

Abstract
Background and aims:
Central to the pathogenesis of nonalcoholic fatty liver disease (NAFLD) is the accumulation of lipids in the liver and various fat tissues. We aimed to elucidate the mechanisms by which lipid droplets (LDs) in the liver and adipocytes are degraded by the autophagy-lysosome system and develop therapeutic means to modulate lipophagy, i.e., autophagic degradation of LDs.
Methods:
We monitored the process in which LDs are pinched off by autophagic membranes and degraded by lysosomal hydrolases in cultured cells and mice. The autophagic receptor p62/SQSTM-1/Sequestosome-1 was identified as a key regulator and used as a target to develop drugs to induce lipophagy. The efficacy of p62 agonists was validated in mice to treat hepatosteatosis and obesity.
Results:
We found that the N-degron pathway modulates lipophagy. This autophagic degradation initiates when the molecular chaperones including BiP/GRP78, retro-translocated from the endoplasmic reticulum, is N-terminally (Nt-) arginylated by ATE1 R-transferase. The resulting Nt-arginine (Nt-Arg) binds the ZZ domain of p62 associated with LDs. Upon binding to Nt-Arg, p62 undergoes self-polymerization and recruits LC3+ phagophores to the site of lipophagy, leading to lysosomal degradation. Liver-specific Ate1 conditional knockout mice under high fat diet developed severe NAFLD. The Nt-Arg was modified into small molecule agonists to p62 that facilitate lipophagy in mice and exerted therapeutic efficacy in obesity and hepatosteatosis of wild-type but not p62 knockout mice.
Conclusions:
Our results show that the N-degron pathway modulates lipophagy and provide p62 as a drug target to treat NAFLD and other diseases related with metabolic syndrome.
Keywords: N-terminal arginylation, obesity, lipid droplet, p62/SQSTM1/Sequestosome-1, hepatosteatosis, the autophagy-lysosome system
Graphical Abstract
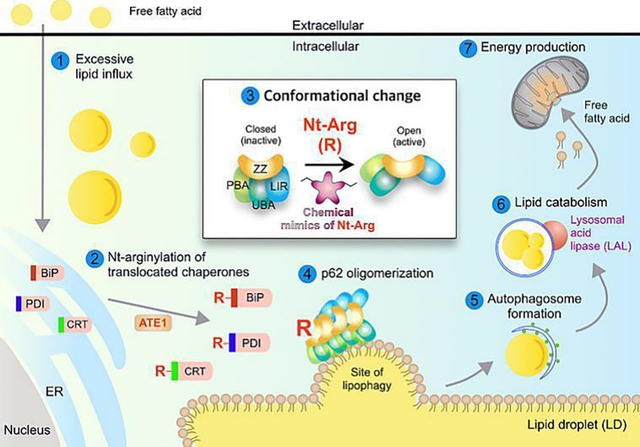
1. INTRODUCTION
NAFLD, a condition in which excess lipids are stored in the liver, is a prevalent cause of chronic liver diseases [1]. As humans in the modern society are surrounded by surplus nutrients, obesity associated with ectopic accumulation of lipids emerged as a strong risk factor for NAFLD. Lipid droplets (LDs) are dynamic organelles for lipid storage consisting of a hydrophobic core of neutral lipids [2]. Under energy deprivation, triglycerides (TGs) in LDs are enzymatically hydrolyzed to free fatty acids to generate energy through mitochondrial β-oxidation [3]. Recent studies showed that LDs, as a whole or smaller droplets, can be sequestered by autophagic membranes and degraded by lysosomal hydrolases through an autophagic process, called lipophagy [4], yet the underlying mechanism remains largely murky.
The N-degron pathway is a degradative system, in which a single N-terminal amino acid of a short-lived protein functions as an N-degron that modulates the protein’s half-life [5]. In mammals, a set of N-recognins (UBR1, UBR2, UBR4, and UBR5) recognize various N-degrons to induce ubiquitination and proteolysis via the Ub-proteasome system [5]. Our recent studies showed that p62 is an N-recognin that binds Nt-Arg and other N-degrons to promote autophagic proteolysis [6–8]. The N-degron Nt-Arg can be generated when the amino acid L-Arg is conjugated to the N-termini of ER-residing molecular chaperones such as BiP/GRP78 by ATE1-encoded R-transferases [9]. The Nt-Arg binds the ZZ domain of p62 and induces its conformational change, exposing PB1 domain, which facilitates cargo condensation through self-polymerization of p62 in complex with Nt-Arg-carrying substrates. In parallel, Nt-Arg-bound p62 interacts with LC3 on phagophores, leading to lysosomal degradation of p62 cargo complexes. This mode-of-action driven by Nt-Arg mediates the degradation of misfolded protein aggregates and the ER via p62-dependent autophagy [6, 8].Here, we show that the N-degron pathway modulates lipophagy. In this autophagic process, Nt-arginylated proteins such as R-BiP are associated with the sites of lipophagy and recruit p62. The chemical mimics of Nt-Arg induced lipophagy in mice and demonstrated their efficacy in obesity and hepatosteatosis. We suggest that the N-degron pathway plays a central role in lipophagy and provides a drug target to treat NAFLD and diseases caused by excessive lipids.
2. MATERIALS AND METHODS
2.1. Animals
Experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Seoul National University (SNU-190618-3-3). All animals received humane care and were housed in a specific-pathogen-free barrier facility at Seoul National University according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals. Mice were acclimatized for 7 days prior to the initiation of experiment. Eight-week-old male C57BL/6 mice were fed either high fat diet (HFD) with 60% kcal fat (D12492) or 45% kcal fat (D12451) purchased from Research Diets Inc. (New Brunswick, NJ). As a control, mice were also fed chow diet (D12450K, Research Diets Inc.). To investigate the preventive effects of p62-ZZ ligands for obesity and hepatosteatosis, mice, fed HFD, were also injected i.p. with 20 mg/kg YTK-1105 three times a week (t.i.w) for 10 weeks. To examine whether p62-ZZ ligands alleviate obesity and hepatosteatosis in progressed condition, mice received i.p. injection of 20 mg/kg YTK-1105 t.i.w. for 7 weeks when their body weight reached 40.0 ± 2.0 g. Alternatively, mice received i.p. injections of 20 mg/kg YTK-2205 for 11 weeks after they were fed HFD for 4 weeks.
To compare the efficacy of non-selective autophagy with p62-mediated lipophagy, mice were i.p. injected with 2 mg/kg rapamycin t.i.w. for 7 weeks once their body weights reached 40.0 ± 2.0 g. To investigate the in vivo therapeutic target of YTK-1105, p62−/− provided by Joonsung Hwang (Korea Research Institute of Bioscience and Biotechology, Ochang, Korea) were i.p. injected with 20 mg/kg YTK-1105 in comparison with +/+ mice. To measure in vivo autophagic flux, mice received 100 mg/kg Chloroquine (CQ) via i.p. injection for 5 h.
To generate liver-specific Ate1 knockout mice, Ate1-floxed mice, previously generated in our group [10], were crossed with Alb-Cre transgenic mice expressing Cre recombinase under the control of the mouse albumin enhancer/promoter (The Jackson Laboratory strain #003574). These mice and their littermate controls were fed HFD (45% kcal fat) for 16 weeks. Total 5–8 mice were randomized and treated per experimental group based on the resource equation method [11]. The number of mice for each experimental group are described in figure legends. To minimize potential confounders, control mice were age- and sex-matched and injected with vehicle. No animals were excluded during experiments.
2.2. Other methods
The information for other materials and methods is provided in Supplementary Information.
3. RESULTS
3.1. p62 is associated with LDs and required to recruit autophagic membranes to the sites of lipophagy.
Although it has been reported that cellular lipids can be selectively degraded via autophagy [12], the mechanism by which LDs are targeted by autophagic machinery remains elusive. Autophagic adaptors such as NBR1 and p62 recognize and recruit cargoes to the autophagosome through the interaction with LC3 [13]. We therefore examined whether p62 mediates the autophagic degradation of LDs. Immunostaining analyses of HepG2 cells exposed to a mixture of saturated and unsaturated fatty acids (oleic acid : palmitic acid = 2 : 1) revealed the association of p62+ and LC3+ membranes with LDs, which became more prominent when the degradation of otherwise metabolically stable LDs was stimulated by serum starvation (Fig. 1A and 1B). Furthermore, whereas lipid loading in mouse embryonic fibroblasts (MEFs) induced the localization of LC3-positive membranes to the surface of LDs, the colocalization was much weaker in p62−/− MEFs (Fig. 1C). These results suggest that p62 is responsible for autophagic lipolysis. Consistently, p62 deficient cells were highly sensitive to lipid challenge, resulting in enlarged LDs (Fig. 1D). A similar accumulation of TG was obvious in p62−/− MEFs as compared with +/+ cells (Fig.1E). In addition, siRNA-mediated p62 gene silencing in 3T3L1 adipocytes drastically increased the size of LDs as determined by Oil Red O staining (Fig. 1F) and the level of perilipin 2 (PLIN2), a protein marker for LDs (Fig. 1G). These results suggest that p62 is the major receptor that brings phagophores to the sites of LDs destined for lysosomal degradation.
Fig. 1. p62 is required for autophagic degradation of lipid droplets.
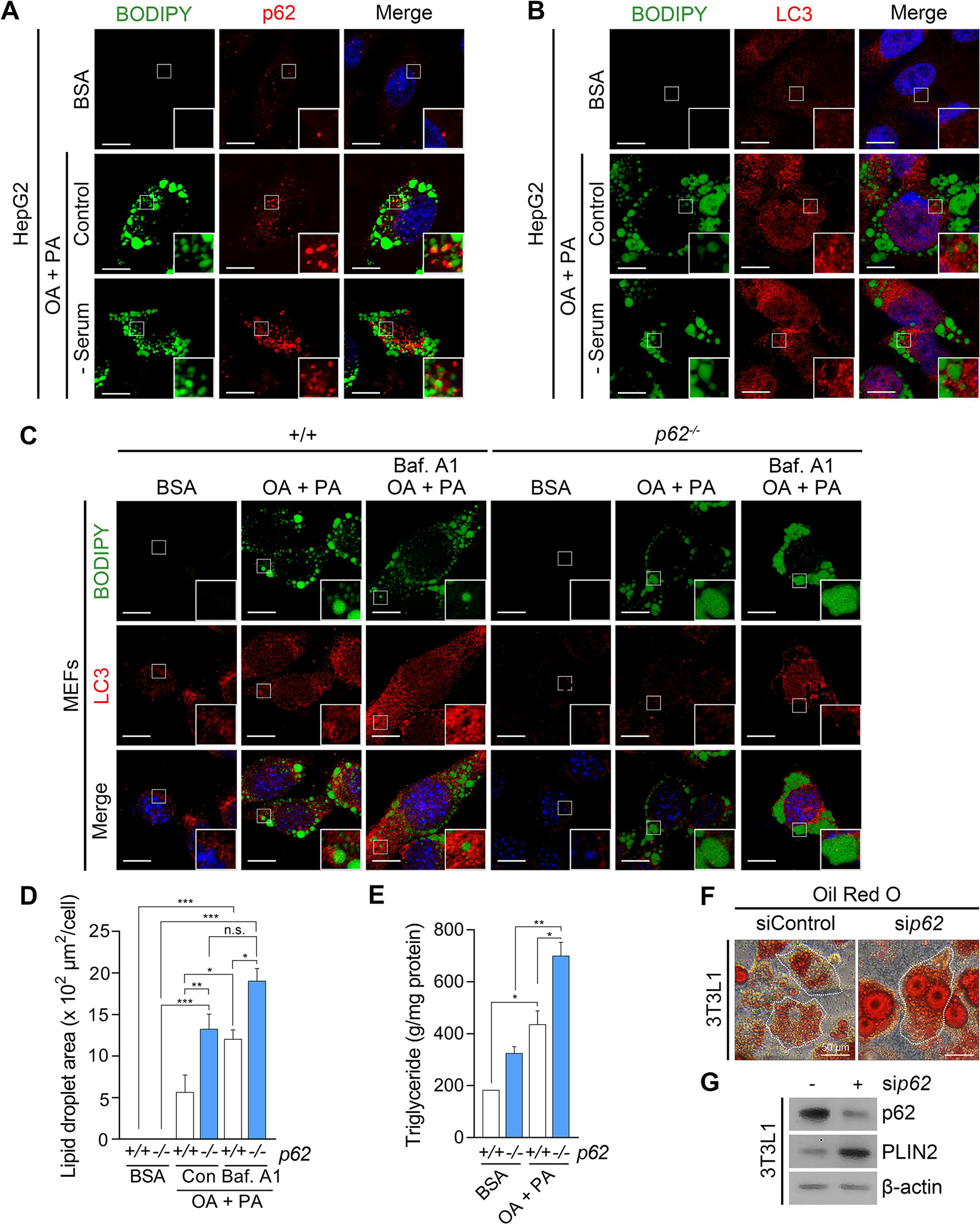
(A) Immunocytochemistry (ICC) of p62 and BODIPY staining of lipid droplets (LDs) in HepG2 cells exposed to oleic acid (OA) (1 mM, 24 h) and palmitic acid (PA) (500 μM, 24 h) with or without sequential serum starvation (24 h). Scale bar, 10 μm. (B) ICC of LC3 and BODIPY signals in HepG2 cells exposed to OA/PA (1 mM/500 μM, 24 h) with or without sequential serum starvation. Scale bar, 10 μm. (C) ICC of LC3 and BODIPY signals in WT and p62−/− MEFs exposed to OA/PA (1 mM/500 μM, 24 h), with or without sequential treatment of bafilomycin A1 (Baf. A1) (200 nM, 4 h). Scale bar, 10 μm. (D) Quantification of LD area by ImageJ (n = 50 cells). (E) Enzyme-linked immunosorbent assay (ELISA) for triglyceride (TG) quantification (n = 4). (F) Oil Red O staining of 3T3L1 cells under siRNA-mediated knockdown of p62 (80 nM, 48 h). Scale bar, 50 μm. (G) Western blotting (WB) of 3T3L1 cells following RNA interference of p62 (80 nM, 48 h). *P < 0.05, **P < 0.01, ***P < 0.001, n.s., not significant.
3.2. The activity of p62 in lipophagy is modulated by the N-degron Nt-Arg
Given our earlier finding that p62 is an N-recognin of the N-degron pathway [6], we hypothesized that the Nt-Arg of proteins may act as an activating ligand to p62 during lipophagy. To this end, fatty acid-loaded HepG2 cells were subject to immunostaining analyses of the Nt-arginylated form of known substrates such as ER-residing molecular chaperones including BiP, protein disulfide isomerase (PDI), and calreticulin (CRT). This screening showed that the Nt-arginylation was induced by fatty acid treatment especially after 6 h (Fig. 2A and 2B), which was strongly inhibited by ATE1 knockdown (Fig. 2C), suggesting that ATE1-dependent biogenesis of the Nt-Arg of proteins is stimulated by fatty acids.
Fig. 2. Nt-Arg of proteins generated by ATE1 propagates lipophagy.

(A) WB of HepG2 cells following treatment of OA/PA (1 mM/500 μM, 9 h). (B) WB of HepG2 cells following treatment of OA/PA (1 mM/500 μM) for indicated time. (C) WB of HepG2 cells under siRNA-mediated knockdown of ATE1 (80 nM, 48 h) with sequential treatment of OA (1 mM) and PA (500 μM) for indicated time. (D) HepG2 cells treated with OA/PA (1 mM/500 μM) for 9 h were fractionated. Whole cell lysates (Total) in comparison with microsome (Micro.), cytosol (Cyto.) and LD fractions were analyzed by WB. Coomassie brilliant blue (CBB) staining of the gel was used to confirm the protein loading. (E) ICC of p62, N-terminal arginylated BiP (R-BiP) and BODIPY signals in Hep3B cells exposed to OA/PA (1 mM/500 μM, 24 h). Scale bar, 10 μm. (F) ICC of p62 and BODIPY signals and (G) quantification of LD area (n = 50 cells) in WT and Ate1−/− MEFs exposed to OA (2 mM, 24 h) and PA (1 mM, 24 h), with or without sequential treatment of Baf. A1 (200 nM, 4 h). Scale bar, 10 μm. (H) TG quantification of WT and Ate1−/− MEFs exposed to conjugating vehicle, bovine serum albumin (BSA), alone or OA/PA (2 mM/1 mM) for 24 h (n = 4). (I) ICC of p62 and BODIPY signals (left panel) and TG quantification (right panel, n = 4) in MEFs treated with tannic acid (TA, 10 μM, 24 h), OA/PA (1 mM/500 μM), or both. Scale bar, 10 μm. (J) TG quantification of WT and Ate1−/− MEFs exposed to OA (2mM, 24 h) and PA (1 mM, 24 h), with or without sequential treatment of Lalistat-1 (30 μM, 24 h). *P < 0.05, **P < 0.01, ***P < 0.001, n.s., not significant.
We therefore determined whether the Nt-Arg is required for the recruitment of p62 to LDs, as well as the turnover of LDs. LD fractionation revealed the localization of R-BiP in LDs, which was accompanied by cytosolic translocation and arginylation of BiP during lipotoxic stress (Fig. 2D). When visualized using immunostaining analyses, R-BiP induced by fatty acids significantly colocalized with p62 puncta on the surface of LDs (Fig. 2E). Moreover, p62 failed to associate with LDs in Ate1−/− MEFs (Fig. 2F), resulting in the enlargement of LDs (Fig. 2G) and the increased levels of TG (Fig. 2H). A similar dysregulation in lipophagy was reproduced when ATE1 was chemically inhibited with tannic acid [14] (Fig. 2I). To confirm that the lysosomal hydrolysis of TG is activated by Nt-Arg, Lalistat-1 (30 μM), a lysosomal acid lipase inhibitor [15], was treated in WT and Ate1−/− MEFs. ATE1 deficiency abrogated TG elevation caused by lysosomal acid lipase inhibition shown in WT MEFs (Fig. 2J), confirming the induction of lysosomal TG hydrolysis by Nt-Arg. These results indicate that R-BiP and possibly other Nt-arginylated proteins associated with LDs act as an activating ligand to the N-recognin p62 to initiate lipophagy.
To further define the role of Nt-Arg in lipophagy, HepG2 cells loaded with fatty acids were transfected with X-BiP-RFP plasmids (X = Arg–Glu (permanently arginylated), Glu (native) or Val (control)) as shown in Figure 3A. In addition to R-BiP, E-BiP permissive to Nt-arginylation was able to recruit endogenous p62 to LDs (Fig. 3A). In sharp contrast, V-BiP showed no such an activity (Fig. 3A), suggesting that R-BiP binding to p62 is required for p62 to associate with LDs. Moreover, in contrast to full-length p62, ZZ-deleted p62 mutant failed to colocalize with LDs in Hep3B cells exposed to fatty acids (Fig. 3B and 3C). These results collectively show that the Nt-Arg exposed on LDs acts as an activating ligand to p62 during lipophagy (Fig. 3D).
Fig. 3. The N-terminal arginylated BiP triggers lipophagy via binding to p62-ZZ domain.
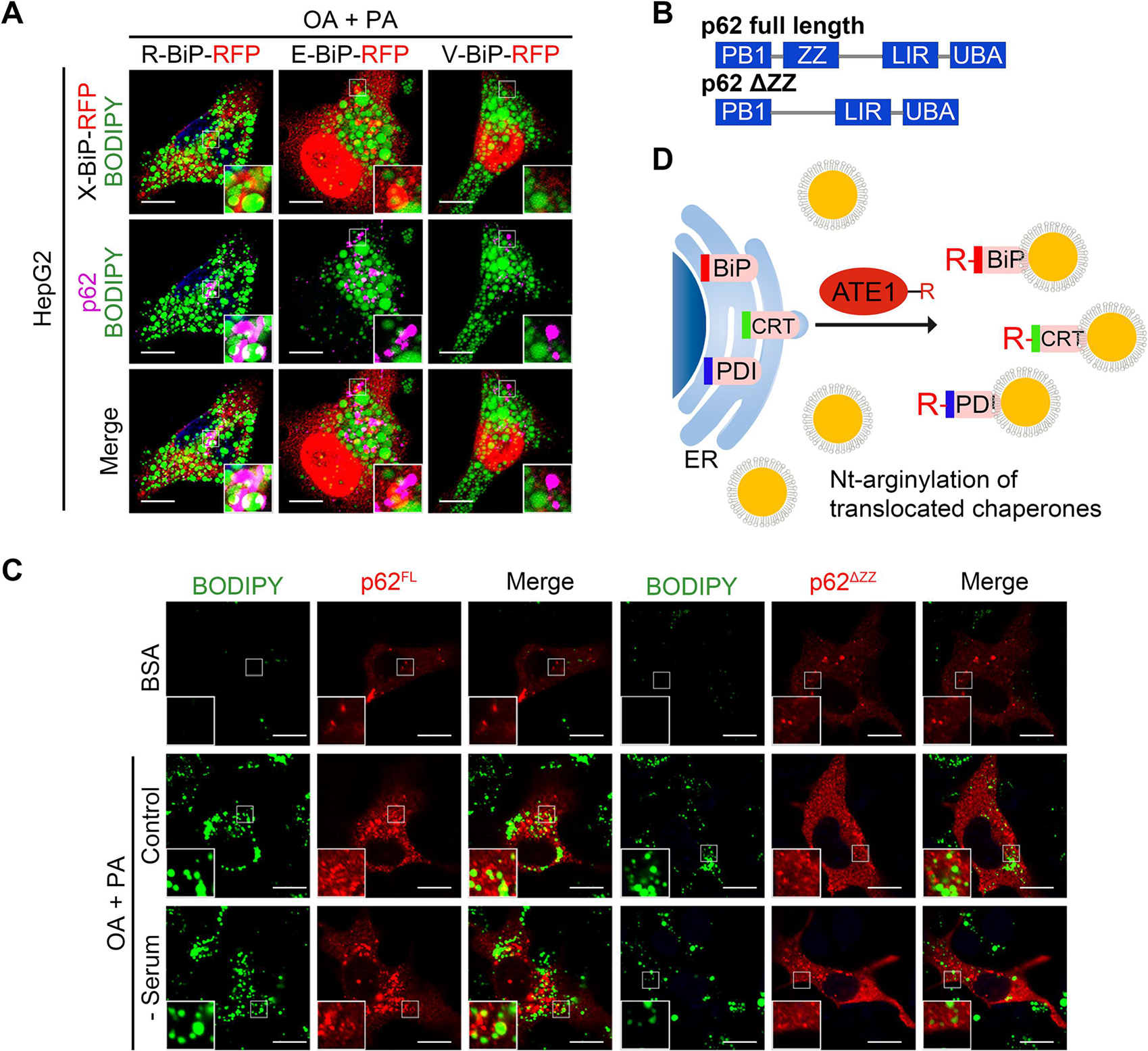
(A) Fluorescence of R-BiP-RFP, E-BiP-RFP, V-BIP-RFP, p62 and BODIPY in HepG2 cells upon OA/PA (1 mM/500 μM) loading for 24 h. (B) Schematic of recombinant p62 constructs used. (C) ICC of p62 and BODIPY signals in Hep3B cells expressing p62-full length (p62FL, left panel) or p62-ZZ mutants (p62ΔZZ, right panel), with sequential treatment of OA/PA (1 mM/500 μM, 24 h) and serum starvation (24 h). Scale bar, 10 μm. (D) Scheme of the formation of Nt-arginylated proteins during lipotoxic stress.
3.3. Liver-specific Ate1 knockout mice develop severe NAFLD upon HFD feeding
Excessive accumulation of LDs is a risk factor for NAFLD [16]. To validate the physiological importance of Nt-arginylation by ATE1 in LD degradation, we generated liver-specific Ate1−/− mice by crossing Ate1flox/flox mice with mice expressing Cre recombinase under the albumin enhancer/promoter (Fig. 4A). The conditional ablation of Ate1 was confirmed by immunoblotting and immunostaining tissue sections (Fig. 4B and 4C). To induce NAFLD, 8 week-old Ate1flox/flox;Alb-Cre+ mice were fed 45% HFD for 16 weeks in comparison with Ate1flox/flox mice. Immunostaining analyses of liver sections revealed that HFD feeding facilitated the formation of R-BiP, R-CRT, R-PDI, and p62 puncta localized on the surface of LDs (Fig. 4D and 4E). In sharp contrast, such an induction of R-BiP was markedly reduced upon Ate1 deletion (Fig. 4F). Moreover, histological analyses of ATE1-deficient livers exhibited the excessive accumulation of LDs in hepatocytes, the hallmark of steatosis, which correlated with the increased hepatic TG levels (Fig. 4G). Finally, not only the infiltration of F4/80+ hepatic macrophages, but also the inflammatory cytokines were elevated in ATE1-deficient livers (Fig. 4G). These results demonstrate that Nt-arginylation plays a central role in lipophagy in the murine liver, and thus possibly counteracts the progression of steatosis into steatohepatitis.
Fig. 4. Liver-specific Ate1 ablation aggravates non-alcoholic hepatosteatosis and its progression to steatohepatitis.
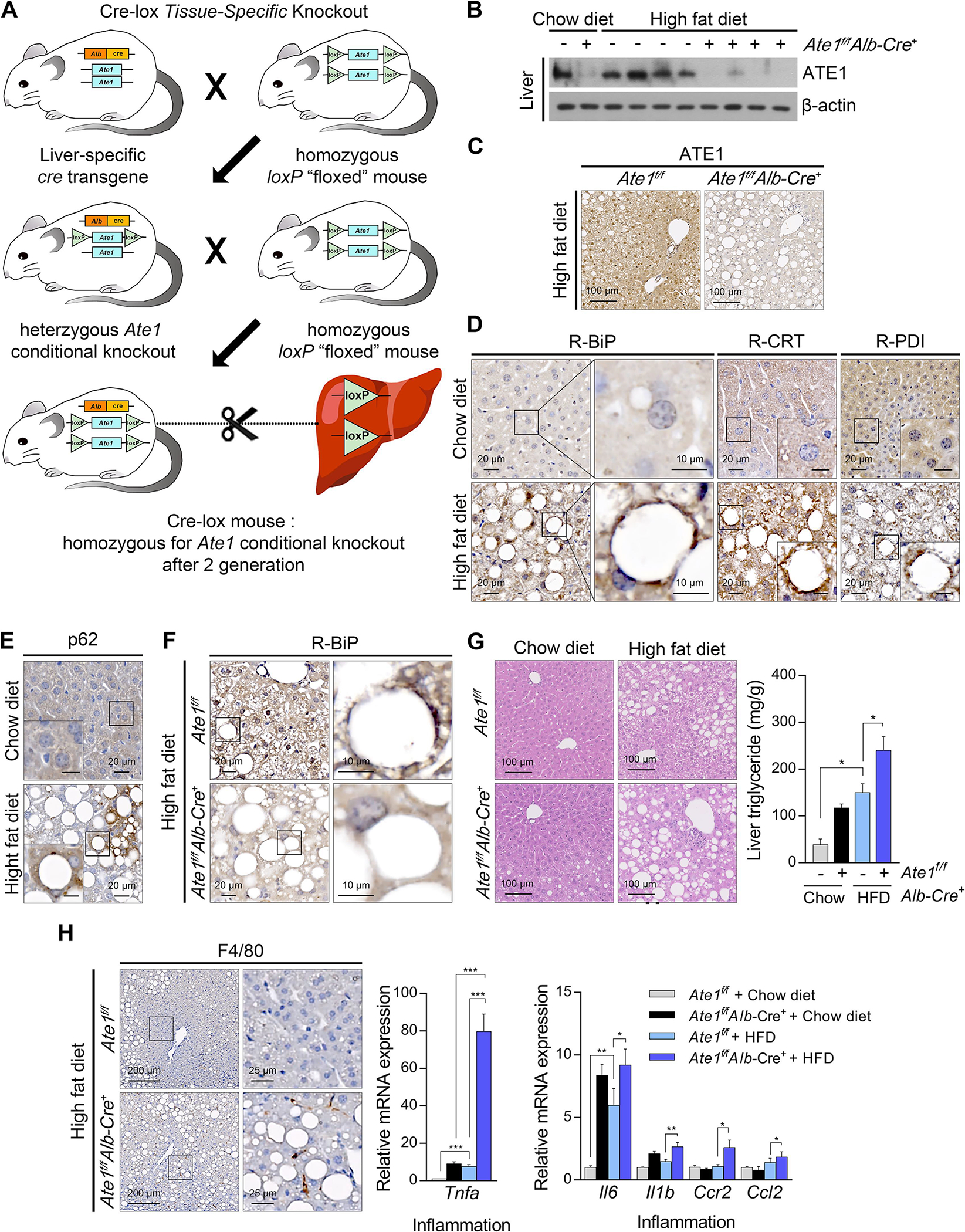
(A) Scheme of generating liver-specific Ate1 knockout mouse. Liver-specific Ate1 knockout mice and their littermate controls were fed with HFD (45% kcal fat) for 16 weeks. (B) WB and (C) IHC staining of hepatic ATE1 in liver-specific Ate1 knockout mice. Scale bars, 100 μm. IHC staining of hepatic (D) R-BiP, R-CRT, R-PDI, and (E) p62 upon HFD feeding. Scale bars, 20 μm or 10 μm (right column or inside box). (F) IHC staining of hepatic R-BiP in liver-specific Ate1 knockout mice. Scale bars, 20 μm (left column), 10 μm (right column). (G) Hematoxylin & eosin (H & E) staining (left panel) and TG quantification (right panel, n = 5) of liver-specific Ate1 knockout liver and their littermate control liver. Scale bars, 100 μm. (H) IHC staining of F4/80 (left panel) and mRNA levels of Tnfa (middle panel), Il6, Il1b, Ccr2, and Ccl2 (right panel) in liver-specific Ate1 knockout mice and their littermate controls. Scale bars, 200 μm (left column), 25 μm (right column). *P < 0.05, **P < 0.01, ***P < 0.001.
3.4. Chemical mimics of the Nt-Arg promotes lipophagy by activating p62-dependent autophagy
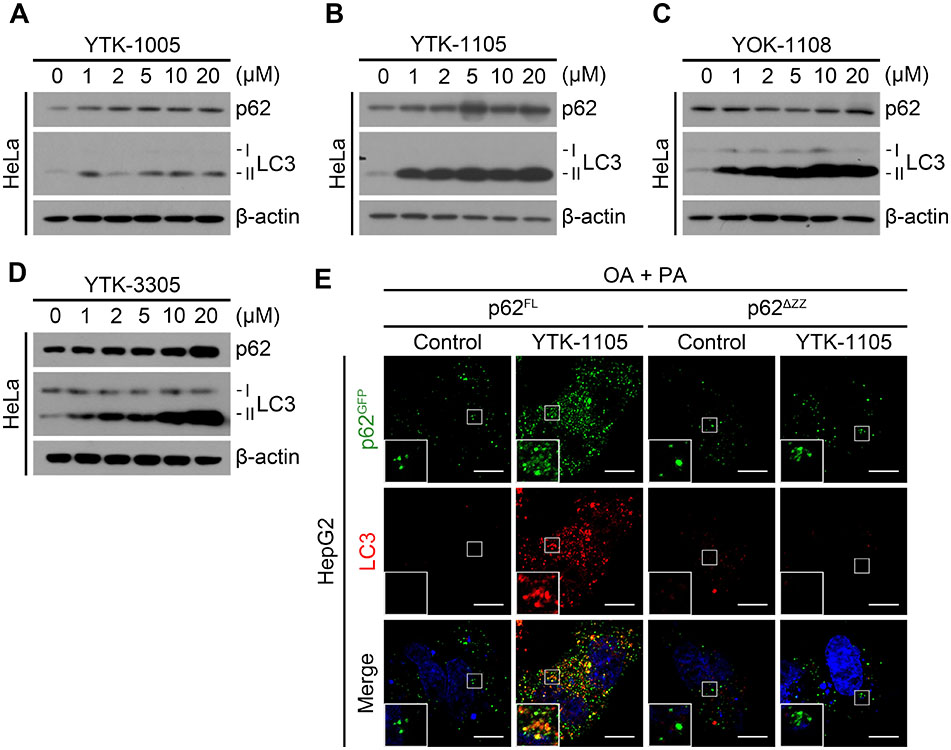
We have previously developed the chemical mimics of Nt-Arg that bind to the ZZ domain of p62 [17]. These chemical ligands to p62-ZZ domain were demonstrated to induce self-polymerization of p62 and the biogenesis of autophagosomes. Immunoblotting analyses of HeLa cells confirmed that YTK-1005, YTK-1105, YOK-1108, and YTK-3305 (Fig. 5A) induced the lipidation of LC3-I into LC3-II, an active form that can be anchored to autophagic membranes (Fig. S1A–D). When HepG2 cells were exposed to fatty acids, these compounds efficiently promoted the degradation of LDs as measured by Oil Red O staining (Fig. 5B). The efficacy to induce autophagic degradation of LDs was demonstrated using autophagic flux assays, in which otherwise short-lived LDs were rapidly stabilized in response to bafilomycin A1 (Fig. 5C). Co-immunostaining analyses showed that YTK-1105 enhanced the levels of p62+ and LC3+ puncta on the surface of LDs in a manner synergistic with autophagic inhibition by bafilomycin A1 (Fig. 5D and 5E). Furthermore, TG levels were significantly reduced by YTK-1105, which was reversed by either hydroxychloroquine or Lalistat-1 treatment, confirming the lysosomal degradation of LDs by YTK-2205 (Fig. 5F and 5G). These results demonstrate that p62 agonists induce the autophagic degradation of LDs.
Fig. 5. Chemical mimicry of the N-degron Arg modulates lipophagy.
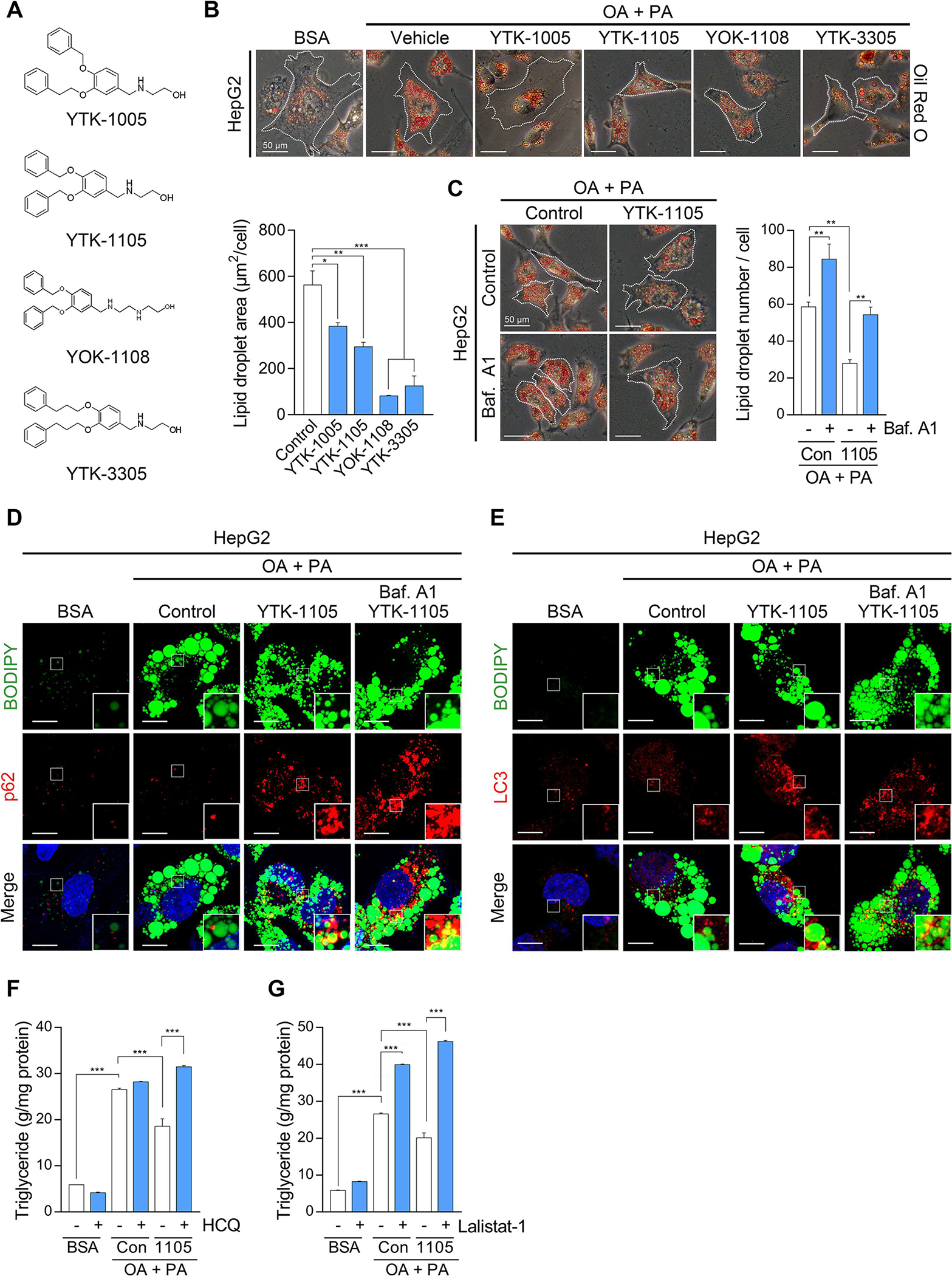
(A) Chemical structures of p62-ZZ ligands. (B) Oil Red O staining of fatty acid-loaded HepG2 cells treated with p62-ZZ ligands (5 μM, 24 h, upper panel) and quantification of LD area by ImageJ (n = 50 cells, lower panel). Scale bar, 50 μm. (C) Oil Red O staining of HepG2 cells treated with YTK-1105 (5 μM, 24 h), Baf. A1 (200 nM, 4 h), or both (left panel), and quantification of LD area (right panel, n = 50 cells). Scale bar, 50 μm. (D) ICC of p62 and BODIPY signals in HepG2 cells treated with YTK-1105 (5 μM, 24 h), Baf. A1 (200 nM, 4 h), or both. Scale bar, 10 μm. (E) ICC of LC3 and BODIPY signals in HepG2 cells treated with YTK-1105 (5 μM, 24 h), Baf. A1 (200 nM, 4 h), or both. Scale bar, 10 μm. TG quantification of HepG2 cells exposed to OA (600 μM, 24 h) and PA (300 μM, 24 h), with or without treatment of (F) hydroxychloroquine (HCQ, 25 μM, 18 h) or (G) Lalistat-1 (80 μM, 24 h). *P < 0.05, **P < 0.01, ***P < 0.001.
Next, we determined whether p62 agonists stimulate lipophagy via p62-ZZ domain by transiently expressing p62 in p62 knockdown HepG2 cells in comparison with ZZ-deleted p62 mutant. In the cells expressing wild-type p62, p62+ and LC3+ puncta were readily induced by YTK-1105 (Fig. S1E). In sharp contrast, YTK-1105 failed to induce either the formation of p62+ and LC3+ puncta or their colocalization in p62 knockdown cells expressing ZZ-deleted p62 mutant (Fig. S1E). These results demonstrate that p62 agonists target LDs to the lysosome by binding the ZZ domain of p62.
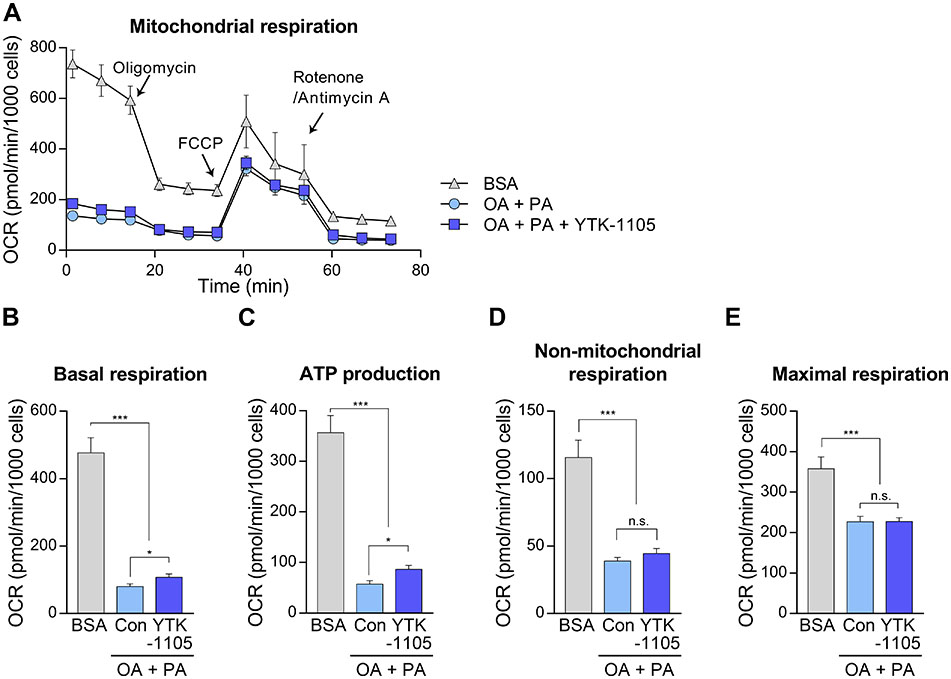
To explore the consequence of increased lipophagy by YTK-1105 on metabolic function of HepG2 cells, we then measured mitochondrial energy consumption using a Seahorse XFe96 analyzer (Agilent, Santa Clara, CA, USA) (Fig. S2A). The oxygen consumption rate was significantly reduced by fatty acid loading, implicating the mitochondrial dysfunction caused by lipotoxic stress (Fig. S2A–E). YTK-1105 treatment slightly increased both basal oxygen consumption rate (Fig. S2B) and mitochondrial ATP production (Fig. S2C), while non-mitochondrial (Fig. S2D) or maximal respiration (Fig. S2E) was not altered. These results indicate that YTK-1105-induced lipophagy may supply free fatty acids to fuel oxidative phosphorylation and energy production in mitochondria.
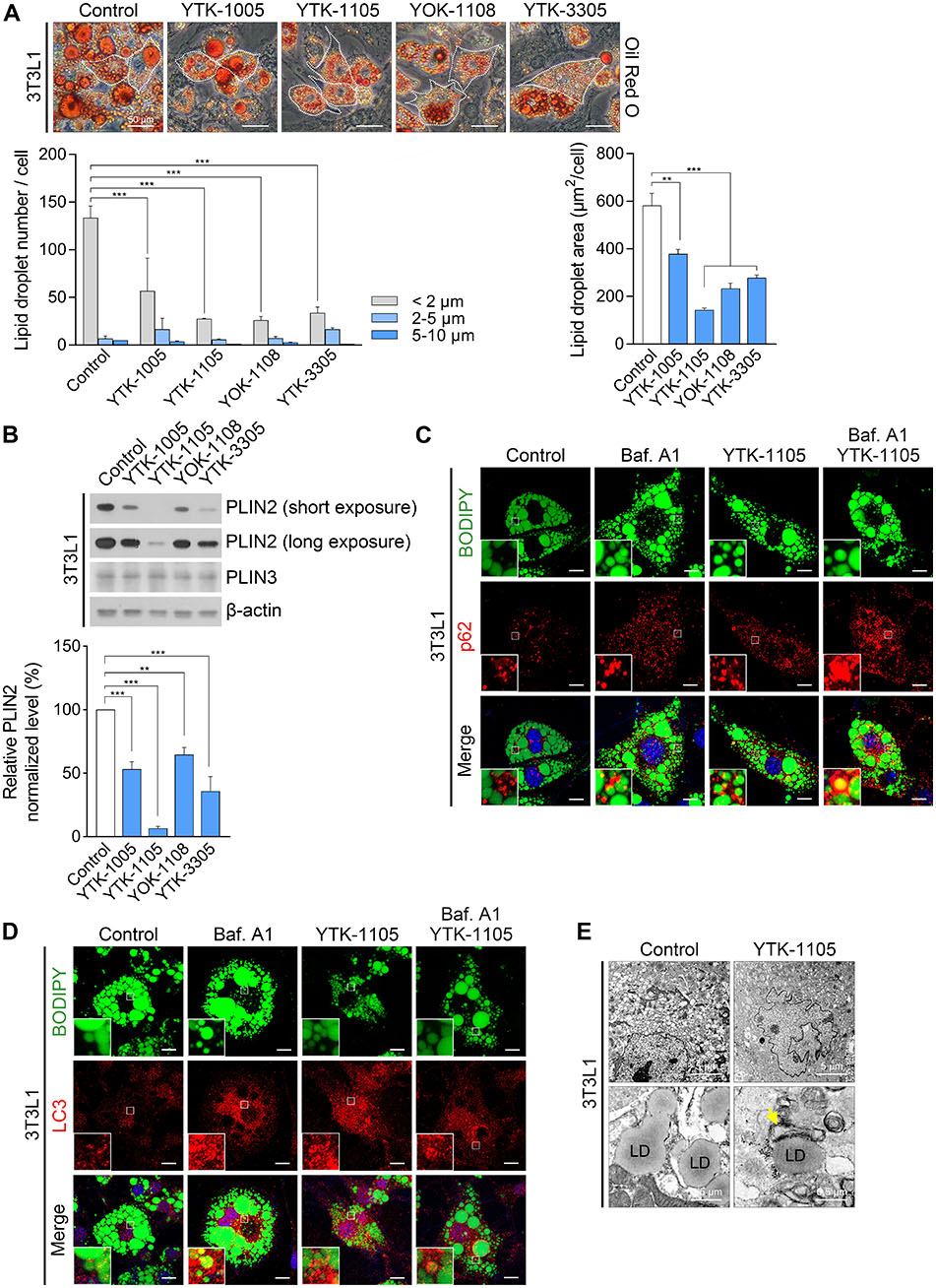
We also characterized the efficacy of p62 agonists to induce lipophagy in adipocytes. Indeed, p62 agonists demonstrated a strong activity to reduce the number and size of LDs in 3T3L1 cells (Fig. S3A), which correlated to the decreased level of PLIN2, an adipose differentiation-related protein localized in LDs (Fig. S3B). Immunostaining analyses of the cells treated with YTK-1105 visualized the marking and enrichment of p62+ and LC3+ membranes on LDs (Fig. S3C and S3D). Autophagic flux assays showed that p62 and LC3 were co-degraded with LDs by the lysosome (Fig. S3C and S3D). Transmission electron microscopy confirmed that LD fragments were targeted to autophagosomes, forming autolysosomes (Fig. S3E). We therefore determined whether the observed efficacy was dependent on p62. RNA interference assays of 3T3L1 cells treated with YTK-1105 confirmed that p62 knockdown strongly inhibited the degradation of LDs (Fig. S4A) as well as PLIN2 (Fig. S4B). Consistently, YTK-1105 failed to induce the lipidation of LC3 in p62−/− MEFs (Fig. S4C). These results demonstrate that p62 agonists have efficacy to induce lipophagy in adipocytes via p62-dependenet selective autophagy.
3.5. The p62 agonist YTK-2205 prevents obesity in HFD-fed mice without muscle loss
It is known that excessive dietary fat impairs autophagic flux, which in turn contributes to lipid accumulation in the adipose tissues and liver, leading to a vicious cycle in obesity and NAFLD [18]. To develop p62 agonists that enable the targeted degradation of fats in vivo with satisfactory physicochemical properties, we employed 3D structure modelling of p62 ZZ domain associated with structure-activity relationship approaches. Approximately 100 derivatives targeting p62 ZZ domain were synthesized based on two different scaffolds, YTK-1105 and YOK-1104. Screening of these derivatives identified YTK-2205 with the efficacy to activate p62 in lipophagy (Fig. 6A). In the docking simulation study, YTK-2205 located in the position where the first and second residues of the N-degron bind (Fig. 6B and 6C). Hydrogen bond between the amine moiety of YTK-2205 and the side chain of Asp147 plays an important role in the binding and activity of YTK-2205. In addition, the hydroxyl groups are hydrogen bonded to the side chain of Asp149. A T-shaped π-π interaction was observed between the phenyl ring and Phe168 (Fig. 6D).
Fig. 6. A novel p62-ZZ ligand, YTK-2205 administration reduces fat pad size without loss of muscle.
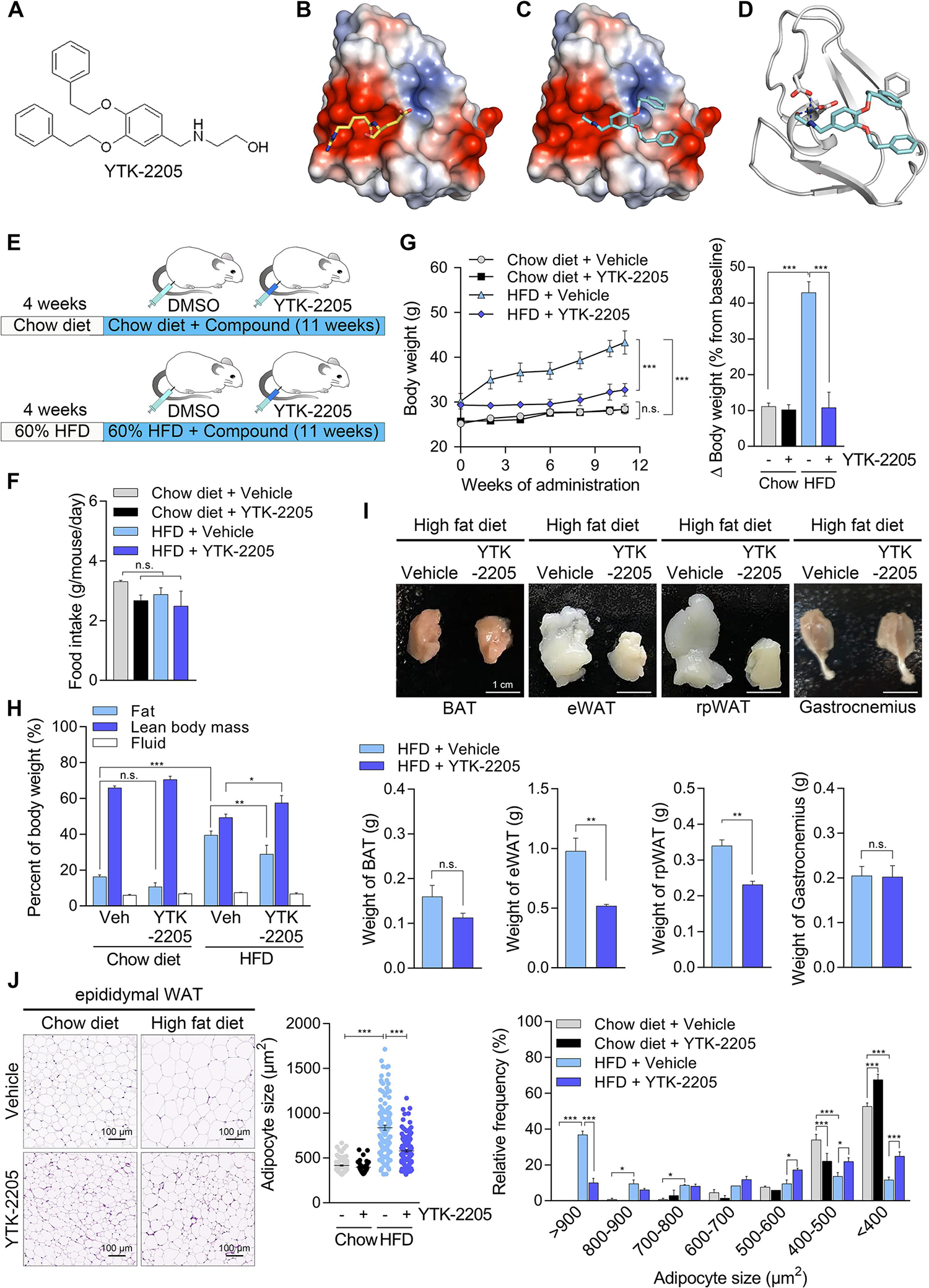
(A) Chemical structure of YTK-2205. (B and C) Docking analysis of YTK-2205 with p62. (D) 3D binding modes and interaction of YTK-2205 with p62. (E) Scheme of YTK-2205 injection in high fat diet (HFD)-induced overweight murine model. (F) Food intake during compound administration (n = 8). (G) Body weight changes during compound administration (left panel, n = 8). Percent body weight changes compared with baseline at the time of sacrifice (right panel, n = 8). (H) Body composition measured by TD-NMR after 11 weeks of compound administration (n = 8). (I) Gross morphology (upper panel) and organ weights (lower panel) of fat pads and gastrocnemius muscle after 11 weeks of compound administration (n = 8). BAT, brown adipose tissue; eWAT, epididymal white adipose tissue; rpWAT, retroperitoneal white adipose tissue. (J) H & E stained eWAT (left panel) and quantification of adipocyte size (middle and right panels, n = 50) treated with YTK-2205 or vehicle. Scale bar, 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001, n.s., not significant.
To evaluate the in vivo efficacy of YTK-2205 in lipophagy, 8 weeks old C57/BL6 male mice were fed HFD (60% kcal fat) for 4 weeks and injected i.p. with YTK-2205 (20 mg/kg) three times a week for another 11 weeks (Fig. 6E). Whereas there were no significant differences in food intake (Fig. 6F), the YTK-2205 group gained body weights (10.8 ± 1.3%) dramatically less than vehicle-treated mice (43.0 ± 3.0%, P < 0.001) during 11 weeks (Fig. 6G). Notably, HFD-fed mice treated with YTK-2205 gained body weights as little as control mice (11.1 ± 1.0%) fed a chow diet (10 kcal% fat), indicating an almost complete anti-obesity efficacy while keeping 60% fat diet (Fig. 6G). Moreover, when the body composition of HFD-fed mice was examined (Fig. 6H), the weight reduction driven by YTK-2205 was mostly attributed to reduced fat contents (28.9 ± 5.0% vs. 39.5 ± 2.4%, P < 0.01). We therefore assessed the relative weights of various fat tissues. Whereas no significant differences were observed with brown adipose tissue, YTK-2205 preferentially induced LD degradation in WAT, as evidenced by weight reductions in epididymal WAT (eWAT, 47.0 ± 1.4%) and retroperitoneal WAT (32.0 ± 2.8%) (Fig. 6I). As expected, histological examination revealed a marked hypertrophy of LDs in eWAT from control mice (Fig. 6J). By sharp contrast, LDs exposed to YTK-2205 remained their normal sizes and morphology (Fig. 6J). These data demonstrate that YTK-2205 induces the autophagic degradation of fats preferentially in WAT amongst various fat tissues.
Despite extensive studies on the methodology to reduce body weights, the efficacy of anti-obesity drugs currently available in the market suffer from lean mass loss as a side effect [19, 20], necessitating the development of therapeutic means to degrade LDs without muscle loss. Importantly, when the percentage of fat and lean mass were measured, mice treated with YTK-2205 contained lean mass 8.3% higher (57.6 ± 4.0% vs.49.3 ± 1.9%) as compared with control mice (Fig. 6H). Similarly, the weight of gastrocnemius muscle was not altered by YTK-2205 treatment (Fig. 6I). These data indicate that YTK-2205 induces lipid degradation without loss of lean body mass mainly caused by muscle loss.
3.6. P62 agonists prevent the development of NAFLD in HFD-fed mice
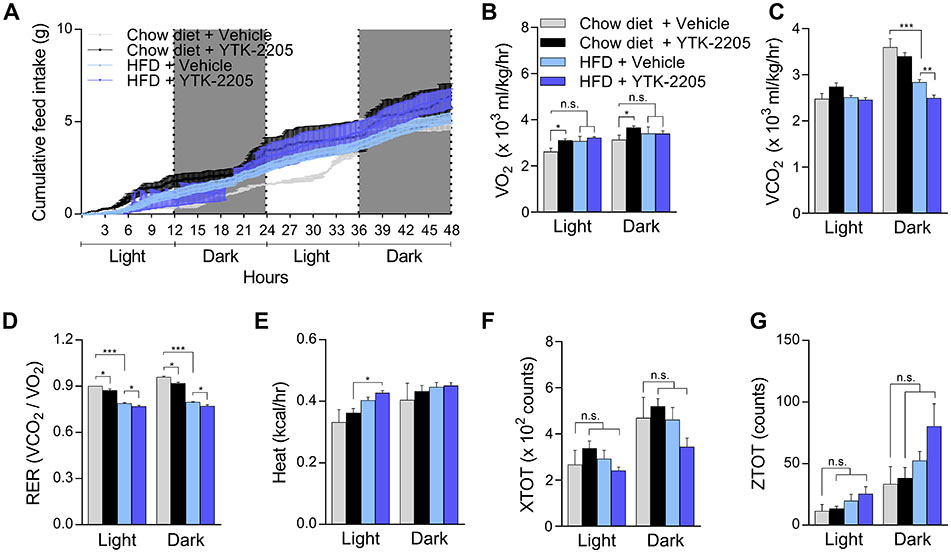
Given that YTK-2205 induced lipophagy in the adipocytes of HFD-fed mice (Fig. 6H–J), we examined whether YTK-2205 also exhibits the therapeutic efficacy in NAFLD. The livers from these HFD-fed mice were subject to histological and immunostaining analyses, which exhibited significant improvements in hepatosteatosis associated with a reduced level of TG upon YTK-2205 administration (Fig. 7A). We therefore determined whether their efficacy in NAFLD is attributed to the activity of the ATE1-p62 circuit in lipophagy. Consistent with R-BiP production stimulated by fatty acid influx (Fig. 2A–D), HFD feeding induced R-BiP biogenesis in the liver as assessed by immunoblotting (Fig. 7B). Immunohistochemical analyses of the livers revealed that hepatocytes exposed to YTK-2205 contained higher levels of both p62+ and LC3+ autophagic membranes, which in turn surrounded or in a juxtaposition with LDs (Fig. 7C and 7D). In vivo LC3 turnover assay with chloroquine also showed increased autophagic flux by YTK-2205, indicating in vivo induction of autophagy (Fig. 7E). Furthermore, among parameters of metabolic cage experiments (Fig.S5A–G), the respiratory exchange ratio was decreased by YTK-2205 injection (Fig. S5D), suggesting a higher lipid oxidation. These results suggest that the degradation of LDs in hepatocytes (and adipocytes as well) is responsible for the efficacy of p62 agonists in NAFLD.
Fig. 7. Intraperitoneal injection of YTK-2205 mitigates NAFLD in high fat diet fed mice.
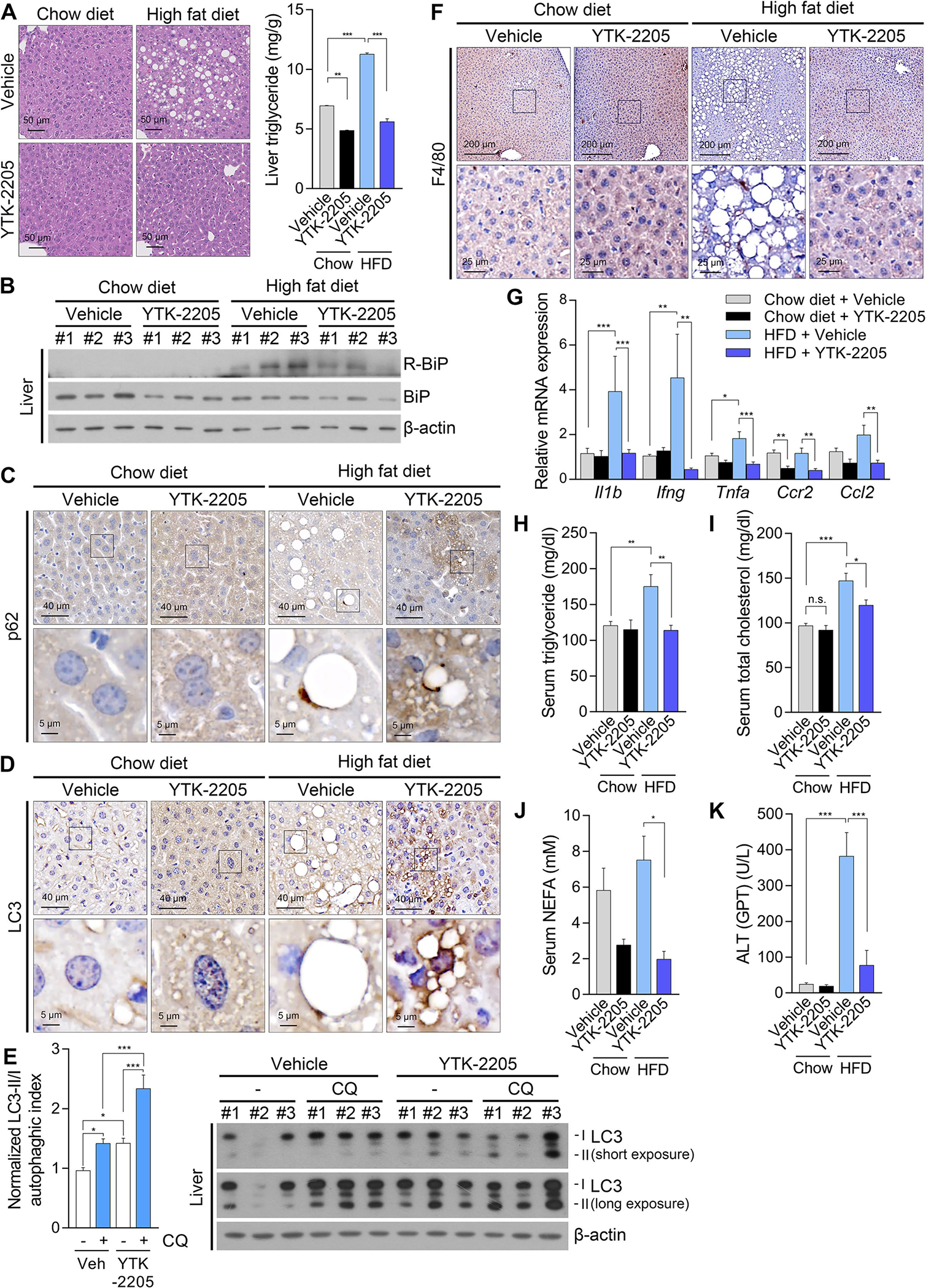
(A) H & E staining (left panel) and TG levels (right panel, n = 8) of liver treated with YTK-2205 or vehicle. Scale bars, 50 μm. (B) WB of liver samples treated with YTK-2205 or vehicle. IHC staining of (C) p62 and (D) LC3 in liver treated with YTK-2205 or vehicle. Scale bars, 40 μm (upper row), 5 μm (lower row). (E) WB of liver samples treated with YTK-2205 (20 mg/kg) or vehicle, with or without chloroquine (100 mg/kg, 5 h). (F) IHC staining of F4/80 in liver treated with YTK-2205 or vehicle. Scale bars, 200 μm (upper row), 25 μm (lower row). (G) Relative mRNA levels of inflammatory cytokines in liver treated with YTK-2205 or vehicle (n = 8). Serum levels of (H) triglyceride, (I) total cholesterol, (J) non-esterified fatty acids (NEFA), and (K) alanine aminotransferase in mice treated with YTK-2205 or vehicle (n = 8). *P < 0.05, **P < 0.01, ***P < 0.001, n.s., not significant.
In addition, immunohistochemical analyses revealed that YTK-2205 inhibited hepatic inflammation, as evidenced by reduced infiltration of macrophages (Fig. 7F) and inflammatory gene expression (Fig. 7G). These results suggest that YTK-2205 has potential to prevent steatohepatitis in HFD-fed mice. In addition, serum levels of TG, total cholesterol, non-esterified fatty acids, and alanine aminotransferase were drastically decreased by YTK-2205 (Fig. 7H–K), which complies with anti-obesity and anti-fatty liver effects.
3.7. P62 is a potential drug target for metabolic syndrome caused by lipid accumulation in adipose tissues and livers.
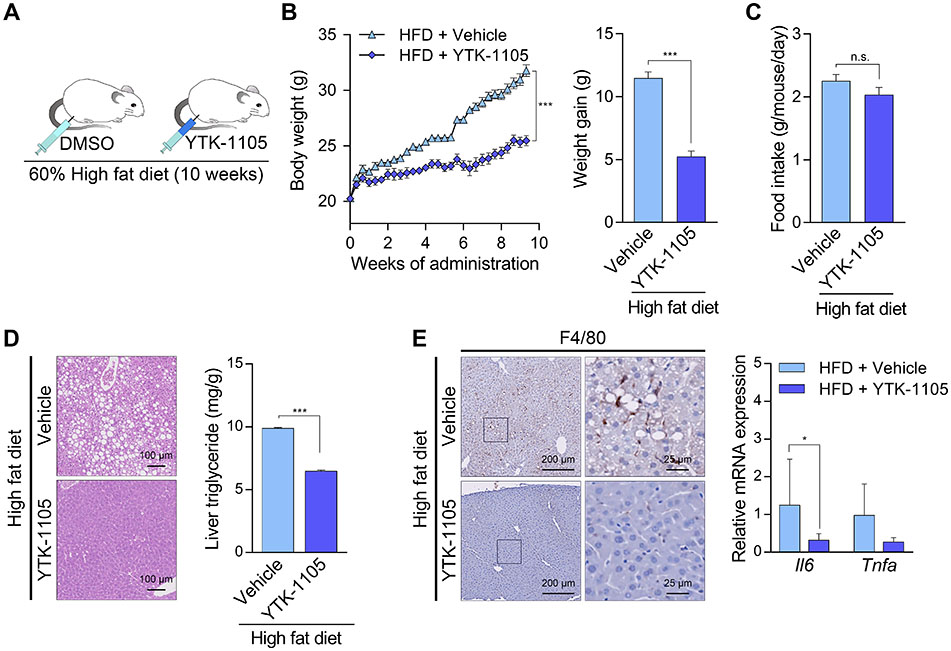
To generalize the therapeutic efficacy of YTK-2205, we further validated p62 as a drug target for lipophagy, NAFLD by using another p62 agonist, YTK-1105 [6, 8]. Male C57/BL6 mice fed HFD (60% kcal fat) were injected i.p. with YTK-1105 (20 mg/kg) three times a week for 10 weeks (Fig. S6A). The weight gain of YTK-1105-treated mice were 54.6 ± 3.8% lower than those of control mice, without a significant difference in food intake (Fig. S6B and S6C). These results suggest that YTK-1105 induces lipid catabolic process without changes of appetite. Similar with YTK-2205, YTK-1105 exhibited the efficacy in hepatosteatosis and inflammation (Fig. S6D and S6E).
To confirm the in vivo mechanism that YTK-1105 exerts therapeutic efficacy via p62, YTK-1105 was administered in p62−/− mice. Male WT and p62−/− mice were fed HFD up to 40.8 ± 0.2 g and injected i.p. with YTK-1105 three times a week for 7 weeks while maintaining HFD feeding (Fig. 8A). Whereas control HFD-fed WT mice gained weights by 6.9 ± 0.3 g (17.1 ± 0.8%) over seven weeks, weight gaining was nearly absent in YTK-1105-treated WT mice, resulting in a difference of 0.02 ± 0.7 g (0.1 ± 1.8%) (Fig. 8B). In sharp contrast, the anti-obesity efficacy of YTK-1105 was not observed in p62−/− mice (Fig. 8B). Similarly, liver weight, hepatic fat accumulation and TG levels were significantly reduced in WT mice, but not in p62−/− mice, by YTK-1105 injection (Fig. 8C and 8D). Immunoblotting and immunostaining analyses also showed that p62 is required for YTK-1105 to induce autophagy in vivo (Fig. 8E) and LC3 recruitment to LDs (Fig. 8F). These data validate that p62 agonists could be developed into drugs to treat metabolic syndrome caused by lipid accumulation.
Fig. 8. YTK-2205 exerts therapeutic efficacy in obesity and hepatosteatosis via p62.
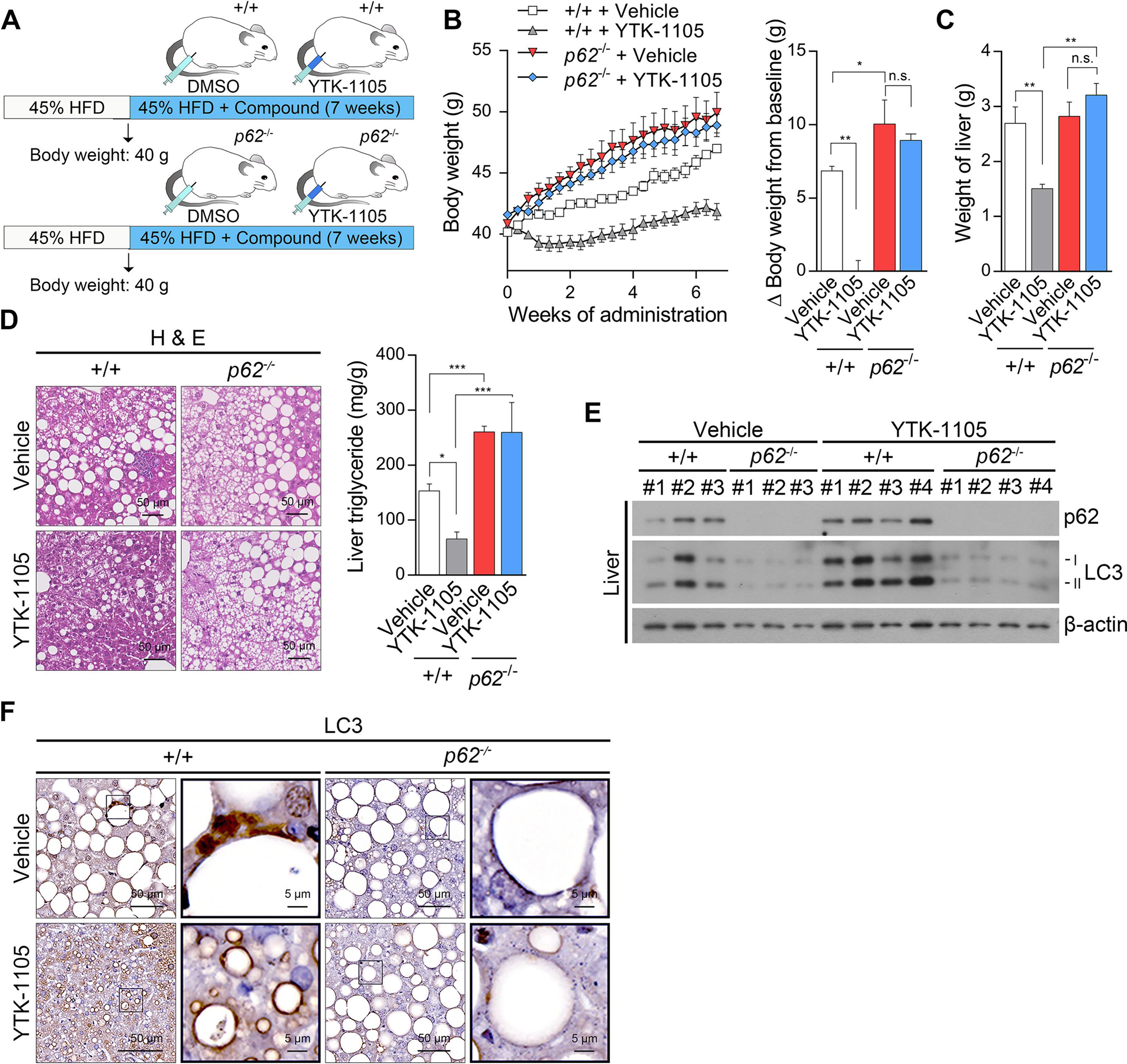
(A) Injection timeline and details of YTK-1105 in HFD-induced obese murine model of p62−/− and +/+ mice. (B) Body weight changes during compound administration in p62−/− and +/+ mice. (left panel, n = 5). Percent body weight changes compared with baseline at the time of sacrifice (right panel, n = 5). (C) Weight of compound treated livers in p62−/− and +/+ mice (n = 5). (D) H & E staining (left panel) and TG levels (right panel, n = 5) of p62−/− and +/+ liver treated with YTK-2205 or vehicle. Scale bars, 50 μm. (E) WB of p62−/− and +/+ liver samples treated with YTK-2205 or vehicle. (F) IHC staining of LC3 in p62−/− and +/+ liver treated with YTK-2205 or vehicle. Scale bars, 50 μm (left column), 5 μm (right column).
3.8. The N-degron pathway modulates lipophagy in vivo independent of mTOR-mediated macroautophagy
The mammalian target of rapamycin (mTOR) plays a key role in cell growth and metabolic pathways such as macroautophagy [21]. Rapamycin, an inhibitor of mTOR, is a potent inducer of macroautophagy and, therefore, has been explored as a therapeutic agent to booster autophagy in pathophysiological conditions [22]. Given our results with the ATE1-p62 circuit, we compared the efficacy of p62 agonists and rapamycin to cure lipid-related diseases such as obesity and hepatosteatosis back to normal conditions under HFD feeding. Male C57/BL6 mice were fed HFD up to 40.0 ± 2.0 g and injected i.p. with rapamycin or YTK-1105 three times a week for 7 weeks while maintaining HFD feeding (Fig. 9A). Whereas control HFD-fed mice gained weights by 3.6 ± 0.7% over one week, a reduction of 7.0 ± 2.1% was observed with YTK-1105, resulting in a difference of 10.6% (Fig. 9B). The anti-obesity efficacy was maintained over 7 weeks while keeping 45% HFD diet (Fig. 9C), without significant differences in food intake (Fig.9D). Consistently, whereas the livers of HFD-fed mice were enlarged owing to LD accumulation, these fatty livers were almost completely cured by YTK-1105, as determined by their normal sizes, histological parameters, and hepatic TG levels (Fig. 9E and 9F). LDs targeted by autophagosomes were also visualized in YTK-1105-treated liver using transmission electron microscopy (Fig. 9G). The efficacy to reduce body weights and to induce LD degradation in the liver was observed only with YTK-1105 but not rapamycin (Fig. 9C and 9F). YTK-1105 but not rapamycin was able to cure hepatosteatosis as determined by gross morphology (Fig. 9E), the number and sizes of LDs in hepatocytes, and the hepatic levels of TG (Fig. 9F). Immunostaining analyses of the liver sections showed that rapamycin exhibited only a marginal activity to induce the association of LC3+ positive membranes with LDs (Fig. 9H) and recruit p62 on the LDs (Fig. 9I), as compared with YTK-1105. A similar contrasting result was observed in hepatic inflammation as evidenced by the reduction in macrophage infiltration (Fig. 9J) and the monocyte/macrophage chemokine receptor C-C chemokine receptor type 2 [23] in the liver (Fig. 9K). These data demonstrate that p62 mediates lipophagy independent of mTOR-regulated core autophagic pathways.
Fig. 9. YTK-1105, a p62-ZZ ligand, displays a stronger therapeutic efficacy in NAFLD and obesity compared with rapamycin, a bulk autophagy inducer, in diet-induced obese mice.
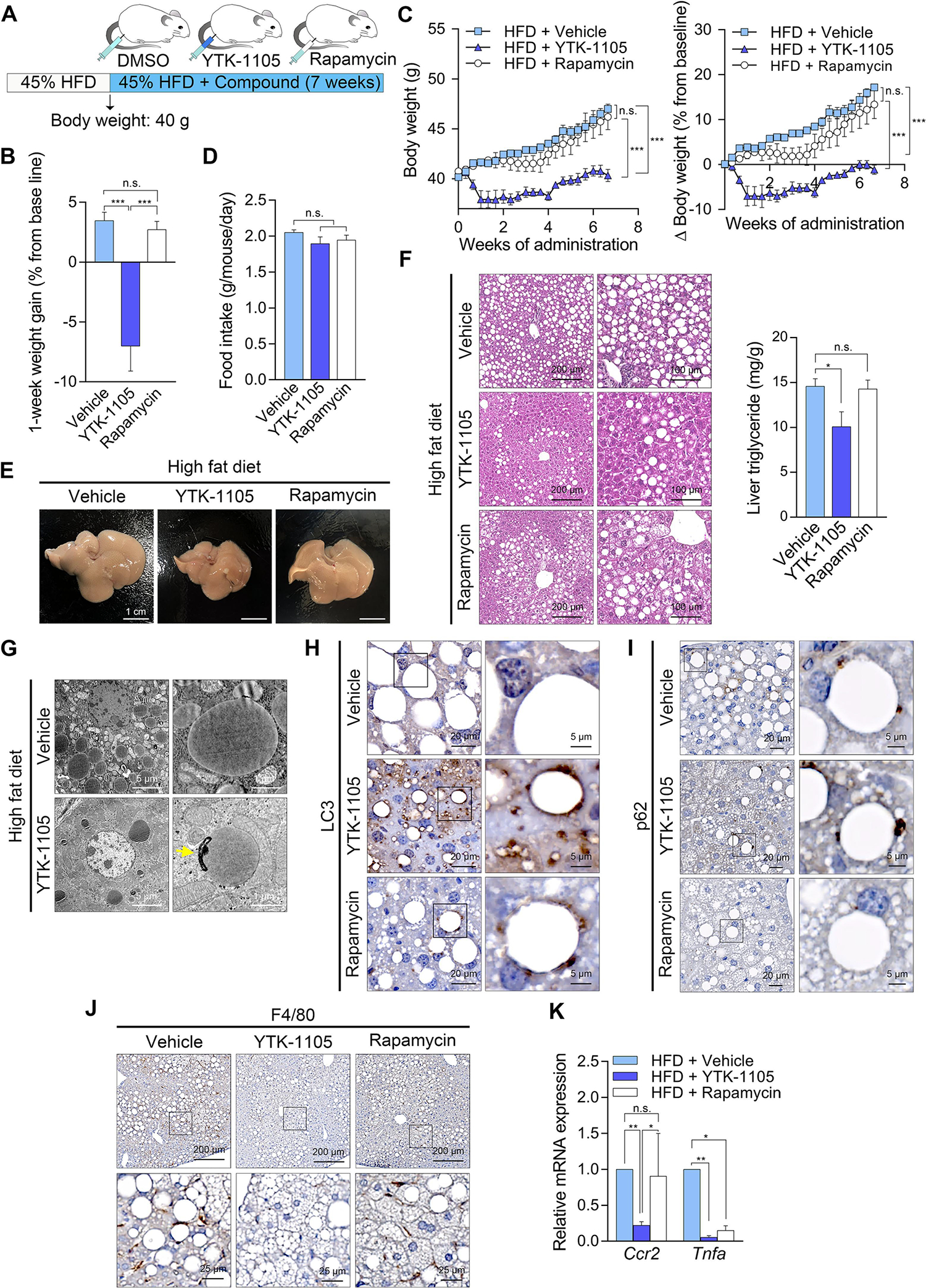
(A) Injection timeline and details of YTK-1105 and rapamycin in HFD-induced obese murine model. (B) Weight gain after 1 week of compound injection (n = 5). (C) Body weight changes during compound administration (left panel, n = 5). Percent body weight changes compared with baseline at the time of sacrifice (right panel, n = 5). (D) Food intake during compound administration (n = 5). (E) Gross morphology of compound treated livers. (F) H & E staining (left panel) and TG levels (right panel, n = 5) in liver treated with YTK-1105, rapamycin, or vehicle. Scale bars, 200 μm (left column), 100 μm (right column). (G) Transmission electron microscopy of liver treated YTK-1105 or vehicle. The autophagosome fused with a LD is indicated by a yellow arrow. Scale bar, 5 μm. IHC staining of (H) LC3 and (I) p62 in liver treated with YTK-1105, rapamycin, or vehicle. Scale bars, 20 μm (left column), 5 μm (right column). (J) IHC staining of F4/80 and (K) mRNA levels of Ccr2 and Tnfa (right panel, n = 5) in liver treated with YTK-1105, rapamycin, or vehicle. Scale bars, 200 μm (upper row), 25 μm (lower row). *P < 0.05, **P < 0.01, ***P < 0.001, n.s., not significant.
4. DISCUSSION
Excessive accumulation of lipids emerges as a major threat to human health in the modern society. Recent studies show that LDs can be degraded via autophagy and that defective lipophagy is a contributing factor underlying metabolic disorders such as obesity, hepatosteatosis, and atherosclerosis [24]. As such, an increasing attention has been paid to the mechanisms underlying LD catabolism and the development of therapeutic means to induce lipophagy. In this study, we identified the p62-mediated N-degron pathway as a central player in lipophagy and developed small molecule ligands to p62, which induce lipophagy in hepatocytes and adipocytes of mice. The physiological ligand to p62 was developed into chemicals that accelerate lipophagy in vitro and in mice and exhibit therapeutic efficacy in obesity and hepatosteatosis. Our results elucidate a novel mechanism underlying targeted degradation of lipids without muscle loss and provide a drug target in NAFLD and other diseases related with metabolic syndrome.
Our earlier studies identified the autophagic receptor p62 as an N-recognin of the N-degron pathway that binds Nt-Arg and other N-degrons to promote autophagic proteolysis of ER-residing molecular chaperons and cytosolic proteins such as R-BiP [25, 26]. Here, we show that the ATE1-p62 branch plays an essential role in lipophagy. In this mechanism, lipophagy initiates when ATE1-dependent Nt-arginylation of proteins such as ER-derived BiP, CRT, and PDI are induced (Fig. 2A–C). The resulting R-BiP and possibly other arginylated proteins are associated with LDs, and their Nt-Arg subsequently recruits and binds p62 to induce self-polymerization of p62 on the sites of lipophagy (Fig. 2D–E and Fig. 3A–C). Our results suggest that LC3 on phagophores are recruited to p62 polymers in complex with Nt-Arg (Fig. 1B–C). As a consequence, a portion of LDs are pinched off and sequestered by phagophores (Fig. S3E), leading to lysosomal degradation (Fig. 2J). As it is known that lipids are degraded by lysosomal hydrolases into fatty acids, fatty acids released from lysosomes may be absorbed by mitochondria and turn to ATP through β-oxidation [24]. In accordance with this concept, the chemical mimic of Nt-Arg, YTK-1105, increased mitochondrial ATP production in HepG2 cells (Fig. S2C). Thus, the ATE1-p62 circuit converts lipids stored in LDs into energy via autophagy.
Our previous studies showed that a set of ER proteins are relocated into the cytosol and Nt-arginylated by ATE1 [9], yet little is known about the role of Nt-arginylated ER proteins. Here, we found that ATE1-dependent Nt-arginylation of proteins such as ER-derived BiP, CRT, and PDI are induced upon fatty acid treatment in vitro and HFD feeding in mice (Fig. 2A–C, 4D and 7B) and that either knockout or inhibition of ATE1 abrogated p62 recruitment to the surface of LDs (Fig. 2F and 2I), highlighting the role of Nt-arginylation in lipophagy. Consistently, liver-specific ATE1-deficiency aggravated hepatosteatosis under HFD feeding compared with littermate controls (Fig. 4G). Despite these findings, the mechanistic details of how Nt-arginylated proteins are targeted to LDs remain to be further investigated.
Although enormous progress has been made in the last half-century in the management of diseases caused by excess body weight, obesity itself has been largely resistant to therapy, which can be attributed to insufficient efficacy and dubious safety after neurological intervention. Given that autophagy is cell-autonomous and minimally regulated by the central nervous system, p62 may be an ideal drug target for anti-obesity. We have previously exploited the N-degron Nt-Arg to develop small molecule ligands to the ZZ domain of p62 [17]. In this study, the prototype chemicals were optimized to develop drugs for obesity and hepatosteatosis. The resulting p62 agonists were demonstrated to induce Nt-Arg/p62-dependent lipophagy in cultured cells, during which p62 as well as LC3+ phagophores were recruited to the sites of lipophagy on LDs, leading to lysosomal hydrolysis of lipids. In 60% HFD-fed mice over 11 weeks, mice injected with YTK-2205 gained only 10.8 ± 1.3% in a striking contrast to vehicle-treated mice (43.0 ± 3.0%) (Fig. 6G). It should be noted that mice fed a chow diet (10 kcal% fat) gained 12.8 ± 1.5%, indicating a robust anti-obesity efficacy under 60% fat diet. The weight reduction was largely attributed to reduced fat contents in WAT but not in BAT (Fig. 6I). Importantly, HFD-fed mice treated with YTK-2205 contained lean mass 8.3% higher as compared with control mice (Fig. 6H), suggesting that YTK-2205 induces lipophagy without loss of lean body mass mainly caused by muscle loss.
Since the cloning of mammalian ATE1 cDNAs [27], the first physiological function of Nt-arginylation was identified from phenotypes of Ate1−/− mouse embryos in cardiovascular development and signaling [28]. Subsequent studies revealed its critical role in diverse processes including neural crest cell migration [29] and chromatin compaction and nuclear architecture [30]. Of relevant to lipid degradation and energy production are the recent findings that ATE1 plays a role in mitochondrial function [29], glycolysis [30], and purine metabolism [31]. In this study, we generated liver-specific Ate1 knockout mice, which developed hepatosteatosis upon HFD feeding (Fig. 4), further supporting the direct role of p62 agonists in lipophagy. Given that the final product of LDs after lysosomal degradation is free fatty acid, one could speculate the lipophagy induced by p62 agonists may influence mitochondrial energy metabolism. Indeed, YTK1105 enhanced the rate of basal oxygen consumption as well as mitochondrial ATP production in HepG2 cells (Fig. S2), suggesting that free fatty acids generated from lipophagy are consumed by mitochondria for ATP production. However, it should be noted that when whole body-knockout of Ate1 was induced in adult mice using tamoxifen-induced conditional knockout system, the mice suffered from pleiotropic phenotypes leading to early death, associated with rapid loss of body weight and WAT [32]. These severe phenotypes can be explained by the loss of diverse biological functions of Ate1 in multiple organs. In that sense, liver-specific knockout of Ate1 provided an opportunity to find its specific role in lipid metabolism.
Excessive fats in the liver are a major cause for NAFLD and other obesity-related diseases. In this study, we demonstrate that p62 agonists induce lipophagy in hepatocytes of mice fed HFD via the ATE1-p62 circuit, leading to therapeutic efficacy in steatosis associated with reduced levels of TG. Such efficacy correlated to the improvements in hepatic inflammation. Comparison with rapamycin demonstrates that these drugs exerted efficacy in obesity and hepatosteatosis through p62-dependent selective macroautophagy as distinguished from mTOR-dependent bulk autophagy. Our results collectively suggest that the p62-dependent N-degron pathway is an ideal target for not only obesity but also NAFLD.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Highlights.
The N-degron pathway mediates lipophagy, i.e., the autophagic degradation of lipids.
The N-terminal arginine (Nt-Arg) conjugated by ATE1 binds p62 during lipophagy.
p62 bound by Nt-Arg recruit phagophores to lipid droplets for lysosomal hydrolysis.
Small molecule ligands to p62 were developed and demonstrated to induce lipophagy.
p62 agonists show robust efficacy in obesity and NAFLD without muscle loss
Funding
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government, the Ministry of Science and ICT [MSIT] (Grant NRF-2020R1A5A1019023 to Y.T.K., NRF-2022R1A2C1006737 to J.W.P., NRF-2019R1A2C1089497 to Y.S.S., NRF-2020R1A5A1019023 and NRF-2021R1F1A1049169 to Y.H.S., NRF-2021R1I1A3A04037479 to S.K.) and the Ministry of Education (Grant NRF-2021R1A2B5B03002614 to Y.T.K.). This work was also supported by the Korea Health Industry Development Institute (HU21C0071 to Y.H.S.) and by the National Institutes of Health (NIH R01NS102435 and R35GM122505 to A.K.).
Abbreviations:
- ATE1
arginyltransferase 1
- BiP
binding immunoglobulin protein
- CRT
calreticulin
- HFD
high fat diet
- mTOR
mammalian target of rapamycin
- Nt
N-terminal
- NAFL
non-alcoholic fatty liver
- NAFLD
non-alcoholic fatty liver disease
- LC3
light chain 3
- LD
lipid droplet
- PDI
protein disulfide isomerase
- PLIN
perilipin
- TG
triglyceride
- WAT
white adipose tissue
Footnotes
Declaration of competing interest
Seoul National University and AUTOTAC Bio, Inc. have filed patent applications based on this study.
CRediT authorship contribution statement
Conceptualization: E.J.J., K.W.S., T.H.B., J-W.P. and Y.T.K. Investigation and data curation: E.J.J., K.W.S., T.H.B. H-Y.K., H.R.C., S.H.K., C.H.J, S.R.M., Y.S.S., S.K. and J-W.P. Formal analysis: E.J.J., K.W.S., T.H.B., Y.H.S., A.K., J-W.P. and Y.T.K. Supervision, J-W.P. and Y.T.K. Writing - original draft: E.J.J., K.W.S., J-W.P. and Y.T.K. Funding acquisition and writing - review and editing: Y.S.S., S.K., Y.H.S., A.K., J-W.P. and Y.T.K. All authors have read and agreed to the final version of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Byrne CD and Targher G, NAFLD: a multisystem disease. J Hepatol, 2015. 62(1 Suppl): p. S47–64. [DOI] [PubMed] [Google Scholar]
- 2.Onal G, et al. , Lipid Droplets in Health and Disease. Lipids Health Dis, 2017. 16(1): p. 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmadian M, et al. , Triacylglycerol metabolism in adipose tissue. Future Lipidol, 2007. 2(2): p. 229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh R, et al. , Autophagy regulates lipid metabolism. Nature, 2009. 458(7242): p. 1131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tasaki T, et al. , The N-end rule pathway. Annu Rev Biochem, 2012. 81: p. 261–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cha-Molstad H, et al. , p62/SQSTM1/Sequestosome-1 is an N-recognin of the N-end rule pathway which modulates autophagosome biogenesis. Nat Commun, 2017. 8(1): p. 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cha-Molstad H, et al. , Regulation of autophagic proteolysis by the N-recognin SQSTM1/p62 of the N-end rule pathway. Autophagy, 2018. 14(2): p. 359–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ji CH, et al. , The N-Degron Pathway Mediates ER-phagy. Mol Cell, 2019. 75(5): p. 1058–1072 e9. [DOI] [PubMed] [Google Scholar]
- 9.Cha-Molstad H, et al. , Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat Cell Biol, 2015. 17(7): p. 917–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leu NA, Kurosaka S, and Kashina A, Conditional Tek promoter-driven deletion of arginyltransferase in the germ line causes defects in gametogenesis and early embryonic lethality in mice. PLoS One, 2009. 4(11): p. e7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charan J and Kantharia ND, How to calculate sample size in animal studies? J Pharmacol Pharmacother, 2013. 4(4): p. 303–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X, et al. , Classical and alternative roles for autophagy in lipid metabolism. Curr Opin Lipidol, 2018. 29(3): p. 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim BW, Kwon DH, and Song HK, Structure biology of selective autophagy receptors. BMB Rep, 2016. 49(2): p. 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saha S, et al. , Small molecule inhibitors of arginyltransferase regulate arginylation-dependent protein degradation, cell motility, and angiogenesis. Biochem Pharmacol, 2012. 83(7): p. 866–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bradic I, et al. , Off-target effects of the lysosomal acid lipase inhibitors Lalistat-1 and Lalistat-2 on neutral lipid hydrolases. Mol Metab, 2022. 61: p. 101510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goh VJ and Silver DL, The lipid droplet as a potential therapeutic target in NAFLD. Semin Liver Dis, 2013. 33(4): p. 312–20. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, et al. , ZZ-dependent regulation of p62/SQSTM1 in autophagy. Nat Commun, 2018. 9(1): p. 4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamamoto T, et al. , High-Fat Diet-Induced Lysosomal Dysfunction and Impaired Autophagic Flux Contribute to Lipotoxicity in the Kidney. J Am Soc Nephrol, 2017. 28(5): p. 1534–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Astrup A, et al. , Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes (Lond), 2012. 36(6): p. 843–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Son JW and Kim S, Comprehensive Review of Current and Upcoming Anti-Obesity Drugs. Diabetes Metab J, 2020. 44(6): p. 802–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glick D, Barth S, and Macleod KF, Autophagy: cellular and molecular mechanisms. J Pathol, 2010. 221(1): p. 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim YC and Guan KL, mTOR: a pharmacologic target for autophagy regulation. J Clin Invest, 2015. 125(1): p. 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dambach DM, et al. , Role of CCR2 in macrophage migration into the liver during acetaminophen-induced hepatotoxicity in the mouse. Hepatology, 2002. 35(5): p. 1093–103. [DOI] [PubMed] [Google Scholar]
- 24.Singh R and Cuervo AM, Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol, 2012. 2012: p. 282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bachmair A, Finley D, and Varshavsky A, In vivo half-life of a protein is a function of its amino-terminal residue. Science, 1986. 234(4773): p. 179–86. [DOI] [PubMed] [Google Scholar]
- 26.Varshavsky A, N-degron and C-degron pathways of protein degradation. Proc Natl Acad Sci U S A, 2019. 116(2): p. 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwon YT, Kashina AS, and Varshavsky A, Alternative splicing results in differential expression, activity, and localization of the two forms of arginyl-tRNA-protein transferase, a component of the N-end rule pathway. Mol Cell Biol, 1999. 19(1): p. 182–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwon YT, et al. , An essential role of N-terminal arginylation in cardiovascular development. Science, 2002. 297(5578): p. 96–9. [DOI] [PubMed] [Google Scholar]
- 29.Jiang C, et al. , Regulation of Mitochondrial Respiratory Chain Complex Levels, Organization, and Function by Arginyltransferase 1. Front Cell Dev Biol, 2020. 8: p. 603688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moorthy BT, et al. , The evolutionarily conserved arginyltransferase 1 mediates a pVHL-independent oxygen-sensing pathway in mammalian cells. Dev Cell, 2022. 57(5): p. 654–669 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang F, et al. , Arginylation regulates purine nucleotide biosynthesis by enhancing the activity of phosphoribosyl pyrophosphate synthase. Nat Commun, 2015. 6: p. 7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brower CS and Varshavsky A, Ablation of arginylation in the mouse N-end rule pathway: loss of fat, higher metabolic rate, damaged spermatogenesis, and neurological perturbations. PLoS One, 2009. 4(11): p. e7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.