

Abstract
The synthesis of di- and trisubstituted vinyl fluorides with high isomeric purity remains a challenge for organic synthesis. While many methods exist to access these compounds, separation of the desired isomer from the minor isomer and/or starting materials often is difficult. Herein we report a practical method to access di- and trisubstituted vinyl fluorides via a selective Horner-Wadsworth-Emmons olefination/hydrolysis, which provides crystalline 2-fluoroacrylic acids in high (>98%) E-isomeric purity. A subsequent silver catalyzed stereoretentive decarboxylation provides the title substances with high isomeric purity and without the need for tedious chromatography to remove the minor isomer. The process was amenable to a variety of aldehydes and ketones and provided a diverse array of di- and trisubstituted vinyl fluorides. The sequence was applied to the synthesis of antibacterial and anti-inflammatory compounds.
Keywords: vinyl fluoride, olefination, decarboxylation
GRAPHICAL ABSTRACT
INTRODUCTION
The incorporation of fluorine into organic molecules often results in advantageous modulation of physical properties.1 This has resulted in the growing presence of fluorine in pharmaceuticals,2 agrochemicals3 and organic materials.4 Among the plethora of fluorine containing functional groups, terminal monofluoroalkenes are of interest due to their applications in materials science, synthetic methodology and medicinal chemistry.5 Several biologically active compounds bearing trisubstituted (1-3) or disubstituted (4-6) terminal vinyl fluorides are shown in Figure 1.6
Figure 1.
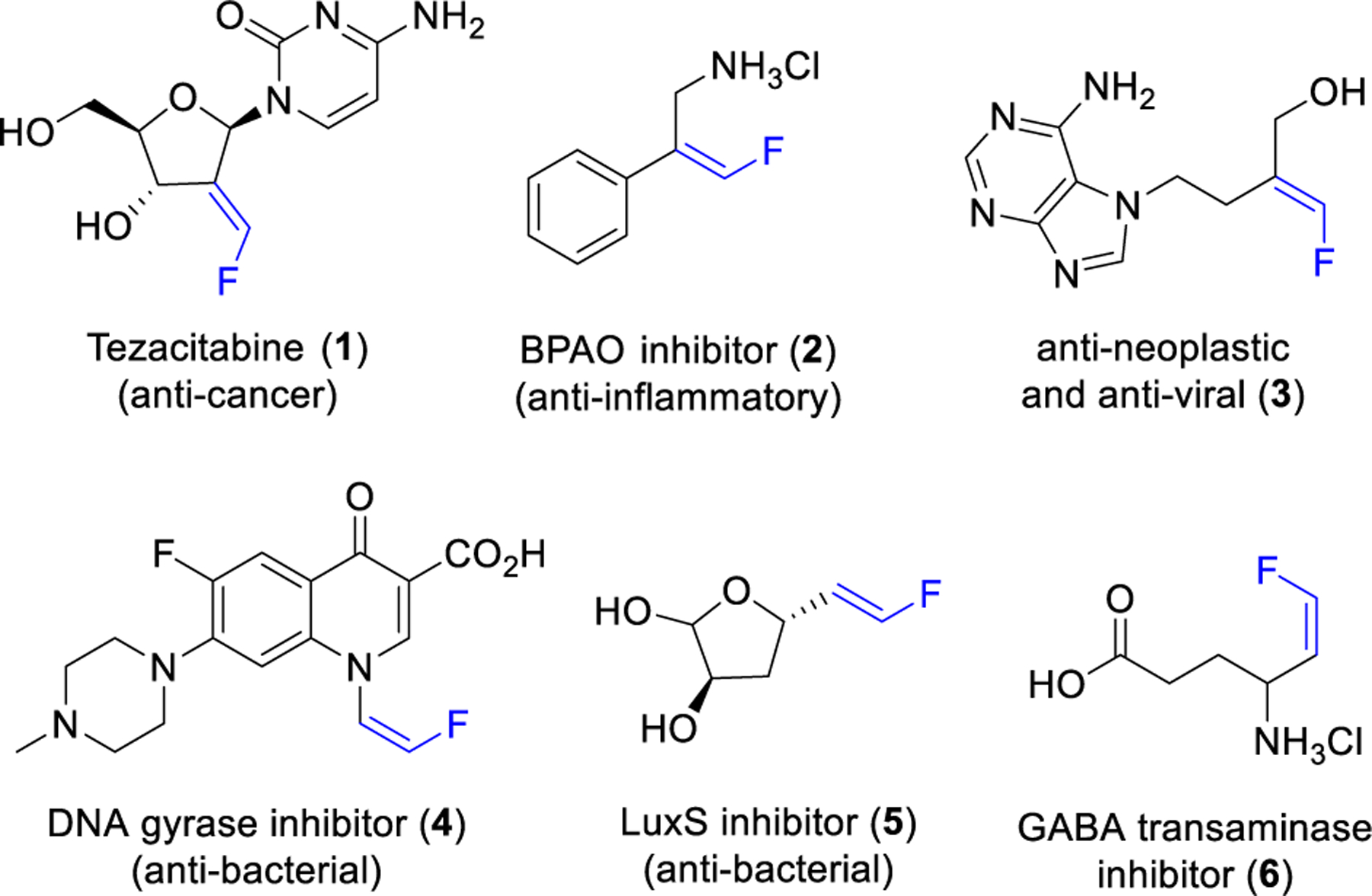
Biologically active compounds containing terminal vinyl fluorides.
The geometry of the vinyl fluoride is often critically important to biological activity, and consequently synthetic methods to access these structures with high isomeric purity are desired.6b To support a recent drug development program, we required a safe and scalable route to trisubstituted terminal vinyl fluorides with high isomeric purity. A survey of the literature revealed a diverse set of approaches, but many raised concerns regarding safety and/or scalability. A selection of published protocols is shown in Figure 2. The Wittig reaction using a fluoromethyl triphenylphosphonium salt was first reported by Schlosser in 1969 (Figure 2A).7 Variations of this reagent were subsequently developed but generally provide low selectivities.8 In 2016, Hoveyda reported cross metathesis approaches to both Z and E disubstituted vinyl fluorides (Figure 2B).9 High selectivities were obtained, but the reaction required a non-commercially available catalyst and was limited to disubstituted products, and in the case of E-vinyl fluorides, a vinyl chloride byproduct was formed which could prove difficult to separate from the desired product.
Figure 2.
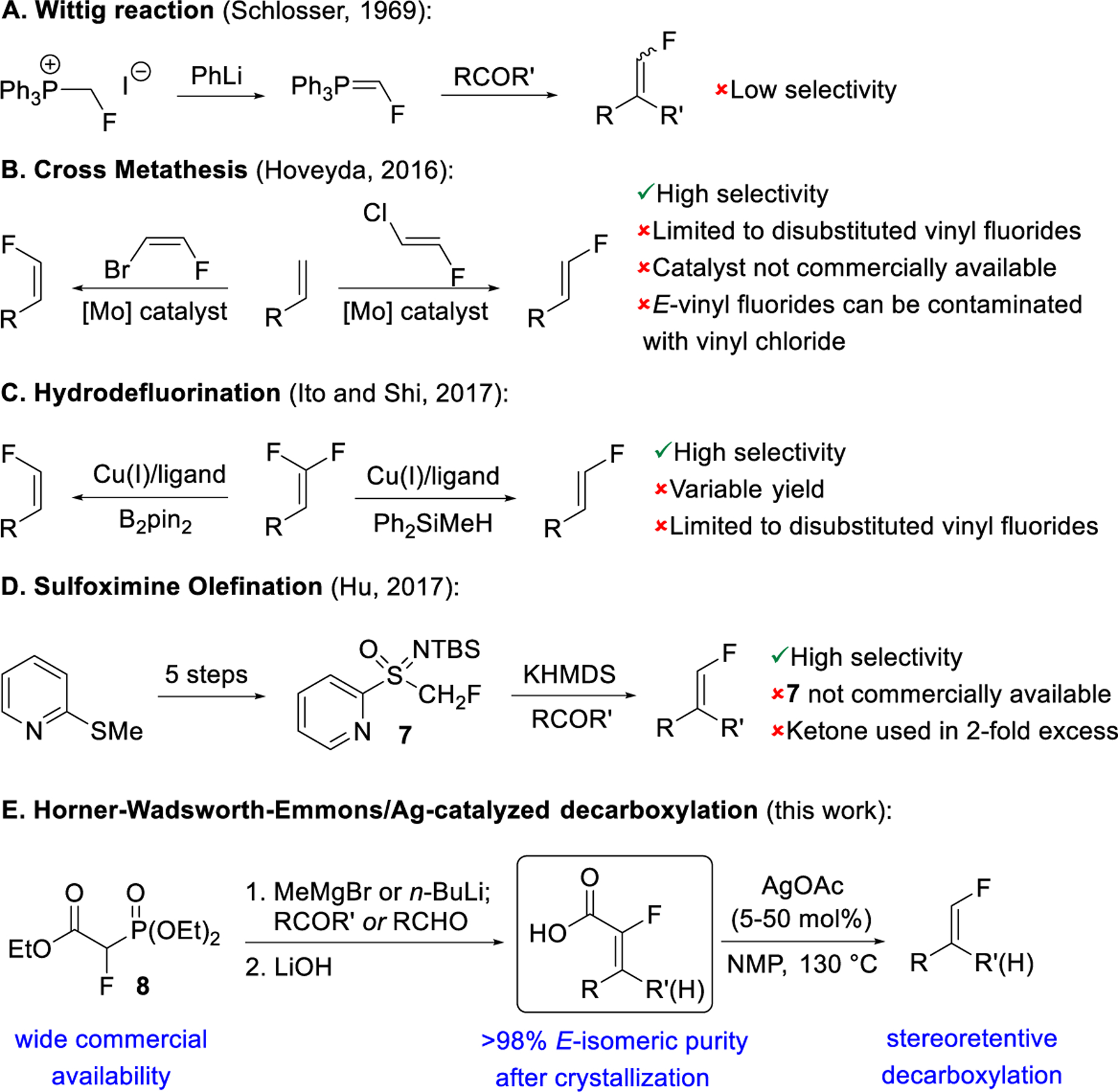
Selected methods for terminal vinyl fluoride synthesis (A-D) and our olefination/decarboxylation approach (E).
The hydrodefluorination of 1,1-difluoroalkenes (Figure 2C) was demonstrated by the groups of Ito and Shi in 2017 and can provide E or Z products depending on reaction conditions but is limited to disubstituted vinyl fluoride products.10 Hu and co-workers reported an olefination using fluoromethylsulfoximine 7 in 2017 (Figure 2D).11 This procedure gave high selectivity for both ketones and aldehydes. The non-commercial availability of the reagent 7 and its 5-step preparation, as well as the requirement to use 7 as the limiting reagent with the ketone in two-fold excess nonetheless presented obstacles to employing this process on large scale.11b The preparation and Horner-Wadsworth-Emmons (HWE) reaction of triethyl 2-fluorophosphonoacetate 8 were first reported by Machleidt and Wessendorf in 1964 (Figure 2E).12 The wide commercial availability and low cost of 8 made it an attractive reagent for introduction of the fluorovinyl group from a ketone or aldehyde starting material. We reasoned that if this olefination could be developed with high selectivity and coupled with stereoretentive decarboxylation of the derived carboxylic acid, it would constitute a practical route to vinyl fluoride products. Herein we report the development of this route to di- and trisubstituted terminal vinyl fluorides by a highly selective olefination of ketones or aldehydes using 8, subsequent hydrolysis to the crystalline α-fluoroacrylic acids, and stereospecific Ag-catalyzed decarboxylation.13
RESULTS AND DISCUSSION
Initial work focused on optimization of the HWE reaction of acetonaphthone 9 with phosphonate 8 (Table 1). The base was screened first, using 2 equiv of 8 in THF at 0 °C. The use of n-BuLi or alkali metal HMDS derivatives gave modest selectivities around 85:15 favoring E-10a (entries 1–4). We next examined Grignard reagents as bases. In 1998, Sano reported the use of i-PrMgBr as base to give Z-selectivity in the reaction of 8 with aldehydes.14 In 2008, Davies described the highly E-selective reactions of non-fluorinated phosphonates with aldehydes using MeMgBr as base.15 When we applied alkyl Grignard bases to the reaction of 8 with ketone 9, a significant improvement in selectivity was observed (entries 5–8). i-PrMgCl gave a 96:4 E/Z ratio of 10a in 88% assay yield. Using MeMgCl gave a higher assay yield of 98% with similar selectivity (95:5, entry 6). The halide counterion had a slight effect on selectivity and yield, with MeMgBr (entry 7) being optimal at 96:4 selectivity and 97% yield. The use of dibutylmagnesium (entry 9) proceeded in high yield but decreased selectivity. The use of an alkylzinc bromide (entry 10) or dimethylzinc (entry 11) was less effective compared to Grignard reagents. A brief examination of other solvents (entries 12–15) showed none to be as effective as THF, although DME did provide high selectivity (97:3, entry 15). Finally, the effect of temperature was examined (entries 16–18). Lowering the temperature to –20 °C resulted in incomplete conversion, although with slightly increased selectivity (97:3, entry 16). Increasing the temperature to 40 °C gave the highest assay yield (99%, entry 17) while maintaining the selectivity observed at 0 °C (96:4) but increasing further to 50 °C resulted in a drop in assay yield and selectivity (entry 18). Thus, the optimal conditions were MeMgBr as base and THF as solvent at a reaction temperature of 40 °C (entry 17).16
Table 1.
Optimization of olefination reaction conditions.

| |||||
|---|---|---|---|---|---|
| entry | base | solvent | T (°C) | yield (%)a | E/Zb,c |
| 1 | n-BuLi | THF | 0 | 81 | 85:15 |
| 2 | LiHMDS | THF | 0 | 85 | 84:16 |
| 3 | NaHMDS | THF | 0 | 90 | 87:13 |
| 4 | KHMDS | THF | 0 | 73 | 86:14 |
| 5 | i-PrMgCl | THF | 0 | 88 | 96:4 |
| 6 | MeMgCl | THF | 0 | 98 | 95:5 |
| 7 | MeMgBr | THF | 0 | 97 | 96:4 |
| 8 | MeMgI | THF | 0 | 88 | 95:5 |
| 9 | Bu2Mg | THF | 0 | 98 | 92:8 |
| 10 | n-PrZnBr | THF | 0 | 44 | 85:15 |
| 11 | Me2Zn | THF | 0 | 81 | 93:7 |
| 12 | MeMgBr | toluene | 0 | 79 | 94:6 |
| 13 | MeMgBr | NMP | 0 | 66 | 91:9 |
| 14 | MeMgBr | 2-MeTHF | 0 | 79 | 93:7 |
| 15 | MeMgBr | DME | 0 | 79 | 97:3 |
| 16 | MeMgBr | THF | −20 | 86d | 97:3 |
| 17 | MeMgBr | THF | 40 | 99 | 96:4 |
| 18 | MeMgBr | THF | 50 | 93 | 95:5 |
Assay yield from 19F NMR using benzotrifluoride as internal standard.
E/Z ratio from 19F NMR of crude product.
Stereochemistry was confirmed by 1H-19F HOESY experiments.
Conversion of 9 to 10a from HPLC (220 nm).
Applying the reaction conditions developed for ketones to the olefination of 4-bromobenzaldehyde 11 showed moderate (80:20) selectivity favoring the Z-isomer (Scheme 1A).13a Alternatively, the use of n-BuLi as base at low temperature (–78 °C) gave the E-isomer with high selectivity (98:2).17 The stereochemistry was confirmed by 1H–19F HOESY experiments and single crystal X-ray structure analysis of the derived acid of Z-12a (vide infra). By contrast, olefination of 11 with triethyl-2-phosphonopropionate 14 gave the product E-13 in >45:1 E-selectivity using either MeMgBr or n-BuLi as the base (Scheme 1B).
Scheme 1.
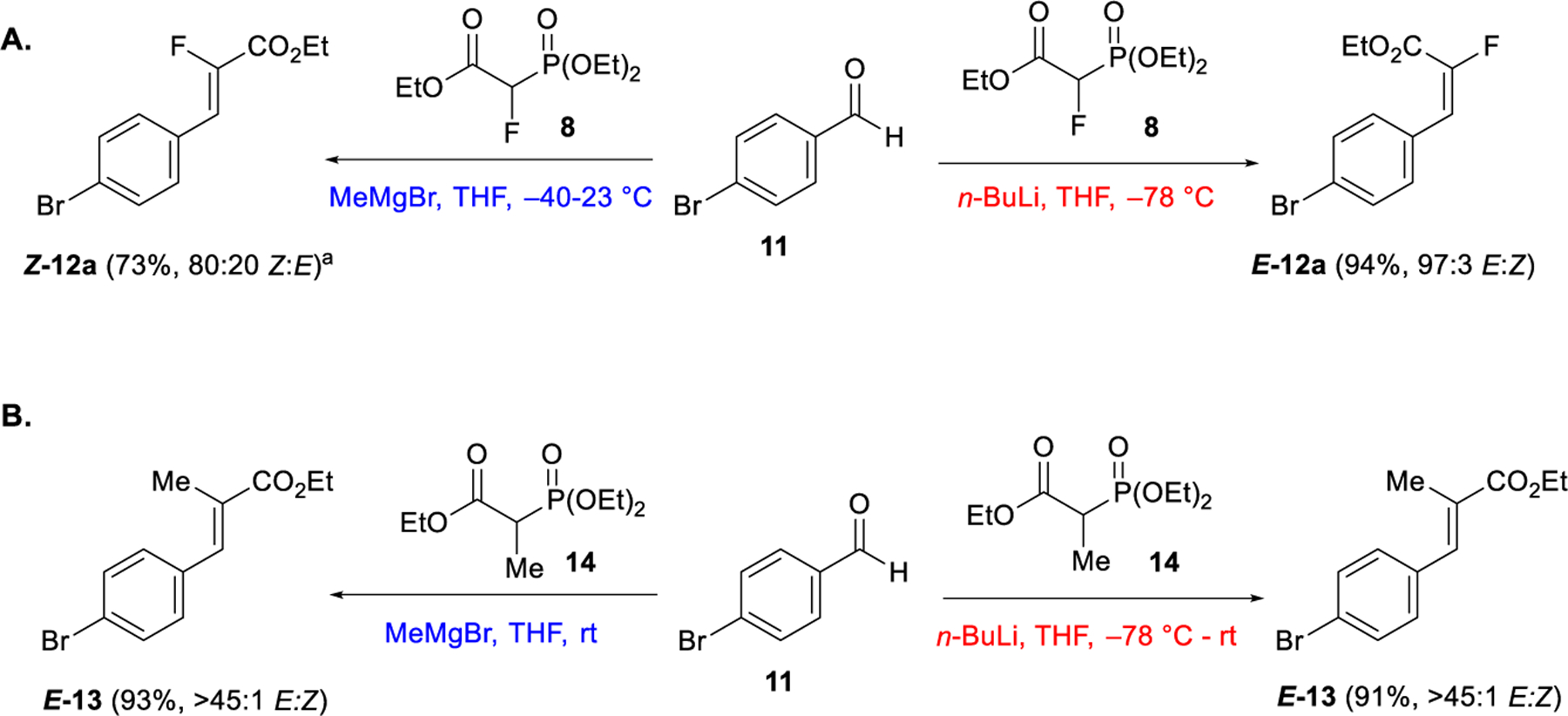
Olefination of aldehyde 11 with 8 and 14 using MeMgBr or n-BuLi as base. aAfter purification >98:2 Z:E.
The basis for the E/Z selectivity, especially the changes with different bases, was investigated computationally. Calculations were performed with Gaussian18 B3LYP-D3/6–31G(d,p)19 with an SMD20 solvation model. The energy diagram for reaction of the lithium and magnesium phosphonates are shown in Figure 3. Coordination of solvent to the Li or BrMg counterions was required to account for the observed trends. Specifically, two THF molecules remained coordinated throughout the reaction. The substrates (A) undergo a precoordination to form B followed by a transition state C for addition of the anion to the aldehyde via a staggered transition state where the metal coordinates the oxygens of the ester, phosphonate, and aldehyde (Scheme 2). The adduct D, then proceeds to a four-membered transition state E (Scheme 2) which gives rise to formation of the alkene.
Figure 3.
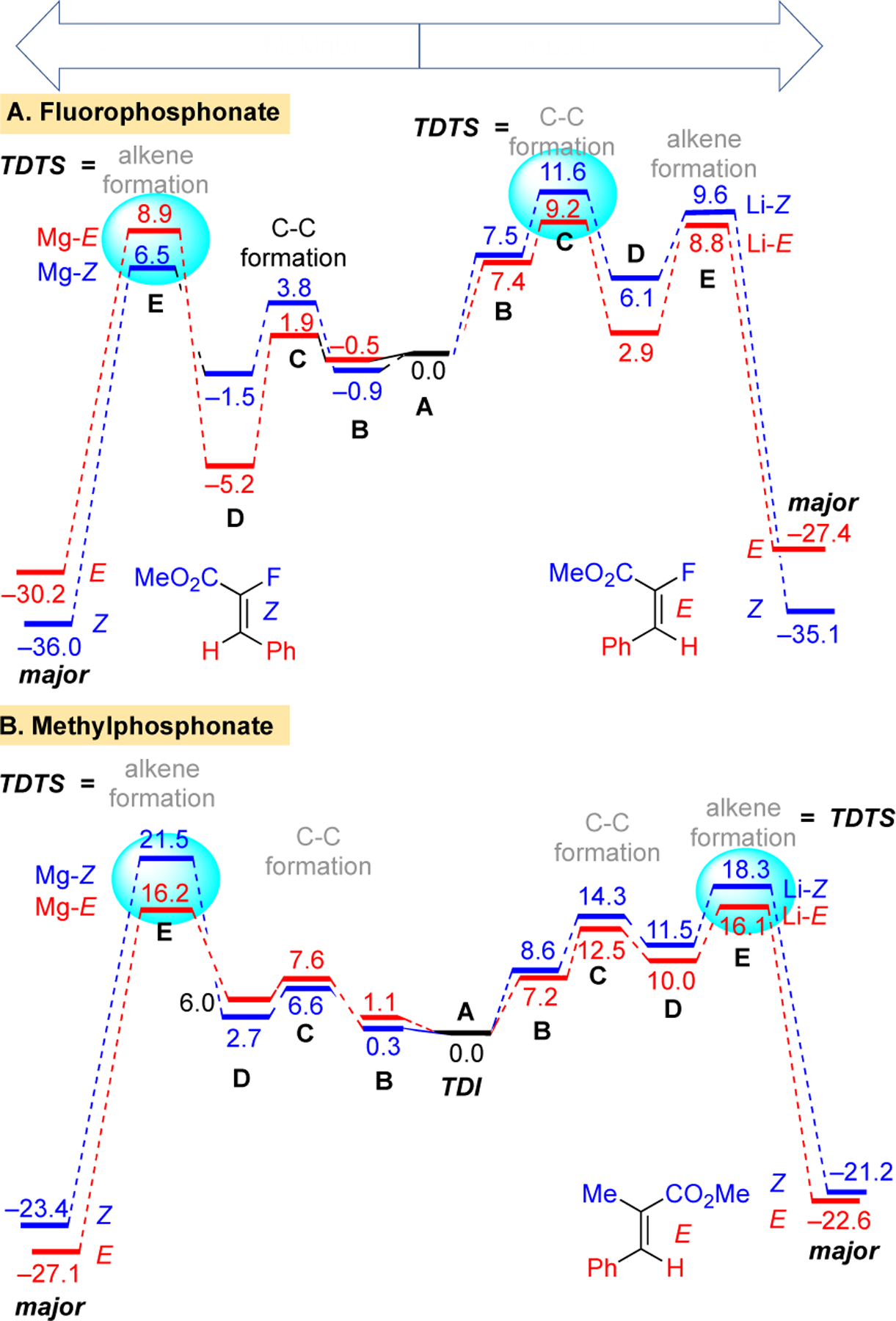
Relative Gibbs free energy (kcal/mol) profile diagram of the formation of olefin from the Mg/Li fluoro- and methyl-phosphonates (SMD(THF)/B3LYP-D3/6–31G**).
Scheme 2.
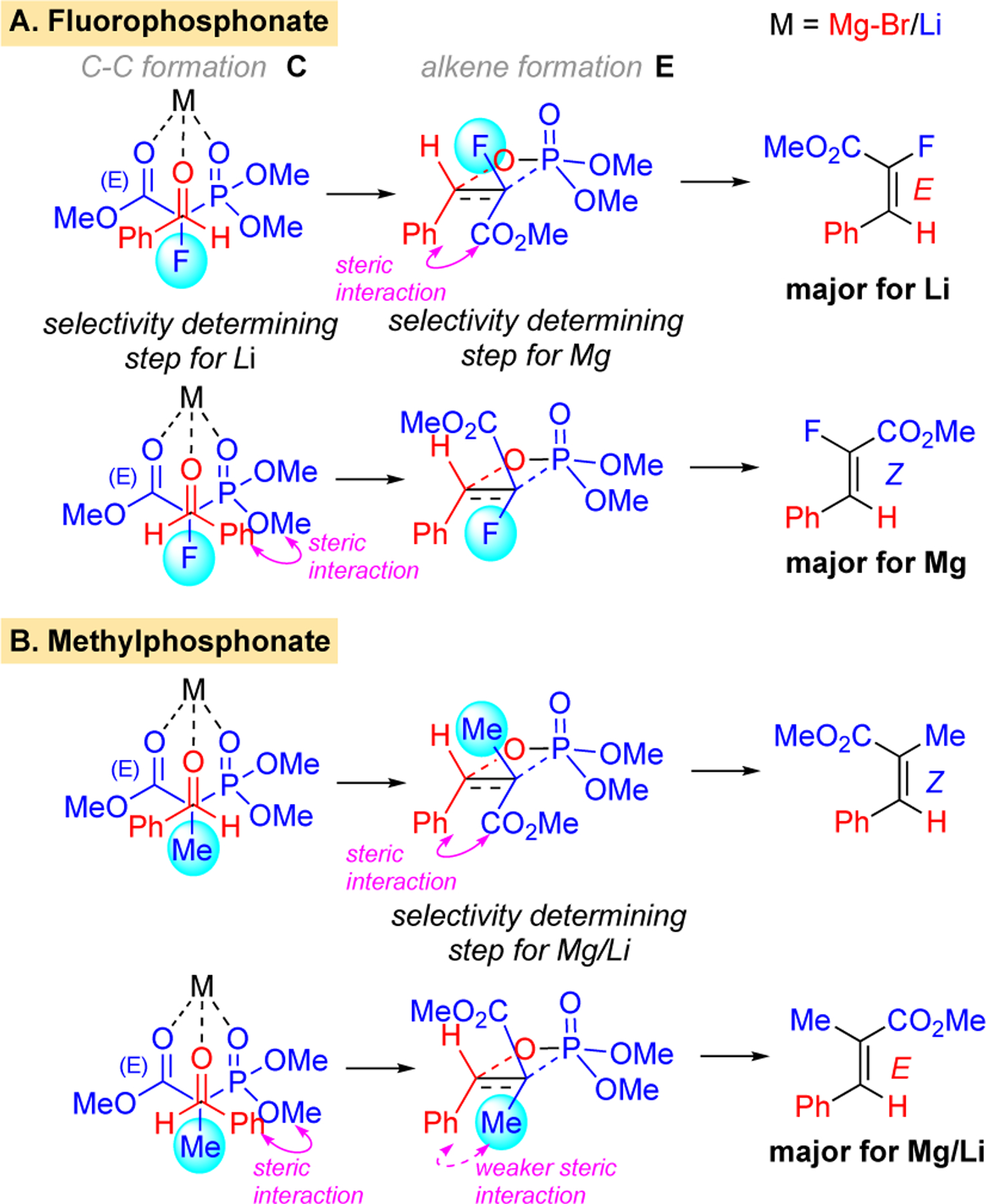
Reaction pathways of PhCHO with 2-fluoro and 2-methyl phosphonoacetates.
For the reactions of stabilized ylides, the initial addition of the anion is reversible and the selectivity-determining step is typically elimination from the oxaphosphetane via transition state E.21 For the α-methylphosphonate 14, this expected trend holds for both the lithium and bromomagnesium phosphonates (Figure 3B) and the E-isomer is the major adduct from each (Scheme 1B). The transition state E has fewer steric interactions en route to the E-isomer as the larger groups (-CO2Me, -Ph) are trans disposed (Scheme 2B). There are, however, some notable differences between the two pathways. The barrier for the addition of the methyl-substituted phosphonate anion to the aldehyde is significantly higher for the lithium case which can be attributed to the lower Lewis acidity of lithium (NBO charge +0.88) vs magnesium (NBO charge +1.69).
For the α-fluorophosphonate 8, the turnover determining transition state (TDTS) differs depending on the metal counterion employed. With MeMgBr, the expected trend holds in that alkene elimination via E is selectivity determining (left side of Figure 3A) and leads to the Z-isomer with the larger groups (-CO2Me, -Ph) trans disposed. In this case, steric interactions are again minimized in the oxaphosphetane transition state E (bottom of Scheme 2A). With n-BuLi, the selectivity determining step for α-fluorophosphonate 8 is transition state C (right side of Figure 3A). Steric interactions are minimized in the staggered approach that ultimately leads to the E-isomer (Scheme 2A).
This change of selectivity determining transition state for the fluoro-substituted phosphonates was investigated further. For the initial addition of the anion to the aldehyde, the barrier is again significantly higher for the lithium vs the magnesium case due to greater Lewis acidity of the latter. However, the barriers are generally lower for the fluoro-substituted phosphonate than for the methyl-substituted phosphonate anion. Apparently, the lower nucleophilicity of the fluoro-substituted phosphonate (NBO charge on enolic carbon: F = –0.32 ± 02, Me = –0.76 ± 01) is offset by the greater steric size of the methyl vs fluoro group. This substituent enters into particularly close proximity to the aldehyde groups due to the non-parallel approach of the phosphonate anion along the Dunitz-Bürgi angle to engage π* of the aldehyde. A distortion interaction analysis highlights this effect (see Supporting Information) where the aldehyde undergoes greater distortion with the methyl-substituted phosphonate anion (average = 14.9 kcal/mol) vs the fluoro analog (average = 7.8 kcal/mol).
The hydrolysis of esters in the products from the above reactions was performed using LiOH in aqueous EtOH or i-PrOH, and upon completion of the reaction, acidification usually resulted in crystallization of the product acids in high yield and often with increased isomeric purity (up to >98:2 E/Z).22 For example, hydrolysis of esters 15a and 26a gave the corresponding acids 15b and 26b as single E-isomers on acidification (Scheme 3). Alternatively, the acids could be recrystallized to achieve the desired purity: acid 18b of 95:5 isomeric purity was upgraded to the pure E-isomer (>98:2) after a recrystallization from heptane. This favorable property of the α-fluoroacrylic acids eliminated the need for chromatography for removal of the minor isomer.
Scheme 3.
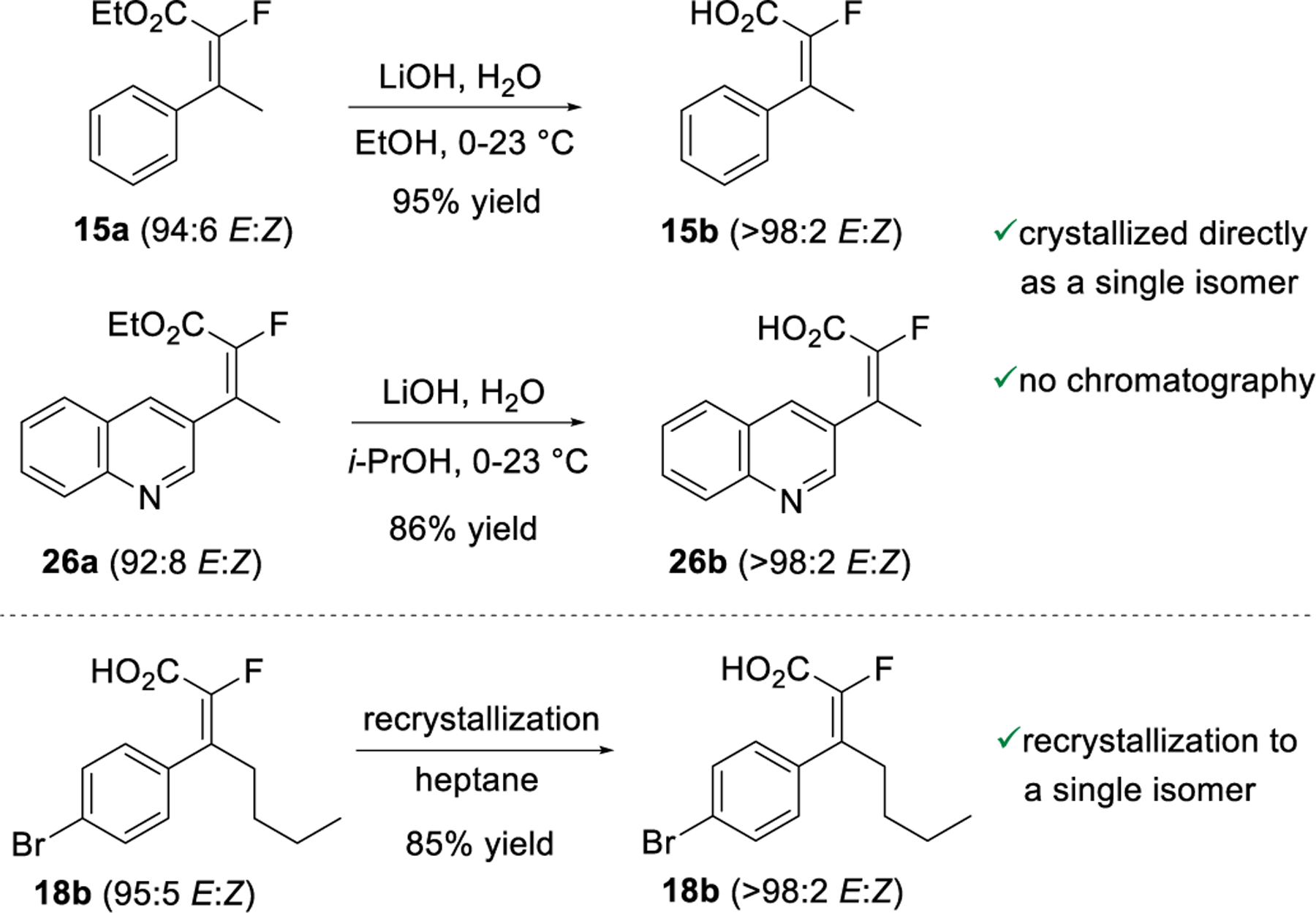
Hydrolysis of esters to crystalline acids and recrystallization to upgrade isomeric purity.
With the optimal reaction conditions in hand, the scope of the olefination/hydrolysis was explored (Table 2). For a variety of aryl alkyl and heteroaryl alkyl ketones, the olefination proceeded with E/Z selectivity of ≥90:10, with the highest selectivity observed for the sterically hindered 24a (single E-isomer). The olefination proceeded with uniformly high selectivity for electron poor (17a and 22a) and electron rich (16a and 21a) substrates. The olefination of vinyl alkyl ketones resulted in dienes 28a and 29a, with lower selectivity for the less sterically differentiated substrate 29a. The acids were often obtained as pure E-isomers (E/Z >98:2 by 19F NMR) after crystallization from the reaction mixture on acidification. For substrates 17b, 19b and 28b, the olefination and hydrolysis steps were telescoped (unpurified ester was taken directly into hydrolysis) and the overall yield of acid from ketone is given.
Table 2.
Scope of olefination/hydrolysis of ketones.a
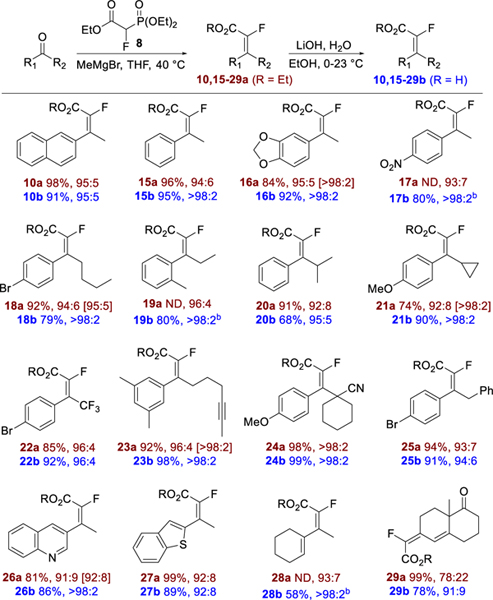
|
For olefination products, yield of purified product, reaction E/Z selectivity, and E/Z isomeric purity of purified product [brackets] are given. When E/Z selectivity was the same for the reaction and purified product, no bracketed ratio is given. For carboxylic acid products, yield of purified product is given followed by E/Z isomeric purity of product. E/Z ratio from 19F NMR of crude product (esters) and isolated product (acids). For detailed reaction conditions, see the Experimental Section.
Unpurified ester taken directly into hydrolysis. Yield of acid is the overall yield from ketone.
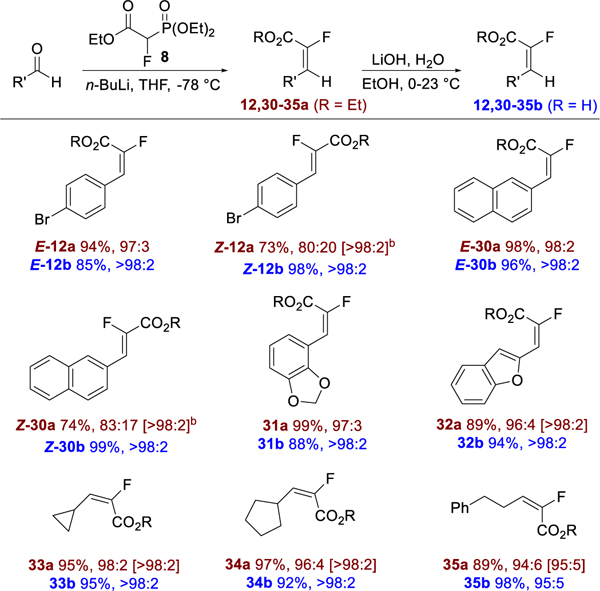
The olefination/hydrolysis sequence was next applied to a selection of aldehydes (Table 3). As observed for olefination of aldehyde 11, the use of n-BuLi gave uniformly high E-selectivity for aryl, heteroaryl and alkyl aldehydes.15 The application of MeMgBr as base for olefination gave complimentary results, favoring the Z-isomers Z-12a and Z-30a and with modest selectivity (80:20 and 83:17, respectively). As observed for the ketone substrates, hydrolysis of the esters usually resulted in single E-isomers after isolation by crystallization directly from the reaction mixtures.
Table 3.
Scope of olefination/hydrolysis of aldehydes.a
|
For olefination products, yield of purified product, reaction E/Z selectivity, and E/Z isomeric purity of purified product [brackets] are given. When E/Z selectivity was the same for the reaction and purified product, no bracketed ratio is given. For carboxylic acid products, yield of purified product is given followed by E/Z isomeric purity of product. E/Z ratio from 19F NMR of crude product (esters) and isolated product (acids). For detailed reaction conditions, see the Experimental Section.
Olefination was performed using MeMgBr as base.
Single crystal X-ray structures were obtained for acids E-16b and Z-12b (Scheme 4) which confirmed the geometry assignments of the olefins.
Scheme 4.
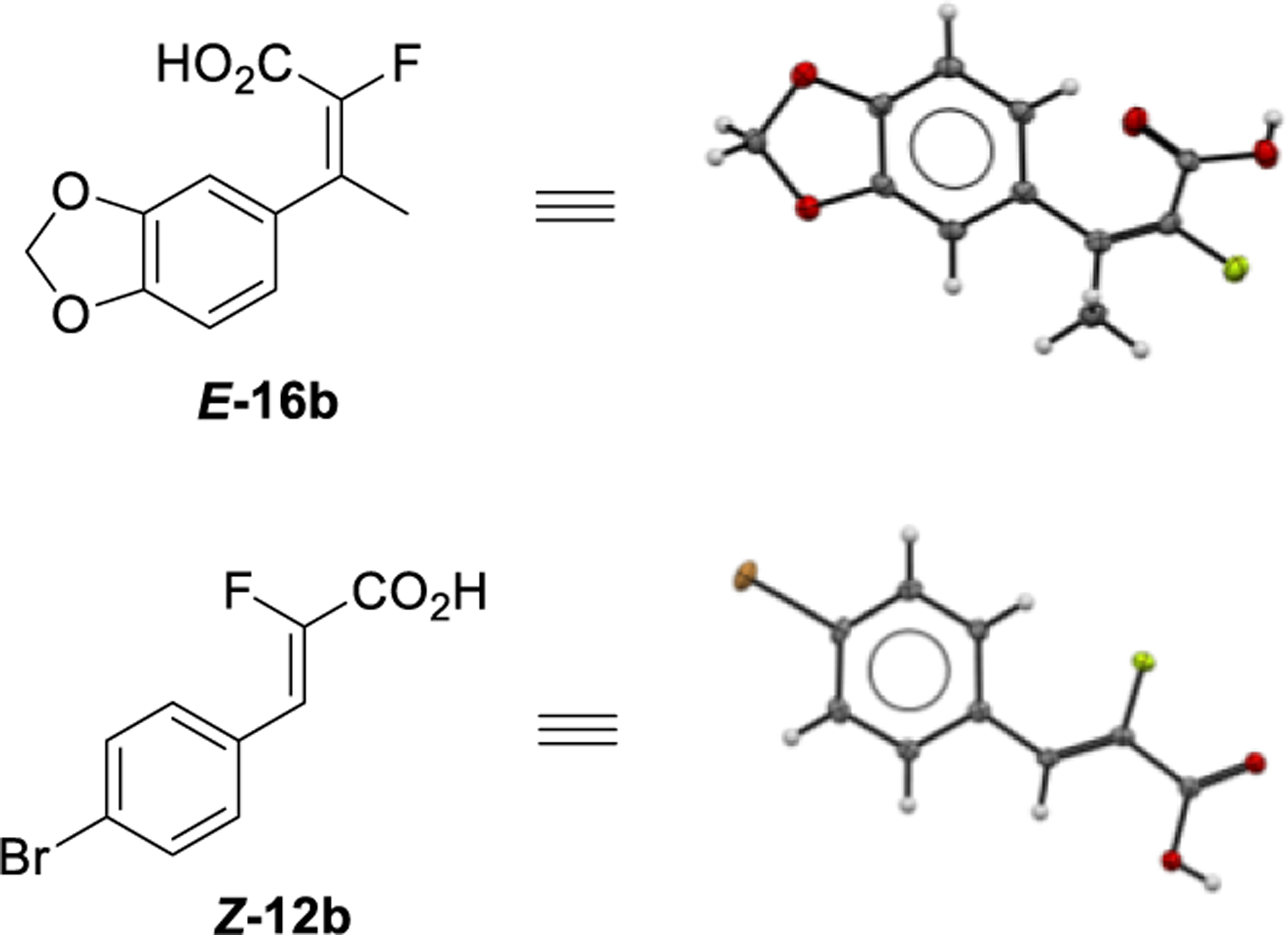
X-ray crystal structures of acids E-16b and Z-12b
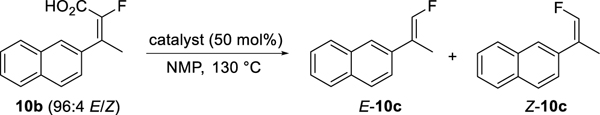
The decarboxylation of three α-fluoroacrylic acids was reported in 1967 by Elkik using a mixture of Cu powder and CuSO4 (36 mol % Cu in total) in quinoline at 205–210 °C.23 Subsequent to Elkik’s pioneering work, only stoichiometric decarboxylations have been reported.24 With the hopes of identifying milder conditions with lower temperature and catalyst loading, we embarked on a screen of catalysts for decarboxylation, using 10b of 96:4 E/Z purity as substrate (Table 4).25 For the initial screen, 50 mol % of catalyst was used in NMP (3 volumes) at 130 °C, with the reaction progress and the E/Z ratio of the product determined by HPLC analysis. Of the Cu catalysts screened, Cu2O was most effective, giving full conversion after 24 h with a slight drop in isomeric purity (94:6, entry 1). Silver salts are well known to promote decarboxylation, and a screen of various Ag salts gave some excellent results.26 Ag(I) salts with low pKa counterions gave low conversions (entries 6–8), but with retention of the starting olefin isomer ratio. AgNO3 gave complete conversion after just 1.5 h, but with significant erosion of isomeric purity (76:24, entry 9). The more basic catalysts Ag2CO3 and Ag2O (entries 10–11) also gave full conversion in 1.5 h and with only slight loss of geometric integrity (95:5 and 94:6, respectively). AgOAc proved to be the optimal silver catalyst, giving full conversion in 1.5 h with no loss of isomeric purity (entry 12). The use of Pd, Fe or Ir catalysts was ineffective (entries 13–15). Interestingly, Rh(nbd)2BF4 gave a moderate conversion after 24 h and with increased isomeric purity (99:1, entry 16). The Wilkinson catalyst, however, gave no reaction (entry 17).
Table 4.
Decarboxylation catalyst screen.
| |||||
|---|---|---|---|---|---|
| entry | catalyst | Conversion (%)a | E/Zb | ||
| 1 h | 4 h | 24 h | |||
| 1 | Cu2O | - | 28 | 100 | 94:6 |
| 2 | CuO | - | 0 | 1 | 87:13 |
| 3 | Cu(OAc)2 | - | 1 | 45 | 94:6 |
| 4 | CuOAc | - | 0 | 0 | - |
| 5 | CuTCc | - | 5 | 50 | 95:5 |
| 6 | AgOTs | - | 8 | 21 | 96:4 |
| 7 | AgBF4 | - | - | 25 | 96:4 |
| 8 | AgOTf | 5 | - | 27 | 96:4 |
| 9 | AgNO3 | 100d | - | - | 76:24 |
| 10 | Ag2CO3 | 100d | - | - | 95:5 |
| 11 | Ag2O | 100d | - | - | 94:6 |
| 12 | AgOAc | 100 d | - | - | 96:4 |
| 13 | Pd(OAc)2 | - | 0 | 0 | - |
| 14 | Fe(acac)3 | - | 0 | 0 | - |
| 15 | [Ir(cod)Cl]2 | - | 0 | 0 | - |
| 16 | Rh(nbd)2BF4 | - | 15 | 55 | 99:1 |
| 17 | Rh(PPh3)3Cl | - | 0 | 0 | - |
Conversion of 10b to 10c as measured by HPLC (220 nm).
E/Z ratio from HPLC.
Copper(I) thiophene-2-carboxylate.
Conversion after 1.5 h.
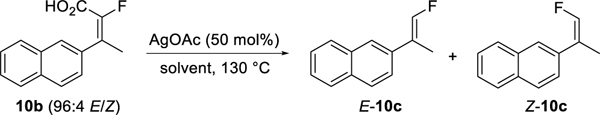
We next performed a solvent screen using the optimal catalyst AgOAc with a 50 mol % loading (Table 5). DMAc and DMSO were effective solvents for the reaction, but with slight reduction in isomeric purity (entries 2 and 3). The use of quinoline and pyridine gave a slower reaction but with retention of isomeric integrity (entries 4 and 5). The reaction proceeded but very slowly in 1-pentanol (entry 6). Decarboxylation was significantly retarded in propionic acid due to competitive binding to Ag (entry 7). Low conversion was observed in xylene due to the poor solubility of the catalyst (entry 8).
Table 5.
Decarboxylation solvent screen.
| |||||
|---|---|---|---|---|---|
| entry | solventa | Conversion (%)b | E/Zc | ||
| 2 h | 4 h | 24 h | |||
| 1 | NMP | 100 | - | - | 96:4 |
| 2 | DMAc | 100 | - | - | 95:5 |
| 3 | DMSO | 100 | - | - | 94:6 |
| 4 | quinoline | 90 | 100 | - | 96:4 |
| 5 | pyridine | 75 | 98 | 100 | 96:4 |
| 6 | 1-pentanol | - | 12 | 51 | 96:4 |
| 7 | propionic acid | 0 | 1 | 7 | 98:2 |
| 8 | p-xylene | 0 | 3 | 3 | 97:3 |
Three volumes of solvent relative to 10b.
Conversion of 10b to 10c as measured by HPLC (220 nm).
E/Z ratio from HPLC.
Finally, the loading of AgOAc was optimized (Table 6). Using 15 or 10 mol % catalyst, the reaction reached complete conversion after 1 h (entries 1 and 2). Lowering further to 5 mol % (entry 3) resulted in high but incomplete conversion after 1 h, with the reaction reaching completion by 2 h. The use of only 2.5 mol % catalyst drastically slowed the reaction, with it stalling at 22% conversion after 1 h (entry 4). Increasing the concentration of the reaction by using two instead of three volumes of NMP did not enable complete conversion with 2.5 mol % catalyst (entry 5) but with 5 mol % complete conversion was achieved in 1 h (entry 6). In all cases, isomeric purity was fully retained.
Table 6.
Catalyst loading optimization for decarboxylation.
| |||||
|---|---|---|---|---|---|
| entry | mol % AgOAc | Conversion (%)a | E/Zb | ||
| 1 h | 2 h | 3 h | |||
| 1 | 15.0 | 100 | - | - | 96:4 |
| 2 | 10.0 | 100 | - | - | 96:4 |
| 3 | 5.0 | 96 | 100 | - | 96:4 |
| 4 | 2.5 | 22 | 22 | 22 | 96:4 |
| 5 | 2.5c | 23 | 23 | 23 | 96:4 |
| 6 | 5.0 c | 100 | - | - | 96:4 |
Conversion of 10b to 10c as measured by HPLC (220 nm).
E/Z ratio from HPLC.
Two volumes of NMP instead of three volumes.
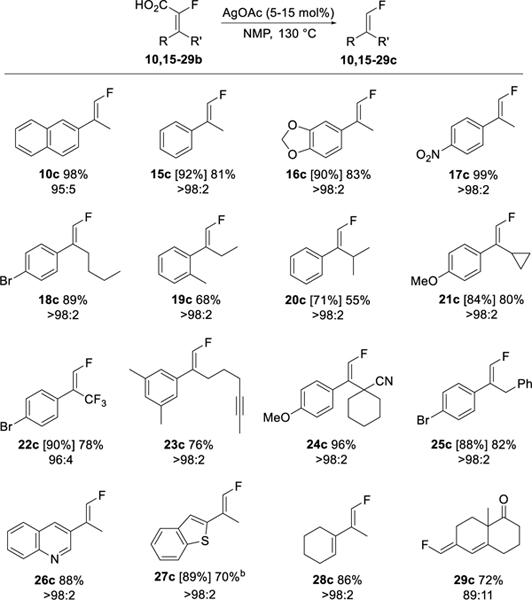
The decarboxylation was then applied to the α-fluoroacrylic acid substrates using the optimized reaction conditions (Table 7). A variety of aryl, heteroaryl and vinyl substituted α-fluoroacrylic acids underwent decarboxylation to yield the corresponding trisubstituted vinyl fluorides in good yields. In all cases except 27c, the isomeric purity of the starting acrylic acid was completely preserved in the trisubstituted vinyl fluoride product. In the case of 27c, the isomeric purity decreased from 92:8 to 81:19 during the decarboxylation. We attribute this anomaly to interaction of the thiophilic silver catalyst with the sulfur atom of the thiophene ring. Notably, the decarboxylation was not perturbed by the presence of other groups known to coordinate or react with silver, such as an aryl bromide (18c, 22c, 25c), an alkyne (23c), a nitrile (24c), and a quinoline nitrogen (26c). The reaction also gave access to unusual terminal monofluoro 1,3-dienes (28-29c).
Table 7.
Silver catalyzed decarboxylation of α-fluoroacrylic acids to trisubstituted vinyl fluorides.a
|
Assay yields from 19F NMR versus PhCF3 are given in brackets, isolated yields are given with no brackets. E/Z isomeric purity of isolated product is given under yields. Isomeric purity determined by 19F NMR.
E/Z ratio degraded from 92:8 to 81:19 during the reaction. The minor isomer was removed by chromatography. For detailed reaction conditions, see the Experimental Section.
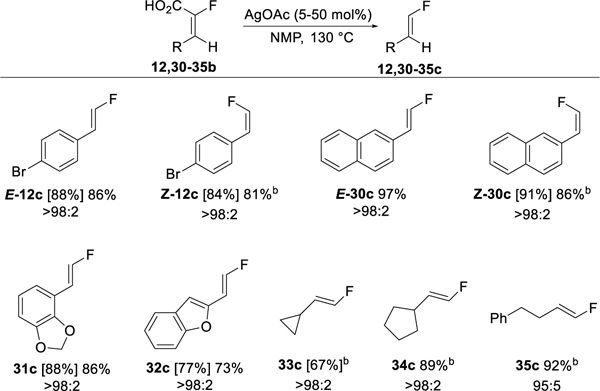
The decarboxylation was applied to aldehyde-derived α-fluoroacrylic acids with similar success (Table 8). Both aryl and alkyl substituted fluoroalkenes were amenable to the decarboxylation. The pure E and pure Z isomers of 12b and 30b were converted to the corresponding vinyl fluorides with the isomeric purity completely maintained. Decarboxylation of E-12b and E-30b was facile with only 5 mol % catalyst loading whereas the reaction of isomeric Z-12b and Z-30b required 50 mol % of AgOAc and higher dilution due to lower solubility of the corresponding Ag carboxylate intermediates. Higher reaction temperature (140–145 °C) and increased catalyst loading was also required for efficient decarboxylation of alkyl substituted fluoroacrylic acids (33–35b).
Table 8.
Silver catalyzed decarboxylation of α-fluoroacrylic acids to disubstituted vinyl fluorides.a
|
Assay yields from 19F NMR versus PhCF3 are given in brackets, isolated yields are given with no brackets. E/Z isomeric purity of isolated product is given under yields. Isomeric purity determined by 19F NMR. For detailed reaction conditions, see the Experimental Section.
AgOAc loading of 50 mol %.
The overall method as described above was applied to the synthesis of the BPAO inhibitor 2 (Scheme 5).6b Olefination of bromoacetophenone 36 gave the ester 37 with a 90:10 Z/E reaction selectivity which was upgraded to a 97:3 Z/E after purification. Displacement of the bromide with di-tert-butyliminodicarboxylate gave 38, which was converted to the mono-Boc acid 39 with aqueous LiOH. The minor E-isomer of 39 was not detectable in the product due to isolation of both 38 and 39 by crystallizations which effectively purged the minor isomer. Decarboxylation of 39 gave a 93% yield of 40 with complete retention of geometric purity. Finally, Boc deprotection yielded the target compound 2 in high yield.
Scheme 5.
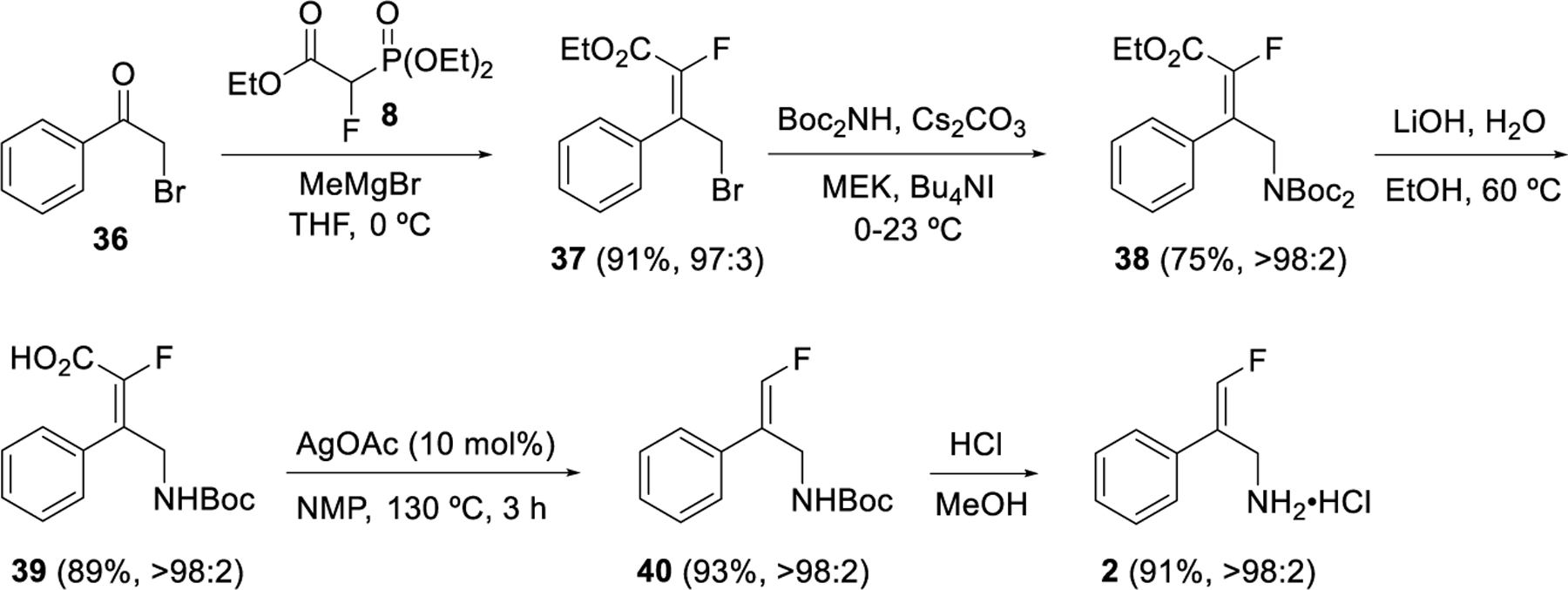
Application of HWE/hydrolysis/decarboxylation to the synthesis of BPAO inhibitor 2.
The protocol was also applied to the synthesis of an intermediate en route to LuxS inhibitor 5 (Scheme 6).6e Periodate cleavage of diol 4127 in EtOH/water gave the aldehyde 42 along with its ethyl hemiacetal. This mixture was taken directly into the olefination with 8 using n-BuLi as base to provide ester 43 with 92:8 E/Z selectivity in 85% yield from 41. Hydrolysis gave the acid 44 (E/Z = 94:6), which upon decarboxylation furnished 45 in 79% yield and as a single isomer after purification. Compound 45 can be converted to 5 by literature methods.6e
Scheme 6.
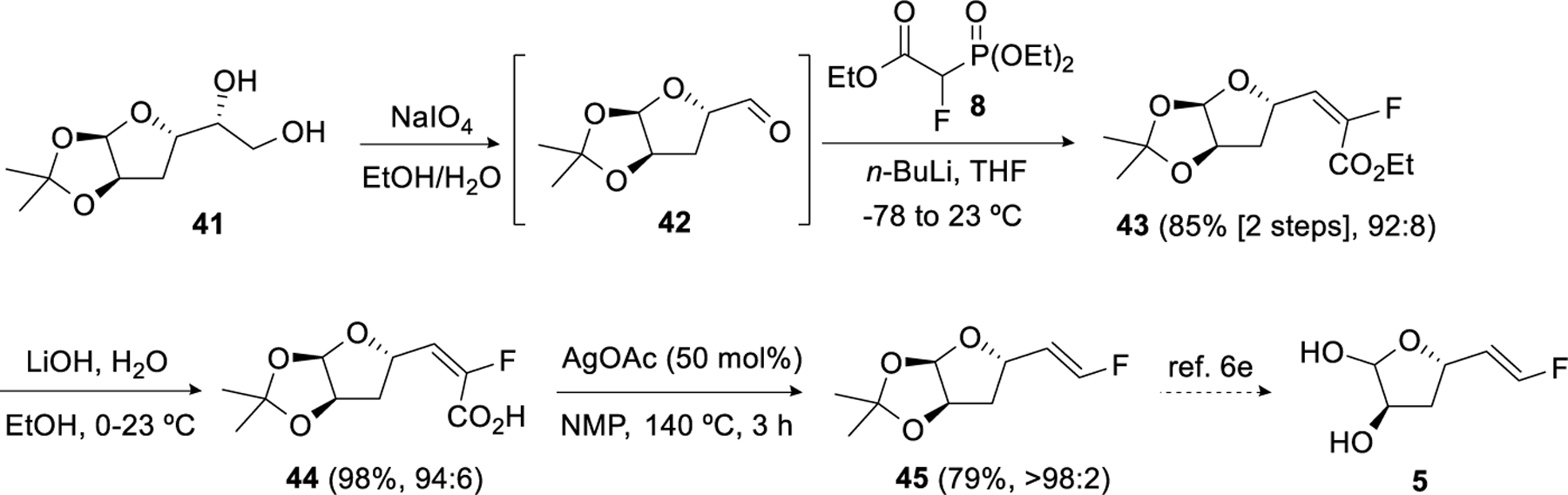
Application of HWE/hydrolysis/decarboxylation to the formal synthesis of LuxS inhibitor 5.
CONCLUSION
In summary, a HWE olefination/hydrolysis/decarboxylation sequence has been developed for the preparation of di- and trisubstituted terminal monofluoroalkenes. The olefination reaction exhibits high E-selectivity for ketone substrates using MeMgBr as base and for aldehyde substrates using n-BuLi as base. Upon hydrolysis, the derived α-fluoroacrylic acids are crystalline solids which may be isolated by direct crystallization from the reaction mixture upon acidification, often with an upgrade in isomeric purity. The acids can also be recrystallized to upgrade isomeric purity if necessary. A silver catalyzed decarboxylation was developed which proceeds with retention of olefin configuration under relatively mild reaction conditions. Importantly, the reagent 8 used for the HWE olefination is inexpensive and widely available. This protocol provides a practical method for accessing α-fluoroacrylic acids and terminal di- and trisubstituted vinyl fluorides with high geometric purity without recourse to difficult chromatography for separation of isomers.
EXPERIMENTAL SECTION
General Information.
All starting materials, reagents and solvents were purchased from commercial sources and used as received unless otherwise noted. For reactions requiring heating, an oil bath was used as the heat source. 1H, 19F and 13C NMR spectra were recorded using Bruker DRX 400 or 500 MHz spectrometers. 1H and 13C chemical shifts are referenced to internal solvent resonances (CHCl3 1H, δ = 7.26 ppm; CDCl3 13C, δ = 77.0 ppm) and reported relative to SiMe4; multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), qn (quintet), m (multiplet) and brs (broad signal). Coupling constants, J, are reported in Hertz. Structural assignments were made with additional information from gCOSY, gHSQC, and gHMBC experiments. Flash chromatography was performed on a Combi-Flash automated system with silica columns. High-resolution accurate mass (HRAM) was performed on both a Thermo LTQ FT-ICR mass spectrometer using DART source ionization in positive/negative ion modes and on a Thermo Scientific Exactive GC/MS using positive ion electron ionization (EI). Melting points were measured using a differential scanning calorimeter TA instrument DSC 2500.
General Procedure for Horner-Wadsworth-Emmons Reaction of Ketones with Triethyl 2-fluoro-2-phosphonoacetate and MeMgBr in THF.
A round bottom flask is charged with triethyl 2-fluoro-2-phosphonoacetate (1.7–4.5 equiv) and THF (0.3 M rel to ketone substrate). The solution is cooled to 0 to −5 °C. MeMgBr (3.4 M in 2-MeTHF, 1.7–4.5 equiv) is then added dropwise maintaining internal temperature below 10 °C. The reaction mixture is warmed to rt and stirred for 10 min, then heated to 40 °C and stirred for another 10 min. A solution of ketone (1.0 equiv) in THF (1 M) is added dropwise, and the reaction mixture is stirred for 0.5–3 h. After reaction is complete, judged by HPLC or GC analysis, the mixture is cooled to rt. The reaction mixture is quenched with 10% aq. NH4Cl solution and extracted with EtOAc. Organic phase is dried over MgSO4, filtered, and concentrated under reduced pressure. The residue is purified by silica gel column chromatography.
Ethyl (E)-2-fluoro-3-(naphthalen-2-yl)but-2-enoate (10a).
The reaction between acetonaphthone 9 (10.0 g, 58.75 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (23.8 mL; 117.50 mmol; 2.0 equiv) and MeMgBr (33.8 mL, 117.50 mmol, 3.48 M in 2-MeTHF, 2.0 equiv) was run at 40 °C for 30 min. 19F NMR of the crude: mixture of stereoisomers in 20.6:1 (95:5) dr. The product was obtained as a colorless oil after purification by silica gel column chromatography eluting with 0–5% EtOAc in hexanes (14.90 g, 98% yield, mixture of stereoisomers in 20.9:1 (95:5) dr). 1H NMR (400 MHz, CDCl3) δ 7.90–7.79 (m, 3H), 7.69 (d, JHF = 1.0 Hz, 1H), 7.55–7.47 (m, 2H), 7.35 (dd, J = 8.4 Hz, J = 1.7 Hz, 1H), 4.06 (q, J = 7.1 Hz, 2H), 2.27 (d, J = 4.5 Hz, 3H), 0.97 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.5 (d, J = 36.1 Hz), 144.5 (d, J = 252.3 Hz), 135.8 (d, J = 5.5 Hz), 132.9, 132.6, 131.2 (d, J = 17.2 Hz), 127.8, 127.59, 127.52, 126.2 (d, J = 3.3 Hz), 126.12, 126.07, 125.7 (d, J = 3.0 Hz), 60.9, 19.2 (d, J = 6.5 Hz), 13.5; 19F NMR (376.5 MHz, CDCl3) δ −123.1 (d, JHF = 4.4 Hz); Z-isomer: −125.3 (s). HRMS (DART) calcd for C16H16O2F [M+H]+: 259.1129, found 259.1129.
Ethyl (Z)-3-(4-bromophenyl)-2-fluoroacrylate (Z-12a).
The reaction between 4-bromobenzaldehyde (4.25 g, 22.97 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (7.0 mL; 34.46 mmol; 1.5 equiv) and MeMgBr (9.90 mL, 34.46 mmol, 3.40 M in 2-MeTHF, 1.5 equiv) was run at −40 °C for 1 h. 19F NMR of the crude: mixture of stereoisomers in 4.0:1 (80:20) dr. The product Z-12a was obtained as a white solid after purification by silica gel column chromatography eluting with 0–2% MTBE in hexanes (4.60 g, 73% yield, single Z-isomer (>98:2 dr)). mp 62.1–63.6 °C; 1H NMR (400 MHz, CDCl3): δ 7.56–7.46 (m, 4H), 6.85 (d, JHF = 34.7 Hz, 1H), 4.35 (q, J = 7.1 Hz, 2H), 1.38 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.1 (d, J = 34.2 Hz), 147.4 (d, J = 269.1 Hz), 132.0, 131.6 (d, J = 8.3 Hz), 130.0 (d, J = 4.4 Hz), 124.0 (d, J = 3.9 Hz), 116.3 (d, J = 4.7 Hz), 62.0, 14.2; 19F NMR (376.5 MHz, CDCl3) δ −124.0 (d, JHF = 34.7 Hz). NMR spectra matched reported data.30
Ethyl (E)-3-(4-bromophenyl)-2-fluoroacrylate (E-12a).
The reaction between 4-bromobenzaldehyde (3.0 g, 16.21 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (4.93 mL; 24.32 mmol; 1.5 equiv) and n-BuLi (9.0 mL, 24.32 mmol, 2.71 M in hexanes, 1.5 equiv) was run at −78 °C to −20 °C for 1 h. 19F NMR of the crude: mixture of stereoisomers in 35.5:1 (97:3) dr. The product E-12a was obtained as a colorless oil after purification by silica gel column chromatography eluting with 0–2% MTBE in hexanes (4.14 g, 94% yield, mixture of stereoisomers in 34.5:1 (97:3) dr). 1H NMR (400 MHz, CDCl3) δ 7.50–7.44 (m, 2H), 7.38–7.31 (m, 2H), 6.81 (d, JHF = 21.9 Hz, 1H), 4.26 (q, J = 7.1 Hz, 2H), 1.26 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.3 (d, J = 35.7 Hz), 147.2 (d, J = 257.6 Hz), 131.3 (d, J = 3.1 Hz), 131.2, 129.9 (d, J = 9.7 Hz), 122.9, 120.4 (d, J = 26.8 Hz), 61.8, 13.9; 19F NMR (376.5 MHz, CDCl3) δ −115.9 (d, JHF = 21.9 Hz), Z-isomer: −124.0 (d, JHF = 34.7 Hz). NMR spectra matched reported data.28
Reaction of triethyl 2-phosphonopropionate (14) with 4-bromobenzaldehyde. Ethyl (E)-3-(4-bromophenyl)-2-methylacrylate (E-13).
Using MeMgBr as base:
The reaction between 4-bromobenzaldehyde (1.00 g, 5.40 mmol, 1 equiv) and reagent prepared from triethyl 2-phosphonopropionate (1.74 mL; 8.11 mmol; 1.5 equiv) and MeMgBr (2.33 mL, 8.11 mmol, 3.48 M in 2-MeTHF, 1.5 equiv) was run at −40 °C with warming to rt. 1H NMR of the crude: >45:1 dr. The product E-13 was obtained as a colorless oil after purification by silica gel column chromatography eluting with 2–5% EtOAc in hexanes (1.35 g, 93% yield). Using n-BuLi as base: The reaction between 4-bromobenzaldehyde (1.00 g, 5.40 mmol, 1 equiv) and reagent prepared from triethyl 2-phosphonopropionate (1.74 mL; 8.11 mmol; 1.5 equiv) and n-BuLi (3.24 mL, 8.11 mmol, 2.50 M in hexanes, 1.5 equiv) was run at −78 °C with warming to rt. 1H NMR of the crude: >45:1 dr. The product E-13 was obtained as a colorless oil after purification by silica gel column chromatography eluting with 2–5% EtOAc in hexanes (1.33 g, 91% yield). NMR spectra for E-13 prepared by both methods matched reported data.29
Ethyl (E)-2-fluoro-3-phenylbut-2-enoate (15a).
A round bottom flask was charged with triethyl 2-fluoro-2-phosphonoacetate (591 μL, 2.91 mmol, 1.7 equiv) and THF (4.9 mL). The solution was cooled to 0 to −5 °C. MeMgBr (985 μL, 2.91 mmol, 2.96 M in Et2O, 1.7 equiv) was added dropwise maintaining internal temperature below 10 °C. The reaction mixture was warmed to rt and stirred for 10 min, then heated to 40 °C and stirred for another 10 min. A solution of acetophenone (200 μL, 1.71 mmol, 1.0 equiv) in THF (1.7 mL) was added dropwise, and the reaction mixture was stirred for 15 min. The mixture was cooled to rt, quenched with sat. aq. NH4Cl solution (10 mL) and extracted with EtOAc (2 × 10 mL). The organic phase was dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was analyzed by quantitative 19F NMR which showed a mixture of stereoisomers in 15.5:1 (94:6) dr. The residue was purified by silica gel column chromatography eluting with 0–2% EtOAc in hexanes to afford 15a as a colorless liquid. Mixture of stereoisomers in 15.9: 1 (94:6) dr (341.6 mg, 96% yield). 1H NMR (500 MHz, CDCl3) δ 7.40–7.30 (m, 3H), 7.23–7.14 (m, 2H), 4.05 (q, J = 7.2 Hz, 2H), 2.15 (d, JHF = 4.4 Hz, 3H), 1.03 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (125 MHz, CDCl3) δ 160.5 (d, J = 36.2 Hz), 144.2 (d, J = 251.7 Hz), 138.5 (d, J = 5.5 Hz), 131.3 (d, J = 17.0 Hz), 128.0, 127.7, 127.3 (d, J = 3.1 Hz), 60.9, 19.2 (d, J = 6.6 Hz), 13.5; 19F NMR (470 MHz, CDCl3) δ −123.9 (s). NMR spectra matched reported data.30
Ethyl (E)-3-(benzo[d][1,3]dioxol-5-yl)-2-fluorobut-2-enoate (16a).
The reaction between 1-(benzo[d][1,3]dioxol-5-yl)ethan-1-one (3.0 g, 18.28 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (8.15 mL; 40.21 mmol; 2.2 equiv) and MeMgBr (11.6 mL, 40.21 mmol, 3.48 M in 2-MeTHF, 2.2 equiv) was run at 40 °C for 2 h. 19F NMR of the crude: mixture of stereoisomers in 17.9:1 (95:5) dr. The product 16a was obtained as a colorless liquid after purification by silica gel column chromatography eluting with 2–8% EtOAc in hexanes (3.88 g, 84% yield, single stereoisomer (>98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 6.78 (d, J = 7.9 Hz, 1H), 6.70–6.60 (m, 2H), 5.96 (s, 2H), 4.10 (q, J = 7.1 Hz, 2H), 2.10 (d, JHF = 4.5 Hz, 3H), 1.12 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.5 (d, J = 35.8 Hz), 147.3, 147.2, 144.3 (d, J = 252.6 Hz), 131.9 (d, J = 5.8 Hz), 131.0 (d, J = 17.6 Hz), 121.0 (d, J = 3.2 Hz), 108.2 (d, J = 3.3 Hz), 108.0, 101.1, 70.0, 19.3 (d, J = 6.4 Hz), 13.7; 19F NMR (376.5 MHz, CDCl3) δ −123.4 (q, JHF = 4.5 Hz). HRMS (DART) calcd for C15H19O2BrF [M+H]+: 329.0547, found 329.0550.
Ethyl (E)-3-(4-bromophenyl)-2-fluorohept-2-enoate (18a).
The reaction between 1-(4-bromophenyl)pentan-1-one (3.0 g, 12.44 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (6.31 mL; 31.10 mmol; 2.5 equiv) and MeMgBr (9.15 mL, 31.10 mmol, 3.40 M in 2-MeTHF, 2.5 equiv) was run at 40 °C for 1 h. 19F NMR of the crude: mixture of stereoisomers in 17.1:1 (94:6) dr. The product 18a was obtained as a colorless liquid after purification by silica gel column chromatography eluting with 5% MTBE in hexanes (3.77 g, 92% yield, mixture of stereoisomers in 20.5:1 (95:5) dr). 1H NMR (400 MHz, CDCl3) δ 7.50–7.45 (m, 2H), 7.06–6.99 (m, 2H), 4.05 (q, J = 7.1 Hz, 2H), 2.60–2.40 (m, 2H), 1.41–1.23 (m, 4H), 1.06 (t, J = 7.1 Hz, 3H), 0.93–0.79 (m, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.3 (d, J = 36.2 Hz), 144.2 (d, J = 254.1 Hz), 136.1 (d, J = 5.8 Hz), 134.4 (d, J = 17.1 Hz), 131.1, 129.5 (d, J = 3.1 Hz), 121.6, 61.0, 32.0 (d, J = 4.8 Hz), 28.7 (d, J = 2.0 Hz), 22.2, 13.59, 13.56; 19F NMR (376.5 MHz, CDCl3) δ −124.3 (t, JHF = 3.5 Hz); Z-isomer: −124.2 (s). HRMS (DART) calcd for C15H19O2BrF [M+H]+: 329.0547, found 329.0550.
Ethyl (E)-2-fluoro-4-methyl-3-phenylpent-2-enoate (20a).
The reaction between isobutyrophenone (3.0 g, 20.2 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (10.26 mL, 50.6 mmol, 2.5 equiv) and MeMgBr (17.1 mL, 50.6 mmol, 2.96 M in Et2O, 2.5 equiv) was run at 40 °C for 30 min. 19F NMR of the crude: mixture of stereoisomers in 11.6:1 (92:8) dr. The product 20a was obtained as a colorless oil after purification by silica gel column chromatography eluting with 0–5% EtOAc in hexanes (4.37 g, 11.6:1 (92:8) dr, 91% yield). 1H NMR (400 MHz, CDCl3) δ 7.41–7.29 (m, 3H), 7.11–7.03 (m, 2H), 3.98 (q, J = 7.1 Hz, 2H), 3.31 (dsept, J = 7.0 Hz, JHF = 0.5 Hz 1H), 1.02 (d, J = 7.0 Hz, 6H), 0.96 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.7 (d, J = 36.9 Hz), 143.7 (d, J = 253.0 Hz), 139.8 (d, J = 14.0 Hz), 134.7 (d, J = 5.6 Hz), 128.3 (d, J = 3.2 Hz), 127.5, 127.2, 60.8, 28.9 (d, J = 5.1 Hz), 20.1 (d, J = 2.0 Hz), 13.4; 19F NMR (376.5 MHz, CDCl3) δ −126.2 (d, JHF = 5.0 Hz); Z-isomer: −121.3 (s). NMR spectra matched reported data.30
Ethyl (E)-3-cyclopropyl-2-fluoro-3-(4-methoxyphenyl)acrylate (21a).
The reaction between cyclopropyl(4-methoxyphenyl) ketone (4.0 g, 22.70 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (19.6 mL; 96.48 mmol; 4.25 equiv) and MeMgBr (27.7 mL, 96.48 mmol, 3.48 M in 2-MeTHF, 4.25 equiv) was run at 70 °C overnight. 19F NMR of the crude: mixture of stereoisomers in 11.0:1 (92:8) dr). The product 21a was obtained as a white solid after purification by silica gel column chromatography eluting with 1–5% EtOAc in hexanes (4.46 g, 74% yield, single stereoisomer (>98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 6.95–6.87 (m, 2H), 6.87–6.79 (m, 2H), 3.99 (q, J = 7.1 Hz, 2H), 3.79 (s, 3H), 2.29–2.10 (m, 1H), 1.00 (t, J = 7.1 Hz, 3H), 0.83–0.70 (m, 2H), 0.44–0.31 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.3 (d, J = 35.1 Hz), 159.0, 145.2 (d, J = 251.6 Hz), 136.9 (d, J = 13.4 Hz), 129.9 (d, J = 3.2 Hz), 124.6 (d, J = 5.1 Hz), 113.1, 60.6, 55.0, 13.6, 11.7 (d, J = 9.4 Hz), 4.8 (d, J = 1.2 Hz); 19F NMR (376.5 MHz, CDCl3) δ −131.2 (s). HRMS (DART) calcd for C15H18O3F [M+H]+: 265.1235, found 265.1235.
Ethyl (Z)-3-(4-bromophenyl)-2,4,4,4-tetrafluorobut-2-enoate (22a).
The reaction between 1-(4-bromophenyl)-2,2,2-trifluoroethan-1-one (4.0 g, 15.81 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (8.0 mL; 39.52 mmol; 2.5 equiv) and MeMgBr (11.6 mL, 39.52 mmol, 3.40 M in 2-MeTHF, 2.5 equiv) was run at 40 °C for 30 min. 19F NMR of the crude: mixture of stereoisomers in 25.8:1 (96:4) dr. The product 22a was obtained as a colorless liquid after purification by silica gel column chromatography eluting with 5% MTBE in hexanes (4.56 g, 85% yield, mixture of stereoisomers in 25.8:1 (96:4) dr). 1H NMR (400 MHz, CDCl3) δ 7.59–7.52 (m, 2H), δ 7.18–7.09 (m, 2H), 4.10 (q, J = 7.1 Hz, 2H), 1.06 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 158.7 (d, J = 34.5 Hz), 149.6, (dq, J = 285.1 Hz, J = 2.4 Hz), 131.6, 131.0 (d, J = 3.4 Hz), 127.5 (d, J = 2.5 Hz), 123.9, 121.6 (q, J = 275.4 Hz), 120.5 (dq, J = 32.9 Hz, J = 9.4 Hz), 62.6, 13.3; 19F NMR (376.5 MHz, CDCl3) δ −107.3 (q, J = 24.3 Hz), −60.5 (d, J = 24.3 Hz); Z-isomer: δ −105.7 (q, J = 10.0 Hz), −58.5 (d, J = 10.0 Hz). HRMS (DART) calcd for C12H10O2BrF4 [M+H]+: 340.9795, found 340.9796.
1-(3,5-Dimethylphenyl)hept-5-yn-1-one.
To a suspension of Mg turnings (489.5 mg, 20.13 mmol, 1.04 equiv) in THF (5 mL) was added 1,2-dibromoethane (63 µL, 0.73 mmol, 0.04 equiv) and the mixture was heated to 50 °C and stirred for 15 min. A solution of 1-bromo-3,5-dimethylbenzene (2.63 mL, 19.36 mmol, 1 equiv) in THF (20 mL) was then added dropwise maintaining steady solvent boiling. After complete addition the reaction mixture was stirred at 50 °C for 30 min. The resultant Grignard reagent was titrated with salicylaldehyde phenylhydrazone: 0.765 M solution was obtained. Grignard reagent (13.8 mL, 10.56 mmol, 1.0 equiv.) was added dropwise to a solution of 2-methyl-3-oxocyclohex-1-en-1-yl trifluoromethanesulfonate31 (3.0 g; 11.62 mmol; 1.1 equiv) in THF (20 mL) at 0 °C. The mixture was stirred at 0 °C for 10 min and then warmed to room temperature and stirred for additional 30 min. The reaction mixture was quenched with 10% aq. NH4Cl solution and extracted with MTBE (2 × 30 mL). Combined organic extracts were dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography eluting with 5–30% EtOAc in hexanes to afford the title ketone as a pale yellow oil (1.56 g, 63% yield). 1H NMR (400 MHz, CDCl3) δ 7.58 (s, 2H), 7.19 (s, 1H), 3.06 (t, J = 7.3 Hz, 2H), 2.37 (s, 6H), 2.29–2.21 (m, 2H), 1.90 (apparent quintet, J = 7.0 Hz, 2H), 1.78 (t, J = 2.5 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 200.3, 138.1, 137.1, 134.5, 125.8, 78.5, 76.3, 37.4, 23.5, 21.2, 18.3, 3.4. HRMS (DART) calcd for C15H19O [M+H]+: 215.1430, found 215.1431.
Ethyl (E)-3-(3,5-dimethylphenyl)-2-fluoronon-2-en-7-ynoate (23a).
The reaction between 1-(3,5-dimethylphenyl)hept-5-yn-1-one (1.50 g, 7.00 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (3.3 mL; 16.10 mmol; 2.3 equiv) and MeMgBr (4.6 mL, 16.10 mmol, 3.48 M in 2-MeTHF, 2.3 equiv) was run at 40 °C for 2 h. 19F NMR of the crude: mixture of stereoisomers in 22.0:1 (96:4) dr. The product 23a was obtained as a colorless liquid after purification by silica gel column chromatography eluting with 0–2% MTBE in hexanes (1.95 g, 92% yield, single stereoisomer (>98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 6.94 (s, 1H), 6.74 (s, 2H), 4.04 (q, J = 7.1 Hz, 2H), 2.63–2.53 (m, 2H), 2.31 (s, 6H), 2.18–2.08 (m, 2H), 1.76 (t, J = 2.5 Hz, 3H), 1.62–1.48 (m, 2H), 1.03 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.6 (d, J = 36.6 Hz), 144.3 (d, J = 252.4 Hz), 137.3, 136.7 (d, J = 5.3 Hz), 134.8 (d, J = 15.7 Hz), 129.3, 125.5 (d, J = 3.0 Hz), 78.2, 75.9, 60.8, 31.7 (d, J = 5.2 Hz), 26.3 (d, J = 2.1 Hz), 21.1, 18.5, 13.5, 3.3; 19F NMR (376.5 MHz, CDCl3) δ −124.9 (s). HRMS (DART) calcd for C19H24O2F [M+H]+: 303.1755, found 303.1755.
Ethyl (Z)-3-(1-cyanocyclohexyl)-2-fluoro-3-(4-methoxyphenyl)acrylate (24a).
The reaction between 1-(4-methoxybenzoyl)cyclohexane-1-carbonitrile32 (1.0 g, 4.11 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (3.75 mL, 18.5 mmol, 4.5 equiv) and MeMgBr (5.31 mL, 18.5 mmol, 3.48 M in 2-MeTHF, 4.5 equiv) was run at 40 °C for 18 h. 19F NMR of the crude: single stereoisomer (>98:2 dr). The product 24a was obtained as a colorless oil after purification by silica gel column chromatography eluting with EtOAc in hexanes (1.33 g, 98% yield, single stereoisomer (>98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 7.01 (d, J = 8.5 Hz, 2H), 6.90 (d, J = 8.5 Hz, 2H), 4.01 (q, J = 7.1 Hz, 2H), 3.82 (s, 3H), 2.12–1.97 (m, 2H), 1.83–1.64 (m, 5H), 1.49–1.33 (m, 2H), 1.18–1.03 (m, 1H), 1.02 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 159.9 (d, J = 35.3 Hz), 159.6, 147.0 (d, J = 269.0 Hz), 132.2 (d, J = 9.5 Hz), 129.9 (d, J = 3.5 Hz), 125.1 (d, J = 5.8 Hz), 120.6, 113.6, 61.5, 55.1, 41.6 (d, J = 1.7 Hz), 34.3 (d, J = 2.1 Hz), 24.6, 22.7, 13.5; 19F NMR (376.5 MHz, CDCl3) δ −111.4 (s). HRMS (DART) calcd for C19H23O3NF [M+H]+: 332.1657, found 332.1657.
Ethyl (E)-3-(4-bromophenyl)-2-fluoro-4-phenylbut-2-enoate (25a).
The reaction between 1-(4-bromophenyl)-2-phenylethan-1-one (4.0 g, 14.54 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (5.9 mL; 29.08 mmol; 2.0 equiv) and MeMgBr (8.4 mL, 29.08 mmol, 3.48 M in 2-MeTHF, 2.0 equiv) was run at 40 °C for 1 h. 19F NMR of the crude: mixture of stereoisomers in 13.2:1 (93:7) dr. The product 25a was obtained as a colorless liquid after purification by silica gel column chromatography eluting with 0–5% MTBE in hexanes (4.94 g, 94% yield, mixture of stereoisomers in 13.3:1 (93:7) dr). 1H NMR (400 MHz, CDCl3) δ 7.40–7.31 (m, 2H), 7.27–7.13 (m, 3H), 7.06–6.97 (m, 2H), 6.84–6.75 (m, 2H), 4.05 (q, J = 7.1 Hz, 2H), 3.79 (d, JHF = 3.6 Hz, 2H), 1.04 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (125 MHz, CDCl3) δ 160.4 (d, J = 36.2 Hz), 144.3 (d, J = 255.8 Hz), 136.3 (d, J = 2.5 Hz), 135.6 (d, J = 5.3 Hz), 132.8 (d, J = 17.0 Hz), 131.0, 129.7 (d, J = 3.1 Hz), 129.1 (d, J = 1.1 Hz), 128.5, 126.7, 121.8, 61.3, 38.6 (d, J = 5.1 Hz), 13.6; 19F NMR (376.5 MHz, CDCl3) δ −123.3 (t, JHF = 3.2 Hz); Z-isomer: −122.6 (s). HRMS (DART) calcd for C18H17O2BrF [M+H]+: 363.0391, found 363.0391.
Ethyl (E)-2-fluoro-3-(quinolin-3-yl)but-2-enoate (26a).
The reaction between 1-(quinolin-3-yl)ethan-1-one (4.0 g, 23.37 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (8.1 mL; 39.72 mmol; 1.7 equiv) and MeMgBr (11.7 mL, 39.72 mmol, 3.40 M in 2-MeTHF, 1.7 equiv) was run at 40 °C for 30 min. 19F NMR of the crude: mixture of stereoisomers in 9.8:1 (91:9) dr. The product 26a was obtained as a light brown oil after purification by silica gel column chromatography eluting with 40% MTBE in hexanes (4.88 g, 81% yield, mixture of stereoisomers in 11.3:1 (92:8) dr). 1H NMR (400 MHz, CDCl3) δ 8.67 (d, JHF = 2.2 Hz, 1H), 8.04 (d, J = 8.5 Hz, 1H), 7.91 (d, JHF = 2.1 Hz, 1H), 7.72 (d, J = 8.1 Hz, 1H), 7.63 (ddd, J = 8.5 Hz, J = 6.9 Hz, J = 1.4 Hz, 1H), 7.47 (ddd, J = 8.1 Hz, J = 6.9 Hz, J = 1.0 Hz, 1H), 3.96 (q, J = 7.1 Hz, 2H), 2.17 (d, JHF = 4.5 Hz, 3H), 0.88 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 159.9 (d, J = 35.7 Hz), 149.6 (d, J = 2.9 Hz), 147.1, 145.0 (d, J = 255.4 Hz), 133.7 (d, J = 3.5 Hz), 131.3 (d, J = 5.4 Hz), 129.4, 129.0, 127.9 (d, J = 5.4 Hz), 127.6, 127.1, 126.7, 61.1, 19.0 (d, J = 6.4 Hz), 13.4; 19F NMR (376.5 MHz, CDCl3) δ −120.4 (q, JHF = 4.5 Hz); Z-isomer: −123.7 (s). HRMS (DART) calcd for C15H15O2NF [M+H]+: 260.1081, found 260.1082.
Ethyl (E)-3-(benzo[b]thiophen-2-yl)-2-fluorobut-2-enoate (27a).
The reaction between 1-(benzo[b]thiophen-2-yl)ethan-1-one (4.0 g, 22.70 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (9.2 mL; 45.39 mmol; 2.0 equiv) and MeMgBr (13.0 mL, 45.39 mmol, 3.48 M in 2-MeTHF, 2.0 equiv) was run at 40 °C for 30 min. 19F NMR of the crude: mixture of stereoisomers in 10.9:1 (92:8) dr. The product 27a was obtained as a pale yellow oil after purification by silica gel column chromatography eluting with 2–8% EtOAc in hexanes (5.97 g, 99% yield, mixture of stereoisomers in 10.9:1 (92:8) dr). 1H NMR (400 MHz, CDCl3) δ 7.84–7.79 (m, 1H), 7.78–7.72 (m, 1H), 7.40–7.29 (m, 2H), 7.25 (s, 1H), 4.16 (q, J = 7.1 Hz, 2H), 2.25 (d, JHF = 4.7 Hz, 3H), 1.11 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.2 (d, J = 35.7 Hz), 145.5 (d, J = 257.6 Hz), 140.1, 139.3, 138.9 (d, J = 6.7 Hz), 124.5, 124.3, 123.9 (d, J = 21.6 Hz), 123.64, 123.63 (d, J = 3.5 Hz), 122.0, 61.4, 19.3 (d, J = 6.4 Hz), 13.7; 19F NMR (376.5 MHz, CDCl3) δ −116.9 (q, JHF = 4.7 Hz); Z-isomer: −116.4 (q, JHF = 3.2 Hz). HRMS (DART) calcd for C14H14O2FS [M+H]+: 265.0693, found 265.0694.
Ethyl (E)-2-fluoro-2-(4a-methyl-5-oxo-4,4a,5,6,7,8-hexahydronaphthalen-2(3H)-ylidene)acetate (29a).
The reaction between Wieland-Miescher ketone (4.0 g, 22.44 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (8.2 mL; 40.40 mmol; 1.8 equiv) and MeMgBr (11.6 mL, 40.40 mmol, 3.48 M in 2-MeTHF, 1.8 equiv) was run at −40 °C for 30 min. 19F NMR of the crude: mixture of double bond isomers in 3.47:1 (78:22) dr. The product 29a was obtained as a colorless oil after purification by silica gel column chromatography eluting with 3–10% EtOAc in hexanes (5.98 g, 99% yield, mixture of double bond isomers in 3.44:1 (78:22) dr). 1H NMR (400 MHz, CDCl3) δ 7.09 (s, 1H), 4.26 (q, J = 7.1 Hz, 2H), 2.84–2.71 (m, 1H), 2.71–2.56 (m, 2H), 2.49–2.34 (m, 2H), 2.34–2.20 (m, 1H), 2.11–1.99 (m, 1H), 1.92–1.74 (m, 2H), 1.71–1.56 (m, 1H), 1.38–1.25 (m, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 212.7, 161.2 (d, J = 33.7 Hz), 151.2 (d, J = 12.4 Hz), 142.0 (d, J = 252.9 Hz), 129.5 (d, J = 18.1 Hz), 118.6 (d, J = 2.5 Hz), 61.0, 50.2, 37.9, 32.0, 28.7 (d, J = 1.6 Hz), 24.0, 23.6 (d, J = 1.4 Hz), 19.5 (d, J = 8.1 Hz), 14.1; 19F NMR (376.5 MHz, CDCl3) δ −129.6 (m); Z-isomer: −132.5 (d, JHF = 4.2 Hz). HRMS (DART) calcd for C15H20O3F [M+H]+: 267.1391, found 267.1392.
Ethyl (Z)-2-fluoro-3-(naphthalen-2-yl)acrylate (Z-30a).
A round bottom flask was charged with triethyl 2-fluoro-2-phosphonoacetate (7.79 mL, 38.42 mmol, 1.5 equiv) and THF (80 mL). The solution was cooled to −10 °C. MeMgBr (11.0 mL, 38.42 mmol, 3.48 M in 2-MeTHF, 1.5 equiv) was added dropwise maintaining internal temperature below 10 °C. The reaction mixture was warmed to rt and stirred for 10 min, then cooled to −78 °C. A solution of 2-naphthaldehyde (4.0 g, 25.61 mmol, 1.0 equiv) in THF (23 mL) was added dropwise, and the reaction mixture was linearly warmed to rt over 2 h. The mixture was quenched with sat. aq. NH4Cl solution (50 mL) and extracted with MTBE (2 × 30 mL). Organic phase was dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was analyzed by quantitative 19F NMR: mixture of stereoisomers in 4.82:1 (83:17) dr. The residue was purified by silica gel column chromatography eluting with 0–2% MTBE in hexanes to afford the product Z-30a as a white solid (4.64 g, 74% yield, single Z-isomer (>98:2 dr)). mp 68.4–70.5 °C; 1H NMR (400 MHz, CDCl3) δ 8.10 (s, 1H), 7.91–7.75 (m, 4H), 7.57–7.46 (m, 2H), 7.09 (d, JHF = 35.3 Hz, 1H), 4.38 (q, J = 7.1 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.4 (d, J = 34.2 Hz), 147.1 (d, J = 267.8 Hz), 133.5 (d, J = 1.9 Hz), 133.1, 130.7 (d, J = 8.1 Hz), 128.7 (d, J = 4.6 Hz), 128.5, 128.4, 127.6, 127.2, 126.8 (d, J = 8.5 Hz), 126.5, 117.6 (d, J = 4.6 Hz), 61.9, 14.2; 19F NMR (376.5 MHz, CDCl3) δ −125.2 (d, JHF = 35.3 Hz). Both 1H and 13C NMR spectra matched reported data.33
General Procedure for Horner-Wadsworth-Emmons Reaction of Aldehydes with Triethyl 2-fluoro-2-phosphonoacetate and n-BuLi in THF. Ethyl (E)-2-fluoro-3-(naphthalen-2-yl)acrylate (E-30a).
A round bottom flask was charged with triethyl 2-fluoro-2-phosphonoacetate (5.84 mL, 28.81 mmol, 1.5 equiv) and THF (50 mL). The solution was cooled to −78 °C. n-BuLi (10.63 mL, 28.81 mmol, 2.71 M in hexanes, 1.5 equiv) was added dropwise maintaining internal temperature below 0 °C. The reaction mixture was warmed to rt and stirred for 10 min, then cooled to −78 °C. A solution of 2-naphthaldehyde (3.0 g, 19.21 mmol, 1.0 equiv) in THF (26 mL) was added dropwise, and the reaction mixture was linearly warmed to 10 °C over 2 h. The mixture was quenched with sat. aq. NH4Cl solution (50 mL) and extracted with MTBE (2 × 30 mL). The organic phase was dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was analyzed by quantitative 19F NMR: mixture of stereoisomers in 40.0:1 (98:2) dr. The residue was purified by silica gel column chromatography eluting with 0–2% MTBE in hexanes to afford product E-30a as a white solid (4.59 g, 98% yield, mixture of stereoisomers in 40.0:1 (98:2) dr. mp 31.3–32.7 °C; 1H NMR (400 MHz, CDCl3) δ 7.95 (s, 1H), 7.89–7.79 (m, 3H), 7.60 (dd, J = 8.5 Hz, J = 1.6 Hz, 1H), 7.56–7.47 (m, 2H), 7.08 (d, JHF = 22.4 Hz, 1H), 4.28 (q, J = 7.1 Hz, 2H), 1.25 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.5 (d, J = 36.1 Hz), 147.0 (d, J = 255.1 Hz), 133.1, 132.8, 129.5 (d, J = 3.5 Hz), 128.3 (d, J = 9.5 Hz), 128.2, 127.6, 127.4, 127.0 (d, J = 2.1 Hz), 126.7, 126.3, 121.5 (d, J = 26.3 Hz), 61.6, 13.8; 19F NMR (376.5 MHz, CDCl3) δ −116.5 (d, JHF = 22.4 Hz); Z-isomer: −125.2 (d, JHF = 35.3 Hz). Both 1H and 13C NMR spectra matched reported data.33
Ethyl (E)-3-(benzo[d][1,3]dioxol-4-yl)-2-fluoroacrylate (31a).
The reaction between benzo[d][1,3]dioxole-4-carbaldehyde (3.0 g, 19.98 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (6.08 mL; 29.97 mmol; 1.5 equiv) and HexLi (12.5 mL, 29.97 mmol, 2.40 M in hexanes, 1.5 equiv) was run at −78 °C to −30 °C for 1 h. 19F NMR of the crude: mixture of stereoisomers in 34.8:1 (97:3) dr. The product 31a was obtained as a colorless oil after purification by silica gel column chromatography eluting with 5–7% EtOAc in hexanes (4.90 g, >99% yield, mixture of stereoisomers in 36.1:1 (97:3) dr). 1H NMR (400 MHz, CDCl3) δ 7.03–6.97 (m, 1H), 6.84–6.73 (m, 3H), 5.94 (s, 2H), 4.26 (q, J = 7.1 Hz, 2H), 1.25 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.3 (d, J = 36.0 Hz), 148.1 (d, J = 257.1 Hz), 147.1, 145.7 (d, J = 2.7 Hz), 122.6 (d, J = 2.6 Hz), 121.1, 113.9 (d, J = 27.9 Hz), 113.3 (d, J = 9.9 Hz), 108.7, 100.9, 61.6, 13.8; 19F NMR (376.5 MHz, CDCl3) δ −115.4 (d, JHF = 20.7 Hz), Z-isomer: −123.9 (d, JHF = 35.5 Hz). HRMS (DART) calcd for C12H12O4F [M+H]+: 239.0714, found 239.0715.
Ethyl (E)-3-(benzofuran-2-yl)-2-fluoroacrylate (32a).
The reaction between benzo[b]furan-2-carboxaldehyde (5.0 g, 34.21 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (10.4 mL; 51.31 mmol; 1.5 equiv) and n-BuLi (18.9 mL, 51.31 mmol, 2.71 M in hexanes, 1.5 equiv) was run at −78 °C to −20 °C for 1 h. 19F NMR of the crude: mixture of stereoisomers in 21.6:1 (96:4) dr, 98% yield. The product 32a was obtained as a pale yellow oil after purification by silica gel column chromatography eluting with 0–3% MTBE in hexanes (7.1 g, 89% yield, single stereoisomer (>98:2 dr). 1H NMR (400 MHz, CDCl3) δ 7.80 (s, 1H), 7.62 (d, J = 7.7 Hz, 1H), 7.46 (dd, J = 8.3 Hz, J = 0.8 Hz, 1H), 7.34 (ddd, J = 8.3 Hz, J = 7.2 Hz, J = 1.3 Hz, 1H), 7.28–7.21 (m, 1H), 6.89 (d, JHF = 21.9 Hz, 1H), 4.41 (q, J = 7.1 Hz, 2H), 1.42 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.3 (d, J = 33.7 Hz), 154.9 (d, J = 2.3 Hz), 146.9 (d, J = 262.0 Hz), 147.5 (d, J = 10.6 Hz), 128.8, 125.8, 123.1, 121.9, 111.8 (d, J = 7.1 Hz), 111.5 (d, J = 33.5 Hz), 111.1, 61.9, 14.1; 19F NMR (376.5 MHz, CDCl3) δ −116.5 (d, JHF = 21.9 Hz). HRMS (DART) calcd for C13H12O3F [M+H]+: 235.0765, found 235.0766.
Ethyl (E)-3-cyclopropyl-2-fluoroacrylate (33a).
The reaction between cyclopropane carboxaldehyde (2.13 mL, 28.53 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (8.7 mL; 42.80 mmol; 1.5 equiv) and HexLi (17.8 mL, 42.80 mmol, 2.40 M in hexanes, 1.5 equiv) was run at −78 °C to −30 °C for 1 h. 19F NMR of the crude: mixture of stereoisomers in 40.7:1 (98:2) dr. The product 33a was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with 4–7% Et2O in pentane (4.28 g, 95% yield, single stereoisomer (>98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 5.27 (dd, JHF = 20.3 Hz, J = 10.9 Hz, 1H), 4.31 (q, J = 7.1 Hz, 2H), 2.52–2.39 (m, 1H), 1.34 (t, J = 7.1 Hz, 3H), 1.02–0.89 (m, 2H), 0.56–0.43 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.5 (d, J = 34.4 Hz), 146.6 (d, J = 246.3 Hz), 129.6 (d, J = 21.8 Hz), 61.2, 14.1, 8.4 (d, J = 7.2 Hz), 8.1 (d, J = 1.1 Hz); 19F NMR (376.5 MHz, CDCl3) δ −127.1 (dd, JHF = 20.3 Hz, JHF = 1.8 Hz). HRMS (DART) calcd for C8H12O2F [M+H]+: 159.0816, found 159.0816.
Ethyl (E)-3-cyclopentyl-2-fluoroacrylate (34a).
The reaction between cyclopentane carboxaldehyde (2.72 mL, 25.47 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (7.75 mL; 38.21 mmol; 1.5 equiv) and HexLi (15.9 mL, 38.21 mmol, 2.40 M in hexanes, 1.5 equiv) was run at −78 °C to −30 °C for 1 h. 19F NMR of the crude: mixture of stereoisomers in 23.6:1 (96:4) dr. The product 34a was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with 4–7% EtOAc in hexanes (4.58 g, 97% yield, single stereoisomer (>98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 5.81 (dd, JHF = 21.8 Hz, J = 10.4 Hz, 1H), 4.27 (q, J = 7.1 Hz, 2H), 3.47–3.32 (m, 1H), 1.97–1.83 (m, 2H), 1.74–1.53 (m, 4H), 1.39–1.18 (m, 5H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.1 (d, J = 35.7 Hz), 146.2 (d, J = 250.7 Hz), 128.6 (d, J = 16.5 Hz), 61.2, 36.2 (d, J = 4.8 Hz), 33.6 (d, J = 2.2 Hz), 25.3, 14.0; 19F NMR (376.5 MHz, CDCl3) δ −124.4 (d, JHF = 21.8 Hz), Z-isomer: −132.0 (d, JHF = 33.5 Hz). HRMS (DART) calcd for C10H16O2F [M+H]+: 187.1129, found 187.1129.
Ethyl (E)-2-fluoro-5-phenylpent-2-enoate (35a).
The reaction between 3-phenylpropionaldehyde (2.97 mL, 22.36 mmol, 1 equiv) and reagent prepared from triethyl 2-fluoro-2-phosphonoacetate (6.8 mL; 33.54 mmol; 1.5 equiv) and HexLi (14.0 mL, 33.54 mmol, 2.40 M in hexanes, 1.5 equiv) was run at −78 °C to −30 °C for 1 h. 19F NMR of the crude: mixture of stereoisomers in 15.1:1 (94:6) dr. The product 35a was obtained as a pale yellow liquid after purification by silica gel column chromatography eluting with 5–7% EtOAc in hexanes (4.44 g, 89% yield, mixture of stereoisomers in 17.6:1 (95:5) dr). 1H NMR (400 MHz, CDCl3) δ 7.35–7.27 (m, 2H), 7.25–7.18 (m, 3H), 5.94 (dt, JHF = 21.3 Hz, J = 8.0 Hz, 1H), 4.30 (q, J = 7.1 Hz, 2H), 2.91–2.82 (m, 2H), 2.81–2.74 (m, 2H), 1.35 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.8 (d, J = 35.8 Hz), 147.3 (d, J = 252.2 Hz), 140.6, 128.4, 126.1, 122.3 (d, J = 18.6 Hz), 61.3, 35.2 (d, J = 2.3 Hz), 27.1 (d, J = 5.1 Hz), 14.1; 19F NMR (376.5 MHz, CDCl3) δ −121.7 (d, JHF = 21.3 Hz), Z-isomer: −129.6 (d, JHF = 33.1 Hz). NMR spectra matched reported data.30 HRMS (DART) calcd for C13H16O2F [M+H]+: 223.1129, found 223.1130.
General Procedure for Ester Saponfication.
To a solution of ethyl ester (1 equiv) in EtOH (~0.5 M) was added a solution of LiOH•H2O (2.0 equiv) in water (volume ratio EtOH/H2O = 2:1) at 0 °C. The reaction mixture was warmed to r.t and stirred until completion as determined by HPLC or GC. The reaction mixture was diluted with water and cooled to 10 °C. Conc. HCl (2.2 equiv) was added dropwise maintaining internal temperature below 25 °C. The resultant slurry was warmed to rt and diluted further with water. The slurry was filtered and the solid was washed with 25 wt.% EtOH in H2O. The solid was dried to afford the carboxylic acid product.
(E)-2-Fluoro-3-(naphthalen-2-yl)but-2-enoic acid (10b).
Saponification of ester 10a (15.1 g, 58.07 mmol, 20.9:1 (95:5) dr) in EtOH/water afforded carboxylic acid 10b as a white solid (12.1 g, 21.1:1 (95:5) dr, 91% yield). mp 170.3–171.6 °C; 1H NMR (400 MHz, MeOH-d4) δ 7.85–7.77 (m, 3H), 7.68 (s, 1H), 7.49–7.42 (m, 2H), 7.31 (dd, J = 8.5 Hz, J = 1.7 Hz, 1H), 5.13–4.75 (brs, 1H), 2.20 (d, J = 4.5 Hz, 3H); 13C{1H} NMR (100 MHz, MeOH-d4) δ 163.7 (d, J = 36.5 Hz), 146.2 (d, J = 250.5 Hz), 137.5 (d, J = 5.6 Hz), 134.6, 134.3, 132.8 (d, J = 17.6 Hz), 129.1, 128.7 (2C), 127.6 (d, J = 3.2 Hz), 127.3 (2C), 127.1 (d, J = 2.9 Hz), 19.6 (d, J = 6.9 Hz); 19F NMR (376.5 MHz, MeOH-d4) δ −123.5 (q, JHF = 4.4 Hz); Z-isomer: −124.6 (s). HRMS (DART) calcd for C14H12O2F [M+H]+: 231.0816, found 231.0816.
(E)-3-(4-Bromophenyl)-2-fluoroacrylic acid (E-12b).
Saponification of ester E-12a (1.05 g, 3.85 mmol, 34.5:1 (97:3) dr) in EtOH/water afforded carboxylic acid E-12b as a white solid (0.80 g, > 98:2 dr, 85% yield). mp 149.1–150.6 °C; 1H NMR (400 MHz, CDCl3) δ 10.05–8.94 (brs, 1H); 7.55–7.43 (m, 2H), 7.40–7.29 (m, 2H), 6.97 (d, JHF = 21.8 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 164.7 (d, J = 36.8 Hz), 145.9 (d, J = 253.9 Hz), 131.45, 131.43 (d, J = 3.6 Hz), 129.1 (d, J = 9.2 Hz), 123.6; 123.5 (d, J = 26.8 Hz); 19F NMR (376.5 MHz, CDCl3) δ −116.3 (d, JHF = 21.8 Hz). HRMS (DART) calcd for C9H7O2BrF [M+H]+: 244.9608, found 244.9609.
(Z)-3-(4-Bromophenyl)-2-fluoroacrylic acid (Z-12b).
Saponification of ester Z-12a (4.53 g, 16.59 mmol, > 98:2 dr) in EtOH/water afforded carboxylic acid Z-12b as a white solid (3.97 g, > 98:2 dr, 98% yield). mp 226.5–227.5 °C; 1H NMR (400 MHz, MeOH-d4) δ 7.56 (apparent s, 4H), 6.94 (d, JHF = 35.0 Hz, 1H) 5.32–4.69 (brs, 1H); 13C{1H} NMR (100 MHz, MeOH-d4) δ 164.0 (d, J = 35.3 Hz), 149.1 (d, J = 265.5 Hz), 133.2, 133.0 (d, J = 8.3 Hz), 131.8 (d, J = 4.3 Hz), 124.9 (d, J = 3.9 Hz), 117.3 (d, J = 4.8 Hz); 19F NMR (376.5 MHz, MeOH-d4) δ −124.7 (d, JHF = 35.0 Hz). NMR spectra matched reported data.34
(E)-2-Fluoro-3-phenylbut-2-enoic acid (15b).
To a solution of ethyl ester 15a (5.85 g, 28.09 mmol; 1 equiv) in EtOH (60 mL) was added a solution of LiOH•H2O (2.36 g; 56.19 mmol; 2.0 equiv) in water (40 mL) at 0 °C. The reaction mixture was warmed to r.t and stirred for 2 h. After reaction completion the reaction mixture was diluted with water (50.0 mL) and cooled to 10 °C. Conc. HCl (5.1 mL; 61.81 mmol; 2.2 equiv) was added dropwise maintaining internal temperature below 25 °C. The resultant slurry was warmed to rt. Water (50.0 mL) was added dropwise, and the slurry was stirred for another 0.5 h. The slurry was filtered and the solid was washed with 25 wt.% EtOH in H2O (2 × 15 mL). The solid was dried on a Buchner funnel for 2 h and then at 50 °C in a vacuum oven overnight to afford carboxylic acid 15b as a white solid (4.81 g, 95% yield, >98:2 dr). mp 131.8–133.4 °C; 1H NMR (400 MHz, CDCl3) δ 10.46–9.19 (brs, 1H), 7.40–7.30 (m, 3H), 7.38–7.28 (m, 3H), 7.20–7.11 (m, 2H), 2.16 (d, JHF = 4.5 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 165.2 (d, J = 36.6 Hz), 143.3 (d, J = 248.6 Hz), 137.7 (d, J = 5.1 Hz), 135.0 (d, J = 17.0 Hz), 128.2, 128.1, 127.3 (d, J = 3.1 Hz), 19.9 (d, J = 6.6 Hz); 19F NMR (376.5 MHz, CDCl3) δ −124.1 (q, JHF = 4.5 Hz). HRMS (DART) calcd for C10H10O2F [M+H]+: 181.0659, found 181.0660.
(E)-3-(Benzo[d][1,3]dioxol-5-yl)-2-fluorobut-2-enoic acid (16b).
Saponification of ester 16a (3.7 g, 14.67 mmol, >98.2 dr) in 2-PrOH/water afforded carboxylic acid 16b as a white solid (3.0 g, >98.2 dr, 92% yield). mp 161.2–163.1 °C; 1H NMR (400 MHz, CDCl3) δ 6.76 (d, J = 8.0 Hz, 1H), 6.73–6.63 (m, 2H), 5.91 (s, 2H), 5.44–4.64 (brs, 1H), 2.08 (d, JHF = 4.6 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 163.9 (d, J = 36.6 Hz), 148.9, 148.8, 146.0 (d, J = 250.0 Hz), 133.6 (d, J = 5.9 Hz), 132.5 (d, J = 17.9 Hz), 122.4 (d, J = 3.2 Hz), 109.5 (d, J = 3.2 Hz), 109.0, 102.6, 19.6 (d, J = 6.8 Hz); 19F NMR (376.5 MHz, CDCl3) δ −123.5 (q, JHF = 4.3 Hz). HRMS (DART) calcd for C11H10O4F [M+H]+: 225.0558, found 225.0558.
General Procedure for Telescoped Horner-Wadsworth-Emmons Reaction and Ester Saponification. (E)-2-Fluoro-3-(4-nitrophenyl)but-2-enoic acid (17b).
A round bottom flask was charged with triethyl 2-fluoro-2-phosphonoacetate (1.04 mL, 5.15 mmol, 1.7 equiv) and THF (8 mL). The solution was cooled to 0 to −5 °C. MeMgBr (1.74 mL, 5.15 mmol, 2.96 M in Et2O, 1.7 equiv) was then added dropwise maintaining internal temperature below 10 °C. The reaction mixture was warmed to rt and stirred for 10 min, then heated to 40 °C and stirred for another 10 min. A solution of 4-nitroacetophenone (500 mg, 3.03 mmol, 1.0 equiv) in THF (4 mL) was added dropwise, and the reaction mixture was stirred for 15 min. The reaction mixture was cooled to rt and concentrated under reduced pressure to minimum volume. 2-PrOH (10 mL) was added, and the mixture was concentrated again. The reaction mixture was diluted with 2-PrOH (5 mL) and cooled to 10 °C. A solution of NaOH (726.6 mg; 18.17 mmol; 6 equiv) in H2O (1.2 mL) was added, and resultant slurry was warmed to rt and stirred at rt for 1 h. Conc. HCl (1.6 mL; 19.70 mmol; 6.5 equiv) was added, and the reaction mixture was slowly concentrated under reduced producing a yellow precipitate of the product. The mixture was filtered, and the filter cake was washed twice with water (5 mL) and then with heptane (5 mL). The solid was dried on a Buchner funnel for 1 h then at 50 °C in a vacuum oven overnight to afford crude acid 17b (yellow solid, 636.7 mg, 93% yield). Crude acid was obtained as 12.7:1 (93:7) mixture of E/Z isomers (19F NMR). Its stereochemical purity was increased by recrystallization as follows. Crude acid 17b (970.0 mg) was dissolved in 2-PrOH (3.00 mL) at 70 °C. Heptane (18 mL) was slowly added at 70 °C. The reaction mixture was gradually cooled to rt and the resultant slurry was stirred for 2 h. The slurry was filtered, and the filter cake was washed with a minimum amount of 2-PrOH/heptane mixture (6:1 v/v), then with heptane (5 mL). The solid was dried in high vacuum to afford the acid (yellow solid, 609.7 mg, single isomer (>98:2 dr)). The mother liquor was concentrated and the residue was recrystallized from 2-PrOH (800 μL) and heptane (6 mL) following the same procedure to afford the second crop of product (yellow solid, 220 mg, >98:2 dr). Combined mass: 829.7 mg, 86% yield, 80% overall yield from 4-nitroacetophenone. mp 192.9–194.4 °C; 1H NMR (400 MHz, MeOH-d4) δ 8.20 (d, J = 8.2 Hz, 2H), 7.46 (d, J = 8.2 Hz, 2H), 5.63–4.34 (brs, 1H), 2.15 (d, JHF = 3.8 Hz, 3H); 13C{1H} NMR (100 MHz, MeOH-d4) δ 162.9 (d, J = 35.8 Hz), 148.8, 146.4 (d, J = 255.0 Hz), 147.3 (d, J = 5.9 Hz), 130.9 (d, J = 19.2 Hz), 130.2 (d, J = 3.2 Hz), 124.4, 19.1 (d, J = 6.8 Hz); 19F NMR (376.5 MHz, MeOH-d4) δ −122.2 (q, JHF = 3.8 Hz). HRMS (DART) calcd for C10H7O4NF [M-H]–: 224.0365, found 224.0364.
(E)-3-(4-Bromophenyl)-2-fluorohept-2-enoic acid (18b).
Saponification of ester 18a (3.0 g, 9.11 mmol, 20.5:1 (95:5) dr) in 2-PrOH/water afforded crude carboxylic acid as an off-white solid (2.56 g, 93% yield). The crude acid was recrystallized from heptane to afford 18b as a single stereoisomer (2.18 g, 85% recovery, 79% overall yield, >98:2 dr). 1H NMR (400 MHz, CDCl3) δ 11.44–10.75 (brs, 1H), 7.52–7.40 (m, 2H), 7.07–6.92 (m, 2H), 2.72–2.31 (m, 2H), 1.53–1.09 (m, 4H), 1.05–0.67 (m, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 165.5 (d, J = 36.9 Hz), 143.2 (d, J = 250.5 Hz), 138.2 (d, J = 17.0 Hz), 135.4 (d, J = 5.4 Hz), 131.3, 129.3 (d, J = 3.0 Hz), 122.2, 32.7 (d, J = 4.8 Hz), 28.7 (d, J = 1.9 Hz), 22.3, 13.6; 19F NMR (376.5 MHz, CDCl3) δ −124.8 (t, JHF = 3.4 Hz); Z-isomer: −124.6 (s). HRMS (DART) calcd for C13H15O2BrF [M+H]+: 301.0234, found 301.0234.
(E)-2-Fluoro-3-(o-tolyl)pent-2-enoic acid (19b).
A round bottom flask was charged with triethyl 2-fluoro-2-phosphonoacetate (6.8 mL, 33.63 mmol, 2.5 equiv) and THF (50 mL). The solution was cooled to 0 to −5 °C. MeMgBr (11.4 mL, 33.63 mmol, 2.96 M in Et2O, 2.5 equiv) was then added dropwise maintaining internal temperature below 10 °C. The reaction mixture was warmed to rt and stirred for 10 min, then heated to 40 °C and stirred for another 10 min. A solution of 2-methylpropiophenone (2.0 mL, 13.45 mmol, 1.0 equiv) in THF (10 mL) was added dropwise, and the reaction mixture was stirred for 30 min. An aliquot of reaction mixture was analyzed by quantitative 19F NMR: ester 19a was formed as a mixture of stereoisomers in 96:4 dr. The reaction mixture was cooled to rt and concentrated under reduced pressure to a minimum volume. 2-PrOH (20 mL) was added, and the mixture was concentrated again. The reaction mixture was diluted with 2-PrOH (10 mL) and cooled to 10 °C. A solution of NaOH (4.30 g; 107.6 mmol; 8 equiv) in H2O (10 mL) was added, and resultant slurry was warmed to rt and stirred at rt overnight. conc. HCl (9.4 mL; 114.3 mmol; 8.5 equiv) was added, and the reaction mixture was slowly concentrated under reduced pressure giving a biphasic mixture (water/oil). EtOAc (30 mL) was added and the layers were separated. The aqueous phase was extracted with EtOAc (2 × 10 mL). Combined organic extracts were dried over MgSO4 and concentrated under reduced pressure to an oil. The oil was treated with 3 M NaOH (9.0 mL; 26.9 mmol; 2 equiv.) and the mixture was extracted with MTBE (20 mL). The aqueous phase was acidified with 3 M HCl (9.9 mL; 29.6 mmol; 2.2 equiv) at 10 °C which resulted in formation of a slurry. The slurry was filtered and the filter cake was washed with water. The solid was dried on a Buchner funnel for 2 h then at 50 °C in vacuum oven overnight to afford crude acid 19b as an off white solid (2.78 g, 99% yield). Crude acid was obtained as 96.5:3.5 mixture of E/Z isomers (19F NMR). The crude acid was recrystallized from a hot (65 °C) mixture of heptane (10 mL) and IPAc (0.5 mL) to afford 19b as a white solid (2.25 g, 81% recovery, 80% overall yield from 2-methylpropiophenone, single stereoisomer (>98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 11.57–9.26 (brs, 1H), 7.25–7.07 (m, 3H), 6.93 (d, J = 7.4 Hz, 1H), 2.78–2.56 (m, 1H), 2.52–2.28 (m, 1H), 2.19 (s, 3H), 1.00 (t, J = 7.6 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 165.2 (d, J = 37.0 Hz), 143.3 (d, J = 249.3 Hz), 139.5 (d, J = 15.3 Hz), 135.8 (d, J = 5.1 Hz), 134.7 (d, J = 2.6 Hz), 130.0, 127.8, 127.4 (d, J = 3.3 Hz), 125.4, 25.8 (d, J = 5.5 Hz), 19.3, 11.1 (d, J = 2.0 Hz); 19F NMR (376.5 MHz, CDCl3) δ −126.8 (s). HRMS (DART) calcd for C12H14O2F [M+H]+: 209.0972, found 209.0973.
(E)-2-Fluoro-3-(o-tolyl)pent-2-enoic acid (19b).
A round bottom flask was charged with triethyl 2-fluoro-2-phosphonoacetate (6.8 mL, 33.63 mmol, 2.5 equiv) and THF (50 mL). The solution was cooled to 0 to −5 °C. MeMgBr (11.4 mL, 33.63 mmol, 2.96 M in Et2O, 2.5 equiv) was then added dropwise maintaining internal temperature below 10 °C. The reaction mixture was warmed to rt and stirred for 10 min, then heated to 40 °C and stirred for another 10 min. A solution of 2-methylpropiophenone (2.0 mL, 13.45 mmol, 1.0 equiv) in THF (10 mL) was added dropwise, and the reaction mixture was stirred for 30 min. An aliquot of reaction mixture was analyzed by quantitative 19F NMR: ester 19a was formed as a mixture of stereoisomers in 96:4 dr. The reaction mixture was cooled to rt and concentrated under reduced pressure to a minimum volume. 2-PrOH (20 mL) was added, and the mixture was concentrated again. The reaction mixture was diluted with 2-PrOH (10 mL) and cooled to 10 °C. A solution of NaOH (4.30 g; 107.6 mmol; 8 equiv) in H2O (10 mL) was added, and resultant slurry was warmed to rt and stirred at rt overnight. conc. HCl (9.4 mL; 114.3 mmol; 8.5 equiv) was added, and the reaction mixture was slowly concentrated under reduced pressure giving a biphasic mixture (water/oil). EtOAc (30 mL) was added and the layers were separated. The aqueous phase was extracted with EtOAc (2 × 10 mL). Combined organic extracts were dried over MgSO4 and concentrated under reduced pressure to an oil. The oil was treated with 3 M NaOH (9.0 mL; 26.9 mmol; 2 equiv.) and the mixture was extracted with MTBE (20 mL). The aqueous phase was acidified with 3 M HCl (9.9 mL; 29.6 mmol; 2.2 equiv) at 10 °C which resulted in formation of a slurry. The slurry was filtered and the filter cake was washed with water. The solid was dried on a Buchner funnel for 2 h then at 50 °C in vacuum oven overnight to afford crude acid 19b as an off white solid (2.78 g, 99% yield). Crude acid was obtained as 96.5:3.5 mixture of E/Z isomers (19F NMR). The crude acid was recrystallized from a hot (65 °C) mixture of heptane (10 mL) and IPAc (0.5 mL) to afford 19b as a white solid (2.25 g, 81% recovery, 80% overall yield from 2-methylpropiophenone, single stereoisomer (>98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 11.57–9.26 (brs, 1H), 7.25–7.07 (m, 3H), 6.93 (d, J = 7.4 Hz, 1H), 2.78–2.56 (m, 1H), 2.52–2.28 (m, 1H), 2.19 (s, 3H), 1.00 (t, J = 7.6 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 165.2 (d, J = 37.0 Hz), 143.3 (d, J = 249.3 Hz), 139.5 (d, J = 15.3 Hz), 135.8 (d, J = 5.1 Hz), 134.7 (d, J = 2.6 Hz), 130.0, 127.8, 127.4 (d, J = 3.3 Hz), 125.4, 25.8 (d, J = 5.5 Hz), 19.3, 11.1 (d, J = 2.0 Hz); 19F NMR (376.5 MHz, CDCl3) δ −126.8 (s). HRMS (DART) calcd for C12H14O2F [M+H]+: 209.0972, found 209.0973.
(E)-2-Fluoro-4-methyl-3-phenylpent-2-enoic acid (20b).
Saponification of ester 20a (4.37 g, 18.49 mmol, 11.6:1 (92:8) dr) in EtOH/water afforded carboxylic acid 20b as a white solid (3.38 g, 11.6:1 (92:8) dr, 88% yield). The product was recrystallized from IPAc (1V) and heptane (15 V) to improve stereochemical purity to 18.4:1 (95:5) dr (2.61 g, 77% recovery, 68% overall yield). E-isomer: 1H NMR (400 MHz, CDCl3) δ 11.15–9.85 (brs, 1H), 7.36–7.27 (m, 3H), 7.06–6.97 (m, 2H), 3.29 (dsept, J = 7.0 Hz, JHF = 0.8 Hz, 1H), 1.00 (d, J = 7.0 Hz, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.5 (dd, J = 37.5 Hz, J = 7.1 Hz), 142.8 (d, J = 250.1 Hz), 143.3 (d, J = 14.0 Hz), 133.9 (d, J = 5.3 Hz), 128.0 (d, J = 3.1 Hz), 127.7, 127.6, 29.4 (d, J = 5.1 Hz), 20.0 (d, J = 2.0 Hz); 19F NMR (376.5 MHz, CDCl3) δ −126.7 (s); Z-isomer: −121.3 (s). HRMS (DART) calcd for C12H14O2F [M+H]+: 209.0972, found 209.0973.
(E)-3-Cyclopropyl-2-fluoro-3-(4-methoxyphenyl)acrylic acid (21b).
Saponification of ester 21a (4.36 g, 16.50 mmol, >98.2 dr) in EtOH/water afforded carboxylic acid 21b as a white solid (3.51 g, >98.2 dr, 90% yield). mp 157.2–158.9 °C; 1H NMR (400 MHz, CDCl3) δ 12.06–10.81 (brs, 1H), 6.96–6.84 (m, 2H), 6.84–6.74 (m, 2H), 3.79 (s, 3H), 2.45–1.96 (m, 1H), 0.94–0.71 (m, 2H), 0.53–0.30 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 165.2 (d, J = 35.9 Hz), 159.2, 144.3 (d, J = 247.2 Hz), 140.7 (d, J = 13.3 Hz), 129.8 (d, J = 3.1 Hz), 123.8 (d, J = 4.8 Hz), 113.3, 55.1, 12.3 (d, J = 9.4 Hz), 5.4; 19F NMR (376.5 MHz, CDCl3) δ −131.2 (s). HRMS (DART) calcd for C13H14O3F [M+H]+: 237.0922, found 237.0922.
(Z)-3-(4-Bromophenyl)-2,4,4,4-tetrafluorobut-2-enoic acid (22b).
Saponification of ester 22a (3.0 g, 8.80 mmol, 25.8:1 (96:4) dr) in 2-PrOH/water afforded carboxylic acid 22b as an off-white solid (2.52 g, 25.8:1 (96:4) dr, 92% yield). 1H NMR (400 MHz, MeOH-d4) δ 7.60–7.53 (m, 2H), δ 7.24–7.16 (m, 2H), 5.46–4.94 (brs, 1H); 13C{1H} NMR (100 MHz, MeOH-d4) δ 161.3 (d, J = 34.3 Hz), 152.2, (dq, J = 284.3 Hz, J = 2.6 Hz), 132.8, 132.7 (d, J = 3.4 Hz), 129.4 (d, J = 3.2 Hz), 124.8, 123.5 (q, J = 274.7 Hz), 121.0 (dq, J = 32.4 Hz, J = 9.7 Hz); 19F NMR (376.5 MHz, MeOH-d4) δ −107.5 (q, J = 24.0 Hz), −61.6 (d, J = 24.0 Hz); Z-isomer: δ −105.6 (q, J = 10.3 Hz), −59.8 (d, J = 10.3 Hz). HRMS (DART) calcd for C10H4O2BrF4 [M-H]–: 310.9336, found 310.9336.
(E)-3-(3,5-Dimethylphenyl)-2-fluoronon-2-en-7-ynoic acid (23b).
Saponification of ester 23a (1.90 g, 6.28 mmol, >98.2 dr) in EtOH/water afforded carboxylic acid 23b as a white solid (1.68 g, >98.2 dr, 98% yield). mp 88.3–90.3 °C; 1H NMR (400 MHz, CDCl3) 6.95 (s, 1H), 6.74 (s, 2H), 2.66–2.54 (m, 2H), 2.30 (s, 6H), 2.17–2.06 (m, 2H), 1.75 (t, J = 2.5 Hz, 3H), 1.58–1.47 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 165.2 (d, J = 37.0 Hz), 143.3 (d, J = 248.8 Hz), 138.5 (d, J = 15.7 Hz), 137.6, 136.0 (d, J = 5.0 Hz), 129.8, 125.4 (d, J = 2.8 Hz), 78.1, 76.2, 32.4 (d, J = 5.2 Hz), 26.3 (d, J = 1.7 Hz), 21.2, 18.6, 3.4; 19F NMR (376.5 MHz, CDCl3) δ −125.1 (s). HRMS (DART) calcd for C17H18O2F [M-H]–: 273.1296, found 273.1295.
(Z)-3-(1-Cyanocyclohexyl)-2-fluoro-3-(4-methoxyphenyl)acrylic acid (24b).
Saponification of ester 24a (1.1 g, 3.32 mmol, >98.2 dr) in EtOH/water afforded carboxylic acid 24b as a white solid (1.0 g, >98.2 dr, 99% yield). mp 156.5–157.6 °C; 1H NMR (400 MHz, CDCl3) δ 10.73–8.78 (brs, 1H), 6.96 (d, J = 8.6 Hz, 2H), 6.87 (d, J = 8.6 Hz, 2H), 3.82 (s, 3H), 2.12–1.92 (m, 2H), 1.82–1.61 (m, 5H), 1.48–1.30 (m, 2H), 1.18–0.97 (m, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 163.9 (d, J = 36.2 Hz), 159.8, 146.0 (d, J = 266.2 Hz), 135.3 (d, J = 9.3 Hz), 129.6 (d, J = 3.4 Hz), 124.4 (d, J = 5.4 Hz), 120.3, 113.8, 55.2, 42.0 (d, J = 1.7 Hz), 34.2 (d, J = 1.9 Hz), 24.5, 22.7; 19F NMR (376.5 MHz, CDCl3) δ −111.9 (s). HRMS (DART) calcd for C17H19O3NF [M+H]+: 304.1344, found 304.1344.
(E)-3-(4-Bromophenyl)-2-fluoro-4-phenylbut-2-enoic acid (25b).
Saponification of ester 25a (4.75 g, 13.08 mmol, 13.3:1 (93:7) dr) in EtOH/water afforded carboxylic acid 25b as a white solid (3.95 g, 14.9:1 (94:6) dr, 91% yield). mp 139.1–141.5 °C; 1H NMR (400 MHz, CDCl3) 9.57–8.49 (brs, 1H), 7.40–7.33 (m, 2H), 7.27–7.15 (m, 3H), 7.04–6.96 (m, 2H), 6.83–6.74 (m, 2H), 3.81 (d, JHF = 3.6 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 164.9 (d, J = 37.0 Hz), 143.3 (d, J = 252.6 Hz), 136.2 (d, J = 16.7 Hz), 135.9 (d, J = 2.4 Hz), 134.8 (d, J = 5.0 Hz), 131.2, 129.6 (d, J = 3.1 Hz), 129.1, 128.6, 126.9, 122.3, 39.1 (d, J = 5.0 Hz); 19F NMR (376.5 MHz, CDCl3) δ −123.7 (t, JHF = 3.3 Hz); Z-isomer: −122.9 (s). HRMS (DART) calcd for C16H13O2BrF [M+H]+: 335.0078, found 335.0078.
(E)-2-Fluoro-3-(quinolin-3-yl)but-2-enoic acid (26b).
Saponification of ester 26a (4.70 g, 58.07 mmol, 11.3:1 (92:8) dr) in 2-PrOH/water afforded carboxylic acid 26b as a white solid (3.60 g, >98:2 dr, 86% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.76 (d, JHF = 2.0 Hz, 1H), 8.30 (d, JHF = 1.4 Hz, 1H), 8.02 (d, J = 8.4 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.82–7.71 (m, 1H), 7.67–7.57 (m, 1H), 4.65–2.66 (brs, 1H), 2.21 (d, JHF = 4.4 Hz, 3H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 161.1 (d, J = 35.4 Hz), 150.2 (d, J = 2.6 Hz), 146.6, 145.2 (d, J = 253.8 Hz), 134.0 (d, J = 3.4 Hz), 131.7 (d, J = 5.8 Hz), 129.7, 128.6, 128.2, 127.6 (d, J = 19.1 Hz), 127.1, 126.9, 19.0 (d, J = 6.4 Hz); 19F NMR (376.5 MHz, DMSO-d6) δ −119.2 (q, JHF = 4.4 Hz); Z-isomer: −121.6 (s). HRMS (DART) calcd for C13H11O2NF [M+H]+: 232.0768, found 232.0769.
(E)-3-(Benzo[b]thiophen-2-yl)-2-fluorobut-2-enoic acid (27b).
Saponification of ester 27a (4.97 g, 18.80 mmol, 10.9:1 (92:8) dr) in EtOH/water afforded carboxylic acid 27b as a pale yellow solid (3.96 g, 10.9:1 (92:8) dr, 89% yield). mp 140.6–142.7 °C; 1H NMR (400 MHz, MeOH-d4) δ 7.81–7.77 (m, 1H), 7.75–7.70 (m, 1H), 7.34–7.27 (m, 2H), 7.26 (s, 1H), 5.45–4.58 (brs, 1H), 2.20 (d, JHF = 4.7 Hz, 3H); 13C{1H} NMR (100 MHz, MeOH-d4) δ 163.2 (d, J = 36.1 Hz), 147.4 (d, J = 256.3 Hz), 141.7, 141.1, 140.3 (d, J = 6.8 Hz), 125.8, 125.6, 125.1 (d, J = 3.1 Hz), 124.9, 123.1, 19.6 (d, J = 6.8 Hz); 19F NMR (376.5 MHz, MeOH-d4) δ −117.2 (q, JHF = 4.7 Hz); Z-isomer: −115.3 (q, JHF = 3.1 Hz). HRMS (DART) calcd for C12H8O2FS [M-H]–: 235.0235, found 235.0234.
(E)-3-(Cyclohex-1-en-1-yl)-2-fluorobut-2-enoic acid (28b).
A round bottom flask was charged with triethyl 2-fluoro-2-phosphonoacetate (3.94 mL, 19.45 mmol, 2.5 equiv) and THF (25 mL). The solution was cooled to 0 to −5 °C. MeMgBr (6.57 mL, 19.45 mmol, 2.96 M in Et2O, 2.5 equiv) was added dropwise maintaining internal temperature below 10 °C. The reaction mixture was warmed to rt and stirred for 10 min, then heated to 40 °C and stirred for another 10 min. A solution of 1-cyclohexenyl methyl ketone (966 mg, 1.0 mL, 7.78 mmol, 1.0 equiv) in THF (6 mL) was added dropwise, and the reaction mixture was stirred for 30 min. An aliquot of reaction mixture was analyzed by quantitative 19F NMR: ester 28a was formed as a mixture of stereoisomers in 12.7:1 (93:7) dr. The reaction mixture was cooled to rt and concentrated under reduced pressure to a minimum volume. 2-PrOH (20 mL) was added, and the mixture was concentrated again. The reaction mixture was diluted with 2-PrOH (35 mL) and cooled to 10 °C. A solution of NaOH (2.81 g; 70.01 mmol; 9 equiv) in H2O (15 mL) was added, and resultant slurry was warmed to rt and stirred at rt for 3 h. conc. HCl (6.07 mL; 73.90 mmol; 9.5 equiv) was added dropwise at 5–10 °C, and the reaction mixture was slowly concentrated under reduced pressure producing a sticky solid. EtOAc (30 mL) was added and the layers were separated. The aqueous phase was extracted with EtOAc (2 × 10 mL). Combined organic extracts were dried over MgSO4 and concentrated under reduced pressure to a brown oil. To this oil was added sat. aq. NaHCO3 solution (10 mL). The reaction mixture was stirred until gas evolution ceased, and then extracted twice with EtOAc. 2-PrOH (3 mL) was added to the aqueous phase, and the resultant solution was concentrated under vacuum to ~10 mL volume. To this solution was added conc. HCl dropwise at 5–10 °C until pH ~2. A pale yellow solid was filtered, washed with a small amount of water, then with heptane. The solid was dried in high vacuum to give the acid 28b in >98:2 dr (830.9 mg, 58% overall yield from 1-cyclohexenyl methyl ketone). mp 103.1–105.5 °C; 1H NMR (400 MHz, CDCl3) δ 12.41–11.18 (brs, 1H), 5.71–5.24 (m, 1H), 2.23–1.97 (m, 4H), 1.93 (d, JHF = 4.2 Hz, 3H), 1.75–1.62 (m, 2H), 1.62–1.49 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 166.1 (d, J = 36.7 Hz), 142.6 (d, J = 247.0 Hz), 137.7 (d, J = 14.0 Hz), 135.3 (d, J = 4.2 Hz), 124.7 (d, J = 2.9 Hz), 27.2 (d, J = 2.6 Hz), 25.1, 22.5, 21.6, 17.6 (d, J = 6.2 Hz); 19F NMR (376.5 MHz, CDCl3) δ −128.6 (s). HRMS (DART) calcd for C10H14O2F [M+H]+: 185.0972, found 185.0973.
(E)-2-Fluoro-2-(4a-methyl-5-oxo-4,4a,5,6,7,8-hexahydronaphthalen-2(3H)-ylidene)acetic acid (29b).
Saponification of ester 29a (4.85 g, 18.20 mmol, 3.44:1 (78:22) dr) in EtOH/water afforded carboxylic acid 29b as a white solid (3.39 g, 9.97:1 (91:9) dr, 78% yield). 1H NMR (400 MHz, CDCl3) δ 7.09 (s, 1H), 6.24–4.95 (brs, 1H), 2.92–2.76 (m, 1H), 2.76–2.57 (m, 2H), 2.55–2.39 (m, 2H), 2.39–2.24 (m, 1H), 2.17–2.02 (m, 1H), 1.99–1.76 (m, 2H), 1.75–1.57 (m, 1H), 1.34 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 212.9, 165.5 (d, J = 35.4 Hz), 152.6 (d, J = 12.4 Hz), 141.3 (d, J = 247.6 Hz), 132.3 (d, J = 18.0 Hz), 118.6 (d, J = 2.2 Hz), 50.3, 37.9, 32.1, 28.6 (d, J = 1.2 Hz), 23.9, 23.7 (d, J = 1.1 Hz), 19.8 (d, J = 7.9 Hz); 19F NMR (376.5 MHz, CDCl3) δ −129.5 (m); Z-isomer: −132.7 (d, JHF = 3.9 Hz). HRMS (DART) calcd for C13H16O3F [M+H]+: 239.1078, found 239.1078.
(E)-2-Fluoro-3-(naphthalen-2-yl)acrylic acid (E-30b).
Saponification of ester E-30a (4.59 g, 18.79 mmol, 98:2 dr) in EtOH/water afforded carboxylic acid E-30b as a white solid (3.91 g, > 98:2 dr, 96% yield). mp 151.1–152.7 °C; 1H NMR (400 MHz, MeOH-d4) δ 7.97 (s, 1H), 7.84–7.72 (m, 3H), 7.63 (dd, J = 8.6 Hz, J = 1.6 Hz, 1H), 7.49–7.39 (m, 2H), 7.10 (d, JHF = 23.0 Hz, 1H), 5.23–4.76 (brs, 1 H); 13C{1H} NMR (100 MHz, MeOH-d4) δ 163.5 (d, J = 36.7 Hz), 149.0 (d, J = 253.6 Hz), 134.7, 134.5, 130.8 (d, J = 3.6 Hz), 130.2 (d, J = 9.7 Hz), 129.4, 128.7, 128.5, 128.4 (d, J = 2.2 Hz), 127.8, 127.4, 122.4 (d, J = 27.2 Hz); 19F NMR (376.5 MHz, MeOH-d4) δ −116.3 (d, JHF = 23.0 Hz). HRMS (DART) calcd for C13H8O2F [M-H]–: 215.0514, found 215.0513.
(Z)-2-Fluoro-3-(naphthalen-2-yl)acrylic acid (Z-30b).
Saponification of ester Z-30a (4.34 g, 17.77 mmol, > 98:2 dr) in EtOH/water afforded carboxylic acid Z-30b as a white solid (3.80 g, > 98:2 dr, 99% yield). mp 187.9–190.0 °C; 1H NMR (400 MHz, MeOH-d4) δ 8.04 (s, 1H), 7.86–7.76 (m, 3H), 7.73 (dd, J = 8.7 Hz, J = 1.6 Hz, 1H), 7.54–7.41 (m, 2H), 7.09 (d, JHF = 35.6 Hz, 1H), 5.33–4.71 (brs, 1H); 13C{1H} NMR (100 MHz, MeOH-d4) δ 164.4 (d, J = 35.3 Hz), 148.8 (d, J = 264.1 Hz), 135.1 (d, J = 1.9 Hz), 134.7, 131.8 (d, J = 7.9 Hz), 130.2 (d, J = 4.6 Hz), 129.7, 129.6, 128.8, 128.5, 127.80 (d, J = 7.4 Hz), 127.77, 118.7 (d, J = 4.7 Hz); 19F NMR (376.5 MHz, MeOH-d4) δ −126.1 (d, JHF = 35.6 Hz). NMR spectra matched reported data.24
(E)-3-(Benzo[d][1,3]dioxol-4-yl)-2-fluoroacrylic acid (31b).
Saponification of ester 31a (4.56 g, 9.14 mmol, 34.5:1 (97:3) dr) in EtOH/water afforded carboxylic acid 31b as a white solid (3.54 g, > 98:2 dr, 88% yield). mp 149.0–150.1 °C; 1H NMR (400 MHz, MeOH-d4) δ 7.02–6.96 (m, 1H), 6.82–6.73 (m, 3H), 5.92 (s, 2H), 5.14–4.90 (brs, 1H); 13C{1H} NMR (100 MHz, MeOH-d4) δ 163.4 (d, J = 36.6 Hz), 150.2 (d, J = 255.1 Hz), 148.8, 147.3 (d, J = 2.5 Hz), 123.8 (d, J = 2.7 Hz), 122.3, 115.0 (d, J = 10.2 Hz), 114.6 (d, J = 28.5 Hz), 109.7, 102.3; 19F NMR (376.5 MHz, MeOH-d4) δ −115.8 (d, JHF = 21.0 Hz). HRMS (DART) calcd for C10H6O4F [M-H]–: 209.0256, found 209.0255.
(E)-3-(Benzofuran-2-yl)-2-fluoroacrylic acid (32b).
Saponification of ester 32a (7.1 g, 30.31 mmol, > 98:2 dr) in EtOH/water afforded carboxylic acid 32b as a white solid (5.86 g, > 98:2 dr, 94% yield). mp 183.8–185.2 °C; 1H NMR (400 MHz, MeOH-d4) δ 7.69 (s, 1H), 7.53 (d, J = 7.7 Hz, 1H), 7.38 (d, J = 8.2 Hz, 1H), 7.31–7.21 (m, 1H), 7.21–7.10 (m, 1H), 6.84 (d, JHF = 22.1 Hz, 1H); 13C{1H} NMR (100 MHz, MeOH-d4) δ 163.1 (d, J = 34.7 Hz), 156.3 (d, J = 2.2 Hz), 149.2 (d, J = 260.7 Hz), 149.3 (d, J = 11.0 Hz), 130.2, 126.9, 124.3, 122.8, 112.5 (d, J = 7.1 Hz), 112.0, 111.7 (d, J = 34.5 Hz); 19F NMR (376.5 MHz, MeOH-d4) δ −115.0 (d, JHF = 22.1 Hz). HRMS (DART) calcd for C11H8O3F [M+H]+: 207.0452, found 207.0452.
(E)-3-Cyclopropyl-2-fluoroacrylic acid (33b).
To a solution of ester 33a (4.18 g; 26.43 mmol, 98:2 dr) in EtOH (45 mL) was added a slurry of LiOH•H2O (2.22 g; 52.85 mmol; 2 equiv) in H2O (45 mL) at 0 °C. The reaction mixture was warmed to rt and stirred for 3.5 h. H2O (56 mL) was added, and the mixture was extracted with hexanes (20 mL). Layers were separated, and the aqueous phase was concentrated under reduced pressure to remove almost all EtOH. The solution was acidified with conc. HCl (4.77 mL; 58.14 mmol; 2.2 equiv) which resulted in separation of the product as an oil. The aqueous phase was then saturated with NaCl and reaction mixture was extracted twice with MTBE (2 × 25 mL). MTBE extracts were dried over Na2SO4. Solid was filtered, and the filtrate was concentrated under reduced pressure to afford the product 33b as a white solid. The solid was dried at 40 °C in vacuum oven overnight (3.25 g, > 98:2 dr, 95% yield). mp 101.4–102.5 °C; 1H NMR (400 MHz, CDCl3) δ 12.11–11.54 (brs, 1H), 5.46 (dd, JHF = 19.8 Hz, J = 11.1 Hz, 1H), 2.55–2.37 (m, 1H), 1.09–0.92 (m, 2H), 0.63–0.49 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 167.1 (d, J = 35.6 Hz), 145.7 (d, J = 241.7 Hz), 133.1 (d, J = 21.5 Hz), 8.9 (d, J = 6.9 Hz), 8.6 (d, J = 1.1 Hz); 19F NMR (376.5 MHz, CDCl3) δ −127.9 (dd, JHF = 19.8 Hz, JHF = 1.7 Hz). HRMS (DART) calcd for C6H6O2F [M-H]–: 129.0357, found 129.0357.
(E)-3-Cyclopentyl-2-fluoroacrylic acid (34b).
To a solution of ester 34a (4.49 g; 24.11 mmol; 96:4 dr) in EtOH (45 mL) was added a slurry of LiOH•H2O (2.02 g; 48.22 mmol; 2 equiv) in H2O (45 mL) at 0 °C. The reaction mixture was warmed to rt and stirred for 4 h. H2O (56 mL) was added, and the mixture was extracted with hexanes (20 mL). Layers were separated, and the aqueous phase was concentrated under reduced pressure to remove almost all EtOH. The solution was acidified with conc. HCl (4.36 mL; 53.04 mmol; 2.2 equiv) which resulted in formation of a slurry. The slurry was filtered, and the solid was washed with water. It was then dried on a Buchner funnel for 2 h followed by at 40 °C in a vacuum oven overnight. This yielded a first crop of 34b as a white solid (3.361 g), >98:2 dr. The combined filtrate and wash were concentrated under reduced pressure to a volume of ~30 mL which resulted in precipitation of a second crop of product. The solid was filtered and washed with water. It was then dried on a Buchner funnel for 2 h followed by at 40 °C in a vacuum oven overnight. This yielded a second crop of 34b as a white solid (163.7 mg), >98:2 dr. Combined yield of 34b: 3.53 g; 92% yield, single stereoisomer (>98:2 dr). 1H NMR (400 MHz, CDCl3) δ 12.21–11.88 (brs, 1H), 6.00 (dd, JHF = 21.3 Hz, J = 10.5 Hz, 1H), 3.52–3.32 (m, 1H), 2.02–1.81 (m, 2H), 1.80–1.48 (m, 4H), 1.41–1.16 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 166.7 (d, J = 37.1 Hz), 145.4 (d, J = 246.2 Hz), 131.8 (d, J = 16.5 Hz), 36.4 (d, J = 4.5 Hz), 33.6 (d, J = 2.1 Hz), 25.3; 19F NMR (376.5 MHz, CDCl3) δ −124.6 (dd, JHF = 21.3 Hz, JHF = 1.2 Hz). HRMS (DART) calcd for C8H10O2F [M-H]–: 157.0670, found 157.0670.
(E)-2-Fluoro-5-phenylpent-2-enoic acid (35b).
To a solution of ester 35a (4.34 g; 19.53 mmol; 94:6 dr) in EtOH (45.0 mL) was added a slurry of LiOH•H2O (1.64 g; 39.05 mmol; 2 equiv) in H2O (45.0 mL) at 0 °C. The reaction mixture was warmed to rt and stirred for 2 h. H2O (58.0 mL) was added, and the mixture was extracted with hexanes (20 mL). Layers were separated, and the aqueous phase was concentrated under reduced pressure to remove almost all EtOH. The solution was then acidified with conc. HCl (3.53 mL; 42.96 mmol; 2.2 equiv) which resulted in separation of the product as an oil. The mixture was extracted twice with EtOAc (2 × 25 mL). EtOAc extracts were dried over Na2SO4 and filtered. The filtrate was concentrated under reduced pressure to afford 35b as a pale yellow oil. The oil solidified after standing overnight in the freezer. It was then dried at 40 °C in a vacuum oven overnight (3.72 g, 98% yield, mixture of stereoisomers in 18.3:1 (95:5) dr). 1H NMR (400 MHz, CDCl3) δ 12.07–10.87 (brs, 1H), 7.41–7.31 (m, 2H), 7.31–7.20 (m, 3H), 6.16 (dt, JHF = 20.7 Hz, J = 8.0 Hz, 1H), 2.98–2.88 (m, 2H), 2.88–2.78 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 166.3 (d, J = 37.1 Hz), 146.4 (d, J = 248.6 Hz), 140.3, 128.5, 128.4, 126.3, 125.8 (d, J = 18.2 Hz), 35.1 (d, J = 2.2 Hz), 27.3 (d, J = 4.7 Hz); 19F NMR (376.5 MHz, CDCl3) δ −122.1 (d, JHF = 20.7 Hz), Z-isomer: −130.7 (dd, JHF = 32.6 Hz, JHF = 2.2 Hz). HRMS (DART) calcd for C11H10O2F [M-H]–: 193.0670, found 193.0670.
General Procedure for Ag-Catalyzed Decarboxylation.
A reaction vial was charged with acid (1 equiv), AgOAc (5 mol%), and NMP (2V). The vial was inerted and placed into a preheated 130 °C oil bath. The reaction mixture was stirred for 4 h under positive Ar pressure. It was then cooled to 0 °C and PhCF3 was added as an internal standard. The mixture was diluted with CDCl3 and analyzed by quantitative 19F NMR to obtain the assay yield. The reaction mixture was directly loaded on a silica gel column using minimum amount of CH2Cl2. It was purified by column chromatography and the fractions containing product were concentrated under reduced pressure with a cold 0–4 °C rotovap bath (ice/water) to afford the product.
(E)-2-(1-Fluoroprop-1-en-2-yl)naphthalene (10c).
Decarboxylation of 10b (2.0 g, 8.69 mmol, 21.1:1 (95:5) dr) was carried out with 5 mol% of AgOAc (72.5 mg, 0.43 mmol) in NMP (4 mL, 2V) at 130 °C for 4 h. The product 10c was obtained as a white solid after purification by silica gel column chromatography eluting with 0–2% EtOAc in hexanes (1.59 g, 98% yield, mixture of stereoisomers in 21.4:1 (95:5) dr). mp 36.8–39.2 °C; 1H NMR (400 MHz, CDCl3) δ 7.88–7.78 (m, 3H), 7.75 (s, 1H), 7.53–7.42 (m, 3H), 7.07 (d, JHF = 84.9 Hz, 1H), 2.22–2.11 (m, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 146.4 (d, J = 258.4 Hz), 134.8 (d, J = 8.8 Hz), 133.4, 132.7, 128.1, 127.9, 127.5, 126.3, 125.8, 124.5 (d, J = 4.4 Hz), 123.8 (d, J = 2.0 Hz), 120.0 (d, J = 10.0 Hz), 12.1 (d, J = 6.1 Hz); 19F NMR (376.5 MHz, CDCl3) δ −130.4 (dq, JHF = 84.9 Hz, JHF = 3.6 Hz); Z-isomer: −128.3 (dq, JHF = 84.7 Hz, JHF = 4.6 Hz). NMR spectra matched reported data.11a HRMS (EI) calcd for C13H11F [M]+: 186.0839, found 186.0839.
(E)-1-Bromo-4-(2-fluorovinyl)benzene (E-12c).
Decarboxylation of acid E-12b (250 mg, 1.02 mmol, > 98:2 dr) was carried out with 5 mol% of AgOAc (8.51 mg, 0.051 mmol) in NMP (0.5 mL, 2V) at 130 °C for 2.5 h. 19F NMR of the crude with PhCF3 as an internal standard: m(prod) = 181.0 mg, 88% NMR yield, single stereoisomer (> 98:2 dr). The product E-12c was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with 0–1% Et2O in pentane (176.3 mg, 86% yield, single stereoisomer (> 98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 7.47–7.39 (m, 2H), 7.16 (dd, JHF = 82.6 Hz, J = 11.4 Hz, 1H), 7.15–7.08 (m, 2H), 6.34 (dd, JHF = 18.6 Hz, J = 11.4 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 150.4 (d, J = 260.7 Hz), 131.9, 131.6 (d, J = 12.1 Hz), 127.6 (d, J = 3.1 Hz), 121.2 (d, J = 2.1 Hz), 113.0 (d, J = 16.7 Hz); 19F NMR (376.5 MHz, CDCl3) δ −128.2 (dd, JHF = 82.6 Hz, JHF = 18.6 Hz). NMR spectra matched reported data.10a HRMS (EI) calcd for C8H6BrF [M]+: 199.9631, found 199.9631.
(Z)-1-Bromo-4-(2-fluorovinyl)benzene (Z-12c).
Decarboxylation of acid Z-12b (250 mg, 1.02 mmol, > 98:2 dr) was carried out with 50 mol% of AgOAc (85.1 mg, 0.51 mmol) in NMP (2.0 mL, 8V) at 140 °C for 8 h. 19F NMR of the crude with PhCF3 as an internal standard: m(prod) = 171.22 mg, 84% NMR yield, single stereoisomer (> 98:2 dr). The product Z-12c was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with 0–1% Et2O in pentane (167.0 mg, 81% yield, single stereoisomer (> 98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 7.34–7.26 (m, 2H), 7.24–7.16 (m, 2H), 6.50 (dd, JHF = 82.4 Hz, J = 5.4 Hz, 1H), 5.40 (dd, JHF = 44.1 Hz, J = 5.4 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.7 (d, J = 271.5 Hz), 131.6, 131.5 (d, J = 1.4 Hz), 130.3 (d, J = 7.3 Hz), 121.3 (d, J = 3.6 Hz), 109.8; 19F NMR (376.5 MHz, CDCl3) δ −120.9 (dd, JHF = 82.4 Hz, JHF = 44.1 Hz). NMR spectra matched reported data.10a HRMS (EI) calcd for C8H6BrF [M]+: 199.9631, found 199.9633.
(E)-(1-Fluoroprop-1-en-2-yl)benzene (15c).
A reaction vial was charged with acid 15b (200.0 mg; 1.11 mmol; 100.0 mol%, >98:2 dr), AgOAc (9.26 mg; 0.06 mmol; 5 mol%), and NMP (0.40 mL, 2V). The vial was inerted and placed into a preheated 130 °C oil bath. The reaction mixture was stirred for 4 h under positive Ar pressure. It was then cooled to 0 °C and PhCF3 (34.8 mg) was added as an internal standard. The mixture was diluted with CDCl3 and analyzed by quantitative 19F NMR: m(prod) = 138.3 mg, 92% NMR yield, single stereoisomer (>98:2 dr). The reaction mixture was directly loaded on a silica gel column using minimum amount of CH2Cl2. It was purified by column chromatography eluting with pentane. Fractions containing product were concentrated under reduced pressure with a cold 0–4 °C rotovap bath (ice/water) to afford 15c as a colorless volatile liquid (123.0 mg, 81% yield, >98:2 dr). 1H NMR (400 MHz, CDCl3) δ 7.37–7.25 (m, 5H), 6.90 (dq, JHF = 85.1 Hz, JHH = 1.5 Hz, 1H), 2.05 (dd, JHF = 3.7 Hz, JHH = 1.5 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) 146.0 (d, J = 257.8 Hz), 137.6 (d, J = 8.9 Hz), 128.5, 127.4, 125.9 (d, J = 3.2 Hz), 120.0 (d, J = 9.8 Hz), 12.2 (d, J = 6.0 Hz); 19F NMR (376.5 MHz, CDCl3) δ −131.2 (dq, JHF = 85.1 Hz, JHF = 3.7 Hz). NMR spectra matched reported data.35 HRMS (EI) calcd for C9H9F [M]+: 136.0683, found 136.0683.
(E)-5-(1-Fluoroprop-1-en-2-yl)benzo[d][1,3]dioxole (16c).
Decarboxylation of acid 16b (250.0 mg, 1.12 mmol, >98:2 dr) was carried out with 5 mol% of AgOAc (9.31 mg, 0.06 mmol) in NMP (0.5 mL, 2V) at 130 °C for 4.0 h. 19F NMR of the crude with PhCF3 as an internal standard: m(prod) = 179.81 mg, 90% NMR yield, single stereoisomer (>98:2 dr). The product 16c was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with 1–5% MTBE in hexanes (165.9 mg, 83% yield, single stereoisomer (>98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 6.84 (dq, JHF = 85.2 Hz, J = 1.5 Hz, 1H), 6.82–6.75 (m, 3H), 5.96 (s, 2H), 2.01 (dd, JHF = 3.7 Hz, J = 1.5 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 147.8, 147.0, 145.4 (d, J = 257.1 Hz), 131.6 (d, J = 8.8 Hz), 119.8 (d, J = 10.2 Hz), 119.3 (d, J = 1.8 Hz), 108.3, 106.4 (d, J = 3.0 Hz), 101.0, 12.5 (d, J = 5.8 Hz); 19F NMR (376.5 MHz, CDCl3) δ −132.1 (dq, JHF = 85.2 Hz, JHF = 3.7 Hz). HRMS (DART) calcd for C10H9O2F [M]+: 180.0581, found 180.0582.
(E)-1-(1-Fluoroprop-1-en-2-yl)-4-nitrobenzene (17c).
Decarboxylation of acid 17b (250.0 mg, 0.89 mmol, >98:2 dr) was carried out with 5 mol% of AgOAc (7.41 mg, 0.04 mmol) in NMP (0.5 mL, 2V) at 130 °C for 4.0 h. The product 17c was obtained as a yellow solid after purification by silica gel column chromatography eluting with 0–2% EtOAc in hexanes (159.0 mg, 99% yield, single stereoisomer (>98:2 dr)). mp 59.8–61.3 °C; 1H NMR (400 MHz, CDCl3) δ 8.26–8.11 (m, 2H), 7.50–7.42 (m, 2H), 7.03 (dq, JHF = 83.0 Hz, J = 1.4 Hz, 1H), 2.08 (dd, JHF = 3.7 Hz, J = 1.4 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 147.9 (d, J = 263.7 Hz), 147.0, 144.3 (d, J = 9.5 Hz), 126.3 (d, J = 3.3 Hz), 123.9, 119.0 (d, J = 11.4 Hz), 11.9 (d, J = 5.9 Hz); 19F NMR (376.5 MHz, CDCl3) δ −124.9 (dq, JHF = 83.0 Hz, JHF = 3.7 Hz). 1H and 19F NMR spectra matched reported data.11a HRMS (DART) calcd for C9H9O2NF [M+H]+: 182.0612, found 182.0612.
(E)-1-Bromo-4-(1-fluorohex-1-en-2-yl)benzene (18c).
Decarboxylation of acid 18b (250.0 mg, 0.83 mmol, >98:2 dr) was carried out with 5 mol% of AgOAc (6.93 mg, 0.05 mmol) in NMP (0.5 mL, 2V) at 130 °C for 5.5 h. The product 18c was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with hexanes (190.6 mg, 89% yield, single stereoisomer (>98:2 dr)). mp 102.9–104.4 °C; 1H NMR (400 MHz, CDCl3) δ 7.52–7.41 (m, 2H), 7.21–7.09 (m, 2H), 6.77 (d, JHF = 85.0 Hz, 1H), 2.73–2.33 (m, 2H), 1.53–1.15 (m, 4H), 1.07–0.69 (m, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 145.9 (d, J = 259.9 Hz), 135.7 (d, J = 9.6 Hz), 131.7, 128.4 (d, J = 3.1 Hz), 124.4 (d, J = 9.9 Hz), 121.3, 29.8 (d, J = 2.1 Hz), 26.1 (d, J = 3.9 Hz), 22.3, 13.8; 19F NMR (376.5 MHz, CDCl3) δ −130.0 (dt, JHF = 85.0 Hz, JHF = 2.8 Hz). HRMS (EI) calcd for C12H14BrF [M]+: 256.0257, found 256.0261.
(E)-1-(1-Fluorobut-1-en-2-yl)-2-methylbenzene (19c).
Decarboxylation of acid 19b (650.0 mg, 3.12 mmol, >98:2 dr) was carried out with 15 mol% of AgOAc (78.5 mg, 0.47 mmol) in NMP (1.3 mL, 2V) at 130 °C for 18 h. The product 19c was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with pentane (346.2 mg, 68% yield, single stereoisomer (>98:2 dr)). mp 107.6–109.1 °C; 1H NMR (400 MHz, CDCl3) δ 7.26–7.12 (m, 3H), 7.12–7.06 (m, 1H), 6.45 (d, JHF = 86.4 Hz, 1H), 2.47 (ddq, J = 7.6 Hz, JHF = 2.7 Hz, J = 0.9 Hz, 2H), 2.31 (s, 3H), 0.96 (t, J = 7.6 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 145.4 (d, J = 259.8 Hz), 136.8 (d, J = 2.9 Hz), 135.9 (d, J = 9.9 Hz), 130.3 (d, J = 3.0 Hz), 130.1, 127.5, 125.8 (d, J = 7.9 Hz), 125.4, 21.5 (d, J = 3.8 Hz), 19.7, 12.0 (d, J = 2.2 Hz); 19F NMR (376.5 MHz, CDCl3) δ −131.7 (dt, JHF = 86.4 Hz, JHF = 2.0 Hz); HRMS (EI) calcd for C11H13F [M]+: 164.0996, found 164.0996.
(E)-(1-Fluoro-3-methylbut-1-en-2-yl)benzene (20c).
Decarboxylation of acid 20b (250 mg, 1.20 mmol, 18.4:1 (95:5) dr) was carried out with 10 mol% of AgOAc (20.0 mg, 0.12 mmol) in NMP (0.5 mL, 2V) at 130 °C for 5 h. 19F NMR of the crude with PhCF3 as an internal standard: m(prod) = 139.5 mg, 71% NMR yield, 17.9:1 (95:5) dr. The product 20c was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with 0–1% Et2O in pentane (108.6 mg, 55% yield, >98:2 dr). 1H NMR (400 MHz, CDCl3) δ 7.34–7.25 (m, 3H), 7.23–7.16 (m, 2H), 6.47 (d, JHF = 85.6 Hz, 1H), 3.07 (sept, J = 7.0 Hz, 1H), 1.13 (d, J = 7.0 Hz, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 145.4 (d, J = 261.4 Hz), 136.2 (d, J = 10.7 Hz), 130.8 (d, J = 6.6 Hz), 129.0 (d, J = 3.1 Hz), 128.0, 127.3, 28.1 (d, J = 2.1 Hz), 21.2 (d, J = 2.6 Hz); 19F NMR (376.5 MHz, CDCl3) δ −130.5 (d, JHF = 85.6 Hz), Z-isomer: −138.2 (dd, JHF = 85.3 Hz, JHF = 4.1 Hz); HRMS (EI) calcd for C11H13F [M]+: 164.0996, found 164.0996.
(E)-1-(1-Cyclopropyl-2-fluorovinyl)-4-methoxybenzene (21c).
Decarboxylation of acid 21b (250.0 mg, 0.55 mmol, >98:2 dr) was carried out with 10 mol% of AgOAc (8.83 mg, 0.05 mmol) in NMP (0.5 mL, 2V) at 130 °C for 4.5 h. 19F NMR of the crude with PhCF3 as an internal standard: m(prod) = 170.33 mg, 84% NMR yield, single stereoisomer (>98:2 dr). The product 21c was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with 1–4% Et2O in hexanes (162.8 mg, 80% yield, single stereoisomer (>98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 7.22–7.14 (m, 2H), 6.90–6.83 (m, 2H), 6.69 (d, JHF = 84.8 Hz, 1H), 3.82 (s, 3H), 1.95–1.77 (m, 1H), 0.86–0.72 (m, 2H), 0.57–0.42 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 159.1, 146.4 (d, J = 260.7 Hz), 129.8 (d, J = 3.2 Hz), 127.3 (d, J = 9.1 Hz), 125.3 (d, J = 6.0 Hz), 113.5, 55.2, 9.2 (d, J = 4.6 Hz), 4.9 (d, J = 3.1 Hz); 19F NMR (376.5 MHz, CDCl3) δ −133.8 (d, JHF = 84.8 Hz). HRMS (DART) calcd for C12H14OF [M+H]+: 193.1023, found 193.1024.
(Z)-1-Bromo-4-(1,3,3,3-tetrafluoroprop-1-en-2-yl)benzene (22c).
Decarboxylation of acid 22b (250.0 mg, 0.80 mmol, 25.8:1 (96:4) dr) was carried out with 5 mol% of AgOAc (6.66 mg, 0.04 mmol) in NMP (0.5 mL, 2V) at 130 °C for 4.5 h. 19F NMR of the crude with PhCF3 as an internal standard: m(prod) = 193.2 mg, 90% NMR yield, mixture of stereoisomers in 25.2:1 (96:4) dr. The product 22c was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with 1% Et2O in pentane (167.4 mg, 78% yield, mixture of stereoisomers in 26.2:1 (96:4) dr). The minor stereoisomer can be separated by silica gel column chromatography eluting with pentane. 1H NMR (400 MHz, CDCl3) δ 7.57–7.51 (m, 2H), δ 7.23–7.17 (m, 2H), 6.81 (dq, JHF = 77.0 Hz, JHF = 0.6 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 151.9, (dq, J = 288.4 Hz, J = 3.1 Hz), 132.0, 130.8 (d, J = 3.0 Hz), 127.7 (dq, J = 6.1 Hz, J = 1.2 Hz), 123.8, 122.0 (q, J = 273.8 Hz), 116.8 (dq, J = 32.2 Hz, J = 1.2 Hz); 19F NMR (376.5 MHz, CDCl3) δ −111.3 (dq, JHF = 77.0 Hz, J = 23.7 Hz), −59.7 (d, J = 23.7 Hz); Z-isomer: δ −123.6 (dq, JHF = 77.6 Hz, J = 7.7 Hz), −63.4 (d, J = 7.7 Hz). HRMS (EI) calcd for C9H5BrF4 [M]+: 267.9505, found 267.9509.
(E)-1-(1-Fluorooct-1-en-6-yn-2-yl)-3,5-dimethylbenzene (23c).
Decarboxylation of acid 23b (150.0 mg, 0.55 mmol, >98:2 dr) was carried out with 10 mol% of AgOAc (9.13 mg, 0.05 mmol) in NMP (0.3 mL, 2V) at 130 °C for 3.0 h. The product 23c was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with 0–2% MTBE in hexanes (96.2 mg, 76% yield, single stereoisomer (>98:2 dr)). 1H NMR (400 MHz, CDCl3) 6.94 (s, 1H), 6.92–6.65 (m, 3H), 2.68–2.55 (m, 2H), 2.32 (s, 6H), 2.19–2.08 (m, 2H), 1.79 (t, J = 2.5 Hz, 3H), 1.65–1.52 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 145.9 (d, J = 259.0 Hz), 138.0, 136.1 (d, J = 9.0 Hz), 129.1, 124.5 (d, J = 3.0 Hz), 124.4 (d, J = 8.7 Hz), 78.8, 75.7, 27.2 (d, J = 2.2 Hz), 25.7 (d, J = 4.3 Hz), 21.3, 18.4, 3.4; 19F NMR (376.5 MHz, CDCl3) δ −131.1 (d, JHF = 85.7 Hz). HRMS (DART) calcd for C16H20F [M+H]+: 231.1544, found 231.1544.
(Z)-1-(2-Fluoro-1-(4-methoxyphenyl)vinyl)cyclohexane-1-carbonitrile (24c).
Decarboxylation of acid 24b (250 mg, 0.82 mmol, >98:2 dr) was carried out with 5 mol% of AgOAc (6.88 mg, 0.04 mmol) in NMP (0.5 mL, 2V) at 130 °C for 4.5 h. The product 24c was obtained as a white solid after purification by silica gel column chromatography eluting with 1–6% EtOAc in hexanes (204.9 mg, 96% yield, single stereoisomer (>98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 10.73–8.78 (brs, 1H), 7.17–7.07 (m, 2H), 6.92–6.80 (m, 2H), 6.47 (d, JHF = 82.8 Hz, 1H), 3.79 (s, 3H), 2.15–2.00 (m, 2H), 1.79–1.63 (m, 5H), 1.63–1.47 (m, 2H), 1.24–1.04 (m, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 159.5, 147.8 (d, J = 275.4 Hz), 131.5 (d, J = 3.3 Hz), 125.2 (d, J = 9.7 Hz), 124.6 (d, J = 1.7 Hz), 121.6, 113.4, 55.1, 40.1 (d, J = 2.6 Hz), 34.7 (d, J = 2.9 Hz), 24.6, 22.7; 19F NMR (376.5 MHz, CDCl3) δ −117.1 (d, JHF = 82.8 Hz). HRMS (DART) calcd for C16H19ONF [M+H]+: 260.1445, found 260.1446.
(E)-1-Bromo-4-(1-fluoro-3-phenylprop-1-en-2-yl)benzene (25c).
Decarboxylation of acid 25b (250.0 mg, 0.55 mmol, 14.9:1 (94:6) dr) was carried out with 10 mol% of AgOAc (12.44 mg, 0.07 mmol) in NMP (0.5 mL, 2V) at 130 °C for 4.0 h. 19F NMR of the crude with PhCF3 as an internal standard: m(prod) = 191.2 mg, 88% NMR yield, mixture of stereoisomers in 15.2:1 (94:6) dr. The product 25c was obtained as a colorless liquid after purification by silica gel column chromatography eluting with 0–2% MTBE in hexanes (177.0 mg, 82% yield, single stereoisomer (>98:2 dr)). 1H NMR (400 MHz, CDCl3) 7.41–7.36 (m, 2H), 7.27–7.21 (m, 2H), 7.20–7.13 (m, 3H), 7.12–7.08 (m, 2H), 6.97 (d, JHF = 84.5 Hz, 1H), 3.85 (d, JHF = 2.9 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 146.6 (d, J = 261.9 Hz), 138.2 (d, J = 2.5 Hz), 135.0 (d, J = 8.7 Hz), 131.6, 128.5, 128.4, 128.3 (d, J = 4.1 Hz), 126.2, 122.8 (d, J = 9.6 Hz), 121.4, 32.6 (d, J = 4.7 Hz); 19F NMR (376.5 MHz, CDCl3) δ −128.9 (dt, JHF = 84.5 Hz, JHF = 2.8 Hz). HRMS (EI) calcd for C15H12BrF [M]+: 290.0101, found 290.0103.
(E)-3-(1-Fluoroprop-1-en-2-yl)quinoline (26c).
Decarboxylation of acid 26b (250.0 mg, 1.08 mmol, >98:2 dr) was carried out with 15 mol% of AgOAc (27.1 mg, 0.16 mmol) in NMP (0.75 mL, 3V) at 130 °C for 6 h. 19F NMR of the crude with PhCF3 as an internal standard: m(prod) = 175.8 mg, 87% NMR yield, single stereoisomer (>98:2 dr). The product 26c was obtained as a white solid after purification by silica gel column chromatography eluting with 5–10% EtOAc in hexanes (178.3 mg, 88% yield, single stereoisomer (>98:2 dr)). mp 58.0–59.9 °C; 1H NMR (400 MHz, CDCl3) δ 8.88 (d, JHF = 2.3 Hz, 1H), 8.07 (d, J = 8.4 Hz, 1H), 7.97 (d, JHF = 2.2 Hz, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.66 (ddd, J = 8.4 Hz, J = 7.0 Hz, J = 1.4 Hz, 1H), 7.52 (ddd, J = 8.0 Hz, J = 7.0 Hz, J = 1.0 Hz, 1H), 7.06 (dq, JHF = 83.6 Hz, J = 1.5 Hz, 1H), 2.13 (dd, JHF = 3.6 Hz, J = 1.5 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.4 (d, J = 2.2 Hz), 147.4, 146.9 (d, J = 261.2 Hz), 132.1 (d, J = 4.8 Hz), 130.4 (d, J = 8.7 Hz), 129.3, 129.2, 127.8, 127.7, 127.0, 117.6 (d, J = 11.2 Hz), 12.0 (d, J = 5.9 Hz); 19F NMR (376.5 MHz, CDCl3) δ −127.1 (dq, JHF = 83.6 Hz, JHF = 3.6 Hz); Z-isomer: −126.1 (dq, JHF = 83.7 Hz, JHF = 4.8 Hz). NMR spectra matched reported data.36 HRMS (DART) calcd for C12H11NF [M+H]+: 188.0870, found 188.0870.
(E)-2-(1-Fluoroprop-1-en-2-yl)benzo[b]thiophene (27c).
Decarboxylation of acid 27b (250.0 mg, 1.06 mmol, 10.9:1 (92:8) dr) was carried out with 5 mol% of AgOAc (8.83 mg, 0.05 mmol) in NMP (0.5 mL, 2V) at 130 °C for 2.0 h. 19F NMR of the crude with PhCF3 as an internal standard: m(prod) = 181.6 mg, 89% NMR yield, mixture of stereoisomers in 4.3:1 (81:19) dr. The isomers were separated by silica gel column chromatography eluting with 0–1% MTBE in hexanes (E-27c: 142.4 mg, white solid, 70% yield, >98:2 dr; Z-27c: 32.5 mg, white solid, 16% yield, 43.7:1 dr). E-isomer: mp 79.9–80.5 °C; 1H NMR (400 MHz, CDCl3) δ 7.85–7.78 (m, 1H), 7.78–7.73 (m, 1H), 7.43–7.33 (m, 2H), 7.25 (dq, JHF = 82.9 Hz, J = 1.4 Hz, 1H), 7.26 (s, 1H), 2.20 (dd, JHF = 3.6 Hz, J = 1.4 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 146.9 (d, J = 262.6 Hz), 140.1 (d, J = 8.5 Hz), 139.8, 138.1, 124.6, 124.4, 123.1, 122.0, 120.6 (d, J = 8.0 Hz), 115.7 (d, J = 13.8 Hz), 11.9 (d, J = 5.7 Hz); 19F NMR (376.5 MHz, CDCl3) δ −129.2 (dq, JHF = 82.9 Hz, JHF = 3.6 Hz); HRMS (EI) calcd for C11H9FS [M]+: 192.0404, found 192.0406. Z-isomer: 1H NMR (400 MHz, CDCl3) δ 7.85–7.79 (m, 1H), 7.79–7.73 (m, 1H), 7.39–7.28 (m, 3H), 6.73 (dq, JHF = 82.7 Hz, J = 1.5 Hz, 1H), 2.04 (dd, JHF = 4.4 Hz, J = 1.5 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 145.1 (d, J = 269.2 Hz), 139.9 (d, J = 9.0 Hz), 138.9, 137.9 (d, J = 4.7 Hz), 124.5, 124.3, 123.5 (d, J = 1.8 Hz), 122.0, 121.9 (d, J = 5.1 Hz), 112.4 (d, J = 2.1 Hz), 15.3 (d, J = 7.5 Hz); 19F NMR (376.5 MHz, CDCl3) δ −120.3 (dq, JHF = 82.7 Hz, JHF = 4.4 Hz). HRMS (EI) calcd for C11H9FS [M]+: 192.0404, found 192.0402.
(E)-1-(1-Fluoroprop-1-en-2-yl)cyclohex-1-ene (28c).
Decarboxylation of acid 28b (650.0 mg, 3.53 mmol, >98:2 dr) was carried out with 5 mol% of AgOAc (29.45 mg, 0.18 mmol) in NMP (1.3 mL, 3V) at 130 °C for 4 h. The product 28c was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with 0–1% Et2O in pentane (427.8 mg, 86% yield, single stereoisomer (>98:2 dr)). 1H NMR (400 MHz, CDCl3) 6.73 (d, JHF = 86.3 Hz, 1H), 5.90–5.64 (m, 1H), 2.21–2.01 (m, 4H), 1.77 (dd, JHF = 3.6 Hz, J = 1.4 Hz, 3H), 1.71–1.63 (m, 2H), 1.63–1.53 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 145.5 (d, J = 253.2 Hz), 132.7 (d, J = 6.9 Hz), 124.0 (d, J = 9.3 Hz), 120.9 (d, J = 7.8 Hz), 25.8, 25.3, 22.6, 22.3, 9.5 (d, J = 8.1 Hz); 19F NMR (376.5 MHz, CDCl3) δ −137.6 (d, JHF = 86.3 Hz, 1H). HRMS (EI) calcd for C9H13F [M]+: 140.0996, found 140.0997.
(E)-6-(Fluoromethylene)-8a-methyl-3,4,6,7,8,8a-hexahydronaphthalen-1(2H)-one (29c).
Decarboxylation of acid 29b (250 mg, 1.05 mmol, 9.97:1 (91:9) dr) was carried out with 25 mol% of AgOAc (43.8 mg, 0.26 mmol) in NMP (0.5 mL, 2V) at 130 °C for 4 h. 19F NMR of the crude with PhCF3 as an internal standard: m(prod) = 154.5 mg, 76% NMR yield, mixture of stereoisomers in 8.93:1 (90:10) dr. The product 29c was obtained as a colorless liquid after purification by silica gel column chromatography eluting with 3–8% EtOAc in hexanes (146.7 mg, 72% yield, mixture of stereoisomers in 7.89:1 (89:11) dr). 1H NMR (400 MHz, CDCl3) δ 6.53 (d, JHF = 84.2 Hz, 1H), 5.70 (s, 1H), 2.71–2.49 (m, 3H), 2.40–2.23 (m, 2H), 2.18–2.06 (m, 1H), 2.05–1.93 (m, 1H), 1.86–1.75 (m, 1H), 1.75–1.65 (m, 1H), 1.65–1.51 (m, 1H), 1.26 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 213.6, 145.2 (d, J = 257.2 Hz), 142.6 (d, J = 13.9 Hz), 119.1 (d, J = 11.6 Hz), 118.3 (d, J = 9.2 Hz), 50.2, 38.1, 31.2, 29.0, 24.5 (d, J = 0.8 Hz), 24.0 (d, J = 1.4 Hz), 17.1 (d, J = 4.8 Hz); 19F NMR (376.5 MHz, CDCl3) δ −136.5 (m, JHF = 84.2 Hz); Z-isomer: −138.7 (d, JHF = 84.4 Hz). HRMS (EI) calcd for C12H15OF [M]+: 194.1101, found 194.1103.
(E)-2-(2-Fluorovinyl)naphthalene (E-30c).
Decarboxylation of acid E-30b (250 mg, 1.16 mmol, > 98:2 dr) was carried out with 5 mol% of AgOAc (9.65 mg, 0.58 mmol) in NMP (0.5 mL, 2V) at 130 °C for 3 h. The product E-30c was obtained as a white solid after purification by silica gel column chromatography eluting with 0–1% Et2O in hexanes (193.8 mg, 97% yield, single stereoisomer (> 98:2 dr). mp 72.0–72.4 °C; 1H NMR (400 MHz, CDCl3) δ 7.86–7.74 (m, 3H), 7.66 (s, 1H), 7.52–7.36 (m, 3H), 7.32 (dd, JHF = 83.1 Hz, J = 11.4 Hz, 1H), 6.56 (dd, JHF = 19.5 Hz, J = 11.4 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 150.5 (d, J = 259.3 Hz), 133.6, 132.7 (d, J = 1.3 Hz), 130.1 (d, J = 11.7 Hz), 128.5, 127.70, 127.68, 126.5, 125.9, 125.7 (d, J = 5.0 Hz), 123.3 (d, J = 1.4 Hz), 114.1 (d, J = 16.2 Hz); 19F NMR (376.5 MHz, CDCl3) δ −129.4 (dd, JHF = 83.1 Hz, JHF = 19.5 Hz). NMR spectra matched reported data.37 HRMS (EI) calcd for C12H9F [M]+: 172.0683, found 172.0682.
(Z)-2-(2-Fluorovinyl)naphthalene (Z-30c).
Decarboxylation of acid Z-30b (250 mg, 1.16 mmol, > 98:2 dr) was carried out with 50 mol% of AgOAc (96.5 mg, 0.58 mmol) in NMP (2.0 mL, 8V) at 145 °C for 10 h. The product Z-30c was obtained as a white solid after purification by silica gel column chromatography eluting with 0–1% Et2O in hexanes (171.9 mg, 86% yield, single stereoisomer (> 98:2 dr)). mp 59.8–61.4 °C; 1H NMR (400 MHz, CDCl3) δ 7.96 (s, 1H), 7.88–7.79 (m, 3H), 7.75–7.68 (m, 1H), 7.54–7.45 (m, 2H), 6.75 (dd, JHF = 82.7 Hz, J = 5.4 Hz, 1H), 5.79 (dd, JHF = 44.8 Hz, J = 5.4 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.5 (d, J = 270.9 Hz), 133.4, 132.6 (d, J = 1.8 Hz), 130.1 (d, J = 1.7 Hz), 128.1, 128.0, 127.9 (d, J = 7.2 Hz), 127.6, 126.7 (d, J = 7.2 Hz), 126.2, 126.1, 110.9; 19F NMR (376.5 MHz, CDCl3) δ −121.7 (dd, JHF = 82.7 Hz, JHF = 44.8 Hz). NMR spectra matched reported data.10a HRMS (EI) calcd for C12H9F [M]+: 172.0683, found 172.0682.
(E)-4-(2-Fluorovinyl)benzo[d][1,3]dioxole (31c).
Decarboxylation of acid 31b (250 mg, 1.19 mmol, > 98:2 dr) was carried out with 5 mol% of AgOAc (9.9 mg, 0.06 mmol) in NMP (0.5 mL, 2V) at 130 °C for 3 h. 19F NMR of the crude with PhCF3 as an internal standard: m(prod) = 173.4 mg, 88% NMR yield, single stereoisomer (> 98:2 dr). The product 31c was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with 3–6% Et2O in pentane (169.4 mg, 86% yield, single stereoisomer (> 98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 7.48 (dd, JHF = 84.1 Hz, J = 11.3 Hz, 1H), 6.83–6.77 (m, 1H), 6.75–6.71 (m, 1H), 6.71–6.66 (m, 1H), 6.29 (dd, JHF = 20.5 Hz, J = 11.3 Hz, 1H), 6.00 (s, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 153.0 (d, J = 260.1 Hz), 147.4, 144.0 (d, J = 2.0 Hz), 121.8, 121.2 (d, J = 3.8 Hz), 115.1 (d, J = 12.3 Hz), 109.3 (d, J = 18.3 Hz), 107.3 (d, J = 2.1 Hz), 100.8; 19F NMR (376.5 MHz, CDCl3) δ −123.9 (dd, JHF = 84.1 Hz, JHF = 20.5 Hz). HRMS (EI) calcd for C9H7O2F [M]+: 166.0425, found 166.0425.
(E)-2-(2-Fluorovinyl)benzofuran (32c).
Decarboxylation of acid 32b (200 mg, 0.97 mmol, > 98:2 dr) was carried out with 5 mol% of AgOAc (8.1 mg, 0.049 mmol) in NMP (0.4 mL, 2V) at 130 °C for 4 h. 19F NMR of the crude with PhCF3 as an internal standard: m(prod) = 121.6 mg, 77% NMR yield, single stereoisomer (> 98:2 dr). The product 32c was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with 0–1% Et2O in pentane (114.0 mg, 73% yield, single stereoisomer (> 98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 7.52–7.48 (m, 1H), 7.45 (ddd, JHF = 82.0 Hz, J = 11.1 Hz, J = 0.6 Hz, 1H), 7.44–7.39 (m, 1H), 7.29–7.24 (m, 1H), 7.23–7.17 (m, 1H), 6.55 (s, 1H), 6.34 (ddd, JHF = 17.1 Hz, J = 11.1 Hz, J = 0.4 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 154.6, 152.1 (d, J = 264.4 Hz), 150.2 (d, J = 13.1 Hz), 128.6, 124.5 (d, J = 0.8 Hz), 122.9, 120.5, 110.8, 104.9 (d, J = 9.9 Hz), 104.2 (d, J = 20.6 Hz); 19F NMR (376.5 MHz, CDCl3) δ −126.1 (dd, JHF = 82.0 Hz, JHF = 17.1 Hz). HRMS (EI) calcd for C10H7OF [M]+: 162.0475, found 162.0476.
(E)-(2-Fluorovinyl)cyclopropane (33c).
Note: due to the low b.p. of the product two reactions were run side by side: one to calculate NMR assay yield, and one to isolate the product as a solution in CDCl3 for product characterization. A reaction vial was charged with acid 33b (1.00 g; 7.69 mmol, 1 equiv), AgOAc (641.38 mg; 3.84 mmol; 50.0 mol%), and NMP (2.00 mL; 2.00 V). The vial was inerted with argon, sealed, and placed to a preheated to 145 °C oil bath. The reaction mixture was stirred at 145 °C for 4 h. The reaction mixture was cooled to 0 °C, and the vial was opened to release CO2. To the first reaction vial was added PhCF3 (77.77 mg) as internal standard, and the mixture was diluted with CDCl3. The mixture was analyzed by quantitative 19F NMR: m(prod) = 111.0 mg, 67% NMR assay yield; >98:2 dr. The second reaction mixture was subjected to distillation. The product 33c was isolated as a colorless volatile liquid by bulb-to-bulb distillation from the reaction mixture (40–50 °C bath / 50 torr) using liquid nitrogen to cool the receiver. The distillation receiver was warmed to 5 °C and the product 33c was dissolved in CDCl3 to avoid product loss as it has a low b.p. 1H NMR (400 MHz, CDCl3) δ 6.59 (ddd, JHF = 85.1 Hz, J = 11.0 Hz, J = 0.8 Hz, 1H), 5.08 (ddd, JHF = 18.4 Hz, J = 11.0 Hz, J = 8.3 Hz, 1H), 1.27–1.16 (m, 1H), 0.72–0.63 (m, 2H), 0.37–0.24 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.3 (d, J = 250.4 Hz), 115.4 (d, J = 12.5 Hz), 6.8 (d, J = 13.6 Hz), 5.6 (d, J = 1.5 Hz); 19F NMR (376.5 MHz, CDCl3) δ −134.4 (dd, JHF = 85.1 Hz, JHF = 18.4 Hz). Attempts to obtain HRMS data were not successful for this compound.
(E)-(2-Fluorovinyl)cyclopentane (34c).
A reaction vial was charged with acid 34b (250.0 mg; 1.58 mmol, 1 equiv), AgOAc (131.91 mg; 0.79 mmol; 50.00 mol%), and NMP (0.50 mL; 2.00 V). The vial was inerted with argon, sealed, and placed to a preheated to 140 °C oil bath. The reaction mixture was stirred at 140 °C for 3 h. The reaction mixture was cooled to 0 °C, and the vial was opened to release CO2. Product 34c was isolated as a colorless volatile liquid by bulb-to-bulb distillation from the reaction mixture (40–50 °C bath / 10 torr) using liquid nitrogen to cool the receiver (160.5 mg, 89% yield, single stereoisomer (> 98:2 dr)). 1H NMR (400 MHz, CDCl3) δ 6.51 (ddd, JHF = 86.1 Hz, J = 11.1 Hz, J = 1.0 Hz, 1H), 5.35 (ddd, JHF = 19.7 Hz, J = 11.1 Hz, J = 8.7 Hz, 1H), 2.40–2.26 (m, 1H), 1.85–1.73 (m, 2H), 1.72–1.50 (m, 4H), 1.32–1.20 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 147.7 (d, J = 252.0 Hz), 116.2 (d, J = 8.0 Hz), 36.5 (d, J = 8.8 Hz), 33.4 (d, J = 2.2 Hz), 24.9; 19F NMR (376.5 MHz, CDCl3) δ −133.4 (dd, JHF = 86.1 Hz, JHF = 19.7 Hz). HRMS (EI) calcd for C7H11F [M]+: 114.0839, found 114.0841.
(E)-(4-Fluorobut-3-en-1-yl)benzene (35c).
Decarboxylation of acid 35b (250 mg, 1.29 mmol, 18.3:1 (95:5) dr) was carried out with 50 mol% of AgOAc (107.4 mg, 0.64 mmol) in NMP (0.5 mL, 2V) at 145 °C for 6 h. The product 35c was obtained as a colorless volatile liquid after purification by silica gel column chromatography eluting with 2–4% Et2O in pentane (177.3 mg, 92% yield, mixture of stereoisomers in 18.1:1 (95:5) dr). 1H NMR (400 MHz, CDCl3) δ 7.37–7.28 (m, 2H), 7.27–7.16 (m, 3H), 6.52 (dd, JHF = 85.6 Hz, J = 11.1 Hz, 1H), 5.41 (ddt, JHF = 18.9 Hz, J = 11.1 Hz, J = 7.7 Hz, 1H), 2.75–2.67 (m, 2H), 2.31–2.20 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 166.3 (d, J = 37.1 Hz), 146.4 (d, J = 248.6 Hz), 140.3, 128.5, 128.4, 126.3, 125.8 (d, J = 18.2 Hz), 35.1 (d, J = 2.2 Hz), 27.3 (d, J = 4.7 Hz); 19F NMR (376.5 MHz, CDCl3) δ −129.8 (dd, JHF = 85.6 Hz, JHF = 18.9 Hz). HRMS (EI) calcd for C10H11F [M]+: 150.0839, found 150.0840.
Ethyl (Z)-4-bromo-2-fluoro-3-phenylbut-2-enoate (37).
A round bottom flask was charged with triethyl 2-fluoro-2-phosphonoacetate (30.6 mL, 150.72 mmol, 2.0 equiv) and THF (250 mL). The solution was cooled to 0 to −5 °C. MeMgBr (43.3 mL, 150.72 mmol, 3.48 M in 2-MeTHF, 2.0 equiv) was added dropwise maintaining internal temperature below 10 °C. The reaction mixture was warmed to rt and stirred for 10 min, then cooled to 0 °C. A solution of 2-bromoacetophenone (15.0 g, 75.36 mmol, 1.0 equiv) in THF (50 mL) was added dropwise over 30 min. The reaction mixture was warmed to rt and stirred for 20 min. The mixture was quenched with 10% aq. NH4Cl solution (200 mL), and phases were separated. Aqueous phase was extracted with MTBE (2 × 50 mL). The combined organic phases were dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was analyzed by quantitative 19F NMR: mixture of stereoisomers in 8.6:1 dr. The residue was purified using silica gel column chromatography eluting with 1–4% EtOAc in hexanes to afford two fractions: less polar fraction (colorless liquid, 1.88 g, E-37) and more polar fraction (Z-37, colorless liquid, mixture of stereoisomers in 30.6:1 (97:3) dr (19.79 g, 91% yield). Polar isomer was used directly in the next step without further purification. Z-37, major: 1H NMR (400 MHz, CDCl3) δ 7.42–7.34 (m, 3H), 7.30–7.23 (m, 2H), 4.29 (d, JHF = 3.5 Hz, 2H), 4.05 (q, J = 7.1 Hz, 2H), 1.01 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 159.8 (d, J = 35.7 Hz), 145.9 (d, J = 265.1 Hz), 134.6 (d, J = 4.0 Hz), 129.8 (d, J = 13.9 Hz), 128.5, 128.3 (d, J = 3.1 Hz), 128.1, 61.6, 28.8 (d, J = 8.4 Hz), 13.5; 19F NMR (376.5 MHz, CDCl3) δ −117.0 (t, JHF = 3.5 Hz); HRMS (DART) calcd for C12H13O2BrF [M+H]+: 287.0078, found 287.0078. E-37, minor: 1H NMR (400 MHz, CDCl3) δ 7.49–7.33 (m, 5H), 4.81 (d, JHF = 1.8 Hz, 2H), 4.39 (q, J = 7.1 Hz, 2H), 1.40 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.7 (d, J = 33.8 Hz), 145.4 (d, J = 266.9 Hz), 134.1, 130.6 (d, J = 15.1 Hz), 129.1, 128.5, 128.2 (d, J = 3.7 Hz), 62.1, 29.5 (d, J = 5.2 Hz), 14.0; 19F NMR (376.5 MHz, CDCl3) δ −118.9 (s).
Ethyl (Z)-4-(bis(tert-butoxycarbonyl)amino)-2-fluoro-3-phenylbut-2-enoate (38).
A flask was charged with bromide 37 (17.0 g, 59.21 mmol, 1 equiv; mixture of stereoisomers in 30.6:1 (97:3) dr), Boc2NH (15.4 g, 71.05 mmol, 1.2 equiv), Bu4NI (2.2 g; 5.92 mmol, 10.0 mol%), and MEK (140.0 mL). The solution was cooled to 0 °C and Cs2CO3 (28.9 g, 88.81 mmol, 1.5 equiv) was added. The reaction mixture was warmed to rt and stirred for 3 h 15 min. The reaction mixture was cooled to 0 °C and quenched with 10% aq. NH4Cl solution (100 mL). The layers were separated and the organic phase was distilled to 2–3 volumes (~40–50 mL) under vacuum at 60 °C. A heptane/MEK mixture (100 mL, 2:1 v/v) was added. The solution was distilled to about 2 volumes (35 mL) and the resultant solution was diluted with heptane (70 mL) at 60 °C. The mixture was linearly cooled to 0 °C over 1 h. The slurry was filtered. Solid was washed with cold (~ 5 °C) heptane/MEK (50:1 v/v) solvent mixture. It was then dried in vacuum oven at 40 °C overnight to afford the first crop of the product. Crop 1: 17.80 g, white solid, > 98:2 dr. The mother liquor was concentrated to a volume of ~20 mL and the residue was recrystallized from heptane/MEK (50:1 v/v) to afford a second crop of the product. Crop 2: 4.44 g, white solid, > 98:2 dr. Combined weight of 38: 22.24 g, 84.3 wt.% purity, >98:2 dr, 75% yield. The product was used in next step without further purification. A 100 mg sample of crop 1 was purified by silica gel column chromatography for analytical purposes. Eluent: 8–10% EtOAc in hexanes. White solid. 1H NMR (400 MHz, CDCl3) δ 7.35–7.25 (m, 3H), 7.15–7.06 (m, 2H), 4.77 (d, JHF = 2.7 Hz, 2H), 4.00 (q, J = 7.1 Hz, 2H), 1.37 (s, 18H), 0.96 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.1 (d, J = 36.0 Hz), 151.7, 144.2 (d, J = 257.3 Hz), 133.3 (d, J = 4.9 Hz), 131.4 (d, J = 12.8 Hz), 128.4 (d, J = 3.1 Hz), 127.8, 127.7, 82.5, 61.0, 45.1 (d, J = 9.0 Hz), 27.7, 13.4; 19F NMR (376.5 MHz, CDCl3) δ −123.4 (s). HRMS (DART) calcd for C18H23O6NF [M+H-C4H8]+: 368.1504, found 368.1505.
(Z)-4-((tert-Butoxycarbonyl)amino)-2-fluoro-3-phenylbut-2-enoic acid (39).
To a solution of 38 (21.74 g, 84.3 wt.%, 43.33 mmol, 1 equiv) in EtOH (170.0 mL) was added a solution of LiOH•H2O (8.60 g; 204.97 mmol; 4.73 equiv) in H2O (131.0 mL) at 0 °C. The reaction mixture was warmed to rt and stirred for 1 h (complete saponification). The reaction mixture was heated to 60 °C and stirred for 13.5 h (complete Boc-cleavage). The reaction mixture was cooled to 10 °C and 1 M HCl (205.0 mL; 204.97 mmol; 4.73 equiv) was added dropwise. A solvent mixture consisting of EtOH (30.00 mL) and H2O (66.00 mL) was added. The resultant slurry was stirred for 0.5 h and H2O (71.0 mL) was added dropwise over 30 min (final dilution 25 wt.% EtOH in H2O). The slurry was stirred for 0.5 h and then filtered. The solid was washed with aq. EtOH (25 wt.% EtOH). It was then dried on a Buchner funnel for 1 h and then in a vacuum oven at 40 °C overnight. Crop 1: white solid (8.09 g, > 98:2 dr). The mother liquor was slowly acidified to pH = 3 with 1 M HCl at 10 °C and extracted with EtOAc. Combined extracts were dried over MgSO4, filtered, and concentrated under reduced pressure. Crude product was recrystallized from EtOH/H2O solvent system (final dilution 25 wt.% EtOH in H2O) to obtain the second crop. Crop 2: white solid (3.32 g, > 98:2 dr). Combined weight of 39: 11.41 g, 89% yield. The compound exists as a mixture of Boc rotamers. mp 138.0–139.0 °C; 1H NMR (400 MHz, CD3CN) δ 7.38–7.27 (m, 3H), 7.21–7.10 (m, 2H), 5.38–5.14 (brs, 1H), 4.17–4.06 (m, 2H), 1.29 (s, 9H); 13C{1H} NMR (100 MHz, CD3CN) δ 161.9 (d, J = 36.7 Hz), 157.0, 145.7 (d, J = 255.3 Hz), 136.0 (d, J = 5.2 Hz), 134.0 (d, J = 14.1 Hz), 129.9 (d, J = 2.8 Hz), 129.23, 129.19, 80.0, 41.4 (d, J = 9.1 Hz), 28.9; 19F NMR (376.5 MHz, CD3CN) δ −123.5 (s). HRMS (DART) calcd for C15H17O4NF [M-H]–: 294.1147, found 294.1147.
tert-Butyl (Z)-(3-fluoro-2-phenylallyl)carbamate (40).
A reaction flask was charged with 39 (2.0 g, 6.77 mmol, 1 equiv, >98:2 dr), AgOAc (113.0 mg, 0.68 mmol, 10 mol%), and NMP (4.0 mL, 2V). The flask was inerted and placed into preheated to 130 °C oil bath. The reaction mixture was stirred for 3 h under positive Ar pressure. After this time the reaction mixture was cooled to rt Celite was added, and the mixture was diluted with EtOAc (20 mL). The mixture was filtered, and the filter pad was washed with EtOAc. Combined filtrates were washed twice with water (10 mL). The organic phase was separated, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography eluting with 0–1% EtOAc in CH2Cl2 to afford 40 as a beige solid (1.59 g, 93% yield, > 98:2 dr). mp 78.4–80.8 °C; 1H NMR (400 MHz, CDCl3) δ 7.57–7.07 (m, 5H), 6.84 (d, JHF = 83.7 Hz), 4.83–4.37 (brs, 1H), 4.37–4.06 (m, 2H), 1.40 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) 155.6, 147.4 (d, J = 264.2 Hz), 134.1 (d, J = 8.0 Hz), 128.7, 127.9, 126.9, 122.5 (d, J = 7.1 Hz), 79.4, 36.3 (d, J = 5.1 Hz), 28.3; 19F NMR (376.5 MHz, CDCl3) δ −128.4 (dt, JHF = 83.7 Hz, JHF = 2.5 Hz). NMR spectra matched reported data.11a HRMS (EI) calcd for C10H10O2NF [M-C4H8]+: 195.0690, found 195.0692.
(Z)-3-Fluoro-2-phenylprop-2-en-1-amine hydrochloride (2)6b.
To a solution of 40 (851 mg, 3.39 mmol, 1 equiv) in MeOH (5.00 mL) was added HCl (4.0 M in dioxane, 4.2 mL; 16.93 mmol, 5 equiv), and the mixture was stirred at rt overnight. Reaction mixture was concentrated under reduced pressure and the residue was re-dissolved in EtOH (3 mL). MTBE (13 mL) was added dropwise over 30 min, and the slurry was stirred for 1 h. The slurry was filtered, and the filter cake was washed with MTBE. The main crop was obtained as a white solid (482.2 mg). The mother liquor was concentrated under reduced pressure to a minimal volume and the residue was diluted with MTBE. The slurry was stirred for 30 min and then filtered. The second crop was obtained as a white solid (92.4 mg). Combined weight of 2: 575.2 mg, 91% yield, > 98:2 dr. mp 160.6–162.0 °C; 1H NMR (400 MHz, MeOH-d4) δ 7.51–7.35 (m, 5H), 7.20 (d, JHF = 82.5 Hz), 5.08–4.58 (brs, 3H), 4.13 (dd, JHF = 2.9 Hz, J = 0.7 Hz, 2H); 13C{1H} NMR (100 MHz, MeOH-d4) 151.6 (d, J = 268.6 Hz), 133.7 (d, J = 7.1 Hz), 130.4, 130.0, 128.3 (d, J = 3.1 Hz), 119.9 (d, J = 6.5 Hz), 36.2 (d, J = 7.6 Hz); 19F NMR (376.5 MHz, MeOH-d4) δ −124.0 (d, JHF = 82.5 Hz). HRMS (EI) calcd for C9H9NF [M-H]+: 150.0714, found 150.0714.
(3aR,5S,6aR)-2,2-Dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carbaldehyde (42).
To a solution of diol 41 (446.0 mg, 2.18 mmol, 1 equiv) in EtOH (5.00 mL) and H2O (2.50 mL) was added NaIO4 (583.9 mg, 2.73 mmol, 1.25 equiv) at 0 °C. The reaction mixture was warmed to rt and stirred for 1 h. The mixture was filtered through a plug of celite and the celite plug was washed with EtOH. The filtrate was concentrated under reduced pressure to remove EtOH and water. The residue was chased twice with anhydrous THF. The crude product was suspended in anhydrous THF and was filtered through a plug of celite. The celite plug was washed with THF. Combined filtrates were passed through a 0.45 micron filter and concentrated under reduced pressure to a minimal volume. The crude product solution (a mixture of aldehyde 42 and its ethyl hemiacetal) was used in the next step without further purification.
Ethyl (E)-3-((3aR,5S,6aR)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)-2-fluoroacrylate (43).
A round bottom flask was charged with triethyl 2-fluoro-2-phosphonoacetate (1.19 mL, 5.89 mmol, 2.70 equiv) and THF (10 mL). The solution was cooled to −78 °C. n-BuLi (2.47 mL, 5.89 mmol, 2.39 M in hexanes, 2.70 equiv) was added dropwise. The reaction mixture was warmed to 0 °C and stirred for 10 min, then cooled to −78 °C. A solution of aldehyde 42 (~376 mg, ~2.18 mmol, 1.0 equiv), which was prepared as described above, was diluted with THF (2.0 mL) and added dropwise at −78 °C. The reaction mixture was linearly warmed to 0 °C over 1 h. The mixture was quenched with 10 wt.% aq. NH4Cl solution (20 mL) and extracted with EtOAc (2 × 20 mL). Organic phase was dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was analyzed by quantitative 1H NMR: mixture of stereoisomers in 12.8:1 (93:7) dr. The residue was purified by silica gel column chromatography eluting with 15–18% EtOAc in hexanes to afford 43 as a colorless oil (481.9 mg, 85% yield, mixture of stereoisomers in 12.0:1 (92:8) dr (from integration of quantitative 1H NMR spectrum. Note: the ratio of stereoisomers by integration of quantitative 19F NMR spectrum is higher due to an overlapping signal of an impurity)). 1H NMR (400 MHz, CDCl3) δ 5.90 (dd, JHF = 19.4 Hz, J = 8.1 Hz, 1H), 5.83 (d, J = 3.8 Hz, 1H), 5.48 (ddd, J = 10.9 Hz, J = 8.1 Hz, J = 4.5 Hz, 1H), 4.76–4.72 (m, 1H), 4.29 (dq, J = 7.1 Hz, J = 0.5 Hz, 2H), 2.34 (ddd, J = 13.3 Hz, J = 4.5 Hz, J = 4.5 Hz, 1H), 1.58 (ddd, J = 13.3 Hz, J = 10.9 Hz, J = 4.7 Hz, 1H), 1.51 (s, 3H), 1.33 (t, J = 7.1 Hz, 3H), 1.30 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) 160.1 (d, J = 35.6 Hz), 147.6 (d, J = 258.2 Hz), 121.8 (d, J = 20.6 Hz), 111.3, 105.5, 80.4, 72.3 (d, J = 8.8 Hz), 61.9, 39.1 (d, J = 2.9 Hz), 26.7, 26.1, 14.0; 19F NMR (376.5 MHz, CDCl3) δ −119.6 (d, JHF = 19.4 Hz), Z-isomer: −124.5 (d, JHF = 33.1 Hz). HRMS (DART) calcd for C12H18O5F [M+H]+: 261.1133, found 261.1134.
(E)-3-((3aR,5S,6aR)-2,2-Dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)-2-fluoroacrylic acid (44).
To a solution of ethyl ester 43 (890.6 mg, 3.42 mmol; 1 equiv, mixture of stereoisomers in 12.0:1 (92:8) dr) in EtOH (10 mL) was added a slurry of LiOH•H2O (315.9 mg; 7.53 mmol; 2.20 equiv) in water (5.0 mL) at 0 °C. The reaction mixture was warmed to r.t and stirred overnight. The reaction mixture was diluted with water (10.0 mL) and concentrated under reduced pressure to remove almost all EtOH. It was then extracted with MTBE (10 mL) and diluted with brine (20.0 mL). The mixture was cooled to 10 °C and conc. HCl (620 µL; 7.53 mmol; 2.20 equiv) was added dropwise. The mixture was extracted twice with EtOAc (20 mL). EtOAc extracts were dried over Na2SO4 and filtered. The filtrate was concentrated under reduced pressure to afford 44 as a colorless viscous oil. The oil solidified after standing overnight in a freezer. The solid was dried under vacuum for 2 h (775.5 mg, 98% yield, mixture of stereoisomers in 15.4:1 (94:6) dr (from integration of quantitative 1H NMR spectrum. Note: the ratio of stereoisomers by integration of quantitative 19F NMR spectrum is higher due to an overlapping signal of an impurity)). 1H NMR (400 MHz, CDCl3): δ 11.05–10.51 (brs, 1H), 6.03 (dd, JHF = 19.1 Hz, J = 8.1 Hz, 1H), 5.86 (d, J = 3.7 Hz, 1H), 5.50 (ddd, J = 10.9 Hz, J = 8.1 Hz, J = 4.5 Hz, 1H), 4.82–4.74 (m, 1H), 2.38 (ddd, J = 13.4 Hz, J = 4.5 Hz, J = 4.5 Hz, 1H), 1.62 (ddd, J = 13.4 Hz, J = 10.9 Hz, J = 4.7 Hz, 1H), 1.51 (s, 3H), 1.32 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) 163.9 (d, J = 36.4 Hz), 147.1 (d, J = 256.6 Hz), 123.7 (d, J = 20.6 Hz), 111.7, 105.5, 80.5, 72.4 (d, J = 8.6 Hz), 38.9 (d, J = 2.7 Hz), 26.6, 26.1; 19F NMR (376.5 MHz, CDCl3) δ −119.1 (d, JHF = 19.1 Hz), Z-isomer: −125.1 (d, JHF = 32.7 Hz). HRMS (DART) calcd for C10H14O5F [M+H]+: 233.0820, found 233.0820.
(3aR,5S,6aR)-5-((E)-2-Fluorovinyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole (45).
A reaction vial was charged with acid 44 (250.0 mg, 1.08 mmol, 1 equiv, mixture of stereoisomers in 15.4:1 (94:6) dr), AgOAc (89.9 mg, 0.54 mmol, 50 mol%), and NMP (0.50 mL, 2V). The vial was inerted and placed into preheated to 140 °C oil bath. The reaction mixture was stirred for 3 h under positive Ar pressure. It was then cooled to 0 °C and PhCF3 (53.9 mg) was added as an internal standard. The mixture was diluted with CDCl3 and analyzed by quantitative 19F NMR: m(prod) = 178.2 mg, 88% NMR yield, mixture of stereoisomers in 15.3:1 (94:6) dr). The reaction mixture was directly loaded on a silica gel column using a minimum amount of CH2Cl2. It was purified by column chromatography eluting with 20–30% MTBE in hexanes to afford two fractions: major less polar isomer (white foam, 160.2 mg, 79% yield, >98:2 dr), minor isomer (white foam, 8.9 mg, 4.4% yield, >98:2 dr). Analytical data of the major isomer: 1H NMR (400 MHz, CDCl3) δ 6.74 (ddd, JHF = 82.7 Hz, J = 11.1 Hz, J = 0.8 Hz, 1H), 5.79 (d, J = 3.7 Hz, 1H), 5.36 (ddd, JHF = 16.9 Hz, J = 11.1 Hz, J = 8.2 Hz, 1H), 4.75–4.69 (m, 1H), 4.57 (ddd, J = 10.9 Hz, J = 8.2 Hz, J = 4.3 Hz, 1H), 2.14 (ddd, J = 13.4 Hz, J = 4.3 Hz, J = 4.3 Hz, 1H), 1.60 (ddd, J = 13.4 Hz, J = 10.9 Hz, J = 4.7 Hz, 1H), 1.49 (s, 3H), 1.29 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) 151.7 (d, J = 260.9 Hz), 111.0, 110.8 (d, J = 11.9 Hz), 105.1, 80.3, 73.0 (d, J = 14.4 Hz), 39.8 (d, J = 2.3 Hz), 26.5, 25.9; 19F NMR (376.5 MHz, CDCl3) δ −125.7 (dd, JHF = 82.7 Hz, JHF = 16.9 Hz). NMR spectra matched reported data.6e.9b HRMS (EI) calcd for C8H10O3F [M-CH3]+: 173.0608, found 173.0609.
Supplementary Material
Acknowledgements
M.C.K. thanks the NIH (R35 GM131902) for financial support and XSEDE (TG-CHE120052) for computational support.
Footnotes
ASSOCIATED CONTENT
Supporting Information. 1H, 19F, and 13C NMR spectra of all compounds (PDF). Crystallographic data for Z-12b and E-16b (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
CCDC 2250577 and 2251003 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interests.
Data Availability Statement.
The data underlying this study are available in the published article and its Supporting Information.
REFERENCES
- 1.(a) Hiyama T in Organofluorine Compounds: Chemistry and Applications (Ed.: Yamamoto H), Springer: New York, 2000, and references cited therein. [Google Scholar]; (b) O’Hagan D Understanding Organofluorine Chemistry. An Introduction to the C–F Bond. Chem. Soc. Rev 2008, 37, 308–319. [DOI] [PubMed] [Google Scholar]
- 2.(a) Wilkinson JA Recent Advances in the Selective Formation of the Carbon-Fluorine Bond. Chem. Rev 1992, 92, 505–519. [Google Scholar]; (b) Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications (Ed.: Filler R; Kobayashi Y; Yagupolskii LM), Elsevier: Amsterdam, 1993. [Google Scholar]; (c) Purser S; Moore P; Sawllow S; Gouverneur V Fluorine in Medicinal Chemistry. Chem. Soc. Rev 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]; (d) Hagmann WK The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem 2008, 51, 4359–4369. [DOI] [PubMed] [Google Scholar]; (e) Wang J; Sánchez-Roselló M; Aceña JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev 2014, 114, 2432–2506. [DOI] [PubMed] [Google Scholar]; (f) Zhou Y; Wang J; Gu Z; Wang S; Zhu W; Aceña JL; Soloshonok VA; Izawa K; Liu H Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev 2016, 116, 422–518. [DOI] [PubMed] [Google Scholar]
- 3.(a) Jeschke P The Unique Role of Fluorine in the Design of Active Ingredients for Modern Crop Protection. ChemBioChem 2004, 5, 570–589. [DOI] [PubMed] [Google Scholar]; (b) Jeschke P The Unique Role of Halogen Substituents in the Design of Modern Agrochemicals. Pest Manage. Sci 2010, 66, 10–27. [DOI] [PubMed] [Google Scholar]; (c) Jeschke P in Modern Methods in Crop Protection Research (Eds.: Jeschke P; Kraemer W; Schirmer U; Witschel M), Wiley-VCH: Weinheim, 2012, pp. 73–128. [Google Scholar]; (d) Fujiwara T; O’Hagan D Successful Fluorine-Containing Herbicide Agrochemicals. J. Fluorine Chem 2014, 167, 16–29. [Google Scholar]
- 4.(a) Ameduri B; Boutevin B Copolymerization of Fluorinated Monomers: Recent Developments and Future Trends. J. Fluorine Chem 2000, 104, 53–62. [Google Scholar]; (b) Berger R; Resnati G; Metrangolo P; Weber E; Hulliger J Organic Fluorine Compounds: A Great Opportunity for Enhanced Materials Properties. Chem. Soc. Rev 2011, 40, 3496–3508. [DOI] [PubMed] [Google Scholar]; (c) Vincent J-M Recent Advances of Fluorous Chemistry in Material Sciences. Chem. Commun 2012, 48, 11382–11391. [DOI] [PubMed] [Google Scholar]
- 5.(a) van Steenis JH; van der Gen A Synthesis of Terminal Monofluoro Olefins. J. Chem. Soc., Perkin Trans 1 2002, 2117–2133. [Google Scholar]; (b) Landelle G; Bergeron M; Turcotte-Savard M-O; Paquin J-F Synthetic Approaches to Monofluoroalkenes. Chem. Soc. Rev 2011, 40, 2867–2908. [DOI] [PubMed] [Google Scholar]; (c) Yanai H; Taguchi T Synthetic Methods for Fluorinated Olefins. Eur. J. Org. Chem 2011, 30, 5939–5954. [Google Scholar]; (d) Champagne PA; Desroches J; Paquin J-F Monofluorination of Organic Compounds: 10 Years of Innovation. Chem. Rev 2015, 115, 9073–9174. [DOI] [PubMed] [Google Scholar]; (e) Drouin M; Hamel J-D; Paquin J-F Synthesis of Monofluoroalkenes: A Leap Forward. Synthesis 2018, 50, 881–955. [Google Scholar]
- 6.(a) McCarthy JR; Matthews DP; Stemerick DM; Huber EW; Bey P; Lippert BJ; Snyder RD; Sunkara PS Stereospecific Method to (E) and (Z) Terminal Fluoroolefins and its Application to the Synthesis of 2’-Deoxy-2’-Fluoromethylenenucleosides as Potential Inhibitors of Ribonucleoside Diphosphate Reductase. J. Am. Chem. Soc 1991, 113, 7439–7440. [Google Scholar]; (b) Kim J; Zhang Y; Ran C; Sayre LM Inactivation of Bovine Plasma Amine Oxidase by Haloallylamines. Bioorg. Med. Chem 2006, 14, 1444–1453. [DOI] [PubMed] [Google Scholar]; (c) Miller SC; McCarthy JR; Sabol JS Terminal Fluoroolefins. The Synthesis of Novel Carboacyclic Nucleosides. Nucleosides Nucleotides, 1998, 17, 1099–1113. [DOI] [PubMed] [Google Scholar]; (d) Asahina Y; Iwase K; Iinuma F; Hosaka M; Ishizaki T Synthesis and Antibacterial Activity of 1-(2-Fluorovinyl)-7-substituted-4-quinolone-3-carboxylic Acid Derivatives, Conformationally Restricted Analogues of Fleroxacin. J. Med. Chem 2005, 48, 3194–3202. [DOI] [PubMed] [Google Scholar]; (e) Wnuk SF; Lalama J; Garmendia CA; Robert J; Zhu J; Pei D S-Ribosylhomocysteine Analogues with the Carbon-5 and Sulfur Atoms Replaced by a Vinyl or (Fluoro)vinyl Unit. Bioorg. Med. Chem 2008, 16, 5090–5102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kolb M; Barth J; Heydt J-G; Jung MJ Synthesis and Evaluation of Mono-, Di-, and Trifluoroethenyl-GABA Derivatives as GABA-T Inhibitors. J. Med. Chem 1987, 30, 267–272. [DOI] [PubMed] [Google Scholar]
- 7.Schlosser M; Zimmermann M Fluor-olefine durch Fluormethylenierung von Carbonylverbindungen. Synthesis 1969, 75–76. [Google Scholar]
- 8.(a) Cox DG; Gurusamy N; Burton DJ Surprising Stereochemical Control of Wittig Olefination Involving Reaction of Fluorine-containing Phosphoranium Salt and Aldehydes J. Am. Chem. Soc 1985, 107, 2811–2812. [Google Scholar]; (b) Tsai H-J Synthesis of Phenyl Substituted Fluoro-olefins. Tetrahedron Lett 1996, 37, 629–632. [Google Scholar]
- 9.(a) Koh MJ; Nguyen TT; Zhang H; Schrock RR; Hoveyda AH Direct Synthesis of Z-Alkenyl Halides Through Catalytic Cross-Metathesis. Nature 2016, 531, 459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nguyen TT; Koh MJ; Shen X; Romiti F; Schrock RR; Hoveyda AH Kinetically Controlled E-Selective Catalytic Olefin Metathesis. Science 2016, 352, 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Kojima R; Kubota K; Ito H Stereodivergent Hydrodefluorination of gem-Difluoroalkenes: Selective Synthesis of (Z)- and (E)-Monofluoroalkenes. Chem. Commun 2017, 53, 10688–10691. [DOI] [PubMed] [Google Scholar]; (b) Hu J; Han X; Yuan Y; Shi Z Stereoselective Synthesis of Z Fluoroalkenes through Copper-Catalyzed Hydrodefluorination of gem-Difluoroalkenes with Water. Angew. Chem. Int. Ed 2017, 56, 13342–13346. [DOI] [PubMed] [Google Scholar]
- 11.(a) Liu Q; Shen X; Ni C; Hu. J. Stereoselective Carbonyl Olefination with Fluorosulfoximines: Facile Access to Z or E Terminal Monofluoroalkenes. Angew. Chem. Int. Ed 2017, 56, 619–623. [DOI] [PubMed] [Google Scholar]; (b) For the first 3 steps in the synthesis of reagent 7, see: Mancheno OG; Bistri O; Bolm C Iodinane- and Metal-Free Synthesis of N-Cyano Sulfilimines: Novel and Easy Access of NH-Sulfoximines. Org. Lett 2007, 9, 3809–3811. Steps 4–5 can be found in the Supporting Information of ref. 11a. [DOI] [PubMed] [Google Scholar]
- 12.Machleidt H; Wessendorf R Organische Fluorverbindungen, IV. Carbonyl Fluorolefinierungen. Justus Liebigs Ann. Chem 1964, 674, 1–10. [Google Scholar]
- 13.:(a) Volchkov IN; Reeves JT; Dang MTQ; Hampel TA; Koch S; Nordstrom FL; Reichel C; Schoerer M; Stange C; Zhong L; Zimmermann UJ A Process for the Preparation of Halo Olefin Derivatives. Pct. Int. Appl. WO/2018/158140, 2018.(b) Dang MTQ; Hampel TA; Koch S; Nordstrom FL; Reeves JT; Reichel C; Schoerer M; Stange C; Volchkov IN; Zhong L; Zimmermann UJ Transition Metal-Catalyzed Protodecarboxylation of α-Halo-Acrylic Acid Derivatives. US 10,252,994, 2019.(c) Dang MTQ; Hampel TA; Koch S; Nordstrom FL; Reeves JT; Reichel C; Schoerer M; Stange C; Volchkov IN; Zhong L; Zimmermann UJ Transition Metal-Catalyzed Protodecarboxylation of α-Halo-Acrylic Acid Derivatives. US 10,654,803, 2020.(d) Dang MTQ; Hampel TA; Koch S; Nordstrom FL; Reeves JT; Reichel C; Schoerer M; Stange C; Volchkov IN; Zhong L; Zimmermann UJ Transition Metal-Catalyzed Protodecarboxylation of α-Halo-Acrylic Acid Derivatives. US 11,046,648, 2021.
- 14.(a) Sano S; Ando T; Yokoyama K; Nagao Y New Reaction Mode of the Horner-Wadsworth-Emmons Reaction for the Preparation of α-Fluoro-α,β-unsaturated Esters. Synlett, 1998, 777–779. [Google Scholar]; (b) Sano S; Teranishi R; Nagao Y Toward (Z)-Selective Horner–Wadsworth–Emmons Reaction of Aldehydes with 2-Fluoro-2-Diethylphosphonoacetic Acid. Tetrahedron Lett 2002, 43, 9183–9186. [Google Scholar]; (c) Park J-H; Lee YR; Chun MW; Jeong LS; Lee C-K; Kim H-D Synthesis of (Z)-(1-Fluoro-2-Hydroxymethylcyclopropylmethyl)purines. Nucleosides Nucleotides, 2003, 22, 659–661. [DOI] [PubMed] [Google Scholar]; (d) Sano S; Matsumoto T; Nanataki H; Tempaku S; Nakao M Z-Selective Horner–Wadsworth–Emmons Reaction of 2-TOM-Cyclopentanone for the Synthesis of rac-N-Cbz-Gly-Ψ[(Z)-CF=C]-Pro-OH Dipeptide Isostere. Tetrahedron Lett 2014, 55, 6248–6251. [Google Scholar]
- 15.Claridge TDW; Davies SG; Lee JA; Nicholson RL; Roberts PM; Russell AJ; Smith AD; Toms SM Highly (E)-Selective Wadsworth−Emmons Reactions Promoted by Methylmagnesium Bromide. Org. Lett 2008, 10, 5437–5440. [DOI] [PubMed] [Google Scholar]
- 16. The olefination using stoichiometric MgBr2 with DBU or Et3N as base gave low conversion and low selectivity. [Google Scholar]
- 17.Etemad-Moghadam G; Seyden-Penne J Synthese stereoselective d’esters α,β−3-ethyleniques α-fluores E par reaction de Wittig-Horner a partir du diethyl phosphono α-fluoroacetate de methyle etude comparative avec le diphenyl phosphonoxy α-fluoroacetate de methyle. Bull. Soc. Chim. Fr 1985, 3, 448–454. [Google Scholar]
- 18.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam NJ; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas O; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision D.01; Gaussian, Inc., Wallingford, CT, 2013. [Google Scholar]
- 19.Grimme S; Antony J; Ehrlich S; Krieg H A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 20.Marenich AV; Cramer CJ; Truhlar DG Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- 21.(a) Maryanoff BE; Reitz AB The Wittig Olefination Reaction and Modifications Involving Phosphoryl-Stabilized Carbanions. Stereochemistry, Mechanism, and Selected Synthetic Aspects. Chem. Rev 1989, 89, 863–927. [Google Scholar]; (b) Boutagy J; Thomas R Olefin Synthesis with Organic Phosphonate Carbanions. Chem. Rev 1974, 74, 87–99. [Google Scholar]
- 22.Selectivity of >98:2 refers to the sensitivity limit of quantitative 19F NMR. When we list an isomeric ratio of >98:2 it means only one isomer was detected [Google Scholar]
- 23.(a) Elkik E Acides α-fluorocinnamiques et β-fluorostyrenes. Bull. Soc. Chim. Fr 1967, 5, 1569–1571. [Google Scholar]; (b) Elkik E; Francesch C Esters α-fluorocinnamiques et β-fluorostyrenes. Etude par RMN des isomeres cis et trans. Bull. Soc. Chim. Fr 1968, 4, 1371–1374. [Google Scholar]
- 24.Rousée K; Schneider C; Couve-Bonnaire S; Pannecoucke X; Levacher V; Hoarau C Pd- and Cu-Catalyzed Stereo- and Regiocontrolled Decarboxylative/C-H Fluoroalkenylation of Heteroarenes. Chem. Eur. J 2014, 20, 15000–15004. [DOI] [PubMed] [Google Scholar]
- 25.During the preparation of this manuscript, Tan and co-workers published the decarboxylation of fluoroacrylic acids using Ag2CO3 as catalyst in NMP: Liu X; Shi F; Jin C; Liu B; Lei M; Tan J Stereospecific Synthesis of Monofluoroalkenes and Their Deuterated Analogues via Ag-Catalyzed Decarboxylation. J. Catal 2022, 413, 1089–1097. [Google Scholar]
- 26.(a) Cornella J; Sanchez C; Banawa D; Larrosa I Silver-Catalysed Protodecarboxylation of ortho-Substituted Benzoic Acids. Chem. Commun 2009, 46, 7176–7178. [DOI] [PubMed] [Google Scholar]; (b) Lu P; Sanchez C; Cornella J; Larrosa I Silver-Catalyzed Protodecarboxylation of Heteroaromatic Carboxylic Acids. Org. Lett 2009, 11, 5710–5713. [DOI] [PubMed] [Google Scholar]; c The mechanism of the α-fluoroacrylic acid decarboxylation with Ag salts has been studied computationally; see reference 25 [Google Scholar]
- 27.Spiegel D The total synthesis of glucosepane and compounds obtained therefrom. Pct. Int. Appl WO/2017/053776A1, 2017. [Google Scholar]
- 28.Qian J; Yi W; Lv M; Cai C A Facile and Mild Approach for Stereoselective Synthesis of α-Fluoro-α,β-unsaturated Esters from α-Fluoro-β-keto Esters via Deacylation. Synlett 2015, 26, 127–132. [Google Scholar]
- 29.Ando K; Isomura W; Uchida N; Mori K Highly E-Selective Solvent-Free Horner-Wadsworth-Emmons Reaction for the Synthesis of α-Methyl-α,β-Unsaturated Esters Using Either LiOH·H2O or Ba(OH)2·8H2O. Bull. Chem. Soc. Jpn 2022, 95, 928–934. [Google Scholar]
- 30.Lemonnier G; Zoute L; Dupas G; Quirion J-C; Jubault P Diethylzinc-Mediated One-Step Stereoselective Synthesis of α-Fluoroacrylates from Aldehydes and Ketones. Two Different Pathways Depending on the Carbonyl Partner. J. Org. Chem 2009, 74, 4124–4131. [DOI] [PubMed] [Google Scholar]
- 31.Lisboa MP; Hoang TT; Dudley GB Tandem Nucleophilic Addition/Fragmentation of vinylogous Acyl Triflates: 2-Methyl-2-(1-Oxo-5-Heptynyl)-1,3-Dithiane. Org. Synth 2011, 88, 353–363. [Google Scholar]
- 32.Malapit CA; Caldwell DR; Luvaga IK; Reeves JT; Volchkov I; Gonnella NC; Han ZS; Busacca CA; Howell AR; Senanayake CH Rhodium-Catalyzed Addition of Aryl Boronic Acids to 2,2-Disubstituted Malononitriles. Angew. Chem. Int. Ed 2017, 56, 6999–7002. [DOI] [PubMed] [Google Scholar]
- 33.Sano S; Kuroda Y; Saito K; Ose Y; Nagao Y Tandem Reduction–Olefination of Triethyl 2-Acyl-2-Fluoro-2-Phosphonoacetates and a Synthetic Approach to Cbz-Gly-Ψ[(Z)-CF=C]-Gly Dipeptide Isostere. Tetrahedron 2006, 62, 11881–11890. [Google Scholar]
- 34.Xie S-L; Cui X-Y; Gao X-T; Zhou F; Wu H-H; Zhou J Stereoselective Defluorinative Carboxylation of gem-Difluoroalkenes with Carbon Dioxide. Org. Chem. Front 2019, 6, 3678–3682. [Google Scholar]
- 35.Poutrel P; Pannecoucke X; Jubault P; Poisson T Stereoselective Synthesis of Terminal Monofluoroalkenes from Trifluoromethylated Alkenes. Org. Lett 2020, 22, 4858–4863. [DOI] [PubMed] [Google Scholar]
- 36.Zhang J; Yang J-D; Cheng J-P Chemoselective Catalytic Hydrodefluorination of Trifluoromethylalkenes Towards mono-/gem-di-fluoroalkenes Under Metal-free Conditions. Nat. Commun 2021, 12, 2835–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang H; Zhou C-B; Chen Q-Y; Xiao J-C; Hong R Monofluorovinyl Tosylate: A Useful Building Block for the Synthesis of Terminal Vinyl Monofluorides via Suzuki−Miyaura Coupling. Org. Lett 2011, 13, 560–563. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.