

Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder influenced by a complex interplay of environmental, epigenetic, and genetic factors. DNA methylation (5mC) and hydroxymethylation (5hmC) are DNA modifications that serve as tissue-specific and temporal regulators of gene expression. TET family enzymes dynamically regulate these epigenetic modifications in response to environmental conditions, connecting environmental factors with gene expression. Previous epigenetic studies have identified 5mC and 5hmC changes associated with AD. In this study, we performed targeted resequencing of TET1 on a cohort of early-onset AD (EOAD) and control samples. Through gene-wise burden analysis, we observed significant enrichment of rare TET1 variants associated with AD (p = 0.04). We also profiled 5hmC in human postmortem brain tissues from AD and control groups. Our analysis identified differentially hydroxymethylated regions (DhMRs) in key genes responsible for regulating the methylome: TET3, DNMT3L, DNMT3A, and MECP2. To further investigate the role of Tet1 in AD pathogenesis, we used the 5xFAD mouse model with a Tet1 KO allele to examine how Tet1 loss influences AD pathogenesis. We observed significant changes in neuropathology, 5hmC, and RNA expression associated with Tet1 loss, while the behavioral alterations were not significant. The loss of Tet1 significantly increased amyloid plaque burden in the 5xFAD mouse (p = 0.044) and lead to a non-significant trend towards exacerbated AD-associated stress response in 5xFAD mice. At the molecular level, we found significant DhMRs enriched in genes involved in pathways responsible for neuronal projection organization, dendritic spine development and organization, and myelin assembly. RNA-Seq analysis revealed a significant increase in the expression of AD-associated genes such as Mpeg1, Ctsd, and Trem2. In conclusion, our results suggest that TET enzymes, particularly TET1, which regulate the methylome, may contribute to AD pathogenesis, as the loss of TET function increases AD-associated pathology.
Keywords: 5-hydroxymethylcytosine, Alzheimer’s disease, Tet1 protein, Gene expression
1. Introduction
Alzheimer’s disease (AD) is a fatal neurological disorder that is the primary cause of dementia and the 7th leading cause of death in the US. There is currently no cure for AD and the underlying causes of the disease remain to be elucidated. AD is a complex disorder characterized by the progressive deterioration of neuronal connections and the eventual death of neurons. Many cell types within the brain promote proper neuronal health and function and can contribute to AD pathogenicity when dysregulated. Based on the age of onset, there are two general classifications for Alzheimer’s disease; late onset Alzheimer’s disease (LOAD, >65 years of age) and early onset Alzheimer’s disease (EOAD, <65 years of age), which is also commonly referred to as young-onset Alzheimer’s disease. LOAD is highly polygenic whereas EOAD is not, and up to 10% of EOAD appears to be inherited in an autosomal dominant fashion (Wingo et al., 2012). Among families with this pattern of inheritance, mutations in APP, PSEN1, and PSEN2 make up the known genetic causes for AD. Mutations in these genes lead to the improper processing of the APP proteins, leading to an increase in the production of toxic Aβ42 monomers and amyloid plaque pathology (Ayodele et al., 2021; Cacace et al., 2016; Mendez, 2017). The causes of SAD are much more uncertain. The strongest known genetic factor contributing to SAD development is the presence of APOE-ε4 alleles (Jansen et al., 2015; Knopman et al., 2021; Serrano-Pozo et al., 2021). The presence of one APOE-ε4 allele only confers a threefold increased risk while two alleles confer an 8-fold risk of developing AD (Fernandez et al., 2019). Within the two classifications of FAD and SAD, there are three physiological components linked to AD: Tau neurofibrillary tangles, amyloid beta (Aβ) plaques, and neuroinflammation, where Aβ plaque development is the first pathology to develop (Dorszewska et al., 2016; Lanoiselée et al., 2017). These pathologies are intertwined, with alterations in one pathology influencing the others (Akiyama et al., 2000; Heneka et al., 2015; Knopman et al., 2021). Prior research has characterized genetic variants associated with AD; however, few variants have been discovered and only explain a proportion of AD pathogenicity, hinting at the involvement of additional factors (Cacace et al., 2016; Dorszewska et al., 2016; Lanoiselée et al., 2017).
Through their functions as genomic regulators, epigenetic modifications work as mediators of gene and environment interactions. These modifications are dynamic and environmental changes often lead to alterations in the abundance and location of modifications, which in turn can impart changes in gene expression through several different mechanisms (Angeloni and Bogdanovic, 2019; Lawrence et al., 2016; Zeng and Chen, 2019). Thus, epigenetic modifications are important governors of spatiotemporal and cell-specific gene expression (Armstrong et al., 2019). Methylation at a cytosine residue, 5-methylcytosine (5mC), is the most common DNA modification and has been linked to transcriptional repression when located at promoters but is not repressive when found in the gene body (Schmitz et al., 2019). 5mC is first placed onto DNA by the DNA methyltransferases DNMT1, DNMT3A, DNMT3B, and DNMT3L. This modification can be oxidized by the ten-eleven translocation (TET) family of proteins including TET1, TET2, and TET3, producing the dynamic 5-hydroxymethylcytosine (5hmC) modification (Kohli and Zhang, 2013; Liu et al., 2021; Rasmussen and Helin, 2016; Wu and Zhang, 2017). The balance of genome-wide 5mC/5hmC modifications is established and maintained by DNMT and TET proteins, and as individuals age, a global decrease in 5mC and an increase in 5hmC occurs in the brain.
In addition to their function as DNA methylation and hydroxymethylation regulators, TET proteins also function through their direct interaction with other proteins, including transcription factors (TF). Prior studies have isolated many proteins directly interact with and or complex with TET proteins. These studies have unearthed a plethora of Tet1 and protein binding activity, including direct interactions between Tet1, chromatin remodeling complexes, and transcription factors (e.g., HDAC1/6/7, OLIG2, CDC5L, SMAD3, EZH2. LSD1, OGT, SIN3A, HIF1α, MECP2, PRC2, NANOG, and LIN28A (Cartron et al., 2013; Cheng et al., 2018; Costa et al., 2013; Neri et al., 2013; Okashita et al., 2014; Stolz et al., 2022; Zeng et al., 2016; Zhang et al., 2020a; Zheng et al., 2016).
Our lab and others have shown that 5hmC is altered in AD mouse models and human post-mortem brain samples, and that the genes with altered 5hmC could modulate Tau-mediated neuronal toxicity (Al-Mahdawi et al., 2014; Cadena-del-Castillo et al., 2014; Coppieters et al., 2014; Pellegrini et al., 2021; van den Hove et al., 2012; Zhang et al., 2020b). Data from human AD patients show a global increase of 5hmC in the middle frontal gyrus (MFG) and the middle temporal gyrus (MTG) and a positive correlation between 5hmC abundance and the severity of AB, tau, and ubiquitin levels. In mouse models of AD, the perturbation of 5hmC profiles is tissue-specific, with cell types such as neurons losing 5hmC with no apparent change in astrocytes (Coppieters et al., 2014). Furthermore, in the 3xTg AD mouse model, overexpression of human TET catalytic domains reduces Aβ accumulation, tau hyperphosphorylation, and synaptic dysfunction (Zhang et al., 2020b). Additionally, targeted re-sequencing data from young-onset dementia cases and healthy control samples have found an enrichment of rare TET2 loss of function (LoF) variants predominantly located within functional regions and additional mutants located in regulatory in the AD relative to controls (Cochran et al., 2020). Taken together, these data support the idea that the methylome is dynamically regulated and perturbations to the methylome may explain a portion of the missing pathogenicity seen in AD. As the hydroxymethylome regulates chromatin accessibility and gene expression, aberrant alterations in the 5hmC landscape are associated with AD, and the TET enzymes responsible for regulating the hydroxymethylome have been implicated in AD, it is critical to investigate the regulation, location, and role of TET1 and 5hmC in AD pathology.
Prior studies largely focused on global levels of 5hmC modifications in the brain and few have explored the relationship between the knockout of TET enzymes and AD-associated outcomes. Due to the dynamic regulation of 5mC and 5hmC modifications by TET family enzymes, we are interested in how these enzymes, specifically TET1, regulate the methylome and how manipulation of TET1 alters the methylome, gene expression, and AD pathogenicity. Here, we explore the association between TET1 variants and AD in humans, as well as looking within individuals with AD to examine how RNA expression and 5hmC landscapes differ between individuals that contain a large number of predicted AD-associated CpG sites with those that contain few. Additionally, we explore whether the partial loss of TET1 alters the transcriptome, hydroxymethylome, behavior, and neuropathology of the 5xFAD mouse model.
2. Experimental procedures
2.1. Study populations
Samples were collected from two distinct human study groups for this project. Individuals from both populations were recruited from U.S. ADRCs or ADCs using the NINCDS-ADRDA diagnostic criteria. For all individuals involved in this study, informed consent was given by the participant or their legal guardian. The first study group contained individuals selected based on probable or definite EOAD diagnoses and cognitively normal individuals. EOAD is defined as individuals displaying symptoms beginning on or before the age of 65, and controls were cognitively normal individuals over the age of 60. The frozen blood plasma was provided by Emory University and UCSF ADRCs. The plasma collected from this study group was to perform the targeted resequencing analysis. The second study group is comprised of individuals recruited by the Rush Alzheimer’s Disease Center for a longitudinal cohort study of aging and AD. These participants will be referred to as the ROS/MAP cohorts. Individuals in this study were given annual assessments and consented to allow their data and tissue to be studied. The annual assessment consisted of detailed medical, neurological, and neuropsychiatric evaluations. When an individual in the cohort passes, they undergo a brain autopsy which includes a neuropathological examination. The ROS/MAP cohorts contain a significant overlap of core data and metrics which allows the efficient merging of the datasets. Both ROS and MAP are approved by the Institutional Review Board of Rush University Medical Center.
2.2. Targeted resequencing
Following the manufacturer’s protocol, the Gentra Puregene kit (Qiagen, Hilden, Germany) was used to extract genomic DNA from the white blood cells of 356 EOAD and 962 control patients. The primers used to perform targeted re-sequencing were designed using the MPD multiplex primer design software (Wingo et al., 2017) and generated using the Access Array Multiplex Target-Specific system (Fluidigm, San Francisco, CA, US). The first set of primers was designed with coverage exceeding 90% for each gene (TET1, TET2, TET3), and the second set was designed for 400 ancestrally informative genes and 25 common X chromosome markers. With respect to disease status, sex, and age, the samples were randomly plated to reduce batch effects. According to the manufacturer’s protocol, libraries were generated and barcoded using the Access Array with 48 samples per run. Following library preparation, 250 bp paired-end sequencing was performed with the Illumina MiSeq platform.
2.3. Base calling and quality control
The first 27 bp were trimmed from the 5′ end of reads to remove the primer sequence. The trimmed reads were then mapped to the human reference assembly (GRCh38) with PEMapper. Variant calling was performed with the default settings using PECaller, and Bystro was used to perform filtering and variant annotation (Kotlar et al., 2018). There were two stages of quality control performed. In the first stage, the standard deviation (SD) of missing sites was calculated within batches and amplicons with >3 SD of missing sites were dropped. In the second stage, batches with >3 SD of sample failures were dropped, >3 SD of missing data, or > 3 SD of excess heterozygosity were removed. Genotyping information from the remaining batches were merged and variant sites with >5% missing data and those that failed Hardy-Weinberg filtering at 10−7 were dropped from the analysis.
2.4. Burden analysis statistics
Targeted resequencing data were analyzed and visualized using the EPACTS (Efficient and Parallelizable Association Container Toolbox; http://genome.sph.umich.edu/wiki/EPACTS) software. Single variant and Gene-wise tests were performed with sex and race as covariates, MAF <0.05, and filtered for nonsynonymous mutations. A logistic score test was used to perform the single variant analysis and the optimal unified test (SKAT-O) was used to compute the gene-wise association test. Solely missense coding variants were included in the gene-wise test. EPACTS results were lifted over from hg38 to hg19 using Picard LiftoverVcf.
2.5. Predictive SNP analysis
PolyPhen-2 was used to predict the deleteriousness of the nonsynonymous TET1 SNPs identified via the targeted re-sequencing analysis. PolyPhen-2 uses a naïve Bayes classifier and incorporates sequence, phylogenetic and structural information changes to predict the impact of amino acid substitutions on both the structure and function of human proteins.
2.6. Animals
The C57BL/6 WT and Tet1 KO lines were obtained from The Jackson Laboratory (stock #000664, and stock #017358, B6;129S4-Tet1tm1.1Jae/J (Dawlaty et al., 2011). The Tet1 KO line has been bred and maintained with the C57BL/6 line. These lines were used for the, Fear Conditioning, Tail suspension, and Forced Swim assays. All procedures performed on animals were done according to Emory University Institutional Animal Care and Use Committee-approved protocols.
2.7. Forced swimming test
The Forced Swimming Test (FST) was performed as outlined previously by Porsolt et al., 1977.(Porsolt et al., 1977). Mice were placed in a glass cylinder (20 cm high, 15 cm diameter) filled with water (23–26 °C, 14 cm in depth) for 6 min. Water was replaced prior to performing each assay. The immobility in the last 4 min of each assay was scored. Following their performance in the assay, mice were gently dried off and returned to their home cages set on top of a heating pad.
2.8. Tail suspension test
The Tail suspension test (TST) was performed according to the protocol outlined by Steru et al., 1985 with slight modifications (Steru et al., 1985). Mice were suspended with adhesive tape by the tip of their tail and suspended roughly 45 cm above a surface for 6 min. The time immobile was recorded and scored.
2.9. Genomic DNA preparation
Mouse cortex was isolated from the mouse brain. Genomic DNA was isolated by homogenizing the samples in 600 μL digestion buffer (100 mM Tris-HCl, pH 8.5, 5 mM EDTA, 0.2% SDS, 200 mM NaCl), Proteinase K (Thermo Fisher, cat# EO0491) treatment at 55 °C for overnight. Following digestion, 600 μL of Phenol:Chloroform:Isoamyl Alcohol (25:24:1 Saturated with 10 mM Tris, pH 8.0, 1 mM EDTA) (Sigma-Aldrich, cat# P-3803) was added and mixed thoroughly prior to centrifugation for 10 min at 12,000 rpm.
2.10. EWASplus sample selection
The EWASplus algorithm was used as described previously (Huang et al., 2021) to classify samples into groups based on their number of predicted AD-associated DNA methylation sites. The ~3100 sites predicted by EWASplus were filtered down to 200 loci that have corresponding 450 k array data. These sites were further filtered down to 178 loci that have beta value information from array-based DNAm AD association testing. Samples with the largest number of AD-associated loci are grouped as ‘AD’, and samples with the lowest number of AD-associated sites are grouped as ‘Control’.
2.11. Human RNA analysis
RNA sequencing was performed on dorsolateral prefrontal cortex samples. Sequencing reads were aligned to the GRCh38 reference genome using STAR v.2.4. Genes with <1 count per million (CPM) in at least 50% of samples per diagnosis (normal, AD, or other), missing gene length, or missing percentage GC content were removed. Samples that were outliers based on principal component analysis of raw CPM values were removed. After these steps, raw counts for 15,582 genes from 632 individuals were available. We applied the variance stabilizing transformation (function ‘vst’) from R package DESeq2 (v. 1.26.0) to normalize for library size, reduce heteroskedasticity, and transform to log2 scaled counts. The design formula used for VST was: ~sex + age at death + cogdxcat + Batch + RIN + pmi + neurons + astrocytes + microglia + oligodendrocytes, where ‘codgxcat’ is a binary version of the ROS/MAP cogdx variable (1 v. 2:6) and the four cell types are estimates of the cell type proportion for each sample. Samples missing cell type or RIN scores were removed.
2.12. 5hmC capture
5hmC enrichment was performed according to the previously described 5hmC-specific chemical labeling and affinity purification protocol (Song et al., 2011). DNA libraries were prepared using 2 μg of captured DNA or input DNA, and performed by following the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® manufacturers protocol. Prior to sequencing, the quality of prepared libraries was checked using Bioanalyzer and TapeStation. Sequencing was performed using the Next Generation Sequencing HiseqX 2 × 150 platform.
2.13. 5hmC capture analysis
Paired end reads were trimmed using Trimmomatic/0.36. Illumina adapters were removed from reads, bases with a pred score below 20 were trimmed off the end of the read, and a sliding window set at 4:15 was used to remove remaining low-quality bases. Bowtie2 was used to align the paired-end reads to NCBI37v1/mm9 genome for mice samples, and GRCh37/hg19 for human samples. Samtools/1.9 was used to remove duplicate reads, sort, and index the reads mapped. 5hmC peaks were called using the MACS2 peak calling software with 5hmC capture and non-pulldown input libraries. Differentially hydroxymethylated regions were identified with bdgdiff within the MACS2 software. The differential hydroxymethylation analysis were performed according to the sample batches for the human 5hmC data prior to merging DhMR results for subsequent hydroxymethylation analyses.
2.14. RNA isolation
Mouse cortex was isolated and homogenized in 1 mL of TRIzol (Invitrogen, cat# 15596026) and samples were incubated at RT for 5 min. Following incubation, 200 μL chloroform was added and samples were vigorously shaken for 15 s, then incubated at RT for 15 min. Samples were then centrifuged at 12,000 RPM for 15 min at 4 °C. The top aqueous layer (RNA) was isolated and added to a new tube. 600 μL of Isopropyl Alcohol was added and samples were incubated for 10 min prior to centrifugation at 12,000 RPM for 10 min at 4 °C. The supernatant was removed, and the RNA pellet was washed with 75% Ethanol. RNA was dissolved in RNase-free water. RNA quality was assessed by RNA ScreenTape with Tapestation (Agilent Technologies Inc., California, USA) and concentration was checked via the Qubit 2.0 RNA HS assay (ThermoFisher, Massachusetts, USA).
2.15. RNA-Seq libraries
Poly(A) + transcripts were isolated with the NEBNext Poly(A) mRNA Magnetic Isolation Module kit according to the manufacturer’s protocol (New England BioLabs Inc., Massachusetts, USA). RNA samples were randomly primed (5′ d(N6) 3′ [N = A,C,G,T]) and fragmented. The first strand was synthesized with Protoscript II Reverse Transcription with a modified extension period as to what is stated in the manufacturer’s protocol (30 min at 42 °C). The remaining steps of library generation were carried out according to the manufacturer’s protocol NEBNext® Ultra™ II Directional RNA Library Prep Kit for Illumina® (New England BioLabs Inc., Massachusetts, USA). The quantity and quality of libraries was assessed with Qubit 2.0 (ThermoFisher, Massachusetts, USA) and TapeStation HSD1000 ScreenTape (Agilent Technologies Inc., California, USA), respectively. The final libraries which were roughly 450 bp in size with an insert size of 300 bp, were indexed with Illumina® 8-nt dual-indices. Libraries were then pooled equimolarly and sequenced on an Illumina® NovaSeq 6000 S4 (Illumina, California, USA) with a read length configuration of 150 PE for 40 M PE reads per sample (20 M in each direction).
2.16. Murine RNA-Seq analysis
RNA sequencing reads were trimmed using Trimmomatic/0.36. Illumina adapters were removed from the reads. The leading and trailing ends of reads were trimmed if the base quality dropped below a Phred score of 20, and a sliding window 4:15 was used to remove remaining low-quality bases from the ends of reads. Following trimming, sequenced reads were aligned to the mouse genome GRCm38/mm10 using STAR/2.7.0 f. DESeq2 was used to analyze RNA sequencing data. Genes with <20 reads were dropped from the analysis.
2.17. Fear conditioning
The fear conditioning assay was adapted from (Curzon et al., 2009) and comprises three separate components: fear habituation, contextual fear, and cued fear. For fear habituation, mice were placed in a 7×7×12” plexiglass fear conditioning enclosure (Coulbourn Instruments, Whitehall, PA) and allowed to habituate uninterrupted to the novel environment for the first 3 min. After habituation, mice were exposed to a conditioned stimulus (tone, 20s, 10 kHz, 85 dB, 1-min inter-trail interval) immediately followed by an unconditioned stimulus (foot shock, 2 s, 0.5 mA; Precision Animal Shocker Coulbourn Instruments, Whitehall, PA) three times. One minute after the last round of stimulus, the mice were returned to their home cage for the remainder of the day. On day 2, the contextual fear portion of the assay, mice were placed in the same fear conditioning apparatus as the previous day for 7 min. The mice were not shocked, and the amount of time spent freezing was recorded and scored. On the third day of the assay, mice were placed in a novel enclosure not previously associated with the conditioned stimulus. The first two minutes of the assay allow the mice to habituate to the novel environment before the shock is administered. The mice remained in the enclosure for the following 6 min prior to being returned to their home cage. The amount of time the mice spent freezing was recorded and scored. Performance in the fear conditioning assay was measured using the FreezeFrame motion detection software provided by Coulbourn Instruments.
2.18. Neuropathological staining
Following a trasncardial perfusion with 0.9% NaCl in PBS and 4% PFA, brains were post-fixed in 10 mL of 4% PFA overnight at 4 °C prior to transfer to 30% sucrose in PBS for 3 days. Fixed brains were embedded in FSC 22 frozen section media (Leica Biosystems, Richmond, IL, USA) and stored at − 80 °C. Using a cryostat, 18 μm coronal sections were collected and stored in 0.1% sodium azide in PBS at 4 °C.
2.19. Tissue fixation and immunohistochemistry
Coronal sections were rinsed 3 times for 5 min with 0.25% Triton X-100 in PBST. Following the wash, sections were blocked with 250ul 0.25% Triton X-100 in PBST and 10% NGS for 1 h at room temperature. The sections were then incubated in primary antibody solutions (ab126649; 1:100 ab7260; 1:50, in 0.25% Triton X-100 in PBST and 5% NGS) overnight at 4 °C before a 3× wash with PBST. After the wash, the sections were incubated in the secondary antibody solution (Alexa Fluor™ 647 A-21236, Alexa Fluor™ 488 A-11008 in PBST and 5% NGS) for 1 h at room temperature in darkness. The secondary antibody solution was removed and sections were washed 3× with PBST. Stained sections were then mounted on slides with VECTASHIELD® Antifade Mounting Medium with DAPI (H–1200–10).
2.20. Imaging
Images of stained sections were gathered using ZEISS ApoTome optical and fluorescence imaging microscope, ZEISS AxioCam MRM microscope camera, and the X-Cite mini+Compact LED illumination system. The images were collected at 2.5× magnification with an exposure time of 1500 ms for Dapi, 250 ms for Alexa Fluor™ 488 A-11008, and 1500 ms for Alexa Fluor™ 647 A-21236) with the AxioVision/4.8 software.
2.21. Image quantification
Raw, unprocessed images were quantified with using the machine learning STARDIST algorithm in the Image J Fiji package. Stardist returned values for the number of amyloid plaques detected and the size of the plaques in pixels. The images were traced using ImageJ to measure the total number of pixels occupied by the brain. This total was then used to calculate the total area of the brain that is occupied by plaques in order to normalize for differences in brain size between samples.
2.22. NCRAD sample agreement
With government support in the form of funding under the cooperative agreement grant U24 AG21886 from the NIA, samples from the National Cell Repository for Alzheimer’s Disease (NCRAD) were used in this study. In addition, the NIA/NIH funds The National Alzheimer’s Coordinating Center (NACC) database with grant U01 AG016976. The NACC data is provided by Alzheimer’s disease centers with the following grants from the NIA: P30 AG019610 (principal investigator [PI], Eric Reiman, MD), P30 AG013846 (PI, Neil Kowall, MD), P50 AG008702 (PI, Scott Small, MD), P50 AG025688 (PI, Allan Levey, MD, PhD), P50 AG047266 (PI, Todd Golde, MD, PhD), P30 AG010133 (PI, Andrew Saykin, PsyD), P50 AG005146 (PI, Marilyn Albert, PhD), P50 AG005134 (PI, Bradley Hyman, MD, PhD), P50 AG016574 (PI, Ronald Petersen, MD, PhD), P50 AG005138 (PI, Mary Sano, PhD), P30 AG008051 (PI, Thomas Wisniewski, MD), P30 AG013854 (PI, M. Marsel Mesulam, MD), P30 AG008017 (PI, Jeffrey Kaye, MD), P30 AG010161 (PI, David Bennett, MD), P50 AG047366 (PI, Victor Henderson, MD, MS), P30 AG010129 (PI, Charles DeCarli, MD), P50 AG016573 (PI, Frank LaFerla, PhD), P50 AG005131 (PI, James Brewer, MD, PhD), P50 AG023501 (PI, Bruce Miller, MD), P30 AG035982 (PI, Russell Swerdlow, MD), P30 AG028383 (PI, Linda Van Eldik, PhD), P30 AG053760 (PI, Henry Paulson, MD, PhD), P30 AG010124 (PI, John Trojanowski, MD, PhD), P50 AG005133 (PI, Oscar Lopez, MD), P50 AG005142 (PI, Helena Chui, MD), P30 AG012300 (PI, Roger Rosenberg, MD), P30 AG049638 (PI, Suzanne Craft, PhD), P50 AG005136 (PI, Thomas Grabowski, MD), P50 AG033514 (PI, Sanjay Asthana, MD, FRCP), P50 AG005681 (PI, John Morris, MD), and P50 AG047270 (PI, Stephen Strittmatter, MD, PhD).
3. Results
3.1. Tet1 genetic variants are associated with EOAD
To assess whether TET1 variants are associated with AD, we performed TET1 targeted resequencing on 356 individuals with EOAD and 962 cognitively normal control. To capture polymorphisms of different allele frequencies and effect sizes, both single-variant and gene-based burden analyses were conducted. The single variant analysis found a significant association between two TET1 variants chr10:70332477_G/T (p.V128F, p = 0.030, CADD 22) and chr10:70450792_A/C (p.T1878P, p = 0.038, CADD 12.54) and EOAD (Fig. 1A). Both these SNPs are non-synonymous TET1 variants (Fig. 1B). The T1878P variant is located within the low complexity insert of the core catalytic domain in TET1 (Akahori et al., 2015). Low complexity inserts regions are often important for the recognition and binding of nucleic acid sequences or protein partners (Coletta et al., 2010; Jarnot et al., 2022; Kastano et al., 2021). Analysis conducted with PolyPhen-2 predicts the two identified variants to be possibly damaging with a score of 0.845 (sensitivity: 0.83; specificity 0.93) and 0.856 (sensitivity:0.83; specificity: 0.93), respectively (Table 1). Additionally, gene-based burden analysis of missense variants (MAF < 0.05) identified significant enrichment in TET1 variants associated with AD (p = 0.04). These data implicate TET1 loss-of-function variants as a potential contributing factor in AD, in addition to the previously implicated TET2 as well as DNA modification profiles (Cochran et al., 2020; Coppieters et al., 2014; Pellegrini et al., 2021).
Fig. 1.
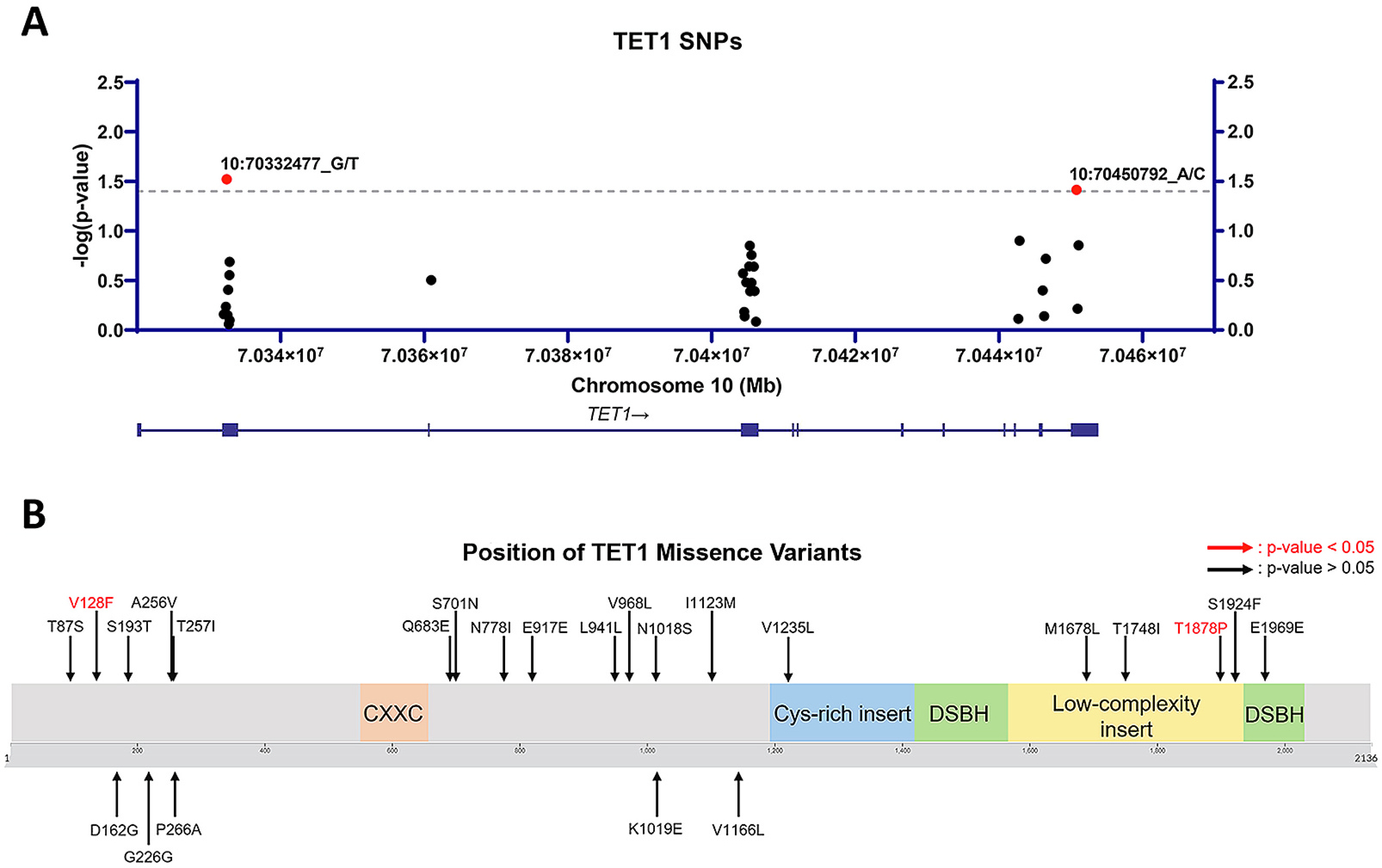
Tet1 Genetic Variants Are Associated with EOAD.
A) Location of detected missense variants within the TET1 gene. B) Location of amino acid changes within the TET1 protein. Amino acid changes due to significant SNPs (p < 0.05; single-variant logistic score test) between AD and control are labeled in red.
3.2. 5hmC and gene expression dysregulation of key AD and DNA modification regulator genes are observed in LOAD
To explore the relationship between TET and AD, given the heterogeneity of AD brain tissues, we utilized the EWASplus algorithm to categorize ROSMAP AD samples prior to performing gene expression and 5-hydroxymethylation analysis (Fig. 2A) (Huang et al., 2021). The samples with the highest and lowest epigenetic signatures of AD cognitive decline, neurofibrillary tangles were compared in RNA expression and 5hmC analysis.
Fig. 2.
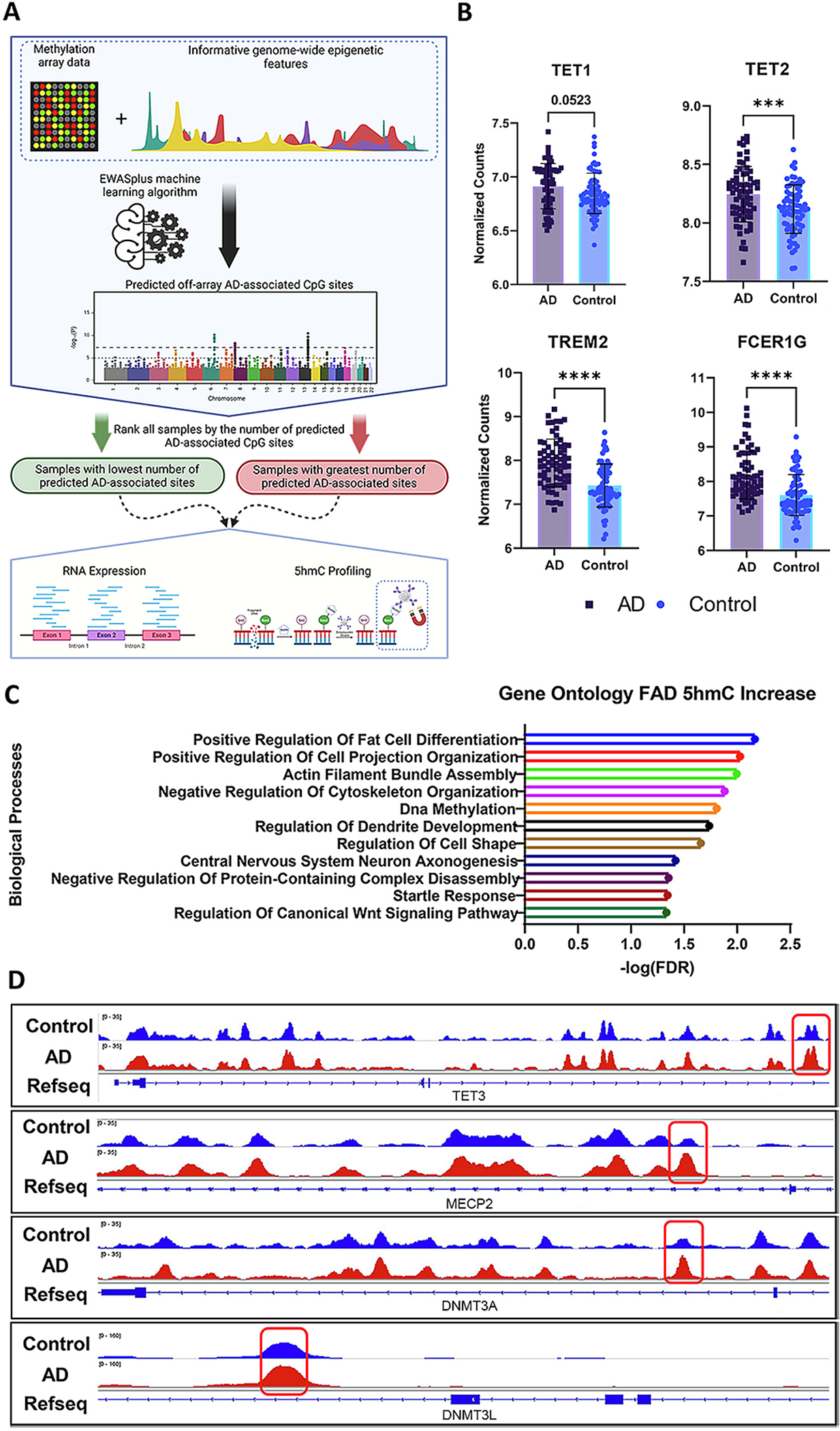
5hmC and Gene Expression Dysregulation of Key AD and DNA Modification Regulator Genes Are Observed in LOAD.
A) Diagram of the experimental design for categorizing samples by the number of EWASplus predicted AD-associated sites. B) VST normalized read counts for expression of TET1 (p = 0.0532), TET2 (p = 0.0009, TREM2 (p < 0.0001), and FCER1G (P < 0.0001) between AD (n = 68) and control (n = 72); two-tailed unpaired t-test. C) Gene ontology analysis (GO) revealed a significant enrichment of DEGs in pathways responsible for DNA methylation, axonogenesis, and dendrite development. D) IGV plots of 5hmC peaks showcase differentially methylated regions within genes responsible for regulating DNA methylation and hydroxymethylation. Error bars indicate mean ± SD; **** p < 0.0001, *** p = 0.0001–0.001, ** p = 0.001–0.01, ** p = 0.01–0.05, ns p ≥ 0.05.
Using the AD-associated epigenetic signature defined groups, the RNA expression comparison between the AD (n = 68) and control (n = 72) sample groups identified a significant increase in the expression of TET2 (p = 0.00090) and a trend for significant increase in expression of TET1 (p = 0.052) (Fig. 2B). We did not observe a significant change in the expression of TET3 or CXXC4, a protein that functions as TET2’s DNA binding domain through the recognition and direct binding of unmethylated CpG sites and TET2, between AD and control (Fig. S1A). Surprisingly, we also identified a significant increase in the expression of two hallmark genes, Fc-Epsilon Receptor Ig (FCER1G, p < 0.0001) and Triggering receptor expressed on myeloid cells-2 (TREM2, p ≤0.0001), both of which are strongly linked with AD (Guerreiro et al., 2013; Mukherjee et al., 2019; Sierksma et al., 2020). Prior studies in humans and mice found that FCER1G expression is upregulated in microglia in response to the presence of APP (Castillo et al., 2017; Matarin et al., 2015; Park et al., 2021; Sierksma et al., 2020). Specifically, increased expression of FCER1G in microglia is thought to promote an immune response in the presence of APP (Castillo et al., 2017; Zhang et al., 2013). TREM2 is one of the most highly expressed receptors in microglia and is critical for mediating immune response and phagocytosis, and, of non-familial AD-associated genes, is among the most strongly linked with AD, where rare mutations in TREM2 strongly increase the risk of developing AD (Del-Aguila et al., 2018; Guerreiro et al., 2013). Increased expression of the cell surface receptor protein, TREM2, is strongly linked with increased AD pathology as TREM2 functions in the recruitment of microglia to amyloid plaques (Cheng-Hathaway et al., 2018; Colonna and Wang, 2016; Slattery et al., 2014; Wang et al., 2015).
To further investigate the link between the regulation of DNA hydroxymethylation changes and AD, we performed 5hmC capture on AD (n = 11) and control (n = 11) samples to identify AD-associated DhMRs. In the human 5hmC data, we identified 169 peaks proximal to 123 genes elevated in the control samples and an increase in 5hmC at 753 peaks proximal to 669 genes. The abundance of 5hmC within different classes of genomic regions is significantly altered between AD and control samples (p = 0.021), although there was a notable increase (13%) in DhMRs located within introns and a slight decrease (6%) in 5hmC located in distal intergenic regions (Fig. S1B). Gene ontology analysis performed with PANTHER on DhMR gain in AD identified significant enrichment in pathways involved in the regulation of DNA methylation, and pathways critical to healthy neuronal function including actin filament bundle assembly, axonogenesis, and dendrite development (Fig. 2C). Within the group of enriched DNA methylation pathway genes were several genes including TET3, DNMT3L, DNMT3A, and MECP2 which are critical to the establishment and maintenance of 5mC and 5hmC profiles (Fig. 2D). We observed differential peaks in intronic region of each of these genes, where there was a significant increase of 5hmC in the AD group. We also observed an identical abundance of 5hmC at the TSS and slightly elevated levels of 5hmC in the transcriptional end sites (TES) and transcript splice sites in control versus AD (Fig. S1B & S1C). The signature of transcription factor binding motifs is also perturbed where the individuals with AD have reduced DNA hydroxymethylation at n-Myc, bHLHE40, and CLOCK binding motifs (Table 4). Combined, our RNA expression and 5hmC data suggest aberrant regulation of the methylome may be linked to increased expression of TREM2 and AD severity, supporting EWASplus as a tool to predict novel AD-associated CpG sites.
3.3. Loss of Tet1 increases AD-associated pathology in 5xFAD mice
The progression of AD is characterized by an increase in the formation of synaptotoxic Aβ plaques and neurofibrillary tangles (NFTs), causing synaptic dysfunction, and ultimately synaptic loss (Breijyeh and Karaman, 2020; Knopman et al., 2021). These pathologies are believed to cause neurodegeneration and cause cognitive and behavior dysfunction. The 5xFAD mouse model recapitulates the Aβ, synaptic loss, neuronal loss, and cognitive/behavioral pathology seen in human AD (Oakley et al., 2006; Oblak et al., 2021). To examine the effect loss of Tet1 has on AD-associated cognitive and behavioral trajectory in mice, the forced swim (FST) and tail-suspension tests (TST) assays were performed to profile behavior, and fear conditioning (FC) was used to profile cognition (Can et al., 2012a; Can et al., 2012b; Carter et al., 2003). Lastly, Immunohistochemical (IHC) staining of Aβ and GFAP marker abundance were used to measure hallmark AD pathologies.
Prior studies utilizing the FST assay revealed an increase in baseline swimming behavior that is strongly linked to AD mouse models (Bachiller et al., 2022; Luo et al., 2017; Torres-Lista and Gimenez-Llort, 2014; Yamazaki et al., 2015). In the TST assay, reduced immobility is similarly observed in the 5xFAD mouse model relative to WT (Yamazaki et al., 2015). We observed similar findings in the FST assay with significantly reduced immobility in 5xFAD mice compared to WT (p < 0.0001) (Fig. 3A). In the FST assay, the loss of Tet1 in mice appears to enhance AD-associated increased mobility with a near significant difference between 5xFAD vs 5xFAD/Tet1(+/−) (p = 0.0637) (Fig. 3A). The tail-suspension test (TST) revealed no significant difference in the performance between WT and 5xFAD mice (p = 0.1679), however, when comparing WT and 5xFAD/Tet1(+/−), we observe a significant difference in immobility (p = 0.0434) (Fig. 3B).
Fig. 3.
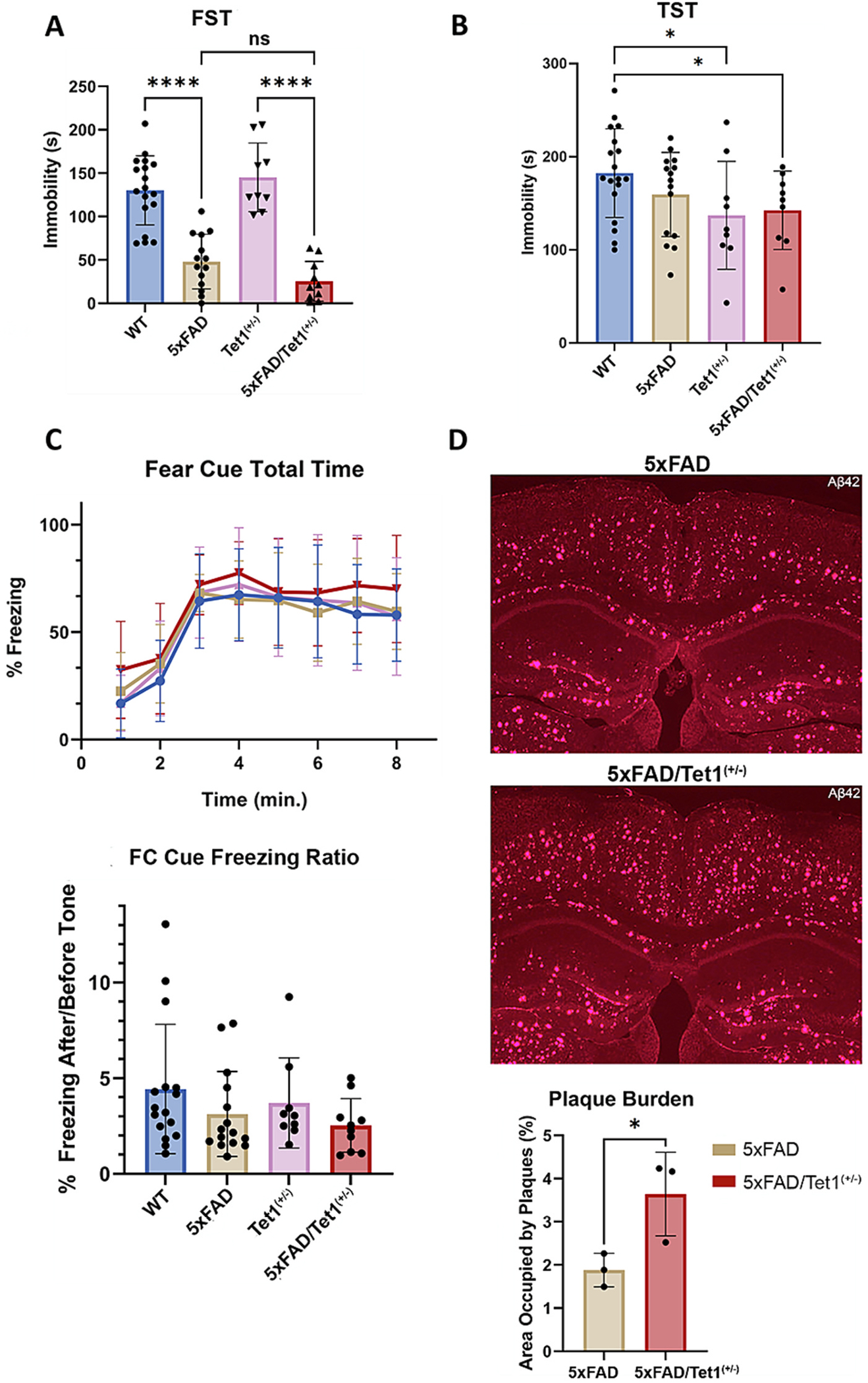
Loss of Tet1 Increases AD-associated Pathology in 5×FAD Mice.
A) Results from the forced-swim assay (FST). We measured a significant difference in immobility between 5×FAD (n = 14) mice relative to WT (n = 18; p < 0.0001) and Tet1(+/−) (n = 9; p < 0.0001) mice relative to 5xFAD/Tet1(+/−) (n = 10; p < 0.0001). A near significant difference between 5xFAD and 5xFAD/Tet1(+/−) was measured (p = 0.0637); two-tailed unpaired t-test. B) The TST identified significant performances between the WT (n = 18) and Tet1(+/−) (n = 9; p = 0.0397) and 5xFAD/Tet1(+/−) (n = 9; p = 0.0434) mice; two-tailed unpaired t-test. C) The fear cue portion of the fear conditioning assay found no significant difference in fear memory between conditions across the total time (p = 0.54; two-way RM ANOVA) and their performance normalized for baseline freezing behavior, but a non-significant towards reduced cognition was noted between 5xFAD (n = 15; p = 0.2178; two-tailed unpaired t-test) and 5xFAD/Tet1(+/−) (n = 10; p = 0.1055; two-tailed unpaired t-test) mice relative to WT (n = 18). The performance between 5xFAD/Tet1(+/−) relative to 5xFAD is not significantly different. D) Anti-Aβ42 staining in mouse brain (2.5x magnification). The image brightness and contrast were adjusted to improve visibility for the image sections displayed in this figure. Using raw, unprocessed image files, the plaque burden was calculated with the STARDIST machine learning algorithm and identified a significant difference in Aβ42 abundance between 5xFAD (n = 3) and 5xFAD/Tet1(+/−) (n = 3) mice (p = 0.044; two-tailed nested t-test). Error bars indicate mean ± SD; **** p < 0.0001, *** p = 0.0001–0.001, ** p = 0.001–0.01, ** p = 0.01–0.05, ns p ≥ 0.05.
With respect to learning, we observe non-significant but lower learning in the fear conditioning paradigm for 5xFAD and FAD/Tet1(+/−) mice versus control measured by total fear cue time or freezing ratio (Fig. 3C. The day 1 acquisition and contextual fear portion of the assay identified no variation in learning and memory performance (Fig. S2A, Fig. S2B). The fear cue portion of the fear conditioning assay results appear consistent with the trend towards increased AD-associated behavior (FST) and pathologies observed in other areas and indicate that the loss of Tet1 increases AD pathophysiology. It is important to note that synaptic loss and neuronal loss begin to occur at 6 months of age in the 5xFAD model and the mice studied were studied at ~7 months of age. Perhaps in an older cohort of mice or examination with additional cognitive and behavioral assays, the difference in cognitive and behavioral performance between 5xFAD and 5xFAD/Tet1(+/−), if any, will be more pronounced.
The relative abundance of Aβ was measured and quantified using the Stardist machine-learning algorithm in ImageJ. The number and size of Aβ plaques in the entire coronal section were measured and the area of the number of pixels occupied by the plaques was recorded. The pixel count of the entire coronal section was used to normalize the number and size of amyloid plaques to yield the plaque burden. As Aβ plaque development is not native to mice, the WT and Tet1(+/−) mouse models displayed no plaque burden, and only the transgenic 5xFAD model exhibited plaque pathology. Relative to the 5xFAD mice, the loss of Tet1 in the 5xFAD model significantly increased the Aβ plaque burden when adjusted for the total area of the brain sectioned (p = 0.044; Fig. 3D, Fig. S2C). The presence of GFAP, a marker of astrocytosis which is strongly associated with AD, is enriched in 5xFAD versus 5xFAD/Tet1(+/−) (Fig. S2D). The combination of the behavioral, cognitive, and neuropathological data suggests the partial loss of Tet1 in the 5xFAD mouse model is sufficient to increase the severity of AD-associated pathologies.
3.4. Tet1 deficiency alters 5xFAD-associated epigenetic landscapes
To examine how the loss of TET1 alters the genome-wide distribution of DNA hydroxymethylation in 5xFAD mice, we performed 5hmC capture on WT, Tet1(+/−) KO, 5xFAD, and 5xFAD/Tet1(+/−) mice. Peaks were called and DhMRs were identified using the MACS2 peak calling software (FDR = 0.05). Previous reports identify a significant reduction in 5hmC in AD in both humans and the 3xTg AD mouse model, and we observed the same pattern in the 5xFAD model (Cadena-del-Castillo et al., 2014; Zhang et al., 2020b). Overall, there is a loss of 3977 DhMRs and a gain of 40 DhMRs in 5xFAD/Tet1(+/−) when compared against 5xFAD mice (Fig. 4A). These DhMRs were associated with 2618 and 40 genes, respectively. Between 5xFAD and 5xFAD/Tet1(+/−) mice, there was an alteration in the genomic distribution of 5hmC, with the largest magnitude of changes occurring in distal intergenic (+12.3%), intronic (−3.4%), and exonic (−4.45%) regions (p < 0.0001; Fig. S3A). When focusing in on enhancer, transcription start sites (TSS), splice sites, and transcription end sites (TES), there is a clear difference between each condition and 5hmC prevalence (Fig. 4B). Aside from the TSS, there is an increase in 5hmC abundance in mice containing familial AD transgenes (5xFAD and 5xFAD/Tet1(+/−) compared to the non-5xFAD mice (WT and Tet1(+/−)).
Fig. 4.
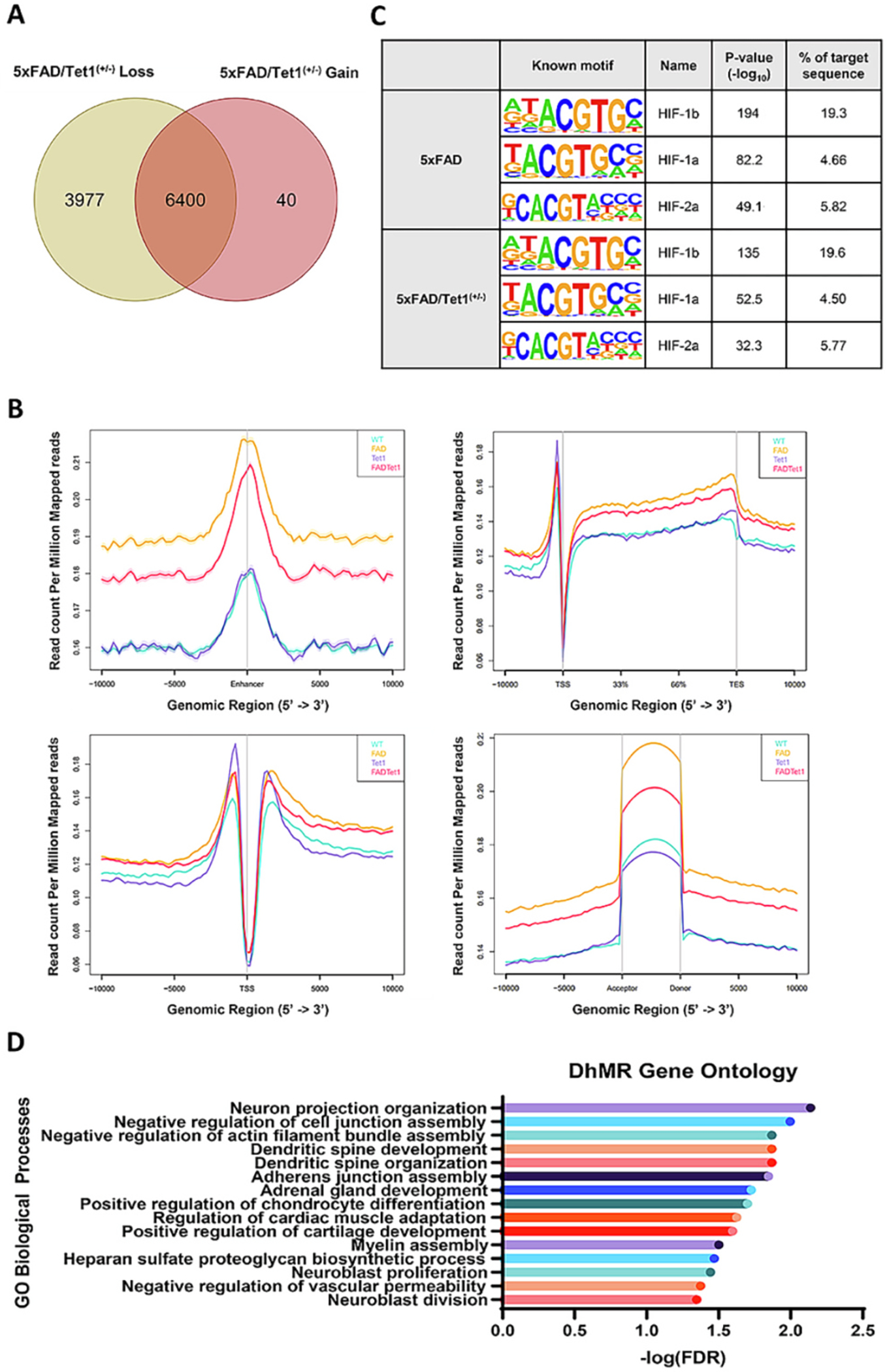
Tet1 Deficiency Alters 5xFAD-associated Epigenetic Landscapes.
A) Variation in the loss and gain of DhMR between 5xFAD (n = 2) and 5xFAD/Tet1(+/−) (n = 4) mice. There is a loss of 3977 DhMRs and a gain of 40 DhMRs in the 5xFAD/Tet1(+/−) mice relative to 5xFAD. B) RPM normalized abundance of 5hmC modifications at Enhancer, Gene body, TSS, and Exon acceptor and donor sites. C) DNA binding motif analysis for HIF-1b, HIF-1a, and HIF-2a for all DhMRs. D) Gene ontology analysis of all DhMRs between 5xFAD and 5xFAD/Tet1(+/−).
The stark increase in hydroxymethylation at gene regulatory regions in the 5xFAD-containing mice highlights a relationship between AD and the hydroxymethylome. It is unclear whether the dysregulation of 5hmC profiles is driving AD-associated outcomes or if the dysregulation is a byproduct of other AD-associated molecular changes. Furthermore, the partial KO of Tet1 may be altering AD-related outcomes not through loss of Tet1 catalytic function, but through its loss of function as a transcription factor recruiter and binding partner (Cheng et al., 2018; Pantier et al., 2020; Zhang et al., 2010). To explore this possibility, we performed a known motif analysis of 5xFAD and 5xFAD/Tet1(+/−) peaks to identify if there is an enrichment of DNA binding motifs (Table 3). Previous work from our lab had discovered a direct interaction between Tet1 and the transcription factor HIF1α (hypoxia-induced factor alpha) (Cheng et al., 2018). The physical interaction between Tet1 and HIF1α is responsible for mediating differential hydroxymethylation of thousands of genomic loci in response to environmental factors. As per our previous findings, we were interested if alterations in the prevalence of HIF binding sites within hydroxymethylated regions could be contributing to 5xFAD and 5xFAD/Tet1(+/−) AD-associated changes. We identified an enrichment of HIF1a, HIF1b, and HIF2a binding motifs in both 5xFAD and 5xFAD/Tet1(+/−), however, the over-representation of these binding motifs is nearly identical in the two conditions (Fig. 4C, Table 3). Although the prevalence of HIF binding motifs remained the same, there was a change in DNA binding motif profiles. The partial loss of Tet1 in the 5xFAD model leads to a reduction in representation for 29 known transcription factor binding motifs and a gain of 0 motifs when compared against 5xFAD mice (Fig. S3C, Table 3). Several TF binding motifs that lost 5hmC signal in the 5xFAD/Tet1(+/−) mice include binding site motifs for many forkhead box family proteins (FOXA1, FOXO1, FOXP1, FOXK1, FOXK2, FOXA2, and FOXO3), as well as FLI1, MYB, and P53. These TFs are involved in nervous system development, cell senescence, cell proliferation, cell death, tumorigenesis, and metabolic homeostasis. Furthermore, six of these TFs, including FOXO1, FOXA2, FLI1, MYB, and P53, function as pioneer transcription factors (Lemma et al., 2022). Additionally, in the TF binding motifs lost in 5xFAD/Tet1(+/−), we observe several ETS family TFs including ETS1, ELF1, and ERG which have been previously shown to have methylation and hydroxymethylation sensitive binding (Song et al., 2021). Inhibition of binding at their canonical genomic locations could contribute to the increased AD-associated pathology seen in the 5xFAD/Tet1(+/−) mice. With respect to Tet1’s function as a hydroxymethylation modifier, we performed a gene ontology (GO) analysis using PANTHER. Gene ontology analysis revealed enrichment in pathways critical for neuronal projection, dendritic spine development dendritic spine organization, and myelin assembly (Fig. 4D). In AD, the buildup of amyloid plaques is known to activate microglia which release tumor necrosis factor alpha (TNFα), leading downstream to excitotoxicity and dendritic spine damage (Centonze et al., 2009; Ou et al., 2021; Yang et al., 2013; Zhou et al., 2020). This damage and loss of dendritic spines and axons are directly correlated to the loss of synaptic function as they are responsible for receiving and communicating neuronal signals and eventually leads to neuronal loss (Knobloch and Mansuy, 2008; van de Weijer et al., 2020; Zhou et al., 2019).
3.5. Partial loss of Tet1 modulates 5xFAD-associated gene expression
With clear dysregulation of the methylome at key regulatory and TF binding sites, we were interested in how these changes impact gene expression. Overall, we discovered significant expression changes in 40 genes with (padj <0.1) with 36 genes upregulated and 4 genes down-regulated in 5xFAD relative to 5xFAD/Tet1(+/−) (Fig. 5A, Fig. S3B). Interestingly, the elevated expression of TREM2 (p = 0.0596) and FCER1G (p = 0.0485) in 5xFAD/Tet1(+/−) mice relative to 5xFAD mice mirrors that of our human AD data. These findings are consistent with what is observed in the literature, with prior studies finding strong associations between TREM2 and FCER1G variants and expression in data from human and murine AD model systems (Castillo et al., 2017; Efthymiou and Goate, 2017; Guerreiro et al., 2013; Lee and Lee, 2020; Mukherjee et al., 2019; Sierksma et al., 2020). GO analysis of the differentially expressed genes (DEGs) revealed an overrepresentation of genes involved in pathways regulating dendritic cell activation, synaptic pruning microglial and astrocyte cell migration, and microglial cell activation (Fig. 5C). Among these genes with significantly increased expression were genes with a strong AD association such, including Mpeg1, Trem2, and Fcer1g (Fig. 5A, B). STRING protein analysis network of DEGs reveals a direct interaction between Trem2 and Fcer1g, and the majority of the significant DEGs are a part of the same core interactome, including additional AD-associated genes such as Mpeg1 (confidence = 0.70) (Fig. 5D). The importance of these three genes largely lies in their function for microglial activation and clearance of amyloid plaques. Increased expression of these microglial genes is consistent with previous works as the Tet1(+/−) KO 5xFAD mice display increased amyloidosis and prior studies link increased Fcer1g, Trem2, and Mpeg1 expression with increased AD plaque burden and additional AD pathologies (Mukherjee et al., 2019; Sierksma et al., 2020). When examining the lost TF binding motifs within 1000 basepair promoter regions of the DEGs, we observe an enrichment of binding motifs for FLI1, ETS1, ERG, and ELF1 (Table 5). Previously, Song et al., 2021 identified variation in binding affinity due to the methylation and hydroxymethylation status of the target DNA. ETS1, ERG, and ELF1 exhibit sensitivity where the loss of 5mC/5hmC may suppresses binding affinity, increasing the expression of the the AD-associated DEGs in 5xFAD/Tet1(+/−) relative to 5xFAD mice.
Fig. 5.
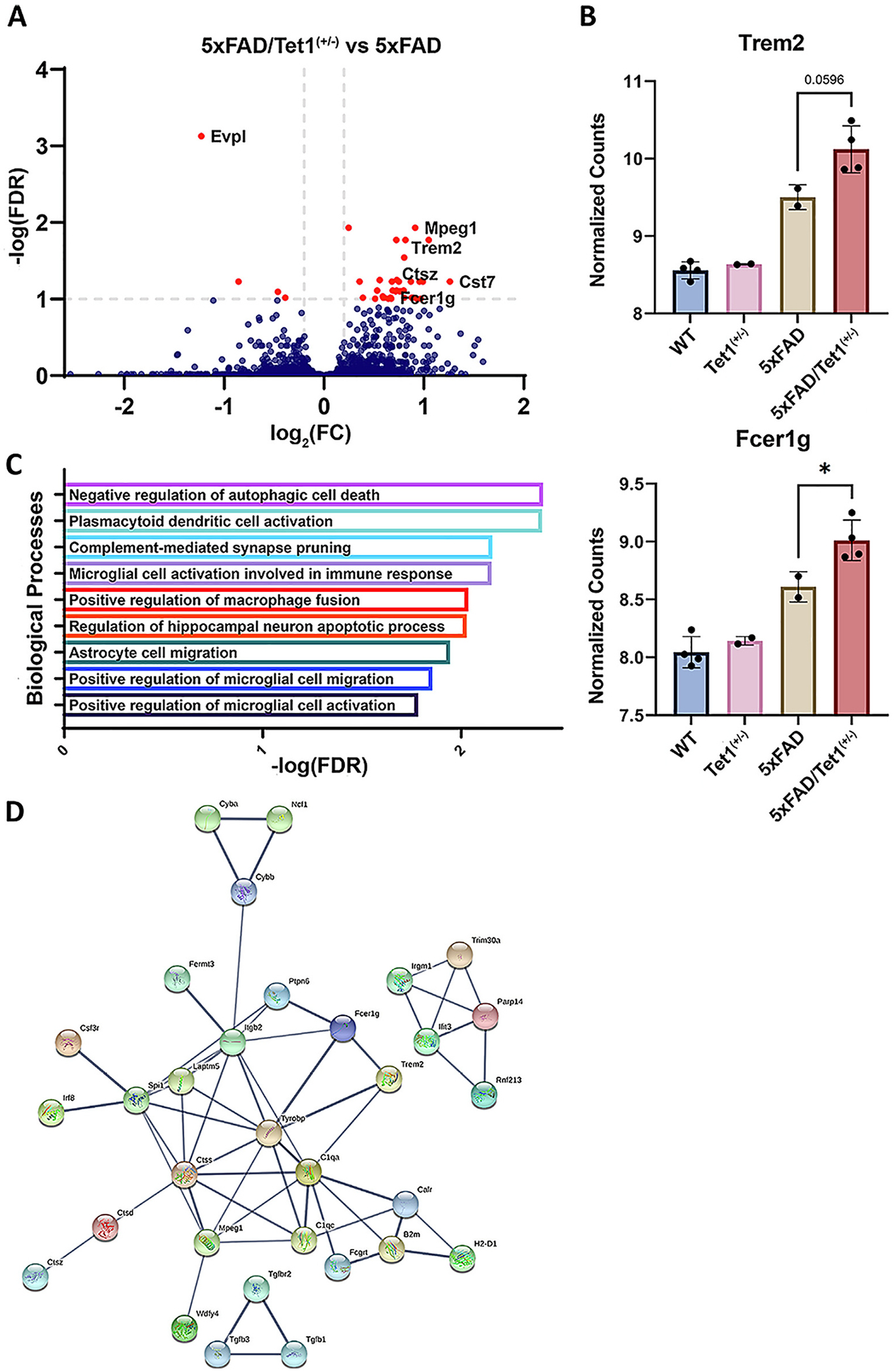
Partial Loss of Tet1 Modifies 5xFAD-associated Gene Expression.
Expression data from the 5xFAD (n = 2) and 5xFAD/Tet1(+/−) (n = 4) mouse models. A) Volcano plot of the 40 differentially expressed genes between 5xFAD (n = 2) and 5xFAD/Tet1(+/−) (n = 4). B) VST normalized read counts of Trem2 and Fcer1g. Near significant difference between Trem2 (p = 0.0596) in 5xFAD and 5xFAD/Tet1(+/−) and a significant difference in expression of Fcer1g (p = 0.0485). C) Gene ontology analysis of the 40 identified DEGs. E) String protein (high confidence 0.700) network analysis of the DEGs indicates strong interactions between the protein products of the identified DEGs, including a direct interaction between Trem2 and Fcer1g. Error bars indicate mean ± SD; **** p < 0.0001, *** p = 0.0001–0.001, ** p = 0.001–0.01, ** p = 0.01–0.05, ns p ≥ 0.05.
4. Discussion
Here we highlight a strong link between regulation of the methylome and neurodegeneration-related outcomes (Al-Mahdawi et al., 2014; Armstrong et al., 2019). Loss of the key regulator of the methylome and hydroxymethylome, TET1, alters the location and abundance of DNA methylation and hydroxymethaltion profiles. This dysregulation can lead to aberrant gene expression and ultimately impact AD-associated outcomes (Pellegrini et al., 2021; van den Hove et al., 2012; Zhang et al., 2020b). Targeted sequencing of TET1 identified two SNPs within the coding region that are associated with AD in humans. Analysis of these variants with PolyPhen-2 predicted both variants to be deleterious and reduce the function of TET1. Work from Cochran et al., found a similar association between TET2 variants and early onset AD (Cochran et al., 2020).
Furthermore, we analyzed human 5hmC capture data and found 753 DhMRs gained and 169 DhMRs lost in AD compared to control brain. Most of the the DhMRs were found in distal intergenic, promoter, and intronic regions. Within DhMRs, we observed less hydroxymethylation at n-Myc, bHLHE40, and CLOCK binding motifs. All of which recognize the E-box sequence within promoter regions, whose 5hmC content is predominantly regulated by Tet1, to regulate gene expression (Freeman et al., 2019; Hardin, 2004; Perini et al., 2005; St-Pierre et al., 2002). The n-Myc TF is a member of the Myc family of proteins and regulates a diverse range of biological processes ranging from cell proliferation and differentiation to apoptosis. CLOCK and BHLHE40 are both TFs involved in the regulation of circadian rhythm which is dysregulated in AD (Boudjelal et al., 1997; Honma et al., 2002; Thome et al., 2011). Interestingly, there were DhMRs in genes critical for the regulation of the methylome, including TET3, DNMT3L, DNMT3A, and MECP2. Of note, we did not observe RNA expression changes in these four genes, however, we did observe a significant increase in TET2, TREM2, FCER1G, and a near significant increase in TET1 expression. Thus, our data suggest that methylome, hydroxymethylome, and the genes that regulate them, potentially play an important role in AD pathogenesis.
When examining the partial loss of Tet1 in the 5xFAD mouse model, we observed a near-significant increase in AD-associated cognitive and behavioral phenotypes. In the pathological data, reduction of Tet1 significantly increased Aβ42 pathology in the 5xFAD mice. The hydroxymethylation and RNA expression changes portray a similar story where the loss of Tet1 exacerbates AD-associated molecular changes, with aberrant regulation of 5hmC profiles and key TF binding sites and genomic loci. We cannot yet be sure whether the change in hydroxymethylation of these TF motifs is due to Tet1 having reduced affinity for the 5xFAD exclusive regions or if the 5xFAD exclusive TFs are less easily able to recruit Tet1 than the shared TFs. In short, it is unclear whether Tet1 is being recruited to these TF sites or if Tet1 is doing the recruiting. We believe that it is likely the ‘loss of 5xFAD’ TF motifs and not the ‘shared’ TF motifs contributing to the molecular and pathological differences observed between 5xFAD and 5xFAD/Tet1(+/−) mice. As for the mechanistic role Tet1 plays in augmenting AD pathology, it is difficult to decipher whether it is the ability of Tet1 to recognize and bind different genomic loci and TFs, or if Tet1’s catalytic function as a hydroxymethylase is driving the differential binding and methylation of TF binding sites. We would not expect to see significant differences between 5xFAD and 5xFAD/Tet1(+/−) except for regions with high Tet1 affinity or accessibility if Tet1’s localization was driven by its own CXXC DNA binding domain. However, we observe that partial loss of Tet1 in the 5xFAD mice leads to the loss of 5hmC at many pioneer transcription factor motif sites (Foxo1, Fli1, Myb, Foxa1, Foxa2, and p53). It could be that at these loci, Tet1 is being recruited primarily by TFs and to a lesser extent, through Tet1’s DNA binding domain. We believe that in the context of the 5xFAD mice, Tet1 is both being recruited to genomic loci through the CXXC domain, and through recruitment by additional TFs. Previous work with collaborators characterizing the influence of DNA modification status on TF binding affinity, hints a potential mechanism for how the loss Tet1 impacts AD-associated gene expression. The large decrease in 5hmC observed genome-wide and the narrow overlap between DhMRs and the 40 DEGs, suggests that the decrease in 5hmC at 3977 DhMRs in the 5xFAD/Tet1(+/−) mice could act as a molecular sponge or free up TFs depending on the context and the specific TF DNA modification sensitivity. An example is ETS1 which exhibits suppressed binding for symmetric 5mC/5hmC and the conversion to hemi5hmC alleviates the reduced binding affinity. In this instance, the loss of Tet1 in 5xFAD mice reduces the conversion of 5mC to 5hmC, leading to a global increase in 5mC and reduced ETS1 binding affinity. Genomic loci that would normally have 5hmC and ETS1 bound no longer do, increasing ETS1 availability. Although our data is unable to distinguish between the exact mechanism behind Tet1’s contribution to AD pathology, in the 5xFAD/Tet1(+/−) mice, it appears the previous mechanism could be responsible. Alternatively, there could be less recruitment of Tet1 to genomic loci by TFs, including the pioneer transcription factors, and the CXXC domain is playing a more outstanding role in Tet1 recruitment. It would be very interesting to test the effects of catalytically dead Tet1 or mutant forms of Tet1 that alter either protein-protein interactions or Tet1’s DNA binding ability to get a better understanding of the role of Tet1 in AD.
Through analyzing gene expression profiles of 5xFAD/Tet1(+/−) KO mice, we observed that Tet1-KO-associated changes that parallel those associated with more severe AD pathology from human samples, with significant increases in Trem2 and Fcer1g. Increased expression of FCER1G in microglia is thought to promote an immune response in the presence of APP (Castillo et al., 2017; Zhang et al., 2013). TREM2 is one of the most highly expressed receptors in microglia and is critical for mediating immune response and phagocytosis, and, of non-familial AD-associated genes, is among the most strongly linked with AD, where rare mutations in TREM2 strongly increase the risk of developing AD (Del-Aguila et al., 2018; Guerreiro et al., 2013). Increased expression of the cell surface receptor protein, TREM2, is strongly linked with increased AD pathology as TREM2 functions in the recruitment of microglia to amyloid plaques (Cheng-Hathaway et al., 2018; Colonna and Wang, 2016; Slattery et al., 2014; Wang et al., 2015). The increased expression of these two microglial AD-link genes in the mouse model could be driving the increase in AD-associated behavioral and neuropathological changes observed through modulating neuroinflammation in response to Aß plaques. Our findings are consistent with the observation that an increase of TREM2 signaling is associated with increased microglial activation which can be protective in the early stages of AD but propagates neuroinflammation as AD progress (Gratuze et al., 2018; Qu and Li, 2020). In the 5xFAD/Tet1(+/−) KO mouse model, there were also significant changes in 5hmC abundance in many genes required for the branching of dendritic spines and myelination of neuronal cells. It remains uncertain whether the dysregulation of hydroxymethylation in pathways critical for neuronal viability is contributing to the increase in the observed Aß deposition or if these changes are a byproduct of an increase in toxic amyloid plaques. Regardless of the cause, changes in DNA methylation and hydroxymethylation are an important component in regulating gene expression, and aberrant hydroxymethylation is likely contributing to altered AD-associated gene expression (Fagiolini et al., 2009; Pellegrini et al., 2021; van den Hove et al., 2012). Additionally, it is unclear whether the localization of Tet1, the recruitment of specific TFs by Tet1, or the Tet1-related disruption of methylation profiles at regulatory regions is the cause of the differential expression of bona fide AD genes. However, the loss of Tet1 in the 5xFAD mouse model increases the severity of AD pathology and increases the expression of the hallmark AD genes Mpeg1, Fcer1g, and Trem2.
Taken together, our results indicate that TET1 variants are nominally associated with EOAD, and that TET1 can modify the severity of AD pathology. We have shown that dysregulation of TET1, and subsequently the methylome, alters AD-associated hydroxymethylation and expression of key genes involved in the maintenance of the epigenome, neuronal health, and microglial activation and function. Similarly, a recent study examining the effects of space radiation exposure in mice identified DNA hydroxymethylation and RNA expression changes enriched in molecular pathways associated with neurodegeneration, further highlighting the importance of 5mC and 5hmC profiles as a universal key regulatory mechanism for the maintenance of neuronal health (Impey et al., 2016). The loss of TET1 has a negative impact on AD-associated outcomes, and the next step is to determine whether the loss of TET1 contributes to AD disease states primarily through TF binding/recruitment or through TET1’s catalytic function in 5mC/5hmC regulation. The strength of this study comes from the combination of many different forms of data, including both human samples and the 5xFAD mouse model. The convergence of which highlights consistencies between the molecular and physiopathology between the two and implicates Tet1 as a mediator of AD pathogenicity. However, there are several limitations in the work presented which include a limited sample size for the human data, meaning that the molecular findings may not generalize across all or diverse human populations. Additionally, the data generated from the 5xFAD model recapitulates the amyloid pathology, but not the tau neurofibrillary tangle pathology. Furthermore, although there is a trend towards increased AD-associated behavioral outcomes, these trends are not significant. Perhaps behavioral tests that measure additional behavioral metrics may be better suited to characterize the AD-associated behavioral outcomes. It is also possible that genes associated with behavioral phenotypes are less sensitive to the loss of Tet1 than the observed neuroinflammatory response genes with robust expression changes. A portion of the differential expression could be explained by context-specific compensatory regulatory mechanisms by Tet2 and Tet3, although additional studies would be required to support this possibility. It would also be interesting to explore whether the loss of Tet1 has a similar influence on tau neurofibrillary tangle pathology, as well as how the overexpression of Tet1 could impact AD progression and both Aβ and tau pathology. All in all, advances in understanding the role TET1 has on methylation in AD will illuminate AD pathophysiology and could potentially lead to improved screening methods for identifying individuals at greater risk of developing AD,AD diagnosis, or potential avenues for the treatment of AD.
Supplementary Material
Acknowledgements
The authors would like to thank Quinn Eastman for critical reading of the manuscript. This work was supported in part by NIH grants (AG072907, AG056533, AG072120, AG075827, AG078937, and NS111602).
Footnotes
Declaration of Competing Interest
The authors declare no competing interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2023.106257.
Data availability
Summary statistics of the targeted resequencing are shared as part of the manuscript reporting. The raw and processed targeted sequencing will be made available through NIAGADS after publication. We have deposited the 5hmC and RNA-seq data into the Gene Expression Omnibus at https://www.ncbi.nlm.nih.gov/geo/. The accession number is GSE226056.
References
- Akahori H, Guindon S, Yoshizaki S, Muto Y, 2015. Molecular evolution of the TET gene family in mammals. Int. J. Mol. Sci. 16, 28472–28485. 10.3390/ijms161226110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, et al. , 2000. Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421. 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mahdawi S, Virmouni SA, Pook MA, 2014. The emerging role of 5-hydroxymethylcytosine in neurodegenerative diseases. Front. Neurosci. 8, 397. 10.3389/fnins.2014.00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeloni A, Bogdanovic O, 2019. Enhancer DNA methylation: implications for gene regulation. Essays Biochem. 63, 707–715. 10.1042/ebc20190030. [DOI] [PubMed] [Google Scholar]
- Armstrong MJ, Jin Y, Allen EG, Jin P, 2019. Diverse and dynamic DNA modifications in brain and diseases. Hum. Mol. Genet. 28, R241–r253. 10.1093/hmg/ddz179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayodele T, Rogaeva E, Kurup JT, Beecham G, Reitz C, 2021. Early-onset Alzheimer’s disease: what is missing in research? Curr. Neurol. Neurosci. Rep. 21, 4. 10.1007/s11910-020-01090-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachiller S, Hidalgo I, Garcia MG, Boza-Serrano A, Paulus A, Denis Q, Haikal C, Manouchehrian O, Klementieva O, Li JY, et al. , 2022. Early-life stress elicits peripheral and brain immune activation differently in wild type and 5xFAD mice in a sex-specific manner. J. Neuroinflammation 19, 151. 10.1186/s12974-022-02515-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudjelal M, Taneja R, Matsubara S, Bouillet P, Dolle P, Chambon P, 1997. Overexpression of Stra13, a novel retinoic acid-inducible gene of the basic helix-loop-helix family, inhibits mesodermal and promotes neuronal differentiation of P19 cells. Genes Dev. 11, 2052–2065. 10.1101/gad.11.16.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breijyeh Z, Karaman R, 2020. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules 25. 10.3390/molecules25245789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacace R, Sleegers K, Van Broeckhoven C, 2016. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 12, 733–748. 10.1016/j.jalz.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Cadena-del-Castillo C, Valdes-Quezada C, Carmona-Aldana F, Arias C, Bermúdez-Rattoni F, Recillas-Targa F, 2014. Age-dependent increment of hydroxymethylation in the brain cortex in the triple-transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 41, 845–854. 10.3233/jad-132285. [DOI] [PubMed] [Google Scholar]
- Can A, Dao DT, Arad M, Terrillion CE, Piantadosi SC, Gould TD, 2012a. The mouse forced swim test. J. Vis. Exp e3638 10.3791/3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Can A, Dao DT, Terrillion CE, Piantadosi SC, Bhat S, Gould TD, 2012b. The tail suspension test. J. Vis. Exp e3769 10.3791/3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RM, Hofstotter C, Tsuchiya N, Koch C, 2003. Working memory and fear conditioning. Proc. Natl. Acad. Sci. U. S. A. 100, 1399–1404. 10.1073/pnas.0334049100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartron PF, Nadaradjane A, Lepape F, Lalier L, Gardie B, Vallette FM, 2013. Identification of TET1 partners that control its DNA-demethylating function. Genes Cancer 4, 235–241. 10.1177/1947601913489020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo E, Leon J, Mazzei G, Abolhassani N, Haruyama N, Saito T, Saido T, Hokama M, Iwaki T, Ohara T, et al. , 2017. Comparative profiling of cortical gene expression in Alzheimer’s disease patients and mouse models demonstrates a link between amyloidosis and neuroinflammation. Sci. Rep. 7, 17762. 10.1038/s41598-017-17999-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Muzio L, Rossi S, Cavasinni F, De Chiara V, Bergami A, Musella A, D’Amelio M, Cavallucci V, Martorana A, et al. , 2009. Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J. Neurosci. 29, 3442–3452. 10.1523/jneurosci.5804-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Sun M, Chen L, Li Y, Lin L, Yao B, Li Z, Wang Z, Chen J, Miao Z, et al. , 2018. Ten-eleven translocation proteins modulate the response to environmental stress in mice. Cell Rep. 25, 3194–3203.e3194. 10.1016/j.celrep.2018.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng-Hathaway PJ, Reed-Geaghan EG, Jay TR, Casali BT, Bemiller SM, Puntambekar SS, von Saucken VE, Williams RY, Karlo JC, Moutinho M, et al. , 2018. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol. Neurodegener. 13, 29. 10.1186/s13024-018-0262-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran JN, Geier EG, Bonham LW, Newberry JS, Amaral MD, Thompson ML, Lasseigne BN, Karydas AM, Roberson ED, Cooper GM, et al. , 2020. Non-coding and loss-of-function coding variants in TET2 are associated with multiple neurodegenerative diseases. Am. J. Hum. Genet. 106, 632–645. 10.1016/j.ajhg.2020.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coletta A, Pinney JW, Solís DY, Marsh J, Pettifer SR, Attwood TK, 2010. Low-complexity regions within protein sequences have position-dependent roles. BMC Syst. Biol. 4, 43. 10.1186/1752-0509-4-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, Wang Y, 2016. TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 17, 201–207. 10.1038/nrn.2016.7. [DOI] [PubMed] [Google Scholar]
- Coppieters N, Dieriks BV, Lill C, Faull RL, Curtis MA, Dragunow M, 2014. Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain. Neurobiol. Aging 35, 1334–1344. 10.1016/j.neurobiolaging.2013.11.031. [DOI] [PubMed] [Google Scholar]
- Costa Y, Ding J, Theunissen TW, Faiola F, Hore TA, Shliaha PV, Fidalgo M, Saunders A, Lawrence M, Dietmann S, et al. , 2013. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 495, 370–374. 10.1038/nature11925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curzon P, Rustay NR, Browman KE, 2009. Frontiers in neuroscience cued and contextual fear conditioning for rodents. In: Buccafusco JJ. (Ed.), Methods of Behavior Analysis in Neuroscience. CRC Press/Taylor & Francis Copyright © 2009, Taylor & Francis Group, LLC. [PubMed] [Google Scholar]
- Dawlaty MM, Ganz K, Powell BE, Hu YC, Markoulaki S, Cheng AW, Gao Q, Kim J, Choi SW, Page DC, Jaenisch R, 2011. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 9, 166–175. 10.1016/j.stem.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del-Aguila JL, Fernández MV, Schindler S, Ibanez L, Deming Y, Ma S, Saef B, Black K, Budde J, Norton J, et al. , 2018. Assessment of the genetic architecture of Alzheimer’s disease risk in rate of memory decline. J. Alzheimers Dis. 62, 745–756. 10.3233/jad-170834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorszewska J, Prendecki M, Oczkowska A, Dezor M, Kozubski W, 2016. Molecular basis of familial and sporadic Alzheimer’s disease. Curr. Alzheimer Res. 13, 952–963. 10.2174/1567205013666160314150501. [DOI] [PubMed] [Google Scholar]
- Efthymiou AG, Goate AM, 2017. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 12, 43. 10.1186/s13024-017-0184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagiolini M, Jensen CL, Champagne FA, 2009. Epigenetic influences on brain development and plasticity. Curr. Opin. Neurobiol. 19, 207–212. 10.1016/j.conb.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez CG, Hamby ME, McReynolds ML, Ray WJ, 2019. The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer’s disease. Front. Aging Neurosci. 11, 14. 10.3389/fnagi.2019.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman SL, Kwon H, Portolano N, Parkin G, Venkatraman Girija U, Basran J, Fielding AJ, Fairall L, Svistunenko DA, Moody PCE, et al. , 2019. Heme binding to human CLOCK affects interactions with the E-box. Proc. Natl. Acad. Sci. U. S. A. 116, 19911–19916. 10.1073/pnas.1905216116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratuze M, Leyns CEG, Holtzman DM, 2018. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 13, 66. 10.1186/s13024-018-0298-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, et al. , 2013. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin PE, 2004. Transcription regulation within the circadian clock: the E-box and beyond. J. Biol. Rhythm. 19, 348–360. 10.1177/0748730404268052. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, et al. , 2015. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. 10.1016/s1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma S, Kawamoto T, Takagi Y, Fujimoto K, Sato F, Noshiro M, Kato Y, Honma K, 2002. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature 419, 841–844. 10.1038/nature01123. [DOI] [PubMed] [Google Scholar]
- Huang Y, Sun X, Jiang H, Yu S, Robins C, Armstrong MJ, Li R, Mei Z, Shi X, Gerasimov ES, et al. , 2021. A machine learning approach to brain epigenetic analysis reveals kinases associated with Alzheimer’s disease. Nat. Commun. 12, 4472. 10.1038/s41467-021-24710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Jopson T, Pelz C, Tafessu A, Fareh F, Zuloaga D, Marzulla T, Riparip LK, Stewart B, Rosi S, et al. , 2016. Short- and long-term effects of (56)Fe irradiation on cognition and hippocampal DNA methylation and gene expression. BMC Genomics 17, 825. 10.1186/s12864-016-3110-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FR, Visser PJ, Aalten P, Aarsland D, Alcolea D, et al. , 2015. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. Jama 313, 1924–1938. 10.1001/jama.2015.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarnot P, Ziemska-Legiecka J, Grynberg M, Gruca A, 2022. Insights from analyses of low complexity regions with canonical methods for protein sequence comparison. Brief. Bioinform. 23, bbac299. 10.1093/bib/bbac299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastano K, Mier P, Andrade-Navarro MA, 2021. The role of low complexity regions in protein interaction modes: an illustration in huntingtin. Int. J. Mol. Sci. 22 10.3390/ijms22041727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, Mansuy IM, 2008. Dendritic spine loss and synaptic alterations in Alzheimer’s disease. Mol. Neurobiol. 37, 73–82. 10.1007/s12035-008-8018-z. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT, Nixon RA, Jones DT, 2021. Alzheimer disease. Nat. Rev. Dis. Primers 7, 33. 10.1038/s41572-021-00269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli RM, Zhang Y, 2013. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479. 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlar AV, Trevino CE, Zwick ME, Cutler DJ, Wingo TS, 2018. Bystro: rapid online variant annotation and natural-language filtering at whole-genome scale. Genome Biol. 19, 14. 10.1186/s13059-018-1387-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoiselée HM, Nicolas G, Wallon D, Rovelet-Lecrux A, Lacour M, Rousseau S, Richard AC, Pasquier F, Rollin-Sillaire A, Martinaud O, et al. , 2017. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: a genetic screening study of familial and sporadic cases. PLoS Med. 14, e1002270 10.1371/journal.pmed.1002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Daujat S, Schneider R, 2016. Lateral thinking: how histone modifications regulate gene expression. Trends Genet. 32, 42–56. 10.1016/j.tig.2015.10.007. [DOI] [PubMed] [Google Scholar]
- Lee T, Lee H, 2020. Prediction of Alzheimer’s disease using blood gene expression data. Sci. Rep. 10, 3485. 10.1038/s41598-020-60595-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemma RB, Fleischer T, Martinsen E, Ledsaak M, Kristensen V, Eskeland R, Gabrielsen OS, Mathelier A, 2022. Pioneer transcription factors are associated with the modulation of DNA methylation patterns across cancers. Epigenetics Chromatin 15, 13. 10.1186/s13072-022-00444-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Wu G, Xiong F, Chen Y, 2021. Advances in the DNA methylation hydroxylase TET1. Biomark. Res. 9, 76. 10.1186/s40364-021-00331-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Shui Y, Wang F, Yamamoto R, Kato N, 2017. Impaired retention of depression-like behavior in a mouse model of Alzheimer’s disease. IBRO Rep. 2, 81–86. 10.1016/j.ibror.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matarin M.Salih, Dervis A., Yasvoina M, Cummings, Damian M, Guelfi., Liu W, Solim, Nahaboo, Muzammil A, Moens, Thomas G., Paublete, Rocio M, Ali, Shabinah S, et al. , 2015. A genome-wide gene-expression analysis and database in transgenic mice during development of amyloid or tau pathology. Cell Rep. 10, 633–644. 10.1016/j.celrep.2014.12.041. [DOI] [PubMed] [Google Scholar]
- Mendez MF, 2017. Early-onset Alzheimer disease. Neurol. Clin. 35, 263–281. 10.1016/j.ncl.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Klaus C, Pricop-Jeckstadt M, Miller JA, Struebing FL, 2019. A microglial signature directing human aging and neurodegeneration-related gene networks. Front. Neurosci. 13, 2. 10.3389/fnins.2019.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri F, Incarnato D, Krepelova A, Rapelli S, Pagnani A, Zecchina R, Parlato C, Oliviero S, 2013. Genome-wide analysis identifies a functional association of Tet1 and Polycomb repressive complex 2 in mouse embryonic stem cells. Genome Biol. 14, R91. 10.1186/gb-2013-14-8-r91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al. , 2006. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. 10.1523/jneurosci.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oblak AL, Lin PB, Kotredes KP, Pandey RS, Garceau D, Williams HM, Uyar A, O’Rourke R, O’Rourke S, Ingraham C, et al. , 2021. Comprehensive evaluation of the 5XFAD mouse model for preclinical testing applications: a MODEL-AD study. Front. Aging Neurosci. 13, 713726 10.3389/fnagi.2021.713726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okashita N, Kumaki Y, Ebi K, Nishi M, Okamoto Y, Nakayama M, Hashimoto S, Nakamura T, Sugasawa K, Kojima N, et al. , 2014. PRDM14 promotes active DNA demethylation through the ten-eleven translocation (TET)-mediated base excision repair pathway in embryonic stem cells. Development 141, 269–280. 10.1242/dev.099622. [DOI] [PubMed] [Google Scholar]
- Ou W, Yang J, Simanauskaite J, Choi M, Castellanos DM, Chang R, Sun J, Jagadeesan N, Parfitt KD, Cribbs DH, Sumbria RK, 2021. Biologic TNF-α inhibitors reduce microgliosis, neuronal loss, and tau phosphorylation in a transgenic mouse model of tauopathy. J. Neuroinflammation 18, 312. 10.1186/s12974-021-02332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantier R, Mullin N, Hall-Ponsele E, Chambers I, 2020. TET1 interacts directly with NANOG via independent domains containing hydrophobic and aromatic residues. J. Mol. Biol. 432, 6075–6091. 10.1016/j.jmb.2020.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YH, Pyun J-M, Hodges A, Jang J-W, Bice PJ, Kim S, Saykin AJ, Nho K, for the AddNeuroMed, c., and the Alzheimer’s Disease Neuroimaging, I, 2021. Dysregulated expression levels of APH1B in peripheral blood are associated with brain atrophy and amyloid-β deposition in Alzheimer’s disease. Alzheimers Res. Ther. 13, 183. 10.1186/s13195-021-00919-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini C, Pirazzini C, Sala C, Sambati L, Yusipov I, Kalyakulina A, Ravaioli F, Kwiatkowska KM, Durso DF, Ivanchenko M, et al. , 2021. A meta-analysis of brain DNA methylation across sex, age, and Alzheimer’s disease points for accelerated epigenetic aging in neurodegeneration. Front. Aging Neurosci. 13, 639428 10.3389/fnagi.2021.639428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perini G, Diolaiti D, Porro A, Della Valle G, 2005. In vivo transcriptional regulation of N-Myc target genes is controlled by E-box methylation. Proc. Natl. Acad. Sci. U. S. A. 102, 12117–12122. 10.1073/pnas.0409097102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsolt RD, Bertin A, Jalfre M, 1977. Behavioral despair in mice: a primary screening test for antidepressants. Arch. Int. Pharmacodyn. Ther. 229, 327–336. [PubMed] [Google Scholar]
- Qu W, Li L, 2020. Loss of TREM2 confers resilience to synaptic and cognitive impairment in aged mice. J. Neurosci. 40, 9552–9563. 10.1523/jneurosci.2193-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen KD, Helin K, 2016. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 30, 733–750. 10.1101/gad.276568.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RJ, Lewis ZA, Goll MG, 2019. DNA methylation: shared and divergent features across eukaryotes. Trends Genet. 35, 818–827. 10.1016/j.tig.2019.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A, Das S, Hyman BT, 2021. APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 20, 68–80. 10.1016/s1474-4422(20)30412-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierksma A, Lu A, Mancuso R, Fattorelli N, Thrupp N, Salta E, Zoco J, Blum D, Buée L, De Strooper B, Fiers M, 2020. Novel Alzheimer risk genes determine the microglia response to amyloid-β but not to TAU pathology. EMBO Mol. Med. 12 10.15252/emmm.201910606 e10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery CF, Beck JA, Harper L, Adamson G, Abdi Z, Uphill J, Campbell T, Druyeh R, Mahoney CJ, Rohrer JD, et al. , 2014. R47H TREM2 variant increases risk of typical early-onset Alzheimer’s disease but not of prion or frontotemporal dementia. Alzheimers Dement. 10, 602–608 e604. 10.1016/j.jalz.2014.05.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, et al. , 2011. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 29, 68–72 nbt.1732 [pii]. 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G, Wang G, Luo X, Cheng Y, Song Q, Wan J, Moore C, Song H, Jin P, Qian J, Zhu H, 2021. An all-to-all approach to the identification of sequence-specific readers for epigenetic DNA modifications on cytosine. Nat. Commun. 12, 795. 10.1038/s41467-021-20950-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steru L, Chermat R, Thierry B, Simon P, 1985. The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology 85, 367–370. 10.1007/bf00428203. [DOI] [PubMed] [Google Scholar]
- Stolz P, Mantero AS, Tvardovskiy A, Ugur E, Wange LE, Mulholland CB, Cheng Y, Wierer M, Enard W, Schneider R, et al. , 2022. TET1 regulates gene expression and repression of endogenous retroviruses independent of DNA demethylation. Nucleic Acids Res. 50, 8491–8511. 10.1093/nar/gkac642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre B, Flock G, Zacksenhaus E, Egan SE, 2002. Stra13 homodimers repress transcription through class B E-box elements *. J. Biol. Chem. 277, 46544–46551. 10.1074/jbc.M111652200. [DOI] [PubMed] [Google Scholar]
- Thome J, Coogan AN, Woods AG, Darie CC, Häßler F, 2011. CLOCK genes and circadian rhythmicity in Alzheimer disease. J. Aging Res. 2011, 383091 10.4061/2011/383091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Lista V, Gimenez-Llort L, 2014. Persistence of behaviours in the forced swim test in 3xTg-AD mice at advanced stages of disease. Behav. Process. 106, 118–121. 10.1016/j.beproc.2014.05.001. [DOI] [PubMed] [Google Scholar]
- van de Weijer MP, Jansen IE, Verboven AHA, Andreassen OA, Posthuma D, 2020. Chapter 22 - Genomics of Alzheimer’s disease. In: Baune BT. (Ed.), Personalized Psychiatry. Academic Press, pp. 275–283. 10.1016/B978-0-12-813176-3.00022-5. [DOI] [Google Scholar]
- van den Hove DL, Chouliaras L, Rutten BP, 2012. The role of 5-hydroxymethylcytosine in aging and Alzheimer’s disease: current status and prospects for future studies. Curr. Alzheimer Res. 9, 545–549. 10.2174/156720512800618008. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, et al. , 2015. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingo TS, Lah JJ, Levey AI, Cutler DJ, 2012. Autosomal recessive causes likely in early-onset Alzheimer disease. Arch. Neurol. 69, 59–64. 10.1001/archneurol.2011.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingo TS, Kotlar A, Cutler DJ, 2017. MPD: multiplex primer design for next-generation targeted sequencing. BMC Bioinform. 18, 14. 10.1186/s12859-016-1453-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zhang Y, 2017. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 18, 517–534. 10.1038/nrg.2017.33. [DOI] [PubMed] [Google Scholar]
- Yamazaki H, Jin Y, Tsuchiya A, Kanno T, Nishizaki T, 2015. Adipose-derived stem cell-conditioned medium ameliorates antidepression-related behaviors in the mouse model of Alzheimer’s disease. Neurosci. Lett. 609, 53–57. 10.1016/j.neulet.2015.10.023. [DOI] [PubMed] [Google Scholar]
- Yang G, Parkhurst CN, Hayes S, Gan WB, 2013. Peripheral elevation of TNF-α leads to early synaptic abnormalities in the mouse somatosensory cortex in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U. S. A. 110, 10306–10311. 10.1073/pnas.1222895110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Chen T, 2019. DNA methylation reprogramming during mammalian development. Genes (Basel) 10. 10.3390/genes10040257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Yao B, Shin J, Lin L, Kim N, Song Q, Liu S, Su Y, Guo JU, Huang L, et al. , 2016. Lin28A binds active promoters and recruits Tet1 to regulate gene expression. Mol. Cell 61, 153–160. 10.1016/j.molcel.2015.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhang X, Clark E, Mulcahey M, Huang S, Shi YG, 2010. TET1 is a DNA-binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res. 20, 1390–1393. 10.1038/cr.2010.156. [DOI] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, et al. , 2013. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720. 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhang K, Wang J, Liu Y, Liu G, Jin W, Wu S, Zhao X, 2020a. Immunoprecipitation and mass spectrometry define TET1 interactome during oligodendrocyte differentiation. Cell Biosci. 10, 110. 10.1186/s13578-020-00473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang Z, Li L, Xu K, Ma Z, Chow HM, Herrup K, Li J, 2020b. Selective loss of 5hmC promotes neurodegeneration in the mouse model of Alzheimer’s disease. FASEB J. 34, 16364–16382. 10.1096/fj.202001271R. [DOI] [PubMed] [Google Scholar]
- Zheng L, Zhai Y, Li N, Ma F, Zhu H, Du X, Li G, Hua J, 2016. The modification of Tet1 in male germline stem cells and interact with PCNA, HDAC1 to promote their self-renewal and proliferation. Sci. Rep. 6, 37414. 10.1038/srep37414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou CN, Chao FL, Zhang Y, Jiang L, Zhang L, Fan JH, Wu YX, Dou XY, Tang Y, 2019. Fluoxetine delays the cognitive function decline and synaptic changes in a transgenic mouse model of early Alzheimer’s disease. J. Comp. Neurol. 527, 1378–1387. 10.1002/cne.24616. [DOI] [PubMed] [Google Scholar]
- Zhou M, Xu R, Kaelber DC, Gurney ME, 2020. Tumor necrosis factor (TNF) blocking agents are associated with lower risk for Alzheimer’s disease in patients with rheumatoid arthritis and psoriasis. PLoS One 15, e0229819. 10.1371/journal.pone.0229819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary statistics of the targeted resequencing are shared as part of the manuscript reporting. The raw and processed targeted sequencing will be made available through NIAGADS after publication. We have deposited the 5hmC and RNA-seq data into the Gene Expression Omnibus at https://www.ncbi.nlm.nih.gov/geo/. The accession number is GSE226056.