

Abstract
The gut microbiome is an important modulator of the host immune system. Here, we found that altering the gut microbiome by oral vancomycin increases liver iNKT cell function. Enhanced iNKT cytokine production and activation marker expression were observed in vancomycin-treated mice following both antigen-specific and antigen-independent in vivo iNKT stimulations, with a more prominent effect in liver as compared to the spleen. Fecal transplantation studies demonstrated that the iNKT functional regulation is mediated by altering the gut microbiome but uncoupled from the modulation of iNKT cell population size. Interestingly, when stimulated in vitro, iNKT cells from vancomycin-treated mice did not show increased activation, suggesting an indirect regulation. iNKT cells expressed high levels of IL-18 receptor, and vancomycin increased the expression of IL-18 in the liver. Blocking IL-18 by neutralizing antibody or using genetically deficient mice attenuated the enhanced iNKT activation. Liver macrophages were identified as a major source of IL-18. General macrophage depletion by clodronate abolished this iNKT activation. Using anti-CSF-1R depletion or LyzCrexCSF-1RLsL-DTR mice identified CSF-1R+ macrophages as a critical modulator of iNKT function. Vancomycin treatment had no effect on iNKT cell function in vivo in IL-18 KO macrophage reconstituted mice. Together, our results demonstrate that the gut microbiome controls liver iNKT function via regulating CSF-1R+ macrophages to produce IL-18.
Introduction
The gut microbiome has a significant impact on the host immune system, and plays a fundamental role in the induction, training, and functional regulation of host immunity (1). The liver is exposed to the gut microbiome since more than 70% of the blood supply to the liver is through the portal vein. The portal vein drains blood from the intestine and not only carries nutrients but also is rich in gut microbial antigens and metabolites (2). The liver harbors abundant immune cells including a large macrophage population to defend the continuous threat of gut microbial pathogens. The close cross-talk between the gut microbiome and local immune environment of the liver, and its implications for liver diseases and systemic immune regulation are starting be partially understood.
Natural killer T (iNKT) cells are innate-like lymphocytes which recognize lipid antigens presented on CD1d molecules (3–7). Unlike conventional T cells recognizing a peptide-MHC complex, iNKT cells are selectively activated by lipid antigens presented by CD1d molecules, of which α-galactosylceramide (α-GalCer) is the most well characterized (3, 7, 8). With the capability of initiating diverse immune responses by rapid release of cytokines after stimulation, iNKT cells are considered as a critical bridge between innate and adaptive immunity (9). iNKT cells are enriched in the liver and are key components of the liver microenvironment (10). Different studies including a very recent report suggest that liver iNKT cells are critical for shaping the local immune landscape in the liver (11). Furthermore, numerous reports have found that iNKT cells play a critical role in various liver physiopathological processes such as liver regeneration, non-alcoholic fatty liver disease, autoimmune liver disease, and liver cancer (12–15). Clinical trials of iNKT-based immunotherapy are also under investigation (16).
Emerging evidence suggests that the gut microbiome regulates iNKT cells (11, 14, 17), which can recognize bacterial glycolipids (18). Recently, we reported that gut commensal bacteria modulate liver cancer through recruiting iNKT cells to liver by regulating chemokine CXCL16 expression in liver sinusoidal endothelial cells (17). Altering gut bacteria using oral vancomycin is sufficient to increase iNKT frequency in the liver (17). Here, we extended the work and examined how gut microbes regulate iNKT cell function. Enhanced iNKT function was observed in mice receiving oral vancomycin. This effect was caused by altering the gut microbiome and independent of our previously reported bile acid-mediated mechanism. Interestingly, the gut microbiome-dependent enhancement of iNKT cell function required the presence of CSF1R+ macrophages in the liver where they were the main source of IL-18. The enhanced iNKT cell activity diminished after blocking IL-18. Our study reveals that the gut microbiome-liver axis shapes iNKT cell function via CSF1R+ macrophages.
Material and Methods
Mouse strains and reagents
C57BL/6 and BALB/c mice at the age of 6~10 weeks were purchased from Charles River. CD45.1 C57BL/6 mice (strain#002014), IL-18−/− (strain# 004130), LyzCre (strain# 004781) and CSF-1RLsL-DTR (strain# 024046) mice were purchased from the Jackson Laboratory and maintained at NIH Bethesda CRC animal facility. A20 cells (Cat# TIB-208) were purchased from ATCC, and EL4 cells were used as described(17). Vancomycin (Hospira Inc) was purchased form NIH veterinary pharmacy. α-galactosylceramide (KRN7000), concanavalin A (Cat# C2010), diphtheria toxin (Cat# D0564), and brefeldin A (Cat# B6542) were purchased from Sigma. Clodronate liposome and control liposome were purchased from LIPOSOMA. In vivo anti-IL18 neutralizing antibody (clone YIGIF74–1G7) and anti-CSF-1R depletion antibody (clone AFS98) were purchased from BioXcell. All experiments were conducted according to local institutional guidelines and approved by the Animal Care and Use Committee of the National Institutes of Health, Bethesda, USA.
Oral vancomycin treatment
Mice were kept on vancomycin containing drinking water (0.5g/L) or control H2O (17). Vancomycin treatment was started in adult mice of age between 7 to 10 weeks and lasted for 3 weeks. Vancomycin drinking water was replaced every other day.
iNKT stimulations
Antigen-specific iNKT stimulation was performed by using α-GalCer-loaded tumor cells (17). EL4 or A20 tumor cells were incubated with α-GalCer (0.5μg/ml) in culture medium overnight, and free α-GalCer was removed by washing cells 3 times with sterile PBS. For in vivo stimulation, mice were i.v. injected with 106 α-GalCer -loaded tumor cells together with 250 μg brefeldin A in 300 μl PBS(17, 19). Some mice received free α-GalCer (0.5 μg/mouse) i.v. together with 250 μg brefeldin A in 300 μl PBS for in vivo stimulation. Mice were euthanized 2 hours following in vivo stimulation. For in vitro stimulation, 5×105 liver mononuclear cells or splenocytes were incubated with α-GalCer- loaded tumor cells at 1:1 ratio in V-bottom 96 well plate in complete RPMI culture medium at 37°C. Two hours later the reaction was stopped by washing cells with PBS and the cells were studied by to flow cytometry. Antigen non-specific iNKT in vivo stimulation was performed by i.v. injecting mice with 50 μg concanavalin A together with 250 μg brefeldin A in total 300 μl PBS. Two hours later, mice were euthanized for cell preparation.
Germ-free mice and fecal transplantation
Germfree BALB/c and C57BL/6 mice were bred at the Gnotobiotics Facility at the National Laboratory for Cancer Research (FNLCR). Animal studies were approved by the Animal Care and Use Committee (ACUC) of the FNLCR. To generate conventionalized mice germ-free mice, male BALB/c mice were transferred to an SPF facility and housed with bedding from SPF mice for 4 weeks before antibiotic treatment. Both female BALB/c and C57BL/6 germ-free mice were used for FMT studies. Vanco or control feces were collected from cecum of SPF mice given 3 weeks of oral vancomycin treatment and mixed with sterile 50% glyceryl to store at −80°C until use. Germ free mice were given oral gavage of 150 μl vanco feces once per day for continuous 4 days. Three weeks after fecal transplantation, mice were given i.v. injection of α-GalCer-loaded tumor cells.
Macrophage depletion
Clodronate liposome (LIPOSOMA) was used to deplete general macrophage population. Mice were given i.v. injection of 200 μl clodronate or control liposome 72 and 24 hours before in vivo iNKT stimulation. Anti-CSF-1R antibody clone AFS98 (BioxCell) was used to deplete CSF1+ macrophage. Mice were given i.v. injection of 200 μg AFS98 or control IgG antibody 72 and 24 hours before in vivo iNKT stimulation. MMDTR mice were used to selectively remove CSF-1R+ macrophage while sparing dendric cells as reported (20). MMDTR mice were generated by crossing LyzCre and CSF-1RLDL-DTR mice. Mice with correct genotyping were identified by using service from TransnetYX. To deplete CSF-1R+ macrophage, MMDTR mice were given s.c. injection of 200 ng diphtheria toxin one day before in vivo iNKT stimulation. Littermates also received diphtheria toxin to rule out toxin related effects.
Liver macrophage reconstitution
Female CD45.1 C57BL/6 mice at the age of 6 weeks were exposed to 900 rads irradiation followed by i.v. transfer of ~2×107 freshly prepared donor bone marrow cells. Bone marrow cells were collected by flushing the bone marrow from femur and tibia bones from either CD45.2 WT or IL-18 KO donor mice (both under C57BL/6 background) followed by removing RBC cells with ACK lysis buffer. Two weeks after bone marrow cell injection, mice received 200 μl clodronate liposome i.v.. Oral vancomycin treatment was started two weeks after clodronate treatment and lasted for 3 weeks. At the experimental end point mice were injected i.v. with 106 α-GalCer-loaded EL4 cells together with 250 μg brefeldin A in 300 μl PBS. Two hours later, mice were euthanized, and liver mononuclear cells were prepared for flow cytometry analysis.
Isolation of hepatocyte, macrophage and hepatic stellate cell
Different types of cells from the same liver were isolated (21). Mice were euthanized with CO2. Then, livers were perfused with 5 mM HEPES and 0.5 mM EDTA in HBSS at 37°C for 5 mins, followed by perfusion with 0.05% collagenase IV (Sigma, C5138) in HBSS supplemented with 5 mM HEPES and 0.5 mM CaCl2 at 37°C for 5 mins. The livers were excised and homogenized, and then passed through 100 μm filter. The suspension was centrifuged at 50xg for 3 min to separate hepatocytes (pellet) from non-parenchymal cells (NPC, supernatant). Hepatocytes were further enriched by magnetic bead depletion of anti-CD45 and anti-CD146 to deplete most immune cells and endothelial cells respectively. The NPC-fraction was then submitted to a 15% Optiprep density gradient, and then centrifuged at 1500 × g for 25 min at room temperature. The well-defined interface of cells was carefully collected, and then centrifuged at 500 × g for 5 mins at 4°C. The NPCs were submitted to cell staining and cell sorting. Antibodies used for staining were as follows: anti-CD146-APC (clone ME-9F1, Biolegend), anti-Tie2-PE (clone TEK4, Biolegend), anti-F4/80-FITC (clone BM8, Biolegend), anti-CD3-PE/Cy7 (clone 17A2, Biolegend). Macrophages were sorted as F4/80+CD146−Tie2−CD3− cells, hepatic stellate cells were negatively sorted as CD3− CD146−Tie2−F4/80−CD11b− cells. Cell sorting was carried out with a FACS Aria II (BD Biosciences). All experiments were performed with 95% yield of purity for each subset.
Flow Cytometry
Mouse livers were collected after euthanasia and homogenized. Large debris were removed by passing 40 μm mesh. After centrifugation, cell pellets were resuspended in 8 ml PBS containing 5% FCS, and mixed with 4.5 ml Percol and subject to gradient centrifuge at 850xg 25 minutes without deceleration. The red blood cells were lysed by ACK buffer. Flow cytometry assay was performed using a CytoFLEX LX platform (Beckman Coulter). Cells were stained with live/dead fixable near-IR dead cells stain kit (ThermoFisher) to gate out dead cells. Fc block reagent TruStain FcX (biolegend, Cat#101320) was used to reduce unspecific staining. Cell surface markers were stained at 4°C in dark with the following antibodies: anti-CD45 (clone 30-F11, biolegend), anti-CD45.1 (clone A20, biolegend), anti-CD45.2 (clone 104, biolegend), anti-TCRβ (clone H57–597, biolegend). CD1d-teramer (PBS57, NIH tetramer core), anti-CD3 (clone 17A2, biolegend), anti-CD4 (clone GK1.5, biolegend), anti-CD8 (clone 53–6.7, biolegend), anti-CD11b (clone M1/70, biolegend), anti-F4/80 (clone BM8, biolegend), anti-CD69 (clone H1.2F3, biolegned), anti-IL18Ra (clone A17071D, biolegend). Intracellular cytokine staining was performed using the eBioscience Foxp3/transcription factor staining buffer set (Cat# 00552300, Thermofisher) with the anti-IFNγ (clone XMG1.2, biolegend), anti-IL-4 (clone 11B11, biolegend).
Real-time PCR
Total RNA was isolated using RNeasy Mini Kit (Cat# 74104, Qiagen) according to manufacturer’s instructions. cDNA synthesis was performed using iScriptTM cDNA synthesis kit (Cat# 170–8891, BIO-RAD). RT-PCR was performed using iQ SYBR Green Supermix (Cat# 1708882, BIO-RAD). The following primers were used for RT-PCR, IL-18: Forward 5’-GGCTGCCATGTCAGAAGACT-3’, Reverse 5’-ACAGTGAAGTCGGCCAAAGT-3’; F4/80: Forward, 5’-TCTGGGGAGCTTACGATGGA-3’, Reverse 5’-GAATCCCGCAATGATGGCAC-3’, GAPDH: Forward 5’-CCTGCACCACCAACTGCTTA-3’, Reverse 5’-TCATGAGCCCTTCCACAATG-3’. The relative gene expression levels were determined by 2-^ĈT methods. GAPDH expression level was used as control.
Statistical analysis
Sample size for animal studies were guided by previous study in our laboratory. Statistical analysis was performed with GraphPad Prism 8 (GraphPad Software). Significance of the difference between groups was calculated by Student’s unpaired t-test, one-way or two-way ANOVA (with Tukey’s and Bonferroni’s multiple comparison test). P<0.05 was considered as statistically significant.
Results
Modulating the gut microbiome with oral vancomycin increases liver iNKT activity.
C57BL/6 mice were treated with vancomycin-containing drinking water for 3 weeks to modulate the gut microbiome. Although the expression of the activation marker CD69 on liver iNKT cells did not change (Fig.S1A), vancomycin treatment reduced the relative frequency of iNKT2 and iNKT17 subsets which led to increased iNKT1/iNKT2 and iNKT1/iNKT17 ratios (Fig.S1B–E). To assess changes in liver iNKT cell function, antigen-specific iNKT cell responses were induced by in vivo challenge with i.v. injected α-GalCer-loaded EL4 tumor cells as previously described (17, 22). iNKT cell activity was assessed by measuring cytokine production and activation marker expression using flow cytometry 2 hours after stimulation (Fig.1A). A distinct IFNγ signal was detected in iNKT cells (Fig.S1F). Vancomycin-treated mice exhibited a marked increase of IFNγ+ iNKT cells as compared to control mice (Fig.1B&C). Consistently, higher median fluorescence intensity (MFI) of IFNγ in liver iNKT cells was found in the vancomycin-treated group (Fig.S1G). iNKT cells can produce both Th1 and Th2 cytokines (23) and higher IL-4 production was also observed in liver iNKT cells (Fig.1A&D). Interestingly, almost all IL-4+ iNKT cells co-expressed IFNγ, whereas the majority of IFNγ+ iNKT cells did not produce IL-4 and the IFNγ+IL4− iNKT population expanded after vancomycin treatment (Fig.1E, Fig.S1H). Consistent with a higher cytokine production, vancomycin treatment also increased the expression of CD69, an activation maker on liver iNKT cells (Fig.1F), supporting a more activated status. Unlike the liver, only a non-significant increase of IFNγ, IL-4 or CD69 was found in splenic iNKT cells (Fig.S1I–K). Similar findings were also found in BALB/c mice following i.v. injection of α-GalCer-loaded A20 cells (Fig.S1L&M), ruling out a mouse strain-specific effect.
Figure.1. Oral vancomycin treatment increases liver iNKT activity.
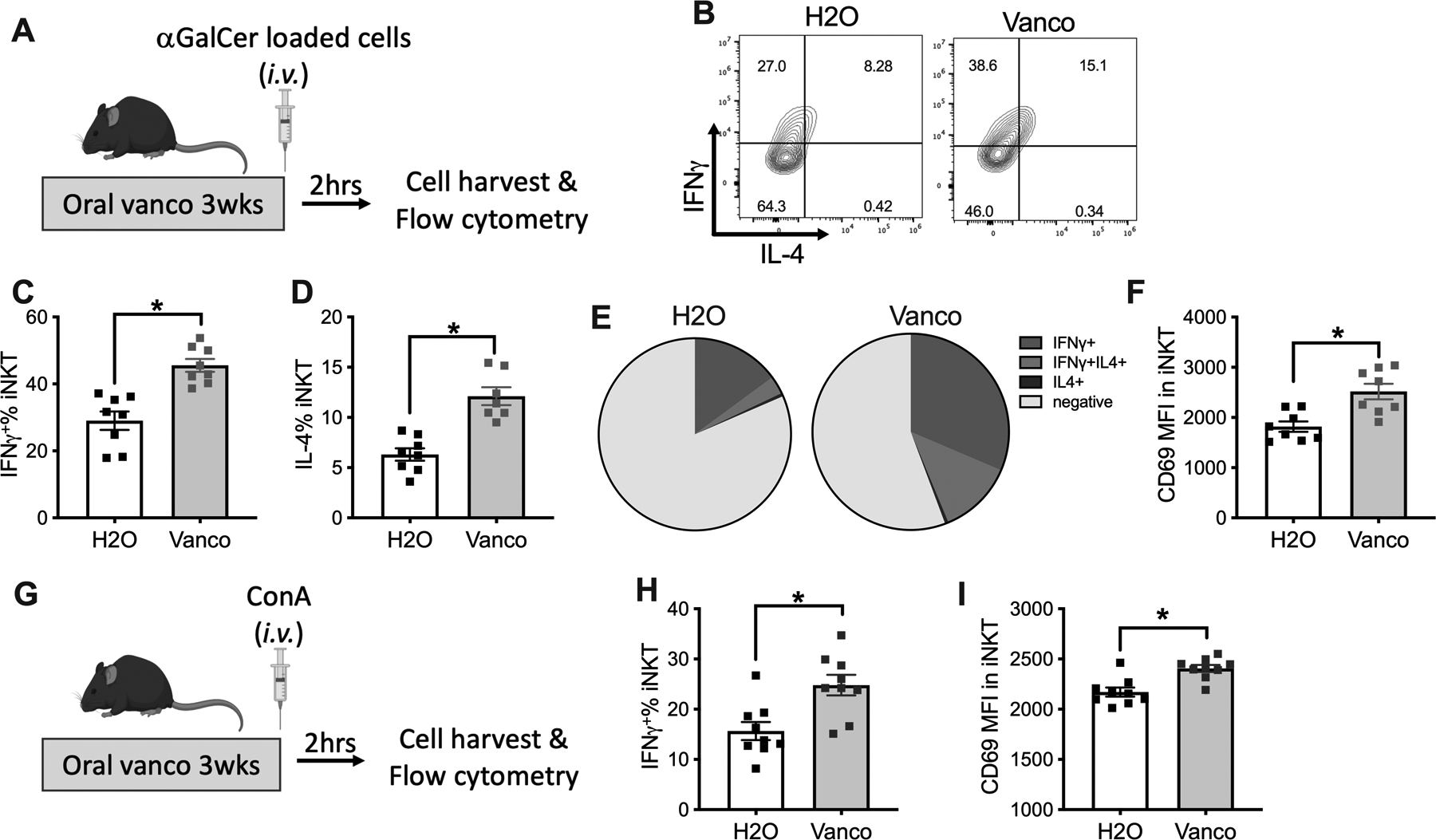
(A-F) C57BL/6 mice (age 7 to 10 weeks) were kept on vancomycin or control H2O for 3 weeks. 106 αGalCer-loaded EL4 cells plus brefeldin A (250ug/mouse) were injected i.v. in mice. Two hours after injection, mice were euthanized, and liver mononuclear cells were prepared for flow cytometry. (A) Experimental setup. (B) Representative IFNγ and IL-4 staining of liver iNKT cells gated on TCRβ+CD1d-tetramer+. Liver iNKT cells IFNγ+% (C), IL-4+% (D), composition of iNKT cells expressing IFNγ or IL-4 (E), and CD69 MFI (F) are shown. Results are presented as mean+/− SEM of two independent experiments. n=8, p<0.05, student t test.
(G-I). Concanavalin A (ConA) 50 ug/mouse plus brefeldin A (250ug/mouse) was injected into mice kept on vancomycin or H2O. Two hours later, liver mononuclear cells were prepared. (G) Experimental setup. Liver iNKT cells IFNγ+% (H) and CD69 MFI (I) are shown. Results are presented as mean+/− SEM of two independent experiments. n=9, p<0.05, student t test.
Antigen-independent iNKT activation has been described upon i.v. injection of a non-hepatotoxic low-dose of concanavalin (Fig.1G). This protocol has been used to fully activate iNKT cells but not conventional T cells (24). Again, vancomycin treatment further increased IFNγ and CD69 expression in liver iNKT cells as compared to control mice following stimulation (Fig.1H&I), but this was not seen in spleen iNKT cells (Fig.S1N&O). Together, using both antigen-specific and antigen-independent stimulation models, our results demonstrate that vancomycin enhances iNKT cell function, and the effect is limited to iNKT cells in the liver.
Altered gut microbiome mediates the enhanced iNKT function.
Orally administered vancomycin is poorly absorbed with bioavailability less than 10% (25), and its iNKT regulatory effect is likely through changing the gut microbiome. We decided to study the role of gut microbiome by comparing germ-free mice with mice kept under SPF condition and germ-free mice transferred to a SPF mouse facility to colonize mice with commensal microbiota. Some of the conventionalized mice were further treated with oral vancomycin. Liver iNKT cell function was assessed following i.v. injection of α-GalCer-loaded tumor cells (Fig.2A). Consistent with our prior findings (17), the frequency of iNKT cells was higher in livers from germ-free mice as compared to conventionalized mice (Fig.S2A) and oral vancomycin treatment also increased the number of liver iNKT cells in conventionalized mice (Fig.S2A, Vanco GF->SPF vs H2O GF->SPF). Interestingly, no differences in IFNγ or IL-4 expression in liver iNKT cells was found comparing germ-free mice with conventionalized mice (Fig.2B&C). However, oral vancomycin still enhanced cytokine production by liver iNKT cells in the conventionalized mice (Fig.2B&C). These results clearly indicates that gut microbiota control iNKT cell function and frequency in the liver through a different mechanism, and alteration of the gut microbiota by vancomycin is important for changing iNKT cell function. As expected, 16S rRNA sequencing demonstrated robust changes of fecal bacterial phyla composition after vancomycin treatment. Oral vancomycin dramatically reduced many bacteria such as Bacteroidetes (Fig.S2B) and caused a significant increase of other bacteria especially Proteobacteria and Verrucomicrobia (Fig.S2B). Akkermansia muciniphila is the major component of the Verrucomicrobia phylum in the human fecal microbiota and has been shown to be associated with anti-PD1 efficacy in patients with lung and kidney cancer (26). Consistent with previous reports (27), vancomycin treatment increased the abundance of Akkermansia muciniphia (Fig.S2C).
Figure.2. Vancomycin altered gut microbiota causes the enhanced iNKT function.
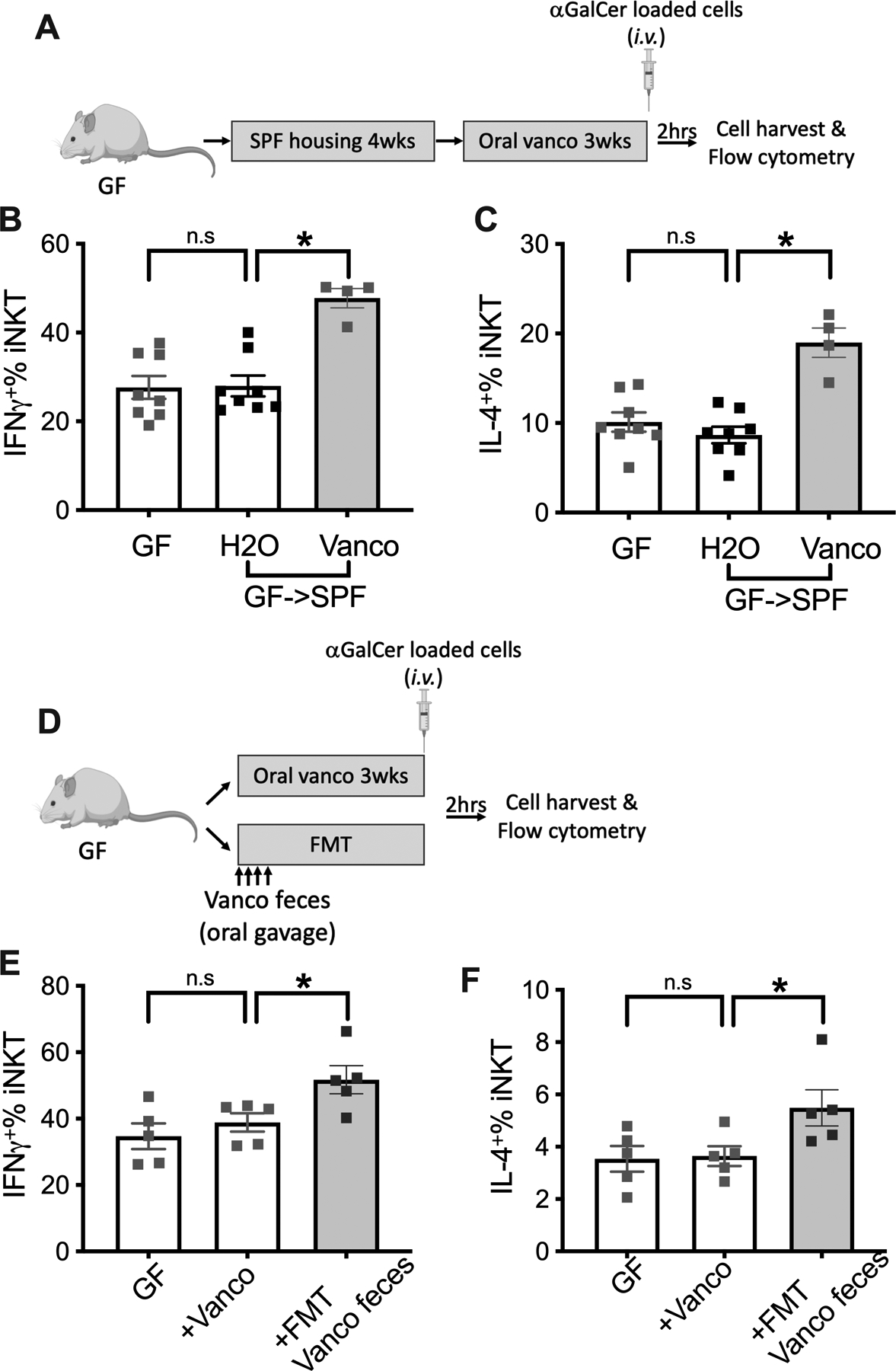
(A-C) Male germ-free BALB/c mice (age ~6 weeks) were kept under germ-free condition or transferred to a SPF facility to colonize commensal microbiome (GF->SPF). Four weeks later, the GF->SPF mice received vancomycin or control H2O for 3 weeks. iNKT cell stimulation was performed by i.v. injection of 106 αGalCer-loaded A20 cells plus brefeldin A (250ug/mouse). Two hours later, liver mononuclear cells were prepared for flow cytometry. (A) Experimental setup. Liver iNKT cells IFNγ+ % (B) and IL-4+ % (C) are shown. Results are presented as mean+/− SEM of two independent experiments. n=8 for GF, 8 for GF->SPF H2O, 4 for GF->SPF Vanco, p<0.05, one-way ANOVA.
(D-F) Germ-free BALB/c mice (age 7~10 weeks) received vancomycin in their drinking water or FMT of feces from vancomycin treated SPF mice by oral gavage. Three weeks later mice were challenged by i.v. injection of 106 αGalCer-loaded A20 cells plus brefeldin A (250ug/mouse). (D) Experimental setup. Liver iNKT cells IFNγ+% (E) and IL-4+% (F) are shown. Results are presented as mean+/− SEM of one experiment. n=5, p<0.05, one-way ANOVA.
To confirm the role of vancomycin- altered gut microbiota in regulating iNKT cell function, we performed fecal microbial transfer (FMT) studies. Germ-free mice were treated with oral vancomycin or feces from mice treated with oral vancomycin (Fig.2D). No changes in the number of iNKT cells in the liver was found in germ-free mice treated with oral vancomycin or after transplantation of feces from mice treated with vancomycin (Fig.S2D). In the absence of the gut microbiota, oral vancomycin failed to affect IFNγ production by liver iNKT cells in germ-free mice upon in vivo challenge with α-GalCer-loaded cells (Fig.2E), which confirms that the regulation was not directly caused by vancomycin. However, as expected, transplantation of feces from SPF mice treated with vancomycin markedly increased IFNγ and IL-4 production by liver iNKT cells (Fig.2E&F). To rule out a nonspecific effect of FMT, the experiment was repeated using feces from either control mice or mice treated with vancomycin (Fig.S2E). Consistent with our previous finding, FMT control feces but not vanco feces reduced the frequency of liver iNKT cells (Fig.S2F&G). Again, iNKT cells showed higher IFNγ and CD69 expression in mice who had been colonized with feces from vanco treated mice than in mice colonized with control feces (Fig.S2H&I). These results show that the iNKT cell function is regulated by vancomycin through altering the gut microbiota.
IL-18 mediates the gut microbiome dependent enhancement of iNKT function in the liver
iNKT cell activity was further assessed in vitro. Isolated liver mononuclear cells from vancomycin- or H2O-treated mice were exposed to the same antigen-specific stimulation using aGalCer-loaded tumor cells. Two hours later, cytokine production by iNKT cells was measured (Fig.3A). Strikingly, in contrast to the finding following in vivo challenge, no increase in IFNγ production was found in liver iNKT cells from vancomycin-treated mice as compared to control (Fig.3B). Similarly, IL-4 levels of in vitro stimulated liver iNKT cells did not increase in vancomycin treated group as compared to the control group (Fig.3C). These data suggest that the observed iNKT regulation by gut microbiome was potentially caused by an altered immune environment in the liver which would favor iNKT cell activation after vancomycin treatment, rather than an intrinsic change in the iNKT cells that would persist ex vivo.
Figure.3. IL-18 controls gut microbiota dependent increase in liver iNKT cell function.
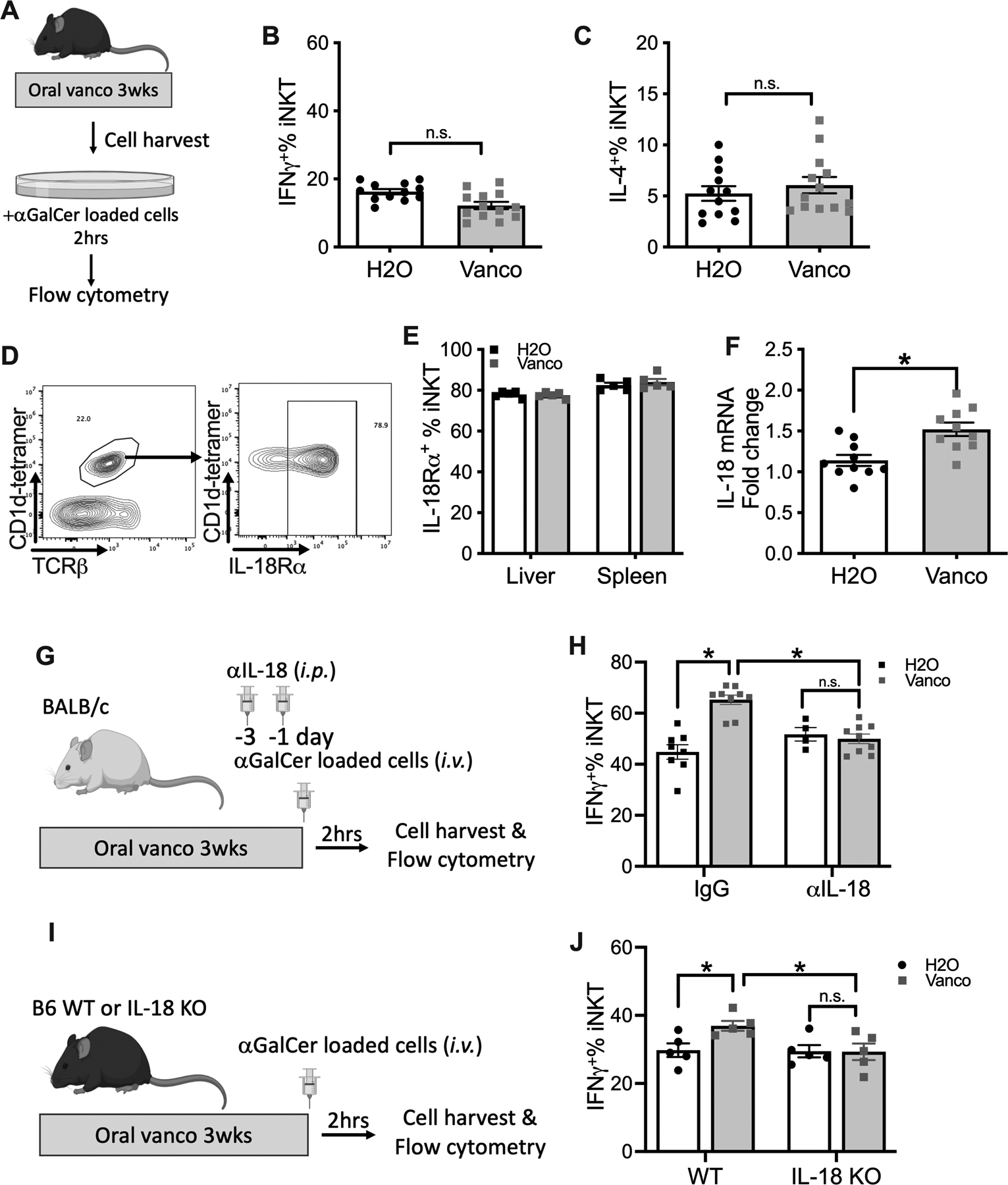
(A-C) Liver mononuclear cells were prepared from C57BL/6 mice kept on vancomycin or H2O, and incubated with αGalCer-loaded EL4 cells in the present of Brefeldin A for two hours. Liver iNKT cell IFNγ+% (B) and IL-4+% (C) are shown. Results are presented as mean+/− SEM of two independent experiments. n=12, p<0.05, student t test.
(D) Representative IL-18 receptor staining of liver iNKT cells. (E) IL-18R+% of liver or spleen iNKT cells. Results are presented as mean+/− SEM of one experiment, n=5. (F) Liver IL-18 mRNA from mice kept on vancomycin or H2O was measured by RT-PCR. Results are presented as mean+/− SEM of two independent experiments. n=10, p<0.05, student t test. (G, H) BALB/c mice (age 7~10 weeks) were kept on vancomycin or H2O for 3 weeks. Anti-IL18 or IgG were given by i.p. injections twice before i.v. injection of αGalCer-loaded A20 cells with brefeldin A. (G) Experimental setup. (H) Liver iNKT cell IFNγ+% is shown. Results are presented as mean+/− SEM from two independent experiments. n=8 for IgG H2O, 9 for IgG vanco, 4 for αIL-18 H2O, 9 for αIL-18 vanco, p<0.05, two-way ANOVA.
(I, J) IL-18KO or Wt C57BL/6 mice (age 7~10 weeks) were kept on vancomycin or H2O for 3 weeks. Then mice were challenged i.v. with αGalCer-loaded EL4 cells plus brefeldin A. (I) Experimental setup. (J) Liver iNKT cell IFNγ+% is shown. Results are presented as mean+/− SEM of one experiment. n=5, p<0.05, two-way ANOVA.
The effect of vancomycin treatment on major liver immune cell populations was studied. Apart from the increase in iNKT cells, oral vancomycin had no notable impact on the frequency of other immune cell populations including CD4+ T cells, CD8+ T cells, B cells, NK, CD11b+F4/80+ macrophage and CD11c+ dendritic cells (Fig.S3A). iNKT cell function can be regulated by different cytokines including IL-18 (28). We found that the majority (60% to 80%) of iNKT cells either from liver or spleen express the IL-18 receptor (IL-18R), and the levels did not change after vancomycin treatment (Fig.3D&E. Fig.S3C). The expression of IL-18 receptor was also observed on T lymphocytes and NK cells (Fig.S3B&D). Following the increase of iNKT cells after vancomycin treatment (Fig.S3A), the contribution of iNKT cells to IL-18R+ liver immune cells increased (Fig.S3E). Importantly, vancomycin treatment increased IL-18 mRNA expression in the liver (Fig.3F), indicating its potential role in gut microbiome- mediated regulation of iNKT cell function.
To confirm the impact of IL-18, we utilized a neutralizing antibody as reported (29). Vancomycin- or H2O-treated mice were injected with IL-18 neutralizing antibody or control IgG and iNKT cell function was tested after i.v. injection of aGalCer-loaded tumor cells (Fig.3G). Blocking IL-18 signaling had no impact on the effect of vancomycin on the frequency of liver iNKT cells (Fig.S3F). However, vancomycin-induced IFNγ upregulation was completely blocked (Fig.3H). Similarly, IL-18 neutralization also blocked IL-4 induction in liver iNKT cells (Fig.S3G). To confirm this finding IL-18 knockout mice were used (Fig.3I). Consistently, oral vancomycin did not enhance IFNγ expression in liver iNKT cells from IL-18 knockout mice upon in vivo challenge with aGalCer-loaded tumor cells (Fig.3J). In summary, our results show that IL-18 mediates the gut microbiome-dependent enhancement of iNKT cell function in the liver.
CSF-1R+ macrophages control liver iNKT function.
Different cell types were isolated from the liver to identify the source of IL-18. Oral vancomycin increased IL-18 expression in both liver macrophages and hepatocytes but not in hepatic stellate cells (Fig.4A), and the increase was stronger in liver macrophages as compared to hepatocytes (Fig.4A). Since liver macrophages acts as the critical defense against microbial pathogens from intestine, we tested their contribution to changes in iNKT cell function. Clodronate liposomes were used to deplete macrophages, and the efficacy was confirmed (Fig.S4A). Indeed, removing macrophages markedly reduced IL-18 expression levels in the liver following oral vancomycin treatment (Fig.4B). Next, the impact of macrophage depletion on iNKT activation was assessed following i.v. injection of αGalCer-loaded tumor cells. Strikingly, although no effect on the liver iNKT population was observed (Fig.S4B), macrophage depletion completely blocked IFNγ production by liver iNKT cells (Fig.4C&D). Importantly, in the absence of liver macrophages, oral vancomycin failed to upregulate IFNγ in liver iNKT cells (Fig.4C&D). Consistently, clodronate treatment abolished the increase of IL-4 production and CD69 expression in liver iNKT cells after vancomycin, which was seen in control mice after liposome treatment (Fig.S4C&D). Clodronate treatment also decreased IFNγ production by splenic iNKT cells, but the effect was much less as compared to the liver (Fig.S4E). The experiment was repeated using free αGalCer (Fig.S4F). Similar to what was seen with αGalCer-loaded cells, free αGalCer enhanced liver iNKT cell activation in vancomycin treated mice (Fig.S4G–I) and clodronate treatment completely blocked liver iNKT activation induced by free αGalCer injection (Fig.S4G–I).
Figure.4. CSF-1R+ macrophages control liver iNKT cell function.
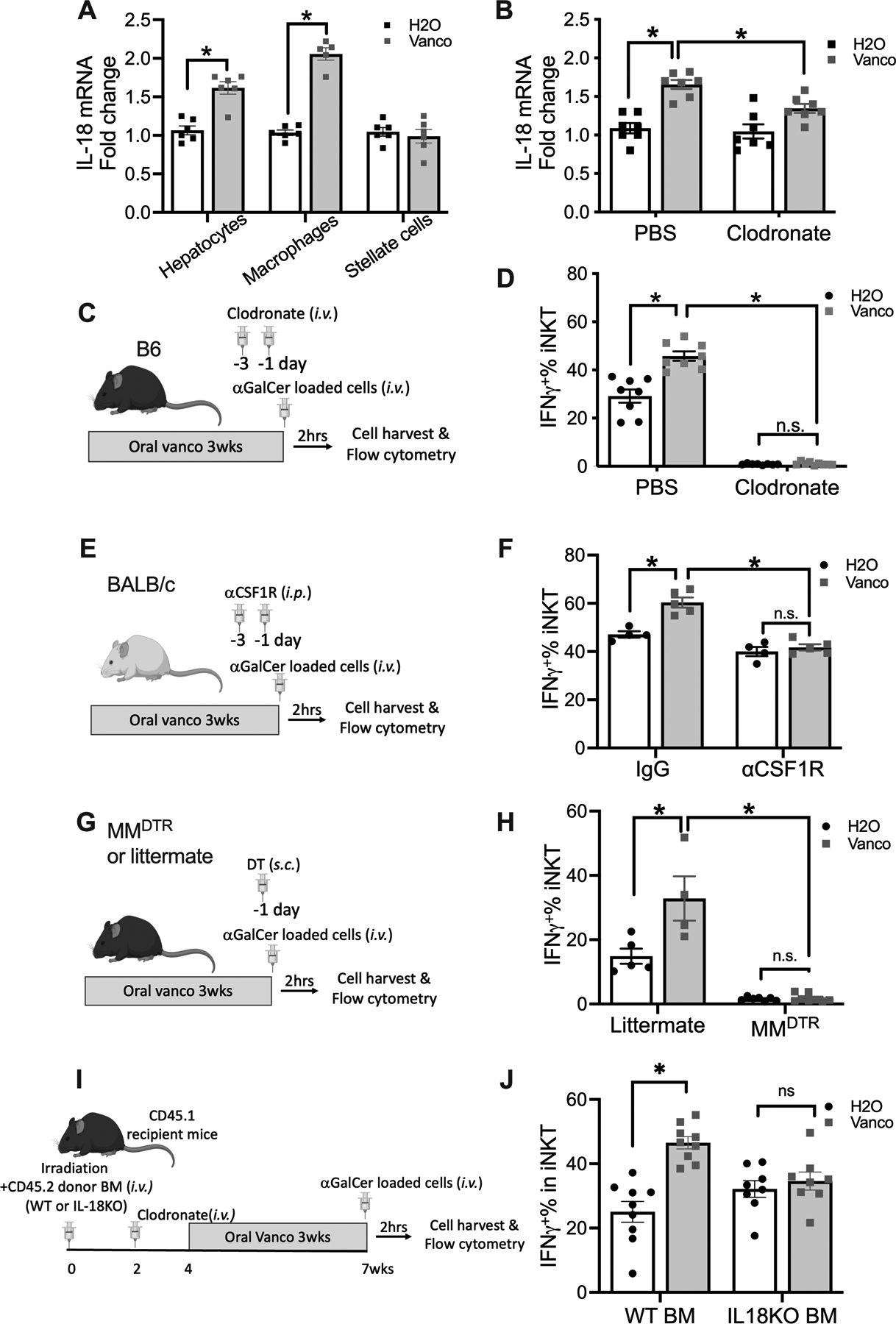
(A) Hepatocytes, macrophages or stellate cells were isolated from C57BL/6 mice kept on vancomycin or H2O. IL-18 mRNA in each cell type was determined by RT-PCR. Results are presented as mean+/− SEM of one experiment. n=5, p<0.05, two-way ANOVA.
(B) Vancomycin or H2O treated C57BL/6 mice were treated with clodronate liposomes to deplete macrophages. One day later, IL-18 mRNA expression in liver tissue was measured by RT-PCR. Results are presented as mean+/− SEM from two independent experiments. n=7, p<0.05, two-way ANOVA.
(C,D) Vancomycin or H2O treated C57BL/6 mice were treated with clodronate liposome to deplete macrophages followed by i.v. injection of αGalCer-loaded EL4 cells plus brefeldin A. (C) Experimental setup. (D) Liver iNKT cell IFNγ+% was measured by flow cytometry. Results are presented as mean+/− SEM from two independent experiments. n=8, p<0.05, two-way ANOVA.
(E,F) BALB/c mice (age 7~10 weeks) were kept on vancomycin or H2O for 3 weeks. Mice received two doses of αCSF-1R or IgG i.p. injections before i.v. injection of αGalCer-loaded A20 cells plus brefeldin A. (E) Experimental setup. (F) Liver iNKT cell IFNγ+% is shown. Results are presented as mean+/− SEM of one experiment, n=4 for IgG H2O and IgG Vanco, n=5 for αCSF-1R H2O and αCSF-1R Vanco, p<0.05, two-way ANOVA.
(G,H) MMDTR mice or littermates (age 7~10 weeks) were kept on vancomycin or H2O for 3 weeks. Then mice were s.c. injected 200 ng diphtheria toxin (DT) to deplete CSF-1R+ macrophage one day before given i.v. injection of αGalCer-loaded EL4 cells with brefeldin A. (G) Experimental setup. (H) Liver iNKT cell IFNγ+% is shown. Results are presented as mean+/− SEM from two independent experiment, n=5 for littermate H2O, 4 for littermate Vanco, 7 for MMDTR H2O, 8 for MMDTR Vanco, p<0.05, two-way ANOVA.
(I, J) CD45.1 recipient mice (age ~6 weeks) were irradiated with 900 rads followed by adoptive transfer of donor bone marrow cells from either CD45.2 WT or IL-18 KO mice, followed by clodronate treatment. Mice were kept on vancomycin or H2O for 3 weeks, and αGalCer-loaded EL4 cells plus brefeldin A was injected i.v.. (G) Experimental setup. (H) Liver iNKT cell IFNγ+% is shown. Results are presented as mean+/− SEM from two independent experiment, n=10, p<0.05, two-way ANOVA.
Colony-stimulating factor-1 (CSF-1) is well known to stimulate the formation of macrophage colonies through its receptor CSF-1R(30). The CSF-1R+ macrophage subset can be targeted by anti-CSF-1R antibody AFS98 (31). Although less potent compared to clodronate, anti-CSF-1R treatment caused a clear reduction of F4/80 expression in mouse liver (Fig.S4J). Next, we used the anti-CSF-1R antibody to deplete CSF-1R+ macrophages and test the influence on liver iNKT activity. Interestingly, anti-CSF-1R treatment was sufficient to block the vancomycin-enhanced IFNγ production in liver iNKT cells (Fig.4E&F) without affecting iNKT cell population size following i.v. injection of aGalCer-loaded tumor cells (Fig.S4K), suggesting the critical role of CSF-1R+ macrophage in iNKT function regulation by gut microbiome.
To confirm the importance of CSF-1R+ macrophages in the functional regulation of iNKT cells, LyzCrexCSF-1RLsL-DTR (MMDTR) mice were used. CSF-1R and Lysozyme M co-expressing macrophages in MMDTR mice can be selectively eliminated by diphtheria toxin (DT) without affecting dendritic cells (20). Again, a drastic reduction of IFNγ production in liver iNKT cells was found in DT- treated MMDTR mice compared to littermates after in vivo stimulation (Fig.4G&H). Similarly, in DT- treated MMDTR mice, oral vancomycin did not enhance IFNγ expression in liver iNKT cells (Fig.4G&H). The depletion of macrophages in MMDTR mice was assessed by flow cytometry using the general mouse macrophage marker F4/80. DT treatment effectively removed liver CD11bloF4/80hi cells in MMDTR mice (Fig.S5A&B). Unexpectedly, an increase of the CD11b+F4/80lo macrophage population was found (Fig.S5A&C). This result suggests that the MMDTR mouse model preferably targets the CD11bloF4/80hi and not the CD11b+F4/80lo macrophage subset. The data also suggests that the CD11bloF4/80hi macrophage subset responsible for regulating iNKT cell function. Interestingly, in the F4/80 expressing liver macrophages, the CD11blo subset has been reported to have a much stronger phagocytic activity compared to the liver CD11b+ subset (32), suggesting that CD11blo macrophages may be better at sensing intestinal microbes. Consistently, lower liver IL-18 mRNA expression was found in DT- treated MMDTR mice (Fig.S5D).
To support our findings, we analyzed published single cell RNA-seq data (33) and looked at liver macrophages. In the CD45+ liver cell compartment, F4/80 expressing cells indeed contain CD11bloF4/80hi and CD11b+F4/80lo clusters (Fig.S5E&F). General expression of CSF-1R and Lysozyme M was found in F4/80 expressing cells, but higher CSF-1R levels were expressed in CD11bloF4/80hi cluster in contrast to the higher Lysozyme M expression by CD11b+F4/80lo cluster (Fig.S5G&H). Since DTR is driven by the Csf-1r promoter in MMDTR mice, the CD11bloF4/80hi population is expected to express more DTR and be more sensitive to DT-induced cell depletion. We also looked at IL-18 transcripts in this data set. Interestingly, IL-18-expressing cells mainly belong to the F4/80 expressing cell subset, especially the CD11bloF4/80hi cluster (Fig.S5I). The single cell RNA-Seq results support our hypothesis that the CSF-1RhiCD11bloF4/80hi macrophage subset was the main target in the MMDTR mice and in anti-CSF1R depletion and this subset is the major source of IL-18 in the liver.
To corroborate the role of macrophage-derived IL-18 in regulating liver iNKT cell function, we established a protocol that allowed us to replace host macrophages with IL-18 KO macrophages. For this we combined bone marrow transplantation using IL-18 KO or WT mice with clodronate treatment (Fig.S5J&K). Using this method, we generated mice reconstituted with either IL-18 KO or WT liver macrophages by transferring CD45.2 IL-18 KO or WT bone marrow into CD45.1 recipient mice followed by clodronate depletion (Fig.4I). Mice were kept on vancomycin or control H2O for 3 weeks, and liver iNKT function was measured after in vivo stimulation. Liver macrophage reconstitution with donor derived macrophages was confirmed by flow cytometry (Fig.S5L). Vancomycin treatment failed to enhance IFNγ production and CD69 expression on iNKT cells in IL-18 KO liver macrophage reconstituted mice but not in WT reconstituted mice (Fig.4J, Fig.S5M&N). This result confirms that macrophage derived IL-18 controls iNKT cell function after vancomycin treatment.
Together, our results demonstrate that CD11bloF4/80hiCSF-1R+ macrophages mediate regulation of liver iNKT function by the gut microbiome. This work suggests that targeting gut microbiome-liver macrophage axis can be potentially used to shape hepatic immune environment.
Discussion
In this study, we identified CSF-1R+ macrophages as the critical mediators of liver iNKT cell function. Using antibiotic treatment, germ-free mice, and fecal transplantation, we demonstrate that altering commensal microbiota can modulate liver iNKT cell function. Interestingly, in vivo and in vitro challenge using the same stimuli led to differential iNKT responses showing an indirect regulation in the liver environment rather than an intrinsic change in the iNKT cells. We also show that liver macrophages mediate gut microbiome-controlled iNKT function through IL-18 by in vivo liver macrophage depletion and reconstitution experiments. The critical role of CD11bloF4/80hi CSF-1R+ macrophage subset was demonstrated by using anti-CSF1R depletion and LyzCrexCSF1R-LsL-DTR mice. Together our results suggest that the gut microbiome/CSF1R+ macrophage/IL-18 pathway is important in regulation of liver iNKT function. CSF-1R+ macrophages can be potentially targeted by modulating gut microbiome to influence liver immunity.
It has been well document that the microbiome can control iNKT cells (11, 14, 17, 34, 35). Bacterial species and microbial products have been reported to activate iNKT cells (18, 36). Gut commensals have been reported to be necessary for maintaining proper iNKT cell function by comparing germ-free and SPF mice (34). Interestingly, iNKT cell function was unchanged in germ-free mice (37). Splenic iNKT cells were used in both studies. In our study, no change of liver iNKT activation was found either after housing germ-free mice in a SPF facility or after FMT with control feces, supporting that unaltered gut commensals are not important for liver iNKT function. Although seemingly controversial, it should be recognized that the control SPF mice can be different. Mice from different SPF facility are known to harbor distinct gut microbiomes. Importantly, these SPF mice can have different immune function, and the same mouse strain from JAX and Taconic responded differently to concanavalin A-caused liver injury (38). The discrepancy of iNKT regulation by gut commensal can be potentially explained by microbial variations among facilities.
Immune cells such as CD8+ T cells have been reported to reduce iNKT cell number through the gut microbiome (39). Macrophages are known to interact with iNKT cells (40) and sense microbial signals which makes them an interesting candidate in the context of gut microbiome-dependent regulation of iNKT function. Importantly, Kupffer cells have been suggested to increase iNKT function by gut microbe changes which modulates liver regeneration (14). However, the study relies on unselective clodronate treatment. Our study revealed that the liver CSF-1R+CD11bloF4/80hi subset but not the CD11b+F4/80lo macrophage subset is important for iNKT function regulation. The presence of CSF-1R+CD11b−F4/80+ macrophages as a major source of IL-18 was confirmed in the published sc-RNA-seq data of mouse liver cells (33). As antigen presenting cells, macrophages express CD1d molecules and can present lipid antigen to activate iNKT cells. Although iNKT cells can be fully activated by cytokines in the absence of TCR engagement (41) and our results show that IL-18 is critical for iNKT function in this setting, we did not rule out the potential requirement of TCR signal for the enhanced iNKT activation, especially when stimulated with aGalCer-loaded tumor cells. Liver CD11blow macrophages have been reported to display higher phagocytic activity compared to the CD11b+F4/80+ counterpart(32), suggesting that they are more likely to uptake and present microbial lipid antigens to stimulate iNKT cells. It is still unknown whether the vancomycin-altered gut microbiome affects CD1d-dependent lipid antigen presentation on macrophages, and subsequently iNKT activation. Mice with selective CD1d deficiency in CSF-1R+CD11bloF4/80hi macrophages are needed to address this question.
Macrophages are critical in regulation of various immune responses including CD8+ T cell activation (42). Macrophages demonstrate high plasticity and can be polarized to M1 pro-inflammatory or M2 anti-inflammatory phenotypes(43), and the CSF-1/CSF1R axis is well recognized in contribution to M2 polarization. Targeting CSF1/CSF-1R macrophages have been shown to reverse suppression of anti-tumor immunity (44). Gut microbiota have been established as a critical modulator of host immune response and are involved in progression and treatment of various diseases including tumor immunotherapy (45, 46). It will be interesting to test whether the observed gut microbiome/CSF-1R+CD11b−F4/80 macrophage axis can affect other immune reactions in various pathological conditions, and its potential role in immunotherapy.
One interesting finding of our study is that the frequency of liver iNKT population and function are differentially regulated by gut microbiota. We previously found that primary-to-secondary bile acid conversion by vancomycin-sensitive intestinal bacteria prevents chemokine CXCL16 expression of liver endothelial cells, thus decreasing accumulation of CXCR6+ iNKT cells in the liver (17). However, here, we found that the absence of bile acid-metabolizing bacteria in germ-free mice increased the iNKT cell number without affecting iNKT activity. Reconstitution of GF mice with SPF fecal microbiota, containing bile processing bacteria reduced the number of iNKT cells whereas reconstitution with fecal microbiota from vancomycin-treated mice, depleted of bile processing bacteria, maintained the high number of iNKT in GF mice while increasing their activity. The dissociation between iNKT cell number and functional regulation was repeatedly observed in IL-18 inhibition and macrophage depletion studies, where liver iNKT function but not frequency was affected. These findings show that the gut microbiome plays a complex role in regulation of iNKT cells. Simply changing iNKT population size by targeting bile acid-metabolizing bacteria may not be enough to interfere with iNKT-mediated biological processes, and iNKT functional interventions may also be needed.
Fecal transplantation and germ-free mouse studies clearly show that the vancomycin-altered gut microbiome is responsible for the enhanced liver iNKT function. It is also still not known how the gut microbiome regulates CSF-1R+CD11b−F4/80+ macrophages. Vancomycin is known to reduce immunoregulatory short chain fatty acids (SCFAs). A recent report suggests that reducing SCFAs contributes to the vancomycin-enhanced tumor radiation therapy by unleashing a strong anti-tumor T cell response (47). The potential role of SCFA in regulating iNKT cell functions in our setting needs further investigation. Microbial metabolites have been found to modulate the host immune system (48–51). Recently c-di-AMP, a microbial STING agonist, has been shown to regulate myeloid cell function (48), and Akkermansia muciniphila, a bacteria shown to improve immunotherapy efficacy, produces c-di-AMP. Interestingly, Akkermansia muciniphila is known to expand upon oral vancomycin treatment. Further detailed investigation of immune regulating metabolite-producing bacteria such as Akkermansia muciniphila will help to better understand regulation of iNKT function.
The effect of age on iNKT cell function and numbers is another interesting topic. During early life microbial exposure has been found to have a profound and long-lasting effect on colon iNKT cells and affects later development of immune mediated diseases such as IBD and asthma (52). It will be interesting to study the impact of early life gut microbiome alteration on liver iNKT cells and immune regulation in later life.
In summary, using three approaches to target macrophages, our study identified IL-18 production by CSF-1R+CD11b−F4/80+ macrophages as critical modulators of gut microbiome-controlled iNKT function. Our study helps to understand the immune regulation through the gut-liver axis, and suggests that modulating gut microbiota can be potentially used to modify liver macrophages with the ultimate goal of regulating the liver immune environment.
Supplementary Material
Key points.
Vancomycin altered gut microbiome enhances liver iNKT cell function.
Vancomycin treatment effects IL-18 production by CSF-1R+ macrophages.
Liver iNKT cell function is controlled by IL-18 producing CSF-1R+ macrophages.
Acknowledgments
We would like to thank the members from Dr. Jay Berzofsky’s lab and Dr. Giorgio Trinchieri’s lab for helpful discussions. We also want to thank Richard Blumberg for advice, NIH tetramer core facility at Emory University for providing Cd1d-tetramer, and Animal Research Technical Support and Gnotobiotics Facility staff for germ-free mouse service.
Grant support
T.F.G. was supported by the Intramural Research Program of the NIH, NCI (ZIA BC 011345). The study was supported by an NCI CCR FLEX Program Synergy Award to G.T. and T.F.G.
Footnotes
Conflict-of-interest statement
The authors declare no conflict of interest.
References
- 1.Rooks MG, and Garrett WS. 2016. Gut microbiota, metabolites and host immunity. Nat Rev Immunol 16: 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albillos A, de Gottardi A, and Rescigno M. 2020. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol 72: 558–577. [DOI] [PubMed] [Google Scholar]
- 3.Cohen NR, Garg S, and Brenner MB. 2009. Antigen Presentation by CD1 Lipids, T Cells, and NKT Cells in Microbial Immunity. Adv Immunol 102: 1–94. [DOI] [PubMed] [Google Scholar]
- 4.Kronenberg M 2005. Toward an understanding of NKT cell biology: progress and paradoxes. Annu Rev Immunol 23: 877–900. [DOI] [PubMed] [Google Scholar]
- 5.Bendelac A, Savage PB, and Teyton L. 2007. The biology of NKT cells. Annu Rev Immunol 25: 297–336. [DOI] [PubMed] [Google Scholar]
- 6.Taniguchi M, Harada M, Dashtsoodol N, and Kojo S. 2015. Discovery of NKT cells and development of NKT cell-targeted anti-tumor immunotherapy. Proc Jpn Acad Ser B Phys Biol Sci 91: 292–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Godfrey DI, Stankovic S, and Baxter AG. 2010. Raising the NKT cell family. Nat Immunol 11: 197–206. [DOI] [PubMed] [Google Scholar]
- 8.Yu KO, Im JS, Molano A, Dutronc Y, Illarionov PA, Forestier C, Fujiwara N, Arias I, Miyake S, Yamamura T, Chang YT, Besra GS, and Porcelli SA. 2005. Modulation of CD1d-restricted NKT cell responses by using N-acyl variants of alpha-galactosylceramides. Proc Natl Acad Sci U S A 102: 3383–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Kaer L, Parekh VV, and Wu L. 2011. Invariant natural killer T cells: bridging innate and adaptive immunity. Cell Tissue Res 343: 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruf B, Heinrich B, and Greten TF. 2021. Immunobiology and immunotherapy of HCC: spotlight on innate and innate-like immune cells. Cell Mol Immunol 18: 112–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leinwand JC, Paul B, Chen R, Xu F, Sierra MA, Paluru MM, Nanduri S, Alcantara CG, Shadaloey SA, Yang F, Adam SA, Li Q, Bandel M, Gakhal I, Appiah L, Guo Y, Vardhan M, Flaminio Z, Grodman ER, Mermelstein A, Wang W, Diskin B, Aykut B, Khan M, Werba G, Pushalkar S, McKinstry M, Kluger Z, Park JJ, Hsieh B, Dancel-Manning K, Liang FX, Park JS, Saxena A, Li X, Theise ND, Saxena D, and Miller G. 2022. Intrahepatic microbes govern liver immunity by programming NKT cells. J Clin Invest 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, Witek RP, Choi SS, Guy CD, Fearing CM, Teaberry V, Pereira FE, Adams DH, and Diehl AM. 2010. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 51: 1998–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santodomingo-Garzon T, and Swain MG. 2011. Role of NKT cells in autoimmune liver disease. Autoimmun Rev 10: 793–800. [DOI] [PubMed] [Google Scholar]
- 14.Wu X, Sun R, Chen Y, Zheng X, Bai L, Lian Z, Wei H, and Tian Z. 2015. Oral ampicillin inhibits liver regeneration by breaking hepatic innate immune tolerance normally maintained by gut commensal bacteria. Hepatology 62: 253–264. [DOI] [PubMed] [Google Scholar]
- 15.Krijgsman D, Hokland M, and Kuppen PJK. 2018. The Role of Natural Killer T Cells in Cancer-A Phenotypical and Functional Approach. Front Immunol 9: 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Exley MA, Friedlander P, Alatrakchi N, Vriend L, Yue S, Sasada T, Zeng W, Mizukami Y, Clark J, Nemer D, LeClair K, Canning C, Daley H, Dranoff G, Giobbie-Hurder A, Hodi FS, Ritz J, and Balk SP. 2017. Adoptive Transfer of Invariant NKT Cells as Immunotherapy for Advanced Melanoma: A Phase I Clinical Trial. Clin Cancer Res 23: 3510–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, Agdashian D, Terabe M, Berzofsky JA, Fako V, Ritz T, Longerich T, Theriot CM, McCulloch JA, Roy S, Yuan W, Thovarai V, Sen SK, Ruchirawat M, Korangy F, Wang XW, Trinchieri G, and Greten TF. 2018. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kinjo Y, Illarionov P, Vela JL, Pei B, Girardi E, Li X, Li Y, Imamura M, Kaneko Y, Okawara A, Miyazaki Y, Gomez-Velasco A, Rogers P, Dahesh S, Uchiyama S, Khurana A, Kawahara K, Yesilkaya H, Andrew PW, Wong CH, Kawakami K, Nizet V, Besra GS, Tsuji M, Zajonc DM, and Kronenberg M. 2011. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat Immunol 12: 966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kovacs SB, Oh C, Aachoui Y, and Miao EA. 2021. Evaluating cytokine production by flow cytometry using brefeldin A in mice. STAR Protoc 2: 100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schreiber HA, Loschko J, Karssemeijer RA, Escolano A, Meredith MM, Mucida D, Guermonprez P, and Nussenzweig MC. 2013. Intestinal monocytes and macrophages are required for T cell polarization in response to Citrobacter rodentium. J Exp Med 210: 2025–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Q, Ma C, Duan Y, Heinrich B, Rosato U, Diggs LP, Ma L, Roy S, Fu Q, Brown ZJ, Wabitsch S, Thovarai V, Fu J, Feng D, Ruf B, Cui LL, Subramanyam V, Frank KM, Wang S, Kleiner DE, Ritz T, Rupp C, Gao B, Longerich T, Kroemer A, Wang XW, Ruchirawat M, Korangy F, Schnabl B, Trinchieri G, and Greten TF. 2021. Gut Microbiome Directs Hepatocytes to Recruit MDSCs and Promote Cholangiocarcinoma. Cancer Discov 11: 1248–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung Y, Qin H, Kang CY, Kim S, Kwak LW, and Dong C. 2007. An NKT-mediated autologous vaccine generates CD4 T-cell dependent potent antilymphoma immunity. Blood 110: 2013–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsuda JL, Gapin L, Baron JL, Sidobre S, Stetson DB, Mohrs M, Locksley RM, and Kronenberg M. 2003. Mouse V alpha 14i natural killer T cells are resistant to cytokine polarization in vivo. Proc Natl Acad Sci U S A 100: 8395–8400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyagi T, Takehara T, Tatsumi T, Suzuki T, Jinushi M, Kanazawa Y, Hiramatsu N, Kanto T, Tsuji S, Hori M, and Hayashi N. 2004. Concanavalin a injection activates intrahepatic innate immune cells to provoke an antitumor effect in murine liver. Hepatology 40: 1190–1196. [DOI] [PubMed] [Google Scholar]
- 25.Patel S, Preuss CV, and Bernice F. 2022. Vancomycin. In StatPearls, Treasure Island (FL). [PubMed] [Google Scholar]
- 26.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, Fidelle M, Flament C, Poirier-Colame V, Opolon P, Klein C, Iribarren K, Mondragon L, Jacquelot N, Qu B, Ferrere G, Clemenson C, Mezquita L, Masip JR, Naltet C, Brosseau S, Kaderbhai C, Richard C, Rizvi H, Levenez F, Galleron N, Quinquis B, Pons N, Ryffel B, Minard-Colin V, Gonin P, Soria JC, Deutsch E, Loriot Y, Ghiringhelli F, Zalcman G, Goldwasser F, Escudier B, Hellmann MD, Eggermont A, Raoult D, Albiges L, Kroemer G, and Zitvogel L. 2018. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359: 91–97. [DOI] [PubMed] [Google Scholar]
- 27.Ray P, Pandey U, and Aich P. 2021. Comparative analysis of beneficial effects of vancomycin treatment on Th1- and Th2-biased mice and the role of gut microbiota. J Appl Microbiol 130: 1337–1356. [DOI] [PubMed] [Google Scholar]
- 28.Leite-De-Moraes MC, Hameg A, Pacilio M, Koezuka Y, Taniguchi M, Van Kaer L, Schneider E, Dy M, and Herbelin A. 2001. IL-18 enhances IL-4 production by ligand-activated NKT lymphocytes: a pro-Th2 effect of IL-18 exerted through NKT cells. J Immunol 166: 945–951. [DOI] [PubMed] [Google Scholar]
- 29.Chudnovskiy A, Mortha A, Kana V, Kennard A, Ramirez JD, Rahman A, Remark R, Mogno I, Ng R, Gnjatic S, Amir ED, Solovyov A, Greenbaum B, Clemente J, Faith J, Belkaid Y, Grigg ME, and Merad M. 2016. Host-Protozoan Interactions Protect from Mucosal Infections through Activation of the Inflammasome. Cell 167: 444–456 e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stanley ER, and Chitu V. 2014. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacDonald KP, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, Kuns R, Pettit AR, Clouston A, Wainwright B, Branstetter D, Smith J, Paxton RJ, Cerretti DP, Bonham L, Hill GR, and Hume DA. 2010. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood 116: 3955–3963. [DOI] [PubMed] [Google Scholar]
- 32.Kinoshita M, Uchida T, Sato A, Nakashima M, Nakashima H, Shono S, Habu Y, Miyazaki H, Hiroi S, and Seki S. 2010. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J Hepatol 53: 903–910. [DOI] [PubMed] [Google Scholar]
- 33.Remmerie A, Martens L, Thone T, Castoldi A, Seurinck R, Pavie B, Roels J, Vanneste B, De Prijck S, Vanhockerhout M, Binte Abdul Latib M, Devisscher L, Hoorens A, Bonnardel J, Vandamme N, Kremer A, Borghgraef P, Van Vlierberghe H, Lippens S, Pearce E, Saeys Y, and Scott CL. 2020. Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct from Kupffer Cells in the Fatty Liver. Immunity 53: 641–657 e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wingender G, Stepniak D, Krebs P, Lin L, McBride S, Wei B, Braun J, Mazmanian SK, and Kronenberg M. 2012. Intestinal microbes affect phenotypes and functions of invariant natural killer T cells in mice. Gastroenterology 143: 418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S, Koh J, Chang Y, Kim HY, and Chung DH. 2019. Invariant NKT Cells Functionally Link Microbiota-Induced Butyrate Production and Joint Inflammation. J Immunol 203: 3199–3208. [DOI] [PubMed] [Google Scholar]
- 36.Wei Y, Zeng B, Chen J, Cui G, Lu C, Wu W, Yang J, Wei H, Xue R, Bai L, Chen Z, Li L, Iwabuchi K, Uede T, Van Kaer L, and Diao H. 2016. Enterogenous bacterial glycolipids are required for the generation of natural killer T cells mediated liver injury. Sci Rep 6: 36365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park SH, Benlagha K, Lee D, Balish E, and Bendelac A. 2000. Unaltered phenotype, tissue distribution and function of Valpha14(+) NKT cells in germ-free mice. Eur J Immunol 30: 620–625. [DOI] [PubMed] [Google Scholar]
- 38.Celaj S, Gleeson MW, Deng J, O’Toole GA, Hampton TH, Toft MF, Morrison HG, Sogin ML, Putra J, Suriawinata AA, and Gorham JD. 2014. The microbiota regulates susceptibility to Fas-mediated acute hepatic injury. Lab Invest 94: 938–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei B, Wingender G, Fujiwara D, Chen DY, McPherson M, Brewer S, Borneman J, Kronenberg M, and Braun J. 2010. Commensal microbiota and CD8+ T cells shape the formation of invariant NKT cells. J Immunol 184: 1218–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cruz MS, Loureiro JP, Oliveira MJ, and Macedo MF. 2022. The iNKT Cell-Macrophage Axis in Homeostasis and Disease. Int J Mol Sci 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leite-De-Moraes MC, Hameg A, Arnould A, Machavoine F, Koezuka Y, Schneider E, Herbelin A, and Dy M. 1999. A distinct IL-18-induced pathway to fully activate NK T lymphocytes independently from TCR engagement. J Immunol 163: 5871–5876. [PubMed] [Google Scholar]
- 42.DeNardo DG, and Ruffell B. 2019. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol 19: 369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yunna C, Mengru H, Lei W, and Weidong C. 2020. Macrophage M1/M2 polarization. Eur J Pharmacol 877: 173090. [DOI] [PubMed] [Google Scholar]
- 44.Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, and Ruttinger D. 2017. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer 5: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu HJ, and Wu E. 2012. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 3: 4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou CB, Zhou YL, and Fang JY. 2021. Gut Microbiota in Cancer Immune Response and Immunotherapy. Trends Cancer 7: 647–660. [DOI] [PubMed] [Google Scholar]
- 47.Uribe-Herranz M, Rafail S, Beghi S, Gil-de-Gomez L, Verginadis I, Bittinger K, Pustylnikov S, Pierini S, Perales-Linares R, Blair IA, Mesaros CA, Snyder NW, Bushman F, Koumenis C, and Facciabene A. 2020. Gut microbiota modulate dendritic cell antigen presentation and radiotherapy-induced antitumor immune response. J Clin Invest 130: 466–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lam KC, Araya RE, Huang A, Chen Q, Di Modica M, Rodrigues RR, Lopes A, Johnson SB, Schwarz B, Bohrnsen E, Cogdill AP, Bosio CM, Wargo JA, Lee MP, and Goldszmid RS. 2021. Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell 184: 5338–5356 e5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agus A, Clement K, and Sokol H. 2021. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut 70: 1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, Paik S, Stagg J, Groves RA, Gallo M, Lewis IA, Geuking MB, and McCoy KD. 2020. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 369: 1481–1489. [DOI] [PubMed] [Google Scholar]
- 51.Hezaveh K, Shinde RS, Klotgen A, Halaby MJ, Lamorte S, Ciudad MT, Quevedo R, Neufeld L, Liu ZQ, Jin R, Grunwald BT, Foerster EG, Chaharlangi D, Guo M, Makhijani P, Zhang X, Pugh TJ, Pinto DM, Co IL, McGuigan AP, Jang GH, Khokha R, Ohashi PS, O’Kane GM, Gallinger S, Navarre WW, Maughan H, Philpott DJ, Brooks DG, and McGaha TL. 2022. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity 55: 324–340 e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, and Blumberg RS. 2012. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336: 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.