

e-TOC blurb:
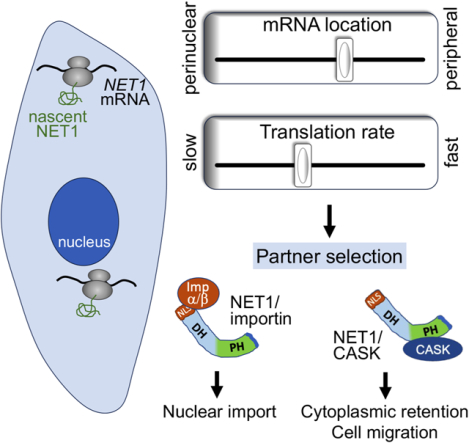
Protein targeting between destinations can be modulated post-translationally. Using NET1 as a model, Gasparski et al. show that, additionally, mRNA location in the cytoplasm and its translation elongation rate robustly influence protein distribution and function. This effect results from modulating the selection of partners that bind to distinct NET1 domains.
Keywords: RNA localization, local translation, elongation rate, ribosome stalling, NET1, CASK, nuclear import, cell migration, RhoA activity
Summary
Numerous proteins are targeted to two or multiple subcellular destinations where they exert distinct functional consequences. The balance between such differential targeting is thought to be determined post-translationally, relying on protein sorting mechanisms. Here, we show that mRNA location and translation rate can also determine protein targeting, through modulating protein binding to specific interacting partners. Peripheral localization of the NET1 mRNA and fast translation lead to higher cytosolic retention of the NET1 protein, through promoting its binding to the membrane-associated scaffold protein CASK. By contrast, perinuclear mRNA location and/or slower translation rate favor nuclear targeting, through promoting binding to importins. This mRNA location-dependent mechanism is modulated by physiological stimuli and profoundly impacts NET1 function in cell motility. These results reveal that the location of protein synthesis and the rate of translation elongation act in coordination as a ‘partner-selection’ mechanism that robustly influences protein distribution and function.
Introduction
Numerous and diverse eukaryotic proteins are targeted to two or multiple subcellular destinations1. Proteins with dual localization include metabolic enzymes, signaling factors and apoptosis regulators2–6. Alterations in the balance of protein targeting affect important physiological outcomes7–9. Dual targeting is often regulated through post-translational mechanisms that favor the action of one out of two or more competing targeting signals or promote processes or interactions that retain the polypeptide in a specific compartment1,10,11.
NET1 is a guanine nucleotide exchange factor, which activates the RhoA GTPase, and is distributed in both the nucleus and cytoplasm. Cytoplasmic NET1 regulates RhoA and controls the cytoskeleton and cell migration, while nuclear NET1 is thought to control cellular responses to DNA damage12–14. Post-translational modifications can alter NET1 distribution and function15,16. Interestingly however, NET1 gene expression exhibits an additional level of regulation at the level of mRNA localization. The NET1 mRNA is prominently targeted to peripheral protrusive cytoplasmic regions of migrating cells17,18. While compartmentalized distribution in the cytosol has now been described for a large fraction of mammalian mRNAs, the precise functional consequences of this prevalent mode of regulation are poorly understood19,20. Peripheral NET1 mRNA localization is observed in in vivo tumors and is important for cancer cell invasion21. However, how peripheral mRNA localization and translation influence NET1 protein function is unknown. Here, we show that NET1 mRNA location in combination with the rate of its translation can influence the ability of competing domains to determine the targeting of the newly synthesized protein, through favoring binding to specific partners. This RNA-based ‘partner-selection’ mechanism provides a robust means of controlling protein function.
Results
Altering NET1 mRNA location between peripheral and perinuclear regions
Localization of the NET1 mRNA to the cell periphery is an active process that requires sequences present in the 3’UTR of the transcript. Specifically, NET1 and co-regulated mRNAs are trafficked to the periphery through the KIF1C kinesin, and contain GA-rich regions in their 3’UTRs, which are necessary for localization21–23. Antisense phosphorodiamidate morpholino oligonucleotides (PMOs) specific to these regions can interfere with mRNA targeting21,22. We tiled 25nt-long PMOs across most of the length of the human NET1 3’UTR (Figure S1A) and tested for their ability to interfere with peripheral NET1 mRNA localization (Figure S1B) as well as for their effects on overall NET1 mRNA and NET1 protein levels (Figure S1C–D). This allowed us to identify PMOs that result in an almost diffuse NET1 mRNA distribution (Figure S1B; indicated by a PDI (Peripheral Distribution Index) of 124) and which have no apparent effect on NET1 mRNA levels or translation efficiency. Consistent with prior observations21, targeting the region highest in GA-content with a combination of two PMOs (921 and 975) significantly reduced peripheral NET1 mRNA localization (Figures 1A, S1B, S2A). Additional PMOs in distant regions produced a similar effect, suggesting the presence of multiple necessary elements or the involvement of long-range, tertiary interactions. Regardless of the exact mechanism, PMOs921+975 and PMO1067 provided us with tools that allow the conversion of the NET1 mRNA distribution from a peripheral one to mostly diffuse or perinuclear (Figure 1A and S2A). Significantly, this alteration was not accompanied by changes in NET1 mRNA or protein levels (Figure 1B and S2B) and was specific for NET1 since the distribution of the co-regulated RAB13 mRNA was not impacted (Figure S2C).
Figure 1: Altering NET1 mRNA localization between peripheral and perinuclear regions.
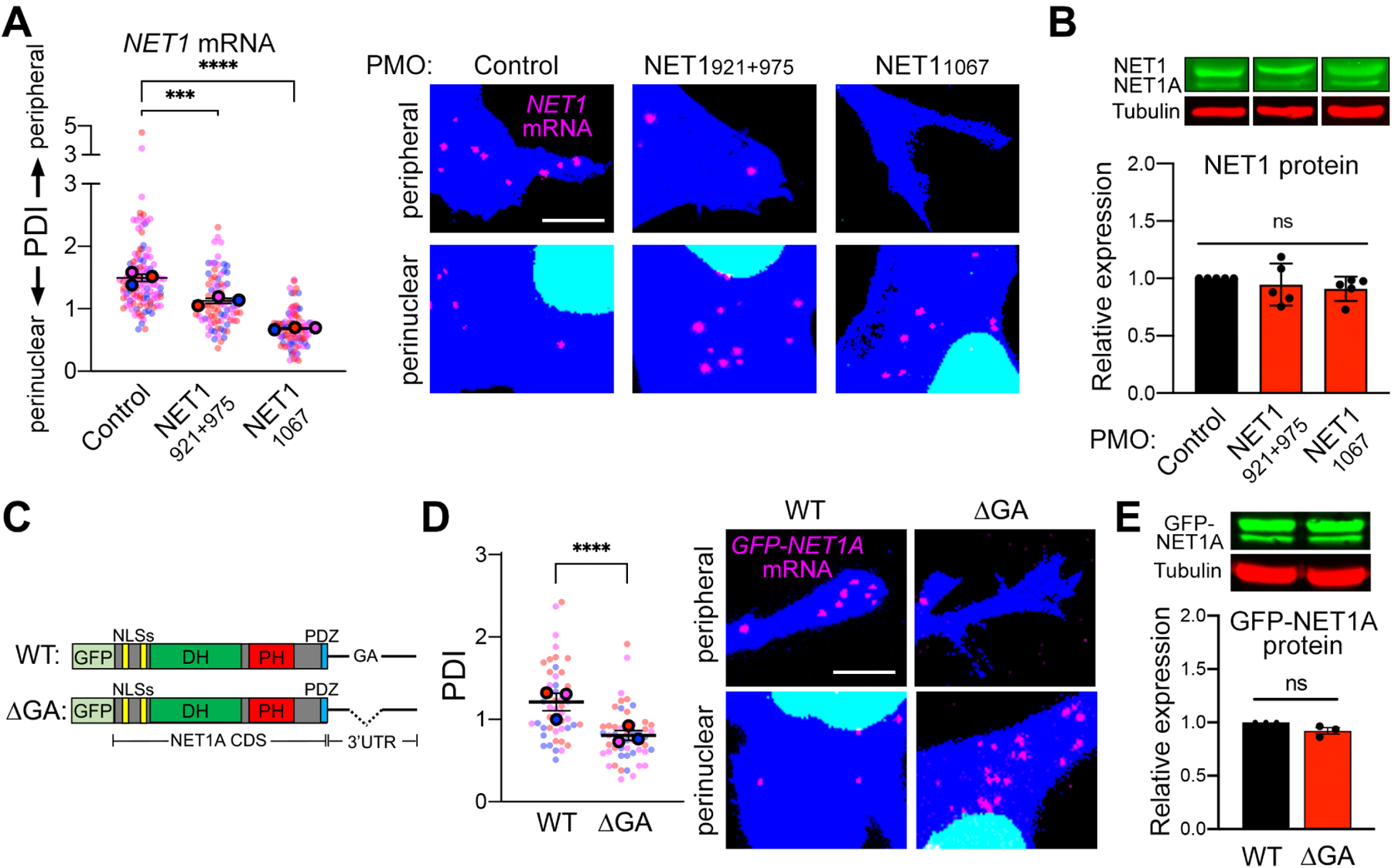
(A) FISH images of MDA-MB-231 cells treated with the indicated PMOs. Zoomed in perinuclear and peripheral regions are shown (whole images are shown in Figure S2A). The graph presents PDI quantification of NET1 mRNA distribution, with higher values indicating a more peripheral mRNA distribution. PDI=1 indicates a random, diffuse distribution. n=84–118 cells from 3 independent experiments. (B) Protein levels of NET1 and NET1A isoforms, by Western blot, in cells treated with the indicated PMOs. n=5. (C) Schematic of the GFP-NET1A constructs used for generation of stably expressing cell lines. Coding sequences (CDS) are identical. The ΔGA construct carries a 78nt deletion of a GA-rich region within the 3’UTR. (D) FISH images and corresponding PDI values of GFP-NET1A mRNA in WT and ΔGA expressing cell lines. Zoomed in perinuclear and peripheral regions are shown (whole images are shown in Figure S2D). n=50 cells from 3 independent experiments. (E) GFP-NET1A protein levels, by Western blot, in WT and ΔGA expressing cell lines. n=3. In superplots, data points from individual replicates are color coded, and large, outlined color dots indicate the mean of each replicate. Error bars: SEM. P-values: ***<0.001, ****<0.0001, ns: not significant by one-way ANOVA (A, B) or unpaired t-test (D, E). Scale bars: 4μm.
To independently manipulate NET1 mRNA localization, we generated stable cell lines that express constructs encoding a fusion protein of GFP with NET1A, the NET1 isoform mostly implicated with cell motility phenotypes15,25. The sequence encoding this fusion protein was attached to either the full-length wild type (WT) NET1 3’UTR, or a deletion mutant with the region highest in GA-content removed (ΔGA) (Figure 1C). Consistent with the above results, the ΔGA mutant mRNA exhibited a perinuclear distribution that was significantly different from the peripheral localization imparted by the WT 3’ UTR (Figure 1D and S2D). Importantly, the overall GFP-NET1A mRNA and GFP-NET1A protein levels produced from these constructs were indistinguishable (Figure 1E and S2E, F), suggesting that the NET1 mRNA is equally stable and translated with similar efficiency in peripheral and perinuclear locations. Overall, PMO delivery or 3’UTR deletion specifically alters the distribution of endogenous or exogenous NET1 mRNA respectively, and thus allows us to further understand the molecular and functional consequences imparted by changing the site of NET1 protein synthesis.
mRNA location specifies NET1 protein targeting
The NET1 protein is dually targeted to both the nucleus and the cytosol. We thus tested whether altering NET1 mRNA location impacts the overall targeting of the protein between these two destinations. We assessed, in live cells, the nucleo-cytoplasmic distribution of GFP-NET1A produced from either a peripheral mRNA (GFP-NET1A/WT UTR) or from a perinuclear mRNA, upon treatment with mis-localizing antisense PMOs (PMOs921+975 and PMO1067; Figure 2A) or deletion of the GA-rich region within the 3’ UTR (GFP-NET1A/ΔGA UTR; Figure 2B). Interestingly, perinuclearly produced NET1A partitioned to a higher extent within the nucleus (Figure 2A, B). Of note, the amount of total protein produced under all conditions is the same, as shown above (Figure 1B, E and S2F), indicating that the reduction in cytoplasmic protein is due to increased import into the nucleus and not due to increased degradation in the cytosol. Furthermore, nuclear accumulation is abolished when the two N-terminal basic NLSs are mutated (Figure 2B), suggesting that NET1A nuclear import involves the action of nuclear transport receptors of the importin/karyopherin superfamily.
Figure 2: NET1 mRNA location determines NET1-importin binding and nucleocytoplasmic distribution.
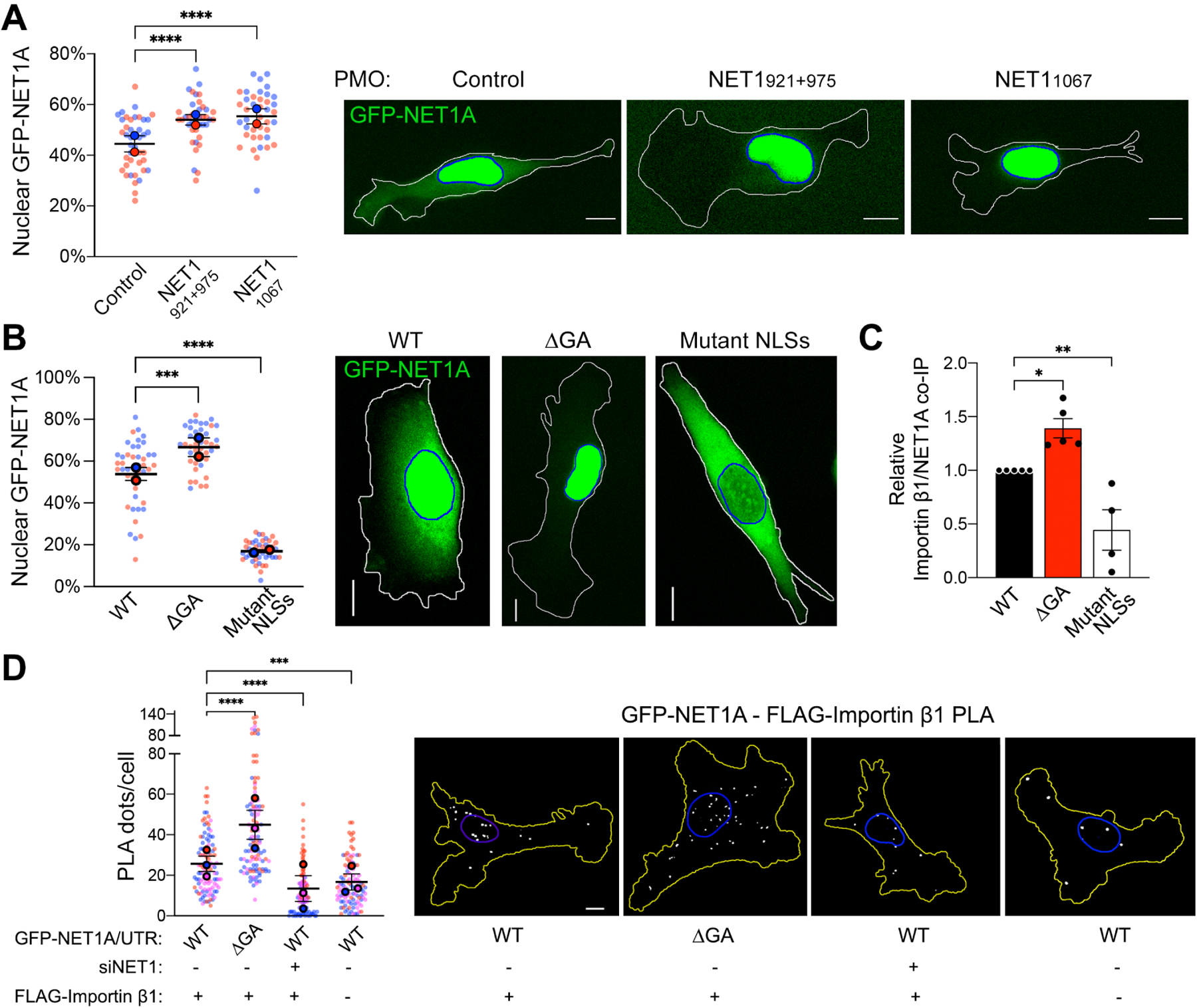
(A) Live GFP fluorescence imaging of cells expressing GFP-NET1A/WT UTR treated with the indicated PMOs. The percent GFP-NET1A signal within the nucleus is quantified. n=39–41 in 2 independent experiments. (B) Live GFP fluorescence imaging of the indicated cell lines (see Figure 1c) and quantification of the percent GFP-NET1A signal within the nucleus. Blue outline: nuclear boundary; white outline: cell boundary. n=43–44 in 2 independent experiments. (C) Relative importin β1 binding to GFP-NET1A from co-immunoprecipitation (co-IP) experiments of the indicated cell lines. GFP trap beads were used for IP and GFP-NET1A and FLAG-importin β1 proteins were detected by Western blot. n=5. (D) In situ detection of interaction between GFP-NET1A and FLAG-importin β1, by PLA in the indicated cell lines. White dots: PLA signal; blue outline: nuclear boundary; yellow outline: cell boundary. n=104–113 in 3 independent experiments. In superplots, data points from individual replicates are color coded, and large, outlined color dots indicate the mean of each replicate. Error bars: SEM. p-values: *<0.05, **<0.01, ***<0.001, ****<0.0001, ns: not significant by one-way ANOVA. Scale bars: 10μm.
To test whether the site of NET1A synthesis affects its interaction with transport receptors, we assessed NET1A association with importin β1/KPNB1, the main transport receptor involved in import of basic NLSs, in a heterodimer with an importin α family member. We detected NET1A-importin β1 interaction by co-immunoprecipitation (Figure 2C) or proximity ligation amplification (PLA), which allows the detection of complex formation between two partners in situ (Figure 2D). Indeed, importin β1 specifically co-IPs with GFP-NET1A in a manner dependent on the presence of functional NLSs (Figure 2C). Furthermore, PLA can specifically report the in situ presence of NET1A-importin β1 complex, in both the cytosol and nucleus, since the observed signal is significantly reduced when either partner is knocked-down (Figure 2D; +siNET1) or missing (Figure 2D; -FLAG-importin β1). Both assays revealed that GFP-NET1A produced perinuclearly (ΔGA UTR) exhibits an increased interaction with importin β1, consistent with its higher nuclear partitioning. Of note, most of the NET1A-importin β1 complex is observed perinuclearly, in agreement with reports showing that importin α and β subunits are preferentially enriched towards the perinuclear cell body rather than peripheral protrusive regions26.
Newly synthesized NET1 interacts with importin
The observation that the location of the NET1 mRNA affected the degree with which the encoded NET1 protein interacted with importin β1, prompted us to address whether mRNA translation is also involved. We thus assessed NET1A-importin β1 interaction after a brief (20 min) treatment with cycloheximide or puromycin to block translation. Interestingly, translation inhibition abolished the increased interaction with importin β1 observed for perinuclearly produced NET1A (ΔGA UTR) (Figure 3A, B), suggesting that importin β1 largely interacts with newly synthesized NET1A. We interpret these data to indicate that perinuclearly synthesized NET1A rapidly and preferentially interacts with nuclear import receptors. Given the presence of NLSs at the very N-terminus this interaction could likely occur co-translationally. Import into the nucleus would then result in dissociation of the transport receptors. By contrast, peripherally synthesized NET1A interacts with importin β1 less efficiently and independent of active translation, potentially reflecting a low level of nucleocytoplasmic shuttling of mature NET1A protein.
Figure 3: The NLS and PH domains competitively determine nuclear import of newly-synthesized NET1A protein.
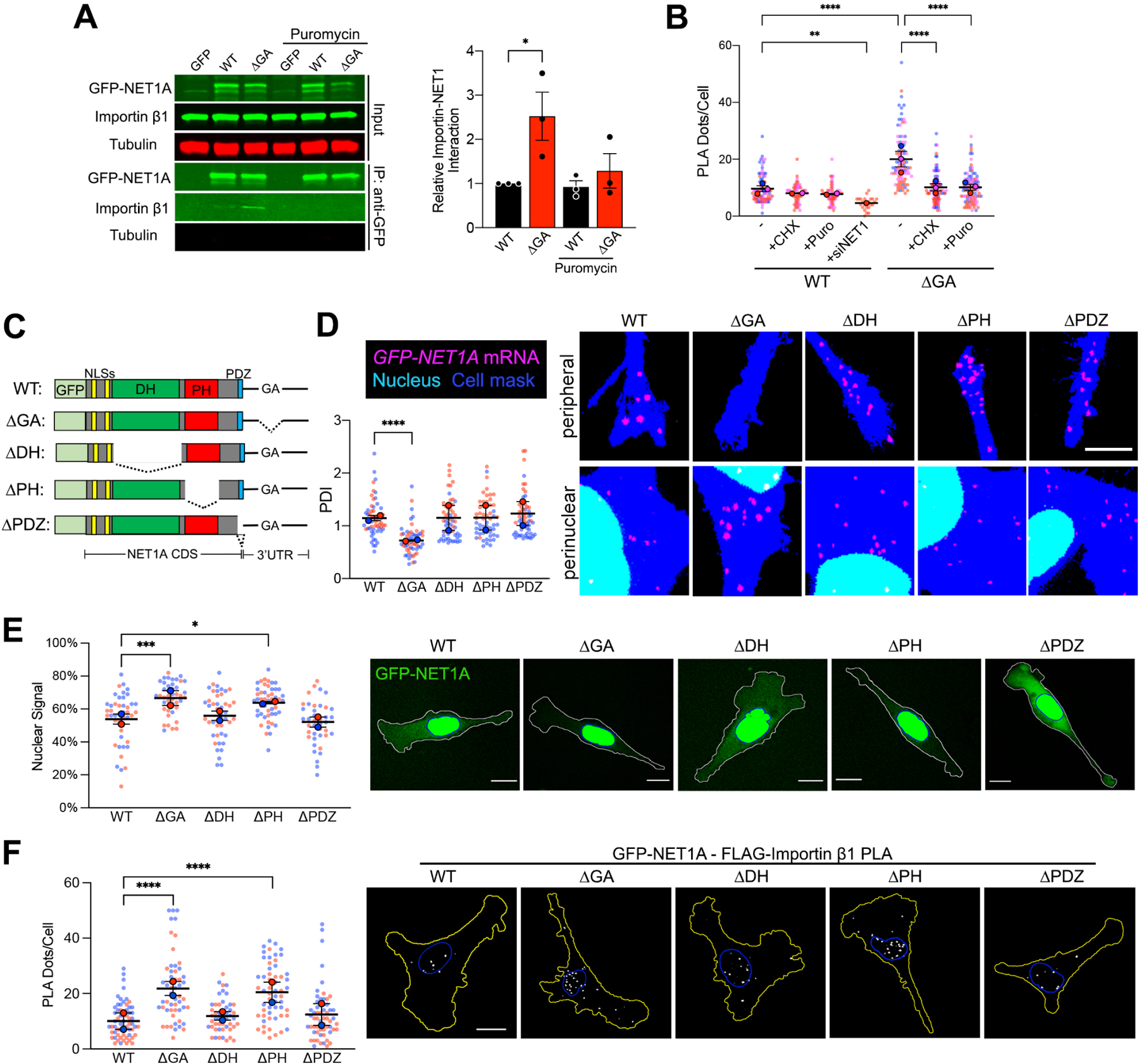
(A) Importin β1 binding to GFP or GFP-NET1A from co-immunoprecipitation experiments of the indicated MDA-MB-231 cell lines with 20min puromycin treatment. Quantifications are shown relative to the WT untreated sample. n=3. (B) Quantification of in situ interaction between GFP-NET1A and FLAG-importin β1, by PLA of the indicated cell lines, with 20min cycloheximide (CHX) or puromycin (Puro) treatment, or NET1 knockdown (siNET1). n=55–98 in 2–3 independent experiments. For siNET1 n=25. (C) Schematic of the GFP-NET1A constructs used for generation of stably expressing MDA-MB-231 cell lines. Dotted lines indicate deleted regions. (D) FISH images and corresponding PDI values of GFP-NET1A mRNA distribution in the indicated stable cell lines. Zoomed in perinuclear and peripheral regions are shown. n=55–58 cells from 2 independent experiments. (E) Live GFP fluorescence imaging of the indicated cell lines and quantification of the percent GFP-NET1A signal within the nucleus. Blue outline: nuclear boundary; white outline: cell boundary. n=40–50 in 2 independent experiments. (F) In situ detection of interaction between GFP-NET1A and FLAG-importin β1, by PLA in the indicated cell lines. White dots: PLA signal; blue outline: nuclear boundary; yellow outline: cell boundary. n=49–59 in 2 independent experiments. In superplots, data points from individual replicates are color coded, and large, outlined color dots indicate the mean of each replicate. Error bars: SEM. p-values: *<0.05, **<0.01, ***<0.001, ****<0.0001 by unpaired t-test (A) or one-way ANOVA (B, D, E, F). Scale bars: 4μm (D); 15μm (E, F).
Competition between NET1 protein domains specifies targeting
Polypeptides that can be targeted to dual destinations often contain distinct signals or domains that direct targeting to one or the other location. In the case of NET1A, as detailed above, nuclear targeting is mediated through the action of the N-terminal NLSs. Other recognizable domains could function to promote cytoplasmic retention. These include: a Dbl-homology (DH) domain, characteristic of proteins that activate Rho family GTPases by functioning as guanine nucleotide exchange factors (GEFs); a Plekstrin-homology (PH) domain, usually involved in protein-protein or protein-membrane interactions; and a C-terminal PDZ motif recognized by PDZ domain-containing partners (Figure 3C). To determine the involvement of these domains in cytosolic retention of NET1A, we generated stable cell lines expressing deletion mutants (Figure 3C and S3). All mutants were expressed from constructs carrying the WT NET1 UTR, which as expected resulted in peripheral mRNA localization similar to that observed for the full-length protein (Figure 3D). Deletion of the DH domain or of the PDZ motif did not alter NET1A association with importin β1, or the fraction partitioning in the nucleus (Figure 3E, F). Interestingly, however, deletion of the PH domain promoted NET1A-importin β1 interaction and increased nuclear accumulation to a degree similar to that seen when full-length NET1A is produced from a perinuclear mRNA (Figure 3E, F). Therefore, the PH domain appears to be in competition with the NLSs to determine the eventual nucleo-cytoplasmic balance of NET1A targeting. Given that deletion of the DH domain does not exhibit a similar effect, we conclude that the GEF activity per se is not necessary for cytoplasmic retention, but that rather some other factor recognized by the PH domain functions to suppress NET1A nuclear import.
NET1 mRNA location determines protein targeting through partner selection
To identify this factor, we sought, through mass spectrometry analysis, for GFP-NET1A binding partners whose interaction is promoted by peripheral localization of the NET1 mRNA. We focused on one candidate, the membrane-associated scaffold protein CASK27,28 (Figure S4). Indeed, CASK interacts with GFP-NET1A in co-immunoprecipitation experiments and this interaction is minimized when NET1A is produced perinuclearly (Figure 4A; ΔGA). Furthermore, CASK-NET1A interaction requires the PH domain of NET1A (Figure 4A; ΔPH). The CASK-NET1A interaction can also be visualized by PLA which further revealed that the complex resides to a large extent close to the cell periphery (potentially reflecting membrane association) and verified that its formation depends on the location of the NET1 mRNA (Figure 4B). Overall, the NET1A-CASK complex is regulated by the location of the NET1 mRNA in a manner opposite to the NET1A-importin β1 complex and resides in spatially distinct regions in the cell.
Figure 4: NET1A-CASK interaction competes with NET1A-importin binding and is regulated in an opposite manner by NET1 mRNA location.
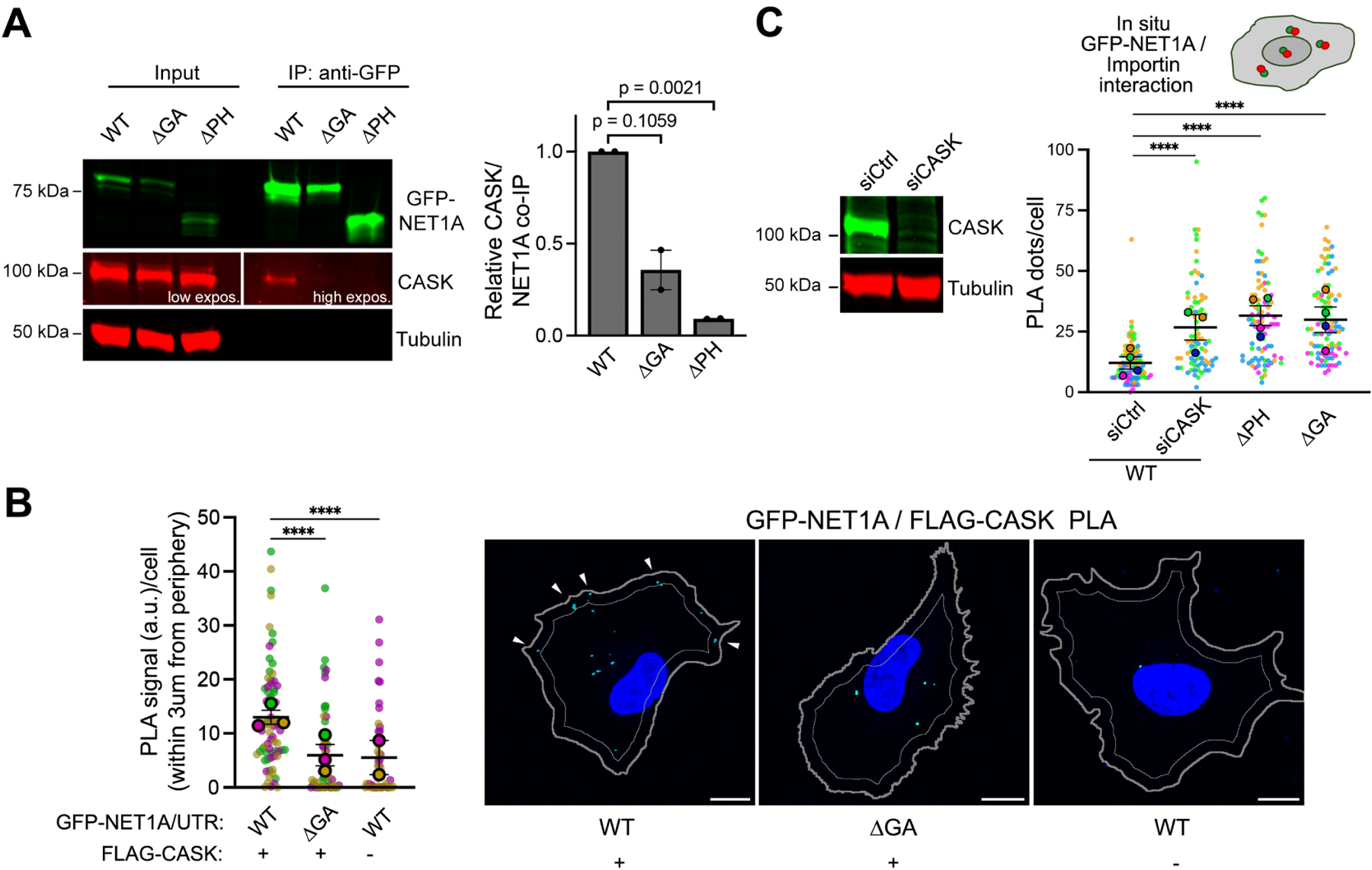
(A) Representative Western blot and quantification of relative CASK binding to GFP-NET1A from co-immunoprecipitation experiments of the indicated MDA-MB-231 cell lines. n=2. (B) In situ detection of interaction between GFP-NET1A and FLAG-CASK, by PLA in the indicated cell lines. Cyan dots: PLA signal; blue: DAPI; thick gray outline: cell boundary; thin gray outline: inner boundary 3μm from periphery. n=45–72 in 3 independent experiments. (C) Western blot showing efficiency of CASK knockdown upon siRNA treatment and quantification of in situ interaction between GFP-NET1A and FLAG-importin β1, by PLA of the indicated cell lines (see Figure 3C). n=78–92 in 4 independent experiments. In superplots, data points from individual replicates are color coded, and large, outlined color dots indicate the mean of each replicate. Error bars: SEM. p-values:, ****<0.0001 by one-way ANOVA (A, C) or Kruskal-Wallis test (B). Scale bars: 10μm.
To directly test whether CASK is the factor that suppresses importin β1 binding and NET1A nuclear import, we knocked-down CASK expression with siRNAs. Indeed, absence of CASK led to increased NET1A-importin β1 interaction, even though the NET1 mRNA is peripheral, and mimicked the effect seen upon deletion of the PH domain or upon expression of NET1A from perinuclear mRNA (Figure 4C).
Modulating the translation elongation rate through the NET1A coding sequence
Since recognition of the NLSs by importins occurs during, or shortly after, translation (Figure 3A, B), we reasoned that the ability of CASK to antagonize with NLS-importin binding would likely occur in a similar time frame. In this regard, the rate of translation elongation could influence the kinetics of appearance and/or folding of competing domains and thereby modulate their ability to direct eventual protein targeting. To assess the potential existence of any distinctive features associated with NET1 mRNA translation, we examined monosome and disome ribosome footprint profiling data from human HEK293 cells29. These cells also exhibit peripheral NET1 mRNA localization that is dependent on GA-rich sequences in the 3’UTR (Figure S5). Intriguingly, we observed higher monosome peaks, believed to reflect slower local elongation rates30, at two sites within the region encoding the DH domain (Figure 5A, arrows). These monosome peaks are also observed in other conditions and backgrounds (Figure S6A) and are detected around proline codons (Figure 5A), which have been described as a major contributor to ribosome stalling31. Disome profiling revealed disome peaks upstream of these high monosome density sites (Figure 5A, S6B), further supporting the notion that they reflect regions of slower elongation leading to ribosome pile-up at upstream sequences. While this pattern is unlikely to occur by chance (p<0.002), the disome sequencing depth we could achieve is relatively low. Therefore, further investigation would be needed to understand the extent of formation of disomes, as well as whether they represent ribosome collision events with additional roles.
Figure 5. The rate of translation elongation determines partner selection and NET1 protein targeting.
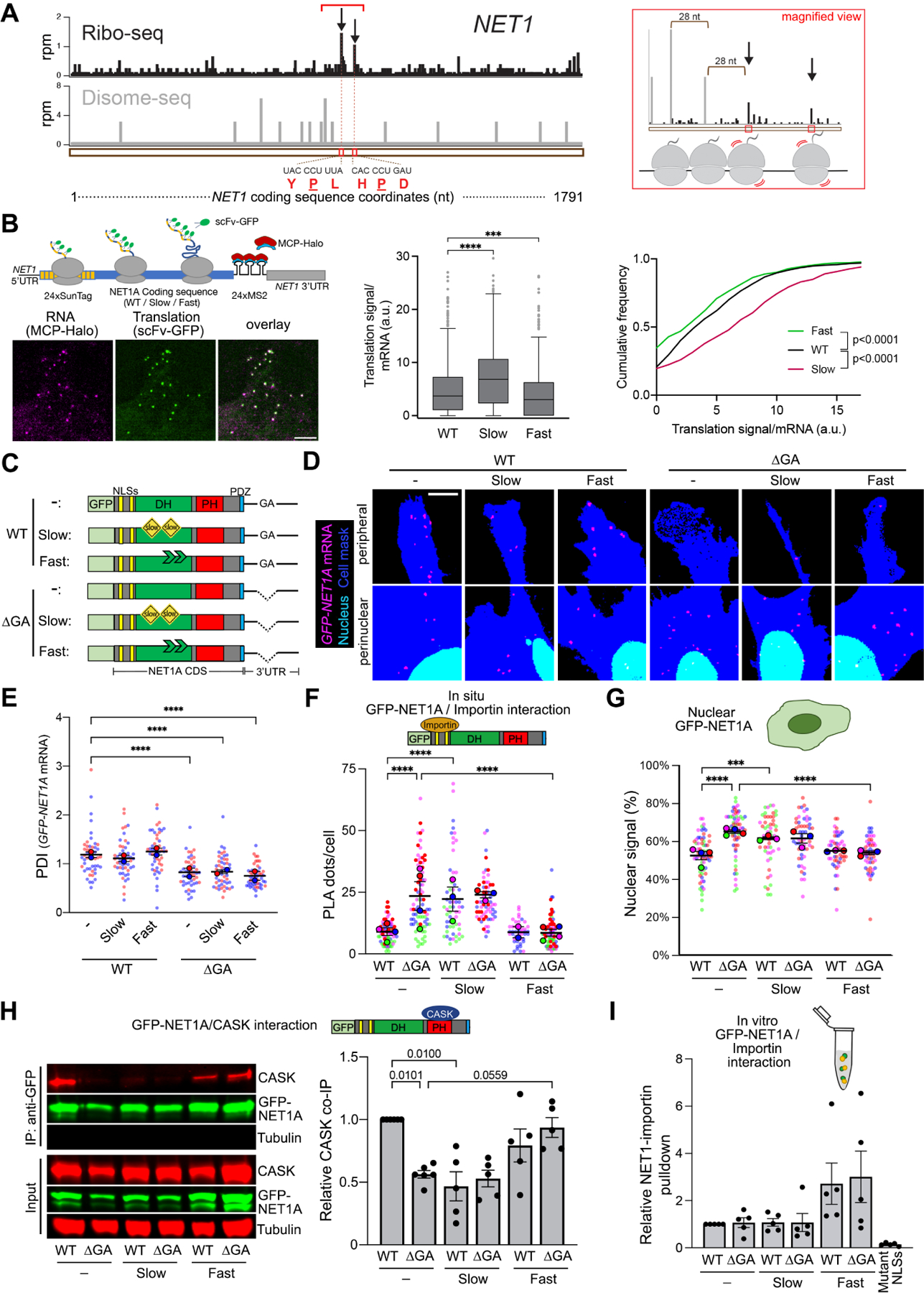
(A) Left panel: Snapshot of Ribo-seq (black) and Disome-seq (gray) reads mapped to the NET1 transcript. For Ribo-seq, reads are plotted to correspond to the approximate P-site of the ribosome. For Disome-seq, reads are plotted to correspond to the approximate P-site of the trailing ribosome in the disome complex. Stalling peaks are indicated with arrows. The amino acid and nucleotide sequence of the stalling motifs corresponding to E, P, A sites of the ribosome are shown below the snapshot, with the P site amino acids underlined. Right panel: magnified snapshot of the Ribo-seq (black) and Disome-seq (gray) peaks within the region indicated by a red bracket in the left panel. Disome peaks ~28nt (a typical ribosome footprint length) apart from each other and ~28nt upstream of the first stalling site indicate the possibility of queued ribosomes in this region. (B) Schematic of NET1-based reporter constructs for single molecule translation site imaging. Examples of typical imaging snapshots are shown (left panel). Graphs present Tukey’s box-plot distributions (middle panel) or cumulative frequency plots (right panel) of translation signals/mRNA of the indicated constructs. n=604–863 mRNA particles in 40–42 cells. (C) Schematic of the GFP-NET1A constructs used for generation of stably expressing MDA-MB-231 cell lines. ‘Slow’ constructs include multiple suboptimal codons within the DH domain. ‘Fast’ constructs include mutations of the identified stall sites, also within the DH domain. (D, E) FISH images (D) and corresponding PDI values (E) of GFP-NET1A mRNA distribution in the indicated stable cell lines. Zoomed in perinuclear and peripheral regions are shown. n=46–52 cells from 2 independent experiments. (F) Quantification of in situ interaction between GFP-NET1A and FLAG-importin β1, by PLA of the indicated cell lines. n=44–86 in 3–4 independent experiments. (G) Quantification of the percent GFP-NET1A signal within the nucleus from live GFP fluorescence imaging of the indicated cell lines. n=51–65 in 3–4 independent experiments. (H) Representative Western blot and quantification of relative CASK binding to GFP-NET1A from co-immunoprecipitation experiments of the indicated cell lines. n=5. (I) Quantification of relative pulldown efficiency of GFP-NET1A from lysates of the indicated cell lines with GST-importin α5. n=5. In superplots, data points from individual replicates are color coded, and large, outlined color dots indicate the mean of each replicate. Error bars: SEM. p-values: ***<0.001, ****<0.0001, ns: not significant by Kruskal-Wallis and Kolmogorov-Smirnov test (B) or one-way ANOVA (E-I). Scale bar: 3μm (B), 4μm (D).
The potential for ribosome stalling between the sequences encoding the NLSs and the PH domain raised the possibility of a mechanism whereby the rate of translation elongation could coordinate partner binding by these two targeting domains. To directly test this idea, we aimed to experimentally alter the translation elongation rate within the intervening region encoding the DH domain. To slow down translation, we replaced DH domain-encoding codons with synonymous rare codons. Approximately 20% of codons were replaced leading to a decrease of codon adaptation index from 0.73 to 0.60 (slow mutant). To speed up translation, we mutated the two potential stall sites described above by replacing the proline and one adjacent amino acid with glycine (fast mutant). The presence of slow or fast mutations altered the amount of GFP-NET1A protein produced as expected, i.e. lower amount of protein was produced from the slow mutant, while higher amount was produced from the fast mutant (Figure S7A).
To further validate the impact of these mutations on translation rate, we generated reporter constructs carrying the WT or mutant NET1A coding sequences together with the wild type NET1 5’- and 3’-UTRs (Figure 5B and S7B). 24 copies of the binding site for the MS2 bacteriophage coat protein (MCP) were introduced at the beginning of the 3’UTR to allow visualization of single mRNAs in live cells, through recruitment of multiple copies of an MCP-Halo fusion protein. Furthermore, at the beginning of the coding sequence, a series of SunTag peptide epitopes were introduced, which are recognized by a single chain antibody fragment fused to superfolder-GFP (scFv-GFP), thus allowing detection of new protein chains (Figure 5B and S7B). The wild type, slow or fast variants were expressed under a doxycycline-inducible promoter and scFv signal at translation sites, i.e. in association with individual mRNA molecules, was quantified. This translation signal intensity is proportional to the number of ribosomes bound to any given mRNA. Given equivalent initiation rates, slower or faster elongation would be expected to respectively increase or decrease translation signal intensity (Figure S7B)32,33. Indeed, compared to the wild type, the fast mutant exhibits overall lower translation signal intensities, while the slow mutant exhibits the opposite trend (Figure 5B). Taken together, the above data indicate that, for the NET1 mRNA, translation elongation is limiting and that the introduced mutations alter elongation rates in opposite ways.
The rate of translation elongation determines partner selection and NET1 protein targeting
We generated stable cell lines that express GFP-NET1A variants (WT, slow and fast) from constructs that carry either the WT or the ΔGA 3’UTR (Figure 5C). The presence of coding sequence mutations did not alter the localization of the expressed mRNAs. Localization was determined by the 3’UTR sequence, such that all mRNAs carrying the WT 3’UTR were distributed peripherally, while mRNAs carrying the ΔGA 3’UTR were perinuclear (Figure 5D, E).
We then assessed how the introduction of these mutations affected binding of GFP-NET1A to its partners and nucleo-cytoplasmic partitioning. Strikingly, slow translation, regardless of mRNA location, led to high interaction with importin and nuclear targeting (Figure 5F, G), and reduced binding to CASK (Figure 5H). In contrast, increasing the rate of translation had the opposite effects, decreasing importin binding and nuclear targeting, while promoting binding to CASK (Figure 5F–H). These data thus show that the location of the NET1 mRNA and the rate of its translation act in concert to specify NET1 protein interactions and targeting. Perinuclear mRNA and/or slower translation favor NET1-importin interaction and nuclear import, while peripheral mRNA and/or faster translation promote NET1-CASK association and cytoplasmic retention.
As an alternative method to slow down translation we treated cells briefly (30min) with sub-inhibitory concentrations of anisomycin. Under these conditions a subset of ribosomes is expected to be stochastically inhibited, effectively mimicking ribosome stalling. Of note, the concentrations we use here (0.01–0.1 mg/L) are below those leading to peak eIF2α phosphorylation, which is likely induced through a stress response due to ribosome collisions (Figure S8A;34,35). In agreement with the above results, we find that slowing translation with sub-inhibitory concentrations of anisomycin leads to increased interaction of peripherally translated NET1A with importin β1 (Figure S8B).
The impact of translation rate suggested that the kinetics and thus likely the order of appearance of NET1 domains could be important. To further test this idea, we generated variants of NET1A in which the NLSs were moved to the C-terminus, i.e. after, rather than before, the PH domain. Interestingly, this placement led to low NET1-importin β1 interaction, even for NET1 translated perinuclearly, and abolished the effect of the mRNA location (Figure S9A, B). These results therefore further support the notion that kinetic competition underlies NET1A partner selection.
It was still possible, however, that the location, rate, or order of protein synthesis alters NET1A-importin interaction by affecting the inherent accessibility of the NLSs, for example through local modifications or altered folding. To assess this, we tested the ability of the various NET1A protein variants within cell lysates to interact with importin in vitro. We first identified importin α5/KNPA1 as the importin α member that binds more efficiently to NET1A NLSs (Figure S10) and then tested for its ability to associate with the various NET1A variants in in vitro pulldown assays. Interestingly, the increased, or decreased, importin interaction observed in vivo was not reflected in corresponding changes of NET1A-importin binding in vitro (compare Figure 5F to 5I, and Figure S9B to S9C). Therefore, the impact of the site, rate and order of translation does not appear to be due to alterations in the inherent ability of NET1A variants to recognize importins. Our overall data rather indicate that, in vivo, the specific local microenvironment encountered by the nascent, or newly synthesized, polypeptide (likely presenting different concentrations of binding partners) together with the rate at which competing domains appear, favor certain interactions over others and specify the eventual partner selection and target destination.
mRNA location affects NET1 protein function
Cytoplasmic NET1A is involved in activation of the RhoA GTPase and controls cell adhesion and migration. According to the mechanism described above, the subcellular location of the NET1 mRNA modulates the nucleo-cytoplasmic distribution of NET1A protein and would thus be expected to influence its function in either compartment. To assess the functional consequences of endogenous NET1 mRNA localization, we used antisense PMOs to alter its predominantly peripheral distribution to a perinuclear one (Figure 1A). To assess RhoA GTPase activity, we used a FRET-based RhoA activity biosensor36 and fluorescence lifetime imaging (FLIM) (Figure 6A and S11). Treatment with NET1 PMOs resulted in a reduction in FRET efficiency which was more pronounced in peripheral protrusive regions (Figure 6A). Therefore, perinuclear NET1 mRNA localization suppresses the cytosolic NET1 function towards RhoA activation, consistent with the observed increased NET1 import into the nucleus. RhoA is a central regulator of the cytoskeleton that influences focal adhesion assembly and maturation. In line with the observed reduced RhoA activity, treatment with NET1 PMOs also led to a significant reduction in the size of paxillin-containing focal adhesions, indicative of a reduced degree of maturation (Figure 6B).
Figure 6. NET1 mRNA location robustly affects NET1 function in cell migration, and controls TGFβ-induced NET1 activity.
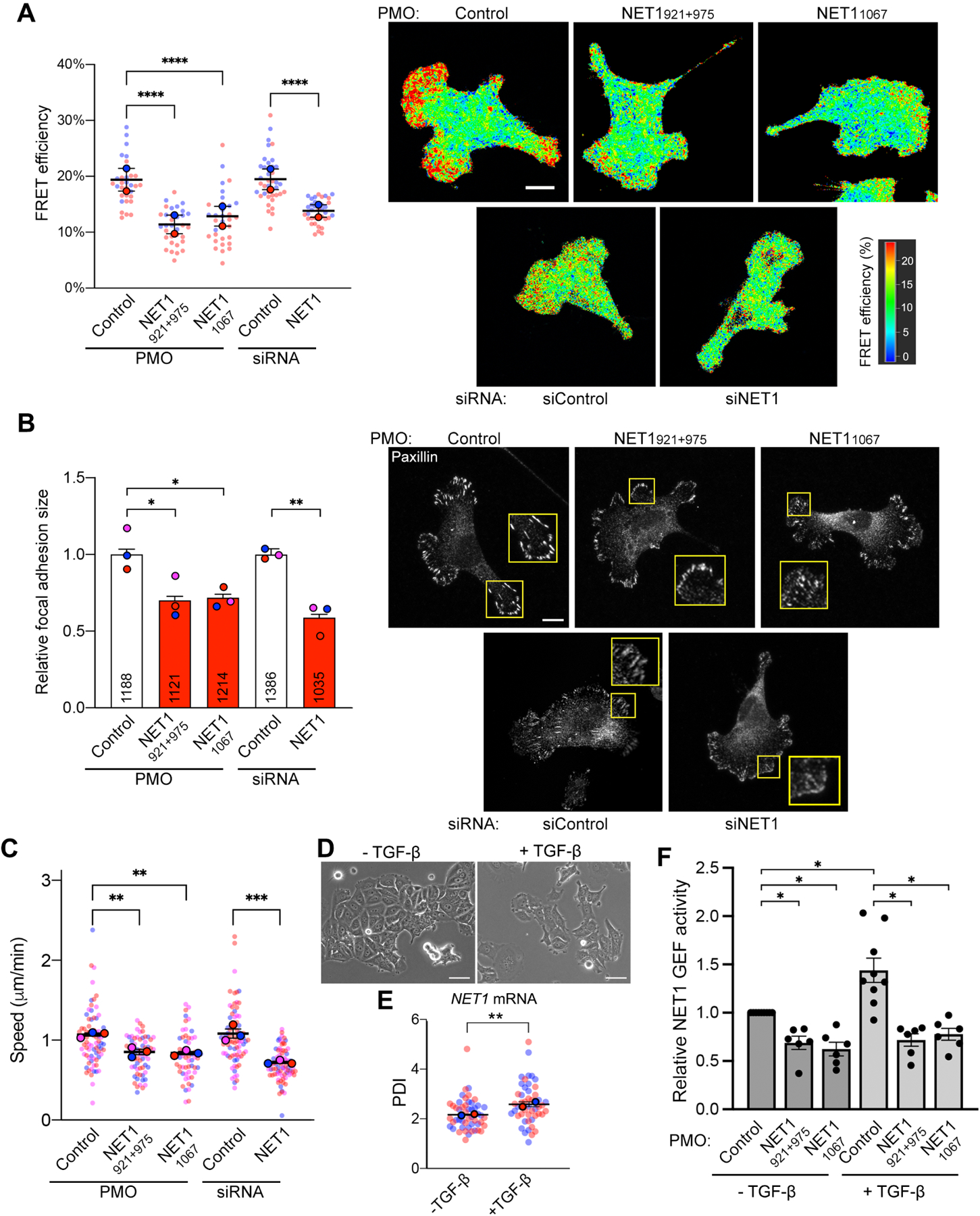
(A) FLIM imaging of a RhoA activity biosensor in MDA-MB-231 cells treated with the indicated PMOs or siRNAs. Graph shows FRET efficiency in protrusive regions. N=32–38 cells from 2 independent experiments. (B) Measurement of focal adhesion size based on paxillin immunofluorescence staining of cells treated with the indicated PMOs or siRNAs. Numbers within each bar indicate the number of focal adhesions measured in 3 independent experiments. (C) Average migration speeds measured by time lapse imaging of mCherry-NLS expressing cells treated with the indicated PMOs or siRNAs. N=57–78 cells from 3 independent experiments. (D) Phase contrast images of MCF7 cells with or without TGF-β treatment. (E) PDI of NET1 mRNA in MCF7 cells with and without TGF-β. n=50 cells from two independent experiments. (F) Relative NET1 binding to nucleotide-free RhoA (RhoA G17A), as a measure of GEF activity, in MCF7 cells with and without TGF-β, and treated with the indicated PMOs. n=3. In superplots, data points from individual replicates are color coded, and large, outlined color dots indicate the mean of each replicate. Error bars: SEM. p-values: *<0.05, **<0.01, ***<0.001, ****<0.0001 by one-way ANOVA. Scale bars: 15μm (A, B); 50μm (D).
To determine whether this alteration in NET1 function was sufficient to cause detectable effects in cell motility, we used cells stably expressing GFP-Lifeact and assessed the speed of protrusive or retracting dynamics of the cell periphery over 1 min intervals (Figure S12A). We further tracked individual cells over long-term time lapse imaging and measured the average cell migration speed (Figure 6C). In both assays, treatment with NET1 PMOs significantly reduced peripheral dynamics and overall cell migration speed, suggesting that peripheral NET1 mRNA localization is important for efficient cell movement.
NET1 mRNA location is altered by physiological stimuli
The potential of modulating RhoA activity and cell migration through altering NET1 mRNA localization, raised the possibility that this mechanism could be utilized under physiological circumstances. We focused on epithelial-mesenchymal transition (EMT) induced by TGF-β, which requires RhoA signaling37 We utilized MCF7 breast epithelial cells, which are known to undergo EMT upon treatment with TGF-β. Indeed, TGF-β induces dissolution and scattering of tightly adherent MCF7 colonies (Figure 6D). Interestingly, TGF-β also significantly increased the peripheral localization of the NET1 mRNA (Figure 6E) as well as the amount of NET1 that can bind to nucleotide-free RhoA, indicative of elevated GEF activity (Figure 6F). Importantly, antisense NET1 PMOs that prevent peripheral NET1 mRNA localization also suppress the TGF-β-induced NET1 GEF activity. Therefore, NET1 mRNA distribution is altered by a physiological stimulus and is responsible for functional changes linked to RhoA signaling, supporting the biological relevance of the described mechanism.
NET1 mRNA localization robustly affects NET1 protein functions
To determine the extent to which this mRNA location-dependent mechanism can control NET1 function, we compared the phenotypic effects observed upon forcing a perinuclear NET1 mRNA distribution to the effects observed upon an almost complete NET1 protein knockdown (Figure S12B). Strikingly, all measured parameters (RhoA activity, focal adhesion size, peripheral dynamics and migration speed) showed a reduction of similar magnitude when NET1 was produced perinuclearly as when its expression was knocked down (Figure 6A–C and S12A). Therefore, even though perinuclearly translated NET1 is expressed at normal levels (Figure 1E and S2E,F), its efficient nuclear import strongly suppresses its ability to function in the cytoplasm, phenocopying acute NET1 protein loss. We conclude that altering NET1 mRNA distribution does not simply provide a subtle regulatory mechanism but can robustly influence partner selection and consequently the targeting of the protein and its function between its two subcellular destinations.
Discussion
Numerous proteins are targeted to multiple subcellular destinations where they can exert distinct moonlighting functions or promote diverse functional outcomes11,38. The mechanisms specifying the balance between distinct functions or destinations are not always clear but are largely thought to involve post-translational events. For example, the distribution of NET1 between the nucleus and the cytoplasm can be controlled post-translationally through protein modification15,16. We show here that a distinct level of control operates during, or early after, protein synthesis and plays a major role in determining the functional potential of the protein. Specifically, the location of the mRNA in the cytoplasm and the rate of its translation act in combination to specify the targeting, and thereby the functional potential, of the encoded NET1 protein. NET1 targeting between the nucleus and cytoplasm is determined through antagonistic binding of importins or CASK to the NLS and PH domain respectively. Our model (Figure 7) suggests that partner selection is determined by kinetic competition and is influenced either by the rate of appearance of the interacting domains or by presenting the newly synthesized protein into different local environments that likely differ in the concentration of binding partners. In this way, NET1 mRNA translation at the periphery disfavors NLS-importin interaction and instead promotes PH domain-CASK binding, thus retaining the protein in the cytoplasm. This balance can be shifted either by moving the mRNA to a perinuclear location, where the NLSs are more efficiently recognized by the higher importin concentration, or by slowing down translation thereby allowing more time for NLS-importin interaction.
Figure 7: Proposed model.
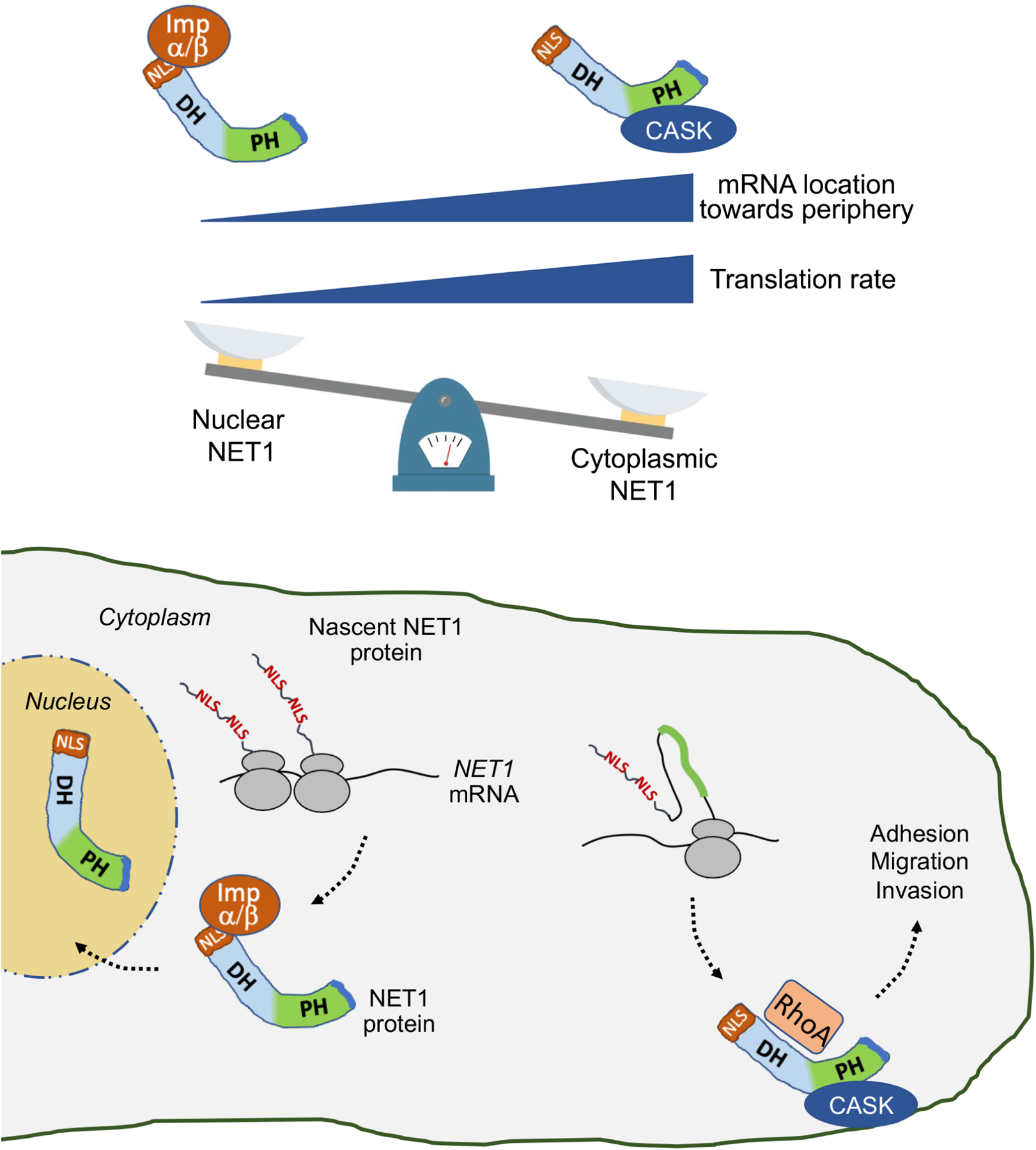
Binding of NET1 to importins or to CASK, and NET1 targeting to the nucleus or cytoplasm, is determined by the location of the NET1 mRNA and the rate of its translation. Perinuclear mRNA and/or slower translation rate favor interaction of the N-terminal NLSs with import receptors and targeting to the nucleus. Peripheral mRNA and/or faster translation rate favor CASK binding through the PH domain to competitively retain NET1 in the cytoplasm. Cytoplasmic NET1 can activate RhoA and promote cell adhesion and migration.
Our data indicate that, in the case of NET1A, a choice for eventual protein targeting occurs during or shortly after translation. It is well appreciated that protein folding and association with partners can occur as soon as the nascent protein chain emerges from the ribosome and such co-translational events can ensure proper protein folding and prevent promiscuous and non-productive interactions39. Co-translational events can be influenced by the rate of translation and by ribosome pausing to ensure proper complex assembly40. They have also been proposed to play a role towards directing interactions of proteins when multiple potential functional options are possible41. It is quite intuitive that the potential co-translational associations that a nascent chain can engage in will be shaped by the immediate microenvironment in which protein synthesis takes place. Supporting this idea, local co-translational interactions, determined by mRNA location, have been shown to direct the functional potential of signaling proteins22. The evidence presented here lends experimental support to the idea that co-translational events serve not only a quality control role, but in combination with mRNA targeting can be used as a selection mechanism for proteins that engage in multiple potential functional complexes.
Such a partner selection mechanism based on mRNA targeting could be widely utilized. A large fraction of cellular mRNAs are not randomly distributed but adopt complex and diverse distribution patterns in the cytoplasm. While the functional roles served by these targeting events have been studied in only a handful of cases, large-scale correlations offer some interesting insights. Proteins expressed by mRNAs localized to distal locations tend to have a larger number of interaction partners compared to proteins expressed from non-targeted mRNAs42. Exclusively mitochondrial proteins are more likely to be expressed from mRNAs that are targeted to mitochondria compared to mitochondrial proteins that exhibit dual localization43. We suggest that mRNA localization and the kinetics of co-translational events, driven by translation rate and the local concentrations of binding partners, can provide a widely-used mechanism for partner selection and thus for specifying protein distribution and function.
Limitations of the study
Our model would predict that at least to some extent the interaction of the NLSs with importin occurs co-translationally. We have tried to experimentally address this by immunoprecipitating importin and assessing whether we can detect a translation-dependent association with the NET1 mRNA. Unfortunately, technical limitations (i.e. high background signal) has prevented a conclusive demonstration of this prediction.
One line of evidence supporting our conclusion that coding sequence changes (i.e. introduction of suboptimal codons or mutations of putative stall sites) predictably change the translation elongation rate relies on steady state ribosome occupancy measurements (through SunTag-scFv-GFP fluorescence). We believe this provides a reasonable reflection of elongation rates, given that the translation initiation context and all regulatory sequences (5’- and 3’-UTRs) are identical between the constructs being compared. Nevertheless, a more direct demonstration could be provided by measuring ribosome run-off rates (SunTag-scFv-GFP fluorescence decay over time) upon blocking translation at the initiation stage with inhibitors such as harringtonine or lactimidomycin. Such an experiment, however, would necessitate observing individual mRNA molecules for several minutes, which we currently cannot achieve due to RNAs eventually moving out of the focal plane.
The presence of disome peaks upstream of high monosome density sites is consistent with ribosome queuing as a result of stalling events. We note, however, that the disome sequencing depth we could achieve is relatively low. Further work would be needed to determine the extent of formation of disomes, as well as whether they represent ribosome collision events that could mediate additional responses.
Finally, our data cannot currently discern whether mRNA location and translation rate are independently modulated or whether they are coupled. Determining whether there are differences in translation elongation rates at different cellular locations and the underlying mechanisms will be an intriguing area for future investigation.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stavroula Mili (voula.mili@nih.gov).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
All Ribo-seq, Disome-seq and Ribosome profiling data generated as a part of this study are deposited at GEO (GSE207146, GSE158044, GSE133393). All imaging data have been deposited to Mendeley Data (DOI: 10.17632/f4mr55trf6.1). All data are publicly available as of the date of publication. Accession numbers and DOI are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-GFP | ThermoFisher | Cat#A-11122 |
| rabbit anti-NET1 | Abcam | Cat#ab 113202 |
| rabbit anti-Importin beta 1 | Cell Signaling Technology | Cat#60769 |
| mouse anti-CASK | Santa Cruz | Cat#sc-13158 |
| rabbit anti-CASK | Invitrogen | Cat#PA1544 |
| Mouse anti-tubulin | Sigma | Cat#T6199 |
| rabbit anti-GAPDH | Cell Signaling Technology | Cat#2118L |
| IR Dye 680RD donkey anti-mouse | Li-Cor | Cat#926–68072 |
| IR Dye 800CW donkey anti-rabbit | Li-Cor | Cat#926–32213 |
| mouse anti-paxillin | BD Transduction Laboratories | Cat#610619 |
| Alexa48 8-Phalloidin | Invitrogen | Cat#A12379 |
| anti-mouse Alexa647 | Invitrogen | Cat#A28181 |
| mouse anti-FLAG M2 | Sigma | Cat#F1804 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine RNAiMAX | ThermoFisher | Cat#13778–150 |
| PolyJet In Vitro DNA Transfection Reagent | SignaGen | Cat# SL100688 |
| Leibovitz L-15 | Gibco | Cat#11415064 |
| Penicillin-Streptomycin | Gibco | Cat#15070063 |
| DMEM, high glucose, pyruvate | Invitrogen | Cat#11995073 |
| Trypsin-EDTA (0.05%) | Gibco | Cat#25300054 |
| Blasticidin | ThermoFisher | Cat#A1113903 |
| Geneticin | Gibco | Cat#10131035 |
| Doxycycline Hydrochloride | ThermoFisher | Cat#BP26535 |
| Puromycin Dihydrochloride | Gibco | Cat#A1113803 |
| Recombinant Human TGF-B1 | Peprotech | Cat#100–21 |
| Endoporter | Genetools LLC | Cat#OT-EP-PEG-1 |
| Collagen IV | Sigma | Cat#C5533–5MG |
| Fibronectin bovine plasma | Sigma | Cat#F1141 |
| GFP-Trap magnetic agarose beads | Chromotek | Cat#gtma-10 |
| Paraformaldehyde | Electron Microscope Sciences | Cat#15710 |
| Intercept Blocking Buffer | Li-Cor | Cat#927–60001 |
| ProLong Gold Antifade reagent with DAPI | Invitrogen | Cat#P36931 |
| Triton X-100 | Sigma | Cat#93443–100ML |
| Halt Protease and Phosphatase Inhibitor Cocktail | ThermoFisher | Cat#78444 |
| HCS Green CellMask Stain | ThermoFisher | Cat# H32714 |
| GST-KPNA1 | Abnova | Cat#H00003836-P01 |
| GST-KPNA2 | Abnova | Cat#H00003838-P01 |
| GST-KPNA4 | Abnova | Cat#H00003840-P01 |
| JFX554 HaloTag ligand | Janelia Research Campus | N/A |
| DRAQ5 | ThermoFisher | Cat#62254 |
| RNase I | ThemorFisher | Cat#AM2295 |
| Superase-In | ThermoFisher | Cat#AM2694 |
| TRIzol | ThermoFisher | Cat#15596026 |
| Critical Commercial Assays | ||
| DuoLink In Situ Red PLA Kit | Sigma | Cat#DUO92008 |
| ViewRNA ISH Cell Assay Kit | ThermoFisher | Cat#QVC0001 |
| Quant-it RiboGreen RNA Assay Kit | ThermoFisher | Cat#R11490 |
| Qiagen FastSelect Kit | Qiagen | Cat#334386 |
| High Sensitivity DNA Kit | Agilent | Cat#5067–4626 |
| Deposited Data | ||
| Ribo-seq and Disome-seq-HEK293 cells | This study | GEO: GSE207146 |
| Ribosome profiling-A549 cells | Karasik et al, 202144 | GEO: GSE158044 |
| Ribosome profiling-HEK293 cells | Han et al, 20 2045 | GEO: GSE133393 |
| Imaging data | This study | http://dx.doi.org/10.17632/f4mr55trf6.1 |
| Experimental Models: Cell Lines | ||
| MDA-MB-231 | ATCC | Cat#CRM-HTB-26 |
| MCF7 | ATCC | Cat#HTB-22 |
| Flp-In T-REx HEK293 | Gibco | Cat#R780–07 |
| NIH/3T3 | ATCC | Cat#CRL-1658 |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A_WT UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A WT UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A WT UTR + pLV-PGK-hCA SK-FLAG | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A_WT UTR + pLV-PGK-hCA SK-FLAG-mCherry | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A_AGA UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A AGA UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A AGA UTR + pLV-PGK-hCASK-FLAG | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A_ AGA UTR + pLV-PGK-hCASK-FLAG-mCherry | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1 A_slow2_WT UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A slow2 WT UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1 A_slow2_AGA UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A slow2 AGA UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A_stallmutant(fast)_WT UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A stallmutant(fast) WT UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A_stallmutant(fast)_ AGA UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A stallmutant(fast) AGA UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A C-term NLS_WT UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A C-term NLS WT UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A C-term NLS_AGA UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A C-term NLS AGA UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-mutantNLS_WT UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-mutantNLS WT UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-ADH_hNET1 UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-ADH WT UTR + pCDH-CMV-hKPNBl | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-APH_WT UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-APH WT UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-APDZ_WT UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-APDZ WT UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-CMV-mTFP 1 | This study | N/A |
| MDA-MB-231 + pIND20-RhoA-WT-biosensor | This study | N/A |
| MDA-MB-231 + pIND20-RhoA-Q63L-biosensor | This study | N/A |
| MDA-MB-231 + pIND20-RhoA-T19N-biosensor | This study | N/A |
| NIH/3T3 + NLS-HA-stdMCP-stdHalo + scFv-GCN4-sfGFP + pIND20-hNet1A 5UTR-24xGCN4-hNet1A CDS-24xMS2v7_hNet1 3UTR | This study | N/A |
| NIH/3T3 + NLS-HA-stdMCP-stdHalo + scFv-GCN4-sfGFP + pIND20-hNet1A 5UTR-24xGCN4-hNet1A SLOW2 CDS-24xMS2v7_hNet1 3UTR | This study | N/A |
| NIH/3T3 + NLS-HA-stdMCP-stdHalo + scFv-GCN4-sfGFP + pIND20-hNet1A 5UTR-24xGCN4-hNet1A STALLMUT(fast) CDS-24xMS2v7 hNet1 3UTR | This study | N/A |
| Oligonucleotides | ||
| Morpholino oligonucleotides (PMOs) | Table S2 | GeneTools LLC |
| human NET1 FISH probes | ThermoFisher | Cat# VA6–3169338 |
| human RHOA FISH probes | ThermoFisher | Cat#VA6–14829-01 |
| GFP FISH probes | ThermoFisher | Cat#VF6–16198 |
| AllStars siRNA Negative Control | Qiagen | Cat#1027281 |
| human siNET1 #4 – 5’-ACGGAAAGAGACTTTGGTGTA-3’ | Qiagen | Cat#SI00082040 |
| human siCASK #5 – 5’-AACCAATGGGAATCACTTTAA-3’ | Qiagen | Cat#SI02223368 |
| human siCASK #10 – 5’-CAGACCGGTTTGCGTACCCTA-3’ | Qiagen | Cat#SI04437720 |
| Recombinant DNA | ||
| KPNB1 (NM_002265) Human Tagged ORF Clone | Origene | Cat#RC200659 |
| pInducer20 | Addgene | Cat#44012 |
| pET263-pUC57 24xMS2V7 | Addgene | Cat#140705 |
| pLentiRhoA2G | Addgene | Cat#40179 |
| pcDNA4TO-24xGCN4_v4-kif18b-24xPP7 | Addgene | Cat#74928 |
| phage UbiC NLS HA stdMCP stdHalo | Addgene | Cat#104999 |
| pHR-scFv-GCN4-sfGFP-GB1-NLS-dWPRE | Addgene | Cat#60906 |
| pCDH-CMV-EF1-MCS-Puro | System Biosciences | Cat#CD510B-1 |
| pCDH-CMV-hKPNB1 | This study | N/A |
| pCDH-PGK-GFP-hNET1A-ADH WT UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A-APH WT UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A-APDZ WT UTR | This study | N/A |
| pCDH-CMV-mTFP1 | This study | N/A |
| pIND20-RhoA-WT-biosensor | This study | N/A |
| pIND20-RhoA-Q63 L-biosensor | This study | N/A |
| pIND20-RhoA-T19N-biosensor | This study | N/A |
| pCDH-PGK-GFP-hNET1A_WT UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A_ AGA UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A-slow2 WT UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A-slow2 AGA UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A_stallmutant(fast)_WT UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A_stallmutant(fast)_ AGA UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A C-term NLS / WT UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A C-term NLS_AGA UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A-mutantNLS hNET1 UTR | This study | N/A |
| pIND20-hNet1A 5UTR-24xGCN4-hNet1A CDS-24xMS2v7_hNet1 3UTR | This study | N/A |
| pIND20-hNet1A 5UTR-24xGCN4-hNet1A CDS-24xMS2v7 hNeti A(GA) 3UTR | This study | N/A |
| pIND20-hNet1A 5UTR-24xGCN4-hNet1A SLOW2 CDS-24xMS2v7 hNet1 3UTR | This study | N/A |
| pIND20-hNet1A 5UTR-24xGCN4-hNet1A STALLMUT(fast) CDS-24xMS2v7_hNet1 3UTR | This study | N/A |
| pLV-PGK-hCASK-FLAG-mCherry | VectorBuilder | N/A |
| pLV-PGK-hCASK-FLAG | VectorBuilder | N/A |
| Software and Algorithms | ||
| Fiji | Schindelin et al., 201246 | https://fiji.sc |
| Imaris | Oxford Instruments | https://imaris.oxinst.com |
| RibofootPrinter | Guydosh, 202147 | https://github.com/guydoshlab/ribofootPrinter |
| Matlab | MathWorks | https://www.mathworks.com/products/matlab.html |
| Prism | GraphPad | https://www.graphpad.com |
| DiPer Macro | Gorelik and Gautreau, 201448 | N/A |
| ADAPT ImageJ plugin | Barry et al., 201549 | N/A |
| LAS X | Leica | https://www.leica.com |
| NIS-Elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| Other | ||
| 35mm glass-bottom dishes | MatTek Corporation | Cat# P35G-1.5–14-C |
| TetraSpeck™ Fluorescent microspheres sampler kit | Fisher Scientific | Cat#T7284 |
| Coverslip, #1.5 thickness | Thomas Scientific | Cat#1217N81 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell culture
MDA-MB-231 cells were obtained from ATCC (cat # HTB-26) and cultured in Leibovitz’s L-15 medium (Gibco) supplemented with 10% FBS and 1% Penicillin-Streptomycin at 37°C in atmospheric air in a humidified cell culture incubator. MCF7 (obtained from ATCC; cat# HTB-22) and 293 TREx cells (ThermoFisher, cat# R780–07) were cultured in Dulbecco’s modified eagle medium (DMEM; Gibco) supplemented with 10% FBS and 1% Penicillin-Streptomycin at 37°C and 5% CO2. NIH/3T3 mouse fibroblast cells were cultured in DMEM supplemented with 10% calf serum, sodium pyruvate and 1% Penicillin-Streptomycin at 37°C and 5% CO2. Cells were passaged by trypsinization using 0.05% trypsin (Gibco). Cells used in this study have tested negative for mycoplasma.
Cell lines
To generate stable cell lines expressing GFP-tagged NET1A and various mutants, MDA-MB-231 cells were infected with the corresponding lentiviruses and selected with 6μg/mL blasticidin (ThermoFisher). Cells were sorted by FACS to select for a low level of expression.
To generate stable cell lines expressing the doxycycline-inducible RhoA FRET biosensor, MDA-MB-231 cells were infected with the corresponding lentivirus and selected with 0.6mg/mL geneticin (ThermoFisher). To prevent leaky expression of the biosensor, these cells were maintained in media supplemented with 10% Tet-system approved FBS (ThermoFisher). To induce RhoA FRET biosensor expression, cells were treated overnight with 1μg/mL doxycycline.
To generate stable cell lines expressing FLAG-KPNB1, LifeAct-GFP, or Cherry-NLS, MDA-MB-231 cells were infected with the corresponding lentiviruses and selected with 1μg/mL puromycin (ThermoFisher) or fluorescent cells were sorted by FACS.
To generate stable cell lines expressing translation reporters, NIH/3T3 cells were sequentially infected with lentiviruses expressing stdMCP-stdHalo (Addgene #104999, modified to remove the Kozak sequence and ATG initiating codon from the sequence of the first HaloTag) and scFv-GFP (derived from Addgene #60906 after introduction of a stop codon before the NLS sequence). A uniformly expressing cell population was selected by fluorescence activated cell sorting. Reporter constructs expressing NET1A variants (WT, slow or fast) were then introduced through lentiviral infection, and stably expressing lines were selected with Geneticin (Thermo Fisher Scientific). Expression of the reporters was induced by addition of 1 μg/ml doxycycline approximately 3–4 hrs before imaging.
For protein synthesis inhibitor studies, cells were incubated with either cycloheximide (100μg/mL) or puromycin (100μg/mL) for 20 min at 37°C. For TGF-β treatment, cells were plated in 1% FBS-containing DMEM for 72 hrs with or without 4ng/mL human recombinant TGF-β (Peprotech; cat #100–21).
Plasmid constructs and lentivirus production
To express N-terminally GFP-tagged NET1A with different UTRs, the coding sequence of EGFP was ligated to the coding sequence of human NET1A. This fusion was then ligated to fragments corresponding to the 3’UTR of human NET1 (NM_001047160.3; wt UTR) or the 3’UTR of NET1 that is missing 78 nucleotides (921–999) (ΔGA UTR). These fragments were then cloned into the pCDH-PGK lentiviral vector. To generate NET1A deletion mutants, the coding sequence of human NET1A, lacking the DH or PH domain, or the PDZ motif, was synthesized as a gBlock gene fragment (Integrated DNA Technologies). Each fragment was ligated to an N-terminal GFP-tag and to the wt NET1 3’UTR into the pCDH-PGK lentiviral vector. To generate the NET1A with mutated NLSs or the slow or fast mutants, gBlock gene fragments (Integrated DNA Technologies) carrying the corresponding mutations were synthesized. To generate NET1A with C-terminal NLSs, the N-terminal NET1A region encompassing the NLSs was PCR amplified from the gBlock fragment and ligated in frame at the C-terminus of a fragment containing the DH-PH domains and PDZ motif of NET1A. Each fragment was ligated to an N-terminal GFP-tag and to the wt or ΔGA NET1 3’UTR into the pCDH-PGK lentiviral vector. See Supplementary Table S1 for exact sequence information of fragments used.
To generate the NET1 translation reporter cell lines the 5’UTR sequence of human NET1A was synthesized as a gBlock gene fragment (Integrated DNA Technologies). A fragment containing 24xGCN4_v4 repeats was derived from Addgene plasmid #74928. Those were cloned upstream of gBlock gene fragments containing the human wild type NET1A coding sequence or slow and fast mutant variants. A fragment with 24xMS2v7 repeats was obtained from Addgene plasmid #140705 and cloned downstream of the coding sequence, followed by the wild type human NET1 3’UTR. Fragments were cloned into the pInducer 20 lentiviral vector (Addgene plasmid #44012) for doxycycline-inducible expression.
To generate the RhoA FRET biosensor, the RhoA biosensor fragment was PCR amplified from the pLentiRhoA2G plasmid (Addgene #40179) to add BamHI and XhoI sites at the ends. Site directed mutagenesis was used to introduce either a T19N (dominant negative) or Q63L (constitutively active) mutation. The wild type or mutant fragments were cloned into the pENTR-1A vector and a recombination reaction using the Gateway LR Clonase II enzyme mix (ThermoFisher) was performed to move the RhoA FRET sensor fragment into the doxycycline-inducible pInducer20 vector (Addgene plasmid # 44012).
To express FLAG-tagged importin β (KPNB1), the KPNB1 coding sequence was PCR amplified from the KPNB1 ORF clone plasmid (Origene, cat# RC200659) and XbaI and BamHI restriction sites were added to the ends of the fragment. This fragment was cloned into the pCDH-CMV-EF1-MCS-Puro lentiviral vector.
To express FLAG-tagged CASK, a lentiviral plasmid that contained the human CASK CDS (NM_001367721.1) with an N-terminal FLAG tag driven by the human PGK promoter with a puromycin resistance gene driven by the CMV promoter was purchased from VectorBuilder. The mCherry-FLAG-tagged CASK construct (VectorBuilder) has mCherry at the C-terminus of the FLAG-CASK CDS.
Lentiviruses were produced in HEK293T cells. The cells were transfected with the lentiviral vectors together with the pMD2.G and psPAX2 packaging plasmids using PolyJet In Vitro DNA Transfection Reagent (SignaGen) for 48 hrs. Harvested virus was precipitated with polyethylene glycol overnight at 4°C.
METHOD DETAILS
Morpholino and siRNA transfection
For knockdown experiments, 40 pmoles of siRNA were transfected into cells by Lipofectamine RNAiMAX (ThermoFisher, cat# 13778–150) according to the manufacturer’s protocol. Cells were assayed 72 hours post-transfection. The following siRNAs were used: AllStars negative control (Qiagen cat# 1027281) and siNET1 #4 (Qiagen cat# SI00082040; target sequence: 5’-ACGGAAAGAGACTTTGGTGTA-3’), siCASK #5 (Qiagen cat# SI02223368; target sequence: 5’-AACCAATGGGAATCACTTTAA-3’) and siCASK #10 (Qiagen cat# SI04437720; target sequence: 5’-CAGACCGGTTTGCGTACCCTA-3’).
Morpholino oligos (Gene Tools, LLC) used are listed in Supplementary Table S2. Morpholinos were transfected using Endoporter (Gene Tools, LLC). Cells were assayed 72 hours post-transfection.
Western blot
The following primary antibodies were used: rabbit anti-GFP (1:1000; ThermoFisher cat# A-11122), rabbit anti-NET1 (1:1000, Abcam cat# ab113202), rabbit anti-importin β1 (1:1000, Cell Signaling Technology cat# 60769), mouse anti-CASK (1:1000, Santa Cruz cat# sc-13158), rabbit anti-CASK (1:1000; Invitrogen cat# PA1544), mouse anti-tubulin (1:2000; Sigma cat# T6199), rabbit anti-GAPDH (1:2000; Cell Signaling cat# 2118L). Anti-rabbit and anti-mouse secondary antibodies from Li-Cor were used at 1:10,000. Membranes were scanned using an Odyssey fluorescent scanner (Li-Cor) and bands were quantified using ImageStudioLite (Li-Cor).
RNA Fluorescence in situ hybridization (FISH)
MDA-MB-231 and MCF7 cells were plated on collagen IV-coated (10μg/mL) glass coverslips for 2 hours and fixed with 4% paraformaldehyde for 20 mins at room temperature. FISH was performed using the ViewRNA ISH Cell Assay kit (ThermoFisher, cat# QVC0001) according to the manufacturer’s protocol. The following probes were used in this study: human NET1 (cat # VA6–3169338), human RHOA (cat # VA6–14829-01) and GFP (cat # VF6–16198). Green cell mask (ThermoFisher) was used to identify the cell border and samples were mounted with ProLong Gold antifade with DAPI.
Focal adhesion immunofluorescence
MDA-MB-231 cells plated on collagen IV-coated (10μg/mL) coverslips were fixed with 4% paraformaldehyde in PBS for 15 mins at room temperature then permeabilized with 0.2% Triton X-100 in PBS for 5 mins at room temperature. Coverslips were blocked for 1 hr in 5% goat serum in PBS for 1 hr at room temperature. Mouse anti-paxillin primary antibody was then added at 1:300 for 1.5 hrs. After washing, Alexa Fluor-488 Phalloidin (1:800; Invitrogen cat# A12379) and anti-mouse Alexa Fluor-647 secondary (1:500; Invitrogen) were added for 1 hr. Samples were mounted in ProLong Antifade mounting media with DAPI.
Co-Immunoprecipitation
Cells were lysed in an ice-cold buffer containing 50mM Tris pH 7.4, 1% NP-40, 150mM NaCl, 10mM MgCl2, 10% glycerol and Halt protease and phosphatase inhibitor cocktail (ThermoFisher). Lysates were cleared by centrifugation at 4°C and added to GFP-Trap Magnetic agarose beads (Chromotek, cat #gtma-10) for 1 hr at 4°C with rotation. After washing with lysis buffer, immobilized complexes were eluted with Laemmli’s buffer by boiling for 5 minutes. The supernatant was separated from the beads by a magnet and then analyzed by SDS-PAGE and immunoblotting.
For the endogenous CASK co-immunoprecipitation experiments, cells were lysed in an ice-cold buffer containing 50mM Tris pH 7.4, 1% TritonX-100, 75mM NaCl, 10mM MgCl2, 10% glycerol and Halt protease and phosphatase inhibitor cocktail (ThermoFisher). After 5 minutes of lysis, lysates were sonicated for 10 seconds on intensity setting #2 using a sonicator (Misonix Inc. Sonicator XL). Lysates were gently spun for 5 mins at 4,000 rpm. The supernatant was then incubated as described above for the other co-immunoprecipitation experiments.
Proximity Ligation Assay (PLA)
Cells plated on collagen IV-coated (10μg/mL) coverslips were washed in PBS and fixed with 4% paraformaldehyde in PBS for 15 min at room temperature then permeabilized in 0.2% Triton X-100 for 5 min at room temperature. The DuoLink In Situ Red Kit (Sigma, cat# DUO92008) was used for PLA and the manufacturer’s protocol was followed. Briefly, cells were blocked in the provided blocking buffer for 1 hr at 37°C in a humidified chamber. Primary antibodies were diluted in the provided DuoLink antibody diluent solution and incubated on the cells for 1.5 hrs at room temperature in a humidified chamber. The following primary antibodies were used: rabbit anti-GFP (1:200; ThermoFisher cat# A11122) and mouse anti-FLAG M2 (1:800; Sigma cat# F1804). After washing, the PLA probes supplied with the kit were used at a 1:10 dilution in the antibody diluent buffer provided and incubated for 1 hr at 37°C. The ligation step was performed for 30 min at 37°C then amplification for 100 min at 37°C. After these steps, the coverslips were washed and fixed with 4% paraformaldehyde in PBS for 10 min at room temperature then stained with Alexa Fluor 488 Phalloidin (1:500; Invitrogen cat# A12379) in blocking buffer for 20 min at room temperature and mounted in DuoLink PLA mounting medium with DAPI. Slides were kept at 4°C in the dark until imaging the next day.
Active NET1 pull down assay
MCF7 cells transfected with control or NET1-targeting morpholinos, with or without TGF-β, were rinsed with ice cold HBS buffer (20mM HEPES pH 7.5, 150mM NaCl) and lysed with ice cold HBS lysis buffer (20mM HEPES pH 7.5, 150mM NaCl, 5mM MgCl2, 1% TritonX-100) with Halt protease and phosphatase inhibitor for 10 mins then centrifuged at 4°C to clear the lysate. Cleared lysate was added to a GST-RhoA(G17A) bead slurry for 1 hr at 4°C with rotation. The beads were washed x5 with cold HBS lysis buffer and proteins were eluted with Laemmli’s buffer by boiling for 5 minutes before running on an SDS-PAGE gel.
In vitro pull down assay
Cells stably expressing GFP-NET1A variants were rinsed with ice cold PBS and lysed in 1mL of HEGMN buffer (25mM HEPES, 100mM KCl, 12.5mM MgCl2, 0.1mM EDTA, 10% glycerol, 0.1% NP-40) supplemented with Halt phosphatase and protease inhibitor on ice for 5 mins. For pull down, 100uL of Pierce glutathione magnetic agarose beads (ThermoFisher, cat#78601) were incubated with the lysates and with 1.0μg, or with the indicated amounts, of GST-KPNA recombinant human protein (Abnova, GST-KPNA1 (cat#H00003836-P01), GST-KPNA2 (cat#H00003838-P01), GST-KPNA4 (cat#H00003840-P01)) for 1 hour with rotation at room temperature. Beads were washed x5 with lysis buffer, proteins were eluted with 35μl reducing buffer, boiled for 5 mins, and analyzed by Western blot.
Microscopy and Image Analysis
Focal adhesion size, RNA FISH and PLA experiments were imaged using a Leica SP8 confocal microscope with an HC PL APO 63x oil immersion objective. Z-stacks through the cell volume were obtained and maximum intensity projections were used for all analysis.
For FISH images, calculation of the PDI index was performed using a previously published custom Matlab script24.
For GFP-NET1A/FLAG-importin PLA experiments, an ImageJ script based on the Analyze Particles plugin was used to calculate the number of PLA dots inside each cell. Equal thresholding among all images was used during analysis. For GFP-NET1A/FLAG-CASK PLA, a series of custom ImageJ macros were used to define an inner boundary 3μm from the cell edge and to obtain PLA signal intensity within this 3μm peripheral zone, after background subtraction.
For live cell imaging of GFP-NET1A nuclear and cytoplasmic ratios, cells were plated on glassbottom dishes and DRAQ5 (ThermoFisher) was added to the cell culture media at a concentration of 1μM for 30 minutes prior to imaging. Cells were placed inside an incubation chamber mounted on the microscope stage to keep cells at 37°C. Z-stack images through the cell volume were taken and imported into Imaris (Bitplane AG) where DRAQ5 staining was used to determine the nuclear volume. Surfaces were created within the software to segment out the nuclear region from the whole cell and the GFP intensities were calculated for both regions.
For cell migration experiments, an Olympus IX81 inverted microscope was used with an incubation chamber around the microscope to keep the cells at 37°C for the duration of the experiment. To facilitate tracking, cells expressing Cherry-NLS were used. A 10x objective was used for imaging and time lapse images were taken at 10 min intervals for a duration of 8 hours. Images were analyzed in ImageJ software using the Manual Tracking plugin. The raw tracking coordinates were imported into the DiPer macro48 for cell migration speed analysis in Microsoft Excel.
To analyze the size of focal adhesions, images were converted to binary masks in ImageJ by thresholding and the analyze particles plugin was used to measure focal adhesion size. Equal thresholding among all images were used during analysis.
To analyze cellular membrane dynamics, cells expressing LifeAct-GFP were plated on collagen IV-coated glass bottomed dishes and imaged on a heated stage incubator chamber on a Leica SP8 confocal microscope. Time lapse Z-stack images were taken throughout the whole Z-plane of the cell every 1 minute for 1 hr total. To measure the speed of membrane edge protrusion and retraction, the ADAPT ImageJ plugin was used49. The images were thresholded within the plugin until the background outside the cell was subtracted out and the outline of the cell remained. The curvature window setting was kept at the default of 10.
To measure RhoA FRET activity, cells expressing the biosensor were plated on collagen IV-coated glass bottomed dishes and imaged on a heated stage incubator chamber on a Leica SP8 confocal microscope. The LAS X Falcon FLIM module was used to analyze the data. To measure FRET efficiency in protrusive and non-protrusive areas, an ROI was made to include all the cell’s protrusions and compared to the FRET value of the donor-only expressing cells in a phasor plot. To measure FRET efficiency in non-protrusive areas, an ROI box was made along the cell membrane in an area without membrane protrusions. This FRET value was compared to the donoronly cell FRET value in a phasor plot.
Live imaging of cells expressing translation reporters was done using a Nikon Eclipse Ti2-E inverted microscope, equipped with a motorized stage, a Yokogawa CSU-X1 spinning disk confocal scanner unit, and operated using NIS-elements software. Acquisitions were performed using an Apochromat TIRF 100× oil immersion objective (N.A. 1.49, W.D. 0.12 mm, F.O.V. 22 mm) and Hamamatsu ORCA-Fusion BT Gen III back-illuminated sCMOS cameras. Constant 37°C temperature and 5% CO2 were maintained using a Tokai Hit incubation system. To label MCP-Halo proteins, cells were supplemented with 200nM of JFX554 HaloTag ligand, obtained from Janelia Research Campus for 2 hrs. The medium was then replaced, and 1 μg/ml doxycycline was added to induce expression of reporter mRNAs for 3 hrs. Cells were plated on fibronectin (5 mg/mL)-coated 35 mm glass bottom dishes for ~1 hr, and samples were simultaneously excited using a 488 nm and 561nm laser lines. Emissions were recorded on two separate cameras, which were aligned prior to every imaging session using fluorescent TetraSpeck beads (0.1, 0.5 and 4 μm in diameter). Individual mRNA and translation particles were identified using the TrackMate plugin in ImageJ/Fiji, and a custom Matlab script was used to assign translation spots to individual mRNA particles (within a 400nm radius) and correct for cell-specific background.
Ribo-seq and Disome-seq experiments
HEK293 cells grown to a confluency of ~80% in a T-75 flask were washed with ice-cold DPBS and flask frozen in liquid nitrogen. 400 μL of lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 100 μg/mL cycloheximide, 1% Triton X-100, 25 U/mL Turbo DNase) was added to the frozen cells and the cells in lysis buffer were thawed on ice. Cells were scraped from the flasks and lysates were transferred to Eppendorf tubes, followed by incubation on ice for 10 min. After passing the lysates through 25 G needles ten times, lysates were clarified by centrifugation for 10 minutes at 21,000 g at 4°C and supernatants were kept for library preparation. The total RNA concentration in the lysates were measured by using Quant-it RiboGreen RNA Assay Kit (ThermoFisher, cat#R11490) and the lysate containing 30 μg of RNA was digested with 60 U of RNase I (ThermoFisher, cat#AM2295) for 1 hour at room temperature to capture both monosome and disome footprints as previously described50. RNase I digestion was stopped by the addition of 10 μL Superase-In (ThermoFisher, cat#AM2694) and lysates were centrifuged for at 21,000 g at 4°C. Supernatants were loaded onto a 1 M sucrose cushion and then pelleted by ultracentrifugation by using a TLA100.3 rotor (Beckman Coulter, cat#349490) for 1 hour at 100,000 rpm at 4°C. The ribosome pellet was resuspended in 700 μL TRIzol (ThermoFisher, cat#15596026) and RNA was extracted as recommended by the manufacturer. Concentration of the RNA was determined by a NanoDrop UV spectrophotometer and 2 μg of RNA was loaded onto 15% Criterion-TBE-Urea polyacrylamide gel (Biorad, cat#3450093) for size selection. For Ribo-seq and Disome-seq libraries, gel regions corresponding to 25–34 nucleotides and 50–70 nucleotides, respectively, were cut. RNA was eluted overnight from the gel slice (0.3 M sodium acetate, 1 mM EDTA and 0.25 % SDS) and isopropanol precipitated the next day. The extracted footprints were dephosphorylated and pre-adenylated linkers were ligated as described previously51. rRNAs were removed by Qiagen FastSelect kit (cat#334386) during reverse transcription step as described by the manufacturer. Quality of the final libraries were assessed by Bioanalyzer 2100 (Agilent, cat#G2939BA) using the High Sensitivity DNA kit (Agilent, cat#5067–4626). Sequencing was performed on a NovaSeq sequencer at the NIH/NHLBI DNA Sequencing and Genomics Core.
Processing of Ribo-seq/Disome-seq data
Sequencing data was processed as described previously44. Briefly, the fastq files of Ribo-seq and Disome-seq data were trimmed to remove linkers and demultiplexed to isolate individual samples from pooled data using Cutadapt. Contaminating tRNAs and rRNAs were filtered out by Bowtie1 allowing two mismatches in -v mode and -y option to increase sensitivity of the alignment. The noncoding RNA fasta file was created by downloading rRNA sequences from the SILVA project (release 128)52 and tRNA sequences from GtRNAdb (H. sapiens release 16)53. After filtering out the non-coding RNAs, a custom python script was used to remove PCR duplicates by comparing the 7-nucleotide unique molecular identifiers (UMIs) in libraries. The UMIs were then trimmed by Cutadapt and reads were aligned to a transcriptome with bowtie1 using the parameters -v 1 (one mismatch allowed), -y and -p 12. The transcriptome, RefSeq Select+MANE (ncbiRefSeqSelect) was downloaded from UCSC on April 14, 2020 and used for alignment after removal of duplicates on alternative chromosomes. The footprint length distribution was obtained by using FastQC, version 0.11.7 (Babraham Bioinformatics). For Ribo-seq and Disome-seq data, only the reads between 25–34 and 58–65 nucleotides were analyzed, respectively.
We used custom python3 scripts (https://github.com/guydoshlab/ribofootPrinter)47 to create files with normalized mapped reads and used writegene2 script to visualize ribosome and disome reads mapped to the NET1 gene.
To compute the p-value that the 4 disome footprint reads (1 read = 3.2 rpm) in a region upstream of NET1 stall sites (Fig. 5A, magnified view) occur by chance, we used a simulation to ask how many reads would be expected in this region (CDS positions 870–925, inclusive) if the 16 reads in total that mapped to the gene were randomly distributed. We found that 4 or more reads fell into this region in <0.2% of the simulations.
Quantification and Statistical Analysis
All statistical analysis was performed using GraphPad Prism software using the appropriate statistical tests as indicated within the text and figure legends. Normally distributed datasets were analyzed using parametric statistical tests. Datasets deviating from a normal distribution were analyzed using non-parametric tests. Follow up tests were included, as appropriate, to adjust for multiple comparisons.
Supplementary Material
Table S1: Sequences of coding region fragments used for generation of plasmid constructs. Related to STAR Methods.
Table S2: Sequences of morpholino oligonucleotides (PMOs) used in the study. Related to STAR Methods.
Figure S1: Antisense oligonucleotide tiling across the 3’ UTR of the NET1 mRNA. Related to Figure 1.
Figure S2: Specificity and effect of altering NET1 mRNA localization on mRNA levels and protein expression. Related to Figure 1.
Figure S3: Characterization of GFP-NET1A mutant variants. Related to Figure 3.
Figure S4: Intracellular CASK distribution. Related to Figure 4.
Figure S5: NET1 mRNA localization in HEK293 cells depends on GA-rich sequences in the 3’UTR. Related to Figure 5.
Figure S6: Ribosome profiling data of the NET1 transcript. Related to Figure 5.
Figure S7: NET1A mutants with altered translation elongation rates. Related to Figure 5.
Figure S8: Sub-inhibitory concentrations of anisomycin increase interaction of NET1A with importin. Related to Figure 5.
Figure S9: Order of NLS appearance influences NET1-importin interaction. Related to Figure 5.
Figure S10: In vitro GFP-NET1A-importin α interaction. Related to Figure 5.
Figure S11: Validation of RhoA FRET biosensor. Related to Figure 6.
Figure S12: Peripheral NET1 mRNA localization is essential for NET1 function in membrane edge dynamics. Related to Figure 6.
Highlights:
NET1 mRNA location determines binding of NET1 protein to CASK or importins
The rate of NET1 mRNA translation elongation also influences NET1 partner selection Partner binding to distinct domains specifies nucleo-cytoplasmic distribution of NET1
NET1 mRNA location controls NET1 function in cell migration and is regulated by TGFb
Acknowledgments
We thank the CCR Genomics Core of the National Cancer Institute, NIH for ddPCR analysis. This work was funded by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute (NCI), National Institutes of Health (NIH) (1ZIA BC011501 to S. Mili); by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NIH (DK075132 to N.R.G.); and by the Postdoctoral Research Associate Training Program (PRAT) at the National Institute of General Medical Sciences (NIGMS) (1FI2GM137845 to S. Meydan).
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.Karniely S, and Pines O (2005). Single translation--dual destination: mechanisms of dual protein targeting in eukaryotes. EMBO Rep 6, 420–425. 10.1038/sj.embor.7400394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yogev O, Naamati A, and Pines O (2011). Fumarase: a paradigm of dual targeting and dual localized functions. FEBS J 278, 4230–4242. 10.1111/j.1742-4658.2011.08359.x. [DOI] [PubMed] [Google Scholar]
- 3.Popgeorgiev N, Jabbour L, and Gillet G (2018). Subcellular Localization and Dynamics of the Bcl-2 Family of Proteins. Front Cell Dev Biol 6, 13. 10.3389/fcell.2018.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, and Levy DE (2009). Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324, 1713–1716. 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, et al. (2009). Function of mitochondrial Stat3 in cellular respiration. Science 323, 793–797. 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, and Moll UM (2003). p53 has a direct apoptogenic role at the mitochondria. Molecular cell 11, 577–590. 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 7.Knockaert L, Fromenty B, and Robin MA (2011). Mechanisms of mitochondrial targeting of cytochrome P450 2E1: physiopathological role in liver injury and obesity. FEBS J 278, 4252–4260. 10.1111/j.1742-4658.2011.08357.x. [DOI] [PubMed] [Google Scholar]
- 8.Raza H (2011). Dual localization of glutathione S-transferase in the cytosol and mitochondria: implications in oxidative stress, toxicity and disease. FEBS J 278, 4243–4251. 10.1111/j.1742-4658.2011.08358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Jesus A, Keyhani-Nejad F, Pusec CM, Goodman L, Geier JA, Stoolman JS, Stanczyk PJ, Nguyen T, Xu K, Suresh KV, et al. (2022). Hexokinase 1 cellular localization regulates the metabolic fate of glucose. Molecular cell 82, 1261–1277 e1269. 10.1016/j.molcel.2022.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Avadhani NG, Sangar MC, Bansal S, and Bajpai P (2011). Bimodal targeting of cytochrome P450s to endoplasmic reticulum and mitochondria: the concept of chimeric signals. FEBS J 278, 4218–4229. 10.1111/j.1742-4658.2011.08356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Avadhani NG (2011). Targeting of the same proteins to multiple subcellular destinations: mechanisms and physiological implications. FEBS J 278, 4217. 10.1111/j.1742-4658.2011.08355.x. [DOI] [PubMed] [Google Scholar]
- 12.Dubash AD, Guilluy C, Srougi MC, Boulter E, Burridge K, and Garcia-Mata R (2011). The small GTPase RhoA localizes to the nucleus and is activated by Net1 and DNA damage signals. PLoS One 6, e17380. 10.1371/journal.pone.0017380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Srougi MC, and Burridge K (2011). The nuclear guanine nucleotide exchange factors Ect2 and Net1 regulate RhoB-mediated cell death after DNA damage. PLoS One 6, e17108. 10.1371/journal.pone.0017108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt A, and Hall A (2002). The Rho exchange factor Net1 is regulated by nuclear sequestration. J Biol Chem 277, 14581–14588. 10.1074/jbc.M111108200. [DOI] [PubMed] [Google Scholar]
- 15.Song EH, Oh W, Ulu A, Carr HS, Zuo Y, and Frost JA (2015). Acetylation of the RhoA GEF Net1A controls its subcellular localization and activity. Journal of cell science 128, 913–922. 10.1242/jcs.158121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ulu A, Oh W, Zuo Y, and Frost JA (2018). Stress-activated MAPKs and CRM1 regulate the subcellular localization of Net1A to control cell motility and invasion. Journal of cell science 131. 10.1242/jcs.204644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mili S, Moissoglu K, and Macara IG (2008). Genome-wide screen reveals APC-associated RNAs enriched in cell protrusions. Nature 453, 115–119. 10.1038/nature06888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang T, Hamilla S, Cam M, Aranda-Espinoza H, and Mili S (2017). Extracellular matrix stiffness and cell contractility control RNA localization to promote cell migration. Nat Commun 8, 896. 10.1038/s41467-017-00884-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das S, Vera M, Gandin V, Singer RH, and Tutucci E (2021). Intracellular mRNA transport and localized translation. Nat Rev Mol Cell Biol 22, 483–504. 10.1038/s41580-021-00356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gasparski AN, Mason DE, Moissoglu K, and Mili S (2022). Regulation and outcomes of localized RNA translation. Wiley Interdiscip Rev RNA, e1721. 10.1002/wrna.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chrisafis G, Wang T, Moissoglu K, Gasparski AN, Ng Y, Weigert R, Lockett SJ, and Mili S (2020). Collective cancer cell invasion requires RNA accumulation at the invasive front. Proceedings of the National Academy of Sciences of the United States of America 117, 27423–27434. 10.1073/pnas.2010872117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moissoglu K, Stueland M, Gasparski AN, Wang T, Jenkins LM, Hastings ML, and Mili S (2020). RNA localization and co-translational interactions control RAB13 GTPase function and cell migration. The EMBO journal 39, e104958. 10.15252/embj.2020104958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pichon X, Moissoglu K, Coleno E, Wang T, Imbert A, Robert MC, Peter M, Chouaib R, Walter T, Mueller F, et al. (2021). The kinesin KIF1C transports APC-dependent mRNAs to cell protrusions. RNA 27, 1528–1544. 10.1261/rna.078576.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stueland M, Wang T, Park HY, and Mili S (2019). RDI Calculator: An Analysis Tool to Assess RNA Distributions in Cells. Scientific reports 9, 8267. 10.1038/s41598-019-44783-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carr HS, Morris CA, Menon S, Song EH, and Frost JA (2013). Rac1 controls the subcellular localization of the Rho guanine nucleotide exchange factor Net1A to regulate focal adhesion formation and cell spreading. Mol Cell Biol 33, 622–634. 10.1128/MCB.00980-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mardakheh FK, Paul A, Kumper S, Sadok A, Paterson H, McCarthy A, Yuan Y, and Marshall CJ (2015). Global Analysis of mRNA, Translation, and Protein Localization: Local Translation Is a Key Regulator of Cell Protrusions. Dev Cell 35, 344–357. 10.1016/j.devcel.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LaConte LE, Chavan V, Liang C, Willis J, Schonhense EM, Schoch S, and Mukherjee K (2016). CASK stabilizes neurexin and links it to liprin-alpha in a neuronal activity-dependent manner. Cell Mol Life Sci 73, 3599–3621. 10.1007/s00018-016-2183-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porter AP, White GRM, Mack NA, and Malliri A (2019). The interaction between CASK and the tumour suppressor Dlg1 regulates mitotic spindle orientation in mammalian epithelia. Journal of cell science 132. 10.1242/jcs.230086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meydan S, and Guydosh NR (2020). Disome and Trisome Profiling Reveal Genome-wide Targets of Ribosome Quality Control. Molecular cell 79, 588–602 e586. 10.1016/j.molcel.2020.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brar GA, and Weissman JS (2015). Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat Rev Mol Cell Biol 16, 651–664. 10.1038/nrm4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi J, Grosely R, Prabhakar A, Lapointe CP, Wang J, and Puglisi JD (2018). How Messenger RNA and Nascent Chain Sequences Regulate Translation Elongation. Annu Rev Biochem 87, 421–449. 10.1146/annurev-biochem-060815-014818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldman DH, Livingston NM, Movsik J, Wu B, and Green R (2021). Live-cell imaging reveals kinetic determinants of quality control triggered by ribosome stalling. Molecular cell 81, 1830–1840 e1838. 10.1016/j.molcel.2021.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vedula P, Kurosaka S, MacTaggart B, Ni Q, Papoian G, Jiang Y, Dong DW, and Kashina A (2021). Different translation dynamics of beta- and gamma-actin regulates cell migration. Elife 10. 10.7554/eLife.68712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu CC, Peterson A, Zinshteyn B, Regot S, and Green R (2020). Ribosome Collisions Trigger General Stress Responses to Regulate Cell Fate. Cell 182, 404–416 e414. 10.1016/j.cell.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juszkiewicz S, Chandrasekaran V, Lin Z, Kraatz S, Ramakrishnan V, and Hegde RS (2018). ZNF598 Is a Quality Control Sensor of Collided Ribosomes. Molecular cell 72, 469–481 e467. 10.1016/j.molcel.2018.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fritz RD, Letzelter M, Reimann A, Martin K, Fusco L, Ritsma L, Ponsioen B, Fluri E, Schulte-Merker S, van Rheenen J, and Pertz O (2013). A versatile toolkit to produce sensitive FRET biosensors to visualize signaling in time and space. Sci Signal 6, rs12. 10.1126/scisignal.2004135. [DOI] [PubMed] [Google Scholar]
- 37.Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, and Moses HL (2001). Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Molecular biology of the cell 12, 27–36. 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh N, and Bhalla N (2020). Moonlighting Proteins. Annu Rev Genet 54, 265–285. 10.1146/annurev-genet-030620-102906. [DOI] [PubMed] [Google Scholar]
- 39.Kramer G, Shiber A, and Bukau B (2019). Mechanisms of Cotranslational Maturation of Newly Synthesized Proteins. Annu Rev Biochem 88, 337–364. 10.1146/annurev-biochem-013118-111717. [DOI] [PubMed] [Google Scholar]
- 40.Collart MA, and Weiss B (2020). Ribosome pausing, a dangerous necessity for co-translational events. Nucleic Acids Res 48, 1043–1055. 10.1093/nar/gkz763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwarz A, and Beck M (2019). The Benefits of Cotranslational Assembly: A Structural Perspective. Trends Cell Biol 29, 791–803. 10.1016/j.tcb.2019.07.006. [DOI] [PubMed] [Google Scholar]
- 42.Weatheritt RJ, Gibson TJ, and Babu MM (2014). Asymmetric mRNA localization contributes to fidelity and sensitivity of spatially localized systems. Nat Struct Mol Biol 21, 833–839. 10.1038/nsmb.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ben-Menachem R, Tal M, Shadur T, and Pines O (2011). A third of the yeast mitochondrial proteome is dual localized: a question of evolution. Proteomics 11, 4468–4476. 10.1002/pmic.201100199. [DOI] [PubMed] [Google Scholar]
- 44.Karasik A, Jones GD, DePass AV, and Guydosh NR (2021). Activation of the antiviral factor RNase L triggers translation of non-coding mRNA sequences. Nucleic Acids Res 49, 6007–6026. 10.1093/nar/gkab036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han P, Shichino Y, Schneider-Poetsch T, Mito M, Hashimoto S, Udagawa T, Kohno K, Yoshida M, Mishima Y, Inada T, and Iwasaki S (2020). Genome-wide Survey of Ribosome Collision. Cell Rep 31, 107610. 10.1016/j.celrep.2020.107610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guydosh NR (2021). ribofootPrinter: A precision python toolbox for analysis of ribosome profiling data. BioRxiv doi: 10.1101/2021.07.04.451082. [DOI] [Google Scholar]
- 48.Gorelik R, and Gautreau A (2014). Quantitative and unbiased analysis of directional persistence in cell migration. Nature protocols 9, 1931–1943. 10.1038/nprot.2014.131. [DOI] [PubMed] [Google Scholar]
- 49.Barry DJ, Durkin CH, Abella JV, and Way M (2015). Open source software for quantification of cell migration, protrusions, and fluorescence intensities. The Journal of cell biology 209, 163–180. 10.1083/jcb.201501081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mito M, Mishima Y, and Iwasaki S (2020). Protocol for Disome Profiling to Survey Ribosome Collision in Humans and Zebrafish. STAR Protoc 1, 100168. 10.1016/j.xpro.2020.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McGlincy NJ, and Ingolia NT (2017). Transcriptome-wide measurement of translation by ribosome profiling. Methods 126, 112–129. 10.1016/j.ymeth.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, and Glockner FO (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–596. 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chan PP, and Lowe TM (2009). GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res 37, D93–97. 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Sequences of coding region fragments used for generation of plasmid constructs. Related to STAR Methods.
Table S2: Sequences of morpholino oligonucleotides (PMOs) used in the study. Related to STAR Methods.
Figure S1: Antisense oligonucleotide tiling across the 3’ UTR of the NET1 mRNA. Related to Figure 1.
Figure S2: Specificity and effect of altering NET1 mRNA localization on mRNA levels and protein expression. Related to Figure 1.
Figure S3: Characterization of GFP-NET1A mutant variants. Related to Figure 3.
Figure S4: Intracellular CASK distribution. Related to Figure 4.
Figure S5: NET1 mRNA localization in HEK293 cells depends on GA-rich sequences in the 3’UTR. Related to Figure 5.
Figure S6: Ribosome profiling data of the NET1 transcript. Related to Figure 5.
Figure S7: NET1A mutants with altered translation elongation rates. Related to Figure 5.
Figure S8: Sub-inhibitory concentrations of anisomycin increase interaction of NET1A with importin. Related to Figure 5.
Figure S9: Order of NLS appearance influences NET1-importin interaction. Related to Figure 5.
Figure S10: In vitro GFP-NET1A-importin α interaction. Related to Figure 5.
Figure S11: Validation of RhoA FRET biosensor. Related to Figure 6.
Figure S12: Peripheral NET1 mRNA localization is essential for NET1 function in membrane edge dynamics. Related to Figure 6.
Data Availability Statement
All Ribo-seq, Disome-seq and Ribosome profiling data generated as a part of this study are deposited at GEO (GSE207146, GSE158044, GSE133393). All imaging data have been deposited to Mendeley Data (DOI: 10.17632/f4mr55trf6.1). All data are publicly available as of the date of publication. Accession numbers and DOI are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-GFP | ThermoFisher | Cat#A-11122 |
| rabbit anti-NET1 | Abcam | Cat#ab 113202 |
| rabbit anti-Importin beta 1 | Cell Signaling Technology | Cat#60769 |
| mouse anti-CASK | Santa Cruz | Cat#sc-13158 |
| rabbit anti-CASK | Invitrogen | Cat#PA1544 |
| Mouse anti-tubulin | Sigma | Cat#T6199 |
| rabbit anti-GAPDH | Cell Signaling Technology | Cat#2118L |
| IR Dye 680RD donkey anti-mouse | Li-Cor | Cat#926–68072 |
| IR Dye 800CW donkey anti-rabbit | Li-Cor | Cat#926–32213 |
| mouse anti-paxillin | BD Transduction Laboratories | Cat#610619 |
| Alexa48 8-Phalloidin | Invitrogen | Cat#A12379 |
| anti-mouse Alexa647 | Invitrogen | Cat#A28181 |
| mouse anti-FLAG M2 | Sigma | Cat#F1804 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine RNAiMAX | ThermoFisher | Cat#13778–150 |
| PolyJet In Vitro DNA Transfection Reagent | SignaGen | Cat# SL100688 |
| Leibovitz L-15 | Gibco | Cat#11415064 |
| Penicillin-Streptomycin | Gibco | Cat#15070063 |
| DMEM, high glucose, pyruvate | Invitrogen | Cat#11995073 |
| Trypsin-EDTA (0.05%) | Gibco | Cat#25300054 |
| Blasticidin | ThermoFisher | Cat#A1113903 |
| Geneticin | Gibco | Cat#10131035 |
| Doxycycline Hydrochloride | ThermoFisher | Cat#BP26535 |
| Puromycin Dihydrochloride | Gibco | Cat#A1113803 |
| Recombinant Human TGF-B1 | Peprotech | Cat#100–21 |
| Endoporter | Genetools LLC | Cat#OT-EP-PEG-1 |
| Collagen IV | Sigma | Cat#C5533–5MG |
| Fibronectin bovine plasma | Sigma | Cat#F1141 |
| GFP-Trap magnetic agarose beads | Chromotek | Cat#gtma-10 |
| Paraformaldehyde | Electron Microscope Sciences | Cat#15710 |
| Intercept Blocking Buffer | Li-Cor | Cat#927–60001 |
| ProLong Gold Antifade reagent with DAPI | Invitrogen | Cat#P36931 |
| Triton X-100 | Sigma | Cat#93443–100ML |
| Halt Protease and Phosphatase Inhibitor Cocktail | ThermoFisher | Cat#78444 |
| HCS Green CellMask Stain | ThermoFisher | Cat# H32714 |
| GST-KPNA1 | Abnova | Cat#H00003836-P01 |
| GST-KPNA2 | Abnova | Cat#H00003838-P01 |
| GST-KPNA4 | Abnova | Cat#H00003840-P01 |
| JFX554 HaloTag ligand | Janelia Research Campus | N/A |
| DRAQ5 | ThermoFisher | Cat#62254 |
| RNase I | ThemorFisher | Cat#AM2295 |
| Superase-In | ThermoFisher | Cat#AM2694 |
| TRIzol | ThermoFisher | Cat#15596026 |
| Critical Commercial Assays | ||
| DuoLink In Situ Red PLA Kit | Sigma | Cat#DUO92008 |
| ViewRNA ISH Cell Assay Kit | ThermoFisher | Cat#QVC0001 |
| Quant-it RiboGreen RNA Assay Kit | ThermoFisher | Cat#R11490 |
| Qiagen FastSelect Kit | Qiagen | Cat#334386 |
| High Sensitivity DNA Kit | Agilent | Cat#5067–4626 |
| Deposited Data | ||
| Ribo-seq and Disome-seq-HEK293 cells | This study | GEO: GSE207146 |
| Ribosome profiling-A549 cells | Karasik et al, 202144 | GEO: GSE158044 |
| Ribosome profiling-HEK293 cells | Han et al, 20 2045 | GEO: GSE133393 |
| Imaging data | This study | http://dx.doi.org/10.17632/f4mr55trf6.1 |
| Experimental Models: Cell Lines | ||
| MDA-MB-231 | ATCC | Cat#CRM-HTB-26 |
| MCF7 | ATCC | Cat#HTB-22 |
| Flp-In T-REx HEK293 | Gibco | Cat#R780–07 |
| NIH/3T3 | ATCC | Cat#CRL-1658 |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A_WT UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A WT UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A WT UTR + pLV-PGK-hCA SK-FLAG | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A_WT UTR + pLV-PGK-hCA SK-FLAG-mCherry | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A_AGA UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A AGA UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A AGA UTR + pLV-PGK-hCASK-FLAG | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A_ AGA UTR + pLV-PGK-hCASK-FLAG-mCherry | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1 A_slow2_WT UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A slow2 WT UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1 A_slow2_AGA UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A slow2 AGA UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A_stallmutant(fast)_WT UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A stallmutant(fast) WT UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A_stallmutant(fast)_ AGA UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A stallmutant(fast) AGA UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A C-term NLS_WT UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A C-term NLS WT UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A C-term NLS_AGA UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A C-term NLS AGA UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-mutantNLS_WT UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-mutantNLS WT UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-ADH_hNET1 UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-ADH WT UTR + pCDH-CMV-hKPNBl | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-APH_WT UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-APH WT UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-APDZ_WT UTR | This study | N/A |
| MDA-MB-231 + pCDH-PGK-GFP-hNET1A-APDZ WT UTR + pCDH-CMV-hKPNB1 | This study | N/A |
| MDA-MB-231 + pCDH-CMV-mTFP 1 | This study | N/A |
| MDA-MB-231 + pIND20-RhoA-WT-biosensor | This study | N/A |
| MDA-MB-231 + pIND20-RhoA-Q63L-biosensor | This study | N/A |
| MDA-MB-231 + pIND20-RhoA-T19N-biosensor | This study | N/A |
| NIH/3T3 + NLS-HA-stdMCP-stdHalo + scFv-GCN4-sfGFP + pIND20-hNet1A 5UTR-24xGCN4-hNet1A CDS-24xMS2v7_hNet1 3UTR | This study | N/A |
| NIH/3T3 + NLS-HA-stdMCP-stdHalo + scFv-GCN4-sfGFP + pIND20-hNet1A 5UTR-24xGCN4-hNet1A SLOW2 CDS-24xMS2v7_hNet1 3UTR | This study | N/A |
| NIH/3T3 + NLS-HA-stdMCP-stdHalo + scFv-GCN4-sfGFP + pIND20-hNet1A 5UTR-24xGCN4-hNet1A STALLMUT(fast) CDS-24xMS2v7 hNet1 3UTR | This study | N/A |
| Oligonucleotides | ||
| Morpholino oligonucleotides (PMOs) | Table S2 | GeneTools LLC |
| human NET1 FISH probes | ThermoFisher | Cat# VA6–3169338 |
| human RHOA FISH probes | ThermoFisher | Cat#VA6–14829-01 |
| GFP FISH probes | ThermoFisher | Cat#VF6–16198 |
| AllStars siRNA Negative Control | Qiagen | Cat#1027281 |
| human siNET1 #4 – 5’-ACGGAAAGAGACTTTGGTGTA-3’ | Qiagen | Cat#SI00082040 |
| human siCASK #5 – 5’-AACCAATGGGAATCACTTTAA-3’ | Qiagen | Cat#SI02223368 |
| human siCASK #10 – 5’-CAGACCGGTTTGCGTACCCTA-3’ | Qiagen | Cat#SI04437720 |
| Recombinant DNA | ||
| KPNB1 (NM_002265) Human Tagged ORF Clone | Origene | Cat#RC200659 |
| pInducer20 | Addgene | Cat#44012 |
| pET263-pUC57 24xMS2V7 | Addgene | Cat#140705 |
| pLentiRhoA2G | Addgene | Cat#40179 |
| pcDNA4TO-24xGCN4_v4-kif18b-24xPP7 | Addgene | Cat#74928 |
| phage UbiC NLS HA stdMCP stdHalo | Addgene | Cat#104999 |
| pHR-scFv-GCN4-sfGFP-GB1-NLS-dWPRE | Addgene | Cat#60906 |
| pCDH-CMV-EF1-MCS-Puro | System Biosciences | Cat#CD510B-1 |
| pCDH-CMV-hKPNB1 | This study | N/A |
| pCDH-PGK-GFP-hNET1A-ADH WT UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A-APH WT UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A-APDZ WT UTR | This study | N/A |
| pCDH-CMV-mTFP1 | This study | N/A |
| pIND20-RhoA-WT-biosensor | This study | N/A |
| pIND20-RhoA-Q63 L-biosensor | This study | N/A |
| pIND20-RhoA-T19N-biosensor | This study | N/A |
| pCDH-PGK-GFP-hNET1A_WT UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A_ AGA UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A-slow2 WT UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A-slow2 AGA UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A_stallmutant(fast)_WT UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A_stallmutant(fast)_ AGA UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A C-term NLS / WT UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A C-term NLS_AGA UTR | This study | N/A |
| pCDH-PGK-GFP-hNET1A-mutantNLS hNET1 UTR | This study | N/A |
| pIND20-hNet1A 5UTR-24xGCN4-hNet1A CDS-24xMS2v7_hNet1 3UTR | This study | N/A |
| pIND20-hNet1A 5UTR-24xGCN4-hNet1A CDS-24xMS2v7 hNeti A(GA) 3UTR | This study | N/A |
| pIND20-hNet1A 5UTR-24xGCN4-hNet1A SLOW2 CDS-24xMS2v7 hNet1 3UTR | This study | N/A |
| pIND20-hNet1A 5UTR-24xGCN4-hNet1A STALLMUT(fast) CDS-24xMS2v7_hNet1 3UTR | This study | N/A |
| pLV-PGK-hCASK-FLAG-mCherry | VectorBuilder | N/A |
| pLV-PGK-hCASK-FLAG | VectorBuilder | N/A |
| Software and Algorithms | ||
| Fiji | Schindelin et al., 201246 | https://fiji.sc |
| Imaris | Oxford Instruments | https://imaris.oxinst.com |
| RibofootPrinter | Guydosh, 202147 | https://github.com/guydoshlab/ribofootPrinter |
| Matlab | MathWorks | https://www.mathworks.com/products/matlab.html |
| Prism | GraphPad | https://www.graphpad.com |
| DiPer Macro | Gorelik and Gautreau, 201448 | N/A |
| ADAPT ImageJ plugin | Barry et al., 201549 | N/A |
| LAS X | Leica | https://www.leica.com |
| NIS-Elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| Other | ||
| 35mm glass-bottom dishes | MatTek Corporation | Cat# P35G-1.5–14-C |
| TetraSpeck™ Fluorescent microspheres sampler kit | Fisher Scientific | Cat#T7284 |
| Coverslip, #1.5 thickness | Thomas Scientific | Cat#1217N81 |