

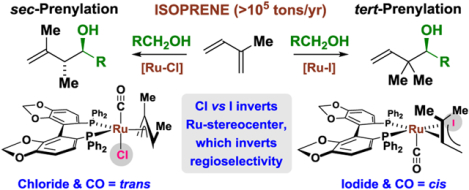
Abstract
The first correlation between metal-centered stereogenicity and regioselectivity in a catalytic process is described. Alternate pseudo-diastereomeric chiral-at-ruthenium complexes of the type RuX(CO)[η3-prenyl][(S)-SEGPHOS] form in a halide-dependent manner, and display divergent regioselectivity in catalytic C-C couplings of isoprene to alcohol proelectrophiles via hydrogen auto-transfer. Whereas the chloride-bound ruthenium-SEGPHOS complex prefers a trans-relationship between the halide and carbonyl ligands and delivers products of carbonyl sec-prenylation, the iodide-bound ruthenium-SEGPHOS complex prefers a cis-relationship between the halide and carbonyl ligands and delivers products of carbonyl tert-prenylation. The chloride- and iodide-bound ruthenium-SEGPHOS complexes were characterized in solution and the solid phase by 31P NMR and X-ray diffraction. DFT calculations of the iodide-bound catalyst implicate a Curtin-Hammett-type scenario in which the transition states for aldehyde coordination from an equilibrating mixture of sec- and tert-prenylruthenium complexes is rate- and product-determining. Control of metal-centered diastereoselectivity has unlocked the first catalytic enantioselective isoprene-mediated carbonyl tert-prenylations.
Graphical Abstract
Introduction
Halide counterions can profoundly influence metal-catalyzed reactions, yet the precise nature of their effect is seldom understood.1 In recent work,2 experimental and computational analyses of the halide-bound ruthenium-JOSIPHOS complexes, RuX(CO)(η3-C3H5)(JOSIPHOS), where X = Cl, Br, I, revealed that the enhanced enantioselectivities of iodide-containing catalysts emanate from iodide’s capacity to promote selective formation of a chiral-at-ruthenium octahedral stereocenter,2,3 and engage in formyl CH···I hydrogen bonding in the transition state for carbonyl addition.4 By exploiting iodide counterions in combination with C1-symmetric JOSIPHOS ligands, the intervention of predominantly one of 10 possible diastereomeric transition structures5 could be achieved, enabling highly enantioselective ruthenium-catalyzed 1-aryl-1-propyne-mediated carbonyl (α-aryl)allylations,2 2-butyne-mediated carbonyl vinylations,6 and butadiene/methylallene-mediated crotylations (Figure 1).7
Figure 1.
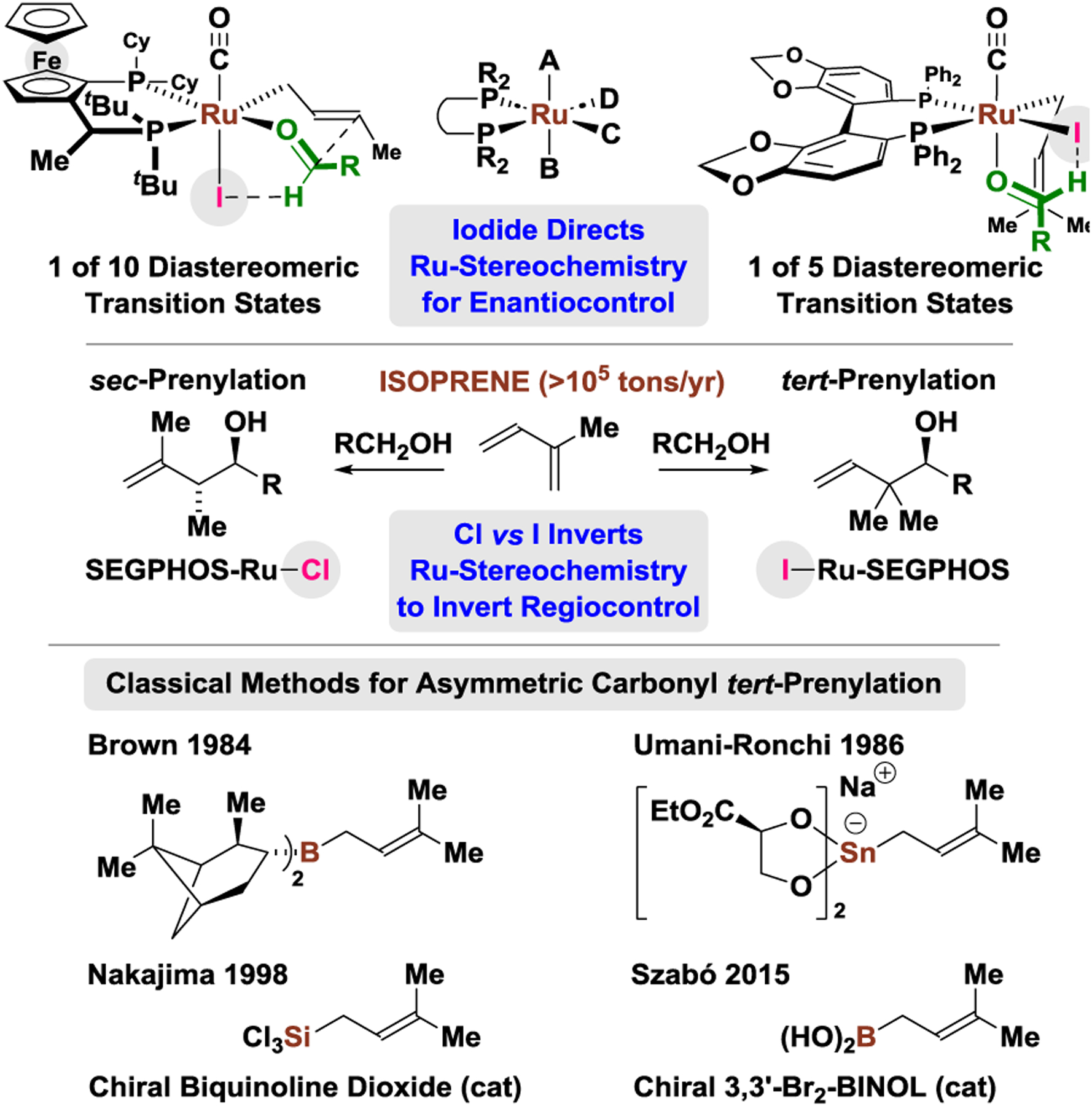
Halide-directed regio- and enantioselectivity unlocks isoprene-mediated carbonyl tert-prenylation.
These data impelled a survey of halide counterions in ruthenium-SEGPHOS-catalyzed reactions of isoprene (>105 tons/yr)8 with primary alcohol proelectrophiles via hydrogen auto-transfer. Not only was a halide-dependent partitioning of diastereomeric chiral-at-ruthenium-SEGPHOS complexes observed, but it was also found that the diastereomeric complexes display regiodivergent reactivity:9 chloride- and bromide-bound ruthenium-SEGPHOS catalysts preferentially promote carbonyl sec-prenylation, whereas the diastereomeric iodide-bound ruthenium-SEGPHOS catalyst delivers products of carbonyl tert-prenylation. These data represent the first correlation between metal-centered stereogenicity and regioselectivity in metal catalysis, which, in turn, has enabled a method for catalytic enantioselective isoprene-mediated carbonyl tert-prenylation beyond premetalated reagents.10–15
Results and Discussion
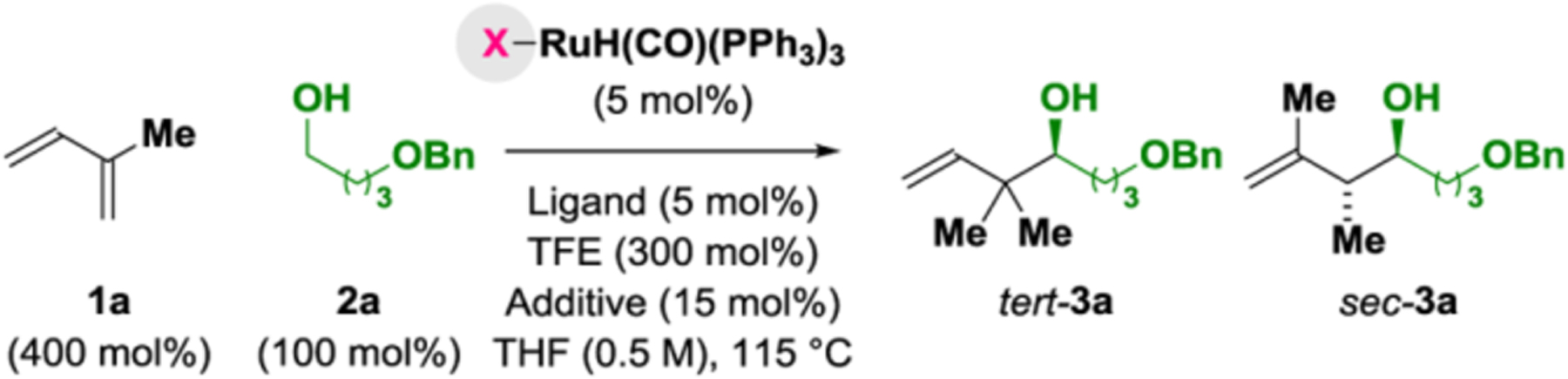
In 2008, our laboratory disclosed the first use of dienes as allylmetal pronucleophiles in late transition metal-catalyzed carbonyl addition.16a This study encompassed racemic examples of isoprene-mediated sec-prenylation and involved the use of a chloride-bound ruthenium catalyst. Subsequent efforts to develop asymmetric variants of this process focused on butadiene,16b-d as reactions of isoprene often led to regioisomeric mixtures of sec-prenylation and tert-prenylation products. Our recent finding that halide counterions can guide formation of pseudo-diastereomeric chiral-at-metal ruthenium complexes led us to probe the question of whether regioisomerism observed in couplings of isoprene occurred in response to metal-centered diastereogenicity. Toward this end, alcohol 2a (100 mol%) and isoprene 1a (400 mol%) were exposed to RuH2(CO)(PPh3)3 (5 mol%) and (S)-SEGPHOS (5 mol%) in THF (0.5 M) and trifluoroethanol (TFE) (300 mol%) at 115 °C in the absence and presence of the halide additives LiX, where X = Cl, Br and I (Table 1, entries 1–4). Whereas the chloride-modified catalyst exclusively promoted formation of the sec-prenylated product sec-3a, an increasing proportion of the tert-prenylated adduct tert-3a was observed using the catalysts modified by bromide and iodide. Remarkably, in the case of the iodide-modified catalyst, a 12:1 regioisomeric mixture favoring tert-3a was formed in 65% yield and 91% enantiomeric excess (Table 1, entry 4), thus representing a halide-dependent inversion in regioselectivity. This trend persisted in reactions conducted using the precatalysts RuHCl(CO)(PPh3)3 and RuHBr(CO)(PPh3)3 in the absence of added lithium halides (Table 1, entries 5 & 6), and in reactions using RuHCl(CO)(PPh3)3 with added LiBr and LiI (Table 1, entries 7 & 8). Higher yields and regioselectivities in the formation of the tert-3a could be achieved using KI instead of LiI, perhaps due to the increased solubility of KI in ethereal solvent (Table 1, entry 9). Further variation of ligand (Table 1, entries 10–14) did not improve conversion or selectivity. Reactions conducted with the precatalyst Ru(CF3CO2)2(CO)(PPh3)2 favored formation of sec-3a (35% yield, 12:1 rr, 3:1 dr, 21% ee), as did the arylsulfonate-modified catalyst generated from the acid-base reaction16c of RuH2(CO)(PPh3)3 with p-toluenesulfonic acid (42% yield, 3:1 rr, 5:1 dr, 18% ee).
Table 1.
Halide-dependent tert- and sec-prenylation in ruthenium-catalyzed C-C couplings of isoprene 1a with alcohol 2a via hydrogen auto-transfer.a
| ||||||
|---|---|---|---|---|---|---|
| Entry | X | Additive | Ligand | Yield | tert-3a (ee) | 3a (tert:sec) |
| 1 | H | — | (S)-SEGPHOS | 54% | - | 1:3 |
| 2b | H | LiCI | (S)-SEGPHOS | 42% | - | >1:20 |
| 3 | H | LiBr | (S)-SEGPHOS | 40% | - | 1:6 |
| 4 | H | Lil | (S)-SEGPHOS | 65% | 91% | 12:1 |
| 5c | Cl | — | (S)-SEGPHOS | 74% | - | 1:16 |
| 6 | Br | — | (S)-SEGPHOS | 72% | - | 1:5 |
| 7 | Cl | LiBr | (S)-SEGPHOS | 64% | - | 1:4 |
| 8 | Cl | Lil | (S)-SEGPHOS | 66% | 91% | 12:1 |
| → 9 | Cl | Kl | (S)-SEGPHOS | 75% | 91% | 17:1 |
| 10 | Cl | Kl | (S)-DM-SEGPHOS | 59% | 89% | 12:1 |
| 11 | Cl | Kl | (S)-BINAP | 83% | 65% | 15:1 |
| 12 | Cl | Kl | (S)-tol-BINAP | 74% | 72% | 13:1 |
| 13 | Cl | Kl | (S)-CI.MeO-BIPHEP | 45% | 58% | 9:1 |
| 14 | Cl | Kl | (S,S)-DIOP | 73% | 85% | 12:1 |
Yields of material isolated by silica gel chromatography as isomeric mixtures. Enantioselectivities were determined by HPLC analysis.
sec-3a (3:1 dr, 11% ee).
sec-3a (3:1 dr, 11% ee). See Supporting Information for further details on reaction optimization.
To assess whether the observed halide-dependent inversion in regioselectivity was due to intervention of pseudo-diastereomeric chiral-at-ruthenium-SEGPHOS complexes, crystals of the indicated chloride- and iodide-bound π-allylruthenium complexes were subjected to X-ray diffraction analysis (Figure 2). A trans-relationship between the carbonyl and chloride ligands, which reside in apical coordination sites, was observed for the π-allylruthenium chloride complex. In contrast, the π-allylruthenium iodide complex exhibited a cis-relationship between the carbonyl and iodide ligands in apical and equatorial coordination sites, respectively. The diastereomeric preference observed in the solid state may reflect crystal packing forces. Hence, it was essential to determine whether this preference persisted in solution. A comparison of the solid and solution phase 31P NMR spectra17 of the chloride and iodide complexes suggested this to be the case. The respective solid and solution phase spectra matched and a single (but distinct) diastereomer was observed for both the chloride and iodide complexes. Notably, for the chloride complex, two signals with very similar chemical shifts were observed in solution (δ = 33.3, 31.7), which is consistent with two phosphorus atoms in similar chemical environments. In contrast, the phosphorus atoms of the iodide complex appeared at very different chemical shifts (δ = 32.7, 48.7) with a low-field doublet consistent with that of a phosphine atom that is trans to a halide.18 As the haptomeric equilibria of π-allylruthenium complexes is dynamic,19 enabling interconversion of diastereomeric chiral-at-ruthenium-SEGPHOS complexes via Berry pseudorotation via pentacoordinate σ-allyl complexes,20 the collective data suggest the diastereomeric preferences of the chloride- and iodide-bound complexes reflect a thermodynamic bias.
Figure 2.
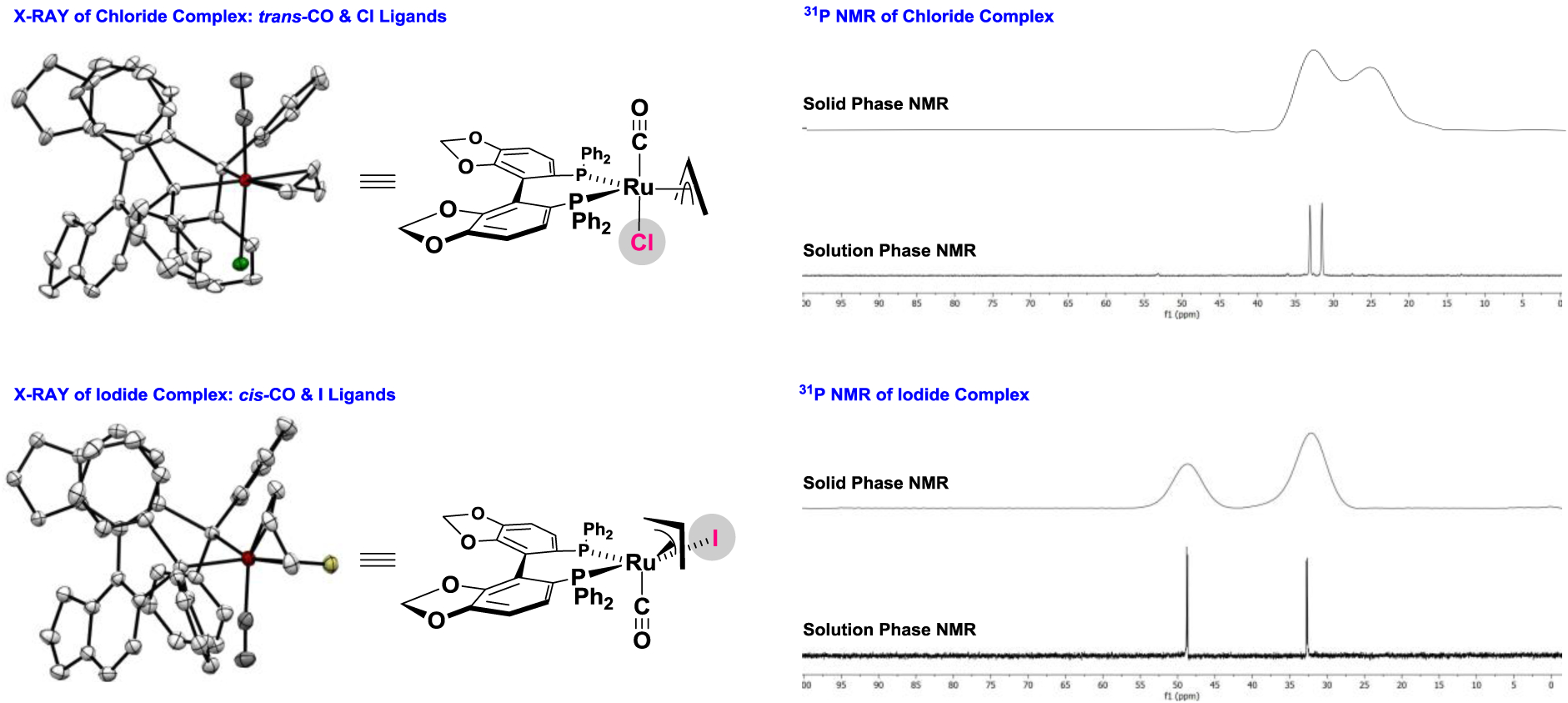
Single crystal X-ray diffraction data for chloride-vs iodide-bound ruthenium-SEGPHOS complexes and their solution and solid phase 31P NMR spectra.a
aDisplacement ellipsoids are scaled to the 50% probability level. Hydrogen atoms have been omitted for clarity. See Supporting Information for complete crystallographic and 31P NMR data.
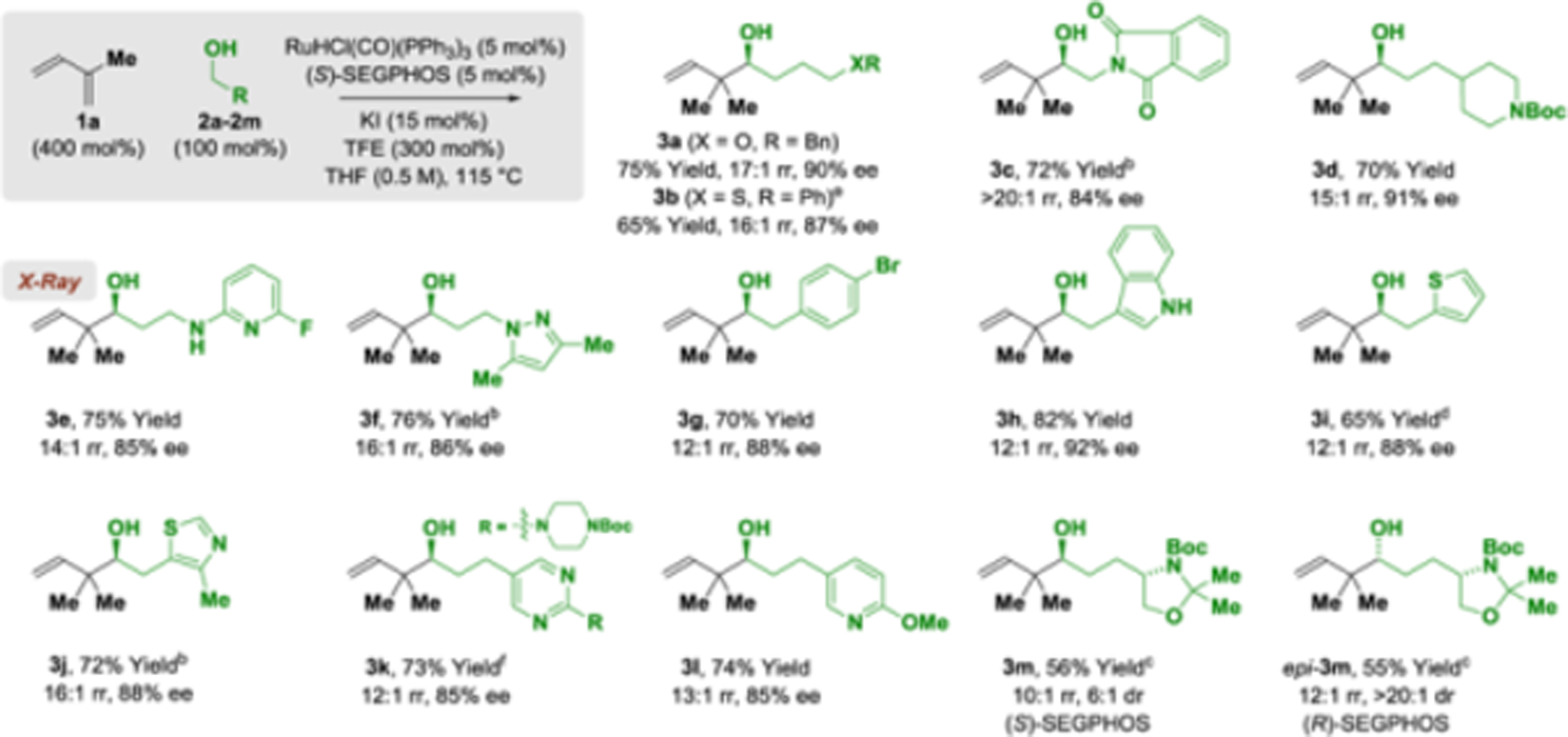
To assess reaction scope, optimal conditions identified for the isoprene-mediated tert-prenylation of alcohol 2a were applied to primary alcohols 2b–2m (Table 2). The homoallylic alcohols 3b–3m and epi-3m were formed in good to excellent yields, regioselectivities and enantioselectivities. Diverse N-heterocycles were tolerated, including piperidines (3d), pyridines (3e, 3l), pyrazoles (3f), unprotected indoles (3h), thiophenes (3i), thiazoles (3j), and pyrimidines (3k). The chiral δ-stereogenic alcohol 2m underwent tert-prenylation with good levels of catalyst-directed diastereoselectivity. Attempted reactions of primary benzylic and allylic alcohols led to low yields of sec-prenylated adduct. The reaction of myrcene 1b with alcohol 2a provides the product tert-geranylation 4a, thus forming an acyclic quaternary carbon stereocenter (eq. 1).21 Finally, using 2-propanol as reductant, isoprene-mediated tert-prenylation of aldehydes dehydro-2a and dehydro-2b occurred with good regio- and stereocontrol (Scheme 1). The absolute stereochemical assignments of adducts 3a-3m and 4a were made in analogy to 3e, which was established via single crystal X-ray diffraction.
Table 2.
Regio- and enantioselective isoprene-mediated tert-prenylation of alcohol proelectrophiles 2a–2m via ruthenium-SEGPHOS-catalyzed hydrogen auto-transfer.a
|
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary-phase HPLC analysis.
120 °C.
110 °C.
DME (0.5M).
Ru-ligand (7.5 mol%), KI (20 mol%).
RuH2(CO)(PPh3)3 (5 mol%), LiI (10 mol%). See Supporting Information for further details.
Scheme 1.

Regio- and enantioselective isoprene-mediated tert-prenylation of aldehydes dehydro-2a and dehydro-2b.a
aYields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary-phase HPLC analysis. See Supporting Information for further details.
 |
(eq.1) |
To gain insight into the reaction mechanism, deuterium la-beling experiments were performed (Scheme 2). Exposure of the alcohol deuterio-2a to isoprene 1a under standard conditions for tert-prenylation using the iodide-bound catalyst led to formation of deuterio-3a, which incorporates deuterium at the primary and secondary vinylic positions (Hc = 12% 2H, Hd/e = 15% 2H) and the methyl groups (Hb = 15% 2H). A substantial loss of deuterium is observed at the carbinol position (Ha = 54% 2H). Omitting potassium iodide, but under otherwise identical conditions, deuterio-2a and isoprene 1a are converted to the sec-prenylation product deuterio-sec-3a, which retains significantly more deuterium at the carbinol position (Ha = 89% 2H). Notably, deuterium is not transferred to the vinylic positions of deuterio-sec-3a (Hd/e = 0% 2H). Reexposure of deuterio-3a or deuterio-sec-3a to the reaction conditions does not alter deuterium content at the carbinol position. These data corroborate reversible hydrogen transfer between alcohol deuterio-2a and isoprene 1a prior to carbonyl addition. For the iodide-bound catalyst, formation of a more highly substituted 3°-4° C-C bond, lowers the rate of carbonyl addition, which increases H/D-exchange between deuterio-2a and isoprene 1a and results in greater deuterium loss at the carbinol position. Hydroruthenation of the less substituted olefin of isoprene 1a is kinetically preferred.22b,c Hydroruthenation of the more substituted olefin of isoprene 1a, only occurs when the rate of carbonyl addition is retarded via formation of the more highly substituted 3°-4° C-C bond. Incomplete deuterium transfer is attributed to H/D-exchange with isoprene, as well H/D-exchange between the ruthenium deuteride and the hydroxyl hydrogens of the alcohol reactant or TFE.23 Thus, a catalytic cycle is envisioned wherein reversible alcohol dehydrogenation via ruthenium alkoxide I delivers aldehyde and ruthenium hydride II. Hydroruthenation of isoprene22 favors formation of the n-prenylruthenium species IIIa (and haptomers), which engages the aldehyde in carbonyl addition to form the homoallylic alkoxide IV. TFE-assisted exchange of the homoallylic ruthenium alkoxide IV2 with alcohol reactant releases the product of tert-prenylation and reforms ruthenium alkoxide I to close the cycle (Scheme 3).
Scheme 2.
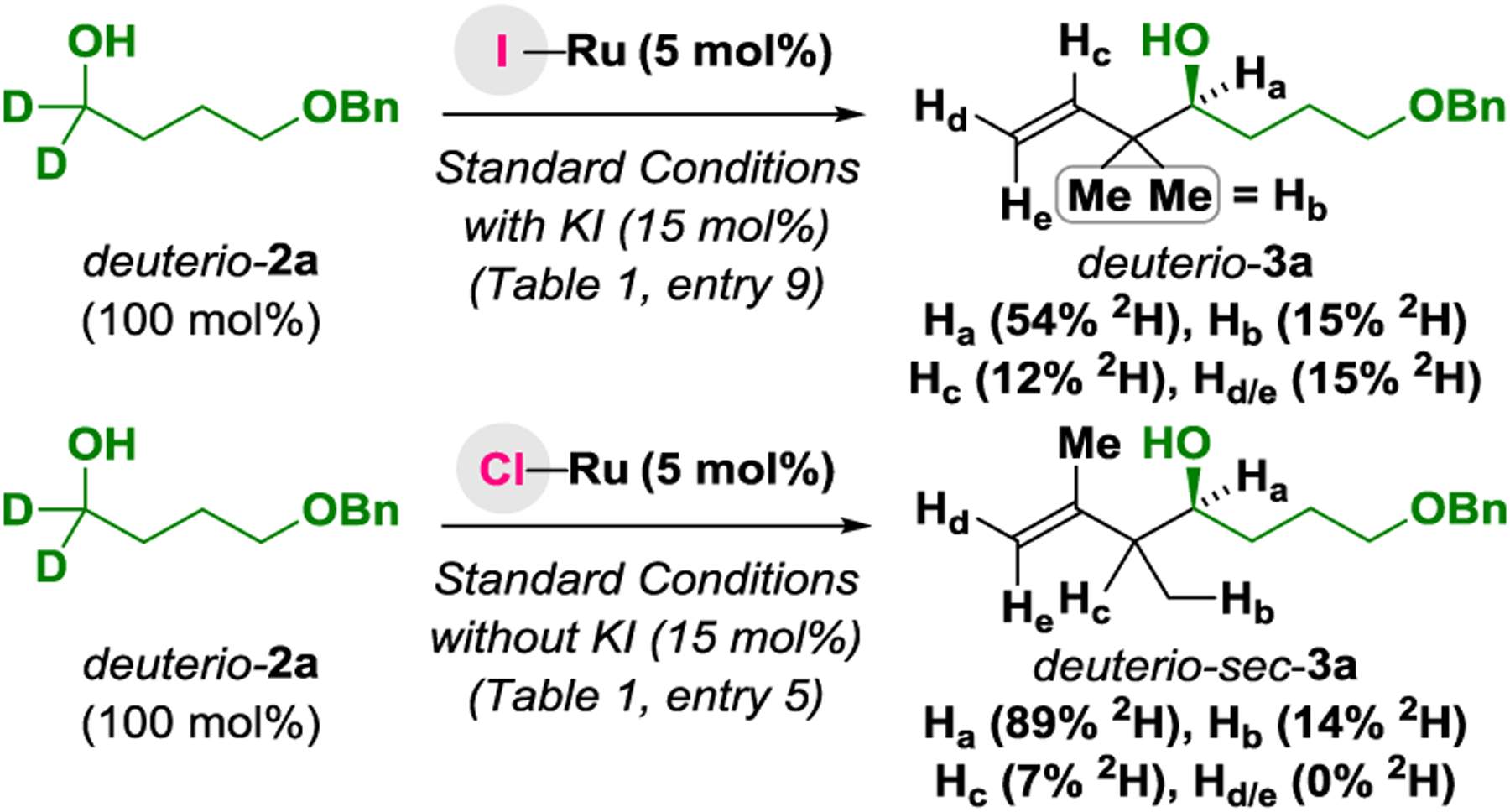
Deuterium labelling experiments.a
aDeuterium incorporation was assessed via 1H, 2H NMR and HRMS. See Supporting Information for further details.
Scheme 3.
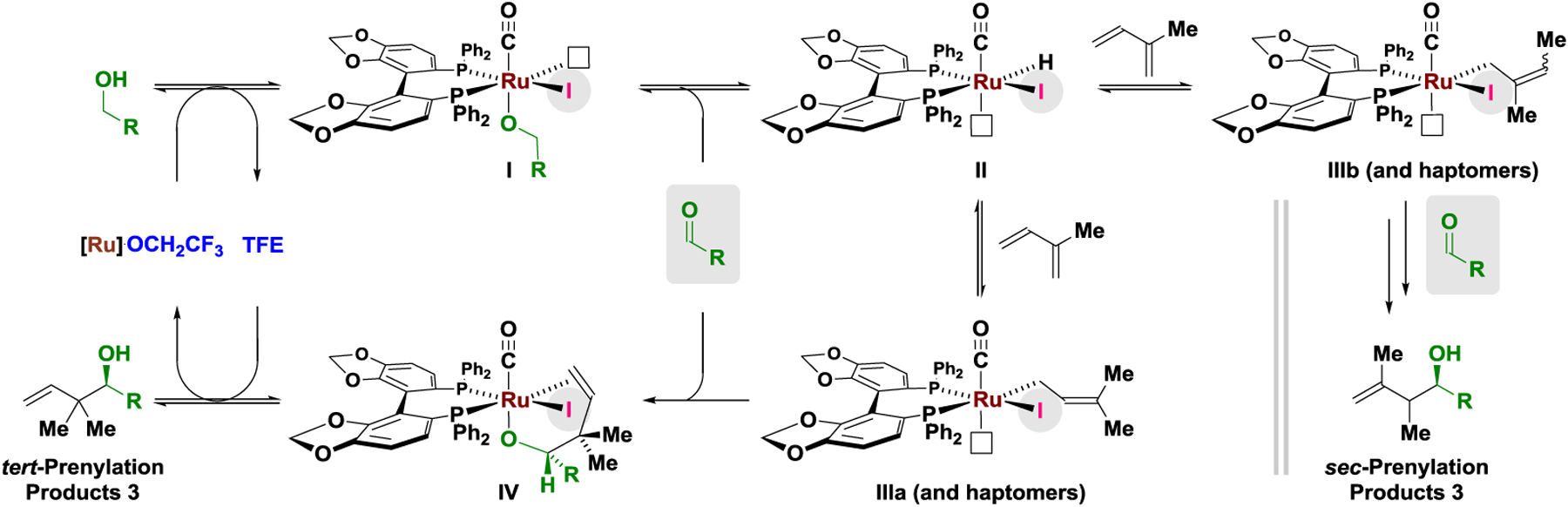
Proposed mechanism for regio- and enantioselective ruthenium-SEGPHOS-catalyzed isoprene-mediated tert-prenylation of primary alcohols as corroborated by deuterium labelling.a
aThe extent of deuterium incorporation was corroborated 1H, 2H NMR and HRMS. See Supporting Information for further details.
To gain deeper insight into how halide counterions bias the regioisomeric C-C coupling pathways in isoprene-mediated carbonyl addition via hydrogen auto-transfer, density functional theory (DFT) calculations were conducted.24 First, the structures of the diastereomeric chloride- and iodide-bound π-allyl- and π-prenylruthenium complexes in which the halide counterions reside cis- or trans- to the carbonyl ligand were computed (Figure 3). The trans-diastereomers were determined to be lower in energy for the chloride-bound complexes. For the iodide complexes, the cis-diastereomers were thermodynamically preferred. These data are consistent with the experimentally observed diastereomeric preferences of the chloride- and iodide-bound π-allylruthenium complexes in the solid state and in solution. For the iodide complexes, steric interactions between the large iodide counterion and the phenyl groups of the SEGPHOS ligand overcome the innate preference for a trans-relationship between the π-donating and π-accepting halide and carbonyl ligands.2,22c,25
Figure 3.
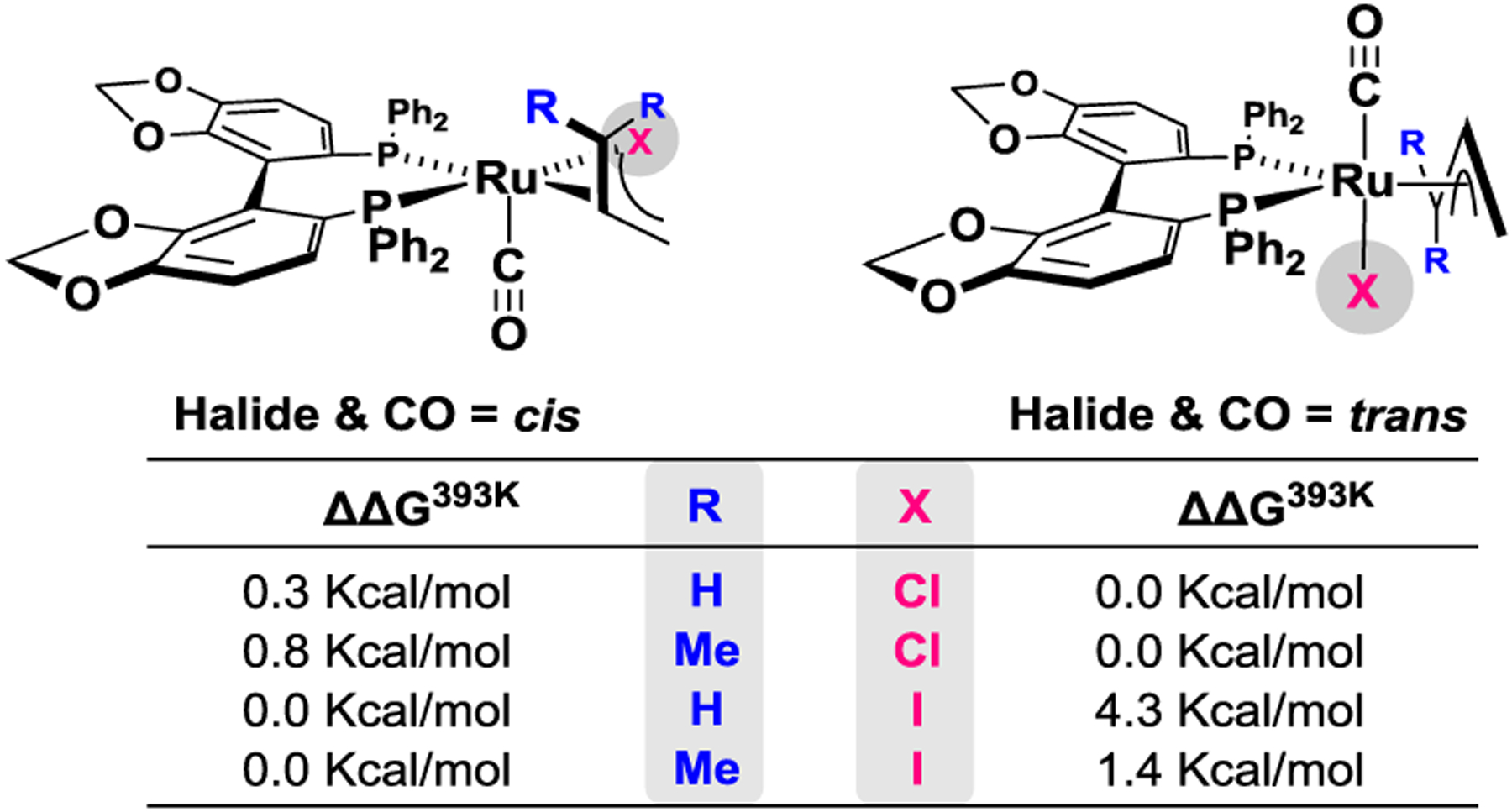
Calculated stabilities of diastereomeric chloride- and iodide-bound π-allyl- and π-prenylruthenium (S)-SEGPHOS complexes.a
aSee supporting information for further details.
Next, the energy profiles for the kinetic pathways leading to the regioisomeric tert- and sec-prenylation products were computed (Figure 4). The reaction pathways diverge from the common intermediate ruthenium-hydride A, which hydrometalates isoprene to form the regioisomeric π-allyl complexes B and F, which interconvert with their respective σ-allyl species C and H. Remarkably, the transition states for subsequent aldehyde coordination (TS1A and TS2A) are rate-determining in both pathways and, hence, represent the regio-determining event. The transition state TS2A, which leads to the sec-prenylation product, is higher in energy than TS1A by 2.1 kcal/mol due to a non-bonded interaction between the iodide and the vinylic methyl of the σ-allyl in TS2A (Figure 5). The transition states for the following C-C bond formations (TS1B and TS2B) are lower in energy than TS1A and TS2A, respectively. TS1B is 0.8 kcal/mol higher in energy than TS2B due to steric interactions between the methyl group of acetaldehyde and the southern methyl substituent of the σ-allyl, which is absent in TS2B. Although not explicitly shown, a non-classical CH···I formyl hydrogen bond between the aldehyde and iodide counterion stabilizes the computed transition states for carbonyl addition.2,4 Thus, the DFT calculations corroborate a scenario in which regioselectivity is determined in a two-fold manner: first by the cis-diastereomeric preference of the iodide and carbonyl ligands at ruthenium, and then by a Curtin-Hammett-type scenario in which the relative energies of the transition states for aldehyde coordination (TS1A and TS2A) to the regioisomeric prenylruthenium complexes is rate- and product-determining.
Figure 4.
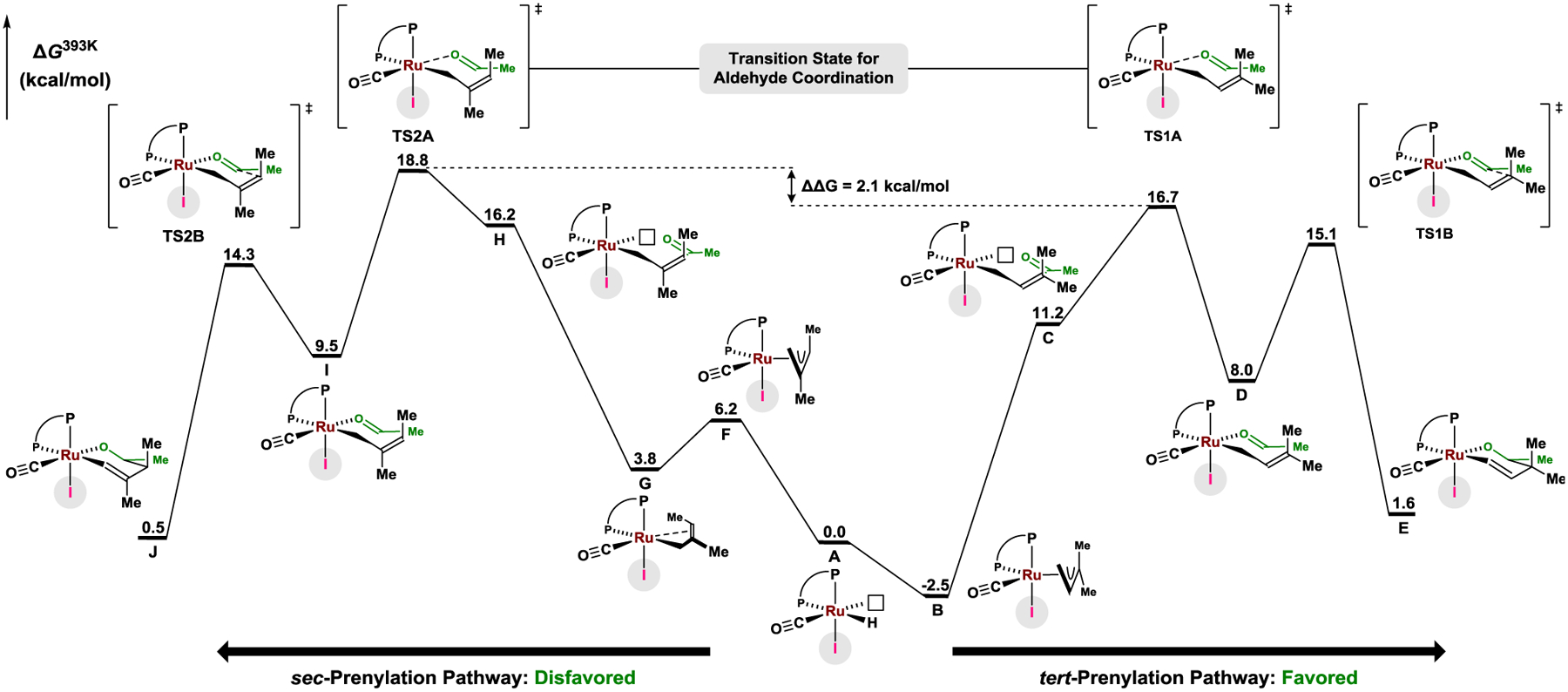
Computed energy profiles of the kinetic pathways leading to the regioisomeric tert- and sec-prenylation products.
Figure 5.
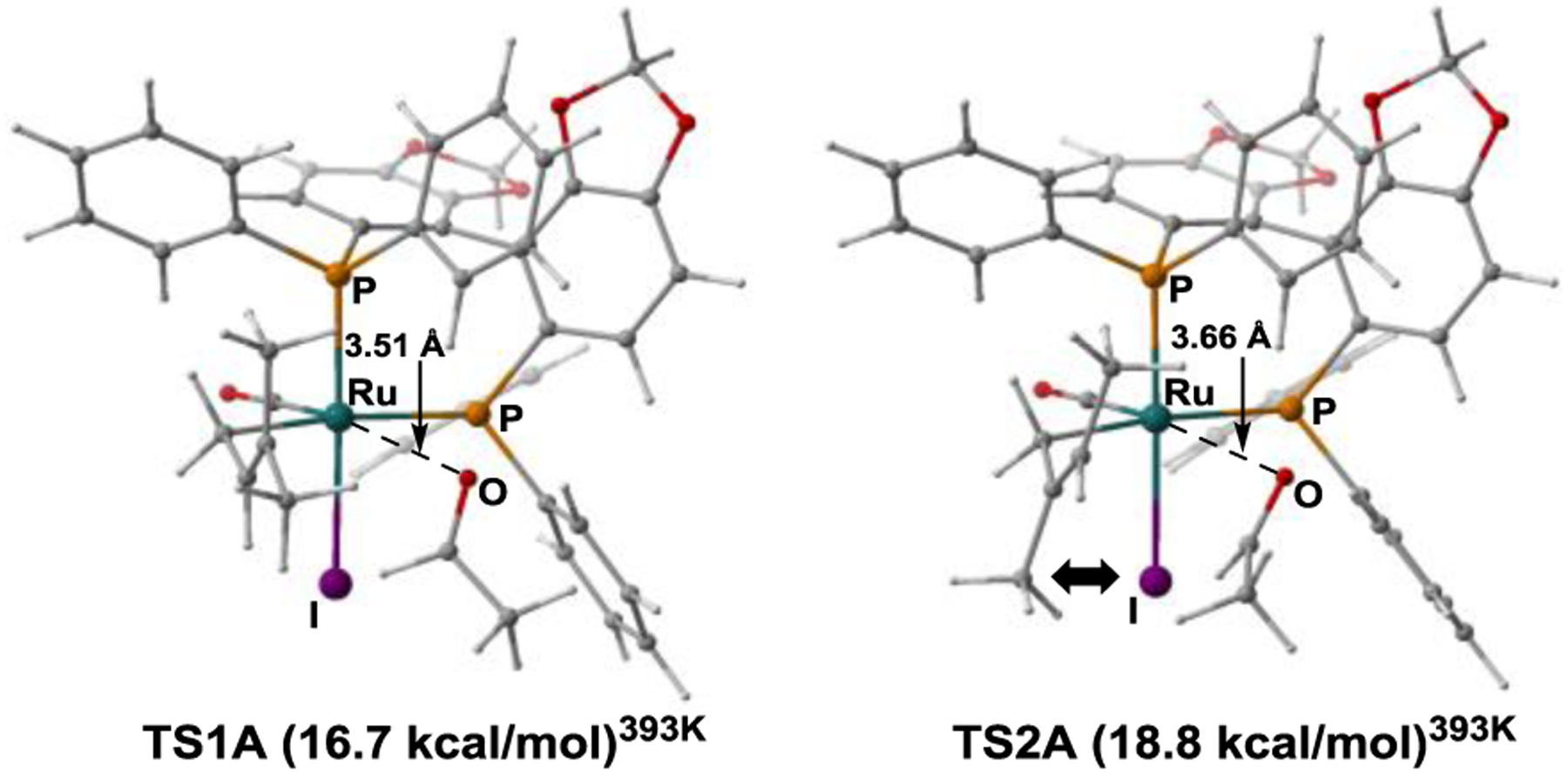
Calculated competing transitions states TS1A and TS2A for rate- and regio-determining aldehyde binding.a
aSee supporting information for further details.
In summary, we describe the first correlation of metal-centered stereogenicity to regioselectivity in metal catalysis, as demonstrated by regiodivergent carbonyl sec- vs tert-prenylations of alcohol proelectrophiles mediated by isoprene. Specifically, whereas chloride-bound ruthenium-SEGPHOS complexes prefer a trans-relationship between the halide and carbonyl ligands and deliver products of carbonyl sec-prenylation, iodide-bound ruthenium-SEGPHOS complexes prefer a cis-relationship between the halide and carbonyl ligands and deliver products of carbonyl tert-prenylation. DFT calculations of the iodide-bound catalyst implicate a Curtin-Hammett-type scenario in which the transition states for aldehyde coordination from an equilibrating mixture of sec- and tert-prenylruthenium complexes is rate- and product-determining. These studies, which underscore the profound influence of both halide counterions1 and metal-centered diastereoselectivity3 vis-à-vis selectivity in metal catalysis, have unlocked a practical method for catalytic enantioselective carbonyl tert-prenylation mediated by the abundant feedstock diene isoprene.
Supplementary Material
ACKNOWLEDGMENT
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445) are acknowledged for partial support of this research.
Footnotes
Supporting Information. Experimental procedures and spectroscopic data for all new compounds (1H NMR, 13C sNMR, 31P NMR, IR, HRMS), including images of NMR spectra and HPLC traces for racemic and enantiomerically enriched compounds. Single-crystal X-ray diffraction data for compound 3e and RuX(CO)(SEGPHOS)(η3-C3H5), where X = Cl or I.
Accession Codes. CCDC 2260272, 2260273 and 2260274 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
REFERENCES
- (1).For selected reviews on halide counterion effects in transition metal catalysis, see:; (a) Maitlis PM; Haynes A; James BR; Catellani M; Chiusoli GP Iodide Effects in Transition Metal Catalyzed Reactions. Dalton Trans. 2004, 3409–3419. [DOI] [PubMed] [Google Scholar]; (b) Fagnou K; Lautens M Halide Effects in Transition Metal Catalysis. Angew. Chem. Int. Ed 2002, 41, 26–47. [DOI] [PubMed] [Google Scholar]
- (2).Ortiz E; Shezaf JZ; Chang Y-H; Goncçalves TP Huang K-W; Krische MJ Understanding Halide Counterion Effects in Enantioselective Ruthenium-Catalyzed Carbonyl (α-Aryl)allylation: Alkynes as Latent Allenes and Trifluoroethanol-Enhanced Turnover in the Conversion of Ethanol to Higher Alcohols via Hydrogen Auto-Transfer. J. Am. Chem. Soc 2021, 143, 16709–16717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).For selected reviews on enantioselective catalysis via chiral-at-metal complexes, see:; (a) Knight PD; Scott P Predetermination of Chirality at Octahedral Centres with Tetradentate Ligands: Prospects for Enantioselective Catalysis. Coord. Chem. Rev 2003, 242, 125–143. [Google Scholar]; (b) Bauer EB Chiral-at-Metal Complexes and Their Catalytic Applications in Organic Synthesis. Chem. Soc. Rev 2012, 41, 3153–3167. [DOI] [PubMed] [Google Scholar]; (c) Gong L; Chen L-A; Meggers E Asymmetric Catalysis Mediated by the Ligand Sphere of Octahedral Chiral-at-Metal Complexes. Angew. Chem. Int. Ed 2014, 53, 10868–10874. [DOI] [PubMed] [Google Scholar]; (d) Steinlandt PS; Zhang L; Meggers E Metal Stereogenicity in Asymmetric Transition Metal Catalysis. Chem. Rev 2023, 123, 4764–4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For selected studies of formyl CH hydrogen bonding, see:; (a) Corey EJ; Lee TW The Formyl C–H⋯O Hydrogen Bond as a Critical Factor in Enantioselective Lewis-Acid Catalyzed Reactions of Aldehydes. Chem. Commun 2001, 1321–1329. [Google Scholar]; (b) Thakur TS; Kirchner MT; Bläser D; Boese R; Desiraju GR Nature and Strength of C–H⋯O Interactions Involving Formyl Hydrogen Atoms: Computational and Experimental Studies of Small Aldehydes. Phys. Chem. Chem. Phys 2011, 13, 14076–14091. [DOI] [PubMed] [Google Scholar]; (c) Grayson MN; Krische MJ; Houk KN Ruthenium-Catalyzed Asymmetric Hydrohydroxyalkylation of Butadiene: The Role of the Formyl Hydrogen Bond in Stereochemical Control. J. Am. Chem. Soc 2015, 137, 8838–8850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).For an octahedral metal complex of the type M(AB)(CD)ef, 10 enantiomeric pairs are possible. For an octahedral M(AA)(BC)de metal complex, 5 enantiomeric pairs are possible. See Miessler GL; Fischer PJ; Tarr DA Inorganic Chemistry, 5th ed.; Pearson: Upper Saddle River, New Jersey, 2014; pp. 326. [Google Scholar]
- (6).Ortiz E; Chang Y-H; Shezaf JZ; Shen W; Krische MJ Stereo- and Site-Selective Conversion of Primary Alcohols to Allylic Alcohols via Ruthenium-Catalyzed Hydrogen Auto-Transfer Mediated by 2-Butyne. J. Am. Chem. Soc 2022, 144, 8861–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ortiz E; Spinello BJ; Cho Y; Wu J; Krische MJ Stereo- and Site-Selective Crotylation of Alcohol Proelectrophiles via Ruthenium-Catalyzed Hydrogen Auto-Transfer Mediated by Methylallene and Butadiene. Angew. Chem. Int. Ed 2022, 61, e202212814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).For isoprene production volumes, see:; (a) Weissermel K; Arpe HJ Industrial Organic Chemistry, 4th ed.; Wiley-VCH, 2003; pp 117. [Google Scholar]; (b) Morais ARC; Dworakowska S; Reis A; Gouveia L; Matos CT; Bogdal D; Lukasik RB Chemical and Biological-Based Isoprene Production: Green Metrics. Catal. Today 2015, 239, 38–43. [Google Scholar]
- (9).For selected reviews on regiodivergent metal-catalyzed reactions, see:; (a) Funken N; Zhang Y-Q; Gansäuer A Regiodivergent Catalysis: A Powerful Tool for Selective Catalysis. Chem. Eur. J 2017, 23, 19–32. [DOI] [PubMed] [Google Scholar]; (b) Najera C; Beletskaya IP; Yus M Metal-Catalyzed Regiodivergent Organic Reactions Chem. Soc. Rev 2019, 48, 4515–4618. [DOI] [PubMed] [Google Scholar]
- (10).For prenyl-boron reagents in enantioselective carbonyl tert-prenylation, see:; (a) Brown HC; Jadhav PK 3,3-Dimethylallyldiisopinocampheylborane: A Novel Reagent for Chiral Isoprenylation of Aldehydes. Synthesis of (+)- and (−)-Artemisia Alcohol in Exceptionally High Enantiomeric Purity. Tetrahedron Lett. 1984, 25, 1215–1218. [Google Scholar]; (b) Jadhav PK; Bhat KS; Perumal PT; Brown HC Chiral Synthesis via Organoboranes. 6. Asymmetric Allylboration via Chiral Allyldialkylboranes. Synthesis of Homoallylic Alcohols with Exceptionally High Enantiomeric Excess. J. Org. Chem 1986, 51, 432–439. [Google Scholar]; (c) Roush WR; Marron TG Toward the Synthesis of Mycalamides A, B and Onnamide A: A Highly Stereoselective Synthesis of the Trioxadecalin Ring System. Tetrahedron Lett. 1993, 34, 5421–5424. [Google Scholar]; (d) Alam R; Vollgraff T; Eriksson L; Szabó KJ Synthesis of Adjacent Quaternary Stereocenters by Catalytic Asymmetric Allyboration. J. Am. Chem. Soc 2015, 137, 11262–11265. [DOI] [PubMed] [Google Scholar]
- (11).For prenyl-indium reagents in enantioselective carbonyl tert-prenylation, see:; (a) Loh T-P; Zhou JR; Li X-R An Enantioselective Indium-Mediated Allylation Reaction of Aldehydes and Ketones in Dichloromethane. Tetrahedron Lett. 1999, 40, 9333–9336. [Google Scholar]; (b) Loh T-P; Zhou J-R; Yin Z A Highly Enantioselective Indium-Mediated Allylation Reaction of Aldehydes. Org. Lett 1999, 1, 1855–1857. [Google Scholar]
- (12).For prenyl-silicon reagents in enantioselective carbonyl tert-prenylation, see:; (a) Nakajima M; Saito M; Shiro M; Hashimoto S-I (S)-3,3’-Dimethyl-2,2’-biquinoline N,N’-Dioxide as an Efficient Catalyst for Enantioselective Addition of Allyltrichlorosilanes to Aldehydes. J. Am. Chem. Soc 1998, 120, 6419–6420. [Google Scholar]; (b) Nakajima M; Kotani S; Ishizuka T; Hashimoto S Chiral Phosphine Oxide BINAPO as a Catalyst for Enantioselective Allylation of Aldehydes with Allyltrichlorosilanes. Tetrahedron Lett. 2005, 46, 157–159. [Google Scholar]; (c) Denmark SE; Fu J Catalytic, Enantioselective Addition of Substituted Allylic Trichlorosilanes Using a Rationally-Designed 2,2’-Bispyrrolidine-Based Bisphosphoramide. J. Am. Chem. Soc 2001, 123, 9488–9489. [DOI] [PubMed] [Google Scholar]; (d) Denmark SE; Fu J; Lawler MJ Chiral Phosphoramide-Catalyzed Enantioselective Addition of Allylic Trichlorosilanes to Aldehydes. Preparative Studies with Bidentate Phosphorus-Based Amides. J. Org. Chem 2006, 71, 1523–1536. [DOI] [PubMed] [Google Scholar]
- (13).For prenyl-tin reagents in enantioselective carbonyl tert-prenylation, see:; (a) Boldrini GP; Tagliavini E; Trombini C; Umani-Ronchi A Enantioselective Synthesis of Homo-Allylic Alcohols from Chiral Allylic Tin(IV)(+)-Diethyl Tartrate Complexes and Aldehydes. J. Chem. Soc., Chem. Commun 1986, 685−686. [Google Scholar]; (b) Boldrini GP; Lodi L; Tagliavini E; Tarasco C; Trombini C; Umani-Ronchi A Synthesis of Enantiomerically Enriched Homoallylic Alcohols and of 1,2-dien-1-ols Using Chiral Tin(IV) Complexes Containing Diethyl Tartrate as an Auxiliary Ligand. J. Org. Chem 1987, 52, 5447–5452. [Google Scholar]
- (14).For selected reviews on the use of dienes as allylmetal pronucleophiles in enantioselective transition metal-catalyzed carbonyl addition, see:; (a) Holmes M; Schwartz LA; Krische MJ Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes and Enynes with Carbonyl Compounds and Imines. Chem. Rev 2018, 118, 6026–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xiang M; Pfaffinger DE; Krische MJ Allenes and Dienes as Chiral Allylmetal Pronucleophiles in Catalytic Enantioselective C=X Addition: Historical Perspective and State-of-The-Art Survey. Chem. Eur. J 2021, 27, 13107–13116. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ortiz E; Saludares C; Wu J; Cho Y; Santana CG; Krische MJ Carbonyl Allylation and Crotylation: Historical Perspective, Relevance to Polyketide Synthesis and Evolution of Enantioselective Ruthenium-Catalyzed Hydrogen Auto-Transfer Processes. Synthesis 2023, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).For enantioselective iridium-catalyzed tert-prenylations mediated by 1,1-dimethyl allene, see:; Han SB; Kim IS; Han H; Krische MJ Enantioselective Carbonyl Reverse Prenylation from the Alcohol or Aldehyde Oxidation Level Employing 1,1-Dimethylallene as the Prenyl Donor. J. Am. Chem. Soc 2009, 131, 6916–6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).(a) Shibahara F; Bower JF; Krische MJ Ruthenium Catalyzed C-C Bond Forming Transfer Hydrogenation: Carbonyl Allylation from the Alcohol or Aldehyde Oxidation Level Employing Acyclic 1,3-Dienes as Surrogates to Preformed Allyl Metal Reagents. J. Am. Chem. Soc 2008, 130, 6338–6339. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zbieg JR; Moran J; Krische MJ Diastereo- and Enantioselective Ruthenium Catalyzed Hydrohydroxyalkylation of 2-Silyl-Butadienes: Carbonyl syn-Crotylation from the Alcohol Oxidation Level. J. Am. Chem. Soc 2011, 133, 10582–10586. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zbieg JR; Yamaguchi E; McInturff EL; Krische MJ Enantioselective C-H Crotylation of Primary Alcohols via Hydrohydroxyalkylation of Butadiene. Science 2012, 336, 324–327. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) McInturff EL; Yamaguchi E; Krische MJ Chiral-Anion-Dependent Inversion of Diastereo- and Enantioselectivity in Carbonyl Crotylation via Ruthenium-Catalyzed Butadiene Hydrohydroxyalkylation. J. Am. Chem. Soc 2012, 134, 20628–20631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).MacFarlane KS; Joshi AM; Rettig SJ; James BR Characterization of Five-Coordinate Ruthenium(II) Phosphine Complexes by X-ray Diffraction and Solid-State 31P CP/MAS NMR Studies and Their Reactivity with Sulfoxides and Thioethers. Inorg. Chem 1996, 35, 7304–7310. [DOI] [PubMed] [Google Scholar]
- (18).Diesveld JW; Menger EM; Edzes HT; Veeman WS High-Resolution Solid-State Phosphorus-31 Nuclear Magnetic Resonance of Some Triphenylphosphine Transition-Metal Complexes J. Am. Chem. Soc 1980, 102, 7935–7936. [Google Scholar]
- (19).Zbieg JR; McInturff EL; Leung JC; Krische MJ Amplification of Anti-Diastereoselectivity via Curtin−Hammett Effects in Ruthenium-Catalyzed Hydrohydroxyalkylation of 1,1-Disubstituted Allenes: Diastereoselective Formation of All-Carbon Quaternary Centers. J. Am. Chem. Soc 2011, 133, 1141–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).For Berry pseudorotation of a pentacoordinate ruthenium(II) carbonyl complex, see:; Porter TM; Wang J; Li Y; Xiang B; Salsman C; Miller JS; Xiong W; Kubiak CP Direct Observation of the Intermediate in an Ultrafast Isomerization. Chem. Sci 2019, 10, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Feng J; Holmes M; Krische MJ Acyclic Quaternary Carbon Stereocenters via Enantioselective Transition Metal Catalysis. Chem. Rev 2017, 117, 12564–12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).For the stoichiometric reaction of HXRu(CO)(PR3)3 (X = Cl, Br) with allenes and dienes to form isolable π-allylruthenium species, see:; (a) Hiraki K; Ochi N; Sasada Y; Hayashida H; Fuchita Y; Yamanaka S Organoruthenium(II) Complexes Formed by Insertion Reactions of Some Vinyl Compounds and Conjugated Dienes into a Hydrido–Ruthenium Bond. J. Chem. Soc., Dalton Trans 1985, 873–877. [Google Scholar]; (b) Hill AF; Ho CT; Wilton-Ely JDET The Coupling of Methylene and Vinyl Ligands at a Ruthenium(II) Centre. Chem. Commun 1997, 2207–2208. [Google Scholar]; (c) Xue P; Bi S; Sung HHY; Williams ID; Lin Z; Jia G Isomerism of [Ru(η3-allyl)Cl(CO)(PPh3)2] Organometallics 2004, 23, 4735–4743. [Google Scholar]
- (23).(a) Tse SKS; Xue P; Lin Z; Jia G Hydrogen/Deuterium Exchange Reactions of Olefins with Deuterium Oxide Mediated by the Carbonylchlorohydrido-tris(triphenylphosphine)ruthenium(II) Complex. Adv. Synth. Catal 2010, 352, 1512–1522. [Google Scholar]; (b) Isbrandt ES; Vandavasi JK; Zhang W; Jamshidi MP; Newman SG Catalytic Deuteration of Aldehydes with D2O. Synlett 2017, 28, 2851–2854. [Google Scholar]
- (24).DFT calculations were performed at the ωB97X-D(IEF-PCM)/Def2-TZVPP//ωB97X-D/(SDD, 6–311G(d,p)) level of theory. See Supporting Information for further details. [Google Scholar]
- (25).An unvarying preference for a trans-relationship between chloride and carbonyl ligands is observed across diverse π-allylruthenium complexes. See reference 2, 22c and the following:; (a) Esteruelas MA; Liu F; Oñate E; Sola E; Zeier B Carbon-Carbon Coupling of two Alkenyl Fragments on a Saturated Compound. Organometallics 1997, 16, 2919–2928. [Google Scholar]; (b) Sasabe H; Nakanishi S; Takata T Preparation of Ru(η3–2-alkenylallyl)(CO)Cl(PPh3)2 Complexes via Carbometallation of Allenes with Alkenyl Ruthenium Complexes. Inorg. Chem. Commun 2002, 5, 177–180. [Google Scholar]; (c) Bai T; Zhu J; Xue P; Sung HH-Y; Williams ID; Ma S; Lin Z; Jia G Ligand Effect on the Insertion Reactions of Allenes with MHCl(CO)(PPh3)3 and MHCl(PPh3)3 (M = Ru, Os). Organometallics 2007, 26, 5581–5589. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.