

Abstract
Pathogenic microbes invade the human body and trigger a host immune response to defend against the infection. In response, host-adapted pathogens employ numerous virulence strategies to overcome host defense mechanisms. As a result, the interaction between the host and pathogen is a dynamic process that shapes the evolution of the host’s immune response. Among the immune responses against intracellular bacteria, pyroptosis, a lytic form of cell death, is a crucial mechanism that eliminates replicative niches for intracellular pathogens and modulates the immune system by releasing danger signals. This review focuses on the role of pyroptosis in combating intracellular bacterial infection. We examine the cell type specific roles of pyroptosis in neutrophils and intestinal epithelial cells. We discuss the regulatory mechanisms of pyroptosis, including its modulation by autophagy and interferon-inducible GTPases. Furthermore, we highlight that while host-adapted pathogens can often subvert pyroptosis, environmental microbes are effectively eliminated by pyroptosis.
Keywords: pyroptosis, intracellular bacteria, host-adapted pathogen, environmental pathogen, autophagy, guanylate-binding protein
1. Introduction
Pathogens are categorized into two main groups according to their dominant location relative to host cells: extracellular and intracellular1. Further classification of intracellular pathogens results in two subtypes: vacuolar and cytosolic2. Intracellular bacterial pathogens invade host cells to facilitate their replication and spread. This location offers intracellular bacterial pathogens advantages, such as protection from the host’s humoral immunity (e.g., complement, secreted antimicrobial peptides, antibodies), sequestration from neutrophils, and access to nutrients that may be scarce extracellularly2. As a result, intracellular bacterial pathogens manipulate host cells to access their preferred niches within targeted cells. After invasion, bacteria are contained within a plasma membrane-derived vacuole in host cells, such as phagosomes or endosomes. Vacuolar bacteria, such as Salmonella enterica serovar Typhimurium, remain within the vacuole, while cytosolic bacteria, for example, Listeria monocytogenes, rupture the vacuole and reside within the host cytosol. Notably, recent studies suggested that whether a bacterium is vacuolar or cytosolic is more context-dependent, such as the host cell types, tissue, metabolic status, and cellular environment2–4.
The virulence strategies of host-adapted pathogens exert selective pressure on the host immune system, leading to the evolution of new host defense mechanisms. In turn, host defense mechanisms impose evolutionary pressure on the virulence strategies of host-adapted pathogens. Together, these selective pressures create a continuous cycle of adaptation and counter-adaptation, resulting in a constant arms race between the host immune system and pathogens’ virulence strategies that shapes the evolution of both host and pathogens5,6. As a result, both hosts and host-adapted pathogens are constantly evolving to maintain their respective advantages in the arms race, an example of the Red Queen hypothesis7. The net result is that the host-pathogen interface is maintained, preserving the ability of host-adapted pathogens to survive, replicate, and transmit to new hosts. In contrast, environmental (opportunist) pathogens, are microorganisms commonly found in a habitat that are typically avirulent to immunocompetent individuals despite encoding potent virulence factors. Environmental pathogens often fail to evade the host’s innate immune defenses and are therefore efficiently eliminated8.
Regulated cell death pathways, including apoptosis, pyroptosis, and necroptosis, are innate immune defense mechanisms that remove the replicative niche of intracellular pathogens while simultaneously recruiting immune cells to the site of infection9,10. Pyroptosis, a type of cell death characterized by cell lysis, is typically initiated by the pyroptotic caspases (caspase-1/4/5/11). Among these, caspase-4/5 (human) and caspase-11 (mouse) are directly activated by their LPS ligand. Activated caspase-4/5/11 cleave the linker region of gasdermin D (GSDMD) to release its N-terminal pore-forming domain, which oligomerizes and forms pores in the plasma membrane, ultimately leading to pyroptosis11–14. In parallel, caspase-1 is activated by inflammasomes, which are cytosolic multiprotein complexes made up of an inflammasome sensor and often an adaptor protein called ASC15. Inflammasomes, including NLRPs, NLRC4, AIM2, and pyrin, detect the presence of pathogen-associated molecular patterns (PAMPs) or other patterns of pathogenesis, leading to the activation of caspase-116. Once activated, caspase-1 also cleaves and activates GSDMD11–14. It should be noted that many genes in these pathways are often duplicated or contracted between humans and mice, however the core functions that we cover in this review are all conserved.
Here, we describe the mechanisms of pyroptosis in defending against intracellular bacterial pathogens. In general, host-adapted bacterial pathogens often have evolved strategies to evade or suppress pyroptotic cell death. We therefore contrast muted host defenses against host-adapted bacterial pathogens with the effective clearance of environmental bacterial pathogens by pyroptosis.
2. Pyroptosis
Regulated cell death pathways are critical for defending against intracellular infections by eliminating the replicative niche of pathogens17. These pathways, including apoptosis and lytic cell death, are distinguished by their morphological and immunological features18. Apoptosis is characterized by DNA fragmentation, cell blebbing, and the formation of apoptotic bodies. The apoptotic bodies are rapidly cleared by phagocytes through efferocytosis, which is not intrinsically inflammatory19. In contrast, regulated lytic cell death, including pyroptosis and necroptosis, leads to the release of cellular contents that cause a pro-inflammatory response. The utility of different cell death pathways can depend upon the cell type that is infected as well as the nature of the infecting pathogen. Sometimes regulated cell death pathways act redundantly, whereas in other cases one specific pathway is required to mediate clearance of a pathogen20,21.
Pyroptosis is typically induced by the activation of caspase-1/4/5/1110 (Figure 1). The signals that activate cytosolic sensors called inflammasomes upstream of caspase-1 or the nature of direct activation of caspase-4/5/11 by LPS are reviewed elsewhere10. Pyroptosis is characterized by the rupture of the plasma membrane, which is facilitated by gasdermins, a family of pore-forming proteins. Among the gasdermins, GSDMD is considered the prototype22. Recent studies have shown that other members of the gasdermin family, including gasdermin A, B, C, and E, also possess pore-forming activities (note that mice lack GSDMB and have expanded Gsdma and Gsdmc genes). These proteins consist of two domains that are connected by a cleavable linker region: an N-terminal pore-forming domain and a C-terminal autoinhibitory domain. Upon cleavage of the linker region, the activated N-terminal domain recognizes membrane lipids and undergoes oligomerization, resulting in the formation of a soluble prepore. This prepore subsequently undergoes a conformational change as it inserts into the membrane, forming large pores with an approximate diameter of 21 nm23,24. These pores allow cellular contents to exit the cell, including proinflammatory cytokines (e.g., IL-1β and IL-18), ATP, and other alarmins11,13,14,24,25. The full dispersion of any particular molecule probably takes several minutes to reach equilibrium. GSDMD-independent release of the proinflammatory cytokines has also been reported26,27.
Figure 1. Inflammasome activation induces pyroptosis.
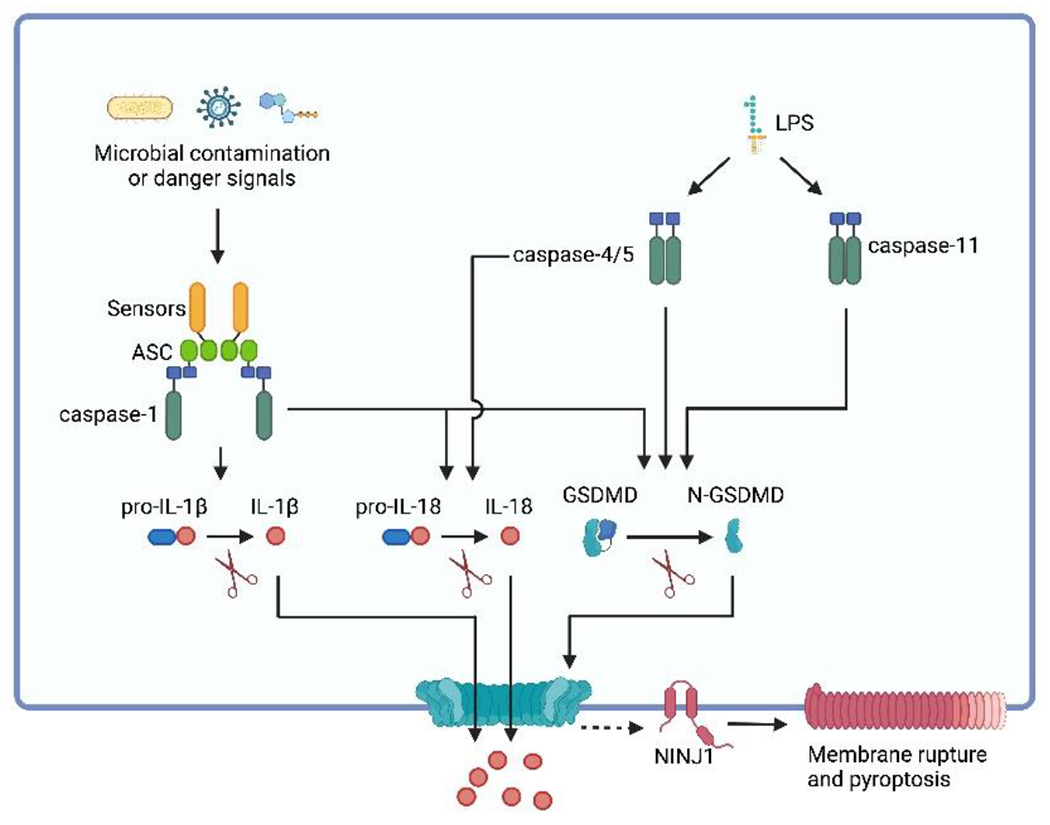
Inflammasomes are multiprotein complexes located in the cytosol that trigger the activation of caspase-1. Typically, an inflammasome comprises an inflammasome sensor and often the protein adaptor ASC. Inflammasome sensors are responsible for detecting various cellular disturbances, including microbial contaminants or danger signals. Upon polymerization, inflammasome sensors often recruit ASC, leading to the formation of ASC specks. Inflammasomes either directly or via ASC recruit pro-caspase-1 and trigger its activation. Once activated, caspase-1 cleaves GSDMD into its active form, N-terminal GSDMD (N-GSDMD). N-GSDMD polymerizes and forms pores in the plasma membrane, resulting in pyroptosis. NINJ1, a transmembrane protein, facilitates plasma membrane rupture in pyroptosis and other types of lytic cell death. Furthermore, caspase-1 also cleaves pro-IL-1β and pro-IL-18, generating active IL-1β and IL-18, respectively. These pro-inflammatory cytokines can be released from the GSDMD pores, along with other inflammatory cellular components. In parallel, caspase-4/5/11, are activated by cytosolic LPS. This activation leads to the cleavage of GSDMD, ultimately inducing pyroptosis. Notably, caspase-4/5 but not caspasae-11 can cleave pro-IL-18 into its mature form156.
After the GSDMD pore opens, the subsequent membrane rupture is an active process mediated by NINJ128. The mechanism by which NINJ1 is activated downstream of the gasdermin pore remains unclear, but once activated, NINJ1 polymerizes in the membrane resulting in rupture of the membrane that immediately disperses all soluble cytosolic molecules, including large proteins such as the lactate dehydrogenase tetramer that is often used as a marker for cell lysis. One possible clue to the NINJ1 activation mechanism is the long-known property of extracellularly applied glycine (or alanine) to inhibit pyroptosis29. Glycine does not inhibit the opening of the gasdermin pore, but does significantly delay rupture of the membrane by several hours, which is similar to the phenotype of a Ninj1-knockout cell. High extracellular glycine inhibits NINJ1 clustering30, which was hypothesized to explain the mechanism of glycine cytoprotection. Interestingly, the very high 5-10 mM concentrations of glycine that must be applied to cells matches the cytosolic concentration of glycine31, suggesting a possible mechanism whereby NINJ1 activates in response to the loss of cytosolic glycine. Although speculative, this would provide a common mechanism whereby NINJ1 can activate in response to multiple forms of cell death that all converge upon a common cytosolic event.
2.1. The role of cell lysis in intracellular bacteria-induced pyroptosis
Pyroptosis plays a crucial role in fighting intracellular bacterial infections, and it has been extensively studied in macrophages using both host-adapted and environmental pathogens8. This lytic form of cell death removes the replicative niche for intracellular pathogens by inducing the formation of large pores in the plasma membrane. This process releases highly inflammatory cytoplasmic contents, including proinflammatory cytokines and damage-associated molecular patterns (DAMPs), which recruit and activate immune cells to combat the infection. In addition, pyroptosis also helps combat bacterial infections by converting the pyroptotic cells into pore-induced intracellular traps (PITs)32. These PITs trap bacteria inside the dead cells, and simultaneously facilitate the recruitment and efferocytic properties of neutrophils through complement and scavenger receptors. These neutrophils then efferocytose the trapped bacteria and subsequently kill the bacteria. It has also been shown that GSDMD can forms pores on the bacterial membrane and directly kill bacteria33, however, intracellular bacteria do survive pyroptosis in macrophages in vitro32. We previously speculated that pyroptosis damages bacteria, which may involve GSDMD pores or mitochondrial ROS34. Indeed, caspase-1 activation results in mitochondrial ROS production35,36, which could explain how intracellular bacteria trapped in PITs become damaged during the process of pyroptosis. Release of bacterial damaging mitochondrial ROS has also been demonstrated after inhibition of TAK1 that results in apoptotic/necroptotic cell death37, suggesting a generalized defense mechanism during regulated cell death.
Ninj1-deficient mice were found to be more susceptible to Citrobacter rodentium infection28. Additionally, the expression of NINJ1 was shown to be beneficial to the host during Y. pseudotuberculosis infection38. Although the exact mechanism of NINJ1-mediated protection during bacterial infection remains unclear, it was speculated that DAMPs released by NINJ1-mediated plasma membrane rupture play a role in defending against bacterial infection28. Furthermore, whether NINJ1-driven membrane rupture will be generally required downstream of GSDMD remains to be established.
2.2. Pyroptosis in neutrophils
Pyroptosis has been predominantly studied in macrophages, but recent research is revealing its role in other cell types during bacterial infection. Neutrophils, for example, are among the first immune cells to respond to infection and accumulate in large numbers at the site of infection. Although neutrophils express inflammasome sensors, inflammatory caspases (caspase-1/4/5/11), and GSDMD39,40, their capacity to undergo pyroptosis seems to be more tightly regulated than that of macrophages. Neutrophils can release bio-active IL-1β upon inflammasome activation39,41–43, but the release of IL-1β in some contexts occurs without membrane rupture39,41, possibly to preserve the antibacterial function of neutrophils. For instance, neutrophils infected with Burkholderia thailandensis undergo caspase-11 dependent pyroptosis44. In contrast, NLRC4 and NLRP3 inflammasome activation in neutrophils did not induce pyroptosis39,45. However, recent studies using extracellular bacterial infections showed that NLRC4 and pyrin inflammasome activation could induce pyroptosis in neutrophils depending upon the nature of the agonist46,47. The difference between lysis or non-lytic caspase activation was caused by the cellular location through which inflammasome activating stimuli entered the cytosol – across the plasma membrane compared to endosomal, however, the mechanism by which this is achieved remains unknown. Additionally, GSDMD activation in neutrophils induces NETosis48,49, which is the release of decondensed nuclear DNA into extracellular compartments. Specifically, cytosolic Gram-negative bacteria, such as Salmonella ΔsifA50 and Citrobacter rodentium, active caspase-11 and GSDMD. Caspase-11 and GSDMD coordinately facilitate nuclear membrane permeabilization and histone degradation in neutrophils, ultimately leading to NETosis. This response contributes to defense against Salmonella ΔsifA infection in mouse models48.
2.3. Pyroptotic caspase-driven extrusion in intestinal epithelial cells
Pyroptosis is not limited to immune cells and can also be activated in epithelial cells to combat bacterial infections51. The invasion of the intestinal epithelium by Salmonella drives its pathogenesis in the gut. The host has evolved mechanisms to fight back against invasion of intestinal epithelial cells (IECs) by extruding the infected IECs into the intestinal lumen. Extrusion can be triggered via several pathways, including by activation of the NAIP-NLRC4 inflammasome; thus pyroptotic signaling causes IEC extrusion. The mechanisms by which NLRC4 activation causes extrusion are still being elucidated, but they include redundant pathways via GSDMD or the ASC to caspase-8 backup pathway51–55. The extrusion of infected IECs removes infected cells from the tissue and prevents further spread of the pathogen to deeper sites56. Apoptotic signaling also causes IEC extrusion, and also limits bacterial burdens in the intestine21. Successfully completed extrusion also limits tissue damage caused by Salmonella infection57. We recently proposed that extrusion is one of the specific effector programs that a cell must execute before the cell loses all functional capacity. These effector programs can be thought of as a “bucket list” of tasks that must be completed before the cell dies58.
The discovery that the host successfully detects the S. Typhimurium SPI1 T3SS as it attempts to invade IECs, and successfully extrudes IECs in response appears to contradict the idea that host-adapted pathogens should evade innate defenses. This is particularly confounding in the case of S. Typhimurium, where the primary intestinal virulence strategy seems to be invasion of IECs, yet the host seems to have a potent and direct countermeasure to extrude the invaded cells. This raises the possibility that the “failure” of S. Typhimurium to evade IECs extrusion may be a deliberate attempt by the bacteria to provoke an inflammatory response. Indeed, S. Typhimurium benefits from the inflammatory response that generates molecules that the bacteria use as alternate electron acceptors in respiration59. It could be that the bacteria “have their cake and eat it too” by simultaneously benefiting from triggering wide scale IEC extrusion that causes inflammation while the few IECs that fail to extrude become replicative niches for the bacteria and promote their dissemination to macrophages in the lamina propria or draining lymph nodes. We propose that these phenotypes be considered carefully in the context of the Red Queen’s race, where S. Typhimurium and NLRC4 compete, sometimes resulting in a victory for Salmonella and invasion, and sometimes in a victory for host and extrusion (but a victory that the bacteria may simultaneously exploit).
2.4. Host-adapted bacterial pathogens subvert pyroptosis
As an innate defense mechanism, pyroptosis exerts selective pressure on pathogens. To enhance their own survival, replication, and transmission, these pathogens are compelled to develop strategies to evade, suppress or even exploit the pyroptotic pathway at all levels, including evading detection or inhibiting inflammasomes, caspases, and gasdermins60,61. For example, during their intracellular phases, S. Typhimurium and L. monocytogenes evade NLRC4 inflammasome by suppressing flagellin expression8. In other pathogens, variant LPS structures allow bacteria to evade cytosolic LPS detection. For example, Francisella species have tetraacylated LPS allowing evasion of murine caspase-1162,63, but not human casapse-464. Shigella species overcome cytosolic LPS detection by direct inhibition, translocating the T3SS effector OspC3, which inactivates human caspase-4 and murine caspase-11 by covalently modifying the caspases via ADP riboxanation. Consequently, S. flexneri ospC3 mutants are recognized and cleared by caspase-11 in a mouse infection model65–67. S. flexneri can also target the executioners of pyroptosis, the gasdermins. The S. flexneri ubiquitin-ligase virulence factor IpaH7.8 targets human GSDMD for proteasome destruction. Interestingly, IpaH7.8 cannot target mouse GSDMD due to amino acid substitutions68–71, which could partially explain the natural resistance of mice to S. flexneri infection. S. flexneri also uses IpaH7.8 to target GSDMB for proteasome destruction (discuss further below)69–72. Mycobacterium tuberculosis has also developed strategies to overcome pyroptosis73. The pore-forming domain of GSDMD has a strong affinity for cell membrane lipids. This allows GSDMD to bind to the inner leaflet of plasma membrane to form pores and induce pyroptosis13,74. M. tuberculosis effector protein PtpB, a phospholipid phosphatase, hijacks ubiquitin to mediate PtbB activation. Activated PtbB dephosphorylates plasma membrane lipids that are targets of the pore-forming domain of GSDMD to disrupt GSDMD membrane localization and inhibit pyroptosis75. M. tuberculosis also inhibits inflammasome detection via PknF through mechanisms that remain to be fully elucidated76.
While inflammasomes are generally believed to be beneficial during bacterial infection, in vivo studies have shown incremental rather than definitive differences in phenotype between wild-type and inflammasome-deficient mice infected with host-adapted bacteria. For example, S. Typhimurium has been extensively studied in the inflammasome field53,77–84. Studies on S. Typhimurium have shown that although WT mice have lower bacterial burdens compared to inflammasome-deficient mice, they still succumb to infection, albeit more slowly. As a result, the lethal dose difference (ΔLD100) between the two types of mice remains unchanged (ΔLD100 = 1-fold), indicating that inflammasomes are unable to prevent death during S. Typhimurium infection. The mild phenotype observed in inflammasome-deficient mice infected with S. Typhimurium may be due to the pathogen’s ability to evade inflammasome detection. Our previous research has demonstrated that S. Typhimurium can evade detection by the NLRC4 inflammasome during systemic infection by repressing flagellin83 and expressing a SPI2 version of the T3SS rod protein that carries evasive point mutations84. However, when these evasion strategies are eliminated in engineered S. Typhimurium, inflammasomes efficiently and completely eradicate the pathogen84.
2.5. Environmental pathogens are efficiently cleared by pyroptosis
Environmental pathogens that are unable to evade or inhibit inflammasomes are usually cleared effectively via pyroptosis8. In the case of B. thailandensis, its T3SS apparatus rod/needle proteins and LPS are recognized by the NAIP-NLCR4 inflammasome and caspase-11, respectively85. Notably, WT mice survived infection by 2x107 CFU of B. thailandensis, while inflammasome-deficient mice were susceptible to as few as 100 CFU85. These findings suggest that inflammasomes changed the lethal dose by 2,000,000-fold during B. thailandensis infection. Mechanistically, B. thailandensis is recognized by the NLRC4 inflammasome in macrophages, leading to the secretion of IL-18. This pro-inflammatory cytokine, in turn, stimulates natural killer (NK) cells and T cells to produce interferon gamma (IFN-γ). IFN-γ then primes caspase-11 in neutrophils, which is subsequently activated by LPS release by the cytosol-invasive B. thailandensis, leading to pyroptosis. Notably, pyroptosis is critical for the efficient clearance of B. thailandensis44,85,86.
Chromobacterium violaceum, a Gram-negative bacterium, is widely distributed in freshwater sediment within tropical and subtropical regions87. This environmental organism is known to only infect individuals with compromised immune systems, especially those suffering from chronic granulomatous disease88,89. C. violaceum encodes a T3SS that is similar to SPI1 in S. Typhimurium90. NLRC4 inflammasome detects the C. violaceum T3SS apparatus and drives bacterial clearance in vivo91. While WT mice were able to survive infection with 106 CFU of C. violaceum, inflammasome-deficient mice died after only 100 CFU infection. Therefore, the difference in ΔLD100 between WT and inflammasome-deficient mice was estimated to be 100,000-fold.
The dramatic lethal dose change between Salmonella (1-fold) and these two environmental pathogens (more than 100,000-fold) leads us to further investigate the ΔLD100 between WT and inflammasome-deficient mice during infection by host-adapted pathogens. To accomplish this, we extensively reviewed published literature on infection studies that utilized inflammasome-deficient mice for a lethal challenge. We then estimated the change in lethal dose comparing WT to inflammasome-deficient mice8,9. Our analysis revealed that nearly all host-adapted infection models had a ΔLD100 of less than 8-fold. However, we observed a significant contrast with environmental pathogens such as B. thailandensis and C. violaceum, where inflammasomes provided potent and fully penetrant protection (Table 1). While WT mice survived the challenge by 2x107 and 106 CFU, respectively, inflammasome-deficient mice succumbed to challenge with as few as 100 CFU.
Table 1. ΔLD100 in inflammasome-deficient mice.
The calculation of ΔLD100 requires publications that investigate WT and inflammasome-deficient mice and utilize multiple doses within a single study155. In cases where only a single dose is administered, we rely on estimations based on studies conducted by the Re lab. Specifically, during Burkholderia pseudomallei infection, the Re lab demonstrated that 100% of WT mice survived a very low dose challenge, whereas 0% of inflammasome-deficient mice survived. Notably, when the Re lab increased the dose from 25 CFU to 200 CFU, 0% of WT mice survived. This data indicates an 8-fold change in the lethal dose. Therefore, if a single dose is used and all WT mice survive while all inflammasome-deficient mice succumb, it can only be interpreted as an eight-fold or greater change in the lethal dose. Notably, most published infection studies utilize only a single infectious dose8,9. Consequently, the full extent of the effect of inflammasomes in preventing lethal infection remains to be fully quantitated.
The discrepancies in the lethal dose change between host-adapted (less than 8-fold) and environmental pathogens (more than 100,000-fold) have led us to suggest that the evolutionary role of inflammasomes is to defend against pathogens that typically infect lower hosts, where inflammasomes are not present. For example, NOD-like receptors are absent in insects92. Interestingly, species from the genus Chromobacterium exhibit toxicity towards insects, such as southern green stink bug and corn rootworm93,94. Further research is needed to identify additional environmental pathogens in which inflammasomes are essential for host defense.
3. Interplay of autophagy and pyroptosis
Autophagy is a cellular process of capturing and degrading cytoplasmic materials. Xenophagy refers to the targeting and elimination of invading pathogens by autophagy machinery. Therefore, xenophagy is an important arm of cell-autonomous immunity, which refers to the ability of an individual cell to defend itself against invading pathogens, and to survive the encounter. Essentially every cell in the body is equipped with this ancient defense mechanism95. Autophagy inhibits inflammasome activation and regulates inflammatory responses96–99 (Figure 2A). The first in vivo evidence of autophagy inhibiting inflammasome activation came from studies with mice lacking an autophagy-related protein ATG16L1. Atg16l-deficient mice produced higher amounts IL-1β compared to WT mice in a colitis model100. In vitro studies showed that autophagy inhibits inflammasome signaling by degrading assembled inflammasomes101 or by removing damaged mitochondria102–104. Autophagy also clears cytosolic PAMPs, such as LPS, to reduce inflammasome activation105,106. Besides removing DAMPs and PAMPs, it was also proposed that autophagy directly targets inflammasome pathway components to inhibit inflammasome activation, such as AIM2, NLRP3, and caspase-1101,107,108.
Figure 2. Interplay of autophagy and pyroptosis.
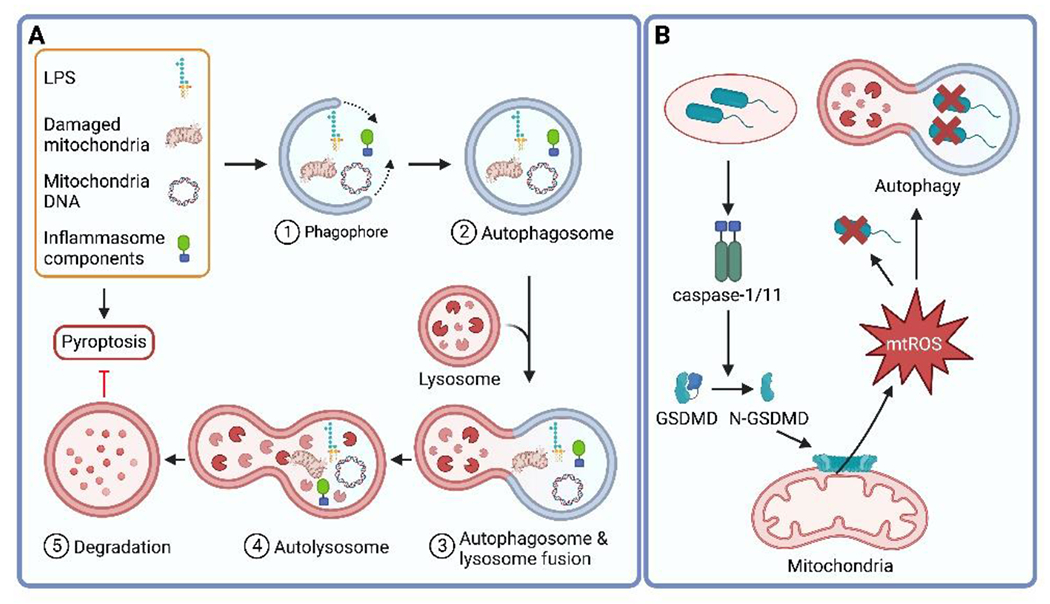
A. Autophagy is a cellular process that captures and degrades a variety of cellular materials, including damaged organelles, self-proteins, and pathogen-derived molecules. This process is initiated by the formation of a double-membrane structure called a phagophore, which expands and engulfs the targeted material to form an autophagosome. The autophagosome fuses with a lysosome to form an autolysosome, where the materials are degraded. Autophagy plays a role in inhibiting inflammasome activation by removing damaged mitochondria, mitochondrial DNA, cytosolic PAMPs, and inflammasome pathway components. B. In response to Burkholderia cenocepacia infection, the inflammasome can be activated by bacterial effectors that are secreted through the bacterial secretion system. Once activated, the inflammasome cleaves GSDMD, which then targets mitochondria to mediate the release of mitochondrial ROS (mtROS). mtROS can directly target cytosolic bacteria or promote the clearance of bacteria through xenophagy, a selective form of autophagy.
Moreover, the relationship between autophagy and inflammasome activation is complex and multifaceted, with evidence suggesting that in certain situations, inflammasome activation can also promote xenophagy. For instance, in macrophages responding to Burkholderia cenocepacia infection, an opportunistic bacterium that causes infections in immunocompromised individuals, caspase-11 activation promotes xenophagy, and caspase-11-deficient macrophages exhibit a defect in xenophagy109,110. Notably, a subsequent investigation demonstrated that upon exposure to B. cenocepacia in macrophages, GSDMD activation led to mitochondrial damage and the release of mitochondrial ROS (mtROS). Subsequently, mtROS facilitated xenophagy, which in turn promoted the elimination of the bacterial pathogen34 (Figure 2B). These findings highlight the intricate mechanisms underlying cell-autonomous immunity and the interconnectedness of cellular processes such as autophagy and inflammasome activation. We recently speculated that successful xenophagy of intracellular bacteria eliminates the need for pyroptosis, which could explain how these mechanisms work in sequence during infection111.
4. Regulation of pyroptosis by interferon-inducible GTPases
Interferon-inducible guanosine triphosphate hydrolyzing enzymes (GTPases) belong to the dynamin-like protein family and play a critical role in cell-autonomous immunity to defend against bacterial infections112,113. Interferon-inducible GTPases are classified into four subfamilies, which include immunity-related GTPases (IRGs), guanylate-binding proteins (GBPs), myxoma resistance proteins, and very large inducible GTPases114. Here, we discuss the role of GBPs and IRGs in regulating pyroptosis.
GBPs promote inflammasome activation through several different strategies depending on the type of bacteria present (Figure 3). They can act on bacteria-containing vacuoles, directly bind to cytosolic bacteria, or bind LPS released by bacteria, either in the form of free LPS micelles or packaged in outer membrane vesicles (OMVs). The diverse actions of GBPs allow activation of several different inflammasomes in response to a variety of bacterial pathogens. By targeting either bacteria themselves, pathogen containing vacuoles, or PAMPs released by bacteria, GBPs provide a multifaceted defense against intracellular invasion.
Figure 3. Regulation of pyroptosis by GBPs.
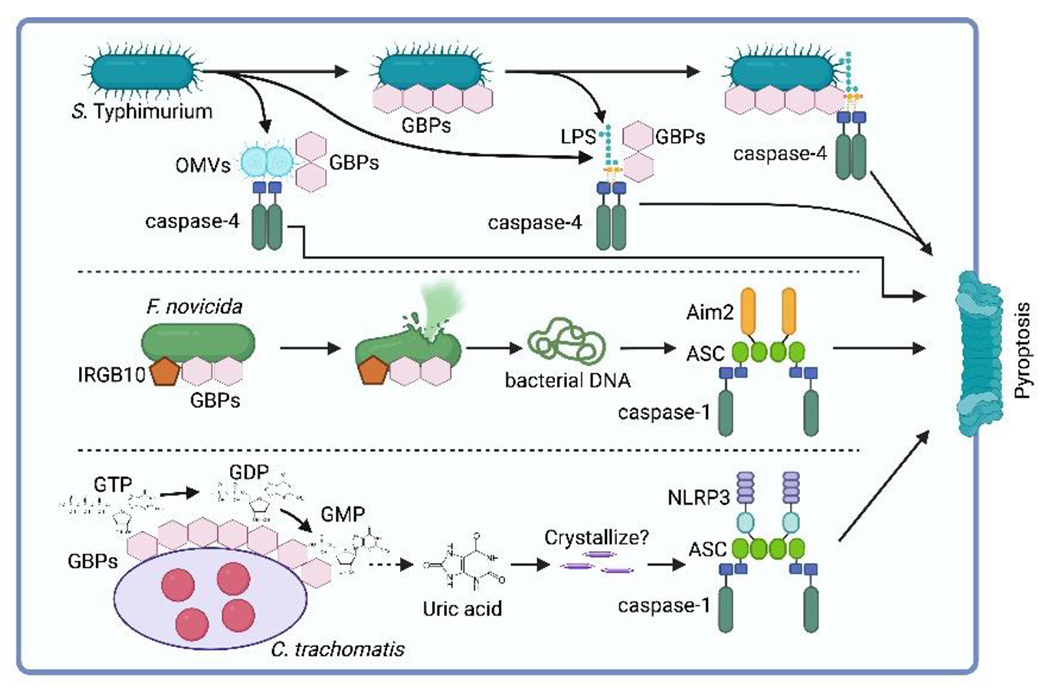
GBPs exhibit diverse mechanisms to activate inflammasomes in response to bacterial infections. GBPs can target bacteria in vacuoles or cytosol, as well as bind to released PAMPs such as LPS. GPBs facilitate caspase-4 activation in response to cytosolic free LPS or LPS packaged in outer membrane vesicles (OMVs). GBP-mediated caspase-4 activation can also occur by recruiting the caspase to the bacterial surface or by promoting the release of LPS into the cytosol. Additionally, GBPs, in conjunction with IRGB10, promote AIM2 inflammasome activation by facilitating bacterial lysis and liberation of bacterial DNA. Furthermore, GBP1 can activate the NLRP3 inflammasome during C. trachomatis infection by hydrolyzing GTP into GMP, which is then catabolized to uric acid that activates the NLRP3 inflammasome. However, it is unclear whether the uric acid concentration is sufficient for crystallization within the infected cell.
GBPs are required for efficient caspase-4/11-dependent pyroptosis in IFN-γ primed cells106,115–124. This was first demonstrated in mouse macrophages, where GBPs were important for caspase-11-dependent pyroptosis induced by intracellular Gram-negative bacteria Legionella pneumophila and S. Typhimurium115. The main clue to GBPs’ role in caspase-11 activation came from an experiment showing GBPs were also able to promote pyroptosis when purified LPS was transfected into the cytosol115. Subsequent studies further demonstrated that in the absence of infection, both free LPS and OMVs containing LPS were able to induce GBP-dependent caspase-11 activation both in macrophages and in an in vivo sepsis model117,125. Three of the seven human GBPs (and 3 of 11 mouse GBPs) contain C-terminal prenylation motifs that direct the attachment of lipid farnesyl or geranylgeranyl groups. The prenylated human GBP1 and GBP2 proteins directly bind LPS and promote caspase-4 activation118,123,124. It is likely, but has not yet been proven, that mouse GBP2, the closest ortholog of human GBP1 and GBP2, also directly binds LPS. Recombinant GBP1 or GBP2 cluster LPS micelles into larger aggregates and enhance the LPS-dependent activity of caspase-4 in vitro, suggesting that GBP-mediated LPS aggregation is responsible for GBP-dependent caspase-4 activation123,124. GBP1 binds to LPS on the surface of Gram-negative bacteria, coating the bacterium with a dense layer of GBP protein118,119,121,124,126,127. This GBP coatomer has direct consequences for the health of the bacterium and can disrupt bacterial virulence traits. GBPs act as a surfactant that extracts LPS molecules from the bacterial surface into the host cell cytosol128 and exposes the lipid A portion of bacterial membrane-embedded LPS for improved cytosolic access124. Consequently, caspase-4 is recruited to the surface of GBP1-coated bacteria118,119,121,124. However, this GBP1-dependent caspase-4 recruitment is dispensable for the induction of pyroptosis123,124. Thus, GBPs likely promote caspase-4/11 activation through multiple mechanisms by allowing caspase-4/11 to deposit upon the bacterial surface, by promoting the release of LPS from the bacterial surface, and by acting directly upon LPS that has been released from the bacterium into the cytosol.
GBPs, in concert with IRG family member IRGB10, also promote activation of the DNA sensing AIM2 inflammasome129–133. Bacterial DNA is normally sequestered within the bacterial cell, shielded from cytosolic detection by AIM2. GBPs recruit IRGB10 to the surface of cytosolic Francisella novicida, resulting in bacterial lysis and liberation of bacterial DNA that then is detected by the AIM2 inflammasome129–132. GBPs similarly facilitate the liberation of DNA from cytosol-exposed Legionella pneumophila, thereby mediating AIM2 inflammasome activation133.
GBPs were also shown to promote NLRP3 inflammasome activation134. Human GBP1 was found to activate the NLRP3 inflammasome during C. trachomatis infection. Mechanistically, hGBP1 is responsible for the hydrolysis of guanosine triphosphate (GTP) into guanosine monophosphate (GMP). The resulting GMP is then catabolized to uric acid, which activates the NLRP3 inflammasome134. Monosodium urate (MSU) crystals are a long-established activator of NLRP3 after macrophages phagocytose the crystal135. Whether uric acid produced by GBP enzymatic activity is in high enough concentration to crystalize within the C. trachomatis-infected cell was not yet determined.
While GBPs promote pyroptosis, conversely, IRGs within the IRGM subset impede pyroptosis. IRGM2 cooperates with GATE16 to inhibit caspase-11 activation in response to various Gram-negative bacteria136,137. Interestingly, extracellular LPS, which does not typically reach the cytosol to activate caspase-11, also triggered inflammasome activation in Irgm2-knockout macrophages137. IRGM2 did not affect caspase-11 activation when LPS was electroporated or transfected into the cytosol. This suggests IRGM2 and GATE16 may suppress caspase-11 activation by preventing aberrant vacuolar instability that could release LPS in the cytosol. The knockout of the sole human IRGM did not phenocopy Irgm2-knockout in mice, but GATE16 knockout in human cells did enhance pyroptosis, suggesting a similar pathway exists in humans though it is likely regulated by different proteins136.
5. Inflammasome-independent activation of pyroptosis
While pyroptosis is frequently associated with inflammasome activation, recent studies have revealed mechanisms of inflammasome-independent activation of pyroptosis (Figure 4). For example, GSDMB, expressed in humans but not in mice, is activated by granzyme A from cytotoxic lymphocytes (i.e., cytotoxic T cells and NK cells) to trigger pyroptosis in target cells138. Although GSDMB has been explored as a potential target for cancer immunotherapy, further investigation is required to understand its role in antibacterial infection. This will be complicated by the natural absence of GSDMB from the mouse genome. Intriguingly, GSDMB can be degraded by IpaH7.8, a virulence factor of Shigella69–72. This suggests the possibility of GSDMB-driven pyroptosis playing a role in defending against bacterial infections.
Figure 4. Activation mechanisms of gasdermins.
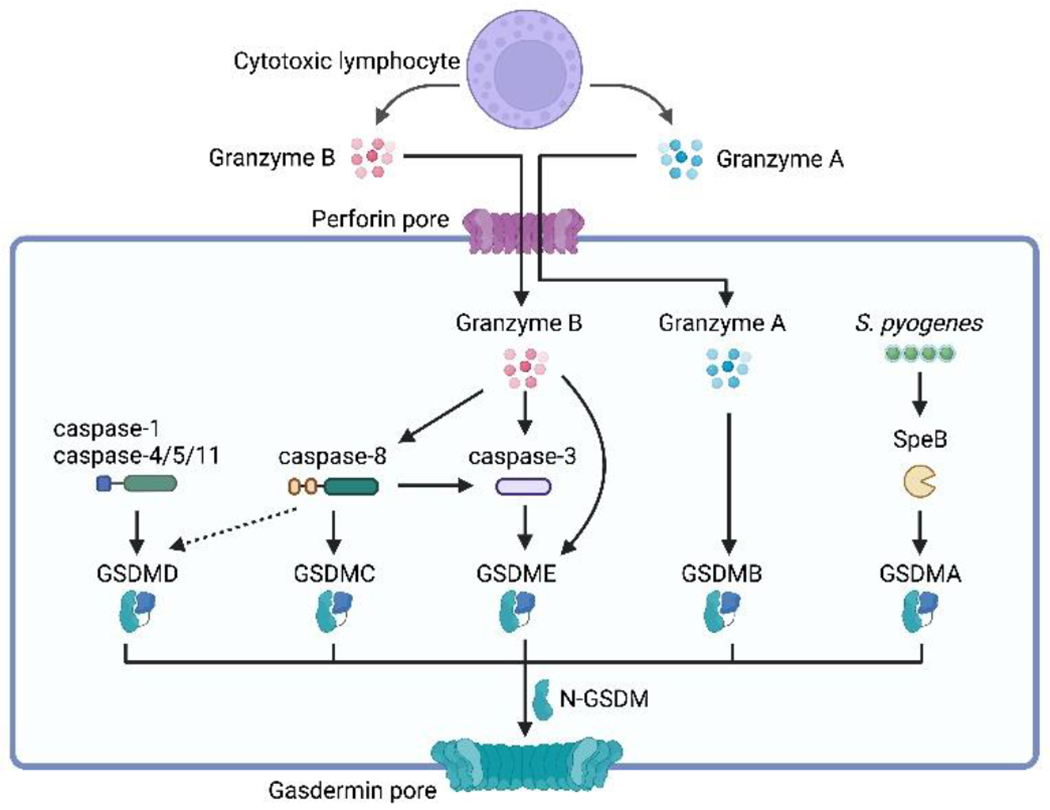
Pyroptosis is triggered through the cleavage of gasdermin proteins, which involves both inflammasome-dependent and inflammasome-independent pathways. Gasdermin D (GSDMD), the prototype of gasdermins, can be activated by inflammatory caspases, including caspase-1/4/5/11, through the inflammasome-dependent pathway. Gasdermins can also divert apoptotic signaling towards pyroptosis through their cleavage. Caspase-8, traditionally known as an initiator of apoptosis, can induce pyroptosis by cleaving GSDMD under specific conditions. Caspase-8 can also cleave GSDMC, which is primarily expressed in epithelial tissues, leading to pyroptosis. Caspase-3 cleaves GSDME, transitioning from caspase-3-mediated apoptosis to pyroptosis. Furthermore, granzyme B, produced by cytotoxic lymphocytes, can directly cleave GSDME, resulting in pyroptosis independent of caspase-3. Granzyme A, another protease produced by cytotoxic lymphocytes, activates GSDMB, triggering pyroptosis in target cells. In addition to host-derived proteases, pathogen-derived factors can also activate gasdermins. For instance, the cysteine protease virulence factor SpeB produced by Streptococcus pyogenes cleaves GSDMA, inducing pyroptosis in keratinocytes.
Apoptotic signaling pathways can be diverted towards pyroptosis through the cleavage of gasdermins. One such gasdermin, GSDME, undergoes cleavage by caspase-3, the primary executor of apoptosis, resulting in the transition from caspase-3-mediated apoptosis to pyroptosis139,140. Unlike pyroptosis, apoptosis is typically non-inflammatory and does not elicit an immune response. Consequently, the activation of GSDME by caspase-3 must be tightly controlled to ensure normal apoptosis can occur. The expression level of GSDME may play a role in determining whether cells undergo apoptosis or pyroptosis upon caspase-3 activation. Furthermore, in caspase-3-activated cells, any low-level gasdermin pores that form are likely to be promptly repaired to facilitate the progression of apoptosis58. Granzyme B could also achieve GSDME activation indirectly by activating caspase-3141. However, GSDME was cleaved even in caspase-3 deficient HeLa cells when co-cultured with NK cells. Furthermore, in a cell-free assay, recombinant granzyme B but not granzyme A cleaved GSDME. These findings indicate that granzyme B can directly cleave and activate GSDME142.
Caspase-8, traditionally known as an initiator of apoptosis, exhibits the ability to trigger pyroptosis as well. This might occur when caspase-8 activates caspase-3 in settings of high GSDME. Additionally, this can occur through the cleavage of GSDMD in response to Yersinia infection or TNF-α signaling combined with the inhibition of transforming growth factor beta-activated kinase (TAK1)143–145. We presume that this caspase-8-mediated pathway involving GSDMD either operates at a slower pace compared to typical apoptotic signaling or is only licensed in a specific manner within cells experiencing TAK1 inhibition.
Caspase-8 can also induce pyroptosis through the cleavage of GSDMC146,147. For example, caspase-8 cleaves GSDMC in response to a metabolite known as α-ketoglutarate (α-KG)147. This process is initiated by α-KG, which induces an elevation in reactive oxygen species (ROS), leading to the oxidation of the death receptor DR6 present on the plasma membrane. Consequently, DR6 is internalized through endocytosis. Upon entry into the cell, DR6 recruits both pro-caspase-8 and GSDMC, resulting in the cleavage of GSDMC by caspase-8, ultimately culminating in pyroptosis. It is important to note that the expression of GSDMC is primarily limited to epithelial tissues, such as stomach and intestine22. As a result, the activation of GSDMC by caspase-8 could be confined by the expression pattern of GSDMC.
In addition to host-derived proteases, pathogen-derived factors can also activate gasdermins148–150. For instance, in keratinocytes, pyroptosis can be induced by SpeB, a cysteine protease virulence factor produced by Streptococcus pyogenes, which cleaves GSDMA and possible other GSDMs148,149. Notably, the expression of GSDMA is restricted to stratified epithelial cells, including skin keratinocytes151,152. In a skin infection model, GSDMA restricted the dissemination of S. pyogenes into inner organs148. SpeB accessed the cytosol during a cytosolic-invasive phase of the bacterial infection. However, GSDMA is not involved in skin infection by Staphylococcus aureus148 or herpes simplex virus type 1152. These studies highlight the dual role of GSDMA as both a direct sensor of pathogen-derived proteases and an executor of pyroptosis.
6. Conclusions
The immune response to intracellular bacteria involves a complex interplay between host and pathogen. Host-adapted pathogens can evade host immune defenses, whereas environmental microbes are effectively eliminated by host defense mechanisms. As their first line of defense, most cells rely on cell-autonomous immunity (including xenophagy), in which the infected host cell eliminates the pathogen, survives, and returns to homeostasis. Pyroptosis is another mechanism used to combat intracellular bacteria, as it eliminates replication niches for these pathogens and releases danger signals that activate subsequent inflammatory responses. However, for certain intracellular pathogens that cannot be cleared by cell-autonomous immunity or cell death, the granuloma response has been suggested as a solution. This response involves the formation of an organized structure, by various cell types, to wall off such bacteria153.
This review emphasizes the critical role of pyroptosis in combating intracellular bacterial infections, particularly environmental pathogens. The interaction between host and pathogen is dynamic and shapes the evolution of the host’s immune response. To fully understand these immune responses, further studies are needed to unravel the complex molecular mechanisms involved. Additionally, how the multiple gasdermin family members act either together or distinctly in defending against intracellular infection warrants further investigation. Moreover, investigating the crosstalk between regulated cell death pathways in the context of bacterial infection can also shed light on the immune response to these pathogens. To address these questions, the discovery of new environmental pathogens that are unable to inhibit or evade the host immune responses can provide valuable tools for understanding the optimal functioning of our immune system.
Highlights:
Pyroptosis combats intracellular bacterial infection.
The role of pyroptosis is cell-type dependent.
Host-adapted pathogens evade or inhibit pyroptosis.
Environmental pathogens are efficiently cleared by pyroptosis.
Pyroptosis is regulated by autophagy and interferon-inducible GTPases.
Acknowledgements.
This work is supported by NIH grants AI133236 (E.A.M), AI139304 (E.A.M), AR072694 (E.A.M), AI136920 (E.A.M), AI139425 (J.C.), AI103197 (J.C.), and AI148243 (J.C.). J.C. holds an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. The figures were created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest. The authors declare no conflicts of interest.
References
- 1.Girardin SE, Sansonetti PJ & Philpott DJ Intracellular vs extracellular recognition of pathogens – common concepts in mammals and flies. Trends Microbiol 10, 193–199 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Petit TJP & Lebreton A Adaptations of intracellular bacteria to vacuolar or cytosolic niches. Trends Microbiol 30, 736–748 (2022). [DOI] [PubMed] [Google Scholar]
- 3.Anand I, Choi W & Isberg RR The vacuole guard hypothesis: how intravacuolar pathogens fight to maintain the integrity of their beloved home. Curr Opin Microbiol 54, 51–58 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gutierrez MG & Enninga J Intracellular niche switching as host subversion strategy of bacterial pathogens. Curr Opin Cell Biol 76, 102081 (2022). [DOI] [PubMed] [Google Scholar]
- 5.Sironi M, Cagliani R, Forni D & Clerici M Evolutionary insights into host–pathogen interactions from mammalian sequence data. Nat Rev Genet 16, 224–236 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aleru O & Barber MF Battlefronts of evolutionary conflict between bacteria and animal hosts. Plos Pathog 16, e1008797 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Solé R. Revisiting Leigh Van Valen’s “A New Evolutionary Law” (1973). Biological Theory 17, 120–125 (2022). [Google Scholar]
- 8.Maltez VI & Miao EA Reassessing the Evolutionary Importance of Inflammasomes. The Journal of Immunology 196, 956–962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jorgensen I, Rayamajhi M & Miao EA Programmed cell death as a defence against infection. Nat Rev Immunol 17, 151–164 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nozaki K, Li L & Miao EA Innate Sensors Trigger Regulated Cell Death to Combat Intracellular Infection. Annu Rev Immunol 40, 469–498 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Kayagaki N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Ding J. et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116(2016). [DOI] [PubMed] [Google Scholar]
- 14.Liu X. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis BK, Wen H & Ting JP-Y The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu Rev Immunol 29, 707–735 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vance RE, Isberg RR & Portnoy DA Patterns of Pathogenesis: Discrimination of Pathogenic and Nonpathogenic Microbes by the Innate Immune System. Cell Host Microbe 6, 10–21 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Green DR The Coming Decade of Cell Death Research: Five Riddles. Cell 177, 1094–1107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galluzzi L et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25, 486–541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elmore S Apoptosis: A Review of Programmed Cell Death. Toxicol Pathol 35, 495–516 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doerflinger M et al. Flexible Usage and Interconnectivity of Diverse Cell Death Pathways Protect against Intracellular Infection. Immunity 53, 533–547.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abele TJ et al. Apoptotic signaling clears engineered Salmonella in an organ-specific manner. (2023) doi: 10.1101/2023.05.06.539681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kovacs SB & Miao EA Gasdermins: Effectors of Pyroptosis. Trends in Cell Biology 27, 673–684 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruan J, Xia S, Liu X, Lieberman J & Wu H Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557, 62–67 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia S et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 593, 607–611 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He W et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res 25, 1285–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider KS et al. The Inflammasome Drives GSDMD-Independent Secondary Pyroptosis and IL-1 Release in the Absence of Caspase-1 Protease Activity. Cell Reports 21, 3846–3859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saeki A et al. Gasdermin D-independent release of interleukin-1β by living macrophages in response to mycoplasmal lipoproteins and lipopeptides. Immunology 161, 114–122 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kayagaki N et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591, 131–136(2021). [DOI] [PubMed] [Google Scholar]
- 29.Loomis WP, Hartigh A. B. den, Cookson BT & Fink SL Diverse small molecules prevent macrophage lysis during pyroptosis. Cell Death Dis 10, 326 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borges JP et al. Glycine inhibits NINJ1 membrane clustering to suppress plasma membrane rupture in cell death. Elife 11, e78609 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hansen HA & Emborg C Extra- and intracellular amino acid concentrations in continuous Chinese hamster ovary cell culture. Appl Microbiol Biotechnol 41, 560–564 (1994). [DOI] [PubMed] [Google Scholar]
- 32.Jorgensen I, Zhang Y, Krantz BA & Miao EA Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. The Journal of Experimental Medicine 213, 2113–2128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wright JA & Bryant CE The killer protein Gasdermin D. Cell Death Differ 23, 1897–1898 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Estfanous S et al. Gasdermin D restricts Burkholderia cenocepacia infection in vitro and in vivo. Sci Rep-uk 11, 855 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torre-Minguela C, Gómez AI, Couillin I & Pelegrín P Gasdermins mediate cellular release of mitochondrial DNA during pyroptosis and apoptosis. Faseb J 35, e21757 (2021). [DOI] [PubMed] [Google Scholar]
- 36.Du G et al. ROS-dependent palmitoylation is an obligate licensing modification for GSDMD pore formation. Biorxiv 2023.03.07.531538 (2023) doi: 10.1101/2023.03.07.531538. [DOI] [Google Scholar]
- 37.López-Pérez W et al. TAK1 inhibition elicits mitochondrial ROS to block intracellular bacterial colonization. Proc National Acad Sci 118, e2023647118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bjanes E et al. Genetic targeting of Card19 is linked to disrupted NINJ1 expression, impaired cell lysis, and increased susceptibility to Yersinia infection. Plos Pathog 17, e1009967 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen KW et al. The Neutrophil NLRC4 Inflammasome Selectively Promotes IL-1β Maturation without Pyroptosis during Acute Salmonella Challenge. Cell Reports 8, 570–582 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Bakele M et al. Localization and Functionality of the Inflammasome in Neutrophils*. J Biol Chem 289, 5320–5329 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karmakar M, Katsnelson MA, Dubyak GR & Pearlman E Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATP. Nat Commun 7, 10555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldberg EL et al. β-Hydroxybutyrate Deactivates Neutrophil NLRP3 Inflammasome to Relieve Gout Flares. Cell Reports 18, 2077–2087 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heilig R et al. The Gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur. J. Immunol 48, 584–592 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Kovacs SB et al. Neutrophil Caspase-11 Is Essential to Defend against a Cytosol-Invasive Bacterium. Cell Reports 32, 107967 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karmakar M et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nat Commun 11, 2212 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santoni K et al. Caspase-1-driven neutrophil pyroptosis and its role in host susceptibility to Pseudomonas aeruginosa. Plos Pathog 18, e1010305 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oh C et al. Neutrophil inflammasomes sense the subcellular delivery route of translocated bacterial effectors and toxins. Cell Reports 41, 111688 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen KW et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Science immunology 3, eaar6676 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Sollberger G et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Science immunology 3, eaar6689 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Aachoui Y et al. Caspase-11 Protects Against Bacteria That Escape the Vacuole. Science 339, 1230751–978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Churchill MJ, Mitchell PS & Rauch I Epithelial Pyroptosis in Host Defense. J Mol Biol 434, 167278 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Knodler LA et al. Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc National Acad Sci 107, 17733–17738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sellin ME et al. Epithelium-Intrinsic NAIP/NLRC4 Inflammasome Drives Infected Enterocyte Expulsion to Restrict Salmonella Replication in the Intestinal Mucosa. Cell Host and Microbe 16, 237–248 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Rauch I et al. NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and -8. Immunity 46, 649–659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sellin ME, Müller AA & Hardt W-D Consequences of Epithelial Inflammasome Activation by Bacterial Pathogens. J Mol Biol 430, 193–206 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Hausmann A et al. Intestinal epithelial NAIP/NLRC4 restricts systemic dissemination of the adapted pathogen Salmonella Typhimurium due to site-specific bacterial PAMP expression. Mucosal Immunol 13, 530–544 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nozaki K et al. Caspase-7 activates ASM to repair gasdermin and perforin pores. Nature 606, 960–967 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nozaki K & Miao EA Bucket lists must be completed during cell death. Trends Cell Biol (2023) doi: 10.1016/j.tcb.2023.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Winter SE et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467, 426–429 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brokatzky D & Mostowy S Pyroptosis in host defence against bacterial infection. Dis Model Mech 15, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wanford JJ, Hachani A & Odendall C Reprogramming of Cell Death Pathways by Bacterial Effectors as a Widespread Virulence Strategy. Infect Immun 90, e00614–21 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hagar JA, Powell DA, Aachoui Y, Ernst RK & Miao EA Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 341, 1250–1253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kayagaki N et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 341, 1246–1249 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Lagrange B et al. Human caspase-4 detects tetra-acylated LPS and cytosolic Francisella and functions differently from murine caspase-11. Nat Commun 9, 242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kobayashi T et al. The Shigella OspC3 Effector Inhibits Caspase-4, Antagonizes Inflammatory Cell Death, and Promotes Epithelial Infection. Cell Host Microbe 13, 570–583 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Oh C, Verma A, Hafeez M, Hogland B & Aachoui Y Shigella OspC3 suppresses murine cytosolic LPS sensing. Iscience 24, 102910 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Z et al. Shigella evades pyroptosis by arginine ADP-riboxanation of caspase-11. Nature 599, 290–295 (2021). [DOI] [PubMed] [Google Scholar]
- 68.Luchetti G et al. Shigella ubiquitin ligase IpaH7.8 targets gasdermin D for degradation to prevent pyroptosis and enable infection. Cell Host Microbe 29, 1521–1530.e10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yin H et al. Insights into the GSDMB-mediated cellular lysis and its targeting by IpaH7.8. Nat Commun 14, 61 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhong X et al. Structural mechanisms for regulation of GSDMB pore-forming activity. Nature 616, 598–605 (2023). [DOI] [PubMed] [Google Scholar]
- 71.Wang C et al. Structural basis for GSDMB pore formation and its targeting by IpaH7.8. Nature 616, 590–597 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hansen JM et al. Pathogenic ubiquitination of GSDMB inhibits NK cell bactericidal functions. Cell 184, 3178–3191.e18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rastogi S & Briken V Interaction of Mycobacteria With Host Cell Inflammasomes. Front Immunol 13, 791136 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen X et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res 26, 1007–20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chai Q et al. A bacterial phospholipid phosphatase inhibits host pyroptosis by hijacking ubiquitin. Science 378, eabq0132 (2022). [DOI] [PubMed] [Google Scholar]
- 76.Rastogi S, Ellinwood S, Augenstreich J, Mayer-Barber KD & Briken V Mycobacterium tuberculosis inhibits the NLRP3 inflammasome activation via its phosphokinase PknF. Plos Pathog 17, e1009712 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lara-Tejero M et al. Role of the caspase-1 inflammasome in Salmonella typhimuriumpathogenesis. The Journal of Experimental Medicine 203, 1407–1412 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Raupach B, Peuschel SK, Monack DM & Zychlinsky A Caspase-1-Mediated Activation of Interleukin-1 (IL-1 ) and IL-18 Contributes to Innate Immune Defenses against Salmonella enterica Serovar Typhimurium Infection. Infection and Immunity 74, 4922–4926 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Broz P et al. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. The Journal of Experimental Medicine 207, 1745–1755 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Franchi L et al. NLRC4-driven interleukin-1β production discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nature Immunology 13, 449–456 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Broz P et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–291 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Knodler LA et al. Noncanonical Inflammasome Activation of Caspase-4/Caspase-11 Mediates Epithelial Defenses against Enteric Bacterial Pathogens. Cell Host Microbe 16, 249–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miao EA et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11, 1136–1142 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miao EA et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proceedings of the National Academy of Sciences 107, 3076–3080 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aachoui Y et al. Canonical Inflammasomes Drive IFN-γ to Prime Caspase-11 in Defense against a Cytosol-Invasive Bacterium. Cell Host & Microbe 18, 320–332 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang J, Deobald K & Re F Gasdermin D Protects from Melioidosis through Pyroptosis and Direct Killing of Bacteria. J Immunol 202, 3468–3473 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Durán N & Menck CFM Chromobacterium violaceum: A Review of Pharmacological and Industiral Perspectives. Crit Rev Microbiol 27, 201–222 (2001). [DOI] [PubMed] [Google Scholar]
- 88.Macher AM, Casale TB & Fauci AS Chronic Granulomatous Disease of Childhood and Chromobacterium violaceum Infections in the Southeastern United States. Ann Intern Med 97, 51 (1982). [DOI] [PubMed] [Google Scholar]
- 89.Yang C-H & Li Y-H Chromobacterium violaceum infection: a clinical review of an important but neglected infection. J Chin Medical Assoc Jcma 74, 435–41 (2011). [DOI] [PubMed] [Google Scholar]
- 90.Miki T et al. Chromobacterium pathogenicity island 1 type III secretion system is a major virulence determinant for Chromobacterium violaceum-induced cell death in hepatocytes. Mol Microbiol 77, 855–872 (2010). [DOI] [PubMed] [Google Scholar]
- 91.Maltez VI et al. Inflammasomes Coordinate Pyroptosis and Natural Killer Cell Cytotoxicity to Clear Infection by a Ubiquitous Environmental Bacterium. Immunity 43, 987–997 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Franchi L, Warner N, Viani K & Nuñez G Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev 227, 106–128 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martin PAW, Hirose E & Aldrich JR Toxicity ofChromobacterium subtsugae to Southern Green Stink Bug (Heteroptera: Pentatomidae) and Corn Rootworm (Coleoptera: Chrysomelidae). J. Econ. Éntomol 107, 680–684 (2007). [DOI] [PubMed] [Google Scholar]
- 94.Martin PAW, Gundersen-Rindal D, Blackburn M & Buyer J Chromobacterium subtsugae sp. nov., a betaproteobacterium toxic to Colorado potato beetle and other insect pests. Int. J. Syst. Evol. Microbiol 57, 993–999 (2007). [DOI] [PubMed] [Google Scholar]
- 95.Brinkworth JF & Alvarado AS Cell-Autonomous Immunity and The Pathogen-Mediated Evolution of Humans: Or How Our Prokaryotic and Single-Celled Origins Affect The Human Evolutionary Story. Q Rev Biology 95, 215–246 (2020). [Google Scholar]
- 96.Cadwell K Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol 16, 661–675 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Deretic V & Levine B Autophagy balances inflammation in innate immunity. Autophagy 14, 243–251 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guo R, Wang H & Cui N Autophagy Regulation on Pyroptosis: Mechanism and Medical Implication in Sepsis. Mediat Inflamm 2021, 9925059 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pant A et al. Interactions of Autophagy and the Immune System in Health and Diseases. Autophagy Reports 1, 438–515 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Saitoh T et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456, 264–268 (2008). [DOI] [PubMed] [Google Scholar]
- 101.Shi C-S et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13, 255–263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nakahira K et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12, 222–230 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhong Z et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 164, 896–910 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sumpter R et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 165, 867–881 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Suzuki T et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathogens 3, e111 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Meunier E et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509, 366–370 (2014). [DOI] [PubMed] [Google Scholar]
- 107.Kimura T et al. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol 210, 973–989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu T et al. TRIM11 Suppresses AIM2 Inflammasome by Degrading AIM2 via p62-Dependent Selective Autophagy. Cell Reports 16, 1988–2002 (2016). [DOI] [PubMed] [Google Scholar]
- 109.Mahenthiralingam E, Baldwin A & Dowson CG Burkholderia cepacia complex bacteria: opportunistic pathogens with important natural biology. J Appl Microbiol 104, 1539–1551 (2008). [DOI] [PubMed] [Google Scholar]
- 110.Krause K et al. CASP4/caspase-11 promotes autophagosome formation in response to bacterial infection. Autophagy 14, 1928–1942 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Harvest CK & Miao EA Autophagy May Allow a Cell to Forbear Pyroptosis When Confronted With Cytosol-Invasive Bacteria. Front Immunol 13, 871190 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pilla-Moffett D, Barber MF, Taylor GA & Coers J Interferon-Inducible GTPases in Host Resistance, Inflammation and Disease. J Mol Biol 428, 3495–3513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rafeld HL, Kolanus W, Driel I. R. van & Hartland EL Interferon-induced GTPases orchestrate host cell-autonomous defence against bacterial pathogens. Biochem Soc T 49, 1287–1297 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim B-H, Shenoy AR, Kumar P, Bradfield CJ & MacMicking JD IFN-Inducible GTPases in Host Cell Defense. Cell Host Microbe 12, 432–444 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pilla DM et al. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc National Acad Sci 111, 6046–6051 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cerqueira DM et al. Guanylate-binding protein 5 licenses caspase-11 for Gasdermin-D mediated host resistance to Brucella abortus infection. Plos Pathog 14, e1007519 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Santos JC et al. LPS targets host guanylate-binding proteins to the bacterial outer membrane for non-canonical inflammasome activation. Embo J 37, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Santos JC et al. Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat Commun 11, 3276 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wandel MP et al. Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat Immunol 282, 2085–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Park E-S et al. A hierarchical GBP network promotes cytosolic LPS recognition and sepsis. Biorxiv 2021.08.25.457662 (2021) doi: 10.1101/2021.08.25.457662. [DOI] [Google Scholar]
- 121.Fisch D et al. Human GBP1 is a microbe-specific gatekeeper of macrophage apoptosis and pyroptosis. Embo J 38, e100926 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fisch D et al. Human GBP1 Differentially Targets Salmonella and Toxoplasma to License Recognition of Microbial Ligands and Caspase-Mediated Death. Cell Reports 32, 108008 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dickinson MS et al. LPS-aggregating proteins GBP1 and GBP2 are each sufficient to enhance caspase-4 activation both in cellulo and in vitro. Proc National Acad Sci 120, e2216028120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kutsch M et al. Direct binding of polymeric GBP1 to LPS disrupts bacterial cell envelope functions. Embo J 39, e104926 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Finethy R et al. Inflammasome Activation by Bacterial Outer Membrane Vesicles Requires Guanylate Binding Proteins. Mbio 8, e01188–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhu S et al. Cryo-ET of a human GBP coatomer governing cell-autonomous innate immunity to infection. Biorxiv 2021.08.26.457804 (2021) doi: 10.1101/2021.08.26.457804. [DOI] [Google Scholar]
- 127.Kuhm T et al. Structural basis of membrane targeting and coatomer assembly by human GBP1. bioRxiv 2023.03.28.534355 (2023) doi: 10.1101/2023.03.28.534355. [DOI] [Google Scholar]
- 128.Goers L. et al. Shigella IpaH9.8 limits GBP1-dependent LPS release from intracytosolic bacteria to suppress caspase-4 activation. Proc. Natl. Acad. Sci 120, e2218469120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Man SM et al. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat Immunol 16, 467–475 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Meunier E et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol 16, 476–484 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wallet P et al. IFN-γ extends the immune functions of Guanylate Binding Proteins to inflammasome-independent antibacterial activities during Francisella novicida infection. Plos Pathog 13, e1006630 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Man SM et al. IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell 167, 382–396.e17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liu BC et al. Constitutive Interferon Maintains GBP Expression Required for Release of Bacterial Components Upstream of Pyroptosis and Anti-DNA Responses. Cell Reports 24, 155–168.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xavier A, Al-Zeer MA, Meyer TF & Daumke O hGBP1 Coordinates Chlamydia Restriction and Inflammasome Activation through Sequential GTP Hydrolysis. Cell Reports 31, 107667 (2020). [DOI] [PubMed] [Google Scholar]
- 135.Mariathasan S et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 (2006). [DOI] [PubMed] [Google Scholar]
- 136.Eren E et al. Irgm2 and Gate-16 cooperatively dampen Gram-negative bacteria-induced caspase-11 response. Embo Rep 21, e50829 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Finethy R et al. Dynamin-related Irgm proteins modulate LPS-induced caspase-11 activation and septic shock. Embo Rep 21, e50830 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhou Z et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368, (2020). [DOI] [PubMed] [Google Scholar]
- 139.Rogers C et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 8, 14128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang Y et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017). [DOI] [PubMed] [Google Scholar]
- 141.Lord SJ, Rajotte RV, Korbutt GS & Bleackley RC Granzyme B: a natural born killer. Immunol Rev 193, 31–38 (2003). [DOI] [PubMed] [Google Scholar]
- 142.Zhang Z et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Orning P et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 30, eaau2818–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sarhan J et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersiniainfection. Proceedings of the National Academy of Sciences 67, 201809548–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chen KW et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. Embo J 38, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hou J et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol 22, 1264–1275 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhang J et al. The metabolite α-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res 31, 980–997 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Deng W et al. Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis. Nature 602, 496–502 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.LaRock DL et al. Group A Streptococcus induces GSDMA-dependent pyroptosis in keratinocytes. Nature 605, 527–531 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Panganiban RA, Mwase C, Park J & Lu Q Direct cleavage and activation of gasdermin B by allergens. Allergy (2023) doi: 10.1111/all.15763. [DOI] [PubMed] [Google Scholar]
- 151.Tamura M et al. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics 89, 618–629 (2007). [DOI] [PubMed] [Google Scholar]
- 152.Li L et al. Role of Caspases and Gasdermin A during HSV-1 Infection in Mice. Viruses 14, 2034 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Harvest CK et al. An innate granuloma eradicates an environmental pathogen using Gsdmd and Nos2. Biorxiv 2023.03.07.531568 (2023) doi: 10.1101/2023.03.07.531568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tsuji NM Roles of caspase-1 in Listeria infection in mice. International Immunology 16, 335–343 (2004). [DOI] [PubMed] [Google Scholar]
- 155.Ceballos-Olvera I, Sahoo M, Miller MA, Barrio L. del & Re F Inflammasome-dependent Pyroptosis and IL-18 Protect against Burkholderia pseudomallei Lung Infection while IL-1β Is Deleterious. PLoS Pathog. 7, e1002452 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Exconde PM et al. The tetrapeptide sequence of IL-1β regulates its recruitment and activation by inflammatory caspases. Biorxiv 2023.02.16.528859 (2023) doi: 10.1101/2023.02.16.528859. [DOI] [PMC free article] [PubMed] [Google Scholar]