been hypothesized that HAS may also play a secondary role in CcO assembly. For example, the absence of a confirmed heme a chaperone in many prokaryotes and the potential danger associated with releasing free heme a into the membrane (where it could have undesirable reactivity) could indicate that HAS helps transfer heme a to subunit 1 of CcO as it matures [43]. Similarly, in eukaryotes, there is evidence to suggest that HAS plays a role in the maturation of Cox1, the heme a-containing structural CcO subunit, possibly by interacting with the Cox1-containing sub-assembly complexes that form prior to heme a insertion [44–47]. Some studies also indicate that Saccharomyces cerevisiae HAS (ScHAS/Cox15) might have a secondary function that is not directly related to cytochrome c oxidase biosynthesis per se, such as HOS regulation [48, 49].
The majority of known HAS sequences, including all eukaryotic versions of HAS, are comprised of eight transmembrane (TM) helices, although certain archaea have a truncated, 4-TM HAS variant and some 9-TM variants have recently been identified in acidophilic bacteria [50–52]. Sequence homology between the N-terminal half of HAS (helices 1–4) and the C-terminal half (helices 5–8) indicates that an ancient gene encoding a 4-TM HAS precursor was duplicated and fused to produce the more common 8-TM version of the enzyme [23, 52, 53]. In addition to the TM domains, HAS contains an elongated loop between TM helices 1 and 2, and 8-TM HAS sequences typically feature an analogous elongated C-terminal loop between TM helices 5 and 6 (Figure 2A). The most common types of HAS, type 1 and type 2, are distinguished by the presence (type 1) or absence (type 2) of a cysteine pair in the elongated N-terminal loop [52, 54]. A second cysteine pair is also present in the elongated C-terminal loop in certain type 1 sub-types, such as Bacillus subtilis HAS (BsHAS/CtaA) [52]. Type-1 HAS sequences are widely distributed among prokaryotes, while all eukaryotic nuclear-encoded HAS sequences and certain prokaryotic HAS sequences are type-2 enzymes [54]. The functional difference between these two types of HAS is poorly understood at present.
Figure 2. Topology and structure of Bacillus subtilis HAS.
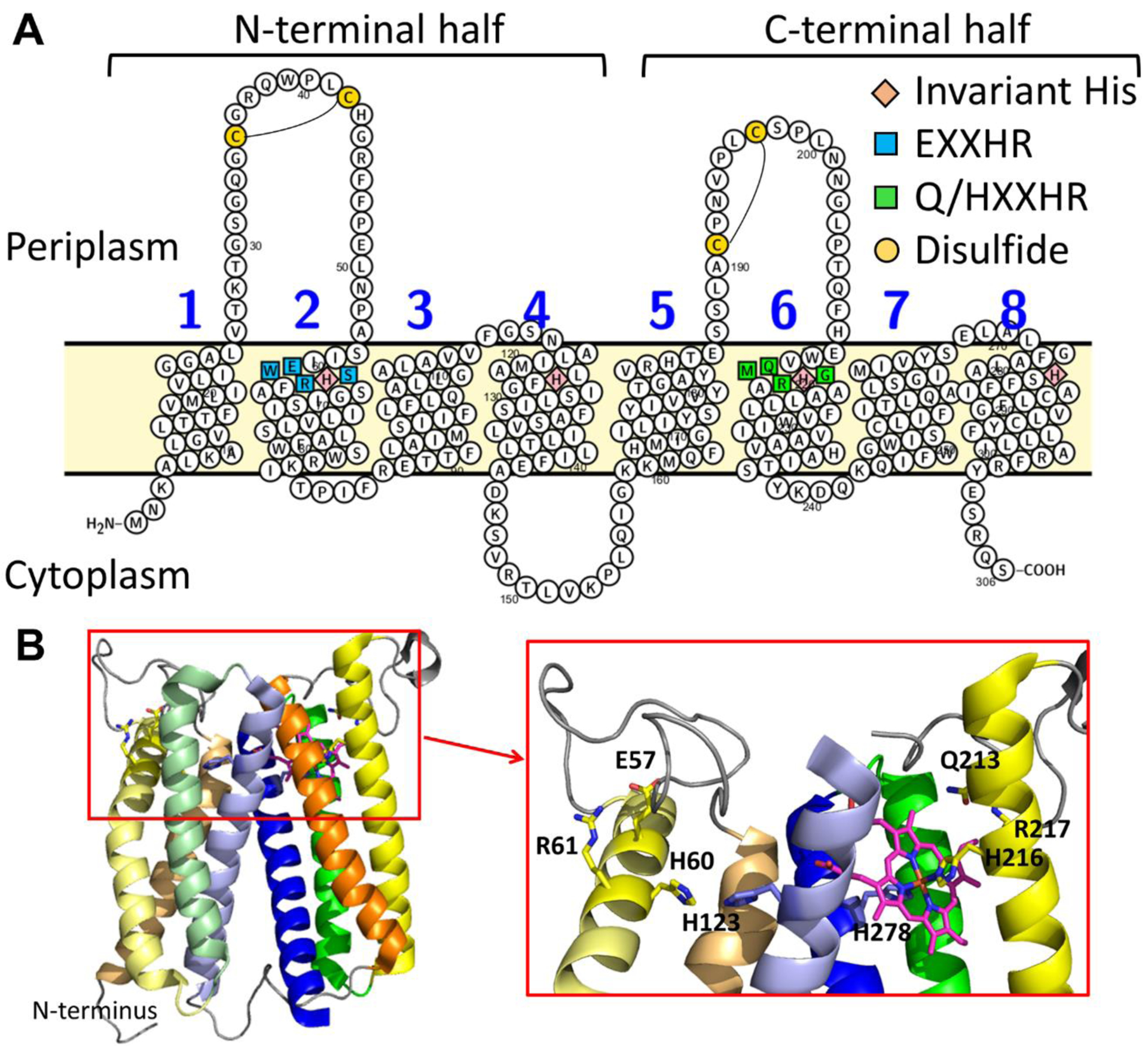
A. The topology of the eight transmembrane (TM) helices in B. subtilis HAS (BsHAS) showing the position of the conserved residues discussed in the text. TM helix 2 harbors a conserved EXXHR motif (cyan squares), and TM helix 6 has a similar Q/HXXHR motif (green squares). The four invariant histidine residues that serve (or are predicted to serve) as heme ligands are shown as pink diamonds. The disulfide bonds (formed between cysteines shown as yellow circles) between the conserved N-terminal cysteine pair (found in all type-1 HAS) and semi-conserved C-terminal cysteine pair (found in type-1.1 and type-1.2 HAS) are also shown in the loops between TM helices 1–2 and 5–6, respectively [52]. Figure prepared using Protter [61]. B. The crystal structure of BsHAS prepared from PDB 6ied [59]. The side chains of the transmembrane residues depicted in panel A are shown as sticks, and heme b is shown with carbon atoms in pink in the C-terminal heme-binding site. TM helices 3 and 5 have been removed in the inset for clarity.
There are four highly conserved histidine residues in all 8-TM (or putative 9-TM) versions of HAS, two located in the N-terminal half of the protein (located on TM helix 2 and TM helix 4) and two located at analogous positions in the C-terminal half (located on TM helix 6 and TM helix 8) (Figure 2A, Table 1) [50, 55, 56]. All four histidines are important for activity in type-1 and type-2 HAS [44, 50, 55–58], and it has long been thought that each of these residues serves as a heme ligand [22, 23, 39, 43, 50, 55, 56]. This hypothesis is supported by structural data for BsHAS and Aquifex aeolicus HAS, which show that the N-and C-terminal halves of the enzyme form two structurally homologous heme-binding sites [59, 60]. The crystal structure for BsHAS reveals that the two conserved histidines in the C-terminal heme-binding site ligate heme b [59]. The N-terminal heme-binding site of that structure was empty, but the position of the conserved N-terminal histidines suggests these residues could serve as ligands to heme o [59], although this has not been demonstrated experimentally. Therefore, the currently available structural and biochemical data indicate that HAS contains two heme-binding sites that, despite being structurally very similar, seem to be specific for either binding heme o (substrate) or heme b (presumably a cofactor). However, biochemical data that conclusively demonstrate the function of each heme-binding site is lacking.
Table 1:
HAS residues discussed in this study.
| Location | BsHAS1 (CtaA) residue | SoHAS2 (CtaA) residue | ScHAS3 (Cox15) residue | Predicted function | References | |
|---|---|---|---|---|---|---|
| Heme-binding site | TM helix | |||||
| N-terminal site | 2 | E57 | E724 | E1664 | Carbocation stabilization | This study |
| 2 | H60 | H754 | H169 | Heme o ligand | [55, 56, 59] | |
| 4 | H123 | H1344 | H245 | Heme o ligand | [55, 56, 59] | |
| C-terminal site | 6 | Q213 | H2414 | Q3654 | This study | |
| 6 | H216 | H2444 | H368 | Heme b ligand | [55, 56, 59] | |
| 8 | H278 | H3024 | H431 | Heme b ligand | [55, 56, 59] | |
B. subtilis heme a synthase (type 1)
S. oneidensis heme a synthase (type 1)
S. cerevisiae heme a synthase (type 2)
Residues substituted in this study.
Other than the four histidine ligands, there are only a few residues that are highly conserved across all known HAS sequences. Of these, one particularly intriguing residue is a glutamate that precedes the first highly conserved histidine as part of the short EXXHR motif located within the N-terminal heme-binding site (TM helix 2). This glutamate is the only highly conserved negatively charged residue within the transmembrane heme-binding domain. The residue at the corresponding position in the C-terminal site is either glutamine (most frequently) or histidine, forming the motif Q/HXXHR (TM helix 6) (Figure 2, Table 1). Because of its location and high degree of conservation, one intriguing possibility is that the glutamate in the N-terminal site plays a role in stabilizing a carbocation that forms transiently at the C8 position of heme o as part of the reaction mechanism (Figure 1) [28, 59], similar to the proposed autocatalytic mechanisms of mammalian peroxidases and certain members of the CYP4 family of cytochrome P450 enzymes (Figure 1) [62–67].
Herein, we systematically probe the function of each heme and the nearby conserved residues via mutagenesis of the N- and C-terminal halves of HAS from representative prokaryotic, type-1 (Shewanella oneidensis) and eukaryotic, type-2 (S. cerevisiae) models (Table 1). Homologous expression of S. oneidensis HAS (SoHAS) in S. oneidensis does not suffer from some of the heterogeneity we have observed previously when expressing BsHAS variants in Escherichia coli. Our results for SoHAS support the hypothesis that the C-terminal four-helical bundle specifically functions as a heme b binding site. Furthermore, analysis of the in vivo activity of SoHAS variants suggests that the highly conserved glutamate (E72) in the N-terminal heme-binding site plays a unique role in catalysis, unlike the corresponding C-terminal residue (H241). Substituting the analogous N-terminal residue (E166) in S. cerevisiae HAS (ScHAS) results in a complete loss of enzyme activity, while substitution of the analogous C-terminal residue (Q365) yields a partially active enzyme. Neither of these substitutions alter the overall structure of ScHAS, as judged by the ability of these variants to form discrete high-molecular-mass complexes similar to the wild-type (WT) enzyme. Interestingly, overexpression of inactive HAS variants results in heme o accumulation in both S. oneidensis and S. cerevisiae, which does not occur in the absence of HAS, suggesting that the enzyme can influence heme o biosynthesis or stability even in the absence of its catalytic activity. Altogether, our results shed light on the catalytic mechanism of HAS and indicate that HAS may positively regulate HOS activity.
Materials and methods
Cloning
The gene encoding S. oneidensis HAS (ctaA) was inserted into the vector pBAD202/D-TOPO (Invitrogen) with the sequence encoding the N-terminal thioredoxin tag removed. Specifically, ctaA was inserted downstream of an arabinose-inducible promoter (araBAD). The ctaA gene was amplified from S. oneidensis MR-1 genomic DNA with primers that added NcoI and HindIII restriction sites to the 5′ and 3′ ends of the gene (Table S1), respectively, and then ligated into pBAD. This produced the vector pBAD So ctaA-V5-His6, in which the encoded HAS had (a) a V5 epitope tag and a hexahistidyl (His6) tag appended to the C-terminus and (b) the amino acid sequence MG added to the N-terminus preceding the native amino acid sequence (Table 2). This plasmid was used as the template for site-directed mutagenesis to produce SoHAS variants. Variants with a single substitution (E72A, E72Q, E72D, H241A, or H241E) were produced by phosphorylating the appropriate PCR product (Table S1) with T4 polynucleotide kinase (New England BioLabs) prior to ligation and transformation. The H75A/H134A and H244A/H302A variants were prepared by site-directed mutagenesis (Table S1) using the QuikChange Site-Directed mutagenesis method (Agilent). All plasmids were verified by DNA sequencing.
Table 2.
Bacterial and yeast plasmids and strains used in this study.
| Strain | Description | Source or reference |
|---|---|---|
| E. coli WM3064 | thrB1004 pro thi rpsL hsdS lacZAM15 RP4–1360 Δ(araBAD)567 ΔdapA 1341:: [erm pir] | William Metcalf, University of Illinois |
| S. oneidensis MR-1 | WT strain | [73] |
| S. cerevisiae DY5113 | MATa, ade2-1, can1-100, leu2-3,112, ura3-1, trp1-1, his3-11,15 | Dennis Winge, University of Utah |
| S. cerevisiae DY5113 cox15S | MATa, ade2-1, can1-100, leu2-3,112, ura3-1, trp1-1, his3-11,15, cox15Δ::KanMX6 | [46] |
| S. cerevisiae DY5113 Pet117::Myc | MATa, ade2-1, can1-100, leu2-3,112, ura3-1, trp1-1, his3-11,15, PET117-Myc13:: TRP1 | [45] |
| Plasmid | Description | Source or reference |
| pBAD So ctaA-V5-His6 | pBAD with S. oneidensis ctaA-V5-His6; araBAD promoter | This study |
| pBAD So ctaA-V5-His6 E72A | pBAD So ctaA-V5-His6with E72A substitution | This study |
| pBAD So ctaA-V5-His6 E72D | pBAD So ctaA-V5-His6with E72D substitution | This study |
| pBAD So ctaA-V5-His6 E72Q | pBAD So ctaA-V5-His6with E72Q substitution | This study |
| pBAD So ctaA-V5-His6 H241A | pBAD So ctaA-V5-His6with H241A substitution | This study |
| pBAD So ctaA-V5-His6 H241E | pBAD So ctaA-V5-His6with H241E substitution | This study |
| pBAD So ctaA-V5-His6 H75A/H134A | pBAD So ctaA-V5-His6with H75A/H134A substitutions | This study |
| pBAD So ctaA-V5-His6 H244A/H302A | pBAD So ctaA-V5-His6with H244A/H302A substitutions | This study |
| pBBR1 MCS3 | Vector | [74] |
| pBBR1-RBS So ctaB | pBBR1-MCS3 with S. oneidensis ctaB preceded by a new RBS; lac promoter (constitutive expression in S. oneidensis) | This study |
| pRS426 | Vector | [75] |
| pRS426 COX15-FLAG | pRS426 with S. cerevisiae COX15-FLAG; MET25 promoter, CYC1 terminator | [57] |
| pRS426 COX15-FLAG E166A | pRS426 COX15-FLAG with E166A substitution | This study |
| pRS426 COX15-FLAG E166D | pRS426 COX15-FLAG with E166D substitution | This study |
| pRS426 COX15-FLAG Q365A | pRS426 COX15-FLAG with Q365A substitution | This study |
The gene encoding HOS from S. oneidensis MR-1 (ctaB) was amplified from genomic DNA by PCR and inserted into the KpnI/SacI site of pBBR1 MCS3 (Table 2, Table S1). A ribosomal binding site (RBS) was added just upstream of the start codon as previously described [68] to allow for better expression in S. oneidensis. The pBAD constructs were introduced into S. oneidensis MR-1 by electroporation [69, 70] and selected for by resistance to 50 μg/mL kanamycin on Luria-Bertani (LB) agar plates. The pBBR1 construct was introduced into S. oneidensis MR-1 via conjugation with donor strain WM3064 [71, 72] and selected for by resistance to 5 μg/mL tetracycline on LB agar plates.
The E166A, E166D, and Q365A variants of ScHAS-FLAG (Cox15-FLAG) were prepared by site-directed mutagenesis (Table S1) from the WT version of the plasmid, pRS426 Cox15-FLAG [57] using the Q5 (New England BioLabs) site-directed mutagenesis kit. All plasmids were verified by DNA sequencing.
Protein expression in S. oneidensis
For purification of His-tagged SoHAS, a 5-mL inoculum of transformed S. oneidensis culture (grown overnight) was used to inoculate 500 mL of LB in a 2-L baffled flask. When necessary, the medium was supplemented with kanamycin (50 μg/mL) to select for pBAD and tetracycline (1 μg/mL) to select for pBBR1. After the cultures reached an OD600 of approximately 1, protein expression was induced by the addition of 0.2% (w/v) arabinose followed by incubation overnight (16–20 h) at 30 °C. The equivalent of 10–20 mL of cell culture at OD600 = 1 was collected by centrifugation and flash frozen for heme extraction. The remaining culture was harvested by low-speed centrifugation (12,000 x g) at 4 °C.
Small-scale S. oneidensis cultures were also grown for the purpose of whole cell heme extraction. For this type of experiment, approximately 0.25 mL of a transformed S. oneidensis culture grown overnight was used to inoculate 25 mL of LB in a 250-mL Erlenmeyer flask. Expression of SoHAS was induced as described above, and the equivalent of 10 mL or, more commonly, 20 mL of culture at OD600 = 1 was harvested for heme extraction. The remaining culture was also harvested so that expression levels of SoHAS could be evaluated. Cell pellets were flash frozen and stored at ‒80 °C.
Protein purification from S. oneidensis
Samples were kept cold (at 4 °C or on ice) for the entire procedure. The cell pellet from a large-scale (500-mL) S. oneidensis culture was resuspended in 10 mL of Buffer 1 (25 mM Tris-HCl, pH 8.0, 50 mM NaCl, 10% (v/v) glycerol, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM EDTA, 0.02% (w/v) NaN3). At this point, the cell suspension was either immediately used for protein purification or frozen and stored at ‒80 °C. Cells were lysed via sonication ten times for 10 s each with a Tekmar High Intensity Ultrasonic Processor (Model 501) equipped with a microtip, with the duty cycle set at 60% and the output control set at 8. The lysate was then centrifuged at 22,000 x g for 30 min. The supernatant was transferred to a new centrifuge tube and centrifuged at 100,600 x g for 60 min. to pellet the membrane fraction. The membrane pellet was resuspended in 9 mL of Buffer 2 (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10% (v/v) glycerol, 1 mM PMSF, 0.02% (w/v) NaN3) and homogenized (ten strokes). n-dodecyl-β-D-maltopyranoside (DDM) was added to a final concentration of 1% (w/v), and the samples were incubated on a rocking platform 1–2 h and centrifuged again for 30 min. at 100,600 x g. The supernatant was supplemented with 10 mM imidazole and incubated with 0.5 mL of nickel-nitrilotriacetic acid (Ni-NTA) resin. Following 1–2 h of incubation, the resin was transferred to a column and washed with 10 column volumes of Buffer 2 supplemented with 10 mM imidazole and 0.02% (w/v) DDM. Finally, the His-tagged protein was eluted with 200 mM imidazole and 0.02% (w/v) DDM. Following purification, the UV-Vis absorbance spectrum of as-isolated (air-oxidized) SoHAS-V5-His6 was measured using an HP 8453 spectrophotometer to determine which elution fractions had the highest levels of co-purifying heme.
Lysis of small-scale S. oneidensis cultures for SDS-PAGE
To assess the steady-state levels of SoHAS variants in small-scale S. oneidensis cultures, frozen cell pellets were thawed and then resuspended and homogenized in 100–500 μL of Buffer 2 supplemented with 1 mM EDTA. (To compensate for the low wet-cell pellet weight of the strain expressing the H244A/H302A SoHAS-V5-His6 variant, the cell pellets from two 25-mL cultures expressing this variant were typically combined.) An aliquot (typically 200 μL) of cell suspension was transferred to a new tube and diluted with Buffer 2 supplemented with Benzonase (Sigma-Aldrich, 1 μL/10 mL), 1 mM EDTA, and 1% (w/v) SDS until the suspension cleared (final SDS concentration 0.4%−0.9% (w/v)). Following determination of total protein concentration by Bradford assay [76], an equal amount of total protein from each sample was loaded onto an SDS-PAGE gel (1–15 μg).
Yeast growth, mitochondrial isolation, heme spectroscopy, co-immunoprecipitation, and respiratory competence tests
Most experiments with S. cerevisiae were performed in a cox15Δ DY5113 background (MATa, ade2-1, can1-100, leu2-3,112, ura3-1, trp1-1, his3-11,15, cox15Δ::KanMX6) in which the COX15 gene was replaced by the KanMX6 cassette (Table 2) [46, 77, 78]. The cox15Δ strain was transformed with the episomal plasmid pRS426 containing either the gene for WT ScHAS-FLAG (Cox15-FLAG) or the gene for a ScHAS-FLAG variant (E166A, E166D, or Q365A) under the control of the S. cerevisiae MET25 promoter. An empty pRS426 vector lacking COX15 (“Vector” in figures) was used as a negative control. WT cultures were DY5113.
For mitochondrial heme extraction and analysis of steady-state expression levels of the ScHAS-FLAG (Cox15-FLAG) variants, transformed yeast cultures were grown in synthetic medium (2% (w/v) glucose) lacking uracil (to select for the presence of pRS426). A small (250-mL) culture was grown for 20 h and used to inoculate a larger (1-L) culture (initial OD600 = 0.1), which was grown for 2 days (~44 h) at 30 °C. The cells were harvested by centrifugation for 10 min. at 4696 x g, washed once with 40 mL of ice-cold water, and flash frozen. For additional SDS-PAGE experiments and for BN-PAGE and heme spectroscopy, cox15Δ cells transformed with empty vector (pRS426) or the plasmids encoding genes for the E166A, E166D, or Q365A Cox15 variants were cultured in synthetic medium (2% (w/v) galactose, 0.1% (w/v) glucose) lacking uracil at 30 °C. Starter cultures (50 mL) were grown overnight and used to inoculate 1-L cultures, which were in turn grown overnight, harvested by centrifugation (8 min. at 2900 x g), washed in 10 mM Tris-HCl, pH 7.4, and frozen. For co-immunoprecipitation, cultures were treated the same, except the cells were either WT or Pet117-13Myc strains transformed with either WT ScHAS-FLAG (Cox15-FLAG) or the E166A variant. Co-immunoprecipitation was performed as previously described [45]. Crude mitochondria produced by lysis with glass beads or isolated mitochondria produced by gentle spheroplast lysis and differential centrifugation were isolated following previously published procedures [79, 80]. Heme-pyridine redox difference spectra were determined from SDS-solubilized mitochondrial lysates according to the method of Berry and Trumpower [81].
For respiratory competence tests, cells were pre-cultured overnight in synthetic selective medium lacking uracil and containing 2% (w/v) glucose. Cultures were normalized to OD600 = 1 in sterile water, serially diluted, and dropped onto synthetic selective medium plates containing 2% (w/v) glucose or 2% (w/v) glycerol/lactate as a carbon source and lacking uracil. Growth was assessed and documented after 1–2 (glucose plates) or 2–4 (glycerol/lactate plates) days of incubation at 30 °C. Both the overnight cultures and the plates lacked uracil to maintain plasmid selection.
Heme extraction
Extraction of non-covalently bound hemes was performed using a modified version of previously described protocols [82]. Briefly, a hydrochloric acid-acetone mixture (HCl-acetone) was prepared by mixing 0.5 mL of concentrated (~37% (w/w) or 12 M) HCl with 9.5 mL acetone (final HCl concentration ~0.6 M). An aqueous, heme-containing sample (purified protein, S. oneidensis cell suspension, or S. cerevisiae crude mitochondria) was then combined with HCl-acetone in a 2:3 ratio. This mixture was incubated on ice for 20 min. with brief vortexing at 0, 10, and 20 min. The sample was then centrifuged at 21,130 x g for 10 min. at room temperature. The supernatant was transferred to a new 1.7-mL microfuge tube and centrifuged again to remove any remaining insoluble material. Finally, the supernatant was removed and analyzed by reverse-phase HPLC.
For the data shown in Figure S3, 0.03–0.2 mg (200 μL) of purified SoHAS was used in an extraction. To analyze the non-covalently bound hemes in S. oneidensis, frozen cell pellets were thawed and resuspended in 200 μL of water. A 100-μL aliquot was then transferred to a 1.7-mL microfuge tube containing 150 μL of the HCl-acetone solution. To analyze the heme content in S. cerevisiae crude mitochondrial extracts, 0.25–0.5 mg of total protein (suspended in 20–50 μL of 600 mM sorbitol, 20 mM HEPES buffer, pH 7.4) was added to an appropriate volume of HCl-acetone.
Heme analysis by HPLC
Hemes were separated and analyzed on an Agilent HPLC system (1260 Infinity) equipped with a diode array detector (Agilent G1315D) using a modified version of a previously published reverse-phase HPLC procedure [18, 83]. The extracted hemes were injected onto a reverse-phase C18 column (Agilent 699975–902) and resolved using a gradient of solvent A (water, 0.1% (v/v) TFA) and solvent B (acetonitrile, 0.1% (v/v) TFA). The chromatography conditions were as follows: 1.00–2.67 min.: 25% B, 2.67–12.67 min.: gradient from 25% B to 100% B, 12.67–20.00 min.: 100% B, 20.00–23.00 min.: gradient to return to starting conditions of 25% B, with a constant 1.18 mL/min. flow rate throughout. Each heme type was identified by its retention time and Soret peak [27]. Retention times were determined by the analysis of known standards (Figure S5). Hemin (heme b chloride) (Sigma H9039) and protoporphyrin IX (PPIX) (Frontier Scientific Porphyrin Products P562–9) were commercially available. Heme o was isolated from E. coli expressing the bo3 quinol oxidase while heme a was isolated directly from bovine heart, as previously described [27]. For quantitative analysis of hemes extracted from lysate or mitochondria, 10-μL injection volumes were used. Higher injection volumes were sometimes used to analyze samples with low heme concentrations extracted from purified SoHAS. For quantitative heme analysis, the approximate percentage of each heme relative to the total amount of all extractable hemes was calculated using the area under the curve for each elution peak in the chromatogram (Table 3). The average percentage and standard deviation for n samples are shown in Table 3, where n is the number of biological replicates. When technical replicates were performed, these percentages differed by ≤ 1%. The concentration of heme b in the S. oneidensis lysate samples ranged from 6–21 μM, and the average heme b concentration in mitochondrial samples ranged from 4–11 μM. For our HPLC setup, the detection limit for heme b was 0.5 μM. Based on the similarity of the extinction coefficients of heme b, heme o, and heme a [81, 84, 85], we estimate that the detection limits for heme o and heme a were also approximately 0.5 μM.
Table 3.
Hemes extracted from S. oneidensis cells expressing SoHAS-V5-His6 variants or S. cerevisiae mitochondria isolated from cells expressing ScHAS/Cox15-FLAG variants.
| Sample | Heme b (%) | Heme o (%) | Heme I (%) | Heme a (%) | n1 |
|---|---|---|---|---|---|
| S. oneidensis MR-1 whole cell | |||||
| No plasmid | 100 ± 0 | — | — | — | 5 |
| SoHOS2 | 100 ± 0 | — | — | — | 4 |
| SoHAS3 | 100 ± 0 | — | — | — | 3 |
| SoHOS SoHAS WT | 96 ± 2 | — | — | 4 ± 2 | 8 |
| SoHOS SoHAS E72A | 92 ± 1 | 8 ± 1 | — | — | 3 |
| SoHOS SoHAS E72D | 90 ± 3 | 3 ± 3 | 5 ± 1 | 2 ± 1 | 4 |
| SoHOS SoHAS E72Q | 88 ± 1 | 12 ± 1 | — | — | 3 |
| SoHOS SoHAS H75A/H134A | 94 ± 2 | 6 ± 2 | — | — | 4 |
| SoHOS SoHAS H241A | 96 ± 1 | — | — | 4 ± 1 | 4 |
| SoHOS SoHAS H241E | 96 ± 2 | 1 ± 2 | 4 | 3 ± 45 | 3 |
| SoHOS SoHAS H244A/H302A | 100 ± 0 | — | — | — | 4 |
| S. cerevisiae cox15Δ mitochondria | |||||
| ScHAS-FLAG6 WT | 90 ± 4 | — | — | 10 ± 4 | 3 |
| pRS426 empty vector | 100 ± 0 | — | — | — | 2 |
| ScHAS-FLAG E166A | 83 ± 7 | 17 ± 7 | — | — | 2 |
| ScHAS-FLAG E166D | 79 ± 2 | 21 ± 2 | — | — | 2 |
| ScHAS-FLAG Q365A | 97 ± 4 | — | — | 3 ± 4 | 2 |
Number of biological replicates.
Plasmid-borne S. oneidensis heme o synthase (CtaB)
Plasmid-borne S. oneidensis heme a synthase (CtaA)
In one sample, a heme I peak was visible but too small to integrate.
In one sample, the heme a peak was visible but too small to integrate. This was the same sample where a small heme I peak was present. Doubling the injection volume for this sample revealed that the heme a peak area was greater than the heme I peak area. (The other two samples had heme a peaks large enough to integrate and no detectable heme I.)
Plasmid-borne S. cerevisiae heme a synthase (Cox15-FLAG)
SDS-PAGE, BN-PAGE, Coomassie staining, and immunodetection
Protein samples were separated on 10% or 15% acrylamide SDS-PAGE gels (self-made) or Any kD Mini-PROTEAN TGX gels (BioRad). Because of the different types of SDS-PAGE gels and molecular weight markers used, the apparent molecular weight of SoHAS-V5-His6 and ScHAS-FLAG varied. BN-PAGE separation of mitochondrial protein complexes solubilized with 1% (w/v) digitonin was performed as previously described [57] using 5–13% gradient polyacrylamide gels (self-made) or 3–12% gradient polyacrylamide gels (Life Technologies). Following SDS-PAGE or BN-PAGE separation, proteins were either visualized by Coomassie staining or transferred to a polyvinylidene difluoride (PVDF) membrane. The PVDF membranes were immunodecorated with the appropriate primary and secondary antibodies. The following primary antibodies were used: mouse anti-His (Lifetein LT0426); rabbit anti-FLAG (Sigma F7425); mouse anti-Cox1 (Abcam ab110270); mouse anti-Cox2 (Abcam ab110271); mouse anti-Cox3 (Abcam ab110259); mouse anti-porin (Thermo 459500); rabbit anti-c-Myc (Santa Cruz Biotechnology sc-789); rabbit anti-Atp2 (reactive towards the Atp2 β subunit of F1F0 ATPase) kindly provided by Dr. Alexander Tzagoloff; and rabbit anti-Cyt1, rabbit anti-Sdh2, and mouse anti-Rip1 kindly provided by Dr. Dennis Winge. The secondary antibodies used were HRP-coupled goat anti-mouse or goat anti-rabbit (Pierce 31430 or Abcam 6721 or Santa Cruz Biotechnology sc-2005 or sc-2030). Blots were imaged with a Chemi-Doc MP imager (Bio-Rad) or an Amersham Imager 600 (GE Healthcare) or by exposure to X-ray film (Thomas Scientific).
Results
Substitution of the conserved glutamate in the N-terminal heme-binding site of S. oneidensis HAS reveals that a negatively charged residue is necessary at this position
To understand the role of conserved residues in HAS, we expressed WT and amino acid substituted SoHAS (CtaA) variants in WT S. oneidensis MR-1 and assessed the in vivo activity of each variant. Although the genome of S. oneidensis includes genes for HOS (ctaB) and HAS (ctaA), as well as the structural genes for an aa3-type CcO, under normal growth conditions S. oneidensis does not express these genes and therefore does not produce detectable levels of heme o or heme a (Table 3) [86, 87]. Thus, the activity of overexpressed SoHAS could be assessed by the presence of heme a in the cell pellet. Heme o, the substrate for HAS, was provided by coexpressing S. oneidensis HOS (SoHOS) with SoHAS. When we expressed SoHAS appended with C-terminal V5 and His6 epitope tags, SoHAS-V5-His6 could be isolated from the cell membrane (Figure S1), confirming that the enzyme was expressed and inserted into the membrane. Importantly, a small amount of heme a (4 ± 2%) was reproducibly detectable in the cells, indicating that both overexpressed proteins, SoHOS and SoHAS-V5-His6, were active (Table 3, Figure S2A). The relative amounts of hemes b, o, I, and a are expressed as a percentage of the total extractable heme, i.e., all non-covalently bound heme in the soluble and membrane fractions, although the farnesylated hemes (hemes o, I, and a) are quite hydrophobic and likely confined to the membrane. More specifically, analysis of purified WT SoHAS-V5-His6 (see below) indicates that at least a fraction of heme a binds to overexpressed SoHAS. After confirming that our coexpression system was functional, we proceeded with the investigation of SoHAS-V5-His6 amino acid variants.
To study the role of the highly conserved glutamate in the N-terminal heme-binding site (E72 in SoHAS), this residue was substituted with alanine (E72A), glutamine (E72Q), or aspartate (E72D). Our goal was to determine if the glutamate was important for catalysis, and if so, if another negatively charged residue (aspartate) or a residue of a similar size and shape (glutamine) could perform the role of E72 in the catalytic mechanism. Replacing E72 with alanine decreased steady-state HAS levels, while the E72Q and E72D substitutions did not significantly affect the expression level of SoHAS-V5-His6 (Figure 3A, Table S2). The E72A and E72Q variants were inactive, as judged by the lack of heme a or the heme I intermediate in the cells (Table 3, Figure S2B). Heme o (the substrate of HAS) was detectable in the strains expressing these inactive HAS variants but was undetectable in the control cells, suggesting that the presence of HAS increased in vivo heme o levels. In contrast to the E72A and E72Q HAS variants, when the E72D variant was expressed, small amounts of heme I (5 ± 1%) and heme a (2 ± 1%) were detectable in the cells, indicating this variant was partially active. Notably, heme I did not accumulate when WT SoHAS-V5-His6 was expressed. The observation that only the E72D variant and WT SoHAS had any level of activity suggests that the presence of a negatively charged residue at this position within the N-terminal heme-binding site is necessary for HAS catalytic activity.
Figure 3. Expression levels of all HAS variants in this study and respiratory growth of S. cerevisiae cox15Δ cells expressing ScHAS-FLAG variants.
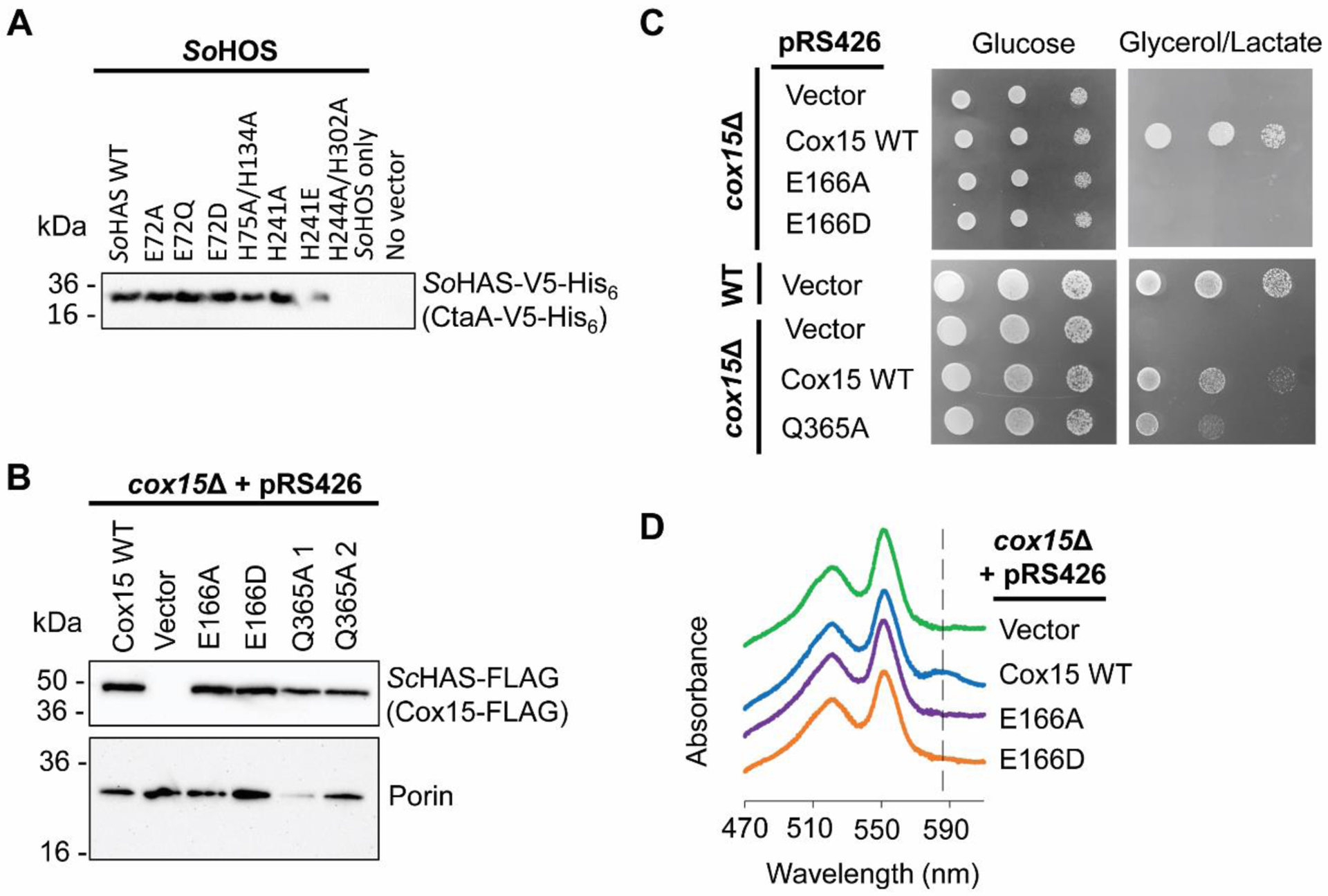
A. Immunoblot analysis of an SDS-PAGE gel of S. oneidensis lysate (~15 μg total protein per lane) showing steady-state expression levels from cells expressing either WT SoHAS-V5-His6 or an indicated HAS variant. The immunoblot shown is representative of three biological replicates. B. Immunoblot of an SDS-PAGE gel of S. cerevisiae mitochondria (10 μg total protein per lane) containing ScHAS-FLAG (Cox15-FLAG) variants expressed from the pRS426 plasmid in a cox15Δ background. A second immunoblot probed with anti-porin is also shown as a loading control. Two different isolates for the Q365A variant are shown (labeled Q365A 1 and Q365A 2, respectively). The immunoblots shown are representative of 2 biological replicates. C. Respiratory competence tests of each plasmid-borne ScHAS-FLAG (Cox15-FLAG) variant in a cox15Δ background. Also shown are cox15Δ cells and WT cells expressing an empty vector. Each cell type was grown in synthetic selective medium (2% (w/v) glucose), normalized, serially diluted, and spotted onto a synthetic medium plate with either 2% (w/v) glucose or 2% (w/v) glycerol + 2% (w/v) lactate. Growth on the non-fermentable carbon source (glycerol/lactate) indicates in vivo respiratory competence. The growth tests shown are representative of at least three biological replicates. D. Heme-pyridine redox difference spectra of mitochondrial lysates isolated from yeast cells expressing ScHAS-FLAG (Cox15-FLAG) variants in a cox15Δ background. Spectra are offset along the y-axis for clarity. The dashed line indicates the absorbance at ~588 nm characteristic of heme a, which is only observed for the Cox15 WT control. Spectra shown are representative of 3 biological replicates.
Because the structures of the N- and C-terminal heme-binding sites are similar, the conserved glutamate in the N-terminal domain has a C-terminal counterpart. This corresponding C-terminal residue is less conserved but is usually glutamine or, less commonly, histidine. In SoHAS, H241 is the residue in the C-terminal domain that occupies the position analogous to the conserved glutamate (E72) in the N-terminal domain. To test the hypothesis that the N-terminal heme-binding site and C-terminal binding site have distinct roles, H241 was substituted with alanine or glutamate, and the resulting variants were coexpressed with SoHOS to allow for analysis of in vivo activity (Figure 3A, Table 3, Figure S2C). While steady-state levels of the H241A variant were typically lower than WT SoHAS-V5-His6 (Table S2), this variant exhibited in vivo activity similar to the WT enzyme (heme a comprised 4 ± 1% of the total extractable heme). Similarly, levels of the H241E variant were decreased compared to WT SoHAS, but this variant was also active in vivo, although in one biological replicate an accumulation of heme o and heme I indicated that HAS activity might be partially compromised. We conclude that unlike its N-terminal analogue, H241 can be replaced by residues with different sizes and chemical properties without significantly altering the catalytic activity of SoHAS.
Substitutions in the C-terminal heme-binding site destabilize prokaryotic HAS (SoHAS) to a greater extent than substitutions in the N-terminal heme-binding site
To test the hypothesis that the N- and C-terminal heme-binding sites have distinct functions, we substituted each of the four invariant, heme-ligating histidines in S. oneidensis HAS with alanine. Structural data indicate that the C-terminal histidines ligate heme b, a cofactor [59], while the N-terminal histidines are presumed to ligate the substrate, heme o, although this has not been demonstrated experimentally. To better understand the roles of these residues, we constructed doubly substituted SoHAS-V5-His6 variants that either had both N-terminal histidines (H75 and H134) or both C-terminal histidines (H244 and H302) substituted with alanine. Consistent with previous studies of BsHAS and ScHAS [44, 55–57], both variants were inactive in vivo (Table 3). A striking difference between the two variants, however, was their apparent stability. While the expression levels of both doubly substituted variants were decreased relative to WT SoHAS (Figure 3A and Table S2), the decrease in the level of the H224A/H302A variant was especially severe, and purification yields for this variant were consistently lower than those of the H75A/H134A variant (Figure S1). Furthermore, heme o was detected in the strain expressing the N-terminal double variant but not in cells expressing the C-terminal double variant (Table 3), suggesting that heme o accumulation is correlated with HAS abundance in S. oneidensis.
The instability of the C-terminal double histidine variant when compared to its N-terminal counterpart supports the hypothesis that the C-terminal half of HAS binds heme b while the N-terminal half binds heme o. Because the heme b cofactor is presumably a prosthetic group that is not released, the C-terminal four-helical bundle is likely relatively static during turnover. On the other hand, the N-terminal half of HAS is expected to be more dynamic since it must bind substrate (heme o) and release product (heme a). Thus, it is logical that substitutions in the N-terminal heme-binding site do not affect HAS stability to the same extent as mutations in the C-terminal heme-binding site, as the C-terminal substitutions likely disrupt cofactor binding and the overall structure of the enzyme.
In an attempt to confirm that the conserved histidine residues in the N-terminal heme-binding site ligate heme o, we analyzed the hemes that co-purified with the double H75A/H134A SoHAS-V5-His6 variant and the double H244A/H302A SoHAS-V5-His6 variant. WT HAS typically co-purifies with heme a and heme b [22, 39, 55, 88], leading to the hypothesis that each heme-binding site ligates a specific heme type. Because previous work individually substituting each conserved histidine with a non-ligating residue did not clarify which histidine coordinates which heme [50, 55, 56], we purified doubly substituted SoHAS-V5-His6 variants that were coexpressed with SoHOS. The rationale behind this approach was that substitution of both histidine ligands in a heme-binding site could sufficiently disrupt the site to prevent heme from binding at that location. Ideally, this would allow us to verify which type of heme binds to each heme-binding site, as any heme that co-purifies with the H75A/H134A variant must be bound to the C-terminal heme-binding site, and vice versa.
The estimated heme b/protein (mol/mol) ratios for preparations of SoHAS-V5-His6 isolated from a nickel-nitrilotriacetic acid (NiNTA) column showed that both WT HAS and the doubly substituted variants co-purified with substoichiometric amounts of heme (i.e., approximately ≤ 0.2 mol heme b/mol protein) (Figures S1A, S3). Although these ratios may be artificially low due to the presence of contaminating proteins in our preparations of SoHAS-V5-His6 (Figure S1B), the data clearly indicate that neither heme-binding site was fully occupied, similar to previous reports of BsHAS-His6 isolated from B. subtilis [22, 55].
WT SoHAS-V5-His6 co-purified with heme b and heme a when coexpressed with SoHOS, as expected (Figures S1A, S3). The H75A/H134A variant co-purified with heme b and, in some cases, a small amount of heme o. Although poor expression of the H244A/H302A variant made it difficult to analyze co-purifying hemes, a small amount of heme b was detected in some preparations of this variant (Figure S3). Overall, these data suggest that replacing both heme-ligating histidines with alanine does not alter the SoHAS heme-binding sites sufficiently to completely prevent heme binding.
Substitution of the conserved glutamate in the N-terminal heme-binding site of S. cerevisiae HAS (ScHAS/Cox15) indicates that this residue is critical for the activity of eukaryotic HAS
We also investigated the significance of the highly conserved glutamate (E166) in the N-terminal heme-binding site in eukaryotic HAS using S. cerevisiae, a model eukaryote that has been instrumental in the study of mitochondrial CcO biogenesis. To assess the in vivo activity of ScHAS (Cox15) variants, plasmids encoding the genes for WT, E166A, or E166D ScHAS-FLAG were expressed in a cox15Δ genetic background (in which cells lack the COX15 gene), followed by extraction of non-covalently bound hemes from isolated mitochondria. As expected, expression of WT ScHAS-FLAG restored respiratory competence in the cox15Δ strain (Figure 3C) [57] and produced detectable levels of heme a (10 ± 4%) (Table 3) (Figure S4). In contrast, both the E166A and E166D variants were inactive, with no detectable heme a, despite generally having steady-state levels (Figure 3B and Table S2) and oligomerization properties (see below) similar to that of the WT enzyme. Interestingly, although heme o was not detectable in the parent cox15Δ strain, heme o accumulated to detectable levels in the cox15Δ cells overexpressing either the E166A or E166D ScHAS variant, thereby mirroring the effect inactive but stably expressed SoHAS variants have on in vivo heme o levels in S. oneidensis. Taken together, these data suggest that 1) inactive HAS must be present for heme o to accumulate to detectable levels in S. cerevisiae mitochondria, and 2) the presence of the conserved glutamate in the N-terminal heme-binding site is important for catalysis in eukaryotic HAS.
In ScHAS, the residue Q365 in the C-terminal domain is analogous to the highly conserved glutamate (E166) from the N-terminal heme-binding site. To assess the importance of this residue in eukaryotic HAS, the Q365A variant of ScHAS-FLAG (Cox15-FLAG) was expressed in S. cerevisiae in a cox15Δ genetic background, producing steady-state levels of the Q365A variant similar to that of WT ScHAS-FLAG (Table S2). Expression of this variant partially rescued the respiratory deficiency of the cox15Δ strain (Figure 3C), and analysis of the extractable mitochondrial hemes revealed a small amount of heme a (3 ± 4%). This evidence of in vivo HAS activity indicates that, unlike the analogous position in the N-terminal heme-binding site, the identity of the residue at position 365 is not critical in S. cerevisiae HAS.
Oligomerization of yeast HAS is unaffected by substitution of E166
WT ScHAS (Cox15) forms several high-mass complexes [44, 46, 57]. These oligomers are readily observed when mitochondrial complexes are separated by non-denaturing blue native polyacrylamide gel electrophoresis (BN-PAGE), where ScHAS-FLAG partitions into complexes with sizes ranging from approximately 232 kDa to over 440 kDa (Figure 4A, 4B), which is much greater than the expected molecular weight of monomeric ScHAS-FLAG (~49 kDa). While these ScHAS-containing complexes appear to be primarily homo-oligomeric [46, 57], they may also include substoichiometric quantities of other proteins, as interactions between ScHAS and various CcO subunits and assembly factors have been reported [44–46]. In particular, the CcO assembly factor Pet117 has been shown to physically interact with ScHAS and help stabilize ScHAS oligomers [45]. While the significance of ScHAS oligomerization remains incompletely understood, it appears to be necessary for proper CcO maturation/hemylation [45].
Figure 4. Substitutions of the conserved glutamate (E166) within the N-terminal heme-binding site do not affect the formation of stable ScHAS-FLAG (Cox15-FLAG) oligomers.
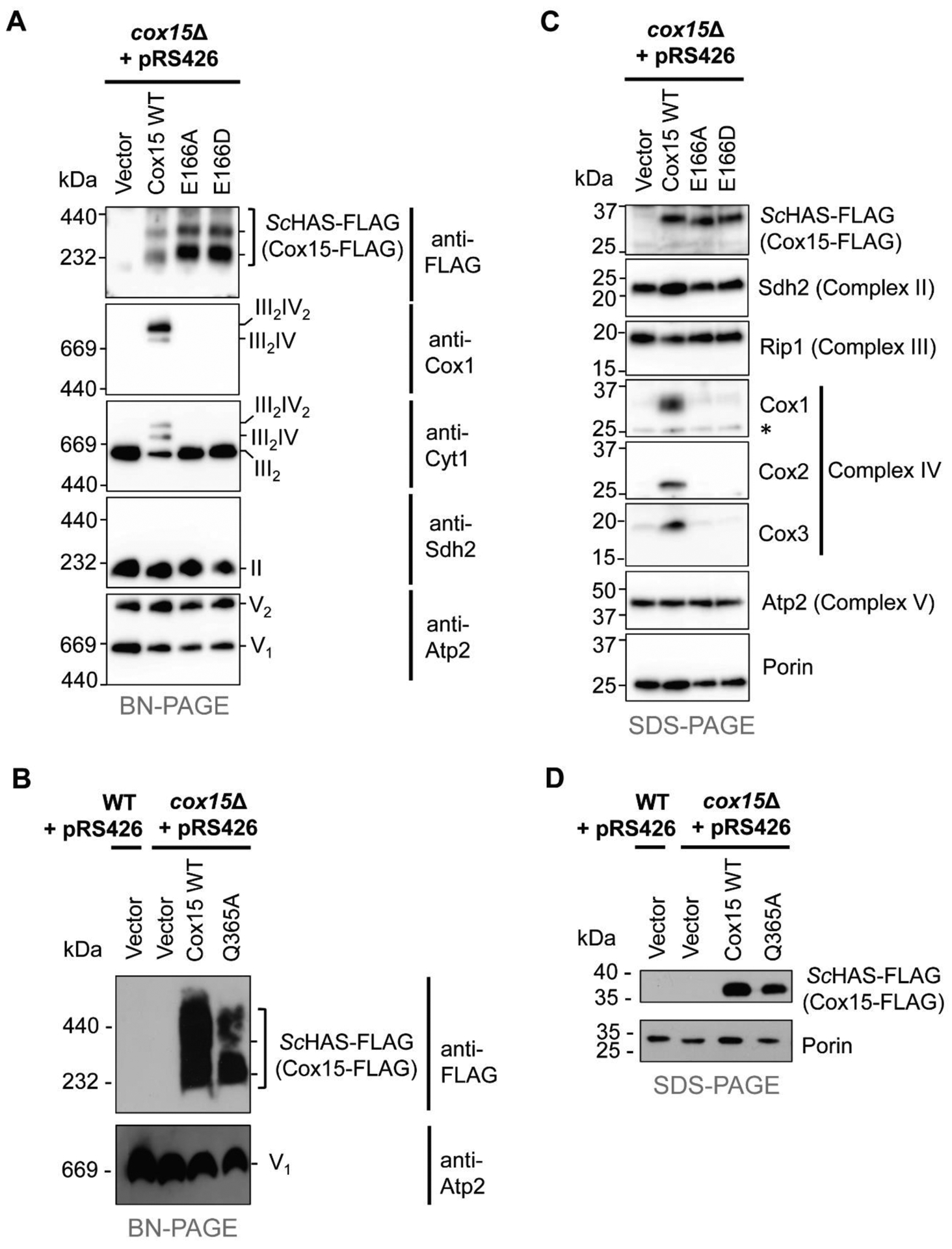
A. BN-PAGE of ScHAS-FLAG (Cox15-FLAG) high-mass complexes in Cox15 WT and the E166A and E166D variants (anti-FLAG immunoblot). Formation of respiratory Complexes II, III, and V, and supercomplexes containing Complexes III and IV are also indicated in the appropriate anti-Cox1, anti-Cytl, anti-Sdh2, and anti-Atp2 immunoblots with relevant stoichiometry of the complexes shown on the right side of the blots. B. BN-PAGE of ScHAS-FLAG (Cox15-FLAG) high-mass complexes in Cox15 WT and the Q365A variant (anti-FLAG immunoblot) with Complex V as a loading control (anti-Atp2 immunoblot). Gels for BN-PAGE in panels A and B were loaded with mitochondrial protein solubilized in digitonin (15 μg total protein per lane) from the same types of cells shown in Figure 3C. C. SDS-PAGE (30 μg total protein per lane) of ScHAS-FLAG (Cox15-FLAG) monomers in Cox15 WT and the E166A and E166D variants (top immunoblot). Representative subunit(s) from respiratory Complexes II, III, IV, and V are also shown in the appropriate immunoblots with porin as the loading control. Asterisk marks a non-specific band. D. SDS-PAGE (50 μg total protein per lane) of ScHAS-FLAG (Cox15-FLAG) monomers in Cox15 WT and the Q365A variant with porin as the loading control. Gels for SDS-PAGE in panels C and D were loaded with mitochondrial protein from the same types of cells shown in Figure 3C. The immunoblots shown in each panel of this figure are representative of three biological replicates.
To determine whether substitution of the highly conserved E166 residue affects the ability of ScHAS to oligomerize, the high-mass complexes formed by ScHAS in mitochondrial samples isolated from cox15Δ cells expressing plasmid-borne ScHAS-FLAG E166A or E166D variants were analyzed by BN-PAGE (Figure 4A). Neither the abundance nor the distribution of ScHAS in high-mass ensembles was affected by substituting E166 (Figure 4A, anti-FLAG immunoblot; while this blot shows slightly higher levels of Cox15 oligomers in E166 variants compared to WT, this difference was not observed in other biological replicates). The high-mass distribution of the Q365A variant was also similar to that of WT ScHAS, although the abundance of the Q365A complexes was reduced (Figure 4B). Because both the E166A and E166D ScHAS variants are inactive in vivo, preventing the hemylation of Cox1 and subsequent CcO assembly, the steady-state levels of the core CcO subunits Cox1, Cox2, and Cox3 are significantly reduced (Figure 4C) when these variants are the only form of ScHAS present, and CcO (Complex IV) does not assemble (Figure 4A, anti-Cox1 immunoblot). As a result, the respiratory supercomplexes, which in S. cerevisiae consist of the Complex III dimer associated with either one (III2IV) or two (III2IV2) CcO complexes, do not form (Figure 4A, anti-Cox1 and anti-Cyt1 immunoblots), leaving Complex III to accumulate in its dimeric form (III2). In contrast, succinate dehydrogenase (Complex II, anti-Sdh2 immunoblot) and monomeric and dimeric ATP synthase (Complex V, anti-Atp2 immunoblot) still assemble in the absence of CcO, as has been observed previously in cases of CcO-specific assembly defects [44, 45].
Pet117 physically interacts with WT ScHAS and is important for its oligomerization [45]. To determine if this interaction persists when E166 is substituted, co-immunoprecipitation experiments were performed to probe for an interaction between chromosomally tagged Pet117 with a C-terminal tag comprised of 13 copies of the Myc epitope (Pet117-13Myc) and the plasmid-borne E166A variant of ScHAS-FLAG. When Pet117-13Myc was immunoprecipitated with anti-Myc resin, the E166A variant co-immunoprecipitated (Figure 5A), analogous to what is observed with WT ScHAS-FLAG (Figure 5B). Together, our native gel electrophoresis and co-immunoprecipitation experiments demonstrate that although substitutions at the E166 position render ScHAS catalytically inactive in vivo, E166 variants are still able to form oligomers stabilized by Pet117. Thus, our data suggest that substitution of E166 alters the substrate heme-binding site and/or catalysis without affecting the global structural features of ScHAS that are critical for its oligomerization.
Figure 5. The E166A variant of ScHAS-FLAG (Cox15-FLAG) co-immunoprecipitates with Pet117-13Myc.
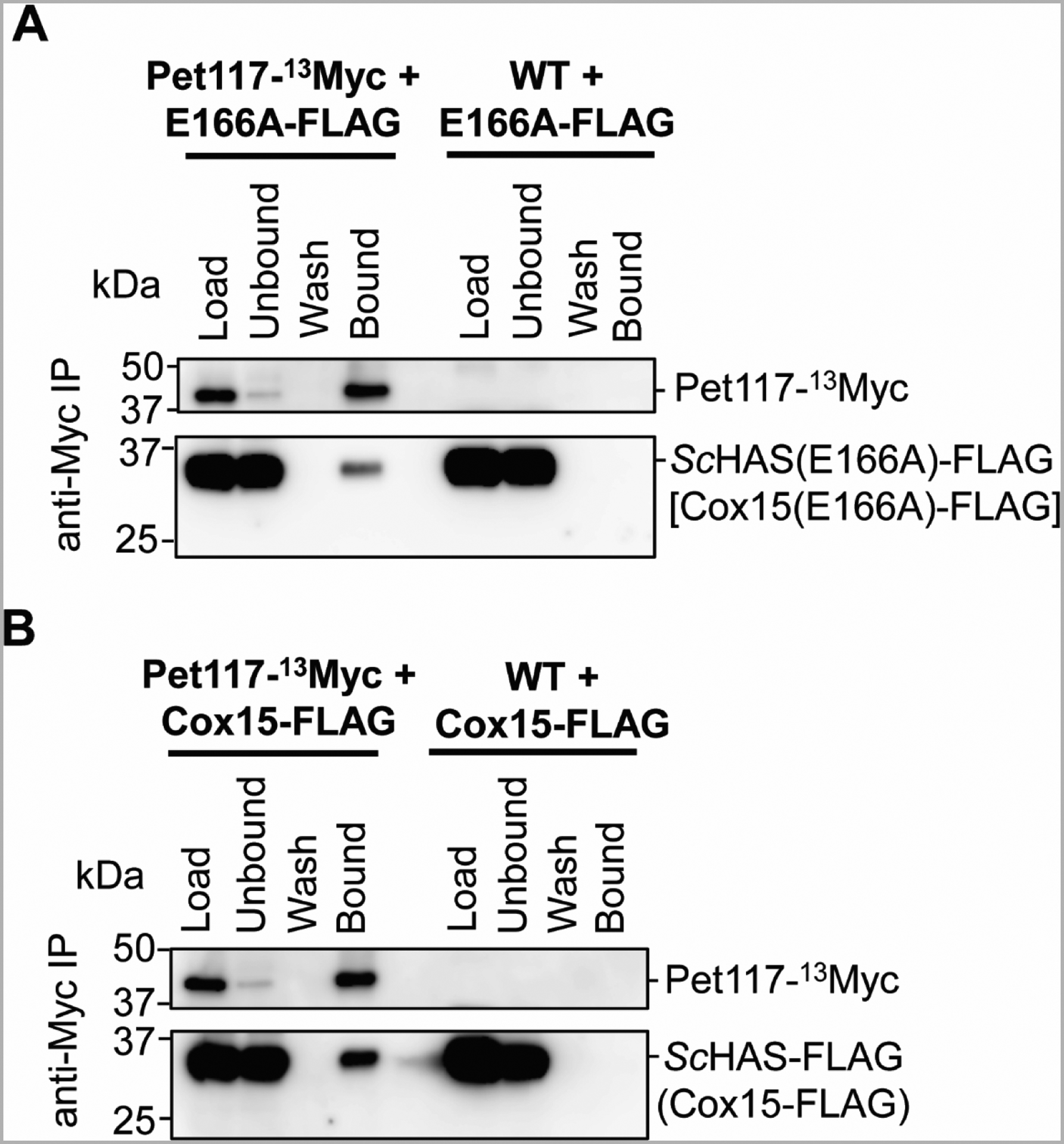
A-B. Immunoblots of SDS-PAGE gels showing the results of co-immunoprecipitation experiments probing for the interaction between chromosomally tagged Pet117-13Myc and the plasmid-borne E166A variant of ScHAS-FLAG [Cox15(E166A)-FLAG or E166-FLAG] (A) or WT ScHAS-FLAG (Cox15-FLAG) (B). Digitonin-solubilized mitochondrial lysates (500 μg total protein per experiment) were incubated with anti-Myc resin. After washing, proteins that were specifically bound to the resin were eluted with Laemmli buffer. Control experiments were performed using strains with unlabeled (WT) Pet117 to demonstrate that ScHAS-FLAG does not bind the anti-Myc resin in the absence of Pet117-13Myc. Blots show 1% of mitochondrial lysates prior to (Load) and after (Unbound) the incubation with affinity resin, the whole fraction of protein precipitated from the final wash (Wash), and half of the eluate (Bound). The immunoblots shown are representative of three biological replicates. IP, immunoprecipitation.
Discussion
Substitution of the conserved, heme-ligating histidines in prokaryotic HAS is consistent with the hypothesis that the N-terminal domain binds heme o and the C-terminal domain binds heme b
It has long been hypothesized that HAS contains two functionally distinct heme-binding sites [22]. The crystal structure of BsHAS, which revealed heme b in the C-terminal site and an unoccupied N-terminal site, strongly supports the idea that the C-terminal heme-binding center is dedicated to cofactor (heme b) binding, while the N-terminal four-helical bundle presumably forms the binding site for the substrate heme [59]. In this study, we attempted to confirm the site of heme o binding by substituting both heme-ligating histidines in either the N- or C-terminal domains of SoHAS. We hypothesized that replacing both histidine ligands in one domain with alanine might disrupt that site enough to prevent heme binding. However, the presence of a small amount of heme o in some of the H75A/H134A variant preparations suggests that substituting both N-terminal histidines does not completely abolish the heme-binding capability of the N-terminal heme-binding site. Similarly, the observation that some preparations of the H244A/H302A variant co-purified with a small amount of heme b could either indicate that some heme b can bind the N-terminal site or that a small portion of heme b can bind the disrupted C-terminal heme-binding site. Because the crystal structure of BsHAS indicates that the C-terminal heme-binding site is occupied by heme b [59], we favor the second scenario, i.e., that a small amount of heme b is still capable of binding to the disrupted C-terminal heme-binding site of the H244A/H302A SoHAS variant. Although our mutagenesis data did not provide clear biochemical evidence demonstrating that heme o binds the N-terminal domain, our mutagenesis data on the stability and catalytic activity of HAS support the idea that the N-terminal four-helical bundle is the site of substrate binding.
We observed that substitution of the conserved, heme-ligating histidine residues in the C-terminal heme-binding site severely reduces the steady-state expression levels of SoHAS (Figure 3A, Figure S1, and Table S2), presumably due to a decrease in stability. In contrast, substitution of analogous residues in the N-terminal heme-binding site does not decrease protein stability to the same extent, even though these substitutions render SoHAS inactive (Table 3). Similarly, studies with BsHAS from our group (data not shown) and others also demonstrated that substitutions of C-terminal residues, such as the C-terminal histidine ligands, seem to decrease the stability of the variants in question relative to WT BsHAS, regardless of the location of the His6 tag (i.e., N-terminal vs. C-terminal) or expression host [55, 56]. Moreover, the core of the stable A. aeolicus HAS trimers is formed by two C-terminal helices from each monomer, and each C-terminal heme-binding site appears to be occupied by heme b, which also indicates that the C-terminal half of prokaryotic HAS binds heme b and remains relatively static [60]. Overall, the observation that substitutions in the C-terminal heme-binding site generally reduce the stability of prokaryotic, type-1 HAS is consistent with the hypothesis that this site is occupied by heme b, as structural changes to the cofactor heme-binding center are likely to have a greater effect on protein stability than analogous alterations in the more dynamic substrate heme-binding site.
A conserved glutamate in the N-terminal heme-binding site is important for the catalytic activity of HAS
A recently published model of heme o binding to BsHAS suggests that the conserved glutamate in the N-terminal domain is positioned near the C8 methyl group of heme o when this substrate binds [59]. We found this prediction intriguing because it suggests that this glutamate could stabilize a carbocation intermediate that is proposed to form transiently at the C8 position during catalysis (Figures 1, 6) [28]. To probe the function of this highly conserved glutamate in the N-terminal heme-binding site, we made substitutions at this position in both prokaryotic HAS (SoHAS-V5-His6) and eukaryotic HAS (ScHAS-FLAG) and assessed the in vivo activity of each variant.
Figure 6. Proposed mechanism of successive heme oxidation steps by HAS.
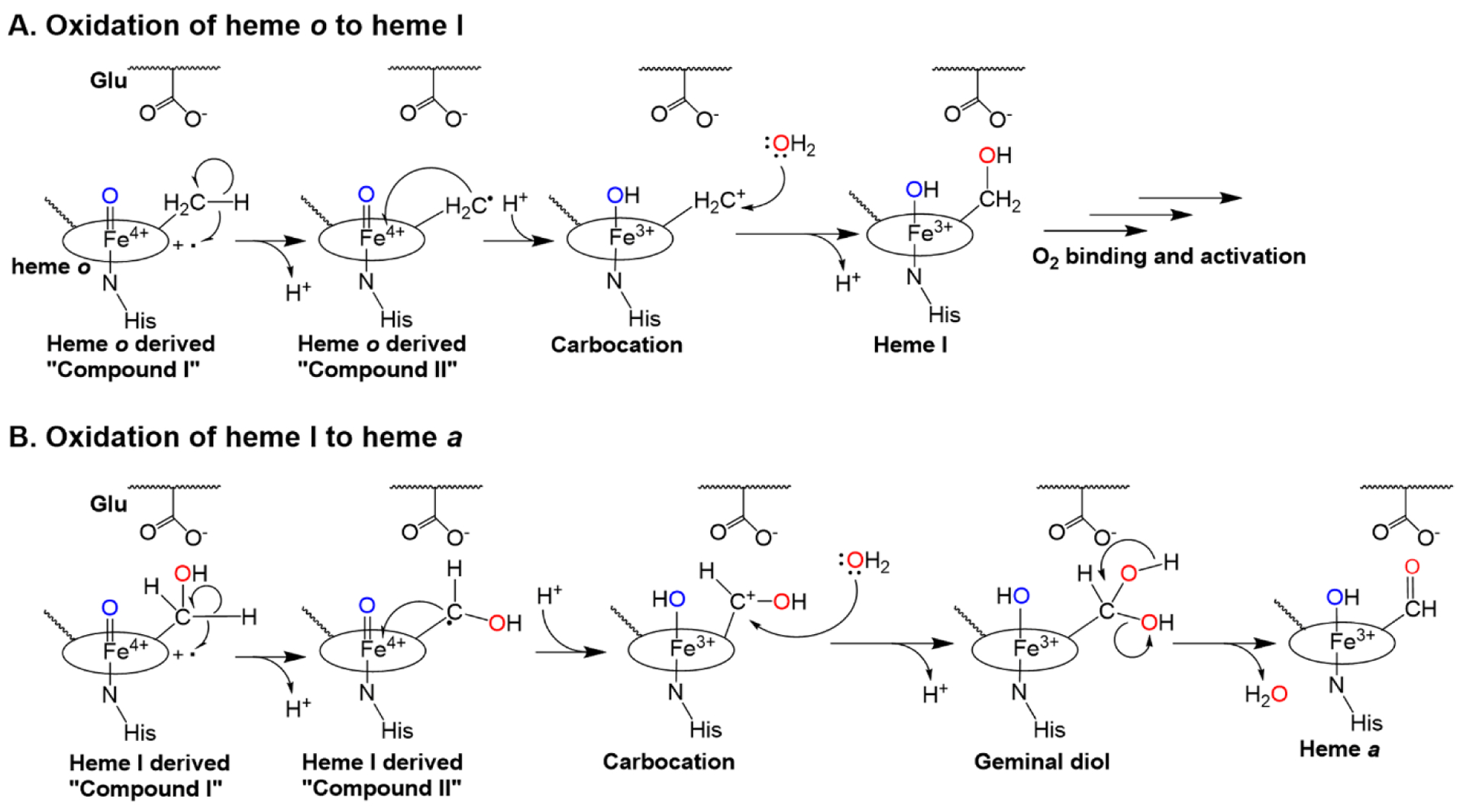
Oxygen activation may occur at heme b (outer-sphere electron transfer, not shown) or heme o (autoxidation, panels A-B), as previously proposed [43]. Either mechanism would likely require the displacement of a histidine ligand to allow O2 to bind. A. Oxidation of heme o to heme I. O2 activation leads to the formation of an Fe(IV)=O porphyrin cation radical (“compound I,” far left), which removes an electron from the C8 methyl group of heme o. This leads to the formation of a radical intermediate (a compound II-like species). The iron ion then removes another electron from the C8 radical, forming a carbocation intermediate that can be trapped by water to form heme I, a stable hydroxylated intermediate. B. In a second autoxidation step, heme I activates a second O2 molecule, forming another compound I-like species (far left). The oxidation process outlined above is repeated to form a geminal diol at C8, which readily dehydrates to form the aldehyde in heme a, the final product. The conserved glutamate positioned near the heme o binding site is proposed to stabilize the carbocation intermediates that form during both oxidation steps.
Our results indicate that replacing the conserved glutamate (E72) in SoHAS with alanine or glutamine abolishes the catalytic activity of this prokaryotic (type-1) enzyme. Analogous substitutions in BsHAS (E57A and E57Q) have also been reported to inactivate BsHAS without affecting the enzyme’s ability to bind heme o [50, 56]. In contrast to the E72A and E72Q variants of SoHAS, the E72D variant is partially active in vivo, as judged by the presence of detectable levels of heme I and heme a in S. oneidensis cells expressing this variant, suggesting a carboxylate is necessary in this position for catalytic activity. In line with our model, we also found that this conserved glutamate is essential for the activity of eukaryotic (type-2) HAS, as both the E166A and E166D variants of ScHAS are inactive and fail to rescue respiration in the cox15Δ mutant. Notably, substitution of the C-terminal counterpart of this highly conserved glutamate in either type of HAS (H241 in SoHAS, Q365 in ScHAS) produces a fully or partially functional enzyme, thereby suggesting that the C-terminal residue is less critical for catalysis. Overall, our results indicate that the conserved glutamate in the N-terminal heme-binding site is important for the catalytic activity of HAS and suggest that this importance stems from its negative charge. The necessity of an anionic residue near the heme o substrate and the accumulation of heme I in the case of the E72D SoHAS variant are both consistent with the proposed catalytic mechanism of HAS, which is discussed below.
The catalytic mechanism of HAS may involve the successive formation of two carbocation intermediates
The putative catalytic mechanism of HAS (Figure 6) [43] is similar to the proposed mechanism for mammalian peroxidases and certain members of the CYP4 family of cytochrome P450 enzymes [63–67]. Enzymes from these families are proposed to form a heme oxyferryl species (similar to compound I in plant heme peroxidases [40, 89, 90]) that oxidizes a methyl substituent of the heme to a carbocation [67]. This autocatalytic oxidation requires a carboxylate residue positioned near the heme [62, 64–67]. Due to the similarities between these enzymes and HAS [59], we previously proposed that the glutamate in the N-terminal domain positioned near heme o may also be involved in stabilizing a carbocation intermediate during each HAS-catalyzed oxidation step (Figure 6) [28, 43]. In this study, we directly tested this hypothesis by substituting E72 with aspartate in SoHAS.
Substitution of E72 with aspartate in SoHAS likely decreases the electrostatic stabilization of the putative carbocation intermediate because the shorter side chain of aspartate is farther away from the C8 carbocation. Although both oxidation steps are expected to involve a carbocation intermediate, carbocation formation during the second oxidation would be more difficult due to the presence of the electron-withdrawing hydroxyl group on the C8 carbon. Thus, the E72D substitution is expected to have a more significant impact on the rate of the second oxidation if both oxidation steps require carbocation formation. This observation may explain why the product of the first oxidation step, heme I, accumulates in cells expressing the E72D variant of SoHAS. Overall, our mutagenesis data are consistent with a mechanism consisting of two consecutive oxidations, both involving formation of carbocation intermediates that are stabilized by a highly conserved glutamate in the N-terminal heme-binding site.
The N-terminal heme-binding site may also function as the site of oxygen activation
Another interesting aspect of the proposed mechanism of HAS is the question of which heme binds O2 [43]. In other classes of heme enzymes that form compound I, one of the heme’s axial coordination sites is vacant, making it obvious where O2 or peroxide binds. Purified WT SoHAS (Figure S3) and WT BsHAS, however, contain both heme b and heme a, and, at least in BsHAS, these hemes are bis-histidyl axially ligated in the as-isolated state [22], making it unclear where O2 binds. As discussed in our recent review, there is literature precedent for O2 activation at either heme b (outer-sphere electron transfer) or heme o (autoxidation) [43]. We have attempted to determine the site of O2 binding using the O2 mimic CO, but the results thus far have been inconclusive. HAS typically co-purifies with sub-stoichiometric amounts of both heme b and heme o/heme a, and the CO-binding capabilities of purified HAS vary significantly depending on the affinity tag(s) present, the type of HAS being expressed (BsHAS versus SoHAS), and the expression host (data not shown). Thus, interpretation of these results would be tenuous at best.
Although the site of O2 activation in HAS remains unclear, we favor the autoxidation mechanism, in which O2 binds heme o within the N-terminal domain. The N-terminal half of HAS contains more conserved residues than the C-terminal half, and the conservation levels of these N-terminal residues are generally higher than their C-terminal counterparts (where present) [50, 54]. The greater number of conserved residues in the N-terminal domain suggests that this domain is dedicated to O2 activation. Other heme enzymes that are capable of O-O bond cleavage, such as peroxidases and cytochromes P450, also require a set of highly conserved residues in the heme-binding pocket [40, 89–94]. Substitution of these residues typically has a severe effect on catalytic activity [40, 89, 90, 92–95]. In contrast, substitutions in electron transfer hemoproteins are often better tolerated [96–99]. Similarly, replacing one of the N-terminal histidine ligands in BsHAS (H60 and H123) with alanine or leucine completely abolishes in vivo activity, while substituting either of the C-terminal histidines with a non-ligating residue often results in a HAS variant that retains partial activity [50, 55, 56, 100]. Previous work has also shown that deletion of seven consecutive residues in the C-terminal heme-binding site of BsHAS, including H216, produces a functional enzyme, indicating that the identity of the amino acids in this region are not critical for the enzyme’s function [101]. These data suggest that the N-terminal heme center is directly involved in oxygen activation while the C-terminal heme center is only involved in electron transfer.
We propose that the N-terminal histidine ligands in HAS participate in a push-pull method of oxygen activation similar to that of peroxidases [40, 43, 89–91]. This mechanism would explain why both N-terminal histidines are necessary for full catalytic activity in HAS. Based on the available experimental data for BsHAS, we further speculate that the invariant histidine on TM helix 2 serves as the “distal” histidine (H60 in BsHAS), while the invariant histidine on TM helix 4 serves as the “proximal” histidine (H123 in BsHAS) that ligates the heme iron and provides the electron-donating “push” necessary for O-O bond cleavage. This hypothesis would explain why all H123 BsHAS variants tested are completely inactive, while a small amount of activity is retained if any of the other three histidine ligands is substituted with methionine [55]. Additionally, in the crystal structure of BsHAS, TM helix 2 is bent, positioning H60 away from the center of the heme-binding site and suggesting that this residue could easily dissociate from the heme o iron and allow O2 to bind [59]. A similar mechanism has been proposed for diheme cytochrome c peroxidase, an enzyme that is activated by conformational changes that cause the distal histidine to dissociate from the active site heme [102, 103]. Future structural and biochemical analyses of HAS will be needed to confirm the location of the O2 binding site and the role of the histidine ligands in O2 activation.
Local perturbations to the heme-binding sites of yeast HAS do not affect the global structure of ScHAS
In addition to allowing us to assess the catalytic importance of conserved (and semi-conserved) residues in eukaryotic (type-2) HAS, our studies with ScHAS allowed us to probe how substitutions in each heme-binding site affect the overall structure of ScHAS, which may impact its proposed secondary functions. Consistent with previous studies, our data show that substitutions in either heme-binding site of ScHAS do not appreciably decrease the steady-state levels of the enzyme, suggesting that alterations to either site do not result in destabilizing global structural changes (Figures 3B, 4). Substitutions at the E166 or Q365 positions also do not alter the ability of ScHAS to form oligomers with molecular weight distributions similar to the WT complexes (Figure 4A, 4B). Moreover, the catalytically inactive E166A variant retains the ability to interact with Pet117-13Myc (Figure 5), indicating that Pet117-mediated stabilization is preserved for the E166A oligomers. Although it is currently unclear precisely which regions of ScHAS are necessary for oligomerization, the fact that substitutions in the N- or C-terminal heme-binding sites do not alter oligomerization suggests that these local changes in ScHAS structure are not propagated globally. Thus, any secondary functions of HAS that are not strictly dependent on catalytic activity are likely to be retained by the inactive E166A and E166D variants.
The in vivo accumulation of heme o is dependent on the presence of HAS
In addition to its obvious function to synthesize heme a, HAS may also play a role in regulating the activity of HOS, its upstream biosynthetic partner. In WT (respiratory-competent) S. cerevisiae, heme o levels are so low that heme o is undetectable in our HPLC analysis of extracted mitochondrial hemes (Table 3), although another group has reported detection of a very small amount of heme o (0.37% of heme b, which is below our detection limit of 0.5 μM heme) in WT S. cerevisiae [48]. Deletion of ScHAS (cox15Δ) prevents the conversion of heme o to heme a, as demonstrated by the lack of heme a in the vector-only cox15Δ control (Table 3, Figure S4). Although the deletion of HAS might be expected to lead to an accumulation of heme o, the substrate for HAS, no heme o accumulation is observed under these circumstances. This lack of heme o accumulation is consistent with the previous observation that deletion of ScHAS in the parental S. cerevisiae strain W303a does not result in significant heme o accumulation [48]. It was also found that heme o accumulation that was detected when other assembly factors were knocked out could be suppressed by knocking out HAS, leading the authors of that study to suggest that either the product of HAS (heme I or heme a) or the HAS enzyme itself could positively regulate HOS activity [48]. However, in the present study, we have shown that the presence of catalytically inactive ScHAS (E166A or E166D variants) allows heme o to accumulate to detectable levels. Thus, it seems unlikely that heme I or heme a molecules are regulating HOS activity. Instead, the ScHAS protein itself seems to effect some change in vivo that allows heme o levels to increase.
The presence of inactive HAS could theoretically regulate in vivo heme o levels by (i) increasing HOS steady-state levels, (ii) increasing HOS activity, or (iii) decreasing heme o degradation. Although there are currently no known degradation pathways for either heme o or heme a in any organism, it could be imagined that inactive, overexpressed HAS binds heme o and protects it from degradation. However, it is not obvious how this scenario would explain the results from the study by Barros and Tzagoloff [48], which show heme o accumulation in certain cytochrome c oxidase assembly factor knockout strains even when ScHAS is expressed at WT levels from its native promoter. Additionally, in S. cerevisiae, attenuation of HAS expression (due to transcriptional repression) does not prevent HOS expression, and deletion of the gene encoding HAS (cox15Δ) does not affect steady-state HOS levels [49]. Therefore, the possibility that HAS increases the steady-state levels of HOS can be ruled out for S. cerevisiae. Thus, one reasonable scenario is that inactive ScHAS increases heme o production by ScHOS, either directly or indirectly. The fact that the distribution of ScHAS in high-mass complexes is unaffected by the E166A or E166D substitutions indicates that these variants maintain the same protein-protein interactions as WT ScHAS, suggesting that these inactive variants could maintain any regulatory role WT ScHAS possesses. Although the mechanism of regulation remains unclear and the presence of an unanticipated pleiotropic effect cannot be ruled out, our results suggest that ScHAS may help control heme o synthesis in S. cerevisiae, potentially by increasing ScHOS (Cox10) activity.
Interestingly, the presence of an inactive but stably expressed HAS variant is also correlated with increased heme o levels in S. oneidensis when SoHOS is coexpressed with SoHAS. When the native SoHOS is constitutively expressed by itself from a low-copy plasmid in S. oneidensis, no significant heme o accumulation is observed (Table 3, Figure S2A). However, when SoHOS and WT SoHAS are coexpressed, heme a is detectable, indicating that both SoHOS and SoHAS are active. Similar to the pattern of heme o accumulation in S. cerevisiae, when inactive but stable SoHAS variants are coexpressed with SoHOS, heme o accumulates to detectable levels, which seems to indicate that the presence of SoHAS also helps increase SoHOS activity in S. oneidensis. However, because these experiments were performed under growth conditions where the native caa3 oxidase was not present (Table 3, Figure S2A) [87], further work is warranted to confirm the physiological relevance of this apparent SoHAS-mediated regulation of SoHOS activity.
Conclusions
In this report, we present data that deepen our understanding of the reaction mechanism of heme a synthase. Our results demonstrate that the conserved glutamate in the N-terminal domain of eukaryotic HAS is essential for catalysis and that a negatively charged residue must occupy this position in prokaryotic HAS. Furthermore, our results support the hypothesis that the N- and C-terminal domains of HAS serve distinct roles as the binding sites for substrate and cofactor heme, respectively. In particular, our results are consistent with a mechanistic model of HAS activity where heme o binding positions the C8 group of heme o near the highly conserved glutamate in the N-terminal heme-binding site, allowing the glutamate to stabilize the carbocation intermediates that are thought to form at the C8 position during both oxidation steps. Surprisingly, expression of inactive HAS variants in S. cerevisiae results in heme o accumulation in vivo, indicating that HAS regulates heme o biosynthesis in a manner that is independent of the catalytic activity of HAS.
Supplementary Material
Highlights.
Type-1 and type-2 heme a synthases have a key glutamate in the N-terminal heme site
For both enzyme types, this glutamate is critical for catalysis but not structure
The type-1 E→D variant is partly active; anionic residue is critical for catalysis
C-terminal domain substitutions decrease type-1 heme a synthase stability
Heme o accumulates in vivo when an inactive heme a synthase variant is expressed
Acknowledgements
We thank Mason Huebsch and Nikki Chen for their experimental contributions to this project. We thank Dr. Alexander Tzagoloff (Columbia University) and Dr. Dennis Winge (University of Utah) for generously providing antibodies. This project was supported by grants from the National Institutes of Health National Institute of General Medical Sciences, R01GM101386 (E. L. H.), R35GM131701–01 (O.K.), T32GM107001 (J.V.D.), and P20GM103499-20. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations and nomenclature
- BN-PAGE
blue native PAGE
- CcO
cytochrome c oxidase
- DDM
n-dodecyl-β-D-maltopyranoside
- HAS
heme a synthase
- HCl
hydrochloric acid
- HOS
heme o synthase
- IP
immunoprecipitation
- LB
Luria-Bertani
- NiNTA
nickel nitrilotriacetic acid
- OD600
optical density at 600 nm
- PPIX
protoporphyrin IX
- PVDF
polyvinylidene difluoride
- RBS
ribosomal binding site
- TM
transmembrane
Footnotes
Declarations of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT statement
Elise D. Rivett: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing.
Hannah G. Addis: Data curation, Formal analysis, Investigation, Validation.
Jonathan V. Dietz: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing.
Jayda A. Carroll-Deaton: Data curation, Formal analysis, Investigation, Validation.
Shipra Gupta: Data curation, Formal analysis, Investigation, Methodology.
Koji L. Foreman: Data curation, Formal analysis, Investigation, Validation.
Minh Anh Dang: Data curation, Formal analysis, Investigation, Validation.
Jennifer L. Fox: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing.
Oleh Khalimonchuk: Conceptualization, Formal analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – review & editing.
Eric L. Hegg: Conceptualization, Formal analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – review & editing
Supporting information
This article contains supporting information.
Data availability
All data in this study are available within the article, supporting information, and/or from the corresponding author(s) on request.
References
- [1].Ferguson-Miller S, Babcock GT, Heme/copper terminal oxidases, Chem Rev 96(7) (1996) 2889–2908. [DOI] [PubMed] [Google Scholar]
- [2].Garcia-Horsman JA, Barquera B, Rumbley J, Ma J, Gennis RB, The superfamily of heme-copper respiratory oxidases, J Bacteriol 176(18) (1994) 5587–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schafer G, Engelhard M, Muller V, Bioenergetics of the Archaea, Microbiol Mol Biol Rev 63(3) (1999) 570–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Refojo PN, Sena FV, Calisto F, Sousa FM, Pereira MM, Chapter six: the plethora of membrane respiratory chains in the phyla of life, in: Poole RK (Ed.), Advances in Microbial Physiology, Academic Press2019, pp. 331–414. [DOI] [PubMed] [Google Scholar]
- [5].Borisov VB, Gennis RB, Hemp J, Verkhovsky MI, The cytochrome bd respiratory oxygen reductases, Biochim Biophys Acta 1807(11) (2011) 1398–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hemp JG, Robert B, Bioenergetics: energy conservation and conversion, in: Schä G.n.P., Harvey S (Ed.) Bioenergetics: Energy Conservation and Conversion, Springer, Berlin, 2008. [Google Scholar]
- [7].Pereira MM, Santana M, Teixeira M, A novel scenario for the evolution of haem-copper oxygen reductases, Biochim Biophys Acta 1505(2–3) (2001) 185–208. [DOI] [PubMed] [Google Scholar]
- [8].Pereira MM, Sousa FL, Veríssimo AF, Teixeira M, Looking for the minimum common denominator in haem-copper oxygen reductases: towards a unified catalytic mechanism, Biochim Biophys Acta 1777(7–8) (2008) 929–34. [DOI] [PubMed] [Google Scholar]
- [9].Quadalti C, Brunetti D, Lagutina I, Duchi R, Perota A, Lazzari G, Cerutti R, Di Meo I, Johnson M, Bottani E, Crociara P, Corona C, Grifoni S, Tiranti V, Fernandez-Vizarra E, Robinson AJ, Viscomi C, Casalone C, Zeviani M, Galli C, SURF1 knockout cloned pigs: Early onset of a severe lethal phenotype, Biochimica et biophysica acta. Molecular basis of disease 1864(6 Pt A) (2018) 2131–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Robinson BH, Human cytochrome oxidase deficiency, Pediatric research 48(5) (2000) 581–5. [DOI] [PubMed] [Google Scholar]
- [11].Fontanesi F, Soto IC, Horn D, Barrientos A, Assembly of mitochondrial cytochrome c-oxidase, a complicated and highly regulated cellular process, American journal of physiology. Cell physiology 291(6) (2006) C1129–47. [DOI] [PubMed] [Google Scholar]
- [12].Kim HJ, Khalimonchuk O, Smith PM, Winge DR, Structure, function, and assembly of heme centers in mitochondrial respiratory complexes, Biochim Biophys Acta 1823(9) (2012) 1604–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Timón-Gómez A, Nývltová E, Abriata LA, Vila AJ, Hosler J, Barrientos A, Mitochondrial cytochrome c oxidase biogenesis: recent developments, Seminars in cell & developmental biology 76 (2018) 163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Antonicka H, Leary SC, Guercin GH, Agar JN, Horvath R, Kennaway NG, Harding CO, Jaksch M, Shoubridge EA, Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency, Human molecular genetics 12(20) (2003) 2693–702. [DOI] [PubMed] [Google Scholar]
- [15].Antonicka H, Mattman A, Carlson CG, Glerum DM, Hoffbuhr KC, Leary SC, Kennaway NG, Shoubridge EA, Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy, American journal of human genetics 72(1) (2003) 101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Oquendo CE, Antonicka H, Shoubridge EA, Reardon W, Brown GK, Functional and genetic studies demonstrate that mutation in the COX15 gene can cause Leigh syndrome, Journal of medical genetics 41(7) (2004) 540–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Khalimonchuk O, Bestwick M, Meunier B, Watts TC, Winge DR, Formation of the redox cofactor centers during Cox1 maturation in yeast cytochrome oxidase, Molecular and cellular biology 30(4) (2010) 1004–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Del Arenal IP, Contreras ML, Svlateorova BB, Rangel P, Lledías F, Dávila JR, Escamilla JE, Haem O and a putative cytochrome bo in a mutant of Bacillus cereus impaired in the synthesis of haem A, Archives of microbiology 167(1) (1997) 24–31. [DOI] [PubMed] [Google Scholar]
- [19].van der Oost J, von Wachenfeld C, Hederstedt L, Saraste M, Bacillus subtilis cytochrome oxidase mutants: biochemical analysis and genetic evidence for two aa3-type oxidases, Molecular microbiology 5(8) (1991) 2063–72. [DOI] [PubMed] [Google Scholar]
- [20].Puustinen A, Wikstrom M, The heme groups of cytochrome o from Escherichia coli, Proceedings of the National Academy of Sciences 88(14) (1991) 6122–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mueller JP, Taber HW, Structure and expression of the cytochrome aa3 regulatory gene ctaA of Bacillus subtilis, Journal of Bacteriology 171(9) (1989) 4979–4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Svensson B, Andersson KK, Hederstedt L, Low-spin heme A in the heme A biosynthetic protein CtaA from Bacillus subtilis, European journal of biochemistry 238(1) (1996) 287–95. [DOI] [PubMed] [Google Scholar]
- [23].Svensson B, Hederstedt L, Bacillus subtilis CtaA is a heme-containing membrane protein involved in heme A biosynthesis, J Bacteriol 176(21) (1994) 6663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Svensson B, Lübben M, Hederstedt L, Bacillus subtilis CtaA and CtaB function in haem A biosynthesis, Molecular microbiology 10(1) (1993) 193–201. [DOI] [PubMed] [Google Scholar]
- [25].Mogi T, Saiki K, Anraku Y, Biosynthesis and functional role of haem O and haem A, Molecular microbiology 14(3) (1994) 391–8. [DOI] [PubMed] [Google Scholar]
- [26].Barros MH, Carlson CG, Glerum DM, Tzagoloff A, Involvement of mitochondrial ferredoxin and Cox15p in hydroxylation of heme O, FEBS letters 492(1–2) (2001) 133–8. [DOI] [PubMed] [Google Scholar]
- [27].Brown KR, Allan BM, Do P, Hegg EL, Identification of novel hemes generated by heme A synthase: evidence for two successive monooxygenase reactions, Biochemistry 41(36) (2002) 10906–13. [DOI] [PubMed] [Google Scholar]
- [28].Brown KR, Brown BM, Hoagland E, Mayne CL, Hegg EL, Heme A synthase does not incorporate molecular oxygen into the formyl group of heme A, Biochemistry 43(27) (2004) 8616–24. [DOI] [PubMed] [Google Scholar]
- [29].Hill J, Goswitz VC, Calhoun M, Garcia-Horsman JA, Lemieux L, Alben JO, Gennis RB, Demonstration by FTIR that the bo-type ubiquinol oxidase of Escherichia coli contains a heme-copper binuclear center similar to that in cytochrome c oxidase and that proper assembly of the binuclear center requires the cyoE gene product, Biochemistry 31(46) (1992) 11435–40. [DOI] [PubMed] [Google Scholar]
- [30].Saiki K, Mogi T, Anraku Y, Heme O biosynthesis in Escherichia coli: the cyoE gene in the cytochrome bo operon encodes a protoheme IX farnesyltransferase, Biochemical and biophysical research communications 189(3) (1992) 1491–7. [DOI] [PubMed] [Google Scholar]
- [31].Saiki K, Mogi T, Ogura K, Anraku Y, In vitro heme O synthesis by the cyoE gene product from Escherichia coli, The Journal of biological chemistry 268(35) (1993) 26041–4. [PubMed] [Google Scholar]
- [32].Glerum DM, Tzagoloff A, Isolation of a human cDNA for heme A:farnesyltransferase by functional complementation of a yeast cox10 mutant, Proceedings of the National Academy of Sciences 91(18) (1994) 8452–8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nobrega MP, Nobrega FG, Tzagoloff A, COX10 codes for a protein homologous to the ORF1 product of Paracoccus denitrificans and is required for the synthesis of yeast cytochrome oxidase, The Journal of biological chemistry 265(24) (1990) 14220–6. [PubMed] [Google Scholar]
- [34].Li W, Bringing bioactive compounds into membranes: the UbiA superfamily of intramembrane aromatic prenyltransferases, Trends in biochemical sciences 41(4) (2016) 356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Poulter CD, Rilling HC, The prenyl transfer reaction: enzymatic and mechanistic studies of the 1’−4 coupling reaction in the terpene biosynthetic pathway, Accounts of Chemical Research 11(8) (1978) 307–313. [Google Scholar]
- [36].Hosfield DJ, Zhang Y, Dougan DR, Broun A, Tari LW, Swanson RV, Finn J, Structural basis for bisphosphonat-emediated inhibition of isoprenoid biosynthesis, The Journal of biological chemistry 279(10) (2004) 8526–9. [DOI] [PubMed] [Google Scholar]
- [37].Kavanagh KL, Dunford JE, Bunkoczi G, Russell RG, Oppermann U, The crystal structure of human geranylgeranyl pyrophosphate synthase reveals a novel hexameric arrangement and inhibitory product binding, The Journal of biological chemistry 281(31) (2006) 22004–12. [DOI] [PubMed] [Google Scholar]
- [38].Saiki K, Mogi T, Hori H, Tsubaki M, Anraku Y, Identification of the functional domains in heme O synthase: site-directed mutagenesis studies on the cyoE gene of the cytochrome bo operon in Escherichia coli, The Journal of biological chemistry 268(36) (1993) 26927–34. [PubMed] [Google Scholar]
- [39].Sakamoto J, Hayakawa A, Uehara T, Noguchi S, Sone N, Cloning of Bacillus stearothermophilus ctaA and heme A synthesis with the CtaA protein produced in Escherichia coli, Bioscience, biotechnology, and biochemistry 63(1) (1999) 96–103. [DOI] [PubMed] [Google Scholar]
- [40].Sono M, Roach MP, Coulter ED, Dawson JH, Heme-containing oxygenases, Chem Rev 96(7) (1996) 2841–2888. [DOI] [PubMed] [Google Scholar]
- [41].Schneegurt MA, Beale SI, Origin of the chlorophyll b formyl oxygen in Chlorella vulgaris, Biochemistry 31(47) (1992) 11677–83. [DOI] [PubMed] [Google Scholar]
- [42].Porra RJ, Schäfer W, Cmiel E, Katheder I, Scheer H, Derivation of the formyl-group oxygen of chlorophyll b from molecular oxygen in greening leaves of a higher plant (Zea mays), FEBS letters 323(1–2) (1993) 31–4. [DOI] [PubMed] [Google Scholar]
- [43].Rivett ED, Heo L, Feig M, Hegg EL, Biosynthesis and trafficking of heme o and heme a: new structural insights and their implications for reaction mechanisms and prenylated heme transfer, Critical reviews in biochemistry and molecular biology 56(6) (2021) 640–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bareth B, Dennerlein S, Mick DU, Nikolov M, Urlaub H, Rehling P, The heme a synthase Cox15 associates with cytochrome c oxidase assembly intermediates during Cox1 maturation, Molecular and cellular biology 33(20) (2013) 4128–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Taylor NG, Swenson S, Harris NJ, Germany EM, Fox JL, Khalimonchuk O, The assembly factor Pet117 couples heme a synthase activity to cytochrome oxidase assembly, The Journal of biological chemistry 292(5) (2017) 1815–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Herwaldt EJ, Rivett ED, White AJ, Hegg EL, Cox15 interacts with the cytochrome bc1 dimer within respiratory supercomplexes as well as in the absence of cytochrome c oxidase, The Journal of biological chemistry 293(42) (2018) 16426–16439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nývltová E, Dietz JV, Seravalli J, Khalimonchuk O, Barrientos A, Coordination of metal center biogenesis in human cytochrome c oxidase, Nature Communications 13(1) (2022) 3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Barros MH, Tzagoloff A, Regulation of the heme A biosynthetic pathway in Saccharomyces cerevisiae, FEBS letters 516(1–3) (2002) 119–23. [DOI] [PubMed] [Google Scholar]
- [49].Wang Z, Wang Y, Hegg EL, Regulation of the heme A biosynthetic pathway: differential regulation of heme A synthase and heme O synthase in Saccharomyces cerevisiae, The Journal of biological chemistry 284(2) (2009) 839–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hederstedt L, Heme A biosynthesis, Biochim Biophys Acta 1817(6) (2012) 920–7. [DOI] [PubMed] [Google Scholar]
- [51].Lewin A, Hederstedt L, Compact archaeal variant of heme A synthase, FEBS letters 580(22) (2006) 5351–6. [DOI] [PubMed] [Google Scholar]
- [52].Degli Esposti M, Moya-Beltrán A, Quatrini R, Hederstedt L, Respiratory heme A-containing oxidases originated in the ancestors of iron-oxidizing bacteria, Frontiers in microbiology 12 (2021) 664216–664216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Degli Esposti M, Garcia-Meza V, Ceniceros Gómez AE, Moya-Beltrán A, Quatrini R, Hederstedt L, Heme A-containing oxidases evolved in the ancestors of iron oxidizing bacteria, bioRxiv (2020) 2020.03.01.968255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].He D, Fu CJ, Baldauf SL, Multiple origins of eukaryotic cox15 suggest horizontal gene transfer from bacteria to jakobid mitochondrial DNA, Molecular biology and evolution 33(1) (2016) 122–33. [DOI] [PubMed] [Google Scholar]
- [55].Hederstedt L, Lewin A, Throne-Holst M, Heme A synthase enzyme functions dissected by mutagenesis of Bacillus subtilis CtaA, J Bacteriol 187(24) (2005) 8361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mogi T, Probing structure of heme A synthase from Bacillus subtilis by site-directed mutagenesis, Journal of biochemistry 145(5) (2009) 625–33. [DOI] [PubMed] [Google Scholar]
- [57].Swenson S, Cannon A, Harris NJ, Taylor NG, Fox JL, Khalimonchuk O, Analysis of oligomerization properties of heme a synthase provides insights into its function in eukaryotes, The Journal of biological chemistry 291(19) (2016) 10411–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Merli ML, Cirulli BA, Menéndez-Bravo SM, Cricco JA, Heme A synthesis and CcO activity are essential for Trypanosoma cruzi infectivity and replication, The Biochemical journal 474(14) (2017) 2315–2332. [DOI] [PubMed] [Google Scholar]
- [59].Niwa S, Takeda K, Kosugi M, Tsutsumi E, Mogi T, Miki K, Crystal structure of heme A synthase from Bacillus subtilis, Proceedings of the National Academy of Sciences of the United States of America 115(47) (2018) 11953–11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zeng H, Zhu G, Zhang S, Li X, Martin J, Morgner N, Sun F, Peng G, Xie H, Michel H, Isolated heme A synthase from Aquifex aeolicus is a trimer, mBio 11(3) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Omasits U, Ahrens CH, Müller S, Wollscheid B, Protter: Interactive protein feature visualization and integration with experimental proteomic data, Bioinformatics (Oxford, England) 30(6) (2014) 884–886. [DOI] [PubMed] [Google Scholar]
- [62].Colas C, Kuo JM, Ortiz de Montellano PR, Asp-225 and Glu-375 in autocatalytic attachment of the prosthetic heme group of lactoperoxidase, The Journal of biological chemistry 277(9) (2002) 7191–200. [DOI] [PubMed] [Google Scholar]
- [63].Ortiz de Montellano PR, Mechanism and role of covalent heme binding in the CYP4 family of P450 enzymes and the mammalian peroxidases, Drug metabolism reviews 40(3) (2008) 405–26. [DOI] [PubMed] [Google Scholar]
- [64].LeBrun LA, Hoch U, Ortiz de Montellano PR, Autocatalytic mechanism and consequences of covalent heme attachment in the cytochrome P4504A family, Journal of Biological Chemistry 277(15) (2002) 12755–12761. [DOI] [PubMed] [Google Scholar]
- [65].LeBrun LA, Xu F, Kroetz DL, Ortiz de Montellano PR, Covalent attachment of the heme prosthetic group in the CYP4F cytochrome P450 family, Biochemistry 41(18) (2002) 5931–5937. [DOI] [PubMed] [Google Scholar]
- [66].Zheng Y-M, Baer BR, Kneller MB, Henne KR, Kunze KL, Rettie AE, Covalent heme binding to CYP4B1 via Glu310 and a carbocation porphyrin intermediate, Biochemistry 42(15) (2003) 4601–4606. [DOI] [PubMed] [Google Scholar]
- [67].Colas C, Ortiz de Montellano PR, Autocatalytic radical reactions in physiological prosthetic heme modification, Chemical Reviews 103(6) (2003) 2305–2332. [DOI] [PubMed] [Google Scholar]
- [68].Sybirna K, Antoine T, Lindberg P, Fourmond V, Rousset M, Méjean V, Bottin H, Shewanella oneidensis: a new and efficient system for expression and maturation of heterologous [Fe-Fe] hydrogenase from Chlamydomonas reinhardtii, BMC biotechnology 8 (2008) 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ozawa K, Yasukawa F, Fujiwara Y, Akutsu H, A simple, rapid, and highly efficient gene expression system for multiheme cytochromes c, Bioscience, biotechnology, and biochemistry 65(1) (2001) 185–9. [DOI] [PubMed] [Google Scholar]
- [70].Corts AD, Thomason LC, Gill RT, Gralnick JA, A new recombineering system for precise genome-editing in Shewanella oneidensis strain MR-1 using single-stranded oligonucleotides, Scientific reports 9(1) (2019) 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Coursolle D, Gralnick JA, Reconstruction of extracellular respiratory pathways for iron(III) reduction in Shewanella oneidensis strain MR-1, Frontiers in microbiology 3 (2012) 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Saltikov CW, Newman DK, Genetic identification of a respiratory arsenate reductase, Proceedings of the National Academy of Sciences of the United States of America 100(19) (2003) 10983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Myers CR, Nealson KH, Bacterial manganese reduction and growth with manganese oxide as the sole electron acceptor, Science (New York, N.Y.) 240(4857) (1988) 1319–21. [DOI] [PubMed] [Google Scholar]
- [74].Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, 2nd, K.M. Peterson, Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes, Gene 166(1) (1995) 175–6. [DOI] [PubMed] [Google Scholar]
- [75].Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P, Multifunctional yeast high-copy-number shuttle vectors, Gene 110(1) (1992) 119–22. [DOI] [PubMed] [Google Scholar]
- [76].Bradford MM, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Analytical Biochemistry 72(1) (1976) 248–254. [DOI] [PubMed] [Google Scholar]
- [77].Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR, Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae, Yeast (Chichester, England) 14(10) (1998) 953–61. [DOI] [PubMed] [Google Scholar]
- [78].Gietz RD, Schiestl RH, High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method, Nature protocols 2(1) (2007) 31–4. [DOI] [PubMed] [Google Scholar]
- [79].Lange H, Kispal G, Lill R, Mechanism of iron transport to the site of heme synthesis inside yeast mitochondria, The Journal of biological chemistry 274(27) (1999) 18989–96. [DOI] [PubMed] [Google Scholar]
- [80].Diekert K, de Kroon AI, Kispal G, Lill R, Isolation and subfractionation of mitochondria from the yeast Saccharomyces cerevisiae, Methods in cell biology 65 (2001) 37–51. [DOI] [PubMed] [Google Scholar]
- [81].Berry EA, Trumpower BL, Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra, Anal Biochem 161(1) (1987) 1–15. [DOI] [PubMed] [Google Scholar]
- [82].Weinstein JD, Beale SI, Separate physiological roles and subcellular compartments for two tetrapyrrole biosynthetic pathways in Euglena gracilis, The Journal of biological chemistry 258(11) (1983) 6799–807. [PubMed] [Google Scholar]
- [83].Lübben M, Morand K, Novel prenylated hemes as cofactors of cytochrome oxidases: archaea have modified hemes A and O, The Journal of biological chemistry 269(34) (1994) 21473–9. [PubMed] [Google Scholar]
- [84].Zhuang J, Reddi AR, Wang Z, Khodaverdian B, Hegg EL, Gibney BR, Evaluating the roles of the heme a side chains in cytochrome c oxidase using designed heme proteins, Biochemistry 45(41) (2006) 12530–8. [DOI] [PubMed] [Google Scholar]
- [85].Matsushita K, Ebisuya H, Adachi O, Homology in the structure and the prosthetic groups between two different terminal ubiquinol oxidases, cytochrome a1 and cytochrome o, of Acetobacter aceti, The Journal of biological chemistry 267(34) (1992) 24748–53. [PubMed] [Google Scholar]
- [86].Le Laz S, Kpebe A, Bauzan M, Lignon S, Rousset M, Brugna M, A biochemical approach to study the role of the terminal oxidases in aerobic respiration in Shewanella oneidensis MR-1, PloS one 9(1) (2014) e86343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Le Laz S, Kpebe A, Bauzan M, Lignon S, Rousset M, Brugna M, Expression of terminal oxidases under nutrient-starved conditions in Shewanella oneidensis: detection of the A-type cytochrome c oxidase, Scientific reports 6 (2016) 19726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hannappel A, Bundschuh FA, Ludwig B, Characterization of heme-binding properties of Paracoccus denitrificans Surf1 proteins, The FEBS journal 278(10) (2011) 1769–78. [DOI] [PubMed] [Google Scholar]
- [89].Hiner AN, Raven EL, Thorneley RN, García-Cánovas F, Rodríguez-López JN, Mechanisms of compound I formation in heme peroxidases, Journal of inorganic biochemistry 91(1) (2002) 27–34. [DOI] [PubMed] [Google Scholar]
- [90].Poulos TL, Thirty years of heme peroxidase structural biology, Archives of biochemistry and biophysics 500(1) (2010) 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Furtmüller PG, Zederbauer M, Jantschko W, Helm J, Bogner M, Jakopitsch C, Obinger C, Active site structure and catalytic mechanisms of human peroxidases, Archives of biochemistry and biophysics 445(2) (2006) 199–213. [DOI] [PubMed] [Google Scholar]
- [92].Denisov IG, Makris TM, Sligar SG, Schlichting I, Structure and chemistry of cytochrome P450, Chem Rev 105(6) (2005) 2253–77. [DOI] [PubMed] [Google Scholar]
- [93].Erman JE, Vitello LB, Yeast cytochrome c peroxidase: mechanistic studies via protein engineering, Biochim Biophys Acta 1597(2) (2002) 193–220. [DOI] [PubMed] [Google Scholar]
- [94].Nicolussi A, Auer M, Sevcnikar B, Paumann-Page M, Pfanzagl V, Zámocký M, Hofbauer S, Furtmüller PG, Obinger C, Posttranslational modification of heme in peroxidases: impact on structure and catalysis, Archives of biochemistry and biophysics 643 (2018) 14–23. [DOI] [PubMed] [Google Scholar]
- [95].Regelsberger G, Jakopitsch C, Rüker F, Krois D, Peschek GA, Obinger C, Effect of distal cavity mutations on the formation of compound I in catalase-peroxidases, The Journal of biological chemistry 275(30) (2000) 22854–61. [DOI] [PubMed] [Google Scholar]
- [96].Matsson M, Tolstoy D, Aasa R, Hederstedt L, The distal heme center in Bacillus subtilis succinate:quinone reductase is crucial for electron transfer to menaquinone, Biochemistry 39(29) (2000) 8617–24. [DOI] [PubMed] [Google Scholar]
- [97].Zaidi S, Hassan MI, Islam A, Ahmad F, The role of key residues in structure, function, and stability of cytochrome-c, Cellular and molecular life sciences : CMLS 71(2) (2014) 229–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Winkler JR, Gray HB, Electron flow through metalloproteins, Chem Rev 114(7) (2014) 3369–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Wen X, Patel KM, Russell BS, Bren KL, Effects of heme pocket structure and mobility on cytochrome c stability, Biochemistry 46(9) (2007) 2537–2544. [DOI] [PubMed] [Google Scholar]
- [100].Lewin A, Hederstedt L, Heme A synthase in bacteria depends on one pair of cysteinyls for activity, Biochim Biophys Acta 1857(2) (2016) 160–168. [DOI] [PubMed] [Google Scholar]
- [101].Lewin A, Hederstedt L, Promoted evolution of a shortened variant of heme A synthase in the membrane of Bacillus subtilis, FEBS letters 582(9) (2008) 1330–4. [DOI] [PubMed] [Google Scholar]
- [102].Hsiao HC, Boycheva S, Watmough NJ, Brittain T, Activation of the cytochrome c peroxidase of Pseudomonas aeruginosa. The role of a heme-linked protein loop: a mutagenesis study, Journal of inorganic biochemistry 101(8) (2007) 1133–9. [DOI] [PubMed] [Google Scholar]
- [103].Dias JM, Alves T, Bonifácio C, Pereira AS, Trincão J, Bourgeois D, Moura I, Romão MJ, Structural basis for the mechanism of Ca2+ activation of the di-heme cytochrome c peroxidase from Pseudomonas nautica 617, Structure (London, England : 1993) 12(6) (2004) 961–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data in this study are available within the article, supporting information, and/or from the corresponding author(s) on request.