

Abstract
MutL family proteins contain an N-terminal ATPase domain (NTD), an unstructured interdomain linker, and a C-terminal domain (CTD), which mediates constitutive dimerization between subunits and often contains an endonuclease active site. Most MutL homologs direct strand-specific DNA mismatch repair by cleaving the error-containing daughter DNA strand. The strand cleavage reaction is poorly understood; however, the structure of the endonuclease active site is consistent with a two- or three-metal ion cleavage mechanism. A motif required for this endonuclease activity is present in the unstructured linker of Mlh1 and is conserved in all eukaryotic Mlh1 proteins, except those from metamonads, which also lack the almost absolutely conserved Mlh1 C-terminal phenylalanine-glutamate-arginine-cysteine (FERC) sequence. We hypothesize that the cysteine in the FERC sequence is autoinhibitory, as it sequesters the active site. We further hypothesize that the evolutionary co-occurrence of the conserved linker motif with the FERC sequence indicates a functional interaction, possibly by linker motif-mediated displacement of the inhibitory cysteine. This role is consistent with available data for interactions between the linker motif with DNA and the CTDs in the vicinity of the active site.
Graphical Abstract
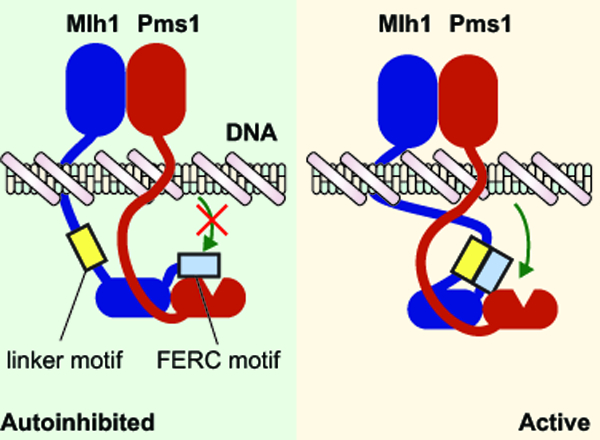
MutL endonucleases target repair to the newly synthesized DNA strand during DNA mismatch repair (MMR). The eukaryotic MutL homolog Mlh1 has two unique evolutionary innovations: a conserved motif in the unstructured interdomain Mlh1 linker, which is required for MMR and a C-terminal FERC (Phe-Glu-Arg-Cys) motif. Here we hypothesize that the FERC motif mediates autoinhibition which is alleviated by interactions with the linker motif.
INTRODUCTION
DNA mismatch repair (MMR) corrects mispairs due to DNA replication errors, mispairs present in heteroduplex recombination intermediates, and some forms of chemically modified DNA.[19, 67] MMR defects cause increased mutation rates and underlie inherited and spontaneous cancers.[18, 49] During MMR, mispairs are recognized by MutS homologs: MutS homodimers in bacteria and archaea and Msh2-Msh6 or Msh2-Msh3 in eukaryotes.[19, 67] ATP binding converts mispair-bound MutS homologs into sliding clamps that recruit MutL homologs: MutL homodimers in bacteria and archaea and Mlh1-Pms1 (called MLH1-PMS2 in humans) in eukaryotes,[19, 67] although other MutL homologs including Mlh1-Mlh2 and Mlh1-Mlh3 have been suggested to play less important roles in MMR.[1, 12, 20, 24] Once recruited to DNA, MutL homologs can also form sliding clamps. Most MutL homologs have endonuclease active sites[65] that appear to direct repair by cleavage (nicking) of the newly synthesized DNA strand in double-stranded DNA[41–42, 63, 66]. In bacteria containing MutL endonucleases, these nicks appear to direct strand displacement by the UvrD helicase.[60] In eukaryotes, these nicks direct nascent strand-specific excision by Exo1 or Rad27 (called FEN1 in humans) and have also been hypothesized to mediate direct mispair removal by successive rounds of nicking.[3, 11, 28] Nicking of DNA by MutL homologs is absolutely required for MMR and is highly regulated, as non-specific nicking could target the incorrect strand and cause mutations, double-stranded DNA breaks, genomic instability, and/or cell death.
MutL homologs can be divided into three regions: an N-terminal domain (NTD), a C-terminal domain (CTD), and an intrinsically disordered intervening linker that can be hundreds of amino acids long (Figure 1A). The CTDs contain the endonuclease active site and constitutively dimerize,[21–23, 59, 63] whereas the NTDs dimerize after binding ATP and dissociate after ATP hydrolysis (Figure 1B).[5] Consistent with this, different ATP induced conformations have been observed by atomic force microscopy (AFM) that are driven with different interdomain interactions and accommodated by linker flexibility.[13, 69] MutL homologs can be loaded onto DNA in the form of sliding clamps in which the DNA is threaded through the channel formed by the unstructured linkers joining the dimerized NTDs and CTDs; however, the exact mechanistic role of these clamps is unclear. It has been suggested that in response to mispairs in the DNA, MutS homologs load multiple MutL homologs that gives rise to a cleavage-proficient MutL homolog polymer,[50] consistent with observed cytological Mlh1-Pms1 foci that have substoichiometric Msh2-Msh6 [38]. Single molecule experiments have shown that MutL homolog clamps can slide along the DNA both by themselves and while interacting with MutS homolog clamps (Figure 1C);[30, 47, 57] the length of the disordered linkers appears to allow MutL homolog clamps to bypass blocks on DNA molecules.[30, 45, 47, 51, 64] In contrast, AFM has suggested that Mlh1-Pms1 and Mlh1-Mlh3 form localized complexes surrounding the mispair, which have been proposed to prevent nucleosome assembly to promote MMR.[8] It remains possible that each of these seemly disparate observations correspond to different mechanistic steps leading to or following DNA nicking.
Figure 1. The architecture of MutL homologs promotes DNA loading by MutS homologs.
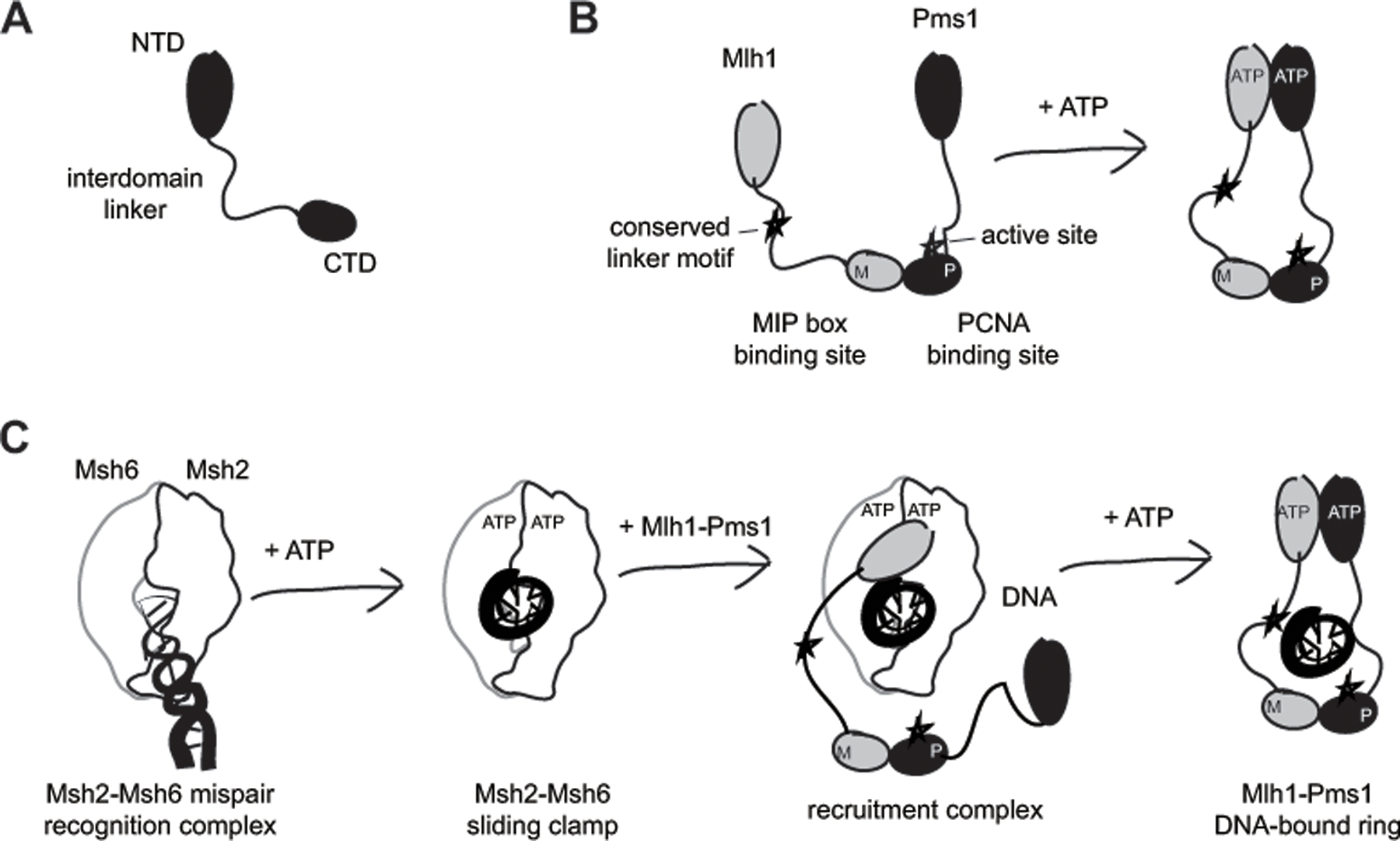
A. MutL homologs can be divided into an N-terminal domain (NTD) belonging to the GHKL ATPase family, a C-terminal domain (CTD) and an unstructured interdomain linker. B. The CTDs are constitutively dimerized, whereas the NTD dimerization is driven by ATP binding. In the heterodimeric S. cerevisiae Mlh1-Pms1, the Mlh1 subunit lacks the CTD endonuclease active site and PCNA binding site, but has a site for binding MIP (Mlh1 Interacting Peptide) box proteins and a conserved linker motif required for efficient DNA nicking. C. DNA loading involves sequential mispair and ATP binding by Msh2-Msh6 to form a sliding clamp that is capable of recruiting Mlh1-Pms1 to DNA. Mlh1-Pms1 ring formation is mediated by Mlh1 NTD binding by Msh2-Msh6[17] and ATP-mediated dimerization of the Mlh1 and Pms1 NTDs,[5] and is facilitated by the flexible interdomain linker.
We recently identified a conserved sequence motif in the unstructured linker of Mlh1 required for Mlh1-Pms1 endonuclease activity in vitro and for MMR in vivo (Figure 1B).[73] Remarkably, the linker motif was required for the function of the distal endonuclease active site, which can be located up to 750 Å away if the linker is fully extended. In addition, the motif could support the endonuclease reaction when it was moved within the Mlh1 linker and when transplanted to the Pms1 linker. But how does the linker motif promote endonuclease activity? Because mutation or deletion of the linker motif inhibits a reaction requiring Mlh1-Pms1, RFC-loaded PCNA, and a DNA substrate,[73] it likely interacts with one or more of these components. The challenge in deciphering the role of this motif, however, highlights that the endonuclease reaction catalyzed by Mlh1-Pms1 remains poorly understood. Here we use analysis of the linker motif combined with available experimental data to propose models for how Mlh1-Pms1 cleaves DNA.
RESULTS AND DISCUSSION
The Mlh1 linker motif is conserved in most eukaryotic clades.
To better understand the structure of the Mlh1 linker motif, we analyzed its evolutionary conservation. We retrieved Mlh1 sequences from more than 1,200 eukaryotes from the NCBI[70] and Ensembl databases[53] and assembled Mlh1 transcripts for key species not present in these databases using RNA sequencing data from the Sequence Read Archive. Mlh1 sequences from RNA sequencing data were generated by: (1) read trimming with Trimmomatic version 0.40[6], (2) de novo assembly with Trinity version 2.15.0[31], (3) translation with Transdecoder version 5.5.0[35], and (4) homolog identification with blastp version 2.13.0[2]. Sequences were aligned with MAFFT version 3.313[44] and analyzed for the presence of the Mlh1 linker motif and the Mlh1 C-terminal FERC sequence (Figure 2).
Figure 2. The Mlh1 conserved linker motif is conserved with two sequence patterns in eukaryotes.
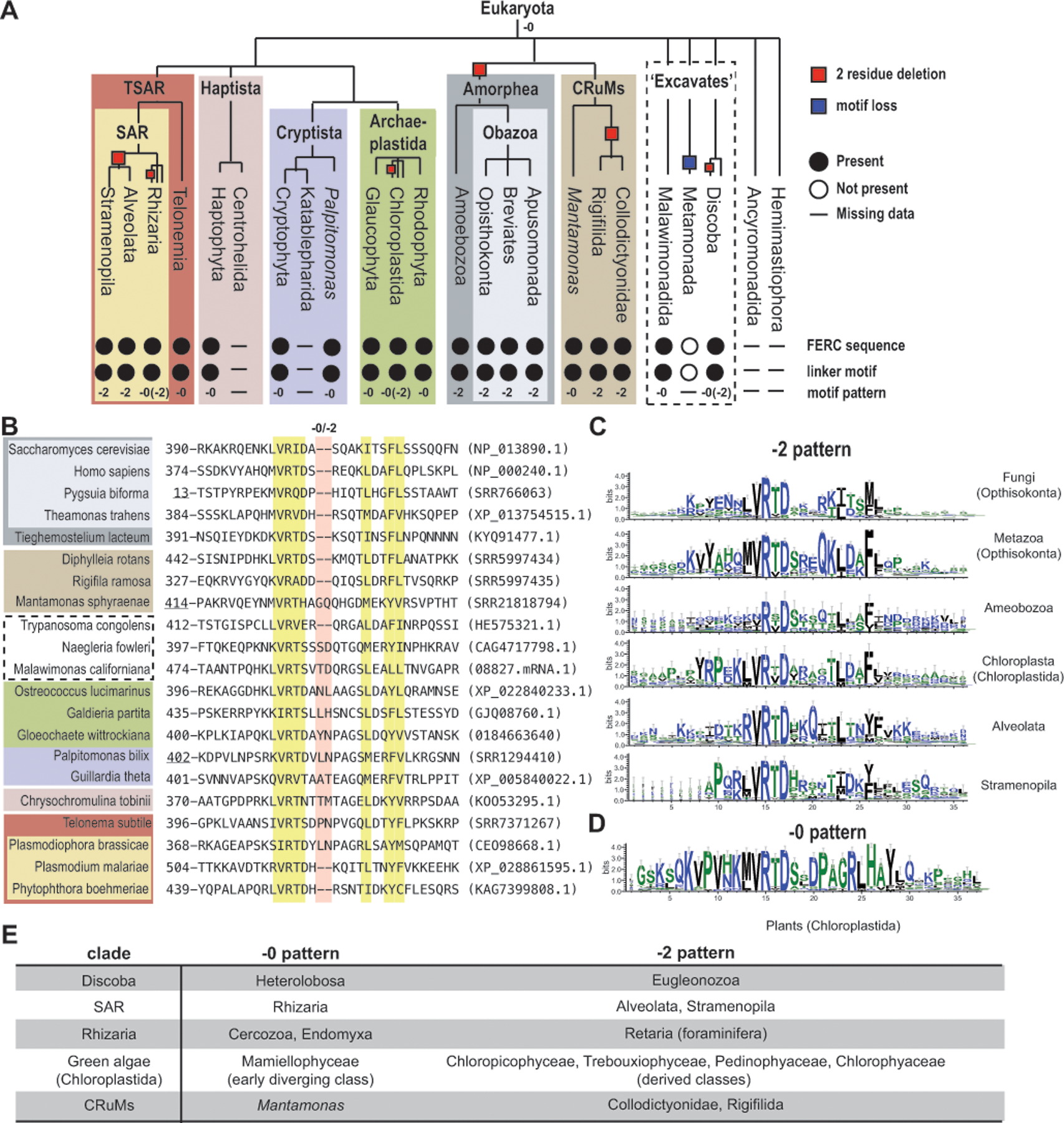
A. The presence (filled circles), absence (empty circles), or no data (horizontal line) of the conserved Mlh1 linker motif and C-terminal FERC sequence in different eukaryotic clades is indicated; the −0 and −2 labels indicate which motif pattern is present. The cladogram is based on previous analyses.[10] B. The conserved linker motif from the alignment of selected Mlh1 sequences; yellow highlighted residues are conserved and red highlighted residues are at −0/−2 sequence difference. Accessions are in parentheses with SRR numbers corresponding to RNA sequence datasets; Mlh1 sequences from other sources include M. californiana (https://megasun.bch.umontreal.ca/papers/T2SS-2020/ sequenceMalawimonas_californiana_08827.mRNA.1), G. wittrockiana (MMETSP0308 sequence CAMPEP_0184663640), C. tobinii (KOO53295.1 where two in-frame introns were merged with the coding sequence). Underlined positions are from partial sequences. C-D. Motif sequence logos were generated with Weblogo3[14]. E. Clades with both −0 and −2 patterns have patterns that segregate by taxonomic group.
The linker motif is conserved in all eukaryotes, except for the metamonads, which are a group of flagellated unicellular protists lacking mitochondria (Figure 2A). Similar motifs were not found in MutL endonucleases from approximately 400 archaea (including representatives from the Euryarchaeota, DPANN, TACK, and Asgard groups) and approximately 2,100 bacteria (including representatives from all bacterial phyla recognized in the NCBI Taxonomy database). Thus, the conserved linker motif likely arose in the last eukaryotic common ancestor (LECA, e.g. the organism all eukaryotes descended from) when MutL duplication and specialization generated Mlh1, as it is missing even in the closely related Asgard group archaea[75].
The linker motif has two distinct sequence patterns. The “−0 pattern”, which is found in plants and other groups, separates the conserved R(T/I/V)D sequence from the conserved FL sequence with ten amino acids (Figure 2B,D). The “−2 pattern”, which is the pattern we first identified in fungi and animals,[73] separates these sequences with eight amino acids (Figure 2B,C). Several clades have taxa with both the −0 and −2 patterns (Figure 2E). The current eukaryotic phylogeny[10] suggests that the LECA had an Mlh1 with a −0 pattern and that the −2 pattern arose six times in lineages leading to Amorphea, Euglenozoa (Discoba), Alveolata and Stramenopila, Collodictyonidae and Rigifilida, derived classes of green algae (Chloroplastida), and foraminifera (Rhizaria), assuming no horizontal gene transfer events (Figure 2A).
Structural predictions of linker motif peptide conformations do not explain the observed evolutionary-conserved motif sequence constraints.
Only the −0 and −2 motif patterns, but not other potential insertion or deletion patterns, have arisen during the ~1 billion years of eukaryotic evolution. This high degree of conservation suggests that the linker motif could adopt a specific conformation by itself or when bound to an interaction partner. To investigate if the linker motif has a stable intrinsic conformation, we used molecular dynamic simulations. Molecular dynamics is a computational tool that uses physical simulations to explore the structure and dynamics of biological molecules, a key aspect of which is that changes of conformation in the simulation over time can be used to describe the molecules at equilibrium.[43] Thus, if the evolutionary sequence constraints are dictated by the linker motif peptide conformations, the motifs from different organisms should have similar lowest free energy structures in the simulations.
Molecular dynamics simulation of three motif-containing peptides (−0 pattern, Arabidopsis thaliana; −2 pattern, S. cerevisiae and human) were performed employing a well-established approach with GROMACS version 2022.3 software[74] using the OPLS-AA forcefield in 2 fs steps for 500 ns. The simulations of the A. thaliana, S. cerevisiae, and human simulations included the peptides, explicit water molecules (5,646, 3,992, and 4,092 molecules, respectively), and ions to neutralize the net charge of the entire system (1 chloride ion in the A. thaliana and S. cerevisiae simulations). Conformations were sampled every 10 ps (50,000 per simulation). To identify the peptide conformations corresponding to the most stable structures, we analyzed the sampled conformations in three steps. First, we calculated all informative Cα-Cα distances for each conformation; these are all distances between different Cα atoms within the peptide, except for those between adjacent residues, as these distances are always around 3.8 Å. Second, we projected the high dimensional Cα-Cα distance space (210 distances for S. cerevisiae and human peptides, 253 distances for A. thaliana peptide) onto two dimensions so that each conformation could be plotted as a single point using Uniform Manifold Approximation and Projection (UMAP) in R version 4.1.1 (Figure 3A–C).[55] Third, we clustered these UMAP plots using k-nearest neighbor clustering as implemented in R to identify clusters with the most number of sampled conformations.
Figure 3. Linker peptide conformations do not explain sequence constraints.
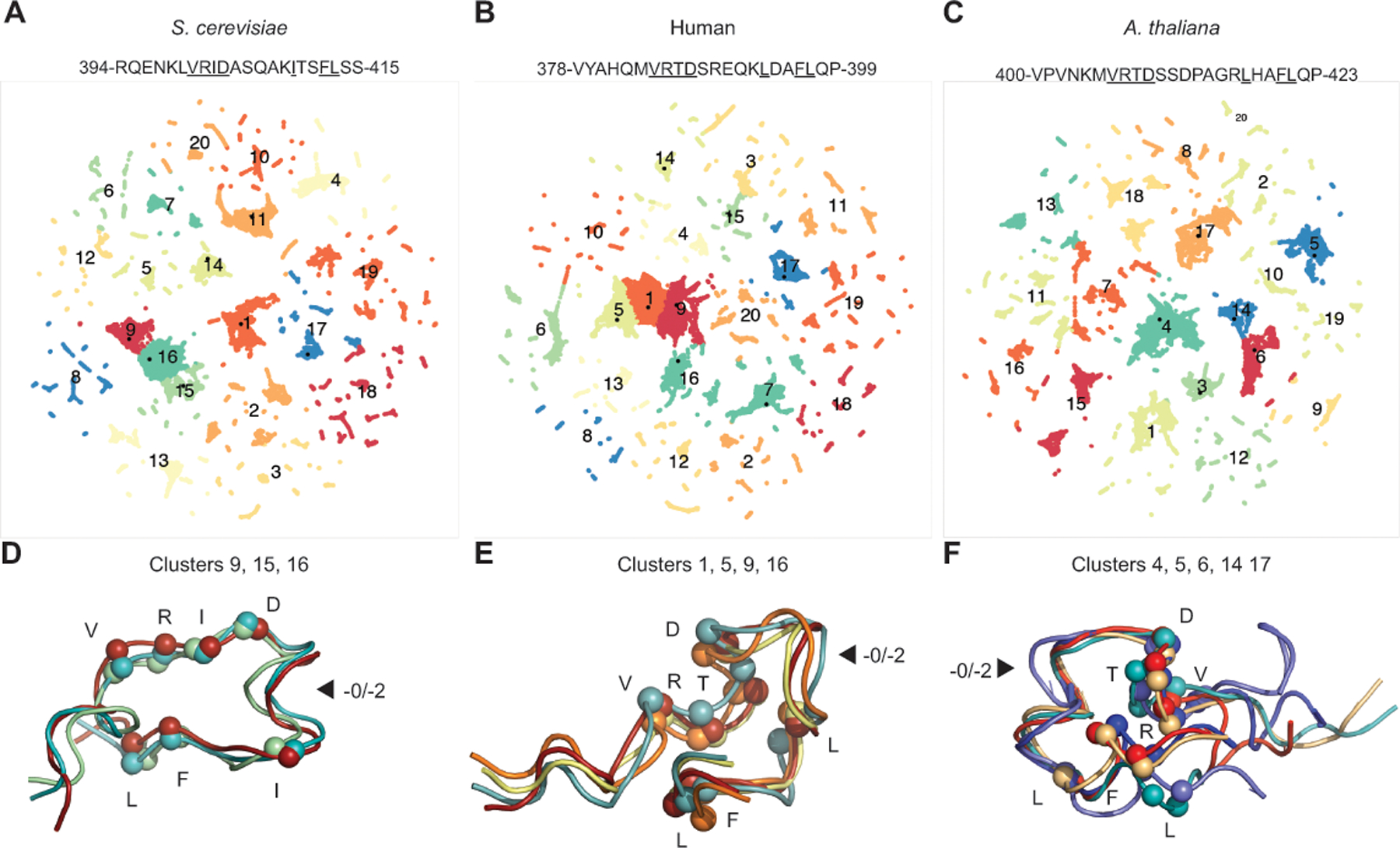
A-C. Mlh1 motif-containing peptides were analyzed by molecular dynamics and related conformations (clusters) were identified by two-dimensional UMAP projections. D-F. The most common conformations are displayed as ribbons with Cα atoms of the conserved residues (underlined in sequence) displayed as spheres. Arrows indicate where the length difference between the −0 and −2 patterns lies. D. The major S. cerevisiae peptide conformation is stabilized by interactions between R401, I402, and F412 and capping of a helix with the D403 side chain. E. The major human peptide conformation is stabilized by interactions between 384-VRTD-387, L393, and 396-FL-397. F. The major A. thaliana peptide conformation contains a helix promoted by the non-conserved P413 and is stabilized by interactions between R407, L417, and F420.
The peptides adopted multiple conformations during the molecular dynamics simulation, as revealed by the presence of multiple clusters (Figure 3A–C). Analysis of representative structures showed that several low energy clusters had very similar conformations (Figure 3D–F), most of which were stabilized by partial burial of hydrophobic side chains or key backbone interactions. Remarkably, the low energy conformations were not stable enough to trap peptides; peptides readily adopted and lost these low energy conformations during the simulation. Moreover, the low energy conformations were different for each peptide analyzed (Figure 3D–F). Thus, the strong sequence constraints on the motif sequences are not driven by internal peptide interactions but most likely are driven by external interactions with other regions of MMR proteins or the DNA substrate.
Does the linker motif interact with a protein component of MMR?
We next investigated if the strong sequence constraints on the evolution of the motif peptide could be due to an interaction with a specific region of an MMR protein. Previous MMR protein crosslinking studies have provided data that could be used to identify these interactions. Reanalysis of lysine crosslinking of S. cerevisiae Mlh1-Pms1 in the absence of ATP and DNA (Figure 4A)[26] suggests that the linker motif interacts with the Mlh1-Pms1 CTDs. Several sets of crosslinks can be explained by motif-CTD interactions combined with Mlh1 CTD wrapping by the Pms1 linker as observed using in silico modeling (Figure 4B):[73] (1) K398 of the Mlh1 linker motif crosslinks with the N-terminal ends of the Mlh1 and Pms1 CTDs; (2) the C-terminal ends of the Mlh1 and Pms1 linkers crosslink with each other; and (3) C-terminal, but not N-terminal, Mlh1 linker lysines show extensive intra-protein crosslinking to the conserved Mlh1 linker motif due to linker looping (Figure 4C). The requirement for a motif-CTD interaction is also consistent with the results of an experiment in which the Mlh1 linker motif was isolated in a loop (termed “hand-cuffing” in the study) induced by rapamycin-induced dimerization of FKBP (FK506-binding protein) and FRB (FKBP and rapamycin binding domain) inserted into Mlh1 linker. In this construct, rapamycin addition caused a complete MMR defect in vivo and a strong endonuclease defect in vitro.[26]
Figure 4. The conserved linker motif likely interacts with the CTD dimer near the active site.
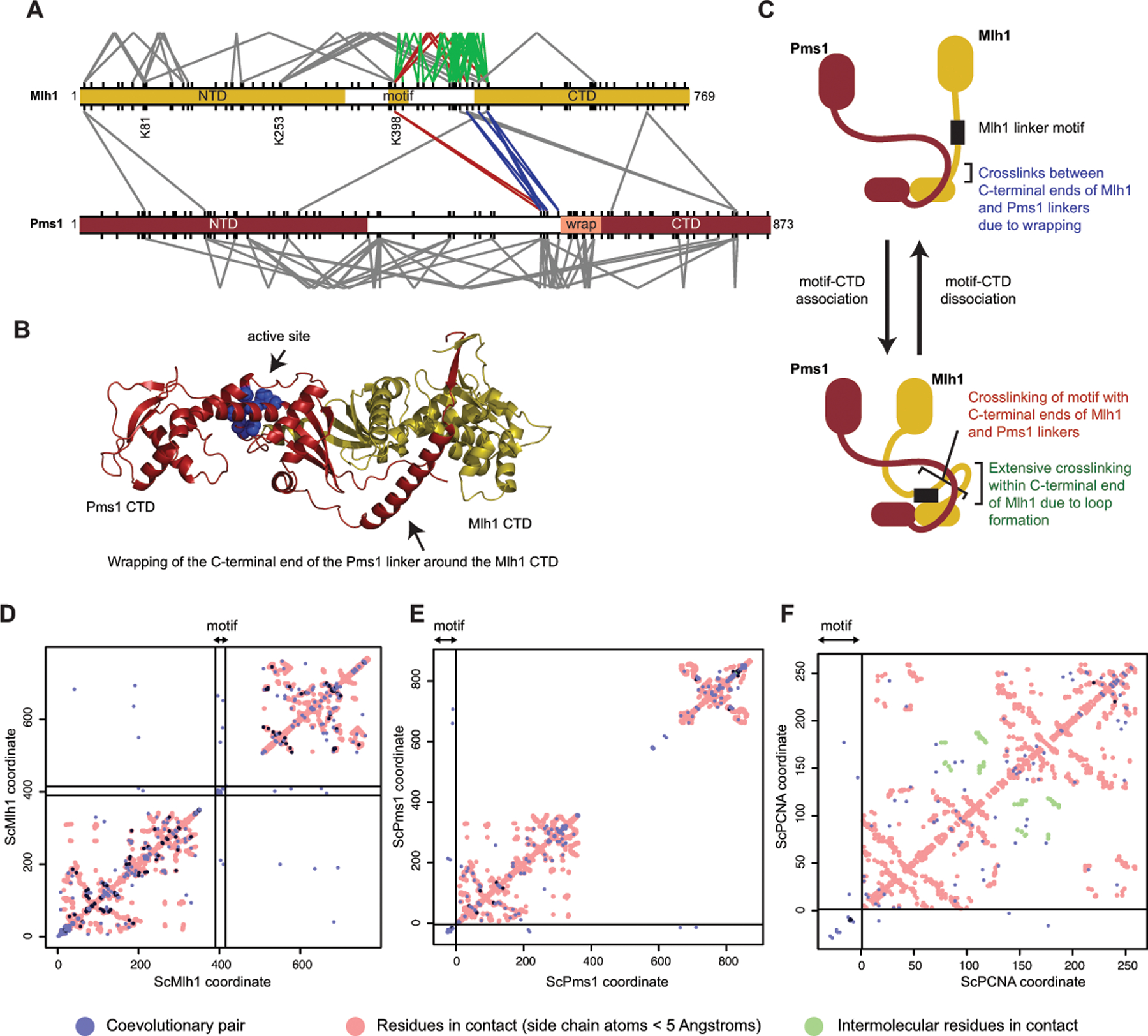
A. Lysine crosslinks of S. cerevisiae Mlh1 (top) and Pms1 (bottom) in the absence of ATP and DNA[26] are depicted as V-shapes for intramolecular crosslinks and lines for intermolecular crosslinks. Red are crosslinks between the conserved linker motif and the Mlh1 and Pms1 CTDs; blue are crosslinks between the N-terminal ends of the Mlh1 and Pms1 CTDs; green are crosslinks within the C-terminal end of the Mlh1 linker. All lysines are shown as tic marks. B. Wrapping of the C-terminus of the Pms1 linker around the Mlh1 domain as predicted by Alphafold2[40] is consistent with crosslinking data (blue lines in panel A). C. A model consistent with many of the observed crosslinks that can be explained by Pms1 wrapping (blue lines in panel A), crosslinking of the motif to the CTD (red lines in panel A), and extensive crosslinking of lysines in the C-terminal half of the Mlh1 linker (green lines in panel A). D. Evolutionary coupling of residues in Mlh1 with GREMLIN[61]. Blue and dark blue points are evolutionary coupled sites; red points are side-chain contacts calculated from an Alphafold2 model of S. cerevisiae Mlh1-Pms1 [40]. Residues in the conserved motif are within the region demarked by black lines; off-diagonal points within these lines indicate coupling to motif residues. E. Evolutionary coupling of the Mlh1 linker motif placed at the beginning of the alignment (left and bottom of graph, separated by black lines) with Pms1. F. Evolutionary coupling of the Mlh1 motif with PCNA shown as for panel E; red and green points are intra- and intersubunit side-chain contacts in the S. cerevisiae PCNA trimer calculated from the PDB accession 1plq.
A complementary computational strategy to identify candidate interactions is to determine evolutionary coupling between the Mlh1 linker motif and other protein regions involved in DNA cleavage by the Mlh1-Pms1 endonuclease. Evolutionary couplings between pairs and groups of amino acids arise from constraints from the protein structure and function that require protein sequence changes at one site to be accommodated by changes at adjacent sites.[39, 52, 61] Coevolving sites have been successfully used to determine residue-residue proximity, protein folds, and protein-protein interaction surfaces; however, not all sites implicated as coevolving are in direct contact as correlations can be noisy.[39, 52, 61] Previous studies have also detected coevolution between regions of the Mlh1, Pms1 and Mlh3 proteins consistent with functional or direct interactions between domains;[25] however, these analyses were not performed at the resolution of single amino acid residues. We therefore used GREMLIN version 2.01[61] to analyze coevolution of the Mlh1 linker at a residue resolution by analyzing alignments of Mlh1 sequences with the −2 motif pattern (1,110 sequences aligned using MAFFT version 3.313[44]), Pms1 (702 sequences) and PCNA (702 sequences), the latter being critical to the activity of the Mlh1-Pms1 endonuclease. In the case of coevolution of the Mlh1 linker motif with Pms1 and PCNA, we prepended a Mlh1 linker motif alignment to the alignments of Pms1 and PCNA prior to GREMLIN analysis. Candidate Mlh1 co-evolving sites were found in the NTD (S200 and D211, S. cerevisiae numbering), which are surface-exposed residues, and the CTD (N537, D577, L652, and K665), of which N537 is surface exposed and near the active site (Figure 4D). In contrast, coevolution analyses did not provide strong evidence for direct interactions with Pms1 or PCNA, as coevolving residues are mostly (Pms1 N214, V218, and F711; PCNA I16 and L139) or partially (Pms1 L664; PCNA G176) buried (Figure 4E,F). Together, the available crosslinking data,[26] evidence that the motif promotes enzymatic activity,[73] and coevolution analyses are most consistent with a Mlh1 linker motif-CTD interaction that occurs in the vicinity of the endonuclease active site (Figure 4C).
Does the linker motif interact with DNA?
There is evidence that the MutL proteins bind to DNA. The N-terminus of MutL and Mlh1 bind weakly to DNA at low ionic strength.[5, 33, 56, 68] A Mlh1-Pms1 complex with DNA has not been structurally characterized, but genetic and biochemical studies suggest that predicted DNA contacting residues are important for MMR and loading of MutL proteins onto DNA as sliding clamps.[5, 7, 36, 68] In addition, potential contacts between Mlh1-Pms1 and DNA can be inferred from the recently determined structure of E. coli MutL with a primer/template DNA,[7] mapping of DNA-adjacent amino acid residues by FeBABE footprinting of S. cerevisiae Mlh1-Mlh3,[13] and mutagenesis of charged Mlh1 amino acid residues.[4, 13, 37, 73] The data generated using these methods are remarkably concordant, and the FeBABE footprinting data directly implicates the conserved Mlh1 linker motif as being adjacent to DNA (Figure 5A). Consistently, S. cerevisiae Mlh1 linker mutagenesis[4, 13, 73] and analysis of cancer-associated human MLH1 linker mutations[48, 73] indicates that the only Mlh1 linker amino acid residues required for MMR are those within the conserved motif; however, the interaction between these linker motif amino acid residues and DNA have not been directly examined (Figure 5A–D). Linker motif-DNA interactions could be consistent with linker motif-CTD interactions and the strong evolutionary constraints on the motif, as the conformation of the DNA in the active site likely has not changed during evolution.
Figure 5. The conserved linker likely interacts with the DNA substrate.
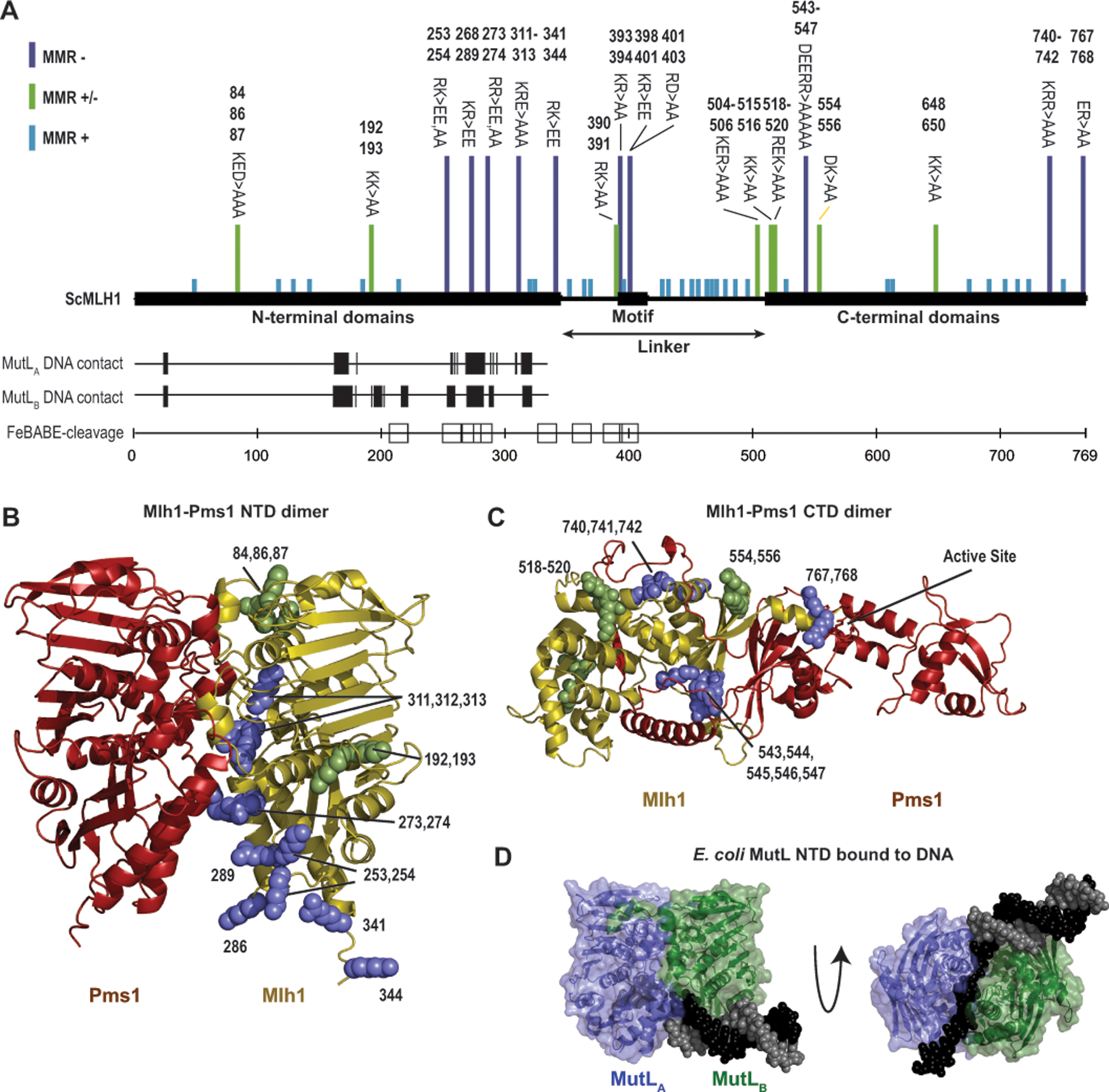
A. Summary of the data indicating regions of S. cerevisiae Mlh1 that interact with DNA provide a consistent view of DNA-interacting residues. Bars above indicate mutations causing full (purple), partial (green) or no (blue) MMR defects.[4, 13, 37, 73] Vertical hashes indicate residues equivalent to DNA contacting residues in each subunit (MutLA and MutLB) of the E. coli MutL-DNA structure.[7] Sites of S. cerevisiae Mlh1 adjacent to DNA in a Mlh1-Mlh3 bound to a Holliday junction[13] are indicated by squares. B. Sites of mutations in Mlh1 that cause complete (purple) or partial (green) MMR defects mapped to a Mlh1-Pms1 NTD dimer model generated with Alphafold2.[40] C. Similar analysis as panel B with a Mlh1-Pms1 CTD dimer model. D. Structure of E. coli MutL (green and blue cartoon) with a DNA containing a single-stranded overhang (black and gray spheres) shows extensive ssDNA-MutL NTD contacts (PDB id 7p8v).[7]
DNA cleavage by a two- or three-metal ion mechanism?
Understanding the role of the linker motif role in MMR requires a detailed understanding of the endonuclease mechanism that the linker motif is required for. The bacterial MutL endonuclease active site contains two tetrahedral zinc ions, ZnA and ZnB, in sites separated by ~4 Å (Figure 6A).[21–23, 59, 63] These ions are ligated by four cysteine and histidine residues and a bridging glutamate. One unoccupied binding site bridges ZnA and ZnB. This geometry is reminiscent of enzymes using the classic two-ion mechanism[72] where the two ions stabilize a pentacovalent phosphate during phosphoryl transfer and hydrolysis reactions (Figure 6A). Consistent with this mechanism, Mlh1-Mlh3 cleavage products are contain nicks with a 3’-hydroxyl and a 5’-phosphate and are consequently religatable.[50]
Figure 6. The active site of eukaryotic Mlh1-Pms1 and Mlh1-Mlh3 is blocked by the Mlh1 C-terminus.
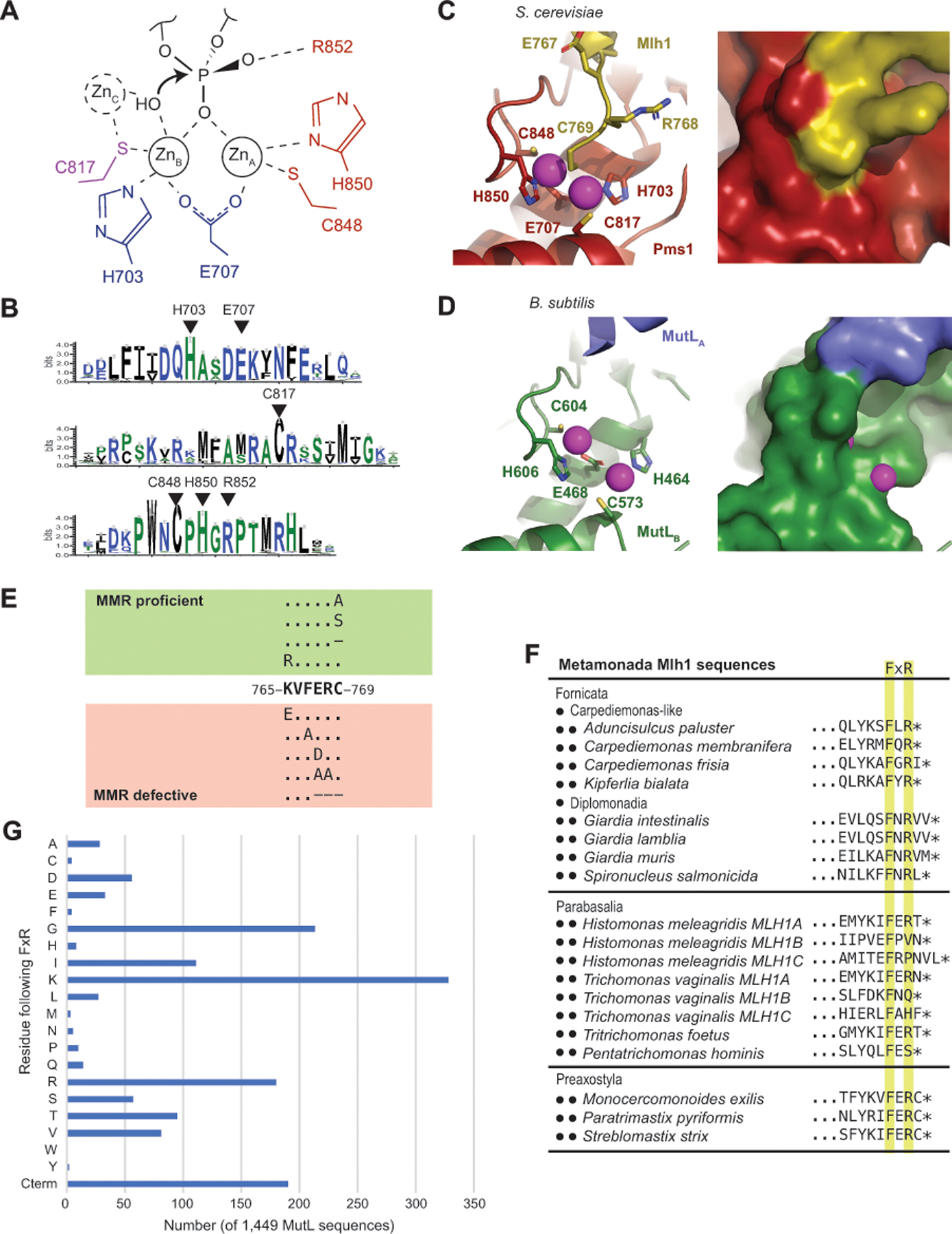
A. The proposed endonuclease reaction of MutL-family proteins involves coordination of a backbone phosphate by ZnA and ZnB, stabilization of the phosphate by Pms1 R852 and potential activation of a hydroxyl nucleophile by a transiently bound ZnC; residues identified with S. cerevisiae Pms1 numbering. B. Sequence logo of the conserved active site motifs in eukaryotic Pms1 sequences with residues identified in panel A indicated with triangles. C. Ribbon diagrams (left and middle) of the S. cerevisiae Mlh1 (yellow) – Pms1 (red) active site (PDB id 4e4w; [34]) showing the metal coordination and binding of the Mlh1 C769 residue at the bridging position between ZnA and ZnB (magenta). Surface view of the active site shows that not only is the fourth coordination sphere blocked by C769, but the entire active site is sequestered from solvent. D. Ribbon diagram (left) and surface (right) of the B. subtilis active site (PDB id 3kdk; [63]) showing accessibility of the metals (magenta) to solvent. E. Summary of the mutagenesis of the conserved S. cerevisiae Mlh1 C-terminus; dashes indicate deletions of the specified residue. F. Two of the three phyla within Metamonada lack the C-terminal Mlh1 FERC sequence but tend to retain a FxR sequence. G. Histogram of the residues that immediately follow the FxR sequence from a sample of 1,449 bacterial and archaeal MutL sequences shows a bias towards small and positively charged residues and a relative lack of cystine residues as in the eukaryotic Mlh1 FERC motif. “Cterm” indicates that the arginine in the FxR sequence is at the C-terminus of the protein.
Zinc binding of a DNA backbone phosphate would allow the arginine in the highly conserved CPHGRP motif of the endonuclease active site to further stabilize the pentacovalent phosphate (Figure 6A,B). However, it is not clear how MutL activates a hydroxyl nucleophile. Remarkably, many two-ion mechanism enzymes, including DNA polymerases η[27], utilize a third stably or transiently bound ion during catalysis. One Aquifex aeolicus MutL CTD structure has a third metal ion, ZnC,[22] which shares its single protein ligand with ZnB and could be accommodated in the Bacillus subtilis and Neisseria gonorrhoeae MutL CTD structures (Figure 6A).[59, 63] A hydroxyl group bridging ZnB and ZnC would be positioned to attack a phosphate coordinated by ZnA and ZnB, which may suggest that a transiently bound ZnC is a common feature of MutL endonucleases.
Autoinhibition at the eukaryotic endonuclease active site?
The active sites of S. cerevisiae Pms1 (human PMS2) and Mlh3 share many of the features with bacterial MutL, including the binding of two Zn ions.[15, 34] Unlike in bacterial MutL, the fourth ligand binding site bridging ZnA and ZnB is not accessible. Instead, the C-terminal cysteine of Mlh1 (C769, S. cerevisiae Mlh1 numbering) is bound at this site (Figure 6C). The C-termini of eukaryotic Mlh1 proteins exclusively have a Phe Glu Arg Cys (FERC) motif (also called the C-terminal homology[58]) or a FERC motif followed by a short extension (e.g. FERCGT in Caenorhabditis elegans). This motif is a Mlh1-specific eukaryotic innovation; it is lacking in other eukaryotic MutL homologs (including Mlh2/PMS1, Pms1/PMS2, and Mlh3) and in both endonuclease-proficient and endonuclease-deficient MutL homologs from bacteria and archaea.[46] Despite its absolute conservation, this terminal cysteine is not required for catalysis; the S. cerevisiae mlh1-C769A, mlh1-C769S, and mlh1-C769stp mutations do not cause MMR defects and where tested do not affect the endonuclease activity in vitro, unlike mutations affecting zinc coordination by Pms1 (Figure 6E).[4, 15, 58, 62, 71]
Although the C-terminal Mlh1 FERC cysteine has been suggested to increase zinc affinity,[32] we hypothesize that a major role of this cysteine is autoinhibition. This amino acid residue appears to sequester the active site from the substrate (compare Figure 6C and Figure 6D), and coordination of a DNA phosphate by ZnA and ZnB necessarily requires cysteine displacement. Consistently, the isolated FERC-containing CTD heterodimer of S. cerevisiae Mlh1 and Pms1 is not an active endonuclease,[34] whereas the isolated FERC-lacking CTDs of the bacterial A. aeolicus and N. gonorrheae MutL proteins are active endonucleases.[16, 54] Also consistent with this proposal, the S. cerevisiae Mlh1-C769stp-Pms1 and Mlh1-E767stp-Pms1 complexes retain significant levels of endonuclease activity.[71] It is possible that autoinhibition in eukaryotic MutL homologs may help prevent promiscuous nicking by DNA-bound Mlh1-Pms1 complexes.
Does the conserved linker motif alleviate autoinhibition?
Like the conserved Mlh1 FERC motif, the conserved Mlh1 linker motif is a eukaryotic Mlh1-specific innovation that appears to have arisen in the ancestral eukaryotic Mlh1 and is present in almost all extant eukaryotes (Figure 2A). The only outliers in eukaryotes belong to Metamonada, which contains phyla that have lost the conserved linker motif and only partially conserve the FERC motif as a FxR sequence that lacks a cysteine (Figure 6F). The loss of these motifs in Metamonada is reminiscent of bacterial and archaeal MutLs, which lack the linker motif and the FERC motif even though they are endonucleases.[46] This suggests that the conserved FERC and linker motifs are functionally related. Moreover, although bacteria and archaea MutL proteins conserve a related FxR sequence, there is a remarkable lack of FxRC sequences, suggesting that displacement of the FERC cysteine from the active site is a challenging mechanistic step. A sample of 1,449 full-length MutL sequences from all major groups of bacteria and archaea reveal that a cysteine residue almost never follows the conserved FxR sequence, except for 4 MutL sequences belonging to Candidatus Woesearchaeota archaeons with a C-terminal FKRCG sequence identified in metagenomic assemblies (Figure 6G). Because of the evolutionary co-occurrence of the Mlh1 FERC and linker motifs in eukaryotes and fact that FxRC sequences essentially do not occur in bacterial and archaeal MutL proteins that lack the linker motif, we hypothesize that the linker motif promotes Mlh1-Pms1 activity by displacing the inhibitory cysteine. Cysteine displacement could be mediated by charged interactions between the Mlh1 linker motif and the glutamate, the arginine, or even the C-terminal carboxylate group of the Mlh1 FERC motif, consistent with evidence for interactions of the conserved linker motif with the CTD in the vicinity of the active site. Consistent with a role in promoting but not in performing the endonucleolytic cleavage, mutation of the conserved linker motif causes an endonuclease defect that is not quite as defective as that caused by an active site mutation.[73]
CONCLUSIONS
To decipher the role of the conserved Mlh1 linker motif, we have reviewed features of the MutL family of endonucleases and presented mechanistic hypotheses for how these features participate in the endonuclease activity. Previous studies of the unstructured interdomain linker of MutL homologs have found evidence for a length-dependence, which has been interpreted as allowing MutL homolog sliding clamps to stably interact with DNA to facilitate MutL homolog activation as well as bypass roadblocks while sliding along the DNA;[9, 29–30, 45, 47, 51] cleavage of these linkers in vivo causes a MMR defect suggesting that MutL homolog sliding clamps are critical to MMR.[64] We recently have identified that the Mlh1 linker contains a conserved motif required for its endonuclease activity and MMR, and that the motif can be functionally moved within the Mlh1 linker and to the Pms1 linker;[73] however, the mechanism by which this motif functions remains poorly understood. Here we have reviewed the available literature and performed computational analyses that have allowed us to make hypotheses for how MutL homologs cleave DNA and how this distal linker motif may contribute to this catalysis in eukaryotic enzymes.
The hypotheses proposed here are readily testable. Potential autoinhibition by the FERC motif can be investigated by testing if removal of the C-terminal cysteine of additional residues of the FERC motif promote DNA nicking by a heterodimer made up of Mlh1-Pms1 CTDs. Alternatively, addition of a C-terminal cysteine to the C-terminal FxR sequence of a bacterial MutL might potentially causes inhibition of the endonuclease activity of bacterial CTDs that normally have a C-terminal FxR sequence. A potential role for the linker motif in alleviating inhibition of endonuclease activity by the FERC sequence can be analyzed by testing if loss of the Mlh1 C-terminal cysteine can rescue the MMR and endonuclease defects of versions of Mlh1-Pms1 in which the conserved linker motif is mutated or deleted. Alternatively, if adding a C-terminal cysteine to a bacterial MutL inhibits the endonuclease activity, insertion of the linker motif in the linker could potentially restore activity. Finally, studies of whether mutations that eliminate Mlh1 linker motif function alter the patterns of protein crosslinking and FeBABE footprinting or alter interactions with DNA such as the ability to act as sliding clamps on DNA would provide insights into linker motif function. Despite the dramatic increases in our understanding of MMR in general and the crucial role of DNA nicking in directing repair, how MutL homologs cleave DNA remains a challenging question. Deciphering this mechanism likely will require additional biochemical, biophysical, and structural studies of MutL homologs in general and eukaryotic-specific innovations, such as the evolutionary co-occurring Mlh1 FERC and linker motifs.
ACKNOWLEDGEMENTS.
We thank Drs. Dayana Salas and Andrew Roger (Dalhousie University) for assistance in obtaining some Metamonada proteomes. This work was supported by NIH grant GM50006 and the Ludwig Institute for Cancer Research.
REFERENCES
- [1].Abdullah MF, Hoffmann ER, Cotton VE, & Borts RH (2004). A role for the MutL homologue MLH2 in controlling heteroduplex formation and in regulating between two different crossover pathways in budding yeast. Cytogenet Genome Res, 107(3–4), 180–190. doi: 10.1159/000080596 [DOI] [PubMed] [Google Scholar]
- [2].Altschul SF, Gish W, Miller W, Myers EW, & Lipman DJ (1990). Basic local alignment search tool. J Mol Biol, 215(3), 403–410. doi: 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- [3].Amin NS, Nguyen MN, Oh S, & Kolodner RD (2001). exo1-Dependent mutator mutations: model system for studying functional interactions in mismatch repair. Mol Cell Biol, 21(15), 5142–5155. doi: 10.1128/MCB.21.15.5142-5155.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Argueso JL, Kijas AW, Sarin S, Heck J, Waase M, & Alani E (2003). Systematic mutagenesis of the Saccharomyces cerevisiae MLH1 gene reveals distinct roles for Mlh1p in meiotic crossing over and in vegetative and meiotic mismatch repair. Mol Cell Biol, 23(3), 873–886. doi: 10.1128/MCB.23.3.873-886.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ban C, Junop M, & Yang W (1999). Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell, 97(1), 85–97. doi: 10.1016/s0092-8674(00)80717-5 [DOI] [PubMed] [Google Scholar]
- [6].Bolger AM, Lohse M, & Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. doi: 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Borsellini A, Lebbink JHG, & Lamers MH (2022). MutL binds to 3’ resected DNA ends and blocks DNA polymerase access. Nucleic Acids Res, 50(11), 6224–6234. doi: 10.1093/nar/gkac432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bradford KC, Wilkins H, Hao P, Li ZM, Wang B, Burke D, … Erie DA (2020). Dynamic human MutSalpha-MutLalpha complexes compact mismatched DNA. Proc Natl Acad Sci U S A, 117(28), 16302–16312. doi: 10.1073/pnas.1918519117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brown MW, Kim Y, Williams GM, Huck JD, Surtees JA, & Finkelstein IJ (2016). Dynamic DNA binding licenses a repair factor to bypass roadblocks in search of DNA lesions. Nat Commun, 7, 10607. doi: 10.1038/ncomms10607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Burki F, Roger AJ, Brown MW, & Simpson AGB (2020). The New Tree of Eukaryotes. Trends Ecol Evol, 35(1), 43–55. doi: 10.1016/j.tree.2019.08.008 [DOI] [PubMed] [Google Scholar]
- [11].Calil FA, Li BZ, Torres KA, Nguyen K, Bowen N, Putnam CD, & Kolodner RD (2021). Rad27 and Exo1 function in different excision pathways for mismatch repair in Saccharomyces cerevisiae. Nat Commun, 12(1), 5568. doi: 10.1038/s41467-021-25866-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Campbell CS, Hombauer H, Srivatsan A, Bowen N, Gries K, Desai A, … Kolodner RD (2014). Mlh2 is an accessory factor for DNA mismatch repair in Saccharomyces cerevisiae. PLoS Genet, 10(5), e1004327. doi: 10.1371/journal.pgen.1004327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Claeys Bouuaert C, & Keeney S (2017). Distinct DNA-binding surfaces in the ATPase and linker domains of MutLgamma determine its substrate specificities and exert separable functions in meiotic recombination and mismatch repair. PLoS Genet, 13(5), e1006722. doi: 10.1371/journal.pgen.1006722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Crooks GE, Hon G, Chandonia JM, & Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res, 14(6), 1188–1190. doi: 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dai J, Sanchez A, Adam C, Ranjha L, Reginato G, Chervy P, … Charbonnier JB (2021). Molecular basis of the dual role of the Mlh1-Mlh3 endonuclease in MMR and in meiotic crossover formation. Proc Natl Acad Sci U S A, 118(23). doi: 10.1073/pnas.2022704118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Duppatla V, Bodda C, Urbanke C, Friedhoff P, & Rao DN (2009). The C-terminal domain is sufficient for endonuclease activity of Neisseria gonorrhoeae MutL. Biochem J, 423(2), 265–277. doi: 10.1042/BJ20090626 [DOI] [PubMed] [Google Scholar]
- [17].DuPrie ML, Palacio T, Calil FA, Kolodner RD, & Putnam CD (2022). Mlh1 interacts with both Msh2 and Msh6 for recruitment during mismatch repair. DNA Repair (Amst), 119, 103405. doi: 10.1016/j.dnarep.2022.103405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Durno CA, Sherman PM, Aronson M, Malkin D, Hawkins C, Bakry D, … International BC (2015). Phenotypic and genotypic characterisation of biallelic mismatch repair deficiency (BMMR-D) syndrome. Eur J Cancer, 51(8), 977–983. doi: 10.1016/j.ejca.2015.02.008 [DOI] [PubMed] [Google Scholar]
- [19].Fishel R (2015). Mismatch repair. J Biol Chem, 290(44), 26395–26403. doi: 10.1074/jbc.R115.660142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Flores-Rozas H, & Kolodner RD (1998). The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc Natl Acad Sci U S A, 95(21), 12404–12409. doi: 10.1073/pnas.95.21.12404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fukui K, Baba S, Kumasaka T, & Yano T (2016). Structural Features and Functional Dependency on beta-Clamp Define Distinct Subfamilies of Bacterial Mismatch Repair Endonuclease MutL. J Biol Chem, 291(33), 16990–17000. doi: 10.1074/jbc.M116.739664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fukui K, Baba S, Kumasaka T, & Yano T (2018). Multiple zinc ions maintain the open conformation of the catalytic site in the DNA mismatch repair endonuclease MutL from Aquifex aeolicus. FEBS Lett, 592(9), 1611–1619. doi: 10.1002/1873-3468.13050 [DOI] [PubMed] [Google Scholar]
- [23].Fukui K, Iino H, Baba S, Kumasaka T, Kuramitsu S, & Yano T (2017). Crystal structure and DNA-binding property of the ATPase domain of bacterial mismatch repair endonuclease MutL from Aquifex aeolicus. Biochim Biophys Acta Proteins Proteom, 1865(9), 1178–1187. doi: 10.1016/j.bbapap.2017.06.024 [DOI] [PubMed] [Google Scholar]
- [24].Furman CM, Elbashir R, & Alani E (2021). Expanded roles for the MutL family of DNA mismatch repair proteins. Yeast, 38(1), 39–53. doi: 10.1002/yea.3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Furman CM, Elbashir R, Pannafino G, Clark NL, & Alani E (2021). Experimental exchange of paralogous domains in the MLH family provides evidence of sub-functionalization after gene duplication. G3 (Bethesda), 11(6). doi: 10.1093/g3journal/jkab111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Furman CM, Wang TY, Zhao Q, Yugandhar K, Yu H, & Alani E (2021). Handcuffing intrinsically disordered regions in Mlh1-Pms1 disrupts mismatch repair. Nucleic Acids Res, 49(16), 9327–9341. doi: 10.1093/nar/gkab694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gao Y, & Yang W (2016). Capture of a third Mg(2)(+) is essential for catalyzing DNA synthesis. Science, 352(6291), 1334–1337. doi: 10.1126/science.aad9633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Goellner EM, Smith CE, Campbell CS, Hombauer H, Desai A, Putnam CD, & Kolodner RD (2014). PCNA and Msh2-Msh6 activate an Mlh1-Pms1 endonuclease pathway required for Exo1-independent mismatch repair. Mol Cell, 55(2), 291–304. doi: 10.1016/j.molcel.2014.04.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gorman J, Chowdhury A, Surtees JA, Shimada J, Reichman DR, Alani E, & Greene EC (2007). Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol Cell, 28(3), 359–370. doi: 10.1016/j.molcel.2007.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gorman J, Plys AJ, Visnapuu ML, Alani E, & Greene EC (2010). Visualizing one-dimensional diffusion of eukaryotic DNA repair factors along a chromatin lattice. Nat Struct Mol Biol, 17(8), 932–938. doi: 10.1038/nsmb.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, … Regev A (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol, 29(7), 644–652. doi: 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Guarne A, & Charbonnier JB (2015). Insights from a decade of biophysical studies on MutL: Roles in strand discrimination and mismatch removal. Prog Biophys Mol Biol, 117(2–3), 149–156. doi: 10.1016/j.pbiomolbio.2015.02.002 [DOI] [PubMed] [Google Scholar]
- [33].Guarne A, Ramon-Maiques S, Wolff EM, Ghirlando R, Hu X, Miller JH, & Yang W (2004). Structure of the MutL C-terminal domain: a model of intact MutL and its roles in mismatch repair. EMBO J, 23(21), 4134–4145. doi: 10.1038/sj.emboj.7600412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gueneau E, Dherin C, Legrand P, Tellier-Lebegue C, Gilquin B, Bonnesoeur P, … Charbonnier JB (2013). Structure of the MutLalpha C-terminal domain reveals how Mlh1 contributes to Pms1 endonuclease site. Nat Struct Mol Biol, 20(4), 461–468. doi: 10.1038/nsmb.2511 [DOI] [PubMed] [Google Scholar]
- [35].Haas BJ (2022). TransDecoder
- [36].Hall MC, Shcherbakova PV, Fortune JM, Borchers CH, Dial JM, Tomer KB, & Kunkel TA (2003). DNA binding by yeast Mlh1 and Pms1: implications for DNA mismatch repair. Nucleic Acids Res, 31(8), 2025–2034. doi: 10.1093/nar/gkg324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hoffmann ER, Shcherbakova PV, Kunkel TA, & Borts RH (2003). MLH1 mutations differentially affect meiotic functions in Saccharomyces cerevisiae. Genetics, 163(2), 515–526. doi: 10.1093/genetics/163.2.515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hombauer H, Campbell CS, Smith CE, Desai A, & Kolodner RD (2011). Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell, 147(5), 1040–1053. doi: 10.1016/j.cell.2011.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hopf TA, Colwell LJ, Sheridan R, Rost B, Sander C, & Marks DS (2012). Three-dimensional structures of membrane proteins from genomic sequencing. Cell, 149(7), 1607–1621. doi: 10.1016/j.cell.2012.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, … Hassabis D (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. doi: 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kadyrov FA, Dzantiev L, Constantin N, & Modrich P (2006). Endonucleolytic function of MutLalpha in human mismatch repair. Cell, 126(2), 297–308. doi: 10.1016/j.cell.2006.05.039 [DOI] [PubMed] [Google Scholar]
- [42].Kadyrov FA, Holmes SF, Arana ME, Lukianova OA, O’Donnell M, Kunkel TA, & Modrich P (2007). Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. J Biol Chem, 282(51), 37181–37190. doi: 10.1074/jbc.M707617200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Karplus M, & McCammon JA (2002). Molecular dynamics simulations of biomolecules. Nat Struct Biol, 9(9), 646–652. doi: 10.1038/nsb0902-646 [DOI] [PubMed] [Google Scholar]
- [44].Katoh K, Misawa K, Kuma K, & Miyata T (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res, 30(14), 3059–3066. doi: 10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kim Y, Furman CM, Manhart CM, Alani E, & Finkelstein IJ (2019). Intrinsically disordered regions regulate both catalytic and non-catalytic activities of the MutLalpha mismatch repair complex. Nucleic Acids Res, 47(4), 1823–1835. doi: 10.1093/nar/gky1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kosinski J, Plotz G, Guarne A, Bujnicki JM, & Friedhoff P (2008). The PMS2 subunit of human MutLalpha contains a metal ion binding domain of the iron-dependent repressor protein family. J Mol Biol, 382(3), 610–627. doi: 10.1016/j.jmb.2008.06.056 [DOI] [PubMed] [Google Scholar]
- [47].Liu J, Hanne J, Britton BM, Bennett J, Kim D, Lee JB, & Fishel R (2016). Cascading MutS and MutL sliding clamps control DNA diffusion to activate mismatch repair. Nature, 539(7630), 583–587. doi: 10.1038/nature20562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].London J, Martin-Lopez J, Yang I, Liu J, Lee JB, & Fishel R (2021). Linker domain function predicts pathogenic MLH1 missense variants. Proc Natl Acad Sci U S A, 118(9). doi: 10.1073/pnas.2019215118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lynch HT, Snyder CL, Shaw TG, Heinen CD, & Hitchins MP (2015). Milestones of Lynch syndrome: 1895–2015. Nat Rev Cancer, 15(3), 181–194. doi: 10.1038/nrc3878 [DOI] [PubMed] [Google Scholar]
- [50].Manhart CM, Ni X, White MA, Ortega J, Surtees JA, & Alani E (2017). The mismatch repair and meiotic recombination endonuclease Mlh1-Mlh3 is activated by polymer formation and can cleave DNA substrates in trans. PLoS Biol, 15(4), e2001164. doi: 10.1371/journal.pbio.2001164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mardenborough YSN, Nitsenko K, Laffeber C, Duboc C, Sahin E, Quessada-Vial A, … Lebbink JHG (2019). The unstructured linker arms of MutL enable GATC site incision beyond roadblocks during initiation of DNA mismatch repair. Nucleic Acids Res, 47(22), 11667–11680. doi: 10.1093/nar/gkz834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Marks DS, Colwell LJ, Sheridan R, Hopf TA, Pagnani A, Zecchina R, & Sander C (2011). Protein 3D structure computed from evolutionary sequence variation. PLoS One, 6(12), e28766. doi: 10.1371/journal.pone.0028766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Martin FJ, Amode MR, Aneja A, Austine-Orimoloye O, Azov AG, Barnes I, … Flicek P (2023). Ensembl 2023. Nucleic Acids Res, 51(D1), D933–D941. doi: 10.1093/nar/gkac958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mauris J, & Evans TC (2009). Adenosine triphosphate stimulates Aquifex aeolicus MutL endonuclease activity. PLoS One, 4(9), e7175. doi: 10.1371/journal.pone.0007175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].McInnes L, Healy J, & Melville J (2018). UMAP: uniform manifold approximation and projection for dimension reduction. arXiv, http://arxiv.org/abs/1802.03426.
- [56].Mechanic LE, Frankel BA, & Matson SW (2000). Escherichia coli MutL loads DNA helicase II onto DNA. J Biol Chem, 275(49), 38337–38346. doi: 10.1074/jbc.M006268200 [DOI] [PubMed] [Google Scholar]
- [57].Mendillo ML, Mazur DJ, & Kolodner RD (2005). Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end-blocking system. J Biol Chem, 280(23), 22245–22257. doi: 10.1074/jbc.M407545200 [DOI] [PubMed] [Google Scholar]
- [58].Mohd AB, Palama B, Nelson SE, Tomer G, Nguyen M, Huo X, & Buermeyer AB (2006). Truncation of the C-terminus of human MLH1 blocks intracellular stabilization of PMS2 and disrupts DNA mismatch repair. DNA Repair (Amst), 5(3), 347–361. doi: 10.1016/j.dnarep.2005.11.001 [DOI] [PubMed] [Google Scholar]
- [59].Namadurai S, Jain D, Kulkarni DS, Tabib CR, Friedhoff P, Rao DN, & Nair DT (2010). The C-terminal domain of the MutL homolog from Neisseria gonorrhoeae forms an inverted homodimer. PLoS One, 5(10), e13726. doi: 10.1371/journal.pone.0013726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Oliver A, Baquero F, & Blazquez J (2002). The mismatch repair system (mutS, mutL and uvrD genes) in Pseudomonas aeruginosa: molecular characterization of naturally occurring mutants. Mol Microbiol, 43(6), 1641–1650. doi: 10.1046/j.1365-2958.2002.02855.x [DOI] [PubMed] [Google Scholar]
- [61].Ovchinnikov S, Kinch L, Park H, Liao Y, Pei J, Kim DE, … Baker, D. (2015). Large-scale determination of previously unsolved protein structures using evolutionary information. Elife, 4, e09248. doi: 10.7554/eLife.09248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Pang Q, Prolla TA, & Liskay RM (1997). Functional domains of the Saccharomyces cerevisiae Mlh1p and Pms1p DNA mismatch repair proteins and their relevance to human hereditary nonpolyposis colorectal cancer-associated mutations. Mol Cell Biol, 17(8), 4465–4473. doi: 10.1128/MCB.17.8.4465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Pillon MC, Lorenowicz JJ, Uckelmann M, Klocko AD, Mitchell RR, Chung YS, … Guarne A (2010). Structure of the endonuclease domain of MutL: unlicensed to cut. Mol Cell, 39(1), 145–151. doi: 10.1016/j.molcel.2010.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Plys AJ, Rogacheva MV, Greene EC, & Alani E (2012). The unstructured linker arms of Mlh1-Pms1 are important for interactions with DNA during mismatch repair. J Mol Biol, 422(2), 192–203. doi: 10.1016/j.jmb.2012.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Putnam CD (2016). Evolution of the methyl directed mismatch repair system in Escherichia coli. DNA Repair (Amst), 38, 32–41. doi: 10.1016/j.dnarep.2015.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Putnam CD (2021). Strand discrimination in DNA mismatch repair. DNA Repair (Amst), 105, 103161. doi: 10.1016/j.dnarep.2021.103161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Reyes GX, Schmidt TT, Kolodner RD, & Hombauer H (2015). New insights into the mechanism of DNA mismatch repair. Chromosoma, 124(4), 443–462. doi: 10.1007/s00412-015-0514-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Robertson A, Pattishall SR, & Matson SW (2006). The DNA binding activity of MutL is required for methyl-directed mismatch repair in Escherichia coli. J Biol Chem, 281(13), 8399–8408. doi: 10.1074/jbc.M509184200 [DOI] [PubMed] [Google Scholar]
- [69].Sacho EJ, Kadyrov FA, Modrich P, Kunkel TA, & Erie DA (2008). Direct visualization of asymmetric adenine-nucleotide-induced conformational changes in MutL alpha. Mol Cell, 29(1), 112–121. doi: 10.1016/j.molcel.2007.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Schuler GD, Epstein JA, Ohkawa H, & Kans JA (1996). Entrez: molecular biology database and retrieval system. Methods Enzymol, 266, 141–162. doi: 10.1016/s0076-6879(96)66012-1 [DOI] [PubMed] [Google Scholar]
- [71].Smith CE, Mendillo ML, Bowen N, Hombauer H, Campbell CS, Desai A, … Kolodner RD (2013). Dominant mutations in S. cerevisiae PMS1 identify the Mlh1-Pms1 endonuclease active site and an exonuclease 1-independent mismatch repair pathway. PLoS Genet, 9(10), e1003869. doi: 10.1371/journal.pgen.1003869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Steitz TA, & Steitz JA (1993). A general two-metal-ion mechanism for catalytic RNA. Proc Natl Acad Sci U S A, 90(14), 6498–6502. doi: 10.1073/pnas.90.14.6498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Torres KA, Calil FA, Zhou AL, DuPrie ML, Putnam CD, & Kolodner RD (2022). The unstructured linker of Mlh1 contains a motif required for endonuclease function which is mutated in cancers. Proc Natl Acad Sci U S A, 119(42), e2212870119. doi: 10.1073/pnas.2212870119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, & Berendsen HJ (2005). GROMACS: fast, flexible, and free. J Comput Chem, 26(16), 1701–1718. doi: 10.1002/jcc.20291 [DOI] [PubMed] [Google Scholar]
- [75].Zaremba-Niedzwiedzka K, Caceres EF, Saw JH, Backstrom D, Juzokaite L, Vancaester E, … Ettema TJ (2017). Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature, 541(7637), 353–358. doi: 10.1038/nature21031 [DOI] [PubMed] [Google Scholar]