

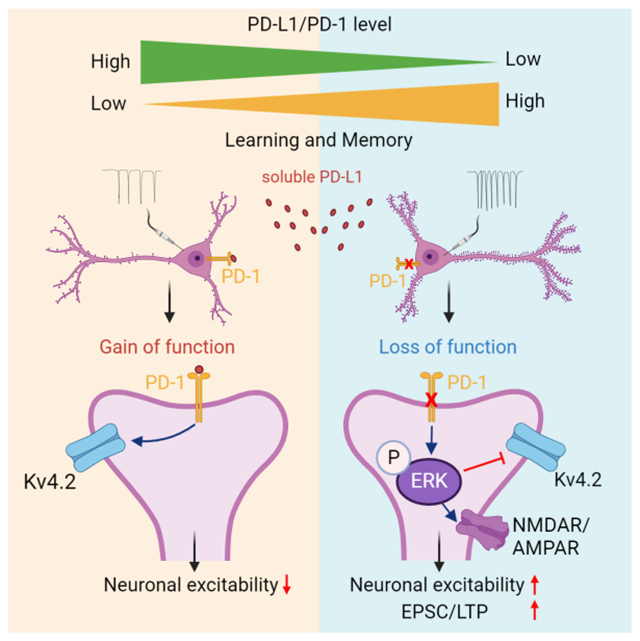
SUMMARY
Programmed death protein 1 (PD-1) and its ligand PD-L1 constitute an immune checkpoint pathway. We report that neuronal PD-1 signaling regulates learning/memory in health and disease. Mice lacking PD-1 (encoded by Pdcd1) exhibit enhanced long-term potentiation (LTP) and memory. Intraventricular administration of anti-mouse PD-1 monoclonal antibody (RMP1-14) potentiated learning and memory. Selective deletion of PD-1 in excitatory neurons (but not microglia) also enhanced LTP and memory. Traumatic brain injury (TBI) impairs learning and memory, which is rescued by Pdcd1 deletion or intraventricular PD-1 blockade. Conversely, re-expression of Pdcd1 in PD-1 deficient hippocampal neurons suppresses memory and LTP. Exogenous PD-L1 suppressed learning/memory in mice and the excitability of mouse and NHP hippocampal neurons through PD-1. Notably, neuronal activation suppressed PD-L1 secretion, and PD-L1/PD-1 signaling is distinctly regulated by learning and TBI. Thus, conditions that reduce PD-L1 levels or PD-1 signaling could promote memory in both physiological and pathological conditions.
Keywords: programmed cell death protein 1, programmed cell death ligand 1, hippocampal neurons, immunotherapy, long-term potentiation, microglia, mice, nonhuman primate, traumatic brain injury
Graphical Abstract
eTOC Blurb
In this study, Zhao et al. reported that PD-L1/PD-1 axis in hippocampal excitatory neurons regulates neuronal excitability, synaptic plasticity, and learning/memory. Blockade of PD-L1 or PD-1 is a new strategy to improve learning and memory in health and disease.
INTRODUCTION
Programmed cell death protein 1 (PD-1), also known as CD279, is a surface molecule expressed by immune cells such as T cells. As an immune-checkpoint regulator, PD-1 down-regulates the immune system and suppresses T-cell activities, serving as a primary target of immunotherapy for cancer.1,2 PD-1 also plays an active role in the central and peripheral nervous system (CNS and PNS).3 Systemic PD-1 blockade was shown to promote the clearance of cerebral amyloid-β plaques and improve memory in mouse models of Alzheimer’s disease (AD)4,5, by recruiting peripheral macrophages into the CNS;6 but there are also contradictory reports.7–9 Notably, these studies focused on immunoregulation of PD-1; direct involvement of neuronal PD-1 was not addressed.
We recently demonstrated that neurons in dorsal root ganglion and spinal cord also express Pdcd1 mRNA and functional PD-1 protein.10–12 Notably, PD-1 activation suppressed neuronal activity, as Pdcd1-deficient mice exhibit hyperexcitability of nociceptive sensory neurons and pain hypersensitivity11 and impaired morphine analgesia.10 Single-cell analysis revealed low expression of Pdcd1 mRNA in cortical and hippocampal excitatory neurons.13 To determine the specific role of PD-1 in neurons and glia, we removed PD-1 function selectively in neurons or microglia by generating Pdcd1 conditional knockout (cKO) mice. Our findings show that the PD-L1/PD-1 pathway plays a critical role in regulating synaptic transmission and cognitive behavior via specific neuronal signaling in the hippocampus. We also demonstrate that deletion of PD-1 or infusion of anti-mouse PD-1 antibody improved learning and memory after traumatic brain injury (TBI). Hence, targeting the PD-L1/PD-1 axis has potential to improve memory as a neurotherapy.
RESULTS
PD-1 regulates learning and memory in physiological conditions
The hippocampus plays a crucial role in learning and memory.14,15 Using sensitive RNAscope in situ hybridization, we previously demonstrated Pdcd1 expression in cortical and thalamic neurons.12 In situ hybridization and double staining showed that 55-65% of CA1 neurons (Nissl+) expressed Pdcd1 in wild-type (WT) mice, but the expression was lost in global knockout mice lacking Pdcd1 (PD-1 KO, Figures 1A). Characterization of PD-1 Cre; Ai9 reporter mice further revealed PD-1 expression in pyramidal neurons (NeuN+) and microglia (IBA-1+) in the CA1 region (Figure 1B). Immunostaining also revealed PD-1 expression in hippocampal CA1 and CA3 neurons (Nissl+), which was blocked by a blocking peptide and absent in KO mice (Figures S1A–S1B).
Figure 1. PD-1 loss or blockade enhances learning and memory.
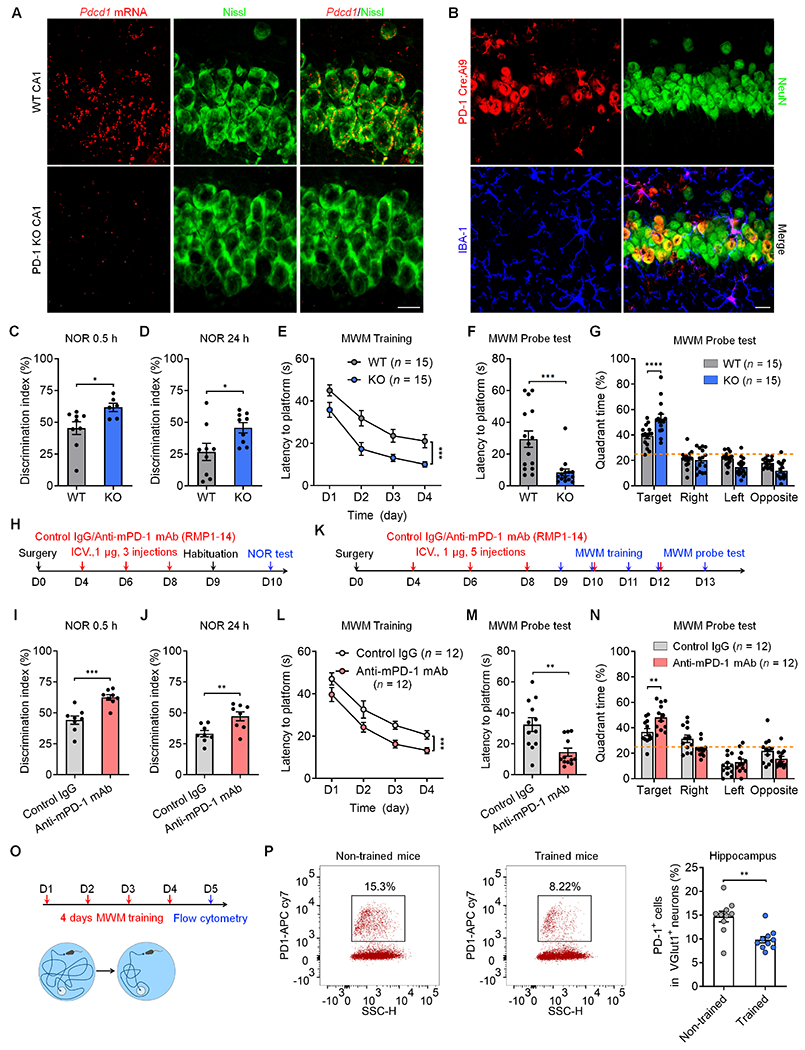
(A) Representative images of Pdcd1 in situ hybridization showing Pdcd1 mRNA expression in WT neurons (labeled by Nissl staining) but not in PD-1 KO neurons. Scale bar, 20 μm
(B) Representative images of NeuN and IBA1 immunostaining in PD-1 Cre;Ai9 reporter mouse. Scale bar, 20 μm.
(C,D) NOR testing showing discrimination index of WT and PD-1 KO mice at 0.5 h (C) and 24 h (D).
(E) MWM testing for spatial learning curves during training in WT and KO mice, measured as latency to find the hidden platform.
(F) MWM probe testing for latency to platform in WT and PD-1 KO mice.
(G) MWM probe test showing percent time spent in different quadrants of WT and PD-1 KO mice.
(H) Schematic of experimental design for ICV surgery, ICV administration of control IgG and anti-mPD-1 mAb (RMP1-14) and NOR testing.
(I, J) NOR test showing discrimination index of control IgG and RMP1-14 treated mice at 0.5 h (I) and 24 h (J).
(K) Schematic of experimental design for ICV surgery, ICV administration of control IgG and anti-mPD-1 mAb and MWM testing.
(L) MWM testing for spatial learning curves of WT mice treated with control IgG and RMP1-14.
(M) MWM probe testing for latency to platform in WT mice treated with control IgG and anti-mPD-1 mAb RMP1-14.
(N) MWM probe test showing percent time spent in different quadrants of WT mice treated with control IgG and RMP1-14.
(O) Timeline for 4-day MWM training and flow cytometry analysis on Day 5.
(P) Flow cytometry images (left) and quantification (right) showing decreased percentage of PD-1+ cells in trained vs. non-trained mice.
Data are represented as mean ± SEM. Also see Figure S1 and Figure S2. Sample size and statistical tests are reported in detail in Tables S1 and S5.
To determine whether PD-1 regulates learning and memory, we tested novel object recognition (NOR) to compare the discrimination index in WT and PD-1 KO mice (8-12 weeks old, Figure S2A). Compared with age-matched WT mice, KO mice spent more time exploring the novel object and exhibited a higher discrimination index at 0.5h and 24h (Figures 1C and 1D). We also examined spatial learning and memory using the Morris Water maze (MWM) test (Figure S2B). During the hidden-platform training phase, KO mice spent less time navigating to the platform than WT mice (Figure 1E), suggesting enhanced spatial learning ability. In the MWM probe test, KO mice showed reduced latency to find the platform and increased number of platform crossings, with slightly increased swimming speed (Figure 1F and Figures S2C–S2D, vs. WT). KO mice also spent more time in the target quadrant than WT mice (Figure 1G). Notably, PD-1 deficiency did not alter the motor function in the rotarod test or anxiety-related behavior in the open-field or elevate-plus maze tests, but KO mice were less immobile in the tail suspension test (Figures S2E–S2H).
To circumvent the possibility of developmental compensation in KO mice, we examined whether central PD-1 blockade in WT adult mice could recapitulate KO phenotypes. RMP1-14, an anti-mouse PD-1 monoclonal antibody (mAb), was shown to produce functional blockade in mouse behavioral assays.11,16–18 We tested RMP1-14 and control IgG in several assays. Intraventricular (ICV) injection of RMP1-14 increased the discrimination index in NOR tests at 0.5h and 24h (Figures 1I and 1J, vs. control IgG). We also examined spatial learning and memory after ICV drug injections (Figure 1K). RMP1-14 treated mice exhibited a shorter latency to find the platform (Figure 1L, vs. control IgG). The mAb-treated mice also showed greater performance in platform location and crossings but comparable swimming speed (Figure 1M and Figures S2I–S2J). RMP1-14 treated mice also spent more time in the target quadrant (Figure 1N). Together, these data indicate that both genetic deletion of Pdcd1 in global KO mice and PD-1 blockade with anti-mPD-1 mAb in WT mice are sufficient to enhance learning and memory.
To investigate whether PD-1 signaling is altered after learning, we tested PD-1 expression in hippocampal tissue 4 days after water maze training (Figure 1O). We examined cell surface expression of PD-1 using biotinylation analysis and found that hippocampal surface PD-1 levels decreased after the training (Figure S2K). Furthermore, flow cytometry analysis revealed that learning and memory training decreased the population of PD-1+ excitatory neurons (Vglut1+, Figure 1P and Figure S2L). Thus, hippocampal neuronal PD-1 expression is correlated with learning and memory behavior.
PD-1 regulates excitability of hippocampal neurons
We examined neuronal excitability in CA1 neurons of WT and KO mice using ex vivo whole-cell, current-clamp patch recording in brain slices (Figure 2A). Compared to WT neurons, CA1 pyramidal neurons from KO mice had increased resting membrane potential (RMP), indicating a cell membrane depolarization (Figure 2B). CA1 pyramidal neurons of KO mice also displayed increased number of action potentials (APs), evoked by 40 pA current (Figure 2C) and injection of increasing currents (Figure 2D). Intriguingly, we observed spontaneous discharges in 40% of KO neurons (8/20) but none in WT neurons (0/20, Figure 2E). By blocking synaptic input, we further confirmed that the increased neuronal excitability in KO mice is driven by intrinsic mechanism of CA1 neurons (Figure S3A). Thus, loss of PD-1 is sufficient to increase excitability of CA1 neurons of brain slices.
Figure 2. Loss of PD-1 increases neuronal excitability in mouse hippocampal neurons.
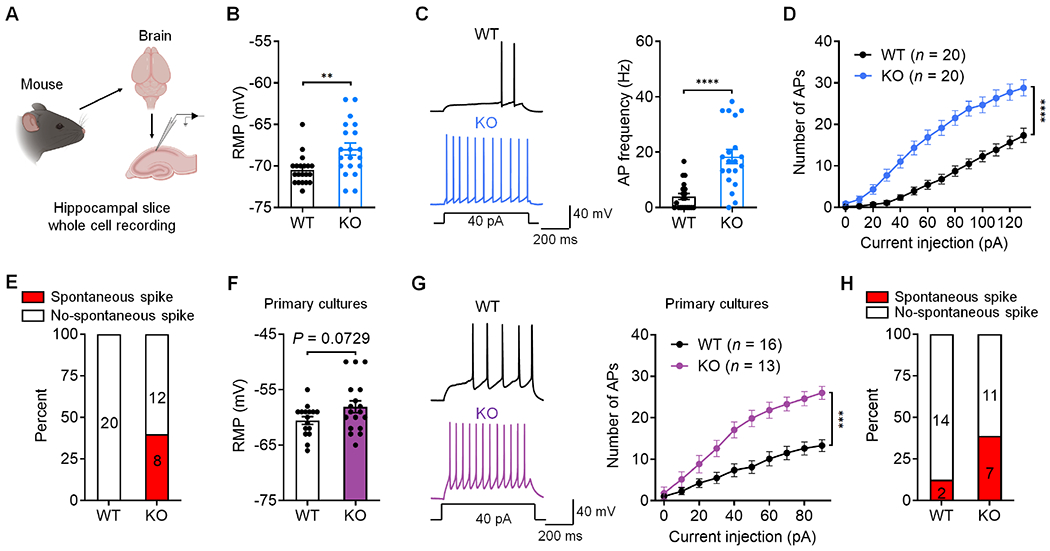
(A) Schematic of experimental design for whole-cell patch clamp recordings in mouse brain slices.
(B) RMP recorded from WT and PD-1 KO CA1 neurons.
(C) Representative AP traces (left) and frequency (right) in WT and PD-1 KO CA1 neurons.
(D) Number of APs evoked by step current injection in WT and KO neurons.
(E) Spontaneous discharges in CA1 neurons of PD-1 KO (8/20) but not in WT neurons (0/20).
(F) RMP recorded from WT and PD-1 KO neurons.
(G) Representative AP traces (left) and AP number (right) evoked by step current injection in WT and PD-1 KO neurons from primary cultures.
(H) Percentage of neurons showing spontaneous discharges in WT (2/16) and PD-1 KO (7/18) neurons from primary cultures.
Data are represented as mean ± SEM. Also see Figure S3. Sample size and statistical tests are reported in detail in Tables S1 and S5.
To further determine the role of PD-1 in CA1 neurons, we tested dissociated hippocampal neurons in primary cultures from E17-E19 mice. Immunocytochemistry revealed PD-1 expression in hippocampal neurons (NeuN+, Figure S3B). Patch-clamp recordings in these KO neurons (7-8 days in vitro, DIV) showed a moderate increase in RMP but marked increases in spontaneous discharge and AP firing rate following current injection, compared to WT neurons (Figures 2F–2H). Thus, PD-1 on individual CA1 neurons may be sufficient to dampen excitability of these neurons.
PD-1 regulates hippocampal synaptic function and dendritic spine density
We used additional electrophysiological and histochemical approaches to study how PD-1 affects hippocampal synaptic functions in WT and KO mice. Patch-clamp recording in brain slices of KO mice revealed increases in the frequency and amplitude of spontaneous miniature excitatory postsynaptic current (mEPSC) in CA1 neurons (Figures 3A–3C, vs. WT).
Figure 3. PD-1 loss in mouse brain slices regulates synaptic transmission, LTP, spine formation, ERK activation, A type K+ currents and NMDAR/AMPAR currents in the CA1 region.
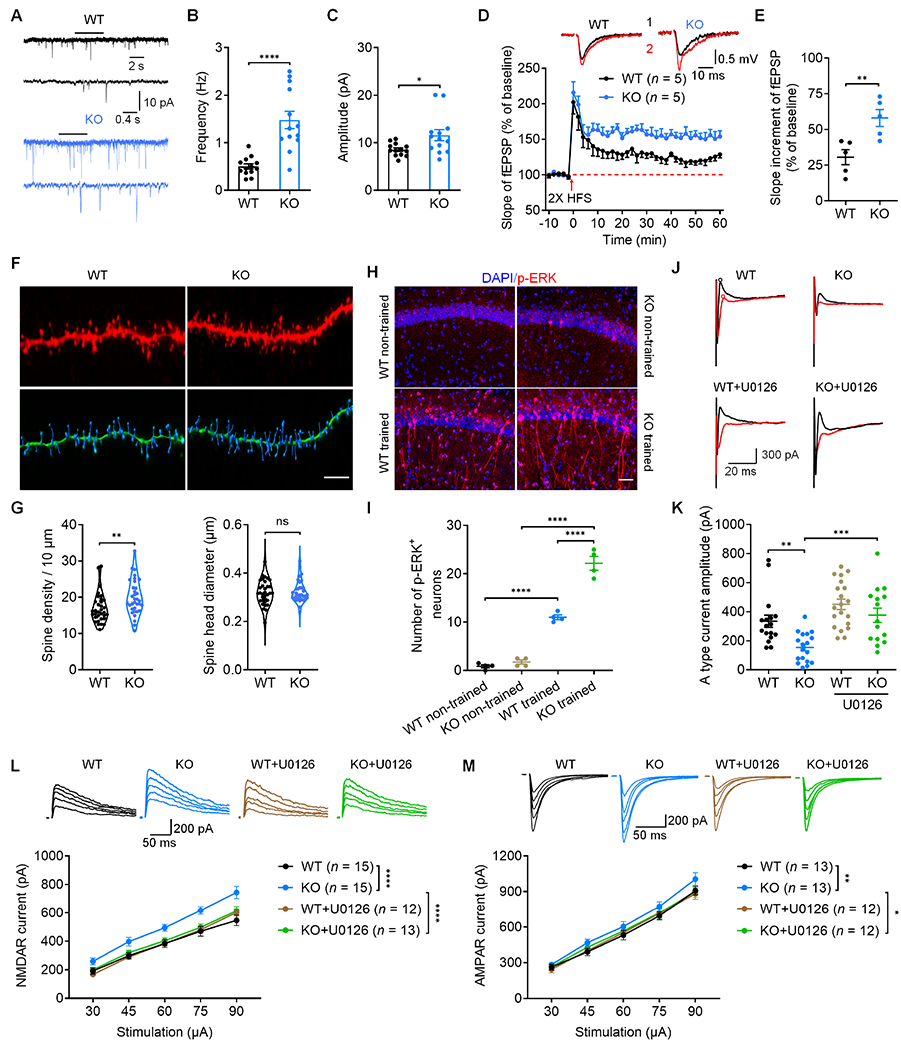
(A) Representative traces mEPSCs recorded from WT and PD-1 KO neurons.
(B-C) Quantification of mEPSC frequency (B) and amplitude (C) recorded from WT and PD-1 KO neurons.
(D-E) Summary plots (D) and average slope (E) of LTP induced by 2 × HFS in CA1 region of WT and PD-1 KO slices. Top: Black traces (1) and red trace (2) represent the baseline fEPSP and post-induction fEPSP, respectively.
(F) Representative confocal z stack and three-dimensional reconstruction images of the apical dendrites of CA1 neurons obtained from WT and PD-1 KO slices. Scale bar, 5 μm.
(G) Spine density (left) and spine size (right) in WT mice and PD-1 KO mice.
(H) Confocal microscopy images of p-ERK immunostaining in the CA1 region. Scale bar, 50 μm.
(I) Quantification of p-ERK+ neurons in CA1 of WT and PD-1 KO mice with and without training.
(J) Representative traces of outward A-type potassium currents from WT and PD-1 KO mice. A type current was isolated from the subtraction of red to black circles.
(K) Quantification of A type potassium current amplitudes from WT and PD-1 KO neurons and the effects of U0126.
(L-M) NMDAR and AMPAR mediated currents in CA1 pyramidal neurons of brain slices of WT and PD-1 KO mice and the effects of U0126 (ERK inhibition) following electrical stimulation of the Schaffer collaterals.
(L) Representative traces of evoked NMDAR currents (top) and summary data of current amplitude (bottom).
(M) Representative traces of evoked AMPAR currents (top) and summary data of current amplitude (bottom).
Data are represented as mean ± SEM. Also see Figure S3. Sample size and statistical tests are reported in detail in Tables S1 and S5.
Long-term potentiation (LTP) is an important synaptic mechanism underlying learning and memory19 and can be induced by high-frequency stimulation (HFS) of Schaffer collaterals in the CA1 region.20 We recorded LTP from brain slices of WT and PD-1 KO mice following 2 × HFS (100 Hz, 2 trains with 30s interval, revealed by field excitatory postsynaptic potential, fEPSP). We found LTP by 2 × HFS was greatly enhanced in KO slices compared to WT slices (Figures 3D and 3E). LTP induced by 1 × HFS (weak stimulation) and 4 × HFS (saturated stimulation) was also enhanced in KO slices (Figures S3C–S3F, vs. WT). Therefore, loss of PD-1 can promote LTP in all the induction conditions.
LTP is also associated with dendritic morphological changes in hippocampal CA1 pyramidal neurons.21 Loss of PD-1 resulted in increased dendritic spine density but unaltered spine size in CA1 neurons of KO mice (Figures 3F and 3G). Pdcd1 deficiency did not affect neuronal survival in the hippocampus (Figures S3G–S3I). Together, these results suggest that increases in synaptic transmission, LTP, and synaptogenesis in hippocampal neurons may contribute to enhanced learning and memory in PD-1 KO mice.
PD-1 regulates the functions of immune cells and sensory neurons via the activation of tyrosine phosphatase SHP-1.11,22 Spinal PD-1 blockade of PD-1 resulted in SHP-mediated hyper-phosphorylation of extracellular signal-regulated kinase (ERK) in dorsal horn neurons.12 We examined ERK phosphorylation (p-ERK) in WT and PD-1 KO mice, as this process is critical for LTP, learning, and the hippocampus-dependent memory.23–26 Hippocampal p-ERK was examined in mice following 4 days of Morris water maze training. Compared to non-trained mice, both WT and KO trained mice had increased p-ERK expression. Strikingly, KO mice with training exhibited more p-ERK+ neurons than WT mice with training (Figures 3H–3I). These results demonstrate that PD-1 regulates ERK signaling during learning and memory.
ERK regulates dendritic A-type K+ currents via direct phosphorylation of the Kv4.2 potassium channels.27,28 Thus, we tested whether PD-1 could regulate neuronal excitability via altering A-type K+ currents in CA1 neurons of WT and KO slices. We observed a substantial reduction in the amplitude of A-type currents in KO neurons compared to WT neurons; but this reduction was completely restored by the MEK (ERK kinase) inhibitor U0126 (Figures 3J and 3K). These results indicate that ERK-mediated suppression of A-type K+ currents in CA1 neurons drives neuronal hyperactivity after loss of PD-1.
Given an important role of NMDA and AMPA receptors (NMDAR and AMPAR) in synaptic transmission and plasticity, we also recorded NMDAR and AMPAR mediated evoked excitatory postsynaptic currents (EPSCs) in CA1 pyramidal neurons in WT and PD-1 KO mice by electrically stimulating Schaffer collaterals. PD-1 deletion enhanced the evoked NMDAR currents and AMPAR currents; but the enhancement of these currents was blocked by U0126 (Figures 3L and 3M). These results indicate that ERK-mediated enhancement of NMDAR and AMPAR activity in CA1 neurons may be responsible for synaptic plasticity after loss of PD-1.
Conditional deletion of Pdcd1 in excitatory neurons enhances memory and LTP
To directly investigate the neuron-specific role of PD-1 in synaptic functions and memory, we generated Pdcd1-floxed mice (Pdcd1fl/fl) by flanking exon 2 with loxP sites (Figure S4); this exon encodes a critical function of PD-1.29 Since PD-1 was expressed in hippocampal pyramidal layers, we next used two strategies to define the function of neuronal PD-1 in hippocampal excitatory neurons. First, we performed bilateral stereotactic injections of an AAV-Camk2a-mCherry:Cre virus into the hippocampus of Pdcd1fl/fl mice (Figure 4A), which lead to robust AAV expression in CA1 and CA3 neurons (Figure 4B) and specific depletion of PD-1 expression in hippocampal excitatory neurons (Figure 4C). Mice that received AAV-Camk2a-mCherry:Cre injections exhibited enhanced learning and memory in the NOR test (Figure 4D). The hippocampus-specific knockout of PD-1 phenocopied the effects of global knockout in the MWM tests, resulting in improved spatial learning and memory in the probe tests but no change in swimming speed (Figures 4E–4G and Figures S5A–S5B).
Figure 4. Selective deletion of PD-1 in hippocampal excitatory neurons enhances memory and LTP.
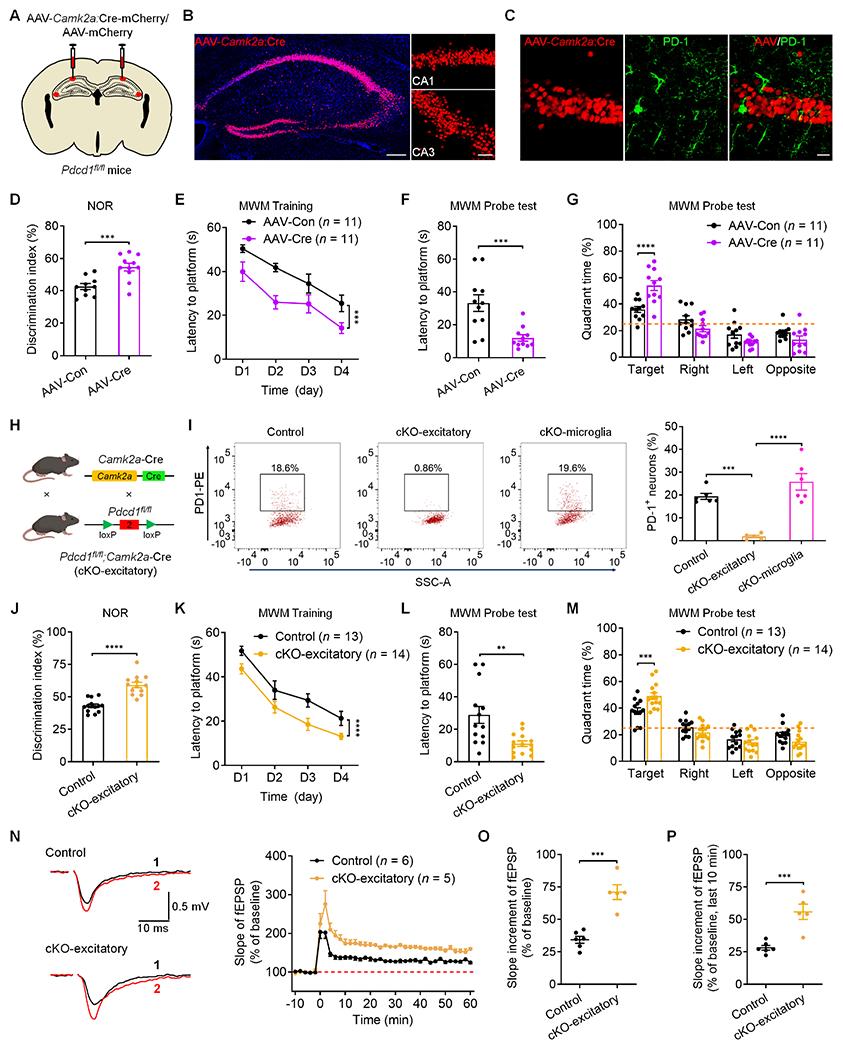
(A) Illustration of bilateral virus (AAV-Camk2a:Cre or AAV-control) injections into the hippocampus of Pdcd1fl/fl mice for selective deletion of Pdcd1 in hippocampal excitatory neurons.
(B) Representative confocal images of the mouse hippocampus show the AAV-infected neurons in the CA1 and CA3 regions. Blue: DAPI staining. Scale bar, 200 μm. Right, higher magnification images showing the infected CA1 and CA3 excitatory neurons. Scale bar, 50 μm.
(C) Representative confocal images showing selective loss of PD-1 in CA1 excitatory neurons labeled with AAV-Camk2a:Cre. Scale bar, 20 μm.
(D) Discrimination index of Pdcd1fl/fl mice injected with AAV-control and AAV-Camk2a:Cre in NOR testing.
(E) Spatial learning curves during MWM training in Pdcd1fl/fl mice show better performance in mice injected with AAV-Camk2a:Cre vs. AAV-control.
(F) MWM probe tests showing latency to platform in Pdcd1fl/fl mice injected with AAV-control and AAV-Camk2a:Cre.
(G) Quadrant percent time of Pdcd1fl/fl mice injected with AAV-control and AAV-Camk2a:Cre in the MWM probe test.
(H) Schematic of conditional deletion of Pdcd1 in excitatory neurons (cKO-excitatory).
(I) Flow cytometry images (left) and quantification (right) showing the percentage of PD-1+ neurons in the hippocampi of control littermates, cKO-excitatory mice, and cKO-microglia mice.
(J) Discrimination index of control littermates and cKO-excitatory mice in NOR testing.
(K) Spatial learning curve during MWM training in control littermates and cKO-excitatory mice.
(L) MWM probe tests for latency to platform in control littermates and cKO-excitatory mice.
(M) Quadrant percent time of control littermates and cKO-excitatory mice in the MWM probe test.
(N) Representative traces (left) and summary plots (right) of LTP induced by 2 × HFS in CA1 region of control littermates and cKO-excitatory slices. Black traces (1) and red trace (2) represent the baseline fEPSP and post-induction fEPSP, respectively.
(O-P) Quantification of average slope of LTP in CA1 region of control littermate and cKO-excitatory slices in entire phase (O) and late phase (P).
Data are represented as mean ± SEM. Also see Figures S4–S7. Sample size and statistical tests are reported in detail in Tables S1 and Table S5.
Next, we crossed Pdcd1fl/fl mice with Camk2a-Cre mice to generate conditional knockout of Pdcd1 in excitatory neurons, described as cKO-excitatory (Figure 4H). Flow cytometry analysis revealed that the percentage of PD-1+ excitatory neurons was substantially decreased in the hippocampus of cKO-excitatory mice, indicating a loss of PD-1 in hippocampal excitatory neurons (Figures 4I and S4C). Genotyping and in situ hybridization validated a loss of Pdcd1 mRNA expression in CA1 neurons of the cKO-excitatory mice (Figures S5C and S5D). The cKO-excitatory mice displayed greater learning and memory capacity than control littermates in the NOR testing (Figure 4J). They also showed enhanced learning in the MWM hidden-platform training phase and improved memory in the probe tests but unaltered swimming speed (Figures 4K–4M and Figure S5E–S5F). The cKO-excitatory mice had normal motor function and emotional states (Figures S5G–S5J). We also recorded LTP in hippocampal neurons from brain slices of cKO-excitatory and littermate control mice following HFS. cKO mice had enhanced LTP compared to controls; as indicated by summary plots and average slope of fEPSP during the entire 60 min and just the last 10 min (Figures 4N–4P). These results confirm the observations from the global KO mice and reveal that PD-1 effects on learning and memory are largely restricted to the hippocampus and excitatory neurons.
PD-1 is expressed by microglia (Figure 1B) and may play a role in neurodegenerative diseases.6,13,30–32 We crossed the Pdcd1fl/fl mice with Cx3cr1CreER mice to generate conditional knockout of Pdcd1 in microglia and monocytes, described as cKO-microglia (Figure S6A). FACS analysis in cKO-microglia mice showed a great reduction in PD-1+ microglia in the hippocampus, as compared with that in control littermate mice or cKO-excitatory mice (Figure S6B). Genotyping and in situ hybridization validated the loss of Pdcd1 mRNA in hippocampus of cKO-microglia mice (Figures S6C–S6D). Furthermore, cKO-microglia mice and control littermates displayed comparable learning and memory performance in NOR and MWM tests (Figures S6E–S6H), as well as comparable swimming speed (Figure S6I), motor function, and general emotional states (Figures S6J–S6M). Together, these data suggest that PD-1 expression in microglia/monocytes may not impact learning and memory under these testing conditions.
We also generated cKO mice in inhibitory neurons by crossing the Pdcd1fl/fl mice with Vgat-Cre mice, described as cKO-inhibitory (Figure S7A). Pdcd1 was expressed by hippocampal inhibitory interneurons, but this expression was lost in cKO-inhibitory mice (Figure S7B). cKO-inhibitory mice and control littermates displayed comparable learning and memory performance in NOR and MWM tests (Figures S7C–S7F), as well as comparable swimming speed (Figure S7G), motor function, and anxiety level (Figures S7H–S7J). However, cKO-inhibitory mice exhibited less immobility in the tail suspension test, as in global knockout mice (Figure S7K). Together, these data suggest that PD-1 in inhibitory neurons does not regulate learning and memory under these testing conditions.
Selective PD-1 re-expression in hippocampal excitatory neurons impairs memory and LTP
To investigate whether re-expression of PD-1 in the hippocampus of PD-1 KO mice would be sufficient to reverse learning and memory behaviors changes, we constructed AAV expressing either Pdcd1 driven by Camk2a promoter (AAV-Pdcd1) or control fluorescent protein eGFP (AAV-control) and delivered the AAV specifically to the hippocampus by bilateral stereotactic injections (Figure 5A). This strategy resulted in selective re-expression of PD-1 in the targeted region including CA1 and CA3 neurons (Figure 5B and Figures S8A and S8B). In situ hybridization analysis confirmed the expression of Pdcd1 in hippocampal CA1 and CA3 neurons (Figures 5C and 5D). No neuronal loss was observed in the hippocampus after the PD-1 re-expression (Figure S8C). Four weeks after the AAV infection, we conducted learning and memory behavioral tests and recorded LTP in hippocampal slices. Viral expression of PD-1 in the KO mice retarded object learning in NOR test (Figure 5E) and impaired learning/memory in MWM tests (Figures 5F–5I), showing no effects on motor activities or anxiety and depression-like behaviors (Figures S8D–S8G). PD-1 re-expression in the KO background also suppressed LTP (Figure 5J). Notably, the average slope of fEPSP in the viral-infected slices were markedly reduced, especially in the late-phase (Figures 5K and 5L). Thus, PD-1 re-expression in hippocampal neurons can is sufficient to reverse both the cognitive and synaptic changes in PD-1 KO mice.
Figure 5. Selective re-expression of PD-1 in the hippocampus excitatory neurons impairs memory and LTP.
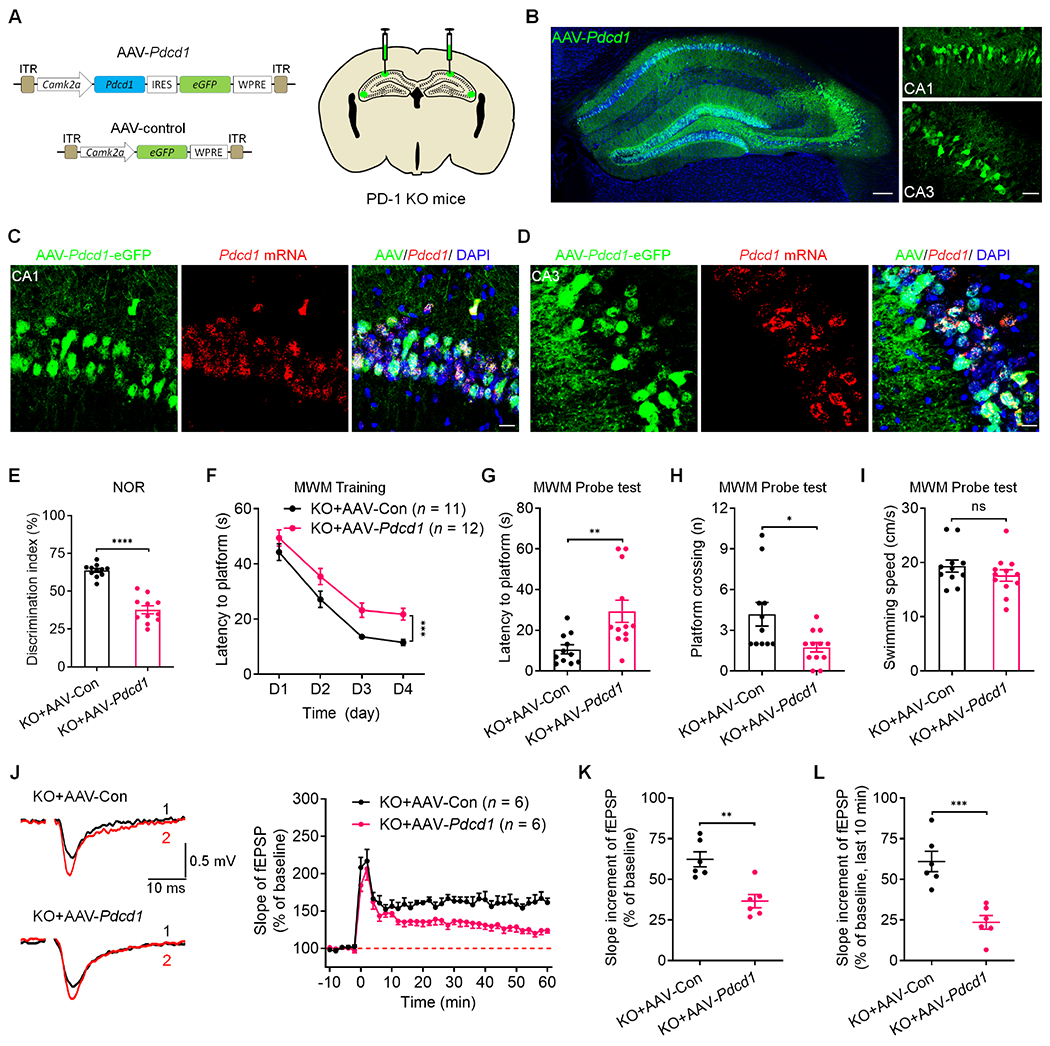
(A) Schematics of AAV constructs of Pdcd1 (AAV-Pdcd1) or control (AAV-control) (left) and bilateral viral injections into the mouse hippocampus (right).
(B) Representative confocal images of mouse hippocampus (left) and infected CA1 and CA3 neurons (right) after the AAV injections. Blue: DAPI. Scale bars, 200 μm (left), 50 μm (right).
(C-D) Representative confocal images of the infected CA1 (C) and CA3 (D) neurons coexpressing eGFP (AAV-Pdcd1) and Pdcd1 mRNA (revealed by RNAscope). Scale bar, 20 μm.
(E) Discrimination index of PD-1 KO mice injected with AAV-control and AAV-Pdcd1 in NOR testing.
(F) Spatial learning curves during MWM training in PD-1 KO mice injected with AAV-control and AAV-Pdcd1, measured as latency to the hidden platform.
(G-H) MWM probe test shows deficits of PD-1 KO mice injected with AAV-Pdcd1 vs. AAV-control in locating the platform (left) and crossing the platform zone (right).
(I) MWM probe test shows comparable swimming speed in PD-1 KO mice injected with AAV-control and AAV-Pdcd1.
(J) Representative traces (left) and summary plots (right) of LTP induced by 2 × HFS in the CA1 region of PD-1 KO mice injected with AAV-control or AAV-Pdcd1. Left: Black traces (1) and red trace (2) represent the baseline fEPSP and post-induction fEPSP, respectively.
(K-L) Quantification of average slope of LTP in the CA1 region of PD-1 KO mice injected with AAV-control and AAV-Pdcd1 in the entire phase (K) and late phase (L).
Data are represented as mean ± SEM. Also see Figure S8. Sample size and statistical tests are reported in detail in Tables S1 and S5.
Suppression of PD-1 improves cognitive decline after traumatic brain injury
Traumatic brain injury (TBI) causes significant cognitive decline.33,34 To define the role of PD-1 in TBI, we tested a mouse model of the closed-head injury.33 Cell-surface biotinylation and ELISA analysis revealed increased membrane PD-1 expression in hippocampal tissue after TBI (Figure S9A). Similarly, flow cytometry experiments showed increased PD-1 expression in hippocampal excitatory neurons following TBI (Figure S9B). Next, we tested motor recovery after TBI in WT and PD-1 KO mice (Figures 6A–6C). Rotarod testing revealed similar motor impairment in both WT and KO mice after TBI, with a recovery in 3 weeks (Figure 6C). NOR test at 3 weeks after TBI showed lower discrimination index in WT mice, but TBI-KO mice had a higher discrimination index than TBI-WT mice, indicating Pdcd1 deletion protects against the TBI-induced cognitive decline (Figure 6D). TBI also resulted in a cognitive deficit in MWM test in WT mice, but KO mice were protected from TBI (Figure 6E). In the MWM tests, TBI-KO mice also shower better performance and higher swimming speed, compared to TBI-WT mice (Figure 6F–6H). We also tested whether anti-mPD-1 mAb (RMP1-14) or control IgG treatment could modulate cognitive deficits after TBI following ICV injections and behavioral tests when animals recovered from motor deficits (Figures 6I–6K). RMP1-14 treated TBI mice exhibited marked improvements in learning and memory capacity in the NOR test (Figure 6L) and spatial learning in the MWM training and probe tests (Figures 6M and 6N) but unaltered platform crossing number and swimming activity (Figures 6O and 6P). We further tested whether cKO-excitatory mice also have a protective effect against TBI-induced memory deficits. Compared to control littermates, cKO-excitatory mice with TBI exhibited a higher discrimination index in NOR test (Figure 6Q) and shorter latency in the MWM hidden-platform training (Figure 6R). In the MWM probe tests, cKO TBI mice exhibited marked improvements in locating the platform zone (Figure 6S) but unaltered platform crossing number and swimming activity (Figures 6T and 6U). Together, suppression of PD-1 in excitatory neurons confers protection against TBI-induced spatial memory deficits.
Figure 6. Suppression of PD-1 protects cognitive function after traumatic brain injury.
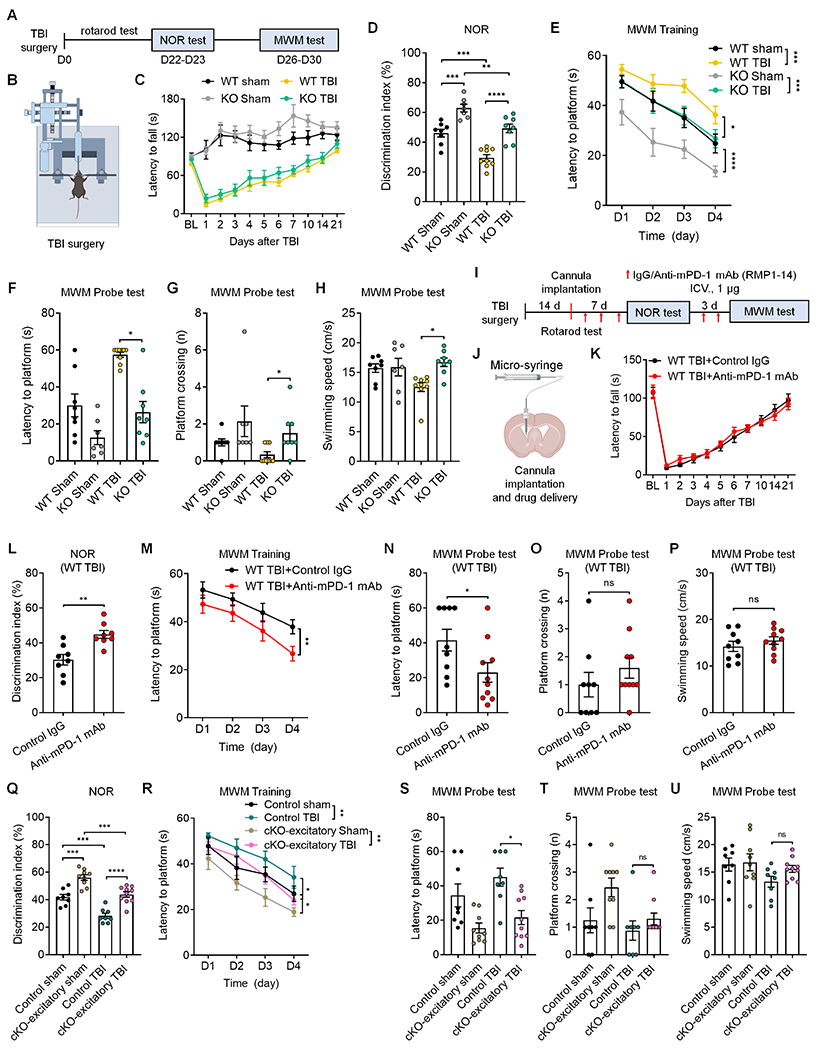
(A-C) Experimental paradigm (A), TBI illustration (B) and rotarod test (C) in WT mice and PD-1 KO mice with TBI or sham surgery.
(D) NOR testing shows cognitive deficits in both WT TBI mice and PD-1 KO TBI mice; but PD-1 KO TBI mice have a higher discrimination index than WT TBI mice.
(E) MWM training curves show deficits in both WT mice and PD-1 KO; but PD-1 KO mice with TBI spend less time on navigating the hidden platform location than WT mice with TBI.
(F-G) MWM probe tests for latency to platform (F) and number of platform zone crossings (G).
(H) MWM probe test shows higher swimming velocity in PD-1 KO TBI mice than WT TBI mice.
(I-K) Experimental paradigm (I), drug delivery (J) and rotarod test (K) in WT TBI mice treated with control IgG or anti-mPD-1 mAb.
(L) Discrimination index of WT TBI mice treated with control IgG and anti-mPD-1 mAb (RMP1-14) in NOR testing.
(M) Spatial learning curves during MWM training show significant improvement in WT TBI mice treated with RMP1-14 than control IgG.
(N-O) MWM probe test for latency to platform (N) and number of platform crossings (O) in control IgG and RMP1-14 treated mice.
(P) MWM probe test shows comparable swimming speed between control IgG and RMP1-14 treated TBI mice.
(Q) Discrimination index of control littermates and cKO-excitatory mice with TBI or sham surgery in NOR testing.
(R) Spatial learning curve during MWM training in control littermates and cKO-excitatory mice with TBI or sham surgery.
(S-U) MWM probe tests for latency to platform (S) and number of platform crossings (T) and swimming speed (U) in control littermates and cKO-excitatory mice with TBI or sham surgery.
Data are represented as mean ± SEM. Also see Figure S9. Sample size and statistical tests are reported in detail in Tables S1 and S5.
PD-L1 is detected in the CSF of different species and regulates neuronal excitability and learning and memory
Since PD-L1 is a major ligand of PD-1, we measured PD-L1 levels in CSF vs. serum/plasma in mouse, non-human primate (NHP), and human (Figure 7A and 7B). ELISA analysis revealed remarkably high PD-L1 levels in the CSF of all three species, ranging from 105 to 591 pg/ml, with the highest levels in mice. Furthermore, PD-L1 levels in the CSF were 12.9-fold (mouse), 3.7-fold (NHP), and 3.5-fold (human) higher than that in serum/plasma collected from the same donors (Figure 7A and 7B). PD-L1 may be derived from myeloid cells in the meninges, glial cells (e.g., microglia), and neurons in the brain (Figure S10A–S10C).3 Using a mouse brain slice preparation, we found a constitutive release of PD-L1, detected within 15 min (Figure 7C). Strikingly, PD-L1 release was suppressed by neuronal depolarization with KCl. Blockade of neuronal activity with TTX had no effect on PD-L1 release (Figure 7D). Treatment of brain slices with the microglial/monocyte inhibitor minocycline, but not the astroglial inhibitor L-β-aminoadipate, also reduced PD-L1 secretion (Figure 7D). Subcutaneous injection of capsaicin produces intense pain in NHP and human.35 Interestingly, subcutaneous capsaicin reduced PD-L1 levels in the spinal CSF of NHPs (Figure 7E). These results indicate that PD-L1 release is regulated by neuronal activity and pain.
Figure 7. Secreted PD-L1 regulates neuronal excitability and memory via PD-1.
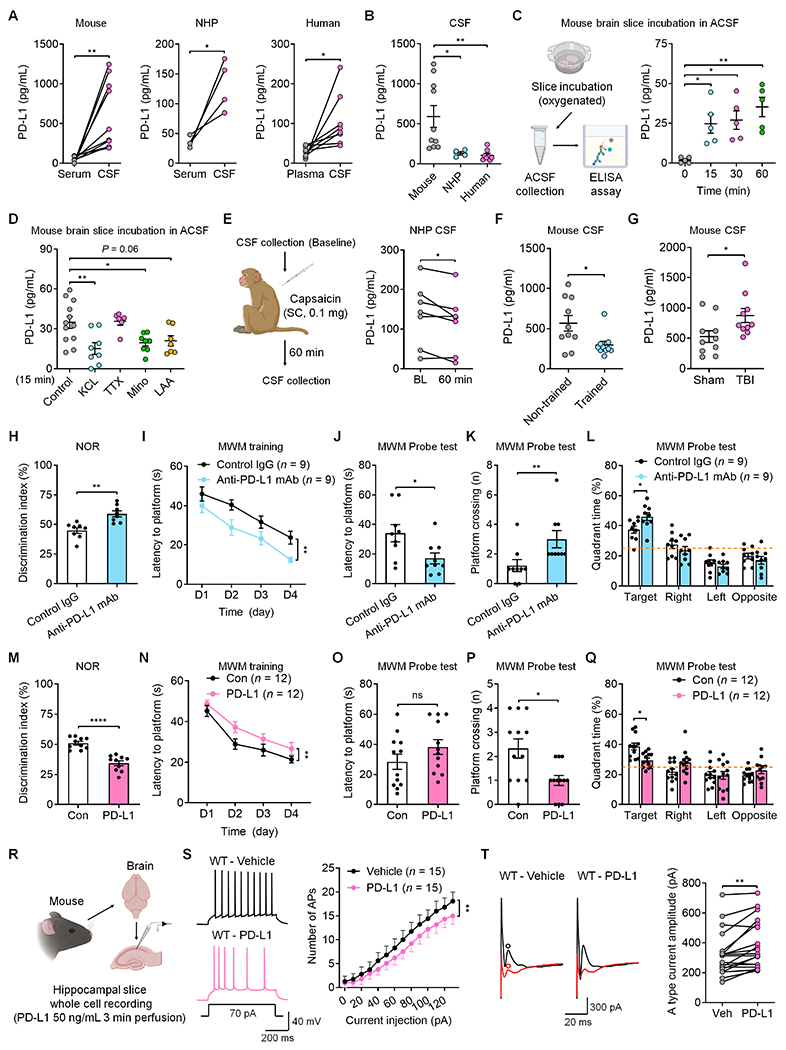
(A-B) Basal levels of PD-L1 in serum/plasma and CSF samples of mice, non-human primates (NHPs) and humans.
(C) Time course of PD-L1 release from mouse brain slices incubated with ACSF.
(D) Effects of control, KCL, TTX, minocycline, and L-α-aminoadipate on PD-L1 release at 15 min.
(E) Schematic of CSF collection in NHP (left) and secreted PD-L1 levels before and 60 min after capsaicin treatment (right).
(F) CSF PD-L1 levels in mice with and without training.
(G) CSF PD-L1 levels in mice with TBI and sham surgery.
(H) Discrimination index of WT mice treated with ICV control IgG or anti-mouse PD-L1 mAb in NOR testing.
(I) Spatial learning curve during MWM training in WT mice treated with ICV control IgG or anti-mouse PD-L1 mAb.
(J-K) MWM probe tests for latency to platform (J) and number of platform crossings (K) in WT mice treated with ICV control IgG or anti-mouse PD-L1 mAb.
(L) Quadrant percent time of WT mice treated with ICV control IgG or anti-mouse PD-L1 mAb for the MWM probe test.
(M) Discrimination index of WT mice treated with ICV administration of recombinant mouse PD-L1 or control (Con) protein in NOR testing.
(N) Spatial learning curve during MWM training in WT mice treated with ICV recombinant PD-L1 or control protein.
(O-P) MWM probe tests for latency to platform (O) and number of platform crossings (P) in WT mice treated with ICV control protein or recombinant mouse PD-L1.
(Q) Quadrant percent time of WT mice treated with ICV PD-L1 and control protein in the MWM probe test.
(R) Schematic of whole-cell patch clamp recordings in WT mouse hippocampal neurons before and after PD-L1 perfusion.
(S) Representative AP traces (left) and quantification of AP numbers (right), evoked by step current injection in WT CA1 neurons before and after perfusion of recombinant mouse PD-L1.
(T) Representative outward A-type potassium currents and quantification of A-type K+ current amplitude in WT CA1 neurons before and after PD-L1 perfusion. Black trace: nonconditioned current; red trace: conditioned current. A type current was isolated from the subtraction of red to black circles.
Data are represented as mean ± SEM. Also see Figure S10. Sample size and statistical tests are reported in detail in Tables S1 and S5.
Next, we tested whether water maze training and TBI would alter membrane-bound and soluble PD-L1 levels in mouse brain and CSF samples. Water maze training caused a marked reduction in soluble PD-L1 in the CSF without altering membrane-localized PD-L1 in the hippocampus (Figure 7F and Figure S10D, vs. non-trained). In contrast, TBI increased both soluble PD-L1 and membrane-localized PD-L1 levels (Figure 7G and Figure S10E, vs. sham surgery). Thus, PD-L1 upregulation is correlated with memory deficits after TBI.
We further tested a causative role of PD-L1 in cognitive behavior using both loss-of-function (anti-PD-L1 mAb treatment) and gain-of-function (exogenous PD-L1 treatment) approaches. NOR testing in non-injured mice showed that ICV administration of anti-PD-L1 antibody (mouse) significantly improved learning and memory (Figure 7H). The mAb treated mice also showed enhanced learning in the MWM tests (Figures 7I–7L). In contrast, exogenous PD-L1 suppressed learning and memory in NOR testing and MWM training phase (Figures 7M–7N). In the MWM probe tests, mice with exogenous PD-L1 injection failed to remember the platform location and the target quadrant (Figures 7O–7Q). Perfusion of brain slices with PD-L1 decreased AP firing rate and increased A-type K+ currents in CA1 neurons of WT mice (Figures 7R–7T). All the effects of PD-L1 were abolished in PD-1 KO mice (Figures S10F–S10H), suggesting that PD-L1 regulates learning and memory and CA1 neuronal excitability via PD-1.
To demonstrate the translational relevance of our findings, we examined PD-1 expression in human hippocampi from two donors. We found widespread expression of PDCD1 mRNA in most CA1 and CA3 neurons (Figure 8A and 8B). Moreover, nivolumab (anti-human PD-1 mAb), but not human IgG, was able to bind CA1 neurons (Figure S11A). In situ hybridization also revealed PDCD1 expression in majority of hippocampal CA1 and CA3 neurons from NHP (Figure 8C and 8D). The specificity of PDCD1 signal was tested by the negative-control probe in NHP and human brain sections (Figures S11B and S11C). Collectively, these data suggest that PDCD1 mRNA and PD-1 protein are extensively expressed by hippocampal neurons of primates.
Figure 8. PDCD1 is widely expressed by human and monkey hippocampal neurons and regulates neuronal excitability in NHP slices.
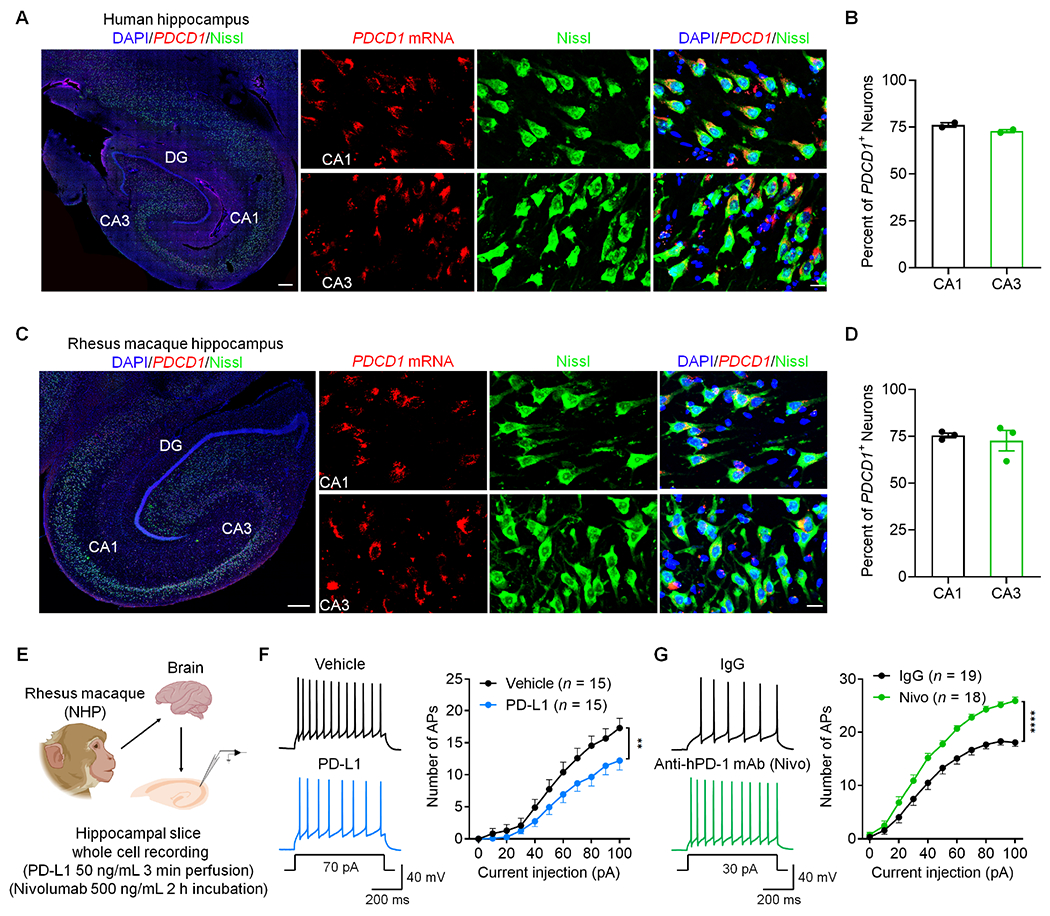
(A) PDCD1 mRNA expression in human hippocampal neurons as shown by in situ hybridization. Left, representative image of the whole hippocampus (Scale bar, 500 μm). Right panels, high magnification images showing PDCD1 mRNA expression in Nissl-labeled neurons (Scale bar, 20 μm).
(B) Quantification of PDCD1+ neurons in the CA1 and CA3 regions of humans.
(C) PDCD1 mRNA expression in NHP hippocampal neurons as shown by in situ hybridization. Left, representative image of the whole hippocampus. Scale bar, 500 μm. Right panels, high magnification images showing PDCD1 mRNA expression in Nissl-labeled neurons. Scale bar, 20 μm.
(D) Quantification of PDCD1+ neurons in the CA1 and CA3 regions of NHPs.
(E) Schematic of experimental design for whole-cell patch clamp recordings in NHP brain slices.
(F) Representative AP traces (left) and quantification of AP firing rate (right) before and after treatment of recombinant monkey PD-L1.
(G) Representative AP traces (left) and quantification of current-evoked APs (right) in NHP hippocampal neurons treated with control IgG and anti-human PD-1 mAb (nivolumab).
Data are represented as mean ± SEM. Also see Figure S11. Sample size and statistical tests are reported in detail in Tables S1 and S5.
Finally, we investigated the functionality of PD-1 in NHP neurons. We prepared brain slices (350-μm thick) for patch-clamp recordings in CA1 neurons from 3 NHPs within 2 h of euthanasia. We treated the brain slices with PD-L1 or nivolumab (Figure 8E) and induced action potentials by current injection. We found that a brief incubation of PD-L1 rapidly suppressed AP firing in monkey CA1 neurons (Figure 8F). Conversely, nivolumab incubation increased AP firing (Figure 8G, vs. IgG control). Thus, the PD-L1/PD-1 axis also regulates excitability of hippocampal neurons in primates.
DISCUSSION
We have revealed a previously unrecognized physiological role of PD-L1 as a neuromodulator that inhibits neuronal activity and excitability in CA1 neurons through PD-1, leading to altered cognitive function in mice. We provided several lines of evidence to support this notion. First, loss of PD-1 in global knockout mice enhanced learning and memory in both NOR and MWM tests and increased LTP in hippocampal CA1 neurons. Second, selective Pdcd1 deletion in excitatory neurons (cKO-excitatory) reproduced behavioral and cellular phenotypes in the global knockout mice. Consistently, injections of AAV-Camk2a-mCherry:Cre virus in the hippocampus of Pdcd1fl/fl mice enhanced spatial memory, whereas re-expression of Pdcd1 in hippocampal neurons in the global knockout mice suppressed spatial memory. In contrast, cKO mice lacking Pdcd1 in microglia/monocytes (cKO-microglia) and cKO mice lacking Pdcd1 in inhibitory neurons (cKO-inhibitory) showed no changes in these memory tests. Third, ICV administration of anti-PD-1 mAb (RMP1-14) enhanced learning and memory in adult WT mice and further improved memory in TBI mice. Fourth, neuronal PD-L1/PD-1 signaling is conservative across different species, as PDCD1 mRNA and PD-1 are extensively expressed in mouse, NHP and human CA1 neurons. Loss of PD-1 function was sufficient to increase action potential firing and decrease A-type potassium currents in CA1 neurons while exogenous PD-L1 suppressed action potential firing and increased A-type potassium currents in mouse CA1 neurons. We also uncovered a dynamic and activity-dependent mechanism governing PD-L1/PD-1 signaling in the nervous system. Under steady-state conditions, PD-L1 can be produced by both glial cells (microglia/astrocyte) and neurons. Intriguingly, neuronal activation by depolarization and intense noxious stimulation suppressed PD-L1 levels in the CSF and brain slice perfusate. Furthermore, PD-L1 and PD-1 expression, including surface expression of PD-1 in excitatory hippocampal neurons, is downregulated after learning (water maze training) and upregulated after TBI, suggesting that PD-L1/PD-1 signaling can be altered by different cognition states.
PD-1 based immunotherapies have achieved success in treating many cancer patients.2,36–39 PD-1 is widely expressed in various cell types, including T cells, macrophages, microglia, and melanoma cells.30,40–42 Increasing evidence suggests PD-1 is expressed by neurons, including retinal ganglion neurons43, primary sensory neurons of dorsal root ganglion11,44, and spinal cord, thalamic, and cortical neurons.12 Single-cell RNA-seq revealed low-level of expression of Pdcd1 mRNA in cortical and hippocampal neurons.13,32 However, single-cell RNA-seq may detect ony the most abundant mRNAs.45 Using more sensitive RNAscope in situ hybridization, our results showed that Pdcd1 mRNA is present in the majority of CA1/CA3 neurons of mice, NHPs, and humans. Since cellular concentrations of proteins and the abundance of their corresponding mRNAs are not necessarily correlated46, it was essential for us to observe specific PD-1 protein expression in hippocampal neurons in different species. Most importantly, we demonstrated that the neuronal PD-1 is functional at both cellular and behavioral levels.
At the cellular level, loss of PD-1 resulted in increased neuronal excitability in CA1 neurons, and strikingly, 40% of Pdcd1-deficient CA1 neurons exhibited spontaneous discharges. Loss of PD-1 also enhanced synaptic plasticity, as indicated by increased EPSC and increased slope of LTP (induced by 1×, 2×, and 4× HFS), associated with increased number of dendritic spines. These activity changes appear to be neuron intrinsic, as PD-1 KO neurons with synaptic input blockade or dissociated neurons in primary cultures of PD-1 KO mice showed increased excitability. However, neuronal survival was not altered in adult PD-1 KO mice. Intriguingly, perfusion of nivolumab but not control IgG also increased excitability of CA1 neurons of NHP, supporting an active role of neuronal PD-1 signaling in primates.
Mechanistically, our results suggest that PD-1-mediated ERK phosphorylation and A-type K+ current modulation may underlie neuronal and synaptic changes in CA1 neurons after PD-1 deficiency (Figure S12). Tyrosine phosphatase SHP-1 was implicated in mediating PD-1’s biological actions in immune cells22 and primary sensory neurons11,44. PD-1 blockade in spinal cord slices resulted in SHP-mediated hyper-phosphorylation of ERK in spinal cord neurons12. ERK activation by phosphorylation is essential for the induction of hippocampal LTP and the formation of the hippocampus-dependent memory.23–26 Our data showed that daily water maze learning training for 4 days was sufficient to induced p-ERK expression in WT CA1 neurons. We also found that PD-1 KO mice exhibited stronger p-ERK expression than control mice following the learning-associated training. Since SHP1/2 is known to mediate the PD-1’s signaling, anti-PD-1 treatment may act as a phosphatase inhibitor to enhance ERK activation. p-ERK enhances learning and memory by inducing dendritic spine formation and stabilization as well as CREB-mediated transcription.47–50 ERK also regulates neuronal excitability through Kv4.2-mediated dendritic A-type K+ currents.28,51 Loss of Kv4.2 was found to eliminate dendritic A-type K+ currents and enhance LTP in hippocampal CA1 neurons.52 A-type K+ currents are at high density in CA1 dendrites and rapid voltage-dependent activation of these channels can limit the peak amplitude of back-propagating action potentials and exert profound effects on hippocampal synaptic functions and memory.27,52,53 Importantly, loss of PD-1 resulted in increased p-ERK expression in CA1 dendrites and marked reduction in Kv4.2-mediated A-type currents, and the latter can be reversed by ERK inhibition. ERK activation following PD-1 suppression also upregulated NMDAR and AMPAR function, as both evoked NMDAR and AMPAR currents were enhanced in CA1 neurons from PD-1 KO mice. Thus, we postulate that ERK activation and subsequent inhibition of A-type K+ currents and activation of NMDAR/AMPAR currents in CA1 neurons may drive the cellular phenotypes (altered neuronal excitability, spine formation, synaptic transmission, and LTP) in PD-1 KO mice (Figure S12). Additionally, anti-PD1-mediated inhibition of GABAergic signaling may also be responsible for neuronal hyperactivity.12
At behavioral levels, we employed loss-of-function approaches in excitatory neuron-specific cKO mice and AAV-treated mice with selective neuronal infection, which demonstrated that lack of excitatory neuronal PD-1 is sufficient to enhance memory. Conversely, our gain-of-function approach through PD-1 re-expression in PD-1-deficient hippocampal neurons impaired memory. By contrast, memory was unaltered in cKO mice lacking PD-1 in microglia/monocytes or inhibitory neurons. Our data strongly suggest a physiological role of PD-1 in memory through direct neuronal modulation. Furthermore, we found a marked improvement in memory by deleting PD-1 in global KO and cKO-excitatory mice, or by intraventricular treatment of anti-PD-1 mAb (RMP1-14) in a mouse model of TBI. Previous studies showed that systemic anti-PD-1 treatment enhanced learning/memory behavior in mouse AD models. This is achieved by increasing the CNS recruitment of immune cells (e.g., macrophages) that can promote the clearance of amyloid-β plaques and alleviate tauopathy.4–6 However, contradictory results were also reported showing no effective removal of amyloid-β plaques by anti-PD-1 treatments.7–9 While immunotherapy is appealing for treating AD, future studies are needed to test whether monotherapy is sufficient or polytherapy is required to increase “immunity” as in some cancer conditions.2,6 Increasing evidence suggests that neuroinflammation after TBI and surgery contributes to cognitive decline, in part through production of pro-inflammatory cytokines.54–56 Millions of cancer survivors suffer from neurological deficits including cognitive decline due to chemotherapy-induced microglial activation, neuroinflammation, and demyelination.57,58 Thus, immunotherapy could be both beneficial and detrimental for cognitive function in a context-dependent manner. Our results showed that intraventricular treatment with anti-PD-1 (RMP1-14) for a short period (a few days) not only enhanced learning and memory in naïve animals but also restored cognitive capability in mice with TBI.
In summary, we have demonstrated a critical role of the PD-L1/PD-1 immune checkpoint pathway in physiological and pathological conditions in which this pathway regulates cognitive behaviors through neuronal modulations. Thus, this immune checkpoint pathway may also serve as a neuronal checkpoint.3,11,12 Expression of PD-1 in hippocampal excitatory neurons suppresses their activity, which dampens learning and memory ability of mice. Consistently, PD-L1/PD-1 signaling is down-regulated in the hippocampus after learning-associated training and upregulated after TBI-induced cognitive decline. Thus, this checkpoint brake provides an opportunity for enhanced learning ability under special conditions by reducing PD-L1/PD-1 levels or function. Recently, it was found that PD-L1+ non-immune cells accumulate in the lung, liver and kidney with age, and furthermore, blocking the PD-L1/PD-1 pathway improves senescence surveillance and ageing phenotypes.59 Similarly, we recently found PD-L1 levels are drastically increased in the CSF of aged mice in our pilot study. Future studies are needed to investigate the role of PD-L1/PD-1 axis in aging-associated dementia.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ru-Rong Ji (ru-rong.ji@duke.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
Adult mice (8-16 wk old) of both sexes were used for behavioral tests, unless specifically described. We did no find sex differences in all the mouse experiments. Mice (5-8 weeks of both sexes) were used for electrophysiological studies. Pdcd1 knockout mice (Stock No:021157), Camk2a-Cre mice (Stock No: 005359), Cx3cr1CreER mice (Stock No: 020940), Vgat-Cre mice (Stock No:028862) and wild-type (WT) mice (Stock No: 000664) were purchased from Jackson Laboratory and maintained at Duke University Animal facilities. Pdcd1-Cre mice were generated by Cyagen Biosciences Inc. Pdcd1fl/fl mice were generated by Dr. Richard Palmiter at the University of Washington. All transgenic mice were maintained on a C57BL/6 background. All the mouse procedures were approved by the Institutional Animal Care & Use Committee of Duke University. Mice were housed under a 12-hour light/dark cycle with food and water available ad libitum. Animals were randomly assigned to each experimental group. Two to five mice were housed in each cage. All behavioral measurements were conducted in a blinded manner and during daytime (light cycle), normally starting at 9 AM. Animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The number of animals and sample size of each experiment were also described in Table S1. If not specified, experiments in this study were conducted in mice. The experiments in this study were done and analyzed by researchers who are blinded to genotype or treatment.
Generation of Pdcd1fl/fl mice and Pdcd1 conditional knockout mice
Pdcd1fl/fl mice were generated at University of Washington. LoxP sites were engineered to flank to second exon of Pdcd1. The 5′ arm (~4 kb with PacI and SalI sites at 5’ and 3’ ends, respectively) and 3′ arm (~6.5 kb with PmeI and NotI sites at 5’ and 3’ ends, respectively) of the Pdcd1 gene were PCR amplified with Q5 polymerase from a C57Bl/6 BAC clone. A loxP site was inserted into the Nhe1 site proximal to exon 2, the correct orientation verified by sequencing, and then the two arms were cloned into polylinkers of a targeting vector (4600C) that contained a loxP site, an frt-flanked Sv40-Neo gene for positive selection, HSV thymidine kinase and pgk-diphtheria toxin A chain genes for negative selection (Figure S4A). The construct was linearized with Asc1, electroporated into G4 ES cells (C57Bl/6 × 129 Sv hybrid) and correct targeting was determined by Southern blot of DNA digested with Xba1 using a 32P-labeled probe upstream of the 5’ arm of the targeting construct. Of the 62 clones analyzed 7 were correctly targeted. One clone that retained the loxP site in the 5’ arm was injected into blastocysts and resulted in good chimeras that transmitted the targeted allele through the germline. Progeny were bred with Gt(Rosa)26Sor-FLP recombinase mice (Stock No: 012930) to remove the frt-flanked SV-Neo gene. Mice were then continuously backcrossed to C57Bl/6 mice. Routine genotyping is performed with 2 primers flanking the loxP site: 5′-CCCTCTCCAAGTCTGAGGTTT-3′ (Pdcd1 F) and 5′-CCTTCCCACTCCTGTTCAAG-3′ (Pdcd1 R). The wild-type allele gives a band of ~150 bp, while the targeted allele with loxP site gives a band of ~ 190 bp. Sometimes there is an additional band ~220 bp that represents a heteroduplex (Figure S4B).
Pdcd1fl/fl; Camk2a-Cre conditional knockout mice (cKO-excitatory neuron) were generated by crossing Pdcd1fl/fl mice with Camk2a-Cre mice to generate the heterozygotes Pdcd1fl/−; Camk2a-Cre and then crossed with Pdcdf1fl/fl mice. Pdcd1fl/fl; Vgat-Cre conditional knockout mice (cKO-inhibitory neuron) were generated by crossing Pdcd1fl/fl mice with Vgat-Cre mice to generate the heterozygotes Pdcd1fl/−; and then crossed with Pdcd1fl/fl mice. Pd1flfl; Cx3cr1CreER conditional knockout mice (cKO-microglia) were generated by crossing Pd1fl/fl mice with Cx3cr1CreER mice to generate the heterozygotes Pdcd1fl/-; Cx3cr1CreER and then crossed with Pdcd1fl/fl mice. Pd1fl/fl; Cx3cr1CreER mice were injected with tamoxifen (75 mg/kg, i.p.) daily for 5 days to delete Pdcd1 in microglial cells. After the final injection, mice were kept for another 7 days before any behavioral or biochemical tests. Pdcd1 cKO mice were validated by flow cytometry, genotyping and immunostaining.
Mouse model of traumatic brain injury (TBI)
The closed-head TBI model33 was established using WT or KO/cKO mice. After anesthesia induction with 4.0% isoflurane, the trachea was intubated, and the lungs were mechanically ventilated with 1.5% isoflurane in 30% O2/70% N2. The mouse head was secured in a stereotactic device and a middle-line scalp incision was made to identify anatomical landmarks. A concave 3-mm metallic disc was adhered to the skull immediately caudal to bregma. A 2.0-mm diameter pneumatic impactor (Air-Power Inc.) was used to deliver a single midline impact to the center of the disc surface. The impactor was discharged at 6.8 ± 0.2 m/s with a head displacement of 2.5 mm. After impact, the animals could recover spontaneous ventilation before the tracheas were extubated. Mice were allowed free access to food and water after recovery. Sham mice were treated identically except for the absence of impact.
Mouse serum and CSF collection
Mouse whole blood was collected from WT mice by submandibular vein puncture. The blood samples were left at room temperature for 20 min and the clot was removed by centrifuging at 2,000 g for 15 min in a refrigerated centrifuge. The supernatant (serum) was stored in −80°C for ELISA tests. Mouse CSF was collected from the cisterna magna by glass pipettes. The inner diameter of the glass tip is10-20 μm.60
Human Hippocampus
Human hippocampus was obtained from donors through National Disease Research Interchange (NDRI) with permission of exemption from the Duke University Institutional Review Board (IRB). Postmortem whole hippocampi were from two donors: a 59-year-old female and a 60-year-old male. Tissues were kept in fixative (10% formalin) upon delivery. There was no sex difference in this study. The donors information was described in Table S2.
Human plasma and CSF samples
Human plasma and CSF samples were collected from a prospective randomized trial registered with http://www.clinicaltrials.gov (NCT01640275) on June 20, 2012, by Miles Berger, the study PI, with approval by the Duke University Institutional Review Board.61 Human plasma was collected before surgery and human CSF samples (10 ml) were obtained from the lumbar drain (AccuDrain INS-8400; Integra Neurosciences, Plainsboro, NJ, USA) at the time of drain placement (0 h).61 We included plasma and CSF samples from the same patients (n = 8) for ELISA analysis of basal PD-L1 levels. No sex difference was noticed. The participants information was described in Table S2.
Monkey hippocampus and CSF samples
NHP hippocampi were obtained from Rhesus macaques (Macaca mulatto) housed in the Wake Forest School of Medicine. Whole live NHP hippocampi were collected from 6 health monkeys for electrophysiology. NHP serum and CSF were collected from four of these monkeys (n = 4). CSF was also collected from 7 monkeys before and after capsaicin treatment (0.1 mg, subcutaneously, 60 min). The details of NHP experiments were described in Table S3. No sex difference was noticed.
Primary neuronal cultures
Hippocampal neuron primary cultures were prepared from embryonic day 17-19 (E17-19) WT and PD-1 KO mice. Embryos were removed from the dams, which were anesthetized with isoflurane and euthanized by decapitation. Hippocampi were dissected and placed in Ca2+- and Mg2+-free Hank’s balanced salt solution (HBSS, GIBCO) and digested at 37°C in a humidified O2 incubator for 30 min with collagenase type II (Worthington, 285 units/mg, 12 mg/mL final concentration) and dispase II (Roche, 1 unit/mg, 20 mg/mL) in HBSS (pH 7.3). Digestion was stopped by DMEM (GIBCO) containing fetal bovine serum. Hippocampi were mechanically dissociated using fire-polished pipettes, filtered through a 100-μm nylon mesh and centrifuged (1,000 g, 5 min). The pellet was resuspended and plated on Poly-D-Lysine/Laminin coated glass coverslips (CORNING), and cells were plated in a DMEM containing 5% fetal bovine serum, 1% premixed penicillin and streptomycin. After a brief culture for 5-6 h, primary cultures were switched to Neurobasal Plus medium containing 2% B27 supplement, 1% GlutaMAX-I, 1% premixed penicillin and streptomycin (GIBCO). Three days after plating, cytosine arabinoside was added to a final concentration of 10 μM to curb glial proliferation. Cultures were kept at 37°C in a 5% CO2-humidified incubator and half of the media was replaced every three days. Whole-cell electrophysiological recording and immunocytochemistry experiments were conducted on DIV 7-8.
METHOD DETAILS
Reagents
Nivolumab (OPDIVO®), a humanized anti-PD-1 antibody, was purchased from Bristol-Myers Squibb (Cat# NDC 0003-3772-11). Human IgG (Cat# ab90286) was purchased from Abcam. Anti-mouse PD-1 monoclonal antibody (RMP1-14, Cat# BE0146) and control IgG (Cat# BE0089), anti-mouse PD-L1 monoclonal antibody (Cat# BE0101) and control IgG (Cat # BE0090) were purchased from Bio X Cell. Recombinant mouse PD-L1 protein (Cat# ab221310), Fc (mouse) IgG2a mimetic protein (inactive control, Cat# ab300223) and recombinant Rhesus monkey PD-L1 (Cat# ab201421) were purchased from Abcam. All drugs and regents for electrophysiology were purchased from Sigma or Tocris. Please find more details in STAR★METHODS and KEY RESOURCES TABLE.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivoMAb anti-mouse PD-1 (CD279, clone: RMP1-14) | Bio X Cell | Cat# BE0146; RRID:AB_10949053 |
| InVivoMAb rat IgG2a isotype control | Bio X Cell | Cat# BE0089; RRID:AB_1107769 |
| InVivoMAb anti-mouse PD-L1 (B7-H1) | Bio X Cell | Cat# BE0101; RRID:AB_10949073 |
| InVivoMAb rat IgG2b isotype control | Bio X Cell | Cat# BE0090; RRID:AB_1107780 |
| Rabbit anti-PD-1 | Sigma | Cat# PRS4065; RRID:AB_1855098 |
| Mouse anti-NeuN | Millipore | Cat# MAB377; RRID:AB_2298772 |
| Guinea pig anti-NeuN | Sigma | Cat# ABN90P; RRID:AB_2341095 |
| Goat anti-tdTomato | Origene | Cat# AB8181-200; RRID:AB_2722750 |
| Rabbit anti-Iba1 | Wako | Cat# 019-19741; RRID:AB_839504 |
| Mouse anti-GFAP | Millipore | Cat# MAB360; RRID:AB_11212597 |
| Anti-mouse CD16/32 | Biolegend | Cat# 156603; RRID:AB_2783137 |
| CD45 Monoclonal Antibody (30-F11), FITC | eBioscience | Cat# 11-0451-82; RRID:AB_465050 |
| PE anti-mouse/human CD 11b Antibody | Biolegend | Cat# 101207; RRID:AB_312790 |
| Monoclonal Anti-Vglut1 - APC antibody | Sigma | Cat# SAB5200268-100UG |
| APC/Cyanine7 anti-mouse CD279 (PD-1) Antibody | BioLegend | Cat# 135223; RRID:AB_2563522 |
| APC/Cy7 anti-mouse/human CD11b | Biolegend | Cat# 101225; RRID:AB_830641 |
| PE anti-mouse CD279 (PD-1) | Biolegend | Cat# 135205; RRID:AB_1877232 |
| CaMKII alpha | Thermo Fisher Scientific | Cat# PA5-19128; RRID:AB_10986857 |
| Nivolumab (human PD-1) | Bristol-Myers Squibb | Cat# NDC 0003-3772-11 |
| Rabbit anti-PD-L1 | Abcam | Cat# ab233482; RRID:AB_2811045 |
| Anti-mouse IgG (H+L), Biotinylated Antibody | Cell Signaling Technology | Cat# 14709; RRID:AB_2798582 |
| Anti-Human vivo 680 | Sigma | Cat# I2511; RRID:AB_1163604 |
| FITC Rat IgG2a, κ Isotype Control | BD Phanningen | Cat# 557228; RRID:AB_396607 |
| Isotype Control Antibody, rat IgG1, PE | Miltenyl Biotech | Cat# 130-123-746; RRID:AB_2857627 |
| Bacterial and virus strains | ||
| AAV-CamKII-mPDCD1-IRES-eGFP-WPRE | Vector Biolabs | N/A |
| AAV-CamKII-mCherry-Cre | UNC Vector Core | N/A |
| AAV-CamKII-eGFP-WPRE | James M. Wilson lab (University of Pennsylvania) | Cat# 105541-AAV9; RRID:Addgene_105541 |
| Biological samples | ||
| Human hippocampus tissue | National Disease Research interchange (NDRI) See Table S2 for sex and age information | N/A |
| Human plasma and CSF sample | MAD-PIA clinical trial See Table S2 for sex and age information | N/A |
| Monkey hippocampus tissue | Wake Forest University See Table S3 for sex and age information | N/A |
| Monkey serum and CSF sample | Wake Forest University See Table S3 for sex and age information | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Nissl | Thermo Scientific | Cat# N21483 |
| Neurobiotin | Vector Laboratories | Cat# SP-1120 |
| Alexa Fluor® 594 streptavidin | Invitrogen | Cat# S32356 |
| Tetrodotoxin (TTX) | Tocris | Cat# 1078 |
| Picrotoxin | Tocris | Cat# 1128 |
| CNQX | Tocris | Cat# 0190 |
| DL-AP5 | Tocris | Cat# 0105 |
| U0126 | InvivoGen | Cat# tlrl-u0126 |
| Human IgG4 | Abcam | Cat# ab90286 |
| Human PD-1-Biotin | Acrobiosystems | Cat# EP101 |
| Human PD-L1-Biotin | Acrobiosystems | Cat# EP158 |
| Recombinant mouse PD-L1 protein | Abcam | Cat# ab221310 |
| Recombinant Mouse IgG2a protein | Abcam | Cat# ab300223 |
| Recombinant Rhesus monkey PD-L1 protein | Abcam | Cat# ab201421 |
| RNAscope probe mouse Pdcd1 | Advanced Cell Diagnostics | Cat# 416781 |
| RNAscope probe human PDCD1 | Advanced Cell Diagnostics | Cat# 602021 |
| RNAscope probe Mmu PDCD1 | Advanced Cell Diagnostics | N/A |
| RNAscope probe mouse Cd274 | Advanced Cell Diagnostics | Cat# 420501 |
| RNAscope Negative control probe | Advanced Cell Diagnostics | Cat# 700141 |
| Critical commercial assays | ||
| RNAscope® Multiplex Fluorescent Reagent Kit v2 | Advanced Cell Diagnostics | Cat# 323100 |
| TSA™ Cyanine 3&5, TMR, Fluorescein Evaluation Kit | PerkinElmer | Cat# NEL760001KT |
| Zombie Violet™ Fixable Viability Kit | Biolegend | Cat# 423113 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J (Wildtype) | The Jackson Laboratory | Stock No: 000664; RRID:IMSR_JAX:000664 |
| Mouse: Pdcd1−/− | The Jackson Laboratory | Stock No: 021157; RRID:IMSR_JAX:021157 |
| Mouse: Camk2a-Cre | The Jackson Laboratory | Stock No: 005359; RRID:IMSR_JAX:005359 |
| Mouse: Vgat-Cre | The Jackson Laboratory | Stock No: 028862; RRID:IMSR_JAX:028862 |
| Mouse: Cx3cr1CreER | The Jackson Laboratory | Stock No: 020940; RRID:IMSR_JAX:020940 |
| Mouse: Pdcd1fl/fl | Richard Palmiter lab (University of Washington) Generated only for this study | N/A |
| Mouse: Pdcd1-Cre | Cyagen Biosciences Inc. | N/A |
| Mouse: Ai9 | The Jackson Laboratory | Stock No: 007909; RRID:IMSR_JAX:007909 |
| Oligonucleotides | ||
| See Table S4 for Sequences of genotyping primers. | N/A | N/A |
| Software and algorithms | ||
| Image J | NIH | https://imagej.nih.gov/ij/ |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Zen lite | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Leica LAS X | Leica | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| ANY-maze | Stoelting Co. | https://www.anymaze.co.uk/index.htm |
| Imaris | Bitplane | https://imaris.oxinst.com/ |
| Patchmaster | HEKA | https://www.heka.com/downloads/downloads_main.html#down_patchmaster |
| Clampfit | Molecular Devices | https://www.moleculardevices.com/products/axon-patch-clamp-system/acquisition-and-analysis-software/pclamp-software-suite#gref |
| MiniAnalysis | Synaptosoft | http://www.synaptosoft.com/MiniAnalysis/ |
| FACSDiva software v8 | BD Bioscience | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software |
| Flow Jo™ v10 | BD Bioscience | https://www.flowjo.com/solutions/flowjo/downloads |
Stereotaxic surgery and drug delivery
For stereotaxic surgery, mice were anesthetized with isoflurane (4% for induction and 1.5% thereafter for maintenance), and their heads were fixed in a stereotaxic apparatus (David Kopf Instruments). A guide cannula (62004, RWD Life Science) was stereotaxically implanted into the left lateral ventricles (AP: −0.3 mm; ML: +1.3 mm; DV: −2.0 mm) based on the mouse brain atlas.62 After a period of 4-day recovery, an infusion needle (62204, RWD Life Science) was inserted into lateral ventricles through the guide cannula to a depth of 2.5 mm for drug injection. Following the completion of the infusion, the needle was left for an additional 2 min to limit reflux. Control IgG or anti-PD-1 antibody (RMP1-14) was injected for three to six times before behavioral testing. Control IgG or Anti-PD-L1 antibody was injected for three to six times before behavioral testing. The drug was administered at 1 μg per mouse every other day. PD-L1 (1 μg/mouse) or inactive control protein (1 μg/mouse) was injected 1 h before behavioral testing.
In vivo stereotactic viral injections
Mice were anesthetized with isoflurane (4% for induction and 1.5% thereafter for maintenance) for stereotaxic injection of viruses into the CA1 (AP: −1.8 mm, ML: ±1.4 mm, DV: −1.5 mm) and CA3 (AP: −1.8 mm; ML: ±2.2 mm; DV: −2.2 mm) for adult mice and CA1 (AP: −1.7 mm; ML: ±2.0 mm; DV: −2.1 mm) for 4-week-old mice. We injected 500 nL of virus into each location at a rate of 60 nl min−1 using the UltraMicroPump injection system (World Precision Instruments). The targeted hippocampal regions are shown in Figure 5A-5B and Figures S8A-S8B. After each injection, the needle was left in place for an additional 10 min for efficient diffusion of the virus-containing reagent and then slowly withdrawn.
For selective re-expression of PD-1 in hippocampal neurons, PD-1 KO mice were micro-injected in the bilateral CA1 and CA3 with AAV- Camk2a (0.4)-Pdcd1-IRES-eGFP-WPRE virus (Vector Biolabs). For selective knockout of PD-1 in hippocampal neurons, Pdcd1fl/fl mice were micro-injected in the bilateral CA1 and CA3 with AAV-Camk2a-mCherry:Cre virus (UNC Vector Core). AAV-Camk2a-eGFP-WPRE virus was injected as a control. Behavioral experiments or electrophysiological recordings were performed at least 3 weeks after virus injection. Virus infection was examined at the end of all the tests.
Novel object recognition (NOR) test
On the first day of experiment, mice were placed in a 30 × 30 × 30 cm3 square arena (TAP plastics) for habituation. On the second day, two identical objects were placed in two distinct corners of the arena. Mice were then returned to their home cages for a 0.5-h or 24-h retention interval, and one of two identical objects was replaced by a new object for novel object recognition test.63 After the retention interval, animals were placed back into the testing arena for 5 min of exploration. A valid exploration was defined as the mouse touching an object with its nose or focusing attention on the object at < 1 cm. Turning around, climbing, and siting on the object was all considered to be invalid. A discrimination index was used to evaluate the scores of exploration time for the familiar or novel object as DI = (TN − TF)/(TN + TF) × 100%. Animal behaviors were video-recorded and analyzed by an experimenter who was blind to the testing conditions. Data were excluded if the total exploration time was less than 10 s.
Morris water maze (MWM) test
The Morris water maze test was conducted in a circular pool (diameter 120 cm) filled with 40-cm height water maintained at 23 ± 1°C. The water was made opaque with white milk powder. The pool is in an experimental room with several visual cues and divided into four equal quadrants. A circular platform, 8 cm in diameter, was placed in the middle of one fixed quadrant of the pool, just 1 cm underneath the water surface. During the hidden platform training process, mice were trained for four trials each day for four consecutive days. Each mouse could swim for 60 s to find the platform. If the mouse failed to find the platform within the allotted time, it was picked up and placed on the platform for 5 s. The interval between trials was 20 min. In the probe test (day 5), the platform was removed and all mice were given one probe trial consisting of 60 s searching.64 If the mouse failed to cross the platform zone, the latency to platform was defined as 60 s. The swimming speed, latency to platform zone, and number of platform zone crossings, as well as the percentile time spent in different quadrants were automatically recorded and analyzed by ANYmaze (Stoelting Co.).
Rotarod test
A total of six trials for the rotarod test were carried out using an accelerating protocol from 4 to 60 rpm in 300 s with 20-min inter-trial intervals. Three testing sessions every day were performed for 2 days. For TBI mice, the rotarod test was measured before TBI induction and on day1, 2, 3, 4, 5, 6, 7, 10, 14 and 21 after TBI. After falling, the mice were returned to their home cages, and the latency to fall was automatically recorded by the rotarod system (IITC Life Science Inc.). The latency was recorded as 300 s if the mice stayed on the rotarod more than 300 s.
Open filed test
The open field test was performed in a 45 × 45 × 45 cm3 square arena (TAP plastics) over 15 min. The center of the arena was defined as a center area that covered 50% of the total area. The total distance travelled and time spent in the center area were automatically recorded and analyzed by ANY-maze (Stoelting Co.).
Elevated plus maze test
The elevated plus maze apparatus (Stoelting Co.) is comprised of two open arms (35 × 5 cm2) and two closed arms (35 × 5 × 15 cm3) elevated 50 cm above the ground. Mice were placed in the center facing one of the two open arms and allowed to explore for 6 min. Anxiety-like behavior was assessed by time travelled within the open arms. The video and data were automatically recorded and analyzed by ANY-maze (Stoelting Co.).
Tail suspension test
Mice were suspended by their tails with adhesive tape (1 cm from the tail tip) and ~15 cm away from the surface. Plastic tubes were placed over the tails to ensure animals could neither climb nor hang on to their tail. The animals were video recorded for 6 min, and the time spent immobile was quantitated by an experimenter who was blind to the testing conditions. Mice were considered immobile only when they hung passively and motionlessly for at least 2 s.
Mouse brain slice preparation for electrophysiological and ELISA analyses
Mice were anesthetized with isoflurane and then decapitated. The brain was quickly transferred to ice-cold artificial sucrose-based cerebrospinal fluid (ACSF) containing (in mM): Sucrose 75, NaCl 87, KCl 2.5, NaH2PO4 1.25, CaCl2 0.5, MgCl2 7, NaHCO3 26 and glucose 25. Hippocampal slices (200-350 μm) were cut using a vibratome (Leica VT1200S). Subsequently, slices were transferred to normal ACSF solution (in mM): NaCl 124, KCl 3, NaH2PO4 1.25, CaCl2, MgSO4 1, NaHCO3 26, glucose 10 at 34°C. All extracellular solutions were constantly carbogenated (95% O2, 5% CO2). In general, slices were kept in ACSF for at least 1 h before recording. For PD-L1 experiments, slices were perfused with mouse PD-L1 (50 ng/mL) for 3 min. For assessing PD-L1 release in brain slice perfusate, we included three mouse brain slices (350 μm) in a culture well and incubated these slices with 500 μL normal ACSF (0-60 min), or together with KCL (90 mM), TTX (1 μM), minocycline (1 μM) or L-α-aminoadipate (100 μM) for 15 min, and then we collected the ACSF perfusate (200 μL) for PD-L1 ELISA analysis. Brain slices were kept oxygenated during experiment.
NHP hippocampal slice preparation for electrophysiological experiments
The whole hippocampus was dissected from disease-free monkeys and quickly transferred to ice-cold artificial sucrose-based cerebrospinal fluid (ACSF) containing (in mM): Sucrose 248, KCl 1, CaCl2 1, MgCl2 10, NaHCO3, 26 and glucose 10. Hippocampal slices (350 μm) were cut in ACSF with a Leica Vibratome (Leica VT1200S) within 2 hours after surgical resection (tissues were collected at Wake Forest University and immediately transferred to Duke University for slice preparation). The slices were placed in an incubation chamber at room temperature with oxygenated recording solution for 1-5 h. The recording solution had the following composition (in mM): NaCl 124, KCl 4, CaCl2 2, MgCl2 2, NaHCO3 26 and glucose 10. For PD-1 blockade expriments, slices were incubated with 500 ng/mL control IgG or nivolumab for 2 h before recording. For PD-L1 experiments, slices were perfused with monkey PD-L1 (50 ng/mL) for 3 min.
Whole-cell patch clamp recordings in mouse and NHP hippocampal slices and mouse hippocampal neurons of primary cultures
Whole-cell recordings were performed at 34 ± 1°C with the help of an automatic temperature controller (Warner instruments) with an EPC10 amplifier (HEKA). Data were low-pass-filtered at 2 KHz and sampled at 10 KHz. Patch pipettes were filled with a solution containing the following (in mM): K-gluconate 135, KCl 5, CaCl2 0.5, MgCl2 2, HEPE 5, EGTA 5, MgATP 5 (pH 7.3, 290ߝ300 mOsm/L). When filled with the pipette solution, the resistance of the pipettes was 4-8 MΩ. RMPs, spontaneous spikes and action potentials were recorded in current-clamp mode. The action potentials were evoked by current injection steps (0 to 130 pA, 10 pA step). For synaptic input blockade, 50 μM picrotoxin, 50 μM DL-AP5 and 20 μM CNQX were added to block GABAA receptors, AMPA receptors and NMDA receptors. Data were analyzed by Patchmaster software (HEKA). For mEPSC recording, neuron was hold at −70 mV in the presence of 0.5 μM TTX and 50 μM picrotoxin to block Na+ currents and GABAA receptors. mEPSC were analyzed using Mini Analysis (Synaptosoft Inc.). For A type K+ current recording, the membrane voltage was held at −80 mV in the presence of 0.5 μM TTX and 2 mM CoCl2 to block voltage-gated Na+ currents and Ca2+ currents, and transient K+ currents (IA) were isolated by a two-step voltage protocol.51 Briefly, a total outward current was evoked by a command potential of +40 mV from a holding potential of −80 mV. The A-type current was dissected away from the sustained current by a voltage protocol (a 150-ms pre-pulse to −10 mV allowing the transient channels to inactivate, leaving only the sustained current). A-type current is isolated by subtraction of the sustained current from the total current.
For recording evoked EPSCs mediated by NMDAR and AMPAR, patch pipettes were filled with a solution containing the following (in mM, pH 7.3 adjusted with CsOH): CsMeSO4 125, CsCl 20, NaCl 10, MgATP 2, HEPES 10, EGTA 0.2, Na3GTP 0.3, QX314 2.5. Neurons were hold at +40 mV to record NMDAR-mediated EPSCs with addition of 50 μM picrotoxin and 20 μM CNQX to block GABAA and AMPA receptors. Neurons were hold at −70 mV to record AMPAR-mediated EPSCs with addition of 50 μM picrotoxin and 50 μM DL-AP5 to block GABAA and NMDA receptors. EPSCs were evoked with a bipolar tungsten electrode placed in Schaffer collaterals using an isolator (World Precision Instruments). To avoid the induction of action potentials in PD-1 KO neurons, the stimuli were kept in a range of low-intensity ranging from 30 μA to 90 μA (15 μA step).
Extracellular field recordings of LTP in brain slices
To record extracellular field excitatory postsynaptic potentials (fEPSP) in mice, a bipolar tungsten stimulating electrode was placed along the Schaffer collateral fibers in a hippocampus slice to deliver test and conditioning stimuli, and a glass recording electrode (4-8 MΩ, filled with ACSF) was placed in the stratum radiatum of the CA1 region, 150-200 μm away from the stimulating electrode. The intensity of the stimulation was adjusted to produce a fEPSP with an amplitude of 30-40% of the maximum response. Test stimulation was delivered once per 30 s. Once a stable test response was attained, fEPSP baselines were recorded every 30 s for 10 min. Then, the LTP was induced by 1× HFS (100 Hz for 1 s), 2× HFS (100 Hz for 1 s, 30 s interval) or 4 × HFS (100 Hz for 1 s, 5 min interval).20 After the LTP induction, fEPSP was recorded for additional 60-120 min. The slope of fEPSP was analyzed by Patchmaster software (HEKA).
Visualization of dendrites in hippocampal neurons
To visualize the dendritic spines of mouse CA1 neurons, Neurobiotin (0.2%, Vector Laboratories) was dissolved in the intracellular pipette solution. After 30 min of whole-cell patch recording, the slices were fixed with 4% paraformaldehyde (PFA) in PBS and then processed using Alexa Fluor 594 streptavidin (1:500, Life Technologies, Cat# S32356) for visualization. Apical dendrites of CA1 neurons were imaged using a Zeiss LSM 880 with Airyscan Microscope with a 63× oil-immersion objective, and 3× optical zoom set to a Z-stack width of 0.2 mm. Imaris software (v.9.3.0, Bitplane) was used to reconstruct dendritic spines and analyze the spine density and head diameter. The dendrites were imaged from 3-4 mice for each group and analyzed similar to other studies.65,66
In situ hybridization in mouse, NHP, and human brain sections
Mice were deeply anesthetized with isoflurane and perfused with PBS, followed by 4% PFA. Following perfusion, mouse brain was isolated and post-fixed overnight at 4°C in 4% PFA. Human brain tissues (NDRI) were delivered within 48 h of dissection and fixed in 10% formalin for >24 h. For NHP tissues, the whole hippocampus was post-fixed with 10% formalin for >24 h after dissection. After dehydration in 30% sucrose, all the tissues were embedded in OCT medium and cryosectioned to produce 14-μm tissue sections which were mounted onto the charged slides. In situ hybridization was performed using RNAscope® Multiplex Fluorescent Reagent Kit v2 (Advanced Cell Diagnostics, Cat# 323100) according to the manufacturer’s instructions. We used probes directed against mouse Cd274 (PD-L1, Cat# 420501), mouse Pdcd1 (Cat# 416781), human PDCD1 (Cat# 602021) and customized monkey PDCD1. Control probe (Cat# 700141) was used as a negative control. Following the completion of the RNAscope protocol, immunohistochemistry was performed for double staining as described in the next section. To quantify the percentages of mouse Pdcd1 and NHP and human PDCD1 mRNA expression in hippocampal CA1 and CA3 neurons, three randomly selected fields from CA1 and CA3 regions were analyzed from 2-3 sections of 4 mouse, 3 NHP, and 2 human samples.
Immunohistochemistry and imaging
Brain tissue sections (14 μm) and free-floating brain sections (30 μm) were cut in a cryostat (Leica CM1950). Tissue sections were washed several times in PBS and blocked with 0.1% Triton X-100 and 5% donkey serum for 1 h at room temperature. The sections were then incubated overnight at 4°C in a humidified chamber with the following primary antibodies: anti-PD-1 antibody (rabbit, 1:300, Sigma, Cat# PRS4065), anti-NeuN antibody (mouse, 1:1000, Millipore, Cat# MAB377), anti-NeuN antibody (Guinea pig, 1:500, Sigma, Cat# ABN90P), anti-IBA-1 antibody (rabbit, 1:500, Wako, Cat# 019-19741), anti-GFAP antibody (mouse, 1:500, Millipore, Cat# MAB360) and anti-PD-1 antibody (nivolumab vivo 680, 1:1000, Cat# NDC 0003-3772-11), anti-phosphop44/42 MAPK (p-Erk1/2) antibody (rabbit, 1:200, Cell Signaling Technology, Cat# 9101). After washing, the sections were incubated with Nissl/Neuro Tracer 530/615 or 640/660 (1:100, Thermo Scientific, Cat# N21482/N21483) or species-specific secondary antibodies conjugated to 488-nm, 555-nm or 633-nm fluorophores (1:500, Jackson ImmunoResearch) for 2 h at room temperature. Sections were subsequently washed and coverslipped using Fluoroshield™ with DAPI (Sigma, Cat# F6057). The stained sections were examined with a Leica SP5 or Zeiss 880 confocal microscope with Z-stack and Tile Scan. The maximum projections and stitch were produced using the Zeiss Zen software. To confirm the specificity of PD-1 antibody, blocking experiments were conducted in brain sections using a mixture of anti-PD-1 antibody (1:300) and immunizing blocking peptide (1:300, Sigma, Catalog: SBP4065), based on a protocol recommended for blockade with immunizing peptide (www.abcam.com/technical). The specificity of PD-1 antibody was also tested in brain sections of PD-1 KO mice. To determine if there is neuronal loss in mice, NeuN/Nissl-positive neurons were quantified in CA1 and CA3 regions by Image J. Two or three sections were analyzed in each mouse and three or four mice per group were analyzed. All the images in the same experiment were obtained using the same settings and all analyses and quantifications were performed blinded to the experimental condition.
Immunocytochemistry in mouse primary cultures of hippocampal neurons
Immunocytochemistry was performed in hippocampal cultures (DIV 7-8) prepared from E17-19 WT and PD-1 KO embryos. Neurons were grown on coverslips and fixed in 4% PFA for 30 min. After washing, the coverslips with neurons were blocked with a solution containing 0.1% Triton X-100 and 5% donkey serum for 1 h at room temperature and then incubated overnight at 4°C with the following primary antibodies: anti-PD-1 antibody (rabbit, 1:300, Sigma, Cat# PRS4065), and anti-NeuN antibody (mouse, 1:1000, Millipore, Cat# MAB377). Then, the sections were incubated with species-specific secondary antibodies conjugated to 488-nm, 555-nm fluorophores secondary antibodies (1:500; Jackson ImmunoResearch) for 1 h at room temperature. Sections were subsequently washed and coverslipped using Fluoroshield™ with DAPI (Sigma, Cat# F6057). The stained neurons were examined with ZEISS LSM 880 confocal microscope.
Flow cytometry in mouse hippocampi
Flow cytometry was conducted to characterize PD-L1 and PD-1 expression in neurons and microglia in the hippocampus and validate cKO mice. The brain tissues were isolated and digested with 1 mg/mL collagenase (Roche) for 10 min at 4°C. The digested tissue was resuspended with PBS + EDTA at 4°C and 40% Ficoll gradient (Roche) was added to remove lipid debris. To detect intracellular epitope, the cells were fixed with 4% PFA for 10 min at 4°C and incubated with Fc receptor blocking buffer (1 μg/mL anti-mouse CD16/CD32, 2.4 G2, 2% FBS, 5% NRS, and 2% NMS 0.1 % Triton-X 100 in HBSS; BD Bioscience), and then stained with a standard panel of antibodies (see Key resource table). After staining, all cells were suspended in PBS with EDTA. Live flow cytometry was also conducted to characterize the percentage of membrane PD-1+ excitatory neurons in the hippocampus. Isolated hippocampal tissues were digested by mild mechanical dissociation with cold PBS containing 4 mM EDTA. The isolated cells were filtered through a 100 μm cell strainer and incubated with flow cytometry blocking solution (2% BSA+ PBS + 2 mM EDTA + 1 μg/ml CD16/32) in a cold room with shaking (100 RPM). The antibody mixture (1:200) was incubated with the blocking solution for 40 min in a cold room. Dissociated cells were washed 5 times using PBS containing 2 mM EDTA before analysis. Flow cytometry events were acquired using a BD FACS Canto II flow cytometer with the FACSDiva software v8 software (BD Bioscience). The FCS files were analyzed using FlowJo™ v10 (BD bioscience). The gating strategies were shown in Figures S2L and S4C.
Enzyme-linked immunosorbent assay (ELISA)
Mouse PD-L1/B7-H1 ELISA Kit (PicoKine™) was purchased from Boster Biological Technology (Cat# EK1450). Human/Monkey PD-L1/B7-H1 Quantikine ELISA Kit was from R&D system (Cat# DB7H10). To examine the PD-L1 levels from different species, we included 5-10 μL of CSF (5 -10 μL for mouse and 10 μL for NHP and human) and 20 μL of serum (mouse and NHP,) and plasma (human) for each ELISA test. To detect PD-L1 levels from mouse brain slices incubated with different drugs, we collected 200 μL of perfusates for each sample. ELISA assays were conducted according to the manufacturer’s instructions. The absorbance was measured in 450 nm wavelength using a 96-well xMark™ Microplate plate reader (BioRad). The PD-L1 levels were calculated by standard curve and analyzed using microplate manager 6 software (BioRad). Standard curve was included for each measurement.
Biotin detection of plasma membrane proteins and ELISA in mouse hippocampi
EZ-LinkTM Sulfo-NHS-Biotin solution (Thermo Scientific, Cat# 21217) was used to biotinylate membrane proteins in dissociated cells. The biotinylated cells were digested with RIPA buffer (Sigma) containing a protease inhibitor (Sigma). The BCA assay was used to calculate the protein concentrations in tissue lysates, and 1 mg of proteins were added to streptavidin-coated 96-well plates (Thermo Scientific, Cat# 15500). After blocking with 2% BSA, the captured proteins and standard curve proteins (biotinylated PD-1/PD-L1, Acrobiosystems, Cat# EP101/EP158) were incubated with nivolumab (human, 1 μg/ml, Bristol-Myers Squibb, Cat# NDC 0003-3772-11), anti-PD-L1 (rabbit, 1 μg/ml, Abcam, Cat# ab233482), and biotinylated IgG antibody (mouse, 1 μg/ml, Cell signaling, Cat# 14709). The primary antibody was detected by anti-human HRP for PD-1, anti-rabbit HRP for PD-L1, anti-mouse HRP for the control. HRP activity was developed by TMB solution, terminated by stop solution, and detected at O.D. 450. The biotinylated protein levels were calculated by a standard curve.
QUNATIFICAITON AND STATISTICAL ANALYSIS
All data were expressed as the mean ± SEM. The sample size and statistical analysis for each experiment were indicated in the figures and figure legends. Sample sizes were estimated based on our previous studies for similar types of behavioral, biochemical, and electrophysiological analyses.67–70 For electrophysiology data, each data point corresponds to an individual neuron, and neurons were collected from at least 3 separate animals/subjects for analysis in all cases. For behavioral studies, each data point corresponds to an individual animal. For ELISA analyses in CSF and serum/plasma samples and flow cytometry experiments in brain samples, each data point corresponds to an individual animal or NHP/human. Unless otherwise noted in figure legends, behavioral and biochemical data and some electrophysiological data were analyzed using two-way or one-way ANOVA with Bonferroni’s or Tukey’s post-hoc test to account for multiple comparisons. A two-tailed t-test (unpaired or paired) or Two-tailed Mann-Whitney test was utilized to compare differences between two groups. The criterion for statistical significance was defined as P < 0.05. Asterisks indicate the following significance levels: *P < 0.05, **P < 0.01, ***P < 0.001, ****p < 0.0001; ns: no significant. Statistical analyses were completed with Prism GraphPad 8.0. The detailed statistical analyses are also described in Table S5. All the experiments in this study were done and analyzed by researchers who are blinded to genotype or treatment.
Supplementary Material
Highlights.
Adult mice lacking Pdcd1 in hippocampal neurons exhibit enhanced memory and LTP.
Cognitive deficits after traumatic brain injury are improved by anti-PD-1 treatment.
Secreted PD-L1 regulates neuronal excitability and learning/memory through PD-1.
Learning and traumatic brain injury differentially regulate PD-L1/PD-1 signaling.
ACKNOWLEDGMENTS
We thank Ji lab members for helpful discussion and Huiping Ding for assistance to monkey tissue collection. This study was supported by Duke University Anesthesiology Research Funds and partially supported by NIH grant DE17794 and DoD grants W81XWH2110885 and W81XWH2210267. Generation of Pdcd1 floxed mice was supported by HHMI (R.D.P). The NHP study was supported by NIH grant AR069861.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors have no completing financial interests in this study.
REFERENCES
- 1.Ishida Y, Agata Y, Shibahara K, and Honjo T (1992). Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 11, 3887–3895. 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma P, and Allison JP (2015). The future of immune checkpoint therapy. Science 348, 56–61. 348/6230/56 [pii]; 10.1126/science.aaa8172 [doi]. [DOI] [PubMed] [Google Scholar]
- 3.Zhao J, Roberts A, Wang Z, Savage J, and Ji RR (2021). Emerging Role of PD-1 in the Central Nervous System and Brain Diseases. Neurosci Bull. 10.1007/sl2264-021-00683-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baruch K, Deczkowska A, Rosenzweig N, Tsitsou-Kampeli A, Sharif AM, Matcovitch-Natan O, Kertser A, David E, Amit I, and Schwartz M (2016). PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat Med 22, 135–137. 10.1038/nm.4022. [DOI] [PubMed] [Google Scholar]
- 5.Rosenzweig N, Dvir-Szternfeld R, Tsitsou-Kampeli A, Keren-Shaul H, Ben-Yehuda H, Weill-Raynal P, Cahalon L, Kertser A, Baruch K, Amit I, et al. (2019). PD-1/PD-L1 checkpoint blockade harnesses monocyte-derived macrophages to combat cognitive impairment in a tauopathy mouse model. Nat Commun 10, 465. 10.1038/s41467-019-08352-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xing Z, Zuo Z, Hu D, Zheng X, Wang X, Yuan L, Zhou L, Qi F, and Yao Z (2021). Influenza vaccine combined with moderate-dose PD1 blockade reduces amyloid-beta accumulation and improves cognition in APP/PS1 mice. Brain Behav Immun 91, 128–141. 10.1016/j.bbi.2020.09.015. [DOI] [PubMed] [Google Scholar]
- 7.Latta-Mahieu M, Elmer B, Bretteville A, Wang Y, Lopez-Grancha M, Goniot P, Moindrot N, Ferrari P, Blanc V, Schussler N, et al. (2018). Systemic immune-checkpoint blockade with anti-PD1 antibodies does not alter cerebral amyloid-beta burden in several amyloid transgenic mouse models. Glia 66, 492–504. 10.1002/glia.23260. [DOI] [PubMed] [Google Scholar]
- 8.Obst J, Mancuso R, Simon E, and Gomez-Nicola D (2018). PD-1 deficiency is not sufficient to induce myeloid mobilization to the brain or alter the inflammatory profile during chronic neurodegeneration. Brain Behav Immun 73, 708–716. 10.1016/jbbi.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin Y, Rajamohamedsait HB, Sandusky-Beltran LA, Gamallo-Lana B, Mar A, and Sigurdsson EM (2019). Chronic PD-1 Checkpoint Blockade Does Not Affect Cognition or Promote Tau Clearance in a Tauopathy Mouse Model. Front Aging Neurosci 11, 377. 10.3389/fnagi.2019.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Jiang C, He Q, Matsuda M, Han Q, Wang K, Bang S, Ding H, Ko MC, and Ji RR (2020). Anti-PD-1 treatment impairs opioid antinociception in rodents and nonhuman primates. Sci Transl Med 12. 10.1126/scitranslmed.aaw6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen G, Kim YH, Li H, Luo H, Liu DL, Zhang ZJ, Lay M, Chang W, Zhang YQ, and Ji RR (2017). PD-L1 inhibits acute and chronic pain by suppressing nociceptive neuron activity via PD-1. Nat Neurosci 20, 917–926. 10.1038/nn.4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang C, Wang Z, Donnelly CR, Wang K, Andriessen AS, Tao X, Matsuda M, Zhao J, and Ji RR (2020). PD-1 Regulates GABAergic Neurotransmission and GABA-Mediated Analgesia and Anesthesia. iScience 23, 101570. 10.1016/j.isci.2020.101570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van der Zwan J, Haring M, Braun E, Borm LE, La Manno G, et al. (2018). Molecular Architecture of the Mouse Nervous System. Cell 174, 999–1014 e1022. 10.1016/j.cell.2018.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, and Waxham MN (1989). An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature 340, 554–557. 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- 15.Volk LJ, Bachman JL, Johnson R, Yu Y, and Huganir RL (2013). PKM-zeta is not required for hippocampal synaptic plasticity, learning and memory. Nature 493, 420–423. 10.1038/naturell802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park HR, Shiva A, Cummings P, Kim S, Kim S, Lee E, Leong A, Chowdhury S, Shawber C, Carvajal R, et al. (2023). Angiopoietin-2-dependent spatial vascular destabilization promotes T-cell exclusion and limits immunotherapy in melanoma. Cancer Res. 10.1158/0008-5472.CAN-22-2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan H, Ding Z, Zhang C, Yan J, Yang Y, and Li P (2021). The Programmed Cell Death Ligand-1/Programmed Cell Death-1 Pathway Mediates Pregnancy-Induced Analgesia via Regulating Spinal Inflammatory Cytokines. Anesth Analg 133, 1321–1330. 10.1213/ANE.0000000000005737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds AM, et al. (2016). Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med 22, 1402–1410. 10.1038/nm.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin SJ, Grimwood PD, and Morris RG (2000). Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci 23, 649–711. 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- 20.Shi H, Zhang X, Weng YL, Lu Z, Liu Y, Lu Z, Li J, Hao P, Zhang Y, Zhang F, et al. (2018). m(6)A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature 563, 249–253. 10.1038/s41586-018-0666-l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuzaki M, Honkura N, Ellis-Davies GC, and Kasai H (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766. 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hebeisen M, Baitsch L, Presotto D, Baumgaertner P, Romero P, Michielin O, Speiser DE, and Rufer N (2013). SHP-1 phosphatase activity counteracts increased T cell receptor affinity. J Clin.Invest 123, 1044–1056. 65325 [pii]; 10.1172/JCI65325 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.English JD, and Sweatt JD (1996). Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem 271, 24329–24332. 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- 24.English JD, and Sweatt JD (1997). A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem 272, 19103–19106. 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 25.Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, and Sweatt JD (1998). The MAPK cascade is required for mammalian associative learning. Nat Neurosci 1, 602–609. 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- 26.Sindreu C, Palmiter RD, and Storm DR (2011). Zinc transporter ZnT-3 regulates presynaptic Erk1/2 signaling and hippocampus-dependent memory. Proc Natl Acad Sci U S A 108, 3366–3370. 10.1073/pnas.l019166108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams JP, Anderson AE, Varga AW, Dineley KT, Cook RG, Pfaffinger PJ, and Sweatt JD (2000). The A-type potassium channel Kv4.2 is a substrate for the mitogen-activated protein kinase ERK. J Neurochem 75, 2277–2287. 10.1046/j.1471-4159.2000.0752277.x. [DOI] [PubMed] [Google Scholar]
- 28.Schrader LA, Birnbaum SG, Nadin BM, Ren Y, Bui D, Anderson AE, and Sweatt JD (2006). ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. Am J Physiol Cell Physiol 290, C852–861. 10.1152/ajpcell.00358.2005. [DOI] [PubMed] [Google Scholar]
- 29.Keir ME, Freeman GJ, and Sharpe AH (2007). PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. J Immunol 179, 5064–5070. 10.4049/jimmunol.179.8.5064. [DOI] [PubMed] [Google Scholar]
- 30.Yao A, Liu F, Chen K, Tang L, Liu L, Zhang K, Yu C, Bian G, Guo H, Zheng J, et al. (2014). Programmed death 1 deficiency induces the polarization of macrophages/microglia to the M1 phenotype after spinal cord injury in mice. Neurotherapeutics. 11, 636–650. 10.1007/s13311-013-0254-x [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suda Y, Nakashima T, Matsumoto H, Sato D, Nagano S, Mikata H, Yoshida S, Tanaka K, Hamada Y, Kuzumaki N, and Narita M (2021). Normal aging induces PD-1-enriched exhausted microglia and A1-like reactive astrocytes in the hypothalamus. Biochem Biophys Res Commun 541, 22–29. 10.1016/j.bbrc.2020.12.086. [DOI] [PubMed] [Google Scholar]
- 32.Tabula Muris C., Overall, c., Logistical, c., Organ, c., processing, Library, p., sequencing, Computational data, a., Cell type, a., Writing, g., et al. (2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372. 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H, Lynch JR, Song P, Yang HJ, Yates RB, Mace B, Warner DS, Guyton JR, and Laskowitz DT (2007). Simvastatin and atorvastatin improve behavioral outcome, reduce hippocampal degeneration, and improve cerebral blood flow after experimental traumatic brain injury. Exp Neurol 206, 59–69. 10.1016/j.expneurol.2007.03.031. [DOI] [PubMed] [Google Scholar]
- 34.Harrison JL, Rowe RK, Ellis TW, Yee NS, O’Hara BF, Adelson PD, and Lifshitz J (2015). Resolvins AT-D1 and E1 differentially impact functional outcome, post-traumatic sleep, and microglial activation following diffuse brain injury in the mouse. Brain Behav Immun 47, 131–140. 10.1016/jbbi.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simone DA, Baumann TK, and LaMotte RH (1989). Dose-dependent pain and mechanical hyperalgesia in humans after intradermal injection of capsaicin. Pain 38, 99–107. [DOI] [PubMed] [Google Scholar]
- 36.Nishimura H, Nose M, Hiai H, Minato N, and Honjo T (1999). Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11, 141–151. [DOI] [PubMed] [Google Scholar]
- 37.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. (2000). Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192, 1027–1034. 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, and Freeman GJ (2007). Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 27, 111–122. S1074-7613(07)00328-7[pii] ; 10.1016/j.immuni.2007.05.016 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keir ME, Butte MJ, Freeman GJ, and Sharpe AH (2008). PD-1 and its ligands in tolerance and immunity. Annu.Rev.Immunol 26, 677–704. 10.1146/annurev.immunol.26.021607.090331 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. (2017). PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499. 10.1038/nature22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang K, Gu Y, Liao Y, Bang S, Donnelly CR, Chen O, Tao X, Mirando AJ, Hilton MJ, and Ji RR (2020). PD-1 blockade inhibits osteoclast formation and murine bone cancer pain. J Clin Invest. 10.1172/JCI133334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, Elco CP, Lee N, Juneja VR, Zhan Q, et al. (2015). Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 162, 1242–1256. S0092-8674(15)01104-6 [pii]; 10.1016/j.cell.2015.08.052 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sham CW, Chan AM, Kwong JM, Caprioli J, Nusinowitz S, Chen B, Lee JG, Gandhi NM, Francisco LM, Sharpe AH, et al. (2012). Neuronal programmed cell death-1 ligand expression regulates retinal ganglion cell number in neonatal and adult mice. J Neuroophthalmol 32, 227–237. 10.1097/WNO.0b013e3182589589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu BL, Cao QL, Zhao X, Liu HZ, and Zhang YQ (2020). Inhibition of TRPV1 by SHP-1 in nociceptive primary sensory neurons is critical in PD-L1 analgesia. JCI Insight 5. 10.1172/jci.insight.137386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lonnerberg P, and Linnarsson S (2014). Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods 11, 163–166. 10.1038/nmeth.2772. [DOI] [PubMed] [Google Scholar]
- 46.Vogel C, and Marcotte EM (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 13, 227–232. 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sweatt JD (2004). Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14, 311–317. 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 48.Impey S, Obrietan K, and Storm DR (1999). Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron 23, 11–14. [DOI] [PubMed] [Google Scholar]
- 49.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, and Storm DR (1998). Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron 21, 869–883. 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 50.Tang S, and Yasuda R (2017). Imaging ERK and PKA Activation in Single Dendritic Spines during Structural Plasticity. Neuron 93, 1315–1324 e1313. 10.1016/j.neuron.2017.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, and Gereau RW (2006). The kv4.2 potassium channel subunit is required for pain plasticity. Neuron 50, 89–100. S0896-6273(06)00178-4 [pii]; 10.1016/j.neuron.2006.03.010 [doi]. [DOI] [PubMed] [Google Scholar]
- 52.Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, and Johnston D (2006). Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci 26, 12143–12151. 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoffman DA, Magee JC, Colbert CM, and Johnston D (1997). K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387, 869–875. 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- 54.Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, and Kochanek PM (2017). The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol 13, 171–191. 10.1038/nrneurol.2017.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang T, Velagapudi R, and Terrando N (2020). Neuroinflammation after surgery: from mechanisms to therapeutic targets. Nat Immunol 21, 1319–1326. 10.1038/s41590-020-00812-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salvador AF, de Lima KA, and Kipnis J (2021). Neuromodulation by the immune system: a focus on cytokines. Nat Rev Immunol. 10.1038/s41577-021-00508-z. [DOI] [PubMed] [Google Scholar]
- 57.Gibson EM, and Monje M (2021). Microglia in Cancer Therapy-Related Cognitive Impairment. Trends Neurosci. 10.1016/j.tins.2021.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gibson EM, Nagaraja S, Ocampo A, Tam LT, Wood LS, Pallegar PN, Greene JJ, Geraghty AC, Goldstein AK, Ni L, et al. (2019). Methotrexate Chemotherapy Induces Persistent Tri-glial Dysregulation that Underlies Chemotherapy-Related Cognitive Impairment. Cell 176, 43–55 e13. 10.1016/jcell.2018.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang TW, Johmura Y, Suzuki N, Omori S, Migita T, Yamaguchi K, Hatakeyama S, Yamazaki S, Shimizu E, Imoto S, et al. (2022). Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature 611, 358–364. 10.1038/s41586-022-05388-4. [DOI] [PubMed] [Google Scholar]
- 60.Lim NK, Moestrup V, Zhang X, Wang WA, Moller A, and Huang FD (2018). An Improved Method for Collection of Cerebrospinal Fluid from Anesthetized Mice. J Vis Exp. 10.3791/56774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berger M, Nadler JW, Friedman A, McDonagh DL, Bennett ER, Cooter M, Qi W, Laskowitz DT, Ponnusamy V, Newman MF, et al. (2016). The Effect of Propofol Versus Isoflurane Anesthesia on Human Cerebrospinal Fluid Markers of Alzheimer’s Disease: Results of a Randomized Trial. Journal of Alzheimer’s disease : JAD 52, 1299–1310. 10.3233/JAD-151190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.George Paxinos KBJF (2001). The Mouse Brain in Stereotaxic Coordinates. Academic Press. [Google Scholar]
- 63.Zhao J, Wang Y, Xu C, Liu K, Wang Y, Chen L, Wu X, Gao F, Guo Y, Zhu J, et al. (2017). Therapeutic potential of an anti-high mobility group box-1 monoclonal antibody in epilepsy. Brain Behav Immun 64, 308–319. 10.1016/j.bbi.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 64.Vorhees CV, and Williams MT (2006). Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1, 848–858. 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weng FJ, Garcia RI, Lutzu S, Alvina K, Zhang Y, Dushko M, Ku T, Zemoura K, Rich D, Garcia-Dominguez D, et al. (2018). Npas4 Is a Critical Regulator of Learning-Induced Plasticity at Mossy Fiber-CA3 Synapses during Contextual Memory Formation. Neuron 97, 1137–1152 e1135. 10.1016/j.neuron.2018.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tan C, Lu NN, Wang CK, Chen DY, Sun NH, Lyu H, Korbelin J, Shi WX, Fukunaga K, Lu YM, and Han F (2019). Endothelium-Derived Semaphorin 3G Regulates Hippocampal Synaptic Structure and Plasticity via Neuropilin-2/PlexinA4. Neuron 101, 920–937 e913. 10.1016/j.neuron.2018.12.036. [DOI] [PubMed] [Google Scholar]
- 67.Xu ZZ, Kim YH, Bang S, Zhang Y, Berta T, Wang F, Oh SB, and Ji RR (2015). Inhibition of mechanical allodynia in neuropathic pain by TLR5-mediated A-fiber blockade. Nat.Med. nm.3978 [pii]; 10.1038/nm.3978 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Berta T, Park CK, Xu ZZ, Xie RG, Liu T, Lu N, Liu YC, and Ji RR (2014). Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-alpha secretion. J Clin.Invest 124, 1173–1186. 72230 [pii]; 10.1172/JCI72230 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen G, Park CK, Xie RG, and Ji RR (2015). Intrathecal bone marrow stromal cells inhibit neuropathic pain via TGF-beta secretion. J Clin.Invest 125, 3226–3240. 80883 [pii]; 10.1172/JCI80883 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Donnelly CR, Jiang C, Andriessen AS, Wang K, Wang Z, Ding H, Zhao J, Luo X, Lee MS, Lei YL, et al. (2021). STING controls nociception via type I interferon signalling in sensory neurons. Nature. 10.1038/s41586-020-03151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.