

Abstract
Purpose:
Antibodies against IGF-1R have shown meaningful but transient tumor responses in patients with rhabdomyosarcoma (RMS). The SRC family member YES has been shown to mediate IGF-1R antibody acquired resistance, and cotargeting IGF-1R and YES resulted in sustained responses in murine RMS models. We conducted a phase I trial of the anti-IGF-1R antibody ganitumab combined with dasatinib, a multi-kinase inhibitor targeting YES, in RMS patients (NCT03041701).
Patients and Methods:
Patients with relapsed/refractory alveolar or embryonal RMS and measurable disease were eligible. All patients received ganitumab 18 mg/kg intravenously every 2 weeks. Dasatinib dose was 60 mg/m2/dose (max 100 mg) oral once daily [dose level (DL)1] or 60 mg/m2/dose (max 70 mg) twice daily (DL2). A 3+3 dose escalation design was used, and maximum tolerated dose (MTD) was determined based on cycle 1 dose-limiting toxicities (DLTs).
Results:
Thirteen eligible patients, median age 18 years (range 8–29) enrolled. Median number of prior systemic therapies was 3; all had received prior radiation. Of 11 toxicity-evaluable patients, 1/6 had a DLT at DL1 (diarrhea) and 2/5 had a DLT at DL2 (pneumonitis, hematuria) confirming DL1 as MTD. Of nine response-evaluable patients, one had a confirmed partial response (PR) for four cycles, and one had stable disease (SD) for six cycles. Genomic studies from cell-free DNA correlated with disease response.
Conclusions:
The combination of dasatinib 60 mg/m2/dose daily and ganitumab 18 mg/kg every two weeks was safe and tolerable. This combination had a disease control rate of 22% at five months.
Translational Relevance
Outcomes for patients with high-risk rhabdomyosarcoma (RMS) have remained dismal for decades. Chemotherapy remains the mainstay of treatment and few RMS-specific clinical trials testing new approaches exist. The results of this multicenter phase I study in pediatric and young adult patients with relapsed/refractory RMS demonstrated that cotargeting IGF-1R and YES using ganitumab plus dasatinib was safe and tolerable. Disease control rate was 22.2% at five months, despite an overrepresentation of tumors with adverse molecular features. Serially collected cell-free DNA correlated with disease response and demonstrated tumor clonal evolution. Although loss of access to ganitumab prematurely terminated the phase II part of the study, initial signals of activity observed during phase I indicate that this combination is worthy of further study for patients with relapsed RMS. Further, this work highlights the need for focused drug development efforts for pediatric cancers, given the ongoing challenges with drug access for children.
Introduction
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma of childhood with an annual incidence of approximately 4–7 cases per million children.1,2 Embryonal rhabdomyosarcoma (ERMS) and alveolar rhabdomyosarcoma (ARMS) are the two major histologic subtypes of RMS, with PAX gene fusion-positive ARMS carrying a particularly poor prognosis.3 Conventional chemotherapy has remained the standard of care for decades, and patients with metastatic or recurrent disease have a 5-year overall survival of less than 20%. Further, outcomes have only minimally improved over the past several decades.4,5 Thus there is an urgent need for novel, more effective therapies for patients with RMS.
The insulin-like growth factor (IGF) pathway plays an important role in the biology of RMS, and a majority of pediatric RMS highly express the IGF type 1 receptor (IGF-1R).6–10 Prior studies with single agent monoclonal antibodies against IGF-1R showed clinically meaningful responses, including confirmed partial responses (PR), unconfirmed PR, and stable disease (SD), in about 10–15% of patients with RMS. However, nearly all of these responses were short-lived with a rapid onset of resistance.11–13 Studies exploring mechanisms of resistance in models of ARMS and ERMS demonstrated that activation of YES, a member of the SRC family tyrosine kinases, acted as a bypass resistance mechanism to IGF-1R targeting. Furthermore, co-targeting IGF-1R and YES resulted in sustained responses in murine RMS models.14
Ganitumab is a fully human monoclonal antibody that inhibits IGF-1R with an IC50 of 0.6–2.5 nM leading to inhibition of cell survival and proliferative signals.15 Prior early phase clinical trials of ganitumab as a single agent and in combination with cytotoxic chemotherapy and targeted agents have evaluated doses ranging from 12 mg/kg – 20 mg/kg intravenously (IV) every 2 weeks in adult and pediatric patients.16–21 In these studies, ganitumab was well tolerated with most common adverse effects in combination studies being fatigue, nausea, and vomiting. Frequent grade 3 toxicities included neutropenia, thrombocytopenia, and hyperglycemia.18–21 Serum trough levels of >10 μg/mL were achieved with these doses and are sufficient for biochemical blockade of IGF-1R signaling.15,17 Dasatinib is an orally administered drug that potently and selectively inhibits multiple protein tyrosine kinases including YES (IC50 0.41 nM) and other SRC family kinases.22 Dasatinib is FDA approved for the treatment of certain forms of chronic myelogenous and acute lymphoblastic leukemias and has been studied as a single agent and in combination with other agents in many trials. Common adverse effects observed in patients with solid tumors treated with dasatinib included gastrointestinal intolerance, fatigue, dyspnea, anorexia, dehydration, fluid retention, pleural and pericardial effusion, bleeding related risks, and a moderate increase in QTcF. Hematologic toxicity was typically mild.22–25 Dasatinib has been used in combination with other therapies, typically at dose of 60 mg/m2 daily, although the recommended phase II doses (RP2D) were significantly higher both for adult and pediatric patients. Based on in vitro data in RMS cell lines, 60 mg/m2 is expected to produce concentrations high enough to inhibit YES.23,26
Based on the strong preclinical rationale for co-targeting IGF-1R and YES in RMS, we developed a phase I/II study of the combination of ganitumab and dasatinib in patients with relapsed or refractory ERMS and ARMS. The primary objectives were to determine the safe dose of dasatinib when given with ganitumab (phase I) and to determine the activity of this combination (phase II) in this patient population. Additional objectives were to describe the adverse effects of this combination, and to assess the progression free survival (PFS) among the response-evaluable patients treated in both the phase I and II cohorts. Correlative genomic studies of the germline, tumor, and cell-free DNA (cfDNA) were performed. The phase II part of the study was terminated early when ganitumab became unavailable. We report here the results of the completed phase I study.
Patients and Methods
Patient eligibility
Patients of any age with histologically confirmed ERMS or ARMS for whom there were no curative or life prolonging treatments available were eligible to enroll. Patients were required to have measurable disease according to the Response Evaluation Criteria in Solid Tumors (RECIST) v1.1, the ability to swallow tablets and undergo imaging studies required per protocol, adequate performance status (as defined as ECOG performance status ≤2 or Karnofsky/Lansky performance score ≥50%), adequate organ function (as defined by absolute neutrophil count ≥1,000/mcL, platelets ≥75,000/mcL, total bilirubin ≤ 1.5X upper limit of normal (ULN), ALT ≤ 3X ULN, creatinine within normal institutional limits or creatinine clearance ≥60 mL/min/1.73 m2, and QTcF < 480 msec), normal blood glucose for age, not be pregnant or breast-feeding, and to have recovered from acute toxic effects of prior therapy. There was no limit on number of prior therapies, and patients could have received other IGF-1R antibodies or inhibitors.
Patients were not eligible if they had known brain metastases, history of radiation pneumonitis, pre-existing diabetes mellitus, clinically significant cardiovascular disease such as myocardial infarction or ventricular tachyarrhythmia within 6 months, or other uncontrolled intercurrent illness. Participants could not be receiving concurrent treatment with potent inhibitors or inducers of CYP3A4, antithrombotic and/or anti-platelet agents, or drugs with proarrhythmic potential. Complete eligibiliy criteria can be found in Supplemental Appendix A.
Treatment regimen and study design
This multi-institutional investigator initiated NCI CTEP sponsored trial (NCT01709435) was coordinated by the NCI and conducted at the NIH Clinical Center and Children’s Hospital Los Angeles (CHLA). The studies were conducted in accordance with recognized ethical guidelines as per The Belmont Report and the and the HHS Common Rule, and the trial was approved by the NIH Institutional Review Board (IRB) and the CHLA IRB. All patients enrolled at the NIH Clinical Center (n=12) were co-enrolled on a biorepository study for solid tumors (NCT01109394). All patients or their parent/legal guardian signed informed consent. Assent was obtained per institutional guidelines.
Ganitumab was given intravenously (IV) every 2 weeks, on day 0 and day 14 of each cycle, starting from cycle 1 day 0 at a dose of 18 mg/kg. Dasatinib was administered orally on a continuous schedule from day −7 to day 27 during cycle 1 (cycle 1 = 35 days) and then day 0 to day 27 for subsequent cycles. All cycles except cycle 1 were 28 days long. Two dose levels (DL) were evaluated for dasatinib using a 3+3 dose escalation design: DL1 was 60 mg/m2/dose (maximum 100 mg) once daily and DL2 was 60 mg/m2/dose (maximum 70 mg) twice daily. The higher dose is below the RP2D in pediatric patients with solid tumors, and equivalent to the RP2D in adult patients with solid tumors. Two doses were tested given the dearth of data on inhibition of YES with dasatinib in human tumors, although based on in vitro data, it was expected that the lower dose would produce concentrations high enough to inhibit YES.23,26
Dosing was achieved using a dosing nomogram with 5 mg, 20 mg, and 50 mg tablets (Supplemental Appendix B). Patients could receive subsequent cycles if they had no evidence of disease progression and did not experience study-drug related adverse events (AE) requiring therapy discontinuation. Treatment was interrupted for dose-limiting toxicities (DLTs) that were defined in the study protocol. Depending on attribution of toxicity, once the AE improved, treatment could be resumed with a dose reduction of the agent that caused the toxicity, or reduction of both agents if the toxicity could not clearly be attributed to either agent specifically. One dose reduction for ganitumab (to 12 mg/kg/dose) and two dose reductions (approximately 30% dose reduction for each instance) for dasatinib were permitted (Supplemental Appendix C). Pharmacokinetic (PK) studies were not performed because pediatric and adult PK data are available on both agents and drug-drug interactions are not expected between small molecules and naked monoclonal antibodies.
Patients underwent regular physical examinations, laboratory assessments, and electrocardiograms while receiving protocol therapy. Adverse events were graded per Common Terminology Criteria for Adverse Events (CTCAE) v5. Disease evaluations were performed at the end of every two cycles and responses were assessed using Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. For dose escalation, patients were considered evaluable for determination of the maximum tolerated dose (MTD) if they either experienced a DLT in cycle 1 or if they received both ganitumab doses and ≥ 85% of the dasatinib doses in cycle 1. Patients with measurable disease at baseline who received at least one cycle of therapy and had their disease status reevaluated were evaluable for response.
Definition of dose- limiting toxicity and maximum tolerated dose
DLTs were defined as any grade 3 or higher toxicity possibly, probably, or definitely attributed to dasatinib, ganitumab or the combination, with the specific exceptions of grade 3 nausea and vomiting of < 5 days duration; grade 3 fever or infection < 5 days duration; grade 3 ALT/AST elevation that returned to eligibility criteria within 7 days of study drug interruption and did not recur upon re-challenge with study drug; grade 3 hypophosphatemia, hypokalemia, hypocalcemia and/or hypomagnesemia that was responsive to supplementation; grade 3 neutropenia or thrombocytopenia; and grade 3 or 4 infusion reactions. Grade 2 hemorrhage, bleeding, or coagulopathy without thrombocytopenia; QTcF prolongation ≥ 550 msec; or any toxicity of grade 2 or greater that was considered intolerable to the patient and could not be controlled with standard supportive measures were considered to be DLTs. The MTD was defined as the dose level at which no more than 1 of up to 6 toxicity evaluable patients experienced DLT during the first cycle of treatment, and the dose below that at which at least 2 (of ≤ 6) patients experienced a DLT.
Clinical specimen processing and analysis
When available, FFPE unstained slides or blocks were collected from archival patient tumor samples and a germline sample was collected from peripheral blood. DNA and RNA were extracted, and whole genome (2 cases) or whole exome (9 cases) paired with RNAseq was performed. Sequencing reads were aligned to hg19, and somatic variant calls were generated using the CCBR pipeliner analysis solution (https://github.com/CCBR/Pipeliner) using Tumor-Normal somatic variant calling. Described mutations were limited to exonic calls with somatic variant allele frequency >10%.
Venous blood samples (10 ml) were collected in EDTA tubes (BD Biosciences, San Jose, CA) for cfDNA on the first day of each cycle. Whole blood samples were centrifuged at room temperature (1,900 x g for 10 minutes). Isolated plasma was centrifuged a second time at room temperature (15,000 x g for 10 minutes) in low-bind Eppendorf tubes to remove residual cells. Purified plasma was frozen at −80°C. EDTA tubes were processed within 6 hours of collection.
Purified plasma was thawed at room temperature and cfDNA was extracted from 3–5 mL of plasma using the QIAamp Circulating Nucleic Acid kit (Qiagen, Hilden, Germany). Extracted cfDNA concentration and quality were assessed using a Tapestation (Agilent Technologies, Santa Clara, CA). Sequencing libraries were subsequently constructed from cfDNA (5–15 ng) using TruSeq Nano (Illumina, San Diego, CA) per the manufacturers’ instructions. Constructed libraries were balanced, pooled and sequenced using 150 bp paired-end reads on a NovaSeq (Illumina, San Diego, CA).
Generated FASTQ files were demultiplexed and raw reads were quality-filtered using fastp v.0.20.0. Reads were aligned to hg19 using BWA v0.7.17 and deduplicated with Samtools v.1.7. GC content and mappability bias correction, depth-based local copy number estimates, and copy number–based estimation of tumor fraction were then performed using low tumor fraction parameters with the ichorCNA tool (Broad v.0.2.0).27 Samples were considered to have circulating tumor DNA (ctDNA) if tumor fraction was greater than 0.03 per Adalsteinsson et al’s previously described limits of detection.27 For longitudinal assessment of changes in individual patients’ tumor fraction, serial tumor fractions were normalized to each patient’s pre-cycle 1 tumor fraction or their first available sample.
Nucleosome footprints were evaluated using Liquorice28,29 with GC and mappability-corrected coverage profiles calculated across a custom fusion-positive RMS region set (Supplemental Dataset 1) informed by previously published PAX3-FOXO1 enhancer sites.30 YES1 and IGF1R region sets (Supplemental Dataset 1) were derived from candidate regulatory motifis on MotifMap.31,32 Nucleosome coverage signatures were compared between clinically identified FOXO1 fusion-positive and fusion-negative RMS patients and between pre-treatment and post-treatment samples as previously decribed using default parameters.29 Briefly, normal distribution of control samples (fusion-negative and pre-treatment) was assured using a Shapiro-Wilk test, prediction intervals were then calculated as the range of coverage dip-areas and dip-depths in which there is a 95% probability that future observations will fall. Fusion-positive samples and post-treatment samples were classified as significantly different if they fell outside of their control’s prediction intervals. Universal DNase-hypersensitivity sites (DHS) (Supplemental Dataset 1) were used for control conditions.28
Data Availability Statement
Sequencing data are available through dbGAP Accession phs003243.v1.p1. Additional data is available upon request to the corresponding author.
RESULTS
Patients
Between July 2017 and September 2020, 13 eligible patients (six male, seven female) enrolled on the phase I study. The median age was 18 years (range 8–29 years). The race/ethnicity of enrolled patients included: Non-Hispanic White (n=8), Non-Hispanic Black (n=1), Non-Hispanic Asian (n=2), White/Unknown Ethnicity (n=1), and Unknown Race/Hispanic (n=1). Six patients had fusion-negative embryonal RMS and seven patients had confirmed PAX3-FOXO1 fusion-positive alveolar RMS. At diagnosis, seven patients had presented with metastatic disease. At the time of enrollment, 11 patients had metastatic disease and two patients had localized disease in the head and neck region. All patients had previously received multiagent chemotherapy, including two patients who had received high dose chemotherapy with stem cell rescue. Nine patients had undergone treatment with molecularly targeted agents, including four patients who had received tyrosine kinase inhibitors. One patient had received immunotherapy. The median number of prior systemic therapies was three (range 1–6). All patients had received prior radiation therapy (median two courses of radiation therapies, range 1–5). Additionally, seven patients had undergone at least 1 prior surgical resection of either primary or recurrent tumor and one had received magnetic resonance-guided high-intensity focused ultrasound (MR-HIFU) treatment. None of the patients had received prior treatment with IGF-1R-targeting therapy. Patient demographics and disease characteristics are presented in Table 1 and Supplemental Table S1.
Table 1:
Patient characteristics
| Patient | Age (years) | Sex | Performance score | PAX-FOXO1 Fusion status | Disease sites at enrollment | Prior courses of XRT | Prior # systemic therapies | Dose level |
|---|---|---|---|---|---|---|---|---|
| 1 | 8 | Female | 70 | FN | Lung, pleura, abdomen, pelvis | 2 | 4 | 1 |
| 2 | 12 | Female | 70 | FN | Lung, pancreas, pelvis | 2 | 4 | 1 |
| 3 | 19 | Male | 90 | FP | Lung, mediastinal and hilar LN | 1 | 3 | 1 |
| 4 | 8 | Female | 100 | FN | Head and neck (temporal fossa)$ | 1 | 4 | 1 |
| 5^ | 19 | Female | 90 | FP | Lung, pleura, mediastinal and hilar LN | 2 | 2 | 1 |
| 6 | 11 | Female | 90 | FP | Mediastinum, retroperitoneum, pelvis, bone | 5 | 2 | 1 |
| 7 | 19 | Female | 90 | FN | Retroperitoneum (paraaortic LN) | 1 | 1 | 1 |
| 8^ | 8 | Male | 90 | FN | Head and neck (parapharyngeal$ | 1 | 4 | 2 |
| 9 | 13 | Male | 100 | FP | Lung, pleura, mediastinum | 2 | 3 | 2 |
| 10 | 18 | Male | 100 | FN | Pleura, mediastinum | 1 | 1 | 2 |
| 11 | 29 | Male | 80 | FP | Pleura, lung, bone, orbit | 2 | 6 | 2 |
| 12 | 28 | Male | 80 | FP | Pleura, mediastinum, bone marrow, muscle, pancreas, retroperitoneum, subcutaneous | 2 | 3 | 2 |
| 13 | 24 | Female | 80 | FP | Lung, pelvis | 2 | 5 | 2 |
| Summary* | 18 (8–29) | 6 Male/ 7 Female | 90 (70–100) | 6 FN ERMS/ 7 FP ARMS | 2 Localized$/ 11 Metastatic | 2 (1–5) | 3 (1–6) | 7 DL1/ 6 DL2 |
Not evaluable for toxicity;
Median (range) reported for age, performance score, prior courses of XRT, and prior # systemic therapies;
Local relapse of primary tumor. Abbreviations: XRT = radiation, FN = fusion negative, FP = fusion positive; DL= dose level
A total of 28 treatment cycles were administered and the median number of cycles per patient was two (range 1–6). Patients came off protocol therapy for disease progression (n=9), patient’s decision to withdraw from treatment (n=1), and changes in patient’s condition that precluded further treatment per investigator’s judgment (n=3).
Toxicity and MTD
Seven patients were enrolled on DL1, of whom one was not evaluable for MTD determination for receiving <85% of the dasatinib dose for cycle 1. Of the six patients evaluable for MTD determination, one developed a DLT of grade 3 diarhea attributed to dasatinib during cycle 1, and the patient came off protocol therapy per family preference. Six patients were enrolled at DL2, one of whom came off protocol therapy immediately after the first dose of dasatinib due to intratumoral hemorrhage not attributable to protocol therapy. This patient had a rapidly progressing head and neck tumor with an intraoral component that began bleeding due to local trauma from the teeth within several hours of the first dose of dasatinib. Of the five patients who were evaluable for MTD determination at DL2, there were two DLTs. One patient developed grade 3 pneumonitis and hypoxia possibly attributed to ganitumab and dasatinib that required interruption of protocol therapy and treatment with steroids. Of note, this patient had received lung radiation 8 weeks prior to starting study treatment. The patient subsequently resumed treatment with dose reductions of both ganitumab and dasatinib, but discontinued therapy prior to cycle 3 per investigator judgement due to concern for worsening radiographic pneumonitis. The other patient had grade 3 hematuria possibly attributed to dasatinib, for which dasatinib was held, and came off therapy for disease progression without resuming therapy. This patient had a history of pelvic irradiation and recent intermitttent hematuria thought to be related to chemotherapy-induced cystitis. Based on these data, DL1 of once daily dasatinib at 60mg/m2/dose (max 100 mg) combined with 18 mg/kg ganitumab every 2 weeks was determined to be the MTD (Figure 1 and Table 2).
Figure 1:
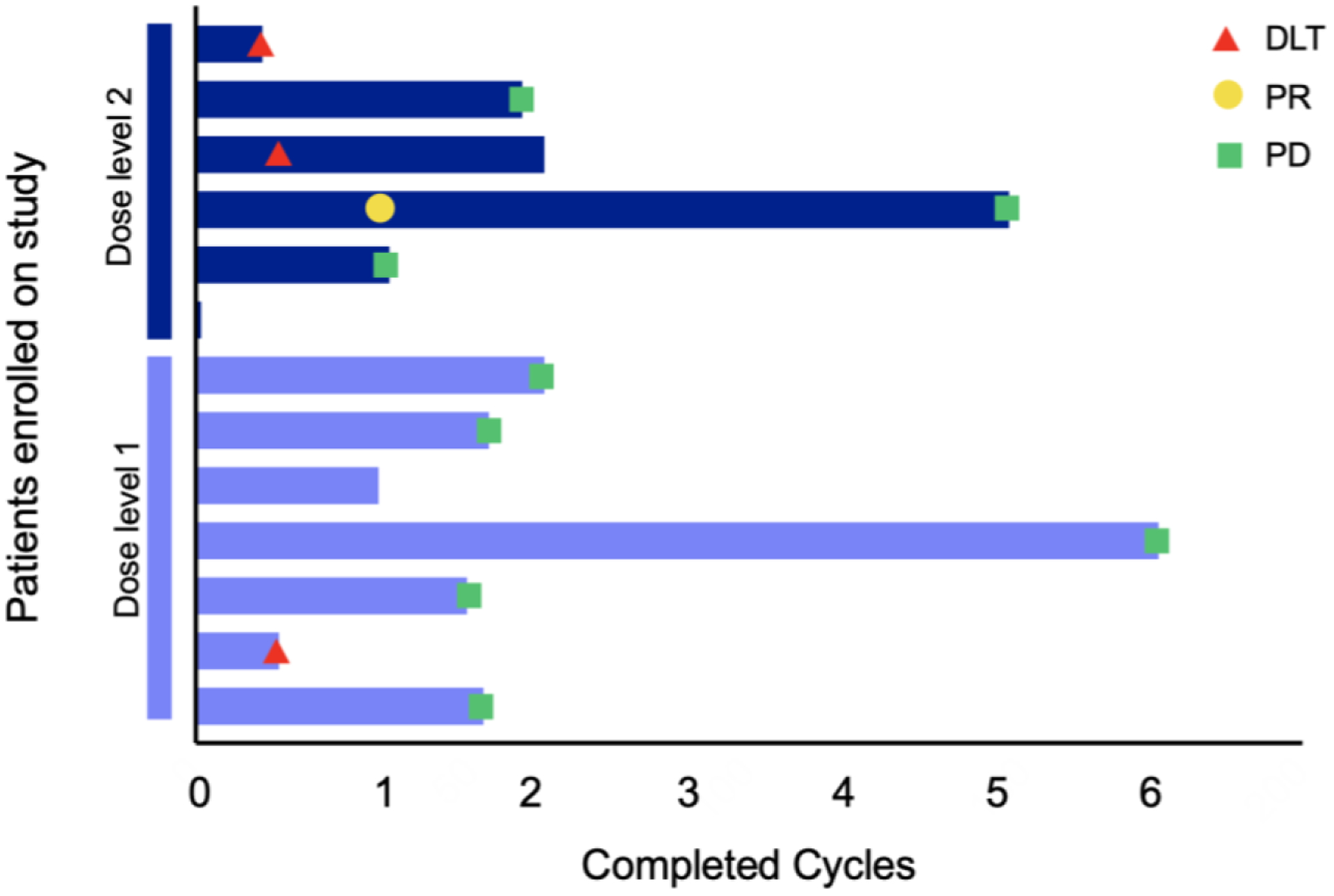
Swimmer plot depicting summary of treatment duration in cycles, timing of dose-limiting toxicities (DLT) (red), and clinical outcomes of partial reponse (PR) (yellow) and progressive disease (PD) (green) for patients treated at dose level 1 (light blue) and dose level 2 (dark blue).
Table 2:
Adverse events (AE) seen in >10% of patients that are possibly, probably, or definitively attributed to at least one of the study drugs (ganitumab or dasatinib) across all 28 cycles in 13 patients (highest grade reported for each patient across all treatment cycles)
| Dose level 1 (N) | Dose level 2 (N) | Total # patients | ||||||
|---|---|---|---|---|---|---|---|---|
| Attributed to | Grade | Grade | ||||||
| 1 | 2 | 3 | 1 | 2 | 3 | |||
| Non- hematological toxicities | ||||||||
| Dasatinib only | Abdominal distension | 2 | 1 | 1 | 4 | |||
| Diarrhea | 2 | 1 | 1 | 4 | ||||
| Edema | 3 | 3 | ||||||
| Hypocalcemia | 1 | 3 | 1 | 1 | 6 | |||
| Hypokalemia | 1 | 1 | 1 | 1 | 4 | |||
| Hypomagnesemia | 2 | 2 | ||||||
| Hypophosphatemia | 2 | 3 | 1 | 1 | 7 | |||
| Pericardial effusion | 2 | 1 | 3 | |||||
| Rash, acneiform | 1 | 2 | 3 | |||||
| Dasatinib and/or ganitumab * | Abdominal pain | 1 | 1 | 2 | ||||
| ALT increase | 3 | 3 | ||||||
| Anorexia | 2 | 2 | 4 | |||||
| AST increase | 3 | 2 | 5 | |||||
| Cough | 2 | 2 | ||||||
| Dyspnea | 1 | 1 | 1 | 3 | ||||
| Fatigue | 2 | 3 | 5 | |||||
| Headache | 1 | 1 | 1 | 3 | ||||
| Hyponatremia | 2 | 1 | 3 | |||||
| Nausea | 3 | 1 | 1 | 1 | 6 | |||
| Pleural effusion | 1 | 2 | 3 | |||||
| Pleuritic pain | 2 | 2 | ||||||
| Vomiting | 3 | 1 | 1 | 5 | ||||
| Hematological toxicities | ||||||||
| Dasatinib and/or ganitumab * | Anemia | 1 | 5 | 2 | 1 | 1 | 10 | |
| Lymphocyte count decrease | 1 | 3 | 1 | 1 | 2 | 8 | ||
| Neutrophil count decrease | 1 | 1 | 2 | |||||
| Platelet count decrease | 6 | 1 | 3 | 1 | 1 | 12 | ||
| White blood cell decrease | 1 | 1 | 2 | 4 | ||||
N= number of patients, AST = aspartate amino transferase, ALT= alanine aminotransferase
At least 1 patient with toxicity attributed to dasatinib only, ganitumab only, or both. AEs only attributed to ganitumab included hypertension, hypotension, hypoxia, and tinnitus (N= 1 for all AEs).
Hematologic and non-hematologic AEs including laboratory abnormalities that were assessed to be possibly, probably or definitely attributed to study agents are summarized in Table 2 and Supplemental Tables S2 and S3. The most common toxicities observed were grade 1–2 hematologic toxicities which, in most cases, did not require transfusion or growth factor support and grade 1 and 2 electrolyte abnormalities. Non-dose-limiting AEs seen in >30% patients that were at least possibly associated with either ganitumab, dasatinib, or both were hematologic (including anemia, thrombocytopenia, and lymphopenia), nausea, vomiting, fatigue, anorexia, and elevated asparatate aminotransferase. Non-dose-limiting AEs seen in >30% patients that were attributed only to dasatinib were abdominal distention, hypocalcemia, hypophosphatemia, and hypokalemia. Hypertension, hypotension, hypoxia, and tinnitus, were the AEs attributed only to ganitumab, and none of these were seen in >30% of patients as each AE occurred in only one patient. Grade 3 non-dose-limiting AEs attributed to both dasatinib and ganitumab were hematologic and seen in three patients (anemia, neutropenia, and lymphopenia in one patient each). Grade 3 AEs attributed to dasatinib alone included thrombocytopenia, lymphopenia, and hypokalemia in one patient, and hypokalemia and hypophosphatemia in another patient. One patient had grade 3 vomiting attributed to ganitumab alone. There were no deaths or grade 4 toxicities that were at least possibly attributed to either study agent.
Response
Nine patients were evaluable for response. One patient had a confirmed PR per RECIST v1.1 that lasted for four cycles. FDG-PET and CT imaging showed a substantial decrease in previously extensive pleural-based disease with only a small focus of residual tumor remaining (Figure 2). A second patient had SD for six cycles. One patient had progressive disease (PD) at the end of cycle 1 and five additional patients had PD during or at the end of cycle 2 (Figure 1). Additionally, one patient had a best response of SD at the end of cycle 2, however came off protocol therapy per investigator judgement given imaging findings concerning for ongoing pneumonitis. By the Kaplan-Meier method, median PFS for the entire cohort was 1.9 months (95% CI: 1.1–2.0 months) and was similar for DL1 and DL2 (1.9 and 1.6 months, respectively). The 3-month PFS probability for the entire cohort was 15.4% (95% CI: 2.5–38.8%) (Supplemental Figure S1).
Figure 2:
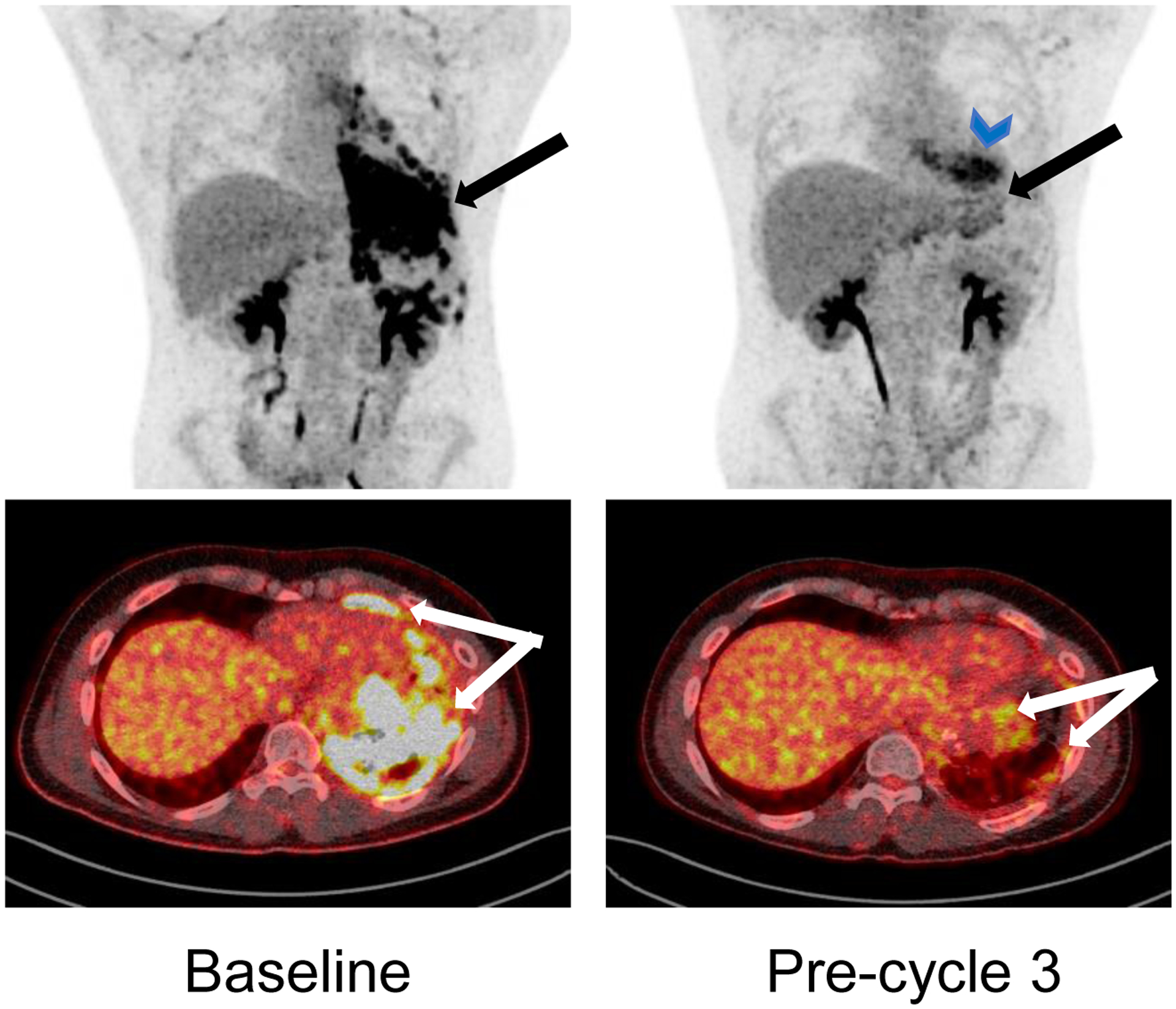
Coronal and axial 18F-Flurodeoxyglucose positron emission tomography (FDG-PET) scan images for patient who had confirmed partial response. Left panel shows the disease extent at baseline (pre-cycle 1) and the right panel shows the response at the pre-cycle 3 timepoint. The large conglomerate of highly FDG avid masses along the pleural surface are significantly reduced. The blue arrowhead symbol shows physiological uptake in cardiac muscle.
Correlative Studies
Summary genomic findings for tumor/normal sequencing (3/12 targeted panel; 9/12 whole exome seq; 2/12 whole genome seq; 10/12 RNAseq) demonstrate that the study population was enriched for patients with unfavorable molecular features, relative to the known incidence of such features at upfront diagnois. PAX3 fusions were identified in seven patients (6/12 patients with PAX3-FOXO1; 1/12 patients with tumor with a PAX3-Cryptic fusion that had been identified as fusion-negative at enrollment) and MYOD1 L122R point mutations were identified in two patients (2/5 fusion-negative patients) (Figure 3a). This is consistent with the known predilection for therapeutic failure in patients with these lesions.33,34 Analysis of target expression revealed focal genomic amplification of the IGF1R gene in one patient, but it was not correlated with response. Gene expression of IGF1R and YES1 were universal but heterogeneous across the population and neither was correlated with response.
Figure 3: Genomic studies of correlative samples.
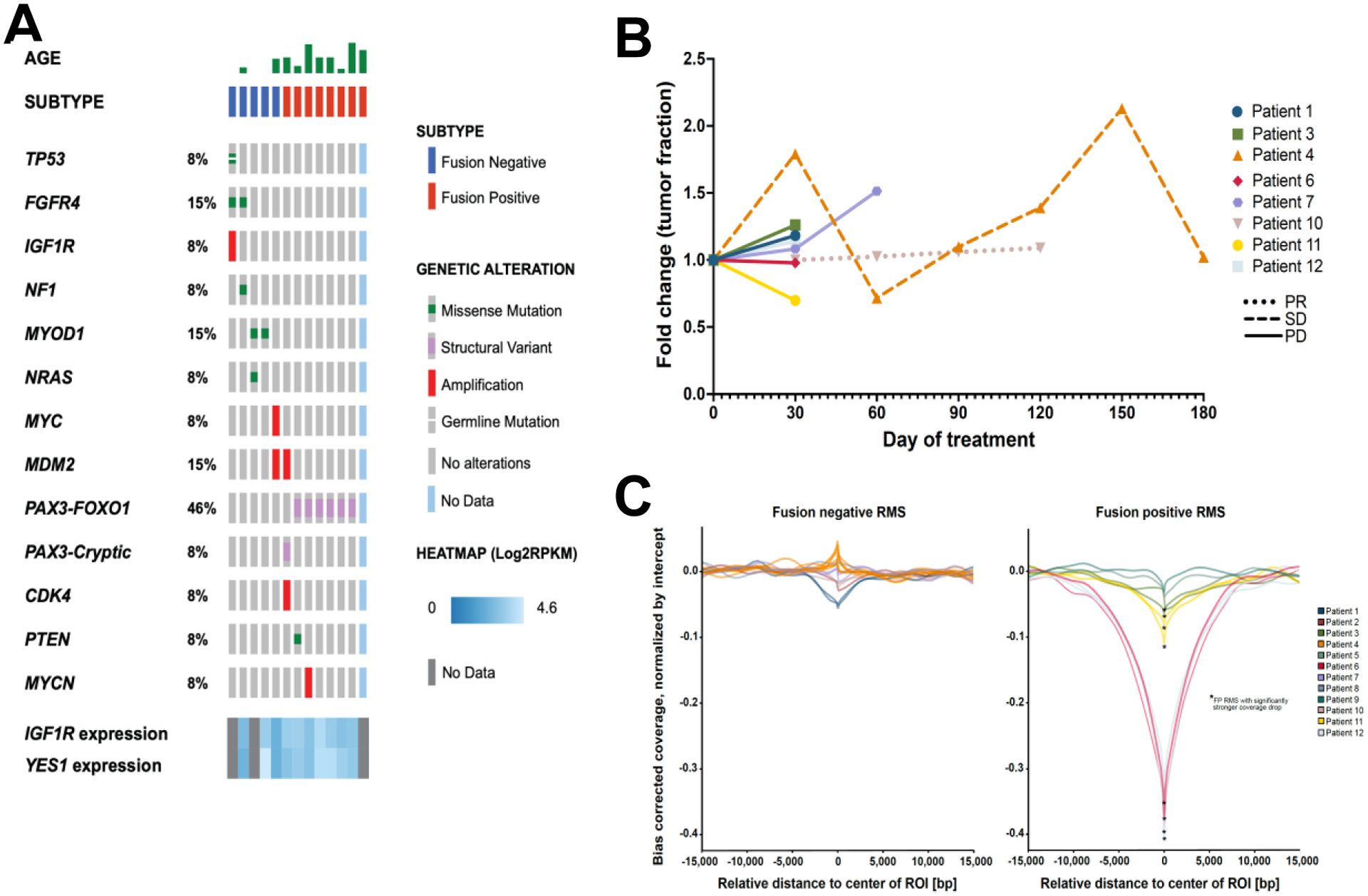
(A) Oncoprint showing the variant genetic alterations identified in the study cases. (B) Calculated tumor fraction fold change detected in cfDNA over time. (C) Comparison of fusion-negative and fusion-positive samples for calculated coverage of known PAX3-FOXO1 binding sites.
ctDNA was detected above the 3% threshold27 by copy number alteration (CNA) in 83% of patients (10/12) and in 71% of all plasma samples (20/28), including 73% of pre-treatment samples (8/11; patient 10’s pre-treatment sample was unavailable for analysis). Median tumor fraction was 13.2% (interquartile range (IQR), 2.7% to 25.2) (Supplemental Table S4). Serial cfDNA samples on treatment showed increasing tumor fractions in 80% (4/5 patients) of patients with PD (Figure 3b). In contrast, patients who experienced SD or PR did not show increases in tumor fractions. Specifically, patient 4, who had SD through cycle 6, had fluctuating tumor fractions but with a mode approximating baseline; patient 10, who had PR through cycle 5, had stable tumor fraction at all available time points where ctDNA was detected; and patient 11, who had SD through cycle 2 but was removed from therapy due to toxicity, had decreasing tumor fraction from baseline. Chromosomal arm-level analysis of CNA in available serial samples demonstrates that both patient 4 (prolonged SD) and patient 10 (prolonged PR) initially maintain relative genomic stability but, preceding their PD, develop new loci with copy number gains (Supplemental Figure S2). Patient 4 developed chromosome 7p (day 90), and 12q (day 120) amplifications and patient 10 developed chromosome 1q (day 120), 8 (day 120) and 20 (day 120) amplifications. Patient 4’s acquired CNA were captured in all subsequent timepoints; patient 10 had no cfDNA timepoints post-day 120.
To interrogate the ability of cfDNA to detect the signature of PAX3 fusions, we inferred nucleosome footprints using cfDNA sequencing coverage signatures28,29 at PAX3-FOXO1 associated super enhancer sites.30 The fusion status of tumors with a PAX3-FOXO1 fusion was correctly classified from the plasma in 67% of patients (4/6, patient 13 did not have plasma samples for analysis) and 80% of cfDNA samples (8/10) (Figure 3c). Four of the patients with fusion-positive disease had serial samples collected, and there was no signficant change in their PAX3-FOXO1 associated signature following treatment (0/4 samples). Finally, there were no sustained detectable changes in nucleosome footprints at YES1 and IGF1R regulatory motifs31,32 between pre-treatment and on-treatment samples (Supplemental Table S5).
Discussion
In this phase I study for patients with relapsed or refractory embryonal or alveolar RMS, we established the recommended phase II dose of the combination of ganitumab (18 mg/kg every two weeks) and dasatinib (60 mg/m2 daily). The most commonly observed toxicities were mild hematologic toxicity, electrolyte disturbances, elevated transaminases, nausea, vomiting, and fatigue. These were expected and consistent with previously reported toxicities of each of these agents from prior early phase studies of single agent ganitumab or dasatinib in solid tumors.16,17,22,23,35–38 In our study, we did not observe hyperglycemia in any patient, although this is a commonly reported AE in studies using ganitumab and other antibodies targeting IGF-1R.16–21,38–43 The absence of this finding in our study may be due to the small number of patients evaluated or the relatively young age of the cohort.
DLTs in our study included diarrhea, hematuria, and pneumonitis. Both diarrhea and bleeding have been reported as DLTs in multiple prior studies of dasatinib in patients with solid tumors.22,23,35,36 Although pneumonitis has not been previously reported in single agent studies of ganitumab, a recent study evaluating the addition of ganitumab in combination with multiagent chemotherapy and radiation in patients with newly diagnosed metastatic Ewing sarcoma reported a five-fold increase in pneumonitis post-radiation in the ganitumab-containing arm compared to the control arm.44 In addition, two other studies using different IGF-1R antibodies as part of combination regimens have reported pneumonitis as an AE. A phase I study of the IGF-1R antibody cixutumumab plus the mTOR inhibitor temsirolimus in patients with metastatic prostate cancer, reported grade 1 or 2 pneumonitis in 73% of patients treated at higher temsirolimus doses.45 Another study testing the combination of the IGF-1R antibody dalotuzumab (D) plus the mTOR inhibitor ridaforolimus (R) and the aromatase inhibitor exemestane (E) in patients with breast cancer, reported pneumonitis in both the D/R/E and R/E arms of the study, although it was more common in the R/E arm (22% vs. 5%).46 Other combination studies using ganitumab have not reported pneumonitis.18–21,39–43,47,48 Pneumonitis has also been reported in patients with solid tumors receiving dasatinib as part of a drug combination, for example, in a study evaluating dasatinib plus gemcitabine and the EGFR inhibitor cetuximab which reported a grade 5 pneumonitis event in a patient treated with the triple regimen.49 Together, these findings suggest that an increased risk for pneumonitis may be a rare class effect of IGF-1R antibodies when combined with other therapies, including TKIs and prior lung radiation. Furthermore, the timing of radiation may play a role in the risk for developing pneumonitis while being treated with this regimen.
Of the nine patients in this heavily pretreated population who were evaluable for response, one had a PR sustained for four cycles and another had SD for six cycles, yielding an objective response rate of 11.1% and a disease control rate of 22.2% at five months. Although there are no prior studies of ganitumab in patients with relapsed RMS, six studies using other IGF-1R antibodies as single agents that included RMS patients have reported similar objective response rates of 0–5% and disease control rates of 0–23.5% in those patients.10–13,50,51 The limited activity of IGF-1R therapies in this population is known, yet a reliable predictive biomarker to identify the likeliest responders remains elusive. Indeed, we did not observe a correlation between gene expression of IGF1R and disease response, which, while consistent with prior studies reporting a lack of correlation between tumor expression of IGF-1R and response to IGF-1R-targeting agents in sarcoma, is a limitation to this study.52,53 An additional limitation is the lack of serial biopsy specimens to track IGF-1R and YES expression over time. Data such as this might have yielded useful insights into pattens of response or resistance. In our cohort, both patients who experienced clinical benefit had fusion-negative ERMS. In the two prior studies that reported at least partial data on RMS subtype,12,50 the single patient with a PR also had embryonal histology. Although the numbers are small, this may suggest that patients with fusion-negative RMS are more likely to respond to IGF-1R targeting than those with fusion-positive disease.
The initial rationale for this study was the preclinical observation that dasatinib prolonged disease control when added to IGF-1R targeting therapies in preclinical models. Of the five prior studies using single agent IGF-1R targeting agents that included patients with relapsed RMS, two reported disease-specific PFS data demonstrating median PFS of 5.7 weeks and 6.1 weeks in the RMS cohort. Among patients who experienced disease control in these studies, the maximum duration of disease control was 25 weeks (n=1) and 42 weeks (n=1) respectively.11,12 In a third study that did not report median PFS, one patient experienced a prolonged PR for 40 weeks and three others had SD for 20, 28 and 88 weeks.13 Median PFS in our study was 1.9 months (~8 weeks) with a maximum disease control duration of 25 weeks. However, we were unable to obtain additional phase II response data due to early study closure from loss of ganitumab availability. Thus, the limited data collected on duration of response in this study is insufficient to determine whether this combination regimen provides longer disease control than single-agent IGF-1R antibody therapy alone.
In this study we used cfDNA sequencing to both monitor disease response and confirm fusion status using a single sequencing assay. To our knowledge, this is the first report of prospective use of cfDNA for these two objectives as part of a clinical trial. cfDNA is a promising, non-invasive tool for cancer detection, risk stratification, and response evaluation.54–70 In pediatric solid tumors, such as sarcomas, the use of cfDNA is of particular interest given the difficulties associated with obtaining serial biopsies.71 In our study, we determine that cfDNA is detectable in plasma from RMS patients, demonstrate that dynamic changes in tumor fraction are correlated with clinical response in most patients, and accurately infer PAX fusion status using changes in sequencing coverage at associated super-enhancers. These findings, along with prior work reporting the detection of ctDNA in RMS patients28,60,69,72,73 and the use of cell free reduced bisulphite sequencing (cfRRBS) and nucleosome footprints to infer RMS fusion status from cfDNA,28,69,74 collectively support the incorporation of cfDNA analysis into future pediatric RMS trials. The use of these assays may provide the foundation for introducing response adaptive therapies for RMS, as is now standard for pediatric hematologic malignancies.75,76
In addition, we demonstrated the feasibility of leveraging cfDNA fragment and sequencing coverage discrepancies to infer transcriptome profiles and, when measured serially, to monitor tumor clonal evolution. Recent studies have demonstrated that non-random regional differences in coverage and cfDNA fragment length reflect nucleosome occupancy and chromatin availability at promoters and transcription factor binding sites.28,77–80 Changes in coverage at regions of interest, therefore, can be used to infer gene expression and transcription factor binding.77,78 Although we did not observe differences in chromatin availability for pre-treatment and on-treatment YES1 and IGF-1R regulatory motifs, we did detect changes in genomic profiles from cfDNA over time. We observed that the patients with prolonged PR or SD developed reproducible copy number gains in specific loci prior to developing radiographic progression, which may represent selection for resistant subclones. For example, chromosome 8 amplification was observed in patient 10, and this has been associated with more aggressive disease in other sarcomas.60,81 Generalization of these findings are limited given this trial’s small sample size, and future studies are required for validation. However, this type of data may help to elucidate mechanisms of drug resistance in RMS, which have remained largely elusive due to difficulties with obtaining serial tissue samples in children.
Finally, this trial serves as an example of how the current system of clinical development for oncology drugs disenfranchises children. This study was originally designed as a phase I/II study with a phase I primary endpoint to determine MTD and phase II primary endpoints to determine the fraction of patients with clinical response (CR and PR) by RECIST and to estimate the fraction of patients that were without progression at 6 months. However, after ganitumab became unavailable mid-study, the phase II part of this study had to be terminated. Hence a full evaluation of this combination will remain incomplete, and we are unable to assess whether the strong preclinical data and some early signals of potential activity observed in the phase I component would translate to meaningful response rate or clinical benefit for patients. Further, since ganitumab was the only remaining IGF-1R antibody available for use as a clinical anticancer agent, there is currently no path forward to conduct follow-up studies testing dasatinib plus other agents blocking IGF-1R, nor studies testing ganitumab plus other agents, despite the established safety and positive preclinical data for such a combination in RMS or other pediatric sarcomas.82,83
Unfortunately, this lack of access to potentially active drugs for pediatric oncology clinical trials is not unique to IGF-1R-targeting agents and is the result of several factors, including a substantial lag time resulting in pediatric trials being initiated only once adult trials are near or at completion.84–86 In the case of IGF-1R-targeting agents, among others, disappointing results in adult trials led to discontinuation of pharmaceutical development programs before the drugs could be adequately tested in specific pediatric settings. This is frequently the case even if there may be evidence of greater activity in a particular pediatric cancer, since the pediatric pharmaceutical market is generally not profitable.87 As a result, there is a dearth of novel agents that have shown sufficient preclinical activity in RMS and are clinically available to study in children.88 This lack of access to potentially effective novel agents remains a major impediment to progress for the field and underscores the need for more emphasis on drug development for targets specific to pediatric cancers.
In summary, the results of this trial suggest that a combinatorial approach to targeting IGF-1R and YES is safe and worthy of further study for patients with rhabdomyosarcoma. Future availability of IGF-1R targeting agents is necessary for further investigation of this and other combinations in pediatric sarcomas.
Supplementary Material
Acknowledgments:
S. Akshintala, R. Sundby, D. Bernstein, J. Glod, R. Kaplan, M. Yohe, A. Gross, J. Derdak, H. Lei., A. Pan, E. Dombi, I. Palacio-Yance, K. Herrera, M. Miettinen, S. Steinberg, B. Widemann, J. Shern, and C. Heske are supported by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute (NCI). This content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or NCI. L. Mascarenhas and F. Navid are supported by the Names Family Foundation and F. Navid is supported by the USC Norris Comprehensive Cancer Center Core Grant (5P30CA014089–45). Drug supply for this trial was supported through a Cooperative Research and Development Agreement between NCI, ImmunityBio, and Bristol Meyers Squibb. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). The authors would like to acknowledge Amanda Carbonell, Melissa Spencer, Kim Walker, Elaine Thomas, and Abrahm Levi for their help with the study conduct. We are extremely grateful to the patients and their families who participated in this trial.
Footnotes
Conflict of Interest Statement: The authors declare no potential conflicts of interest.
References
- 1.Dagher R, Helman L: Rhabdomyosarcoma: An overview. The Oncologist 4:34–44, 1999 [PubMed] [Google Scholar]
- 2.Skapek SX, Ferrari A, Gupta AA, et al. : Rhabdomyosarcoma. Nat Rev Dis Primers 5:1, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davicioni E, Anderson MJ, Finckenstein FG, et al. : Molecular classification of rhabdomyosarcoma--genotypic and phenotypic determinants of diagnosis: a report from the Children’s Oncology Group. Am J Pathol 174:550–64, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pappo AS, Anderson JR, Crist WM, et al. : Survival after relapse in children and adolescents with rhabdomyosarcoma: A report from the Intergroup Rhabdomyosarcoma Study Group. J Clin Oncol 17:3487–93, 1999 [DOI] [PubMed] [Google Scholar]
- 5.Rudzinski ER, Anderson JR, Chi YY, et al. : Histology, fusion status, and outcome in metastatic rhabdomyosarcoma: A report from the Children’s Oncology Group. Pediatr Blood Cancer 64, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El-Badry OM, Minniti C, Kohn EC, et al. : Insulin-like growth factor II acts as an autocrine growth and motility factor in human rhabdomyosarcoma tumors. Cell Growth Differ 1:325–31, 1990 [PubMed] [Google Scholar]
- 7.Kalebic T, Blakesley V, Slade C, et al. : Expression of a kinase-deficient IGF-I-R suppresses tumorigenicity of rhabdomyosarcoma cells constitutively expressing a wild type IGF-I-R. Int J Cancer 76:223–7, 1998 [DOI] [PubMed] [Google Scholar]
- 8.Wang W, Kumar P, Wang W, et al. : Insulin-like growth factor II and PAX3-FKHR cooperate in the oncogenesis of rhabdomyosarcoma. Cancer Res 58:4426–33, 1998 [PubMed] [Google Scholar]
- 9.Cao L, Yu Y, Darko I, et al. : Addiction to elevated insulin-like growth factor I receptor and initial modulation of the AKT pathway define the responsiveness of rhabdomyosarcoma to the targeting antibody. Cancer Res 68:8039–48, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner LM, Fouladi M, Ahmed A, et al. : Phase II study of cixutumumab in combination with temsirolimus in pediatric patients and young adults with recurrent or refractory sarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer 62:440–4, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schoffski P, Adkins D, Blay JY, et al. : An open-label, phase 2 study evaluating the efficacy and safety of the anti-IGF-1R antibody cixutumumab in patients with previously treated advanced or metastatic soft-tissue sarcoma or Ewing family of tumours. Eur J Cancer 49:3219–28, 2013 [DOI] [PubMed] [Google Scholar]
- 12.Pappo AS, Vassal G, Crowley JJ, et al. : A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a Sarcoma Alliance for Research Through Collaboration study. Cancer 120:2448–56, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weigel B, Malempati S, Reid JM, et al. : Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: a report from the Children’s Oncology Group. Pediatr Blood Cancer 61:452–6, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wan X, Yeung C, Heske C, et al. : IGF-1R Inhibition Activates a YES/SFK Bypass Resistance Pathway: Rational Basis for Co-Targeting IGF-1R and Yes/SFK Kinase in Rhabdomyosarcoma. Neoplasia 17:358–66, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calzone FJ, Cajulis E, Chung YA, et al. : Epitope-specific mechanisms of IGF1R inhibition by ganitumab. PLoS One 8:e55135, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tolcher AW, Sarantopoulos J, Patnaik A, et al. : Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1. J Clin Oncol 27:5800–7, 2009 [DOI] [PubMed] [Google Scholar]
- 17.Tap WD, Demetri G, Barnette P, et al. : Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol 30:1849–56, 2012 [DOI] [PubMed] [Google Scholar]
- 18.Rosen LS, Puzanov I, Friberg G, et al. : Safety and pharmacokinetics of ganitumab (AMG 479) combined with sorafenib, panitumumab, erlotinib, or gemcitabine in patients with advanced solid tumors. Clin Cancer Res 18:3414–27, 2012 [DOI] [PubMed] [Google Scholar]
- 19.Kindler HL, Richards DA, Garbo LE, et al. : A randomized, placebo-controlled phase 2 study of ganitumab (AMG 479) or conatumumab (AMG 655) in combination with gemcitabine in patients with metastatic pancreatic cancer. Ann Oncol 23:2834–2842, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Fuchs CS, Azevedo S, Okusaka T, et al. : A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: the GAMMA trial. Ann Oncol 26:921–927, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tabernero J, Chawla SP, Kindler H, et al. : Anticancer activity of the type I insulin-like growth factor receptor antagonist, ganitumab, in combination with the death receptor 5 agonist, conatumumab. Target Oncol 10:65–76, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demetri GD, Lo Russo P, MacPherson IR, et al. : Phase I dose-escalation and pharmacokinetic study of dasatinib in patients with advanced solid tumors. Clin Cancer Res 15:6232–40, 2009 [DOI] [PubMed] [Google Scholar]
- 23.Aplenc R, Blaney SM, Strauss LC, et al. : Pediatric phase I trial and pharmacokinetic study of dasatinib: a report from the children’s oncology group phase I consortium. J Clin Oncol 29:839–44, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franceschi E, Stupp R, van den Bent MJ, et al. : EORTC 26083 phase I/II trial of dasatinib in combination with CCNU in patients with recurrent glioblastoma. Neuro Oncol 14:1503–10, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong DS, Choe JH, Naing A, et al. : A phase 1 study of gemcitabine combined with dasatinib in patients with advanced solid tumors. Invest New Drugs 31:918–26, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Investigator’s Brochure. Dasatinib. BMS-354825. Bristol-Myers Squibb.Version No: 10. Version Date: November 2010, [Google Scholar]
- 27.Adalsteinsson VA, Ha G, Freeman SS, et al. : Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun 8:1324, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peneder P, Stuetz AM, Surdez D, et al. : Multimodal analysis of cell-free DNA whole-genome sequencing for pediatric cancers with low mutational burden. Nature Communications 12, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peneder P, Bock C, Tomazou EM: LIQUORICE: detection of epigenetic signatures in liquid biopsies based on whole-genome sequencing data. Bioinform Adv 2:vbac017, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gryder BE, Yohe ME, Chou HC, et al. : PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov 7:884–899, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie X, Rigor P, Baldi P: MotifMap: a human genome-wide map of candidate regulatory motif sites. Bioinformatics 25:167–74, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daily K, Patel VR, Rigor P, et al. : MotifMap: integrative genome-wide maps of regulatory motif sites for model species. BMC Bioinformatics 12:495, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Missiaglia E, Williamson D, Chisholm J, et al. : PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol 30:1670–7, 2012 [DOI] [PubMed] [Google Scholar]
- 34.Shern JF, Selfe J, Izquierdo E, et al. : Genomic Classification and Clinical Outcome in Rhabdomyosarcoma: A Report From an International Consortium. J Clin Oncol 39:2859–2871, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson FM, Agrawal S, Burris H, et al. : Phase 1 pharmacokinetic and drug-interaction study of dasatinib in patients with advanced solid tumors. Cancer 116:1582–91, 2010 [DOI] [PubMed] [Google Scholar]
- 36.Takahashi S, Miyazaki M, Okamoto I, et al. : Phase I study of dasatinib (BMS-354825) in Japanese patients with solid tumors. Cancer Sci 102:2058–64, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Murakami H, Doi T, Yamamoto N, et al. : Phase 1 study of ganitumab (AMG 479), a fully human monoclonal antibody against the insulin-like growth factor receptor type I (IGF1R), in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol 70:407–14, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strosberg JR, Chan JA, Ryan DP, et al. : A multi-institutional, phase II open-label study of ganitumab (AMG 479) in advanced carcinoid and pancreatic neuroendocrine tumors. Endocr Relat Cancer 20:383–90, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robertson JF, Ferrero JM, Bourgeois H, et al. : Ganitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: a randomised, controlled, double-blind, phase 2 trial. Lancet Oncol 14:228–35, 2013 [DOI] [PubMed] [Google Scholar]
- 40.Cohn AL, Tabernero J, Maurel J, et al. : A randomized, placebo-controlled phase 2 study of ganitumab or conatumumab in combination with FOLFIRI for second-line treatment of mutant KRAS metastatic colorectal cancer. Ann Oncol 24:1777–1785, 2013 [DOI] [PubMed] [Google Scholar]
- 41.Glisson B, Besse B, Dols MC, et al. : A Randomized, Placebo-Controlled, Phase 1b/2 Study of Rilotumumab or Ganitumab in Combination With Platinum-Based Chemotherapy as First-Line Treatment for Extensive-Stage Small-Cell Lung Cancer. Clin Lung Cancer 18:615–625 e8, 2017 [DOI] [PubMed] [Google Scholar]
- 42.Vlahovic G, Meadows KL, Hatch AJ, et al. : A Phase I Trial of the IGF-1R Antibody Ganitumab (AMG 479) in Combination with Everolimus (RAD001) and Panitumumab in Patients with Advanced Cancer. Oncologist 23:782–790, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Konecny GE, Hendrickson AEW, Davidson TM, et al. : Results of TRIO-14, a phase II, multicenter, randomized, placebo-controlled trial of carboplatin-paclitaxel versus carboplatin-paclitaxel-ganitumab in newly diagnosed epithelial ovarian cancer. Gynecol Oncol 163:465–472, 2021 [DOI] [PubMed] [Google Scholar]
- 44.DuBois S, Glade Bender J, Buxton A, et al. : Randomized Phase 3 Trial of Ganitumab Added to Interval Compressed Chemotherapy for Patients with Newly Diagnosed Metastatic Ewing Sarcoma: A Report from the Children’s Oncology Group (COG). . Presented at: Connective Tissue Oncology Society Annual Meeting; November 13–16, 2019; Tokyo, Japan., 2019 [Google Scholar]
- 45.McHugh DJ, Chudow J, DeNunzio M, et al. : A Phase I Trial of IGF-1R Inhibitor Cixutumumab and mTOR Inhibitor Temsirolimus in Metastatic Castration-resistant Prostate Cancer. Clin Genitourin Cancer 18:171–178 e2, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rugo HS, Tredan O, Ro J, et al. : A randomized phase II trial of ridaforolimus, dalotuzumab, and exemestane compared with ridaforolimus and exemestane in patients with advanced breast cancer. Breast Cancer Res Treat 165:601–609, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okusaka T, Ikeda M, Fukutomi A, et al. : Safety, tolerability, pharmacokinetics and antitumor activity of ganitumab, an investigational fully human monoclonal antibody to insulin-like growth factor type 1 receptor, combined with gemcitabine as first-line therapy in patients with metastatic pancreatic cancer: a phase 1b study. Jpn J Clin Oncol 44:442–7, 2014 [DOI] [PubMed] [Google Scholar]
- 48.Van Cutsem E, Eng C, Nowara E, et al. : Randomized phase Ib/II trial of rilotumumab or ganitumab with panitumumab versus panitumumab alone in patients with wild-type KRAS metastatic colorectal cancer. Clin Cancer Res 20:4240–50, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mettu NB, Niedzwiecki D, Rushing C, et al. : A phase I study of gemcitabine + dasatinib (gd) or gemcitabine + dasatinib + cetuximab (GDC) in refractory solid tumors. Cancer Chemother Pharmacol 83:1025–1035, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olmos D, Postel-Vinay S, Molife LR, et al. : Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study. Lancet Oncol 11:129–35, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Malempati S, Weigel B, Ingle AM, et al. : Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol 30:256–62, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwartz GK, Tap WD, Qin LX, et al. : Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol 14:371–82, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chugh R, Griffith KA, Davis EJ, et al. : Doxorubicin plus the IGF-1R antibody cixutumumab in soft tissue sarcoma: a phase I study using the TITE-CRM model. Ann Oncol 26:1459–64, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Theissen J, Boensch M, Spitz R, et al. : Heterogeneity of the MYCN Oncogene in Neuroblastoma. Clinical Cancer Research 15:2085–2090, 2009 [DOI] [PubMed] [Google Scholar]
- 55.Bellini A, Bernard V, Leroy Q, et al. : Deep Sequencing Reveals Occurrence of Subclonal ALK Mutations in Neuroblastoma at Diagnosis. Clinical Cancer Research 21:4913–4921, 2015 [DOI] [PubMed] [Google Scholar]
- 56.Hayashi M, Chu D, Meyer CF, et al. : Highly Personalized Detection of Minimal Ewing Sarcoma Disease Burden from Plasma Tumor DNA. Cancer 122:3015–3015, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chaudhuri AA, Chabon JJ, Lovejoy AF, et al. : Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov 7:1394–1403, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cohen JD, Javed AA, Thoburn C, et al. : Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proceedings of the National Academy of Sciences 114:10202–10207, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cohen JD, Li L, Wang Y, et al. : Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359:926–930, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shulman DS, Klega K, Imamovic-Tuco A, et al. : Detection of circulating tumour DNA is associated with inferior outcomes in Ewing sarcoma and osteosarcoma: a report from the Children’s Oncology Group. Br J Cancer 119:615–621, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dudley JC, Schroers-Martin J, Lazzareschi DV, et al. : Detection and Surveillance of Bladder Cancer Using Urine Tumor DNA. Cancer Discov 9:500–509, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen SY, Singhania R, Fehringer G, et al. : Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 563:579-+, 2018 [DOI] [PubMed] [Google Scholar]
- 63.Chin R-I, Chen K, Usmani A, et al. : Detection of Solid Tumor Molecular Residual Disease (MRD) Using Circulating Tumor DNA (ctDNA). Molecular Diagnosis & Therapy 2019 23:3 23:311–331, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shulman DS, Crompton BD, Ds S, et al. : Using Liquid Biopsy in the Treatment of Patient with OS, Adv Exp Med Biol, 2020, pp 95–105 [DOI] [PubMed] [Google Scholar]
- 65.Cho N-Y, Park J-W, Wen X, et al. : Blood-Based Detection of Colorectal Cancer Using Cancer-Specific DNA Methylation Markers. Diagnostics 2021, Vol. 11, Page 51 11:51–51, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Szymanski JJ, Sundby RT, Jones PA, et al. : Cell-free DNA ultra-low-pass whole genome sequencing to distinguish malignant peripheral nerve sheath tumor (MPNST) from its benign precursor lesion: A cross-sectional study. PLOS Medicine 18:e1003734–e1003734, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Henriksen TVV, Tarazona N, Frydendahl A, et al. : Serial circulating tumor DNA analysis to assess recurrence risk, benefit of adjuvant therapy, growth rate and early relapse detection in stage III colorectal cancer patients. Journal of Clinical Oncology 39:3540–3540, 2021 [Google Scholar]
- 68.Pellini B, Pejovic N, Feng W, et al. : ctDNA MRD Detection and Personalized Oncogenomic Analysis in Oligometastatic Colorectal Cancer From Plasma and Urine. JCO Precision Oncology:378–388, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lak NSM, van Zogchel LMJ, Zappeij-Kannegieter L, et al. : Cell-Free DNA as a Diagnostic and Prognostic Biomarker in Pediatric Rhabdomyosarcoma. JCO Precis Oncol 7:e2200113, 2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chauhan PS, Shiang A, Alahi I, et al. : Urine cell-free DNA multi-omics to detect MRD and predict survival in bladder cancer patients. npj Precision Oncology 7:6, 2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sundby RT, Pan A, Shern JF: Liquid biopsies in pediatric oncology: opportunities and obstacles. Curr Opin Pediatr, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tombolan L, Rossi E, Binatti A, et al. : Clinical significance of circulating tumor cells and cell-free DNA in pediatric rhabdomyosarcoma. Mol Oncol 16:2071–2085, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Abbou S, Klega K, Tsuji J, et al. : Circulating Tumor DNA Is Prognostic in Intermediate-Risk Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J Clin Oncol:JCO2200409, 2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van Paemel R, De Koker A, Vandeputte C, et al. : Minimally invasive classification of paediatric solid tumours using reduced representation bisulphite sequencing of cell-free DNA: a proof-of-principle study. Epigenetics 16:196–208, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kelly KM, Cole PD, Pei Q, et al. : Response‐adapted therapy for the treatment of children with newly diagnosed high risk Hodgkin lymphoma (AHOD0831): a report from the Children’s Oncology Group. British Journal of Haematology 187:39–48, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oeffinger KC, Stratton KL, Hudson MM, et al. : Impact of Risk-Adapted Therapy for Pediatric Hodgkin Lymphoma on Risk of Long-Term Morbidity: A Report From the Childhood Cancer Survivor Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 39:2266–2275, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ulz P, Thallinger GG, Auer M, et al. : Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat Genet 48:1273–8, 2016 [DOI] [PubMed] [Google Scholar]
- 78.Ulz P, Perakis S, Zhou Q, et al. : Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat Commun 10:4666, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zviran A, Schulman RC, Shah M, et al. : Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nature Medicine 2020 26:7 26:1114–1124, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Doebley A-L, Ko M, Liao H, et al. : Griffin: Framework for clinical cancer subtyping from nucleosome profiling of cell-free DNA. medRxiv, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dehner C, Moon CI, Zhou Z, et al. : Chromosome 8 gain is associated with high-grade transformation in MPNST. JCI Insight 6:e146351, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guenther LM, Dharia NV, Ross L, et al. : A Combination CDK4/6 and IGF1R Inhibitor Strategy for Ewing Sarcoma. Clin Cancer Res 25:1343–1357, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hebron KE, Wan X, Roth JS, et al. : The combination of trametinib and ganitumab is effective in RAS-mutated PAX-fusion negative rhabdomyosarcoma models. Clin Cancer Res, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kearns P, Morland B: New drug development in childhood cancer. Curr Opin Pediatr 26:37–42, 2014 [DOI] [PubMed] [Google Scholar]
- 85.Khan T, Stewart M, Blackman S, et al. : Accelerating Pediatric Cancer Drug Development: Challenges and Opportunities for Pediatric Master Protocols. Ther Innov Regul Sci 53:270–278, 2019 [DOI] [PubMed] [Google Scholar]
- 86.Moreno L, DuBois SG, Marshall LV, et al. : How to address challenges and opportunities in pediatric cancer drug development? Expert Opin Drug Discov 15:869–872, 2020 [DOI] [PubMed] [Google Scholar]
- 87.Adamson PC, Houghton PJ, Perilongo G, et al. : Drug discovery in paediatric oncology: roadblocks to progress. Nat Rev Clin Oncol 11:732–9, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pacenta HL, Allen-Rhoades W, Langenau D, et al. : Prioritization of Novel Agents for Patients with Rhabdomyosarcoma: A Report from the Children’s Oncology Group (COG) New Agents for Rhabdomyosarcoma Task Force. J Clin Med 10, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data are available through dbGAP Accession phs003243.v1.p1. Additional data is available upon request to the corresponding author.