

Summary
Patterns of diel activity – how animals allocate their activity throughout the 24hr daily cycle – play key roles in shaping the internal physiology of an animal and its relationship with the external environment1–5. Although shifts in diel activity patterns have occurred numerous times over the course of amniote evolution6, the genomic correlates of such transitions remain unknown. Here, we use the African striped mouse (Rhabdomys pumilio), a species that transitioned from the ancestrally-nocturnal diel niche of its close relatives to a diurnal one7–11, to define patterns of naturally-occurring molecular variation in diel niche traits. First, to facilitate genomic analyses, we generated a chromosome-level genome assembly of the striped mouse. Next, using transcriptomics, we show that the switch to daytime activity in this species is associated with a realignment of daily rhythms in peripheral tissues with respect to the light:dark cycle and the central circadian clock. To uncover selection pressures associated with this temporal niche shift, we perform comparative genomic analyses of closely related rodent species and find evidence of relaxation of purifying selection on striped mouse genes in the rod phototransduction pathway. In agreement with this, electroretinogram measurements demonstrate that striped mice have functional differences in dim light visual responses compared to nocturnal rodents. Taken together, our results show that striped mice have undergone a drastic change in circadian organisation and provide evidence that the visual system has been a major target of selection as this species transitioned to a novel temporal niche.
eTOC Blurb
Richardson et al. study R. pumilio, a rodent that transitioned from the ancestrally-nocturnal diel niche of its close relatives to a diurnal one. Using whole-genome sequencing, transcriptomics, comparative genomics, and retinal measurements of light response, they define patterns of naturally occurring molecular variation in diel niche traits.
Results and Discussion
The set of ecological interactions that define a species’ niche are distributed both spatially across its habitat as well as temporally across seasonal and diel (day-night) cycles. Although it is common for animals to show some flexibility in daily patterns of activity, many species exhibit a preferred diel temporal niche. A species’ diel niche profoundly shapes its ability to acquire information through its senses, its access to resources and concomitant foraging strategies, energy and water consumption, methods of predator avoidance and its means for both intraspecific and interspecific communication1. Given this, diel niche is often reflected by a suite of morphological, physiological, and behavioural adaptations that are well-documented among diverse lineages2–5,12–16. Critically, diel niche preference is fundamentally linked to organismal biology through internal circadian clocks, which establish 24hr rhythms in physiology and behaviour. Implementation and maintenance of diel niche therefore requires correct phasing of these rhythms with respect to the light:dark cycle.
While the molecular basis of some diel temporal niche traits has been studied in traditional model organisms like the laboratory mouse17–19, much less is understood about how these traits have evolved. Evolutionary shifts in diel niche have occurred numerous times among vertebrates, including in the lineage leading to humans. Moreover, shifts to diurnality from nocturnal ancestors represents one of the critical evolutionary transitions that facilitated mammalian dominance of terrestrial niches after the end of the Mesozoic6. Therefore, defining patterns of naturally occurring molecular variation in diel niche traits has the potential to provide fundamental insights into the genomic basis of mammalian diversification.
The African striped mouse, Rhabdomys pumilio (Family Muridae, Subfamily Murinae), a rodent distinguished by its prominent dorsal stripe pattern20, is notable for having transitioned from the ancestrally-nocturnal diel niche of Muridae to a diurnal one (Figure 1A)8. African striped mice (hereafter, striped mice/mouse) show several adaptive features associated with a diurnal lifestyle, including a cone-rich retina and a UV-blocking lens9,10 and have also proven to be a useful model for studying the electrophysiological aspects of circadian timekeeping11. These characteristics, together with the striped mouse’s close phylogenetic relationship with the nocturnal laboratory mouse (Mus musculus)7,21 present an excellent comparative model system in which to unravel the molecular mechanisms and selective pressures underlying diel niche evolution in mammals. Here, we present a high-quality genome assembly for the striped mouse, which we use to facilitate experimental interrogation of diurnality-associated phenotypes.
Figure 1. The striped mouse reference genome.
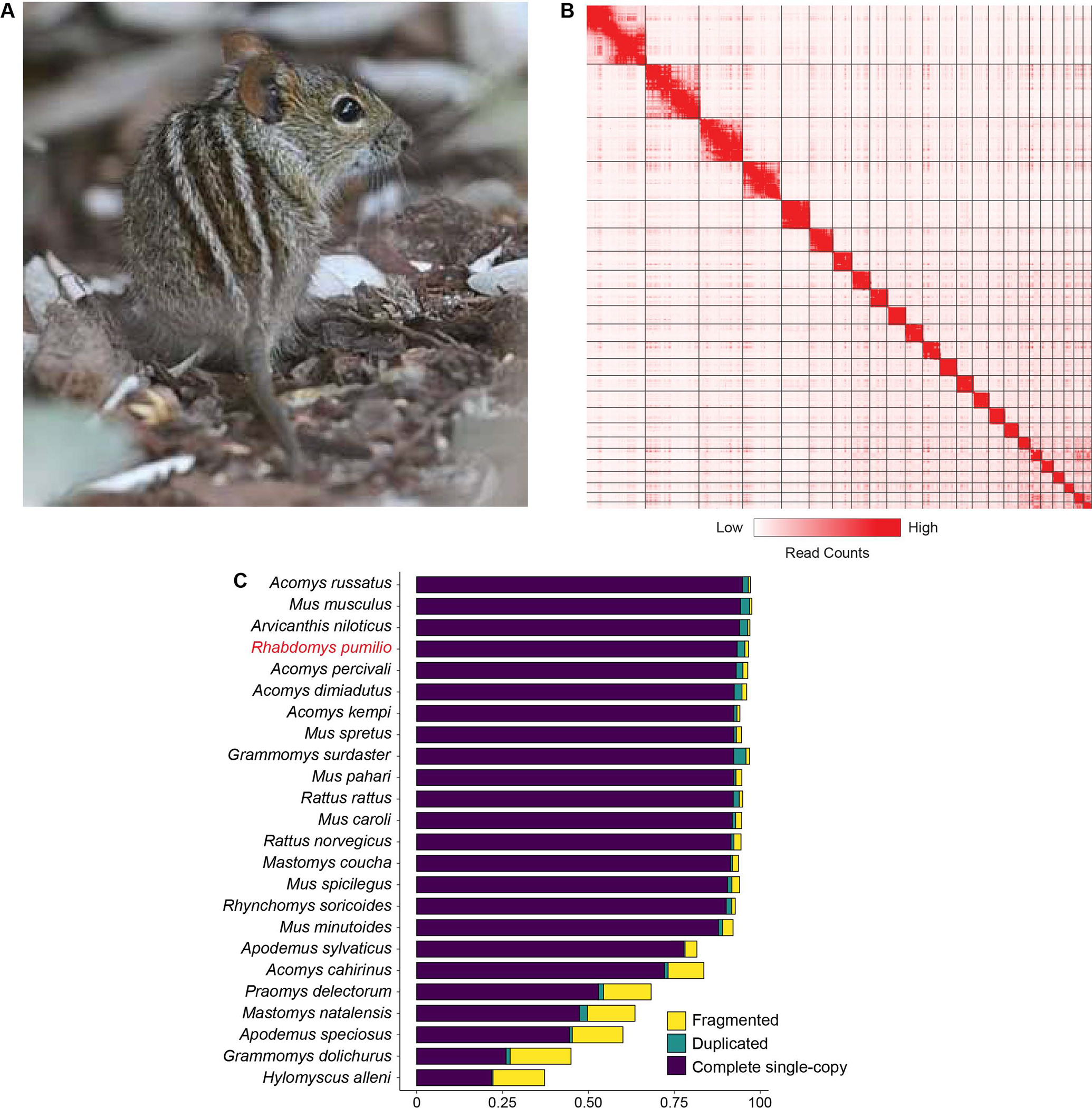
(A) Adult striped mouse photographed during the day, showing its characteristic dorsal stripe pattern. (B) Hi-C contact map of the 24 chromosome-scale scaffolds. Enrichment of long-range contacts is shown in red. Higher pixel intensity (red) represents a greater number of contacts between loci. Strong enrichment along the diagonal demonstrates accurate scaffold assembly. (C) Comparison of BUSCO gene recovery in the striped mouse reference genome compared to that of 23 other species in the family Muridae. The striped mouse shows high recovery of single-copy mammalian benchmark orthologs with low rates of duplication and fragmentation. Photography credit: Trevor Hardaker.
Striped mouse genome
To permit functional and comparative genomic studies of diel niche traits in the striped mouse, we first produced a high-quality reference genome assembly for this species, using a combination of linked-read technology and Hi-C scaffolding. The resulting assembly is ~2.3Gb in length with a G+C content of ~41%, comparable to that of other species in its subfamily (Murinae; Data S1A). N50 scaffold length is 79.8Mb, which exceeds that of most available murine genomes and is approximately equal to that of its close relative the African grass rat, Arvicanthis niloticus (Figure S1). Approximately 95% of the assembly is contained within 24 scaffolds having a length greater than or equal to 25Mb with the remainder distributed across smaller scaffolds (Figure 1B and Data S1A). Given the 2n=48 karyotype found in striped mice22, this indicates that the assembly very closely approaches full chromosome-scale, though some chromosomal segments in the lower megabase range remain unplaced. These metrics, together with a low gap percentage (~1%), demonstrate that our assembly exceeds most published murid genome assemblies in terms of contiguity. Next, to assess the assembly’s completeness and integrity, we annotated benchmarking mammalian orthologs using Benchmarking Universal Single-Copy Orthologue (BUSCO)23. Recovery of complete single-copy BUSCO genes was high at 93.3%, with a few more genes (1.1%) recovered as partial copies and the rate of duplication was low (2.3%; Figure 1C, Data S1B). These values compared favourably with most sequenced murid species (Figure 1C). Next, we sequenced RNA from multiple striped mouse tissues and used these data to produce transcriptome-based gene annotations (see STAR Methods). BUSCO analysis of the 31,928 predicted protein sequences (encoded by 28,594 protein-coding genes) showed that 93.3% of benchmarking genes were present as complete single copies. Taken together, these metrics demonstrate that our striped mouse assembly represents a high-quality resource for genomic studies of this species.
Transcriptomics of circadian rhythm
Circadian clocks are essential for generating and coordinating daily animal rhythms24. These rhythms result from the activity of an intracellular molecular oscillator that operates with a ~24-hour periodicity25,26. The core molecular clock consists of a transcriptional-translational feedback loop (TTFL) in which Bmal1 and Clock genes constitute a ‘positive arm’, activating the transcription of clock-controlled genes (CCGs). These CCGs include the canonical clock genes Period (Per1-3), Cryptochrome (Cry1-2) and Rev-Erbα and β (which then act as negative regulators of Clock and Bmal1 activity), as well as other ‘output’ genes endowing rhythmicity on physiological processes26,27. In mammals, the hypothalamic suprachiasmatic nuclei (SCN) contain a master circadian oscillator, which is synchronised to the light:dark cycle and in turn sets the phase of local oscillators in cells and tissues throughout the body27. Diel niche may alter the SCN oscillator’s experience of the synchronizing light:dark cycle as nocturnal animals typically adopt light avoiding strategies during the day. This could in turn impose a more intermittent exposure than is the case for species active through the day. In addition, as the SCN clock adopts a similar phase relationship with the light:dark cycle irrespective of preferred temporal niche in mammals28–30, it follows that diel niche also alters the phase relationship between the central clock and many overt rhythms.
Given the striped mouse’s evolutionary transition to diurnality, we sought to characterize daily variations of gene expression in the SCN and peripheral tissues to define the features of its circadian organisation. To do this, we first performed RNA-Seq on the SCN, retina, lung and liver of striped mice stably entrained to a 12:12 light-dark cycle, at two time points: 2 hours after lights on (Zeitgeber Time (ZT2); coinciding with high behavioural activity), and 2 hours after lights off (ZT14) during the animal’s resting/sleep stage (Figure 2A). The number of unique transcripts represented in our RNA-Seq datasets ranged from 19,258 for the retina to 14,381 for the SCN (Figure 2B). Of these, substantial fractions showed differential expression between ZT2 and ZT14 in all tissues, making up 23–33% of all expressed genes in retina, lung and liver, and ~7% of the transcripts in the SCN (FDR<0.05) (Figure 2B and C). Functional annotation of differentially expressed genes (DEGs) using the Kyoto Encyclopedia of Genes and Genomes (KEGG) resource revealed that day:night differences extended to numerous pathways (Figure S2), including those involved in core cell activities (including circadian rhythm) as well as more tissue specific functions (e.g., fatty acid biosynthesis).
Figure 2. Rhythmicity of the diurnal African striped mouse transcriptome across central and peripheral tissues.
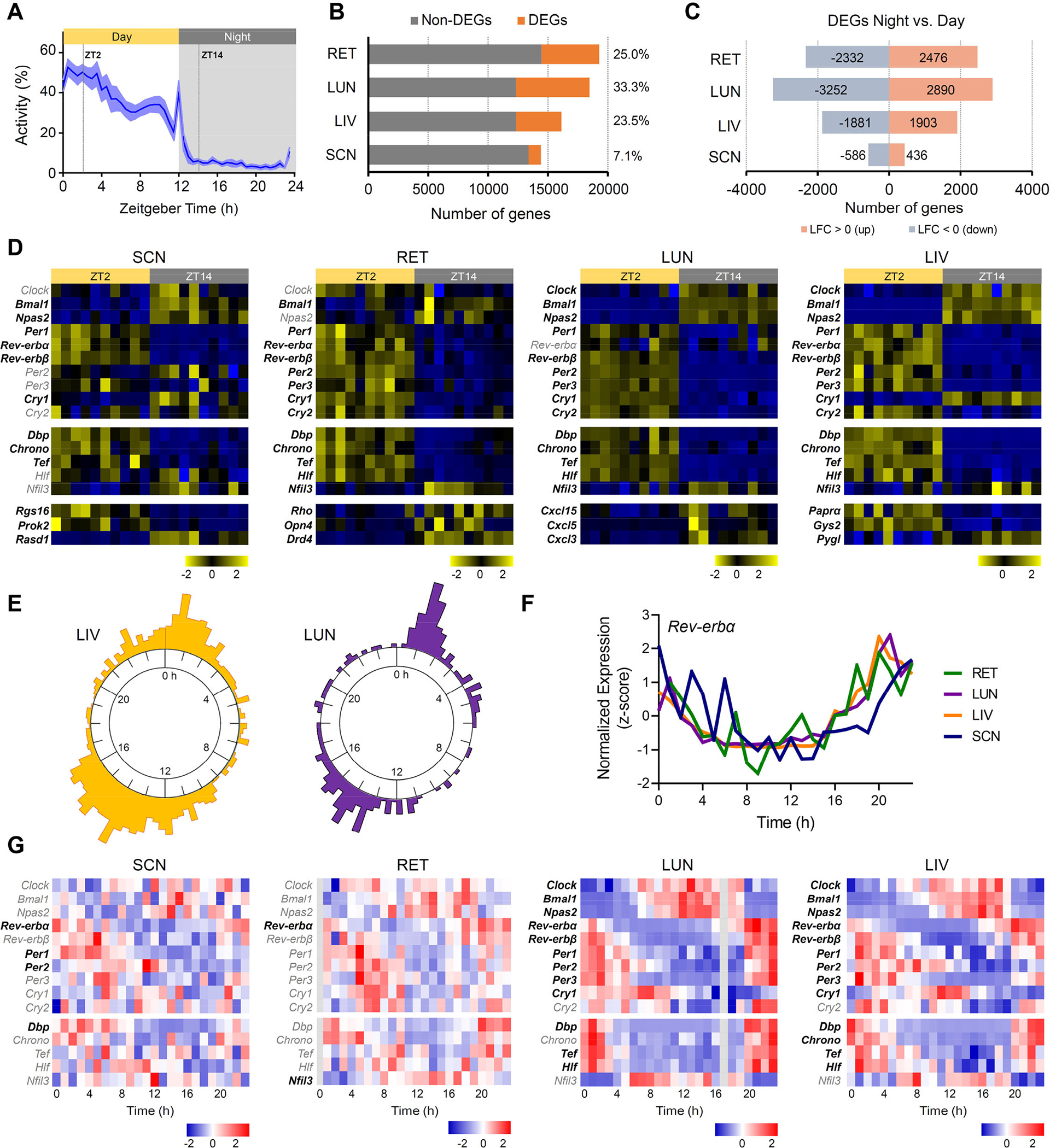
(A) Daily general activity pattern of striped mice under 12h light: 12h dark cycle. Values are expressed as mean ± SEM (n=10). Grey area indicates period of darkness. Zeitgeber time ZT0 corresponds to time of lights on. Dotted lines indicate timing of tissue collection during the day (ZT2) and at night (ZT14). (B) Number of day:night differentially expressed genes (DEGs; orange) and non-DGE (grey) across different tissues (retina (RET), lung (LUN), liver (LIV), and suprachiasmatic nucleus (SCN)). Numbers on the right indicate the percentage (%) of DEGs within each tissue. (C) Number of down- and up-regulated DEGsat night compared to daytime across tissues. Numbers of genes upregulated at night (log2 fold change (LFC>0) are indicated in orange and those downregulated (LFC<0) in blue. Only genes with an adjusted p-value below 0.05 were considered as DEGs. (D) Heatmaps showing expression levels (normalized values, z-score) for core clock genes (Clock, Bmal1, Npas2, Pers, Crys, Rev-erb, Chrono, Dpb, Tef, Hlf and Nfil3) and representative genes key for specific local-tissue function during the day (ZT2) and at night (ZT14). Squares across rows correspond to individual samples (n=10 day and n=10 night). Gene labels in black indicate DEGs. (E) Radial phase plot showing the distribution of times of peak expression relative to prior light:dark cycle (Time 0 = time of lights on) for genes that exhibited circadian rhythmicity in liver (left) and lung (right) under constant dark conditions (841 and 202 genes, respectively). (F) Circadian profile of Rev-erbα expression across the four tissues (Time 0 = clock time of lights on for prior light:dark cycle). (G) Heatmaps showing expression levels (normalized values, z-score) for core clock genes (Clock, Bmal1, Npas2, Pers, Crys, Rev-erb, Chrono, Dpb, Tef, Hlf and Nfil3) in SCN, RET, LIV and LUN collected over 24h in constant dark conditions (at 1h interval). Gene labels in bold indicate cycling genes with a BH.Q <0.1. Grey columns in the RET (Time 0h) and LUN (Time 17h) heatmaps represent missing data.
Turning to the question of the timing of gene expression with respect to the light:dark cycle, we first interrogated the dataset for a small number of core clock genes (elements of the circadian TTFL and clock output transcription factors) (Figure 2D). In all tissues we found that components of the positive arm, including Bmal1 and Npas2 (paralog of Clock) were higher at night, while most negative regulators (Per, Cry2 and Rev-Erb) were highly expressed during the day. The antiphase arrangement of these two groups of genes is consistent with their role as positive vs negative elements of the TTFL. Similarly, in all tissues, expression of clock-controlled transcription factors Chrono (Circadian-associated transcriptional repressor) and the circadian PAR-domain basic leucine zipper transcription factors Dbp, Tef, and Hlf were higher during the day, while Nifl3 expression was higher at night.
The appearance of similarly phased day/night differences in core clock gene expression across all tissues tested represents a stark difference in daily rhythmicity of striped mice compared to that reported in laboratory mice and rats (Rattus sp.), in which genes from the negative limb of the TTFL are low at night in the SCN and retina, but high at night in peripheral tissues including the liver and lung31–36. In accordance with that observation, a selection of DEGs considered critical to tissue-specific functions in striped mice had a similar phase to that reported in laboratory mice in the SCN and retina, but not in liver and lung (Figure 2D). Thus, in the SCN, genes associated with neuronal signaling and excitability (Rgs16, Rasd1, and Prok2) adopted a similar phase in striped mice to that previously shown in laboratory mice37,38. The same was true for photopigment Rho and dopamine signaling (Drd4) genes in the retina39,40. Interestingly, the expression profile of melanopsin (Opn4) in striped mouse was more similar to rats than to mice41,42. In the lung, conversely, we found that members of the chemokine family (associated with inflammatory response) were higher at ZT14 in striped mice, vs published work showing a peak in the early light phase in mice35,43. In the liver, clock gene expression is tightly linked with energy metabolic regulators such as Peroxisome proliferator-activated receptors (PPARs)44. In striped mouse liver, Pparα was differentially expressed between day and night, as were genes encoding proteins defining glycogenolysis (Pygl) and glycogenesis (Gys2). In common with our findings in the lung, the phasing of these processes was opposite to that reported for laboratory mice45.
Taken together, these transcriptome comparisons between day and night suggest a fundamental change in circadian organisation in striped mice compared to laboratory mice. Elements of the molecular clock and key physiological processes in lung and liver in striped mice have adjusted their phase with respect to the light:dark cycle, to match the diurnal pattern of rhythms in overt behaviour and gross physiology (activity and body temperature). This leaves daily patterns of gene expression in phase across SCN and periphery in striped mice (vs out of phase in laboratory mice).
To extend these observations and determine whether these patterns represented a genuine change in circadian organisation, we turned to a description of rhythms in constant darkness. Based upon recommendations to sample at high density when assessing rhythmic gene expression46,47 we culled individual striped mice at 1 h intervals over 24 hrs in constant darkness and collected SCN, retina, liver, and lung tissue for RNAseq analysis. We identified an average of 20,297 expressed genes in these samples (17,443 in SCN; 19361 in liver; 21,824 in lung; 22,558 in retina) and applied the ‘R’ package MetaCycle to identify genes with a significant circadian variation in expression. The number of genes defined as rhythmic using this approach was reduced compared to those showing differential expression between ZT2 and 14, likely due to the smaller number of samples per timepoint (Figure 2B and C). Thus, 841 genes were rhythmic at BH.Q < 0.1 in the liver, but this number was smaller for lung, SCN, and retina (202, 20, and 14 genes, respectively). In liver and lung, acrophyses for rhythmic genes covered the circadian cycle, but in common with reports in some other species29,48, they were especially common at two phases at nearly opposite sides of the cycle (in this case around projected dawn and dusk; Figure 2E). Turning to the phasing of rhythms, only one of the canonical clock genes (Rev-erbα) was significantly rhythmic in all datasets (Figure 2F and G). Consistent with the concept that circadian rhythms have a similar phase across SCN and periphery in striped mice, Rev-erbα expression peaked around the end of the subjective night (subjective time 20–0) in all 4 tissues (Figure 2F). More clock genes showed significant rhythmicity in the lung and liver, and in these cases, elements of the positive limb were higher during the subjective night (Clock, Bmal1, Npas2), while elements of the negative limb were more highly expressed during the subjective day (albeit with slightly phase difference for Cry1). It was harder to detect rhythmicity in the SCN, but all 3 clock genes in this tissue were elements of the negative limb (Rev-erbα, Per1 and Per2) and all were higher during the subjective day consistent with their phasing in lung and liver (Figure 2G). In summary then, our transcriptome analyses suggest that circadian gene expression adopts a similar phase across tissues in striped mice in both light-dark cycle and constant dark. This represents a comprehensive realignment of circadian organisation across the body compared to that described in nocturnal mice and rats25,33.
Shifts in selective regime associated with the evolution of diurnality
Diurnal temporal niche presents numerous ecological challenges distinct from those experienced by nocturnal species. Therefore, we next sought to use our striped mouse genome assembly to identify genomic signatures of selection that might indicate changing selective regimes in response to these factors. To accomplish this, we first identified orthologous protein-coding transcripts across the genomes of the striped mouse and 24 murid species with publicly available assemblies (Figure 3A and Data S1C). After filtering, we constructed 62,879 alignments of orthologous transcripts, each with representative sequences from a minimum of 10 species, which were used for subsequent analyses. Using these ortholog alignments, we performed analysis of relative evolutionary rates (RERs) to identify genes showing accelerated evolution relative to the per-species background rate (Figure S3, see Methods)49.
Figure 3. Selection on phototransduction genes.
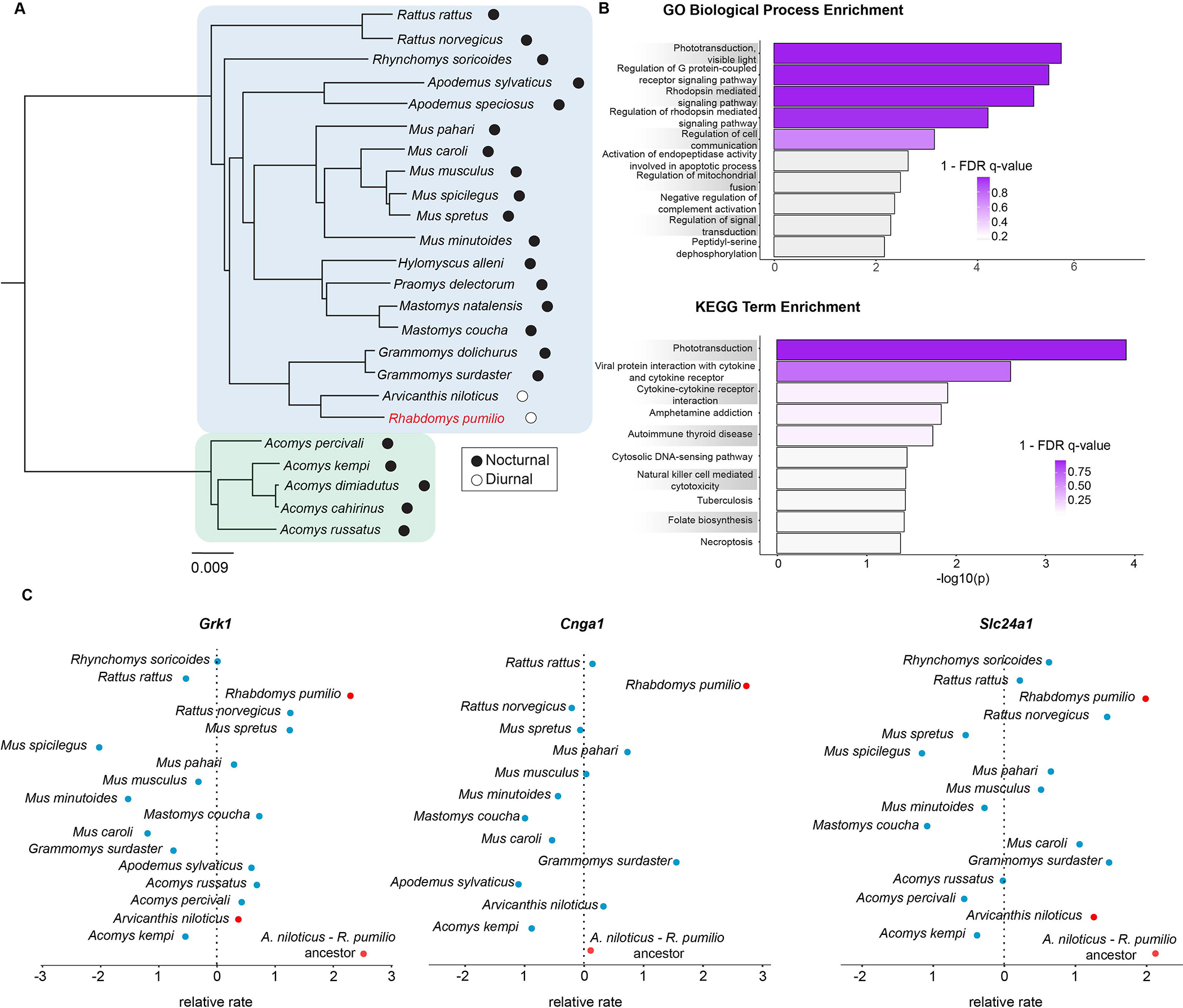
(A) A phylogeny of species used in comparative genomic analyses. Blue colour indicates the subfamily Murinae to which the striped mouse (red font) belongs. Green colour indicates the genus Acomys (subfamily Deomyinae) which served as an outgroup. (B) Enrichment of Gene Ontology Biological Process and KEGG pathway terms among striped mouse genes showing the greatest evolutionary acceleration relative to the background rate. Striped mouse accelerated genes are highly enriched for functions related to phototransduction, particularly those related to the rhodopsin cascade. Colour indicates FDR q-value. (C) Plots showing the relative evolutionary rates (RERs) of three core genes involved in rhodopsin-mediated phototransduction: Rhodopsin Kinase (Grk1), Cyclic Nucleotide Gated Channel Subunit Alpha 1 (Cnga1), and Solute Carrier Family 24 Member 1 (Slc24a1). Compared to closely related murids (blue), phototransduction locus orthologs in the striped mouse (red) show a markedly elevated evolutionary rate.
See also Figures S3, S4 and Data S1C–J.
Given the changes in circadian organisation in the striped mouse identified by our transcriptomic survey, we first examined the RERs of genes encoding the core clock components. These revealed no evidence of strong evolutionary acceleration compared to background rates of coding gene evolution (Data S1D). This suggests that shifts in diel niche do not have strong implications for the coding sequences of proteins responsible for generating circadian rhythms. It remains possible that evolution of the clock proceeds mainly via alterations in cis-regulatory elements but overall, this finding is consistent with the view that functional constraints on the molecular machinery of circadian rhythm generation are not altered by switches in diel niche.
Functional annotation revealed that the Gene Ontology and KEGG terms most significantly enriched among accelerated genes in the African striped mouse were related to the rhodopsin-mediated phototransduction cascade (Figure 3B, Data S1E). These terms were comprised of 8 striped mouse accelerated genes (Grk1, Slc24a1, Rho, Sag, Pde6g, Cnga1, Gna11, and Metap2) (Figure 3C and Figure S4). The majority of striped mouse accelerated genes (i.e., Grk1, Slc24a1, Rho, Sag, Pde6g, Cnga1) are expressed specifically in rods, a type of light-sensitive photoreceptor cell found in the retina that specializes in dim-light vision (i.e., scotopic vision)50. The functional specialization of rods results from the action of genes that participate in the rod phototransduction cascade, a process by which light is converted into an electrical signal. Among the 8 striped mouse accelerated genes, Rho, Cnga1, and Pde6g play key roles in phototransduction activation whereas Grk1, Slc24a1, and Sag are critical for photoresponse recovery. Interestingly, many of these genes have undergone positive selection in other lineages experiencing evolutionary shifts in light conditions. For example, in a comparison among bird species with different diel niches, Sag, Slc24a1, and Cnga1 showed strong positive selection in owls, a nocturnal bird with a visual system tuned for hunting at night51. In falcons, which are characterized by their high-speed pursuit of prey, sometimes under relatively low-light conditions, Grk1 and Slc24a1 also showed evidence of positive selection52,53. As catching prey in flight involves tracking fast-moving objects, having enhanced temporal resolution of vision likely leads to effective hunting. In some cases, the physiological relevance of protein coding changes in dim-light vision genes has been directly established through functional tests54. However, while predatory birds may benefit from enhanced low-light vision mediated by rhodopsin phototransduction, it is less clear whether a species transitioning away from low-light conditions like the striped mouse would require such adaptations. Elevated substitution rates in striped mouse rod phototransduction genes may instead be explained by a reduced reliance on rod vision and a corresponding relaxation of purifying selection. Therefore, to address this uncertainty we re-examined striped mouse rod phototransduction genes using the branch-site model, a Ka/Ks-based test for positive selection. Our analysis indicated that acceleration of striped mouse phototransduction genes was not consistent with positive selection (p > 0.01; Data S1F). This suggests that rather than necessitating adaptive changes in rod function, the importance of rod-based vision was reduced following the transition to diurnality in the striped mouse, leading to relaxed purifying selection on rod phototransduction.
The striped mouse belongs to the tribe Arvicanthini which includes several closely related African murines exhibiting degrees of diurnality7,55,56. This suggests that the initial diel niche shift in the striped mouse lineage occurred in its most recent common ancestor (MRCA) with the other diurnal arvicanthines. This raises the question of whether the observed pattern of relaxed constraint on striped mouse rod phototransduction genes is unique to this species or reflects a common trajectory within its diurnal lineage. As our dataset included the genome assembly for the diurnal African grass rat (A. niloticus), an arvicanthine species closely related to striped mice (Figure 3A), we next examined patterns of sequence evolution in this species as well as its common ancestor with the striped mouse. In stark contrast to the striped mouse, rod phototransduction genes were not found to be enriched among African grass rat accelerated genes, with only Sag showing a strongly elevated relative evolutionary rate (Data S1G–H). Branch-site tests indicated that, as in the striped mouse, acceleration of Sag in the grass rat was consistent with relaxed purifying selection (Data S1F). In the inferred MRCA of the striped mouse and grass rat, rod phototransduction genes showed a more similar pattern to that observed in the striped mouse, with a similar complement of genes in this pathway exhibiting elevated evolutionary rates (Data S1I–J). Overlapping genes included Grk1, Slc24a1, Pd36g, Rho, Gna11 and Sag. A further accelerated gene unique to the ancestor, Gnat2, was also identified (Data S1I). Interestingly, while branch-site tests generally rejected adaptation in most of these ancestral genes, evidence of positive selection was identified in Sag, suggesting that this gene may have experienced and early phase of adaptation during the transition to diurnality (Data S1F).
Taken together, our results indicate that the transition to diurnality in the African striped mouse primarily corresponds with broad relaxation of purifying selection on rod phototransduction genes, likely reflecting an overall reduced requirement for acute low-light vision. Moreover, our examination of the related African grass rat as well as their common ancestor provides molecular evidence supporting an ancestral origin for diurnality prior to the striped mouse-grass rat divergence. The initial shift toward a diurnal niche in this lineage was accompanied by widespread relaxation of selection on rod phototransduction genes, as well as adaptation in a small number of sites in the Sag gene, encoding S-arrestin. Interestingly, our findings also indicate that the selective regimes experienced by African grass rats and striped mice since their divergence from a common ancestor have not been uniform, with the African grass rat lacking the extended period of relaxed selection seen in the striped mouse. While the causes of this difference remain unclear, one possibility is that the striped mouse genus Rhabdomys has proceeded further in its diel niche transition than the grass rat genus Arvicanthis. The presence of dorsal pigment patterns with potential camouflaging effects in Rhabdomys (absent in Arvicanthis) may support this notion57, as do reports that Arvicanthis can readily adopt nocturnality in the laboratory56,58. Interestingly, this pattern of evolutionary acceleration differs markedly with that of circadian clock genes. In contrast to clock genes which function in many tissues across the body, expression of the rhodopsin phototransduction cascade is restricted to rod cells of the retina. Differing patterns of pleiotropy potentially create a scenario under which changes in circadian organization may favour cis-regulatory evolution while coding sequence evolution may be permitted for genes in the rhodopsin cascade.
Dim-light vision in striped mice
Relaxed purifying selection on rod phototransduction genes in the striped mouse suggests that there may be corresponding functional changes in dim-light vision compared to nocturnal relatives. To test this prediction, we applied electroretinography to compare visual performance under scotopic in striped mice vs nocturnal laboratory mice. Animals were dark adapted for 6 hours and exposed to full field, square wave modulations in light intensity (80.5% Michelson contrast) against a dim background light (9.6 log photons/cm2/s, equivalent to 3.58 R*/rod/s) and across a range of frequencies (1–25Hz). Both laboratory and striped mice responded to elements of this stimulus as evidenced by modulations in the electroretinogram (ERG) at the appropriate frequency (Figure 4A). To facilitate comparisons between species, we applied fast Fourier transform (FFT) to objectively quantify ERG oscillation amplitudes occurring at the stimulus frequency (Figure 4B). In both species, FFT amplitude fell away as temporal frequency increased. However, there was a species difference in this relationship (F- test for sigmoidal curve fit p=0.006) with the reduction in amplitude occurring at lower frequencies in striped mice (EC50=2.0 vs 9.5 Hz in striped vs laboratory mice). This deficit in striped mouse response to higher frequency flicker was specific to dim light conditions, as when we presented this same stimulus under a brighter background light we found that in fact striped mouse ERGs had higher FFT amplitude than mice across the higher frequencies (Figure 4C and D). Further work will be required to determine whether there is a mechanistic link between the accelerated evolution of rod phototransduction genes and this change in dim light visual response. The genes showing accelerated evolution encode proteins which collectively define the gain and longevity of the rod photoresponse, raising the possibility of a causative link between accumulated changes in these proteins during striped mouse evolution and reductions in temporal resolution for rod vision. Testing that possibility will require direct recordings of the striped mouse rod response and, ideally, expressing striped mouse proteins in the rod of laboratory mouse. Whether or not such a direct causative relationship exists, the discovery of reduced performance in dim-light vision in striped mice is consistent with a reduction in the selective constraints on rod photoreception in this species.
Figure 4. ERG responses in African striped mice and Laboratory mice.
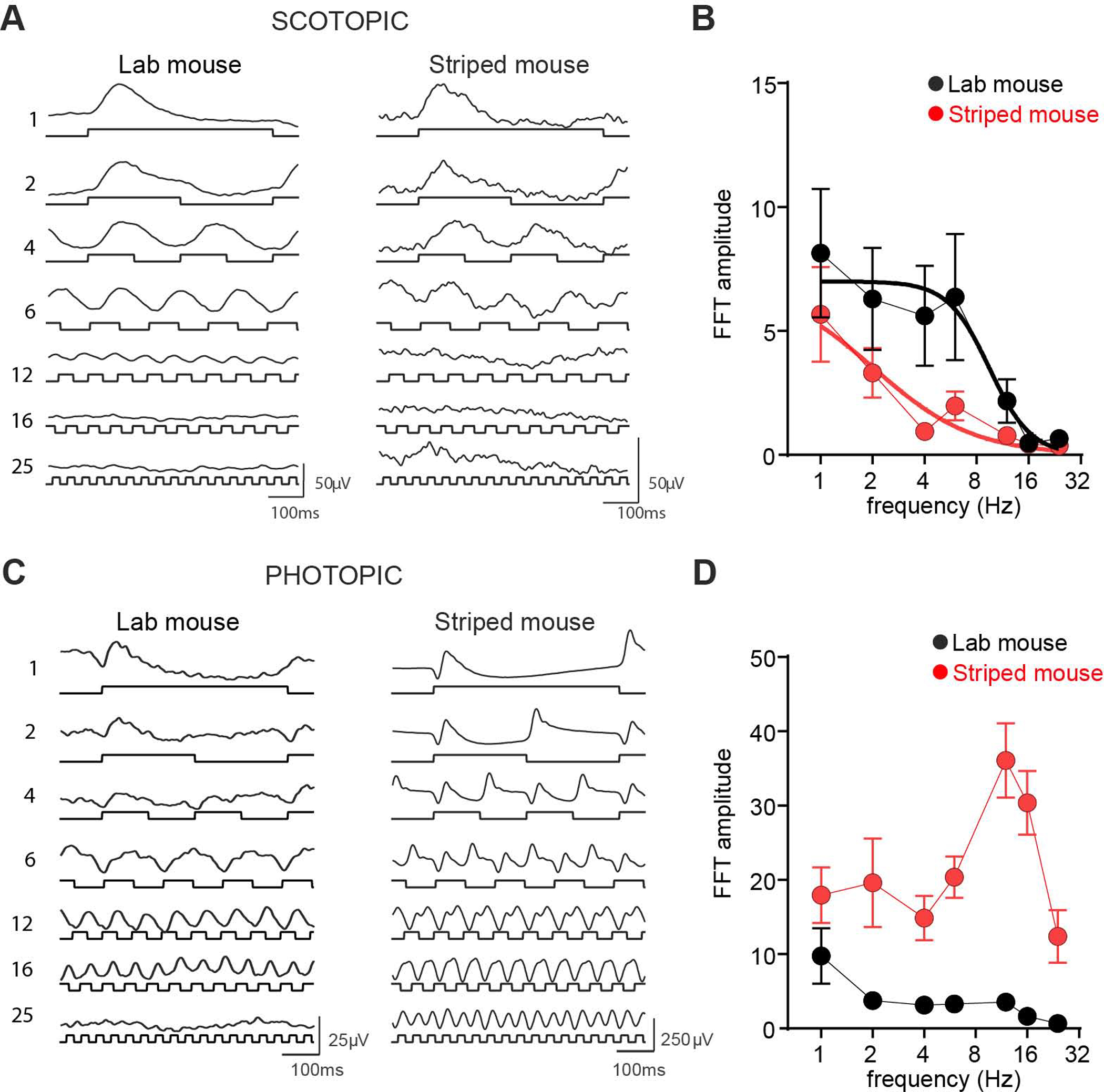
(A) Mean ERG traces from laboratory mice (left) and striped mice (right) in response to square-wave modulations (80.5% Michelson contrast) presented against a dim (9.6 log photons/cm2/s) background. Frequency in Hz shown to left, and light modulation shown below each trace. Scale bar = 100ms (x-axis), 50μV (y axis). (B) Mean±SEM FFT amplitude at modulation frequency for square-wave modulation across frequency range at 9.6 log photons/cm2/s background for laboratory mice (black) and striped mice (red). Data for each species are fitted with a separate sigmoidal curve (F test comparison p<0.001) with variable slope. C and D as A and B but for stimuli presented against a bright background (14.6 log photons/cm2/s). Scale bar = 100ms (x-axis), 25μV or 250μV (y axis) for laboratory and striped mice respectively. n=5 per group.
By taking advantage of naturally evolved, adaptive phenotypes in African striped mice, our work provides insights into the transcriptional changes and genomic patterns of natural selection associated with temporal niche transition in a diurnal rodent. We identify a district pattern of molecular clock reorganization concomitant with the inverted diel activity in this species and raise important new questions about show such phasing is achieved at the regulatory level. Moreover, we find evidence of relaxed selection on rod phototransduction, reflecting a reduced reliance on low-light vision, supported by in vivo retinal measurements. More broadly, our approach exemplifies how integrating interdisciplinary approaches, including whole-genome sequencing, transcriptomics, comparative genomics, and functional experiments, can improve our understanding of the evolutionary processes shaping organismal traits in the wild.
STAR Methods
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ricardo Mallarino (rmallarino@princeton.edu).
Materials availability
This study did not generate new unique reagents
Data and code availability
RNA-Seq data have been deposited in the ArrayExpress database at EMBL-EBI under accession number E-MTAB-12024. The genome assembly, 10X Chromium Linked Reads, and Dovetail Hi-C reads have been deposited in the BioProject database: PRJNA858857. Lastly, we have created a FigShare repository (https://doi.org/10.26188/20321655) containing the following: 1) Rhabdomys_pumilio_final.gff.gz: A raw GFF-formatted gene annotation set for the Rhabdomys pumilio genome produced by Funannotate and used in transcriptomic analyses; 2) Rhabdomys_pumilio.mouse_gene_name_final.gff.gz: A copy of the above GFF-formatted gene annotation set for the Rhabdomys pumilio genome, in which gene symbols from the laboratory mouse (Mus musculus) have been assigned to their predicted R. pumilio orthologs; 3) CompGenAnno.tar.gz: A folder of GFF-formatted annotations used in comparative genomic analyses, produced by directly lifting-over gene annotations from the M. musculus genome (annotation: GCF_000001635.27_GRCm39_genomic.gff, assembly: GCF_000001635.27_GRCm39_genomic.fna) onto each of 23 other murid genome assemblies. Additionally, a manifest of each reference genome can be found in .tsv format (manifest_of_genome_assemblies_and_liftover_annotations.txt) along with a file with locus trees (locus_trees.txt) for each orthologous group of genes used in comparative genomic analyses are provided; 4) Table_of_RER_data_for_examined_species.xlsx: A large table showing relative evolutionary rates measurements for each orthologous group of gene sequences (referenced against a M. musculus transcript), for each species examined. Each species may be listed in multiple columns, reflecting different species representation for each orthologous group (i.e., representing cases in which the branch to a given leaf node originates at a different ancestral node due to sister species not represented in that alignment); and 5) Rhabdomys_pumilio_Princeton_asm1.0_preNCBI.fasta.gz: A copy of the genome assembly prior to any re-formatting that NCBI performs after upload.
This paper does not report original code.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
Experimental model
All experiments done at the University of Manchester were performed in accordance with the UK Animals, Scientific Procedures Act of 1986, and the study was approved by the UK Home Office under the animal license PP3176367. All experiments done at Princeton University were performed with authorisation from Princeton University’s IACUC.
Rhabdomys pumilio:
F10 descendants of wild-derived striped mice (R. pumilio, originating from Goegap Nature Reserve, South Africa, S 29° 41.56′, E 18° 1.60′) were obtained from a captive colony at the University of Zurich (Switzerland) and are maintained at Manchester University and Princeton University. Striped mice are kept at a 12:12 light-dark cycle and given food ad libitum. Both males and female adult striped mice were used for this study.
Mus musculus:
All laboratory mice used in this study were from the C57BL/6 inbred strain, obtained from (Envigo). Importantly, while many inbred laboratory mice harbor mutations in their visual systems that may alter functional measurements, the strain used his has a fully functional visual system. Both males and female adult laboratory mice were used for this study
Methods Details
Genome assembly and annotation
We collected 50mg of thigh muscle tissue from a euthanized female African striped mouse from the captive colony housed at Princeton University for genome sequencing. High molecular weight DNA was extracted with the Qiagen MagAttract HMW kit (NEB 67563), quantified with the Qubit DNA HS kit (Thermofisher Q32851) and DNA fragment size distribution was visualized using the BioAnalyzer 2100 High Sensitivity chip. We next prepared a single genome library using the 10X Genomics Chromium system with the Genome v2 Library Prep kit and performed sequencing on an Illumina HiSeq 2500 Rapid Flowcell in 2×150bp format. In total, ~275Gb of sequence data were generated, corresponding to approximately 119X haploid genome coverage. A primary assembly was generated using the Supernova2 pipeline (10X Genomics). Next, additional tissue samples were provided to Dovetail Genomics who generated Hi-C libraries, which were used to scaffold the primary assembly with the HiRise pipeline. Genome assembly metrics were calculated using the stats.sh script included in the BBMap package v37.9359. BUSCO v5.2.223 was used to annotate benchmarking mammalian orthologs from the mammalia_odb10 database across murid genome assemblies.
Gene prediction and annotation of the assembled genome was carried out using Funannotate v1.8.160. RNA data generated from peripheral tissues (see below) was used as evidence for gene prediction. Prior to gene prediction, RNA data was quality and adapter trimmed using TrimGalore v0.6.661. Repeat models were generated using Red62. These repeats were used as BLASTX queries against the UniProt/Swiss-Prot database of curated proteins. Sequences that significantly matched with any protein and the flanking 50bp were removed from the repeat models using ProtExcluder63. Filtered repeat models were applied to the genome using RepeatMasker64. BUSCO was used to estimate annotation completeness (93.3% complete single copy, 1.1% recovered as partial copies).
General activity monitoring
General activity rhythms were recorded for 2 weeks in single housed striped mice kept under a 12:12 light:dark cycle (Zeitgeber Time (ZT) 0 corresponds to the time of lights on, and ZT12 to lights off) using a passive infrared motion sensor system, as previously described. Data were acquired in 10s bins and expressed as % of the ceiling activity reading of these sensors in that time window, in this way we calculate activity as % of the maximum amount of activity detectable with the sensor65. Daily 24h-profile of general activity was calculated for each animal using 30min binned data across a period of 7 days and averaged from a group of 10 animals.
RNA extraction, library preparation and sequencing
ZT2 and ZT14 analysis
Tissue samples from animals (age ~13mo; male and female) kept under 12:12 light:dark cycle were collected at ZT2 and ZT14 (10 animals per time point). Animals were sedated with isoflurane and immediately culled by cervical dislocation.
Retina, lung, and liver samples:
Total RNA was extracted from < 5 mg frozen tissue using the Qiagen RNeasy Mini kit (Qiagen 74104) according to the manufacturer’s instructions. Total RNA was submitted to the Genomic Technologies Core Facility (GTCF, University of Manchester, UK). Quality and integrity of the RNA samples were assessed using a 4200 TapeStation (Agilent Technologies) and then libraries generated using the Illumina® Stranded mRNA Prep. Ligation kit (Illumina, Inc. 20040532) according to the manufacturer’s protocol. Briefly, total RNA (typically 1ug) was used as input material from which polyadenylated mRNA was purified using poly-T, oligo-attached, magnetic beads. Next, the mRNA was fragmented under elevated temperature and then reverse transcribed into first strand cDNA using random hexamer primers and in the presence of Actinomycin D (thus improving strand specificity whilst mitigating spurious DNA-dependent synthesis). Following removal of the template RNA, second strand cDNA was then synthesized to yield blunt-ended, double-stranded cDNA fragments. Strand specificity was maintained by the incorporation of deoxyuridine triphosphate (dUTP) in place of dTTP to quench the second strand during subsequent amplification. Following a single adenine (A) base addition, adapters with a corresponding, complementary thymine (T) overhang were ligated to the cDNA fragments. Pre-index anchors were then ligated to the ends of the double-stranded cDNA fragments to prepare them for dual indexing. A subsequent PCR amplification step was then used to add the index adapter sequences to create the final cDNA library. The adapter indices enabled the multiplexing of the libraries, which were pooled prior to cluster generation using a cBot instrument. The loaded flow-cell was then paired-end sequenced (76 + 76 cycles, plus indices) on an Illumina HiSeq4000 instrument. Finally, the output data was demultiplexed and BCL-to-Fastq conversion performed using Illumina’s bcl2fastq software, version 2.20.0.422.
Suprachiasmatic nucleus (SCN) samples:
Punches were collected from 1 mm thick brain slices containing the SCN using a sample corer. RNA was extracted from snap-frozen brain punches using the Qiagen RNeasy Micro kit (Qiagen 74004) according to the manufacturer’s instructions and quantified using a 4200 TapeStation (Agilent Technologies). Three nanograms of RNA (RIN ≥ 8.7) was used as input material and libraries were prepared by following the SMARTer Stranded Total RNA-Seq Kit V3 - Pico Input Mammalian (Takara Bio 634485) user manual. In brief, samples were fragmented at 94 °C for 4 min prior to first-strand synthesis. Illumina adaptors and indexes were added to single-stranded cDNA via 5 cycles of PCR. Libraries were hybridized to R-probes for fragments originating from ribosomal RNA to be cleaved by ZapR. The resulting ribo-depleted library fragments were amplified with 15 cycles of PCR. Samples were pooled and sequenced as described above.
24-hr circadian transcriptomic profiling
For circadian transcriptome profiling, animals (age ~ 3mo; males and females) were housed in constant darkness, with tissue samples were taken at hourly intervals (one animal per time point) starting at circadian time 18 (6 hours after lights went off for the last time) and continuing for a further 23-hour. Samples were snap frozen and stored at −80C until RNA extraction.
Retina, lung, and liver samples:
Total RNA was extracted from 5–10mg of snap frozen tissue using the Qiagen RNeasy Fibrous Tissue Mini Kit (Qiagen 74704) according to the manufacturer’s instructions. RNA concentration was measured via Qubit 2.0 (ThermoFisher) and RNA quality was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies). mRNA was isolated using the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs E7490) and libraries were prepped using the NEBNext Ultra II Directional RNA library prep kit for Illumina (New England Biolabs E7760) and NEBNext Multiplex Oligos for Illumina (New England Biolabs E6440), following the manufacturer’s instructions. In brief, mRNA was isolated using OligodT beads and fragmented at 94°C for 15 minutes. cDNA was synthesized, adaptors were added, and the adaptor-ligated DNA was amplified with unique barcodes using 10–12 PCR cycles. Samples were combined into a single pool and sequenced on an Illumina NovaSeq S1 100nt Flowcell (2 × 69-bp format). To increase read depth, a second round of sequencing was performed on an Illumina NovaSeq SP 100nt Flowcell (2 × 69-bp format). Raw reads from both sequencing runs were concatenated prior to downstream processing/analyses.
Suprachiasmatic nucleus (SCN) samples:
Punches were collected from 1 mm thick brain slices containing the SCN using a sample corer. RNA was extracted from snap-frozen brain punches using the Qiagen RNeasy Micro kit (Qiagen 74004) according to the manufacturer’s instructions. RNA concentration was measured via Qubit 2.0 (ThermoFisher) and RNA quality was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies). Libraries were prepared using the SMARTer Stranded Total RNA-Seq Kit V3 - Pico Input Mammalian (Takara Bio 634485) following the manufacturer’s instructions, including the RNA fragmentation step. In brief, samples were fragmented at 94 °C for 4 min prior to first-strand synthesis. Illumina adaptors and barcodes were added to single-stranded cDNA (5 PCR cycles), and ribosomal cDNA was depleted using ZapR v3 and R-Probes v3. Libraries were then amplified using 12 cycles of PCR. Samples were pooled with the retina, lung, and liver samples and sequenced as described above for retina, liver, and lung samples.
Quantification and Statistical Analysis
Differential gene expression analysis:
The quality of the stranded paired-end RNA-Seq reads was assessed using FastQC (v0.11.3)66 and FastQ Screen (v0.14.0)67, and adapter and low-quality base were trimmed using BBDuk (BBMap suite v38.93)59. Processed reads were then mapped against the Rhabdomys pumilio genome and in-house gene annotation (available from the cited FigShare repository) using STAR (v2.7.9a)68. The “--quantMode GeneCounts” option was used to obtain read counts per gene from STAR.
In the R environment (v3.6.3), differential gene expression analysis was performed using Bioconductor package DESeq2 (v1.26.0)69, and Benjamini-Hochberg (BH) procedure was used to controls false discovery rate (FDR) at 5%. Additionally, the lfcShrink function (with apeglm method) was applied to generate a more accurate log2 fold change estimate. The counts function (with normalized = TRUE) was used to generate normalised counts by the median-of-ratios method as described in ref70.
Pathway enrichment analysis:
Pathway enrichment analysis of DEGs was performed using the R package enrichR (v3.0)71. The KEGG 2019 Mouse gene-set library defined by the Enrichr web server71 was chosen for gene ontology analysis. Pathway results were ranked by the combined scores (calculated as log(P-value) multiplied by z-score) and pathways with an FDR-adjusted p-value < 0.05 were considered significant.
Rhythmic analysis:
The time-course RNA-seq datasets from SCN, retina, lung and liver were processed using BBDuk and STAR as described above with the exception that the transcript alignments were obtained by using “--quantMode TranscriptomeSAM” when running STAR. RSEM (v1.3.1)72 was used to perform gene expression quantification. The transcriptomic data from all four tissues were checked for the presence of outliers using principal component analysis. Two samples, ZT0 from retina and ZT17 from lung, were excluded from the circadian rhythmicity analysis on this basis. Furthermore, genes that were not expressed in any samples were also excluded.
The gene-level transcripts per million mapped reads (TPM) values were used as input to detect rhythmic signals using MetaCycle (v1.2.0)73 in R environment. The “meta2d” function was run with the following parameters: minimum period length of 20, maximum period length of 28, expected period length of 24, phase adjusted with predicted period length, ARSER, JTK_CYCLE and Lomb-Scargle algorithms were used (ARSER was disabled automatically in lung and retina when there were missing values) and the Fisher’s method was used to integrate p-values from the chosen methods. Rhythmic genes were identified with statistical significance BH q-values (meta2d_BH.Q) < 0.1.
Analysis of relative evolutionary rates and positive selection
To permit comparative analysis of relative evolutionary rates, we use the following procedure to predict orthologous genes across 24 murid species. We first downloaded the reference genome for the laboratory mouse M. musculus (GRCm39) and its corresponding RefSeq gene annotations74. The mouse genome is of excellent quality and is rigorously annotated using transcriptome data from numerous tissues, thus it was used as the reference species for evolutionary analyses. To extract orthologous transcripts suitable for comparative evolutionary analysis from other murids, we used liftoff v1.6.175 to lift-over gene models from M. musculus to a total of 23 other murid species including the African striped mouse. 17 additional species within the subfamily Murinae were examined, as this group includes the African striped mouse and its diurnal sister genus Arvicanthis, as well as the reference species M. musculus. 5 additional species belonging to the genus Acomys (subfamily Deomyinae) were included as an outgroup (Data S1C).
Next, coding sequences (CDS) were extracted from each murid genome using gffread v0.12.4 and collated into orthologous groups in multifasta files76. Each orthologous group was first filtered to remove sequences differing in total length from the M. musculus reference gene by more than 10%. Next, orthologous groups were filtered such that only those with a representative sequence for the African striped mouse and at least one Acomys outgroup species were retained. Next, files in which fewer than 75% of sequences began with an ATG were excluded, as were files containing sequences from fewer than 10 total species. Next, orthologous sequences were aligned using mafft v7.407 with parameters --localpair and --maxiterate 100077. Resulting ortholog alignments were then re-filtered by the criteria above to remove low-quality orthologs and then re-aligned.
To infer a species tree topology for subsequent evolutionary analyses, alignments containing representative orthologs for all 24 murid species were filtered to retain only one representative transcript per gene based on highest alignment score, were concatenated and were then provided to RAxML v8.2.1278 with parameters: -f a -x 58744 -p 58744 -# 1000 -m GTRGAMMA, flagging the genus Acomys the outgroup (parameter -o) and using 1000 bootstrap replicates (parameter -# 1000). The topology of the best maximum likelihood tree was found to be consistent with a recent, large-scale phylogeny of Muroids21 and was thus taken as the species tree for subsequent evolutionary analyses. RAxML was used to estimate branch lengths on the fixed species tree topology for each ortholog alignment with parameter -f e and -m GTRGAMMA. We chose to use RAxML because it is a commonly used and computationally efficient tool to generate branch length estimates from multiple sequence alignments, which are required as input for our downstream analyses using RER (see below).
Several approaches exist to define evolutionarily accelerated loci. For instance, PhyloP implements multiple tests for evolutionary acceleration relative to a background model of neutral evolutionary rates, usually calculated from 4-fold degenerate sites. The growing recognition that selection is ubiquitous across the genome and that phenomena such as linked-selection can markedly alter the evolutionary rates of 4-fold degenerate sites79 calls into question the degree to which such models accurately reflect neutral rates. Moreover, when examining evolution of protein-coding sequences, neutral evolutionary rates may not represent an appropriate null model. Protein-coding genes are generally considered to experience evolutionary constraint as a product of purifying selection. Thus, their expected rate of sequence evolution should be slower than would be expected under neutrality. Given this, we took an alternate approach, by calculated relative evolutionary rates for each sequence in each ortholog alignment using the getAllResiduals function (parameters: transform = “sqrt”, weighted = T, scale = T) implemented in the package RERConverge v0.3.049. Rather than comparing evolutionary rates against a neutral model, RERConverge generates a consensus or “average” tree from a set of input locus trees (i.e., those calculated by RAxML above). Each locus tree is then scaled to the master tree, and residual branch lengths are taken as the relative evolutionary rate (RER) for each ortholog with the locus tree. In this way, shifts in evolutionary rate are relative to a background expectation based on the class of elements being tested. Moreover, given the close evolutionary relationships and similar genome compositions of species being examined in our analyses, plots of ortholog RERs (such as those shown in Figure 3C and Figure S4) can be used to rule out alternate explanations for acceleration based on genomic region (such as GC-biased gene conversion80), as acceleration driven by such effects would be present across orthologs from most or all species for a given locus with relative uniformity. After calculating RERs for each group of orthologs, we defined highly accelerated sequences in the African striped mouse as the 95th percentile of genes with a positive relative evolutionary rate (Figure S3) and focused on these in downstream examinations. It is worth noting that, while the branch lengths for the initial gene trees calculated for each group of orthologous loci represent mean substitutions per site, after scaling the gene tree for comparison with the master/consensus tree, that is no longer strictly the case. Therefore, the RER value is best understood at being unitless.
Highly accelerated genes were examined for enrichment of KEGG Pathway and GO Biological Process terms using the Enrichr web server81. Dot plots of per-species, per-gene relative evolutionary rates were generated using the foreground2Paths and plotRers functions in RERconverge. Results from the RERconverge analysis for striped mice are listed in Data S1D.
The branch-site model was used to test for evidence of positive selection. Briefly, sequence files for rhodopsin cascade genes were first re-aligned using MACSE v2.06 and branch-lengths recalculated with RAxML (parameter -f e and -m GTRGAMMA)78. These alignments and trees were then provided to EasyCodeML v1.082 to perform branch-site tests on the striped mouse, grass rat, and their most recent common ancestor. Multiple testing correction was performed using the Benjamini-Hochberg procedure83.
ERG measurements
ERGs were recorded in adult M. musculus (C57/BL6, aged 4–5 months) and adult male R. pumilio (aged 7–8 months). All ERGs were recorded at subjective midday following dark adaptation for a period of 6 hours. Animals were anaesthetised under isoflurane in a 95/5% Oxygen/CO2 mix at a flow rate of 0.5 – 1.0L/min. Isoflurane concentrations of 5% and 1.5–3.5% were used for induction and maintenance of anaesthesia, respectively. A topical mydriatic (tropicamide 1%; Bausch and Lomb) and hypromellose eye drops were applied to the recording eye before placement of a corneal contact-lens–type electrode. A needle reference electrode (Ambu, Neuroline) was inserted approximately 5mm from the base of the contralateral eye, and a second subcutaneous needle in the scruff acted as a ground. Electrodes were connected to a Windows PC via a signal conditioner (Model 1902 Mark III, CED) that differentially amplified (X3000) and filtered (band-pass filter cut off 0.5 to 200Hz) the signal, and a digitizer (Model 1401, CED). Core body temperature was maintained at 37°C throughout recording with a homeothermic heat mat (Harvard Apparatus).
Visual stimuli were generated with a combination of violet, blue and cyan elements of a multispectral LED light source (Lumencor). Intensities were modulated via pulse width modulations via an Arduino Uno. Light from the light engine passed through a filter-wheel containing neutral-density filters (reducing the light by between 101 and 105) and focused onto opal diffusing glass (5mm diameter; Edmund Optics Inc.) positioned <5mm from the eye. All LED intensities were controlled dynamically with a PC. Stimuli were measured at the corneal plane using a spectroradiometer (SpectroCAL II, Cambridge Research Systems, UK) between 350–700nm. Stimuli were presented either as square-wave modulations from background at 80.5% Michelson contrast across a range of frequencies (1, 2, 4, 6, 12, 16, 25 Hz) and at two backgrounds (9.6, 14.6 log photons/cm2/s). ERG responses were analysed in MATLAB (release 2018a, Mathworks), using a fast Fourier Transform (FFT) to analyse ERG responses in the frequency domain. This allowed us to use an objective measure to quantify variations in the ERG signal at the frequency of the visual stimulus presented.
Supplementary Material
(A) African striped mouse genome assembly metrics; (B) African striped mouse BUSCO recovery; (C) Genomes accessed in the present study; (D) Evolutionarily accelerated African striped mouse genes; (E) Summary of KEGG and GO Biological Process terms enriched among African striped mouse (Rhabdomys pumilio) accelerated genes; (F) Branch-site tests of positive selection in Arvicanthine accelerated genes; (G) Evolutionarily accelerated genes in the African grass rat (Arvicanthis niloticus); (H) Summary of KEGG and GO Biological Process terms enriched among African grass rat (Arvicanthis niloticus) accelerated genes; (I) Evolutionarily accelerated genes in the R. pumilio and A. niloticus most recent common ancestor; (J) Summary of KEGG and GO Biological Process terms enriched among accelerated genes in the R. pumilio and A. niloticus most recent common ancestor.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Critical commercial assays | ||
| Deposited data | ||
| RNA Sequencing Data | This paper | ArrayExpress (EMBL-EBI) under accession number E-MTAB-12024 |
| The genome assembly, 10X Chromium Linked Reads, and Dovetail Hi-C reads | This paper | BioProject database: PRJNA858857 |
| Rhabdomys_pumilio_final.gff.gz: A raw GFF-formatted gene annotation set for the Rhabdomys pumilio genome produced by Funannotate and used in transcriptomic analyses | This paper | FigShare repository (https://doi.org/10.26188/20321655 |
| Rhabdomys_pumilio.mouse_gene_name_final.gff.gz: A copy of the above GFF-formatted gene annotation set for the Rhabdomys pumilio genome, in which gene symbols from the laboratory mouse (Mus musculus) have been assigned to their predicted R. pumilio orthologs. | This paper | FigShare repository (https://doi.org/10.26188/20321655 |
| CompGenAnno.tar.gz: A folder of GFF-formatted annotations used in comparative genomic analyses, produced by directly lifting-over gene annotations from the M. musculus genome (annotation: GCF_000001635.27_GRCm39_genomic.gff, assembly: GCF_000001635.27_GRCm39_genomic.fna) onto each of 23 other murid genome assemblies. Additionally, a manifest of each reference genome can be found in .tsv format (manifest_of_genome_assemblies_and_liftover_annotations.txt) along with a file with locus trees (locus_trees.txt) for each orthologous group of genes used in comparative genomic analyses are provided. | This paper | FigShare repository (https://doi.org/10.26188/20321655 |
| Table_of_RER_data_for_examined_species.xlsx: A large table showing relative evolutionary rates measurements for each orthologous group of gene sequences (referenced against a M. musculus transcript), for each species examined. Each species may be listed in multiple columns, reflecting different species representation for each orthologous group (i.e., representing cases in which the branch to a given leaf node originates at a different ancestral node due to sister species not represented in that alignment). | This paper | FigShare repository (https://doi.org/10.26188/20321655 |
| Rhabdomys_pumilio_Princeton_asm1.0_preNCBI.fasta.gz: A copy of the genome assembly prior to any re-formatting that NCBI performs after upload | This paper | FigShare repository (https://doi.org/10.26188/20321655 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Rhabdomys pumilio | Mallarino and Lucas laboratory | F10 descendants of wild-derived striped mice (R. pumilio, originating from Goegap Nature Reserve, South Africa, S 29° 41.56′, E 18° 1.60′) were obtained from a captive colony at the University of Zurich (Switzerland) and are maintained at Manchester University and Princeton University |
| Mus musculus | Lucas laboratory | C57BL/6 inbred strain, obtained from (Envigo) |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and algorithms | ||
| BBMap suite v38.93 | Buschnell et al.56 | https://sourceforge.net/projects/bbmap/ |
| BUSCO v5.2.2 | Seppey et al.22 | https://mybiosoftware.com/busco-assessing-genome-assembly-and-annotation-completeness-with-single-copy-orthologs.html |
| Funannotate v1.8.1 | Palmer57 | https://github.com/nextgenusfs/funannotate/tree/v1.8.1 |
| TrimGalore v0.6.6 | Kruger58 | https://github.com/FelixKrueger/TrimGalore |
| ProtExcluder | Campbell et al.60 | https://github.com/NBISweden/ProtExcluder |
| RepeatMasker | Smit et al.61 | https://www.repeatmasker.org/ |
| FastQC (v0.11.3) | Andrews63 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| FastQ Screen (v0.14.0) | Wingett and Andrews64 | https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/_build/html/index.html |
| STAR (v2.7.9a) | Dobin et al.65 | https://github.com/alexdobin/STAR |
| DESeq2 (v1.26.0) | Lewis et al.66 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| enrichR (v3.0) | Chen et al.68 | https://cran.r-project.org/web/packages/enrichR/index.html |
| RSEM (v1.3.1) | Li and Dewey70 | https://deweylab.github.io/RSEM/ |
| MetaCycle (v1.2.0) | Wu et al.72 | https://github.com/gangwug/MetaCycle |
| liftoff v1.6.1 | Shumate and Salzberg71 | https://github.com/agshumate/Liftoff |
| RAxML v8.2.12 | Stamakis74 | https://cme.h-its.org/exelixis/web/software/raxml/ |
| RERConverge v0.3.0 | Pertea and Pertea75 | https://github.com/nclark-lab/RERconverge |
| EasyCodeML v1.0 | Gao et al.79 | https://github.com/BioEasy/EasyCodeML |
| Other | ||
LIFE SCIENCE TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB_2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Bacterial and virus strains | ||
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry | Krashes et al.1 | Addgene AAV5; 44361-AAV5 |
| AAV5-EF1a-DIO-hChR2(H134R)-EYFP | Hope Center Viral Vectors Core | N/A |
| Cowpox virus Brighton Red | BEI Resources | NR-88 |
| Zika-SMGC-1, GENBANK: KX266255 | Isolated from patient (Wang et al.2) | N/A |
| Staphylococcus aureus | ATCC | ATCC 29213 |
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC 700294 |
| Biological samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children’s Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, peptides, and recombinant proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350–13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433–59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124–87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14–486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical commercial assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | PerkinElmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202–1012 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Nanog STILT inference | This paper; Mendeley Data | http://dx.doi.org/10.17632/wx6s4mj7s8.2 |
| Affinity-based mass spectrometry performed with 57 genes | This paper; Mendeley Data | Table S8; http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Experimental models: Cell lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09–0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental models: Organisms/strains | ||
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl: y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard3 | ZFIN: ZDB-GENO-060207–1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al.4 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Oligonucleotides | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106–5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper | N/A |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl0130415 |
| AAV2/1-hsyn-GCaMP6- WPRE | Chen et al.5 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al.6 | Addgene Plasmid #21339 |
| Software and algorithms | ||
| ImageJ | Schneider et al.7 | https://imagej.nih.gov/ij/ |
| Bowtie2 | Langmead and Salzberg8 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al.9 | http://samtools.sourceforge.net/ |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al.10 | https://github.com/ChristophRau/wMICA |
| ICS algorithm | This paper; Mendeley Data | http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
PHYSICAL SCIENCE TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| QD605 streptavidin conjugated quantum dot | Thermo Fisher Scientific | Cat#Q10101MP |
| Platinum black | Sigma-Aldrich | Cat#205915 |
| Sodium formate BioUltra, ≥99.0% (NT) | Sigma-Aldrich | Cat#71359 |
| Chloramphenicol | Sigma-Aldrich | Cat#C0378 |
| Carbon dioxide (13C, 99%) (<2% 18O) | Cambridge Isotope Laboratories | CLM-185–5 |
| Poly(vinylidene fluoride-co-hexafluoropropylene) | Sigma-Aldrich | 427179 |
| PTFE Hydrophilic Membrane Filters, 0.22 mm, 90 mm | Scientificfilters.com/Tisch Scientific | SF13842 |
| Critical commercial assays | ||
| Folic Acid (FA) ELISA kit | Alpha Diagnostic International | Cat# 0365–0B9 |
| TMT10plex Isobaric Label Reagent Set | Thermo Fisher | A37725 |
| Surface Plasmon Resonance CM5 kit | GE Healthcare | Cat#29104988 |
| NanoBRET Target Engagement K-5 kit | Promega | Cat#N2500 |
| Deposited data | ||
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Structure of compound 5 | This paper; Cambridge Crystallographic Data Center | CCDC: 2016466 |
| Code for constraints-based modeling and analysis of autotrophic E. coli | This paper | https://gitlab.com/elad.noor/sloppy/tree/master/rubisco |
| Software and algorithms | ||
| Gaussian09 | Frish et al.1 | https://gaussian.com |
| Python version 2.7 | Python Software Foundation | https://www.python.org |
| ChemDraw Professional 18.0 | PerkinElmer | https://www.perkinelmer.com/category/chemdraw |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al.2 | https://github.com/ChristophRau/wMICA |
| Other | ||
| DASGIP MX4/4 Gas Mixing Module for 4 Vessels with a Mass Flow Controller | Eppendorf | Cat#76DGMX44 |
| Agilent 1200 series HPLC | Agilent Technologies | https://www.agilent.com/en/products/liquid-chromatography |
| PHI Quantera II XPS | ULVAC-PHI, Inc. | https://www.ulvac-phi.com/en/products/xps/phi-quantera-ii/ |
Highlights.
A chromosome-level genome assembly of the diurnal rodent R. pumilio was generated
R. pumilio shows a realigned circadian organisation compared to nocturnal rodents
There has been a relaxation of purifying selection in phototransduction genes
R. pumilio and nocturnal rodents have differences in dim light visual responses
Acknowledgements:
We thank Princeton LAR and the University of Manchester Biological Services Facility for help with striped mouse husbandry; the Genomic Technology Core Facility at the University of Manchester for carrying out sequencing of RNA samples; Forrest Rogers for logistical support; Rebecca Hughes for technical assistance; and Elise Ireland for proofreading. This work was supported by an NIH grant R35GM133758 (RM) and by a Wellcome Investigator Award 210684/Z/18/Z (RJL) and BBSRC project grant BB/V011111/1(RJL). CF is funded by NIH F32 GM139240-01; MRJ is funded by NIH F32 GM139253; AEA is funded by a Sir Henry Dale Fellowship, jointly funded by the Wellcome Trust and the Royal Society (Grant Number 218556/Z/19/Z).
Inclusion and diversity:
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of interests:
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kronfeld-Schor N, and Dayan T (2003). Partitioning of Time as an Ecological Resource. Annu. Rev. Ecol. Evol. Syst. 34, 153–181. [Google Scholar]
- 2.Penteriani V, del Mar Delgado M, Alonso-Alvarez C, and Sergio F (2006). The importance of visual cues for nocturnal species: eagle owls signal by badge brightness. Behav. Ecol. 18, 143–147. [Google Scholar]
- 3.Schmitz L, and Motani R (2010). Morphological differences between the eyeballs of nocturnal and diurnal amniotes revisited from optical perspectives of visual environments. Vision Res. 50, 936–946. [DOI] [PubMed] [Google Scholar]
- 4.Barton RA, Purvis A, and Harvey PH (1995). Evolutionary radiation of visual and olfactory brain systems in primates, bats and insectivores. Philos. Trans. R. Soc. Lond. B Biol. Sci. 348, 381–392. [DOI] [PubMed] [Google Scholar]
- 5.Gerkema MP, Davies WIL, Foster RG, Menaker M, and Hut RA (2013). The nocturnal bottleneck and the evolution of activity patterns in mammals. Proc. Biol. Sci. 280, 20130508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox DTC, Gardner AS, and Gaston KJ (2021). Diel niche variation in mammals associated with expanded trait space. Nat. Commun. 12, 1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mallarino R, Pillay N, Hoekstra HE, and Schradin C (2018). African striped mice. Curr. Biol. 28, R299–R301. [DOI] [PubMed] [Google Scholar]
- 8.Schumann DM, Cooper HM, Hofmeyr MD, and Bennett NC (2005). Circadian rhythm of locomotor activity in the four-striped field mouse, Rhabdomys pumilio: a diurnal African rodent. Physiol. Behav. 85, 231–239. [DOI] [PubMed] [Google Scholar]
- 9.Allen AE, Mouland JW, Rodgers J, Baño-Otálora B, Douglas RH, Jeffery G, Vugler AA, Brown TM, and Lucas RJ (2020). Spectral sensitivity of cone vision in the diurnal murid Rhabdomys pumilio. J. Exp. Biol. 223. 10.1242/jeb.215368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Merwe I, Lukáts Á, Bláhová V, Oosthuizen MK, Bennett NC, and Němec P (2018). The topography of rods, cones and intrinsically photosensitive retinal ganglion cells in the retinas of a nocturnal (Micaelamys namaquensis) and a diurnal (Rhabdomys pumilio) rodent. PLoS One 13, e0202106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bano-Otalora B, Moye MJ, Brown T, Lucas RJ, Diekman CO, and Belle MD (2021). Daily electrical activity in the master circadian clock of a diurnal mammal. Elife 10. 10.7554/eLife.68179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katti C, Stacey-Solis M, Coronel-Rojas NA, and Davies WIL (2019). The Diversity and Adaptive Evolution of Visual Photopigments in Reptiles. Frontiers in Ecology and Evolution 7. 10.3389/fevo.2019.00352. [DOI] [Google Scholar]
- 13.Schott RK, Van Nynatten A, Card DC, Castoe TA, and S W Chang B (2018). Shifts in Selective Pressures on Snake Phototransduction Genes Associated with Photoreceptor Transmutation and Dim-Light Ancestry. Mol. Biol. Evol. 35, 1376–1389. [DOI] [PubMed] [Google Scholar]
- 14.Borges R, Fonseca J, Gomes C, Johnson WE, O’Brien SJ, Zhang G, Gilbert MTP, Jarvis ED, and Antunes A (2019). Avian Binocularity and Adaptation to Nocturnal Environments: Genomic Insights from a Highly Derived Visual Phenotype. Genome Biol. Evol. 11, 2244–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y, Wang H, Wang H, and Hadly EA (2017). Rethinking the Origin of Primates by Reconstructing Their Diel Activity Patterns Using Genetics and Morphology. Sci. Rep. 7, 11837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hut RA, Kronfeld-Schor N, van der Vinne V, and De la Iglesia H (2012). In search of a temporal niche: environmental factors. Prog. Brain Res. 199, 281–304. [DOI] [PubMed] [Google Scholar]
- 17.Lucas RJ, Freedman MS, Lupi D, Munoz M, David-Gray ZK, and Foster RG (2001). Identifying the photoreceptive inputs to the mammalian circadian system using transgenic and retinally degenerate mice. Behav. Brain Res. 125, 97–102. [DOI] [PubMed] [Google Scholar]
- 18.Ripperger JA, Jud C, and Albrecht U (2011). The daily rhythm of mice. FEBS Lett. 585, 1384–1392. [DOI] [PubMed] [Google Scholar]
- 19.Doyle SE, Yoshikawa T, Hillson H, and Menaker M (2008). Retinal pathways influence temporal niche. Proc. Natl. Acad. Sci. U. S. A. 105, 13133–13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mallarino R, Henegar C, Mirasierra M, Manceau M, Schradin C, Vallejo M, Beronja S, Barsh GS, and Hoekstra HE (2016). Developmental mechanisms of stripe patterns in rodents. Nature 539, 518–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steppan SJ, and Schenk JJ (2017). Muroid rodent phylogenetics: 900-species tree reveals increasing diversification rates. PLoS One 12, e0183070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castiglia R, Solano E, Makundi RH, Hulselmans J, Verheyen E, and Colangelo P (2012). Rapid chromosomal evolution in the mesic four- striped grass ratRhabdomys dilectus(Rodentia, Muridae) revealed by mtDNA phylogeographic analysis. Journal of Zoological Systematics and Evolutionary Research 50, 165–172. [Google Scholar]
- 23.Seppey M, Manni M, and Zdobnov EM (2019). BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods in Molecular Biology, 227–245. [DOI] [PubMed] [Google Scholar]
- 24.Dibner C, Schibler U, and Albrecht U (2010). The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu. Rev. Physiol. 72, 517–549. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi JS (2017). Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 18, 164–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ko CH, and Takahashi JS (2006). Molecular components of the mammalian circadian clock. Hum. Mol. Genet. 15 Spec No 2, R271–7. [DOI] [PubMed] [Google Scholar]
- 27.Reppert SM, and Weaver DR (2002). Coordination of circadian timing in mammals. Nature 418, 935–941. [DOI] [PubMed] [Google Scholar]
- 28.Mrosovsky N, Edelstein K, Hastings MH, and Maywood ES (2001). Cycle of period gene expression in a diurnal mammal (Spermophilus tridecemlineatus): implications for nonphotic phase shifting. J. Biol. Rhythms 16, 471–478. [DOI] [PubMed] [Google Scholar]
- 29.Mure LS, Le HD, Benegiamo G, Chang MW, Rios L, Jillani N, Ngotho M, Kariuki T, Dkhissi-Benyahya O, Cooper HM, et al. (2018). Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 359. 10.1126/science.aao0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwartz WJ, Reppert SM, Eagan SM, and Moore-Ede MC (1983). In vivo metabolic activity of the suprachiasmatic nuclei: a comparative study. Brain Res. 274, 184–187. [DOI] [PubMed] [Google Scholar]
- 31.Wen S, Ma D, Zhao M, Xie L, Wu Q, Gou L, Zhu C, Fan Y, Wang H, and Yan J (2020). Spatiotemporal single-cell analysis of gene expression in the mouse suprachiasmatic nucleus. Nat. Neurosci. 23, 456–467. [DOI] [PubMed] [Google Scholar]
- 32.Ruan G-X, Zhang D-Q, Zhou T, Yamazaki S, and McMahon DG (2006). Circadian organization of the mammalian retina. Proc. Natl. Acad. Sci. U. S. A. 103, 9703–9708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang R, Lahens NF, Ballance HI, Hughes ME, and Hogenesch JB (2014). A circadian gene expression atlas in mammals: implications for biology and medicine. Proc. Natl. Acad. Sci. U. S. A. 111, 16219–16224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mavroudis PD, DuBois DC, Almon RR, and Jusko WJ (2018). Daily variation of gene expression in diverse rat tissues. PLoS One 13, e0197258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z, Hunter L, Wu G, Maidstone R, Mizoro Y, Vonslow R, Fife M, Hopwood T, Begley N, Saer B, et al. (2019). Genome-wide effect of pulmonary airway epithelial cell-specific Bmal1 deletion. FASEB J. 33, 6226–6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sukumaran S, Jusko WJ, Dubois DC, and Almon RR (2011). Light-dark oscillations in the lung transcriptome: implications for lung homeostasis, repair, metabolism, disease, and drug action. J. Appl. Physiol. 110, 1732–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morris EL, Patton AP, Chesham JE, Crisp A, Adamson A, and Hastings MH (2021). Single-cell transcriptomics of suprachiasmatic nuclei reveal a Prokineticin-driven circadian network. EMBO J. 40, e108614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng AH, Bouchard-Cannon P, Hegazi S, Lowden C, Fung SW, Chiang C-K, Ness RW, and Cheng H-YM (2019). SOX2-Dependent Transcription in Clock Neurons Promotes the Robustness of the Central Circadian Pacemaker. Cell Rep. 26, 3191–3202.e8. [DOI] [PubMed] [Google Scholar]
- 39.von Schantz M, Lucas RJ, and Foster RG (1999). Circadian oscillation of photopigment transcript levels in the mouse retina. Brain Res. Mol. Brain Res. 72, 108–114. [DOI] [PubMed] [Google Scholar]
- 40.Storch K-F, Paz C, Signorovitch J, Raviola E, Pawlyk B, Li T, and Weitz CJ (2007). Intrinsic circadian clock of the mammalian retina: importance for retinal processing of visual information. Cell 130, 730–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hannibal J, Georg B, Hindersson P, and Fahrenkrug J (2005). Light and darkness regulate melanopsin in the retinal ganglion cells of the albino Wistar rat. J. Mol. Neurosci. 27, 147–155. [DOI] [PubMed] [Google Scholar]
- 42.Hughes S, Welsh L, Katti C, González-Menéndez I, Turton M, Halford S, Sekaran S, Peirson SN, Hankins MW, and Foster RG (2012). Differential expression of melanopsin isoforms Opn4L and Opn4S during postnatal development of the mouse retina. PLoS One 7, e34531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gibbs J, Ince L, Matthews L, Mei J, Bell T, Yang N, Saer B, Begley N, Poolman T, Pariollaud M, et al. (2014). An epithelial circadian clock controls pulmonary inflammation and glucocorticoid action. Nat. Med. 20, 919–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang X, Downes M, Yu RT, Bookout AL, He W, Straume M, Mangelsdorf DJ, and Evans RM (2006). Nuclear receptor expression links the circadian clock to metabolism. Cell 126, 801–810. [DOI] [PubMed] [Google Scholar]
- 45.Koronowski KB, Kinouchi K, Welz P-S, Smith JG, Zinna VM, Shi J, Samad M, Chen S, Magnan CN, Kinchen JM, et al. (2019). Defining the Independence of the Liver Circadian Clock. Cell 177, 1448–1462.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hughes ME, Abruzzi KC, Allada R, Anafi R, Arpat AB, Asher G, Baldi P, de Bekker C, Bell-Pedersen D, Blau J, et al. (2017). Guidelines for Genome-Scale Analysis of Biological Rhythms. J. Biol. Rhythms 32, 380–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu G, Ruben MD, Lee Y, Li J, Hughes ME, and Hogenesch JB (2020). Genome-wide studies of time of day in the brain: Design and analysis. Brain Science Advances 6, 92–105. [Google Scholar]
- 48.Sancar C, Sancar G, Ha N, Cesbron F, and Brunner M (2015). Dawn- and dusk-phased circadian transcription rhythms coordinate anabolic and catabolic functions in Neurospora. BMC Biol. 13, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kowalczyk A, Meyer WK, Partha R, Mao W, Clark NL, and Chikina M (2019). RERconverge: an R package for associating evolutionary rates with convergent traits. Bioinformatics 35, 4815–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fain GL, Hardie R, and Laughlin SB (2010). Phototransduction and the evolution of photoreceptors. Curr. Biol. 20, R114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu Y (2019). Widespread nocturnality of living birds stemming from their common ancestor. BMC Evol. Biol. 19, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu Y, Hadly EA, Teng W, Hao Y, Liang W, Liu Y, and Wang H (2016). Corrigendum: Retinal transcriptome sequencing sheds light on the adaptation to nocturnal and diurnal lifestyles in raptors. Sci. Rep. 6, 35895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.White ND, Batz ZA, Braun EL, Braun MJ, Carleton KL, Kimball RT, and Swaroop A (2022). A novel exome probe set captures phototransduction genes across birds (Aves) enabling efficient analysis of vision evolution. Mol. Ecol. Resour. 22, 587–601. [DOI] [PubMed] [Google Scholar]
- 54.Yokoyama S (2008). Evolution of dim-light and color vision pigments. Annu. Rev. Genomics Hum. Genet. 9, 259–282. [DOI] [PubMed] [Google Scholar]
- 55.Denys C, Lalis A, Aniskin V, Gerbault-Seureau M, Delapre A, Gilissen E, Merker S, and Nicolas V (2020). Integrative taxonomy of Guinean Lemniscomys species (Rodentia, Mammalia). fozo.1 69, 20008.1. [Google Scholar]
- 56.Redlin U, and Mrosovsky N (2004). Nocturnal activity in a diurnal rodent (Arvicanthis niloticus): the importance of masking. J. Biol. Rhythms 19, 58–67. [DOI] [PubMed] [Google Scholar]
- 57.Hughes AE, Troscianko J, and Stevens M (2014). Motion dazzle and the effects of target patterning on capture success. BMC Evol. Biol. 14, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blanchong JA, McElhinny TL, Mahoney MM, and Smale L (1999). Nocturnal and diurnal rhythms in the unstriped Nile rat, Arvicanthis niloticus. J. Biol. Rhythms 14, 364–377. [DOI] [PubMed] [Google Scholar]
- 59.Bushnell B (2014). BBMap: A fast, accurate, splice-aware aligner (Lawrence Berkeley National Lab. (LBNL; ), Berkeley, CA (United States)). [Google Scholar]
- 60.Palmer JM ASJ (2020). Funannotate v1.8.1: Eukaryotic genome annotation (v1.8.1). 10.5281/zenodo.4054262. [DOI] [Google Scholar]
- 61.Krueger F (2019). Trim Galore. https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
- 62.Girgis HZ (2015). Red: an intelligent, rapid, accurate tool for detecting repeats de-novo on the genomic scale. BMC Bioinformatics 16, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Campbell MS, Law M, Holt C, Stein JC, Moghe GD, Hufnagel DE, Lei J, Achawanantakun R, Jiao D, Lawrence CJ, et al. (2014). MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 164, 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smit AFA, Hubley R & Green P (2004). Repeat-Masker Open-4.0. http://www.repeatmasker.org.
- 65.Brown LA, Hasan S, Foster RG, and Peirson SN (2016). COMPASS: Continuous Open Mouse Phenotyping of Activity and Sleep Status. Wellcome Open Res 1, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andrews S (2010). FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 67.Wingett SW, and Andrews S (2018). FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res. 7, 1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu G, Anafi RC, Hughes ME, Kornacker K, and Hogenesch JB (2016). MetaCycle: an integrated R package to evaluate periodicity in large scale data. Bioinformatics 32, 3351–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shumate A, and Salzberg SL (2021). Liftoff: accurate mapping of gene annotations. Bioinformatics 37, 1639–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pertea G, and Pertea M (2020). GFF Utilities: GffRead and GffCompare. F1000Res. 9. 10.12688/f1000research.23297.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Katoh K, Misawa K, Kuma K-I, and Miyata T (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stamatakis A (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kern AD, and Hahn MW (2018). The Neutral Theory in Light of Natural Selection. Mol. Biol. Evol. 35, 1366–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kostka D, Hubisz MJ, Siepel A, and Pollard KS (2012). The role of GC-biased gene conversion in shaping the fastest evolving regions of the human genome. Mol. Biol. Evol. 29, 1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gao F, Chen C, Arab DA, Du Z, He Y, and Ho SYW (2019). EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 9, 3891–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) African striped mouse genome assembly metrics; (B) African striped mouse BUSCO recovery; (C) Genomes accessed in the present study; (D) Evolutionarily accelerated African striped mouse genes; (E) Summary of KEGG and GO Biological Process terms enriched among African striped mouse (Rhabdomys pumilio) accelerated genes; (F) Branch-site tests of positive selection in Arvicanthine accelerated genes; (G) Evolutionarily accelerated genes in the African grass rat (Arvicanthis niloticus); (H) Summary of KEGG and GO Biological Process terms enriched among African grass rat (Arvicanthis niloticus) accelerated genes; (I) Evolutionarily accelerated genes in the R. pumilio and A. niloticus most recent common ancestor; (J) Summary of KEGG and GO Biological Process terms enriched among accelerated genes in the R. pumilio and A. niloticus most recent common ancestor.
Data Availability Statement
RNA-Seq data have been deposited in the ArrayExpress database at EMBL-EBI under accession number E-MTAB-12024. The genome assembly, 10X Chromium Linked Reads, and Dovetail Hi-C reads have been deposited in the BioProject database: PRJNA858857. Lastly, we have created a FigShare repository (https://doi.org/10.26188/20321655) containing the following: 1) Rhabdomys_pumilio_final.gff.gz: A raw GFF-formatted gene annotation set for the Rhabdomys pumilio genome produced by Funannotate and used in transcriptomic analyses; 2) Rhabdomys_pumilio.mouse_gene_name_final.gff.gz: A copy of the above GFF-formatted gene annotation set for the Rhabdomys pumilio genome, in which gene symbols from the laboratory mouse (Mus musculus) have been assigned to their predicted R. pumilio orthologs; 3) CompGenAnno.tar.gz: A folder of GFF-formatted annotations used in comparative genomic analyses, produced by directly lifting-over gene annotations from the M. musculus genome (annotation: GCF_000001635.27_GRCm39_genomic.gff, assembly: GCF_000001635.27_GRCm39_genomic.fna) onto each of 23 other murid genome assemblies. Additionally, a manifest of each reference genome can be found in .tsv format (manifest_of_genome_assemblies_and_liftover_annotations.txt) along with a file with locus trees (locus_trees.txt) for each orthologous group of genes used in comparative genomic analyses are provided; 4) Table_of_RER_data_for_examined_species.xlsx: A large table showing relative evolutionary rates measurements for each orthologous group of gene sequences (referenced against a M. musculus transcript), for each species examined. Each species may be listed in multiple columns, reflecting different species representation for each orthologous group (i.e., representing cases in which the branch to a given leaf node originates at a different ancestral node due to sister species not represented in that alignment); and 5) Rhabdomys_pumilio_Princeton_asm1.0_preNCBI.fasta.gz: A copy of the genome assembly prior to any re-formatting that NCBI performs after upload.
This paper does not report original code.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.