

SUMMARY
Pancreatic islets are endocrine organs that depend on their microvasculature to function. Along with endothelial cells, pericytes comprise the islet microvascular network. These mural cells are crucial for microvascular stability and function, but it is not known if/how they are affected during the development of type 1 diabetes (T1D). Here, we investigate islet pericyte density, phenotype, and function using living pancreas slices from donors without diabetes, donors with a single T1D-associated autoantibody (GADA+), and recent onset T1D cases. Our data show that islet pericyte and capillary responses to vasoactive stimuli are impaired early on in T1D. Microvascular dysfunction is associated with a switch in the phenotype of islet pericytes toward myofibroblasts. Using publicly available RNA sequencing (RNA-seq) data, we further found that transcriptional alterations related to endothelin-1 signaling and vascular and extracellular matrix (ECM) remodeling are hallmarks of single autoantibody (Aab)+ donor pancreata. Our data show that microvascular dysfunction is present at early stages of islet autoimmunity.
In brief
Mateus Gonçalves et al. found that islet pericytes and capillaries are dysfunctional at early stages of islet autoimmunity. Several alterations related to endothelin-1 signaling and vascular and ECM remodeling are present in single Aab+ donor pancreata, supporting the involvement of the vasculature in type 1 diabetes pathogenesis.
Graphical Abstract
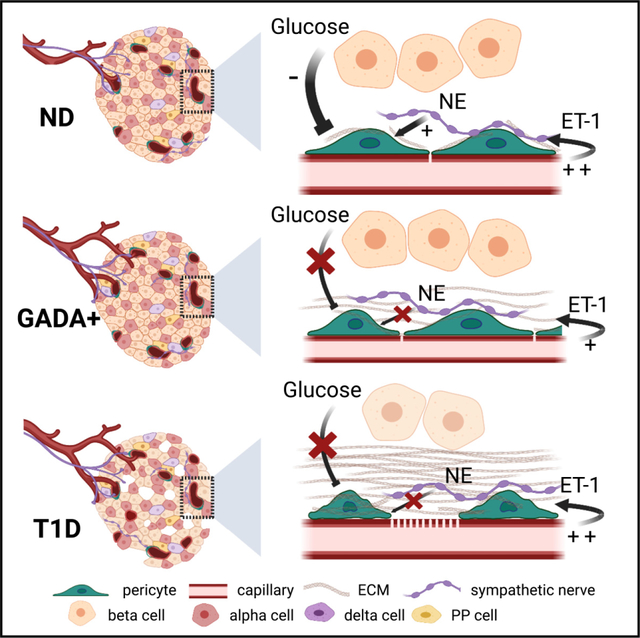
INTRODUCTION
Type 1 diabetes (T1D) is a chronic autoimmune disease that currently has no cure. It is caused by the autoimmune destruction of insulin-producing beta cells in the pancreas, leading to impaired glucose homeostasis and chronic hyperglycemia.1 While the prominent pathological features of T1D pancreata are a marked loss of beta cells and lymphocytic infiltrates in islets,2,3 additional pathological abnormalities have been described in endocrine compartments. These relate, in particular, to the structure and function of the islet microvasculature. Not only have changes in islet blood vessel diameter, density, and composition of the extracellular matrix (ECM) been reported,4–8 but also functional defects such as increased vascular leakage9–11 and abnormal blood flow dynamics are observed in pre-symptomatic stages of T1D.12,13 Because some vascular alterations have been detected during stages 1 and 2 of T1D, when beta cell autoimmunity is present but symptomatic disease has not yet developed,14 elucidating the specific mechanisms of dysfunction will shed new light onto our understanding of T1D pathogenesis.
Vascular networks in pancreatic islets are essential for adequate nutrient sensing by endocrine cells, efficient release of hormones, and timely responses to changes in glycemia.15,16 Islets are densely vascularized by capillaries made of endothelial cells and covered by pericytes.17,18 In mice, pericytes provide direct trophic support for beta cells, contributing to their proliferation, maturation, and insulin secretion,19–22 or indirectly by participating in ECM synthesis.23 Pericytes may also impact beta cell health and function by maintaining proper blood supply to endocrine cells, as they are important regulators of islet capillary diameter and blood flow.18,24,25 Importantly, pericyte-mediated changes in islet blood flow are required for proper islet hormone secretion and glucose homeostasis in mice.26 Changes in islet pericyte density, morphology, and function occur under pathophysiological conditions. In mouse models of type 2 diabetes, for instance, islet pericytes hypertrophy and adopt a smooth muscle cell-like appearance,16,27,28 which may affect their capacity to regulate islet blood flow.25 Although pericytes have important physiological and pathophysiological functions in islets, it is not known what happens to this cell population during T1D development.
Circulating islet autoantibodies (Aabs) are the most robust biomarkers of T1D and are found in 95% of individuals that develop clinical symptoms.29 Single Aab positivity (pre-stage 1 or “stage 0” of T1D) represents the earliest phase in the natural history of islet autoimmunity,2 and antibodies that recognize glutamic acid decarboxylase 65 (GADA) are the most common Aab in single Aab+ donors.30 For these reasons, in this study, we compared pericyte and vascular responses in islets from Aab−, non-diabetic (control) donors with those in single Aab+ (GADA+; “stage 0”) and recent onset T1D organ donors (stage 314) using time-lapse confocal microscopy and living pancreas slices. Our study revealed major changes in the phenotype and function of islet pericytes and, consequently, impaired islet capillary responses to vasoactive stimuli such as high glucose and norepinephrine. We further found abnormal vascular responses to the potent vasoconstrictor endothelin-1 in islets from Aab+ donors, along with altered expression of endothelin-1 receptors in stellate and endothelial cells isolated from Aab+ donor pancreata. Our study shows that pericyte dysfunction and impaired vasomotion occur at early stages of islet autoimmunity, potentially interfering with the islet response to different metabolic challenges.
RESULTS
Pericyte coverage of capillaries is decreased in islets from donors with T1D
Pericytes are crucial for microvascular homeostasis throughout the body,31 but whether they are affected as T1D progresses has not been examined. Here, we assessed changes in the density of pericytes in endocrine and exocrine compartments of the human pancreas and compared the pericyte:endothelial cell ratio, a major determinant of the tightness of the endothelial barrier.32 We immunostained endothelial cells and pericytes in fixed pancreas slices from organ donors without diabetes (non-diabetic [ND]), donors with a single islet Aab (GADA+; Aab+; “stage 0”), and donors with T1D (stage 3; Figures 1A and S2). We used two different markers to identify pericytes: the chondroitin sulfate proteoglycan neuron-glial antigen 2 (NG2) and the cell adhesion molecule CD146.26 Capillaries in islets from ND donors were covered with pericytes that exhibited long cytoplasmic processes that extended along islet endothelial cells (Figure 1B). The average density of pericytes and endothelial cells did not change in islets from Aab+ and T1D donors (Figures 1A, S2, and S3), but there was a significant decrease in the pericyte:endothelial cell ratio in islets from T1D donors compared with ND donors (Figures 1A and 1D). Several capillaries in T1D islets lacked pericytes (Figure 1A, arrows) or were covered by pericytes that had lost their typical shapes and regular surfaces (Figure 1B). The extent of colocalization between NG2 and CD146 did not change at these different stages (Figure S3). Pericytes in the vicinity of T1D islets also had an abnormal morphology, characterized by multiple cytoplasmic processes or stellate shapes (Figure 1C),33 and pericyte coverage of acinar capillaries increased in T1D (Figure 1E).
Figure 1. Pericyte coverage of capillaries is decreased in islets from donors with T1D.
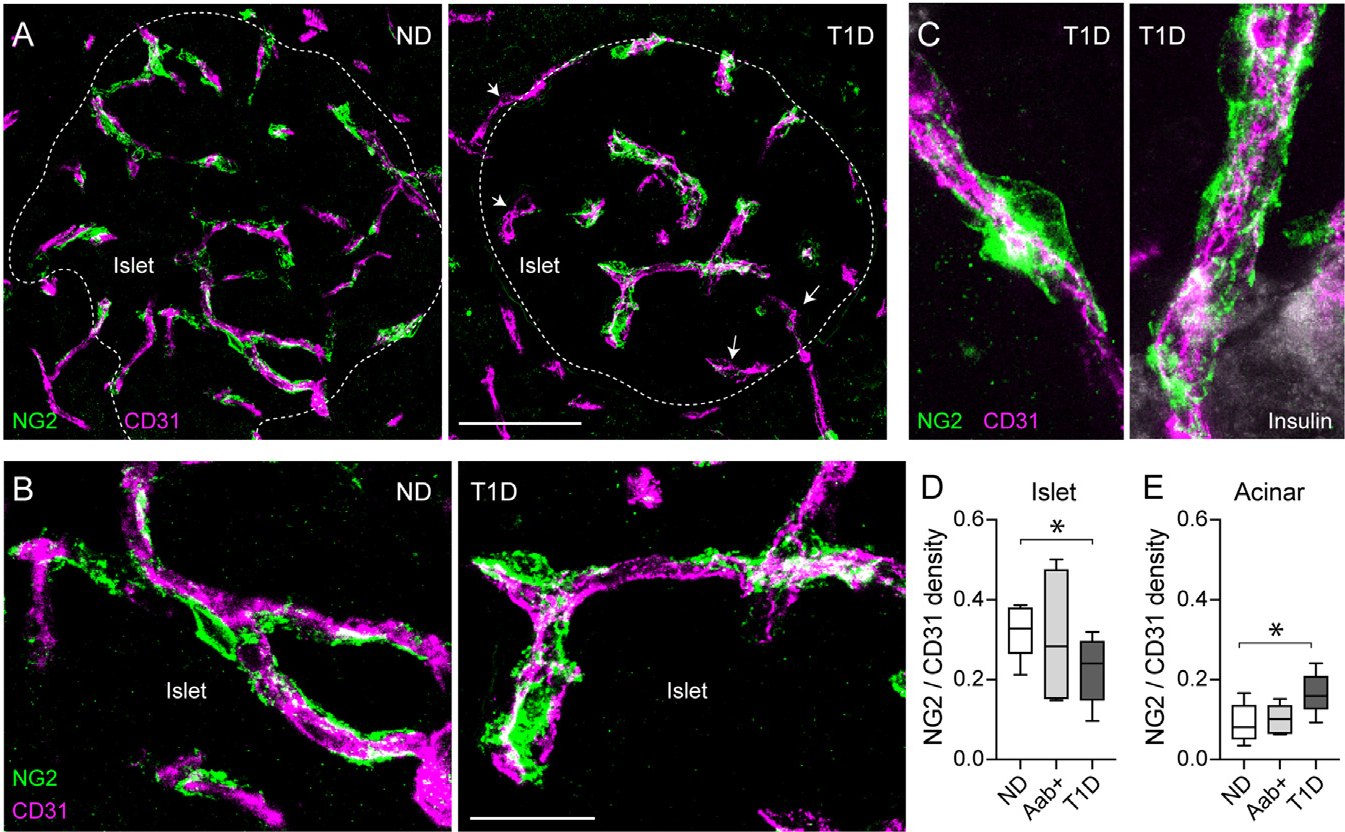
(A and B) z stack of confocal images of the vasculature in islets from a non-diabetic (ND; nPOD6531) and a T1D donor (T1D duration: 1.5 years; nPOD6469) immunostained for endothelial cells (CD31; magenta) and pericytes (NG2; green). Dashed lines indicate islet border (related to Figure S2), and arrows indicate T1D capillaries lacking pericytes.
(C) z stack of confocal images of capillaries at the border of islets in a T1D donor.
(D and E) Ratio of pericyte:endothelial cells quantified as NG2 area:CD31 area in islets (D) and acinar tissue (E). n = 9 ND, 7 Aab+, and 8 T1D organ donors. For each donor, around 5–7 islets were imaged, and an average value was calculated. Box and whiskers plots are shown, and whiskers reflect minimum and maximum values. *p < 0.05 (one-way ANOVA followed by Tukey’s multiple comparisons test). Scale bars: 50 μm (A) and 20 μm (B).
Within this cohort of T1D donors with relatively short disease duration (first 4 years after diagnosis), we did not observe any significant correlation between the density of islet pericytes and either the duration of diabetes, the plasma levels of glycated hemoglobin, or the number of circulating antibodies against islet antigens (Figure S2). In summary, our data show that the pericyte:endothelial cell ratio decreases in individuals recently diagnosed with T1D, although pericyte density is similar in islets from single Aab+ and ND donors. Because remaining pericytes in T1D islets exhibited an abnormal morphology, we then asked whether their function was preserved.
Pericyte calcium responses to glucose are impaired in islets from Aab+ and T1D donors
Previous studies have shown that circulating glucose regulates blood flow in the islet without affecting flow rates in the exocrine tissue.34–36 We have shown that pericytes mediate this vascular response.18 In particular, increasing the extracellular glucose concentration decreased cytosolic calcium levels ([Ca2+]i) in pericytes and dilated islet capillaries.18 To determine whether pericytes have similar functions in human islets, we used living human pancreas slices as previously described.24,26 In slices, pericytes remain in their niches within islets and can be visualized with a fluorescent antibody against their surface marker, NG2 (Figure S4). Slices were incubated with a membrane-permeant calcium indicator (Fluo4) to record changes in [Ca2+]i in islet pericytes induced by increasing the glucose concentration from 3 (3G) to 11 mM (11G). High glucose decreased [Ca2+]i in the majority of pericytes in islets from ND donors (Figures 2A and 2B). After ~7 min of being stimulated with glucose, [Ca2+]i of islet pericytes had decreased by 10% of baseline values (Figure 2F).
Figure 2. Pericyte calcium responses to glucose are impaired in islets from Aab+ and T1D donors.
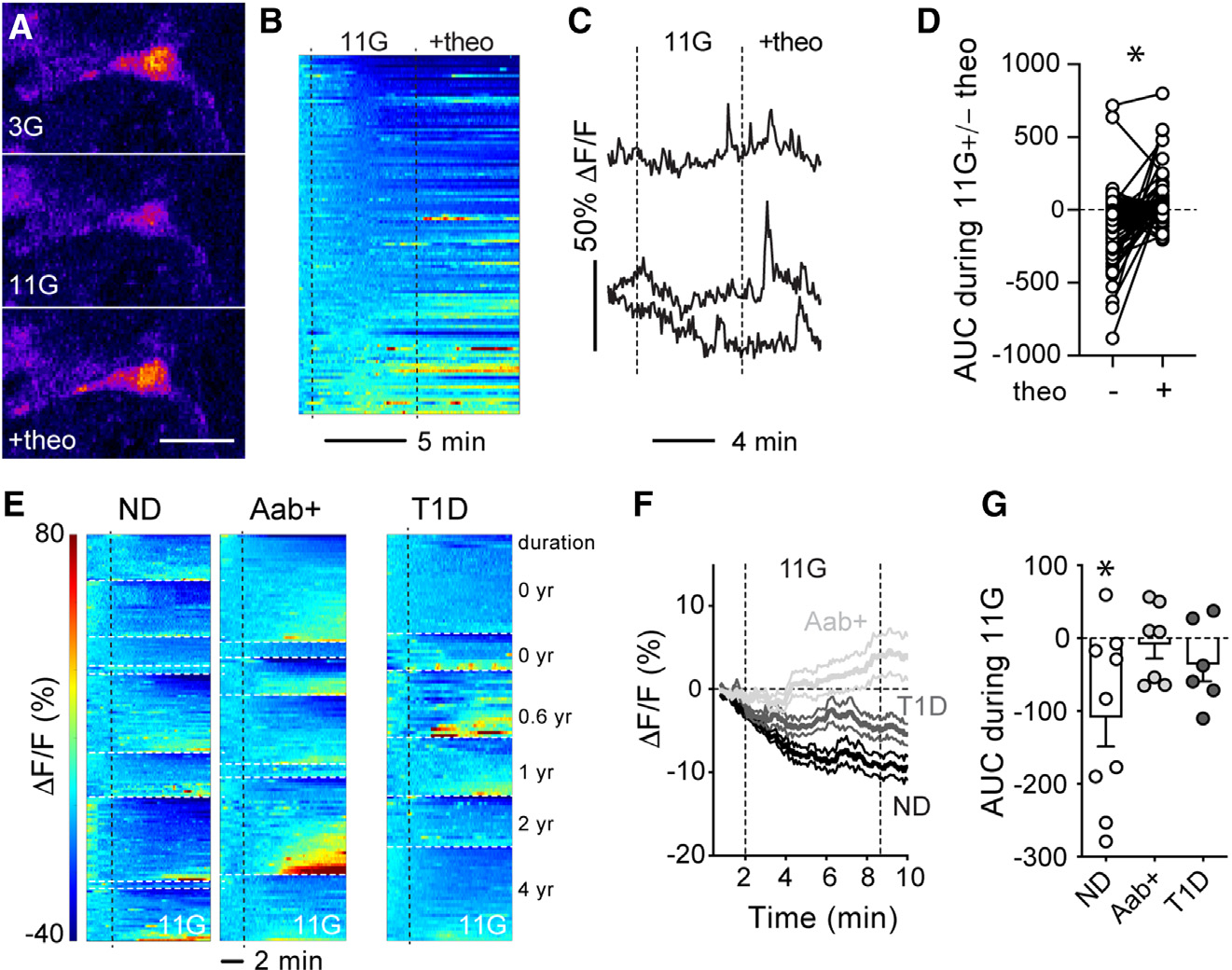
Living pancreas slices were incubated with Fluo4 and NG2-Alexa647.
(A) Confocal image of a pericyte in an islet from a NDdonor (nPOD6516) in basal glucose concentration (3 mM), upon high glucose (11 mM) alone, or in the presence of theophylline (20 μM; 11G + theo). Scale bar: 10 μm.
(B) Heatmap showing changes in Fluo4 fluorescence (DF/F; %) in pericytes in islets from ND donors elicited by 11 mM glucose and then theo (20 μM; in 11G). Each line is a different pericyte.
(C) Representative traces showing changes in Fluo4 fluorescence elicited by glucose and theo.
(D) Net area under the curve (AUC) of fluorescence traces for individual pericytes. *p < 0.05 (paired t test; n = 113 pericytes from 5 ND donors).
(E) Heatmaps showing changes in Fluo4 fluorescence in pericytes from ND (n = 9), Aab+ (n = 7), and T1D donors (n = 6). Donors are separated by white dashed lines. T1D donor data are presented based on T1D duration, while data from ND and Aab+ donors are presented according to Table S1.
(F) Traces showing average changes in Fluo4 fluorescence elicited 11mM glucose for all islet pericytes from each group. Average (thicker lines) ± SEM values are shown.
(G) Net AUC of fluorescence traces during stimulation with 11G (dots: data point for each donor, bars: mean ± SEM). For each donor, responses from around 3–36 pericytes were recorded, and an average value calculated. *p < 0.05 (one-sample t test, theoretical mean = 0).
In mouse islets, adenosine, most likely produced from ATP coreleased with insulin from dense-core granules,37,38 partially mediates the inhibitory effects of glucose on islet pericytes.18 To assess whether adenosine had similar effects in human islets, we applied theophylline, a non-specific adenosine receptor antagonist, in the presence of 11 mM glucose. Theophylline (20 mM) increased [Ca2+]i in a subset of pericytes in islets from ND donors (Figures 2A–2C), reversing the inhibitory effects of high glucose on islet pericytes (Figure 2D).
We then compared pericyte [Ca2+]i responses to high glucose in islets from Aab+ and T1D donors. In Aab+ donors, while a small subset of islet pericytes were still inhibited in every donor analyzed (Figure 2E), the majority were activated by high-glucose stimulation (Figure 2E). Glucose led to an average increase of around 5% of [Ca2+]i levels in islet pericytes in Aab+ donors (Figure 2F). Similarly, responses of pericytes in T1D islets to high glucose were also greatly reduced (Figures 2E–2G). In summary, our data show that pericyte responses to glucose stimulation are impaired at early stages of T1D. In future studies, we will investigate whether endogenous adenosine production and/or sensing are compromised in islets from Aab+ and T1D donors, potentially underlying impaired pericytic responses to glucose.
Vasodilatory responses to glucose are impaired in islets from Aab+ and T1D donors
Because pericytes control capillary diameter in mouse and human islets,18,24,26 we then examined capillary responses (vasomotion) induced by high glucose in slices from donors at different stages of T1D. Living pancreas slices were incubated with a fluorescent lectin (Lycopersicon esculentum lectin 647) to visualize the islet microvasculature as described.24 In most lectin-labeled capillaries, a lumen was visible, and the average islet vessel diameter was around 5 μm in islets from ND and T1D donors (basal diameter = 5 ± 0.2 for ND and 5.7 ± 0.3 for T1D; p = not significant [ns]). Interestingly, in islets from Aab+ donors, the average islet capillary diameter was slightly reduced under basal, non-stimulatory conditions (diameter = 4.1 ± 0.4 for Aab+ donors; p < 0.05 between Aab+ and both ND and T1D donors [one-way ANOVA followed by Tukey’s multiple comparisons test]).
Glucose inhibited pericytes in islets from ND donors (Figure 2). This glucose-dependent decrease in [Ca2+]i in islet pericytes was associated with a significant increase in capillary diameter in islets from ND donors (Figure 3). Although only a subset (% dilating = 60% ± 14%, n = 5 donors) of islet capillaries dilated upon glucose stimulation (Figure 3B), high glucose led to an ~5% average increase in islet capillary diameter (Figure 3D). Importantly, vasodilatory responses to glucose were impaired in islets from Aab+ and T1D donors (Figures 3B and 3C; % dilating = 40% ± 7% for Aab+ [n = 3] and 39% ± 19% for T1D [n = 5]). In summary, not only were pericyte [Ca2+]i responses to glucose impaired (Figure 2), but also glucose failed to elicit efficient vasodilation of capillaries in islets from Aab+ and T1D donors (Figure 3C). Islet vascular responses to glucose are thus impaired at early stages of T1D. This can have metabolic consequences, as in mice, vascular responses to glucose are needed for proper islet hormone secretion upon a glucose challenge.26
Figure 3. Vasodilatory responses to glucose are impaired in islets from Aab+ and T1D donors.
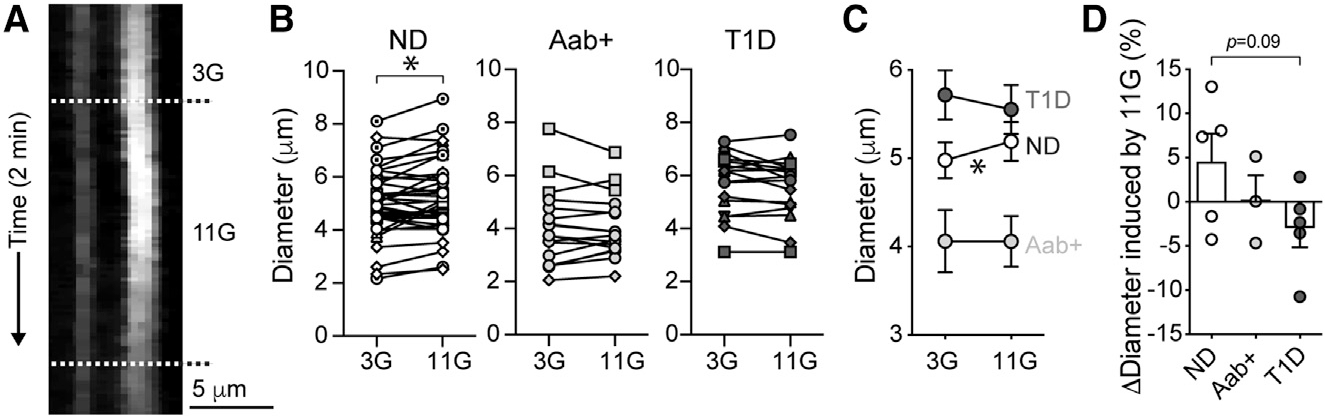
Living human pancreas slices were incubated with Lycopersicon esculentum lectin to label the islet microvasculature.
(A) Temporal projection showing changes in islet capillary diameter in a ND donor (nPOD6555) induced by 11 mM glucose.
(B and C) Quantification of capillary diameters in slices from ND (n = 37 capillaries/5 donors), Aab+ (n = 17 capillaries/3 donors), and T1D donors (n = 17 capillaries/5 donors). Each donor is shown with a different symbol.
(C) Mean ± SEM diameter values in 3G and 11G are shown. *p < 0.05 (paired t test).
(D) Quantification of average relative change in capillary diameter (Δdiameter; % baseline) induced by 11G for each group (dots: data point for each donor, bars: mean ± SEM). For each donor, responses from 3–11 vessels were recorded, and an average value was calculated.
Islet microvascular responses to norepinephrine are impaired in Aab+ and T1D donors
Blood flow in the pancreas is also under the control of local sympathetic and parasympathetic nerve fibers.39,40 Sympathetic nerve fibers in islets, in particular, interact very closely with pericytes, which respond to different sympathetic agonists ex vivo and in vivo.18,24,26 We thus hypothesized that changes in sympathetic tonus to islet blood vessels could underlie impaired vascular responses to glucose in islets from Aab+ and T1D donors. To address this, we first assessed sympathetic innervation patterns in islets at different stages of T1D. Sympathetic nerve fibers were identified with an antibody against the sympathetic nerve marker tyrosine hydroxylase (TH). Sympathetic axons could be seen in both endocrine and exocrine compartments of the pancreas of ND, Aab+, and T1D organ donors (Figure S5). The density of TH+ fibers in islets did not change during these stages of T1D (Figures 4A and 4B), in line with a recent study showing that sympathetic nerves are preserved in T1D islets.41 Around 25% of pericytes in islets from ND individuals were in close contact with sympathetic nerve fibers (Figure 4A). Similarly, sympathetic axons that reached islets in Aab+ and T1D donors also innervated pericytes, and the fractions that were innervated remained around 17% and 18%, respectively (Figures 4A and S5).
Figure 4. Islet microvascular responses to norepinephrine are impaired in Aab+ and T1D donors.
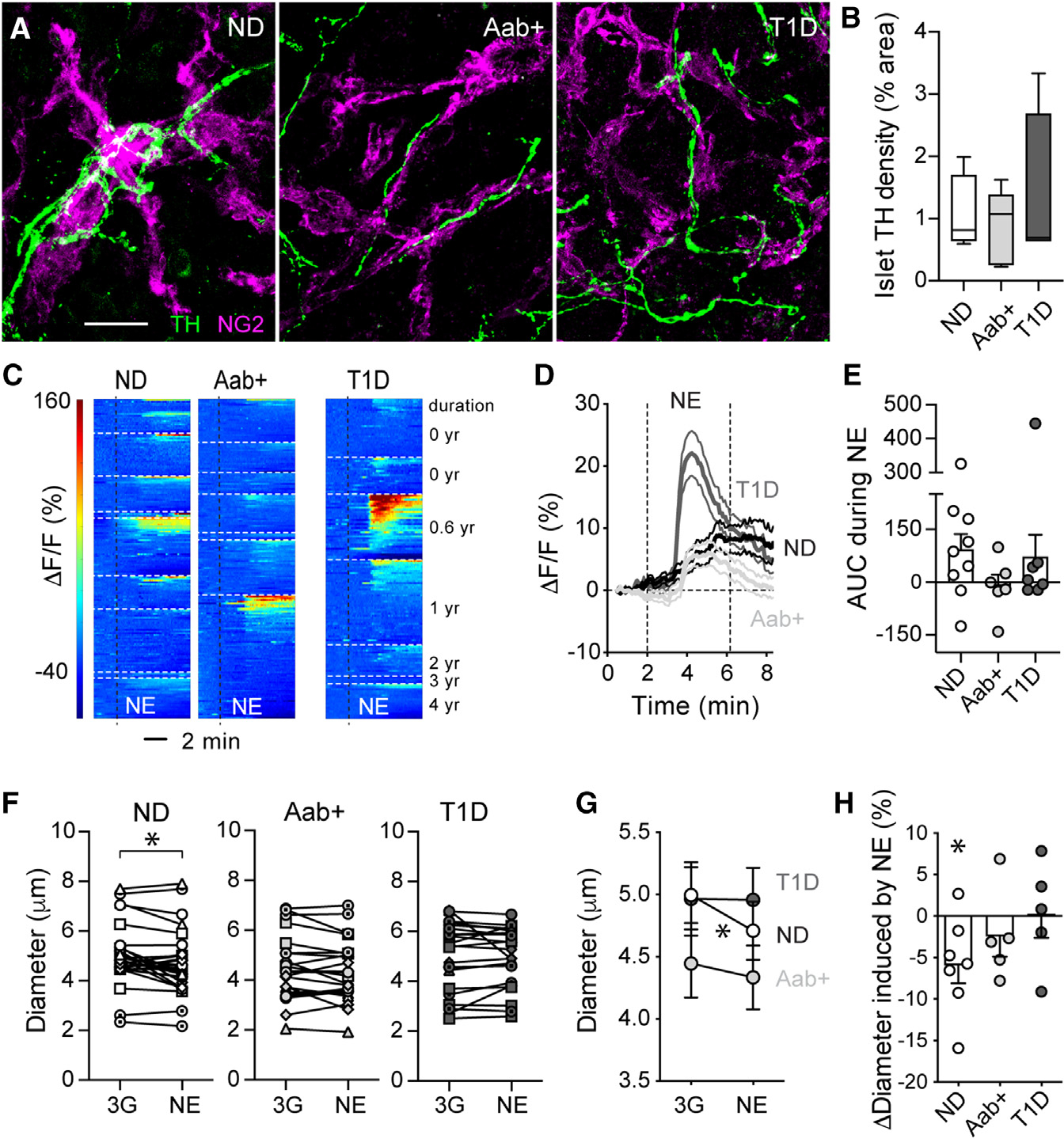
(A) z stack of confocal images of islets in ND (nPOD6546), Aab+ (nPOD6569), and T1D sections (T1D for 2 years; nPOD6566) immunostained for the sympathetic nerve marker tyrosine hydroxylase (TH; green) and NG2 (magenta). Islets were found using insulin or somatostatin (Figure S5). Scalebar: 20μm.
(B) Percentage of islet area immunostained for TH. n = 4–5 donors per group, 5–7 islets per donor. Box and whiskers plot is shown, and whiskers reflect minimum and maximum values for each dataset.
(C) Heatmaps showing changes in Fluo4 fluorescence (ΔF/F; %) elicited by norepinephrine (NE; 20 μM; in 3G) in islet pericytes from ND (n = 9), Aab+ (n = 7), and T1D donors (n = 7 donors). Each donor is shown separately.
(D) Traces showing changes in Fluo4 fluorescence of all pericytes elicited by NE for each group.
(E) Quantification of the average net AUC of fluorescence traces during stimulation with NE (dots: data point for each donor, bars: mean ± SEM). For each donor, responses from 3–73 cells were recorded, and an average value was calculated.
(F and G) Quantification of changes in islet capillary diameter induced by NE in slices from ND (n = 29 capillaries/7 donors), Aab+ (n = 23 capillaries/5 donors), and T1D donors (n = 22 capillaries/5 donors). Mean ± SEM diameter are shown in (G). *p < 0.05 (paired t test comparing3G with NE for all).
(H) Relative changes in capillary diameter (Δdiameter; % baseline) induced by NE for each group (dots: data point for each donor, bars: mean ± SEM). For each donor, responses from 3–8 vessels were recorded, and an average value was calculated. *p < 0.05 (one-sample t test, theoretical mean = 0).
We then examined islet microvascular responses to the sympathetic agonist norepinephrine by confocal imaging of living pancreas slices. Norepinephrine (20 mM) increased [Ca2+]i levels in a subset of pericytes in islets from different ND donors (Figure 4C). Indeed, only around 40% ± 9% of islet pericytes were activated by norepinephrine, and we have previously shown that responding pericytes were those innervated by sympathetic nerve fibers.26 Increases in pericyte [Ca2+]i induced by norepinephrine were accompanied by a significant constriction of a subset of islet capillaries (Figures 4F–4H). Norepinephrine induced a ~5% decrease in islet capillary diameter in ND donors (Figure 4H). Importantly, pericyte [Ca2+]i responses to norepinephrine were diminished in islets from different Aab+ donors (Figures 4C–4E), and norepinephrine stimulation did not constrict islet capillaries (Figures 4F–4H). Some pericytes in islets from different T1D donors still responded to norepinephrine, and this agonist induced a robust increase in pericyte [Ca2+]i, particularly in one T1D donor (nPOD 6551; T1D duration of 0.6 years; Figures 4C–4E). However, norepinephrine stimulation failed to elicit vasoconstriction of capillaries in islets from T1D donors (Figures 4F–4H). Our data show that islet capillaries lose the capacity to constrict upon stimulation with the sympathetic agonist norepinephrine at early stages of T1D. Abnormal islet pericyte function most likely contributed to impaired vasomotive responses, although dysfunctional smooth muscle and endothelial cells (not examined in this study) could have also played a role. In summary, abnormal vascular responses to sympathetic agonists may compromise sympathetic-induced changes in blood flow in human islets at early stages of the disease. This can negatively impact changes in plasma levels of islet hormones, as in mice, activation of islet pericytes with a sympathetic agonist decreases blood flow, which reduces plasma insulin levels.26
Altered endothelin-1 vascular effects and receptor expression in Aab+ and T1D islets
Because capillaries in islets from Aab+ and T1D donors were unresponsive to both glucose or norepinephrine, we then examined vascular responses to endothelin-1, an extremely potent vasoconstrictor peptide that is produced by endothelial cells.42 Exogenous endothelin-1 (10 nM) increased [Ca2+]i levels specifically in islet pericytes in all ND donors analyzed (Figures 5A and 5B) and decreased islet capillary diameter by ~8% (Figures 5E and 5F). Pericyte [Ca2+]i responses to endothelin-1 were diminished in islets from Aab+ donors (Figures 5B and 5C), and endothelin-1 administration did not result in significant vasoconstriction (Figures 5E and 5F). Interestingly, pericytes in islets from Aab+ donors were still very responsive to another potent vasoconstrictor peptide, angiotensin II (Figure 5D). Pericytes in T1D islets were potently activated by endothelin-1 (Figures 5A–5C), but again, [Ca2+]i responses were not associated with significant changes in capillary diameter (Figures 5E and 5F).
Figure 5. Altered endothelin-1 vascular effects and receptor expression in Aab+ and T1D islets.
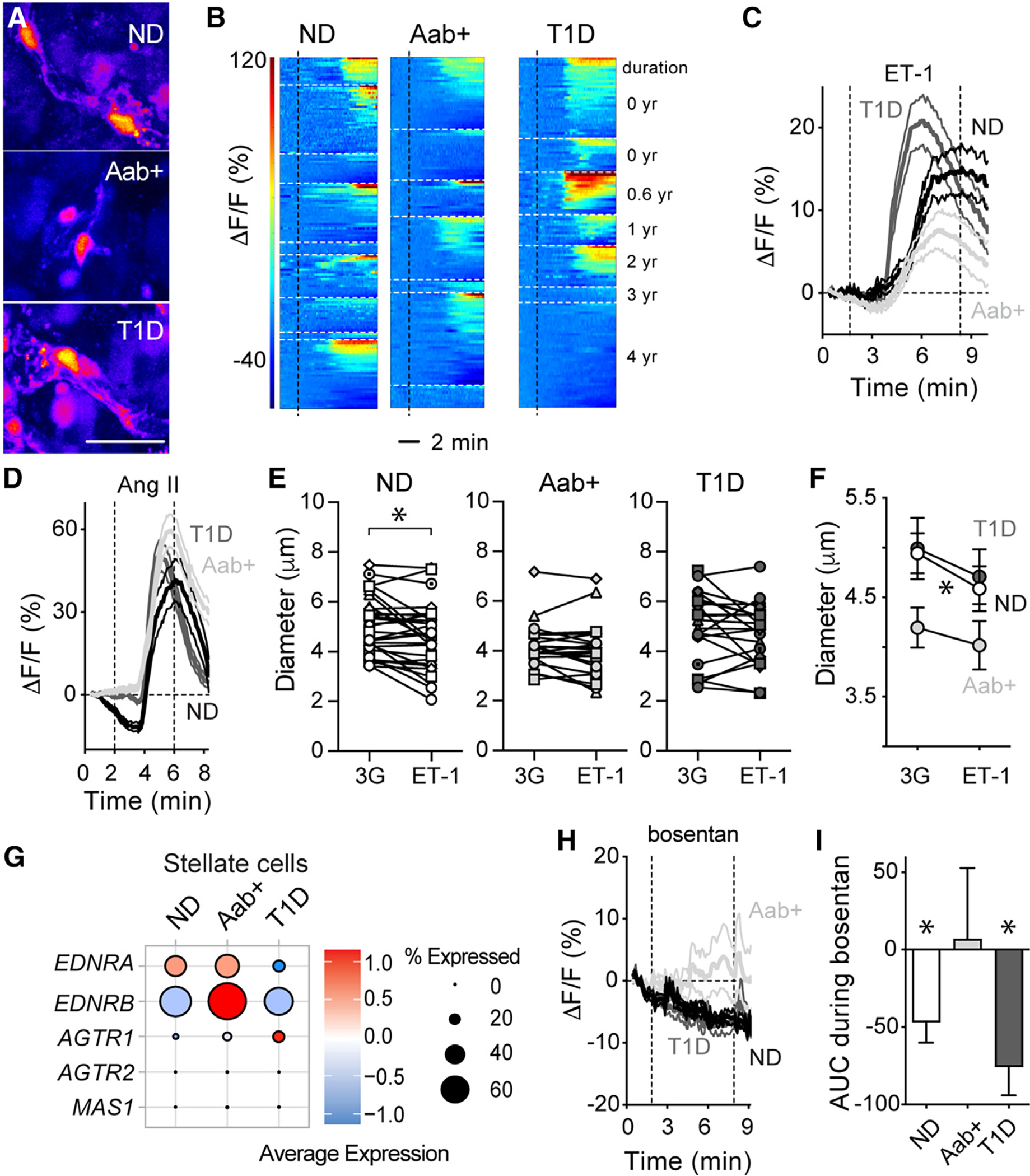
(A) Pericytes in islets from an ND (nPOD6516),Aab+ (nPOD6578), and T1D donor (T1D for 0.6 years; nPOD6551) responding to endothelin-1 (ET-1; 10 nM; in 3G) shown in pseudo-color scale. Scale: 20 μm.
(B) Heatmaps showing changes in Fluo4 fluorescence (ΔF/F; %) elicited by ET-1 in islet pericytes in ND (n = 9), Aab+ (n = 7), and T1D donors (n = 7), organized by donor.
(C) Traces showing changes in Fluo4 fluorescence(ΔF/F; %) of all islet pericytes elicited by ET-1. Mean ± SEM values are shown (for ND, n = 166 pericytes/9 donors; for Aab+, n = 125 pericytes/7 donors; for T1D, n = 116 pericytes/7 donors).
(D) Traces showing changes in Fluo4 fluorescence of islet pericytes elicited by angiotensin II (Ang II; 100 nM; for ND, n = 91 pericytes/3 donors; for Aab+, n = 138 pericytes/5 donors; for T1D, n = 119 pericytes/5 donors).
(E and F) Changes in islet capillary diameter induced by ET-1 for individual vessels in islets from ND (n = 32 capillaries/6 donors), Aab+ (n = 21 capillaries/5 donors), and T1D donors (n = 21 capillaries/5 donors).
(F) Mean ± SEM diameter values, *p < 0.05 (paired t test between 3G and ET-1).
(G) Dot plots showing expression levels of genes encoding ET and Ang II receptors in stellate cells from ND, Aab+, or T1D islets. Data were extracted from HPAP database.
(H) Traces showing changes in Fluo4 fluorescence of islet pericytes elicited by ET receptor antagonist bosentan (100 nM in 3G).
(I) Net AUC of fluorescence traces (mean ± SEM values are shown; ND: n = 25 pericytes/3 donors; Aab+: n = 34 pericytes/3 donors; T1D: n = 56 pericytes/3 donors; *p < 0.05 [one-sample t test, mean = 0]).
Using a publicly available database containing single-cell RNA sequencing (RNA-seq) data from different cells in human pancreatic islets (from the Human Pancreas Analysis Program43), we analyzed potential changes in gene expression of endothelin-1 receptors in stellate cell and endothelial cell clusters at different stages of T1D. Stellate cells, identified by their expression of the marker gene PDGFRB44 and of COL1A1 (Figure S6), can be subdivided into quiescent, standard activated, and immune-activated, with pericytes included within the quiescent stellate cell population.44,45 Indeed, a subcluster of stellate cells expresses pericyte identity genes such as CSPG4, PDGFRB, RGS5, and ACTA2 (Figure S6). Endothelin-1 exerts its effects by binding to two G protein-coupled receptors—ETA and ETB—encoded by EDNRA and EDNRB genes, respectively. Stellate cells in ND islets express mainly EDNRA, which mediates the vasoconstrictor effects of endothelin-1 (Figure 5G).46 Interestingly, there was a significant upregulation (fold change [FC] vs. ND = 1.5) of EDNRB in stellate cells from Aab+ donors, while EDNRA was downregulated in stellate cells from T1D donors (Figure 5G; FC vs. ND = 0.8). There were no significant changes in the expression levels of angiotensin II receptors (Figure 5G) or adenosine or adrenergic receptors in stellate cells (data not shown). Similarly, EDNRB was upregulated in endothelial cells from Aab+ donors (FC vs. ND = 1.3). In line with altered endothelin-1 receptor expression by vascular cells in Aab+ islets, antagonizing endothelin-1 receptor signaling with the dual receptor antagonist bosentan (100 nM) affected differentially [Ca2+]i levels in pericytes in islets from Aab+ donors (Figures 5H and 5I). In particular, while bosentan significantly decreased pericyte [Ca2+]i in islets from ND and T1D donors, it was without effect in islets from Aab+ donors. This differential effect could have been caused by the upregulation of the EDNRB gene in endothelial cells from Aab+ donors, as ET-1 action through ETB receptors in endothelial cells leads instead to vasodilation.46 In summary, our data show that altered endothelin-1 vascular effects and receptor expression occur in Aab+ and T1D donor pancreata.
Altered pericyte and stellate cell phenotypes in islets from Aab+ and T1D donors
Given pericytes’ abnormal function and impaired vasomotive responses in islets from Aab+ and T1D donors, we hypothesized that islet pericytes were turning into ECM-producing myofibroblasts and losing their capacity to control islet capillary diameter. Indeed, conversion of pericytes into myofibroblasts had been previously reported in a mouse model of islet fibrosis.47 To test this hypothesis, we examined by immunohistochemistry the pericytic expression of the ECM protein collagen type IV. The density of this basement membrane component increased significantly in islets from T1D donors (Figures 6A and 6B). Islet pericytes were found embedded within collagen type IV networks (Figure 6C). Importantly, the proportion of colocalization of the pericyte marker NG2 with collagen IV increased significantly in islets from Aab+ and T1D donors (Figure 6D), indicative of an increased contribution of islet pericytes to the synthesis of this ECM protein. We then assessed pericytic expression of myofibroblast markers such as alpha smooth muscle actin (αSMA) and periostin.48 Pericytes could be seen in close proximity to periostin aggregates in islets from Aab+ donors (Figure S7), but this matricellular protein was only detected in a subset of islets from these donors (Figure S7). Interestingly, a similar variability in changes in the composition of the ECM in islets from single Aab+ donors had been previously noted in a study examining another ECM component, hyaluronic acid.6 Moreover, the proportion of NG2-expressing mural cells that also expressed αSMA inside islets or at the islet border also increased from around 30%–40% in ND donors to ~70% in islets from T1D donors (Figure S7).
Figure 6. Altered pericyte and stellate cell phenotypes in islets from Aab+ and T1D donors.
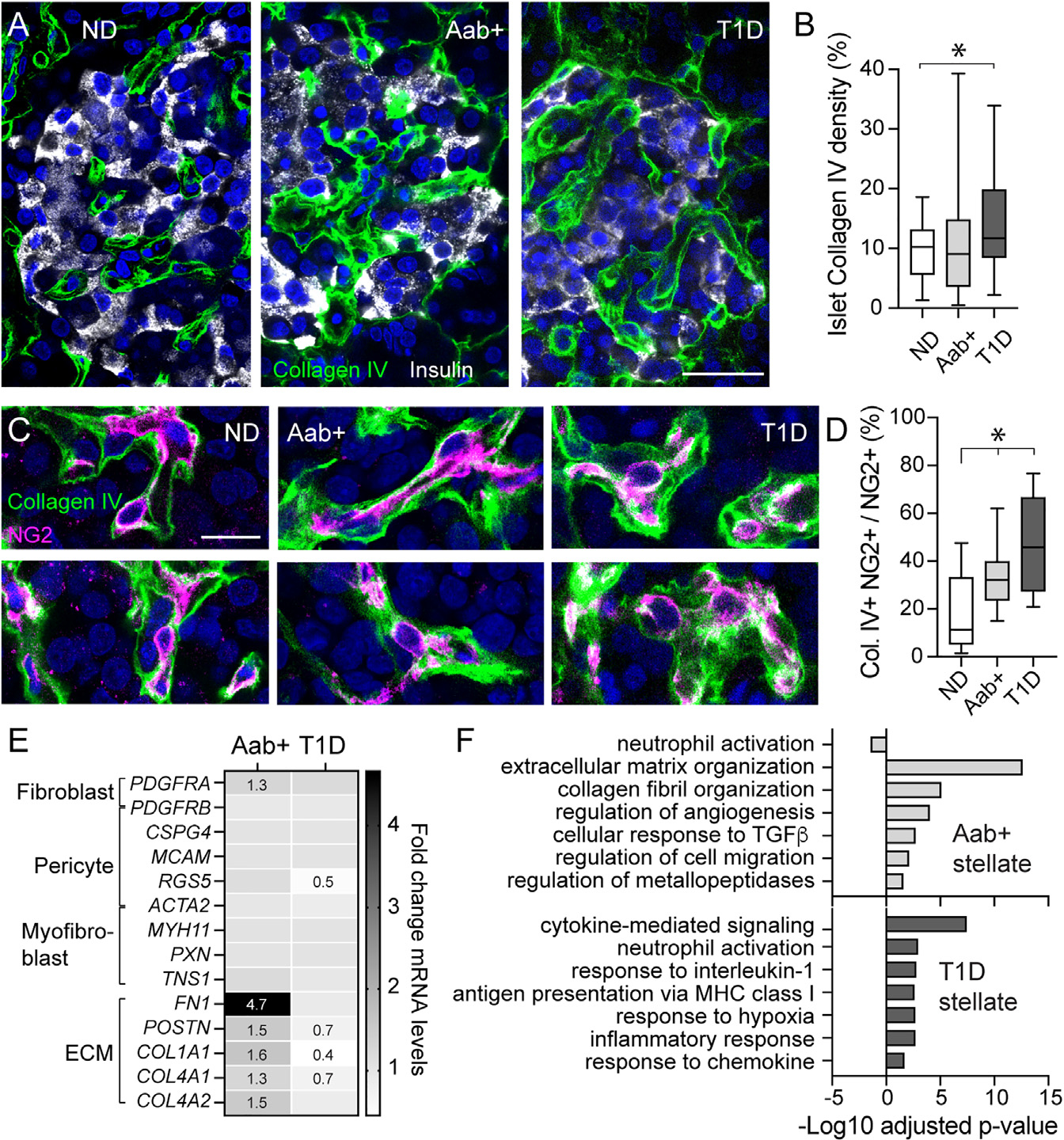
(A) Confocal images of islets in ND (nPOD6539),Aab+ (nPOD6573), and T1D donor (nPOD6563) sections immunostained for insulin (gray) and collagen type IV (green).
(B) Percentage of islet area immunostained with collagen IV in each group (n = 28–36 islets/3–5 donors/group; *p < 0.05 [one-way ANOVA followed by Tukey’s multiple comparisons test]). Box and whiskers plot is shown, and whiskers reflect minimum and maximum values for each dataset.
(C) Confocal images pericytes (magenta) embedded in the basement membrane. Colocalization between NG2 and collagen IV appears in white.
(D) Box and whiskers plot showing Mander’s correlation coefficient estimating colocalization between NG2 and collagen IV in confocal images of islets from each group (n = 17–24 islets from 3–5 donors per group; *p < 0.05 [one-way ANOVA followed by Tukey’s multiple comparisons test]).
(E) Changes in expression of pericyte genes (PDGFRB, CSPG4, MCAM, RGS5, ACTA2), fibroblasts/myofibroblast genes (PDGFRA, MYH11, PXN, and TNS1), and genes encoding ECM proteins (FN1, POSTN, COLA1A1, COL4A1, COL4A2) in stellate cell clusters from Aab+ or T1D donors compared with ND donors. Fold change (FC) values compared to levels in ND are indicated when FC >1.2 or <0.8.
(F) Gene Ontology analysis of top 6 biological processes significantly upregulated in stellate cells from Aab+ (light gray) or T1D pancreata (dark gray) compared with ND. Scale bars: 20 μm (A) and 10 mm (C).
To examine in more detail the changes in the phenotype of pericyte/stellate cells in the pancreas of Aab+ and T1D donors, we used the HPAP single-cell RNA-seq (scRNA-seq) database. We analyzed the expression of different pericyte identity genes (PDGFRB, CSPG4, MCAM, RGS5, ACTA2); genes associated with a myofibroblast phenotype such as smooth muscle myosin heavy chain (MYH11) and periostin (POSTN); genes encoding focal adhesion proteins such as paxillin (PXN) and tensin (TSN1); and the ECM protein fibronectin (FN1) and collagens (e.g., COL1A1, COL4A1, COL4A2) in stellate cell populations from ND, Aab+, and T1D donors (Figure 6E). There were no significant transcriptional alterations of most pericyte genes except a significant downregulation of RGS5 in stellate cells from T1D donors (Figure 6E). RGS5 (regulator of G protein signaling 5) is a pericyte-specific gene whose expression is reduced in vessels with poor pericyte coverage compared to a mature vasculature.49 Genes encoding fibronectin, periostin, and collagen subunits were all significantly upregulated in stellate cells from Aab+ donors but downregulated in T1D stellate cells when compared with levels in ND donors (Figure 6E). Moreover, there was a switch in gene programs significantly upregulated in stellate cells in Aab+ and T1D donors: while stellate cells in Aab+ pancreata upregulated pathways involved in ECM production, collagen organization, and cell migration, those in T1D donors exhibited more immunoregulatory functions (Figure 6F). Our data show that there are major changes occurring in the phenotype of islet pericytes and stellate cells at different stages of T1D, which could have impacted their function.
Strong vascular remodeling occurs in the pancreas of Aab+ and T1D donors
Lastly, we examined transcriptional alterations of pancreatic endocrine, endothelial, and stellate cell populations potentially associated with the vasculature. We hypothesized that microvascular dysfunction could have interfered with islet blood perfusion, creating a hypoxic environment and triggering compensatory vascular and metabolic responses. Using the HPAP database, we looked for changes in the expression of genes known to be involved in tissue responses to hypoxia (e.g., during cancer50,51). These include genes encoding hypoxia-inducible transcription factors (e.g., HIF1A, HIF3A, ARNT) or proteins involved in HIF1α stabilization/degradation (e.g., VHL); genes involved in angiogenesis and vascular remodeling such as adrenomedullin (ADM) and different angiogenic factors (e.g., ANGPTL4, VEGFA) and their receptors (e.g., NRP1, KDR, FLT1); and genes involved in cell migration (e.g., ENPP2, MIF), glucose transport (SLC2A1, SLC2A3), and metabolism (e.g., phosphofructokinase [PFKP], pyruvate dehydrogenase kinase isozyme 4 [PDK4], lactate dehydrogenase A [LDHA], and phosphoglycerate kinase [PGK1]). We noticed that the gene encoding the vasodilator peptide ADM was upregulated in different cell types in Aab+ and T1D pancreata (Figure 7A). In addition, there was a significant upregulation of genes encoding different angiogenic and growth factors and their respective receptors in islet endothelial and stellate cells in Aab+ donors (Figure 7A), indicating potential active vascular remodeling. Vascular alterations were accompanied by metabolic changes indicated by significant upregulation of PDK4 and LDHA in endocrine alpha and beta cells in T1D islets. These data show that transcriptional alterations related to the vasculature are present in the pancreas of single Aab+ donors, similar to what had been described for T1D donors.52
Figure 7. Vascular remodeling occurs in Aab+ and T1D donor pancreata.
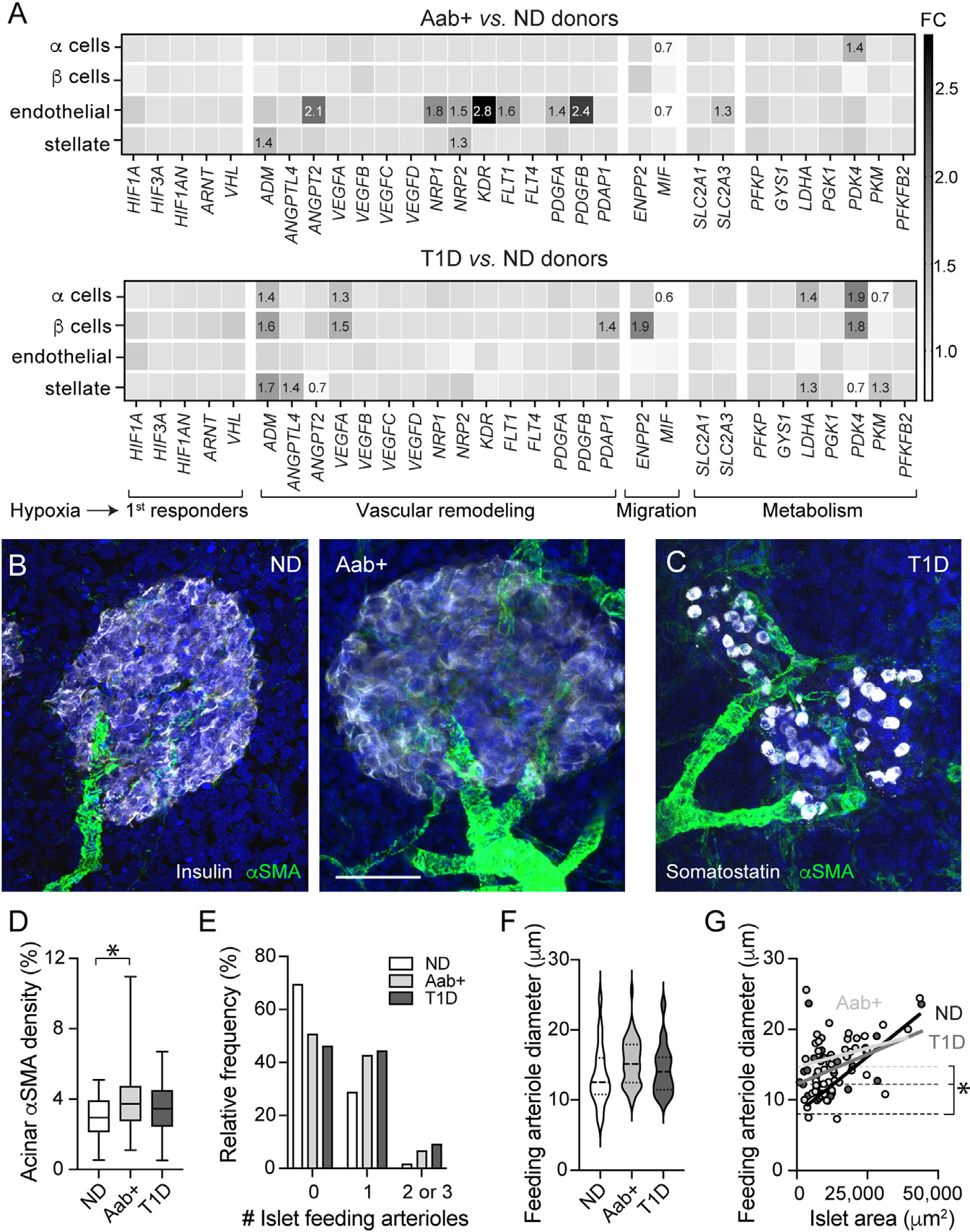
(A) Transcriptional alterations in alpha, beta, endothelial, and stellate cells from ND, Aab+, and T1D donor pancreatic islets. Shown are FC values compared to levels in ND of genes involved in hypoxia response (HIF1A, HIF3A, HIF1AN, ARNT, VHL), vasodilation (ADM), angiogenesis and vascular remodeling (ANGPTL4, ANGPT2, VEGFA-D, NRP1–2, KDR, FLT1, FLT4, PDGFA, PDGFB, PDAP1), cell migration (ENPP2, MIF), and metabolism (SLC2A1, SLC2A3, PFKP, GYS1, LDHA, PGK1, PDK4, PKM, PFKFB2).
(B and C) z stack of confocal images of islets in ND (nPOD6539), Aab+ (nPOD6558), and T1D donor sections (T1D for 2.5 years; nPOD6480) immunostained for aSMA (green) and insulin (gray; ND and Aab+; B) or somatostatin (gray; T1D; C). Scale bar: 40 μm.
(D) Box and whiskers plot (whiskers reflect minimum and maximum values) showing percentage of exocrine tissue immunostained with αSMA in slices from each group (n = 39–64 islets/5–7 donors/group; *p < 0.05 [one-way ANOVA followed by Tukey’s multiple comparisons test]).
(E) Histogram showing percentage of islets containing 0, 1, or 2–3 arterioles.
(F) Volcano plot showing feeding arteriole diameter in islets from ND (n = 20 islets/7 donors), Aab+ (n = 40 islets/6 donors), and T1D donors (n = 35 islets/6 donors).
(G) Correlation between islet area and diameter of feeding arteriole. In ND (black) and T1D (dark gray), there is a linear relationship (R2 = 0.61, p = 0.0001 and R2 = 0.2, p = 0.01, respectively), but not in Aab+ (light gray; R2 = 0.05, p = 0.23).
To visualize whether vascular remodeling occurred in the pancreas, we examined αSMA staining patterns extra islet in pancreatic tissue from Aab+ and T1D donors. αSMA is a contractile protein expressed by mural cells—pericytes and smooth muscle cells—and myofibroblasts (Figure S7,18,47). Smooth muscle cells can be easily distinguished from other cell types given their circular cytoplasmic processes and the multiple rings they form around arterioles or arteries (Figures 7B and 7C). We observed an increase in the number of arterioles irrigating islets in Aab+ and T1D pancreata (Figures 7B–7D). Indeed, while only around ~30% of islets in ND donors had one feeding arteriole, around 50% of islets in Aab+ and T1D pancreata had one or more feeding arterioles (Figure 7E). There was no significant difference in the diameter of islet feeding arterioles (Figure 7F). Interestingly, while in islets from ND donors, there was a significant correlation between the islet area and the diameter of the corresponding feeding arteriole (R2 = 0.6, p = 0.0001), this correlation was lost in islets from Aab+ donors (R2 = 0.05, p = ns; Figure 7G). Moreover, y intercept values (elevations) were significantly higher for both Aab+ and T1D donors, suggesting that Aab+ and T1D islets may be equipped to receive more blood than what would be needed to supply those tissue areas (Figure 7G). This apparent increase in vascular supply of Aab+ and T1D islets could have occurred as a compensatory response to impaired vascular perfusion of endocrine tissues at these stages. In future studies, we will extend our analyses to the exocrine tissue and examine the function of extra-islet blood vessels as the disease progresses.
DISCUSSION
In this study, we inspected islet pericytes and compared their density, phenotype, response profile, and gene expression in ND donors with those in individuals at different stages of T1D (GADA+ or “stage 0” and T1D or stage 3). We found striking changes in the function and phenotype of these vascular cells in islets at early stages of the disease. Previous studies have shown that islet pericytes are needed for proper insulin secretion and glucose homeostasis in mice.26,53 Their dysfunction could compromise insulin release from endocrine beta cells, exacerbating beta cell stress and potential failure. In this study, we have used living pancreas slices to monitor islet pericyte and capillary responses ex vivo. The impact of the changes we observed may be even greater in vivo when important regulators of blood perfusion are present (central nervous system input, blood flow and shear stress, intercellular coupling between pericytes or endothelial cells). However, we believe that our approach reflects the in vivo situation to the highest degree currently possible with humans, giving important insights into the pathophysiology of T1D.
A variety of mechanisms are believed to contribute to the pathogenesis of T1D, which could explain the significant heterogeneity and pathological features of this disease.3 T1D is considered a disease of both the immune system and the beta cell, where not only islet-specific autoreactive T cells mistakenly destroy “healthy” beta cells but stressed beta cells may also trigger an autoimmune attack against themselves.54 A defective islet microvasculature could contribute in both ways, either by facilitating immune cell infiltration into the pancreas (see below) or by compromising vascular perfusion and lowering oxygen levels, exacerbating beta cell stress. Vasculopathy is observed in different tissues throughout the diabetic body, and it is usually considered a consequence of diabetes.55 However, some of the metabolic challenges associated with a diabetic environment (e.g., hyperglycemia, inflammation, and oxidative stress) that damage vascular cells56 may already be present in a prediabetic state in the pancreas (e.g., inflammation in exocrine tissue57). Indeed, different groups have reported significant anatomical and functional alterations of the islet microvasculature before the onset of symptoms,5,7,10,13,58,59 but the cellular and molecular mechanisms underlying these defects were missing.
Pericytes are crucial for microvascular homeostasis and stability throughout the body.31,60 Substantial evidence indicates that pericyte loss (or dysfunction) leads to microvascular instability, disrupting the barrier properties of the endothelium, and to the formation of microaneurysms, microhemorrhages, acellular capillaries, and capillary non-perfusion.61,62 In this study, we observed that some capillaries in islets from T1D donors lacked pericytes (Figure 1). Moreover, we found that pericytes in islets from Aab+ and T1D donors were dysfunctional (Figures 2, 4, and 5), potentially due to a switch in their phenotype toward a myofibroblast-like cell (Figure 6). Pericyte loss and alterations in their function/phenotype, which may involve their detachment form the capillary wall, compromise pericytes’ vascular-stabilizing properties.63 This results in vulnerable capillaries that are prone to instability and increased vascular leakage, which, ultimately, may facilitate immune cell infiltration. Our data are in line with a previous study using magnetic resonance imaging and nanoparticles that revealed abnormal vascular integrity and leukocyte infiltration in the pancreas of patients with T1D within 6 months of diagnosis.58 Besides controlling transendothelial permeability, pericytes have also been shown to actively participate in immunosurveillance in different tissues.64 In response to inflammatory mediators, pericytes express adhesion molecules and chemoattractant factors controlling leukocyte migration in vivo.65,66 Future studies investigating the role of pericytes as active components/mediators of immune responses in islets would be needed to understand the potential impact of their dysfunction in (pre-)diabetes.
Pericytes are also important regulators of capillary blood flow in different tissues such as the brain, the retina, and the islet.18,67–70 Interestingly, there is compelling evidence that islet blood flow is disturbed during conditions of impaired glucose tolerance and overt diabetes, but it is not known if these disturbances are of pathogenic importance.71 Researchers have reported both increases and decreases in islet blood flow in animal models of T1D and T2D. It seems that at early stages of deranged metabolism, blood flow increases in islets, but, as the disease progresses, it is then followed by a decrease in islet blood perfusion.72 A recent study using ultrasound contrast imaging revealed altered islet blood flow dynamics in mice before T1D onset, characterized by increased perfusion velocity but reduced perfusion volume.13 These findings in mouse models are in line with our observations in islets from Aab+ and T1D donors, which have a higher number of feeding arterioles (Figure 7) but either decreased islet capillary diameter (as we show in this article) or decreased vessel density as previously shown.8
In this study, we also found that changes in [Ca2+]i in pericytes in islets from Aab+ and T1D donors, elicited either by high glucose, norepinephrine, or endothelin-1, are not associated with significant changes in islet vessel diameter (Figures 2, 3, 4, and 5). We do not know why islet pericytes become dysfunctional. We hypothesized that, given pericytes’ enormous plasticity and postnatal undifferentiated nature, they have switched their phenotype and turned into ECM-producing myofibroblasts (activated stellate cells; Figure 6). Indeed, human T1D is characterized by a modified islet ECM,73 and islet pericytes are capable of producing ECM proteins under physiological and pathophysiological conditions.23,47 A switch in the pericyte phenotype—from an excitable, electrically coupled mural cell to a non-excitable fibroblast-like cell—would have functional consequences. Electrical coupling between mural cells allows the sequential spread of depolarizations to develop synchronous calcium transients within their network.74 Because these rhythmical contractions of mural cells are required for efficient vasomotion,75 the conversion of islet pericytes, even if only a subset of them, into myofibroblasts could produce barriers for conduction of signals, interfering with effective vasodilation or vasoconstriction. Pericyte dysfunction at early stages of the disease would interfere with the islet’s capacity to induce acute changes in its blood flow, triggered, for instance, upon hyperglycemia or changes in sympathetic tonus.26
Our study further suggests that altered endothelin-1 signaling/action in the pancreas may contribute to vascular dysfunction. Endothelins are among the most potent vasoconstrictors in the body. Previous studies had shown that endothelin-1 was present in the human pancreas and that its expression could be regulated by hypoxia.76,77 Here, we found that pericyte [Ca2+]i responses to a dual endothelin-1 receptor antagonist (bosentan) were altered in islets from Aab+ donors (Figure 5). A potential explanation for these results is that the gene encoding ETB receptors (EDNRB) is significantly upregulated in endothelial cells from Aab+ donors (Figure 5). Upregulation of the EDNRB gene in islets has been previously observed during hypoxia in vitro.77 In different tissues throughout the body (e.g., kidney, liver, and lungs), ETB receptors have also been shown to play a critical role scavenging pathological levels of endothelin-1 from circulation and targeting it to degradation.78,79 Upregulation of ETB receptors by vascular cells in Aab+ islets could have been a compensatory response of the tissue to limit the vasoconstrictive effects of endothelin 1, which, besides its vasoactive functions, also stimulates proliferation of fibroblasts and ECM production.46,76
To summarize, islets are equipped with vascular networks that not only support endocrine cell health and maturation but also their function, as they enable proper glucose sensing and hormone secretion. Our study shows that the islet microvasculature is dysfunctional already in islets from single Aab+ donors. Whether islet vascular dysfunction impairs in vivo endocrine cell responses to different metabolic challenges remains to be determined. In vitro glucose-stimulated insulin secretion from islets isolated from single Aab+ donors is preserved,80 but a case report of a GADA+ donor showed fasting blood glucose and HbA1C in pre-diabetic ranges and marked glucose intolerance.57 Studies have also shown that individuals at greater risk of developing T1D fail to increase insulin/C-peptide secretion with age and have diminished first-phase insulin response to intravenous glucose.81 Whether islet vascular dysfunction potentiates this functional decline, which accelerates as the disease approaches,82 will be the focus of future work.
Limitations of the study
In this study, we have not identified the molecular mechanisms underlying hemodynamic changes in islets. One of the reasons is that we still know very little about how islet cells (endocrine, vascular, immune cells, etc.) work together to maintain homeostasis and proper function. Understanding how different cells in the human islet communicate with each other under normal physiological conditions is crucial to be able to later determine what becomes compromised in a disease state. Another limitation of the study is that the limited amount of living human material we received prevented us from correlating our functional findings with the composition of surrounding microenvironment, in particular with the presence of residual (insulin-positive) beta cells. However, recent studies have shown that a considerable amount of islets still contain beta cells (~40%–60%) at diagnosis in people who developed T1D as teenagers or later.83 We are also aware that only a subset of single Aab+ donors represent true pre-diabetic individuals who would have developed T1D. However, several studies have shown that islets from single Aab+ donors already present some alterations, regarding, for instance, the ECM composition,6 alpha cell function,80 and proinsulin levels,84 among others. It would be interesting to compare responses of single Aab+ with those in donors with multiple Aab+ to examine vascular function at later stages of islet autoimmunity.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources or reagents should be directed to the Lead Contact, Joana Almaça (jalmaca@med.miami.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
All original code has been deposited at Github and Zenodo and is publicly available as of the date of publication. DOIs are listed in key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Neuron-glial antigen 2 (NG2) | R&D | RRID: FAB2585R |
| Platelet-derived growth factor receptor-beta (PDGFRβ) | R&D | RRID: AF385 |
| Alpha smooth muscle actin (αSMA) | Sigma | RRID: A5228 |
| CD31 | Millipore | RRID: 550389 |
| Tyrosine hydroxylase (TH) | Millipore | RRID: AB152 |
| Collagen IV | Abcam | RRID: AB6586 |
| Periostin | R&D | RRID: AF2955 |
| Insulin | Dako | RRID: IR002 |
| Somatostatin | Millipore | RRID: MAB354 |
| CD146 | Abcam | RRID: Ab75769 |
| Alexa Fluor Secondary antibodies | ThermoFischer | |
|
| ||
| Biological samples | ||
|
| ||
| Living human pancreas slices | nPOD | https://npod.org |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Theophylline | Sigma | T1633 |
| Norepinephrine | Tocris | 5169 |
| Endothelin 1 | Sigma | E7764 |
| Angiotensin II | Tocris | 1158 |
| Bosentan | Sigma | 1265 |
| Adenosine | Sigma | A9251 |
| Lycopersicon esculentum Lectin | Vector Labs | DL-1178-1 |
| Fluo-4 | Invitrogen | F14201 |
| Aprotinin | Sigma Aldrich | A6106 |
| Penicillin-Streptomycin-Amphotericin B | Sigma Aldrich | A5955 |
| BrainPhys neuronal medium | Stemcell Technologies | 05790 |
| B27 minus-insulin | Invitrogen | A1895601 |
| Chymostatin | Sigma Aldrich | 11004638001 |
|
| ||
| Deposited data | ||
|
| ||
| Human Pancreas Analysis Program, pancreas single cell RNAseq data repository | HPAP | https://hpap.pmacs.upenn.edu/analysis |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism | GraphPad | Prism9 |
| ImageJ (https://imagej.nih.gov/ij/) | NIH | ImageJ |
| MatLab (www.mathworks.com) | MathWorks | MatLab R2020b |
| Single cell analysis architecture (Github + Zenodo) | JA Lab |
https://github.com/jalmaca/Microvasculature_T1D
https://zenodo.org/record/8122389 |
|
| ||
| Other | ||
|
| ||
| SP5 inverted laser-scanning confocal microscope | Leica | SP5 |
| SP8 upright laser-scanning confocal microscope | Leica | SP8 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Human participants
In this study we used living pancreas slices (150 μm thickness; pancreas pieces were taken from the tail of pancreas) from de-identified cadaveric donors from the Network of Pancreatic Organ Donors with Diabetes (nPOD). We received living pancreas slices from 12 Aab−, non-diabetic donors (ND), 7 single autoantibody positive (GADA+) donors and 12 T1D organ donors, with T1D duration of 0–4 years. Donor age ranged from 11–33 years old, from both genders (details on donor characteristics are included in Table S1). All T1D donors were positive for islet autoantibodies (Table S1). C-peptide levels and glycated hemoglobin values are shown in Figure S1.
Slices were produced by nPOD and 9–11 slices/donor were shipped overnight from Gainesville to Miami and used within 4–36h after arrival. Slices were cultured as previously described.85 Briefly, upon arrival, living slices were placed on perfluorocarbon (PFC) membrane AirHive cell culture dishes containing BrainPhys neuronal medium (Stemcell Technologies, cat. nr. 05790) supplemented with 2% B27 minus-insulin (Invitrogen, cat. nr. A1895601), 1% penicillin-Streptomycin-Amphotericin B solution (Sigma Aldrich, cat. nr. A5955), 1% Glutamax (Invitrogen, cat. nr. 35050061), 5.5 mM D-glucose (Sigma Aldrich, cat. nr. G8644), 100 μg/ml trypsin inhibitor from Glycine max (Sigma Aldrich, cat. nr. T6522), 10 μg/ml aprotinin (Sigma Aldrich, cat. nr. A6106), 10 μg/ml chymostatin (solubilized initially in DMSO; Sigma Aldrich, cat. nr. 11004638001) and 1% HEPES buffer (Invitrogen, cat. nr. 15630080). Dishes containing 3–4 slices and culture medium were placed in a humidified incubator at 30°C, and meedium was changed every 12 h.
METHOD DETAILS
Confocal imaging of living pancreas slices
Living human pancreas slices were incubated with the cytosolic calcium indicator ([Ca2+]i) Fluo4-AM (6 mM, Invitrogen, cat. nr. F14201) in 3 mM glucose solution prepared in HEPES buffer (125 mM NaCl, 5.9 mM KCl, 2.56 mM CaCl2, 1 mM MgCl2, 25 mM HEPES, 0.1% BSA [w/v], pH 7.4), supplemented with aprotinin (25 KIU), at room temperature and in the dark with either (a) DyLight 649 lectin from Lycopersicon Esculentum (3.3 mg/mL, VectorLabs, cat. nr. DL1178) for 1 hour; or with (b) a fluorescent-conjugated antibody against the pericyte marker neuron-glial antigen 2 (NG2-alexa647; 1:50, R&D Systems, cat. nr. Fab2585R) for 2 hours. After incubation, living pancreas slices were placed on a coverslip in an imaging chamber (Warner instruments, Hamden, CT, USA) and imaged under an upright confocal microscope (Leica TCS SP8 upright; Leica Microsystems). The chamber was continuously perfused with HEPES-buffered solution containing 3 mM glucose and confocal images were acquired with LAS AF software using a 40X water immersion objective (NA 0.8). We used a resonance scanner for fast image acquisition to produce time-lapse recordings spanning 50–100 μm of the slice (z-step: 5–10 μm, stack of 10–15 confocal images with a size of 512×512 pixels) at 5 seconds resolution (xyzt imaging). Fluo-4 fluorescence was excited at 488 nm and emission detected at 510–550 nm, DyLight 649 lectin or NG2-alexa647 antibody were excited at 638 nm and emission detected at 648–690 nm.
We recorded changes in islet pericyte [Ca2+]i and capillary diameter induced by exchanging extracellular glucose concentration from 3 mM to 11mM (3G to 11G; 11G applied for 5–7 min), norepinephrine (20 μM; applied for 4 min in 3G), endothelin-1 (10 nM; applied for 5 min in 3G), angiotensin II (100 nM; applied for 4 min in 3G), bosentan (100 nM; applied for 5 min in 3G) and theophylline (20 μM; applied for 5 min in 11G). Islets in slices were identified using the backscatter signal produced by dense-core granules. To quantify changes in pericyte [Ca2+]i, using ImageJ software (http://imagej.nih.gov/ij/), we drew regions of interest around NG2-alexa647 labeled cells in islets as previously described.24 Changes in fluorescence intensity were expressed as percentage over baseline (ΔF/F;%). The baseline was defined as the mean of the first 10 values of the control period of each recording [basal glucose concentration (3 mM) for all stimuli, except for theophylline for which baseline values were calculated during 11 mM glucose]. After subtracting the baseline, we calculated the net area under the curve of fluorescence traces to estimate the magnitude of the effect of each stimulus on cellular [Ca2+]i. MatLab software (https://www.mathworks.com/) was used to generate heatmaps showing changes in fluorescence of all recorded pericytes.
Immunohistochemistry
After physiological recordings, living human pancreas slices from non-diabetic, Aab+ and T1D organ donors were fixed for 1h with 4% PFA, washed 3x in PBS, and stored at 4’C. For immunohistochemistry, slices were incubated in blocking solution (PBS-Triton X-100 0.3% and Universal Blocker Reagent; Biogenex, San Ramon, CA) for 3h. Thereafter, slices were incubated for 48h (20°C) with primary antibodies diluted in blocking solution. To identify pancreatic islets in slices, we immunostained either beta cells (insulin, 1:5) or delta cells (somatostatin; 1:250). We labeled pericytes using an anti-NG2 antibody (1:50–1:100), smooth muscle cells/myofibroblasts using anti-αSMA (1:250), endothelial cells using anti-CD31 (1:25), sympathetic nerve fibers using anti-tyrosine hydroxylase (TH; 1:400), basement membrane using anti-collagen IV antibody (1:250), periostin (1:250). Immunostaining was visualized using conjugated secondary antibodies (1:500 in PBS; 16h at 20°C; ThermoFischer). Cell nuclei were stained with dapi. Slides were mounted with Vectashield mounting medium (Vector Laboratories) and imaged on an inverted laser-scanning confocal microscope (Leica TCS SP5; Leica Microsystems) with LAS AF software using a 63X oil immersion objective (NA 1.4). Image analysis was performed using artificial intelligence and automated macros. Briefly, to quantify the density of each protein/marker in islets, an hormone staining (insulin or somatostatin) was used to locate islets within the image, while DAPI staining was used to outline the whole tissue area. The image was then segmented into islet area and exocrine tissue area, and the immunostained marker was thresholded based on mean image value for that channel. A mask for each immunostaining was created and the corresponding area was measured. To estimate islet versus acinar densities, we divided the immunostained area for each marker by the corresponding tissue area. To quantify colocalization between the pericyte marker NG2 and either CD146 or collagen IV, we calculated Mander’s coefficients in confocal images of islets from different donors using the ImageJ plugin “JACoP: Just Another Co-localization Plugin”.
Single-cell RNAseq analysis
To examine changes in gene expression related to the vasculature of different cell populations in human pancreatic islets, we accessed the single cell RNAseq dataset (PancDB) from the Human Pancreas Analysis Program (HPAP43,86; https://hpap.pmacs.upenn.edu), which is in the form of an archived Seurat R object. This dataset contains 222,077 cells across 67 individuals. We chose human donors that matched the characteristics of those we had used for physiological experimentation (Table S1; Figure S1). Data were subsetted from 20 individuals accounting for 7 single Aab+ (GADA+; 16,784 cells), 9 non-diabetic (17,603 cells) and 4 type-1 diabetic donors (disease duration: 0–3 years; 8,813 cells), collectively represented by 43,200 cells. We employed a comprehensive analytical pipeline using custom designed R scripts similar to that outlined previously.87 Cells clusters were selected based on their specific expression of different gene sets. Briefly, beta cells were identified based on the expression of INS, IAPP, PDX1 and MAFA, alpha cells on GCG, DPP4 and GC genes, endothelial cell on VWF and ENG genes and stellate cells (which include pericytes) on the expression of PDGFRB and COL1A1 genes (Figure S6). The complete analytical architecture is deposited in Github (https://github.com/jalmaca/Microvasculature_T1D) and Zenodo (https://zenodo.org/record/8122389). In this analysis we consider a differentially regulated gene when change in gene expression (FC values) were higher than 1.2 (upregulated gene) or below 0.8 (downregulated gene), having a p value less than 0.05 and being expressed in at least in 25% of cells across a cluster.
QUANTIFICATION AND STATISTICAL ANALYSIS
Vasomotion analysis
Blood vessels in living slices were labeled with DyLight-649 fluorescent lectin. We quantified changes in vessel diameter as previously described.24 Briefly, we drew a straight-line transversal to the blood vessel borders and used the “reslice” z-function in ImageJ to generate a single image showing the changes in vessel diameter over time (xt scan; temporal projection; Figure 3A). xt scan (resliced) images were despeckled, blurred with Gaussian filter sigma = 1, before enhancing contrast, and image sharpened using the following kernel in order to emphasize vertical lines: −10 −5 50 −5 −10; −10 −5 75 −5 −10; −10 −5 50 −5 −10. An horizontal line was drawn on the xt scan (resliced) image (which corresponds to a single time point), and an array of pixel intensity values was sorted and first 2 maxima were considered vessel borders (tolerance = 4). Vessel diameter was calculated by subtracting these 2 values. For each stimulus, an average diameter value was calculated using the 10 last diameter values obtained during stimulus application. To determine the extent of constriction/dilation, we pooled diameter data from different capillaries from different islets for each group of donors and calculated the relative change in diameter (as % of baseline (3G) vessel diameter).
Statistical analyses
For statistical comparisons we used Prism 9 (GraphPad software, La Jolla, CA) and performed one-way ANOVA followed by Tukey’s multiple comparisons test when comparing data obtained from non-diabetic donors, single Aab+ and T1D organ donors. Paired t-tests were used to determine if changes in diameter induced by a certain stimulus for each individual capillary were significantly different from baseline values. One sample t tests were used to determine if average changes in [Ca2+]i or vessel diameter induced by different stimuli were significantly different from 0. p values < 0.05 were considered statistically significant (indicated with an * in figures). Throughout the manuscript we present data as mean ± SEM (bar graphs) or in box and whiskers plots where whiskers reflect minimum and maximum values.
Supplementary Material
Highlights.
Changes in islet pericyte phenotype and function occur during T1D progression
Vascular responses to vasoactive stimuli are impaired in islets from Aab+ and T1D donors
Changes related to ET-1 action/signaling are found in Aab+ and T1D islet vascular cells
Vascular and ECM remodeling occurs in Aab+ and T1D donor pancreata
ACKNOWLEDGMENTS
The authors would like to thank Drs. Alberto Pugliese (City of Hope, CA, USA), Alejandro Caicedo, and Ruy Andrade Louzada (University of Miami, FL, USA) and Profs. Nilda Gallardo and Antonio Andres Hueva (Castilla La Mancha University) for careful revision and discussion of the manuscript. The authors would like to thank the Network for Pancreatic Organ Donors with Diabetes (nPOD), in particular the organ donors, their families, and the nPOD slicing team, under Dr. Irina Kusmartseva’s supervision, for producing living pancreas slices that allowed us to conduct these studies. This manuscript used data acquired from the Human Pancreas Analysis Program (HPAP-RRID: SCR_016202) Database (https://hpap.pmacs.upenn.edu), a Human Islet Research Network (RRID: SCR_014393) consortium (UC4-DK-112217, U01-DK-123594, UC4-DK-112232, and U01-DK-123716). This work has been funded by NIH grants K01DK111757 and R01DK133483 (to J.A.), by the NIDDK-supported Human Islet Research Network (HIRN, RRID: SCR_014393; https://hirnetwork.org; UC4 DK104162) New Investigator Pilot Award (to J.A.), and by the Helmsley Charitable Trust for nPOD team science (2018PG-T1D060 and 2112-04895).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112913.
REFERENCES
- 1.Eisenbarth GS (1986). Type I diabetes mellitus. A chronic autoimmune disease. N. Engl. J. Med. 314, 1360–1368. 10.1056/nejm198605223142106. [DOI] [PubMed] [Google Scholar]
- 2.Rodriguez-Calvo T, Richardson SJ, and Pugliese A (2018). Pancreas Pathology During the Natural History of Type 1 Diabetes. Curr. Diab. Rep. 18, 124. 10.1007/s11892-018-1084-3. [DOI] [PubMed] [Google Scholar]
- 3.Richardson SJ, and Pugliese A (2021). 100 YEARS OF INSULIN: Pancreas pathology in type 1 diabetes: an evolving story. J. Endocrinol. 252, R41–r57. 10.1530/joe-21-0358. [DOI] [PubMed] [Google Scholar]
- 4.Canzano JS, Nasif LH, Butterworth EA, Fu DA, Atkinson MA, and Campbell-Thompson M (2019). Islet Microvasculature Alterations With Loss of Beta-cells in Patients With Type 1 Diabetes. J. Histochem. Cytochem. 67, 41–52. 10.1369/0022155418778546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bogdani M, Johnson PY, Potter-Perigo S, Nagy N, Day AJ, Bollyky PL, and Wight TN (2014). Hyaluronan and hyaluronan-binding proteins accumulate in both human type 1 diabetic islets and lymphoid tissues and associate with inflammatory cells in insulitis. Diabetes 63, 2727–2743. 10.2337/db13-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bogdani M, Speake C, Dufort MJ, Johnson PY, Larmore MJ, Day AJ, Wight TN, Lernmark Å, and Greenbaum CJ (2020). Hyaluronan deposition in islets may precede and direct the location of islet immunecell infiltrates. Diabetologia 63, 549–560. 10.1007/s00125-019-05066-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korpos É, Kadri N, Kappelhoff R, Wegner J, Overall CM, Weber E, Holmberg D, Cardell S, and Sorokin L (2013). The peri-islet basement membrane, a barrier to infiltrating leukocytes in type 1 diabetes in mouse and human. Diabetes 62, 531–542. 10.2337/db12-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang YJ, Traum D, Schug J, Gao L, Liu C, HPAP Consortium; Atkinson MA, Powers AC, Feldman MD, Naji A, Chang KM, and Kaestner KH. (2019). Multiplexed In Situ Imaging Mass Cytometry Analysis of the Human Endocrine Pancreas and Immune System in Type 1 Diabetes. Cell Metab. 29, 769–783.e4. 10.1016/j.cmet.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turvey SE, Swart E, Denis MC, Mahmood U, Benoist C, Weissleder R, and Mathis D (2005). Noninvasive imaging of pancreatic inflammation and its reversal in type 1 diabetes. J. Clin. Invest. 115, 2454–2461. 10.1172/jci25048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramirez DG, Ciccaglione M, Upadhyay AK, Pham VT, Borden MA, and Benninger RKP (2021). Detecting insulitis in type 1 diabetes with ultrasound phase-change contrast agents. Proc. Natl. Acad. Sci. USA 118, e2022523118. 10.1073/pnas.2022523118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdulreda MH, Molano RD, Faleo G, Lopez-Cabezas M, Shishido A, Ulissi U, Fotino C, Hernandez LF, Tschiggfrie A, Aldrich VR, et al. (2019). In vivo imaging of type 1 diabetes immunopathology using eye-transplanted islets in NOD mice. Diabetologia 62, 1237–1250. 10.1007/s00125-019-4879-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlsson PO, Sandler S, and Jansson L (1998). Pancreatic islet blood perfusion in the nonobese diabetic mouse: diabetes-prone female mice exhibit a higher blood flow compared with male mice in the prediabetic phase. Endocrinology 139, 3534–3541. 10.1210/endo.139.8.6153. [DOI] [PubMed] [Google Scholar]
- 13.St Clair JR, Ramirez D, Passman S, and Benninger RKP (2018). Contrast-enhanced ultrasound measurement of pancreatic blood flow dynamics predicts type 1 diabetes progression in preclinical models. Nat. Commun. 9, 1742. 10.1038/s41467-018-03953-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Insel RA, Dunne JL, Atkinson MA, Chiang JL, Dabelea D, Gottlieb PA, Greenbaum CJ, Herold KC, Krischer JP, Lernmark Å, et al. (2015). Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 38, 1964–1974. 10.2337/dc15-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ballian N, and Brunicardi FC (2007). Islet vasculature as a regulator of endocrine pancreas function. World J. Surg. 31, 705–714. 10.1007/s00268-006-0719-8. [DOI] [PubMed] [Google Scholar]
- 16.Richards OC, Raines SM, and Attie AD (2010). The role of blood vessels, endothelial cells, and vascular pericytes in insulin secretion and peripheral insulin action. Endocr. Rev. 31, 343–363. 10.1210/er.2009-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonner-Weir S (1993). The microvasculature of the pancreas, with emphasis on that of the islets of Langerhans. Pancreas: Biology, Pathobiology and Disease. Chapter 39. [Google Scholar]
- 18.Almaça J, Weitz J, Rodriguez-Diaz R, Pereira E, and Caicedo A(2018). The Pericyte of the Pancreatic Islet Regulates Capillary Diameter and Local Blood Flow. Cell Metabol. 27, 630–644.e4. 10.1016/j.cmet.2018.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Epshtein A, Rachi E, Sakhneny L, Mizrachi S, Baer D, and Landsman L (2017). Neonatal pancreatic pericytes support beta-cell proliferation. Mol. Metab. 6, 1330–1338. 10.1016/j.molmet.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Houtz J, Borden P, Ceasrine A, Minichiello L, and Kuruvilla R (2016). Neurotrophin Signaling Is Required for Glucose-Induced Insulin Secretion. Dev. Cell 39, 329–345. 10.1016/j.devcel.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakhneny L, Rachi E, Epshtein A, Guez HC, Wald-Altman S, Lisnyansky M, Khalifa-Malka L, Hazan A, Baer D, Priel A, et al. (2018). Pancreatic Pericytes Support beta-Cell Function in a Tcf7l2-Dependent Manner. Diabetes 67, 437–447. 10.2337/db17-0697. [DOI] [PubMed] [Google Scholar]
- 22.Sasson A, Rachi E, Sakhneny L, Baer D, Lisnyansky M, Epshtein A, and Landsman L (2016). Islet Pericytes Are Required for beta-Cell Maturity. Diabetes 65, 3008–3014. 10.2337/db16-0365. [DOI] [PubMed] [Google Scholar]
- 23.Sakhneny L, Epshtein A, and Landsman L (2021). Pericytes contribute to the islet basement membranes to promote beta-cell gene expression. Sci. Rep. 11, 2378. 10.1038/s41598-021-81774-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mateus Gonçalves L, and Almaça J (2020). Functional Characterization of the Human Islet Microvasculature Using Living Pancreas Slices. Front. Endocrinol. 11, 602519. 10.3389/fendo.2020.602519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michau A, Lafont C, Bargi-Souza P, Kemkem Y, Guillou A, Ravier MA, Bertrand G, Varrault A, Fiordelisio T, Hodson DJ, et al. (2022). Metabolic Stress Impairs Pericyte Response to Optogenetic Stimulation in Pancreatic Islets. Front. Endocrinol. 13, 918733. 10.3389/fendo.2022.918733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamayo A, Gonçalves LM, Rodriguez-Diaz R, Pereira E, Canales M, Caicedo A, and Almaça J (2022). Pericyte Control of Blood Flow in Intraocular Islet Grafts Impacts Glucose Homeostasis in Mice. Diabetes 71, 1679–1693. 10.2337/db21-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakamura M, Kitamura H, Konishi S, Nishimura M, Ono J, Ina K, Shimada T, and Takaki R (1995). The endocrine pancreas of spontaneously diabetic db/db mice: microangiopathy as revealed by transmission electron microscopy. Diabetes Res. Clin. Pract. 30, 89–100. [DOI] [PubMed] [Google Scholar]
- 28.Dai C, Brissova M, Reinert RB, Nyman L, Liu EH, Thompson C, Shostak A, Shiota M, Takahashi T, and Powers AC (2013). Pancreatic islet vasculature adapts to insulin resistance through dilation and not angiogenesis. Diabetes 62, 4144–4153. 10.2337/db12-1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wenzlau JM, and Hutton JC (2013). Novel diabetes autoantibodies and prediction of type 1 diabetes. Curr. Diab. Rep. 13, 608–615. 10.1007/s11892-013-0405-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, Winkler C, Ilonen J, Veijola R, Knip M, et al. (2013). Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 309, 2473–2479. 10.1001/jama.2013.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Armulik A, Genové G, and Betsholtz C (2011). Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 21, 193–215. 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 32.Shepro D, and Morel NM (1993). Pericyte physiology. Faseb. J. : official publication of the Federation of American Societies for Experimental Biology 7, 1031–1038. [DOI] [PubMed] [Google Scholar]
- 33.Baum J, and Duffy HS (2011). Fibroblasts and myofibroblasts: what are we talking about? J. Cardiovasc. Pharmacol. 57, 376–379. 10.1097/FJC.0b013e3182116e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jansson L, and Hellerström C (1983). Stimulation by glucose of the blood flow to the pancreatic islets of the rat. Diabetologia 25, 45–50. 10.1007/bf00251896. [DOI] [PubMed] [Google Scholar]
- 35.Moldovan S, Livingston E, Zhang RS, Kleinman R, Guth P, and Brunicardi FC (1996). Glucose-induced islet hyperemia is mediated by nitric oxide. Am. J. Surg. 171, 16–20. [DOI] [PubMed] [Google Scholar]
- 36.Nyman LR, Ford E, Powers AC, and Piston DW (2010). Glucose-dependent blood flow dynamics in murine pancreatic islets in vivo. Am. J. Physiol. Endocrinol. Metab. 298, E807–E814. 10.1152/ajpendo.00715.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geisler JC, Corbin KL, Li Q, Feranchak AP, Nunemaker CS, and Li C (2013). Vesicular nucleotide transporter-mediated ATP release regulates insulin secretion. Endocrinology 154, 675–684. 10.1210/en.2012-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacques-Silva MC, Correa-Medina M, Cabrera O, Rodriguez-Diaz R, Makeeva N, Fachado A, Diez J, Berman DM, Kenyon NS, Ricordi C, et al. (2010). ATP-gated P2X3 receptors constitute a positive autocrine signal for insulin release in the human pancreatic beta cell. Proc. Natl. Acad. Sci. USA 107, 6465–6470. 10.1073/pnas.0908935107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fontaine AK, Ramirez DG, Littich SF, Piscopio RA, Kravets V, Schleicher WE, Mizoguchi N, Caldwell JH, Weir RFF, and Benninger RKP (2021). Optogenetic stimulation of cholinergic fibers for the modulation of insulin and glycemia. Sci. Rep. 11, 3670. 10.1038/s41598-021-83361-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guyot M, Simon T, Ceppo F, Panzolini C, Guyon A, Lavergne J, Murris E, Daoudlarian D, Brusini R, Zarif H, et al. (2019). Pancreatic nerve electrostimulation inhibits recent-onset autoimmune diabetes. Nat. Biotechnol. 37, 1446–1451. 10.1038/s41587-019-0295-8. [DOI] [PubMed] [Google Scholar]
- 41.Richardson TM, Saunders DC, Haliyur R, Shrestha S, Cartailler JP, Reinert RB, Petronglo J, Bottino R, Aramandla R, Bradley AM, et al. (2023). Human pancreatic capillaries and nerve fibers persist in type 1 diabetes despite beta cell loss. Am. J. Physiol. Endocrinol. Metab. 324, E251–e267. 10.1152/ajpendo.00246.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Houde M, Desbiens L, and D’Orléans-Juste P (2016). Endothelin-1: Biosynthesis, Signaling and Vasoreactivity. Adv. Pharmacol. 77, 143–175. 10.1016/bs.apha.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 43.Kaestner KH, Powers AC, Naji A, and HPAP Consortium; and Atkinson MA. (2019). NIH Initiative to Improve Understanding of the Pancreas, Islet, and Autoimmunity in Type 1 Diabetes: The Human Pancreas Analysis Program (HPAP). Diabetes 68, 1394–1402. 10.2337/db19-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, Ryu JH, Wagner BK, Shen-Orr SS, Klein AM, et al. (2016). A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst. 3, 346–360.e4. 10.1016/j.cels.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coate KC, Cha J, Shrestha S, Wang W, Gonçalves LM, Almaça J, Kapp ME, Fasolino M, Morgan A, Dai C, et al. (2020). SARS-CoV-2 Cell Entry Factors ACE2 and TMPRSS2 Are Expressed in the Microvasculature and Ducts of Human Pancreas but Are Not Enriched in β Cells. Cell Metab. 32, 1028–1040.e4. 10.1016/j.cmet.2020.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maguire JJ, and Davenport AP (2015). Endothelin receptors and their antagonists. Semin. Nephrol. 35, 125–136. 10.1016/j.sem-nephrol.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mateus Gonçalves L, Pereira E, Werneck de Castro JP, Bernal-Mizrachi E, and Almaça J (2020). Islet pericytes convert into profibrotic myofibroblasts in a mouse model of islet vascular fibrosis. Diabetologia 63, 1564–1575. 10.1007/s00125-020-05168-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuppe C, Ibrahim MM, Kranz J, Zhang X, Ziegler S, Perales-Patón J, Jansen J, Reimer KC, Smith JR, Dobie R, et al. (2021). Decoding myofibroblast origins in human kidney fibrosis. Nature 589, 281–286. 10.1038/s41586-020-2941-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitchell TS, Bradley J, Robinson GS, Shima DT, and Ng YS(2008). RGS5 expression is a quantitative measure of pericyte coverage of blood vessels. Angiogenesis 11, 141–151. 10.1007/s10456-007-9085-x. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Q, Huang R, Hu H, Yu L, Tang Q, Tao Y, Liu Z, Li J, and Wang G (2020). Integrative Analysis of Hypoxia-Associated Signature in Pan-Cancer. iScience 23, 101460. 10.1016/j.isci.2020.101460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chi JT, Wang Z, Nuyten DSA, Rodriguez EH, Schaner ME, Salim A, Wang Y, Kristensen GB, Helland A, Børresen-Dale AL, et al. (2006). Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med. 3, e47. 10.1371/journal.pmed.0030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Granlund L, Hedin A, Korsgren O, Skog O, and Lundberg M (2022). Altered microvasculature in pancreatic islets from subjects with type 1 diabetes. PLoS One 17, e0276942. 10.1371/journal.pone.0276942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Almaça J, Caicedo A, and Landsman L (2020). Beta cell dysfunction in diabetes: the islet microenvironment as an unusual suspect. Diabetologia 63, 2076–2085. 10.1007/s00125-020-05186-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roep BO, Thomaidou S, van Tienhoven R, and Zaldumbide A (2021). Type 1 diabetes mellitus as a disease of the b-cell (do not blame the immune system?). Nat. Rev. Endocrinol. 17, 150–161. 10.1038/s41574-020-00443-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.King GL, Shiba T, Oliver J, Inoguchi T, and Bursell SE (1994). Cellular and molecular abnormalities in the vascular endothelium of diabetes mellitus. Annu. Rev. Med. 45, 179–188. 10.1146/annurev.med.45.1.179. [DOI] [PubMed] [Google Scholar]
- 56.Arboleda-Velasquez JF, Valdez CN, Marko CK, and D’Amore PA(2015). From pathobiology to the targeting of pericytes for the treatment of diabetic retinopathy. Curr. Diab. Rep. 15, 573. 10.1007/s11892-014-0573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nomoto H, Gurlo T, Rosenberger M, Girgis MD, Dry S, and Butler PC (2019). Low Grade Islet but Marked Exocrine Pancreas Inflammation in an Adult with Autoimmune Pre-Diabetes. Case Rep. Endocrinol. 2019, 5863569. 10.1155/2019/5863569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gaglia JL, Guimaraes AR, Harisinghani M, Turvey SE, Jackson R, Benoist C, Mathis D, and Weissleder R (2011). Noninvasive imaging of pancreatic islet inflammation in type 1A diabetes patients. J. Clin. Invest. 121, 442–445. 10.1172/jci44339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Denis MC, Mahmood U, Benoist C, Mathis D, and Weissleder R(2004). Imaging inflammation of the pancreatic islets in type 1 diabetes. Proc. Natl. Acad. Sci. USA 101, 12634–12639. 10.1073/pnas.0404307101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.von Tell D, Armulik A, and Betsholtz C (2006). Pericytes and vascular stability. Exp. Cell Res. 312, 623–629. 10.1016/j.yexcr.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 61.Hellström M, Gerhardt H, Kalén M, Li X, Eriksson U, Wolburg H, and Betsholtz C (2001). Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J. Cell Biol. 153, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, et al. (2010). Pericytes regulate the blood-brain barrier. Nature 468, 557–561. 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 63.Schrimpf C, Teebken OE, Wilhelmi M, and Duffield JS (2014). The role of pericyte detachment in vascular rarefaction. J. Vasc. Res. 51, 247–258. 10.1159/000365149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stark K, Pekayvaz K, and Massberg S (2018). Role of pericytes in vascular immunosurveillance. Front. Biosci. 23, 767–781. 10.2741/4615. [DOI] [PubMed] [Google Scholar]
- 65.Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von Brühl ML, Gärtner F, Khandoga AG, Legate KR, Pless R, et al. (2013). Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and ‘instruct’ them with pattern-recognition and motility programs. Nat. Immunol. 14, 41–51. 10.1038/ni.2477. [DOI] [PubMed] [Google Scholar]
- 66.Proebstl D, Voisin MB, Woodfin A, Whiteford J, D’Acquisto F, Jones GE, Rowe D, and Nourshargh S (2012). Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J. Exp. Med. 209, 1219–1234. 10.1084/jem.20111622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Crawford C, Wildman SSP, Kelly MC, Kennedy-Lydon TM, and Peppiatt-Wildman CM (2013). Sympathetic nerve-derived ATP regulates renal medullary vasa recta diameter via pericyte cells: a role for regulating medullary blood flow? Front. Physiol. 4, 307. 10.3389/fphys.2013.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gonzales AL, Klug NR, Moshkforoush A, Lee JC, Lee FK, Shui B, Tsoukias NM, Kotlikoff MI, Hill-Eubanks D, and Nelson MT (2020). Contractile pericytes determine the direction of blood flow at capillary junctions. Proc. Natl. Acad. Sci. USA 117, 27022–27033. 10.1073/pnas.1922755117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O’Farrell FM, Buchan AM, Lauritzen M, and Attwell D (2014). Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508, 55–60. 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hartmann DA, Berthiaume AA, Grant RI, Harrill SA, Koski T, Tieu T, McDowell KP, Faino AV, Kelly AL, and Shih AY (2021). Brain capillary pericytes exert a substantial but slow influence on blood flow. Nat. Neurosci. 24, 633–645. 10.1038/s41593-020-00793-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jansson L, Barbu A, Bodin B, Drott CJ, Espes D, Gao X, Grapensparr L, Källskog Ö, Lau J, Liljebäck H, et al. (2016). Pancreatic islet blood flow and its measurement. Ups. J. Med. Sci. 121, 81–95. 10.3109/03009734.2016.1164769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jansson L, and Carlsson PO (2019). Pancreatic Blood Flow with Special Emphasis on Blood Perfusion of the Islets of Langerhans . Compr. Physiol. 9, 799–837. 10.1002/cphy.c160050. [DOI] [PubMed] [Google Scholar]
- 73.Bogdani M, Korpos E, Simeonovic CJ, Parish CR, Sorokin L, and Wight TN (2014). Extracellular matrix components in the pathogenesis of type 1 diabetes. Curr. Diab. Rep. 14, 552. 10.1007/s11892-014-0552-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mitsui R, and Hashitani H (2020). Synchrony of spontaneous Ca(2+) activity in microvascular mural cells. Journal of smooth muscle research = Nihon Heikatsukin Gakkai kikanshi 56, 1–18. 10.1540/jsmr.56.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cai C, Fordsmann JC, Jensen SH, Gesslein B, Lønstrup M, Hald BO, Zambach SA, Brodin B, and Lauritzen MJ (2018). Stimulation-induced increases in cerebral blood flow and local capillary vasoconstriction depend on conducted vascular responses. Proc. Natl. Acad. Sci. USA 115, E5796–e5804. 10.1073/pnas.1707702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kakugawa Y, Giaid A, Yanagisawa M, Baynash AG, Melnyk P, Rosenberg L, and Duguid WP (1996). Expression of endothelin-1 in pancreatic tissue of patients with chronic pancreatitis. J. Pathol. 178, 78–83. . [DOI] [PubMed] [Google Scholar]
- 77.Kugelmeier P, Nett PC, Züllig R, Lehmann R, Weber M, and Moritz W (2008). Expression and hypoxic regulation of the endothelin system in endocrine cells of human and rat pancreatic islets. JOP : Journal of the pancreas 9, 133–149. [PubMed] [Google Scholar]
- 78.Fukuroda T, Fujikawa T, Ozaki S, Ishikawa K, Yano M, and Nishikibe M (1994). Clearance of circulating endothelin-1 by ETB receptors in rats. Biochem. Biophys. Res. Commun. 199, 1461–1465. 10.1006/bbrc.1994.1395. [DOI] [PubMed] [Google Scholar]
- 79.Kelland NF, Kuc RE, McLean DL, Azfer A, Bagnall AJ, Gray GA, Gulliver-Sloan FH, Maguire JJ, Davenport AP, Kotelevtsev YV, and Webb DJ (2010). Endothelial cell-specific ETB receptor knockout: autoradiographic and histological characterisation and crucial role in the clearance of endothelin-1. Can. J. Physiol. Pharmacol. 88, 644–651. 10.1139/y10-041. [DOI] [PubMed] [Google Scholar]
- 80.Doliba NM, Rozo AV, Roman J, Qin W, Traum D, Gao L, Liu J, Manduchi E, Liu C, Golson ML, et al. (2022). a Cell dysfunction in islets from nondiabetic, glutamic acid decarboxylase autoantibody-positive individuals. J. Clin. Invest. 132, e156243. 10.1172/jci156243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sherry NA, Tsai EB, and Herold KC (2005). Natural history of beta cell function in type 1 diabetes. Diabetes 54, S32–S39. 10.2337/diabetes.54.suppl_2.s32. [DOI] [PubMed] [Google Scholar]
- 82.Sosenko JM, Skyler JS, Beam CA, Krischer JP, Greenbaum CJ, Mahon J, Rafkin LE, Matheson D, Herold KC, and Palmer JP; Type 1 Diabetes TrialNet and Diabetes Prevention Trial–Type 1 Study Groups (2013). Acceleration of the loss of the first-phase insulin response during the progression to type 1 diabetes in diabetes prevention trial-type 1 participants. Diabetes 62, 4179–4183. 10.2337/db13-0656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leete P, Willcox A, Krogvold L, Dahl-Jørgensen K, Foulis AK, Richardson SJ, and Morgan NG (2016). Differential Insulitic Profiles Determine the Extent of b-Cell Destruction and the Age at Onset of Type 1 Diabetes. Diabetes 65, 1362–1369. 10.2337/db15-1615. [DOI] [PubMed] [Google Scholar]
- 84.Rodriguez-Calvo T, Zapardiel-Gonzalo J, Amirian N, Castillo E, Lajevardi Y, Krogvold L, Dahl-Jørgensen K, and von Herrath MG (2017). Increase in Pancreatic Proinsulin and Preservation of b-Cell Mass in Autoantibody-Positive Donors Prior to Type 1 Diabetes Onset. Diabetes 66, 1334–1345. 10.2337/db16-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qadir MMF, Álvarez-Cubela S, Weitz J, Panzer JK, Klein D, Moreno-Hernández Y, Cechin S, Tamayo A, Almaça J, Hiller H, et al. (2020). Long-term culture of human pancreatic slices as a model to study real-time islet regeneration. Nat. Commun. 11, 3265. 10.1038/s41467-020-17040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Patil AR, Schug J, Naji A, Kaestner KH, Faryabi RB, and Vahedi G(2023). Single-cell expression profiling of islets generated by the Human Pancreas Analysis Program. Nat. Metab. 5, 713–715. 10.1038/s42255-023-00806-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Qadir MMF, Álvarez-Cubela S, Klein D, van Dijk J, Muñiz-Anquela R, Moreno-Hernández YB, Lanzoni G, Sadiq S, Navarro-Rubio B, Garcıá MT, et al. (2020). Single-cell resolution analysis of the human pancreatic ductal progenitor cell niche. Proc. Natl. Acad. Sci. USA 117, 10876–10887. 10.1073/pnas.1918314117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
All original code has been deposited at Github and Zenodo and is publicly available as of the date of publication. DOIs are listed in key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Neuron-glial antigen 2 (NG2) | R&D | RRID: FAB2585R |
| Platelet-derived growth factor receptor-beta (PDGFRβ) | R&D | RRID: AF385 |
| Alpha smooth muscle actin (αSMA) | Sigma | RRID: A5228 |
| CD31 | Millipore | RRID: 550389 |
| Tyrosine hydroxylase (TH) | Millipore | RRID: AB152 |
| Collagen IV | Abcam | RRID: AB6586 |
| Periostin | R&D | RRID: AF2955 |
| Insulin | Dako | RRID: IR002 |
| Somatostatin | Millipore | RRID: MAB354 |
| CD146 | Abcam | RRID: Ab75769 |
| Alexa Fluor Secondary antibodies | ThermoFischer | |
|
| ||
| Biological samples | ||
|
| ||
| Living human pancreas slices | nPOD | https://npod.org |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Theophylline | Sigma | T1633 |
| Norepinephrine | Tocris | 5169 |
| Endothelin 1 | Sigma | E7764 |
| Angiotensin II | Tocris | 1158 |
| Bosentan | Sigma | 1265 |
| Adenosine | Sigma | A9251 |
| Lycopersicon esculentum Lectin | Vector Labs | DL-1178-1 |
| Fluo-4 | Invitrogen | F14201 |
| Aprotinin | Sigma Aldrich | A6106 |
| Penicillin-Streptomycin-Amphotericin B | Sigma Aldrich | A5955 |
| BrainPhys neuronal medium | Stemcell Technologies | 05790 |
| B27 minus-insulin | Invitrogen | A1895601 |
| Chymostatin | Sigma Aldrich | 11004638001 |
|
| ||
| Deposited data | ||
|
| ||
| Human Pancreas Analysis Program, pancreas single cell RNAseq data repository | HPAP | https://hpap.pmacs.upenn.edu/analysis |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism | GraphPad | Prism9 |
| ImageJ (https://imagej.nih.gov/ij/) | NIH | ImageJ |
| MatLab (www.mathworks.com) | MathWorks | MatLab R2020b |
| Single cell analysis architecture (Github + Zenodo) | JA Lab |
https://github.com/jalmaca/Microvasculature_T1D
https://zenodo.org/record/8122389 |
|
| ||
| Other | ||
|
| ||
| SP5 inverted laser-scanning confocal microscope | Leica | SP5 |
| SP8 upright laser-scanning confocal microscope | Leica | SP8 |