

Abstract
INTRODUCTION:
European local ancestry (ELA) surrounding APOE4 confers higher risk for Alzheimer Disease (AD) compared to African local ancestry (ALA). We demonstrated significantly higher APOE4 expression in ELA vs ALA in AD brains from APOE4/4 carriers. Chromatin accessibility differences could contribute to these expression changes.
METHODS:
We performed single nuclei Assays for Transposase Accessible Chromatin sequencing from frontal cortex of six ALA and six ELA AD brains, homozygous for local ancestry and APOE4.
RESULTS:
Our results showed an increased chromatin accessibility at the APOE4 promoter area in ELA vs ALA astrocytes. This increased accessibility in ELA astrocytes extended genome wide. Genes with increased accessibility in ELA in astrocytes were enriched for synapsis, cholesterol processing and astrocyte reactivity.
DISCUSSION:
Our results suggest that increased chromatin accessibility of APOE4 in ELA astrocyte contributes to the observed elevated APOE4 expression, corresponding to the increased AD risk in ELA vs ALA APOE4/4 carriers.
Keywords: Alzheimer disease, ancestry, APOE4, chromatin accessibility, gene expression, snATAC-seq, snRNA-seq, European, African
Graphical Abstract
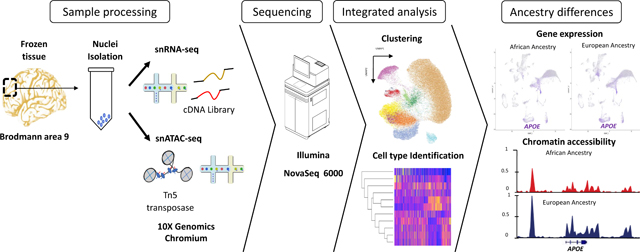
BACKGROUND:
Alzheimer disease (AD) is the most common neurodegenerative disorder and one of the leading causes of disability worldwide for individuals aged 75 and older1. The strongest genetic risk factor for late-onset AD is the apolipoprotein E4 (APOE4) allele located on chromosome 192,3. However, the risk associated with APOE4 differs dramatically between individuals of European vs African ancestries4–7 with APOE4 carriers of European ancestry having substantially increased risk for AD compared to individuals with African ancestry.
This observed difference in risk has recently been shown in three independent studies to be due to differences in the genetic local ancestry (LA) region surrounding APOE rather than overall global European or African ancestry8–10. Using single nucleus RNA sequencing (snRNA-seq) of frontal cortex we have previously demonstrated that APOE4/4 homozygous carriers with European local ancestry (ELA) expressed significantly higher APOE compared to those with African local ancestry (ALA), especially in astrocytes11, suggesting a potential mechanism underlying the differential AD risk seen between ancestries.
Control of gene expression is orchestrated by the integration of cis-regulatory modules, such as enhancer and promoter elements, along with transcription factors12–14. The cis-regulatory modules of actively transcribed genes are generally in ‘accessible’ euchromatin with low nucleosome occupancy and few high-order structures15–17, while ‘closed’ chromatin (heterochromatin) generally associates with transcriptional silencing16,18,19. Accordingly, ancestry-specific changes in accessibility of cis-regulatory elements that modulate the binding of transcription factors is a potential mechanism responsible for the difference in APOE4 expression seen between ancestries.
The single nuclei Assay for Transposase Accessible Chromatin followed by sequencing (snATAC-seq), facilitates the creation of chromatin accessibility maps with single cellular resolution. For example, Corces et al20 demonstrated that open chromatin regions in promoter or enhancer areas are associated with increased gene expression through snATAC-seq. Investigation of chromatin accessibility profiles specifically in AD have mostly focused on pathological hallmarks21. More recently, Morabito et al22 conducted an integrated analysis using snATAC-seq and snRNA-seq from brains of AD and control individuals and reported that cell-type-specific chromatin accessibility changes in regulatory elements may be key to gene expression changes in AD. However, to date, the role of chromatin accessibility in AD between different ancestries, specifically at the APOE4 locus, has not been investigated.
Therefore, we performed snATAC-seq combined with snRNA-seq to investigate the cell specific patterns of chromatin accessibility in ELA and ALA APOE4/4 brains. We observed increased accessibility in APOE ELA relative to ALA in astrocytes. The differentially accessible peaks at the APOE promoter are predicted to bind a subset of transcription factors that exhibit differential gene expression in ELA astrocyte samples. These data support the hypothesis that European ancestry at the APOE promoter and in the area around APOE have a more open chromatin conformation than African ancestry. We speculate that this difference is permissive for transcription factor binding and transcriptional activation of APOE. Increased chromatin accessibility in ELA samples was also observed genome-wide in this astrocytic cell type. Thus, these findings provide novel insights into the molecular mechanisms of ancestry specific differences in AD risk.
METHODS:
Brain Samples
Brain autopsy samples were obtained as part of a multi-center collaboration from four Alzheimer Disease Research Centers (ADRCs; Emory University, Northwestern University, Rush University Medical Center, and the University of Pennsylvania) and from the John P. Hussman Institute for Human Genomics (HIHG) at the University of Miami. All ADRC samples were initially selected from the National Alzheimer Coordinating Center (NACC) and compliant with site-specific approved institutional review board protocols as previously described11.
Local ancestry determination
Genotyping arrays were processed to assess both global and local genetic ancestry (LA) in the APOE region. To determine the LA, we phased independently our datasets with the SHAPEIT tool (version 2) using 1000 Genomes Phase 3 as the reference panel8. The APOE region was defined within 1Mb on either side of APOE, using chr19:44–46Mb as the LA boundaries for ancestry assignments, broad enough to include potential enhancers and topological associated domains. Individuals homozygous for the ELA or ALA haplotype in this region were Whole Genome Sequenced (WGS) at either the Center for Genome Technology (CGT) at the HIHG or The American Genome Center at Uniformed Services University of the Health Sciences (USUHS).
snATAC-seq and snRNA-seq data generation and analysis
Isolation of Nuclei:
Nuclei were isolated from frozen frontal cortex brain tissue (Brodmann area 9) as described previously23,24. A suspension of 65,000 nuclei was prepped according to the 10X Genomics Chromium Next GEM Single Cell ATAC 1.1 protocol. Libraries were sequenced on an Illumina NovaSeq 6000 targeting 75,000 reads per nuclei in paired end 50bp sequencing reactions. FASTQ files were processed using the CellRanger ATAC software package v1.2 (10X Genomics). snRNA-seq was performed as previously described11.
Integrated analysis of the snATAC-seq and snRNA-seq data:
This was performed using the ArchR software package v1.0.125. Clustering of cells was performed with five iterations of Iterative Latent Semantic Indexing (LSI)26,27 followed by batch correction with Harmony28. ATAC clusters were identified using FindClusters in Seurat v4.0 software package29 and refined the cell type definitions by integration with snRNA-seq from the same samples using Seurat’s FindTransferAnchors.
SnATAC-seq peak calling and differential accessibility
We created pseudo-bulk replicates combining all ELA and all ALA samples separately and called peaks for each ancestry within each cluster using the addReproduciblePeakSet function in ArchR utilizing the default parameters of the MACS2 callpeak command v2.2.7.130. Differential accessibility for each peak within each cluster was calculated using ArchR getMarkerFeatures.
ATAC peak annotations
HOMER31 (version 4.1132), was used to annotate peaks closest to transcription start sites (TSS) and to indicate exon, intron, intergenic, or promoter-TSS regions. A peak was assigned to a gene promoter-TSS when the peak location was ±2kb from the TSS. GREAT33 (version 4.0.4, http://great.stanford.edu/public/html/) was used to refine annotations where the peak was near multiple genes or intergenic. Distal enhancers were identified with the ELITE GeneHancer database from UCSC34 of enhancers and promoters. We considered as distal enhancer peaks those annotated by GREAT or HOMER farther than ±2kb from a TSS and with coordinates overlapping one of these ELITE enhancers.
Transcription Factor Analysis
Bedtools getfasta35 was used to extract the sequence of each peak (refdata-cellranger-atac-GRCh38–1.2.0 version). The algorithm FIMO36 within MEME Suite version 5.4.137 was used to identify known transcription factor (TF) binding motifs in the two APOE promoter differentially accessible peaks (DAP). We surveyed defined transcription factor binding site databases JASPAR CORE 2022 (vertebrates non redundant)38, Jolma 2013 (human and mouse)39, and Swiss regulon (human and mouse)40, and selected those with false discovery rate (FDR) adjusted p-value <0.05. In addition, we used all the human specific JASPAR 2022 defined TFs of the UCSC HG38 table browser41 filtering by a JASPAR score ≥ 300.
Functional Enrichment Analysis
Functional analysis was performed using enrichR42. using as reference the human specific gene-set libraries KEGG 202143, the Gene Ontology Biological Process 202144, Reactome 202145 and ENCODE Histone post-translational modifications46. Enrichment for cell type specific markers was performed using the cell type gene-set atlas lists from Azimuth 202147 and PanglaoDB 202148. Disease gene enrichment analysis used GeneSigDb49, HDSigDb50 and MsigDB51. Enrichment in chromosomal location was performed using a Fischer exact test.
RESULTS:
Brain Sample Characteristics
Twelve brain samples were used for the study (Table 1). Six ELA samples (European global ancestry >96%) were obtained from the HIHG Brain Bank (3), Rush University Medical Center (2) and Emory University (1). Six ALA samples (African global ancestry 68% to 88%) were obtained from Emory University (2), Northwestern University (1) and Rush University Medical Center (3). Samples included seven females (four ALA and three ELA) and five males (two ALA and three ELA). All samples had BRAAK staging scores ranging from IV to VI and had a mean age-of-death of 79 years. Whole genome sequencing confirmed the homozygous APOE4/4 genotype and absence of mutations in known Mendelian genes for AD (PSEN1, PSEN2, APP, MAPT) as well as in known rare AD risk variants in ABCA7, TREM2 and SORL1.
Table 1.
Demographic characteristics of the samples
| Sample | Center | Sex | AOD | APOE genotype | Local Ancestry (Chr19: 44–46Mb) | % Of Global Ancestry | BRAAK staging score |
|---|---|---|---|---|---|---|---|
| 1 | Emory | Female | 82 | 4,4 | AF/AF | 87% AF | V |
| 2 | NW | Female | 85 | 4,4 | AF/AF | 86% AF | V |
| 3 | Emory | Female | 80 | 4,4 | AF/AF | 88% AF | VI |
| 4 | Rush | Male | 93 | 4,4 | AF/AF | 68% AF | V |
| 5 | Rush | Male | 83 | 4,4 | AF/AF | 79% AF | VI |
| 6 | Rush | Female | 77 | 4,4 | AF/AF | 85% AF | VI |
| 7 | UM/Duke | Male | 75 | 4,4 | EU/EU | 95% EU | IV |
| 8 | Rush | Male | 71 | 4,4 | EU/EU | 98% EU | VI |
| 9 | UM/Duke | Female | 70 | 4,4 | EU/EU | 95% EU | VI |
| 10 | Rush | Male | 76 | 4,4 | EU/EU | 97% EU | IV |
| 11 | UM/Duke | Female | 76 | 4,4 | EU/EU | 96% EU | V |
| 12 | Rush | Female | 86 | 4,4 | EU/EU | 98% EU | VI |
AOD: Age of Death; UM: University of Miami; NW: Northwestern University; RUSH: RUSH University Medical Center; AF: African; EU: European
Cell type identification
We obtained data from a total of 60,306 nuclei from snATAC-seq and 94,411 nuclei from snRNA-seq. The snATAC-seq libraries had an average of ~13,000 fragments per nucleus and the snRNA-seq libraries had a median depth of ~116,000 reads per nucleus with on average ~1,900 genes/nucleus (Supplementary Table 1). The clusters with more than 500 nuclei in ELA and ALA showed no statistical differences in the number of nuclei per cluster between the ancestry groups (p-value= 0.497). Integration of the snATAC-seq and snRNA-seq resulted in the identification of 11 cell type clusters (Figure 1A) with no sample specific bias (Supplementary Figure 1A and 1B, Supplementary Table 2). The cell type identity of each cluster was determined using top 50 snATAC-seq predicted gene scores for marker genes of known cell types in the frontal cortex (Figure 1B). We used SLC17A7 for excitatory neurons; SLC32A1 for inhibitory neurons; MAG for oligodendrocytes; AIF1 for microglia; GFAP for astrocytes, CSPG4 for oligodendrocyte precursor cell (OPCs) and COL1A2 for vascular leptomeningeal cell (VLMC) (Supplementary Figure 2). We identified one oligodendrocyte cluster (55.6% of total cells), four excitatory neuron clusters (20.8% of the total cells), one inhibitory neuron cluster (8.3% of total cells), one microglia cluster (7.4% of total cells), one OPC cluster (3.9% of total cells), two astrocyte clusters (2.5% of total cells), and one vascular leptomeningeal cell (VLMC) cluster (1.5% of total cells) (Supplementary Table 2).
Figure 1. Visualization of the integrated single nuclei ATAC and RNA sequencing clusters and cell identification across the samples.
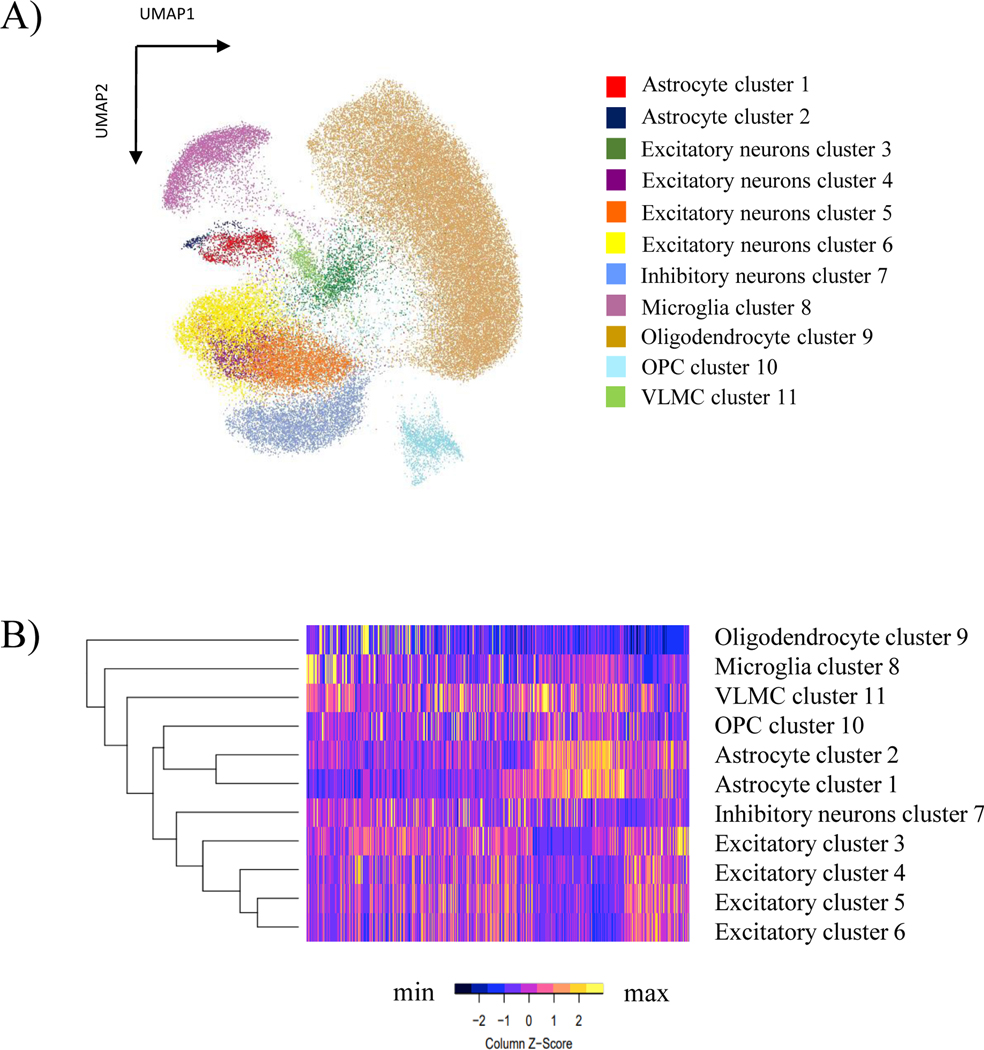
A) UMAP reduction plot using resolution of 0.4 resulting in 11 cell type clusters from the integration of snATAC-seq and snRNA-seq data. B) Heatmap showing cell cluster identification by chromatin accessibility patterns using the top 50 snATAC-Seq predicted gene scores for markers genes of the known cortex cell types.
To further characterize the two astrocyte cell clusters, we determined the genes that identified astrocyte cluster 1 and astrocyte cluster 2 and investigated their expression in the single cell atlas of the Entorhinal Cortex in Human Alzheimer’s Disease (ECHAD)52 and astrocyte transcriptomic data from frontal cortex53. We observed that the genes characterizing astrocyte cluster 1 were primarily expressed in astrocyte subclusters a4 to a8, while the genes from our astrocyte cluster 2 were primarily expressed in astrocyte subclusters a2 and a3 from ECHAD, which correspond to reactive astrocytes subpopulations (Supplementary Figure 3)53.
Accessibility analysis at the APOE locus and LA
We first investigated whether the ancestry-related differential expression of APOE4 previously described11 was recapitulated when additional samples from Rush University Medical Center were included in our analyses. Indeed, APOE4 was differentially expressed when considering expression over all cell types between ancestries with greater expression in ELA (FC= 1.31; p-value=9.66E−219), except one excitatory neurons cluster with higher expression in ALA (FC= 1.28; p-value=5.87E6). This pattern of increased expression in ELA samples was true for four cell types (excitatory and inhibitory neurons, astrocytes and microglia) (Supplementary Table 3). Astrocytes had the highest fold change difference in ELA (FC = 1.56) and most significant p-value (p-value=1.24E−129).
We next determined whether the APOE expression difference in astrocytes is associated with differences in chromatin accessibility. The snATAC-seq and snRNA-seq integrated UMAP showed that APOE expression as well as accessibility was highest in the astrocyte cluster 1 (Figure 2A). Comparison of accessibility between ancestries revealed two peaks with significantly increased accessibility in ELA at the APOE promoter in astrocyte cluster 1 (Figure 2B). These peaks were located at −19bp and −1990bp upstream of the APOE transcription start site (TSS) (FC: 2.57 and 4.52; FDR: 0.001 and 0.02, respectively). Notably, although APOE showed significantly increased expression in several cell types, significant differences in accessibility were only detected in astrocyte cluster 1 (Supplementary Figure 4).
Figure 2. Differential chromatin accessibility and expression of APOE in Astrocyte cluster 1.

A) APOE expression represented in clusters generated by the integrated snATAC-seq and snRNA-seq data; B) Visualization of chromatin differential accessible peaks in the APOE locus between ancestries from Astrocyte cluster 1. *Represents significantly differentially accessible peaks between ancestries (*FDR=0.02; ***FDR=0.001). C) Venn diagram showing transcription factors biding to APOE that are differentially expressed (DEG) and differentially accessible (DAG).
To gain insight into the molecular mechanisms involved in the increased accessibility and increased expression of APOE, we determined which known transcription factor binding sites were present in the differentially accessible peaks and analyzed the accessibility and gene expression of those transcription factors. The two differentially accessible peaks at the APOE promoter overlap predicted binding sites for a set of 15 transcription factors (GLI2, NPAS2, KLF15, HIF1A, LHX2, RXRA, MXI1, FOS, JUNB, KLF2, PURA, SREBF1, TEAD1, KLF6, ZBTB7C) that are also differentially expressed and have differentially accessible genomic region between ancestries (Supplemental Table 4). From these 15 putative APOE binding transcription factors, PURA and SREBF1 have increased accessibility and expression in ELA samples, KLF6 has increased accessibility and expression in ALA samples and the other 12 transcription factors have increased accessibility in ELA and increased expression in ALA.
We identified 32 additional differentially accessible peaks with greater accessibility in ELA than ALA astrocytes in gene promoters (within 2kb from the TSS) in the LA region surrounding APOE (chr19:44–46MB), which corresponds to 19 additional genes, including the APOE proximal genes TOMM40 and APOC1 (Table 2). No genes in the defined LA region were more accessible in the ALA. In addition, by overlapping the differentially accessible peaks in the APOE LA region with previously classified enhancers (ELITE GeneHancer from USCS) we identified 32 additional peaks among 23 LA genes, all with increased accessibility in ELA brains (Supplementary Table 5).
Table 2.
Differentially accessible peaks in promoter regions of Local Ancestry genes in Astrocyte cluster 1.
| Gene | Chromosome | Peak location | Distance to TSS | FC | FDR | Region |
|---|---|---|---|---|---|---|
| CTB-171A8.1 | chr19 | 44718456 | −21 | 3.66 | 0.003 | promoter-TSS |
| BCL3 | chr19 | 44748309 | 12 | 2.77 | 0.007 | promoter-TSS |
| BCAM | chr19 | 44808802 | −7 | 5.11 | 0.043 | promoter-TSS |
| BCAM | chr19 | 44808802 | −51 | 5.11 | 0.043 | promoter-TSS |
| CTB-129P6.4 | chr19 | 44890205 | 421 | 2.70 | 0.002 | promoter-TSS |
| TOMM40 | chr19 | 44890205 | −781 | 2.70 | 0.002 | promoter-TSS |
| APOE* | chr19 | 44903514 | −1990 | 4.52 | 0.022 | promoter-TSS |
| APOE* | chr19 | 44905485 | −19 | 2.57 | 0.001 | promoter-TSS |
| APOC1 | chr19 | 44914897 | 278 | 2.05 | 0.000 | promoter-TSS |
| APOC1 | chr19 | 44914897 | 900 | 2.05 | 0.000 | promoter-TSS |
| APOC1P1 | chr19 | 44926992 | 266 | 2.23 | 0.025 | promoter-TSS |
| CLPTM1 | chr19 | 44955080 | 84 | 3.61 | 0.007 | promoter-TSS |
| CLPTM1 | chr19 | 44955080 | 84 | 3.61 | 0.007 | promoter-TSS |
| CTB-179K24.3 | chr19 | 45091377 | −1236 | 2.19 | 0.027 | promoter-TSS |
| PPP1R37 | chr19 | 45091377 | −1335 | 2.19 | 0.027 | promoter-TSS |
| MARK4* | chr19 | 45294609 | 504 | 3.65 | 0.011 | promoter-TSS |
| ERCC2 | chr19 | 45370425 | −88 | 2.40 | 0.002 | promoter-TSS |
| ERCC2 | chr19 | 45370425 | −57 | 2.40 | 0.002 | promoter-TSS |
| PPP1R13L | chr19 | 45405279 | −491 | 3.68 | 0.004 | promoter-TSS |
| CD3EAP | chr19 | 45405279 | −1162 | 3.68 | 0.004 | promoter-TSS |
| PPP1R13L | chr19 | 45405279 | 820 | 3.68 | 0.004 | promoter-TSS |
| FOSB | chr19 | 45467194 | −551 | 2.61 | 0.024 | promoter-TSS |
| VASP | chr19 | 45506348 | −818 | 3.58 | 0.020 | promoter-TSS |
| OPA3 | chr19 | 45584758 | −166 | 2.16 | 0.000 | promoter-TSS |
| EML2* | chr19 | 45644874 | 505 | 7.63 | 0.032 | promoter-TSS |
| EML2* | chr19 | 45645399 | −20 | 3.28 | 0.002 | promoter-TSS |
| FBXO46 | chr19 | 45730087 | 567 | 4.28 | 0.025 | promoter-TSS |
| FBXO46 | chr19 | 45730692 | −38 | 2.01 | 0.028 | promoter-TSS |
| FBXO46 | chr19 | 45730692 | −38 | 2.01 | 0.028 | promoter-TSS |
| RSPH6A | chr19 | 45815742 | −673 | 3.10 | 0.033 | promoter-TSS |
| IRF2BP1 | chr19 | 45885449 | 471 | 4.72 | 0.025 | promoter-TSS |
| NOVA2 | chr19 | 45973825 | −529 | 5.11 | 0.004 | promoter-TSS |
| CCDC61 | chr19 | 45995053 | −158 | 2.38 | 0.000 | promoter-TSS |
| CCDC61 | chr19 | 45995053 | −164 | 2.38 | 0.000 | promoter-TSS |
FC: Fold change; TSS: Transcription Starting Site; Negative value in Distance to TSS reflects peaks falling upstream of TSS
Genome-wide differential accessibility
Since the global ancestry of the ELA samples were predominantly European and the European local ancestry blocks are uniformly distributed among ALA samples (Supplementary Figure 5), we performed genome-wide differential accessibility analysis. In total we identified 5,154 significant (FDR ≤ 0.05 and FC ≥ 2) differentially accessible peaks between the ancestries in three cell types (astrocytes, excitatory neurons and microglia), representing less than 1% of all called ATAC peaks. 99% of these differentially accessible peaks were seen in the astrocyte cluster 1, with an overall increased chromatin accessibility in the European ancestry blocks compared to African ancestry blocks (more accessible in ELA: 4,546 peaks; more accessible in ALA: 107 peaks) (Figure 3A). This astrocyte specific accessibility difference was widely spread across all chromosomes (Figure 3B). Among all differentially accessible peaks, 25.8% (2,248 peaks) are in promoters and TSS of genes, 33.5 % (2,920 peaks) are intragenic, 34.1% (2,970 peaks) are intergenic and 6.6% (574 peaks) are in distal ELITE enhancers (Figure 3C), altogether corresponding to 6,067 genes.
Figure 3. Chromatin accessibility differences between ancestries genome-wide in Astrocyte cluster 1.

A) Volcano plot representation of global chromatin accessibility difference between ancestries in Astrocyte cluster 1; B) Chromatin accessibility differences across the genome displayed by chromosomes. Blue is increased accessibility in Europeans, Red is increased accessibility in African ancestry; C) Pie chart showing the differentially accessible peak region distribution in Astrocyte cluster 1.
Sixteen percent of the differentially accessible peaks were found on chromosome 19, a significant enrichment as compared to a random chromosomal distribution of peaks (Fischer exact test adjusted p-value=2.501E−31) with 36.4% of these differentially accessible peaks in the promoter regions of chromosome 19 genes (Supplementary Figure 6).
Pathway enrichment of differentially accessible and expressed genes
To gain mechanistic insights from the integrative snRNA-seq and snATAC-seq approach, we determined the genome-wide overlap between DEG in the astrocyte clusters identified by snRNA-seq and genes associated with differentially accessible peaks identified by snATAC-seq (DAG). From the 6,067 genes with differentially accessible peaks, 239 genes were both differentially expressed and accessible (DEG-DAG) between the ancestries (Supplementary table 6). However only 55 genes had concordant differences in expression and accessibility: 48 with increased accessibility and expression in ELA (including APOE) and 7 with reduced expression and accessibility in ELA. This set of 55 DEG-DAG in astrocytes were used to compute a functional enrichment analysis.
KEGG pathways (KEGG) and Gene Ontology (GO) analysis of gene set enrichment consider different category sets. KEGG pathway analyses identify over-representation of genes participating in the same biological process, while GO analyses aim to reduce complexity and focus on the common functional properties of the gene products. Thus, both analyses were performed, as it is expected that they will highlight different aspects of the data. Analysis of KEGG Molecular functional pathways showed enrichment in signaling pathways including calcium signaling and MAPK signaling pathway. The GO gene-sets highlighted alterations in the biological process of cholesterol metabolism, including regulation of cholesterol metabolic process and biosynthetic process, pathways linked to synapses and axonal transport, spine development and pathways linked to glial cell and astrocyte projection (Supplementary Table 7).
Using Human MSigDB database of disease associated genes from published transcriptomic studies revealed the highest overlap with lipopolysaccharide (LPS) treated astrocytes to induce reactivity (adjusted p-value =8.16E−10, GSE75246)54 and with genes showing altered expression in brains (adjusted p-value =2.26E−19, GSE79666)55 and astrocytes (adjusted p-value =2.92E−19)56 of Huntington Disease (HD) individuals. The overlapping genes between the ancestry DEG-DAG and genes up-regulated in astrocytes of HD patients include a molecular signature of reactive astrocyte markers also observed in the HD striatal astroglia data, but also extends to pathways related to glutamatergic synapse and signaling. In addition, the ENCODE database of brain histone post-translational modifications identified an enrichment of DEG-DAG in those genes having H3K27me3 signals in astrocytes (H3K27me3 astrocytes Hg19, adjusted p-value = 0.02) (Supplementary Table 7).
Chromatin accessibility in AD candidate genes
We also assessed chromatin accessibility differences between the ancestries in AD associated genes, including Mendelian genes (APP, PS1, PS2, and MAPT) and genes suggested by GWAS and rare variant association studies across ancestries57,58. 32 differentially accessible peaks between the ancestries were identified in seven out of 76 AD-associated loci as defined by the ADSP Gene Verification Committee (https://adsp.niagads.org/gvc-top-hits-list/)58. The correlation between accessibility and expression of these seven genes with differential accessibility is shown in Table 3. Aside from APOE, nine differentially accessible peaks were observed to fall in the promoter areas of three genes, SORL1, VRK3 and ABCA7, all with increased accessibility in ELA regions. Ten differentially accessible peaks fall in intergenic regions close to five AD genes, all with increase accessibility in ELA. In addition, twelve peaks are found in the gene body of four AD-associated genes (CLU, PTK2B, SORL1, SPHK1), also with increased accessibility in ELA. Nonetheless, none of these genes containing differentially accessible peaks between local ancestries showed significant differential expression between ancestries (Table 3).
Table 3.
Differentially accessible peaks in Astrocyte cluster 1 in Alzheimer disease GWAS candidate genes.
| Gene | Chr. | Peak location | Cell cluster | Distance to TSS | SnATAC FC | SnATAC FDR | SnRNA FC | SnRNA FDR | Region |
|---|---|---|---|---|---|---|---|---|---|
| PTK2B | chr8 | 27340471 | Astrocyte 1 | 29239 | 2.327 | 0.026 | −1.05 | 4.67E-13 | intron |
| CLU | chr8 | 27596089 | Astrocyte 1 | 18692 | 4.235 | 0.019 | −1.15 | 3.39E-73 | intergenic |
| CLU | chr8 | 27597084 | Astrocyte 1 | 17697 | 2.556 | 0.035 | −1.15 | 3.39E-73 | exon |
| CLU | chr8 | 27597084 | Astrocyte 1 | 17697 | 2.556 | 0.035 | −1.15 | 3.39E-73 | intron |
| CLU | chr8 | 27597917 | Astrocyte 1 | 16864 | 18.113 | 0.008 | −1.15 | 3.39E-73 | exon |
| CLU | chr8 | 27597917 | Astrocyte 1 | 16864 | 18.113 | 0.008 | −1.15 | 3.39E-73 | intron |
| CLU | chr8 | 27607577 | Astrocyte 1 | 7204 | 6.695 | 0.032 | −1.15 | 3.39E-73 | intron |
| ACER3 | chr11 | 76799174 | Astrocyte 1 | −61449 | 2.41 | 0.008 | 1.04 | 1.12E-30 | intergenic |
| SORL1 | chr11 | 121566449 | Astrocyte 1 | 114496 | 3.538 | 0.044 | −1.05 | 4.51E-53 | exon |
| SORL1 | chr11 | 121566449 | Astrocyte 1 | 114496 | 3.538 | 0.044 | −1.05 | 4.51E-53 | intron |
| SORL1 | chr11 | 121566449 | Astrocyte 1 | −26 | 3.538 | 0.044 | −1.05 | 4.51E-53 | promoter-TSS |
| SORL1 | chr11 | 121591362 | Astrocyte 1 | 139409 | 7.332 | 0.011 | −1.05 | 4.51E-53 | intron |
| SORL1 | chr11 | 121591362 | Astrocyte 1 | 1193 | 7.332 | 0.011 | −1.05 | 4.51E-53 | promoter-TSS |
| SORL1 | chr11 | 121596337 | Astrocyte 1 | 6168 | 6.485 | 0.046 | −1.05 | 4.51E-53 | intron |
| SORL1 | chr11 | 121596337 | Astrocyte 1 | 144384 | 6.485 | 0.046 | −1.05 | 4.51E-53 | intron |
| SORL1 | chr11 | 121695992 | Astrocyte 1 | 105823 | 3.063 | 0.039 | −1.05 | 4.51E-53 | intergenic |
| SORL1 | chr11 | 121695992 | Astrocyte 1 | 244039 | 3.063 | 0.039 | −1.05 | 4.51E-53 | intergenic |
| SORL1 | chr11 | 121795198 | Astrocyte 1 | 205029 | 3.099 | 0.027 | −1.05 | 4.51E-53 | intergenic |
| SORL1 | chr11 | 121795198 | Astrocyte 1 | 343245 | 3.099 | 0.027 | −1.05 | 4.51E-53 | intergenic |
| SORL1 | chr11 | 121929505 | Astrocyte 1 | 477552 | 3.494 | 0.004 | −1.05 | 4.51E-53 | intergenic |
| SORL1 | chr11 | 121932043 | Astrocyte 1 | 480090 | 11.926 | 0.031 | −1.05 | 4.51E-53 | intergenic |
| SPHK1 | chr17 | 76397939 | Astrocyte 1 | 13539 | 4.176 | 0.041 | −1.01 | 9.98E-43 | intergenic |
| SPHK1 | chr17 | 76397939 | Astrocyte 1 | 12897 | 4.176 | 0.041 | −1.01 | 9.98E-43 | intron |
| SPHK1 | chr17 | 76414406 | Astrocyte 1 | 30006 | 15.903 | 0 | −1.01 | 9.98E-43 | intergenic |
| ABCA7 | chr19 | 1039766 | Astrocyte 1 | −87 | 2.429 | 0.001 | 1.09 | 2.95E-21 | promoter-TSS |
| ABCA7 | chr19 | 1040877 | Astrocyte 1 | −119 | 4.091 | 0.002 | 1.09 | 2.95E-21 | promoter-TSS |
| ABCA7 | chr19 | 1040877 | Astrocyte 1 | 1024 | 4.091 | 0.002 | 1.09 | 2.95E-21 | promoter-TSS |
| VRK3 | chr19 | 50024717 | Astrocyte 1 | 360 | 5.864 | 0.043 | 1.09 | 4.88E-24 | promoter-TSS |
| VRK3 | chr19 | 50024717 | Astrocyte 1 | 979 | 5.864 | 0.043 | 1.09 | 4.88E-24 | promoter-TSS |
| VRK3 | chr19 | 50025268 | Astrocyte 1 | −116 | 3.408 | 0.002 | 1.09 | 4.88E-24 | promoter-TSS |
| VRK3 | chr19 | 50025268 | Astrocyte 1 | 428 | 3.408 | 0.002 | 1.09 | 4.88E-24 | promoter-TSS |
| VRK3 | chr19 | 50050285 | Astrocyte 1 | −24589 | 5.267 | 0.002 | 1.09 | 4.88E-24 | intergenic |
FC: Chr.: Chromosome; FC:Fold change; TSS: Transcription Starting Site; Negative value in Distance to TSS reflects peaks falling upstream of TSS
DISCUSSION:
To understand the complex mechanisms by which APOE4 confers differential risk for AD in the context of genetic ancestry, we evaluated both gene expression and chromatin accessibility profile differences between homozygous APOE4/4 ELA and ALA Alzheimer disease brains at single cell resolution. We confirmed our previous finding11 that APOE is significantly more expressed in astrocytes of ELA compared to ALA. In fact, APOE is the most differentially expressed gene in astrocytes in the 2Mb local ancestry region surrounding APOE. Beyond being the most differentially expressed, the promoter of APOE in astrocytes showed a significant increase in accessibility in ELA compared to ALA. These results suggest that the increased APOE4 expression previously reported in ELA APOE4 carriers11 may be partly due to differences in chromatin remodeling at the promoter of APOE4 between ancestries. We have also shown using Capture Chromatin Conformation analysis and massively parallel reporter assays that specific DNA sequence differences in areas physically interacting with the APOE promoter have functional effects in microglia and astrocytes, with greater expression occurring in ELA sequence variants than ALA59. Thus, multiple mechanisms affecting APOE4 gene expression in astrocytes may be activated at different times in life or by stress and could affect the differences in AD risk seen between ancestral homozygous APOE4 carriers. This emphasizes that APOE loci is under a complex regulatory environment that involves cell type-specific processes and strengthens the primary functional role of APOE in astrocytes60.
Though our principal research question focused on the underlying mechanisms leading to the higher expression of APOE in ELA brains using snATAC-seq, this study also provides a global view on the chromatin landscape of AD APOE4/4 carriers from different genetic ancestries. One challenge is the relative scarcity of available autopsy material for African Americans. This is made further challenging as we required homozygous local ancestry for African local ancestry and homozygosity for the APOE4 genotype. These requirements limited the available number for final analysis. It highlights the need for an effort to understand the concerns of the African American and Hispanic/Latino populations on participating in autopsy studies and work with these populations to increase the number of autopsies in AD family members. Non-Hispanic Europeans are not admixed and thus almost exclusively European local ancestry, so that the only additional limiting criteria is APOE4 homozygosity.
We observed in astrocyte cluster 1 a significantly higher chromatin accessibility in ELA brains genome wide. A recent study using mesenchymal progenitor cells has reported that APOE accumulation in the nucleus can disrupt and destabilize heterochromatin, which would increase chromatin accessibility, similar to what we observe here in the ELA with higher APOE expression61. As astrocytes are the primary cells expressing APOE, this could explain the finding of increased global accessibility primarily in this cell type. Notably, only a small percent of significant peaks of higher chromatin accessibility overlap with the significant differentially expressed genes identified by snRNA-seq. Conversely, only 40% of the significant differentially expressed genes showed significant changes in accessibility between the ancestries. This is in accordance with the growing understanding that gene expression depends on multiple regulatory elements62, many located far from the gene locus itself22. Some of the DEG-DAG show a positive correlation between expression and accessibility, suggesting a functional relationship. However, we also observed an anti-correlation in accessibility-expression pairs. This anti-correlation could represent binding by repressors leading to a decrease in expression as previously described63,64. In addition, highly accessible sites not associated with enhanced expression could represent poised enhancers/promoter with no active transcription occurring65. Besides this known biological partial correlation of accessibility and expression, in this study the snATAC and snRNA were derived from separate, but adjacent, brain tissue. Differences in tissue composition could compound the discordance between these two outcomes. Future studies with paired snATAC-seq and snRNA-seq from the same nuclei such as the 10X Genomics Single Cell Multiome will minimize tissue composition effects if present. In fact, while we do identify differential expression of APOE between ancestries in several cell types (e.g., microglia), we only see significant differential accessibility of its promoter and immediate region in astrocytes.
Astrocytes have well established roles in neuronal metabolic support and neuroprotection, synapse formation and function, ion signaling homeostasis, integrity of the blood brain barrier, tissue repair and complex brain functions such as memory and sleep66–70. Thus, it is not surprising that a relevant role for astrocytes in AD pathology is suggested71. Our data suggest that astrocytes are involved in the modulation of APOE4-afforded AD risk and pinpoint this cell type as one of the most sensitive to differences in genetic ancestry. The significant enrichment of astrocytic ancestry-associated DEG-DAG in genes upregulated in HD astrocytes suggest that the differences associated with diverse ancestry entail functional properties that are potentially common to other neurodegenerative disorders. In addition to this astrocyte-ancestry association, we also evidence a difference in DEG-DAG between our astrocyte clusters 1 and 2. We observed that our astrocyte cluster 1 was transcriptionally most similar to astrocytes subclusters a4 and a8 described by Grubman et al52, clusters where APOE was upregulated in AD52; while our astrocyte subcluster 2 was transcriptionally comparable to astrocytes subcluster a2 where APOE was downregulated.
Interestingly, there was a significant enrichment of differential chromatin accessibility and expression on chromosome 19 relative to the rest of the genome (Supplemental Figure 5) in astrocyte cluster 1. These results correlate with the observation of a comparatively high number of ATAC-seq peaks in chromosome 1972, potentially due to the high gene density, GC content and DNA binding proteins in chromosome 19 compared to other chromosomes73–75. It is possible the high gene density and the structural nature of chromosome 19 could modulate the differential binding of chromatin modifying enzymes, explaining the enrichment in differentially accessible chromatin and gene expression in this specific chromosome observed between ancestries.
Our analysis of transcription factors with binding sites in the differentially accessible peaks at the 5’end of APOE that were both differentially expressed and accessible between ancestries suggests transcriptional modulators of APOE expression differences. Of the 15 transcription factors we identified, only SREBF1 and PURA showed increased accessibility (intergenic and intronic) and expression in ELA samples and KLF6 showed increased accessibility (distal enhancer) and expression in ALA samples. A polymorphism in SREBF1 has been shown to influence the risk of AD specifically in APOE4 carriers76, while PURA has been shown to regulate expression of AD and Aβ clearance related genes77. On the other hand, KLF6 is found to be differentially expressed in AD brains and implicated in Aβ-induced oxidative stress78. Their role with APOE activation has not been previously described, although SREBF1 is a major regulator of lipid metabolism.
Although many genetic risk factors for disease are expected to be shared among populations, genomic diversity among ancestries can provide new opportunities for discoveries regarding disease susceptibility. In AD, understanding why the APOE4 allele confers a lower risk to African ancestry carriers versus European ancestry carriers is instrumental for the identification of potential AD therapeutics. Here we provide chromatin accessibility and gene expression data from AD APOE4 carriers of African and European ancestries at single cell resolution. One of the main challenges in comparing local ancestry effects is the current limitation of available samples from diverse ancestries. The need for both specific genotype and local ancestry criteria further restricted the number of samples available. This highlights the importance of increasing autopsy material from diverse populations. Expansion of this study into other APOE genotypes and more diverse ancestries could provide further insight into the importance of brain astrocytes and chromatin landscape of APOE, as well as genome-wide changes, implicating specific molecular pathways and regulatory proteins in this risk difference. Finally, these findings support the concept of reducing APOE4 expression as a potential therapeutic pathway for AD.
CONCLUSIONS:
Our results provide novel compelling insights to understand the AD risk difference known to exist between European and African APOE4 local ancestry carriers. Here we demonstrate that differences in chromatin accessibility between the African and European APOE locus could explain the expression differences in APOE expression in astrocytes and supports the concept that this contributes to the risk difference between African American and European populations for AD in APOE4 carriers. We found that this increase in accessibility in the region surrounding APOE4 in European astrocytes extended genome-wide, suggesting a wider mechanism of regulatory dysfunction is occurring in these cells. Not only is it important to include diverse ancestries to ensure all individuals are represented in research, but also we have shown here and in previous work that including diversity in research can provide additional windows of opportunity to elucidate disease and biological mechanisms. Understanding regulatory dysfunction in AD has not been well studied and should be and area of future research in AD.
Supplementary Material
Supplementary Figure 1. SnATAC-seq data analysis for sample bias.
Supplementary Figure 2. SnATAC-seq cell marker cluster identification by chromatin accessibility.
Supplementary Figure 3. Astrocyte subtype classification.
Supplementary Figure 4. APOE chromatin accessibility in the other cell clusters where APOE is highly expressed but not differentially accessible between the ancestries.
Supplementary Figure 5. Visualization of local ancestry across all chromosomes per individual sample.
Supplementary Figure 6. Enrichment of differentially accessible peak between ancestries in chromosome 19.
Supplementary Table 1. Summary of snATAC and snRNA sequencing results.
Supplementary Table 2. Total cell count and proportion per cluster in the integrated sequencing analysis.
Supplementary Table 3. snRNA sequencing clusters with significant APOE4 differential expression between ancestries.
Supplementary Table 4. TFs (DEGs-DAGs) binding to differentially accessible peaks at the promoter of APOE.
Supplementary Table 5. APOE Local ancestry accessibility analysis.
Supplementary Table 6. Differentially accessible and expressed genes (DEG-DAG) in astrocyte cluster 1.
Supplementary Table 7. Subset of the differential functional analysis of the astrocyte cluster 1 DEG-DAG with positive correlation between expression and accessibility using EnrichR software.
Acknowledgements
We acknowledge the collaboration of Dr. John Q. Trojanowski in this study, whose untimely death is a great loss to both his family, colleagues and to the field of neurodegeneration research.
Funding
This study was supported by the National Institute of Health [grant numbers R01-AG059018, U01-AG052410, U01-AG057659, U01AG072579, and R01-AG015819], the Alzheimer Disease Center (ADC) networks (NIA) [grant number AG054074], the BrightFocus Foundation and Alzheimer Association [grant number A2018425S]. The work was also funded by the National Institutes of Health [grant number P50-AG0256878 and P30-AG013854] from Emory and Northwestern, as well as from the Alzheimer Disease Core Center grant [P30AG010161] from Rush Alzheimer Disease Center. Genomic and data analysis was provided by the Center for Genome Technology (CGT) from the John P. Hussman Institute for Human Genomics (HIHG).
LIST OF ABBREVIATIONS:
- AD
Alzheimer Disease
- ABCA7
ATP Binding Cassette Subfamily A Member 7
- ACER3
Alkaline Ceramidase 3
- ADRC
Alzheimer Disease Research Centers
- AIF1
Allograft inflammatory factor 1
- ALA
African Local Ancestry
- APOC1
Apolipoprotein C1
- APOE
Apolipoprotein E
- APOE4
Apolipoprotein E allele 4
- APOE4/4
Homozygous for Apolipoprotein E allele 4
- APP
Amyloid Beta Precursor Protein
- ATAC
Assay for Transposase-Accessible Chromatin
- Azimth
App for reference-based single-cell analysis
- Aβ
Amyloid beta
- BRAAK
Braak staging (pathology score for Alzheimer Disease)
- CGT
Center for Genome Technology
- CLU
Clusterin
- COL1A2
Collagen Type I Alpha 2 Chain
- CSPG4
Chondroitin Sulfate Proteoglycan 4
- DAG
Differentially Accessible Genes
- DAP
Differentially Accessible Peaks
- DEG
Differentially Expressed Genes
- DEG-DAG
Differentially Expressed and Accessible Genes
- ECHAD
Entorhinal Cortex in Human Alzheimer’s Disease
- ELA
European Local Ancestry
- ELITE
enhancer gene with both a high likelihood enhancer definition and a strong enhancer–gene association
- ENCODE
Encyclopedia of DNA Elements
- FC
Fold Change
- FDR
false discovery rate
- FOS
Fos Proto-Oncogene, AP-1 Transcription Factor Subunit
- GC
Guanine-Cytosine content
- GeneSigDb
Gene Signature Data Base
- GFAP
Glial Fibrillary Acidic Protein
- GLI2
GLI Family Zinc Finger 2
- GO
Gene Ontology
- GRCh38
Genome Reference Consortium Human Build 38
- GREAT
Genomic Regions Enrichment of Annotations Tool
- H3K27me3
tri-methylation of lysine 27 on histone H3 protein
- HD
Huntington Disease
- HDSigDb
Huntigton Disease Signature Database
- HIF1A
Hypoxia Inducible Factor 1 Subunit Alpha
- HIHG
John P. Hussman Institute for Human Genomics
- HOMER
Hypergeometric Optimization of Motif EnRichment
- JASPAR
transcription factor binding profile database
- JOLMA
Human specific transcription factor binding
- JUNB
JunB Proto-Oncogene, AP-1 Transcription Factor Subunit
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KLF15
Kruppel Like Factor 15
- KLF2
Kruppel Like Factor 2
- KLF6
Kruppel Like Factor 6
- LA
Local Ancestry
- LHX2
LIM Homeobox 2
- LPS
lipopolysaccharide
- LSI
Iterative Latent Semantic Indexing
- MAG
Myelin Associated Glycoprotein
- MAPT
microtubule-associated protein tau
- MB
Million Base pair
- Mb
Mega base
- MsigDB
Molecular Signatures Database
- MXI1
MAX interactor-1
- NACC
National Alzheimer Coordinating Center
- NPAS2
Neuronal PAS Domain Protein 2
- OPCs
oligodendrocyte precursor cells
- PanglaoDB
Single-cell sequencing mouse and human database
- PCR
Polymerase chain reaction
- PSEN1
Presenillin-1
- PSEN2
Presenillin-2
- PURA
Purine Rich Element Binding Protein A
- Reactome
pathway database
- RSB
Resuspension Buffer
- RXRA
Retinoid X Receptor Alpha
- SLC17A7
Solute Carrier Family 17 Member 7
- SLC32A1
Solute Carrier Family 32 Member 1
- snATAC-seq
single nuclei Assay for Transposase-Accessible Chromatin sequencing
- snRNA-seq
single nuclei RNA sequencing
- SORL1
Sortilin Related Receptor 1
- SPHK1
Sphingosine Kinase 1
- SREBF1
Sterol Regulatory Element Binding Transcription Factor 1
- TEAD1
TEA Domain Transcription Factor 1
- TF
Transcription Factor
- TOMM40
Translocase Of Outer Mitochondrial Membrane 40
- TREM2
Triggering Receptor Expressed On Myeloid Cells 2
- TSS
Transcription Starting Site
- TTS
transcription terminating site
- UCSC
University of California Santa Cruz
- USUHS
Uniformed Services University of the Health Sciences
- VLMC
vascular leptomeningeal cell
- VRK3
VRK Serine/Threonine Kinase 3
- ZBTB7C
Zinc Finger And BTB Domain Containing 7C
- WGS
Whole Genome Sequencing
Footnotes
Consent Statement
Individuals included in this study provided informed autopsy consent to the Institutional Review Board from their participating center.
Consent for publication
All authors consent for publication.
Conflicts of Interest
K.C, M.D.M.M, F.R, P.W, K.H, D.M.D, K.N, L.W, M.F, S.W, C.G, M.G, C.L.D, F.J, D.A.B, T.S, M.A.P, A.J.G, J.I.Y and J.M.V have nothing to disclose. The authors declare that they have no competing interests.
Availability of data and material
Data are available through the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS) Data Sharing Service (DSS): https://dss.niagads.org/datasets/ng00067/
REFERENCES:
- 1.. GBD 2019 Diseases and Injuries Collaborators. (2020). Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 10258, 1204–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.. Yamazaki Y, Zhao N, Caulfield TR, Liu CC, and Bu G. (2019). Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol 9, 501–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, and Pericak-Vance MA (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 5123, 921–923. [DOI] [PubMed] [Google Scholar]
- 4.. Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, and van Duijn CM (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 16, 1349–1356. [PubMed] [Google Scholar]
- 5.. Graff-Radford NR, Green RC, Go RCP, Hutton ML, Edeki T, Bachman D, Adamson JL, Griffith P, Willis FB, Williams M, Hipps Y, Haines JL, Cupples LA, and Farrer LA (2002). Association between apolipoprotein E genotype and Alzheimer disease in African American subjects. Arch. Neurol 4, 594–600. [DOI] [PubMed] [Google Scholar]
- 6.. Neu SC, Pa J, Kukull W, Beekly D, Kuzma A, Gangadharan P, Wang LS, Romero K, Arneric SP, Redolfi A, Orlandi D, Frisoni GB, Au R, Devine S, Auerbach S, Espinosa A, Boada M, Ruiz A, Johnson SC, Koscik R, Wang JJ, Hsu WC, Chen YL, and Toga AW (2017). Apolipoprotein E Genotype and Sex Risk Factors for Alzheimer Disease: A Meta-analysis. JAMA Neurol 10, 1178–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.. Tang MX, Stern Y, Marder K, Bell K, Gurland B, Lantigua R, Andrews H, Feng L, Tycko B, and Mayeux R. (1998). The APOE-epsilon4 allele and the risk of Alzheimer disease among African Americans, whites, and Hispanics. JAMA 10, 751–755. [DOI] [PubMed] [Google Scholar]
- 8.. Rajabli F, Feliciano BE, Celis K, Hamilton-Nelson KL, Whitehead PL, Adams LD, Bussies PL, Manrique CP, Rodriguez A, Rodriguez V, Starks T, Byfield GE, Sierra Lopez CB, McCauley JL, Acosta H, Chinea A, Kunkle BW, Reitz C, Farrer LA, Schellenberg GD, Vardarajan BN, Vance JM, Cuccaro ML, Martin ER, Haines JL, Byrd GS, Beecham GW, and Pericak-Vance MA (2018). Ancestral origin of ApoE epsilon4 Alzheimer disease risk in Puerto Rican and African American populations. PLoS Genet. 12, e1007791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.. Blue EE, Horimoto ARVR, Mukherjee S, Wijsman EM, and Thornton TA (2019). Local ancestry at APOE modifies Alzheimer’s disease risk in Caribbean Hispanics. Alzheimers Dement. 12, 1524–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.. Naslavsky MS, Suemoto CK, Brito LA, Scliar MO, Ferretti-Rebustini RE, Rodriguez RD, Leite REP, Araujo NM, Borda V, Tarazona-Santos E, Jacob-Filho W, Pasqualucci C, Nitrini R, Yaffe K, Zatz M, and Grinberg LT (2022). Global and local ancestry modulate APOE association with Alzheimer’s neuropathology and cognitive outcomes in an admixed sample. medRxiv 7/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.. Griswold AJ, Celis K, Bussies PL, Rajabli F, Whitehead PL, Hamilton-Nelson KL, Beecham GW, Dykxhoorn DM, Nuytemans K, Wang L, Gardner OK, Dorfsman DA, Bigio EH, Mesulam MM, Weintraub S, Geula C, Gearing M, McGrath-Martinez E, Dalgard CL, Scott WK, Haines JL, Pericak-Vance MA, Young JI, and Vance JM (2021). Increased APOEε4 expression is associated with the difference in Alzheimer’s disease risk from diverse ancestral backgrounds. Alzheimers Dement. 7, 1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.. Bonn S, and Furlong E. (2008. Dec). cis-Regulatory networks during development: a view of Drosophila. Curr Opin Genet Dev 18(6):513–20. doi: 10.1016/j.gde.2008.09.005. Epub 2008 Oct 16. [DOI] [PubMed] [Google Scholar]
- 13.. Wilczynski B, and Furlong E. (2010. Apr 15). Challenges for modeling global gene regulatory networks during development: insights from Drosophila. Dev Biol 340(2):161–9. doi: 10.1016/j.ydbio.2009.10.032. Epub 2009 Oct 27. [DOI] [PubMed] [Google Scholar]
- 14.. Zinzen R, Girardot C, Gagneur J, Braun M, and Furlong E. (2009. Nov 5). Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature 462(7269):65–70. doi: 10.1038/nature08531. [DOI] [PubMed] [Google Scholar]
- 15.. Gross D, and Garrard W. (1988). Nuclease hypersensitive sites in chromatin. Annu Rev Biochem 57:159–97. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]
- 16.. Kornberg R. (1977). Structure of chromatin. Annu Rev Biochem 46:931–54. doi: 10.1146/annurev.bi.46.070177.004435. [DOI] [PubMed] [Google Scholar]
- 17.. Kornberg R, and Lorch Y. (1999). Chromatin-modifying and -remodeling complexes. Curr Opin Genet Dev 9(2), 148–51. doi: 10.1016/S0959-437X(99)80022-7. [DOI] [PubMed] [Google Scholar]
- 18.. Luger K, Dechassa ML, and Tremethick DJ (2012). New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol 7, 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.. Tremethick DJ (2007). Higher-order structures of chromatin: the elusive 30 nm fiber. Cell 4, 651–654. [DOI] [PubMed] [Google Scholar]
- 20.. Corces MR, Shcherbina A, Kundu S, Gloudemans MJ, Frésard L, Granja JM, Louie BH, Eulalio T, Shams S, Bagdatli ST, Mumbach MR, Liu B, Montine KS, Greenleaf WJ, Kundaje A, Montgomery SB, Chang HY, and Montine TJ (2020). Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet 11, 1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.. Wang Y, Zhang X, Song Q, Hou Y, Liu J, Sun Y, and Wang P. (2020). Characterization of the chromatin accessibility in an Alzheimer’s disease (AD) mouse model. Alzheimers Res. Ther 1, 29–020-00598–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.. Morabito S, Miyoshi E, Michael N, Shahin S, Martini AC, Head E, Silva J, Leavy K, Perez-Rosendahl M, and Swarup V. (2021). Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat. Genet 8, 1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.. Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, Kathiria A, Cho SW, Mumbach MR, Carter AC, Kasowski M, Orloff LA, Risca VI, Kundaje A, Khavari PA, Montine TJ, Greenleaf WJ, and Chang HY (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 10, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.. Corces R, Greenleaf WJ, and Chang HY (2019). Isolation of nuceli from frozen tissue for ATAC-seq and other epigenomic assays V.1. Nat Methods. [Google Scholar]
- 25.. Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, and Greenleaf WJ (2021). ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet 3, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.. Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, Shah P, Bell JC, Jhutty D, Nemec CM, Wang J, Wang L, Yin Y, Giresi PG, Chang ALS, Zheng GXY, Greenleaf WJ, and Chang HY (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol 8, 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.. Granja JM, Klemm S, McGinnis LM, Kathiria AS, Mezger A, Corces MR, Parks B, Gars E, Liedtke M, Zheng GXY, Chang HY, Majeti R, and Greenleaf WJ (2019). Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol 12, 1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.. Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S. (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 12, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R. (2019). Comprehensive Integration of Single-Cell Data. Cell 7, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 4, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.. Duttke SH, Chang MW, Heinz S, and Benner C. (2019). Identification and dynamic quantification of regulatory elements using total RNA. Genome Res. 11, 1836–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.. McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G. (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 5, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.. Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, Rosen N, Kohn A, Twik M, Safran M, Lancet D, and Cohen D. (2017). GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford), 10.1093/database/bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.. Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 6, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.. Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 7, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.. Bailey TL, Johnson J, Grant CE, and Noble WS (2015). The MEME Suite. Nucleic Acids Res. W1, W39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.. Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Lemma RB, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Perez N, Fornes O, Leung TY, Aguirre A, Hammal F, Schmelter D, Baranasic D, Ballester B, Sandelin A, Lenhard B, Vandepoele K, Wasserman WW, Parcy F, and Mathelier A. (2022). JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. D1, D165–D173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.. Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Morgunova E, Enge M, Taipale M, Wei G, Palin K, Vaquerizas JM, Vincentelli R, Luscombe NM, Hughes TR, Lemaire P, Ukkonen E, Kivioja T, and Taipale J. (2013). DNA-binding specificities of human transcription factors. Cell 1-2, 327–339. [DOI] [PubMed] [Google Scholar]
- 40.. Pachkov M, Balwierz PJ, Arnold P, Ozonov E, and van Nimwegen E. (2013). SwissRegulon, a database of genome-wide annotations of regulatory sites: recent updates. Nucleic Acids Res. Database issue, D214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.. Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, and Kent WJ (2004). The UCSC Table Browser data retrieval tool. Nucleic Acids Res. Database issue, D493–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A. (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics, 128–2105-14–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.. Ogata H, Goto S, Fujibuchi W, and Kanehisa M. (1998). Computation with the KEGG pathway database. BioSystems 1-2, 119–128. [DOI] [PubMed] [Google Scholar]
- 44.. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, and Sherlock G. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 1, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.. Joshi-Tope G, Gillespie M, Vastrik I, D’Eustachio P, Schmidt E, de Bono B, Jassal B, Gopinath GR, Wu GR, Matthews L, Lewis S, Birney E, and Stein L. (2005). Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. Database issue, D428–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.. ENCODE Project Consortium. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 7414, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.. Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, and Satija R. (2021). Integrated analysis of multimodal single-cell data. Cell 13, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.. Franzén O, Gan L, and Björkegren J. (2019). PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford) 2019:baz046. doi: 10.1093/database/baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.. Culhane AC, Schwarzl T, Sultana R, Picard KC, Picard SC, Lu TH, Franklin KR, French SJ, Papenhausen G, Correll M, and Quackenbush J. (2010). GeneSigDB--a curated database of gene expression signatures. Nucleic Acids Res. Database issue, D716–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.. Aaronson J, Beaumont V, Blevins RA, Andreeva V, Murasheva I, Shneyderman A, Armah K, Gill R, Chen J, Rosinski J, Park LC, Coppola G, Munoz-Sanjuan I, and Vogt TF (2021). HDinHD: A Rich Data Portal for Huntington’s Disease Research. J. Huntingtons Dis 3, 405–412. [DOI] [PubMed] [Google Scholar]
- 51.. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 12, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.. Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, Simmons RK, Buckberry S, Vargas-Landin DB, Poppe D, Pflueger J, Lister R, Rackham OJL, Petretto E, and Polo JM (2019). A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci 12, 2087–2097. [DOI] [PubMed] [Google Scholar]
- 53.. Sadick JS, O’Dea MR, Hasel P, Dykstra T, Faustin A, and Liddelow SA (2022). Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer’s disease. Neuron 11, 1788–1805.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.. Srinivasan K, Friedman BA, Larson JL, Lauffer BE, Goldstein LD, Appling LL, Borneo J, Poon C, Ho T, Cai F, Steiner P, van der Brug MP, Modrusan Z, Kaminker JS, and Hansen DV (2016). Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun, 11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.. Lin L, Park J, Ramachandran S, Zhang Y, Tseng Y, Shen S, Waldvogel H, Curtis M, Faull R, Troncoso J, Pletnikova O, Ross C, Davidson B, and Xing Y. (2016. Aug 15). Transcriptome sequencing reveals aberrant alternative splicing in Huntington’s disease. Hum Mol Genet 2016 Aug 15;25(16):3454–3466. doi: 10.1093/hmg/ddw187. Epub 2016 Jul 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.. Lee H, Fenster RJ, Pineda SS, Gibbs WS, Mohammadi S, Davila-Velderrain J, Garcia FJ, Therrien M, Novis HS, Gao F, Wilkinson H, Vogt T, Kellis M, LaVoie MJ, and Heiman M. (2020). Cell Type-Specific Transcriptomics Reveals that Mutant Huntingtin Leads to Mitochondrial RNA Release and Neuronal Innate Immune Activation. Neuron 5, 891–908.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.. Kunkle BW, Schmidt M, Klein HU, Naj AC, Hamilton-Nelson KL, Larson EB, Evans DA, De Jager PL, Crane PK, Buxbaum JD, Ertekin-Taner N, Barnes LL, Fallin MD, Manly JJ, Go RCP, Obisesan TO, Kamboh MI, Bennett DA, Hall KS, Goate AM, Foroud TM, Martin ER, Wang LS, Byrd GS, Farrer LA, Haines JL, Schellenberg GD, Mayeux R, Pericak-Vance MA, Reitz C, Writing Group for the Alzheimer’s Disease Genetics Consortium (ADGC), Graff-Radford NR, Martinez I, Ayodele T, Logue MW, Cantwell LB, Jean-Francois M, Kuzma AB, Adams LD, Vance JM, Cuccaro ML, Chung J, Mez J, Lunetta KL, Jun GR, Lopez OL, Hendrie HC, Reiman EM, Kowall NW, Leverenz JB, Small SA, Levey AI, Golde TE, Saykin AJ, Starks TD, Albert MS, Hyman BT, Petersen RC, Sano M, Wisniewski T, Vassar R, Kaye JA, Henderson VW, DeCarli C, LaFerla FM, Brewer JB, Miller BL, Swerdlow RH, Van Eldik LJ, Paulson HL, Trojanowski JQ, Chui HC, Rosenberg RN, Craft S, Grabowski TJ, Asthana S, Morris JC, Strittmatter SM, and Kukull WA (2021). Novel Alzheimer Disease Risk Loci and Pathways in African American Individuals Using the African Genome Resources Panel: A Meta-analysis. JAMA Neurol 1, 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.. Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, Bellenguez C, Frizatti A, Chouraki V, Martin ER, Sleegers K, Badarinarayan N, Jakobsdottir J, Hamilton-Nelson KL, Moreno-Grau S, Olaso R, Raybould R, Chen Y, Kuzma AB, Hiltunen M, Morgan T, Ahmad S, Vardarajan BN, Epelbaum J, Hoffmann P, Boada M, Beecham GW, Garnier JG, Harold D, Fitzpatrick AL, Valladares O, Moutet ML, Gerrish A, Smith AV, Qu L, Bacq D, Denning N, Jian X, Zhao Y, Del Zompo M, Fox NC, Choi SH, Mateo I, Hughes JT, Adams HH, Malamon J, Sanchez-Garcia F, Patel Y, Brody JA, Dombroski BA, Naranjo MCD, Daniilidou M, Eiriksdottir G, Mukherjee S, Wallon D, Uphill J, Aspelund T, Cantwell LB, Garzia F, Galimberti D, Hofer E, Butkiewicz M, Fin B, Scarpini E, Sarnowski C, Bush WS, Meslage S, Kornhuber J, White CC, Song Y, Barber RC, Engelborghs S, Sordon S, Voijnovic D, Adams PM, Vandenberghe R, Mayhaus M, Cupples LA, Albert MS, De Deyn PP, Gu W, Himali JJ, Beekly D, Squassina A, Hartmann AM, Orellana A, Blacker D, Rodriguez-Rodriguez E, Lovestone S, Garcia ME, Doody RS, Munoz-Fernadez C, Sussams R, Lin H, Fairchild TJ, Benito YA, Holmes C, Karamujic-Comic H, Frosch MP, Thonberg H, Maier W, Roshchupkin G, Ghetti B, Giedraitis V, Kawalia A, Li S, Huebinger RM, Kilander L, Moebus S, Hernandez I, Kamboh MI, Brundin R, Turton J, Yang Q, Katz MJ, Concari L, Lord J, Beiser AS, Keene CD, Helisalmi S, Kloszewska I, Kukull WA, Koivisto AM, Lynch A, Tarraga L, Larson EB, Haapasalo A, Lawlor B, Mosley TH, Lipton RB, Solfrizzi V, Gill M, Longstreth WT Jr, Montine TJ, Frisardi V, Diez-Fairen M, Rivadeneira F, Petersen RC, Deramecourt V, Alvarez I, Salani F, Ciaramella A, Boerwinkle E, Reiman EM, Fievet N, Rotter JI, Reisch JS, Hanon O, Cupidi C, Andre Uitterlinden AG, Royall DR, Dufouil C, Maletta RG, de Rojas I, Sano M, Brice A, Cecchetti R, George-Hyslop PS, Ritchie K, Tsolaki M, Tsuang DW, Dubois B, Craig D, Wu CK, Soininen H, Avramidou D, Albin RL, Fratiglioni L, Germanou A, Apostolova LG, Keller L, Koutroumani M, Arnold SE, Panza F, Gkatzima O, Asthana S, Hannequin D, Whitehead P, Atwood CS, Caffarra P, Hampel H, Quintela I, Carracedo A, Lannfelt L, Rubinsztein DC, Barnes LL, Pasquier F, Frolich L, Barral S, McGuinness B, Beach TG, Johnston JA, Becker JT, Passmore P, Bigio EH, Schott JM, Bird TD, Warren JD, Boeve BF, Lupton MK, Bowen JD, Proitsi P, Boxer A, Powell JF, Burke JR, Kauwe JSK, Burns JM, Mancuso M, Buxbaum JD, Bonuccelli U, Cairns NJ, McQuillin A, Cao C, Livingston G, Carlson CS, Bass NJ, Carlsson CM, Hardy J, Carney RM, Bras J, Carrasquillo MM, Guerreiro R, Allen M, Chui HC, Fisher E, Masullo C, Crocco EA, DeCarli C, Bisceglio G, Dick M, Ma L, Duara R, Graff-Radford NR, Evans DA, Hodges A, Faber KM, Scherer M, Fallon KB, Riemenschneider M, Fardo DW, Heun R, Farlow MR, Kolsch H, Ferris S, Leber M, Foroud TM, Heuser I, Galasko DR, Giegling I, Gearing M, Hull M, Geschwind DH, Gilbert JR, Morris J, Green RC, Mayo K, Growdon JH, Feulner T, Hamilton RL, Harrell LE, Drichel D, Honig LS, Cushion TD, Huentelman MJ, Hollingworth P, Hulette CM, Hyman BT, Marshall R, Jarvik GP, Meggy A, Abner E, Menzies GE, Jin LW, Leonenko G, Real LM, Jun GR, Baldwin CT, Grozeva D, Karydas A, Russo G, Kaye JA, Kim R, Jessen F, Kowall NW, Vellas B, Kramer JH, Vardy E, LaFerla FM, Jockel KH, Lah JJ, Dichgans M, Leverenz JB, Mann D, Levey AI, Pickering-Brown S, Lieberman AP, Klopp N, Lunetta KL, Wichmann HE, Lyketsos CG, Morgan K, Marson DC, Brown K, Martiniuk F, Medway C, Mash DC, Nothen MM, Masliah E, Hooper NM, McCormick WC, Daniele A, McCurry SM, Bayer A, McDavid AN, Gallacher J, McKee AC, van den Bussche H, Mesulam M, Brayne C, Miller BL, Riedel-Heller S, Miller CA, Miller JW, Al-Chalabi A, Morris JC, Shaw CE, Myers AJ, Wiltfang J, O’Bryant S, Olichney JM, Alvarez V, Parisi JE, Singleton AB, Paulson HL, Collinge J, Perry WR, Mead S, Peskind E, Cribbs DH, Rossor M, Pierce A, Ryan NS, Poon WW, Nacmias B, Potter H, Sorbi S, Quinn JF, Sacchinelli E, Raj A, Spalletta G, Raskind M, Caltagirone C, Bossu P, Orfei MD, Reisberg B, Clarke R, Reitz C, Smith AD, Ringman JM, Warden D, Roberson ED, Wilcock G, Rogaeva E, Bruni AC, Rosen HJ, Gallo M, Rosenberg RN, Ben-Shlomo Y, Sager MA, Mecocci P, Saykin AJ, Pastor P, Cuccaro ML, Vance JM, Schneider JA, Schneider LS, Slifer S, Seeley WW, Smith AG, Sonnen JA, Spina S, Stern RA, Swerdlow RH, Tang M, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Van Eldik LJ, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Wilhelmsen KC, Williamson J, Wingo TS, Woltjer RL, Wright CB, Yu CE, Yu L, Saba Y, Pilotto A, Bullido MJ, Peters O, Crane PK, Bennett D, Bosco P, Coto E, Boccardi V, De Jager PL, Lleo A, Warner N, Lopez OL, Ingelsson M, Deloukas P, Cruchaga C, Graff C, Gwilliam R, Fornage M, Goate AM, Sanchez-Juan P, Kehoe PG, Amin N, Ertekin-Taner N, Berr C, Debette S, Love S, Launer LJ, Younkin SG, Dartigues JF, Corcoran C, Ikram MA, Dickson DW, Nicolas G, Campion D, Tschanz J, Schmidt H, Hakonarson H, Clarimon J, Munger R, Schmidt R, Farrer LA, Van Broeckhoven C, C O’Donovan M, DeStefano AL, Jones L, Haines JL, Deleuze JF, Owen MJ, Gudnason V, Mayeux R, Escott-Price V, Psaty BM, Ramirez A, Wang LS, Ruiz A, van Duijn CM, Holmans PA, Seshadri S, Williams J, Amouyel P, Schellenberg GD, Lambert JC, Pericak-Vance MA, Alzheimer Disease Genetics Consortium (ADGC), European Alzheimer’s Disease Initiative (EADI), Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium (CHARGE), and Genetic and Environmental Risk in AD/Defining Genetic, Polygenic and Environmental Risk for Alzheimer’s Disease Consortium (GERAD/PERADES),. (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet 3, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.. Nuytemans K, Lipkin Vasquez M, Wang L, Van Booven D, Griswold AJ, Rajabli F, Celis K, Oron O, Hofmann N, Rolati S, Garcia-Serje C, Zhang S, Jin F, Argenziano M, Grant SFA, Chesi A, Brown CD, Young JI, Dykxhoorn DM, Pericak-Vance MA, and Vance JM (2022). Identifying differential regulatory control of APOE varepsilon4 on African versus European haplotypes as potential therapeutic targets. Alzheimers Dement. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.. Fernandez CG, Hamby ME, McReynolds ML, and Ray WJ (2019). The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Front. Aging Neurosci, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.. Zhao H, Ji Q, Wu Z, Wang S, Ren J, Yan K, Wang Z, Hu J, Chu Q, Hu H, Cai Y, Wang Q, Huang D, Ji Z, Li J, Izpisua Belmonte JC, Song M, Zhang W, Qu J, and Liu G. (2022). Destabilizing heterochromatin by APOE mediates senescence. Nat Aging., 303–316. [DOI] [PubMed] [Google Scholar]
- 62.. Narlikar GJ, Fan HY, and Kingston RE (2002). Cooperation between complexes that regulate chromatin structure and transcription. Cell 4, 475–487. [DOI] [PubMed] [Google Scholar]
- 63.. Merrill CB, Montgomery AB, Pabon MA, Shabalin AA, Rodan AR, and Rothenfluh A. (2022). Harnessing changes in open chromatin determined by ATAC-seq to generate insulin-responsive reporter constructs. BMC Genomics 1, 399–022-08637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.. Klemm SL, Shipony Z, and Greenleaf WJ (2019). Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet 4, 207–220. [DOI] [PubMed] [Google Scholar]
- 65.. Klemm SL, Shipony Z, and Greenleaf WJ (2019). Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet 4, 207–220. [DOI] [PubMed] [Google Scholar]
- 66.. Belanger M, and Magistretti PJ (2009). The role of astroglia in neuroprotection. Dialogues Clin. Neurosci 3, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.. Belanger M, Allaman I, and Magistretti PJ (2011). Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell. Metab 6, 724–738. [DOI] [PubMed] [Google Scholar]
- 68.. Gibbs ME, Hutchinson D, and Hertz L. (2008). Astrocytic involvement in learning and memory consolidation. Neurosci. Biobehav. Rev 5, 927–944. [DOI] [PubMed] [Google Scholar]
- 69.. Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, and Frank MG (2009). Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 2, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.. Phatnani H, and Maniatis T. (2015). Astrocytes in neurodegenerative disease. Cold Spring Harb Perspect. Biol 6, 10.1101/cshperspect.a020628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.. Monterey MD, Wei H, Wu X, and Wu JQ (2021). The Many Faces of Astrocytes in Alzheimer’s Disease. Front. Neurol, 619626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.. Liu Y. (2020). Clinical implications of chromatin accessibility in human cancers. Oncotarget 18, 1666–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.. Harris RA, Raveendran M, Worley KC, and Rogers J. (2020). Unusual sequence characteristics of human chromosome 19 are conserved across 11 nonhuman primates. BMC Evol. Biol 1, 33–020-1595–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.. Castresana J, Guigo R, and Alba MM (2004). Clustering of genes coding for DNA binding proteins in a region of atypical evolution of the human genome. J. Mol. Evol 1, 72–79. [DOI] [PubMed] [Google Scholar]
- 75.. Grimwood J, Gordon LA, Olsen A, Terry A, Schmutz J, Lamerdin J, Hellsten U, Goodstein D, Couronne O, Tran-Gyamfi M, Aerts A, Altherr M, Ashworth L, Bajorek E, Black S, Branscomb E, Caenepeel S, Carrano A, Caoile C, Chan YM, Christensen M, Cleland CA, Copeland A, Dalin E, Dehal P, Denys M, Detter JC, Escobar J, Flowers D, Fotopulos D, Garcia C, Georgescu AM, Glavina T, Gomez M, Gonzales E, Groza M, Hammon N, Hawkins T, Haydu L, Ho I, Huang W, Israni S, Jett J, Kadner K, Kimball H, Kobayashi A, Larionov V, Leem SH, Lopez F, Lou Y, Lowry S, Malfatti S, Martinez D, McCready P, Medina C, Morgan J, Nelson K, Nolan M, Ovcharenko I, Pitluck S, Pollard M, Popkie AP, Predki P, Quan G, Ramirez L, Rash S, Retterer J, Rodriguez A, Rogers S, Salamov A, Salazar A, She X, Smith D, Slezak T, Solovyev V, Thayer N, Tice H, Tsai M, Ustaszewska A, Vo N, Wagner M, Wheeler J, Wu K, Xie G, Yang J, Dubchak I, Furey TS, DeJong P, Dickson M, Gordon D, Eichler EE, Pennacchio LA, Richardson P, Stubbs L, Rokhsar DS, Myers RM, Rubin EM, and Lucas SM (2004). The DNA sequence and biology of human chromosome 19. Nature 6982, 529–535. [DOI] [PubMed] [Google Scholar]
- 76.. Spell C, Kolsch H, Lutjohann D, Kerksiek A, Hentschel F, Damian M, von Bergmann K, Rao ML, Maier W, and Heun R. (2004). SREBP-1a polymorphism influences the risk of Alzheimer’s disease in carriers of the ApoE4 allele. Dement. Geriatr. Cogn. Disord 3-4, 245–249. [DOI] [PubMed] [Google Scholar]
- 77.. Shi X, Ren S, Zhang B, Guo S, He W, Yuan C, Yang X, Ig-Lzevbekhai K, Sun T, Wang Q, and Cui J. (2021). Analysis of the role of Puralpha in the pathogenesis of Alzheimer’s disease based on RNA-seq and ChIP-seq. Sci. Rep 1, 12178–021-90982–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.. Fang X, Zhong X, Yu G, Shao S, and Yang Q. (2017). Vascular protective effects of KLF2 on Abeta-induced toxicity: Implications for Alzheimer’s disease. Brain Res., 174–183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. SnATAC-seq data analysis for sample bias.
Supplementary Figure 2. SnATAC-seq cell marker cluster identification by chromatin accessibility.
Supplementary Figure 3. Astrocyte subtype classification.
Supplementary Figure 4. APOE chromatin accessibility in the other cell clusters where APOE is highly expressed but not differentially accessible between the ancestries.
Supplementary Figure 5. Visualization of local ancestry across all chromosomes per individual sample.
Supplementary Figure 6. Enrichment of differentially accessible peak between ancestries in chromosome 19.
Supplementary Table 1. Summary of snATAC and snRNA sequencing results.
Supplementary Table 2. Total cell count and proportion per cluster in the integrated sequencing analysis.
Supplementary Table 3. snRNA sequencing clusters with significant APOE4 differential expression between ancestries.
Supplementary Table 4. TFs (DEGs-DAGs) binding to differentially accessible peaks at the promoter of APOE.
Supplementary Table 5. APOE Local ancestry accessibility analysis.
Supplementary Table 6. Differentially accessible and expressed genes (DEG-DAG) in astrocyte cluster 1.
Supplementary Table 7. Subset of the differential functional analysis of the astrocyte cluster 1 DEG-DAG with positive correlation between expression and accessibility using EnrichR software.
Data Availability Statement
Data are available through the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS) Data Sharing Service (DSS): https://dss.niagads.org/datasets/ng00067/