

Abstract
Introduction:
The burden of chronic hepatitis B virus (HBV) results in almost a million deaths per year. The most common treatment for chronic hepatitis B infection is long-term nucleoside analogs (NUC) or one-year interferon-alpha (pegylated or non-pegylated) therapy before or after NUC therapy. Unfortunately, these therapies rarely result in HBV functional cure because they do not eradicate HBV from the nucleus of the hepatocytes, where the covalently closed circular DNA (cccDNA) is formed and/or where the integrated HBV DNA persists in the host genome. Hence, the search continues for novel antiviral therapies that target different steps of the HBV replication cycle to cure chronically infected HBV individuals and eliminate HBV from the liver reservoirs.
Areas covered:
The authors focus on capsid assembly modulators (CAMs). These molecules are unique because they impact not only one but several steps of HBV viral replication, including capsid assembly, capsid trafficking into the nucleus, reverse transcription, pre-genomic RNA (pgRNA), and polymerase protein co-packaging.
Expert opinion:
Mono- or combination therapy, including CAMs with other HBV drugs, may potentially eliminate hepatitis B infections. Nevertheless, more data on their potential effect on HBV elimination is needed, especially when used daily for 6–12 months.
Keywords: Antiviral, Antiviral therapy, Capsid assembly effector, CAM - Capsid assembly modulator, cccDNA, Core inhibitor, DAA – Directly acting antiviral, Hepatitis B, HBV, drug resistance
1. Introduction: the unique effects of CAMs on the HBV replication cycle
The global prevalence of individuals chronically infected with hepatitis B virus (HBV) infection is estimated to be approximately 316 million, more than the prevalence of HIV and HCV combined [1]. Although anti-HBV therapies and prophylactic vaccines are in use, they do not eliminate HBV from the liver reservoirs [2,3]. Consequently, about one million individuals die annually because of liver diseases associated with HBV infection [4,5]. There is an urgent need to develop new potent drugs to eliminate the virus from the liver and prevent HBV-related liver complications, including cirrhosis, cancer, transplantation, and death [2,6,7].
The covalently closed circular DNA (cccDNA) has a central role in the replication of HBV as, in the nuclei of hepatocyte, is the transcription template for all four viral RNAs that encode seven proteins: large (L), middle (M), surface (S), X, pre-core, core, and polymerase proteins, and the template for the reverse transcription of the pre-genomic RNA (pg-RNA) [8]. The core (capsid) protein (Cp) plays a crucial function in forming the nucleocapsids where viral replication emerges [9]. Once the pg-RNA is released to the cytoplasm, it is soon encapsidated inside the nucleocapsid with the HBV polymerase and reverse-transcribed into minus-strand DNA. Finally, the plus-stranded DNA is synthesized to form the partially double-stranded relaxed circular DNA (rcDNA); at this step of viral replication, the matured nucleocapsid can be either recycled to the nucleus of the hepatocyte for amplification/recycling of cccDNA or enveloped by the envelope proteins and excreted from cells to infect naïve cells [10–13]. The current antiviral therapies (NUC and interferon-alpha (IFN-a) pegylated or non-pegylated) reduce the viral loads of chronically infected individuals. The three mostly recommended antiviral NUC, entecavir (ETV), tenofovir disoproxil fumarate (TDF), or tenofovir alafenamide (TAF), are used worldwide for the treatment of chronic HBV to inhibit reverse transcriptase and HBV DNA replication.
Still, they do not affect the nucleocapsid assembly, eliminate the cccDNA from the nuclei of the hepatocytes, or the integrated HBV DNA from the hepatocyte genome. One consequence is the continuous production of hepatitis B surface antigens (HBsAg) transcribed from integrated DNA or the amplified cccDNA [14]. Several new HBV direct-acting antivirals or immunomodulators are being investigated experimentally and clinically to aim for a functional cure with clearance of HBsAg and HBV DNA in serum after end-of-treatment [15–18]. A liver-targeting prodrug called ATI-2173 is currently in phase II development. Other HBV antivirals include Bulevirtide, which blocks HBV entry receptor sodium taurocholate co-transporting polypeptide (NTCP) and is in phase III development for hepatitis delta. Two nucleic acid polymers, REP-2055 and REP-2139, are in phase II/III, and one oligonucleotide polymer is in phase I (ALG-010133), and they all decrease HBsAg production and secretion. There are also a few translational inhibitors, such as small-interfering RNA (JNJ-3898, AB-729, RG-6346, VIR-2218) in phase II, and antisense oligonucleotides GSK-3228836 and ALG-020572 in phase I/II. In addition, HBV mRNA destabilizers via cellular proteins PPAD5/7 inhibition are being investigated, and one of them is in phase I under EDP-721 [18]. Lastly, several HBV capsid assembly modulators (CAMs) are under investigation (preclinical, phase I/II, or discontinued) that interfere with nucleocapsid assembly, disassembly, and several other steps of the HBV replication cycle. For example, a recent study by Kum et al. examined the effects of a CAM-A-RG7907 on protein aggregation in vitro and in a mouse model with adeno-associated virus (AAV)-HBV. The results showed that the RG7907 caused cell death of infected hepatocytes and led to the clearance of core and HBsAg and AAV-HBV episome from the liver. This suggests that CAMs like RG7907 that induce core aggregates can promote hepatocyte proliferation and loss of cccDNA, which could lead to a functional cure for chronic hepatitis B (CHB) [19].
This review will discuss the different capsid assembly modulators (CAMs) in clinical development, their potential to interfere with mechanisms associated with HBV persistence, and potential HBV resistance mutations in the HBV core protein related to their use.
The envelope of HBV comprises three distinct surface proteins, namely large, middle, and small, which enclose the capsid that houses the double-stranded DNA genome, as illustrated in Fig. 1. A. The HBV replication occurs when the virus attaches to the hepatocyte at the NTCP receptor. The viral particles shed their protective coating upon entry and travel to the cell nucleus as the nucleocapsid particle. The HBV protein-free relaxed circular rcDNA (Pf-rcDNA) then undergoes the conversion into an episomal cccDNA, which serves as the foundation for all four viral RNAs. The viral polymerase and pre-genomic RNA (pgRNA) are encapsidated and reverse-transcribed into viral minus-strand DNA. RNAseH then degrades the RNA. Subsequently, the partially double-stranded rcDNA is formed by synthesizing plus-stranded DNA. After maturation, nucleocapsids have two potential outcomes: they can either be returned to the nucleus to maintain the cccDNA pool or combined with envelope proteins and released as infectious virions to infect other cells. Additionally, nucleocapsids containing pgRNA and empty nucleocapsids are combined with envelope proteins and released. See Fig. 1B for a visual representation.
Figure 1.
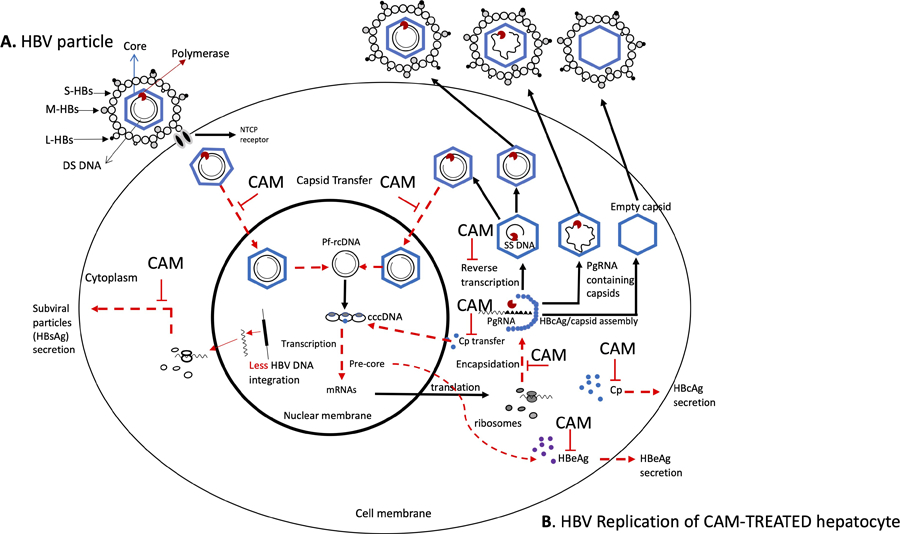
(A & B). HBV replication mechanism and schematic representation of CAM-modulations sites. Panel A represents the HBV particle, and panel B represents the different steps of the HBV replication being affected by CAMs. The dotted arrows in red represent the effects of CAMs on (i) the nuclear transfer of relaxed circular (RC) DNA containing nucleocapsid for covalently closed circular DNA (cccDNA) synthesis and cccDNA recycling/amplification, (ii) core protein (Cp) secretion on plasma or cytoplasm for nucleocapsid assembly, and nuclear transfer for cccDNA formation, (iii) capsid assembly for pre-genomic RNA (pgRNA) encapsidation, (iv) inhibition of reverse transcription to single-stranded (SS) DNA, (v) HBeAg secretion, and (vi) exposure of HBV DNA for integration in the host genome with low levels of HBsAg production. S, small, M, medium, L, large hepatitis B surface (HBs); DS, double stranded; NTCP, sodium taurocholate co-transporting polypeptide; CAM, capsid assembly modulator.
CAMs that disrupt the HBV replication cycle target the HBV core dimer protein and can misdirect capsid formation in several ways. First, they can affect the core protein (Cp), so it loses its ability to be transported to the host nucleus and serve as a tool for cccDNA formation. Second, CAMs can alter the nucleocapsid assembly in size, quantity, and rigidity and cause malformation, affecting the capsid’s capacity to encapsidate pgRNA and DNA synthesis. Thus, empty, aggregated, or aberrant capsids inadequate for the HBV replication to continue are formed. As for the amplification/recycling of cccDNA, with an abundance of malformed capsids, rigid or empty capsids, fewer intact capsids carrying rcDNA, if any, travel back to the nucleus of hepatocytes to release rcDNA to replenish the cccDNA reservoir. Finally, because CAMs directly affect nucleocapsids to pgRNA encapsidation, they will rapidly and significantly reduce the levels of HBV DNA, RNA, and core-related HBV antigen (HBcrAg) levels in serum and reduce the rcDNA recycling to the nucleus and, ultimately, the formation of intact virions released from hepatocytes to infect naïve cells [9,20,21]. In addition, less HBV DNA integration may occur as linear double-stranded HBV DNA will to a lesser extent, be presented to the host hepatocyte genomes when individuals are treated with CAM (Figure 1B) [22]*.
CAMs are categorized into two main classes: 1) Class I (CAM-A or CAM-Aberrant), including the heteroaryldihydropyrimidines (HAP) core protein allosteric modulators (CpAM) that bind to HBV capsids and promote their misassemble to aberrant non-capsid core polymers, and 2) Class II (CAM-E or CAM-Empty), including phenylpropenimides (PP), sulfamoylbenzamides (SBA), glyoxamide (GLP) are CAMs that bind to the capsid to form empty non-functional capsids lacking pgRNA/rcDNA [7,21,23,24].
2. CAMs in Phase I or II clinical development
2.1. Morphothiadin (GLS4)
Morphothiadin (GLS4) is a HAP derivative developed by HEC Pharm that triggers aberrant HBV core particle assembly (Class I or CAM-A). GLS4 exhibits excellent anti-HBV activity in HepG2.2.15 cells. It is active against various polymerase drug-resistant mutants, including nucleoside analog-resistant HBV strains [lamivudine, telbivudine, entecavir (ETV)], with EC50 ranging from 1 to 20 nM [25]. An in vivo study in nude mice inoculated with HepAD38 cells showed that GLS4 at doses greater than 7.5 mg/kg per day significantly suppressed virus replication throughout the treatment period, and GLS4 doses of >15 mg/kg per day suppressed the virus for up to 2 weeks after the end of treatment [26]. However, the bioavailability of GLS4 is minimal (5–15%, depending on species and dose-proportional plasma concentrations) [26]. Cellular pharmacology studies demonstrated that GLS4 is readily metabolized through N-dealkylation of the morpholine ring by CYP3A4 [27]. Therefore, a combination with known CYP inhibitor ritonavir [28] was contemplated to improve GLS4 plasma concentrations and achieve superior antiviral effects. In 2019, a phase Ib study of GLS4 in combination with ritonavir involving chronic HBV-infected individuals was reported [29]. Subjects received a 28-day course of GLS4 (120 or 240 mg, cohort A and B) in combination with 100 mg ritonavir and were followed up for 40 days. The GLS4/ritonavir combination was well tolerated despite 2 (cohort A) and 3 (cohort B) subjects presenting alanine aminotransferase flare. In cohorts A and B, the mean declines in HBV DNA after 28 days of treatment were significant (1.42 and 2.13 log10 IU/mL), while reductions in HBsAg, pgRNA, and hepatitis B core antigen (HBcAg) were minimal (Table 1). Recently, a study was conducted to evaluate the effectiveness and safety of GLS4/ritonavir (RTV) combined with ETV compared to ETV alone in individuals with chronic hepatitis B who are HBeAg-positive. The study’s interim results were published in 2020 [30]. After 12 weeks of treatment, treatment-naïve patients showed a mean reduction in HBV DNA compared to baseline of 5.02 (Table 1) and 3.84 log10 IU/ml. Meanwhile, the decline of pgRNA was 2.63 vs. 0.27 log10 IU/ml in cohort A and cohort B, respectively, who received either 120 mg GLS4/100 mg RTV (TID) combined with 0.5 mg ETV (QD) or 0.5 mg ETV (QD) monotherapy for 96 weeks. For HBsAg, the mean reduction from baseline was 0.43 vs. 0.21 log10 IU/ml, and for HBeAg was 0.49 vs. 0.29 log10 IU/ml, respectively, in cohorts A and B. In cohort A, 28.6% of patients had HBsAg levels decline ≥ 0.5 log10 IU/ml, while only 5.9% of patients in cohort B experienced the same decline. Further, two patients (14.3%) had HBsAg dropped ≥ 1.5 log10 IU/ml in cohort A. In the suppressed group (subjects who took ETV for more than one year and achieved virus suppression, assigned as cohort C and cohort D), the mean declines of HBV pgRNA were 1.59 vs. 0.15 log10 IU/ml, while decreases in HBsAg and HBeAg were 0.11 vs. 0 log10 IU/ml and 0.17 vs. 0.06 log10 IU/ml (Table 1). The combination of GLS4/RTV with ETV was generally safe and well tolerated, with the most common adverse events (AEs) being alanine aminotransferase (ALT) elevation and hypertriglyceridemia. GLS4 is undergoing phase III clinical trials in China (CTR20213273) to establish its effectiveness when combined with ritonavir and a NUC in CHB subjects, but no results have been published yet.
Table 1:
List of HBV capsid assembly modulators in clinical development.
| HBV CAM/Class | Company | Clinical trial phase | Structure | HBV DNA log10 IU/mL reduction (in vivo) | HBsAg/HBeAg log10 IU/mL reduction | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|---|
| Morphothiadin (GLS4)/I | HEC Pharma | II |
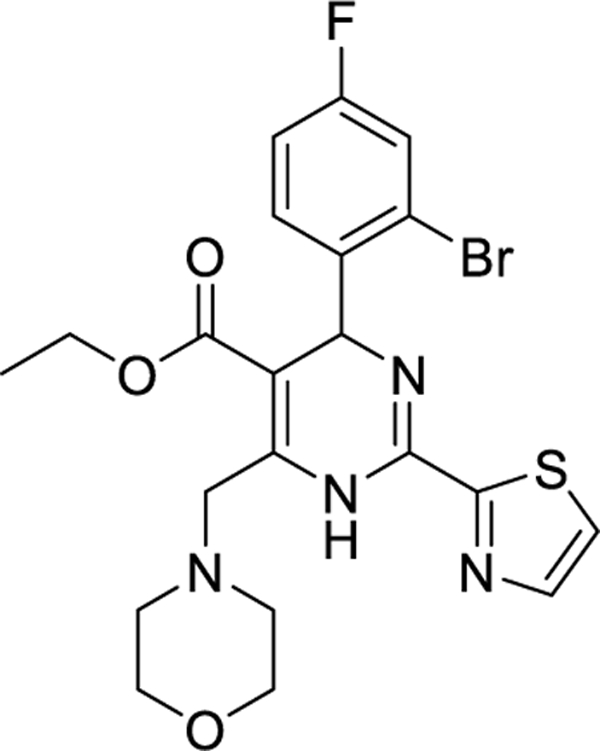
|
1.4 and 2.1 at 28 days of
monotherapy. 5.0 at 12 weeks GLS4/ritonavir combined with ETV |
Minimal with monotherapy or combined with
ETV. 1.59 pgRNA reduction combined with ETV. |
NCT03638076
NCT03662568 NCT04147208 |
| Bersacapavir JNJ 56136379/II | Janssen | II |
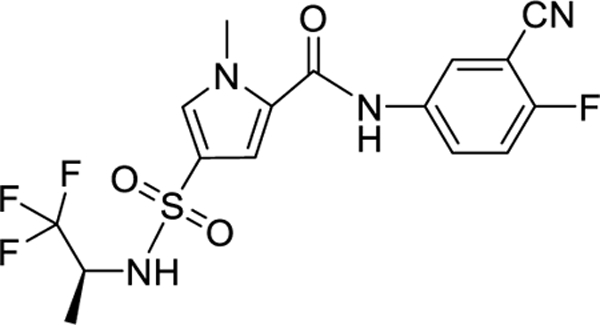
|
5.5 and 5.9 at week 24 of monotherapy or
combined with TDF. Monotherapy resulted in T33N viral resistance. |
Minimal with monotherapy or combined with
TDF. Maximal individual HBsAg and HBeAg reductions were 1.28 and 1.8 at week 24 when combined with TDF in patients with the most pronounced HBV DNA declines at week 24. |
NCT04208399
NCT04474210 NCT02933580 NCT02662712 NCT02662712 NCT03982186 NCT04129554 NCT04439539 NCT04667104 NCT04585789 |
| QL-007 | Qilu Pharmaceutical | II | Not disclosed | Not available | Not available |
NCT03770624
NCT03244085 NCT04157699 NCT04157257 |
| EDP-514/II | Enanta Pharma | I | Not disclosed | > 4 (in chimeric mice). 2.9 to 3.5 after 28 days treatment in CHB, non-cirrhotic, viremic, HBeAg (+). | HBV RNA reduction (up to 2.9) |
NCT04470388
NCT04008004 NCT04783753 NCT04971512 |
| ABI-H3733/II | Assembly Biosciences | I | Not disclosed | 3.1 | Not available |
NCT05414981
NCT04271592 |
| Canocapavir (ZM-H1505R)/ Pirazole | Zhimeng Biopharma | I |
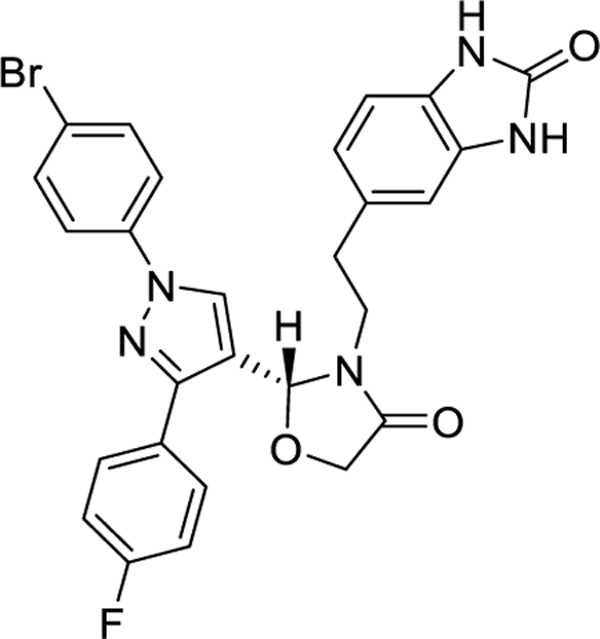
|
2.7 with monotherapy at 200 mg (28 days treatment) | No reduction of surface or e
antigens. pgRNA: 2.3 log10 copies/mL reduction. |
NCT05484466
NCT05470829 NCT04220801 |
| ALG-000184/II | Aligos Therapeutics | I | Not disclosed but related to GLP-26 | 4.2 | 0.8 HBsAg reduction. | NCT04536337 |
| VNRX-9945/I | Venatorx | I-terminated | Not disclosed | EC50 = 3–31 nM* | Not available | NCT04845321 |
| KL060332/I | Kelun-Biotech Biopharmaceuticals Co | I | Not disclosed | ~3 in HBV mouse model | Reduced surface and e antigens (>1.5) | Not available |
| ABI-4334/II | Assembly Biosciences | I | Not disclosed | EC50 = 0.5 nM* | pgRNA reduction*. cccDNA-EC50 = 3.1nM* (PHH). |
NCT05569941 |
In vitro; ETV, entecavir; pgRNA, pre-genomic RNA; TDF, tenofovir disoproxil fumarate; CHB, chronic hepatitis B; HBsAg, hepatitis B surface antigen; HBeAg, hepatitis B e antigen; cccDNA, covalently closed circular DNA; EC50, half maximal effective concentration; PHH, primary human hepatocytes.
2.2. JNJ-56136379 (JNJ-6379, Bersacapavir)
JNJ-56136379 is a class II or CAM-E in phase I clinical trial that displays activity against the majority of HBV A-H genotypes, including those with NUC resistance or pre-core (PC) or basal core promoter (BCP) mutations (mean EC50s comprised between 10–33 nM) [31]**. JNJ-56136379 accelerates the rate of the core assembly resulting in the formation of normal nucleocapsids devoid of genetic material and, at higher concentrations, inhibits de novo formation of cccDNA [32]. In phase I clinical study, JNJ-56136379 was well tolerated with only mild to adverse effects (AEs) in healthy volunteers (dose up to 600 mg) [33]. In CHB patients (dose up to 250 mg), treatment-naïve patients showed a dose-dependent reduction of HBV DNA and RNA when given JNJ-56136379 as monotherapy for four weeks [33,34]. However, no effect on HBsAg was noted, and the viral rebound was observed after the end of treatment. In a phase II study in patients with CHB, treatment with JNJ-56136379 combined with TDF or ETV once daily resulted in greater HBV DNA and RNA reductions from baseline at week 24 compared to NUC treatment alone. However, JNJ-56136379 and/or NUC had little effect on HBsAg and HBeAg levels (Table 1). It is worth noting that monotherapy will not be considered anymore due to cases of viral breakthrough (and emerging resistant variant T33N) during treatment with JNJ-56136379 alone [33]. More recently, a safety and tolerability study performed with JNJ-73763989 (JNJ most advanced liver-targeted siRNAs) in CHB patients receiving JNJ-56136379 (250 mg) and a NUC [either ETV or tenofovir (TNF)] showed that the combination was generally safe and well tolerated [34]. It is worth noting that, even though many patients experienced HBsAg declines ≥1 log10 IU/mL, there was no additive effect between JNJ-73763989 and JNJ-56136379 [35]. Further phase II studies of JNJ-56136379 in combination with JNJ-73763989, NUC, and pegylated IFN-α 2a are ongoing (NCT04439539 and NCT04667104). Because JNJ-56136379 is known to be metabolized via cytochrome P450 (CYP) 3A4, a phase I, open-label trial (NCT03945539) in healthy patients receiving JNJ-56136379 (250 mg) with and without prior exposure to itraconazole (200 mg), a known CYP3A4 inhibitor, was performed. However, this study showed that using a CYP3A4 inhibitor only modestly affected JNJ-56136379 exposure. The same group also reported the results of a study involving healthy women who received one dose of drospirenone/ethinyl estradiol and midazolam; three drugs are known to be metabolized by CYP3A before and after 15 days of JNJ-56136379. JNJ-56136379 appeared to be a mild to moderate inducer of CYP3A4, affecting the metabolism of CYP3A4 metabolized drugs. During that study, the effect of midazolam was minimum. Still, the increase in ethinyl estradiol exposure was of concern, and the authors concluded that JNJ-56136379 should not be administered to patients taking high-dose estrogen-based contraceptives [36].
2.3. EDP-514
EDP-514 (Enanta Pharmaceuticals Inc) is a class II core inhibitor in phase IIb clinical trial that reduces HBV RNA.
It inhibits HBV DNA replication in various cell systems (HepAD38, HepDE19, primary human hepatocytes (PHH), and HepG2 2.2.15 with EC50 value between 6 to 27 nM [37]. At d0 (time of infection) in PHH cells, EDP-514 prevented the formation of cccDNA (EC50 of 35 nM, using HBsAg as a readout). It also showed antiviral activity against HBV genotypes A-H (EC50: 9–32 nM) and known NUC resistance mutations. Combination with ETV, TNF, or a CAM Class I inhibitor, such as GLS4, led to an in vitro synergistic antiviral effect. Treatment of HBV-infected human liver-chimeric mice (PXB mice) with EDP-514 for 12 weeks led to a >4-log viral load reduction (Table 1).
A phase Ia study was initiated in June 2019 to evaluate pharmacokinetics and safety profiles in healthy volunteers. Both single ascending doses (SAD: 50, 100, 200, 400, 600, 800 mg) and multiple ascending doses (MAD: 200, 400, 800 mg under fasted conditions and 400 mg under fed conditions) studies were performed over 14 days [38]. The pharmacokinetics study showed a plasma concentration at 24 h up to 9.3 and 22.1-fold higher than the in vitro DEFINE paEC50 for the SAD and MAD cohorts with a moderate food influence. Overall, EDP-514 was well tolerated at all the doses, with only gastrointestinal (9.4%) and nervous system (18.8%) disorders observed, confirming that the drug was suitable for a single daily oral dosing. A phase Ib study, conducted over a period of 28 days with non-cirrhotic, CHB, HBeAg(+) or (−) patients virologically suppressed under NUC therapy showed that EDP-514 was generally well-tolerated and rapidly absorbed, with PK results similar to the phase Ia for the 800 mg dose (QD) [39]. For patients with detectable HBV RNA at a baseline of 0.6 log10 IU/mL, a mean reduction of 2.0, 1.7, and 1.9 log10 IU/mL were obtained for placebo, 200 mg, 400 mg, and 800 mg after 28 days. A maximum reduction in HBV RNA of 2.3 and 2.8 log10 IU/mL was observed in HBeAg (−) and HBeAg (+) subjects, respectively (compared to a 1.2 for placebo).
EDP-514 was also evaluated in CHB, non-cirrhotic, viremic, HBeAg(+), or (−) volunteers not currently under any treatment over 28 days [40]. In this cohort, the pharmacokinetics profile and safety data obtained were similar to those reported on NUC-suppressed CHB volunteers for the same doses. After 28 days, a mean HBV DNA reduction of 2.9, 3.3, and 3.5 log10 IU/mL was observed for the 200 mg, 400 mg, and 800 mg doses (Table 1). In addition, a mean HBV RNA reduction of 2.9, 2.4, and 2.0 log10 IU/mL was obtained over the same period. No virologic failures in EDP-514 arms have been observed during all phases, Ia and Ib.
2.4. QL-007
After a phase Ib study evaluating the dose-related safety, efficacy, and PK profile of different doses of QL-007 (200, 400, and then 600 mg) in CHB patients, Qilu Pharmaceutical initiated in 2019 (NCT03770624), a phase II clinical trial to evaluate the efficacy and safety of QL-007 in combination with TDF or ETV in patients with CHB who have received nucleoside therapy. No data have been reported yet (NCT04157257).
2.5. ABI-H3733 (3733)
ABI-H3733 is a second-generation class II capsid core protein inhibitor developed by Assembly Bio.
In vitro, H3733 is a potent inhibitor of HBV DNA (EC50 = 5 nM) and cccDNA formation (EC50 = 125 nM) (EASL 2019). H3733 binds to the dimer-dimer interface of the core protein. It triggers the formation of aberrant capsids, preventing the packaging of pgRNA and the production of the virus. It also disrupts the trafficking of nucleocapsid to the nucleus, blocking the generation of cccDNA. The phase Ia studies of a 250 mg liquid formulation of H3733 showed favorable safety, tolerability, and pharmacokinetics profile when administrated to healthy volunteers [41]. However, an initial prototype tablet formulation only achieved 30% exposure compared with the liquid formulation, leading to the evaluation of a new tablet formulation in beagle dogs. This new tablet was rapidly absorbed, and PK results suggest that a 300 mg dose of this new formulation should deliver, in humans, equivalent area-under-the-curve (AUC) compared to the liquid formulation (presented at EASL2022 by Shen M et al.) [42]. In June 2022, Assembly Bio announced the initiation of the phase Ib trial to evaluate the safety, PK, and antiviral activity of ABI-H3733 in adults with chronic HBV infection who are treatment naïve or off treatment with 25, 50 and 100 mg doses (NCT04157257). Interim results show that six of eight HBV patients taking the 50 mg dose reached a lower limit of quantification for HBV DNA by day 21, along with a mean reduction of 3.1 logs in the plasma HBV DNA (Table 1) (NCT04271592) and website: assemblybio.com/.
2.6. ZM-H1505R (Canocapavir)
ZM-H1505R is a pyrazole derivative that has been proven effective in inhibiting the replication of HBV DNA. It does this by preventing formation of cccDNA and encapsidation of pgRNA.
A phase Ia clinical trial evaluated the safety, tolerability, and pharmacokinetic properties of ZM-H1505R following single (SAD) and multiple (MAD) oral escalating dose administration. In this study, healthy subjects receive up to 450 mg of ZM-H1505R or placebo or 75–300 mg of ZM-H1505R or placebo once daily for 14 consecutive days. The results showed that oral doses of 25–450 mg of ZM-H1505R or oral administration of 75, 150, or 300 mg once daily for 14 consecutive days were generally safe. No serious adverse events were reported (The most common TEAEs were gastrointestinal symptoms, all deemed mild). In June 2022, the results of a phase Ib study were disclosed. In a recent study, patients were divided into three groups and administered either a placebo or a daily dose of ZM-H1505R at 50, 100, or 200 mg for 28 days. The findings demonstrated that ZM-H1505R was well-tolerated and safe, and most adverse effects were mild to moderate in severity. Following the 28-day treatment period, there was a significant reduction in serum levels of HBV DNA and HBV pgRNA from baseline. The mean maximum decreases in HBV DNA were −1.54, −2.50, and −2.75 log10 IU/mL in the 50, 100, and 200 mg groups, respectively. Similarly, the mean maximum reductions in HBV pgRNA were −1.53, −2.35, and −2.34 log10 IU/mL in the same groups. Moreover, in the 100 and 200 mg cohorts, the mean maximum declines in HBcrAg were −0.61 and −0.51 log10 IU/mL, respectively. A phase IIa trial to evaluate the effectiveness and safety of ZM-H1505R in combination with ETV compared to ETV monotherapy commenced in August 2022 [43] (NTC05470829; CTR20210686).
2.7. ALG-000184
ALG-000184 (Aligos Therapeutics Inc.), the bioavailable prodrug of ALG-001075, is a pan-genotypic Class II (CAM-E) with picomolar potency and broad antiviral activity covering all genotypes A to J.
It has a dual mechanism of action. ALG-001075 was based on GLP-26 [44–47] and technologies licensed from Emory University. In vitro studies showed that ALG-000184 acts primarily by inhibiting pg-RNA encapsidation and by inhibiting HBsAg production through regulating the de-novo establishment and replenishment of cccDNA (2nd mode of action). ALG-001075/ALG-000184 has a moderate 28-fold loss activity against the known CAM resistance mutation T33N but retained activity against other CAM and NUC resistance mutations. ALG-000184 is highly soluble in water, stable in simulated gastric and intestinal fluids, and readily converted to ALG-001075. An initial evaluation in dogs showed that ALG-000184, in a tablet formulation, led to complete oral absorption and high exposure to ALG-001075. In a phase I study, single oral doses of ALG-000184 (up to 500 mg) and multiple doses for seven days (up to 250 mg) in healthy volunteers were well tolerated with dose-dependent and linear PK[48] (NCT04536337). The daily oral dosing of currently not treated/treatment naïve HBeAg negative or positive CHB patients for 28 days with 10, 50, and 10 mg of ALG-000184 was well tolerated, and all resulted in rapid declines in HBV DNA and RNA levels below the limit of quantification. Furthermore, unlike what was observed for recently abandoned HBV CAM RO7049389 [49,50], no significant difference in exposures by body weight-adjusted doses and antiviral activity was observed between Asian and non-Asian subjects [51].
Importantly, during a multi-part, double-blind, randomized, placebo-controlled Phase I study in HBeAg-positive CHB patients, oral treatment with 300 mg for 28 days led to HBsAg declines up to 0.78 log10 IU/mL (Table 1), suggesting that this higher dose may be triggering ALG-000184 second mechanism of action (MoA). The ability of ALG-000184 to affect HBsAg levels is currently being evaluated over a longer-duration treatment [52]**. Initial results at 20 weeks of treatment with 100 and 300 mg per day ALG-000184 orally are encouraging and will be presented in 2Q2023.
2.8. AB-836
AB-836 is a class II (CAM-E) HBV capsid inhibitor developed by Arbutus Biopharma.
AB-836 displays potent antiviral activity in vitro against all HBV genotypes (A through H) by inhibiting pgRNA encapsidation, resulting in empty capsid formation and interfering with the uncoating step post a viral entry at a higher concentration. AB-836 binds to the core protein dimer-dimer interface and inhibits rcDNA synthesis in HBV-infected PHH (EC50 = 0.002 µM), HepG2-NTCP-4 (EC50 = 0.012 µM), and cccDNA establishment (EC50 = 0.18 µM) in HBV infected cell-systems [53]. In addition, it has been proven active against NUC-resistant HBV variants and other known potential capsid-resistant variants [54]. In a Phase Ia/Ib clinical trial, treatment with AB-836 either as a single dose (175 mg) or multiple doses for QD 10 days in healthy subjects and QD 28 days in CHB subjects was safe and generally well-tolerated, even though three subjects experienced treatment-emergent AE (ALT elevations and dyspepsia). AB-836 showed efficacy with a mean decline in HBV DNA at day 28 of 3.04 and 3.10 log10 at 100 and 200 mg QD, respectively (Table 1) [55]. AB-836 was recently discontinued from clinical trials due to ALT elevation after more than 20 days of dosing in two healthy subjects.
2.9. VNRX-9945
VNRX-9945 is a broadly active and orally bioavailable class I CAM (CAM-A) reported for the first time by Venatorx Pharmaceutical in June 2021 [56].
VNRX-9945 inhibits HBV DNA formation with an EC50 of 2.3 and 10 nM in HEPG2.2.15, and PHH cells, respectively, while inhibiting capsid assembly and cccDNA formation. It shows broad antiviral activities against HBV genotypes A-H (EC50: 3–31 nM) and HBV variants, alone or in combination with one NUC (TNF or ETV). VNRX-9945, combined with TNF or ETV, displayed an additive antiviral effect. In addition, VNRX-9945 remains susceptible to the HBV variant that confers resistance to ETV, while ETV remains susceptible to the HBV variant that confers VNRX-9945 loss of susceptibility. Data on CYP isoforms and human transporters suggested a low potential for drug-drug interactions. PK studies in dogs and cynomolgus monkeys showed that a single 10 mg/kg oral dose would lead, after 24 hr, to plasma concentration superior to both capsid assembly and cccDNA formation EC90s. Compound safety studies in rats and monkeys at doses up to 600 mg/kg daily over 28 days did not lead to any AE. Phase Ia was initiated (NCT04845321) to evaluate: 1) the safety and PK of single and multiple ranges of doses in healthy adults and 2) daily doses of VNRX-9945 for 14 days. It is worth noting that part 2 of the study was terminated (unrelated to safety) (Table 1).
2.10. KL060332
KL060332 is an oral class I or CAM-A HAP developed by Kelun-Biotech Biopharmaceuticals.
KL060332 shows potent antiviral activity (EC50 = 5.18 nM) and low cytotoxicity in HepG2.2.15 cells. In a study using mice, KL060332 (9 mg/kg, taken orally twice a day) reduced viral DNA levels in the plasma and liver by 2.45 and 2.41 log10, respectively, on the seventh day after infection in a hydrodynamic injection HDI-HBV mouse model. When combined with TAF, KL060332 was more effective than either treatment alone. In a recombinant adeno-associated-virus AAV-HBV mouse model, KL060332 (6 mg/kg, taken orally twice a day) led to a reduction in HBV DNA level by approximately 3 log10. It also reduced HBeAg and HBsAg levels in the plasma (by more than 1.5 log10) and liver after 28 days of treatment (see Table 1). In May 2020, a randomized, placebo-controlled, dose-escalating study was conducted to evaluate the safety and tolerability of KL060332 after oral administration in healthy subjects [57]. However, there is currently no available data on the clinical outcomes of this study.
2.11. ABI-4334
ABI-4334 is a novel class II CAM displaying sub-nanomolar potency against pgRNA encapsidation and single-digit nanomolar potency against cccDNA formation in vitro.
ABI-4334 was evaluated in phase Ia clinical trial to determine its safety, tolerability, and PK profile in healthy subjects. A 30 mg single-dose cohort results show that ABI-4334 is relatively safe, as AEs were mild to moderate and considered unrelated to the study treatment. Results also support once-a-day (QD) dosing as Cmin is projected to be >10 times above the protein-adjusted EC50 for antiviral activity and against cccDNA formation. A second cohort using a higher single dose (100 mg) is currently under evaluation (NTC05569941) and website: assemblybio.com. AASLD2022 - Hepatology. 2022;76 (Suppl. 1): S1-S1564 Abstract#1119.
3. CAM resistance
An understanding of HBV CAM resistance still needs to be elucidated. HBV replication has high fidelity compared to other viruses, and Cp mutations are relatively uncommon [58,59]. In contrast, direct-acting anti-HBV reverse transcriptase inhibitors are prone to developing resistance but have a high resistance barrier [60]. For example, first-line NUCs ETV, TDF, and TAF resistance rates are at most 1% after several years of treatment [61]. Identification of clinical resistance mutations caused by CAM treatment is not yet sufficient. Furthermore, the effectiveness of resistance testing in clinical practice is still a topic of discussion.
To achieve resistance while maintaining viral functions, it’s essential to efficiently suppress genome replication and control the production rate of mutations. To do this, it’s crucial to understand the Cp sequence space that supports both resistance and proper nucleocapsid functioning. One way to do this is through structure-guided mutagenesis and characterizing virus variants in cell culture. However, this method can be time-consuming, expensive, and labor-intensive.
Also, a major barrier to capturing the resistance to novel antivirals is the lack of a culture system that can support multiple rounds of high-yield replication under drug pressure. However, the availability of high-resolution structures of HBV capsids bound with CAMs provides detailed information about the key Cp residues binding to these compounds. Therefore, it can be used to predict resistance mutations associated with CAMs. Thus, our group has recently developed a computational approach to predict antiviral drug resistance mutations, successfully implemented in predicting and validating the resistance mutations in HBV Cp for CAM, GLP-26 [62]. Known resistance mutations, identified through in vitro site-directed mutagenesis, in the HBV CAM binding pocket for clinical CAMs including, morphothiadin [63], bersacapavir [31]**, EDP-514 [64], ABI-H3733 [65], ALG-000184 [66], AB-836 [67], and ABI-4334 [68] are summarized in Table 2. They are key resistance mutations as they were reported as resistance to one or more preclinical CAMs. As the clinical trial advances with CAMs, any of these mutants might emerge as CAM-resistant. The binding site residues involved in resistance are highlighted in Figure 2.
Table 2.
Reported resistant mutations in the HBV CAM binding pocket for various CAMs.
| Residue Position | Variant | Morphothiadin (GLS4) | Bersacapavir (JNJ56136379) | EDP-514 | ABI-H3733 | ALG-000184 | AB-836 | ABI-4334 |
|---|---|---|---|---|---|---|---|---|
| 23 | F23Y | ND | + | ND | ND | + | ND | ND |
| 24 | F24Y | ND | − | ND | ND | ND | ND | ND |
| F24L | ND | − | ND | ND | ND | ND | ND | |
| 25 | P25G | ND | + | ND | ND | ND | ND | ND |
| P25A | ND | + | ND | + | ND | ND | ND | |
| P25S | ND | − | ND | ND | ND | ND | ND | |
| 29 | D29G | ND | + | + | + | ND | + | − |
| D29H | ND | − | ND | ND | ND | ND | ND | |
| 30 | L30F | ND | + | ND | ND | ND | + | ND |
| 33 | T33S | ND | + | ND | ND | ND | + | ND |
| T33N | ND | + | + | + | + | + | + | |
| T33P | ND | + | ND | ND | ND | ND | ND | |
| T33Q | ND | ND | ND | ND | ND | + | ND | |
| 37 | L37Q | ND | + | ND | ND | ND | + | ND |
| 38 | Y38H | ND | − | ND | ND | ND | − | ND |
| Y38F | ND | + | ND | + | ND | − | ND | |
| Y38C | ND | ND | ND | − | ND | ND | ND | |
| 105 | I105T | ND | + | ND | + | + | + | + |
| I105V | ND | + | ND | ND | ND | − | ND | |
| I105F | ND | ND | ND | ND | + | ND | ND | |
| I105L | ND | − | ND | − | ND | ND | ND | |
| 106 | S106T | ND | + | − | ND | ND | ND | ND |
| 109 | T109S | − | + | ND | ND | ND | + | ND |
| T109A | ND | − | ND | ND | ND | ND | ND | |
| T109C | − | ND | ND | ND | ND | ND | ND | |
| T109M | + | + | − | − | + | + | ND | |
| T109N | + | ND | ND | ND | ND | ND | ND | |
| T109I | + | − | − | − | ND | − | − | |
| 110 | F110I | ND | + | ND | ND | ND | ND | ND |
| 116 | I116L | ND | ND | ND | − | ND | ND | ND |
| 118 | Y118F | ND | + | + | + | + | − | − |
| 124 | V124A | − | ND | ND | ND | ND | ND | ND |
| V124M | + | ND | ND | ND | ND | ND | ND | |
| V124I | ND | ND | ND | ND | ND | ND | ND | |
| V124F | ND | ND | + | ND | ND | ND | ND | |
| V124G | + | + | ND | ND | ND | ND | ND | |
| 125 | W125F | ND | ND | ND | ND | ND | ND | ND |
| 127 | R127H | ND | + | ND | ND | ND | ND | ND |
| 128 | T128I | ND | + | ND | ND | − | ND | ND |
| 132 | Y132F | ND | + | ND | ND | ND | − | ND |
| 133 | R133K | ND | − | ND | ND | ND | ND | ND |
| 134 | P134T | ND | − | ND | ND | ND | ND | ND |
| 140 | L140I | ND | − | ND | ND | ND | ND | ND |
| 141 | S141P | ND | + | ND | ND | ND | ND | ND |
+ denotes resistance; − denotes no resistance; ND Not determined.
Figure 2.
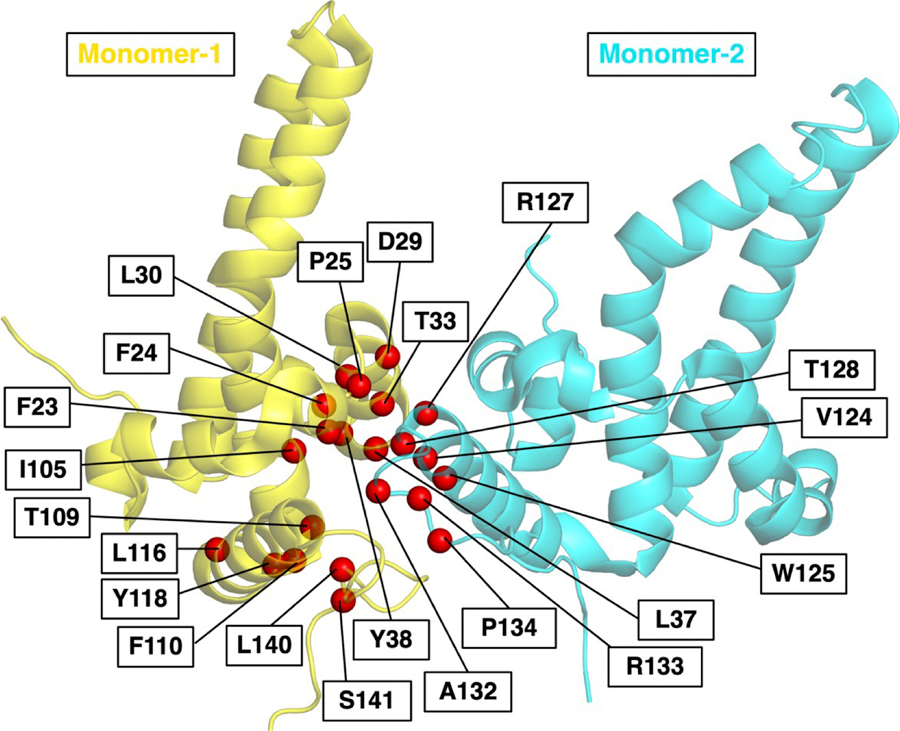
HBV CAM binding site in Cp dimer with the position of resistance mutations shown as red spheres. Cp monomers are shown as yellow and cyan. (PDB ID-5T2P).
4. Conclusion
Current US FDA-approved HBV therapies are mostly safe and effective. However, they have limitations to complete functional cure with loss of HBsAg from chronically infected individuals because they rarely eliminate integrated HBV DNA from the host genome and the cccDNA from the nucleus of the hepatocytes. In addition, they are given indefinitely to suppress virus replication, and it can be unsafe to stop therapy since the virus rebound immediately occurs. Therefore, several novel drugs with different mechanisms of action are being developed, aiming to target critical steps of the HBV replication cycle that can help to eliminate the HBV more efficiently from the reservoirs on the prolonged therapy [17,69]. We focused on reviewing ten pipeline CAMs in clinical development: eight in phase I and two in phase II studies. They all seem promising drugs combined with NUCs, but more data on their potential effect on HBV elimination is needed, especially when used daily for 6–12 months. It is anticipated that long-acting HBV formulations will be developed once the optimum combination of drugs is developed. In addition, studying the resistance profile of these drugs could help understand their mechanism of action to improve the development of novel drugs and strategies to eradicate HBV from the reservoirs completely.
5. Expert Opinion
CAMs are a class of compounds with great potential because they target several steps of the HBV replication cycle, affecting the Cp, capsid modulation/formation, and pg-RNA-encapsidation, drastically affecting the cccDNA formation and amplification/recycling. Unfortunately, these drugs have a limited impact on integrated viral DNA (iDNA). If iDNA plays a vital role in viral persistence, using a CAM alone is not enough to eliminate HBV unless treatment without interruption is conducted for a significant time (months and more likely a year) for the infected cells to be naturally eliminated in the presence of the CAM. The half-life of HBV virions in chronically infected individuals is currently estimated to be about four hr, shorter than was previously estimated (1 day), and similar to HIV and HCV. [70] Recently, Huang et al. determined the half-life of cccDNA in clinical samples and found that the cccDNA turnover occurs in several months, not decades, as previously believed [71]. Fortunately, not all cells in the liver are HBV infected, so the clearance of cccDNA in all cells may take several months rather than years, and the use of CAM with another DAA and NUC or interferon-alpha could clear the viral reservoirs quicker than anticipated. Animal and human data demonstrate that CAMs are remarkably effective at reducing viral load to undetectable levels, but when the drug suppression is stopped, the virus usually rebounds. Studies using long treatment durations with CAMs have yet to be completed, and the results of such studies are anticipated at the end of 2023. The key will be determining how many subjects have virus rebound on stoppage of all therapy and if this rebound is durable. Although many clinicians would like a greater than 90% success rate, a lack of rebound in 30–50% of subjects would be considered a significant achievement. Only one highly potent CAM may be sufficient, but a combination of drugs would be preferable to block the main pathways to virus rebound and reduce or eliminate the chance of selecting a drug-resistant virus. Thus, it would be wise to use two CAMs with different resistance profiles or a CAM combined with another modality, such as a NUC or siRNA approach to prevent the emergence of drug-resistant viruses. As demonstrated with the successful 8–12 weeks of HCV curative therapies, we anticipate that there will be a stepwise increased cure for HBV that may require multiple drugs, and we believe that CAMs will be the backbone of these curative regimens. We anticipate that a long-acting sustained-release formulation or device for highly potent CAMs would ensure compliance and viral suppression for the long-time treatment needed to eliminate all the viral reservoirs in hepatocytes. The first-generation CAMs were effective at low µM levels but proved insufficient to impact HBsAg. Currently, there are CAMs in phases 1 and 2 that are active at low pM levels, and these are expected to improve the probability of affecting the second viral kinetic phase, which should impact cccDNA and HBsAg on prolonged therapy. We are battling a giant killer, and now we are finally on the way to making incremental successes toward HBV elimination.
Article highlights.
Today, the hepatitis B virus (HBV) results in almost a million deaths per year.
Current antiviral therapies are inadequate and do not eliminate HBV from liver reservoirs, leading to lifetime treatment and the potential development of hepatocellular carcinoma (HCC).
Novel HBV direct-acting antivirals targeting different steps of the HBV replication cycle are being studied experimentally and clinically toward a functional cure, including the loss of HBsAg and other HBV markers.
Data on long-term treatment of chronically HBV-infected individuals with new capsid assembly modulators alone or in combination with nucleoside analogs (NUC) are limited.
In the absence of simple cell-based assays, a computational approach to predict HBV resistance mutations to CAMs has been successfully developed and validated.
Acknowledgements:
We thank Drs. James J. Kohler and Lefteris Michailidis for the helpful review of the paper
Funding:
This work was supported by funding from the National Institutes of Health (AI-132833 to RF Schinazi), (AI-148740 to JC Gumbart and L Bassit), and the Emory Center for AIDS Research (5P30-AI-50409 to RF Schinazi).
Footnotes
Declaration of Interest:
RF Schinazi, F Amblard, L Bassit, and Emory University, are entitled to equity and royalties related to anti-HBV products licensed to Aligos Therapeutics, Inc., being further evaluated in the research described in this review. Emory University has reviewed and approved the terms of this arrangement per its conflict-of-interest policies. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures:
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References (endnote)
- 1.Collaborators GHB. Global, regional, and national burden of hepatitis B, 1990–2019: A systematic analysis for the global burden of disease study 2019. Lancet Gastroenterol Hepatol 2022;7:796–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Revill PA PC, Brechot C, Zoulim F. Meeting the challenge of eliminating chronic hepatitis B infection. Genes (Basel) 2019;10(4):260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ligat G, Verrier ER, Nassal M, et al. Hepatitis B virus-host interactions and novel targets for viral cure. Curr Opin Virol 2021. Aug;49:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lavanchy D Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat 2004;11(2):97–107. [DOI] [PubMed] [Google Scholar]
- 5.Organization WH. Global hepatitis report, 2017. Global hepatitis report 2019.
- 6.Nicolini LA, Orsi A, Tatarelli P, et al. A global view to HBV chronic infection: evolving strategies for diagnosis, treatment and prevention in immunocompetent individuals. Int J Environ Res Public Health 2019. Sep 9;16(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dusheiko G, Agarwal K, Maini MK. New approaches to chronic hepatitis B. N Engl J Med 2023. Jan 5;388(1):55–69. [DOI] [PubMed] [Google Scholar]
- 8.Nassal M HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015. Dec;64(12):1972–84. [DOI] [PubMed] [Google Scholar]
- 9.Viswanathan U, Mani N, Hu Z, et al. Targeting the multifunctional HBV core protein as a potential cure for chronic hepatitis B. Antiviral Res 2020. Oct;182:104917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seeger C, Mason WS. Hepatitis B virus biology. Microbiology and Molecular Biology Reviews 2000;64(1):51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prange R Hepatitis B virus movement through the hepatocyte: An update. . Biology of the Cell 2022;114:325–348. [DOI] [PubMed] [Google Scholar]
- 12.Bustamante-Jaramillo LFF J; Blondot M-L; Rydell GE; Kann M Imaging of hepatitis B virus nucleic acids: current advances and challenges. . Viruses 2022;14(557). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prange R Hepatitis B virus movement through the hepatocyte: An update. Biol Cell 2022. Dec;114(12):325–348. [DOI] [PubMed] [Google Scholar]
- 14.Maura Dandri JP. cccDNA maintenance in chronic hepatitis B - targeting the matrix of viral replication. Infection and Drug Resistance 2020;13:3873–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cornberg M, Lok AS, Terrault NA, et al. Guidance for design and endpoints of clinical trials in chronic hepatitis B - Report from the 2019 EASL-AASLD HBV treatment endpoints conference (double dagger). J Hepatol 2020. Mar;72(3):539–557. [DOI] [PubMed] [Google Scholar]
- 16.Jeng WJ PG, Lok ASF. Hepatitis B. Lancet. Hepatitis B. The Lancet 2023;S0140–6736(22)01468–4. [Google Scholar]
- 17.Tsounis EP, Tourkochristou E, Mouzaki A, et al. Toward a new era of hepatitis B virus therapeutics: The pursuit of a functional cure. World J Gastroenterol 2021. Jun 7;27(21):2727–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong GLH, Gane E, Lok ASF. How to achieve functional cure of HBV: Stopping NUCs, adding interferon or new drug development? J Hepatol 2022. Jun;76(6):1249–1262. [DOI] [PubMed] [Google Scholar]
- 19.Kum DB, Vanrusselt H, Acosta Sanchez A, et al. Class a capsid assembly modulator RG7907 clears HBV-infected hepatocytes through core-dependent hepatocyte death and proliferation. Hepatology 2023. Apr 28. [DOI] [PubMed]
- 20.Kim H, Ko C, Lee JY, et al. Current progress in the development of hepatitis B virus capsid assembly modulators: Chemical structure, mode-of-action and efficacy. Molecules 2021. Dec 7;26(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niklasch M, Zimmermann P, Nassal M. The hepatitis B virus nucleocapsid-dynamic compartment for infectious virus production and new antiviral target. Biomedicines 2021. Oct 29;9(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lucifora J, Pastor F, Charles E, et al. Evidence for long-term association of virion-delivered HBV core protein with cccDNA independently of viral protein production. JHEP Rep 2021. Oct;3(5):100330. *This article explains that the hepatitis B core protein can bind to the integrated viral DNA before viral transcription begins. This association can be reduced by drugs such as CAMs, which may also decrease the formation of cccDNA [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zlotnick A, Venkatakrishnan B, Tan Z, et al. Core protein: A pleiotropic keystone in the HBV lifecycle. Antiviral Res 2015. Sep;121:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zoulim F, Zlotnick A, Buchholz S, et al. Nomenclature of HBV core protein-targeting antivirals. Nat Rev Gastroenterol Hepatol 2022. Dec;19(12):748–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren Q, Liu X, Luo Z, et al. Discovery of hepatitis B virus capsid assembly inhibitors leading to a heteroaryldihydropyrimidine based clinical candidate (GLS4). Bioorg Med Chem 2017. Feb 1;25(3):1042–1056. [DOI] [PubMed] [Google Scholar]
- 26.Wu G, Liu B, Zhang Y, et al. Preclinical characterization of GLS4, an inhibitor of hepatitis B virus core particle assembly. Antimicrob Agents Chemother 2013. Nov;57(11):5344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou X, Gao ZW, Meng J, et al. Effects of ketoconazole and rifampicin on the pharmacokinetics of GLS4, a novel anti-hepatitis B virus compound, in dogs. Acta Pharmacol Sin 2013. Nov;34(11):1420–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenblatt DJ, Harmatz JS. Ritonavir is the best alternative to ketoconazole as an index inhibitor of cytochrome P450-3A in drug-drug interaction studies. Br J Clin Pharmacol 2015. Sep;80(3):342–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H, Wang F, Zhu X, et al. Antiviral activity and pharmacokinetics of the hepatitis B virus (HBV) capsid assembly modulator GLS4 in patients with chronic HBV infection. Clin Infect Dis 2021. Jul 15;73(2):175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang M, Zhang J, Tan Y. SAT452-efficacy and safety of GLS4/ritonavir combined with entecavir in HBeAg-positive patients with chronic hepatitis B: Interim results from phase IIb, multi-center study. s878. AASLD: J Hepatology; 2020.
- 31.Verbinnen T, Tan Y, Wang G, et al. Anti-HBV activity of the HBV capsid assembly modulator JNJ-56136379 across full-length genotype A-H clinical isolates and core site-directed mutants in vitro. J Antimicrob Chemother 2020. Sep 1;75(9):2526–2534. **This article has shown detailed site-directed mutagenesis studies for a few class-I and class-II CAMs. [DOI] [PubMed] [Google Scholar]
- 32.Berke JM, Dehertogh P, Vergauwen K, et al. Antiviral properties and mechanism of action studies of the hepatitis B virus capsid assembly modulator JNJ-56136379. Antimicrob Agents Chemother 2020. Apr 21;64(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janssen HLA, Hou J, Asselah T, et al. Randomised phase 2 study (JADE) of the HBV capsid assembly modulator JNJ-56136379 with or without a nucleos(t)ide analogue in patients with chronic hepatitis B infection. Gut 2023. Jan 25. [DOI] [PMC free article] [PubMed]
- 34.Gane E, Yuen MF, Kakuda TN, et al. JNJ-73763989 pharmacokinetics and safety: Liver-targeted siRNAs against hepatitis B virus, in Japanese and non-Japanese healthy adults, and combined with JNJ-56136379 and a nucleos(t)ide analogue in patients with chronic hepatitis B. Antivir Ther; Jun2022. p. 13596535221093856. [DOI] [PubMed]
- 35.Yuen MF, Locarnini S, Lim TH, et al. Combination treatments including the small-interfering RNA JNJ-3989 induce rapid and sometimes prolonged viral responses in patients with CHB. J Hepatol 2022. Nov;77(5):1287–1298. [DOI] [PubMed] [Google Scholar]
- 36.Vandenbossche J, Yogaratnam J, Hillewaert V, et al. Drug-drug interactions with the hepatitis B virus capsid assembly modulator JNJ-56136379 (Bersacapavir). Clin Pharmacol Drug Dev 2022. Dec;11(12):1419–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vaine M, Dellispola V, Clugston S, et al. EDP-514, a novel HBV core inhibitor with potent antiviral activity both in vitro and in vivo. EASL 2019: J Hepathol; 2022. p. e474–475.
- 38.Kaja L, Alaa A, Coakley E, et al. EDP-514, a novel pangenotypic class II hepatitis B virus core inhibitor: preliminary results of a phase I study in healthy adult subjects. S871. J Hepatol; 2020.
- 39.Feld JJ, Lawitz E, Nguyen T, et al. EDP-514 in healthy subjects and nucleos(t)ide reverse transcriptase inhibitor-suppressed patients with chronic hepatitis B. Antivir Ther 2022. Dec;27(6):13596535221127848. [DOI] [PubMed] [Google Scholar]
- 40.Yuen MF, Chuang W-L, Peng C-Y, et al. EDP-514, a potent pangenotypic class II hepatitis B virus core inhibitor, demonstrates significant HBV DNA and HBV RNA reductions in a phase 1b study in viremic, chronic hepatitis B patients. S849. EASL 2022: J Hepatol; 2022.
- 41.Gane E, Schwabe C, Alves K, et al. 842. AASLD Digital International Liver Conference 2021.
- 42.Shen M, Zong Z, Mohammed N, et al. EASL International Liver Congress 2022.
- 43.Jia H, Mai J, Wu M, et al. Safety, tolerability, pharmacokinetics, and antiviral activity of the novel core protein allosteric modulator ZM-H1505R (Canocapavir) in chronic hepatitis B patients: a randomized multiple-dose escalation trial. BMC Med 2023. Mar 16;21(1):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amblard F, Boucle S, Bassit L, et al. Novel hepatitis B virus capsid assembly modulator induces potent antiviral responses in vitro and in humanized mice. Antimicrob Agents Chemother 2020. Jan 27;64(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hurwitz SJ, McBrearty N, Arzumanyan A, et al. Studies on the efficacy, potential cardiotoxicity and monkey pharmacokinetics of GLP-26 as a potent hepatitis B virus capsid assembly modulator. Viruses 2021. Jan 15;13(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pavlova A, Bassit L, Cox BD, et al. The mechanism of action of hepatitis B virus capsid assembly modulators can be predicted from binding to early assembly intermediates. J Med Chem 2022. Mar 24;65(6):4854–4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amblard F, Boucle S, Bassit L, et al. Discovery and structure activity relationship of glyoxamide derivatives as anti-hepatitis B virus agents. Bioorg Med Chem 2021. Feb 1;31:115952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li C, Wu M, Zhang H, et al. Safety, tolerability, and pharmacokinetics of the novel hepatitis B virus capsid assembly modulator GST-HG141 in healthy Chinese subjects: a first-in-human single- and multiple-dose escalation trial. Antimicrob Agents Chemother 2021. Sep 17;65(10):e0122021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu X, Feng S, Zhang J, et al. Evaluation of the safety, tolerability, and pharmacokinetics of RO7049389 in healthy Chinese volunteers. Clin Transl Sci 2022. Jan;15(1):195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tomita Y, Maeda K, Sugiyama Y. Ethnic variability in the plasma exposures of OATP1B1 substrates such as HMG-CoA reductase inhibitors: a kinetic consideration of its mechanism. Clin Pharmacol Ther 2013. Jul;94(1):37–51. [DOI] [PubMed] [Google Scholar]
- 51.APASL, The 31st Conference of the Asian Pacific Association for the Study of the Liver; Seol2022
- 52.Safety, Pharmacokinetics (PK), and Antiviral Activity of the Capsid Assembly Modulator (CAM) ALG-000184 in Subjects with HBeAg Positive Chronic Hepatitis B (CHB) AASLD Liver Meeting. **The first report of a substantial reduction in HBsAg following treatment with a CAM in HBV-infected patients suggesting that the dose/exposure level may be engaging ALG-000184 2nd MoA .
- 53.Manil N, Cole AG, Kulgen SG, et al. Preclinical antiviral profile of AB-836, a potent highly selective hepatitis B virus capsid inhibitor. Hepatitis B: novel therapeutic approaches. International Liver Conference EASL 2–21.
- 54.Sofia MJ. Progress toward an HBV cure combination therapy. HepDart2021
- 55.Gane E, Jucov A, Kolomiichuk L, et al. Safety, tolerability, pharmacokinetics (PK), and antiviral activity of 3rd generation capsid inhibitor AB-836 in healthy subjects (HS) and subjects with chronic hepatitis B (CHB). International Liver Congress EASL; London 2022.
- 56.Coburn G, Benetatos C, Yao J, et al. Discovery and preclinical profile of VNRX-9945, a potent, broadly active core protein inhibitor for the treatment of hepatitis B virus (HBV) infection. . EASL: J Hepatol; 2021. p. S294–803.
- 57.other Z, Tian Q, Zhao X, et al. , editors. Discovery of KL060332, a potential best-in-class capsid inhibitor. . The Liver Meeting Digital Experience™; 2020: AASLD. [Google Scholar]
- 58.Klumpp K, Lam AM, Lukacs C, et al. High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Proc Natl Acad Sci U S A 2015. Dec 8;112(49):15196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gunther S, Fischer L, Pult I, et al. Naturally occurring variants of hepatitis B virus. Adv Virus Res 1999;52:25–137. [DOI] [PubMed] [Google Scholar]
- 60.Tacke F, Kroy DC. Treatment for hepatitis B in patients with drug resistance. Ann Transl Med 2016. Sep;4(18):334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buti M, Marcos-Fosch C, Esteban R. Nucleos(t)ide analogue therapy: The role of tenofovir alafenamide. Liver Int 2021. Jun;41 Suppl 1:9–14. [DOI] [PubMed] [Google Scholar]
- 62.Patel D, Ono SK, Bassit L, et al. Assessment of a computational approach to predict drug resistance mutations for HIV, HBV and SARS-CoV-2. Molecules 2022. Aug 24;27(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang J, Zhang H, Zhang Y, et al. Influences on viral replication and sensitivity to GLS4, a HAP compound, of naturally occurring T109/V124 mutations in hepatitis B virus core protein. J Med Virol 2017. Oct;89(10):1804–1810. [DOI] [PubMed] [Google Scholar]
- 64.Alexopoulou A, Vasilieva L, Karayiannis P. New approaches to the treatment of chronic hepatitis B. J Clin Med 2020. Oct 1;9(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cai D, Connelly E, Kumar R, et al. Amino acid substitutions in the inhibitor binding pocket of HBV core protein confer differential changes in susceptibility to three generations of HBV core inhibitors AASLD; San Francisco, CA, USA2021. [Google Scholar]
- 66.Zhang Q, Jekle A, Serebryany V, et al. Best-in-class preclinical characteristics of Alg-000184, a prodrug of the capsid assembly modulator Alg-001075 for the treatment of chronic hepatitis B. . 503A. AASLD The Liver Meeting: J Hepatol; 2020.
- 67.Mani N, Cole AG, Kultgen SG, et al. Preclinical antiviral profile of AB-836, a potent, highly selective hepatitis B virus capsid inhibitor. EASL International Liver Congress: J Hepatol; 2021. p. S201–293.
- 68.Xu X, Shen M, Guo L, et al. Preclinical characterization of ABI-4334, a novel highly potent core inhibitor for the treatment of chronic hepatitis B virus infection. AASLD: J Hepatology; 2021. p. 1394A–1395A.
- 69.Bassit L, Ono SK, Schinazi RF. Moving fast toward hepatitis B virus elimination. Adv Exp Med Biol 2021;1322:115–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murray JM, Purcell RH, Wieland SF. The half-life of hepatitis B virions. Hepatology 2006. Nov;44(5):1117–21. [DOI] [PubMed] [Google Scholar]
- 71.Huang Q, Zhou B, Cai D, et al. Rapid turnover of hepatitis B virus covalently closed circular DNA indicated by monitoring emergence and reversion of signature-mutation in treated chronic hepatitis B patients. Hepatology 2021. Jan;73(1):41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]