

Summary
Epigenetic lesions that disrupt regulatory elements represent potential cancer drivers. However, we lack experimental models for validating their tumorigenic impact. Here we model aberrations arising in Isocitrate Dehydrogenase-mutant gliomas, which exhibit DNA hypermethylation. We focus on a CTCF insulator near the PDGFRA oncogene that is recurrently disrupted by methylation in these tumors. We demonstrate that disruption of the syntenic insulator in mouse oligodendrocyte progenitor cells (OPCs) allows an OPC-specific enhancer to contact and induce Pdgfra, thereby increasing proliferation. We show that a second lesion, methylation-dependent silencing of the Cdkn2a tumor suppressor, cooperates with insulator loss in OPCs. Coordinate inactivation of the Pdgfra insulator and Cdkn2a drives gliomagenesis in vivo. Despite locus synteny, the insulator is CpG-rich only in human, a feature that may confer human glioma risk but complicate mouse modelling. Our study demonstrates the capacity of recurrent epigenetic lesions to drive OPC proliferation in vitro and gliomagenesis in vivo.
Graphical Abstract
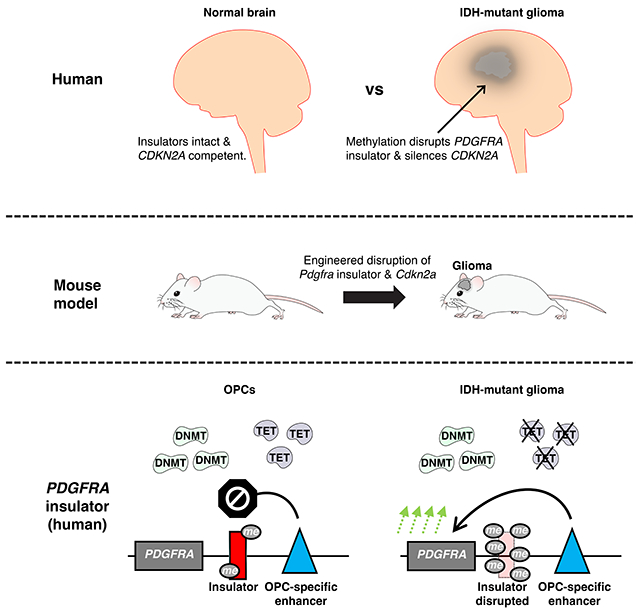
In Brief
A mouse model of IDH-mutant glioma is established by simulating downstream epigenetic lesions that activate the PDGFRA oncogene and silence the CDKN2A tumor suppressor. The study provides insight into glioma mechanism, putative cell-of-origin and disease risk.
Introduction
Although cancer is classically framed as a genetic disease driven by somatic mutations, epigenetic alterations are highly prevalent1–3. Gliomas, leukemias and colorectal tumors frequently present with a ‘methylator phenotype’1,4,5, characterized by hypermethylation of CpG islands and other CpG-rich sequence elements. Excessive DNA methylation has been shown to silence the promoters of tumor suppressor genes, including MLH1, APC, BRCA1, and CDKN2A1,3. It can also disrupt noncoding regulatory elements termed ‘insulators’6–8, resulting in aberrant enhancer-promoter interactions with potential to induce oncogenes9–12. As a bona fide epigenetic mark, DNA methylation can be inherited through cell division and selected for fitness, much like driver mutations1,2,13. However, the epigenetic variability in most tumors hinders our ability to distinguish true driver events from inconsequential passengers14. Furthermore, we currently lack in vitro and in vivo experimental models required to validate and functionally characterize potential driver epigenetic lesions.
Tumors with IDH mutations, which include lower-grade gliomas and certain leukemias, exhibit profound DNA hypermethylation4,15. Mutant IDH yields a neomorphic enzyme that produces high levels of 2-hydroxyglutarate (2HG), an inhibitor of DNA demethylases15. In IDHmut gliomas, excess methylation is presumed to affect tumor suppressor genes, though the precise targets have yet to be clarified4. The hypermethylation also disrupts CTCF insulators16,17, potentially altering topologically associated domain (TAD) organization8,18,19 and allowing enhancers to aberrantly activate proto-oncogenes such as PDGFRA9. Yet despite tantalizing evidence from human tumor profiles, the ability of these epigenetic alterations to augment the proliferation and/or transformation of progenitor cells has yet to be demonstrated. Moreover, IDH mutations also affect histone demethylases and other iron-dependent hydroxylase enzymes15. Hence, the mechanisms by which IDH mutations and the associated hypermethylation drive tumorigenesis remain controversial.
Although IDHmut gliomas have a better prognosis than IDHwt glioblastomas, they are nonetheless incurable and ultimately fatal20. Experimental models to study these lower-grade tumors are woefully inadequate. Their indolent phenotype hinders the establishment of patient-derived models, such that cell lines and xenografts have primarily been derived from advanced malignancies21. IDH mutations have been engineered in cultured human astrocytes, but these in vitro models do not fully recapitulate methylation changes in the human tumors22,23. IDHmut mouse models have also been described but have significant caveats. Conditional expression of mutant IDH in adult mouse subventricular zone led to hypermethylation, increased progenitor cell proliferation, and infiltration of surrounding regions24. Yet other studies that combined mutant IDH with other genetic drivers, including P53 loss or RAS mutation, found that mutant IDH actually reduced proliferation and penetrance in these tumor models25–27. Moreover, none of the modelling studies traced the downstream mechanisms and mediators of mutant IDH.
Here we identify and model recurrent epigenetic lesions that drive IDHmut gliomagenesis, including the methylation-dependent disruption of a CTCF insulator in the PDGFRA locus and silencing of the CDKN2A/p14ARF tumor suppressor. We use CRISPR engineering to disrupt the syntenic CTCF insulator in mouse embryonic stem (ES) cell-derived neural progenitor cells (NPCs) and oligodendrocyte progenitor cells (OPCs), two potent candidate cells-of-origin for gliomas28. Insulator loss allows an OPC-specific enhancer to activate Pdgfra and increase proliferation in OPCs but has no effect in NPCs which lack the enhancer. We then direct DNA methylation to the Cdkn2a promoter, silencing the tumor suppressor and further enhancing OPC proliferation. Coordinate disruption of the Pdgfra insulator and the Cdkn2a tumor suppressor in mouse brain OPC niches drives lower-grade gliomagenesis in vivo. Despite extended synteny over the PDGFRA locus, the mouse insulator region has far fewer CpG dinucleotides than the human element and is thus not responsive to methylation. The inability of the mouse Pdgfra insulator to be regulated by methylation, as a direct result of its lack of CpG sites, could potentially explain the latency and low penetrance of IDHmut mouse models. Our study demonstrates that disruption of an insulator and topological boundary can drive oncogene expression and gliomagenesis in vivo and provides a framework for modeling and functionally characterizing oncogenic regulatory alterations going forward.
Results
A recurrently lost CTCF insulator creates a conserved topological boundary near PDGFRA
The receptor tyrosine kinase PDGFRA is a canonical proto-oncogene that is frequently copy number amplified and over-expressed in glioblastoma29. Although PDGFRA is also highly expressed in IDHmut lower-grade gliomas, the locus typically remains wild-type5. We previously showed that global hypermethylation destabilizes CTCF insulators in IDHmut gliomas and that this was associated with the epigenetic deregulation of PDGFRA expression9. Here we sought to evaluate the functional impact of insulator loss directly in OPCs and NPCs, which represent candidate cells of origin for glioma28 (Figure S1A–B).
We first compared chromatin topology, CTCF binding and the enhancer-associated chromatin mark H3 K27 acetylation (H3K27ac) in human and mouse (Figure 1A–B). The PDGFRA locus shows striking conservation between the species, with over 2 Mb of syntenic sequence. The topological structure of the locus is also highly conserved, as revealed by high-throughput chromosome conformation capture (HiC) maps. The PDGFRA gene is located in between two TADs, each bounded by convergent CTCF sites18 engaged in long-range interactions. The topology of the locus is largely similar across HiC maps for other human and mouse cell types30.
Figure 1. Enhancer landscape and topology of the PDGFRA locus in human gliomas and mouse neural/oligodendrocyte progenitors.
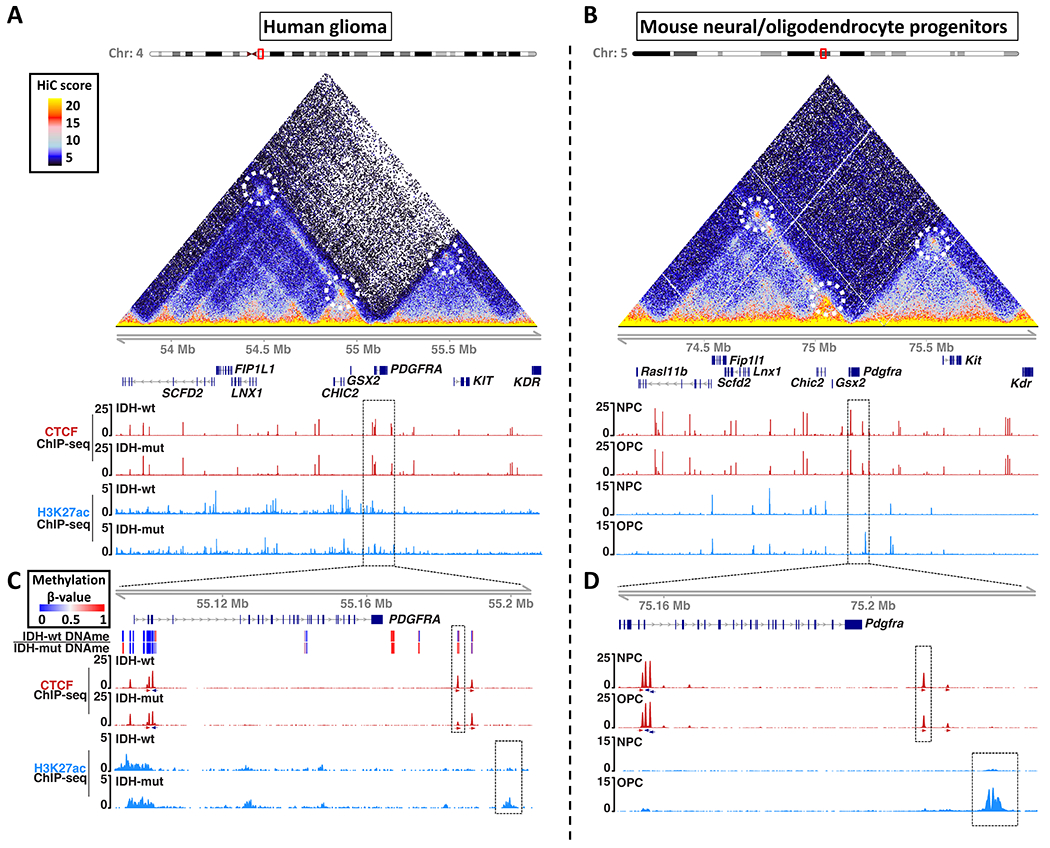
Maps of chromosome topology, CTCF insulator and enhancer-associated H3K27ac reveal synteny and conservation of the PDGFRA locus in (A) human and (B) mouse. HiC interaction maps (triangles) show pairwise contact frequencies in human IMR90 (left) and mouse NPCs (right). Chromatin loops are annotated with dashed circles. ChIP-seq tracks show CTCF (red) and H3K27ac (blue) in IDH1wt and IDH1mut human gliomas (A), and mouse NPCs and OPCs (B). Dashed boxes in (A) and (B) are expanded in (C) and (D) respectively. (C) Expanded genomic view shows DNA methylation (top heatmap), CTCF (red, orientation arrows below), and H3K27ac (blue) around the PDGFRA gene in human gliomas. The CTCF insulator disrupted in IDHmut gliomas (red peak in dashed box) corresponds to a boundary that shields PDGFRA from an enhancer (blue peak in dashed box). (D) Expanded genomic view shows the analogous CTCF site and enhancer in mouse progenitors. All ChIP-seq tracks are RPM normalized. These data (together with Figure S1) identify a conserved CTCF site that insulates PDGFRA from an OPC-specific enhancer, but is recurrently lost in IDH1mut gliomas.
We next investigated the DNA methylation status of 18 CTCF binding sites across the 2 Mb region in IDHwt and IDHmut gliomas, leveraging newly generated (n=8 tumors) and publicly available (n=5 tumors) bisulfite sequencing data5. We identified a significantly methylated CTCF site located ~20 kb downstream of PDGFRA that was specifically methylated in IDHmut tumors (Figure S1C–E,G). The average methylation of the central CpG in the binding site is 77% in IDHmut tumors, but just 18% in IDHwt (Figure S1E). Furthermore, CTCF occupancy is ~3-fold lower in IDHmut tumors, compared to IDHwt, consistent with the established inability of CTCF to bind methylated DNA (Figure S1D,F). Importantly, the disrupted CTCF site precisely coincides with a boundary element that partitions PDGFRA from the downstream TAD (Figure 1A,C, top dashed box in panel C). The orientation of the underlying CTCF motif and its engagement in a chromatin loop with the opposite side of the TAD suggests that it plays a critical role in enforcing the topological boundary.
Modelling insulator disruption and oncogene activation in candidate cells of origin
We next sought to model Pdgfra insulator loss in OPCs and NPCs, which we derived from mouse ES cells using established procedures31 (Figure S1A–B). The topological structure of the locus is highly conserved across cell types, including the downstream TAD and the CTCF site that partitions it from Pdgfra. However, its enhancer landscape varies markedly. Mouse and human OPCs contain a putative enhancer marked by strong H3K27ac within the downstream TAD, ~75 kb from the Pdgfra promoter. The enhancer appears to be OPC-specific as it lacks H3K27ac entirely in NPCs and other cell types. Importantly, the enhancer is highly acetylated and presumably active in IDHmut gliomas, likely due to their OPC-like state (Figure 1C–D, Figure S2A–C).
We hypothesized that disruption of the CTCF insulator would destabilize the TAD boundary and allow the OPC-specific enhancer to contact and activate Pdgfra. Since the syntenic insulator in mouse has just three CpGs in its vicinity, contrasting with nine CpGs in human, we leveraged genetic editing to disrupt the insulator in mouse OPCs. We specifically used CRISPR-Cas9 and a single guide RNA (sgRNA) to mutate the CTCF motif, which completely and selectively abrogated CTCF binding (Figure 2A, dashed box, Figure S3A–D, Table S1). We then assessed integrity of the TAD boundary by using circularized chromatin conformation capture sequencing (4C-seq) to quantify physical contacts between the OPC-specific enhancer and other genomic positions. In control OPCs, enhancer interactions were sharply curtailed by the TAD boundary, which prevented contact with Pdgfra. However, in insulator disrupted cells, the enhancer contacted sequences well beyond the boundary, including the Pdgfra promoter (Figure 2A). Consistently, insulator disruption increased Pdgfra expression and stimulated OPC proliferation. Insulator disruption had no effect on Pdgfra or proliferation in NPCs, which lack the enhancer (Figure 2B–D, Figure S2F).
Figure 2. Insulator disruption in mouse OPCs upregulates PDGFRA and increases proliferation.
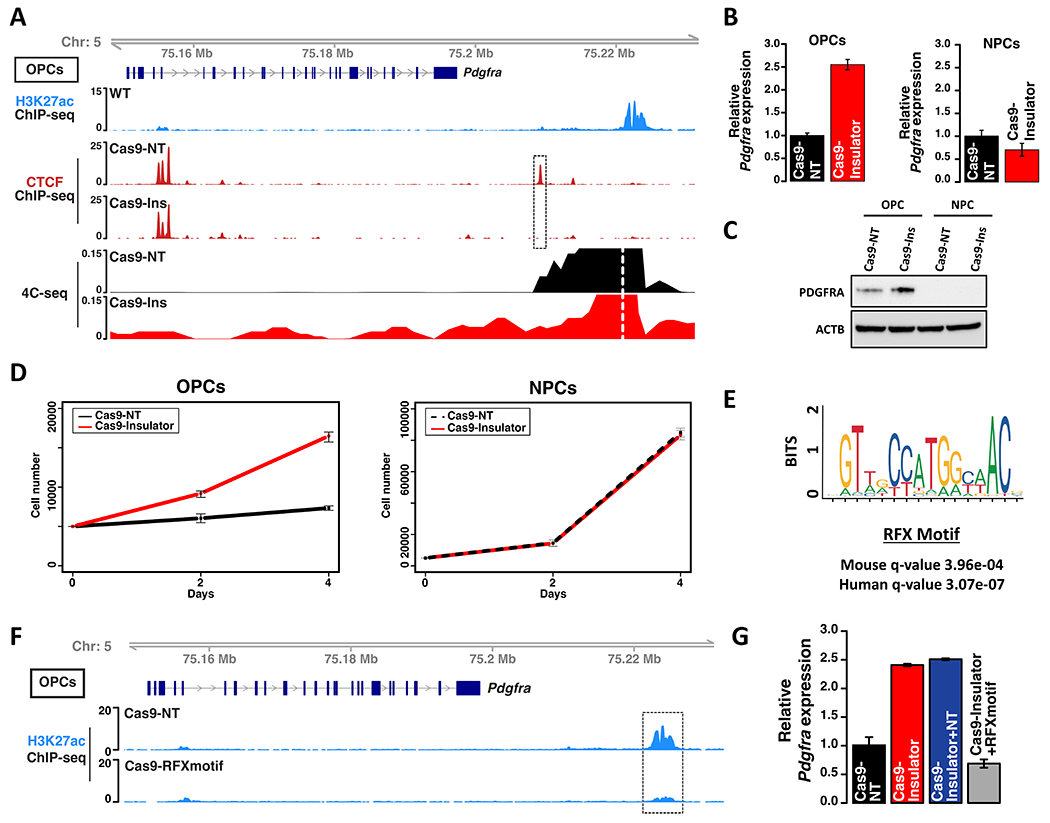
(A) Genomic tracks depict Pdgfra gene structure (top), H3K27ac ChIP-seq (blue), CTCF ChIP-seq (red), and contact frequency with the OPC-specific enhancer viewpoint (dashed white line) per 4C-seq. CTCF binding and contact frequency are shown for OPCs expressing Cas9 and non-targeting (Cas9-NT) sgRNA or Cas9 with a sgRNA targeting the Pdgfra insulator (Cas9-Ins). Dashed box highlights the disrupted insulator. (B) Pdgfra RNA expression levels, relative to 18S control, shown for control and insulator-disrupted OPCs and NPCs from three biologically independent replicates (P values (two-sided t-test) < 0.0001 for the OPC comparison). (C) Western blot shows PDGFRA protein expression for control and insulator-disrupted OPCs and NPCs. (D) Growth curves shown for control and insulator-disrupted OPCs and NPCs from three biologically independent replicates (P values (two-sided t-test) <0.0001 for the OPC comparison. (E) The OPC-specific enhancer downstream of Pdgfra contains a high-scoring RFX motif conserved in mouse and human. (F) Genomic view of the Pdgfra locus shows H3K27ac (blue) in OPCs expressing Cas9-NT or Cas9 with a sgRNA targeting the RFX motif in the OPC-specific enhancer (dashed box). (G) Relative Pdgfra expression for OPCs with combined disruption of insulator and RFX motif by two sgRNAs, compared to insulator-only and NT controls from three biologically independent replicates (P values (one-way ANOVA) <0.0001 for insulator deletions versus control). Error bars in panels B, D, G represent standard deviation. These data indicate that CTCF insulator loss allows an RFX-driven, OPC-specific enhancer to aberrantly activate Pdgfra.
To characterize the OPC-specific enhancer, we scanned its primary sequence. We identified a high-scoring RFX motif conserved in mouse and human (Figure 2E, Figure S2D). The cognate factors Rfx4 and Rfx7 are expressed in OPCs and may account for the specificity of the enhancer (Figure S2E). When we used CRISPR-Cas9 to disrupt the RFX motif in OPCs, the enhancer acetylation was almost completely lost (Figure 2F, Figure S3E–F, Table S2). This provided an opportunity to test whether the enhancer is in fact required for Pdgfra induction upon insulator loss. We found that coordinate disruption of the RFX motif and the insulator using CRISPR-Cas9 and two sgRNAs failed to induce Pdgfra in OPCs (Figure 2G). These results indicate that disruption of a CTCF insulator, which is recurrently lost in IDHmut gliomas, causes an OPC-specific enhancer to activate Pdgfra expression and drive proliferation in OPCs.
Methylation-dependent silencing of the Cdkn2a/p19ARF tumor suppressor
We next considered whether the hypermethylation in IDHmut gliomas also silences specific tumor suppressors, potentially in cooperation with PDGFRA activation. Examination of six prominent glioma tumor suppressors5,32 across tumors from our cohort and TCGA revealed recurrent methylation of the CDKN2A/p14ARF promoter in IDHmut tumors. As expected, the methylated promoter was devoid of active chromatin marks. Consistently, the CDKN2A/p14ARF transcript was expressed at very low levels in IDHmut gliomas, contrasting with its robust expression in IDHwt glioblastoma that retain the CDKN2A locus (Figure 3A–C). The CDKN2A locus is frequently deleted in IDHwt glioblastoma as well as a subset of secondary IDHmut glioblastoma. However, it is rarely lost in lower-grade IDHmut gliomas with a hypermethylator phenotype, likely reflecting the sufficiency of methylation-dependent silencing (Figure S4A).
Figure 3. Modeling CDKN2A promoter silencing.
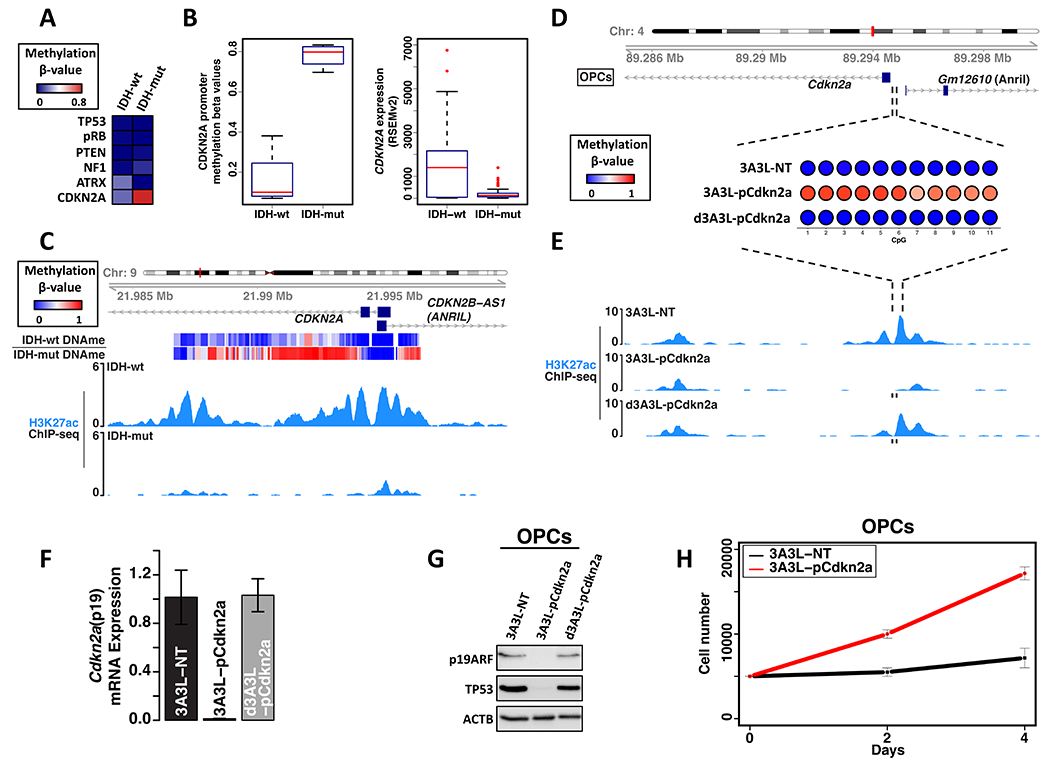
(A) Heatmap depicts mean methylation over tumor suppressor gene promoters in glioma samples stratified by IDH status. (B) Box plots depict distribution of CDKN2A promoter methylation (left, this study and TCGA) and CDKN2A mRNA expression (right, TCGA, RSEMv2 counts) for gliomas (excluding samples with genetic loss of CDKN2A). (C) Genomic tracks over the human CDKN2A promoter show DNA methylation (top heatmap) and H3K27ac (blue) in representative IDHwt and IDHmut glioma samples. (D) Bubble plots show mean methylation of 11 CpGs in the mouse Cdkn2a promoter in OPCs transfected with the DNMT3A3L epigenome editing construct (3A3L) and promoter targeting sgRNAs, or with control constructs (non-targeting guide RNAs or catalytically-dead DNMT3A3L). (E) Genomic tracks show H3K27ac over the mouse Cdkn2a promoter in OPCs transfected with epigenome editing constructs, as in (D). sgRNA locations shown below tracks (black bars). (F) Plot shows normalized Cdkn2a (p19 exon) expression for OPCs transfected as in (D-E) from three biologically independent replicates (P values (one-way ANOVA) <0.0001 for Cdkn2a methylation versus controls). (G) CDKN2A-p19ARF and TP53 protein expression in OPCs transfected with the DNMT3A3L construct and promoter targeting sgRNAs versus controls. (H) Growth curves for OPCs transfected as in (G). Data is from three biologically independent replicates (two-sided t-test P values <0.0001). Error bars in panels F, H represent standard deviation. These data indicate that expression of DNMT3A3L and sgRNA leads to Cdkn2a promoter methylation, p19ARF silencing, and increased OPC proliferation.
To investigate the functional significance of CDKN2A silencing, we used epigenome editing to target DNA methylation to the promoter of Cdkn2a/p19ARF, which is the mouse homolog of human CDKN2A/p14ARF. We transfected mouse OPCs with a construct encoding a fusion between catalytically-inactive Cas9 and the DNMT3A3L methyltransferase (dCas9-3A3L)33 plus a pair of convergent sgRNAs. This led to a robust increase in methylation and a nearly complete loss of acetylation over the promoter. These methylation and chromatin changes were accompanied by robust silencing of Cdkn2a/p19ARF mRNA and protein (Figure 3D–G, Figure S4B–D, Table S3). Like its human homolog, Cdkn2a/p19ARF prevents the degradation of the P53 tumor suppressor by regulating MDM234. Consistently, silencing of Cdkn2a/p19ARF in the OPCs also led to a striking reduction in P53 protein levels (Figure 3G). Importantly, the engineered Cdkn2a methylation also increased OPC proliferation by nearly 2-fold (Figure 3H).
Our findings indicate that recurrent methylation events in IDHmut gliomas deregulate the PDGFRA proto-oncogene and CDKN2A tumor suppressor, and stimulate OPC proliferation. This prompted us to test for cooperation between the respective lesions by combinatorial CRISPR editing. We transfected OPCs with a construct expressing Cas9 plus two sgRNAs targeting the Pdgfra insulator and the p19ARF exon (Cdkn2a exon 1B). This led to robust Pdgfra upregulation and Cdkn2a/p19ARF downregulation (Figure 4A–C), and increased OPC proliferation by ~3-fold (Figure 4D).
Figure 4. Combined Pdgfra insulator disruption and Cdkn2a silencing drives hyperproliferation in vitro and low-grade gliomagenesis in vivo.
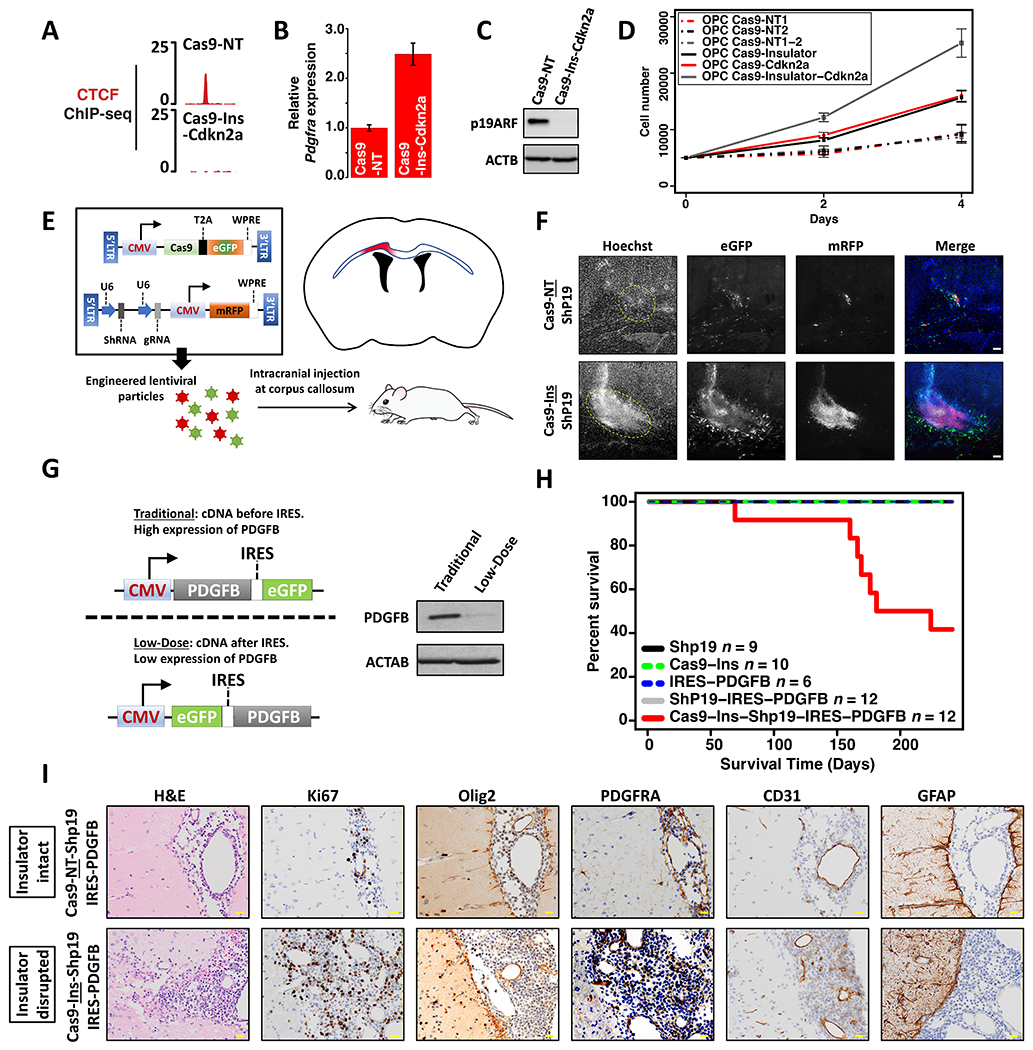
CTCF binding profile (A), Pdgfra mRNA expression (three biologically independent replicates, P values (two-sided t-test) <0.0001) (B), and western blot for Cdkn2a/p19ARF (C) confirm Pdgfra insulator disruption, Pdgfra upregulation, and Cdkn2a silencing in OPCs with dual disruption of the Pdgfra insulator and Cdkn2a (Cas9-Ins-Cdkn2a). (D) Growth curves shown for OPCs transfected with constructs targeting the Pdgfra insulator and/or Cdkn2a, or corresponding controls. Data is from three biologically independent replicates (P values (one-way ANOVA) <0.0001 for deletions versus control). (E) Strategy used to disrupt the Pdgfra insulator and/or Cdkn2a in mouse brain. Lentiviral vectors expressing Cas9, sgRNA, and shRNA were injected into the corpus callosum (blue outline), targeting a small area (red) just above the lateral ventricles (black). (F) Fluorescence images (10X) of the corpus callosum of an adult mouse one week post lentiviral injection. GFP (marking Cas9 expression), mRFP (marking sgRNA and shRNA) and Hoechst (nuclei) channels are shown in grayscale. Rightmost image merges GFP (green), mRFP (red) and Hoechst (blue). (G) Left: Schema shows expression construct used to express low-dose PDGFB (IRES dampened), compared to a traditional CMV driven construct. Right: Western blot shows PDGFB expression in 293T cells transfected with traditional or low-dose constructs. (H) Kaplan-Meier survival curve for mice injected with the indicated lentiviral preparations. The number of mice injected is indicated for each arm in the panel legend. (I) Histological images of stained brain sections from mice injected with lentivirus expressing a shRNA targeting Cdkn2a (p19ARF), IRES-PDGFB and Cas9 with either non-targeting sgRNA (NT) or the Pdgfra insulator (Ins) sgRNA. Yellow scale bar: 20 μm. Error bars in panels B, D represent standard deviation. These data show that combined disruption of Cdkn2a and the Pdgfra insulator drives hyperproliferation in vivo and low grade gliomagenesis in the presence of low level PDGFB ligand.
Combinatorial disruption of the Pdgfra insulator and Cdkn2a drives tumorigenesis in vivo
We investigated the potential of the respective epigenetic lesions to drive gliomagenesis in vivo. We engineered lentiviral constructs expressing Cas9 with sgRNA targeting the CTCF insulator (or a non-targeting control) and an shRNA targeting Cdkn2a/p19ARF (Figure 4E). Lentiviruses were injected into the mouse corpus callosum, an anatomical landmark enriched for OPCs. Examination of mouse brains harvested seven days after lentiviral injection revealed hypercellularity in the group with combined disruption of Pdgfra insulator and p19ARF, but not with either lesion alone (Figure 4F and Figure S4E–F). However, despite a strong induction of hypercellularity, we did not observe further malignant progression in these mice.
We reasoned that these lesions, while powerful in their ability to alter gene expression, might require additional environmental context for efficient tumorigenesis. We therefore sought to combine them with low-level PDGF ligand, a frequently expressed mitogen in gliomas including IDHmut glioma35. We added to our constructs an IRES-PDGFB cassette that expresses PDGFB at a low dose insufficient to drive gliomagenesis on its own36 (Figure 4G). We found that the combination of Pdgfra insulator disruption, Cdkn2a/p19ARF repression and low-dose PDGFB led to malignant gliomas with a median survival of 202 days and ~50% penetrance (Figure 4H). Histological analysis of brain cross-sections showed diffuse hypercellularity and widespread infiltration of brain parenchyma (Figure 4I). Immunohistochemistry (IHC) analysis of brain cross-sections at the injection site confirmed expression of the proliferation marker Ki67, the OPC-markers OLIG2 and PDGFRA, as well as the vasculature marker CD31 (Figure 4I). The tumors lacked expression of GFAP, which marks NPCs and astrocytes37. No single lesion nor the combination of p19ARF repression and low-dose PDGFB showed signs of malignant progression over the duration of our study (Figure 4H). These results establish Pdgfra insulator loss and Cdkn2a promoter silencing as epigenetic drivers of IDHmut gliomagenesis.
The human PDGFRA locus and glioma susceptibility
The PDGFRA locus exhibits striking synteny of gene order and conserved topology in human and mouse (Figure 1A–B; Figure 5A). However, the CpG density of the PDGFRA insulator varies considerably across these and other mammalian species (Figure 5B). The 600 bp window centered on the CTCF binding site contains nine CpGs in human but just three CpGs in mouse. In particular, the mouse insulator lacks the critical CpG closest to the CTCF motif. Furthermore, when we targeted DNMT3A3L to the insulator in mouse OPCs, this led to full methylation of the three CpGs, but failed to impact CTCF binding (Figure S5E–F). Hence, although the mouse CTCF insulator is conserved in location and function, its insensitivity to DNA methylation may explain challenges associated with modeling IDH mutations in mouse.
Figure 5. Human-specific features of the PDGFRA locus and glioma risk.
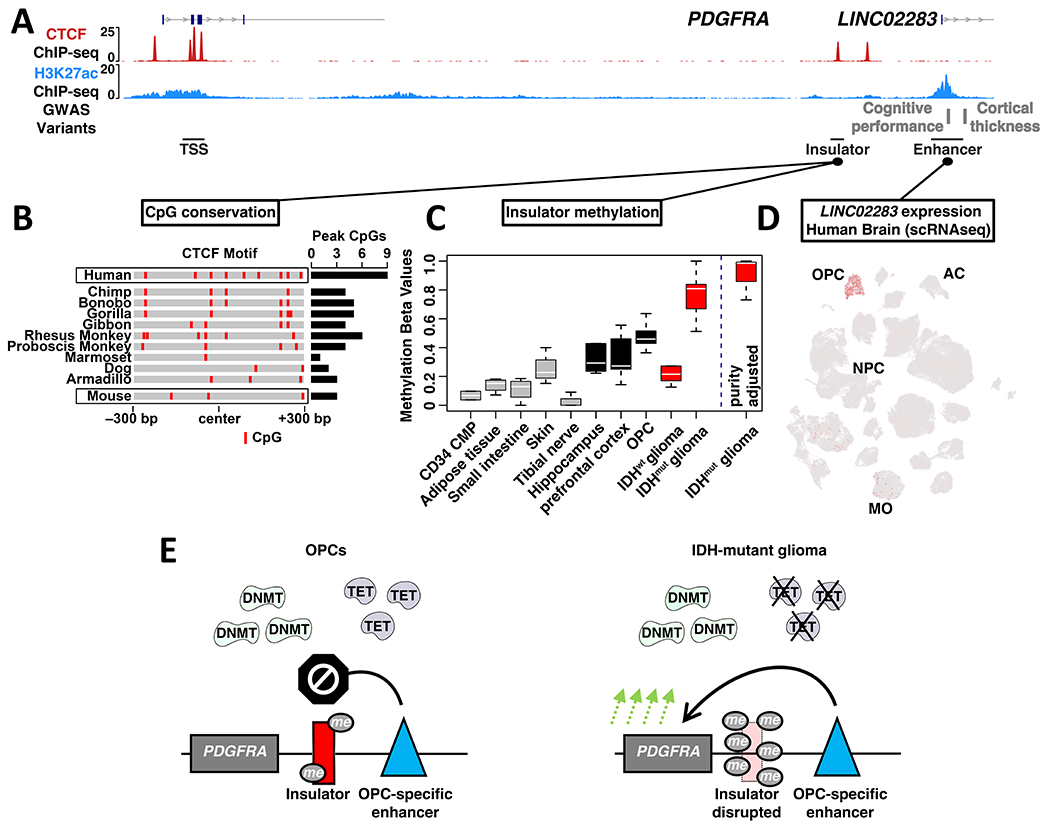
(A) ChIP-seq signals for CTCF (NPCs) and H3K27ac (OPCs) are shown for a ~110 kb region encompassing PDGFRA, insulator and OPC-specific enhancer. Genetic variants associated with cognitive performance and cortical thickness that coincide with the enhancer are indicated (GWAS), along with a corresponding non-coding RNA. (B) Heat depicts CpG dinucleotides over the PDGFRA insulator across representative mammalian species. Insulator intervals were defined based on synteny and conservation to the 600 bp CTCF binding peak called from human ChIP-seq data. The total number of CpGs in the 600 bp intervals is indicated for each species at right. (C) Box plot depicts PDGFRA insulator methylation in selected non-brain human cell and tissue types, human brain compartments, OPC-enriched fractions from human brain, and for IDHwt and IDHmut gliomas. Box plot at right depicts insulator methylation in IDHmut gliomas after correction for sample purity. (D) t-SNE plot generated from scRNA-seq of human brain cells in the middle temporal gyrus is annotated for expression of the non-coding RNA that emanates from the enhancer (LINC02283; MO: mature oligodendrocytes; AC: astrocytes; data from Allen brain map)50. (E) Proposed model contrasts insulator states in normal human OPCs and IDHmut glioma. In OPCs, methyltransferases (DNMT) and demethylases (TET) maintain low to intermediate methylation levels that leave the PDGFRA insulator largely intact. In IDHmut gliomas, inhibition of the demethylases results in hypermethylation and disruption of the insulator, driving aberrant PDGFRA expression and tumorigenesis.
We next assessed whether the human insulator is subject to methylation under physiologic contexts, leveraging public methylation profiles38,39. Insulator methylation was very low in most non-brain tissues and organs (Figure 5C, Figure S1G), but relatively higher in brain samples (Figure 5C). Insulator methylation was also evident in OPC-enriched fractions from human brain40, suggesting that these progenitors may be affected (Figure 5C). The methylation levels in brain and OPCs remain considerably lower than the levels in IDHmut gliomas (~77%, Figure 5C). Given that typical purity estimates for these tumors range from 70 to 80%41, insulator methylation likely approaches 100% in malignant IDHmut cells (Figure 5C; see Methods)42. Nonetheless, intermediate methylation levels in OPCs could predispose the insulator to hypermethylation under conditions where Tet demethylase activity is suppressed by mutant IDH.
We also examined the context-specificity and physiology of the OPC-specific enhancer in the PDGFRA locus. The enhancer coincides with a long intergenic non-coding RNA (LINC02283) that provides a surrogate measure of enhancer activity (Figure 5A; Figure S5A). LINC02283 expression showed remarkable specificity to bulk brain samples (Figure S5B), and OPCs within brain (Figure 5D), consistent with the specific activation of the enhancer in OPCs (Figure S2A–B). Notably, expression of LINC02283 and PDGFRA are highly correlated across IDHmut gliomas (r~0.45), but less correlated in IDHwt gliomas (r~0.3) (Figure S5C–D), consistent with insulator dysfunction in the hypermethylated subtype.
The OPC-specificity of the enhancer and coincident non-coding RNA led us to consider their possible physiologic functions. We found that the enhancer overlaps genetic variants associated with cortical thickness43 and cognitive performance44, both of which represent phenotypes that could be affected by OPCs (Figure 5A). These associations provide further indication that the enhancer has a critical function in OPCs. This function might involve the noncoding RNA, though it is not clear how this transcript could affect OPC biology. Alternatively, it could boost PDGFRA expression in OPCs via long-range interactions that may be facilitated by partial methylation of the insulator.
These analyses indicate that the OPC-specific enhancer has critical physiologic functions in human brain that may involve a non-coding RNA and/or long-range effects on PDGFRA expression. In the setting of IDH mutation and hypermethylation, the CpG-rich human insulator can be fully disrupted, causing the enhancer to activate PDGFRA expression and thereby drive gliomagenesis (Figure 5E).
Discussion
We have modeled the impact of recurrent epigenetic lesions that deregulate oncogene and tumor suppressor gene expression in IDHmut lower-grade gliomas. The respective lesions arise in the setting of global DNA hypermethylation and involve the methylation-dependent disruption of a PDGFRA insulator and silencing of the CDKN2A promoter. We demonstrate that these regulatory alterations can drive progenitor cell proliferation in vitro and tumorigenesis in vivo. Defining epigenetic drivers has been a major challenge due to the inherent variability of epigenetic landscapes and the lack of experimental models for their evaluation. We overcome this challenge by identifying recurrent methylation events in human tumors and validating their functions using contextual mouse progenitor models and targeted in vivo perturbation.
Our study also has implications for tracing glioma cells of origin, which presents a special challenge due to the complexity of neural development. Disruption of the CTCF insulator induces Pdgfra expression and proliferation specifically in OPCs, which harbor an RFX-driven enhancer in the neighboring TAD that is unleashed upon insulator loss. Consistently, engineering of the lesions in vivo drove tumorigenesis in an OPC-rich brain compartment. While the initiating IDH mutation and consequent hypermethylation could arise at earlier developmental stages, our findings suggest that they initiate tumors in the OPC context. Interestingly, clonal IDH mutations were recently described in normal human brain specimens, primarily within NeuN-negative glial populations comprising OPCs, astrocytes and oligodendrocytes45. These data suggest that IDH mutations confer a proliferative advantage to glial cells in human brain, and thus complement our mouse modeling data pinpointing OPCs as a likely cell-of-origin for this glioma subtype.
Modeling lower-grade gliomas in vivo has been complicated by their slow growth and the slow kinetics with which they accumulate hypermethylation. Addition of mutant IDH has even been found to moderate prevailing genetic glioma models25–27. We circumvented this challenge by directly engineering downstream lesions, and thus informing the functions of at least a subset of functional mediators. Nonetheless, our models are limited in that they do not fully recapitulate the mutant IDH phenotype which, in addition to widespread DNA hypermethylation, has also been associated with alterations to histone methylation46, metabolic programs23, differentiation17, and the immune microenvironment26,27. Further studies are needed to discern the functional significance and interactions of these potential mediators. Nevertheless, our framework for modeling and characterizing epigenetic lesions and mediators can guide such studies, while also advancing understanding of hypermethylator phenotypes and epigenetic regulatory alterations across a range of human cancers.
We also document human-specific features of the PDGFRA insulator that can explain challenges associated with IDHmut mouse models and have important implications for human biology. The PDGFRA locus is highly conserved between human and mouse, with extended synteny over >2 Mb and a remarkably concordant topological structure. The constitutive CTCF occupancy of the insulator and the OPC-specificity of the enhancer are also shared between species. Despite this conservation, the CpG density over the insulator differs markedly with nine CpGs in human but just three in mouse (Figure 5B). A technical consequence of this divergence is that the methylation-dependent insulator disruption in human IDHmut gliomas could not be recreated in mice which lack the critical CpGs whose methylation disrupts CTCF binding and insulator function. Although we circumvented this limitation by direct genetic disruption of the CTCF motif, mouse models driven by IDH mutation and hypermethylation may not realize this critical downstream oncogenic event. Indeed, IDH mutations have been associated with inconsistent outcomes in mouse glioma models, promoting proliferation and invasion in certain contexts, but slowing tumor progression in others.
More broadly, our analyses suggest that the CpG-rich insulator and the OPC-specific enhancer that it restrains play critical physiologic roles in human brain. The insulator is moderately methylated in normal brain. The methylation may partially destabilize the insulator in OPCs, allowing the enhancer to boost PDGFRA expression. Enhanced expression of this critical regulator could in turn increase proliferation or otherwise alter OPC biology during human brain development. Importantly, the OPC-specific enhancer coincides with genetic variants associated with cortical thickness and cognitive performance. The associations provide further support for a link between enhancer, PDGFRA and OPC biology – a link that may be strengthened by human-specific features and physiologic methylation that destabilize the intervening insulator. We draw analogy to recent reports that insulator boundaries in the SOX2 and HOX loci become permissive to enhancer-promoter contacts during specific developmental stages19,47,48.
Insulator methylation levels in human brain and OPCs are increased but do not approach the levels in IDHmut gliomas. In the tumors, high concentrations of 2HG inhibit Tet family demethylases leading to their characteristic hypermethylation. Tet enzymes modulate methylation levels over regulatory elements and play critical roles in brain development. The recurrent methylation and disruption of the PDGFRA insulator in tumors likely reflects some combination of its lability in normal development, its dependence on Tet enzymes for homeostatic regulation, and the proliferative fitness conferred by its loss. Thus, the same human-specific insulator features that appear to enhance OPC phenotypes could also create a vulnerability to glioma. We draw analogy to recent studies that implicated a human-specific neural cell type in the initiation of pediatric medulloblastoma brain tumors49. The extent to which other human-specific features or adaptations also confer risk for brain tumors or other cancers is an exciting area for future inquiry.
In conclusion, we have investigated the causality of two epigenetic regulatory lesions that arise in hypermethylated IDHmut gliomas. We validated both lesions as drivers of OPC proliferation and in vivo gliomagenesis. Our engineered mouse tumor models, driven by regulatory alterations downstream of the initiating IDH mutation, can provide a framework for modeling epigenetic cancer drivers going forward. The finding that methylation-dependent disruption of a single CTCF insulator reorganizes locus topology, upregulates the PDGFRA oncogene and represents a bona fide tumor driver is of high interest. Beyond its role in cancer, our analysis suggests that the human insulator may be partially methylated in physiologic contexts with potential effects on PDGFRA expression and OPC phenotypes. Future studies are needed to further characterize the PDGFRA locus and its regulation in developing brain tissue and tumors, as well as to identify and characterize other epigenetic lesions that drive tumorigenesis.
Limitations of the study
Our approach to model IDHmut gliomas by directly perturbing downstream lesions is unlikely to fully recapitulate the pathological roles of mutant IDH. The 2HG neo-metabolite generated by mutant IDH can also impact histone methylation, hypoxia programs, immune function, and other cellular programs which we do not model using our approach15. Incorporation of additional downstream mediators and/or the IDH mutation itself into our model may yield tumors that more closely recapitulate IDHmut glioma pathology. Additionally, we used CRISPR-Cas9 to incur genetic changes that simulate epigenetic lesions discovered in IDHmut gliomas. While this approach effectively induced tumorigenesis, it likely misses specific features associated with the sequencesparing methylation events in the human tumors. Although targeted epigenome remodeling using Cas9m4-3A3L or other tools could address this limitation, such modelling efforts would need to overcome the lack of CpGs in the mouse Pdgfra insulator or leverage other in vivo systems. Finally, we note that our model emulates early stages of gliomagenesis and does not capture other genetic, epigenetic and physiologic changes that arise during the inevitable progression of IDHmut gliomas to higher grade malignancies20.
STAR Methods
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Bradley E. Bernstein (Bradley_Bernstein@DFCI.HARVARD.EDU).
Materials availability
Materials used in this study will be provided upon request and available upon publication.
Experimental model and study participant details
Derivation of NPCs and OPCs from mouse ES cells
V6.5 ES cells were cultured on mouse embryonic fibroblast feeder layers and differentiated to NPCs as described31. Briefly, confluent ES cells were separated from the feeder layer by incubation with a 0.5 mM EDTA solution in PBS for 10 minutes. Cells were then spun down, and plated in low-binding dishes without Leukemia Inhibitory Factor (LIF) for 24 hours, followed by incubation with NPC generation media (DMEM:F12, 1X N2, 1X B27, 1% penicillin/streptomycin, 500 ng/ml Noggin) for four days. The resulting embryoid bodies were then dissociated using accutase and plated with NPC expansion media (same as NPC generation without noggin, but with 20 ng/ml of EGF and FGF2 and 1 μg/ml laminin). NPCs were maintained in NPC expansion media for 5-7 passages from generation. OPCs were generated from NPCs as described31. Briefly, ~4 million NPCs were plated in a 10 cm dish in OPC derivation media (same as NPC generation media, without noggin, with 10 ng/ml PDGFB) for seven days. OPCs were then propagated in OPC proliferation media (same as OPC derivation media with 20 ng/ml FGF2 added). OPCs were only maintained for up to five passages and were closely examined for differentiation under phase contrast microscopy. Only OPCs with bipolar morphology56 were used for experiments. NPCs were differentiated in NPC expansion media supplemented with 4% FBS for four days. OPCs were differentiated in NPC derivation media without Noggin, with 10 ng/ml CNTF, 5 ng/ml NT3, and 40 ng/ml T3 for seven days, as described31.
Genome and epigenome editing
For genome editing, we designed a U6-sgRNA-CMV-Cas9-T2A-eGFP piggyBac plasmid with the following sgRNAs: mouse non-targeting 1: 5’-GCGAGGTATTCGGCTCCGCG-3’; mouse non-targeting 2: 5’-GCTTTCACGGAGGTTCGACG-3’; Pdgfra insulator: 5’-AATTGTT AAAAGTTCCACAA-3’; Cdkn2a/p19ARF exon 1B: 5’-CGGGCCGCCCACTCCAAGAG-3’; RFX motif in OPC enhancer: 5’-CTGCCCCCTTCCCGTTGCCA-3’; sgRNAs were designed to disrupt the NGG portions of the CTCF motif and the RFX motif using the Benchling software. sgRNAs designed to disrupt the Cdkn2a exon 1B were designed using the ChopChop software. In both cases, sgRNAs were designed to have high specificity. Constructs harboring two sequential sgRNAs cassettes were cloned using standard methods. We verified mutations at target loci using Illumina sequencing and Crispresso software (insulator, RFX motif deletion), or verified gene deletion by western blot (CDKN2A). Primers used to amplify loci for sequencing were: Pdgfra-insulator 5’-GTCAGGAGTAGATCCTCGTG-3’ and 5’-GCTGAAGACTGGGAGCTATA-3’. RFX-motif 5’-TCTCCCTGTTTGGTGCCCTT-3’ and 5’-CTCTCCATCAATCATTGCCAAC-3’. For epigenome editing, we designed a U6-sgRNA-U6-sgRNA-CMV-Cas9m4-DNMT3A3L-P2A-eGFP piggyBac plasmid to de novo methylate the Cdkn2a promoter region. sgRNAs were designed to be convergent and have high specificity using the ChopChop software. The non-targeting sgRNAs are the same as the ones described in the genome editing section above. The sgRNAs used to target the Cdkn2a p19ARF promoter were: sgRNAI 5’-CCCCCGAGTCCCAAGGCGCG-3’, sgRNA2 5’-GCGCTGGCTGTCACCGCGAT-3’. The dead version of the epigenome editing construct had two mutations in the DNMT3A3L (C706A and R832E) that were previously shown to disrupt catalytic function without affecting expression33. We verified epigenome editing by locus bisulfite sequencing. Briefly genomic DNA was extracted (DNeasy kit, Qiagen) and subjected to bisulfite conversion (EZ DNA Methylation-Lightning Kit, Zymo Research). Each conversion was split to four independent PCR reactions and the Cdkn2a promoter was then PCR amplified (KAPA HiFi Uracil+ HotStart ReadyMix, KAPA no. KK2800) using the following primers: 5’-GAAAATTTTTTTTTGGAGTGGG-3’ and 5’-CCTCTAAAAAACTTTCC-3’. Sequencing reads (Illumina) were aligned to the bisulfite converted locus and the frequency of methylated to unmethylated Cytosines was calculated.
NPCs or OPCs were transfected with 1:1 ratio of the genome/epigenome editing construct along with the piggyBac transposase construct using LipoD293 per manufacturer’s guidelines. Cells were sorted twice for GFP positivity on a Sony SH800 flow cytometer, once 48 hours after transfection, and another time after the cells reached confluence from the initial sort.
Hybrid selection bisulfite sequencing on glioma tissues
Hybrid selection was performed for four IDHmut and four IDHwt gliomas using a set of probes designed to capture all CTCF sites, promoters and the CDKN2A locus, as described57. Briefly, genomic DNA was isolated using the DNeasy Blood and Tissue Kit (Qiagen), sheared using the Covaris LE220, then size selected to 150–300 bp (Tapestation verified-D1000 tapes, Agilent). One microgram of gDNA was end repaired, 3′ A base tailed (KAPA Hyper Prep Kit no. KK8502) and ligated to sequencing adaptors (Roche SeqCap Epi Enrichment System). Ligated products were purified using AMPure XP beads, bisulfite-converted (EZ DNA Methylation-Lightning Kit, Zymo Research), and then PCR amplified (KAPA HiFi Uracil+ HotStart ReadyMix, KAPA no. KK2800). Sets of four libraries were then combined at equal concentrations with SeqCap Epi universal and indexing oligos and the bisulfite capture enhancer (SeqCap Epi Accessory Kit). Pools were lyophilized (TOMY Micro-Vac, MV100), resuspended in hybridization buffer (SeqCap Epi Hybridization and Wash Kit), and hybridized to the SeqCap Epi Probe Pool (Roche) for 72 hours at 47 °C in a thermocycler. Capture libraries were recovered (SeqCap Pur Capture Bead Kit) at 47 °C in a thermocycler, washed in a 47 °C water bath, and amplified by PCR (SeqCap Epi Accessory Kit). Libraries were sequenced with 10% PhiX addition as 100 base paired-end reads on an Illumina HiSeq2500 in rapid mode. Glioma samples were obtained under an IRB approved protocol.
Stereotactic injections of mice with lentiviruses and survival monitoring
Stereotactic injections of mice with purified lentiviruses were done as described55,58. Lentiviral constructs with CMV-Cas9-T2A-eGFP, U6-ShRNA-U6-sgRNA-CMV-RFP, and U6-ShRNA-U6-sgRNA-CMV-RFP-IRES-PDGFB were cloned using standard methods, transfected into 293FT cells along with pVSV-G and pCMV delta R8.2 using LipoD293 according to the manufacturer’s guidelines. Supernatant was collected 48 hours and 96 hours after transfection and purified as described58. Viral pellets were resuspended in PBS, and frozen at −80 °C. Viruses were titrated by serially infecting 293FT cells cultured with 10 μg/ml polybrene. Fluorescent colonies were counted on an epifluorescent microscope under 20x magnification. Equal amounts (10^8 viruses/ml) of lentiviruses were mixed and injected into 8-12-week-old 129S1/SvlmJ mice (Jackson lab strain 002448). Briefly, mice were anesthetized using 4% isoflurane on a stereotactic frame (Stoelting) and injected with 2.5 μL (total of 4*106 fluorescent colony forming units, rate of 0.5 μL/min) in the corpus callosum using the following coordinates relative to bregma: x = −1.1, y = −1.9, and z = −1.8/−1.7/−1.6, with x representing left(−)/right(+), y: anterior(+)/posterior(−), and z: depth from surface of the skull. All mouse experiments were done following an Institutional Animal Care and Use Committee–approved protocol. Equal number of female and male mice were used. The shRNA sequence targeting the Cdkn2a exon 1B (p19ARF) was previously validated in vivo58. shRNA sequence: 5’-CCGGCGCTCTGGCTTTCGTGAACATCTCGAGATGTTCACGAAAGCCAGAGCGTTTTTG-3’. Mice were monitored for terminal symptoms daily, which included a body condition score of 2 or lower or seizures, at which point the mice were euthanized and tissues were collected for processing.
Method details
Chromatin immunoprecipitation
ChIP-seq was performed as described9. Briefly, cultured cells were crosslinked in 1% formaldehyde for 12 minutes, flash frozen in liquid nitrogen, and then stored at −80 °C. Chromatin was fragmented using a Branson Sonifier calibrated to shear DNA to between 200-600 bp fragment length. CTCF was precipitated using a monoclonal rabbit antibody (Cell signaling clone D31H2, no. 3418). H3K27ac was precipitated using a polyclonal rabbit antibody (Active Motif no. 39133). Eluted ChIP DNA was used to generate sequencing libraries by end repair (End-It DNA repair kit, Epicentre no. ER81050), 3’ A-tailing by using Klenow exo- (NEB no. M0212L), and ligation of barcoded sequencing adapters. Barcoded fragments were amplified by PCR using PfuUltra II Hotstart Mix (Agilent no. 600850) for 16 cycles and double-size selected using AMPure XP beads for fragments between 300-500 bp.
4C-seq
4C-seq was performed as described59. Briefly, ~5 million cells were crosslinked in 1% formaldehyde for 12 minutes. Fixed samples were lysed in lysis buffer containing protease inhibitors and dissociated by pipetting. Nuclei were digested with DpnII (NEB) overnight at 37 °C in a thermomixer at 950 rpm. Reactions were then heat inactivated at 65 °C for 20 minutes, ligated with T4 DNA ligase (NEB) for four hours at room temperature, followed by RNase A and proteinase K digestion. DNA was then purified and digested with Csp6I (NEB) overnight at 37 °C, then ligated with T4 DNA ligase for four hours at room temperature. DNA was then purified, run on an agarose gel to verify circularization, and then amplified/prepared for Illumina sequencing using PCR with the Superfi taq polymerase (16 reactions, each containing 200 ng of purified DNA). PCR product was purified with AMPureXP beads and sequenced on an Illumina NextSeq 500. The 4C primers contained sequencing adaptors and barcodes. The annealing sections of the primers were as follows: OPC-specific enhancer viewpoint: 5’-TGTGGCTTGGCATCCTGATC-3’; OPC-specific enhancer non-viewpoint: 5’-TTGAACTCTCAGAGACCCAC-3’.
4C-seq libraries were sequenced as 38 base paired-end reads on an Illumina NextSeq 500. Only the first read (viewpoint) was used for further processing.
Quantitative real-time PCR
Total RNA was isolated using Trizol/BCP and 1 μg was used for cDNA synthesis using the iScript kit (Bio-Rad). cDNA was analyzed with SYBR master mix (Applied Biosystems) on a 7500 Fast Real Time system. Real-time primers were: Pdgfra forward 5’-GACTTCCTAAAGAGTGACCATCC-3’, Pdgfra reverse 5’-CTTCCCAGTCCTTCAGCTTATC-3’, Cdkn2a/p19ARF forward 5’-CATGTTGTTGAGGCTAGAGAGG-3’, Cdkn2a/p19ARF reverse 5’-CACCGTAGTTGAGCAGAAGAG-3’. All gene expression quantifications were normalized to 18S: 18S forward: 5’-GAGGGAGCCTGAGAAACGG-3’, 18S reverse: 5’-GTCGGGAGTGGGTAATTTGC-3’.
Western blots
Western blot antibodies were: Rabbit anti-PDGFRA (Cell signaling 3174), Rabbit anti-CDKN2A/p19ARF (Millipore 07-543), Mouse anti-TP53 (Cell signaling 2524), and beta-actin-HRP (sigma A3854). Secondary HRP-conjugated antibodies were: anti-rabbit IgG-HRP (Cell signaling 7074), and anti-mouse IgG-HRP (Cell signaling 7076).
Growth curve analysis
5000 cells were plated in each well of a 24 well plate such that each timepoint had three wells. Cells were dissociated with accutase, mixed with trypan blue, and then counted using a hemocytometer on days 0, 2, and 4.
Confocal microscopy
Brain sections were analyzed using direct fluorescence on a Zeiss LSM 980 confocal microscope. Briefly 100-μm sections were obtained from embedded brains (2.5% agarose in PBS with 4% sucrose) and cut on a vibratome (Leica VT 1200S). The sections were then placed in a 24-well plate, screened for GFP/RFP fluorescence using an epifluorescent microscope, and mounted using Vectashield antifade mounting medium (Vector Laboratories) on a microscope slide. The ImageJ (FIJI) suite was used to construct maximal projection images from these sections.
Tissue processing and immunohistochemistry
Mice were sacrificed 45 days post lentiviral injections through an i.p. injection of avertin (2,2,2 tribromoethanol) (25 mg/kg) in PBS followed by intracardiac perfusion first with PBS for five minutes, and second with 10% acetate buffered formalin for five minutes. Brains were then placed in formalin overnight before being processed and embedded in paraffin. Eight-micrometer sections of these paraffin blocks were cut on a microtome and deparaffinized by two sequential xylene incubations followed by rehydration through a decreasing gradient of ethanol. Rehydrated sections were stained on a Leica Bond Max automated staining instrument (Leica Microsystems). The following antibodies were used: Ki67 (Cell signaling 12202, dilution 1:50), OLIG2 (Millipore AB9610, dilution 1:100), PDGFRA (Cell signaling 3174, dilution 1:200), CD31 (Cell signaling 77699, dilution 1:100), and GFAP (Dako Z 0334, dilution 1:2500). Antigen retrieval was done for 20 minutes in 10 mM Tris-Cl (pH=9) with 1 mM EDTA, except for the GFAP antibody for which antigen retrieval was done for 20 minutes in 10mM sodium citrate, (pH=6).
Quantification and statistical analysis
CpG counts in mammalian species
We used the LiftOvertool (https://genome.ucsc.edu/cgi-bin/hgLiftOver) to search for the syntenic sequences to the human PDGFRA insulator across the species listed in the vertebrate 30-way phylogenetic comparison using the 600 bp surrounding the CTCF motif in the human insulator site (hg38). The resulting syntenic sequences were first scanned for the presence of the CTCF motif using the MEME suite (version 4.11.3-1) FIMO command. Species lacking the CTCF motif were removed. The genome locations of the syntenic sequences were then probed to ensure that the location of the syntenic CTCF insulator was present in the PDGFRA locus. Species with poor annotations and in which the syntenic insulator site was not present near PDGFRA were removed. The insulator sequences of the remaining species were centered around the CTCF motif and trimmed to 600 bp. We then counted the CpGs in those sequences and plotted their locations on R using the pheatmap package.
Motif analysis
Motif analysis was done using the MEME suite (version 4.11.3-1) FIMO command using default settings, while scanning for all motifs in the “JASPAR_CORE_2016_vertebrates” database (OPC enhancer) or only the CTCF motif in the same database.
Analysis of ENCODE, TCGA, GTEX, and Allen Brain Atlas data
Normalized ChIP-seq data with the corresponding peak calls were downloaded from ENCODE, or a previously published database51. RNA-seq data for lower-grade glioma (LGG) and glioblastoma (GBM) were downloaded from TCGA and separated by IDH mutation status. Samples with deletions in the CDKN2A locus were excluded from the CDKN2A RNA-seq analysis. Whole genome bisulfite sequencing data was downloaded from the archive GDC data portal (TCGA) or Encode (normal tissues, only samples that passed ENCODE QC were used). ATAC-seq data performed on TCGA samples was downloaded from a previously published database52. GTEX data (GTEX dbGaP Accession phs000424.v8.p2) was downloaded from the GTEX portal and plotted on R. Genome locus figures were plotted using the GVIZ package on R version 4.1.3. Data for the insulator or enhancer sites was extracted from the data matrices on R version 4.1.3. Expression of LINC02283 in scRNA-seq from adult brain cells was plotted from Allen Brain Map, dataset MTG - 10X SEA-AD.
4C-seq analysis
Data were analyzed using 4Cseqpipe60 and median normalized with a main trend resolution set at 500 bp. The sequencing primers were part of a published database60. Data was visualized using R version 4.1.3.
Chip-seq analysis
Libraries were sequenced as paired 38 base end reads on an Illumina NextSeq 500. Reads were then aligned to the mm10, or hg19 reference genomes using BWA aln version 0.7.4, removing reads with MAPQ score lower than 10. PCR duplicates were removed by Picard toolkit 2.9.2. Peaks were called with HOMER 4.9, correcting against input controls. H3K27ac and CTCF peaks were called with the ‘histone’ and ‘factor’ settings respectively. Differential analysis of CTCF peaks and quantification of reads per peak were previously done9. CTCF ChIP-seq on IDHmut and IDHwt gliomas was downloaded from GEO (GSE70991)9 and analyzed. BigWig files normalized for reads per million (RPM) were visualized using the GVIZ package on R version 4.1.3. Processed (RPM normalized BigWig), peak annotation, and raw (fastq) data has been deposited on GEO (accession numbers: GSE225794, GSE225732, GSE225793).
Hybrid-selection bisulfite sequencing analysis
Hybrid-selection bisulfite sequencing (HSBS) data were processed by methylCtools 0.9.438 (filtered by a minimum of 5 reads per locus) using BWA mem version 0.7.12, Picard toolkit 2.9.2 (to remove PCR duplicates), and aligned to human reference hg19. CTCF CpG methylation beta values report the methylation of the CpG in the CTCF motif or the closest CpG to the CTCF motif that is located within the called ChIP-seq peak. Promoter CpG methylation beta values report the average methylation for the promoter region covered.
HiC data analysis and visualization
HiC data was obtained from GEO (Datasets GSE63525 and GSE96107) and visualized at 5 kb resolution using the ggplot package on R version 4.1.3. Loops were called by HICCUPS (juicer tools)61. Data displayed in Figure 1 represent human IMR90 cells18 (~1.3 billion reads, aligned to hg19), and mouse NPCs53 (~3 billion reads, aligned to mm10).
Group comparisons
Spearman r values were computed for all correlations. Two-sided t-test P values were computed when comparing two groups. One-way ANOVA P values were computed when comparing more than 2 groups.
Data and code availability
Raw and processed sequencing data generated for this study have been deposited at the NCBI SRA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
The code supporting the current study is available from the corresponding author on request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti CTCF (clone D31H2) | Cell signaling technologies | 3418 |
| Rabbit anti H3K27ac | Active motif | 39133 |
| Rabbit anti PDGFRA | Cell signaling technologies | 3174 |
| Rabbit anti CDKN2A (p19 ARF) | Millipore | 07-543 |
| Mouse anti TP53 | Cell signaling technologies | 2524 |
| beta-actin-HRP conjugated | Sigma | A3854 |
| anti-rabbit IgG-HRP | Cell signaling technologies | 7074 |
| anti-mouse IgG-HRP | Cell signaling technologies | 7076 |
| Rabbit anti-Ki67 (D3B5) | Cell signaling technologies | 12202 |
| Rabbit anti Olig2 | Millipore | AB9610 |
| Rabbit anti CD31 | Cell signaling technologies | 77699 |
| Rabbit anti Glial Fibrillary acidic protein | Agilent Dako | Z0334 |
| Bacterial and virus strains | ||
| NEB stable competent E. coli | New England Biolabs | C3040I |
| Biological samples | ||
| Human IDH wild-type and IDH mutant glioma samples | Flavahan et al. 2016 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| ESGRO Leukemia Inhibitory Factor (LIF) | Millipore | ESG1107 |
| HyClone Fetal Bovine Serum (U.S.), Embryonic Stem (ES) Cell Screened | Hyclone | SH30070.02E |
| Recombinant Human EGF Protein | R&D Systems | 236-EG-200 |
| Recombinant Human FGF basic/FGF2 | R&D Systems | 4114-TC-01M |
| N-2 Supplement (100X) | GIBCO | 17502048 |
| B-27 Supplement (50X), serum free | GIBCO | 17504044 |
| GlutaMAX Supplement | GIBCO | 35050061 |
| Penicillin-Streptomycin | GIBCO | 15140122 |
| MEM Non-Essential Amino Acids Solution (100X) | GIBCO | 11140050 |
| L-Glutamine (200 mM) | GIBCO | 25030081 |
| 2-Mercaptoethanol | Sigma | 6010 |
| Knockout DMEM | GIBCO | 10829018 |
| DMEM:F12 | GIBCO | 11320-033 |
| Neurobasal | GIBCO | 21103-049 |
| IMDM | GIBCO | 31980-030 |
| Fetal Bovine Serum | GIBCO | 26140079 |
| Recombinant Murine IGF-1 | Peprotech | 250-19 |
| Recombinant Human PDGFB | Peprotech | 100-14B |
| Laminin | Sigma | L2020-1MG |
| Klenow, exo- | New England Biolabs | M0212L |
| Critical commercial assays | ||
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | 20024906 |
| Qubit dsDNA HS Assay kit | Invitrogen | Q32854 |
| QIAquick Gel Extraction Kit | Qiagen | 28706 |
| Qiagen Maxiprep plasmid kit | Qiagen | 12163 |
| Agencourt AMPure XP | Beckman Coulter | A63882 |
| Bioanalyzer D1000 screentapes | Agilent | 5067-5582 |
| LipoD293 | Signagen | SL100668 |
| End-It DNA End-Repair Kit | Biosearch technologies | ER0720 |
| TRIzol™ Reagent | Thermo Fisher | 15596026 |
| EZ DNA Methylation-Lightning Kit | Zymo | D5030 |
| Qiagen DNeasy Blood & Tissue Kit | Qiagen | 69504 |
| Deposited data | ||
| Processed data | GEO | GSE225794 |
| Raw data (Chip-seq) | GEO | GSE225732 |
| Raw data (Capture Bis-seq) | GEO | GSE225793 |
| Experimental models: Cell lines | ||
| mESC (v6.5) | Broad Institute | N/A |
| C57BL/6 MEF 4M IRR | GlobalStem | GSC-6002G |
| Mouse NPCs | This study | N/A |
| Mouse OPCs | This study | N/A |
| 293FT cells | Thermo Fisher | R70007 |
| Human NPCs, BYS012 | ATCC | ACS-5004 |
| Experimental models: Organisms/strains | ||
| 129S1/SvImJ | Jackson laboratories | 002448 |
| Oligonucleotides | ||
| Pdgfra insulator CRISPR locus sequencing: GTCAGGAGTAGATCCTCGTGGCTGAAGACTGGGAGCTATA | IDT | N/A |
| RFX motif CRISPR locus sequencing: TCTCCCTGTTTGGTGCCCTTCTCTCCATCAATCATTGCCAAC | IDT | N/A |
| Cdkn2a-p19 promoter locus bisulfite sequencing: GAAAATTTTTTTTTGGAGTGGGCCTCTAAAAAACTTTCC | IDT | N/A |
| OPC enhancer 4C-seq primers: TGTGGCTTGGCATCCTGATC (viewpoint) TTGAACTCTCAGAGACCCAC | IDT | N/A |
| 18S real time PCR primers: GAGGGAGCCTGAGAAACGG (Forward) GTCGGGAGTGGGTAATTTGC (Reverse) | IDT | N/A |
| Pdgfra real time PCR primers: GACTTCCTAAAGAGTGACCATCC (Forward) CTTCCCAGTCCTTCAGCTTATC (Reverse) | IDT | N/A |
| Cdkn2a real time PCR primers: CATGTTGTTGAGGCTAGAGAGG (Forward) CACCGTAGTTGAGCAGAAGAG (Reverse) | IDT | N/A |
| Recombinant DNA | ||
| pVSV-G | Broad Institute | N/A |
| pCMV delta R8.2 | Broad Institute | N/A |
| CMV-Cas9-T2A-eGFP (lentiviral construct) | This study | N/A |
| U6-ShRNA-U6-sgRNA-CMV-RFP (lentiviral construct) | This study | N/A |
| U6-ShRNA-U6-sgRNA-CMV-RFP-IRES-PDGFB (lentiviral construct) | This study | N/A |
| U6-sgRNA-CMV-Cas9-T2A-eGFP (piggybac construct) | This study | N/A |
| Software and algorithms | ||
| R version 4.1.3 | R Core Team | https://www.r-project.org |
| Benchling (for sgRNA design) | Benchling | https://www.benchling.com |
| ChopChop (for sgRNA design) | ChopChop | https://chopchop.cbu.uib.no |
| Cas off finder (off target analysis) | Cas Off Finder | https://www.rgenome.net/cas-offinder/ |
| Juicer tools | Aiden laboratory | https://github.com/aidenlab/juicer |
| BWA aln version 0.7.4 | Li laboratory | https://github.com/lh3/bwa |
| Picard toolkit 2.9.2 | Broad Institute | https://broadinstitute.github.io/picard/ |
| GVIZ package | Bioconductor | https://bioconductor.org/packages/release/bioc/html/Gviz.html |
| methylCtools 0.9.438 | Hovestadt laboratory | https://github.com/hovestadt/methylCtools |
| MEME suite (version 4.11.3-1) | MEME suite | https://meme-suite.org/meme/doc/download.html |
| LiftOver tool | UCSC genome browser team | https://genome.ucsc.edu/cgi-bin/hgLiftOver |
Supplementary Material
Figure S1. Mouse glial progenitor models, and identification of disrupted insulators in the PDGFRA locus in IDHmut glioma, related to Figure 1
NPCs and OPCs were derived from mouse ES cells using established procedures31. (A) ES cell-derived NPCs have a thick cell body and express NPC markers (NESTIN, SOX2). Serum-based differentiation of NPCs (right panels) gave rise to astrocytes (GFAP), oligodendrocytes (NG2), and neurons (β3-TUBULIN), cell types that were otherwise absent in NPC cultures. (B) ES cell-derived OPCs have a circular cell body with thin projections and express OPC markers (OLIG2, PDGFRA, NG2). Differentiation of OPCs gave rise to mature oligodendrocytes. (C) Heatmaps depict CpG methylation in CTCF motifs (left) and CTCF ChIP-seq binding scores (right) across the 2 Mb region that contains PDGFRA. For each CTCF site, bar plot at right depicts significance of differential occupancy between IDH1wt and IDH1mut gliomas. Red arrow highlights the most significantly disrupted site in IDH1mut tumors. (D) Genomic view (20 kb) over the disrupted CTCF site shows CpG methylation (heatmap; each box corresponds to a single CpG) and CTCF ChIP-seq signal (RPM). (E-F) Box plots show distribution of methylation (E) and CTCF ChIP-seq binding signal (F) over the disrupted insulator site in IDH1wt and IDH1mut glioma samples. (G) Bar plot depicts CpG methylation over the CTCF insulator site across a range of human tissues and cell lines, and in gliomas.
Figure S2. Cell type-specificity, RFX motif conservation and functional impact of the OPC-specific enhancer, related to Figures 1 and 2
Genomic tracks show H3K27ac ChIP-seq signal (blue) over the PDGFRA locus for multiple human (A) and mouse (B) tissue and cell types from ENCODE30, a published database51, and data generated in this study. The OPC-specific enhancer region is highlighted by red box. Black lines below ChIP-seq tracks represent called peaks. (C) ATAC-seq signal (grey, normalized) in TCGA glioma samples52 separated by transcriptomic subtype. NC: not classified. H3K27ac ChIP-seq signal (blue, RPM normalized) in glioma samples (top tracks in blue) and glioma spheroid cell lines (lower tracks in blue). The OPC-specific enhancer region is highlighted by red box. (D) OPC-specific enhancer locus with H3K27ac ChIP-seq (blue) and phastCons (mammalian 46-way, navy blue) signal. The H3K27ac called peak is highlighted as a line under the H3K27ac track. The RFX motif present in the OPC-specific enhancer is highlighted as a line under the phastCons track. (E) RNA-seq expression of Rfx transcription factors in NPCs and OPCs derived from ES cells53,54 (left heatmap, GEO datasets GSM2533845 and GSM1557625) or purified from mouse brains53,54 (right heatmap, GEO datasets GSM2533849 and GSM1557624). (F) Growth curves shown for control, insulator-disrupted, and CDK4 overexpressing NPCs from three biologically independent replicates. Insulator disruption does not affect NPC proliferation even after the addition of PDGF ligand.
Figure S3. Efficacy and specificity of sgRNAs targeting the CTCF insulator and RFX motif, related to Figure 2
(A,B) Locus sequencing data demonstrate efficacy of CRISPR editing of the target CTCF motif in (A) NPCs and (B) OPCs edited with the Pdgfra insulator sgRNA (Cas9-Ins) or control (Cas9-NT). (C,D) Plots show differential CTCF ChIP-seq binding between Cas9-Ins and Cas9-NT NPCs (C) and OPCs (D). The insulator CTCF site is the only peak with robust reduction of ChIP-seq signal. (E) Locus sequencing data demonstrate efficacy of CRISPR editing of the target RFX motif in OPCs. (F) Volcano plot shows differential H3K27ac ChIP-seq signals in cells edited with the RFX motif sgRNA, relative to Cas9-NT control cells. The OPC-specific enhancer is the only peak with robust reduction of ChIP-seq signal. N.D = not detected.
Figure S4. CDKN2A deletion rates in human gliomas, efficacy and specificity of DNMT3A3L sgRNAs targeting CDKN2A in mouse OPCs, and impact of PDGFRA and CDKN2A perturbations in vivo, related to Figures 3 and 4
(A) Pie charts indicate proportion of tumors with homozygous loss of the CDKN2A locus, stratified by CpG island methylator phenotype (G-CIMP). The locus is frequently lost in tumors with lower DNA methylation. (B) Locus bisulfite sequencing data demonstrate efficacy of epigenetic editing at four CpG containing hubs within the target locus in OPCs. Percentage values indicate fraction of alleles with methylation of the corresponding CpG. (C) Differential H3K27ac ChIP-seq signal in OPCs with Cas9m4-3A3L and the Cdkn2a promoter sgRNA (3A3L-pCdkn2a), compared to control cells (3A3L-NT). The Cdkn2a promoter is the only H3K27ac peak with robust reduction of ChIP-seq signal. (D) Bar plot depicts average methylation beta values for the targeted promoter (Cdkn2a-p19) and the nearby promoter (Cdkn2b-p15) in mouse OPCs expressing Cas9m4-DNMT3A3L with non-targeting (3A3L-NT) or Cdkn2a targeting (3A3L-pCdkn2a) sgRNA. The epigenetic editor is specific to the targeted promoter. (E) Schematics of vectors used to interrogate the role of enhanced Pdgfra expression along with Cdkn2a (p19ARF) silencing individually or in combination. Vectors used in (E) linked to the histology observed in (F) where transduced cells are fluorescently tracked in the corpus callosum one week after injection of adult mice. White scale bar: 100 μm.
Figure S5. Cell type-specificity of the OPC-specific enhancer and noncoding RNA in human brain compartments, and impact of methylation on the mouse insulator, related to Figure 5
(A) Scatter plot shows LINC02283 expression (x-axis) versus ATAC signal of the OPC-specific enhancer (y-axis) in TCGA gliomas analyzed by ATAC-seq and RNA-seq. (B) Violin plot shows LINC02283 expression in human bulk tissue samples (GTEX)55. (C,D) Scatter plots compare LINC02283 expression (x-axis) against PDGFRA expression (y-axis) in (C) IDHwt and (D) IDHmut gliomas. (E) Bar plot depicts average methylation beta values in mouse OPCs expressing Cas9m4-DNMT3A3L with non-targeting sgRNAs (3A3L-NT) or insulator targeting sgRNAs (3A3L-Insulator). (F) CTCF ChIP-seq signal at the Pdgfra insulator region for mouse OPCs with 3A3L-NT (solid black) or 3A3L-Insulator (dashed red). Methylation of the insulator region does not affect CTCF binding due to sparsity of CpG dinucleotides in mouse. Spearman r values are computed for all correlations. Shaded areas represent confidence interval in A, C and D.
Highlights.
A PDGFRA insulator and CDKN2A promoter are recurrently methylated in IDH-mutant gliomas
Disruption of the orthologous insulator in mouse induces PDGFRA and OPC proliferation
Mouse glioma model engineered by combining insulator disruption with CDKN2A silencing
Human-specific features of PDGFRA locus may impact glioma risk and brain development
Acknowledgements
The authors thank Mario Suva, Chuck Stiles, Mark Israel, and all Bernstein lab members for their critical feedback on this work. This project received support from the National Cancer Institute, the NIH Common Fund, and the American Cancer Society. G.J.R. is supported by a K99 award from the National Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
B.E.B. discloses financial interests in Fulcrum Therapeutics, HiFiBio, Arsenal Biosciences, Chroma Medicine, Cell Signaling Technologies, and Design Pharmaceuticals.
References
- 1.Baylin SB, and Jones PA (2011). A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 11, 726–734. 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flavahan WA, Gaskell E, and Bernstein BE (2017). Epigenetic plasticity and the hallmarks of cancer. Science 357. 10.1126/science.aa12380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esteller M (2008). Epigenetics in cancer. N Engl J Med 358, 1148–1159. 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 4.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. (2010). Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522. 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research Network, Brat DJ, Verhaak RGW, Aldape KD, Yung WKA, Salama SR, Cooper LAD, Rheinbay E, Miller CR, Vitucci M, et al. (2015). Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med 372, 2481–2498. 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, and Tilghman SM (2000). CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405, 486–489. 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- 7.Bell AC, and Felsenfeld G (2000). Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405, 482–485. 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- 8.Dixon JR, Gorkin DU, and Ren B (2016). Chromatin Domains: The Unit of Chromosome Organization. Mol Cell 62, 668–680. 10.1016/j.molcel.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suvà ML, and Bernstein BE (2016). Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114. 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. (2015). Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025. 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karnuta JM, and Scacheri PC (2018). Enhancers: bridging the gap between gene control and human disease. Hum Mol Genet 27, R219–R227. 10.1093/hmg/ddy167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valton A-L, and Dekker J (2016). TAD disruption as oncogenic driver. Curr Opin Genet Dev 36, 34–40. 10.1016/j.gde.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Margueron R, and Reinberg D (2010). Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet 11, 285–296. 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan H, Renaud L, Chaligne R, Bloehdorn J, Tausch E, Mertens D, Fink AM, Fischer K, Zhang C, Betel D, et al. (2021). Discovery of Candidate DNA Methylation Cancer Driver Genes. Cancer Discov 11, 2266–2281. 10.1158/2159-8290.CD-20-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cairns RA, and Mak TW (2013). Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov 3, 730–741. 10.1158/2159-8290.CD-13-0083. [DOI] [PubMed] [Google Scholar]
- 16.Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K, and Stamatoyannopoulos JA (2015). Role of DNA Methylation in Modulating Transcription Factor Occupancy. Cell Rep 12, 1184–1195. 10.1016/j.celrep.2015.07.024. [DOI] [PubMed] [Google Scholar]
- 17.Modrek AS, Golub D, Khan T, Bready D, Prado J, Bowman C, Deng J, Zhang G, Rocha PP, Raviram R, et al. (2017). Low-Grade Astrocytoma Mutations in IDH1, P53, and ATRX Cooperate to Block Differentiation of Human Neural Stem Cells via Repression of SOX2. Cell Rep 21, 1267–1280. 10.1016/j.celrep.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Misteli T (2020). The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell 183, 28–45. 10.1016/j.cell.2020.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. (2009). IDH1 and IDH2 mutations in gliomas. N Engl J Med 360, 765–773. 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wakimoto H, Tanaka S, Curry WT, Loebel F, Zhao D, Tateishi K, Chen J, Klofas LK, Lelic N, Kim JC, et al. (2014). Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin Cancer Res 20, 2898–2909. 10.1158/1078-0432.CCR-13-3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AWM, Lu C, Ward PS, et al. (2012). IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483. 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McBrayer SK, Mayers JR, DiNatale GJ, Shi DD, Khanal J, Chakraborty AA, Sarosiek KA, Briggs KJ, Robbins AK, Sewastianik T, et al. (2018). Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 175, 101–116.e25. 10.1016/j.cell.2018.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bardella C, Al-Dalahmah O, Krell D, Brazauskas P, Al-Qahtani K, Tomkova M, Adam J, Serres S, Lockstone H, Freeman-Mills L, et al. (2016). Expression of Idh1R132H in the Murine Subventricular Zone Stem Cell Niche Recapitulates Features of Early Gliomagenesis. Cancer Cell 30, 578–594. 10.1016/j.ccell.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pirozzi CJ, Carpenter AB, Waitkus MS, Wang CY, Zhu H, Hansen LJ, Chen LH, Greer PK, Feng J, Wang Y, et al. (2017). Mutant IDH1 Disrupts the Mouse Subventricular Zone and Alters Brain Tumor Progression. Mol Cancer Res 15, 507–520. 10.1158/1541-7786.MCR-16-0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alghamri MS, McClellan BL, Avvari RP, Thalia R, Carney S, Hartlage CS, Haase S, Ventosa M, Taher A, Kamran N, et al. (2021). G-CSF secreted by mutant IDH1 glioma stem cells abolishes myeloid cell immunosuppression and enhances the efficacy of immunotherapy. Sci Adv 7, eabh3243. 10.1126/sciadv.abh3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amankulor NM, Kim Y, Arora S, Kargl J, Szulzewsky F, Hanke M, Margineantu DH, Rao A, Bolouri H, Delrow J, et al. (2017). Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev 31, 774–786. 10.1101/gad.294991.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zong H, Parada LF, and Baker SJ (2015). Cell of origin for malignant gliomas and its implication in therapeutic development. Cold Spring Harb Perspect Biol 7. 10.1101/cshperspect.a020610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110. 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, Kaul R, et al. (2020). Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710. 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kerman BE, Kim HJ, Padmanabhan K, Mei A, Georges S, Joens MS, Fitzpatrick JAJ, Jappelli R, Chandross KJ, August P, et al. (2015). In vitro myelin formation using embryonic stem cells. Development 142, 2213–2225. 10.1242/dev.116517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Costello JF, Berger MS, Huang HS, and Cavenee WK (1996). Silencing of p16/CDKN2 expression in human gliomas by methylation and chromatin condensation. Cancer Res 56, 2405–2410. [PubMed] [Google Scholar]
- 33.Stepper P, Kungulovski G, Jurkowska RZ, Chandra T, Krueger F, Reinhardt R, Reik W, Jeltsch A, and Jurkowski TP (2017). Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3L methyltransferase. Nucleic Acids Res 45, 1703–1713. 10.1093/nar/gkw1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polager S, and Ginsberg D (2009). p53 and E2f: partners in life and death. Nat Rev Cancer 9, 738–748. 10.1038/nrc2718. [DOI] [PubMed] [Google Scholar]
- 35.Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, Agarwalla PK, Chheda MG, Campos B, Wang A, et al. (2012). Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev 26, 756–784. 10.1101/gad.187922.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hede S-M, Hansson I, Afink GB, Eriksson A, Nazarenko I, Andrae J, Genove G, Westermark B, and Nistér M (2009). GFAP promoter driven transgenic expression of PDGFB in the mouse brain leads to glioblastoma in a Trp53 null background. Glia 57, 1143–1153. 10.1002/glia.20837. [DOI] [PubMed] [Google Scholar]
- 37.Garcia ADR, Doan NB, Imura T, Bush TG, and Sofroniew MV (2004). GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci 7, 1233–1241. 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- 38.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. (2013). Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905. 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rizzardi LF, Hickey PF, Rodriguez DiBlasi V, Tryggvadóttir R, Callahan CM, Idrizi A, Hansen KD, and Feinberg AP (2019). Neuronal brain-region-specific DNA methylation and chromatin accessibility are associated with neuropsychiatric trait heritability. Nat Neurosci 22, 307–316. 10.1038/s41593-018-0297-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mendizabal I, Berto S, Usui N, Toriumi K, Chatterjee P, Douglas C, Huh I, Jeong H, Layman T, Tamminga CA, et al. (2019). Cell type-specific epigenetic links to schizophrenia risk in the brain. Genome Biol 20, 135. 10.1186/s13059-019-1747-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, et al. (2016). Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 164, 550–563. 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4, 2612. 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shadrin AA, Kaufmann T, van der Meer D, Palmer CE, Makowski C, Loughnan R, Jernigan TL, Seibert TM, Hagler DJ, Smeland OB, et al. (2021). Vertex-wise multivariate genome-wide association study identifies 780 unique genetic loci associated with cortical morphology. Neuroimage 244, 118603. 10.1016/j.neuroimage.2021.118603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JJ, Wedow R, Okbay A, Kong E, Maghzian O, Zacher M, Nguyen-Viet TA, Bowers P, Sidorenko J, Karlsson Linnér R, et al. (2018). Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet 50, 1112–1121. 10.1038/s41588-018-0147-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ganz J, Maury EA, Becerra B, Bizzotto S, Doan RN, Kenny CJ, Shin T, Kim J, Zhou Z, Ligon KL, et al. (2022). Rates and Patterns of Clonal Oncogenic Mutations in the Normal Human Brain. Cancer Discov 12, 172–185. 10.1158/2159-8290.CD-21-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. (2012). IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478. 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Narendra V, Bulajić M, Dekker J, Mazzoni EO, and Reinberg D (2016). CTCF-mediated topological boundaries during development foster appropriate gene regulation. Genes Dev 30, 2657–2662. 10.1101/gad.288324.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chakraborty S, Kopitchinski N, Zuo Z, Eraso A, Awasthi P, Chari R, Mitra A, Tobias IC, Moorthy SD, Dale RK, et al. (2023). Enhancer-promoter interactions can bypass CTCF-mediated boundaries and contribute to phenotypic robustness. Nat Genet. 10.1038/s41588-022-01295-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith KS, Bihannic L, Gudenas BL, Haldipur P, Tao R, Gao Q, Li Y, Aldinger KA, Iskusnykh IY, Chizhikov VV, et al. (2022). Unified rhombic lip origins of group 3 and group 4 medulloblastoma. Nature 609, 1012–1020. 10.1038/s41586-022-05208-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, Sunkin SM, Li Z, Shin Y, Zhu Y, et al. (2018). Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362. 10.1126/science.aat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, et al. (2019). Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139. 10.1126/science.aay0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al. (2018). The chromatin accessibility landscape of primary human cancers. Science 362. 10.1126/science.aav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot J-P, Tanay A, et al. (2017). Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171, 557–572.e24. 10.1016/j.cell.2017.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Najm FJ, Madhavan M, Zaremba A, Shick E, Karl RT, Factor DC, Miller TE, Nevin ZS, Kantor C, Sargent A, et al. (2015). Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature 522, 216–220. 10.1038/nature14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. (2015). Science 348, 648–660. 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Najm FJ, Zaremba A, Caprariello AV, Nayak S, Freundt EC, Scacheri PC, Miller RH, and Tesar PJ (2011). Rapid and robust generation of functional oligodendrocyte progenitor cells from epiblast stem cells. Nat Methods 8, 957–962. 10.1038/nmeth.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flavahan WA, Drier Y, Johnstone SE, Hemming ML, Tarjan DR, Hegazi E, Shareef SJ, Javed NM, Raut CP, Eschle BK, et al. (2019). Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature 575, 229–233. 10.1038/s41586-019-1668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rahme GJ, Luikart BW, Cheng C, and Israel MA (2018). A recombinant lentiviral PDGF-driven mouse model of proneural glioblastoma. Neuro-oncology 20, 332–342. 10.1093/neuonc/nox129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van de Werken HJG, de Vree PJP, Splinter E, Holwerda SJB, Klous P, de Wit E, and de Laat W (2012). 4C Technology: Protocols and Data Analysis. In Methods in Enzymology (Elsevier; ), pp. 89–112. 10.1016/B978-0-12-391938-0.00004-5. [DOI] [PubMed] [Google Scholar]
- 60.van de Werken HJG, Landan G, Holwerda SJB, Hoichman M, Klous P, Chachik R, Splinter E, Valdes-Quezada C, Oz Y, Bouwman BAM, et al. (2012). Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat Methods 9, 969–972. 10.1038/nmeth.2173. [DOI] [PubMed] [Google Scholar]
- 61.Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, and Aiden EL (2016). Juicer Provides a One-Click System for Analyzing Loop-Resolution HiC Experiments. Cell Syst 3, 95–98. 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Mouse glial progenitor models, and identification of disrupted insulators in the PDGFRA locus in IDHmut glioma, related to Figure 1
NPCs and OPCs were derived from mouse ES cells using established procedures31. (A) ES cell-derived NPCs have a thick cell body and express NPC markers (NESTIN, SOX2). Serum-based differentiation of NPCs (right panels) gave rise to astrocytes (GFAP), oligodendrocytes (NG2), and neurons (β3-TUBULIN), cell types that were otherwise absent in NPC cultures. (B) ES cell-derived OPCs have a circular cell body with thin projections and express OPC markers (OLIG2, PDGFRA, NG2). Differentiation of OPCs gave rise to mature oligodendrocytes. (C) Heatmaps depict CpG methylation in CTCF motifs (left) and CTCF ChIP-seq binding scores (right) across the 2 Mb region that contains PDGFRA. For each CTCF site, bar plot at right depicts significance of differential occupancy between IDH1wt and IDH1mut gliomas. Red arrow highlights the most significantly disrupted site in IDH1mut tumors. (D) Genomic view (20 kb) over the disrupted CTCF site shows CpG methylation (heatmap; each box corresponds to a single CpG) and CTCF ChIP-seq signal (RPM). (E-F) Box plots show distribution of methylation (E) and CTCF ChIP-seq binding signal (F) over the disrupted insulator site in IDH1wt and IDH1mut glioma samples. (G) Bar plot depicts CpG methylation over the CTCF insulator site across a range of human tissues and cell lines, and in gliomas.
Figure S2. Cell type-specificity, RFX motif conservation and functional impact of the OPC-specific enhancer, related to Figures 1 and 2
Genomic tracks show H3K27ac ChIP-seq signal (blue) over the PDGFRA locus for multiple human (A) and mouse (B) tissue and cell types from ENCODE30, a published database51, and data generated in this study. The OPC-specific enhancer region is highlighted by red box. Black lines below ChIP-seq tracks represent called peaks. (C) ATAC-seq signal (grey, normalized) in TCGA glioma samples52 separated by transcriptomic subtype. NC: not classified. H3K27ac ChIP-seq signal (blue, RPM normalized) in glioma samples (top tracks in blue) and glioma spheroid cell lines (lower tracks in blue). The OPC-specific enhancer region is highlighted by red box. (D) OPC-specific enhancer locus with H3K27ac ChIP-seq (blue) and phastCons (mammalian 46-way, navy blue) signal. The H3K27ac called peak is highlighted as a line under the H3K27ac track. The RFX motif present in the OPC-specific enhancer is highlighted as a line under the phastCons track. (E) RNA-seq expression of Rfx transcription factors in NPCs and OPCs derived from ES cells53,54 (left heatmap, GEO datasets GSM2533845 and GSM1557625) or purified from mouse brains53,54 (right heatmap, GEO datasets GSM2533849 and GSM1557624). (F) Growth curves shown for control, insulator-disrupted, and CDK4 overexpressing NPCs from three biologically independent replicates. Insulator disruption does not affect NPC proliferation even after the addition of PDGF ligand.
Figure S3. Efficacy and specificity of sgRNAs targeting the CTCF insulator and RFX motif, related to Figure 2
(A,B) Locus sequencing data demonstrate efficacy of CRISPR editing of the target CTCF motif in (A) NPCs and (B) OPCs edited with the Pdgfra insulator sgRNA (Cas9-Ins) or control (Cas9-NT). (C,D) Plots show differential CTCF ChIP-seq binding between Cas9-Ins and Cas9-NT NPCs (C) and OPCs (D). The insulator CTCF site is the only peak with robust reduction of ChIP-seq signal. (E) Locus sequencing data demonstrate efficacy of CRISPR editing of the target RFX motif in OPCs. (F) Volcano plot shows differential H3K27ac ChIP-seq signals in cells edited with the RFX motif sgRNA, relative to Cas9-NT control cells. The OPC-specific enhancer is the only peak with robust reduction of ChIP-seq signal. N.D = not detected.
Figure S4. CDKN2A deletion rates in human gliomas, efficacy and specificity of DNMT3A3L sgRNAs targeting CDKN2A in mouse OPCs, and impact of PDGFRA and CDKN2A perturbations in vivo, related to Figures 3 and 4
(A) Pie charts indicate proportion of tumors with homozygous loss of the CDKN2A locus, stratified by CpG island methylator phenotype (G-CIMP). The locus is frequently lost in tumors with lower DNA methylation. (B) Locus bisulfite sequencing data demonstrate efficacy of epigenetic editing at four CpG containing hubs within the target locus in OPCs. Percentage values indicate fraction of alleles with methylation of the corresponding CpG. (C) Differential H3K27ac ChIP-seq signal in OPCs with Cas9m4-3A3L and the Cdkn2a promoter sgRNA (3A3L-pCdkn2a), compared to control cells (3A3L-NT). The Cdkn2a promoter is the only H3K27ac peak with robust reduction of ChIP-seq signal. (D) Bar plot depicts average methylation beta values for the targeted promoter (Cdkn2a-p19) and the nearby promoter (Cdkn2b-p15) in mouse OPCs expressing Cas9m4-DNMT3A3L with non-targeting (3A3L-NT) or Cdkn2a targeting (3A3L-pCdkn2a) sgRNA. The epigenetic editor is specific to the targeted promoter. (E) Schematics of vectors used to interrogate the role of enhanced Pdgfra expression along with Cdkn2a (p19ARF) silencing individually or in combination. Vectors used in (E) linked to the histology observed in (F) where transduced cells are fluorescently tracked in the corpus callosum one week after injection of adult mice. White scale bar: 100 μm.
Figure S5. Cell type-specificity of the OPC-specific enhancer and noncoding RNA in human brain compartments, and impact of methylation on the mouse insulator, related to Figure 5
(A) Scatter plot shows LINC02283 expression (x-axis) versus ATAC signal of the OPC-specific enhancer (y-axis) in TCGA gliomas analyzed by ATAC-seq and RNA-seq. (B) Violin plot shows LINC02283 expression in human bulk tissue samples (GTEX)55. (C,D) Scatter plots compare LINC02283 expression (x-axis) against PDGFRA expression (y-axis) in (C) IDHwt and (D) IDHmut gliomas. (E) Bar plot depicts average methylation beta values in mouse OPCs expressing Cas9m4-DNMT3A3L with non-targeting sgRNAs (3A3L-NT) or insulator targeting sgRNAs (3A3L-Insulator). (F) CTCF ChIP-seq signal at the Pdgfra insulator region for mouse OPCs with 3A3L-NT (solid black) or 3A3L-Insulator (dashed red). Methylation of the insulator region does not affect CTCF binding due to sparsity of CpG dinucleotides in mouse. Spearman r values are computed for all correlations. Shaded areas represent confidence interval in A, C and D.
Data Availability Statement
Raw and processed sequencing data generated for this study have been deposited at the NCBI SRA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
The code supporting the current study is available from the corresponding author on request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti CTCF (clone D31H2) | Cell signaling technologies | 3418 |
| Rabbit anti H3K27ac | Active motif | 39133 |
| Rabbit anti PDGFRA | Cell signaling technologies | 3174 |
| Rabbit anti CDKN2A (p19 ARF) | Millipore | 07-543 |
| Mouse anti TP53 | Cell signaling technologies | 2524 |
| beta-actin-HRP conjugated | Sigma | A3854 |
| anti-rabbit IgG-HRP | Cell signaling technologies | 7074 |
| anti-mouse IgG-HRP | Cell signaling technologies | 7076 |
| Rabbit anti-Ki67 (D3B5) | Cell signaling technologies | 12202 |
| Rabbit anti Olig2 | Millipore | AB9610 |
| Rabbit anti CD31 | Cell signaling technologies | 77699 |
| Rabbit anti Glial Fibrillary acidic protein | Agilent Dako | Z0334 |
| Bacterial and virus strains | ||
| NEB stable competent E. coli | New England Biolabs | C3040I |
| Biological samples | ||
| Human IDH wild-type and IDH mutant glioma samples | Flavahan et al. 2016 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| ESGRO Leukemia Inhibitory Factor (LIF) | Millipore | ESG1107 |
| HyClone Fetal Bovine Serum (U.S.), Embryonic Stem (ES) Cell Screened | Hyclone | SH30070.02E |
| Recombinant Human EGF Protein | R&D Systems | 236-EG-200 |
| Recombinant Human FGF basic/FGF2 | R&D Systems | 4114-TC-01M |
| N-2 Supplement (100X) | GIBCO | 17502048 |
| B-27 Supplement (50X), serum free | GIBCO | 17504044 |
| GlutaMAX Supplement | GIBCO | 35050061 |
| Penicillin-Streptomycin | GIBCO | 15140122 |
| MEM Non-Essential Amino Acids Solution (100X) | GIBCO | 11140050 |
| L-Glutamine (200 mM) | GIBCO | 25030081 |
| 2-Mercaptoethanol | Sigma | 6010 |
| Knockout DMEM | GIBCO | 10829018 |
| DMEM:F12 | GIBCO | 11320-033 |
| Neurobasal | GIBCO | 21103-049 |
| IMDM | GIBCO | 31980-030 |
| Fetal Bovine Serum | GIBCO | 26140079 |
| Recombinant Murine IGF-1 | Peprotech | 250-19 |
| Recombinant Human PDGFB | Peprotech | 100-14B |
| Laminin | Sigma | L2020-1MG |
| Klenow, exo- | New England Biolabs | M0212L |
| Critical commercial assays | ||
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | 20024906 |
| Qubit dsDNA HS Assay kit | Invitrogen | Q32854 |
| QIAquick Gel Extraction Kit | Qiagen | 28706 |
| Qiagen Maxiprep plasmid kit | Qiagen | 12163 |
| Agencourt AMPure XP | Beckman Coulter | A63882 |
| Bioanalyzer D1000 screentapes | Agilent | 5067-5582 |
| LipoD293 | Signagen | SL100668 |
| End-It DNA End-Repair Kit | Biosearch technologies | ER0720 |
| TRIzol™ Reagent | Thermo Fisher | 15596026 |
| EZ DNA Methylation-Lightning Kit | Zymo | D5030 |
| Qiagen DNeasy Blood & Tissue Kit | Qiagen | 69504 |
| Deposited data | ||
| Processed data | GEO | GSE225794 |
| Raw data (Chip-seq) | GEO | GSE225732 |
| Raw data (Capture Bis-seq) | GEO | GSE225793 |
| Experimental models: Cell lines | ||
| mESC (v6.5) | Broad Institute | N/A |
| C57BL/6 MEF 4M IRR | GlobalStem | GSC-6002G |
| Mouse NPCs | This study | N/A |
| Mouse OPCs | This study | N/A |
| 293FT cells | Thermo Fisher | R70007 |
| Human NPCs, BYS012 | ATCC | ACS-5004 |
| Experimental models: Organisms/strains | ||
| 129S1/SvImJ | Jackson laboratories | 002448 |
| Oligonucleotides | ||
| Pdgfra insulator CRISPR locus sequencing: GTCAGGAGTAGATCCTCGTGGCTGAAGACTGGGAGCTATA | IDT | N/A |
| RFX motif CRISPR locus sequencing: TCTCCCTGTTTGGTGCCCTTCTCTCCATCAATCATTGCCAAC | IDT | N/A |
| Cdkn2a-p19 promoter locus bisulfite sequencing: GAAAATTTTTTTTTGGAGTGGGCCTCTAAAAAACTTTCC | IDT | N/A |
| OPC enhancer 4C-seq primers: TGTGGCTTGGCATCCTGATC (viewpoint) TTGAACTCTCAGAGACCCAC | IDT | N/A |
| 18S real time PCR primers: GAGGGAGCCTGAGAAACGG (Forward) GTCGGGAGTGGGTAATTTGC (Reverse) | IDT | N/A |
| Pdgfra real time PCR primers: GACTTCCTAAAGAGTGACCATCC (Forward) CTTCCCAGTCCTTCAGCTTATC (Reverse) | IDT | N/A |
| Cdkn2a real time PCR primers: CATGTTGTTGAGGCTAGAGAGG (Forward) CACCGTAGTTGAGCAGAAGAG (Reverse) | IDT | N/A |
| Recombinant DNA | ||
| pVSV-G | Broad Institute | N/A |
| pCMV delta R8.2 | Broad Institute | N/A |
| CMV-Cas9-T2A-eGFP (lentiviral construct) | This study | N/A |
| U6-ShRNA-U6-sgRNA-CMV-RFP (lentiviral construct) | This study | N/A |
| U6-ShRNA-U6-sgRNA-CMV-RFP-IRES-PDGFB (lentiviral construct) | This study | N/A |
| U6-sgRNA-CMV-Cas9-T2A-eGFP (piggybac construct) | This study | N/A |
| Software and algorithms | ||
| R version 4.1.3 | R Core Team | https://www.r-project.org |
| Benchling (for sgRNA design) | Benchling | https://www.benchling.com |
| ChopChop (for sgRNA design) | ChopChop | https://chopchop.cbu.uib.no |
| Cas off finder (off target analysis) | Cas Off Finder | https://www.rgenome.net/cas-offinder/ |
| Juicer tools | Aiden laboratory | https://github.com/aidenlab/juicer |
| BWA aln version 0.7.4 | Li laboratory | https://github.com/lh3/bwa |
| Picard toolkit 2.9.2 | Broad Institute | https://broadinstitute.github.io/picard/ |
| GVIZ package | Bioconductor | https://bioconductor.org/packages/release/bioc/html/Gviz.html |
| methylCtools 0.9.438 | Hovestadt laboratory | https://github.com/hovestadt/methylCtools |
| MEME suite (version 4.11.3-1) | MEME suite | https://meme-suite.org/meme/doc/download.html |
| LiftOver tool | UCSC genome browser team | https://genome.ucsc.edu/cgi-bin/hgLiftOver |