

Summary
By combining mass spectrometry-based proteomics and phosphoproteomics with genomics, epi-genomics, and transcriptomics, proteogenomics provides comprehensive molecular characterization of cancer. Using this approach, the Clinical Proteomic Tumor Analysis Consortium (CPTAC) has characterized over 1,000 primary tumors spanning 10 cancer types, many with matched normal tissues. Here, we present LinkedOmicsKB, a proteogenomics data-driven knowledge base that makes consistently processed and systematically precomputed CPTAC pan-cancer proteogenomics data available to the public through ~40,000 gene-, protein-, mutation-, and phenotype-centric web pages. Visualization techniques facilitate efficient exploration and reasoning of complex, interconnected data. Using three case studies, we illustrate the practical utility of LinkedOmicsKB in providing new insights into genes, phosphorylation sites, somatic mutations, and cancer phenotypes. With precomputed results of 19,701 coding genes, 125,969 phosphosites, and 256 genotypes and phenotypes, LinkedOmicsKB provides a comprehensive resource to accelerate proteogenomics data-driven discoveries to improve our understanding and treatment of human cancer. A record of this paper’s Transparent Peer Review process is included in the Supplemental Information.
Keywords: Proteogenomics, proteomics, phosphoproteomics, cancer, knowledge base, CPTAC, pan-cancer
Graphical Abstract
eTOC blurb
LinkedOmicsKB makes consistently processed and systematically precomputed CPTAC pan-cancer proteogenomics data easily accessible to the public through a web portal. With approximately 40,000 gene-, protein-, mutation-, and phenotype-centric web pages, it enables anyone with internet access to conduct meaningful inquiries into CPTAC data, facilitating data-driven scientific discoveries.
Introduction
Cancer is a disease of genetic aberrations, but there are many molecular processes downstream of the genome that may affect cancer phenotype. Proteins and their modifications connect genotype to phenotype and are central components in understanding cancer and finding effective treatments. In early large-scale cancer multi-omics studies, while genomic, epigenomic, and transcriptomic assays provide unbiased, genome-wide measurements, proteomics data are either missing or generated through antibody-based analysis of a small number of pre-selected proteins1. More recent cancer studies have combined unbiased mass spectrometry (MS)-based proteomics and phosphoproteomics with other genome-wide assays, an approach known as proteogenomics2,3. As the pioneer and a catalyst of cancer proteogenomics, the National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium (CPTAC) has systematically characterized over 1,000 treatment naïve primary tumors spanning 10 cancer types. Published CPTAC studies have demonstrated the value of these proteogenomics datasets for reinforcing existing knowledge, identifying new biological insights, and generating therapeutic hypotheses4–16. Beyond these publications, proteogenomics data generated in these studies also holds great potential to serve as a rich resource for the broad cancer research community to address their own basic and translational research questions for years to come17. However, until computational tools are available for biologists and clinicians to efficiently explore the vast amount of complex, interconnected data, the potential of these data will be severely underexploited.
Previously, we developed LinkedOmics18, which makes proteogenomics data from individual CPTAC studies available through a web portal to enable on the fly association analysis for one cancer type and one omics data type at a time. Despite its quickly increasing popularity, performing one analysis across all cancer types and all omics data types will take hundreds of clicks and many hours. Moreover, because effective methods for integrating and co-visualizing results from multiple cancer types and multiple data types remain severely underdeveloped in proteogenomics, it is challenging for data consumers to gain a holistic understanding of pan-cancer, multi-omics results.
Here, we present LinkedOmicsKB (http://kb.linkedomics.org), a proteogenomics data-driven knowledge base that makes consistently processed and systematically precomputed CPTAC pan-cancer proteogenomics data readily available to the public through a user-friendly web portal. We devised powerful visualization techniques to facilitate efficient exploration and reasoning of these data. Using three case studies, we illustrate the practical utility of LinkedOmicsKB in allowing users to gain new insights into genes, phosphorylation sites, somatic mutations, and cancer phenotypes.
Results
Overview of the proteogenomics data-driven knowledge base
LinkedOmicsKB was built upon omics and clinical data recently harmonized by the CPTAC pan-cancer resource working group (Fig. 1a, Methods). Clinical and histopathological data were carefully curated. Standardized reprocessing of data from all omics platforms and all cancer types using common computational pipelines and the same versions of genome assembly and gene annotation enabled streamlined and accurate downstream pan-cancer multi-omics data integration. Somatic mutation calls and copy number variation (CNV), methylation, mRNA, and protein quantifications were aggregated to gene level, whereas phosphorylation quantifications were aggregated by phosphosites. The complete dataset included mutation, CNV, methylation, mRNA, and protein data from 1,043 cancer patients spanning 10 cancer types for 18,469, 19,688, 12,809, 19,701, and 14,949 genes, respectively, as well as data for 125,969 phosphosites (Fig. 1a, Table S1).
Figure 1. Overview of the LinkedOmicsKB pipeline.
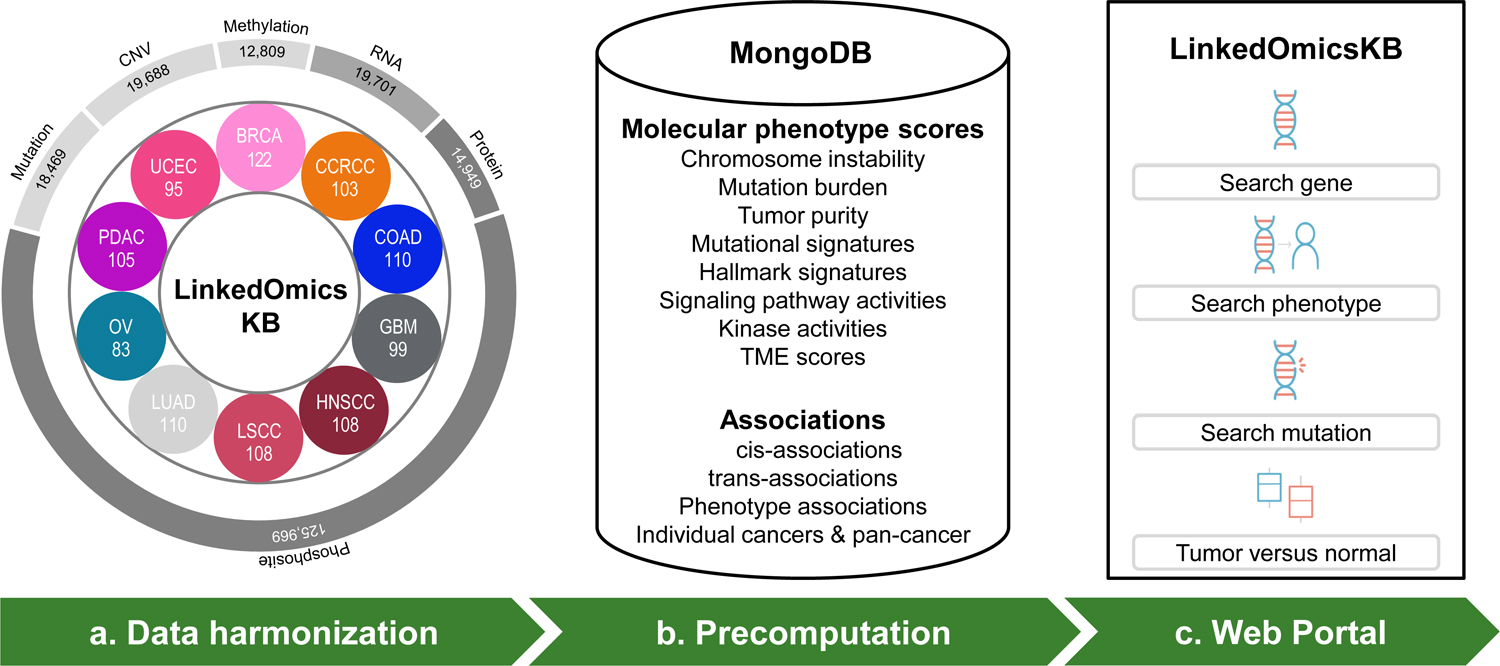
(a) Overview of the pan-cancer proteogenomics data in LinkedOmicsKB, including numbers of patients for each cancer type and total feature numbers of different omics data types. BRCA: breast cancer, CCRCC: clear cell renal cell carcinoma, COAD: colon adenocarcinoma, GBM: glioblastoma, HNSCC: head and neck squamous cell carcinoma, LSCC: lung squamous cell carcinoma, LUAD: lung adenocarcinoma, OV: ovarian cancer, PDAC: pancreatic ductal adenocarcinoma, and UCEC: uterine corpus endometrial carcinoma. (b) Summary of precomputed molecular phenotype scores and associations stored in the MongoDB. (c) The home page of the web portal allows querying with gene, phenotype, or mutation and browsing tumor-normal comparison results.
Based on the above harmonized data at DNA, RNA, protein, and phosphosite levels, we computed various molecular phenotypes, including chromosome instability, mutation burden, tumor purity, mutational signature, hallmark signature, signaling pathway activity, kinase activity, and immune infiltration and tumor microenvironment scores (Methods). To reduce online response time, which is critical for good user experience, we performed extensive offline analyses using carefully selected statistical tests, and meta p-values were calculated to integrate results at the pan-cancer level (Methods). To enable fast retrieval of the vast amount of heterogeneous precomputed data, we used MongoDB to store all relevant data (e.g., for a gene) in a hierarchical document (Fig. 1b). The web portal organizes precomputed analysis results into ~40,000 gene-, protein-, mutation-, and phenotype-centric web pages, which can be easily accessed by querying a gene, mutation, or phenotype of interest from the homepage (Fig. 1c).
Each gene-, protein-, mutation-, and phenotype-centric page includes several sections (see details in Method S1). Information in the portal includes phosphosites detectability across all studies, tumor vs normal difference at mRNA, protein, and phosphosite levels, respectively, mutation and phenotype associations for individual mRNAs, proteins, and phosphosites, cis-association across omics layers, and pairwise trans-associations between mRNAs, proteins, and phosphosites and kinases/phosphatases. Association results are directly fed into WebGestalt19 for pathway and network analysis.
Visualization to facilitate data exploration and reasoning
To address the key challenge of making complex pan-cancer, multi-omics results easily comprehensible to the users, we devised several advanced data visualization approaches. Associations between a gene and all clinical phenotypes, molecular phenotypes, and somatic mutations across all cancer types at copy number, mRNA, and protein levels, respectively, are summarized in an interactive pan-cancer, multi-omics Manhattan plot (Fig. 2a). This plot enables quick identification of cancer types and omics data types with interesting associations, as well as interactive examination of the top, highly significant associations. Detailed association results are presented in a sortable, searchable, filterable, and expandable table that can be switched between protein, mRNA, and copy number views (Fig. 2b). The table was designed to hold four dimensions of information, with columns corresponding to individual cancer types or pan-cancer, primary rows corresponding to associations between a phenotype/mutation and the omics data type of the primary view, expandable rows displaying associations at other omics levels for multi-omics comparison, and pop-up windows with appropriate statistical plots supporting the signed p-values displayed in the table, such as scatter plot, box plot, or Kaplan-Meier plot. Pan-cancer, multi-omics association results between a gene and a phenotype or mutation is depicted in a heatmap (Fig. 2c). Cis-associations between protein, mRNA, copy number, and methylation levels of a gene within each cancer type are visualized in a correlogram (Fig. 2d), in which each circle can be clicked to show the corresponding scatter plot. A correlogram is also used to visualize cis-associations between individual phosphosites and measurements at protein, mRNA, copy number and methylation levels, respectively, which facilitates the prioritization of phosphosites that are regulated independent of protein abundance (Fig. 2e). Zoomable lollipop plot and 3D protein structure viewer are used to visualize all identified phosphosites in the context of protein sequence, domains, and structure (Fig. 2f). In the phenotype and mutation pages, a scatter plot comparing mRNA and protein associations and another comparing protein and phosphosite associations (Fig. 2g) help prioritize protein and phosphosite-specific associations, respectively. Moreover, WebGestalt provides pathway and network visualization for mRNA, protein, and phosphosite-level association results (Fig. 2h).
Figure 2. Visualizations to facilitate efficient exploration and reasoning of pan-cancer, multi-omics data.
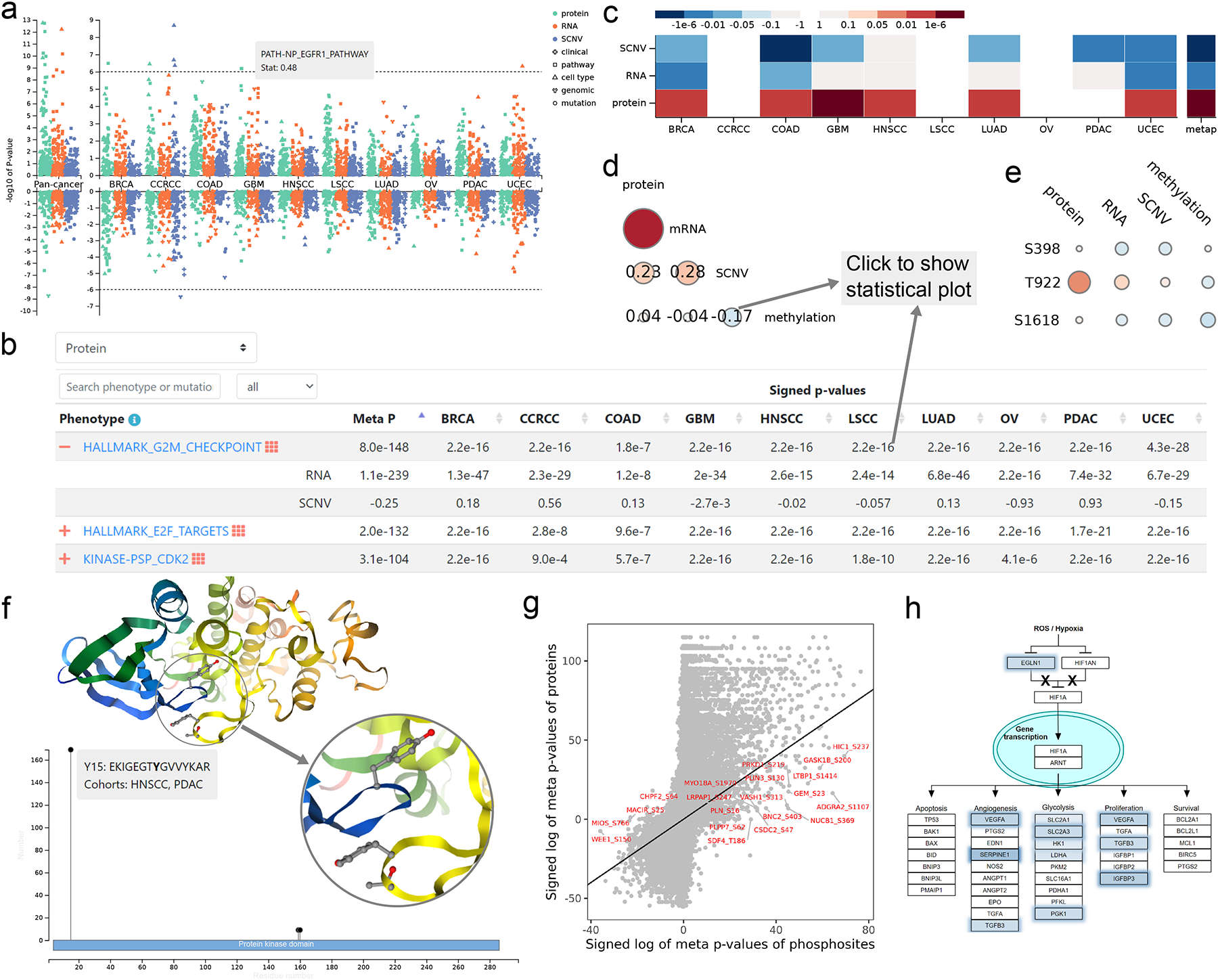
(a) Manhattan plot summarizing associations between a gene and all clinical phenotypes, molecular phenotypes, and somatic mutations across all cancer types at copy number, mRNA, and protein levels, respectively. Significant phenotypes or mutations (p<1.0e-6) can be shown with mouse hovering. (b) Detailed association results are presented in an interactive table that can be switched between protein, mRNA, and copy number views, with columns corresponding to individual cancer types or pan-cancer, primary rows corresponding to associations between a phenotype/mutation and the omics data type of the primary view, expandable rows displaying associations at other omics levels for multi-omics comparison, and pop-up windows with appropriate statistical plots supporting the signed p-values displayed in the table, such as scatter plot, box plot or Kaplan-Meier plot. (c) Heatmap summarizing associations between a phenotype or mutation and measurements of a gene at copy number, mRNA, and protein levels. Red and blue colors indicate positive and negative associations, respectively. (d) Correlogram visualizing pairwise cis-associations between protein, mRNA, copy number, and methylation levels of a gene. (e) Correlogram visualizing pairwise cis-associations between individual phosphosites and measurements at protein, mRNA, copy number and methylation levels, respectively. (f) Experimentally identified phosphosites of a protein are visualized on interactive protein structures and in a zoomable lollipop plot showing detected sample number or cohort number in the context of protein domains. Neighboring sequence and quantified cohorts for each phosphosite can be shown with mouse hovering in the lollipop plot. The zoom-in structure shows spatial proximity of sequentially distant phosphosites. (g) Scatter plot highlighting highly significant phosphosite associations that are independent of corresponding protein associations. (h) Pathway diagram highlighting genes contributing to the enrichment signal.
Proteogenomics insights into an understudied druggable protein
A primary utility of LinkedOmicsKB is to gain insights into any gene or phosphosite of interest. Because LinkedOmicsKB is data-driven and independent of existing knowledge, it is particularly useful for shedding light on understudied genes20 and the dark phosphoproteome21. To illustrate this utility, we analyzed CALHM5 (calcium homeostasis modulator family member 5), one of the 328 understudied druggable proteins nominated by the Illuminating the Druggable Genome (IDG) program22 for increased investigation.
The pan-cancer, multi-omics Manhattan plot highlighted extremely strong correlation between CALHM5 mRNA abundance and the stroma score, the epithelial-mesenchymal transition (EMT) pathway activity score, and the TGFbeta perturbation signature score (Fig. 3a). A closer look at the pan-cancer, multi-omics gene-phenotype association heatmaps showed that both CALHM5 mRNA and protein abundance were significantly associated with TGFbeta signaling (Fig. 3b, Fig. S1) and EMT (Fig. 3c, Fig. S2) in almost all ten cancer types, suggesting a role of CALHM5 in these tumor progression-related biological processes.
Figure 3. Proteogenomics insights into CALHM5.
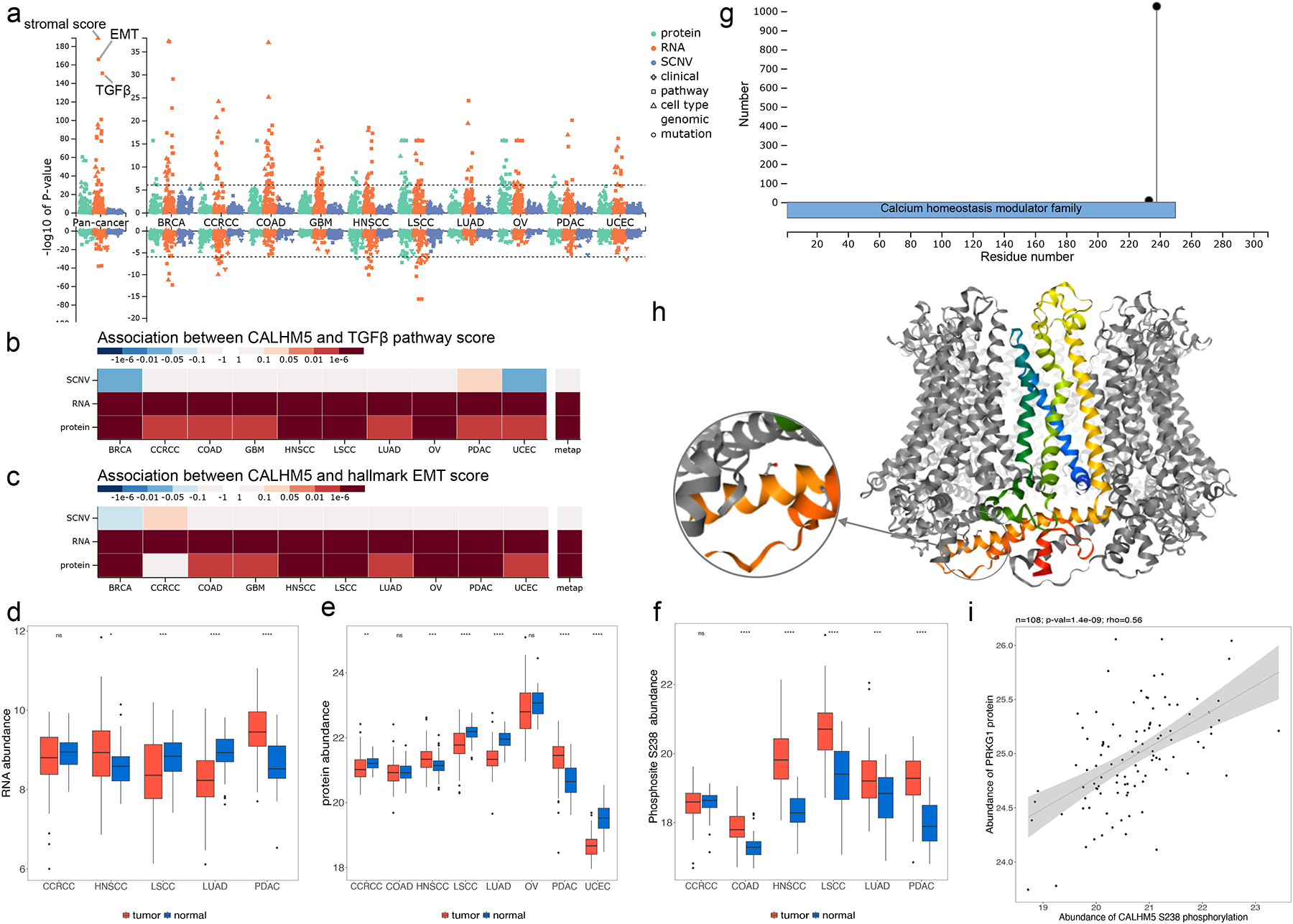
(a) Manhattan plot showing p-values of phenotype and mutation associations of CALHM5 at copy number, mRNA, and protein levels, respectively. The stroma, TGFbeta and epithelial-mesenchymal transition (EMT) scores are labeled in pan-cancer analysis at the mRNA level. (b) P-value heatmap summary of CALHM5 associations with the TGFbeta perturbation signature score computed by the PROGENy algorithm. (c) P-value heatmap summary of CALHM5 associations with the EMT pathway activity score computed by applying single sample gene set enrichment analysis (ssGSEA) to MSigDB Hallmark gene sets. (d) Boxplots depicting tumor and NAT difference of RNA data. (e) Boxplots depicting tumor and NAT difference of protein data. (f) Tumor and NAT difference of CALHM5 S238 phosphosite abundance. S238 phosphorylation abundance is significantly higher in LSCC and LUAD tumors despite significantly decreased mRNA and protein level shown in d and e. (g) Lollipop plot showing phosphosite S328 with high occurrence in samples and its sequence domain location. (h) Experimental structure of the CALHM5 homo-oligomer forming a channel with S238 highlighted (PDB: 7D60). (i) Kinase PRKG1 protein level is significantly positively correlated with S238 in LSCC with a Spearman correlation coefficient of 0.56 and p-value of 1.4e-9. ns: p ≥ 0.05; *p < 0.05; **p< 0.01; ***p < 0.001; ****p < 0.0001.
In tumor versus normal comparison, protein abundance of CALHM5 was significantly decreased in CCRCC, LSCC, LUAD, and UCEC but significantly increased in HNSCC and PDAC (Fig. 3d), and the same trend was observed at the mRNA level when data was available (Fig. 3e). Interestingly, phosphosite abundance of CALHM5 S238 was significantly increased in almost all cohorts where phosphoproteomics measurements are available (Fig. 3f), suggesting strong post-translational regulation of this gene during tumorigenesis. Identified in 1,026 tumor samples, S238 is located at the C terminal of the calcium homeostasis modulator domain (Fig. 3g) and the cytoplasmic region of the protein (Fig. 3h). Based on the kinase association table in LinkedOmicsKB, PRKG1 (protein kinase cGMP-dependent 1) ranked among the top kinases showing highly significant positive correlations with the phosphorylation abundance of CALHM5 S238 in LSCC (Fig. 3i) and other cancer types. PRKG1 is reported to phosphorylate numerous other proteins implicated in modulating cellular calcium in the UniProt database23, and thus LinkedOmicsKB connected CALHM5 S238 to a putative regulator.
Together, a quick analysis in LinkedOmicsKB generated abundant information and multiple testable hypotheses on cancer-associated function of CALHM5 and its regulation, paving the way to further investigation of this understudied druggable protein and its putative kinase regulator as targets for cancer treatment.
Proteogenomics insights into clinical phenotypes
Another important utility of LinkedOmicsKB is to gain insights into clinical and molecular phenotypes of interest. We explored the tumor vs normal comparison page to identify proteins that may play important roles in tumorigenesis. Among the top 10 most significantly elevated proteins in the pan-cancer tumor vs normal comparison (Fig. 4a), only three (FEN1, HSP90AB1, and CBFB) have corresponding encoding genes documented in the Cancer Gene Census24, demonstrating the potential of LinkedOmicsKB proteomics data in revealing putative cancer genes that were missed in genomic studies, such as NSUN2, PLOD2, P4HA1, NRDC, SMARCA5, UTP4, NUP93. The scatter plot comparing pan-cancer tumor vs normal differences at mRNA and protein levels further showed protein-specific elevation for SMARCA5 and UTP4 (Fig. 4b). SMARCA5 protein abundance was poorly correlated with its mRNA abundance in all cancer types (Fig. 4c–d, Fig. S3a); however, it showed the highest correlation with the protein abundance of its interaction partner BAZ1B among all proteins in pan-cancer analysis, with highly significant associations observed in all cancer types (Fig. 4e–f, Fig. S3b). SMARCA5 binds to BAZ1B to form a complex known as WICH (WSTF-ISWI chromatin remodeling complex), which facilitates DNA replication and promotes DNA repair25. The pan-cancer, multi-omics Manhattan plot highlighted extremely strong correlation between SMARCA5 protein abundance and the G2M checkpoint signature score, E2F targets signature score, MYC targets signature score, and chromosomal instability score (Fig. 4g), and these associations were much weaker or missing based on SMARCA5 mRNA measurements (Fig. 4h, Fig. S3c). These data suggest that the protein abundance of SMARCA5 in tumor samples is determined primarily by the abundance of its interaction partner BAZ1B instead of its mRNA abundance, and protein abundance better reflects known function of the gene in DNA replication and DNA repair than mRNA abundance.
Figure 4. Proteogenomics insights into clinical phenotypes.

(a) The top 10 overexpressed proteins in tumors sorted by meta p-values. (b) Scatter plots highlighting tumor-associated protein abundance changes that are independent of mRNA abundance changes. (c) Cis-correlation between protein, mRNA, copy number, and methylation levels of SMARCA5, showing low correlation between protein and mRNA abundance in all cancer cohorts. (d) Scatter plot showing the low correlation between the mRNA and protein abundance of SMARCA in BRCA as an example. (e) Scatter plot showing the correlation between the protein abundance of SMARCA5 and BAZ1B in BRCA as an example. (f) Heatmap summarizing p-values of associations between SMARCA5 and BAZ1B at mRNA, and protein levels. (g) Manhattan plot summarizing p-values of SMARCA5 associations with phenotypes and mutations at copy number, mRNA, and protein levels, respectively. The G2M checkpoint signature score, E2F targets signature score, MYC targets signature score, and chromosomal instability score are labeled in pan-cancer analysis at the protein level. (h) Heatmap summarizing p-values of associations between SMARCA5 and G2M checkpoint signature score and chromosomal instability score, respectively. The SMARCA5 protein levels show stronger associations than mRNA. (i-j) Kaplan-Meier plots of PLOD2 and PLOD1 protein, respectively, in the CCRCC cohort. Hazard ratio from Cox proportional hazards regression model and p-value from logrank test are shown on the top. (k) Heatmap summarizing associations of PLOD1 and PLOD2 with EMT signature score, respectively, at protein, mRNA, copy number levels.
To further explore potential roles of the top 10 tumor-overexpressed proteins in tumor progression, we overlapped them with the top 10 poor prognosis-associated proteins identified through pan-cancer analysis in LinkedOmicsKB (Fig. S4) and found an overlapping protein PLOD2 (Fig. 4i). The top 10 poor prognosis-associated proteins also included its paralogue PLOD1 (Fig. 4j). These procollagen-lysine, 2-oxoglutarate 5-dioxygenases catalyze lysyl hydroxylation to hydroxylysine, which is a critical step in biosynthesis of fibrillar collagens26. mRNA and protein abundance of both PLOD1 and PLOD2 were significantly associated with EMT in tumor samples in almost all ten cancer types (Fig. 4k), consistent with their reported role in cancer cell invasion and migration in model systems27,28. In summary, analysis in LinkedOmicsKB not only identified genes that are important in cancer initiation and progression but also enhanced our understanding of the function and regulation of these genes.
Proteogenomics insights into TP53 mutations
LinkedOmicsKB also provides an effective means to gain new insights into somatic mutations. As an example, TP53 mutants were associated with increased TP53 protein abundance in all cancer types with sufficient sample size for statistical analysis, but TP53 mRNA showed no difference or decreased abundance in TP53 mutants compared to other samples (Fig. 5a), suggesting a direct impact of the mutations on protein translation or stability. Moreover, TP53 mutants showed highly increased phosphorylation levels of multiple phosphosites on TP53BP1 (e.g., S1758, S398, S1618, S1971, and T922), independent of TP53BP1 protein abundance (Fig. 5b). TP53BP1, a tumor suppressor protein with a critical role in DNA double-strand break repair, is an extensively phosphorylated protein with 198 phosphorylation sites identified in the CPTAC pan-cancer data (Fig. 5c). The correlogram further showed moderate or even negative correlations between TP53BP1 protein abundance and the phosphorylation level of these TP53 mutation associated sites in many cancer types (Fig. 5d), such as a negative correlation between S1618 phosphorylation and TP53BP1 protein and RNA abundance in breast cancer (Fig. 5e). Phosphorylation of S1618 was significantly positively associated with G2M checkpoint signature score in breast cancer (Fig. 5f), whereas TP53BP1 protein abundance showed an opposite direction of association (Fig. 5g), suggesting phosphorylation significantly affects TP53BP1 activity. Moreover, the kinase association table connected these phosphorylation events to multiple cell cycle kinases including TTK, PLK1, AURKA, CDK1, and PRKDC (Fig. 5h), among which CDK1 and PLK1 have been previously reported to phosphorylate TP53BP1 within its ubiquitin dependent recruitment (UDR) domain to suppress its function in DNA repair29. Together, this analysis revealed a previously undocumented relationship between TP53 mutation and TP53BP1 hyperphosphorylation, which may underlie reduced DNA repair in TP53 mutants.
Figure 5. Proteogenomics insights into TP53 mutations.
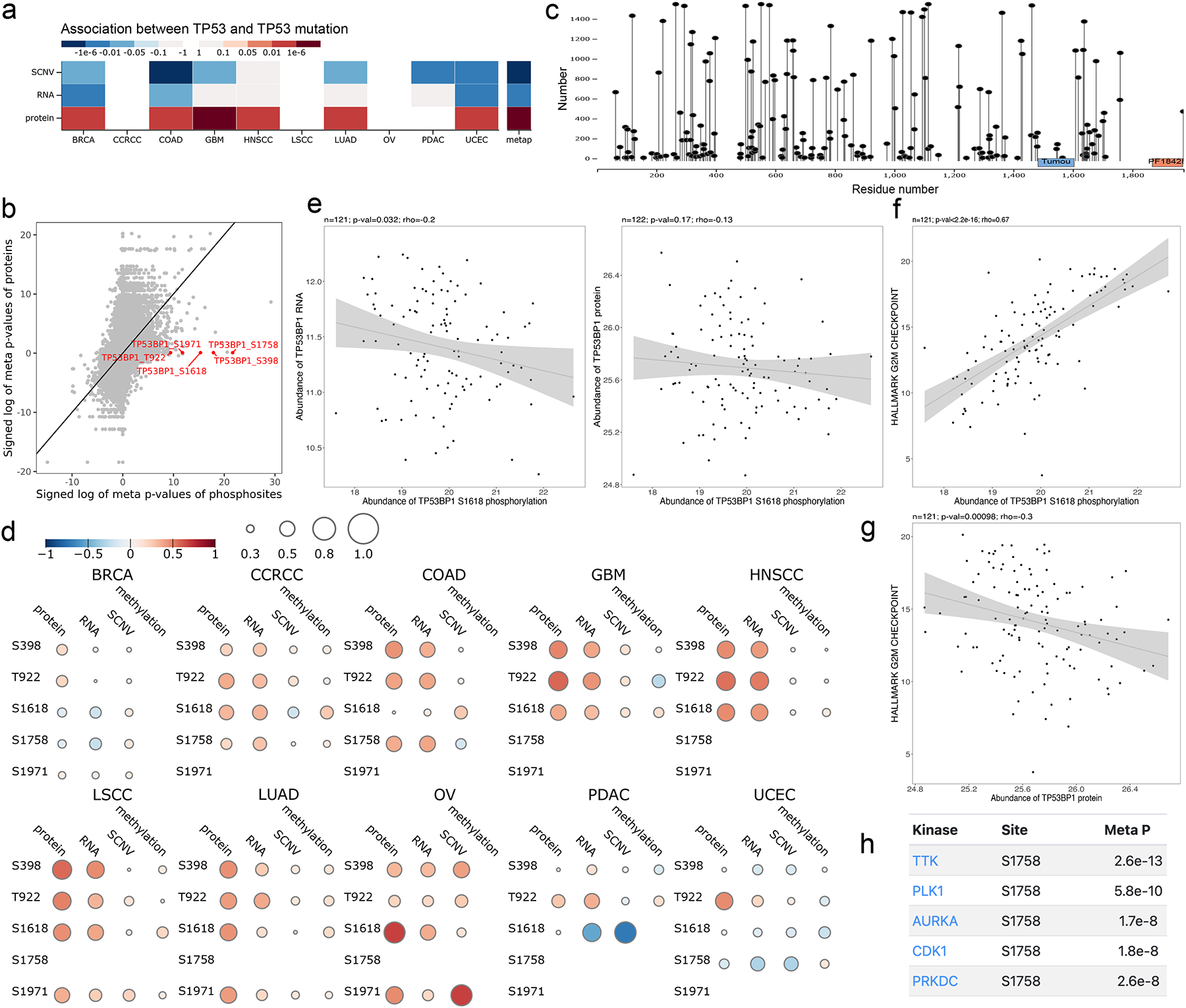
(a) Heatmap showing TP53 mutation is highly associated with TP53 protein but not with mRNA abundance. (b) TP53 mutation is highly associated with the abundance of multiple phosphosites on TP53BP1 but is not significantly associated with TP53BP1 protein abundance. (c) Lollipop plot showing TP53BP1 with the sequence locations and sample numbers of 198 phosphorylation sites identified in the CPTAC pan-cancer data. (d) Correlogram for the TP53 mutation-associated TP53BP1 phosphosites showing moderate or negative associations between these phosphosites and TP53BP1 protein and RNA measurements in all cohorts. (e) Scatter plots showing negative correlation between S1618 phosphorylation and TP53BP1 protein and RNA abundance in breast cancer, respectively. (f) Scatter plot showing significant association between S1618 phosphorylation and G2M checkpoint signature score in breast cancer with a Spearman correlation coefficient of 0.67 and p-value smaller than 2.2e-16. (g) Scatter plot showing negative association between TP53BP1 protein and G2M checkpoint signature score in breast cancer. (h) The top kinase associations for TP53BP1 S1758 p
Discussion
Proteogenomics is becoming a powerful approach to comprehensive molecular understanding of human cancer, but meanwhile, the resulting large multi-omics data have led to an increasing gap between data generation and investigators’ ability to interpret the data. LinkedOmicsKB makes consistently processed and systematically precomputed CPTAC pan-cancer proteogenomics data available through ~40,000 gene-, protein-, mutation-, and phenotype-centric web pages and uses intuitive yet effective visualization techniques to facilitate efficient exploration and reasoning of the complex, interconnected data.
LinkedOmicsKB provides a powerful platform to gain functional insights into proteins and their phosphorylation in an unbiased manner. Proteomics and other omics technologies enable genome-wide molecular profiling; however, there remain many functionally uncharacterized or poorly characterized proteins20. For example, 18% of the 1,878 genes that are essential for proliferation in a human cell line remained uncharacterized as of 201530, and only 5–10% of the potentially druggable proteins are targeted by FDA-approved drugs22. This problem is even worse for protein modifications including phosphorylation21. Among the 125,969 phosphosites identified in the CPTAC studies, only 5% have known regulatory kinase or functional annotation in PhosphositePlus31, the most comprehensive knowledge base of phosphosites. As demonstrated in the case study of the understudied druggable protein CALHM5, analysis in LinkedOmicsKB generated experimentally testable hypotheses on the function of CALHM5 and the regulation of its phosphorylation. Similar analysis can be applied to all genes and phosphosites in the knowledge base, independent of existing knowledge.
LinkedOmicsKB democratizes the investigation of the relationship between proteins as well as their modifications and genotypes or phenotypes, a hallmark of proteogenomics. Moreover, convenient exploration of such relationships across cancer types expedites the discovery of shared and cancer-type-specific associations. One limitation of the CPTAC dataset is that the clinical follow-up time is relatively short because the samples were prospectively collected. Moreover, treatment and response information is also limited. To compensate for the shortage of clinical phenotype information, we quantified a wide range of molecular phenotypes for individual tumor samples, allowing users to associate proteins and phosphorylations to not only clinical phenotypes but molecular phenotypes. Another limitation of LinkedOmicsKB is that molecular subtype information is not specifically considered. This is due to the pan-cancer focus of the resource, which makes it difficult to consider cancer subtypes across all cancer types. Moreover, most of the analyses in LinkedOmicsKB are based on associations, and statistical power would be greatly reduced when analyses are limited to individual cancer subtypes. For the same reason, genotype association analysis was limited to genes with coding mutations in at least 10 samples in a cohort, and we did not further separate the mutations into different categories in association analysis. In the future, we will aggregate mutations to pathway level, which will allow more comprehensive protein-genotype association analysis.
To ensure that LinkedOmicsKB remains up-to-date, we will follow a systematic plan for updating the portal with new data as it becomes available. This plan involves identifying relevant sources of data, assessing their quality, processing the data using our standardized pipelines, and integrating it into the existing database. We will prioritize maintaining the comprehensiveness, quality, and consistency of the portal throughout this process. Regular updates will be scheduled and communicated to users, providing relevant metadata, annotations, and summary statistics to aid in the interpretation of the new data. Additionally, we also plan to make the analysis pipeline and web portal customizable in the future, so that the framework can be applied to other cohort-based proteogenomic studies both within and outside the cancer community. For example, a breast cancer portal can be developed to integrate proteogenomics data generated from multiple independent breast cancer studies.
Despite the above-mentioned limitations and planned updates and developments, the three case studies clearly demonstrated the practical utility of LinkedOmicsKB in generating novel testable hypotheses on genes, phosphorylation sites, somatic mutations, and cancer phenotypes, and they are just a quick glimpse of what can be achieved using the tool. With easily browsable data on 19,701 protein coding genes, 125,969 phosphosites, and 256 genotypes and phenotypes from 10 cancer types and pan-cancer analyses, LinkedOmicsKB provides a user-friendly platform for anyone with internet access to conduct meaningful inquiries into CPTAC data and to make data-driven scientific discoveries.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead author, Bing Zhang (bing.zhang@bcm.edu).
Materials Availability
This study did not generate new materials.
Data and Code Availability
Processed CPATC pan-cancer data matrices used in this study have been deposited at LinkedOmicsKB and are publicly available as of the date of publication. These accession numbers for the datasets are listed in the key resources table. In addition, raw and processed proteomics as well as open access genomic data can be obtained via Proteomic Data Commons (PDC) at https://pdc.cancer.gov/pdc/cptac-pancancer. Raw genomic and transcriptomic data files can be accessed via the Genomic Data Commons (GDC) Data Portal at https://portal.gdc.cancer.gov.
Original code has been deposited at Figshare and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
METHOD DETAILS
Data sets
CPTAC pan-cancer proteogenomics data processed using standardized data processing pipelines by the CPTAC pan-cancer working group and harmonized by the BCM harmonization pipeline were used in this study. Detailed information on data processing and harmonization is described in a companion paper (Li et al., accepted, Cancer Cell, CANCER-CELL-D-22–00603). Briefly, the data included only cases and samples used in the flagship manuscripts4–10,13,16,72. For gene harmonization between omics, all data were processed using a common reference genome annotation, GENCODE V34 basic (CHR)32. A single primary isoform was selected for each gene. For coding genes, MANE Select and SwissProt were used to prioritize isoforms. If a gene did not have a single MANE Select and/or SwissProt isoform, then the isoform was prioritized using the longest protein sequence followed by the longest transcript. Additionally, remaining Swiss-Prot proteins and MANE Plus Clinical isoforms were retained as secondary isoforms for web portal display. For data harmonization across cohorts, standardized pipelines were used to process each omics type. For RNA and proteomics, expression levels were normalized to a common value. Below, we provide a brief overview of the data processing methods used for each omics data type, and further details are available in the companion paper (Li et al, accepted, Cancer Cell, CANCERCELL-D-22–00603).
Identification of significantly mutated genes
The Strelka v248, MUTECT v1.749, VarScan v2.3.850, and Pindel v0.2.551 were used by the Broad Institute and Washington University in St Louis teams in the CPTAC pan-cancer working group to call mutations from whole exome sequencing (WES). Mutations from any two tools with a minimal variant allele frequency (VAF) of 0.05 in tumors were retained and rare mutations in cancer driver genes reported in Bailey et al. 201838 with a VAF of at least 0.015 were rescued. Somatic mutations were converted from the genome assembly hg38 to hg19 by CrossMap (version 0.5.3)52. Unmapped mutations were excluded from downstream analysis. MutSigCV (version 1.41)53 was applied to the converted somatic mutations to identify significantly mutated genes (q value < 0.01) for each cancer type. Synonymous mutations were not included. The default reference files, background coverage (exome full192), mutation types, and gene covariates, were used in this analysis.
DNA methylation processing
We downloaded processed probe level beta values (methylated to unmethylated signal intensities) from the GDC. All loci in the EPIC Manifest file infinium-methylationepic-v-1–0-b5-manifest-file-csv.zip were reannotated using ANNOVAR (v 04.16.2018)54 with the selected gene annotation GENCODE V34 basic (CHR). For downstream integrated analyses, CpG islands annotated as upstream (1kb upstream of the transcription start site) and UTR’5 were included for coding genes. For noncoding genes, only CpG islands annotated as upstream were included. Then, the gene-level methylation was derived by averaging these probe-level methylation beta values.
Copy-number assessment
For somatic copy number alteration quantification, WES bam files were processed by the CopywriteR package55 to derive log2 tumor-to-normal copy number ratios, and the circular binary segmentation (CBS) algorithm73 implemented in the CopywriteR package55 was used for the copy number segmentation, with default parameters. Next, a GISTIC247 reference was built using GENCODE V34 basic (CHR) gene annotation. Then GISTIC2 with this reference and CNV threshold of +/−0.3 was used to identify gene-wise and focal level copy number alterations. Each gene of every sample was assigned a thresholded copy number level and a log2 tumor-to-normal copy number ratio.
RNA quantification
CIRI v2.0.656 with BWA v0.7.17-r118857 was used to call circular RNA with at least 10 supporting reads. RSEM v1.3.158 and Bowtie2 v2.3.359 were used to quantify both linear and circular RNA expression. The upper quantiles of coding gene RSEMs were normalized to 1500 and the same normalization factors were applied to noncoding genes. Then, the normalized RSEM values were log2-transformed.
Proteomics and phosphoproteomics data processing
MSFragger v3.460, the Philosopher v4.0.161 toolkit, and the TMT-Integrator62 pipeline were used by the Michigan University team in the CPTAC pan-cancer working group to process and quantify the mass spectrometry data. Data were normalized by median centering the medians of the reference intensities.
Phosphoproteomics isoform mapping
Single phosphosites were re-annotated to the selected primary and secondary protein isoforms. First, if the original selected isoform protein sequence matched the primary selected isoform sequence, only the protein isoform ID was changed. If the protein sequences did not match, the primary selected sequence was searched for all peptides identified for the phosphorylation site. If at least one peptide matched exactly once to the sequence, that peptide was used to update the site position. Otherwise, for peptides that matched more than one location, the one that matched the fewest locations was selected and the first matching position was used to update the site position. Finally, if no peptides exactly matched the selected protein sequence, all I’s were changed to L’s in both the sequence and peptides and the matching step was performed again. All sites with no peptides that could be mapped to the selected protein sequence were discarded after this step. Because some re-annotated site IDs were no longer unique, the data row with the fewest missing values was selected for that site and all others discarded. The site ID finally consisted of the Ensembl gene ID, Ensembl protein ID, site position based on the selected protein ID, fifteenmer (+/− 7 amino acids) based on the selected protein ID, and a flag for whether the protein is a primary (1) or secondary (2) selected sequence.
Gene ID to gene name mapping
For web portal display, all Ensembl gene IDs for primary selected protein isoforms were mapped uniquely to a gene name. “PAR_Y” was added to the name of all PAR_Y gene names. The following order of priority was used to assign the gene symbol to the gene ID; the Ensembl gene ID was appended to the gene symbol for all following duplicates (e.g., AHRR_ENSG00000063438). First, order by presence of a SwissProt ID assigned to the protein ID, isoforms listed in the MANE plus Clinical annotation, longer CCDS length, longer transcript length, and alphabetic order of the Ensembl gene ID.
Clinical data
The clinical data used in the portal were collected from CPTAC with the May 2022 update. Age was truncated to 90 years. Tumors with a size <=0 were replaced with NA. For overall survival analysis, cases were removed if a death occurred within 30 days of initial diagnosis and for progression free survival analysis, cases were removed with follow up or a new tumor event that occurred within 10 days of the initial diagnosis.
CIN score
The chromosome instability (CIN) score reflects the overall copy number aberration across the whole genome. From the segmentation result, we used a weighted-sum approach to summarize the chromosome instability for each sample10. The absolute segment level log2 ratios of all segments (indicating the copy number aberration of these segments) within a chromosome were weighted by the segment length and summed up to derive the instability score for the chromosome. The genome-wide chromosome instability index was calculated by summing up the instability score of all 22 autosomes. The R package genomicWidgets was used to implement the method (https://github.com/bzhanglab/genomicWidgets).
Mutation burden
Tumor mutation burden (TMB) was extracted from the mutation annotation file (maf) by the maftools R package (V2.10.0)63. It was calculated as the number of non-synonymous variants per million bp.
Immune deconvolution
The R package immunedeconv (V2.0.4)64 was used to perform immune cell deconvolution using RNA expression data (TPM). Among the seven deconvolution methods in immunedeconv, CIBERSORT65 and xCell66 were selected in our analysis. CIBERSORT was performed in the ‘abs’ mode.
ESTIMATE
The ESTIMATE scores reflecting the overall immune and stromal infiltration were calculated by the R package ESTIMATE67 using the normalized RNA expression data (RSEM). We removed genes with 0 expression in >=50% samples of a cohort.
PROGENy score
The PROGENy scores were inferred using the R package progeny (V1.10.0)68 with default parameters using the RNA expression data (FPKM). Genes with mean expression = 0 in a cohort were removed from the analysis.
MSigDB hallmark pathway single sample gene set enrichment analysis (ssGSEA)
ssGSEA was performed for each cancer type using gene-wise Z-scores of the RNA expression data (RSEM) for the MSigDB Hallmark gene sets v7.074 via the ssGSEA2.0 R package40. RNA data were filtered to coding genes with < 50% 0 expression. (Parameters: sample.norm.type=“rank”, weight=0.75, statistic=“area.under.RES”, nperm=1000, min.overlap=10). Pathway activity scores are normalized enrichment scores from ssGSEA.
Phosphosite signature scores
Phosphosite signature scores were calculated using the PTMsigDB v1.9.0 database and the ssGSEA2.0 R package40. The parameters were the same as those used for Hallmark pathway activity (sample.norm.type=“rank”, weight=0.75, statistic=“area.under.RES”, nperm=1000, min.overlap=10). Phosphoproteomics data were filtered to the fifteenmer phosphosites with complete data across all samples within a cohort. If there were multiple rows with complete data for identical fifteenmers, one row was selected at random. Each site was z-score transformed. Activity scores are normalized enrichment scores from ssGSEA.
Mutation signature calling
The R package SigProfilerMatrixGeneratorR69 (version 1.0) was used to call mutation signatures from WES-derived somatic mutation data. All synonymous and non-synonymous mutations were included. The maximum number of signatures was set to 10 and nmf replicates parameter was set to 100. The activity scores of the decomposed solution suggested by SigProfilerMatrixGenerator were used as signature scores.
Tumor purity
The DoAbsolute R package (V2.2)70 with ABSOLUTE (V1.0.6)71 was used to infer tumor purity and ploidy from somatic mutations and WES-based CNV. The parameters min.mut.af and max.as.seg.count were set to 0.02 and 5000, respectively. All other parameters were set as default. These results are referred to as Tumor Purity (ABSOLUTE) in the portal. Additionally, CNVEX16 was used to infer tumor purity using both whole genome sequencing (WGS) and WES data (https://github.com/mctp/cnvex) by the University of Michigan team, and these results are referred to as Tumor Purity (WGS) in the portal.
Statistical associations
For all association tests, at least 10 samples within a group were required to have measurements. The statistical test was tailored to the data type. Spearman’s correlation was used for continuous data, Jonckheere-Terpstra trend test for ordinal data, Student’s T-test for binary data, and Cox regression for time to event data.
Meta p-value calculation
Meta p-values were calculated with the “sumz” method from the R package metap (V1.4). P-values of individual cohorts were first converted to one-sided p-values and the sign for p-values not consistent with the majority were reversed. The calculated meta p-value was converted back to two-sided p-values and then the major sign of association was added.
Tumor versus normal comparison
Paired tumor samples and normal samples derived from 8 cancer types (CCRCC, COAD, HNSCC, LSCC, LUAD, OV, PDAC, and UCEC) for both proteomics and phosphoproteomics and 5 cancer types (CCRCC, HNSCC, LSCC, LUAD and PDAC) for RNASeq were used for differential expression analysis. Proteins were required to be detected in at least 20 tumor samples and 10 normal samples for proteomics and phosphoproteomics datasets. The unpaired Wilcoxon Rank Sum test was used to calculate significance.
Gene annotations
Gene names and descriptions were acquired from RefSeq. Additionally, genes were annotated with important functions and categories. Kinases were defined as those listed in KinBase (http://kinase.com/web/current/kinbase/genes/SpeciesID/9606/)41 or SwissProt (keyword:”Kinase [KW-0418]” AND reviewed:yes AND organism:”Homo sapiens (Human) [9606]”)23. Phosphatases with an “active” status were collected from DEPOD http://www.depod.bioss.uni-freiburg.de/download.php)42. Transcription factors were downloaded from http://humantfs.ccbr.utoronto.ca44 and filtered to those with the ‘Is TF’ = “Yes”. Receptors and ligands were downloaded from CellTalkDB (https://github.com/ZJUFanLab/CellTalkDB/tree/master/database)43 and mouse interactions were excluded. Essential genes were defined as those deemed pan cancer essential genes in cell lines downloaded from DepMap Public 21Q445. Cancer drivers were collated from the Cancer Gene Census37 (Tier 1), Bailey et al, 201838 Table S1 (high confidence calls from 20/20+), and Tokheim et al, 201639 Table S1 (all three tools). Drug targets were collected from DrugBank33 and Guide to Pharmacology34 and potentially druggable genes were collected from the Drug Gene Interaction Database36 and the Cell Surface Protein Atlas35.
Website development
Precomputed data were stored in MongoDB (v4.2). The backend was developed with PHP (v5.6) and Slim framework (v3). The frontend was based on Bootstrap 4 and some extension JavaScript libraries (Bootstrap-table v1.19). The interactive visualizations were built with D3.js (v5).
Supplementary Material
HIGHLIGHTS.
CPTAC proteogenomics data from 1,043 cancer patients across 10 cancer types
40,000 web pages dedicated to genes, proteins, mutations, and phenotypes
User-friendly visualization tools for efficient data exploration and analysis
Practical utility demonstrated through three informative case studies
Acknowledgements
We gratefully acknowledge contributions from the CPTAC and its Pan-Cancer Analysis Working Group. This study was supported by grants U24 CA210954, U24 CA271076, R01 CA245903, and U01 CA271247 from the National Cancer Institute (NCI), the Cancer Prevention & Research Institutes of Texas (CPRIT) award RR160027, and funding from the McNair Medical Institute at The Robert and Janice McNair Foundation. B.Z. is a CPRIT Scholar in Cancer Research and a McNair Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
B.Z. received consulting fees from AstraZeneca.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
References
- 1.Hutter C, and Zenklusen JC (2018). The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 173, 283–285. [DOI] [PubMed] [Google Scholar]
- 2.Zhang B, Whiteaker JR, Hoofnagle AN, Baird GS, Rodland KD, and Paulovich AG (2019). Clinical potential of mass spectrometry-based proteogenomics. Nature Reviews Clinical Oncology 16, 256–268. 10.1038/s41571-018-0135-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mani DR, Krug K, Zhang B, Satpathy S, Clauser KR, Ding L, Ellis M, Gillette MA, and Carr SA (2022). Cancer proteogenomics: current impact and future prospects. Nat. Rev. Cancer 22, 298–313. [DOI] [PubMed] [Google Scholar]
- 4.Cao L, Huang C, Cui Zhou D, Hu Y, Lih TM, Savage SR, Krug K, Clark DJ, Schnaubelt M, Chen L, et al. (2021). Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 184, 5031–5052.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Satpathy S, Krug K, Jean Beltran PM, Savage SR, Petralia F, Kumar-Sinha C, Dou Y, Reva B, Kane MH, Avanessian SC, et al. (2021). A proteogenomic portrait of lung squamous cell carcinoma. Cell 184, 4348–4371.e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L-B, Karpova A, Gritsenko MA, Kyle JE, Cao S, Li Y, Rykunov D, Colaprico A, Rothstein JH, Hong R, et al. (2021). Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 39, 509–528.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang C, Chen L, Savage SR, Eguez RV, Dou Y, Li Y, da Veiga Leprevost F, Jaehnig EJ, Lei JT, Wen B, et al. (2021). Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell 39, 361–379.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krug K, Jaehnig EJ, Satpathy S, Blumenberg L, Karpova A, Anurag M, Miles G, Mertins P, Geffen Y, Tang LC, et al. (2020). Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 183, 1436–1456.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K, Petralia F, Li Y, Liang W-W, Reva B, et al. (2020). Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 182, 200–225.e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen B, Dou Y, Zhang Y, Shi Z, Arshad OA, et al. (2019). Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 177, 1035–1049.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, et al. (2014). Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, et al. (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dou Y, Kawaler EA, Cui Zhou D, Gritsenko MA, Huang C, Blumenberg L, Karpova A, Petyuk VA, Savage SR, Satpathy S, et al. (2020). Proteogenomic Characterization of Endometrial Carcinoma. Cell 180, 729–748.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDermott JE, Arshad OA, Petyuk VA, Fu Y, Gritsenko MA, Clauss TR, Moore RJ, Schepmoes AA, Zhao R, Monroe ME, et al. (2020). Proteogenomic Characterization of Ovarian HGSC Implicates Mitotic Kinases, Replication Stress in Observed Chromosomal Instability. Cell Rep Med 1. 10.1016/j.xcrm.2020.100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou J-Y, Petyuk VA, Chen L, Ray D, et al. (2016). Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 166, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark DJ, Dhanasekaran SM, Petralia F, Pan J, Song X, Hu Y, da Veiga Leprevost F, Reva B, Lih T-SM, Chang H-Y, et al. (2019). Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 179, 964–983.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodriguez H, Zenklusen JC, Staudt LM, Doroshow JH, and Lowy DR (2021). The next horizon in precision oncology: Proteogenomics to inform cancer diagnosis and treatment. Cell 184, 1661–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vasaikar SV, Straub P, Wang J, and Zhang B (2018). LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Research 46, D956–D963. 10.1093/nar/gkx1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao Y, Wang J, Jaehnig EJ, Shi Z, and Zhang B (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Research 47, W199–W205. 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kustatscher G, Collins T, Gingras A-C, Guo T, Hermjakob H, Ideker T, Lilley KS, Lundberg E, Marcotte EM, Ralser M, et al. (2022). Understudied proteins: opportunities and challenges for functional proteomics. Nat. Methods 19, 774–779. [DOI] [PubMed] [Google Scholar]
- 21.Needham EJ, Parker BL, Burykin T, James DE, and Humphrey SJ (2019). Illuminating the dark phosphoproteome. Sci. Signal 12. 10.1126/scisignal.aau8645. [DOI] [PubMed] [Google Scholar]
- 22.Oprea TI, Bologa CG, Brunak S, Campbell A, Gan GN, Gaulton A, Gomez SM, Guha R, Hersey A, Holmes J, et al. (2018). Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discov 17, 377. [DOI] [PubMed] [Google Scholar]
- 23.UniProt Consortium (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49, D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, and Forbes SA (2018). The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oppikofer M, Bai T, Gan Y, Haley B, Liu P, Sandoval W, Ciferri C, and Cochran AG (2017). Expansion of the ISWI chromatin remodeler family with new active complexes. EMBO Rep 18, 1697–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gilkes DM, Semenza GL, and Wirtz D (2014). Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat. Rev. Cancer 14, 430–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SFT, Csiszar K, Giaccia A, Weninger W, et al. (2009). Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, and Keely PJ (2008). Collagen density promotes mammary tumor initiation and progression. BMC Med 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benada J, Burdová K, Lidak T, von Morgen P, and Macurek L (2015). Polo-like kinase 1 inhibits DNA damage response during mitosis. Cell Cycle 14, 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, and Sullivan M (2012). PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res 40, D261–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, et al. (2018). DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46, D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harding SD, Armstrong JF, Faccenda E, Southan C, Alexander SPH, Davenport AP, Pawson AJ, Spedding M, Davies JA, and NC-IUPHAR (2022). The IUPHAR/BPS guide to PHARMACOLOGY in 2022: curating pharmacology for COVID-19, malaria and antibacterials. Nucleic Acids Res 50, D1282–D1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bausch-Fluck D, Hofmann A, Bock T, Frei AP, Cerciello F, Jacobs A, Moest H, Omasits U, Gundry RL, Yoon C, et al. (2015). A mass spectrometric-derived cell surface protein atlas. PLoS One 10, e0121314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freshour SL, Kiwala S, Cotto KC, Coffman AC, McMichael JF, Song JJ, Griffith M, Griffith OL, and Wagner AH (2021). Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res 49, D1144–D1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. (2019). COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 47, D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. (2018). Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tokheim CJ, Papadopoulos N, Kinzler KW, Vogelstein B, and Karchin R (2016). Evaluating the evaluation of cancer driver genes. Proc. Natl. Acad. Sci. U. S. A 113, 14330–14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krug K, Mertins P, Zhang B, Hornbeck P, Raju R, Ahmad R, Szucs M, Mundt F, Forestier D, Jane-Valbuena J, et al. (2019). A Curated Resource for Phosphosite-specific Signature Analysis. Mol. Cell. Proteomics 18, 576–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manning G, Whyte DB, Martinez R, Hunter T, and Sudarsanam S (2002). The protein kinase complement of the human genome. Science 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- 42.Damle NP, and Köhn M (2019). The human DEPhOsphorylation Database DEPOD: 2019 update. Database 2019. 10.1093/database/baz133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shao X, Liao J, Li C, Lu X, Cheng J, and Fan X (2021). CellTalkDB: a manually curated database of ligand-receptor interactions in humans and mice. Brief. Bioinform 22. 10.1093/bib/bbaa269. [DOI] [PubMed] [Google Scholar]
- 44.Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, and Weirauch MT (2018). The Human Transcription Factors. Cell 172, 650–665. [DOI] [PubMed] [Google Scholar]
- 45.Dempster JM, Pacini C, Pantel S, Behan FM, Green T, Krill-Burger J, Beaver CM, Younger ST, Zhivich V, Najgebauer H, et al. (2019). Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat. Commun 10, 5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dewey M (2022). metap: meta-analysis of significance values. [Google Scholar]
- 47.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, Chen X, Kim Y, Beyter D, Krusche P, et al. (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594. [DOI] [PubMed] [Google Scholar]
- 49.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ye K, Schulz MH, Long Q, Apweiler R, and Ning Z (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao H, Sun Z, Wang J, Huang H, Kocher J-P, and Wang L (2014). CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuilman T, Velds A, Kemper K, Ranzani M, Bombardelli L, Hoogstraat M, Nevedomskaya E, Xu G, de Ruiter J, Lolkema MP, et al. (2015). CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol 16, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gao Y, Wang J, and Zhao F (2015). CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol 16, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, and Nesvizhskii AI (2017). MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 14, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.da Veiga Leprevost F, Haynes SE, Avtonomov DM, Chang H-Y, Shanmugam AK, Mellacheruvu D, Kong AT, and Nesvizhskii AI (2020). Philosopher: a versatile toolkit for shotgun proteomics data analysis. Nat. Methods 17, 869–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Djomehri SI, Gonzalez ME, da Veiga Leprevost F, Tekula SR, Chang H-Y, White MJ, Cimino-Mathews A, Burman B, Basrur V, Argani P, et al. (2020). Quantitative proteomic landscape of metaplastic breast carcinoma pathological subtypes and their relationship to triple-negative tumors. Nat. Commun 11, 1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mayakonda A, Lin D-C, Assenov Y, Plass C, and Koeffler HP (2018). Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res 28, 1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sturm G, Finotello F, Petitprez F, Zhang JD, Baumbach J, Fridman WH, List M, and Aneichyk T (2019). Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics 35, i436–i445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aran D, Hu Z, and Butte AJ (2017). xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 18, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schubert M, Klinger B, Klünemann M, Sieber A, Uhlitz F, Sauer S, Garnett MJ, Blüthgen N, and Saez-Rodriguez J (2018). Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bergstrom EN, Huang MN, Mahto U, Barnes M, Stratton MR, Rozen SG, and Alexandrov LB (2019). SigProfilerMatrixGenerator: a tool for visualizing and exploring patterns of small mutational events. BMC Genomics 20, 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang S, Zhang J, He Z, Wu K, and Liu X-S (2019). The predictive power of tumor mutational burden in lung cancer immunotherapy response is influenced by patients’ sex. Int. J. Cancer 145, 2840–2849. [DOI] [PubMed] [Google Scholar]
- 71.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. (2012). Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hu Y, Pan J, Shah P, Ao M, Thomas SN, Liu Y, Chen L, Schnaubelt M, Clark DJ, Rodriguez H, et al. (2020). Integrated Proteomic and Glycoproteomic Characterization of Human High-Grade Serous Ovarian Carcinoma. Cell Rep 33, 108276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Venkatraman ES, and Olshen AB (2007). A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 23, 657–663. [DOI] [PubMed] [Google Scholar]
- 74.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed CPATC pan-cancer data matrices used in this study have been deposited at LinkedOmicsKB and are publicly available as of the date of publication. These accession numbers for the datasets are listed in the key resources table. In addition, raw and processed proteomics as well as open access genomic data can be obtained via Proteomic Data Commons (PDC) at https://pdc.cancer.gov/pdc/cptac-pancancer. Raw genomic and transcriptomic data files can be accessed via the Genomic Data Commons (GDC) Data Portal at https://portal.gdc.cancer.gov.
Original code has been deposited at Figshare and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE