

Abstract
Congenital hypomyelinating polyneuropathy (HPN) restricted to the peripheral nervous system was reported in 1989 in two Golden Retriever (GR) littermates. Recently, four additional cases of congenital HPN in young, unrelated GRs were diagnosed via neurological examination, electrodiagnostic evaluation, and peripheral nerve pathology. Whole-genome sequencing was performed on all four GRs, and variants from each dog were compared to variants found across >1,000 other dogs, all presumably unaffected with HPN. Likely causative variants were identified for each HPN-affected GR. Two cases shared a homozygous splice donor site variant in MTMR2, with a stop codon introduced within six codons following the inclusion of the intron. One case had a heterozygous MPZ isoleucine to threonine substitution. The last case had a homozygous SH3TC2 nonsense variant predicted to truncate approximately one-half of the protein. Haplotype analysis using 524 GR established the novelty of the identified variants. Each variant occurs within genes that are associated with the human Charcot-Marie-Tooth (CMT) group of heterogeneous diseases, affecting the peripheral nervous system. Testing a large GR population (n = >200) did not identify any dogs with these variants. Although these variants are rare within the general GR population, breeders should be cautious to avoid propagating these alleles.
Keywords: animal model, dysmyelination, genetic, genocopies, electrodiagnostic testing, histopathology
1. Introduction
Abnormal myelination of the nervous system is observed in many clinical diseases. Both demyelination and hypomyelination can occur; the former is characterized by the degeneration of existing myelin, whereas the latter is characterized by the underproduction of myelin [1]. Surrounding the axons of neurons, myelin acts to insulate the axons and to increase the rate of electrical pulse transmission along the axon [2]. Myelin is produced by oligodendrocytes in the central nervous system (CNS) and by Schwann cells in the peripheral nervous system (PNS) [3,4]. Typically, hypomyelination affects either the CNS exclusively or the CNS in conjunction with the PNS; though rare, PNS exclusive hypomyelination has been reported in humans and murine models[5–7]. Here, we describe for the first time in a domestic animal the genetic underpinnings of four canine cases of congenital hypomyelinating polyneuropathy (HPN) exclusively affecting the PNS. This rare condition leads to weakness, slowed motor and sensory nerve conduction velocities (NCVs), and hypomyelination of PNS nerve fibers [8,9].
In humans, Charcot-Marie-Tooth (CMT) or hereditary motor and sensory polyneuropathy is the most prevalent category of inherited polyneuropathies [10]. The most common inheritance pattern is autosomal dominant (AD-CMT), however, X-linked and autosomal recessive (AR-CMT) subtypes also exist [11]. The prevalence of CMT varies in different populations, with AD-CMT more frequent in some countries (e.g., Western Europe, the United States, and Japan) and AR-CMT more frequent in other countries (e.g., the Mediterranean Basin) [12]. CMT patients, despite their genetic heterogeneity, present with a slowly progressive, indolent, length-dependent sensorimotor polyneuropathy. The many types of CMT are distinguished by age of onset, mode of inheritance, and severity of symptoms; variants in more than 90 genes are reported to cause CMT [11,13,14]. MPZ variants are representative of AD-CMT and variants in MTMR2 and SH3TC2 are representative of AR-CMT [12,15].
Inherited CNS and PNS disorders have also been described in many breeds of dogs. CMT-analogous or similar conditions have been reported in at least 22 breeds of dogs [16], with the genetic basis described in Miniature Schnauzers [17], Greyhounds [18], Alaskan Malamutes [19], and Saint Bernards and Leonbergers [20]. Hypomyelinating conditions, specifically, have been described in several dog breeds, including Samoyeds, Lurchers, Dalmatians, Welsh Springer Spaniels, Weimaraners, Chow-Chows, Bernese Mountain Dogs, Border Terriers, and German Shepherds [21–31]. In every report, the hypomyelination exclusively affected the CNS. In 1989, the first case of PNS-exclusive hypomyelination reported in any domestic animal was described in two 7-week-old Golden Retriever (GR) littermates [32,33]. Both dogs, one male and one female, presented with pelvic limb ataxia, weakness, mild muscle atrophy, and gait abnormalities. Motor nerve conduction velocities were markedly decreased, and peripheral nerve biopsies were consistent with a hypomyelinating polyneuropathy [32,33].
In the present study, four unrelated GRs presented with early onset neuromuscular weakness, markedly reduced nerve conduction velocities, and peripheral nerve pathology consistent with hypomyelinating polyneuropathy, broadly recapitulating those described more than thirty years ago by Braund et al. [32,33]. Genetic and genomic tools have advanced significantly in the intervening decades since the first description of hypomyelinating polyneuropathy in GRs. Here we used whole-genome sequencing (WGS) data of these four GR cases to determine the variant(s) underlying each dog’s hypomyelinating polyneuropathy. We hypothesized that the variant(s) were likely to be in genes previously known to be involved in hypomyelinating conditions in humans.
2. Methods
2.1. Animals
Four young Golden Retrievers were presented to the William R. Pritchard Veterinary Teaching Hospital, University of California Davis (Cases 1–3, in 2007, 2011, and 2014, respectively) and to a private veterinary specialty hospital (Case 4, in 2016) for evaluation of neuromuscular weakness (Table 1). All dogs were evaluated by board certified veterinary neurologists. For each dog, complete physical and neurological examinations were performed, along with minimum database evaluations including CBC and biochemistry panel. Electrodiagnostic examinations performed included electromyography (EMG) and measurement of motor NCV studies in all dogs, sensory NCV and late wave studies in Cases 1–3, and repetitive nerve stimulation (RNS) in Cases 2 and 3. Peripheral nerve and muscle biopsies were collected from Cases 1–3 under the same anesthetic episode but on the opposite side from the electrodiagnostic studies. Skeletal muscle biopsies containing small distal intramuscular nerve branches were obtained from Case 4. Videos of the gait and neurological examination were also obtained for Case 4 (see Supplemental Video 1). Peroneal nerve biopsies were immersion fixed in 2.5% glutaraldehyde. Unfixed chilled biopsies from the quadriceps and triceps muscles were also collected, flash frozen in isopentane precooled in liquid nitrogen, then stored at −80°C until further processed. IACUC approval was not required as these tissues were submitted for diagnostic purposes under the care of the attending veterinarian.
Table 1.
Clinical and neurological findings in four GRs with hypomyelinating polyneuropathy
| Case 1 | Case 2 | Case 3 | Case 4 | |
|---|---|---|---|---|
| Age at presentation, sex | 5 months, F | 6 months, M | 1.5 years, FS | 6 months, F |
| Body Condition Score | 4/9 | 4/9 | 4/9 | 5/9 |
| Mentation | BAR, appropriate | BAR, appropriate | Quiet to obtunded | BAR, appropriate |
| Posture | Broad based | Broad based in pelvic limbs | Flaccid tetraparesis | Palmigrade, hyperextended tarsal joint bilaterally |
| Gait | Generalized weakness, ataxic | Ataxic, pacing walk | Nonambulatory | Generalized weakness |
| Muscle atrophy | Moderate diffuse | Mild to moderate diffuse | None | Mild to moderate |
| Reflexes: | ||||
| Patellar | Absent | Absent | Absent | Decreased to absent |
| Gastrocnemius | Decreased | Decreased | Absent | Decreased to absent |
| Biceps | Decreased | Absent | Absent | Decreased |
| Triceps | Decreased | Decreased | Absent | Decreased |
| Withdrawal | Decreased in pelvic limbs | Decreased in pelvic limbs | Absent in all 4 limbs | Decreased to absent |
| Gag | Decreased | Decreased | Absent | Decreased |
| Postural reactions: | ||||
| CPs | Absent to decreased in all 4 limbs | Absent in all 4 limbs | Absent in all 4 limbs | Normal thoracic limbs Absent pelvic limbs |
| Mutation | MTMR2 | MTMR2 | MPZ | SH3TC2 |
GR = Golden Retriever; BAR = Bright, alert, and responsive; CPs = conscious proprioception, F = female, M = male, FS = spayed female.
2.2. Electrodiagnostics
All cases underwent standard electrodiagnostic testing for suspected neuromuscular disease while under inhalation anesthesia [34]. Specific details for all electrodiagnostic procedures are in the Supplemental Methods. A thorough EMG survey was performed on numerous appendicular and axial muscles in addition to those of the head, pharynx, and larynx. Insertional activity was scored as normal, prolonged, or decreased. Spontaneous activity was scored as 0 (normal) to 4+ (severely abnormal) based on its distribution within a given muscle and how much of the baseline was obliterated [35].
Motor NCV studies were performed on the sciatic/peroneal nerve in all dogs, with motor and sensory NCV in the ulnar nerve on Cases 1–3. An additional sensory study was performed on the radial nerve in Cases 1–3. Compound muscle action potential (CMAP) and sensory nerve action potential (SNAP) amplitudes were recorded along with NCV values for all sites examined. All data were compared to reference values established at the Clinical Electrophysiology Laboratory, University of California Davis (DC Williams, personal communication).
To evaluate neuromuscular transmission, RNS was performed on the peroneal nerve in Cases 2 and 3 using trains of 10 stimuli at various repetition rates. Amplitude and area were determined for each CMAP in the train with the initial one serving as the baseline to which each subsequent potential was compared. Late wave recordings to evaluate proximal motor and sensory segments were attempted in Cases 1–3 using the sciatic/peroneal and ulnar nerves.
2.3. Histopathology and Electron Microscopy
Frozen sections of muscle biopsies were cut (8 μm thick) and evaluated using a standard panel of histochemical stains and reactions [36], including hematoxylin & eosin, modified Gomori trichrome, periodic acid-Schiff, oil red O, myofibrillar adenosine triphosphatase (ATPase) reactions at pH 9.8 and 4.3, acid and alkaline phosphatase, the reduced form of nicotinamide-adenine dinucleotide-tetrazolium reductase, and succinic dehydrogenase. Peroneal nerves from Cases 1–3 were fixed in 2.5% glutaraldehyde then processed and embedded in araldite resin as previously described [37]. Thick sections (1 μm) were cut with glass knives and stained with toluidine blue-basic fuchsin or paraphenylenediamine prior to light microscopic examination. Thin sections (60–90 nm) were cut with a diamond knife and stained with uranyl acetate and lead citrate before electron microscopic examination.
2.4. Genome Wide Association Study
DNA was extracted from archived, surplus frozen muscle tissue after manual tissue disruption using either the Qiagen DNeasy Blood and Tissue kit or the Qiagen Puregene Blood and Tissue kit following the manufacturer’s protocol. Assuming all affected dogs had the same causal variant, Cases 1, 2, and 3 were genotyped on the Illumina CanineHD BeadChip (n ~ 170,000 SNPs). Standard quality control was performed and a GWAS was conducted via PLINK [38] with data from 34 control GRs from a previous ophthalmic study. Briefly, SNPs with >10% missing genotypes or with a minor allele frequency <0.01 were pruned, as well as individuals with >10% of SNPs with missing genotypes. In total, 122,177 SNPs and 33 dogs (3 cases and 30 controls) remained for GWAS analysis.
2.5. Whole Genome Sequencing
DNA from each dog (Cases 1–4) was subjected to WGS (Illumina HiSeq 2 × 150 bp paired-end reads, Genewiz, South Plainfield, NJ), with an average of 28X coverage per dog. A previously described standardized bioinformatics pipeline was used for variant calling from the WGS data [39]. The sequence reads were trimmed via Trimmomatic [40] and aligned to the CanFam3 reference sequence [41] using Burrows-Wheeler Aligner (BWA) [42]. Genome Analysis Toolkit (GATK) 3.7 was used to prepare the aligned reads for further analysis [43]. Variant calls were made via GATK’s HaplotypeCaller, which identifies both SNPs and indels.
Affymetrix Axiom Canine HD array SNP-level data (n ~ 710,000 SNPs) from 478 GRs (a mix of publicly available data and internal data for different ongoing projects), inclusive of the four HPN-affected dogs, was used to estimate relationships between the affected dogs, as pedigree information was not available. After pruning SNPs with a minor allele frequency of <0.1, 303,027 SNPs remained. A relationship matrix and pi-hats were calculated in PLINK [38]. Inbreeding values were estimated for each affected dog using the --het command in PLINK [38].
VCFtools 0.1.16 [44] was used to determine which variants were private to the four affected GRs compared to WGS from 1,023 dogs, under the assumption that the disease allele is absent in this WGS dog population. The control genomes represented 158 breeds as well as mixed breeds, wild canids, and village dogs. A total of 301 genomes were from the University of Minnesota’s private WGS database [45] and 722 genomes were publicly available [46]. Initially, all four GRs were analyzed together under the assumption of a shared homozygous causative variant, then under the assumption of a shared heterozygous causative variant. Next, each of the four affected dogs had its data analyzed separately, compared to all other dogs in the database while excluding the three other affected GRs. A list of private variants for each of the four affected GRs was generated; these private variants were processed through the Ensembl Variant Effect Predictor (VEP) [47]. The variants predicted to have a high, medium, or low impact were further explored to determine which genes were affected. The function of each gene and any associated diseases were noted via Ensembl BioMart [48]. Each gene with a private variant of high or moderate impact was filtered through VarElect [49], which ranked each gene based on its relation to the phenotypic keywords “hypomyelination,” “peripheral nervous system,” and “polyneuropathy.” The predicted effect of splice site variants in candidate genes was tested with NNSplice 0.9, Spliceator, Human Splice Finder, and maximum entropy software programs [50–53]. All identified likely causative variants had their positions updated to their CanFam4 [54] positions, and as such, all locations listed below are for CanFam4.
2.6. Population Testing
To test the three WGS-identified variants in a larger GR population, both restricted fragment length polymorphism (RFLP) analysis and Sanger sequencing were used. DNA from GRs unrelated at the parent level (n = 255), and originally collected for ongoing, unrelated projects, were either already banked and frozen or were frozen as whole blood in EDTA tubes. For the latter, DNA was extracted via standard phenol-chloroform extraction. Polymerase chain reactions (PCR) were performed using KOD Xtreme Hot Start DNA Polymerase (Millipore-Sigma, Burlington, MA) with standard conditions to amplify the three regions of interest followed by either RFLP or Sanger sequencing to determine each dog’s genotype. The MTMR2 variant was amplified using primers: F-5’- CCATTTCGAGCATCCCATGTC-3’ and R-5’- AGAACACAACTGAACCTACTGC-3’, creating a 730 bp product for Sanger sequence genotyping. For the RFLP, a mismatch forward primer was used in order to differentiate between the wild-type and mutant alleles: F-5’- GTGTCTGGCAAATGACAAGCC-3’ together with the same reverse primer (R-5’- AGAACACAACTGAACCTACTGC-3’), for a 421 bp product. The enzyme MvaI (Thermo Scientific, Waltham, MA) was used, cutting the 421 bp product with the wild-type allele twice (product sizes of 305, 95, and 21 bp) and the mutant allele once (product sizes of 326 and 95 bp). Dogs were genotyped via visualization of RFLP products on 3% agarose gels and/or via visual inspection of Sanger sequence electropherogram in Sequencher 5.4.6 [55]
The MPZ variant was amplified using primers: F-5’-ACAAGCTTCCCCTCATTCCTC-3’ and R-5’-GAGGGAAGAGAAGGACAGGAG-3’ for Sanger sequencing, and with a mismatch reverse primer for RFLP: R-5’-TGTGACCTGAGAAGTCTTGCAC-3’ (together with the original forward primer, F-5’-ACAAGCTTCCCCTCATTCCTC-3’). These created product sizes of 435 and 220 bp, respectively. The AdeI (Thermo Scientific, Waltham, MA) enzyme cut only PCR products with the mutant allele (leaving the wild-type allele products uncut), producing 198 and 22 bp fragments out of the mutant allele product. As before, samples were genotyped via visualization on a 3% agarose gel and/or electropherogram.
Primers were designed for the SH3TC2 variant: F-5’- TTGATGGGGCATTTGAAGACC-3’ and R-5’- GAGCTTGGTGGTGTTCTGAAG-3’, with an overall product size of 491 bp. This product was subjected to the enzyme BseGI (Thermo Scientific, Waltham, MA) in an RFLP; DNA strands with the wild-type allele were cut three times (product sizes of 231, 117, 101, and 42 bp) and those with the mutant allele were cut twice (product sizes of 348, 101, and 42 bp). Dogs were genotyped via visualization of RFLP products on 3% agarose gels. The same primer pair was used for Sanger sequence genotyping (a subset of samples) and each dog was genotyped by visual inspection of electropherogram.
Manufacturer recommended digestion protocols were followed for each enzyme, using the maximum amount of enzyme. The BseGI enzyme digested the PCR product for 16 hours at 55°C, and the MvaI and AdeI enzymes digested the PCR product for 6 hours at 37°C.
Sanger sequencing (Eurofins Genomics, Louisville, KY) confirmed the genotypes of a subset of RFLP samples and was used to genotype additional samples. For these PCRs, primers were used to amplify the regions of interest with standard conditions, followed by ExoSAP-IT PCR Product Cleanup Reagent (Thermo Scientific, Waltham, MA).
2.7. Haplotype Construction
Haplotype analysis of the region surrounding each variant was performed in PLINK 1.9 [38] using existing Illumina CanineHD BeadChip data (n ~ 170,000 SNPs) from 525 GRs (n = 478 GRs from a database combining publicly-available and private internal data, a subset of which were extracted from WGS, and n = 47 GRs from a private internal UK database), inclusive of the four affected dogs. Following removal of SNPs with a minor allele frequency less than 0.05, SNP data was phased with SHAPEIT v2 [56]. An additional dataset (a combination of overlapping samples imputed from the above Illumina data and extracted from WGS) of Affymetrix Axiom Canine HD Array data (n ~ 710,000 SNPs, n = 478 GRs), pruned for a minor allele frequency of 0.05 and phased with SHAPEIT v2 [56], was used for the analysis of the region encompassing the MPZ variant. Haplotype blocks were visualized via Haploview software [57]. Where possible (n = 2 of 3 variants), one dog that shared the haplotype homozygously with the affected dog(s) was genotyped to determine if the haplotype block tagged the variant successfully or if the variant occurred on an existing haplotype within the population.
3. Results
3.1. Animals: Clinical Presentation and Neurological Examination
Clinical presentations and results of neurological examinations in all four dogs are summarized in Table 1. All dogs presented at a young age for chronic histories of variably severe neuromuscular weakness, exercise intolerance, dysphagia, megaesophagus, and respiratory difficulties including laryngeal paralysis. No significant abnormalities were identified on routine blood evaluations. Neurological examinations were consistent with lower motor neuron disease and electrodiagnostic evaluations and tissue biopsies performed on each dog.
3.2. Electrodiagnostic Testing
EMG changes in Cases 1 and 2 (MTMR2 variant) were similar and consisted of mild to severe spontaneous activity, including fibrillation potentials (FP) and positive sharp waves (PSW) in distal appendicular muscles, the thoracic paraspinal muscles, and muscles of the larynx and pharynx (Table 2). In Case 3 (MPZ variant), EMG testing revealed mild changes, consisting of PSW in the thoracic and pelvic interosseous and gluteal muscles only. Electromyographic testing in Case 4 (SH3TC2) revealed mild to moderate spontaneous activity, consisting of FP and PSW, in the thoracic limb and the distal appendicular muscles in the pelvic limb.
Table 2.
Electrodiagnostic findings in four GRs with hypomyelinating polyneuropathy
| Case 1 | Case 2 | Case 3 | Case 4 | |
|---|---|---|---|---|
| EMG | 1+ to 3+ FP, PSW in distal limbs, thoracic paraspinals, larynx and pharynx | Decreased insertion activity, larynx only 1+ to 2+ FP, PSW diffuse axial and appendicular muscles, larynx | 1+ PSW in thoracic and pelvic interossei and gluteal only | 1+ to 3+ FP and PSW thoracic limbs and distal pelvic limbs |
| Peroneal CMAP Amp (ref ≥ 6 mV) | ||||
| Hock | Unobtainable | Unobtainable | 3.1 mV | 3.0 mV |
| Stifle | 3.3 mV | 1.1 mV | 2.6 mV | 1.5 mV |
| Hip | 2.1 mV | 0.6 mV | 2.4 mV | 2.0 mV |
| Current | 9.4 –19.4 mA | 37.6 – 78.8 mA | 4.2 – 7.1 mA | NA |
| Peroneal MNCV (ref ≥ 50 m/s) | ||||
| Hock to stifle | Not calculable | Not calculable | 22 m/s | 15 m/s |
| Stifle to hip | 9 m/s | 10 m/s | 26 m/s | 12 m/s |
| Configuration | Temporal dispersion | Temporal dispersion | Normal | Temporal dispersion |
| Ulnar CMAP Amp (ref ≥ 4 mV) | ||||
| Carpus | 1.2 mV | 0.4 mV | 1.8 mV | Not tested |
| Elbow | 0.1 mV* | 0.1 mV* | 1.4 mV | Not tested |
| Current | 21.3 – 25.0 mA | 74.9–75.6 mA | 9.5 – 10.0 mA | Not tested |
| Ulnar MNCV (ref ≥ 50 m/s) | ||||
| Carpus to elbow | 5 m/s | 7 m/s | 21 m/s | Not tested |
| Configuration | Temporal dispersion | Temporal dispersion | Jagged but normal | Not tested |
| Peroneal SNAP Amp | ||||
| Hock | Unobtainable | Unobtainable | 0.9 pV | Not tested |
| Stifle | Unobtainable | Unobtainable | 0.3 pV | Not tested |
| Hip | Unobtainable | Unobtainable | 0.2 pV | Not tested |
| Current | 100 mA | 100 mA | 15 mA | Not tested |
| Peroneal SNCV (ref ≥ 50 m/s) | ||||
| Stim to hock | Not calculable | Not calculable | 21 m/s | Not tested |
| Hock to stifle | Not calculable | Not calculable | 19 m/s | Not tested |
| Stifle to hip | Not calculable | Not calculable | 24 m/s | Not tested |
| Configuration | None | None | Normal, no CD recorded | Not tested |
| Ulnar SNAP Amp | ||||
| Elbow | Unobtainable | Unobtainable | 0.3 pV | Not tested |
| Current | 100 mA | 100 mA | 15 mA | Not tested |
| Ulnar SNCV (ref ≥ 50 m/s) | ||||
| Stim to elbow | Not calculable | Not calculable | 31 m/s | Not tested |
| Configuration | No potentials obtained at any site | No SNAP, CD or C1, a cortical SEP was present | Temporal dispersion, no CD or C1, a cortical SEP was present | Not tested |
| Radial SNAP Amp | ||||
| Elbow | Unobtainable | Unobtainable | 1.0 mV | Not tested |
| Current | 100 mA | 100 mA | 15 mA | Not tested |
| Radial SNCV (ref ≥ 50 m/s) | ||||
| Stim to elbow | Not calculable | Not calculable | 26 m/s | Not tested |
| Configuration | No potentials obtained at any site | No SNAP, CD or C1, a cortical SEP was present | Temporal dispersion, no CD or C1, a cortical SEP was present | Not tested |
| RNS | Not tested | Pseudofacilitation at 50 Hz only | Pseudofacilitation at 20, 30, 50 Hz | Not tested |
| Late Waves (F, H, A) | Unobtainable | Unobtainable | F waves present at all stimulation sites, in addition H waves likely on ulnar with carpus stimulation, at the stifle increased chronodispersion was apparent | Not tested |
| Electrodiagnostic Summary/Diagnos is | Sensorimotor polyneuropathy, myelin-associated | Sensorimotor polyneuropathy, myelin-associated | Sensorimotor polyneuropathy, myelin-associated | Polyneuropathy |
| Mutation | MTMR2 | MTMR2 | MPZ | SH3TC2 |
GR = Golden Retriever, FP = fibrillation potentials, PSW = positive sharp waves, CMAP = compound motor action potential, MNCV = motor nerve conduction velocity, SNAP = sensory nerve action potential, SNCV = sensory nerve conduction velocity, CD = cord dorsum, C1 = first cervical spinal cord segment, SEP = somatosensory evoked potential, RNS = repetitive nerve stimulation, Amp = amplitude, mV = millivolts, μV = microvolts, mA = milliamps, m/s = meters/second
= conduction block, ref = reference values established for the lab, motor amplitudes are from onset to peak, thus smaller than reports using peak to peak; sensory amplitudes can be quite variable, so no reference values are listed.
The most striking finding in all four dogs was the markedly slowed motor NCVs (Table 2, Fig. 1). In the two MTMR2 variant dogs (Cases 1 and 2), the standard technique for testing motor NCVs in the sciatic/peroneal nerve was unsuccessful, as no potential could be recorded from the extensor digitorum brevis (EDB) muscle after stimulating at the hock [58]. A modified technique utilizing the cranial tibial muscle was applied [59]. The motor NCVs were markedly slowed (9 m/s [Case 1] and 10 m/s [Case 2], reference >50m/s) [59]. In addition, CMAPs were temporally dispersed, and amplitudes were reduced (half that of normal values). The currents needed to stimulate the nerve were mildly (Case 1) to markedly (Case 2) increased. Ulnar motor NCV results were also markedly slowed in both dogs (Table 2, Fig. 1a). Conduction block, whereby the CMAP amplitude following proximal stimulation is reduced by 50% or greater over that recorded from stimulating distally, was noted in Cases 1 and 2 on ulnar nerve stimulation. Motor NCVs in Case 3 were slowed (22 m/s distally, 26 m/s proximally, and 24 m/s overall) and the EDB CMAP amplitudes were decreased (roughly 50% of normal) on peroneal nerve stimulation, although waveforms were normal in configuration [58]. Only slight increases in stimulus currents were required. Similar findings were obtained from stimulation of the ulnar nerve in this dog (Fig. 1b). In Case 4, CMAP amplitudes were decreased on peroneal nerve stimulation and the NCVs were slowed (15 m/s distally, 12 m/s proximally, and 13 m/s overall). Temporal dispersion was apparent.
Fig. 1.
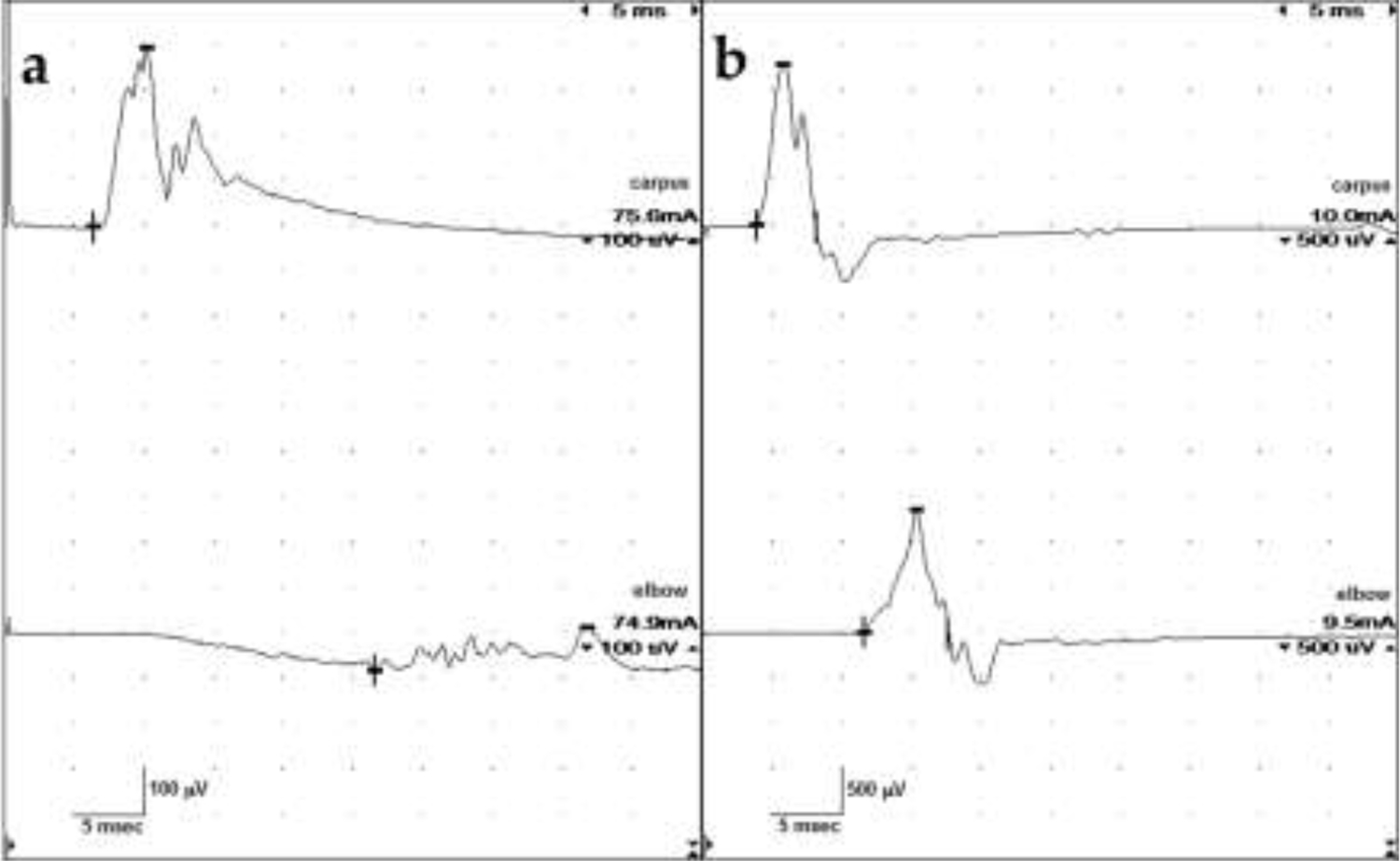
Ulnar motor nerve conduction velocity studies. (a) Case 2. Amplitudes were severely reduced. The stimulus intensity required to obtain these potentials was increased (reference <5 mA). The potentials are temporally dispersed with a conduction velocity of 7 m/s. (b) Case 3. Amplitudes are also reduced but the stimulus applied was only slightly elevated. CMAP configurations were normal but somewhat jagged in appearance, with no definitive temporal dispersion noted. Nerve conduction velocities are slow at 21 m/s.
Attempts at recording sensory NCVs were performed in Cases 1–3 (Fig. 2). None were obtainable from the sciatic/peroneal, ulnar, or radial nerves in Cases 1 and 2, though a cortical somatosensory evoked potential was recorded after ulnar and radial nerve stimulation in the latter (Fig. 2a). In Case 3, sensory NCVs were obtained from all three nerves. Velocities were slow in each (Table 2), but temporal dispersion was more pronounced on stimulation of nerves in the thoracic limb (Fig. 2b).
Fig. 2.
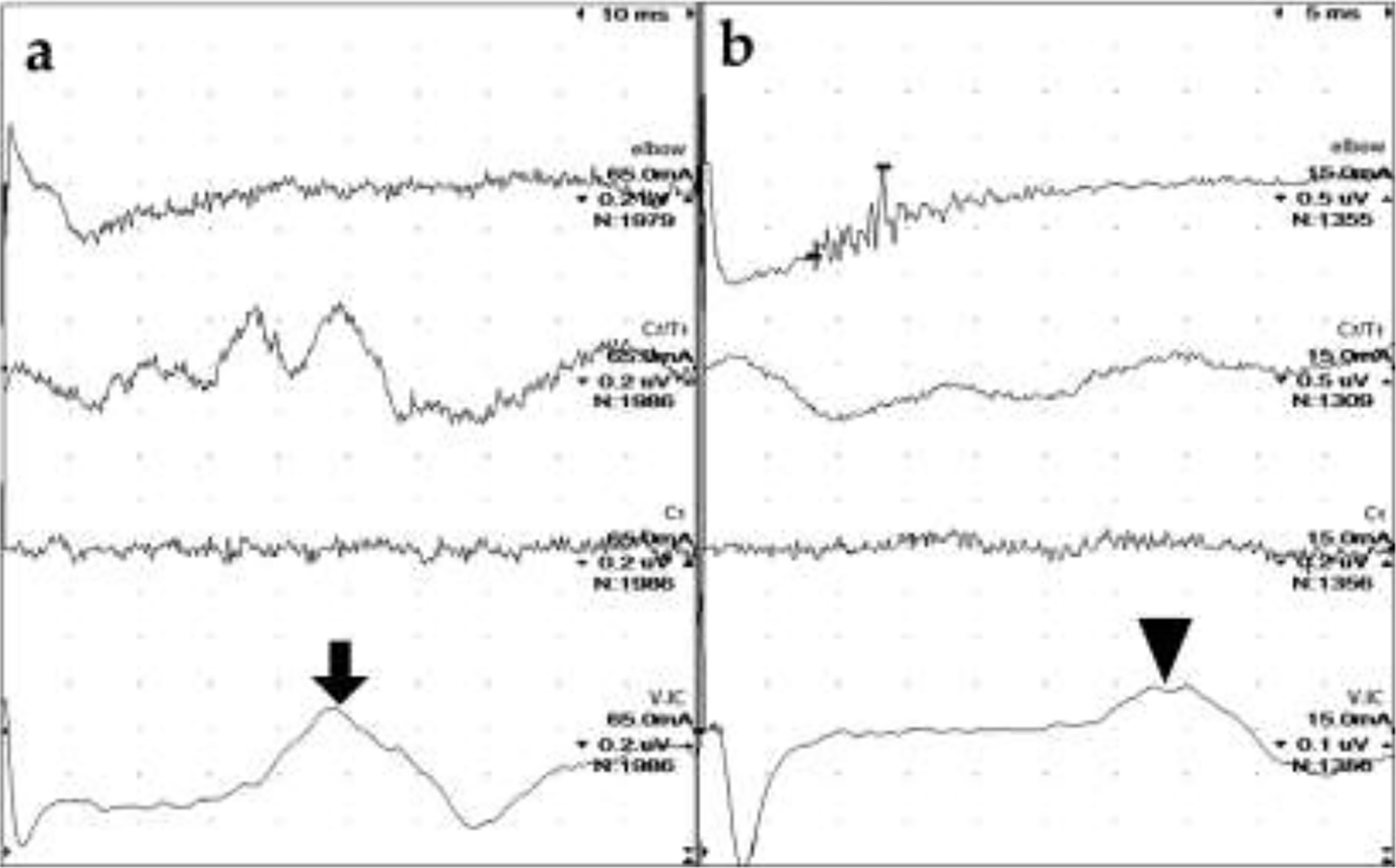
Radial sensory nerve conduction studies. (a) Case 2. No definitive SNAP (elbow), CD (C7/T1) or spinal evoked potential (C1) were apparent despite high sensitivity (0.2 μV/div) and a large number of signals averaged (nearly 2000). A cortical somatosensory response (arrow) was recorded by electrodes placed at the vertex (V) and intercanthus (IC). (b) Case 3. The SNAP is very temporally dispersed with a slow conduction velocity of 31 m/s. No CD or spinal evoked potentials were recorded but a cortical somatosensory response (arrowhead) is present. Note: the sweep speed in (a) is double that of (b) so the cortical SEP is later in Case 2 (nearly 50 ms) compared to Case 3 (nearly 35 ms). Sensitivities and stimulus intensities are also different. CD = cord dorsum
RNS studies were performed in Cases 2 and 3 only. Findings were normal, with pseudofacilitation at 50 Hz only in Case 2 and all high repetition rates (20, 30, and 50 Hz) in Case 3. Late wave studies were attempted in Cases 1–3 (Fig. 3). No obvious potentials (F wave, H waves, or A waves) were obtained from Cases 1 and 2, though presumed FP and PSW made detection difficult. In Case 2, two of the M waves (CMAPs) in the sequence lost their large component with ulnar nerve stimulation at the elbow (Fig. 3a). Case 3 had prolonged latencies on F wave testing (double that expected from a dog of this size) and increased chronodispersion was present after stimulating the peroneal nerve at the stifle. Additional late waves were recorded with ulnar nerve stimulation at the carpus but could not be definitively identified (Fig. 3b).
Fig. 3.
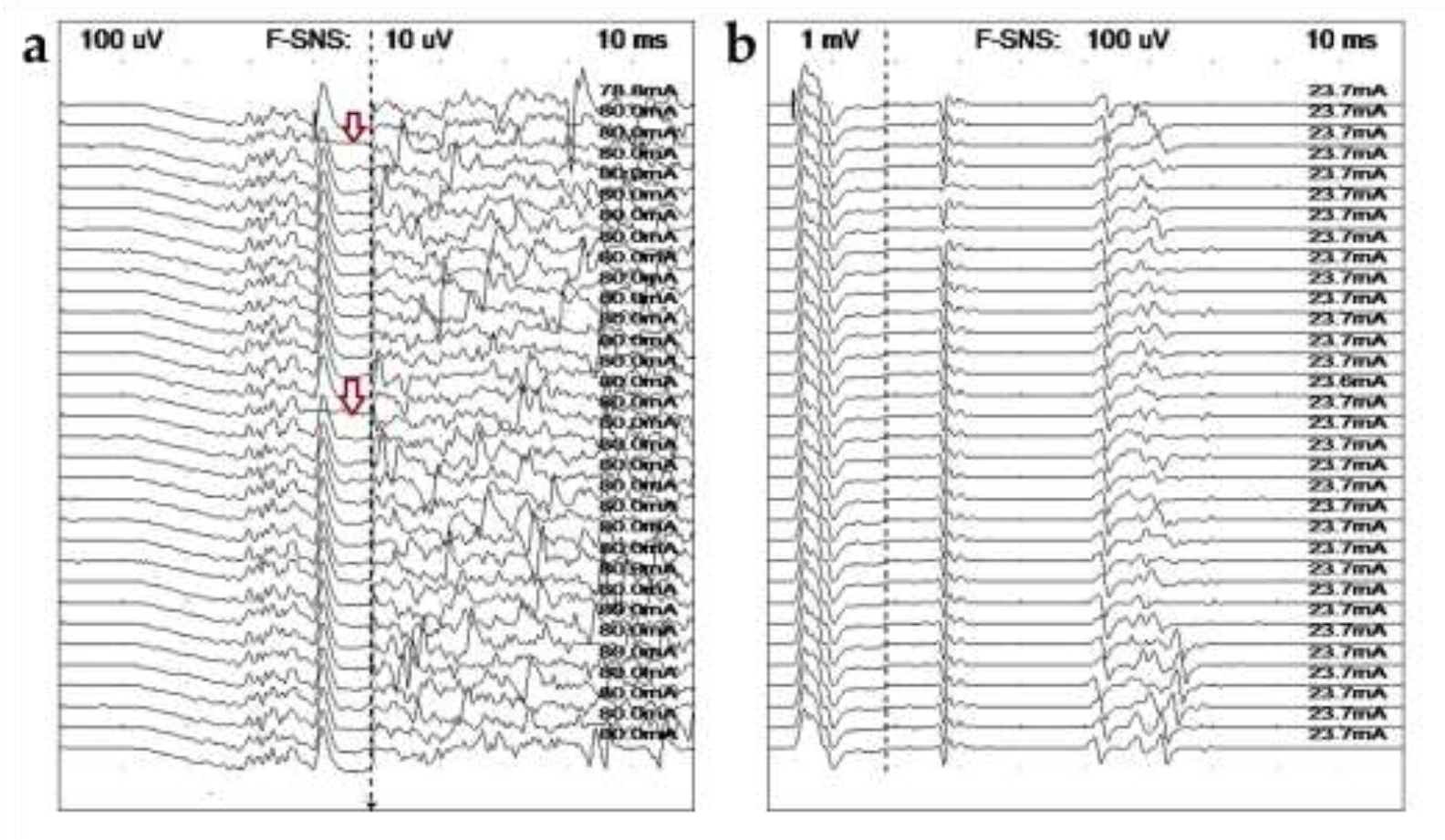
Ulnar late wave studies. (a) Case 2. Recorded from the thoracic interosseous muscle following stimulation at the elbow, no definitive late waves could be identified. Loss of the largest peak in the M-wave (CMAP) can be seen in two tracings (arrows). (b) Same study in Case 3 recorded after stimulating at the carpus, the minimum F wave latencies are prolonged, 50 ms. It is unclear whether the earlier potentials are H waves (most likely) or A waves.
In all four cases, the markedly slowed NCVs were indicative of a myelin-associated neuropathy. Consistent with clinical findings and supported by similar results from multiple nerves studied in three of the dogs, this was a generalized disorder affecting both motor and sensory nerve function. The relatively normal EMG examination in Case 3 was further evidence that motor axons were likely still intact (and innervating muscle) in this dog, whereas, in the others, secondary axonopathies had developed. Those neuromuscular junctions that were intact appeared to have normal function based on the limited RNS results. No potentials were recorded from electrodes placed along the spinal cord in Cases 1–3, despite the presence of SEP (indicating some sensory information was getting through) in the latter two dogs. Late waves were only obtained from Case 3, so no comparisons could be made.
3.3. Peripheral Nerve Light and Electron Microscopy
Sections from the peroneal nerves of Cases 1–3 stained with paraphenylenediamine and toluidine blue-basic fuchsin are shown in Fig. 4. In cross sectional preparations of Case 1, numerous inappropriately thin myelinated fibers, fibers showing myelin splitting and ballooning, and fibers with thickened myelin and redundant myelin loops consistent with tomacula were observed. In staining of longitudinal sections from Case 2, similar inappropriately thin myelinated fibers, redundant myelin loops, and myelin splitting and ballooning were noted. Transverse sections from Case 3 showed numerous inappropriately thin myelinated fibers for the axon diameter with tomacula an infrequent finding. Axonal degeneration was not observed in any of these three cases. Onion bulb formations typical of demyelinating and remyelinating polyneuropathies were not found. A peroneal nerve biopsy was not available for Case 4. However, small intramuscular nerve branches in the skeletal muscle biopsies appeared devoid of myelinated fibers.
Fig. 4.
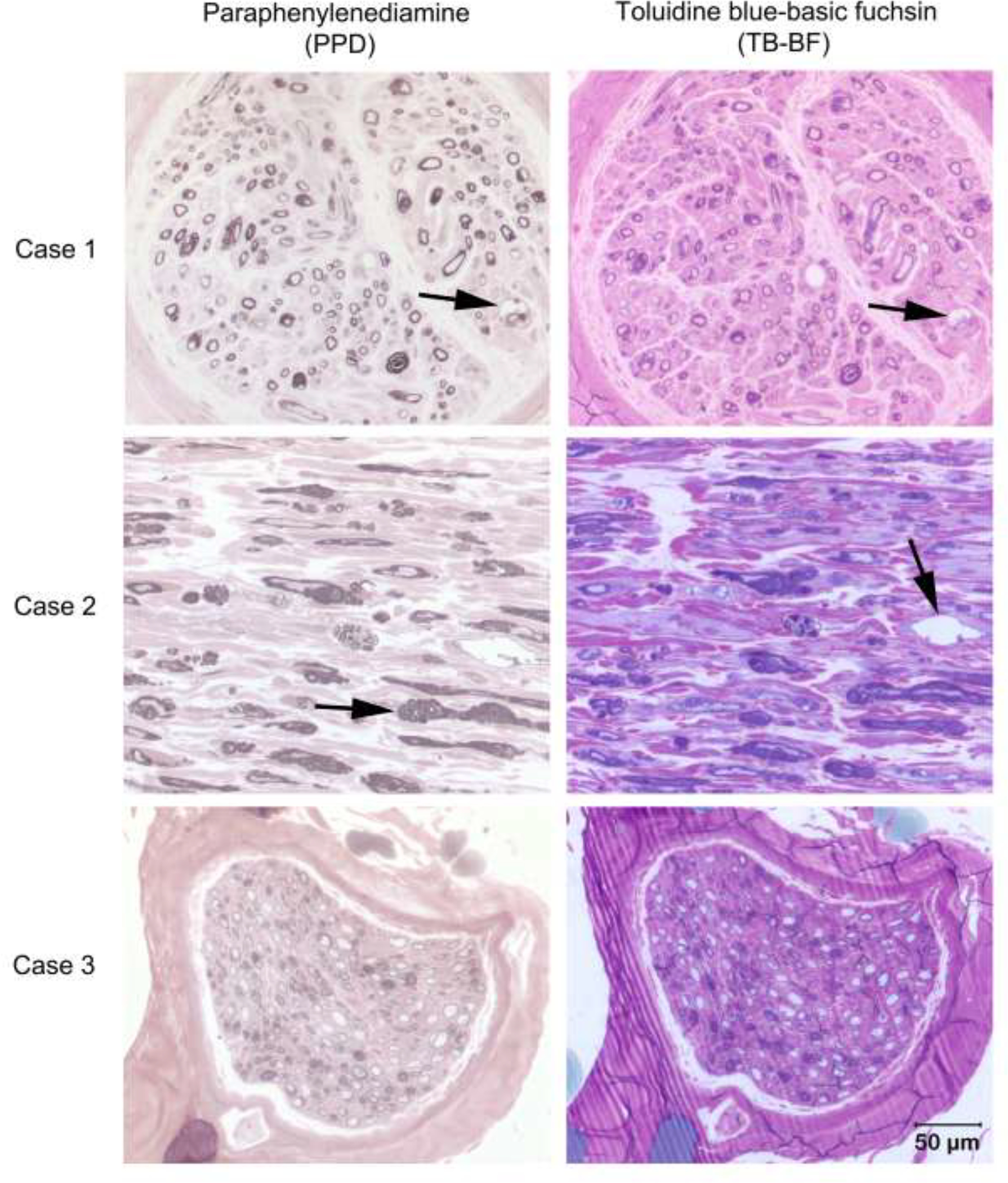
Light Microscopic evaluation of the peroneal nerve from Cases 1–3. Transverse (Cases 1 and 3) and longitudinal (Case 2) sections from resin embedded peroneal nerve stained with paraphenylenediamine (PPD in left column) for myelin and toluidine blue-basic fuchsin (TB-BF in right column). In all three cases most myelin sheaths are inappropriately thin for the axon calibers. In Cases 1 and 2 thickened myelin sheaths (arrow in Case 2 PPD stain) and redundant myelin loops consistent with tomacula are noted along with myelin ballooning (arrows in Cases 1 PPD and TB-BF and 2 TB-BF). Bar in right lower corner = 50 μm for all images.
Ultrastructural analysis of the peroneal nerve was performed on Case 1 (Fig. 5). Multiple outpouchings of myelin and redundant myelin loops were present in addition to inappropriately thin myelin sheaths for the axon diameter, confirming the light microscopic findings. The pathological changes are characteristic of a predominantly hypomyelinating neuropathy without obvious axonal degeneration or onion bulb formation.
Fig. 5.
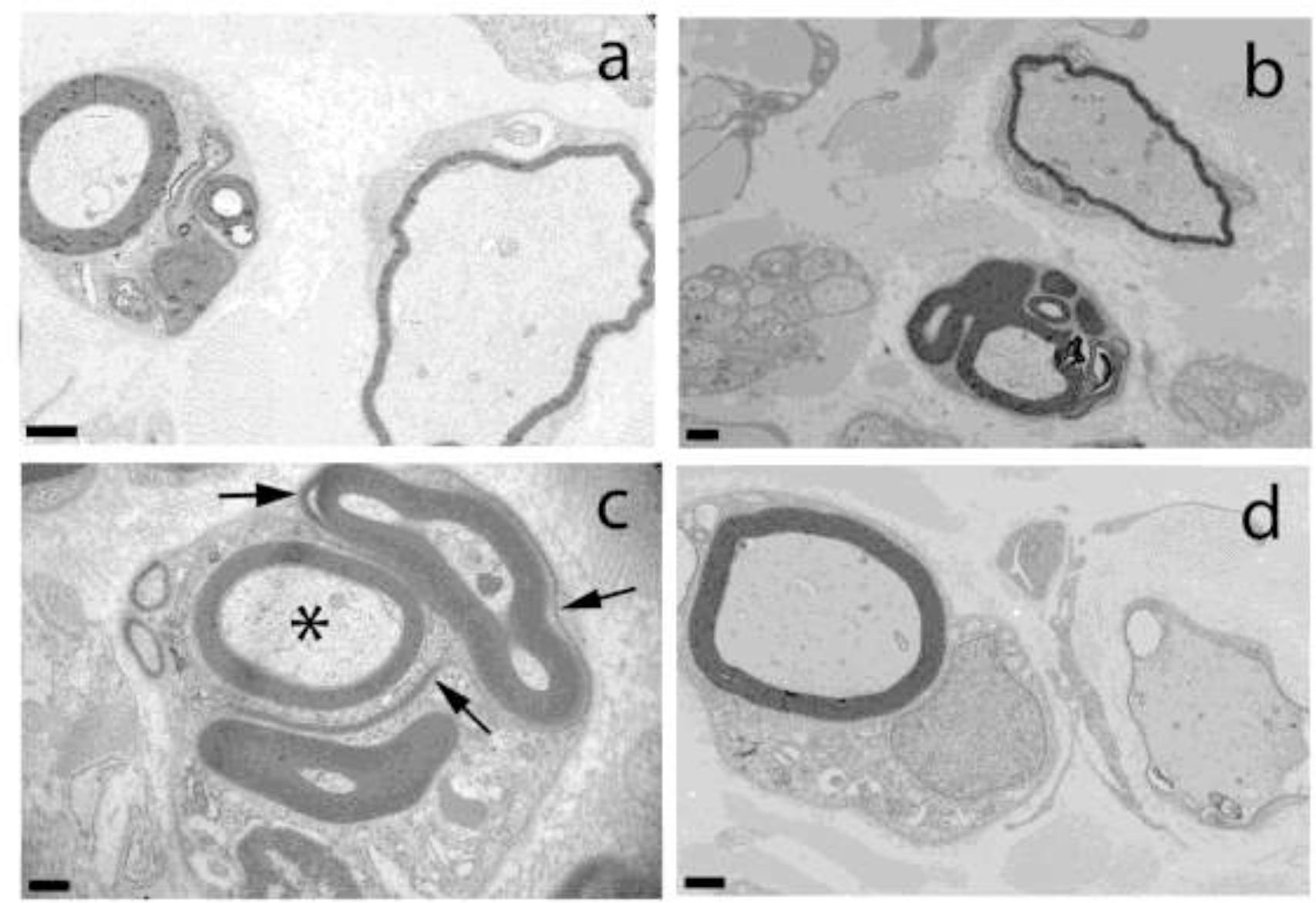
Electron Microscopic evaluation of the peroneal nerve from Case 2. Ultrastructural analysis of the peroneal nerve from Case 2 demonstrating multiple outpouchings of myelin and redundant myelin loops in large and small nerve fibers (a-c). In c, note disintegrating myelin loops (arrows) and intact axon (asterisk). Inappropriately thin myelin sheaths for the axon diameters are shown in a, b, and d. Bar in lower left corner = 1000 nm for a, 1 μm for b, 0.5 μm for c, and 1 μm for d.
Skeletal muscle changes in all cases were minimal and characterized by a mild to moderate variability in myofiber size and scattered angular atrophied fibers. Rare areas of fiber type grouping were noted. There was no evidence of inflammation, fibrosis, or other specific cytoarchitectural abnormalities (not shown).
3.4. Genetic Relationship and Inbreeding
The PLINK [38]-created relationship matrix indicated that the four affected dogs were not any more related to each other than any other GRs in the available data. However, the --genome command indicated that Cases 1 and 3 may be distantly related with a pi-hat of 0.0633. All other pairs of affected dogs shared a pi-hat of 0. Of the four affected dogs, Cases 2 and 4 were the most homozygous, with --het command generated F coefficient estimates of 0.2555 and 0.1587, respectively. Cases 1 and 3 had F coefficients no larger than the average GR in the dataset, with values of −0.04844 and −0.0357, respectively.
3.5. GWAS and WGS
An initial GWAS failed to identify any regions of interest under the assumption that all three cases (Cases 1, 2, and 3) shared a causative variant. Therefore, WGS of all three cases, plus one additional case (Case 4), was performed. WGS data was first analyzed under the assumption that all four cases shared a novel variant that was not in LD with any SNP markers, however this also failed to identify any shared, private variants among the four cases. Finally, each GR HPN case was analyzed individually, excluding the other three HPN GRs, under the assumption that they may not all share the same genetic cause for their disease; indeed, this turned out to be the case.
3.6. Cases 1 and 2
Cases 1 and 2 shared a homozygous MTMR2 (myotubulin related protein 2) variant located within the splice site at the end of exon 12 (Fig. 6); variants in this gene cause Charcot-Marie-Tooth type 4B1 [60,61]. This G to A transition at CFA21: 5,387,227 (XM_038568229.1:c.1479+1G>A) is predicted to result in the loss of the splice donor site. The splice site detection software, NNSplice 0.9, was used to predict the effect of the mutation on splicing, with scores greater than 0.4 indicating a possible splice site [51]. The wild-type sequence generated a donor site prediction score of 0.92, however, replacing the wild-type G allele with the variant A allele led to the donor site no longer being predicted. Similarly, the Spliceator [52], Human Splicing Finder (HSF Pro from Genomnis) [50], and maximum entropy (MaxEnt) [53] algorithms also predicted that the variant would create a broken donor splice site.
Fig. 6.
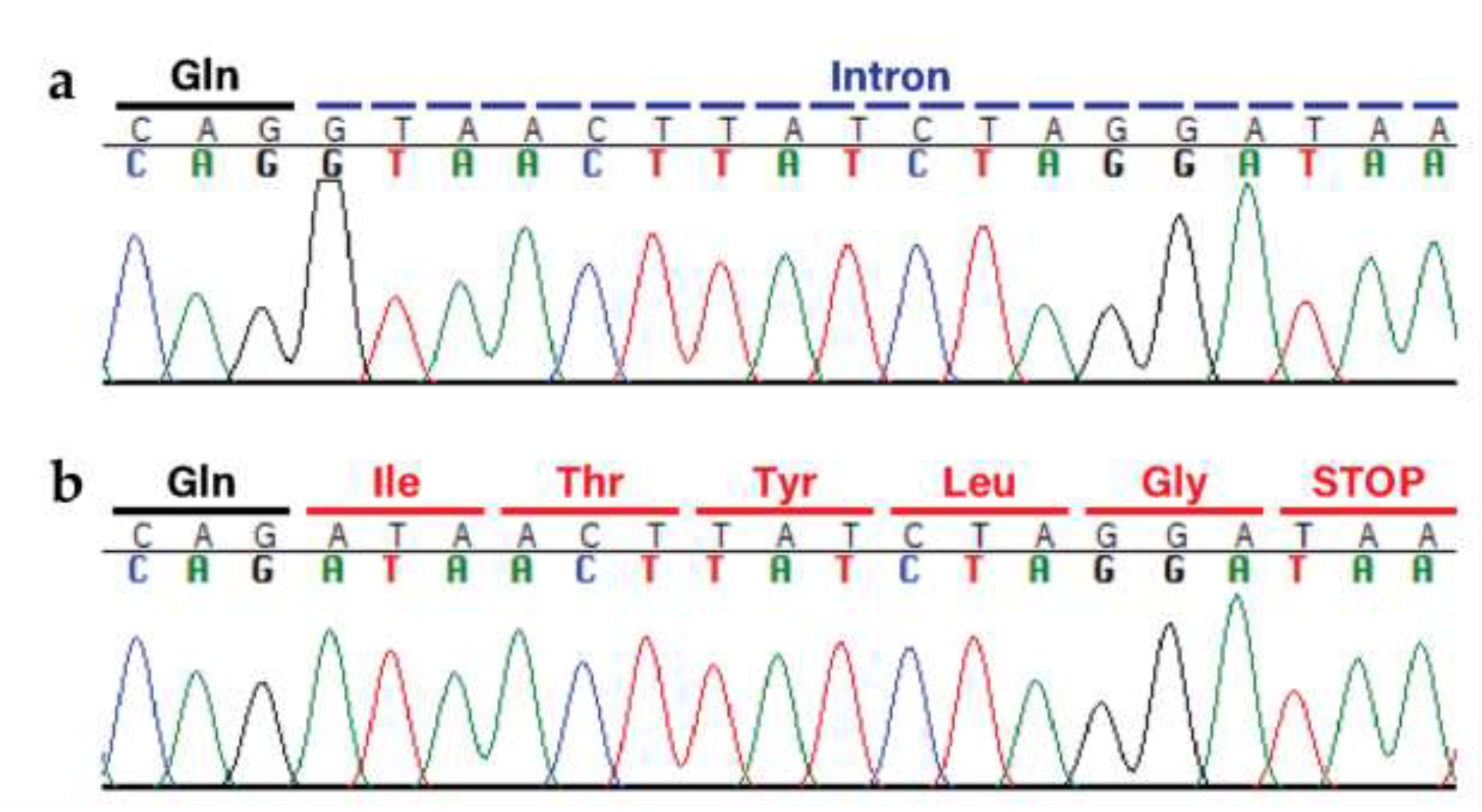
The identified MTMR2 variant XM_038568229.1:c.1479+1G>A. a Wild-type sequence electropherogram from an unaffected dog with the original glutamine at the end of exon 12 (marked in black) and the following intron (marked in blue). b Mutant sequence electropherogram from Case 1 (Case 2 looks identical), demonstrating the single base change, loss of the splice donor site, and subsequent transcription (with translated codons marked in red).
This MTMR2 variant was private to Cases 1 and 2 compared to 1,025 other dogs with available WGS data (Table 3). VarElect analysis assigned MTMR2 a score of 35.71 for both cases (n = 29 and 33 genes in put for Cases 1 and 2, respectively). This was the largest score allotted for both cases.
Table 3.
Variant counts for each HPN-affected GR. Each dog retained >7,000 private variants after excluding all variants found in >1,000 control dogs. All identified variants were processed through VEP, with variants of high or moderate effect, i.e., those that were predicted to alter the amino acid sequence, prioritized. A VarElect score >10 indicates the best disease-causing candidate genes. CFA = Canis lupus familiaris chromosome; VEP = variant effect predictor
| Case 1 | Case 2 | Case 3 | Case 4 | |
|---|---|---|---|---|
|
| ||||
| Variants on CFA 1–39 | 7,718 | 7,568 | 7,870 | 7,454 |
| Variants of High, Moderate, and Low Effect | 80 | 72 | 77 | 86 |
| Variants Altering Protein Sequence | 61 | 57 | 58 | 64 |
| VarElect Score ≥10 | 1 | 1 | 1 | 1 |
Population screening of 225 GRs demonstrated all dogs were homozygous wild-type for this MTMR2 variant. Haplotype analysis using 525 GRs with previously generated Illumina SNP array data built a 12 SNP haplotype, ranging from CFA21: 5,284,640 to 5,502,102, and containing the variant. Cases 1 and 2 shared this haplotype homozygously; four and 103 other GRs had the same haplotype homozygously and heterozygously, respectively. One of the four non-case GRs homozygous for the haplotype was tested for the MTMR2 variant; Sanger sequencing proved that this dog was homozygous wild type. Seventeen GRs included in the SNP data who shared the haplotype heterozygously also had WGS data in the previously described dataset, and all were homozygous wild type for the variant.
3.7. Case 3
Case 3 had a heterozygous missense MPZ (Myelin protein zero) variant, located in exon 3 (Fig. 7). MPZ is a well-recognized causative gene for various Charcot-Marie-Tooth subtypes [62–64]. Case 3’s amino acid change, an isoleucine to threonine (CFA38: 22,037,876; XP_038441854.1:c.Ile145Thr), was predicted to be deleterious via multiple programs (Table 4). This missense variant was private to Case 3 compared to 1,026 other dogs with WGS (Table 3) and VarElect ranked MPZ as the most strongly associated gene (out of 30) for the submitted keywords, assigning a score of 87.93. For comparison, the second largest score was 6.6.
Fig. 7.
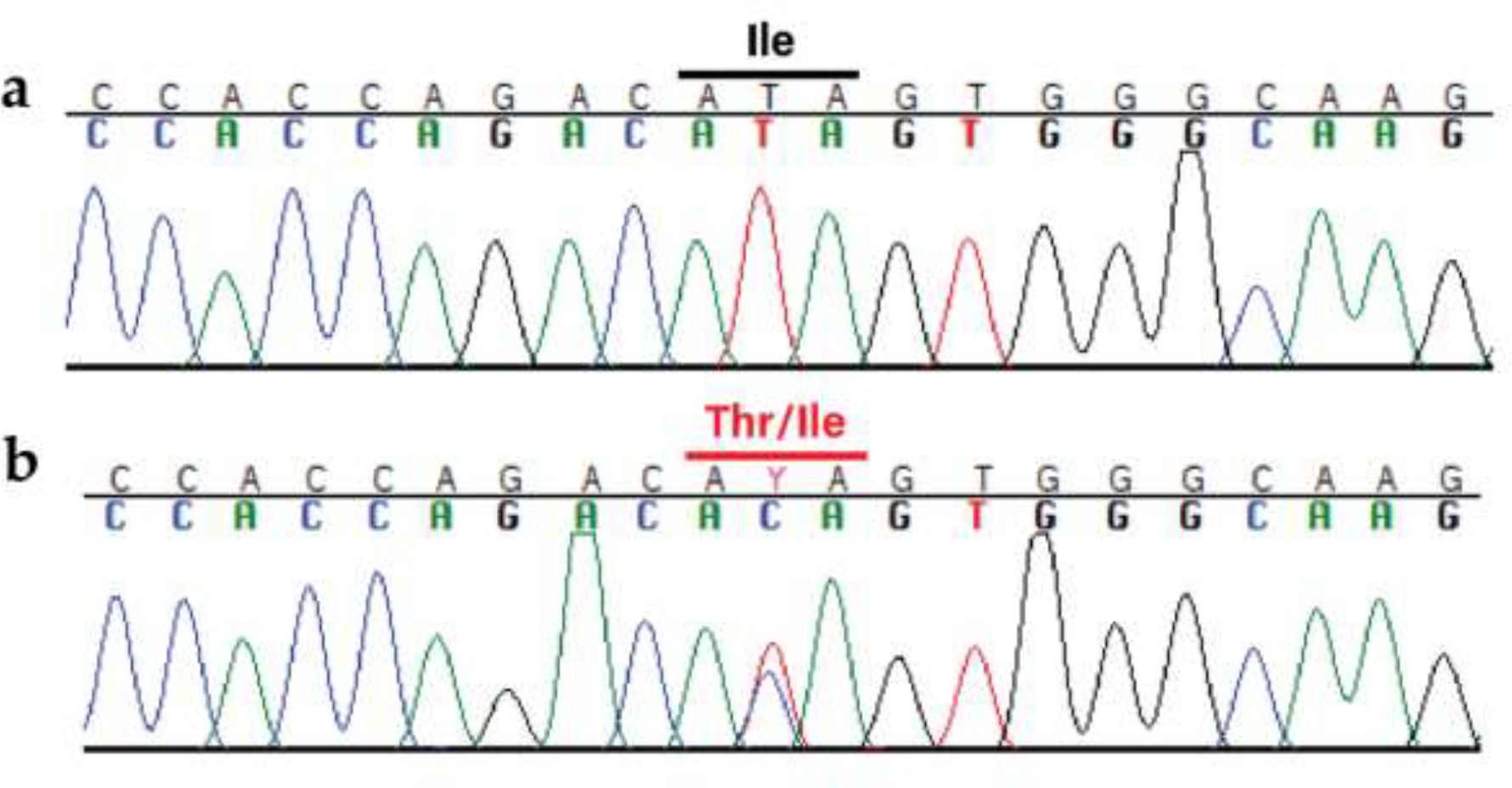
The identified MPZ variant XP_038441854.1:c.Ile145Thr. a Wild-type sequence electropherogram from an unaffected dog, with the isoleucine marked in black. b Mutant sequence electropherogram from the affected GR (Case 3), with the heterozygous codon marked in red. The variant changes the isoleucine on one strand to a threonine.
Table 4.
Protein pathogenicity predictive software results for the Case 3 missense variant. Four of the six programs predicted the variant to be deleterious (D) to the MPZ protein, one program indicated that it is tolerated (T), and one program was uncertain of the effect of the amino acid substitution
| MPZ | |
|---|---|
|
| |
| PON-P2 | 0.806 (Unknown) |
| MutPred2 | 0.293 (T) |
| PROVEAN | −3.927 (D) |
| PANTHER | 0.5 (Possibly D) |
| PolyPhen-2 | 0.997 (Probably D) |
| Mutation Taster 2021 | D |
All screened GR population samples (n = 215) were homozygous wild-type. Haplotype analysis using the population of GRs with Illumina-level SNP data failed to generate any haplotype blocks, as the variant fell between blocks. Subsequent analysis of Affymetrix SNP array data did place the variant within a haplotype block; however, it was a very small region of three SNPs totaling 5kb. This haplotype had the highest frequency of the three in this region (0.448 vs 0.311 and 0.236).
3.8. Case 4
Case 4 possessed a homozygous SH3TC2 (SH3 domain and tetratricopeptide repeats 2) variant in exon 11 of 17 (Fig. 8a,b). This C to T nonsense mutation (CFA4: 60,798,310 (XP_038391009.1:p.Arg642*)) was private to Case 4 compared to 1,026 other dogs with WGS data available (Table 3). Approximately half of the protein is predicted to be truncated (Fig. 8c). VarElect analysis prioritized SH3TC2, assigning it the largest score (49.53) of all genes input (n = 30). Variants in SH3TC2 are associated with Charcot-Marie-Tooth type 4C [65–67].
Fig. 8.
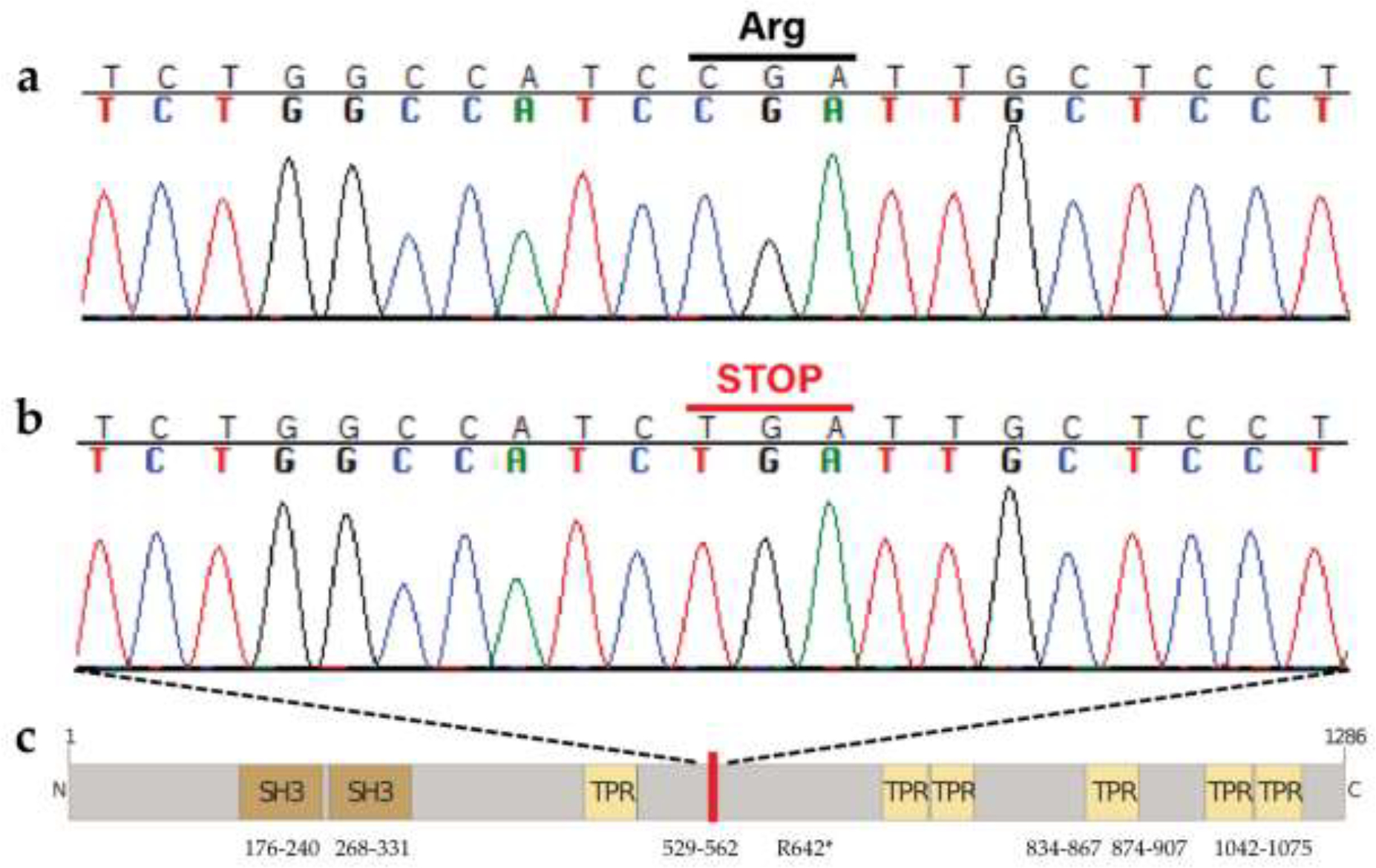
The identified SH3TC2 variant XM_038568229.1:c.1479G>A. a Wild-type sequence electropherogram from an unaffected dog with the original arginine indicated, and b mutant sequence electropherogram from the affected GR (Case 4) with the premature stop codon, introduced halfway through the amino acid sequence, at position 642 out of 1286. c Predicted protein structure and domains, with the red bar marking the location of the stop codon. Amino acid positions are indicated.
Using RFLP with the BseGI enzyme, 248 GRs were homozygous wild-type, inclusive of the three other HPN cases in this study. Haplotype analysis using 525 GRs with previously generated Illumina SNP array data identified a 12 SNP block, spanning from CFA4: 60,684,633 to 60,859,652, which included the variant’s location. Case 4 shared this haplotype homozygously with seven other GRs and heterozygously with 80 GRs. One of the seven dogs who shared the haplotype homozygously was tested for the SH3TC2 nonsense variant, and Sanger sequencing indicated that this dog was homozygous wild type. Additionally, 15 GRs included in the SNP data were also in the previously described WGS dataset and are heterozygous for the haplotype and homozygous wild type for variant.
3.9. Follow-Up on Cases
Case 1 went home after the diagnostic examination at UC Davis but returned to the clinic 17 days later with profound worsening of signs including intractable regurgitation and vomiting. The owner elected to take Case 1 home the same day for euthanasia by the referring veterinarian. Cases 2 and 3 were euthanized by referring veterinarians 40 and 13 days, respectively, after discharge from UC-Davis, due to progressive severe generalized weakness and dysphagia. Case 4 was evaluated for two episodes of aspiration pneumonia secondary to laryngeal paralysis and esophageal dysfunction. Euthanasia was requested twenty-four months after initial presentation. Postmortem examinations were not performed in any of the cases.
4. Discussion
This work describes, for the first time, the genetic causes of a PNS-exclusive HPN in GRs, a phenotype first described clinically in the GR more than 30 years ago [32,33]. Despite the similarities in electrodiagnostic testing, most notably markedly decreased motor and sensory NCVs, and hypomyelination of peripheral nerves evaluated at the light microscopic and ultrastructural levels, the genetic investigations demonstrated that they are not all identical genetically. This explains the failure of the initial GWAS, where all cases were assumed to have the same underlying genetic cause; future studies should be cognizant of the potential for genocopies, even within the same breed, ultimately driven by different mutations. Moreover, although the breed, clinical signs, and peripheral nerve pathology are reminiscent of the decades old first-described cases [32,33], there is no way to ascertain which, if any, of the three described variants these earlier dogs may have had. Indeed, the older GRs may have had none of the variants identified in these novel cases.
Both demyelinating and hypomyelinating disorders are encompassed by CMT disease [11,14,68]. Distinctive “onion bulb” formations in peripheral nerve biopsies aid in determining demyelinating conditions, distinguishing these biopsies from hypomyelinating conditions. Onion bulbs are formed from repetitive attempts by Schwann cells to regenerate lost myelin, and appear as distinctive rings of Schwann cell processes and collagen around the nerve fibers [69]. HPN produces either no onion bulbs or atypical onion bulbs, the latter of which are composed of basement membrane and little or no Schwann cell processes [8]. Onion bulb formations were not identified in the peripheral nerve biopsies of Cases 1–3. In addition, axonal degeneration was also not observed.
All three identified variants were located in candidate genes where known mutations cause myelination disorders of the PNS [12,70]. The homozygous variant in Cases 1 and 2 was located at a splice donor site between MTMR2’s exon 12 and intron 13. While sequencing of mRNA for this gene would allow for definitive confirmation of the loss of the donor site, unfortunately, tissues were not collected in a way to maintain RNA stability and all dogs are now deceased. If intron 13 following the splice site is aberrantly included due to erroneous RNA splicing, then five additional amino acids are added to the protein followed by a premature stop codon, which is predicted to truncate 23% of the mRNA/protein. Both Schwann cells and motor neurons express this protein, however, disruption of the gene in Schwann cells leads to a dysmyelinating phenotype, whereas disruption in motor neurons has no effect [60].
MTMR2 mutations are responsible for CMT type 4B1 in humans [12], where it is an early-onset disease with a recessive mode of inheritance [12,71]. This coincides with the clinical presentation of GR Cases 1 and 2. Laryngeal paralysis, inspiratory stridor, and dyspnea with exercise were noted in Cases 1 and 2. CMT4B1 is likewise associated with laryngeal involvement and vocal cord paralysis in humans [72], demonstrating a strong phenotypic correlation.
Based on electrodiagnostic (EDX) findings, Cases 1 and 2 were the most severely affected dogs in this study. Laryngeal EMG abnormalities were consistent with respiratory dysfunction seen clinically and the presence of numerous FP and PSW in distal limb and axial musculature suggests secondary axonal pathology. The lack of distal motor NCVs in the pelvic limbs, conduction block on ulnar NCVs, and loss of all sensory NCVs in both dogs are consistent with features of severe CMT in people [73]. The loss of parts of the CMAP on Case 2’s late wave study (Fig. 2a) likely indicates a decrease in the safety factor of transmission associated with decreased myelin in the ulnar nerve [35].
A defining CMT4B1 characteristic is the presence of peripheral nerve myelin loops which increase in number as the patient ages [12,60,61,74]; this key finding of myelin loops was likewise noted in peripheral nerve biopsies from both GR Cases 1 and 2. In people, CMT4B1 is characterized as a demyelinating sensorimotor polyneuropathy and not a primary hypomyelinating polyneuropathy [60,61]. In cases 1 and 2, there was no evidence of remyelination including onion-bulb formations. Alternatively, demyelination occurred with suboptimal remyelination.
The majority of CMT4B1 MTMR2 variants are loss-of-function mutations, with most protein truncations occurring within MTMR2’s singular phosphatase domain [61]. This domain’s catalytic function involves dephosphorylation of a variety of phosphatidylinositols involved in response to cellular stress and the recycling of membranes [75–77]. The phosphatase domain is predicted to consist of amino acids 133 to 508 in the dog [78]; the homozygous mutation in Cases 1 and 2 is located after Q421 and therefore located within the canine phosphatase domain, consistent with most CMT4B1 cases. Finally, and most convincingly, a splice site variant identical to the one described in GR Cases 1 and 2 has been associated with CMT4B1in human patients [79].
GR Case 3 possessed the second newly identified variant, a heterozygous missense mutation in MPZ’s third exon. This changes an isoleucine residue to a threonine residue; while isoleucine is a nonpolar, hydrophobic amino acid, threonine is polar and uncharged, a change that could interfere with the normal secondary protein structure. It is important to note that this protein is the most abundant in PNS myelin [80].
Heterozygous MPZ variants are causative of CMT types 1B, 2I, 2J, and DID, as well as Roussy-Levy syndrome and congenital hypomyelinating neuropathy-2 (CHN2) [64,81–91]. Different mutations within the gene affect the age of onset [92,93]. In humans, some variants result in an early-onset neuropathy, while others are associated with later-onset symptoms around age 40 [93]. GR Case 3’s missense isoleucine to threonine change is directly analogous to a previously described I135T MPZ mutation in several human CMT1B patients [63,94,95]. The I135T mutation is classified as an infantile-onset CMT1B mutation, with age of onset between 0 to 5 years, as opposed to childhood- or adult-onset [70]. It is unknown why GR Case 3 was not examined until the age of 1.5 years. It is possible that clinical signs were manifesting at a younger age but were mild enough the owner was not prompted to seek medical evaluation. Despite this dog’s age (three times that of the others), Case 3 was the least affected in terms of EDX results. Mild EMG abnormalities in the interosseous muscles are common in normal dogs and the focal nature of the gluteal changes were thought to be the result of a recent injection in this muscle. Motor NCVs were slow and CMAP amplitudes decreased, but no temporal dispersion or conduction block was observed. Sensory NCV abnormalities were more pronounced with temporal dispersion seen in the ulnar and radial studies, although the peroneal SNAPs were normal. The prolonged F latencies and lack of CDs suggest motor and sensory proximal pathway involvement. Case 3 clearly was the most severe and had progressive clinical signs (Table 1), which contrasts with having the mildest EDX changes. This might be due to older age at presentation and therefore a more advanced condition. However, it is also possible that this observation results from the specific MPZ mutation, as compared to the clinical signs and EDX changes seen with the MTMR2 and SH3TC2 mutations.
Of note, Case 3 exhibited coxofemoral joint laxity and joint pain, clinical signs of hip dysplasia. Human CMT patients are predisposed to hip dysplasia as children, particularly those with CMT1 subtypes [96–98], consistent with the clinical presentation of GR Case 3.
Case 4 had a variant within SH3TC2, which is exclusively expressed in Schwann cells [99], and homozygous knockout mice have a PNS-specific phenotype where the peripheral nerves are noticeably hypomyelinated with decreases in both motor and sensory nerve conduction velocities; intriguingly the axons appeared healthy after one year [100]. These mice exhibited normal development indistinguishable from control mice, were fertile, and had the same lifespan as both their wild-type and heterozygous littermates [100]. In humans, more than 20 different SH3TC2 mutations, including nonsense mutations such as the one observed in Case 4, have been reported in patients with CMT4C [12]. Also similar to Case 4, CMT4C has an early age of onset, a recessive pattern of inheritance [12,101,102] and weakness, particularly of the lower limbs [102,103]. Human CMT4C patients have few classic onion bulbs and many unmyelinated fibers and, interestingly, scoliosis is also observed in the majority of CMT4C patients [12]. While Case 4 did not present with scoliosis, this is consistent with the SH3TC2 knockout mice, which had no notable vertebral abnormalities [100]. GR Case 4 also presented with regurgitation, a clinical sign not typically reported in human CMT4C patients. This may be due to the species differences in muscle composition along the esophagus; in humans, the upper esophagus is skeletal muscle, but transitions to entirely smooth muscle in its lower part [104], whereas the dog has striated skeletal muscle throughout the entire length of the esophagus [105]. This difference may account for the regurgitation seen in this dog. Another possibility may be related to the esophageal anatomical orientation differences in dogs (horizontal) versus humans (vertical); the lack of gravity’s pull in dogs could provide an added reason for the regurgitation.
Case 4 appeared to be moderately affected based on EDX, when compared to the other dogs in this study. EMG changes were similar to that of the MTMR2 dogs, but MCVs were obtained from the EDB upon peroneal nerve stimulation. NCVs were slower and CMAP temporal dispersion was present indicating more severe involvement than that seen in the MPZ dog. Conduction block may be present as the amplitudes drop by 50% between the hock (3.0 mV) and stifle (1.5 mV) recordings, but the hip (2.0 mV) is a confounding feature.
Case 4 was from a litter in which three littermates died shortly after birth and two other littermates showed clinical signs of weakness, one female and one male. It is theoretically possible that the spectrum of clinical signs for the SH3TC2 variant in the GR is yet to be fully characterized. CMT4C in people consists of a broad spectrum of phenotypes, ranging from mild to severe [101–103,106], and the slight dissimilarities between clinical signs in the canine versus human cases can still be explained by SH3TC2 mutations. Due to the nonsense variant and subsequent truncation occurring halfway through the protein in Case 4 (Fig. 8c), it is likely that the mRNA undergoes nonsense-mediated decay, leaving this dog with no functional SH3TC2.
Cases 1–3 exhibited respiratory distress, particularly after periods of exercise or excitement consistent with the human CMT equivalent conditions. Specifically, CMT4 subtypes are associated with respiratory distress in some, although not all, patients [61,107], and CMT1 affected patients have reductions in total lung capacity, forced vital capacity, maximal expiratory pressure, and maximal inspiratory pressures [108]. Case 4 did not have respiratory signs at the time of presentation, but clinical signs of laryngeal paralysis developed prior to euthanasia. All four GR cases exhibited a decreased gag reflex predisposing to dysphagia, likewise seen in human cases of CMT [79,109,110]. Furthermore, MTMR2, MPZ, and SH3TC2 are all expressed in Schwann cells, consistent with perturbations of the PNS [60,100,111]. Taken together, the four affected GRs had genetic variants in genes associated with subtypes of CMT, with strikingly consistent clinical correlations.
The presumptive origin and mode of inheritance varies for the three canine mutations described here. The heterozygous MPZ variant is inherited in an autosomal dominant fashion and was likely a de novo mutation in this single case. The MTMR2 and SH3TC2 variants, conversely, are autosomal recessive. Because it is much less likely that a de novo mutation occurred homozygously, the SH3TC2 and MTMR2 variants are more likely to be present in the GR population at a low level and inherited by these dogs from carrier parents, despite finding no additional carriers in the population screening. This is particularly true for the MTMR2 variant because it was identified in two unrelated GRs. Indeed, neither of the GRs with this variant appear to be related, with a pi-hat of 0, and only one of the dogs, Case 2, had an increased F coefficient of inbreeding. It is possible that this disease arose in Case 2 due to breeding of related individuals, however, there is no evidence of inbreeding in Case 1, which shares the same variant. With these dogs seemingly unrelated, speculation remains whether this variant arose in one distant common ancestor to both GRs or if the variant has spontaneously occurred in more than one dog. The possibility of direct inheritance cannot be tested, as DNA was not available from any parents. It is likely that the MTMR2 and SH3TC2 variants are either present in the breed at a very low frequency or are present in families not represented within this study’s population sample, potentially both. Further supporting the nascent emergence of these mutations is the fact that all three variants arose on existing GR haplotypes. As these haplotypes are not tagging the variants, this further supports the theory that the variants only exist in small pockets of the GR breed.
The original GR cases from more than 30 years ago had not progressed in severity 2.5 years after their initial diagnosis [32,33]. However, neither of the dogs in the original study [32,33] were reported to have dysphagia, esophageal dilation, or laryngeal paresis as affected the four dogs in this current study. Dysphagia and esophageal dilation can result in aspiration pneumonia and early death or euthanasia requested by the owners; as seen in the present four cases, regurgitation, vomiting, dysphagia, and aspiration pneumonia unquestionably contributed to decreased quality of life and decisions to euthanize. Similar to other congenital neuromuscular diseases in dogs, such as X-linked myotubular myopathy (MTM1 gene)[112] and X-linked (Duchenne-like) muscular dystrophy (DMD gene)[113], which recapitulate human diseases and have proven to be useful large animal therapeutic models, these GR diseases could likewise contribute to therapeutic trials for the equivalent human diseases. However, given the rarity of these variants in the GR breed population, it may prove difficult to develop a breeding colony of affected dogs; given its occurrence in two cases, the MTMR2 mutation would be the most likely candidate for such future work.
Together, these three variants are highly likely to explain the clinical syndromes of these four dogs. The mutations are in highly relevant candidate genes and either create highly pathogenic changes to predicted protein structure (with two leading to premature stop codons) or literally recapitulate the exact mutation previously described in human patients. The MTMR2 variant, present homozygously in two affected dogs, is perhaps the most likely to affect additional GRs. Genetic testing of the SH3TC2 and MTMR2 variants in a much larger population is needed to establish the actual frequency of these alleles in the breed. Testing for these two mutations would provide breeders with needed information to prevent the production of affected offspring. Testing for the MPZ variant may not be indicated, as it was likely a de novo mutation. In conclusion, these dogs represent the first confirmed large animal genetic models for PNS-exclusive hypomyelination and for CMT types 4C, 4B1, and 1B.
Supplementary Material
Highlights.
Neurological testing confirmed a hypomyelinating polyneuropathy (HPN) in four dogs
Variants in MTMR2, MPZ, and SH3TC2 were identified by whole-genome sequencing
These variants genetically describe the first peripheral nervous system-exclusive HPNs in dogs
Cognizance of potential genocopies is vital for genetic studies, even in one breed
Acknowledgements:
The authors gratefully thank Dr. Dayna Dreger for her assistance with whole-genome-sequence data management. Drs. Richard A LeCouteur, Beverly Sturges, and Kathryn Winger are thanked for their contributions to clinical case evaluations.
Funding Information:
This work was funded by a Student Canine Research Grant from the Theriogenology Foundation and a Veterinary Student Scholar award from the Morris Animal Foundation. Partial support for KJE was provided by the Office of the Director, National Institutes of Health, under award number K01-OD027051. SGF is supported in part by an NIH Special Emphasis Research Career Award (K01 OD027058) in Pathology and Comparative Medicine sponsored by the Division of Comparative Medicine, Office of Research Infrastructure Programs.
Footnotes
Conflict of Interest: The authors declare they have no conflicts of interest.
Declaration of Interest
None for any authors or co-authors on “Canine models of Charcot-Marie-Tooth: MTMR2, MPZ, and SH3TC2 variants in Golden Retrievers with congenital hypomyelinating polyneuropathy” by Cook, et al. declare any conflicts of interest.
Ethics Statement: IACUC was not required because these dogs were clinical patients at two different veterinary hospital institutions. All other SNP and WGS data derived from publicly available sources or private internal data from other projects.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability:
The whole genome sequence for the four affected Golden Retrievers is available in Sequence Read Archive (SRA) under accession numbers: Cases 1 and 2: SRR23867282 and SRR23867281; Case 3: SRR23628641; and Case 4: SRR23628642.
References
- [1].Bizzi A, Castelli G, Bugiani M, Barker PB, Herskovits EH, Danesi U, et al. Classification of Childhood White Matter Disorders Using Proton MR Spectroscopic Imaging. Am J Neuroradiol 2008;29:1270–5. 10.3174/ajnr.A1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Carroll SL. The Molecular and Morphologic Structures That Make Saltatory Conduction Possible in Peripheral Nerve. J Neuropathol Exp Neurol 2017;76:255–7. 10.1093/jnen/nlx013. [DOI] [PubMed] [Google Scholar]
- [3].Simons M, Nave K-A. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb Perspect Biol 2016;8:a020479. 10.1101/cshperspect.a020479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Salzer JL. Schwann Cell Myelination. Cold Spring Harb Perspect Biol 2015;7:a020529. 10.1101/cshperspect.a020529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Nelis E, Haites N, Van Broeckhoven C. Mutations in the peripheral myelin genes and associated genes in inherited peripheral neuropathies. Hum Mutat 1999;13:11–28. . [DOI] [PubMed] [Google Scholar]
- [6].Fratta P, Ornaghi F, Dati G, Zambroni D, Saveri P, Belin S, et al. A nonsense mutation in myelin protein zero causes congenital hypomyelination neuropathy through altered P0 membrane targeting and gain of abnormal function. Hum Mol Genet 2019;28:124–32. 10.1093/hmg/ddy336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sagane K, Hayakawa K, Kai J, Hirohashi T, Takahashi E, Miyamoto N, et al. Ataxia and peripheral nerve hypomyelination in ADAM22-deficient mice. BMC Neurosci 2005;6:33. 10.1186/1471-2202-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Harati Y, Butler IJ. Congenital hypomyelinating neuropathy. J Neurol Neurosurg Psychiatry 1985;48:1269–76. 10.1136/jnnp.48.12.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Warner LE, Garcia CA MD, Lupski JR MD, PhD. Hereditary Peripheral Neuropathies: Clinical Forms, Genetics, and Molecular Mechanisms. Annu Rev Med 1999;50:263–75. 10.1146/annurev.med.50.1.263. [DOI] [PubMed] [Google Scholar]
- [10].Saporta MA. Charcot-Marie-Tooth disease and other inherited neuropathies. Contin Minneap Minn 2014;20:1208–25. 10.1212/01.CON.0000455885.37169.4c. [DOI] [PubMed] [Google Scholar]
- [11].Morena J, Gupta A, Hoyle JC. Charcot-Marie-Tooth: From Molecules to Therapy. Int J Mol Sci 2019;20:3419. 10.3390/ijms20143419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tazir M, Bellatache M, Nouioua S, Vallat J-M. Autosomal recessive Charcot-Marie-Tooth disease: from genes to phenotypes. J Peripher Nerv Syst JPNS 2013;18:113–29. 10.1111/jns5.12026. [DOI] [PubMed] [Google Scholar]
- [13].Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol 2013;9:562–71. 10.1038/nrneurol.2013.179. [DOI] [PubMed] [Google Scholar]
- [14].Scapin C, Ferri C, Pettinato E, Zambroni D, Bianchi F, Del Carro U, et al. Enhanced axonal neuregulin-1 type-III signaling ameliorates neurophysiology and hypomyelination in a Charcot–Marie–Tooth type 1B mouse model. Hum Mol Genet 2019;28:992–1006. 10.1093/hmg/ddy411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hoyle JC, Isfort MC, Roggenbuck J, Arnold WD. The genetics of Charcot–Marie–Tooth disease: current trends and future implications for diagnosis and management. Appl Clin Genet 2015;8:235–43. 10.2147/TACG.S69969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Granger N Canine inherited motor and sensory neuropathies: an updated classification in 22 breeds and comparison to Charcot-Marie-Tooth disease. Vet J Lond Engl 1997 2011;188:274–85. 10.1016/j.tvjl.2010.06.003. [DOI] [PubMed] [Google Scholar]
- [17].Granger N, Luján Feliu-Pascual A, Spicer C, Ricketts S, Hitti R, Forman O, et al. Charcot-Marie-Tooth type 4B2 demyelinating neuropathy in miniature Schnauzer dogs caused by a novel splicing SBF2 (MTMR13) genetic variant: a new spontaneous clinical model. PeerJ 2019;7:e7983. 10.7717/peerj.7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Drögemüller C, Becker D, Kessler B, Kemter E, Tetens J, Jurina K, et al. A Deletion in the N-Myc Downstream Regulated Gene 1 (NDRG1) Gene in Greyhounds with Polyneuropathy. PLOS ONE 2010;5:e11258. 10.1371/journal.pone.0011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bruun CS, Jäderlund KH, Berendt M, Jensen KB, Spodsberg EH, Gredal H, et al. A Gly98Val Mutation in the N-Myc Downstream Regulated Gene 1 (NDRG1) in Alaskan Malamutes with Polyneuropathy. PLOS ONE 2013;8:e54547. 10.1371/journal.pone.0054547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ekenstedt KJ, Becker D, Minor KM, Shelton GD, Patterson EE, Bley T, et al. An ARHGEF10 Deletion Is Highly Associated with a Juvenile-Onset Inherited Polyneuropathy in Leonberger and Saint Bernard Dogs. PLOS Genet 2014;10:e1004635. 10.1371/journal.pgen.1004635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cummings JF, Summers BA, de Lahunta A, Lawson C. Tremors in Samoyed pups with oligodendrocyte deficiencies and hypomyelination. Acta Neuropathol (Berl) 1986;71:267–77. 10.1007/BF00688049. [DOI] [PubMed] [Google Scholar]
- [22].Greene CE, Vandevelde M, Hoff EJ. Congenital cerebrospinal hypomyelinogenesis in a pup. J Am Vet Med Assoc 1977;171:534–6. [PubMed] [Google Scholar]
- [23].Griffiths IR, Duncan ID, McCulloch M, Harvey MJA. Shaking pups: A disorder of central myelination in the spaniel dog. J Neurol Sci 1981;50:423–33. 10.1016/0022-510X(81)90154-4. [DOI] [PubMed] [Google Scholar]
- [24].Kornegay JN, Goodwin MA, Spyridakis LK. Hypomyelination in Weimaraner dogs. Acta Neuropathol (Berl) 1987;72:394–401. 10.1007/BF00687272. [DOI] [PubMed] [Google Scholar]
- [25].Martin-Vaquero P, da Costa RC, Simmons JK, Beamer GL, Jäderlund KH, Oglesbee MJ. A Novel Spongiform Leukoencephalomyelopathy in Border Terrier Puppies. J Vet Intern Med 2012;26:402–6. 10.1111/j.1939-1676.2011.00873.x. [DOI] [PubMed] [Google Scholar]
- [26].Mayhew IG, Blakemore WF, Palmer AC, Clarke CJ. Tremor syndrome and hypomyelination in Lurcher pups. J Small Anim Pract 1984;25:551–9. 10.1111/j.1748-5827.1984.tb03428.x. [DOI] [Google Scholar]
- [27].Millán Y, Mascort J, Blanco A, Costa C, Masian D, Guil-Luna S, et al. Hypomyelination in three Weimaraner dogs. J Small Anim Pract 2010;51:594–8. 10.1111/j.1748-5827.2010.00997.x. [DOI] [PubMed] [Google Scholar]
- [28].Palmer A, Blakemore W, Wallace M, Wilkes M, Herrtage M, Matic S. Recognition of “trembler”, a hypomyelinating condition in the Bernese mountain dog. Vet Rec 1987;120:609–12. 10.1136/vr.120.26.609. [DOI] [PubMed] [Google Scholar]
- [29].Quitt PR, Brühschwein A, Matiasek K, Wielaender F, Karkamo V, Hytönen MK, et al. A hypomyelinating leukodystrophy in German Shepherd dogs. J Vet Intern Med 2021;35:1455–65. 10.1111/jvim.16085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vandevelde M, Braund KG, Walker TL, Kornegay JN. Dysmyelination of the central nervous system in the Chow-Chow dog. Acta Neuropathol (Berl) 1978;42:211–5. 10.1007/BF00690359. [DOI] [PubMed] [Google Scholar]
- [31].Duncan I. Inherited and Acquired Disorders of Myelin in the Dog and Cat. World Small Anim. Vet. Assoc. World Congr. Proc. 2010, Geneva, Switzerland: 2010. [Google Scholar]
- [32].Braund KG, Mehta JR, Toivio-Kinnucan M, Amling KA, Shell LG, Matz ME. Congenital Hypomyelinating Polyneuropathy in Two Golden Retriever Littermates. Vet Pathol 1989;26:202–8. 10.1177/030098588902600303. [DOI] [PubMed] [Google Scholar]
- [33].Matz ME, Shell L, Braund K. Peripheral hypomyelinization in two golden retriever littermates. J Am Vet Med Assoc 1990;197:228–30. [PubMed] [Google Scholar]
- [34].Niederhauser UB, Holliday TA. Electrodiagnostic studies of muscles and neuromuscular junctions. Semin Vet Med Surg (Small Anim) 1989;4:116–25. [PubMed] [Google Scholar]
- [35].Kimura J Electrodiagnostics in diseases of nerve and muscle: principles and practice. 4th ed. New York: Oxford University Press; 2013. [Google Scholar]
- [36].Dubowitz V, Sewry CA, Oldfors A. Histological and histochemical stains and reactions. Muscle Biopsy Pract. Approach. 4th ed., Philadelphia: Saunders Elsevier; 2013, p. 16–27. [Google Scholar]
- [37].Vanhaesebrouck AE, Couturier J, Cauzinille L, Mizisin AP, Shelton GD, Granger N. Demyelinating polyneuropathy with focally folded myelin sheaths in a family of Miniature Schnauzer dogs. J Neurol Sci 2008;275:100–5. 10.1016/j.jns.2008.07.031. [DOI] [PubMed] [Google Scholar]
- [38].Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–75. 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Friedenberg SG, Meurs KM. Genotype imputation in the domestic dog. Mamm Genome 2016;27:485–94. 10.1007/s00335-016-9636-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–20. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005;438:803–19. 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- [42].Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics 2011;27:2156–8. 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shelton GD, Minor KM, Guo LT, Thomas-Hollands A, Walsh KA, Friedenberg SG, et al. An EHPB1L1 Nonsense Mutation Associated with Congenital Dyserythropoietic Anemia and Polymyopathy in Labrador Retriever Littermates. Genes 2022;13:1427. 10.3390/genes13081427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Plassais J, Kim J, Davis BW, Karyadi DM, Hogan AN, Harris AC, et al. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat Commun 2019;10:1489. 10.1038/s41467-019-09373-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, et al. The Ensembl Variant Effect Predictor. Genome Biol 2016;17:122. 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kinsella RJ, Kähäri A, Haider S, Zamora J, Proctor G, Spudich G, et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database J Biol Databases Curation 2011;2011:bar030. 10.1093/database/bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Stelzer G, Plaschkes I, Oz-Levi D, Alkelai A, Olender T, Zimmerman S, et al. VarElect: the phenotype-based variation prioritizer of the GeneCards Suite. BMC Genomics 2016. 10.1186/s12864-016-2722-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Desmet F-O, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009;37:e67. 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Reese MG. Improved splice site detection in Genie. J. Comput. Biol, vol. 4, Mary Ann Liebert Inc.; 1997, p. 311–23. 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- [52].Scalzitti N, Kress A, Orhand R, Weber T, Moulinier L, Jeannin-Girardon A, et al. Spliceator: multi-species splice site prediction using convolutional neural networks. BMC Bioinformatics 2021;22:561. 10.1186/s12859-021-04471-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. Proc. Seventh Annu. Int. Conf. Res. Comput. Mol. Biol., New York, NY, USA: Association for Computing Machinery; 2003, p. 322–31. 10.1145/640075.640118. [DOI] [PubMed] [Google Scholar]
- [54].Wang C, Wallerman O, Arendt M-L, Sundström E, Karlsson Å, Nordin J, et al. A novel canine reference genome resolves genomic architecture and uncovers transcript complexity. Commun Biol 2021;4:1–11. 10.1038/s42003-021-01698-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sequencher® version 5.4.6 DNA sequence analysis software 2016. [Google Scholar]
- [56].Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods 2012;9:179–81. 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- [57].Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005;21:263–5. 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- [58].Tuler S, Bower J. Measurement of conduction velocity of the peroneal nerve based on recordings from extensor digitorum brevis muscle. J Am Anim Hosp Assoc 1990;26:164–8. [Google Scholar]
- [59].Walker TL, Redding RW, Braund KG. Motor nerve conduction velocity and latency in the dog. Am J Vet Res 1979;40:1433–9. [PubMed] [Google Scholar]
- [60].Bolis A, Coviello S, Bussini S, Dina G, Pardini C, Previtali SC, et al. Loss of Mtmr2 Phosphatase in Schwann Cells But Not in Motor Neurons Causes Charcot-Marie-Tooth Type 4B1 Neuropathy with Myelin Outfoldings. J Neurosci 2005;25:8567–77. 10.1523/JNEUROSCI.2493-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pareyson D, Stojkovic T, Reilly MM, Leonard-Louis S, Laurà M, Blake J, et al. A multicentre retrospective study of Charcot-Marie-Tooth disease type 4B (CMT4B) due to mutations in Myotubularin-related proteins (MTMRs). Ann Neurol 2019;86:55–67. 10.1002/ana.25500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].McMillan HJ, Santagata S, Shapiro F, Batish SD, Couchon L, Donnelly S, et al. Novel MPZ mutations and congenital hypomyelinating neuropathy. Neuromuscul Disord 2010;20:725–9. 10.1016/j.nmd.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Roa BB, Warner LE, Garcia CA, Russo D, Lovelace R, Chance PF, et al. Myelin protein zero (MPZ) gene mutations in nonduplication type 1 Charcot-Marie-Tooth disease. Hum Mutat 1996;7:36–45. . [DOI] [PubMed] [Google Scholar]
- [64].Warner LE, Hilz MJ, Appel SH, Killian JM, Kolodny EH, Karpati G, et al. Clinical Phenotypes of Different MPZ (P0) Mutations May Include Charcot–Marie–Tooth Type 1B, Dejerine–Sottas, and Congenital Hypomyelination. Neuron 1996;17:451–60. 10.1016/S0896-6273(00)80177-4. [DOI] [PubMed] [Google Scholar]
- [65].Duan X, Ma Y, Fan D, Liu X. Characteristics of Clinical and Electrophysiological Pattern in a Large Cohort of Chinese Patients With Charcot-Marie-Tooth 4C. Front Neurol 2021;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lee AJ, Nam SH, Park J-M, Kanwal S, Choi YJ, Lee HJ, et al. Compound heterozygous mutations of SH3TC2 in Charcot–Marie–Tooth disease type 4C patients. J Hum Genet 2019;64:961–5. 10.1038/s10038-019-0636-y. [DOI] [PubMed] [Google Scholar]
- [67].Shin J-Y, Lee J-M. A homozygous SH3TC2 mutation in a Korean patient with Charcot–Marie–Tooth disease type 4C. Neurol Asia 2021;26:835–7. 10.54029/2021vfs. [DOI] [Google Scholar]
- [68].Barisic N, Claeys KG, Sirotkovid-Skerlev M, Löfgren A, Nelis E, De Jonghe P, et al. Charcot-Marie-Tooth Disease: A Clinico-genetic Confrontation. Ann Hum Genet 2008;72:416–41. 10.1111/j.1469-1809.2007.00412.x. [DOI] [PubMed] [Google Scholar]
- [69].Plante-Bordeneuve V, Said G. Dejerine-Sottas disease and hereditary demyelinating polyneuropathy of infancy. Muscle Nerve 2002;26:608–21. 10.1002/mus.10197. [DOI] [PubMed] [Google Scholar]
- [70].Sanmaneechai O, Feely S, Scherer SS, Herrmann DN, Burns J, Muntoni F, et al. Genotype–phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene. Brain 2015;138:3180–92. 10.1093/brain/awv241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Bolino A, Muglia M, Conforti FL, LeGuern E, Salih MAM, Georgiou D-M, et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat Genet 2000;25:17–9. 10.1038/75542. [DOI] [PubMed] [Google Scholar]
- [72].Zambon AA, Natali Sora MG, Cantarella G, Cerri F, Quattrini A, Comi G, et al. Vocal cord paralysis in Charcot–Marie–Tooth type 4b1 disease associated with a novel mutation in the myotubularin-related protein 2 gene: A case report and review of the literature. Neuromuscul Disord 2017;27:487–91. 10.1016/j.nmd.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Deymeer F, Matur Z, Poyraz M, Battaloglu E, Oflazer-Serdaroglu P, Parman Y. Nerve conduction studies in Charcot-Marie-Tooth disease in a cohort from Turkey. Muscle Nerve 2011;43:657–64. 10.1002/mus.21932. [DOI] [PubMed] [Google Scholar]
- [74].Gambardella A, Bono F, Muglia M, Valentino P, Quattrone A. Autosomal recessive hereditary motor and sensory neuropathy with focally folded myelin sheaths (CMT4B). Ann N Y Acad Sci 1999;883:47–55. [PubMed] [Google Scholar]
- [75].Berger P, Bonneick S, Willi S, Wymann M, Suter U. Loss of phosphatase activity in myotubularin-related protein 2 is associated with Charcot–Marie–Tooth disease type 4B1. Hum Mol Genet 2002;11:1569–79. 10.1093/hmg/11.13.1569. [DOI] [PubMed] [Google Scholar]
- [76].Dove SK, Cooke FT, Douglas MR, Sayers LG, Parker PJ, Michell RH. Osmotic stress activates phosphatidylinositol-3,5-bisphosphate synthesis. Nature 1997;390:187–92. 10.1038/36613. [DOI] [PubMed] [Google Scholar]
- [77].Gary JD, Wurmser AE, Bonangelino CJ, Weisman LS, Emr SD. Fab1p is essential for PtdIns(3)P 5-kinase activity and the maintenance of vacuolar size and membrane homeostasis. J Cell Biol 1998;143:65–79. 10.1083/jcb.143.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Consortium UniProt. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 2019;47:D506–15. 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wang H, Bayram AK, Sprute R, Ozdemir O, Cooper E, Pergande M, et al. Genotype-Phenotype Correlations in Charcot-Marie-Tooth Disease Due to MTMR2 Mutations and Implications in Membrane Trafficking. Front Neurosci 2019;13. 10.3389/FNINS.2019.00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Greenfield S, Brostoff S, Eylar EH, Morell P. Protein Composition of Myelin of the Peripheral Nervous System. J Neurochem 1973;20:1207–16. 10.1111/j.1471-4159.1973.tb00089.x. [DOI] [PubMed] [Google Scholar]
- [81].Auer-Grumbach M, Strasser-Fuchs S, Robl T, Windpassinger C, Wagner K. Late onset Charcot-Marie-Tooth 2 syndrome caused by two novel mutations in the MPZ gene. Neurology 2003;61:1435–7. 10.1212/01.wnl.0000094197.46109.75. [DOI] [PubMed] [Google Scholar]
- [82].Chapon F, Latour P, Diraison P, Schaeffer S, Vandenberghe A. Axonal phenotype of Charcot-Marie-Tooth disease associated with a mutation in the myelin protein zero gene. J Neurol Neurosurg Psychiatry 1999;66:779–82. 10.1136/jnnp.66.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].De Jonghe P, Timmerman V, Ceuterick C, Nelis E, De Vriendt E, Löfgren A, et al. The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain J Neurol 1999;122 ( Pt 2):281–90. 10.1093/brain/122.2.281. [DOI] [PubMed] [Google Scholar]
- [84].Fabrizi GM, Pellegrini M, Angiari C, Cavallaro T, Morini A, Taioli F, et al. Gene dosage sensitivity of a novel mutation in the intracellular domain of P0 associated with Charcot-Marie-Tooth disease type 1B. Neuromuscul Disord NMD 2006;16:183–7. 10.1016/j.nmd.2006.01.006. [DOI] [PubMed] [Google Scholar]
- [85].Hayasaka K, Himoro M, Sato W, Takada G, Uyemura K, Shimizu N, et al. Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat Genet 1993;5:31–4. 10.1038/ng0993-31. [DOI] [PubMed] [Google Scholar]
- [86].Kochanski A, Drac H, Kabzioska D, Ryniewicz B, Rowioska-Marcioska K, Nowakowski A, et al. A novel MPZ gene mutation in congenital neuropathy with hypomyelination. Neurology 2004;62:2122–3. 10.1212/01.wnl.0000127606.93772.3a. [DOI] [PubMed] [Google Scholar]
- [87].Mandich P, Mancardi GL, Varese A, Soriani S, Di Maria E, Bellone E, et al. Congenital hypomyelination due to myelin protein zero Q215X mutation. Ann Neurol 1999;45:676–8. . [DOI] [PubMed] [Google Scholar]
- [88].Marrosu MG, Vaccargiu S, Marrosu G, Vannelli A, Cianchetti C, Muntoni F. Charcot-Marie-Tooth disease type 2 associated with mutation of the myelin protein zero gene. Neurology 1998;50:1397–401. 10.1212/wnl.50.5.1397. [DOI] [PubMed] [Google Scholar]
- [89].Mastaglia FL, Nowak KJ, Stell R, Phillips BA, Edmondston JE, Dorosz SM, et al. Novel mutation in the myelin protein zero gene in a family with intermediate hereditary motor and sensory neuropathy. J Neurol Neurosurg Psychiatry 1999;67:174–9. 10.1136/jnnp.67.2.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Misu K, Yoshihara T, Shikama Y, Awaki E, Yamamoto M, Hattori N, et al. An axonal form of Charcot-Marie-Tooth disease showing distinctive features in association with mutations in the peripheral myelin protein zero gene (Thr124Met or Asp75Val). J Neurol Neurosurg Psychiatry 2000;69:806–11. 10.1136/jnnp.69.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Nakagawa M, Suehara M, Saito A, Takashima H, Umehara F, Saito M, et al. A novel MPZ gene mutation in dominantly inherited neuropathy with focally folded myelin sheaths. Neurology 1999;52:1271–5. 10.1212/wnl.52.6.1271. [DOI] [PubMed] [Google Scholar]
- [92].Grandis M, Vigo T, Passalacqua M, Jain M, Scazzola S, La Padula V, et al. Different cellular and molecular mechanisms for early and late-onset myelin protein zero mutations. Hum Mol Genet 2008;17:1877–89. 10.1093/hmg/ddn083. [DOI] [PubMed] [Google Scholar]
- [93].Shy ME, Jáni A, Krajewski K, Grandis M, Lewis RA, Li J, et al. Phenotypic clustering in MPZ mutations. Brain 2004;127:371–84. 10.1093/brain/awh048. [DOI] [PubMed] [Google Scholar]
- [94].Mersiyanova IV, Ismailov SM, Polyakov AV, Dadali EL, Fedotov VP, Nelis E, et al. Screening for mutations the peripheral myelin genes PMP22, MPZ and Cx32 (GJB1) in Russian Charcot-Marie-Tooth neuropathy patients. Hum Mutat 2000;15:340–7. . [DOI] [PubMed] [Google Scholar]
- [95].Tyson J, Ellis D, Fairbrother U, King RH, Muntoni F, Jacobs J, et al. Hereditary demyelinating neuropathy of infancy. A genetically complex syndrome. Brain J Neurol 1997;120 (Pt 1:47–63. 10.1093/brain/120.1.47. [DOI] [PubMed] [Google Scholar]
- [96].Langlais T, Leonard J-C, Ursu C, Morin C. Charcot-Marie-Tooth disease associated with hip dysplasia in an adolescent. Arch Pediatr Organe Off Soc Francaise Pediatr 2017;24:675–81. 10.1016/j.arcped.2017.04.014. [DOI] [PubMed] [Google Scholar]
- [97].Novais EN, Bixby SD, Rennick J, Carry PM, Kim Y-J, Millis MB. Hip Dysplasia Is More Severe in Charcot-Marie-Tooth Disease Than in Developmental Dysplasia of the Hip. Clin Orthop 2014;472:665–73. 10.1007/s11999-013-3127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Pozdnikin IY. Dysfunction in the hip joints in children with Charcot-Marie-Tooth syndrome (literature review). Pediatr Traumatol Orthop Reconstr Surg 2015;3:52–6. 10.17816/PTORS3352-56. [DOI] [Google Scholar]
- [99].Vijay S, Chiu M, Dacks JB, Roberts RC. Exclusive expression of the Rab11 effector SH3TC2 in Schwann cells links integrin-α6 and myelin maintenance to Charcot-Marie-Tooth disease type 4C. Biochim Biophys Acta BBA - Mol Basis Dis 2016;1862:1279–90. 10.1016/j.bbadis.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Arnaud E, Zenker J, de Preux Charles A-S, Stendel C, Roos A, Medard J-J, et al. SH3TC2/KIAA1985 protein is required for proper myelination and the integrity of the node of Ranvier in the peripheral nervous system. Proc Natl Acad Sci 2009;106:17528–33. 10.1073/pnas.0905523106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Azzedine H, Ravisé N, Verny C, Gabrëels-Festen A, Lammens M, Grid D, et al. Spine deformities in Charcot-Marie-Tooth 4C caused by SH3TC2 gene mutations. Neurology 2006;67:602–6. 10.1212/01.wnl.0000230225.19797.93. [DOI] [PubMed] [Google Scholar]
- [102].Gabreëls-Festen A, Beersum S van, Eshuis L, LeGuern E, Gabreëls F, Engelen B van, et al. Study on the gene and phenotypic characterisation of autosomal recessive demyelinating motor and sensory neuropathy (Charcot-Marie-Tooth disease) with a gene locus on chromosome 5q23-q33. J Neurol Neurosurg Psychiatry 1999;66:569–74. 10.1136/jnnp.66.5.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Jerath NU, Mankodi A, Crawford TO, Grunseich C, Baloui H, Nnamdi-Emeratom C, et al. Charcot Marie Tooth Disease type 4C: Novel Mutations, Clinical Presentations, and Diagnostic Challenges. Muscle Nerve 2018;57:749–55. 10.1002/mus.25981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Mittal RK. Motor Function of the Pharynx, Esophagus, and its Sphincters. San Rafael (CA): Morgan & Claypool Life Sciences; 2011. [PubMed] [Google Scholar]
- [105].Singh B, Dyce KM. Dyce, Sack, and Wensing’s Textbook of Veterinary Anatomy. Fifth edition. St. Louis, Missouri: Elsevier; 2018. [Google Scholar]
- [106].Gooding R, Colomer J, King R, Angelicheva D, Marns L, Parman Y, et al. A novel Gypsy founder mutation, p.Arg1109X in the CMT4C gene, causes variable peripheral neuropathy phenotypes. J Med Genet 2005;42:e69. 10.1136/jmg.2005.034132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Senderek J, Bergmann C, Stendel C, Kirfel J, Verpoorten N, De Jonghe P, et al. Mutations in a Gene Encoding a Novel SH3/TPR Domain Protein Cause Autosomal Recessive Charcot-Marie-Tooth Type 4C Neuropathy. Am J Hum Genet 2003;73:1106–19. 10.1086/379525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Aboussouan LS, Lewis RA, Shy ME. Disorders of Pulmonary Function, Sleep, and the Upper Airway in Charcot-Marie-Tooth Disease. Lung 2007;185:1–7. 10.1007/s00408-006-0053-9. [DOI] [PubMed] [Google Scholar]
- [109].de Oliveira CM, Fussiger H, Winckler PB, Saute JAM. Dropped head syndrome as a manifestation of Charcot–Marie–Tooth disease type 4C. Neuromuscul Disord 2019;29:138–41. 10.1016/j.nmd.2018.11.010. [DOI] [PubMed] [Google Scholar]
- [110].Taniguchi T, Ando M, Okamoto Y, Yoshimura A, Higuchi Y, Hashiguchi A, et al. Genetic spectrum of Charcot–Marie–Tooth disease associated with myelin protein zero gene variants in Japan. Clin Genet 2021;99:359–75. 10.1111/cge.13881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Jessen KR, Mirsky R. Schwann Cell Precursors; Multipotent Glial Cells in Embryonic Nerves. Front Mol Neurosci 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Lawlor MW, Dowling JJ. X-linked myotubular myopathy. Neuromuscul Disord 2021;31:1004–12. 10.1016/j.nmd.2021.08.003. [DOI] [PubMed] [Google Scholar]
- [113].Nghiem PP, Kornegay JN. Gene therapies in canine models for Duchenne muscular dystrophy. Hum Genet 2019;138:483–9. 10.1007/s00439-019-01976-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The whole genome sequence for the four affected Golden Retrievers is available in Sequence Read Archive (SRA) under accession numbers: Cases 1 and 2: SRR23867282 and SRR23867281; Case 3: SRR23628641; and Case 4: SRR23628642.