

Summary
Radiation therapy (RT) provides therapeutic benefit for patients with glioblastoma (GBM), but inevitably induces poorly-understood global changes in GBM and its microenvironment (TME) that promote radio-resistance and recurrence. Through a cell surface marker screen, we identified that CD142 (Tissue factor or F3) is robustly induced in the senescence-associated β-galactosidase (SA-βGal)-positive GBM cells after irradiation. F3 promotes clonal expansion of irradiated SA-βGal+ GBM cells and orchestrates oncogenic TME remodeling by activating both tumor-autonomous signaling and extrinsic coagulation pathways. Intratumoral F3 signaling induces a mesenchymal-like cell state transition and elevated chemokine secretion. Simultaneously, F3-mediated focal hypercoagulation states lead to activation of tumor-associated macrophages (TAMs) and extracellular matrix (ECM) remodeling. A newly developed F3-targeting agent potently inhibits the above oncogenic events and impedes tumor relapse in vivo. These findings support F3 as a critical regulator for therapeutic resistance and oncogenic senescence in GBM, opening potential therapeutic avenues.
Keywords: Glioblastoma, Tissue factor, Senescence, Therapeutic resistance, Tumor microenvironment
Graphical Abstract
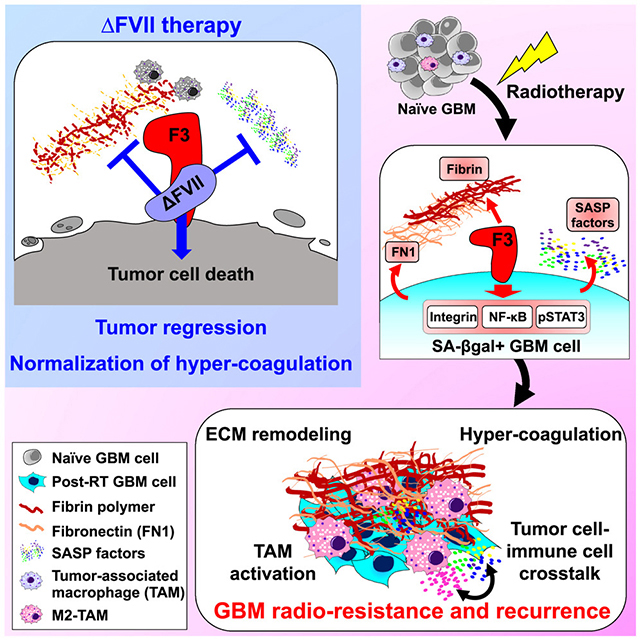
eTOC
Jeon et al. find that Tissue factor (F3) is robustly upregulated in irradiated glioblastoma (GBM) cells. F3 signaling promotes irradiation-induced global remodeling of GBM tumor and its microenvironment, leading to GBM radio-resistance and recurrence. A new F3-targeting agent potently inhibits the above oncogenic events and impedes tumor relapse in vivo.
Introduction
Glioblastoma (GBM) is the most lethal brain cancer with no curative therapies available. Recent anti-angiogenic and immunotherapy approaches have not yet shown durable clinical benefits for GBM patients1,2. Currently, maximal surgical resection followed by radiation therapy (RT) and temozolomide (TMZ) chemotherapy is standard-of-care therapy for newly diagnosed GBM patients3. Limited benefits from current therapies can be attributed to multiple factors, including inherent radio-resistance of GBM tumors, preferential survival of GBM stem-like cells, and treatment-induced activation of pro-tumorigenic adaptive pathways4–6.
RT exerts cytostatic and cytotoxic anti-tumor effects against GBM, but tumor relapse is almost inevitable. Following RT, GBM tumors undergo phenotypic transition via global remodeling of epigenetic and transcriptome landscapes. RT enhances the secretion of various chemokines, cytokines, and extracellular matrix (ECM) molecules by tumor cells7,8. Many of RT-induced chemo/cytokines are known to induce GBM cell state transition and mediate reciprocal crosstalk between GBM cells and immune cells9–11. In addition, RT is a potent inducer of coagulopathy and immune modulation12–14. While the above RT-induced global changes have long been hypothesized to account for tumor evolution and aggressiveness of recurrent tumors, the upstream initiators of these processes are poorly understood.
In many malignant tumors such as GBM, irradiation and targeted inhibitors transiently induce tumor cell senescence, referred to as therapy-induced senescence (TIS)15,16. Because the senescence program involves global epigenomic reprogramming and the elevated secretion of multiple proteins, referred to as the senescence-associated secretory phenotype (SASP), TIS may play a role in RT-induced global changes17,18. Tumor cell senescence remains poorly defined, with key questions unanswered including how senescent tumor cell states are regulated and how TIS affects treatment resistance and tumor recurrence. Here, we used an integrative approach to understand tumor cell senescence in the course of radiation resistance, combining the assays with a modified senescence-associated β-galactosidase (SA-βGal) substrate that allows sorting and live cell fate tracing of the senescent tumor cells, cell surface marker screening, single cell RNA sequencing, and profiling of transcriptome and chromatin landscapes. We demonstrate a causal link between RT-induced senescence and global oncogenic reprogramming and derive new therapeutic strategy to inhibit these processes.
Results
Irradiation-induced SA-βGal+ GBM cells harbor stemness and senescence-like features.
To determine the extent of senescence in GBM tumor, we utilized three different orthotopic patient-derived GBM models (Figure 1A). 827 and 022 GBM cells have homozygous deletion of Cdkn2a, a common genomic alteration found in GBM19. The brains of tumor-bearing mice were irradiated and the activity of SA-βGal were measured with either a colorimetric substrate X-Gal or a cell-permeable fluorogenic substrate C12FDG20,21 (Figures 1A and S1A). Irradiation induced strong SA-βGal activity especially in the tumor regions (Figure 1A). To determine the cell types of SA-βGal+ cells, we implanted red fluorescent protein (RFP)-transduced GBM tumors, irradiated the mice in vivo, and stained with C12FDG (Figure 1B). Flow cytometry analysis revealed that more than 90 % of SA-βGal+ cells in the irradiated brains were positive for RFP, indicating that primarily GBM cells acquire RT-induced SA-βGal activity (Figure 1B). To better cover inter-tumoral GBM genetic heterogeneity and to avoid model-dependent biases, we utilized multiple patient-derived GBM slices and organoids (Details are in Method sections)22–24. Across all GBM samples tested, we found robust induction of SA-βGal activity after RT (Figure S1 B and C).
Figure 1. Radiation-induced SA-βGal+ GBM cells clonally expand in vivo.

(A) Representative images of SA-βGal activity in the brain sections. GBM tumor-bearing mice were not irradiated or irradiated (naïve and RT, respectively) and stained with X-gal or C12FDG five days later. Tumor regions were shown in H&E images. Different patient-derived GBMs are designated as numbers. * p < 0.001 by one-way ANOVA.
(B) Proportion of RFP+ cells (human GBM cells) and RFP− cells (mouse cells) within C12FDG+ populations (n = 3 per each).
(C) Immunoblots of DNA damage response (DDR) proteins in sorted RT-C12FDG+ and C12FDG− GBM subpopulations. β-actin was used as a loading control.
(D) In vitro clonogenic growth of irradiated GBM subpopulations. Single cells from each subpopulation were plated in soft agar, cultured for 3 weeks, and resultant colonies counted. * p < 0.001 by one-way ANOVA with Tukey’s multiple comparison test.
(E) Flow cytometry plot of C12FDG and Nestin staining in naïve and RT GBM cells. Quantitation of C12FDG+ / Nestin+ cells are shown. * p < 0.001 by one-way ANOVA.
(F) Immunofluorescence (IF) staining images of C12FDG and senescence markers (HP1γ and H3K9me3).
(G) Flow cytometry plot of H3K9me3 and Nestin staining in naïve, RT-bulk, and RT-C12FDG+ GBM cells. * p < 0.001 by one-way ANOVA with Tukey’s multiple comparison test.
(H) t-distributed Stochastic Neighbor Embedding (t-SNE) plots of GBM single cells in the naïve, RT-bulk, and RT-C12FDG+ GBM cells (total of 105,653 cells). Color gradient was overlaid with stemness signature scores. * p < 0.001 by Mann-Whitney test.
(I) Immunoblots of H3K9me3 and GBM stemness markers (Nestin and Sox2) proteins in three matched sets of RT-C12FDG + and RT-C12FDG − GBM subpopulations.
(J to L) Barcode-mediated clonal expansion and clonal diversity determination analysis. (J) Experimental schematic. (K) Floating bar plots of RFP-labeled cell populations in vivo. Center line in each bar represents mean value (n=4 animals per group). (L) Numbers of unique barcodes and barcode distribution within the tumors derived from the indicated cell populations. * p < 0.01 by two-way ANOVA. NS, not significant.
(M) In vivo limiting dilution tumor formation results using naïve, RT-bulk, and RT-C12FDG+ GBM cells. P values were determined by two-way ANOVA. Results are presented as mean ± SD. See also Figure S1.
To investigate cellular states of post-irradiated SA-βGal+ GBM cells, we separated SA-βGal+ and SA-βGal− GBM subpopulations by C12FDG staining and subsequent fluorescence-activated cell sorting (FACS). DNA damage repair activity and clonal growth capacity are critical for GBM radio-resistance and subsequent recurrence4. C12FDG+ cells after irradiation (RT-C12FDG+) showed enhanced activities of ATM and DNA checkpoint kinases, relative to matched C12FDGlow/− or mock-sorted bulk tumor cells (Figure 1C). Furthermore, RT-C12FDG+ cells expressed Nestin (a representative GBM stemness marker) and were highly enriched with clonogenic cells, as determined by in vitro colony forming analysis (Figure 1 D and E). Next, we determined the levels of representative senescence markers in GBM tumors with or without irradiation. RT-C12FDG+ cells had high levels of HP1Jand H3K9me3 but little or no expression of CDKN2A (also known as p16, a cell-cycle arrest-associated senescence marker) (Figure 1 F to I and data not shown). Notably, RT increased the number of GBM cells that express both Nestin and H3K9me3, which were further enriched in RT-C12FDG+ cells (Figure 1G).
To examine global transcriptomes of SA-βGal+ GBM cells, we performed single-cell RNA sequencing (scRNA-seq) and bulk RNA seq analysis. In total, we profiled about 100,000 single GBM cells and classified these cells based on the expression levels of stemness and cell cycle progression25–27. Both naïve and irradiated GBM cells contained the cell populations with high expression levels of the cell cycle and/or stemness gene signatures, reflecting their aggressive nature. Notably, RT-C12FDG+ subpopulation harbored most of the single cells that have the highest scores for the stemness gene signature (Figures 1 H and I).
Cellular senescence has been extensively studied in irradiated IMR90 or WI38 cells (non-transformed human fibroblasts). Consistent with a traditional view of senescence, SA-βGal+ IMR90 or WI38 cells did not proliferate (Figure S1 D and E). In contrast, bulk RNA sequencing analysis of matched C12FDG+ and C12FDGlow/− GBM cells after radiation showed that many of the cell cycle signature genes, including PCNA, BUB1, and FoxM1 are highly expressed in C12FDG+ cells28,29 (Figure S1 F to H). These data suggest that SA-βGal+ GBM cells do not exhibit the irreversible cell cycle arrest state.
To formally test whether RT-SA-βGal+ GBM cells can clonally expand and contribute to post-RT tumor growth, we used the lentiviral-mediated barcode/RFP transduction technology to label GBM cells. We isolated C12FDG+ cells from the irradiated, labeled GBM tumors and immediately injected these cells into the brains of new recipient mice (Figure 1 J to M). RT-C12FDG+ GBM cells generated significantly larger tumors in new recipient mice, compared to non-irradiated (naïve) or irradiated bulk tumor cells (Figure 1K). Clonal expansion analysis by barcode sequencing revealed that larger numbers of individual barcodes were detected in the tumors derived from RT-C12FDG+ GBM cells compared to those of naïve or RT-bulk cells (Figure 1L). This trend corroborates well with the current notion of the clonal diversity in cancer following treatment30,31. Lastly, we performed in vivo limiting dilution tumor formation assays using the above cell populations. RT-C12FDG+ cells derived from two different patient GBMs contained higher frequencies of tumor-forming cells than RT-bulk- or C12FDG− cells (Figure 1M). Collectively, these data suggest that irradiation-induced SA-βGal+ GBM cells harbor stemness and some of senescence-like characteristics and that they are a cell population contributing to post-RT tumor growth.
F3, highly expressed in irradiated C12FDG+ GBM cells, is associated with stemness, cell state transition, and an enhanced secretory phenotype.
To investigate cellular states and molecular regulators of SA-βGal+ GBM cells, we profiled levels of cell surface proteins. Matched naïve and irradiated GBM cells (20 million cells each) were stained with C12FDG, split into individual wells, and co-stained with each of 242 human clusters of differentiation (CD) antibodies (Figure 2A). Co-staining results were quantitated by flow cytometry and expression level of each CD marker was calculated. Enriched cell surface receptors in RT-C12FDG+ cells included ABCG2, JAMA, PDGFR, CD109 and integrin proteins, implicated in stemness, GBM mesenchymal transition, and therapeutic resistance (Figure 2B)32–34. The most enriched CD marker in our screen was CD142 (Tissue factor or F3), originally identified as a cell surface receptor that initiates blood coagulation35. Tissue factor/F3 levels were increased over 10-fold in irradiated cells compared to the matched naïve cells, further increased in RT-C12FDG+ cells (Figure 2B). In the irradiated PDX tumor-bearing mice, we found the robust F3 induction, co-localized with C12FDG+ tumor cells (Figure 2C). This trend was further confirmed in GBM spheroids, GBM organoids and patient GBM slices (Figure S2 A to G).
Figure 2. F3, highly expressed in irradiated C12FDG+ GBM cells, is associated with stemness, cell state transition, and an enhanced secretory phenotype.

(A) A schematic of the cell surface marker screen. (B) The lists of the cell surface proteins that were enriched in RT-bulk and RT-C12FDG+ GBM cells compared to naïve cells.
(C) IF staining images of C12FDG and F3 in the brains of PDX tumor-bearing mice. C12FDG+/ F3+ double-positive cells were quantitated (n=3 per each tumor). * p < 0.001 by one-way ANOVA.
(D and E) In vivo proliferation of RT bulk, RT-F3+, and RT-F3− cell populations (D) and in vivo limiting dilution tumor formation results. * p < 0.01 by two-way ANOVA.
(F) Enriched transcription factor-binding motifs in RT-F3+ subpopulations, as determined by ATAC sequencing analysis.
(G and H) Immunoblots of pp65, pIκBα, EZH2, active-β-catenin, Sox2, and HUTS4 proteins in the RT-F3+ and RT-F3− GBM subpopulations. For detection with HUTS4 antibody (specific for the activated form of ITGB1), non-denaturing gels were used.
(I) Quantitation of stemness gene set expression in naïve, RT-bulk, RT-F3+, and RT-F3− GBM single cells (total of 28,890 cells).
(J) Immunoblots of MES GBM transition-associated proteins (pSTAT3, IL6, and YKL-40).
(K) IF images of F3 and CD44 in matched naïve and irradiated GBM tissue slices and quantitation.
(L) Quantitation of SASP factor gene set expression in the above subpopulations.
(M) Heatmap plots and quantitation of the secreted proteins from naïve, RT-bulk, and RT-F3+ GBM cells. Results are mean ± SD. * p < 0.001 by Mann-Whitney test in I and L. See also Figure S2 and Table S1.
Two F3 canonical functions are initiation of blood coagulation and promotion of cell survival via intracellular signaling. Coagulation factor VII (F7, a cognate F3 ligand) is mainly produced in the liver and circulates in the blood stream as an inactive pro-enzyme. Upon interaction with F3, F7 is converted into a protease-active form (FVIIa), which then initiates coagulation by catalyzing a thrombin-producing protease cascade36,37. Intracellular signaling is mediated by activation of integrin and the protease-activated receptor (PAR), leading to activation of MAPK, PI3K, and NFκB38,39. While F3 is expressed in various tumor types—including gliomas—and generally exerts pro-tumor effects40–42, the role(s) of F3 in RT responses and senescence are largely unknown.
To test whether post-irradiated F3high cells are enriched for in vivo tumorigenic capacity, we isolated matched F3high and F3low/− cells from PDX tumors 5 days after in vivo radiation by F3 antibody-based cell sorting, and then injected these cells into immunodeficient mice. Compared to matched F3low/− or mock-sorted bulk tumor cells, post-irradiated F3high subpopulations yielded significantly higher capacities for tumor formation (Figure 2 D and E).
To gain insight into signaling nodes associated with F3, we performed ATAC (assay for transposase-accessible chromatin)-seq analysis to map genome regions with open chromatin structures in matched F3high and F3low/− subpopulations. The most enriched transcription factor-binding motifs in RT-F3+ GBM cells are STAT3 and FOSL2 (master transcriptional factors for the GBM mesenchymal (MES) transcriptional network), RELA (NFκB, a master regulator for senescence and GBM cell survival), as well as Foxo4 and TEAD2 (implicated in tumor dormancy and senescence)43–46, most of which were also enriched in RT-C12FDG+ GBM cells (Figures 2F and S2I). Following RT, GBM tumors undergo phenotypic transition toward MES subtype, which is mediated by activation of NFκB and STAT3 signaling9,45. We then determined the activation status of these pathways by immunoblots, immunostaining, and transcriptome analysis (Figures 2 G to L and S2 A to K). Consistent with ATAC data, irradiated F3high GBM cells have elevated levels of active NFκB and STAT3 as well as stemness-associated proteins (Sox2, EZH2, and active β-catenin and integrin β147,48 (Figures 2 G and H, and S2 H to K). Active integrin signaling in this subpopulation is further supported by additional surface marker screening using anti-F3 antibody and 240 human CD markers. Similar to the C12FDG co-staining data, several different integrin family proteins were highly enriched in irradiated F3high GBM cells (Table S1). In addition, RT-F3+ cells have high levels of GBM mesenchymal markers (YKL-40, CD44, and active STAT3) (Figures 2J, 2K and S2J)49. Lastly, secretome profiling and gene signature analysis showed that irradiated F3high GBM cells secreted significantly higher levels of SASP factors such as IL6, IL8, HGF, and EGF, relative to bulk naïve or irradiated tumor cells (Figure 2 L and M). Together, these data indicate that the RT-F3+ populations have enriched traits of stemness, mesenchymal GBM cell transition, senescence-like epigenomic reprogramming, and SASP.
F3 signaling primes radiation-induced changes both in GBM and the TME.
To probe the roles of F3 in GBM radiation responses in vivo, we performed immunohistochemical staining analyses using human PDX, GBM slice, and syngeneic mouse glioma models (Figure 3). Orthotopic PDX tumor-bearing mice were irradiated, and the brain tissues harvested 5 days later. In regions with a robust upregulation of F3, we found that F3 positively correlated with high levels of fibrin, a key effector in the F3-initiaited coagulation cascade (Figure 3A). F3-positive GBM cells were also positive for YKL-40 and CD44 (MES-like GBM markers) and fibronectin 1 (FN1, an ECM protein and MES-like GBM marker)9,50,51. Fibrin polymer and FN1 are known to form a mesh-like structure together, referred to as an oncogenic provisional ECM. It provides a scaffold for recruitment of macrophages, activated platelets, neutrophil extracellular traps (NETs), and traps various growth factors and chemokines52. Indeed, we found significant increases in the numbers of TAMs and M2-like TAMs (CD163+) in F3-positive, fibrin/FN1 complex-rich regions in the irradiated tumors (Figures 3B, 3C, and S3A).
Figure 3. Radiation-induced F3 prime global changes both in tumor and microenvironment.

(A) Immunohistochemical (IHC) staining images of F3, fibrin, and YKL-40 in 827 GBM tumors. Tumor-bearing mice were irradiated in vivo and tumors were harvested 5 days later.
(B and C) IF images of fibrin, FN1(an ECM molecule), F4/80, CD163 (M2-like TAM) staining. Fibrin+/FN1+ areas and numbers of YKL-40+, F4/80+, and CD163+ cells were quantitated (n = 4 per each). * p < 0.001 by one-way ANOVA.
(D to F) IF images of fibrin, IBA1, CD163, CD44, F3 in syngeneic mouse gliomas and quantitation. * p < 0.001 by one-way ANOVA with Tukey’s multiple comparison test.
(G) Correlation between the levels of fibrin polymers and CD163+ TAMs in the brain sections of tumor-bearing mice with or without RT. r value determined by Pearson’s correlation coefficient analysis. Results are presented as mean ± SD. * p < 0.001. See also Figure S3.
Human GBM slices resected from newly diagnosed GBM patients maintain in vivo tumor architecture and TME including immune cells, endothelial cells, and astrocytes; thus, they can mimic acute responses of human GBM in situ24. We prepared acute GBM slices within 6 hours post-surgery, irradiated them ex vivo, and then processed for immunostaining and cytokine array analysis 3 days later (Figure S3B). Fibrinogen (precursor of fibrin) and F7 were detected in these GBM slices. Irradiation resulted in robust induction of fibrin/FN1 complexes and CD163+ macrophages. Cytokine analysis utilizing 4 sets of matched naïve and irradiated GBM tissue slices consistently showed higher levels of secreted proteins in the conditioned media from irradiated GBM slices (Figure S3C). These data suggest a causal relationship between that RT and fibrin polymerization, an enhanced secretory phenotype, and TAM polarization.
To validate the above findings in the intact immune microenvironment, we employed a PDGFβ-driven, p53-null, syngeneic mouse glioma, a representative proneural subtype tumor53. Naïve tumors showed relatively low basal levels of fibrin, TAMs and CD44+ cells. Upon irradiation, however, we found massive increases in fibrin polymerization and TAM infiltration, as well as in levels of F3 and CD44 (Figure 3 D to F). Notably, most of the CD163+, CD206+ M2-like TAMs were detected in the fibrin polymer-enriched, F3+, CD44+ tumor regions, indicating a strong positive correlation between them (Figure 3G). Together, these data further suggest a link between RT-induced coagulation, oncogenic TAMs, and GBM mesenchymal transition.
Molecular mechanisms of F3 signaling in GBM reprogramming and radio-resistance
Given strong spatiotemporal associations between F3 and RT-induced GBM remodeling (Figure 3), we first determined the roles of F3 in vivo via shRNA-mediated F3 knockdown. Non-targeting shRNA or F3 knockdown (KD) shRNA-expressing GBM cells were transplanted into the brains of nude mice and in vivo radiation was started 20 days later (Figure 4 A and B). Immunostaining analysis of the tumor-bearing brain sections revealed that F3 suppression significantly abrogated RT-induced fibrin polymers and accumulation of IBA1+ and CD44+ cells (Figure 4A). F3 knockdown or irradiation alone extended the survival of tumor-bearing mice compared to the control group. Notably, the group injected with F3 KD cells and given radiation showed far longer survival than all other groups (p <0.001 by log-rank analysis) (Figure 4B).
Figure 4. F3 knockdown suppresses radiation-induced coagulation, SASP factor secretion and TAM activation.

(A) IHC images of fibrin, IBA1, and CD44 in the brains of 827 tumors expressing either non-targeting (NT) or F3 shRNAs (KD) with or without RT.
(B) Kaplan-Meier survival curves of mice in (A). n=8 for each group. p < 0.001 by log-rank analysis.
(C) Immunoblots to determine the activation status of NFκB and STAT3, and integrin in F3 KD GBM cells with or without RT.
(D) Luciferase reporter assays to measure transcriptional activities of NFκB and STAT3 signaling.
(E) Survival of irradiated F3 KD cells with forced activation of STAT3 or NFκB.
(F) Levels of the secreted proteins from GBM cells with or without F3 KD and RT.
(G) Recruitment of macrophage (Mφ)-like U937 cells co-cultured with the above cell groups.
(H and I) Levels of cytokine secretion in the cells with forced activation of STAT3 or NFκB (H) or inhibition of NFκB, STAT3, integrin signaling (I). Details of inhibitors are in Method section. SASP factor index is an arbitrary unit calculated from total amounts of the secreted proteins.
(J) IHC images of doxycycline-inducible F3 shRNA expressing tumors. Tumors were harvested 7 days after doxycycline treatment. * p < 0.001 by one-way ANOVA with Tukey’s multiple comparison test in D, E, G, H, and I. See also Figure S4.
We then determined the effects of F3 knockdown on the activation status of NFκB, STAT3, and integrin signaling (Figures 4 and S4). In the naïve state, F3 knockdown did not induce significant decreases in pp65 and pSTAT3, possibly reflecting low levels of F3. In contrast, F3 knockdown potently impeded RT-induced upregulation of NFκB, STAT3, and integrin activities, as well as mesenchymal traits (YKL-40 and FN1) (Figures 4 C and D, S4 A and B). Conversely, over-expression of constitutively active mutants of NFκB or STAT3 signaling rescued cell survival of RT-F3 KD cells, indicating that both NFκB and STAT3 activities are key downstream effectors of RT-induced F3 signaling (Figure 4E).
As RT-F3+ GBM cells secrete high levels of SASP factors (Figure 2M) and NFκB activity is a major regulator of SASP factor secretion, we determined the levels of cytokines/chemokines secreted by F3 KD GBM cells with or without irradiation. RT-induced upregulation of multiple cytokine/chemokines was significantly reduced by F3 knockdown (Figures 4F and S4C). To investigate the roles of F3 in chemokine-mediated TAM recruitment, we adapted in vitro transwell assays using U937 macrophage-like cells (primed U937)54. Conditioned media from irradiated GBM cells attracted significantly larger numbers of U937 cells compared to conditioned media from matched naïve cells. F3 knockdown reduced the levels of macrophage recruitment by ~80% (Figure 4G). Forced activation of NFκB activity but not STAT3 rescued SASP factor secretion in irradiated F3 knockdown cells and NFκB suppression by p65 shRNA significantly inhibited RT-induced SASP factor secretion, suggesting that RT-induced SASP in GBM is largely dependent on NFκB signaling (Figure 4 H and I).
Conversely, over-expression of F3 in naïve GBM cells showed elevated levels of active NFκB, STAT3 and integrin signaling (Figure S4D). Consistent with this, F3 over-expressing cells were more proliferative in the culture media without the added growth factors and these cells had the enhanced SASP factor secretion compared to the control (Figure S4 E and F). Lastly, the roles of F3 in RT responses in tumor and TME in vivo were further confirmed in a doxycycline-mediated inducible F3 KD system (Figure 4J). These data support that F3 is a critical regulator for the survival and cell state transition of irradiated GBM cells, as well as for SASP factor secretion.
Recombinant ΔFVII protein impeded radiation-induced coagulation, SASP factor secretion, and TAM accumulation in vivo.
The above data collectively suggest that F3 signaling is a critical regulator for post-RT tumor growth and drives an oncogenic TME, making it a potential therapeutic target. Since F3 protein, after binding to its ligand F7, is eventually degraded via the ubiquitin pathway, we hypothesized that specific F7 derivatives may directly trigger F3 degradation without eliciting F3-mediated oncogenic effects55. To test, we over-expressed a series of F7 deletion mutants in GBM cells via lentiviral transduction and determined the proliferation of these GBM cells after irradiation (Figure 5A). Through this screen, we found a deletion mutant that potently impeded the growth of irradiated GBM cells, which we designated as ΔFVII (141-amino acid protein without the F7 protease domain).
Figure 5. ΔFVII treatment impeded RT-induced coagulation, SASP factor secretion, and TAM accumulation in vivo.

(A) Schematic of F7 deletion mutant structures.
(B) Co-immunoprecipitation (IP)-immunoblots of F3 and Ubiquitin (Ub) in naïve and irradiated GBM cells treated with ΔFVII recombinant protein for 1 day. Levels of ubiquitinated F3 proteins were quantitated by densitometry.
(C to E) Immunoblots of F3 (C), pp65 and pSTAT3 (D), and integrin signaling components (E) in irradiated GBM cells treated with ΔFVII for 1 day. (E) Co-IP blots of F3-integrin β1 (ITGB1) immunocomplexes.
(F) Cell survival of normal brain cells and irradiated GBM cells treated with ΔFVII. Irradiated GBM cells, NPCs, and astrocytes were cultured with various concentrations of ΔFVII for 3 days and cell survival was determined by MTT assay.
(G) Live-cell imaging of RT-F3+ 022 GBM cells treated with ΔFVII. Cell growth was monitored in real-time over 12 days.
(H) Quantitation of the secreted proteins using the conditioned media from irradiated GBM (022 and 827) cells treated with ΔFVII for 1 day.
(I) Representative staining images of fibrin, CD163, CCR2, and cleaved-caspase-3 (C-cas3) in the 827 GBM-derived PDX tumors treated with RT, ΔFVII, or both.
(J) Immunoblots of F3, pSTAT3, BBC3, C-cas3, bFGF, HGF, and H3K9me3 proteins using GBM tumor lysates.
(K) Levels of the secreted proteins from the above tumor sets.
(L) Tumor sizes and fibrin staining in the above tumor sets harvested 5 days after RT.
(M) Tail bleeding times of tumor-bearing mice treated with RT, ΔFVII, or both. n=3 for each group.
(N) Tumor volumes of the above groups (n = 6). Results are presented as mean ± SD. * p < 0.001 by one-way ANOVA with Tukey’s multiple comparison test in B, F, G and N. * p < 0.001 by one-way ANOVA in C, H, and M. See also Figure S5.
ΔFVII recombinant protein lacked the pro-coagulation activity unlike wild-type F7 or FVIIa, as determined by in vitro coagulation assays (Figure S5A). Instead, ΔFVII robustly induced ubiquitin-mediated degradation of F3 proteins and significantly reduced the level of F3 proteins in irradiated GBM cells (Figure 5 B and C). Consistent with this, irradiated GBM cells treated with ΔFVII for a day showed a significant decrease in phosphorylated p65 and STAT3 proteins (Figure 5D). Furthermore, we found that ΔFVII decreased the levels of the co-immunoprecipitated F3-integrin complexes, HUTS4 (specific for active integrin β1), and phosphorylated FAK (an immediate downstream effector of integrin signaling) (Figures 5E and S5B). We then determined the effects of ΔFVII on survival and clonogenic growth of GBM cells (Figures 5 F and G). ΔFVII treatment effectively impaired survival of irradiated GBM cells with an IC50 in the 0.1 to 1 nM range (Figure 5F). In contrast, normal neural progenitor cells (NPCs) and primary astrocytes did not exhibit cytotoxicity even at micromolar ΔFVII concentration (Figure 5F). Unlike GBM cells, normal brain cells showed little change in the levels of pFAK and pERK after ΔFVII treatment, partially explaining GBM-specific cytotoxicity of ΔFVII (Figure S5C). Real-time cell imaging analysis demonstrated that ΔFVII treatment potently impaired clonogenic growth of irradiated F3high GBM cells (Figure 5G). GBM cells treated with ΔFVII showed disruption of mitochondrial membrane potential and defective mitochondrial structure (Figure S5 D to F). Lastly, to determine the effects of ΔFVII on SASP factor secretion, we treated irradiated GBM cells with ΔFVII for 1 day and collected the conditioned culture media. ΔFVII treatment significantly diminished the secretion of cytokines (Figure 5H). Together, these data suggest that ΔFVII is a potent anti-GBM agent that mitigates F3-mediated oncogenic signaling, especially in combination with RT.
To determine in vivo effects of ΔFVII, we started with subcutaneous tumor models. We irradiated human PDX tumors focally when the tumors reached ~500 mm3 and administered ΔFVII protein (50 μg/kg body weight) via intravenous injection. Compared to the naïve controls, tumors harvested 5 days after radiation showed marked increases in fibrin/FN1 complexes, tumor-infiltrating CD11b+, F4/80+, CCR2+ immune cells and CD163+ M2-like TAMs, along with massive upregulation of F3 (Figure 5I and data not shown). Notably, ΔFVII treatment potently inhibited all the above changes, with a dramatic increase in the number of cleaved-caspase 3 (C-Cas3)+ cells (Figure 5I). To evaluate in vivo effects of ΔFVII on signaling pathway activation and senescence-like characteristics, we harvested the tumors from each group and processed for further analyses. ΔFVII treatment significantly reduced RT-induced cell survival signaling such STAT3, but increased levels of cell death-associated proteins in tumor (Figure 5J). RT-induced SA-βGal+ reactivities were significantly decreased by ΔFVII treatment and clonogenic capacities of sorted RT-C12FDG+ cells isolated from these tumors were significantly abolished by in vivo ΔFVII treatment (Figure S5 G and H). To analyze the chemokine/cytokine microenvironment within the tumors treated with radiation, ΔFVII, or both agents, we performed cytokine profiling assays using tumor lysates. Irradiation induced significant upregulation of secreted proteins, many of which have well-known functions for macrophage recruitment, M2-like TAM polarization, and cell state transition. Notably, ΔFVII treatment significantly and globally repressed the chemokine/cytokine levels (Figures 5K and S5I).
To determine the effects of tumors and ΔFVII on systemic hemostasis, we measured the blood clotting activities by standard tail bleeding assays (Figure 5M). Compared to the control mice, tumor-bearing mice had shorter blood clotting time, which was further shortened by irradiation. While ΔFVII treatment on non-tumor bearing mice did not affect blood clotting time, the combination of irradiation and ΔFVII reverted hypercoagulation state in the RT-tumor bearing mice to near-normal range (Figure 5M). Studies of tumor growth kinetics showed that irradiation or ΔFVII monotherapy alone suppressed tumor growth rate to about 50 to 60 % of that of naive tumors, but the effect of either agent alone was transient and led to rapid regrowth (Figure 5L and 5N). In contrast, combination therapy yielded near-complete tumor regression (Figure 5N).
ΔFVII therapies radio-sensitize GBM tumors in orthotopic PDX models.
We tested the effects of ΔFVII recombinant protein in orthotopic GBM PDX models using patient-derived 022 or 827 GBM cells. Irradiation was started 20 days (for 827 tumor) or 25 days (for 022 tumor) after tumor cell implantation and ΔFVII protein was delivered via intravenous injection, concurrently with irradiation (Figures 6A and S6A). Five days after irradiation, the brains from each group were harvested for immunostaining (n=3) and immunoblot analysis through tumor dissection (n=3). Combination treatment with irradiation and ΔFVII greatly diminished RT-induced fibrin accumulation, MES cell state transition, TAM polarization, and activities of integrin and NFκB signaling, but increased numbers of C-cas3+ cells (Figure 6 A to D). Notably, histological examination of the brain sections from the mice receiving combined treatment at 38 days after implantation (the time when RT-treated mice were sacrificed) showed few tumor cells and only faint staining for fibrin polymers (Figure 6E). We also determined the effects of orthotopic GBM tumors and ΔFVII on systemic hemostasis by D-dimer and tail bleeding assays (Figure 6 F and G). Similar to subcutaneous tumor results (Figure 5M), ΔFVII treatment potently inhibited the hypercoagulatory state in RT-tumor bearing mice (Figure 6 F and G). Lastly, irradiation or ΔFVII monotherapy alone extended survival of tumor-bearing mice by about 10 days compared to the control group (Figure 6H). Notably, the group that received combination therapy showed a far greater survival extension, with about 50 % of mice surviving and lacking detectable tumors three months later (Figure 6H).
Figure 6. ΔFVII therapies radio-sensitize GBM tumors in orthotopic PDX models.
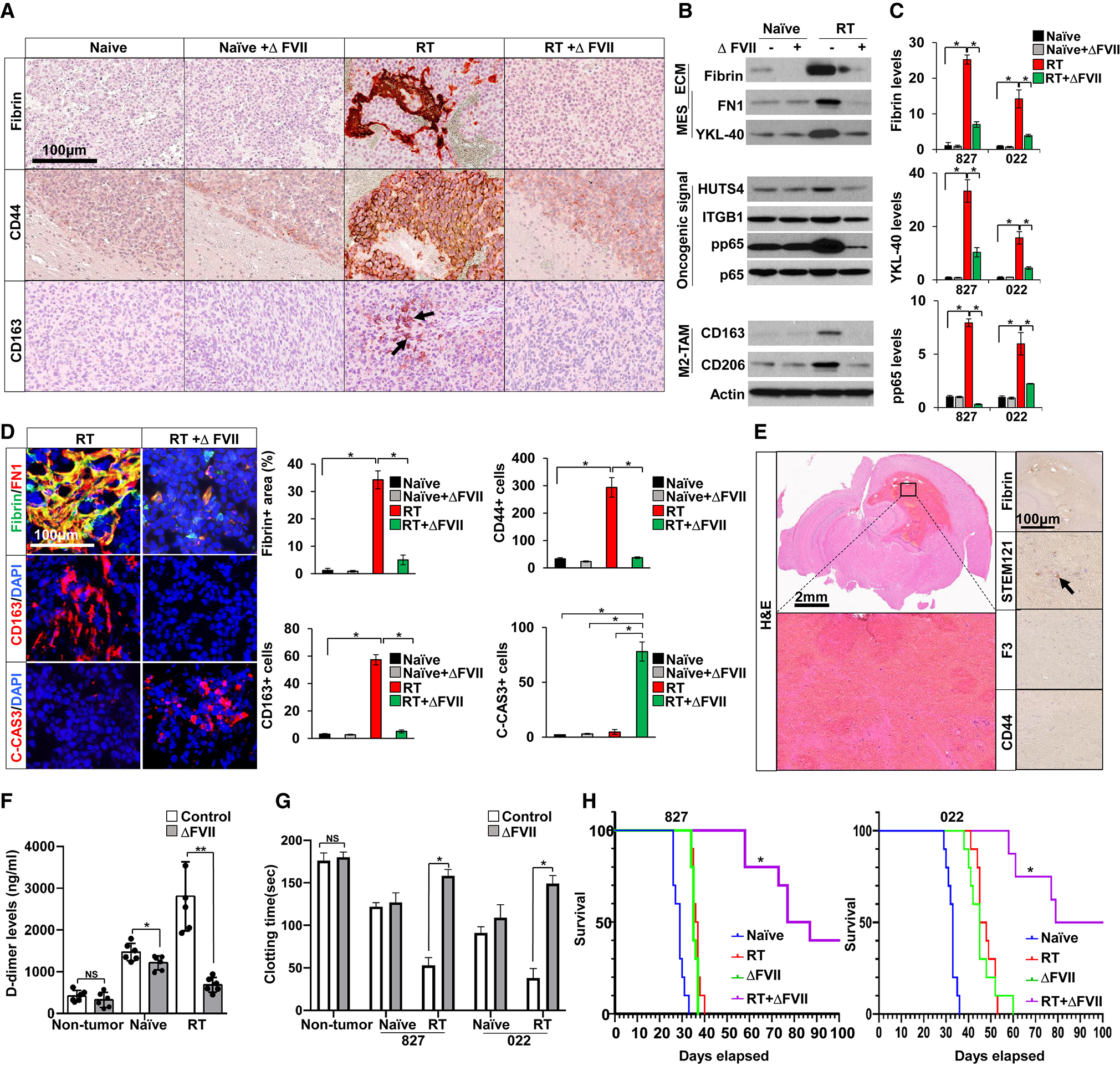
(A) IHC images of fibrin, CD44, and CD163 in orthotopic 827 GBM tumor-bearing mice treated with RT, ΔFVII, or both. Recombinant ΔFVII protein (50 μg/kg body weight) was administered via intravenous injection daily, concurrent with irradiation.
(B and C) Representative immunoblots of Fibrin, FN1, YKL-40, HUTS4, pp65, CD163, and CD206 in GBM tumors (B) and quantitation in (C).
(D) IF images of fibrin/FN1, CD44, CD163, and C-cas3 and quantitation. * p < 0.001 by one-way ANOVA with Tukey’s multiple comparison test in C and D.
(E) H&E staining images of the mouse brain sections from combination treated group. Human tumor cells were stained with STEM121 antibody (human cell-specific marker).
(F and G) D-dimer levels in plasma (F) and tail bleeding time determination (G). N=3 for each group. * p < 0.05, ** p < 0.001 by one-way ANOVA in F. * p < 0.001 by one-way ANOVA in G.
(H) Kaplan-Meier survival curves of tumor bearing mice treated with radiation, ΔFVII, or both. n=10 for each group. Combination-treated group showed a significant survival extension, compared to all other groups. p < 0.001 by log-rank analysis. See also Figure S6.
While several studies have reported that the blood-brain barrier (BBB) is functionally disrupted in some GBM56, the highly infiltrative GBM cells in the neighboring brain parenchyma presents technical challenges in developing effective anti-GBM therapeutics. As an independent but complementary therapeutic approach to ΔFVII recombinant protein, we developed a neural progenitor cell (NPC)-based cellular vector system. NPCs have intrinsic tumor-homing properties and survive well in the brains57,58. Lentivirally transduced ΔFVII-expressing NPCs were viable and maintained stable levels of secreted ΔFVII (Figure S6B). Upon co-culture with irradiated GBM cells, ΔFVII-expressing NPCs but not the control NPCs induced massive GBM cell death (Figure S6B). To mimic a clinical scenario in newly diagnosed human glioma patients, we injected ΔFVII-expressing NPCs into the established PDX tumors, followed by head-only irradiation two days later (Figure S6 C and D). Similar to the RT and ΔFVII combination, co-treatment with RT and ΔFVII-NPCs greatly diminished RT-induced F3 induction and activities of pro-tumorigenic signaling, leading to longer survival of tumor-bearing mice (Figures S6 C to E).
Recurrent tumors from early-relapse GBM patients harbor the upregulated gene signatures for the senescence and coagulation pathways.
Comparison of matching primary and recurrent GBMs can inform therapy-induced phenotypic tumor evolution, including GBM cell state and associations between TME components. We therefore analyzed transcriptional profiles of primary GBMs (n = 25) treated with radiation therapy and separated early relapses (Progression-free survival (PFS) < 6 months, n = 11) from late relapses (PFS >12 months, n = 14) using the dataset from GLASS consortium (Figure 7)50,59. The status of pathway activation in each tumor was inferred by the pathway enrichment scores from representative gene signature sets9,14,60,61 (Figure 7 A and B). We observed no significant differences in signature gene set levels between primary tumors with early and late relapse. When comparing relapsed GBMs, however, we found that early-relapse GBMs showed a significantly higher predicted presence of M2 macrophages and the enrichment of senescence, coagulation, and NFκB signatures compared with late-relapse GBMs50 (Figure 7B). Furthermore, the enrichment scores for senescence, coagulation, and NFκB signatures highly correlated with each other, especially in the early-relapse tumor pairs (Figure 7 C and D). These data are consistent with our findings and may provide clinical relevance for potential translation of ΔFVII-based therapies.
Figure 7. Expression of senescence and coagulation gene sets in matched primary and recurrent human GBM pairs.

(A) Heatmap plots of the senescence, coagulation, NFκB, cell cycling, and stemness gene set expression in early (n = 11) and late relapsed (n =14) GBM pairs.
(B) Quantitation of each gene signature expression in the following tumor sets. EP, early-relapse GBM patients’ primary tumors; ER, early-relapse GBM patients’ recurrent tumors; LP, late-relapse GBM patients’ primary tumors; LR, late-relapse GBM patients’ recurrent tumors. Wilcoxon rank test and two-tailed Student’s t test were performed. * p < 0.001.
(C and D) Scatter plot showing the correlations of coagulation and senescence signature gene expression (C), and NFκB and coagulation (D). r values were determined by Pearson’s correlation coefficient analysis.
(E) Schematic illustration to depict the roles of F3 signaling in RT-induced GBM remodeling. See also Figure S7.
We then determined whether F3 targeting can be combined with other anti-cancer therapeutic approaches. A combination therapy with TMZ and ΔFVII, and triple combination of RT, TMZ, and ΔFVII significantly p rolonged the survival of tumor-bearing mice (Figure S7A). Aberrant activation of the EGFR, MET, and AKT pathways is frequently found in GBM, and each pathway has been established as a potential therapeutic target62,63. We treated 4 different GBM cells with representative EGFR, MET, and AKT inhibitors and ΔFVII (Figure S7B). Combination treatment robustly induced tumor cell death and impaired the clonogenic growth of GBM cells to a much greater degree than monotherapy alone (Figure S7C). Lastly, pre-metastatic MG63 osteosarcoma model revealed RT-induced F3 signaling very similar to GBMs, raising the possibility that our findings on RT-induced F3 signaling may be applicable to other cancer types (Figure S7 D to H).
Our data collectively support the previously unidentified concept that activation of F3 signaling during therapy-induced senescence is a central initiator to trigger global adaptation programs in tumor cells and in the TME, leading to therapy resistance and tumor recurrence (Figure 7E).
Discussion
We show that radiation-induced SA-βGal+ GBM cells robustly contribute to post-RT tumor regrowth by active clonal expansion and global reorganization of immune, ECM, and cytokine landscapes in the TME. F3 proteins are rapidly elevated in the SA-βGal+ GBM cells upon irradiation, and F3 signaling promotes clonal expansion, mesenchymal-like cellular state transition, and secretion of oncogenic SASP factors and ECM proteins. Concurrently, F3 also initiates a hyperactive coagulation cascade including local fibrin polymers, which in turn facilitates TAM accumulation/polarization and ECM remodeling in the tumor regions. These F3-initiated cellular events are functionally linked, and together they constitute an oncogenic feed-forward loop, in which TAMs and SASP factors are critical players. In fact, F3 appears to be a master switch regulating the radiation response in GBM, with striking effects on senescence-like and mesenchymal tumor phenotypes as well as the microenvironment. We also demonstrate a strategy to inhibit oncogenic F3 signaling as an anti-GBM therapeutic approach. These data collectively support that F3 is a critical driver of oncogenic senescence and therapeutic resistance in GBM, opening potential therapeutic avenues for F3 targeting.
We report here the existence of an F3-driven phenotype in GBM sharing some features of senescent cells—including SA-βGal reactivity, senescence marker expression (HP1J and H3K9me3), enrichment of representative senescence gene sets, and SASP-like secretory phenotype—with features of GBM stem cells. On the other hand, robust clonal expansion of irradiated SA-βGal+ GBM cells is different from traditional view of cellular senescence in which irreversible cell cycle arrest is a central tenet. Our findings are highly consistent with recent studies showing that the senescence program activates key stemness signaling pathways such as WNT and YAP44,64 and that the senescent state in cancer is highly dynamic and reversible44. Thus, we propose the cellular states of irradiated SA-βGal+ GBM cells as the senescence-like GBM cell phenotype without stable cell cycle arrest.
At the early stages of tumor initiation, oncogene-induced senescence (OIS) is an essential tumor-suppressive barrier that limits expansion of pre-malignant cells. It has been generally thought that cells under OIS remain arrested due to activation of TP53 and/or Cdkn2a pathways, however, a subset of these cells escapes from OIS and progresses to more aggressive stages65. GBM harbors defects in apoptosis and cell cycle regulators such as homozygous deletion of Cdkn2a and deregulated TP53 pathways, possibly enabling bypass of cell cycle arrest while adopting other pro-tumorigenic features of senescence. It is increasingly clear now that various therapies including irradiation, chemotherapeutics, and targeted inhibitors induce massive numbers of senescent or senescent-like cells in malignant cancers 15,66. In most of these studies, however, senescent-like tumor cells have been identified by colorimetric SA-βGal assay and very limited senescent cell marker staining without cell fate determination. Our findings in GBM may provide a clue to better characterize TIS in other malignancies.
The close relationship between cancer and thrombosis has been recognized by Trousseau since 186567. Hyper-coagulability can lead to serious, life-threatening conditions such as venous thromboembolism (VTE). VTE is frequently found in cancer patients, among whom high-grade glioma patients showed the highest incidence52,68. F3, originally identified as an initiator of a stress-responsive coagulation cascade, has been implicated in hyper-coagulation activities in cancer, pro-proliferative signaling, and tumor dormancy, and metastasis38,40,52,69,70. A more recent study reported that F3 is preferentially expressed in quiescent stem-like GBM cells42. In addition, increased blood clotting activities and aberrant fibrin clots have frequently been observed in patients with senescence-associated pathologies including COVID, tissue injury, and aging71–74. Our findings may suggest the possibility that senescence plays a causal role in hypercoagulation activity in non-cancer pathologies as well.
Various therapeutic approaches that target senescent tumor cells and/or senescence-associated pathways have been developed as potential anticancer approaches66. These approaches can be largely classified into three categories: (i) targeting individual oncogenic SASP factors75,76, (ii) targeting individual stemness/survival pathways that are particularly induced by the TIS program 44,77, and (iii) targeting senescent tumor cells with senolytic approaches78. Similar to the highly overlapping RTK signaling networks found in cancer, post-therapy malignant tumors secrete multiple SASP factors with very similar or redundant functions11,75,79–83. TIS-associated epigenetic reprogramming may reactivate multiple, potentially redundant, oncogenic transcription networks44,64,77. Our data suggest that ΔFVII targeting approach can be developed into a viable therapeutic option. F3 targeting potently inhibited radiation-induced TAM accumulation and oncogenic TAM polarization, indicating a potential intersection with TAM re-education approaches and/or other immune-based anti-cancer approaches. We postulate that F3 signaling is a crucial hub triggering multiple oncogenic pathways in the therapy-induced tumor cell senescence setting.
As senescence in the brain parenchyma and immune landscapes are likely critical for RT-induced remodeling and GBM radio-resistance, extensive studies in syngeneic tumor models can provide deeper understanding of associations between immune, microenvironmental components and treatment response. Most of our data were obtained from PDX-bearing immunocompromised mice models. Despite this caveat, our data reveal that F3 signaling in GBM is an active adaptation program to evade therapeutic pressure and leverage oncogenic aspects of therapy-induced senescence. Thus, inhibiting F3 signaling may represent a promising strategy to markedly enhance otherwise suboptimal anti-GBM therapies. Our findings may provide a step forward to a better understanding of F3 signaling, which may be critical for advancing therapeutic strategies.
STAR METHODS TEXT
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jeongwu Lee (leej7@ccf.org).
Materials availability
Materials generated in this study are available from the lead contact upon request.
Data and code availability
The datasets generated during this study are available at GEO under accession number GEO; GSE162931. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
METHOD DETAILS
Patient-derived glioblastoma specimens and derivatives
Following written informed consent, tumor samples classified as GBM, based on the WHO criteria, were obtained from patients undergoing surgical treatment in accordance with the NIH, Cleveland Clinic Lerner Research Institute, and Samsung Medical Center Institutional Review Boards. Within 1 to 6 hours after surgical removal, tumors were washed in PBS and processed for the following models.
GBM spheroids: Tumor cells were cultured in Neurobasal medium supplemented with N2, B27 and bFGF and EGF (NBE medium; Neurobasal media, N2 and B27 supplements (0.5x each; Invitrogen), and human recombinant bFGF and EGF (25 ng/mL each; R&D Systems)47,84.
GBM organoids: GBM organoids display tumor cell hierarchy and differentiation heterogeneity by oxygen/nutrient gradient. Tissue pieces were cut into 0.5 to 1 mm diameter and cultured in the dishes on top of an orbital shaker rotating at 120 rpm at 37°C in a 5% CO2, 95% humidity incubator22,23,85.
GBM slices: GBM tissue pieces were sliced using a vibrating blade microtome into 3 to 5 mm diameter, 300 μm thick slices and transferred to a culture dish.
Primary cell cultures
Normal NPCs derived from human embryonic stem cells (H9, Invitrogen, Cat # N7800100) and aborted fetal brain tissues (Lonza, Cat # PT-2599) were cultured in NBE media or neural progenitor maintenance media (Lonza, Cat# CC-3209). Primary human astrocytes (Lonza, Cat# CC-2565) were cultured in astrocyte growth media (Lonza, Cat# CC-3186). Human U937 cells from ATCC (CRL-1593.2) were maintained in RPMI 1640 media (Gibco,11875093) with 10% FBS (Gibco,10438026), and 1 % penicillin/streptomycin (Gibco, 15140148). For all co-culture experiments, FBS serum was not used.
Patient-derived xenograft (PDX) and RCAS-TVA mouse glioma models
All mice experiments were performed according with the IACUC approved protocols. For orthotopic tumor implantation, GBM cells were injected intracranially into the striatum of nude mice (BALB/c nu/nu) by using a stereotactic device (Kopf instruments, coordinates: 2 mm anterior, 2 mm lateral from the bregma, 2.5 mm depth from the dura) as previously described47. For F3 inducible knockdown, drinking water containing doxycycline (D9891, Sigma, 2mg/ml) and 5% sucrose was given at 20 days after injection. The water was protected from light and exchanged every 2 days. Syngeneic RCAS-TVA mouse gliomas induced by PDGFB overexpression in p53-null background were generated as previously reported53. When mice develop neurological symptoms (lethargy, ataxia, and seizures) or significant body weight loss, mice were killed and processed for histological analysis.
Lung metastasis model
Naïve or irradiated MG63 cells (5 × 105) were injected into the tail vein of each nude mice (BALB/c nu/nu).
ΔFVII-mediated therapy in animal models
All mice were randomly assigned to appropriate treatment groups. For recombinant ΔFVII protein experiments, ΔFVII proteins (50 μg/kg body weight) were administered via intravenous injection daily, concurrent with irradiation or temozolomide treatment (25 mg/kg body weight, intraperitoneal injection, T2577, Sigma)86. For ΔFVII-expressing NPC experiments, NPCs (1×106 cells per mice) were injected into the brains of tumor-bearing mice, two days before irradiation.
Blood clotting time test
To determine tail-bleeding time, mice were kept under anesthesia and placed on a heating pad. Distal tail was cut at 5 mm from the tip and immediately submerged into 10 ml PBS at 37 °C87.
D-dimer ELISA assays
Plasma was collected in 3.2% buffered sodium citrate tube from each sample for D-dimer detection. The amount of D-dimer was assayed using mouse D-dimer ELISA kit (Novus Biologicals, NBP3-08100) according to manufacturer’s protocol.
Radiation regime
For irradiation of tissues and cells, a single dose of 3Gy, 5Gy, or 10 Gy was used. For in vivo irradiation, the anesthetized mice were placed in a lead shielding device in which the brains or subcutaneous tumors were exposed. Localized radiation was performed with either a single dose of 10 Gy or a fractionated regime (2Gy daily for 5 days).
Senescence-Associated β-Galactosidase (SA-βGal) reactivity assays
For colorimetric SA-βGal assay, we used senescence-β-galactosidase kit (9860, Cell Signaling). Briefly, samples were incubated with β-galactosidase staining solution (pH 6.0) at 37°C in a dry incubator without the added CO2. Staining images were analyzed using an inverted fluorescence microscope (DM4000 B, Leica). For fluorometric SA-βGal staining assay, live cells or tissues were pre-treated with bafilomycin A1 (B1793, Sigma-Aldrich, 100 nM) and then cultured with C12FDG (5-Dodecanoylaminofluorescein Di-β-D-Galactopyranoside, D2893, Invitrogen) as described with minor modifications21. C12FDG intensity was analyzed by LSR II Fortessa flow cytometer (BD) or confocal microscope.
Fluorescence activated cell sorting (FACS)
Cell sorting was performed using BD FACS Aria II. GBM cells were stained with either C12FDG or F3 antibody (BD 550312 1:20), and each subpopulation was sorted based on the levels of staining intensities. A matched isotype antibody was used as a control and propidium iodide (PI, 5 μg/ml) was used for live/dead cell determination. To ensure purity and viability of the sorted subpopulations, we repeated flow cytometry analysis and PI staining after initial sorting. Data were collected and analyzed using FlowJo software.
Flow cytometry analysis
Dissociated GBM cells were incubated with 5% donkey or goat serum, 2mM EDTA in PBS for 30 minutes to block non-specific binding, and then labeled with anti-F3-FITC antibody (BD 550312, 1:20), Nestin (SC-23927, 1:100), H3K9me3 (ab8898, 1:100), or ABCG2 (BD562167, 1:100) in 5% serum containing PBS for 1 hour. For detection of intracellular proteins, cells were permeabilized with 0.1% saponin (S7900, Sigma-Aldrich). After gently wash by cold PBS, the cells then incubated with Alexa Flour secondary antibodies (Invitrogen, 1:400) for additional 30 minutes.
Annexin V staining and mitochondria TMRE assays were performed using standard detection kits (ab113852, Abcam). Flow cytometry assays were performed using at least three independent biological samples.
In vivo clonal analysis using barcode-sequencing
GBM cells were transduced with Clone Tracker 50M lentiviral barcode library (BC13X13V, Cellecta Inc.). For in vivo clonal analysis, transduced GBM cells were injected into the brains of nude mice. Genomic DNA was extracted from the resultant tumor tissues using QIAamp DNeasy Blood and Tissue kit (69504, Qiagen) following manufacturer instructions. Barcodes from the tumor were amplified using sample-specific primer sets provided in the NGS prep kit (LNGS-200, Cellecta Inc.). Library quality and fragment sizes were assessed on a Fragment Analyzer before high-throughput sequencing on a HiSeq. Sequence processing and analysis were performed by using Cellecta NGS Demultiplexing and Alignment software.
Cell surface marker screening
BD lyoplate human cell surface marker screening panel contains 242 purified monoclonal antibodies against human clusters of differentiation (CD) markers (560747, BD). Patient-derived GBM cells (131 and 559) were used for cell surface marker screening. Briefly, GBM cells (about 1.5×108) were dissociated with Accutase (A6964, Sigma-Aldrich) and incubated with bafilomycin A1 (100 nM) and C12FDG (33 μM) for 2 hours at 37°C or anti-F3-FITC for 1 hour at 4°C. After C12FDG or F3 staining, cells were split and incubated with each CD antibody for 1 hour at 4°C. Flow cytometry data were measured using LSRII HTS system and analyzed using FlowJo software (NIH).
Single cell RNA-sequencing (scRNA-seq) and bulk RNA-seq analysis
Cells were dissociated with Accutase and suspended in 1% BSA PBS solution. Live cell FACS sorting was performed with DRAQ5 fluorescent probe (62251, Thermo Fisher Scientific). Cell vitality was determined by trypan blue staining and live cells were diluted to a final concentration of 1000 viable cells/μL in 0.1% BSA PBS solution. Each sample had over 90% viability. ScRNA-seq library preparation and sequencing were performed as previously reported23,26. Briefly, scRNA-seq data were processed through 10x Genomics Chromium Single Cell Platform, and count matrices were generated using their Cell Ranger pipeline (10x Genomics). ScRNA-seq data were analyzed using scanpy. For quality control, genes detected in less than 5 cells and cells with fewer than 1000 genes were excluded. Expression values were corrected to 100,000 reads per cell and transformed. Unbiased clustering was performed by UMAP dimensionality reduction visualization analysis26. Gene signature sets used in this report are; GBM subtype88, stemness25,42, cell cycling26, NFκB9, senescence60,61,89, and coagulation14.
ATAC-sequencing analysis
Cells were stained with either C12FDG or F3 antibody (BD 550312, 1:20), and each subpopulation was sorted using BD FACS Aria II. Propidium iodide (PI, 5 μg/ml) was used for live/dead cell determination. 5 × 105 cells / sample were used for ATAC-Seq Library Preparation (K1157, APExBio). Briefly, cells were resuspended in 50μl ATAC lysis buffer containing (0.5 μl 10% NP40, 0.5 μl 10% Tween 20, and 0.5 μl 2% Digitonin) for 3 min on ice and then cold ATAC lysis buffer containing (0.5 μl 10% Tween 20) was added. Nuclei were centrifuged at 1000 rpm for 10 min at 4°C in a fixed angle. Nuclei were resuspended in 50μl tagmentation master mix containing (5μl transposase, 16.5μl PBS, 0.5μl 2% Digitonin, 0.5 μl 10% Tween 20, 2.5μl water, and 25μl 2x tagmentation buffer). The tagmentation reaction was incubated at 37°C for 30 min in a thermomixer with 1000 rpm. Reactions were cleaned up with DNA purification kit (28104, Qiagen). Libraries were amplified and sequenced on a Nextseq instrument (Illumina).
Bioinformatics data analysis
Analysis for genomic alterations including amplification, deletion, and mutation of key genes and glioma subtype assignment were performed, as described 50,59,62,88,90. Pearson correlation coefficient was calculated by “cor” function of R and Pearson’s Chi-squared Test was conducted using “chisq.test” function of R with default settings.
Chemokine/Cytokine profiling assays
Conditioned media was filtered through 0.2 μm filters (Sartorious Stedium Biotech). Filtered media or cell lysates were incubated with Human Cytokine antibody array kit (AAH-CYT-1000, RayBiotech). Intensity of each spot was measured using ImageJ and analyzed using the RayBiotech analysis tool (AAH-ANG-1000, RayBiotech). Heatmaps were generated by “heatmap.2” function of R package.
Lentivirus
Human HEK293 cells (ATCC) were cultured in DMEM media supplemented with 10% FBS, 1% penicillin and streptomycin. For viral production, 293T cells were co-transfected with the corresponding lentiviral vector and packaging plasmids (psPAX2 and pCMV-VSV-G) using CalPhos Mammalian Transfection Kit (631312, Clontech). Virus-containing supernatants were collected and concentrated by Lenti-X concentrator (631231, Clontech).
All expression vectors were cloned into pLenti6/V5 vector (K495500, Invitrogen) and validated by sequencing and immunoblot analysis. Expression vectors used in this study include wildtype F3, wildtype F7, F7 deletion series, STAT3C mutant, and IKK-2 S177E S181E (IKK2SSEE) mutant (a gift from Anjana Rao, Addgene plasmid # 11105)91. A series of F7 deletion mutants was designed to include and/or exclude domains of F7 proteins. Short hairpin RNA (shRNA)-expressing lentiviral vectors were purchased from Sigma-Aldrich and doxycycline-inducible shRNA plasmids were purchased from Dharmacon. F3 or pp65 shRNA constructs were tested and at least two independent shRNA knockdown vectors were selected for further studies.
ΔFVII recombinant proteins
We designed various recombinant variant FVII proteins including ΔFVII and S404A FVII mutant as well as wild type FVII 92. Recombinant FVII variant proteins including wildtype F7, S404A F7, and ΔFVII (181 amino acid protein without peptidase S1 domain; 141 amino acid protein in a secreted form) were synthesized (Genscript) and validated by immunoblot analysis, in vitro clotting assay (factor 7 human chromogenic activity Assay; ab108830, Abcam), and cysteine bond determination by MASS spectrometry.
Immunofluorescence analysis
Tissue slices were harvested 1 to 3 days after irradiation and fixed in 4% PFA (SC281692, Santa Cruz biotechnology). To prepare the frozen sections of tumors, samples were washed in PBS, cryoprotected in 30% sucrose (S0389, Sigma-Aldrich) at 4°C overnight, embedded in O.C.T compound (4583, SAKURA Tissue Tek), and sectioned using a cryostat (CM3050S, Leica). Tissue sections and cells were blocked using a blocking solution (0.3% Triton X-100, 5% goat or donkey serum, 1% BSA in PBS) for 1 hour at room temperature. For mouse tissue sections, mouse-on-mouse blocking reagent (MKB-2213, Vector Laboratories) was added to the blocking solution. Immunofluorescence images were taken using a Leica TCS SP5 Confocal Microscope.
Immunohistochemistry analysis (IHC)
Paraffin sections were prepared in the Cleveland Clinic Lerner Research Institute imaging core. For immunohistochemistry analysis (IHC), tissue sections were deparaffinized in xylene (214736, Sigma-Aldrich) and rehydrated through ethanol gradient. Antigen retrieval was achieved by microwaving the sections in Unmasking solution (citrated based buffer, pH 6.0, H-3300, Vector Laboratories). Endogenous peroxidase activity was blocked by incubation with BLOXALL blocking solution (SP-6000, Vector Laboratories). All images were taken by slide scanner Leica SCN400 microscope and analyzed by Imagescope and ImageJ.
Immunoblots and co-immunoprecipitation
Cells were lysed in Pierce IP lysis buffer (25 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, and 5% glycerol; #87788, Thermo) supplemented with protease inhibitors cocktail (Complete Mini, 11836153001, Sigma-Aldrich) and phosphatase inhibitor (#78428, Thermo), incubated on ice for 30 minutes and cleared by centrifugation at 4°C for 20 minutes. For immunoprecipitation, protein lysates were incubated with appropriate antibodies for overnight at 4 °C. Protein bands were visualized using ECL Western Blotting Detection Reagents (RPN2232, GE Healthcare) and subjected to densitometry analysis using ImageJ.
Cell growth, viability, drug treatment, and soft agar colony forming assays
Standard methods including cell counting and MTT assays (11465007001, Sigma-Aldrich) were used. Cilengitide (1 to 5 μM, S6387, Selleckchem), integrin blocking antibody (ab24693, Abcam), and cucurbitacin (100 nM, C4493, Sigma-Aldrich) were used to inhibit integrin or STAT3 signaling, respectively.
Soft agar colony forming assays were performed to determine capacity of clonogenic cell growth by counting single cell-driven colonies. GBM cells were mixed with top agar (Neurobasal media, N2 and B27, 0.4% agarose) and layered on top of 0.8% agarose. Medium with fresh ΔFVII (100 ng/ml) was added every 3 days. Three weeks later, colonies were fixed with 4% PFA and stained with 0.5% crystal violet.
Macrophage recruitment assay
Immune cell recruitment capacity of GBM cells and U937 cells was measured using transwell inserts (8 μm pore size, 3422). The cells were subsequently fixed with 4% paraformaldehyde at room temperature for 15 min and stained with crystal violet at room temperature for 10 min.
NFκB, STAT3, and TCF/LEF reporter assays
To determine transcriptional activities of the above pathways, cells were transduced with lentiviral constructs containing NFκB consensus element, STAT3 binding elements, and TCF/LEF transcriptional response elements with the minimal promoter red firefly luciferase reporter gene (Systems Biosciences). Luciferase intensities were measured by ONE-Glo Luciferase Assay System according to manufacturer’s protocol (E6110, Promega).
Transmission electron microscopy (TEM)
Cells were fixed in 2.5% glutaraldehyde / 4% PFA in 0.1M sodium cacodylate buffer at 4°C overnight. Cell suspension samples were washed and treated with 1% osmium tetroxide for 1 hour, stained with 1% uranyl acetate, dehydrated, and then embedded in LX-112. Samples were analyzed using Zeiss EM 10 transmission electron microscope.
Real-time live cell imaging analysis
Cell growth was monitored by using Incucyte live cell analysis system (Essen Bioscience). Confluency of each well was determined every 3 hours for the entire experimental periods.
Quantitation and statistical analysis
All data were expressed as means ± SD from at least three independent experiments. Quantification of immuno-positive cells in immunostaining analyses was carried out using NIH imageJ software (National Institutes of Health, Bethesda, MD). For the animal survival studies, p values were determined by log-rank test. Student’s t-test or ANOVA were used to determine statistical significance. Pearson correlation coefficient was calculated by “cor” function of R.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-F3 (HTF-1) | BD Biosciences | Cat#55025; RRID: AB 393557 |
| Mouse monoclonal anti-F3, Phycoerythrin Conjugated, (HTF-1) | BD Biosciences | Cat#550312; RRID: AB 393602 |
| Rabbit polyclonal anti-F3 | Abcam | Cat#ab104513; RRID: AB 10711603 |
| Mouse monoclonal anti-F3 (10H10) | Invitrogen | Cat#MA1-83495; RRID: AB 2101347 |
| Rabbit anti-phospho-ATM (Ser1981) (D6H9) | Cell Signaling | Cat#5883; RRID: AB 10835213 |
| Rabbit anti-ATM (D2E2) | Cell Signaling | Cat#2873; RRID: AB 2062659 |
| Rabbit anti-phospho-Chk1 (Ser345) (133D3) | Cell Signaling | Cat#2348; RRID: AB 331212 |
| Mouse anti-Chk1 (2G1D5) | Cell Signaling | Cat#2360; RRID: AB 2080320 |
| Rabbit anti-phospho-Chk2 (Thr68) (C13C1) | Cell Signaling | Cat#2197; RRID: AB 2080501 |
| Rabbit anti-Chk2 | Cell Signaling | Cat#2662; RRID: AB 2080793 |
| Rabbit anti-Histone H3 (tri methyl K9) | Abcam | Cat#ab8898; RRID: AB 306848 |
| Rabbit-anti-trimethyl-Histone H3 (Lys9) | Millipore Sigma | Cat#07-442; RRID: AB 310620 |
| Mouse monoclonal anti-HP1γ (14D3.1) | Millipore Sigma | Cat#MABE656; RRID: AB 2722628 |
| Mouse monoclonal anti-PCNA | Santa Cruz Biotechnology | Cat#SC56; RRID: AB 628110 |
| Monoclonal anti-Ki67 (MM1) | Leica Biosystems | Cat# NCL-L-Ki67-MM1; RRID:AB 563841 |
| Goat polyclonal anti-Human Sox2 | R&D systems | Cat#AF2018; RRID:AB 355110 |
| Mouse monoclonal anti-Nestin (10c2) | Santa Cruz Biotechnology | Cat#SC-23927; RRID:AB 627994 |
| Mouse anti-Human CD338 (ABCG2) | BD Biosciences | Cat#562167; RRID: AB 11153672 |
| Mouse monoclonal anti-Fibrin (59D8) | Millipore Sigma | Cat#MAB52155; RRID: AB 2893306 |
| Rabbit polyclonal anti-Fibronectin | Abcam | Cat#ab2413; RRID: AB 2262874 |
| Mouse anti-Fibronectin | BD Biosciences | Cat#610077; RRID: AB 2105706 |
| Rabbit polyclonal anti-CHI3L1 | Abcam | Cat#ab77528; RRID:AB 2040911 |
| Mouse monoclonal anti-CD44 (G44-26) | BD Biosciences | Cat#550392; RRID:AB 2074674 |
| Rat monoclonal anti-CD44 (IM7) | Thermo Fisher Scientific | Cat#14044181; RRID:AB 467245 |
| Rabbit monoclonal anti-Phospho-NFkB p65 (Ser536) (93H1) | Cell Signaling | Cat#3033; RRID:AB 331284 |
| Rabbit monoclonal Anti-NFkB p65 (D14E12) | Cell Signaling | Cat#8242; RRID:AB 10859369 |
| Mouse monoclonal Anti-phospho-lkBα (Ser32/36)(5A5) | Cell Signaling | Cat#9246; RRID:AB 2267145 |
| Mouse monoclonal Anti-IkB-alpha (L35A5) | Cell Signaling | Cat#4814; RRID: AB 390781 |
| Rabbit monoclonal anti-human beta-Catenin, phospho (Ser675) (D2F1) | Cell Signaling | Cat#4176; RRID:AB 1903923 |
| Mouse monoclonal anti-Active-beta-catenin (8E7) | Millipore | Cat# 05-665; RRID:AB 309887 |
| Mouse monoclonal anti-CD42b (42C01) | Thermo Fisher Scientific | Cat#MA5-11642; RRID:AB 10986763 |
| Rabbit polyclonal anti-Histone H3 (CitH3) | Abcam | Cat#ab5103; RRID:AB 304752 |
| Rat monoclonal anti-F4/80 (BM8) | Thermo Fisher Scientific | Cat#MF48000; RRID:AB 10376289 |
| Rat monoclonal anti-CD163 (TNKUPJ) | Thermo Fisher Scientific | Cat#14163182; RRID:AB 2716934 |
| Mouse monoclonal anti-CD163 (10D6) | Thermo Fisher Scientific | Cat#MA5-11458; RRID:AB 10982556 |
| Rabbit recombinant CCR2 (EPR20844-15) | Abcam | Cat#ab273050; RRID: AB 2893307 |
| Rat Monoclonal anti-CD11b | Abcam | Cat#ab8878; RRID:AB 306831 |
| Rabbit polyclonal anti-Iba1 | FUJIFILM Wako | Cat#019-19741; RRID:AB 839504 |
| Rabbit polyclonal anti-Fibrinogen | Abcam | Cat#ab34269; RRID:AB 732367 |
| Rabbit polyclonal anti-Ezh2 (D2C9) | Cell Signaling | Cat#5246; RRID:AB 10694683 |
| Mouse monoclonal anti-Ubiquitin (P4D1): | Santa Cruz Biotechnology | Cat#SC8017; RRID: AB 628423 |
| Rat monoclonal anti-Integrin β1b (AIIB2) | Sigma-Aldrich | Cat#MABT409; RRID:AB 2893323 |
| Rabbit polyclonal anti-Integrin alpha 3 | Abcam | Cat#ab131055; RRID:AB 11156484 |
| Rabbit polyclonal anti-Integrin beta1 | Cell Signaling | Cat#4706; RRID:AB 823544 |
| Goat Polyclonal anti-Human Integrin alpha 5 / cd49e | R&D systems | Cat#AF1864; RRID:AB 355026 |
| Mouse monoclonal anti-Integrin beta 1 (HUTS4) | Millipore Sigma | Cat#MAB2079Z; RRID:AB 2233964 |
| Mouse monoclonal anti-ITGB1 (TS2/16) | Thermo Fisher Scientific | Cat#MA2910; RRID:AB 223515 |
| Mouse monoclonal anti-Integrin beta 1 [P5D2] | Abcam | Cat#ab24693; RRID: AB 448230 |
| Rabbit monoclonal anti-pSTAT3(Tyr705) (D3A7) | Cell Signaling | Cat#9145; RRID:AB 2491009 |
| Mouse monoclonal anti-Stat3 (124H6) | Cell Signaling | Cat#9139; RRID:AB 331757 |
| Rabbit monoclonal anti-pAKT(S473) (D9E) | Cell Signaling | Cat#4060; RRID:AB 2315049 |
| Rabbit monoclonal anti-AKT (C67E7) | Cell Signaling | Cat#4691; RRID:AB 915783 |
| Rabbit monoclonal anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (197G2) | Cell Signaling | Cat#4377; RRID:AB 331775 |
| Rabbit monoclonal p44/42 MAPK (Erk1/2) (137F5) | Cell Signaling | Cat#4695; RRID:AB 390779 |
| Rabbit polyclonal anti-IL6 | Abcam | Cat#ab6672; RRID:AB 2127460 |
| Rabbit polyclonal anti-pFAK (Tyr397) | Cell Signaling | Cat#3283; RRID:AB 2173659 |
| Rabbit monoclonal anti-FAK (D2R2E) | Cell Signaling | Cat#13009; RRID:AB 2798086 |
| Mouse monoclonal anti-PUMAα/β (G-3) | Santa Cruz Biotechnology | Cat#SC-374223; RRID:AB 10987708 |
| Rabbit monoclonal anti-Cleaved Caspase3 (Asp175) (5A1E) | Cell Signaling | Cat#9664; RRID:AB 2070042 |
| Mouse monoclonal anti-STEM121 | Takara | Cat#Y40410; RRID:AB 2801314 |
| Goat polyclonal anti-Factor Vll | R&D systems | Cat#AF2338; RRID:AB 416580 |
| Rabbit polyclonal anti-Lamin B1 | Abcam | Cat#ab16048; RRID:AB 10107828 |
| Rabbit polyclonal anti-phospho-GSK3 β (Ser9) | Cell signaling | Cat#9336; RRID:AB 331405 |
| Rabbit monoclonal anti-GSK-3β (D5C5Z) | Cell signaling | Cat# 2456; RRID:AB 2636978 |
| Goat polyclonal anti-Hepatocyte Growth Factor | R&D systems | Cat#AF-294-NA; RRID:AB 354451 |
| Goat polyclonal anti-Human FGF | R&D systems | Cat#AF-233-NA; RRID:AB 354413 |
| Rabbit anti-Histone H3 (D1H2) | Cell signaling | Cat# 4499; RRID:AB 10544537 |
| Rabbit anti-Flag Tag (D6W5B) | Cell signaling | Cat#14793; RRID:AB 2572291 |
| Mouse monoclonal anti-F3 blocking antibody (5G9) | Absolute Antibody | Cat#ab00516 RRID:AB 2934070 |
| Mouse monoclonal anti-α-tubulin (DM1A) | Millipore Sigma | Cat#T9026; RRID:AB 477593 |
| Mouse monoclonal anti-actin (C-2) | Santa Cruz Biotechnology | Cat#SC-8432; RRID:AB 626630 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary antibody, Alexa Fluor 594 | Invitrogen | Cat#A-21203; RRID:AB 141633 |
| Donkey polyclonal anti-Mouse IgG (H+L) Cross-absorbed secondary antibody, Alexa Fluor 488 | Invitrogen | Cat#A-21202; RRID: AB 141607 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | Invitrogen | Cat#A-31571; RRID: AB 162542 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary antibody, Alexa Fluor 594 | Invitrogen | Cat#A-21207; RRID:AB 141637 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-absorbed secondary antibody, Alexa Fluor 488 | Invitrogen | Cat#A-21206; RRID: AB 2535792 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary antibody, Alexa Fluor 594 | Invitrogen | Cat#A-11058; RRID:AB 2534105 |
| Donkey polyclonal anti-Goat IgG (H+L) Cross-absorbed secondary antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-11055; RRID: AB 2534102 |
| Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary antibody, Alexa Fluor 594 | Invitrogen | Cat#A-21209; RRID:AB 2535795 |
| Donkey polyclonal anti-Rat IgG (H+L) Highly Cross-Absorbed secondary antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-21208; RRID: AB 2535794 |
| Goat anti-Mouse IGG antibody (H+L), Biotinylated | Vector Laboratories | Cat#BA9200; RRID:AB 2336171 |
| Goat anti-Rabbit IgG antibody (H+L), Biotinylated | Vector Laboratories | Cat#BA1000; RRID:AB 2313606 |
| Goat anti-Rat IgG antibody, mouse absorbed (H+L), biotinylated | Vector Laboratories | Cat#BA9401; RRID:AB 2336208 |
| Rabbit anti-goat IgG antibody (H+L), Biotinylated | Vector Laboratories | Cat#BA5000; RRID:AB 2336126 |
| Biological samples | ||
| Human glioblastoma specimen | Cleveland Clinic | |
| Bacterial and Virus strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Thermo Fisher Scientific | C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 4%PFA | Santa Cruz Biotechnology | Cat#Sc281692; CAS: 30525-89-4 |
| A/G agarose beads | Santa Cruz Biotechnology | SC2003 |
| ABT-263 | Selleckchem | Cat#S1001; CAS: 923564-51-6 |
| Accutase Cell detachment solution | Sigma-Aldrich | A6964 |
| Antigen Unmasking solution, citrate based | Vector Laboratories | H3300 |
| B-27 Supplement (50x), minus Vitamin A | Thermo Fisher Scientific | 12587010 |
| Bafilomycin A1 | Sigma-Aldrich | Cat#B1793; CAS: 88899-55-2 |
| Blasticidin | Thermo Fisher Scientific | R21001 |
| BLOXALL Endogenous blocking solution, peroxidase and alkaline phosphatase | Vector Laboratories | SP6000100 |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat#B6917; CAS: 9048-46-8 |
| BYL 719 | ApexBio technology | Cat#A8346; CAS: 1217486-61-7 |
| C12FDG (5-Dodecanoylaminofluorescein Di-β-D-Galactopyranoside) | Invitrogen | D2893 |
| Cilengitide | Selleckchem | Cat#S6387; CAS: 188968-51-6 |
| Cucurbitacin | Sigma-Aldrich | Cat#C4493 CAS:2222-07-3 |
| DAPI | Sigma-Aldrich | Cat#D9542; CAS: 47165-04-8 |
| Dasatinib | Sigma-Aldrich | Cat#SML2589; CAS: 302962-49-8 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat#D2650; CAS: 67-68-5 |
| Doxycycline | Sigma-Aldrich | Cat#D9891; CAS: 564-25-0 |
| DMEM | Gibco | Cat#D5796 |
| DRAQ5 fluorescent probe | Thermo Fisher Scientific | 62251 |
| Dulbecco’s phosphate-buffered saline (DPBS), no calcium, no magnesium | Thermo Fisher Scientific | 14190144 |
| ECL Western Blotting detection reagents | GE Healthcare | RPN2232 |
| Gefitinib | Abcam | Cat#ab142052; CAS: 184475-35-2 |
| Halt Phosphatase Inhibitor Single-Use Cocktail | Thermo Fisher | 78428 |
| Lab Vison Mayer’s Hematoxylin | Vector Laboratories | TA-125-MH |
| Lenti-X concentrator | Clontech | 631231 |
| Matrigel | Corning costar | 354230 |
| Mini Protease inhibitor cocktail | Sigma-Aldrich | 11836153001 |
| Mouse-on-mouse blocking reagent | Vector Laboratories | Cat#MKB-2213; RRID:AB 2336587 |
| N-2 Supplement (100x) | Thermo Fisher Scientific | 17502048 |
| Neurobasal medium | Thermo Fisher Scientific | 12349015 |
| Nuclease-Free Water (not DEPC-treated) | Thermo Fisher Scientific | AM9937 |
| O.C.T compound | SAKURA Tissue Tek | 4583 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | 15070063 |
| PHA 665752 | ApexBio technology | Cat#A2307; CAS: 477575-56-7 |
| ProLong Gold Antifade Mountant | Invitrogen | P36934 |
| Puromycin | Sigma-Aldrich | Cat#P8833; CAS:53-79-2 |
| Saponin | Sigma-Aldrich | Cat#S7900; CAS: 8047-15-2 |
| Sodium citrate | Sigma-Aldrich | Cat#PHR1416; CAS: 6132-04-3 |
| Sucrose | Sigma-Aldrich | Cat#S0389; CAS: 57-50-1 |
| Temozolomide | Sigma-Aldrich | Cat#T2577; CAS: 85622-93-1 |
| Triton X-100 | Sigma-Aldrich | Cat#T9284; CAS: 9002-93-1 |
| Trypan blue stain, 0.4% | Thermo Fisher Scientific | T10282 |
| VectaMount AQ Aqueous Mounting medium | Vector Laboratories | H550160 |
| Xylene | Sigma-Aldrich | Cat#214736; CAS:1330-20-7 |
| Critical Commercial Assays | ||
| ATAC-Seq Library Preparation Kit | APEXBIO | K1157 |
| CalPhos Mammalian Transfection Kit | Clontech | 631312 |
| Cell Proliferation kit (MTT) | Sigma-Aldrich | 11465007001 |
| DAB Substrate Kit | Vector Laboratories | SK4100 |
| DNA purification Kit | Qiagen | 28104 |
| Factor 7 human chromogenic activity Assay | Abcam | ab108830 |
| Human Cytokine antibody array kit | RayBiotech | AAH-CYT-1000 |
| Mouse D-dimer ELISA kit | Novus Biologicals | NBP3-08100 |
| PE Annexin V Apoptosis Detection Kit | BD biosciences | 559763 |
| QIAmp DNeasy Blood and Tissue Kit | Qiagen | 69504 |
| Senescence-β-galactosidase kit | Cell Signaling | 9860 |
| TMRE-Mitochondrial Membrane Potential Assay Kit | Abcam | ab113852 |
| Vectastain elite ABC kit | Vector Laboratories | PK6100 |
| CloneTracker™ 50M Lentiviral Barcode Library | Cellecta | BC13X13-V |
| NGS Prep Kit for Barcode Libraries in pRSI16/17 (QoneTracker™)48x50ugCellectaLNGS-200 | Cellecta | LNGS-200 |
| ONE-Glo Luciferase Assay System | Promega | E6110 |
| Deposited data | ||
| RNA Sequencing data | This study | GEO: GSE162931 |
| Experimental Models: Cell Lines | ||
| NPCs from aborted fetal brain tissues | Lonza | Cat # PT-2599 |
| Embryonic stem cell-driven NPCs | Invitrogen | Cat #N7800100 |
| Human HEK293 | ATCC | CRL-1573 |
| Human HEK293T | ATCC | CRL-3216 |
| Primary human astrocytes | Lonza | Cat# CC-2565 |
| U937 | ATCC | CRL-1593.2 |
| Human glioma cells | N/A | |
| MG63 | Received from NIH | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: BALB/c, nu/nu (C.Cg/AnNTac-Foxn/nu NE9) | TACONIC | BALBNU |
| Mouse: RCAS-TVA, p53 -/-, PDGFB O/E | Hambardzumyan et al.53 | |
| Recombinant DNA | ||
| pCMV-VSV-G | Addgene | 8454 |
| pLenti6/V5 vector | Invitrogen | K495500 |
| psPAX2 | Addgene | 12260 |
| pGreenFire 2.0 NFkB Reporter | Systems Biosciences | TR412PA-P |
| pGreenFire 2.0 TCF/LEF Reporter | Systems Biosciences | TR413PA-P |
| Software and Algorithms | ||
| 10x Genomics Chromium Single Cell Platform | 10x Genomics | https://www.10xgenomics.com/products/single-cell-gene-expression |
| Cell ranger Pipeline | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| FlowJo | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads |
| Gene Set Enrichment Analysis (GSEA) | Subramanian et al.93 | https://www.gsea-msigdb.org/gsea/index.jsp |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Imagescope | Leica Biosystems | https://www.leicabiosystems.com/digital-pathology/manage/aperio-imagescope/ |
| LAS X application suite | Leica Biosystems | https://www.kica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| Imagescope | Leica Biosystems | https://www.leicabiosystems.com/digital-pathology/manage/aperio-imagescope/ |
| LAS X application suite | Leica Biosystems | https://www.kica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| Plotly R package | Sievert C (2020). | https://github.com/ropensci/plotly |
| RayBiotech analysis tool | Raybiotech | AAH-ANG-1000 |
| Scanpy | Wolf et al.94 | https://scanpy.readthedocs.io/en/stable/ |
| UMAP dimensionality reduction visualization | Becht et al.95 | https://github.com/lmcinnes/umap |
| Other | ||
| Cancer Genome Atlas (TCGA) database | ||
| GLASS consortium | ||
| Gene Expression Omnibus (GEO) | ||
| Leica TCS SP5 confocal microscope | Leica Microsystems | |
| Zeiss EM 10 transmission electron microscope | Carl Zeiss | |
| Aperio AT2 Slide Scanner | Leica Microsystems | |
| Leica DM6B (with Leica 7000T camera) | Leica Microsystems | |
| LSR II Fortessa flow cytometer | BD | |
| FACS Aria II | BD | |
| Stereotactic device | Kopf instruments | |
| IncuCyte Live-cell analysis system | Sartorius | |
| Vibratome | Leica | |
Highlights.
Irradiated SA-βGal+ GBM cells clonally expand and promote post-RT tumor regrowth.
RT-induced F3 signaling enhances the enriched features of senescence and stemness.
F3 is a key driver for oncogenic ECM and TME remodeling in GBM radio-resistance.
ΔFVII is a potent anti-GBM agent that mitigates F3-mediated oncogenic signaling.
ACKNOWLEDGEMENTS
This work was supported by NIH grants R01 NS082312, R01 CA223770 and Velosano cancer grant (to J.L).
INCLUSION AND DIVERSITY
All authors of this paper support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
H-M.J. and J.L. are co-inventors on a filed patent application for the use of truncated F7 recombinant protein to treat cancer.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, et al. (2014). A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370, 699–708. 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim M, Xia Y, Bettegowda C, and Weller M (2018). Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol 15, 422–442. 10.1038/s41571-018-0003-5. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352, 987–996. 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, and Rich JN (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760. 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, McKay RM, and Parada LF (2012). Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell 149, 36–47. 10.1016/j.cell.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barker HE, Paget JT, Khan AA, and Harrington KJ (2015). The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer 15, 409–425. 10.1038/nrc3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoo KC., Suh Y., An Y., Lee HJ., Jeong YJ., Uddin N., Cui YH., Roh TH., Shim JK., Chang JH., et al. (2018). Proinvasive extracellular matrix remodeling in tumor microenvironment in response to radiation. Oncogene 37, 3317–3328. 10.1038/s41388-018-0199-y. [DOI] [PubMed] [Google Scholar]
- 8.Winchell HS, Anderson AC, and Pollycove M (1964). Radiation-Induced Hemorrhagic Diathesis in Dogs Unassociated with Thrombocytopenia: Association with an Intravascular Protein-Polysaccharide Particle. Blood 23, 186–192. [PubMed] [Google Scholar]
- 9.Bhat KPL, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, Wani K, Heathcock L, James JD, Goodman LD, et al. (2013). Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 24, 331–346. 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halliday J, Helmy K, Pattwell SS, Pitter KL, LaPlant Q, Ozawa T, and Holland EC (2014). In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proc Natl Acad Sci U S A 111, 5248–5253. 10.1073/pnas.1321014111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hara T, Chanoch-Myers R, Mathewson ND, Myskiw C, Atta L, Bussema L, Eichhorn SW, Greenwald AC, Kinker GS, Rodman C, et al. (2021). Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 39, 779–792 e711. 10.1016/j.ccell.2021.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmed MM, Hodge JW, Guha C, Bernhard EJ, Vikram B, and Coleman CN (2013). Harnessing the potential of radiation-induced immune modulation for cancer therapy. Cancer Immunol Res 1, 280–284. 10.1158/2326-6066.CIR-13-0141. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy AR, Maity A, and Sanzari JK (2016). A Review of Radiation-Induced Coagulopathy and New Findings to Support Potential Prevention Strategies and Treatments. Radiat Res 186, 121–140. 10.1667/RR14406.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magnus N, Gerges N, Jabado N, and Rak J (2013). Coagulation-related gene expression profile in glioblastoma is defined by molecular disease subtype. J Thromb Haemost 11, 1197–1200. 10.1111/jth.12242. [DOI] [PubMed] [Google Scholar]
- 15.Ewald JA, Desotelle JA, Wilding G, and Jarrard DF (2010). Therapy-induced senescence in cancer. J Natl Cancer Inst 102, 1536–1546. 10.1093/jnci/djq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmitt CA, Wang B, and Demaria M (2022). Senescence and cancer - role and therapeutic opportunities. Nat Rev Clin Oncol 19, 619–636. 10.1038/s41571-022-00668-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, et al. (2019). Cellular Senescence: Defining a Path Forward. Cell 179, 813–827. 10.1016/j.cell.2019.10.005. [DOI] [PubMed] [Google Scholar]
- 18.He S, and Sharpless NE (2017). Senescence in Health and Disease. Cell 169, 1000–1011. 10.1016/j.cell.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.TCGA (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068. nature07385 [pii] 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dimri GP., Lee X., Basile G., Acosta M., Scott G., Roskelle C., Medrano EE., Linskens M., Rubelj I., Pereira-Smith O., and et al. (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 92, 9363–9367. 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, and Toussaint O (2009). Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 4, 1798–1806. 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- 22.Hubert CG, Rivera M, Spangler LC, Wu Q, Mack SC, Prager BC, Couce M, McLendon RE, Sloan AE, and Rich JN (2016). A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res 76, 2465–2477. 10.1158/0008-5472.CAN-15-2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacob F, Salinas RD, Zhang DY, Nguyen PTT, Schnoll JG, Wong SZH, Thokala R, Sheikh S, Saxena D, Prokop S, et al. (2020). A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 180, 188–204 e122. 10.1016/j.cell.2019.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.LeBlanc VG, Trinh DL, Aslanpour S, Hughes M, Livingstone D, Jin D, Ahn BY, Blough MD, Cairncross JG, Chan JA, et al. (2022). Single-cell landscapes of primary glioblastomas and matched explants and cell lines show variable retention of inter- and intratumor heterogeneity. Cancer Cell 40, 379–392 e379. 10.1016/j.ccell.2022.02.016. [DOI] [PubMed] [Google Scholar]
- 25.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, and Weinberg RA (2008). An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 40, 499–507. 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM, et al. (2019). An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178, 835–849 e821. 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV, et al. (2014). Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 157, 580–594. 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding Y, Hubert CG, Herman J, Corrin P, Toledo CM, Skutt-Kakaria K, Vazquez J, Basom R, Zhang B, Risler JK, et al. (2013). Cancer-Specific requirement for BUB1B/BUBR1 in human brain tumor isolates and genetically transformed cells. Cancer Discov 3, 198–211. 10.1158/2159-8290.CD-12-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H, Huang H, Xue J, Liu M, Wang Y, et al. (2011). FoxM1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell 20, 427–442. 10.1016/j.ccr.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhang HE, Ruddy DA, Krishnamurthy Radhakrishna V, Caushi JX, Zhao R, Hims MM, Singh AP, Kao I, Rakiec D, Shaw P, et al. (2015). Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med 21, 440–448. 10.1038/nm.3841. [DOI] [PubMed] [Google Scholar]
- 31.Greaves M, and Maley CC (2012). Clonal evolution in cancer. Nature 481, 306–313. 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reya T, Morrison SJ, Clarke MF, and Weissman IL (2001). Stem cells, cancer, and cancer stem cells. Nature 414, 105–111. 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 33.Bleau AM., Hambardzumyan D., Ozawa T., Fomchenko EI., Huse JT., Brennan CW., and Holland EC. (2009). PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 4, 226–235. 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minata M, Audia A, Shi J, Lu S, Bernstock J, Pavlyukov MS, Das A, Kim SH, Shin YJ, Lee Y, et al. (2019). Phenotypic Plasticity of Invasive Edge Glioma Stem-like Cells in Response to Ionizing Radiation. Cell Rep 26, 1893–1905 e1897. 10.1016/j.celrep.2019.01.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morrissey JH, Fakhrai H, and Edgington TS (1987). Molecular cloning of the cDNA for tissue factor, the cellular receptor for the initiation of the coagulation protease cascade. Cell 50, 129–135. 10.1016/0092-8674(87)90669-6. [DOI] [PubMed] [Google Scholar]
- 36.Rao LV, and Rapaport SI (1988). Activation of factor VII bound to tissue factor: a key early step in the tissue factor pathway of blood coagulation. Proc Natl Acad Sci U S A 85, 6687–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mackman N (2004). Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol 24, 1015–1022. 10.1161/01.ATV.0000130465.23430.74. [DOI] [PubMed] [Google Scholar]
- 38.Cimmino G, and Cirillo P (2018). Tissue factor: newer concepts in thrombosis and its role beyond thrombosis and hemostasis. Cardiovasc Diagn Ther 8, 581–593. 10.21037/cdt.2018.10.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van den Berg YW, Osanto S, Reitsma PH, and Versteeg HH (2012). The relationship between tissue factor and cancer progression: insights from bench and bedside. Blood 119, 924–932. 10.1182/blood-2011-06-317685. [DOI] [PubMed] [Google Scholar]
- 40.Bourcy M, Suarez-Carmona M, Lambert J, Francart ME, Schroeder H, Delierneux C, Skrypek N, Thompson EW, Jerusalem G, Berx G, et al. (2016). Tissue Factor Induced by Epithelial-Mesenchymal Transition Triggers a Procoagulant State That Drives Metastasis of Circulating Tumor Cells. Cancer Res 76, 4270–4282. 10.1158/0008-5472.CAN-15-2263. [DOI] [PubMed] [Google Scholar]
- 41.Unruh D, Mirkov S, Wray B, Drumm M, Lamano J, Li YD, Haider QF, Javier R, McCortney K, Saratsis A, et al. (2019). Methylation-dependent Tissue Factor Suppression Contributes to the Reduced Malignancy of IDH1-mutant Gliomas. Clin Cancer Res 25, 747–759. 10.1158/1078-0432.CCR-18-1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie XP, Laks DR, Sun D, Ganbold M, Wang Z, Pedraza AM, Bale T, Tabar V, Brennan C, Zhou X, and Parada LF (2022). Quiescent human glioblastoma cancer stem cells drive tumor initiation, expansion, and recurrence following chemotherapy. Dev Cell 57, 32–46 e38. 10.1016/j.devcel.2021.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, et al. (2017). Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 169, 132–147 e116. 10.1016/j.cell.2017.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurppa KJ, Liu Y, To C, Zhang T, Fan M, Vajdi A, Knelson EH, Xie Y, Lim K, Cejas P, et al. (2020). Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 37, 104–122 e112. 10.1016/j.ccell.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, et al. (2010). The transcriptional network for mesenchymal transformation of brain tumours. Nature 463, 318–325. 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chien Y., Scuoppo C., Wang X., Fang X., Balgley B., Bolden JE., Premsrirut P., Luo W., Chicas A., Lee CS., et al. (2011). Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev 25, 2125–2136. 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, Nakano I, et al. (2013). Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 23, 839–852. 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prager BC, Bhargava S, Mahadev V, Hubert CG, and Rich JN (2020). Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 6, 223–235. 10.1016/j.trecan.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aldape K, Brindle KM, Chesler L, Chopra R, Gajjar A, Gilbert MR, Gottardo N, Gutmann DH, Hargrave D, Holland EC, et al. (2019). Challenges to curing primary brain tumours. Nat Rev Clin Oncol 16, 509–520. 10.1038/s41571-019-0177-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y, et al. (2017). Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 32, 42–56 e46. 10.1016/j.ccell.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, et al. (2006). Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173. 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 52.Galmiche A, Rak J, Roumenina LT, and Saidak Z (2022). Coagulome and the tumor microenvironment: an actionable interplay. Trends Cancer. 10.1016/j.trecan.2021.12.008. [DOI] [PubMed] [Google Scholar]
- 53.Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, and Holland EC (2009). Modeling Adult Gliomas Using RCAS/t-va Technology. Transl Oncol 2, 89–95. 10.1593/tlo.09100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koren HS, Anderson SJ, and Larrick JW (1979). In vitro activation of a human macrophage-like cell line. Nature 279, 328–331. 10.1038/279328a0. [DOI] [PubMed] [Google Scholar]
- 55.Ettelaie C, Collier ME, Featherby S, Greenman J, and Maraveyas A (2016). Oligoubiquitination of tissue factor on Lys255 promotes Ser253-dephosphorylation and terminates TF release. Biochim Biophys Acta 1863, 2846–2857. 10.1016/j.bbamcr.2016.09.005. [DOI] [PubMed] [Google Scholar]
- 56.Arvanitis CD, Ferraro GB, and Jain RK (2020). The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat Rev Cancer 20, 26–41. 10.1038/s41568-019-0205-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bago JR, Okolie O, Dumitru R, Ewend MG, Parker JS, Werff RV, Underhill TM, Schmid RS, Miller CR, and Hingtgen SD (2017). Tumor-homing cytotoxic human induced neural stem cells for cancer therapy. Sci Transl Med 9. 10.1126/scitranslmed.aah6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kauer TM, Figueiredo JL, Hingtgen S, and Shah K (2011). Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat Neurosci 15, 197–204. 10.1038/nn.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Varn FS, Johnson KC, Martinek J, Huse JT, Nasrallah MP, Wesseling P, Cooper LAD, Malta TM, Wade TE, Sabedot TS, et al. (2022). Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell 185, 2184–2199 e2116. 10.1016/j.cell.2022.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coppe JP., Patil CK., Rodier F., Sun Y., Munoz DP., Goldstein J., Nelson PS., Desprez PY., and Campisi J. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6, 2853–2868. 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fridman AL, and Tainsky MA (2008). Critical pathways in cellular senescence and immortalization revealed by gene expression profiling. Oncogene 27, 5975–5987. 10.1038/onc.2008.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462–477. 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liau BB, Sievers C, Donohue LK, Gillespie SM, Flavahan WA, Miller TE, Venteicher AS, Hebert CH, Carey CD, Rodig SJ, et al. (2017). Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 20, 233–246 e237. 10.1016/j.stem.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Milanovic M, Fan DNY, Belenki D, Dabritz JHM, Zhao Z, Yu Y, Dorr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA, et al. (2018). Senescence-associated reprogramming promotes cancer stemness. Nature 553, 96–100. 10.1038/nature25167. [DOI] [PubMed] [Google Scholar]
- 65.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, et al. (2005). Tumour biology: senescence in premalignant tumours. Nature 436, 642. 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 66.Prasanna PG, Citrin DE, Hildesheim J, Ahmed MM, Venkatachalam S, Riscuta G, Xi D, Zheng G, Deursen JV, Goronzy J, et al. (2021). Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J Natl Cancer Inst 113, 1285–1298. 10.1093/jnci/djab064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trousseau A (1865). Phlegmasia alba dolens. Clinique medicule de l’Hotel-Dieu de Paris 3, 94. [Google Scholar]
- 68.Saidak Z, Soudet S, Lottin M, Salle V, Sevestre MA, Clatot F, and Galmiche A (2021). A pan-cancer analysis of the human tumor coagulome and its link to the tumor immune microenvironment. Cancer Immunol Immunother 70, 923–933. 10.1007/s00262-020-02739-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Morrow JJ, Bayles I, Funnell APW, Miller TE, Saiakhova A, Lizardo MM, Bartels CF, Kapteijn MY, Hung S, Mendoza A, et al. (2018). Positively selected enhancer elements endow osteosarcoma cells with metastatic competence. Nat Med 24, 176–185. 10.1038/nm.4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feinauer MJ, Schneider SW, Berghoff AS, Robador JR, Tehranian C, Karreman MA, Venkataramani V, Solecki G, Grosch JK, Gunkel K, et al. (2021). Local blood coagulation drives cancer cell arrest and brain metastasis in a mouse model. Blood 137, 1219–1232. 10.1182/blood.2020005710. [DOI] [PubMed] [Google Scholar]
- 71.Wiley CD., Liu S., Limbad C., Zawadzka AM., Beck J., Demaria M., Artwood R., Alimirah F., Lopez-Dominguez JA., Kuehnemann C., et al. (2019). SILAC Analysis Reveals Increased Secretion of Hemostasis-Related Factors by Senescent Cells. Cell Rep 28, 3329–3337 e3325. 10.1016/j.celrep.2019.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee S, Yu Y, Trimpert J, Benthani F, Mairhofer M, Richter-Pechanska P, Wyler E, Belenki D, Kaltenbrunner S, Pammer M, et al. (2021). Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 599, 283–289. 10.1038/s41586-021-03995-1. [DOI] [PubMed] [Google Scholar]
- 73.Moiseeva V, Cisneros A, Sica V, Deryagin O, Lai Y, Jung S, Andres E, An J, Segales J, Ortet L, et al. (2023). Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature 613, 169–178. 10.1038/s41586-022-05535-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nguyen D, Jeon HM, and Lee J (2022). Tissue factor links inflammation, thrombosis, and senescence in COVID-19. Sci Rep 12, 19842. 10.1038/s41598-022-23950-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Birch J, and Gil J (2020). Senescence and the SASP: many therapeutic avenues. Genes Dev 34, 1565–1576. 10.1101/gad.343129.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406. 10.1038/nature24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang C, Vegna S, Jin H, Benedict B, Lieftink C, Ramirez C, de Oliveira RL, Morris B, Gadiot J, Wang W, et al. (2019). Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 574, 268–272. 10.1038/s41586-019-1607-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, Mansilla-Soto J, Boyer JA, Li X, Giavridis T, et al. (2020). Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132. 10.1038/s41586-020-2403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Coppe JP, Desprez PY, Krtolica A, and Campisi J (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5, 99–118. 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Faget DV, Ren Q, and Stewart SA (2019). Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer 19, 439–453. 10.1038/s41568-019-0156-2. [DOI] [PubMed] [Google Scholar]
- 81.Wang B, Kohli J, and Demaria M (2020). Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer. 10.1016/j.trecan.2020.05.004. [DOI] [PubMed] [Google Scholar]
- 82.De Palma M, and Lewis CE (2013). Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 23, 277–286. 10.1016/j.ccr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 83.DeNardo DG, and Ruffell B (2019). Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol 19, 369–382. 10.1038/s41577-019-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al. (2006). Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 9, 391–403. 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 85.Pine AR, Cirigliano SM, Nicholson JG, Hu Y, Linkous A, Miyaguchi K, Edwards L, Singhania R, Schwartz TH, Ramakrishna R, et al. (2020). Tumor Microenvironment Is Critical for the Maintenance of Cellular States Found in Primary Glioblastomas. Cancer Discov 10, 964–979. 10.1158/2159-8290.CD-20-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mathieu V, De Neve N, Le Mercier M, Dewelle J, Gaussin JF, Dehoux M, Kiss R, and Lefranc F (2008). Combining bevacizumab with temozolomide increases the antitumor efficacy of temozolomide in a human glioblastoma orthotopic xenograft model. Neoplasia 10, 1383–1392. 10.1593/neo.08928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brake MA, Ivanciu L, Maroney SA, Martinez ND, Mast AE, and Westrick RJ (2019). Assessing Blood Clotting and Coagulation Factors in Mice. Curr Protoc Mouse Biol 9, e61. 10.1002/cpmo.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Verhaak RG., Hoadley KA., Purdom E., Wang V., Qi Y., Wilkerson MD., Miller CR., Ding L., Golub T., Mesirov JP., et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110. 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tasdemir N, Banito A, Roe JS, Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, Huang CH, Aksoy O, Bolden JE, Chen CC, et al. (2016). BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov 6, 612–629. 10.1158/2159-8290.CD-16-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M, Rice T, Kosel ML, Smirnov IV, et al. (2015). Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med 372, 2499–2508. 10.1056/NEJMoa1407279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, and Rao A (1997). IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278, 860–866. 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 92.Toso R, Pinotti M, High KA, Pollak ES, and Bernardi F (2002). A frequent human coagulation Factor VII mutation (A294V, c152) in loop 140s affects the interaction with activators, tissue factor and substrates. Biochem J 363, 411–416. 10.1042/0264-6021:3630411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15. 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at GEO under accession number GEO; GSE162931. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.