

Abstract
Multiple sclerosis (MS) is the most common causes of non-traumatic disability in young adults worldwide. MS pathophysiologies include the formation of inflammatory lesions, axonal damage and demyelination, and blood brain barrier (BBB) disruption. Coagulation proteins, including factor (F)XII, can serve as important mediators of the adaptive immune response during neuroinflammation. Indeed, plasma FXII levels are increased during relapse in relapsing-remitting MS patients, and previous studies showed that reducing FXII levels was protective in a murine model of MS, experimental autoimmune encephalomyelitis (EAE). Our objective was to determine if pharmacological targeting of FXI, a major substrate of activated FXII (FXIIa), improves neurological function and attenuates CNS damage in the setting of EAE. EAE was induced in male mice using murine myelin oligodendrocyte glycoprotein peptides combined with heat-inactivated Mycobacterium tuberculosis and pertussis toxin. Upon onset of symptoms, mice were treated every other day intravenously with anti-FXI antibody, 14E11, or saline. Disease scores were recorded daily until euthanasia for ex vivo analyses of inflammation. Compared to the vehicle control, 14E11 treatment reduced the clinical severity of EAE and total mononuclear cells, including CD11b+CD45high macrophage/microglia and CD4+ T cell numbers in brain. Following pharmacological targeting of FXI, BBB disruption was reduced, as measured by decreased axonal damage and fibrin(ogen) accumulation in the spinal cord. These data demonstrate that pharmacological inhibition of FXI reduces disease severity, immune cell migration, axonal damage, and BBB disruption in mice with EAE. Thus, therapeutic agents targeting FXI and FXII may provide a useful approach for treating autoimmune and neurologic disorders.
Keywords: Multiple sclerosis, EAE, Thrombin, Factor XI
Introduction
Multiple sclerosis (MS) is a neurodegenerative, demyelinating disease of the central nervous system (CNS) that affects approximately 2.2 million individuals globally (Collaborators 2019). MS is characterized by inflammatory lesions, axonal damage, and leukocyte trafficking across the blood-brain barrier (BBB). The pathophysiology of MS is believed to be caused in part by increased BBB vascular permeability, leading to the transmigration of peripheral T cells into the brain parenchyma that induce lesions and subsequent demyelination.
Recent evidence suggests that in addition to immune cells, other factors contribute to the pathophysiology of MS. In particular, clinical observations of MS patients and studies using animal models have advanced a connection between the coagulation cascade and inflammatory processes in MS (Jordan et al. 2021). The coagulation cascade is a series of enzymatic reactions in which a zymogen precursor becomes activated to its enzyme form, which then catalyzes the next reaction in the pathway, ultimately generating thrombin and forming a fibrin clot.
Two distinct activation pathways are recognized to lead to the formation of thrombin, with elements of both pathways found to be associated in the pathogenesis of MS. For example, increased levels of thrombin activity have been documented within the plasma of MS patients (Göbel et al. 2016a; Parsons et al. 2017). Likewise, studies using an animal model of MS, experimental autoimmune encephalomyelitis (EAE), suggest that thrombin activity may contribute to MS pathology by influencing BBB permeability and myelination (Ahmed et al. 2019; Davalos et al. 2014; Peeters et al. 2014). Prothrombin, the zymogen precursor of thrombin, and coagulation factor X (FX) have also been found to be elevated in the plasma of patients with MS (Göbel et al. 2016a; Yoon et al. 2015). In addition, relapsing-remitting MS patients experiencing relapse were found to have markedly higher plasma levels of coagulation factor XII (FXII), which was correlated with their disease activity (Göbel et al. 2016b). Further evidence that components of the coagulation system play a role in the MS disease state is described in studies demonstrating the presence of the blood plasma factors fibrinogen, tissue factor, and protein C inhibitor in active MS lesions and plaques (Adams et al. 2007;Gveric et al. 2003; Han et al. 2008; Ryu et al. 2015). These observations from MS patient studies are further supported by data from disease models, in which constituents of the blood coagulation system, including platelets, fibrinogen, activated coagulation factor X (FXa), FXII, as well as the proinflammatory kallikrein-kinin system have been shown to contribute to EAE pathologies (Göbel et al. 2016b, 2019; Langer et al. 2012; Merker et al. 2017).
Therapeutic strategies that inhibit thrombin activity reduce disease severity in both MS patients and EAE models. Historically, the anticoagulant heparin was used to treat MS with mixed success (Courville 1959; Maschmeyer et al. 1961). In experimental models, anticoagulation with heparin, warfarin, or rivaroxaban have been shown to ameliorate the clinical course of EAE (Han et al. 2008; Lider et al. 1989; Stolz et al. 2017). Similarly, treatments that directly target FXa (Merker et al. 2017; Stolz et al. 2017), platelets (Langer et al. 2012), fibrin formation (Davalos et al. 2012), or FXII (Göbel et al. 2011) have been shown to ameliorate EAE. Though effective in disease models, traditional anti-coagulants may pose additional risks to MS patients, in particular increased bleeding. Thus, they may be unsuitable for long-term clinical use. Strategies that target both inflammation and fibrin formation within the CNS without impairing hemostasis significantly may have therapeutic value.
Coagulation factor XI (FXI) is a major substrate of activated FXII (FXIIa), the initiator of the intrinsic pathway of coagulation. In various animal models, decreasing or eliminating FXI activity through gene knockout, pharmacologic inhibition, or antisense oligonucleotide mediated knock-down has been shown to be antithrombotic without impairing hemostasis (Gruber and Hanson 2003; Rosen et al. 2002; Schumacher et al. 2007; Tucker et al. 2009; Zhang et al. 2010). We have previously demonstrated that targeting FXI with 14E11, a mouse monoclonal antibody that selectively inhibits the activation of FXI by FXIIa and reciprocal activation of FXII by FXIa in vitro (Lorentz et al. 2018; Silasi et al. 2019), has antithrombotic effects in multiple disease models. 14E11 improves outcomes in mouse polymicrobial sepsis (Tucker et al. 2012), acute ischemic stroke (Leung et al. 2012), acute myocardial ischemia (Lorentz et al. 2018), atherosclerosis (Ngo et al. 2021), and deep vein thrombosis (Jordan et al. 2022). Additionally, the humanized version of 14E11, AB023, has been shown to reduce markers of thrombin generation and inflammation in renal failure patients undergoing hemodialysis (Lorentz 2021), underscoring the potential utility of pharmacologic inhibition of FXI in conditions where thrombin activity is increased. In the present study, we evaluated if treatment with 14E11 improves neurologic outcomes in a mouse model of EAE. We found that 14E11 improved disease scores and prevented both axonal damage and fibrin accumulation in the spinal cord. Our results show that pharmacologic inhibition of FXI reduces disease severity in EAE and suggests that contact activation of FXI through FXIIa has a pathogenic role in the progression of autoimmune disorders of the CNS.
Methods
Reagents
The anti-FXI monoclonal antibody, 14E11, was generated and purified as previously described (Cheng et al. 2010).
Animals
The animal care and procedures in this study were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the regulations of the Oregon Health & Science University Institutional Animal Care and Use Committee (National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals 2011). The male C57BL/6 mice used in this study were housed at the Portland Veterans Affairs Medical Center in the Animal Resource Facility (Portland, OR, US) according to guidelines set by the National Research Council and the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources.
EAE induction and 14E11 treatment
EAE was induced by inoculating C57BL/6 mice subcutaneously with mouse myelin oligodendrocyte glycoprotein, peptides 35–55 (MOG35–55) combined with complete Freund’s adjuvant containing heat-inactivated M. tuberculosis, as previously described (Sinha et al. 2010). On days 0 and 2 relative to immunization, mice were intraperitoneally injected with 75 ng and 200 ng of pertussis toxin (Ptx), respectively. To quantify the symptoms of EAE, a disease score was assigned to each mice daily. The scale used to assess EAE symptoms was as follows: 0, normal; 1, limp tail or mild hind limb weakness; 2, moderate hind limb weakness or mild ataxia; 3, moderately severe hind limb weakness; 4, severe hind limb weakness or mild forelimb weakness or moderate ataxia; 5, paraplegia with no more than moderate forelimb weakness; and 6, paraplegia with severe forelimb weakness or severe ataxia or moribund condition.
At the onset of clinical signs of EAE (disease score ≥ 2.5, typically between days 10–13), mice were randomized into two groups. The mice were then administered either the anti-FXI antibody, 14E11 (1 mg/kg, n = 9), or vehicle (saline, n = 7) at the same volume intravenously (Fig. 1a). The 14E11 dose level was selected based on previous work showing that administration of 14E11 at similar doses produced a sustained anticoagulant effect, as evidenced by prolonged clotting times(Cheng et al. 2010; Leung et al. 2012; Ngo et al. 2021; Tucker et al. 2012). The mice were monitored for changes in disease score by an investigator blinded to treatment status until mice were euthanized for ex vivo analyses. In accord with efforts to replace, reduce, and refine the use of animals, the saline-treated animal cohort concurrently served as a vehicle control for two treatment arms: (1) 14E11 presented herein and (2) the thrombin mutant W215A/E217A. These three cohorts were run simultaneously; the data from the vehicle cohort was previously published as part of Verbout et al. study comparing the vehicle and the W215A/E217A cohort and thus technically used herein as a historic control.
Fig. 1.
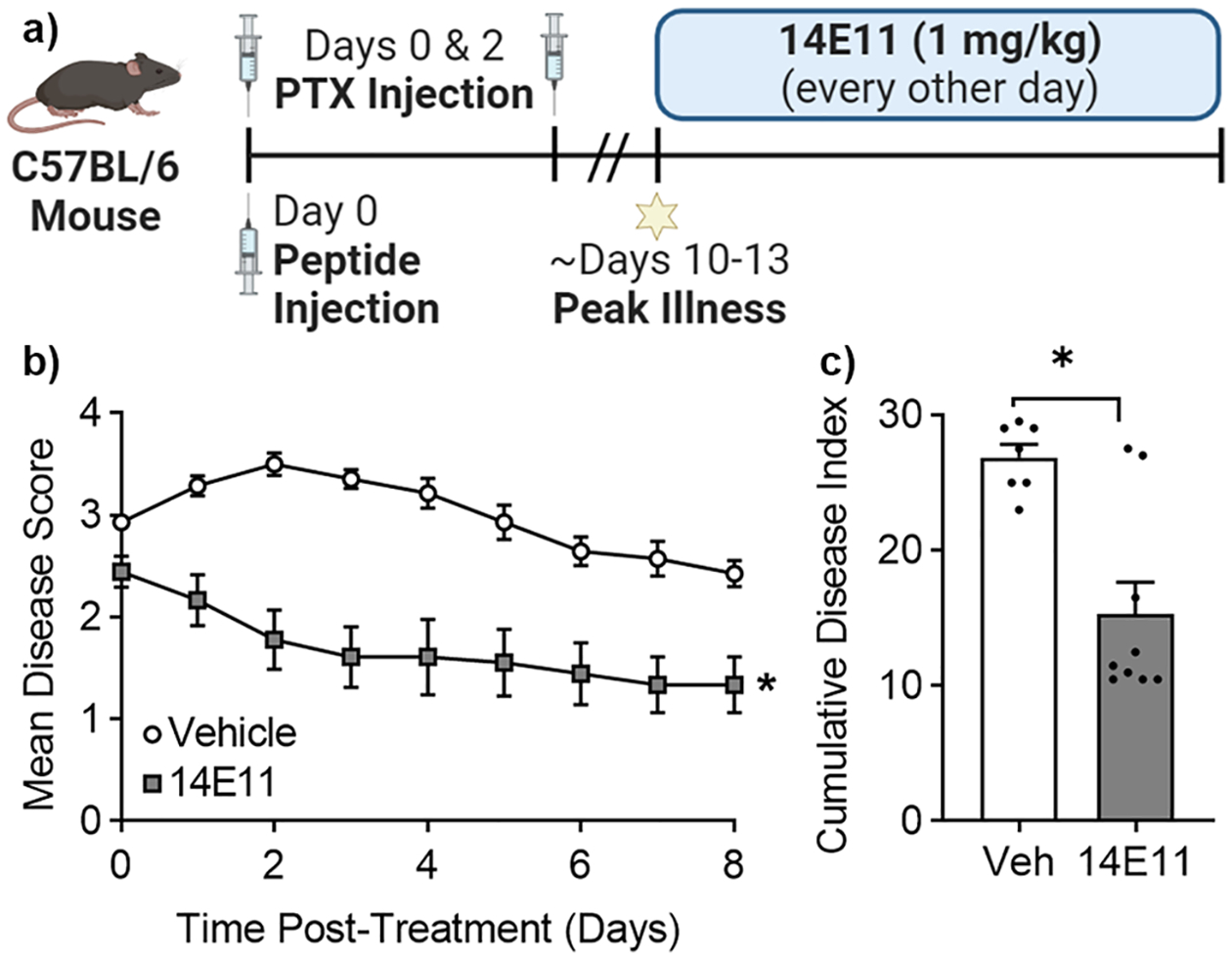
Pharmacologic targeting of FXI with 14E11 attenuates EAE symptoms. Male C57BL/6 mice immunized with MOG/CFA/Ptx were monitored and assigned a disease score daily. At peak disease onset (disease score ≥ 2.5), mAb 14E11 (1 mg/kg, i.v.) was administered every other day for four days (a). The mean disease scores (b) and the cumulative disease index (c) for 14E11-treated mice were significantly lower compared to mice treated with vehicle. Data are presented as mean ± SEM for two independent experiments, where n = 7–9. Statistical analyses were performed using Mann-Whitney tests on GraphPad Prism 9. Statistical significance is indicated by a single asterisk (*) for P ≤ 0.05
Flow cytometry
Flow cytometry was used to measure markers of inflammation in the CNS and periphery after euthanasia. To quantify the population of mononuclear cells in the CNS, Percoll density gradient centrifugation was used to isolate mononuclear cells (Bebo et al. 1996).The CNS cells (n = 3 per group) were then pooled to achieve sufficient numbers for the antibody staining protocol. To measure the activation of the macrophage subpopulation of splenocytes, spleen tissue was homogenized and single cell suspensions were prepared. Cells (1 × 106) were washed with a staining media comprised of PBS with NaN3 (0.1%) and BSA (1%) and stained with combinations of anti-CD4, anti-CD45, anti-ICAM-1 and anti-CD11b antibodies. These antibodies were obtained from sources previously described (Dziennis et al. 2011).
Immunohistochemistry
To perform ex vivo analyses following vehicle or 14E11 treatment, mice were euthanized with an isoflurane overdose until respiration ceased. They were then heparinized, transcardially perfused with 100 mL of 4% paraformaldehyde (mass/volume in sodium phosphate buffer [0.1 M, pH 7.4]), and fixed at 4 °C for 24 h. Mice then underwent necropsy, and intact spinal columns were removed. Spinal cord sections 1–2 mm in length were dissected from the thoracic and lumbar regions from one representative animal per treatment group to analyze CNS pathology and fibrin(ogen) accumulation, respectively.
To assess CNS pathology, thoracic spinal cord tissues were re-fixed in 5% glutaraldehyde (mass/volume in sodium phosphate buffer [0.1 M, pH 7.4]) at 4 °C for 72 h, post-fixed in 1% osmium tetroxide for 3.5 h, rehydrated in ethanol, and embedded in plastic. Then, 0.5 μm semithin sections were obtained with a microtome, mounted onto precleaned microscope slides, and stained with toluidine blue to visualize nerve structures. Spinal sections were imaged at 20× and manually stitched into a complete composite of the section. To assess for axonal damage within the thoracic tissue, composite images were analyzed by a trained, blinded user as described previously (Wang et al. 2006). Briefly, regions of interest within the white matter tracts were segmented for areas of tissue damage, which included demyelinated axons, degenerating axons, and disrupted compact myelin. Percent area of tissue damage was quantified by measuring area of damaged white matter and total area of white matter (damaged and intact).
To assess fibrin(ogen) accumulation within the CNS, lumbar spinal cord tissues were fixed and embedded in paraffin for sectioning. Sections were blocked in 10% normal goat serum containing 1% bovine serum albumin and 0.025% Triton-X at room temperature for 45 min to decrease nonspecific staining. Tissue sections were then incubated with a primary antibody to fibrin(ogen) (1:50 in goat serum, rabbit polyclonal, MP Biomedicals) overnight at 4 °C followed by goat anti-rabbit IgG Alexa Fluor 488 (Molecular Probes). Slides were rinsed, mounted in aqueous media, and imaged at 20× with a Zeiss Axiovert fluorescent microscope. 20× images were processed identically in SlideBook (ver. 5.5) and manually stitched into a composite of the entire section using Adobe Photoshop. Quantitative analysis of fibrin(ogen) accumulation was performed on composite images using ImageJ (ver. 2.3.0). Color channels were split, and the green channel intensity histogram data were obtained and utilized to determine the background threshold value. Mean gray values corresponding to fibrin(ogen) signal were measured above the threshold value. Corrected integrated density was determined by subtracting background and accounting for area of fibrin(ogen).
Statistical analysis
Shapiro-Wilk and F-tests were used to test for normality and compare variances in the data. If the data were normally distributed with equal standard deviations, unpaired t-tests were used to compare vehicle and 14E11-treated mice. If the data were not normally distributed, the data were compared by a Mann-Whitney test. A P value less than 0.05 was considered statistically significant. GraphPad Prism 9 (ver. 9.5.0) was used to perform statistical analyses.
Results
Effect of FXI inhibition on clinical signs of EAE
Patients with MS experience discrete episodes, also referred to as ‘attacks’ or ‘relapses’, during which they present with varying clinical symptoms (e.g. changes in gait, muscle weakness, and incoordination) (Gelfand 2014). Clinical rating scales are commonly used to assess the severity of neurological dysfunction and inform treatment strategies in humans, as well as monitor disease progression in animal models (Hulleck et al. 2022; Noseworthy 1994). To quantify the effects of FXI inhibition on EAE symptoms, EAE mice were administered 14E11 or vehicle control at peak disease every other day for four days (Fig. 1a). Mice treated with 14E11 had an average peak disease score of 2.4, whereas mice in the control cohort had an average peak disease score of 3.5, which represents a 31.4% reduction (Fig. 1b). To further quantify this effect, we calculated cumulative disease index (CDI) by summing daily disease scores for each cohort. Mice treated with 14E11 had a mean CDI of 15.3, while mice in the control group had a mean CDI of 26.9, representing a 43.1% decrease in mean CDI with 14E11 treatment (Fig. 1c). These data confirm that targeting FXI reduced clinical symptom severity in a mouse model of MS. We did not observe any bleeding in mice during this study, nor during hemostatic evaluations in previous mouse studies using 14E11 (Leung et al. 2012; Tucker et al. 2012).
Effect of FXI inhibition on EAE-induced inflammation
Since activated macrophages and microglia have been shown to upregulate demyelination in the early stages of MS (Wang et al. 2019), we next evaluated the role of FXI in mediating the inflammatory response in EAE. To do this, we first quantified the population of activated macrophages/microglia (CD11b/CD45high cells) and CD4+ T-cells in brain samples from vehicle control and 14E11 treated mice. Note, to overcome the lower limit of CNS cells required for the antibody staining protocol, the CNS cells (n = 3 per group) were pooled (n = 1 per group) before flow cytometry analyses. Compared to the vehicle-treated group, total numbers of mononuclear cells recovered from the 14E11 treated group were reduced by ~ 50% (from 3.2 M to 1.6 M cells) and absolute numbers of infiltrating CD11b/CD45high cells in the CNS were reduced by 24% (from 188,800 to 142,880 cells) and CD4+ T cells by 44% (from 115,520 to 65,120 cells), respectively (Fig. 2a–b). Additionally, we measured macrophage subpopulation activation in splenocytes harvested from mice with EAE. No differences were observed for CD11b+ macrophages expressing TNFα or ICAM-1 following treatment with 14E11 (Fig. 2c–d). These results suggest that FXI plays a role in reducing EAE-induced inflammation in the CNS, but not in the peripheral splenocytes.
Fig. 2.
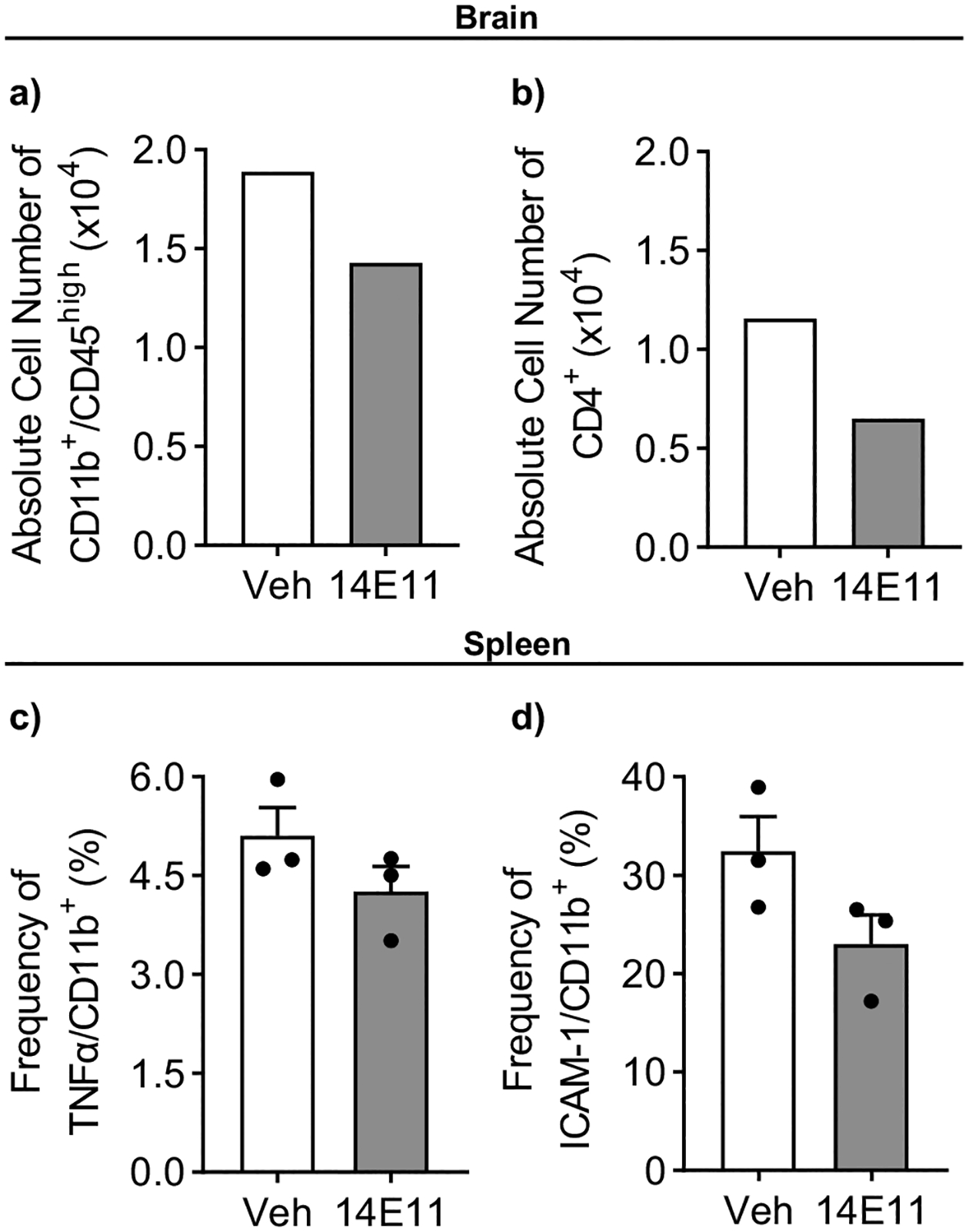
Treatment with 14E11 reduced inflammation caused by EAE in the CNS, but not in splenic macrophages. CNS cells and splenocytes were obtained from mice with EAE treated with either vehicle or 14E11 (1 mg/kg, i.v.). To achieve sufficient cell numbers for antibody staining, CNS cells were pooled (n = 3 per group). Flow cytometry was used to measure the population of activated macrophages/microglia (CD11b+/CD45high) (a) and T cells (CD4+) (b) within the brain. Data are presented as the absolute numbers of infiltrating cells in the pooled CNS samples. To study macrophage activation in the spleen, flow cytometry was used to measure the expression of TNFα (c) and intercellular adhesion molecule-1 (ICAM-1) (d) on CD11b+ macrophages. Data are presented as mean ± SEM from n = 3. Statistical analyses were conducted using unpaired t-tests on GraphPad Prism 9. Statistical significance is denoted by a single asterisk (*) for P ≤ 0.05
Effect of FXI inhibition on demyelination in the thoracic spinal cord
The progression of MS is marked by increasing instances of demyelinated lesions in the CNS that result in the debilitating loss of nervous system function (Gelfand 2014). To determine the effect of FXI inhibition on myelination, we stained spinal cord cross sections with toluidine blue and used light microscopy to assess myelination. As shown in Fig. 3, there appeared to be a notable reduction in demyelination in the spinal cord sections of mice treated with 14E11 compared to mice in the control group. Indeed, we quantified demyelination damage as percent of damaged white matter in a representative spinal cord image (n = 1) from each group. The percent of white matter area damaged was reduced from 13.6% in vehicle treated controls to 2.1% in 14E11 treated mice, representing an 84.3% decrease in damage with 14E11 treatment. Note, due to the limitation of a single sample size per group, cautious interpretation of these findings is warranted. Nonetheless, these results suggest that targeting FXI may preserve myelination in the spinal cords of EAE mice.
Fig. 3.
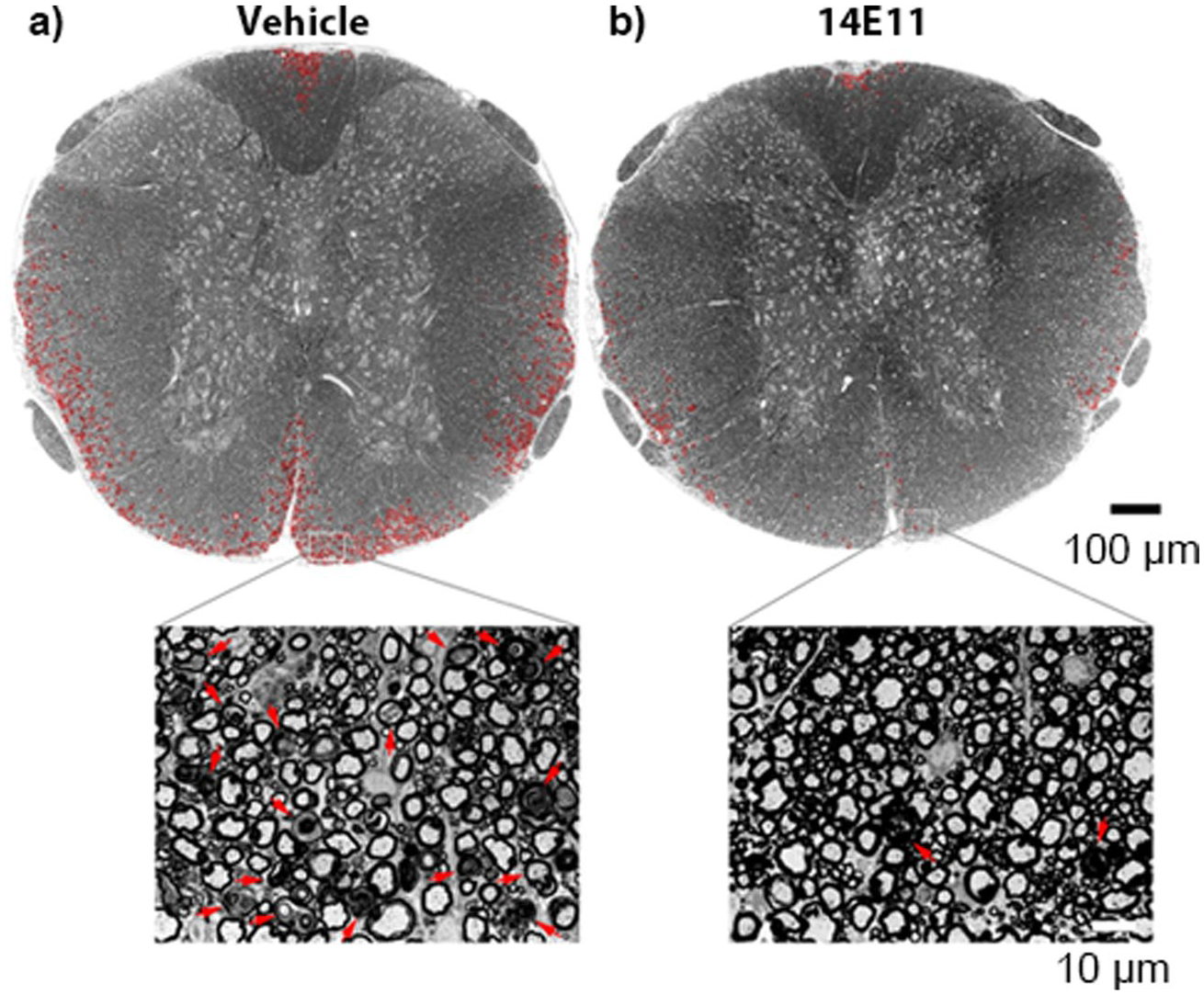
Axonal damage and demyelination in the thoracic spinal cord is reduced following treatment with 14E11. Damage to the myelin in spinal cord sections was assessed by staining the tissues with toluidine blue and using light microscopy to image the semithin sections at 20×. A complete view of the spinal cord cross section was obtained by manually stitching individual images into composites using Adobe Photoshop. The representative images shown are from mice treated with either vehicle (a) or 14E11 (b). Tissue damage in the white matter is denoted in red. The scale bar in the composite image indicates 100 μm, and the scale bar in the magnified view (63×) indicates 10 μm
Fibrin(ogen) accumulation in the CNS
Accumulation of the plasma protein fibrin(ogen) in the CNS is a prominent feature of BBB disruption and MS pathology. Studies have shown that fibrinogen accumulation correlates with axonal damage in the mouse models of EAE (Davalos et al. 2012). Since we observed a reduction in the percent area of damaged white matter, we investigated the effects of FXI inhibition on fibrin(ogen) deposition in the lumbar region of the CNS (Fig. 4a, b). We quantified fibrin(ogen) accumulation as the percent area positive in representative images of the lumbar region (n = 1 per group). The fibrin(ogen) signal was reduced from 4.9% in the vehicle control to 2.6% for 14E11 treatment. Additionally, we found the corrected fluorescent integrated density reduced from 5.48E7 RFUs to 2.59E7 RFUs for vehicle and 14E11 treatments, respectively. This corresponds to a 52.7% reduction in intensity of fibrin(ogen) signal for 14E11 treatment compared to vehicle control. These preliminary findings suggest that pharmacological targeting of FXI may preserve barrier function in the setting of EAE.
Fig. 4.
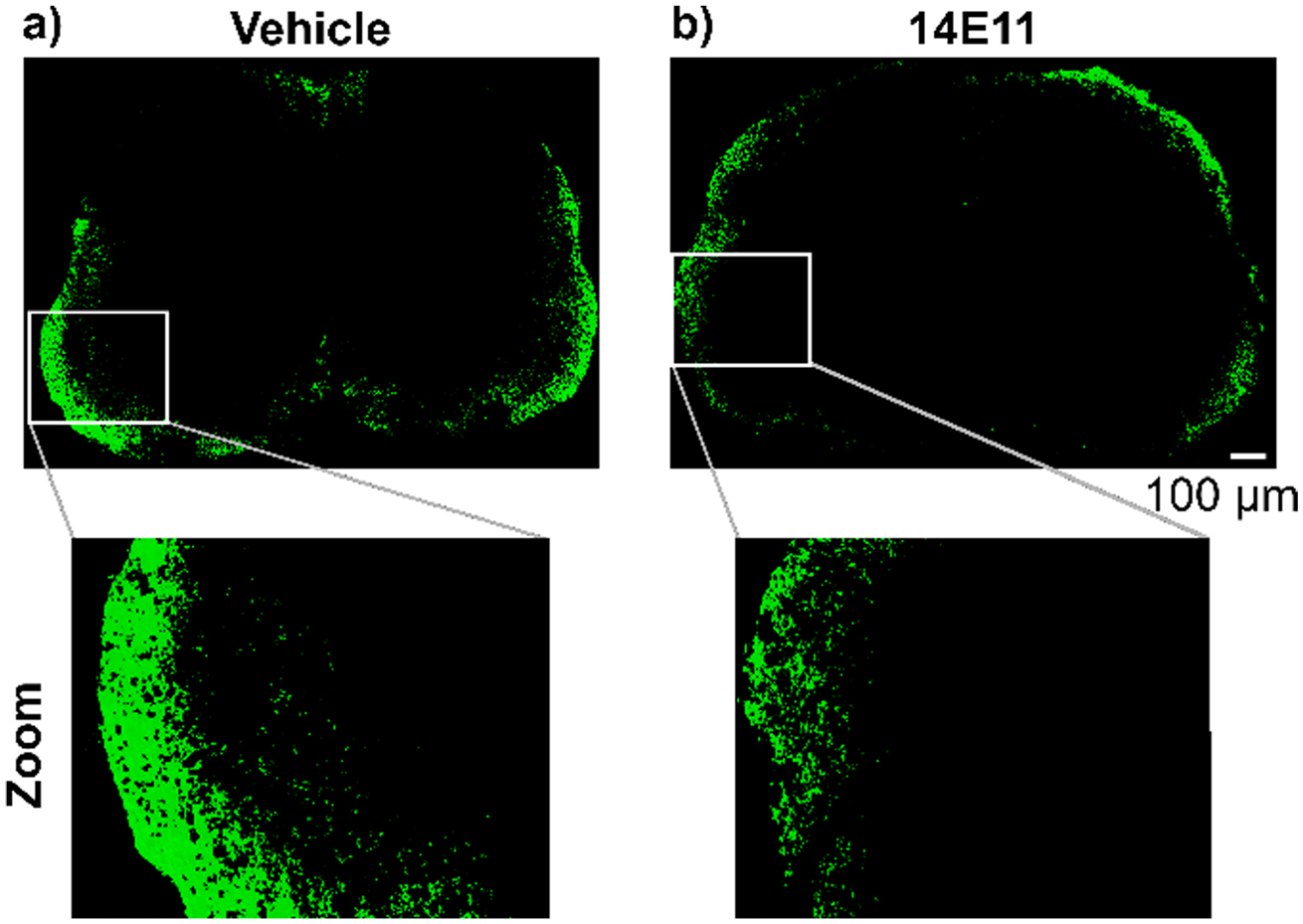
Effects of pharmacologic targeting of FXI by 14E11 treatment on fibrin(ogen) deposition in the spinal cord. Sections of the lumbar region of the spinal cord were stained for fibrin(ogen) deposition from one representative animal per treatment group. 20× images were manually stitched into composites and thresholded for background fluorescence for vehicle (a) or 14E11 treatment (b). Representative images show a reduction in area positive for fibrin(ogen) signal as well as corrected fluorescent integrated density for 14E11 treatment compared to vehicle control. Scale bar indicates 100 μm
Discussion
Clinical and experimental evidence suggest that coagulation components play a role in exacerbating the pathologies associated with MS (Jordan et al. 2021; Miranda Acuña et al. 2017; Plantone et al. 2019). Indeed, the intrinsic pathway of coagulation has been implicated in MS and EAE pathologies, as plasma FXII levels or activity have been shown to be elevated in MS patients (Zamolodchikov et al. 2019), and FXII deficiency is protective in EAE (Ziliotto et al. 2018). However, the mechanism by which the intrinsic pathway of coagulation contributes to EAE through FXII remains ill-defined. Therefore, we evaluated the effects of pharmacologically targeting of FXI on autoimmune responses in EAE. At the onset of symptoms (disease score ≥ 2.5), EAE mice were treated with the anti-FXI monoclonal antibody, 14E11. Following treatment with 14E11, we observed a reduction in the clinical severity of EAE symptoms, axonal damage and demyelination, and fibrin(ogen) accumulation in the CNS. Our findings indicate that FXI may be a potential therapeutic target in treating the progression of autoimmune disorders of the CNS.
Our results indicate that pharmacological targeting of FXI improves disease scores of EAE. Treatment with 14E11 significantly attenuated clinical symptom severity over the course of eight days, which corresponded to approximately a 40% reduction in the cumulative disease index. These data are consistent with other studies in which targeting components of the coagulation cascade improved disease severity in EAE, including FXII (Göbel et al. 2016b), FXa (Merker et al. 2017), thrombin (Han et al. 2008; Verbout et al. 2015), fibrin (Ryu et al. 2018), and activated protein C (APC) (Han et al. 2008). Previous findings by Göbel et al. showed that there were no significant differences in clinical disease scores between FXI-deficient mice and wild-type mice (Göbel et al. 2016b). One possible explanation for the difference between these studies is that Göbel et al. evaluated the effect of complete FXI deficiency, while our study employed a strategy to pharmacologically target FXI at peak disease. Incorporating prophylactic 14E11 treatment as a future direction may resolve the discrepancies between our current study and the prior work using a FXI-null mouse model and provide valuable mechanistic insights.
Treatment with 14E11 also resulted in a trend towards a reduction in EAE-induced inflammation. During the early stages of MS, fibrinogen infiltrates the CNS and activates microglia through the integrin receptor, CD11b/CD18 (Davalos and Akassoglou 2012), resulting in an M1-like activation and pro-inflammatory phenotype (Adams et al. 2007; Davalos and Akassoglou 2012; Ryu et al. 2015). Therefore, CD11b + macrophage expression of TNFα and ICAM-1 were measured as markers of activated macrophages/microglia found to be causative in early stages of EAE. Interestingly, only modest decreases of TNFα or ICAM-1 macrophage expression were observed between 14E11 and vehicle treatment. This observation may be due to an evaluation of cells isolated from peripheral splenic tissue rather than within the CNS itself. Nonetheless, this suggests that inhibition of FXI by 14E11 does not provoke significant attenuation of the primary, peripheral immune response after EAE induction. We next quantified the population of activated macrophages/microglia (CD11b/CD45high cells) and CD4 T cells in pooled murine brain samples. Not only did treatment with 14E11 reduce the total number of mononuclear cells in the brain by 50%, but we also observed reduced percentages of activated CNS macrophages/microglia and T cells. These results strongly support the conclusion that inhibition of FXI plays an important role in limiting EAE-induced inflammation.
Additionally, we found that 14E11-treated mice had reduced demyelination in the white matter of spinal cord tissue compared to the vehicle cohort. Previous studies have found that the inhibition of oligodendrocyte progenitor cell (OPC) maturation contributes to demyelination processes, and that OPC maturation and recruitment to demyelinating lesions is negatively regulated by thrombin in a PAR1-dependent manner (Choi et al. 2018; Yoon et al. 2015). Since thrombin activity is upregulated in the CNS of MS patients, and FXI is positioned upstream of thrombin generation, it is possible that targeting FXI reduces the amount of thrombin generated (Parsons et al. 2017). Indeed, the reduction in demyelination observed in the 14E11-treated mice might be attributed to decreased levels of FXIa-mediated thrombin generation. However, we did not evaluate thrombin activity nor the effect of 14E11 on thrombin generation in this study.
FXI inhibition also reduced BBB disruption, as measured by fibrin(ogen) accumulation in the spinal cord. Under physiological conditions, BBB integrity is modulated by intercellular junction proteins (e.g. gap, adherens, or tight junctions) and matrix metalloproteinases (MMP). These proteins can signal through a number of receptors on the surface of cerebral vascular endothelium, including the thrombin receptors PAR-1 and PAR-4 (Jordan et al. 2021). Previous studies have demonstrated that blocking PAR-1 in the setting of EAE prevents BBB disruption by downregulating MMP9 and attenuating the loss of tight junctions (Brailoiu et al. 2017). Therefore, it is possible that targeting FXI reduces BBB permeability by reducing the amount of thrombin generated. Alternatively, we have recently shown that FXIa activity increases endothelial permeability both in vitro and in vivo by downregulating vascular endothelial-cadherin (VE-cadherin) expression and increasing endothelial permeability (Ngo et al. 2021). Thus, targeting FXIa activity with 14E11 may have elicited a protective effect on barrier function by preventing VE-cadherin inhibition.
There are several limitations of this study. One issue is that limited numbers of mononuclear cells in CNS required pooling of cells from three mice in order to overcome detection thresholds needed for FACs analysis of cell subtypes. Although such pooling gave a greater sampling of smaller subsets, it also precluded biological replicates. Pooling samples can decrease study power, modify the mean values and standard deviations of the data, and mask outliers within a heterogeneous population. Another limitation of this study is that we evaluated treatment with 14E11 at one dose level and at one timepoint, which precludes a full evaluation of the treatment window and effect level. It is conceivable that prophylactic treatment with 14E11, which would more closely mimic therapeutic strategies to combat and treat disease clinically, could have strengthened our findings.
Overall, treatment with 14E11 decreased severity of EAE clinical signins, axonal damage, and fibrin(ogen) deposition within the CNS. However, no significant differences were found in splenic immune responses, indicating that FXI may have a limited role in peripheral EAE-associated inflammatory responses. Future work would benefit from immunohistochemistry or immunofluorescence studies that document potential treatment-dependent differences in the distribution of inflammatory lesions in the CNS. Since 14E11 only inhibits FXIIa activation of FXI and conversely FXII by FXIa in vitro (Puy et al. 2016), future studies would also benefit from investigating the role of feedback activation of FXIa by thrombin in EAE pathology. Doing so will aid in further delineating the function of the intrinsic pathway in EAE pathology and progression. In conclusion, the results reported herein support pharmacological targeting of FXI as a therapeutic strategy to facilitate the treatment of MS and other neurological autoimmune diseases.
Funding
This study was supported by the National Multiple Sclerosis Society (PP1900), the National Institutes of Health (R01HL101972, 1F31HL162467-01A1), the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development Merit Review Award 2I01 BX000226 (AAV), BLR&D Merit Review for Pre-IND studies of Drugs and Biologics Award 5I01 BX005112 (AAV), Senior Research Career Scientist Award 1IK6BX004209 (AAV), the National Institute of Allergy and Infectious Diseases Award R21 AI148490 (HO) and 2R42AI122574 (AAV; HO).
Footnotes
Ethics approval The animal care and procedures in this study were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the regulations of the Oregon Health & Science University Institutional Animal Care and Use Committee (National Research Council Committee for the Update of the Guide for the C and Use of Laboratory A 2011). The male C57BL/6 mice used in this study were housed at the Portland Veterans Affairs Medical Center in the Animal Resource Facility (Portland, OR, US) according to guidelines set by the National Research Council and the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources.
Competing interests E.I. Tucker and N.G. Verbout are employees of Aronora, Inc., a company that may have financial interest in the study results. The Oregon Health & Science University Conflict of Interest in Research Committee has reviewed and managed this potential conflict of interest. The remaining authors declare no competing interests.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
- Adams RA, Bauer J, Flick MJ, Sikorski SL et al. (2007) The fibrin-derived γ377–395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med 204(3):571–582. 10.1084/jem.20061931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed O, Geraldes R, DeLuca GC, Palace J (2019) Multiple sclerosis and the risk of systemic venous thrombosis: a systematic review. Mult Scler Relat Disord 27:424–430. 10.1016/j.msard.2018.10.008 [DOI] [PubMed] [Google Scholar]
- Bebo BF Jr, Vandenbark AA, Offner H (1996) Male SJL mice do not relapse after induction of EAE with PLP 139–151. J Neurosci Res 45(6):680–689. [DOI] [PubMed] [Google Scholar]
- Brailoiu E, Shipsky MM, Yan G, Abood ME, Brailoiu GC (2017) Mechanisms of modulation of brain microvascular endothelial cells function by thrombin. Brain Res 1657:167–175. 10.1016/j.brainres.2016.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Tucker EI, Pine MS, Sisler I et al. (2010) A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood 116(19):3981–3989. 10.1182/blood-2010-02-270918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C-I, Yoon H, Drucker KL, Langley MR, Kleppe L, Scarisbrick IA (2018) The thrombin receptor restricts Subventricular Zone neural stem cell expansion and differentiation. Sci Rep 8(1):9360–9360. 10.1038/s41598-018-27613-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborators GMS (2019) Global, regional, and national burden of multiple sclerosis 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol 18(3):269–285. 10.1016/s1474-4422(18)30443-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courville CB (1959) The effects of heparin in acute exacerbations of multiple sclerosis. Observations and deductions. Bull Los Angel Neuro Soc 24:187–196 [PubMed] [Google Scholar]
- Davalos D, Akassoglou K (2012) Fibrinogen as a key regulator of inflammation in disease. Semin Immunopathol 34(1):43–62. 10.1007/s00281-011-0290-8 [DOI] [PubMed] [Google Scholar]
- Davalos D, Kyu Ryu J, Merlini M, Baeten KM et al. (2012) Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 3(1):1227. 10.1038/ncomms2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Baeten KM, Whitney MA, Mullins ES et al. (2014) Early detection of thrombin activity in neuroinflammatory disease. Ann Neurol 75(2):303–308. 10.1002/ana.24078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziennis S, Akiyoshi K, Subramanian S, Offner H, Hurn PD (2011) Role of dihydrotestosterone in post-stroke peripheral immunosuppression after cerebral ischemia. Brain Behav Immun 25(4):685–695. 10.1016/j.bbi.2011.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand JM (2014) Multiple sclerosis: diagnosis, differential diagnosis, and clinical presentation. Handb Clin Neurol 122:269–290. 10.1016/b978-0-444-52001-2.00011-x [DOI] [PubMed] [Google Scholar]
- Göbel K, Pankratz S, Schneider-Hohendorf T, Bittner S et al. (2011) Blockade of the kinin receptor B1 protects from autoimmune CNS disease by reducing leukocyte trafficking. J Autoimmun 36(2):106–114. 10.1016/j.jaut.2010.11.004 [DOI] [PubMed] [Google Scholar]
- Göbel K, Kraft P, Pankratz S, Gross CC et al. (2016a) Prothrombin and factor X are elevated in multiple sclerosis patients. Ann Neurol 80(6):946–951. 10.1002/ana.24807 [DOI] [PubMed] [Google Scholar]
- Göbel K, Pankratz S, Asaridou C-M, Herrmann AM et al. (2016b) Blood coagulation factor XII drives adaptive immunity during neuroinflammation via CD87-mediated modulation of dendritic cells. Nat Commun 7(1):11626. 10.1038/ncomms11626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göbel K, Asaridou C-M, Merker M, Eichler S et al. (2019) Plasma kallikrein modulates immune cell trafficking during neuroinflammation via PAR2 and bradykinin release. Proc Natl Acad Sci 116(1):271–276. 10.1073/pnas.1810020116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber A, Hanson SR (2003) Factor XI-dependence of surface- and tissue factor-initiated thrombus propagation in primates. Blood 102(3):953–955. 10.1182/blood-2003-01-0324 [DOI] [PubMed] [Google Scholar]
- Gveric D, Herrera B, Petzold A, Lawrence DA, Cuzner ML (2003) Impaired fibrinolysis in multiple sclerosis: a role for tissue plasminogen activator inhibitors. Brain 126(Pt 7):1590–1598. 10.1093/brain/awg167 [DOI] [PubMed] [Google Scholar]
- Han MH, Hwang S-I, Roy DB, Lundgren DH et al. (2008) Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 451(7182):1076–1081. 10.1038/nature06559 [DOI] [PubMed] [Google Scholar]
- Hulleck AA, Menoth Mohan D, Abdallah N, El Rich M, Khalaf K (2022) Present and future of gait assessment in clinical practice: towards the application of novel trends and technologies. Front Med Technol 4:901331. 10.3389/fmedt.2022.901331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan KR, Parra-Izquierdo I, Gruber A, Shatzel JJ et al. (2021) Thrombin generation and activity in multiple sclerosis. Metab Brain Dis 36(3):407–420. 10.1007/s11011-020-00652-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan KR, Wyatt CR, Fallon ME, Woltjer R et al. (2022) Pharmacological reduction of coagulation factor XI reduces macrophage accumulation and accelerates deep vein thrombosis resolution in a mouse model of venous thrombosis. J Thromb Haemost 20(9):2035–2045. 10.1111/jth.15777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer HF, Choi EY, Zhou H, Schleicher R et al. (2012) Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Circ Res 110(9):1202–1210. 10.1161/CIRCRESAHA.111.256370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung PY, Hurst S, Berny-Lang MA, Verbout NG et al. (2012) Inhibition of factor XII-Mediated activation of factor XI provides Protection Against Experimental Acute ischemic stroke in mice. Transl Stroke Res 3(3):381–389. 10.1007/s12975-012-0186-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lider O, Baharav E, Mekori YA, Miller T, Naparstek Y, Vlodavsky I, Cohen IR (1989) Suppression of experimental autoimmune diseases and prolongation of allograft survival by treatment of animals with low doses of heparins. J Clin Invest 83(3):752–756. 10.1172/JCI113953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorentz CU, Verbout NG, Cao Z, Liu L et al. (2018) Factor XI contributes to myocardial ischemia-reperfusion injury in mice. Blood Adv 2(2):85–88. 10.1182/bloodadvances.2017004879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorentz CU, Tucker EI, Verbout NG, Shatzel JJ et al. (2021) The contact activation inhibitor AB023 in heparin free hemodialysis: results of a randomized phase 2 clinical trial. Blood 138(22):2173–2184. 10.1182/blood.2021011725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maschmeyer J, Shearer R, Lonser E, Spindle DK (1961) Heparin potassium in the treatment of chronic multiple sclerosis. Bull Los Angel Neuro Soc 26:165–171 [PubMed] [Google Scholar]
- Merker M, Eichler S, Herrmann AM, Wiendl H, Kleinschnitz C, Göbel K, Meuth SG (2017) Rivaroxaban ameliorates disease course in an animal model of multiple sclerosis. J Neuroimmunol 313. 10.1016/j.jneuroim.2017.08.013 [DOI] [PubMed] [Google Scholar]
- Miranda Acuña J, Hidalgo de la Cruz M, Ros AL, Tapia SP, Martínez Ginés ML, de Andrés Frutos CD (2017) Elevated plasma fibrinogen levels in multiple sclerosis patients during relapse. Mult Scler Relat Disord 18:157–160. 10.1016/j.msard.2017.09.033 [DOI] [PubMed] [Google Scholar]
- National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals (2011) Guide for the Care and Use of Laboratory Animals, 8th edn. National Academies Press, Washington, DC [Google Scholar]
- Ngo ATP, Jordan KR, Mueller PA, Hagen MW et al. (2021) Pharmacological targeting of coagulation factor XI mitigates the development of experimental atherosclerosis in low-density lipoprotein receptor-deficient mice. J Thromb Haemost 19(4):1001–1017. 10.1111/jth.15236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noseworthy JH (1994) Clinical scoring methods for multiple sclerosis. Ann Neurol 36(Suppl):S80–85. 10.1002/ana.410360718 [DOI] [PubMed] [Google Scholar]
- Parsons ME, O’Connell K, Allen S, Egan K et al. (2017) Thrombin generation correlates with disease duration in multiple sclerosis (MS): novel insights into the MS-associated prothrombotic state. Mult Scler J Exp Transl Clin 3(4):2055217317747624. 10.1177/2055217317747624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters PJ, Bazelier MT, Uitdehaag BM, Leufkens HG, De Bruin ML, de Vries F (2014) The risk of venous thromboembolism in patients with multiple sclerosis: the clinical practice research datalink. J Thromb Haemost 12(4):444–451. 10.1111/jth.12523 [DOI] [PubMed] [Google Scholar]
- Plantone D, Inglese M, Salvetti M, Koudriavtseva T (2019) A perspective of Coagulation Dysfunction in multiple sclerosis and in experimental allergic encephalomyelitis. Front Neurol 9(JAN). 10.3389/fneur.2018.01175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puy C, Tucker EI, Ivanov IS, Gailani D et al. (2016) Platelet-derived short-chain polyphosphates enhance the inactivation of tissue factor pathway inhibitor by activated coagulation factor XI. PLoS ONE 11(10):e0165172. 10.1371/journal.pone.0165172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, Gailani D, Castellino FJ (2002) FXI is essential for thrombus formation following FeCl3-induced injury of the carotid artery in the mouse. Thromb Haemost 87(4):774–776 [PubMed] [Google Scholar]
- Ryu JK, Petersen MA, Murray SG, Baeten KM et al. (2015) Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat Commun 6(1):8164–8164. 10.1038/ncomms9164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu JK, Rafalski VA, Meyer-Franke A, Adams RA et al. (2018) Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat Immunol 19(11). 10.1038/s41590-018-0232-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher WA, Seiler SE, Steinbacher TE, Stewart AB et al. (2007) Antithrombotic and hemostatic effects of a small molecule factor XIa inhibitor in rats. Eur J Pharmacol 570(1–3):167–174. 10.1016/j.ejphar.2007.05.043 [DOI] [PubMed] [Google Scholar]
- Silasi R, Keshari RS, Lupu C, Van Rensburg WJ et al. (2019) Inhibition of contact-mediated activation of factor XI protects baboons against S aureus-induced organ damage and death. Blood Adv 3(4):658–669. 10.1182/bloodadvances.2018029983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Subramanian S, Emerson-Webber A, Lindner M et al. (2010) Recombinant TCR ligand reverses clinical signs and CNS damage of EAE induced by recombinant human MOG. J Neuro-immune Pharmacol 5(2):231–239. 10.1007/s11481-009-9175-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz L, Derouiche A, Devraj K, Weber F, Brunkhorst R, Foerch C (2017) Anticoagulation with warfarin and rivaroxaban ameliorates experimental autoimmune encephalomyelitis. J Neuroinflammation 14(1):152. 10.1186/s12974-017-0926-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker EI, Marzec UM, White TC, Hurst S et al. (2009) Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood 113(4):936–944. 10.1182/blood-2008-06-163675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker EI, Verbout NG, Leung PY, Hurst S, McCarty OJ, Gailani D, Gruber A (2012) Inhibition of factor XI activation attenuates inflammation and coagulopathy while improving the survival of mouse polymicrobial sepsis. Blood 119(20):4762–4768. 10.1182/blood-2011-10-386185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbout NG, Yu X, Healy LD, Phillips KG et al. (2015) Thrombin mutant W215A/E217A treatment improves neurological outcome and attenuates central nervous system damage in experimental autoimmune encephalomyelitis. Metab Brain Dis 30(1):57–65. 10.1007/s11011-014-9558-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Gold BG, Kaler LJ, Yu X et al. (2006) Antigen-specific therapy promotes repair of myelin and axonal damage in established EAE. J Neurochem 98(6):1817–1827. 10.1111/j.1471-4159.2006.04081.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang J, Wang J, Yang B, Weng Q, He Q (2019) Targeting microglia and macrophages: a potential treatment strategy for multiple sclerosis. Front Pharmacol 10:286. 10.3389/fphar.2019.00286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H, Radulovic M, Drucker KL, Wu J, Scarisbrick IA (2015) The thrombin receptor is a critical extracellular switch controlling myelination. Glia 63(5):846–859. 10.1002/glia.22788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamolodchikov D, Bai Y, Tang Y, McWhirter JR, Macdonald LE, Alessandri-Haber N (2019) A short isoform of coagulation factor XII mRNA is expressed by neurons in the human brain. Neurosci 413:294–307. 10.1016/j.neuroscience.2019.05.040 [DOI] [PubMed] [Google Scholar]
- Zhang H, Lowenberg EC, Crosby JR, MacLeod AR et al. (2010) Inhibition of the intrinsic coagulation pathway factor XI by antisense oligonucleotides: a novel antithrombotic strategy with lowered bleeding risk. Blood 116(22):4684–4692. 10.1182/blood-2010-04-277798 [DOI] [PubMed] [Google Scholar]
- Ziliotto N, Baroni M, Straudi S, Manfredini F et al. (2018) Coagulation factor XII levels and intrinsic thrombin generation in multiple sclerosis. Front Neurol 9(APR). 10.3389/fneur.2018.00245 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.