

Abstract
GpsB links peptidoglycan synthases to other proteins that determine the shape of the respiratory pathogen Streptococcus pneumoniae (pneumococcus; Spn) and other low-GC Gram-positive bacteria. GpsB is also required for phosphorylation of proteins by the essential StkP(Spn) Ser/Thr protein kinase. Here we report three classes of frequently arising chromosomal duplications (≈21–176 genes) containing murZ (MurZ-family homolog of MurA) or murA that suppress ΔgpsB or ΔstkP. These duplications arose from three different repeated sequences and demonstrate the facility of pneumococcus to modulate gene dosage of numerous genes. Overproduction of MurZ or MurA alone or overproduction of MurZ caused by ΔkhpAB mutations suppressed ΔgpsB or ΔstkP phenotypes to varying extents. ΔgpsB and ΔstkP were also suppressed by MurZ amino-acid changes distant from the active site, including one in commonly studied laboratory strains, and by truncation or deletion of the homolog of IreB(ReoM). Unlike in other Gram-positive bacteria, MurZ is predominant to MurA in pneumococcal cells. However, ΔgpsB and ΔstkP were not suppressed by ΔclpCP, which did not alter MurZ or MurA amounts. These results support a model in which regulation of MurZ and MurA activity, likely by IreB(Spn), is the only essential requirement for StkP-mediated protein phosphorylation in exponentially growing D39 pneumococcal cells.
Keywords: peptidoglycan precursor synthesis, gene duplication and amplification, GpsB peptidoglycan regulator, StkP protein kinase, KhpA/B RNA binding protein
1 |. INTRODUCTION
Bacterial survival depends on the regulation of the synthesis and assembly of the peptidoglycan (PG) cell wall (Rohs & Bernhardt, 2021, Egan et al., 2020, Kumar et al., 2022). PG determines cell shape and morphology and protects against osmotic stress(Booth & Lewis, 2019, Egan et al., 2020, Garde et al., 2021). The proteins that carry out the numerous steps of PG synthesis are major targets for clinically relevant antibiotics, for which widespread resistance has developed (Booth & Lewis, 2019, Egan et al., 2020, Bush & Bradford, 2016). In Gram-positive bacteria, such as Streptococcus pneumoniae (pneumococcus; Spn), the PG cell wall also provides a scaffolding for attachment of capsule, wall teichoic acids, and extracellular proteins and virulence factors (Booth & Lewis, 2019, Briggs et al., 2021, Kumar et al., 2022). S. pneumoniae is a commensal bacterium of the human nasopharynx and a major opportunistic respiratory-tract pathogen that kills millions of people annually worldwide, including following influenza and COVID-19 infections (Sender et al., 2021, Cox et al., 2020, Weiser et al., 2018). S. pneumoniae is continuing to acquire antibiotic resistance to a broad range of antibiotics and is now classified as a “superbug” by the CDC and WHO (WHO, 2017, CDC et al., 2019).
The GpsB protein is a major regulator of PG synthesis in low-GC Gram-positive bacteria (Claessen et al., 2008, Rismondo et al., 2016, Cleverley et al., 2019, Fleurie et al., 2014, Rued et al., 2017). In Bacillus subtilis (Bsu), ΔgpsB results in growth and morphological abnormality in high salt media and synthetic lethality with ΔezrA or ΔftsA (Claessen et al., 2008; Tavares et al., 2008), while in Listeria monocytogenes (Lmo), ΔgpsB causes marked growth and division defects at 37°C and is lethal at 42°C (Rismondo et al., 2016). ΔgpsB mutants of Enterococcus faecalis (Efa) also show growth defects at 45°C, but grow normally at 37°C (Minton et al., 2022). In contrast, in derivatives of serotype-2 S. pneumoniae D39 progenitor strains, gpsB is essential at 37°C, and GpsB depletion leads to drastic cell enlargement and elongation, incomplete closure of septal division rings, and eventual cell lysis (Land et al., 2013, Rued et al., 2017). Depletion of GpsB in Staphylococcus aureus (Sau), however, arrests cell division without coincident cell enlargement and ultimately causes aberrant membrane accumulation (Eswara et al., 2018).
Combined studies indicate that GpsB plays species-specific roles in regulating PG synthesis (Cleverley et al., 2019, Hammond et al., 2019). Based on genetic and biochemical studies, one role shared by GpsB in different bacteria is as an adaptor that docks PG synthases to other cell-wall enzymes and scaffold proteins to form complexes for division and septal and lateral PG synthesis (Rued et al., 2017, Cleverley et al., 2019, Halbedel & Lewis, 2019, Sacco et al., 2022). Binding between GpsB homologs and Class A PBPs, including PBP1(Bsu), PBPA1(Lmo), and aPBP2a(Spn) and Class C PBP4(Sau) occurs by a conserved mechanism, wherein Arg residues in amino-terminal, cytoplasmic microdomains of the PBPs bind to a specific site in the amino-terminal domain of GpsB (Cleverley et al., 2019, Sacco et al., 2022). Species-specific binding to other subsets of PG synthesis and cell division proteins occurs at other surfaces in GpsB homologs (Cleverley et al., 2019). For example, besides interacting with aPBP2a, GpsB(Spn) is in complexes with EzrA, MreC, StkP, and possibly bPBP2x, bPBP2b, and aPBP1a, but not with FtsZ and FtsA (Rued et al., 2017, Cleverley et al., 2019). Unlike other GpsB homologs, GpsB(Sau) binds to a non-conserved C-terminal tail of FtsZ, which affects FtsZ polymerization (Sacco et al., 2022). GpsB(Sau) also interacts with teichoic acid biogenesis proteins through binding motifs that are not widely conserved in GpsB from other bacteria (Eswara et al., 2018, Hammond et al., 2022). The significance of GpsB in maintaining cell wall integrity during antibiotic stress in S. pneumoniae was underscored by a genome-wide association study of clinical isolates that revealed significant correlation of β-lactam resistance and the presence of gpsB variants (Mobegi et al., 2017).
An additional important regulatory function of GpsB is the maintenance of protein phosphorylation mediated by conserved homologues of serine/threonine kinases, StkP(Spn), PrkC(Bsu), and IreK(Efa) (Rued et al., 2017, Pompeo et al., 2015, Fleurie et al., 2014, Minton et al., 2022). In S. pneumoniae, phosphorylation of StkP and other StkP substrates is significantly reduced in ΔgpsB mutants of laboratory strains Rx1, R6, or R800 or upon depletion of GpsB in D39-derived strains (Rued et al., 2017, Fleurie et al., 2014). The link between GpsB function and protein phosphorylation was further supported in D39-derived strains by the finding that ΔgpsB is suppressed by mutations that inactivate the cognate PhpP Ser/Thr protein phosphatase, such as phpP(G229D), which restore protein phosphorylation (Rued et al., 2017). Notably, phpP(G229D), restores the growth and cell morphology of ΔgpsB mutants to nearly those of WT cells (Rued et al., 2017), indicating that GpsB mediates StkP phosphorylation of one or more proteins required for exponential growth of S. pneumoniae.
StkP(Spn) belongs to the subfamily of eukaryotic-type Ser/Thr kinases (ESTKs) and together with cognate PP2C-type phosphatase PhpP(Spn), constitutes a signaling system (Echenique et al., 2004, Novakova et al., 2005). Based on phenotypes of ΔstkP mutants in different genetic backgrounds, StkP has been implicated in the regulation of cell growth and cell division (Beilharz et al., 2012, Giefing et al., 2010, Hirschfeld et al., 2019, Fleurie et al., 2012), competence (Echenique et al., 2004, Saskova et al., 2007, Rued et al., 2017), stress resistance (Saskova et al., 2007)), acidic stress-induced lysis (Pinas et al., 2018), capsule synthesis and virulence (Echenique et al., 2004, Kant et al., 2023), pilus expression and adherence (Herbert et al., 2015), and β-lactam susceptibility (Dias et al., 2009). However, the essentiality of both gpsB(Spn) and stkP(Spn) has been controversial. Based on numerous studies of common laboratory strains R6 (and its derivative R800) and Rx1, gpsB and stkP have generally been classified as non-essential (Rued et al., 2017, Fleurie et al., 2014), despite variations in growth properties and cell morphologies consistent with the presence of suppressor mutations (Beilharz et al., 2012, Fleurie et al., 2012, Massidda et al., 2013, Rued et al., 2017, Ulrych et al., 2021, Vollmer et al., 2019). In contrast, gpsB and stkP are essential in D39-derived strains (Land et al., 2013, Rued et al., 2017), from which the laboratory strains were originally derived (Cuppone et al., 2021, Lanie et al., 2007, Santoro et al., 2019). Depletion and transformation experiments clearly indicate that gpsB is essential in D39 strains and that ΔgpsB mutants accumulate suppressor mutations (Land et al., 2013, Rued et al., 2017). In contrast, ΔstkP mutants are unstable and rapidly acquire suppressor mutations that cause faster growth (Beilharz et al., 2012, Rued et al., 2017, Ulrych et al., 2021). Moreover, the primary cell morphology changes caused by StkP depletion remain unknown, as do mutations in the common laboratory strains that bypass the essentiality of gpsB and stkP.
Multiple proteins phosphorylated by StkP(Spn) have been identified in studies comparing global phosphoproteomes of ΔstkP mutants with that of their isogenic encapsulated D39 (cps+) or unencapsulated D39 (Δcps) parent strains (Hirschfeld et al., 2019, Sun et al., 2010, Ulrych et al., 2021). Several proteins associated with division and PG synthesis are phosphorylated in pneumococcal cells, including DivIVA (Novakova et al., 2010, Fleurie et al., 2012), MapZ (LocZ) (Fleurie et al., 2014, Holeckova et al., 2014), KhpB (Jag/EloR) (Zheng et al., 2017, Ulrych et al., 2016, Stamsas et al., 2017), MacP (Fenton et al., 2018), FtsZ (Ulrych et al., 2021), GpsB (Hirschfeld et al., 2019, Ulrych et al., 2021), MpgA (formerly MltG(Spn) (Hirschfeld et al., 2019, Taguchi et al., 2021, Ulrych et al., 2021), and IreB (Ulrych et al., 2021). In addition, the pattern of protein phosphorylation changes between exponentially growing and antibiotic stressed cells (Ulrych et al., 2021). Nevertheless, the roles of phosphorylation of individual proteins in growing D39 cells remains problematic, because phosphoablative and phosphomimetic mutants of cell division and PG synthesis proteins, such as DivIVA, MapZ, and KhpB, have not consistently shown aberrant phenotypes in exponentially growing cultures (Holeckova et al., 2014, Massidda et al., 2013, Zheng et al., 2017, Manuse et al., 2016, Fleurie et al., 2012, Grangeasse, 2016). It has not yet been determined which StkP-phosphorylation proteins are required for normal exponential growth of D39 strains.
Besides phpP null mutations, ΔgpsB(Spn) was suppressed by two large chromosomal duplications that also contained deletions (Rued et al., 2017). Notably, these duplications contain murZ (Rued et al., 2017, Wamp et al., 2020), which encodes one of two homologs of the UDP-N-acetylglucosamine 1-carboxyvinyltransferase that converts PEP and UDP-GlcNAc to Pi and UDP-N-acetyl-3-O-(1-carboxyvinyl)-alpha-D-glucosamine in the first committed step in the synthesis of the PG precursor Lipid II (Brown et al., 1995, Zhou et al., 2022). Like other low-GC Gram-positive bacteria, S. pneumoniae encodes two distinct homologs of this enzyme (Fig. 1) (Blake et al., 2009, Chan et al., 2022, Du et al., 2000, Kedar et al., 2008, Kock et al., 2004, Mascari et al., 2022, Vesic & Kristich, 2012). The two homologs in S. pneumoniae strains were annotated as MurZ (MurA2) (Spd_0967) and MurA (MurA1) (Spn)(Spd_1764) (Hoskins et al., 2001) (Fig. 1). The MurA-family homolog, which is the sole enzyme present in Gram-negative bacteria (Brown et al., 1995, Du et al., 2000, Hummels et al., 2023, Zhou et al., 2022), often plays a predominant enzymatic role in Gram-positive bacteria and is essential in B. subtilis, B. anthracis, and L. monocytogenes (Kock et al., 2004, Kedar et al., 2008, Rismondo et al., 2017), and required for normal growth of E. faecalis and S. aureus (Vesic & Kristich, 2012, Blake et al., 2009, Mascari et al., 2022). MurZ(Spn) and MurA(Spn) have a synthetic lethal relationship, where one homolog functions in the absence of the other, but both homologs cannot be deleted in the same strain(Du et al., 2000). Absence of MurAA(Efa) and MurAB(Efa) or MurA(Sau) and MurZ(Sau) is also synthetically lethal, where MurA-family MurAA(Efa) or MurA(Sau) is catalytically dominant in cells (Blake et al., 2009, Vesic & Kristich, 2012, Mascari et al., 2022). In contrast, previous biochemical studies demonstrated that MurZ(Spn) purified from strain R6 has a considerably higher (≈3.5-fold) catalytic efficiency (kcat/Km) for UDP-GlcNAc than MurA(Spn) (Du et al., 2000). Consistent with these kinetic results, a ΔmurZ(Spn) mutant substantially reduced the circumferential velocity of the bPBP2x:FtsW septal PG synthase, without changing the rate of FtsZ treadmilling (Perez et al., 2019). However, the relative contributions of MurZ and MurA to pneumococcal growth and physiology remain unknown.
Figure 1. Two evolutionary branches of the MurA-family and MurZ-family homologs of S. pneumoniae and other Gram-positive bacteria.
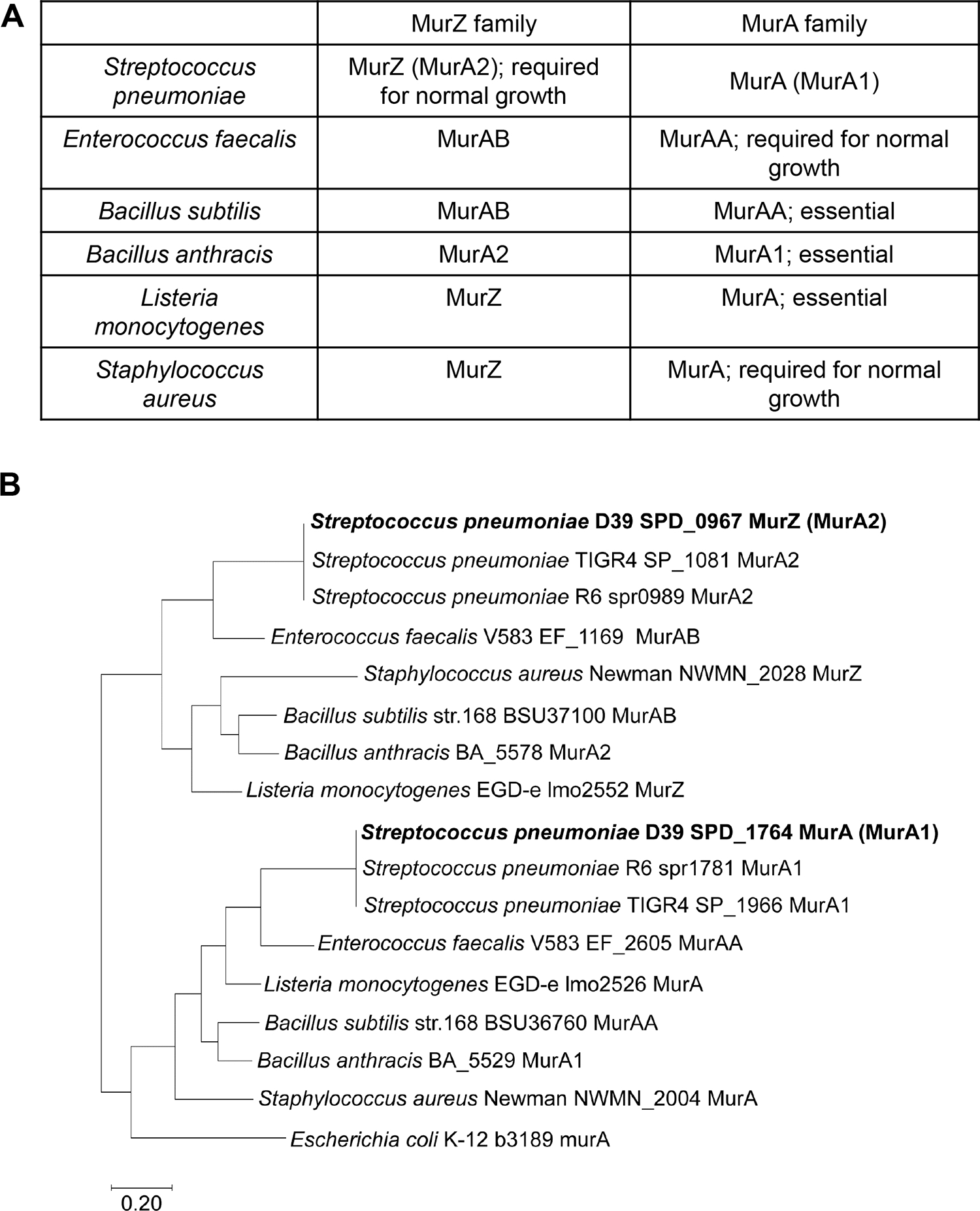
(A) Nomenclature and function of MurA and MurZ homologs from six Gram-positive bacteria S. pneumoniae (Spn) (Du et al.), E. faecalis (Efa) (Vesic & Kristich, 2012), B. subtilis (Bsu) (Kock et al., 2004), B. anthracis (Ban) (Kedar et al., 2008), L. monocytogenes (Lmo) (Rismondo et al., 2017), and S. aureus (Sau) (Blake et al., 2009). (B) Partial evolutionary tree of the MurZ-family and MurA-family homologs from five Gram-positive bacteria S. pneumoniae, E. faecalis, S. aureus, B. subtilis, and L. monocytogenes, and the single MurA-homolog in Gram-negative bacterium E. coli. MurZ(Spn) (Spd_0967)(Spn) is phylogenetically closely related to MurAB(Efa), MurAB(Bsu), MurZ(Sau), and MurZ(Lmo), while MurA(Spn) (Spd_1764) is phylogenetically closely related to MurAA(Efa), MurAA(Bsu), MurA(Sau), and MurA(Lmo). Note that in the original annotation of the S. pneumoniae D39 genome, the MurZ(Spn) homolog was called “MurA1” and the MurA(Spn) homolog was called “MurA2” (Lanie et al., 2007, Slager et al., 2018). For consistency with the field, the revised nomenclature in the table is used.
Concurrent with our previous study (Rued et al., 2017) and the work reported here on suppression of ΔgpsB in S. pneumoniae D39 strains, suppressors of ΔgpsB were isolated in L. monocytogenes (Rismondo et al., 2017, Wamp et al., 2020). Remarkably, these studies by Rismondo, Wamp and colleagues showed that ΔgpsB(Lmo) is suppressed by mutations in genes that encode the following proteins: MurZ(Lmo); ReoY(Lmo) (protein of unknown function in Bacillus and Enterococcus species); ClpC(Lmo) (ATPase subunit of the ClpP protease); ReoM(Lmo) (small protein that is phosphorylated by the PrkA(Lmo) Ser/Thr kinase); and PrpC(Lmo) (cognate phosphatase to PrkA(Lmo)) (Rismondo et al., 2016, Wamp et al., 2020, Wamp et al., 2022). In parallel work, Vesić and Kristich linked MurAA(Efa) function to protein phosphorylation by demonstrating that overexpression of murAA(Efa) restored cephalosporin resistance to a mutant lacking the IreK(Efa) Ser/Thr protein kinase (Vesic & Kristich, 2012).
These and other supporting data have led to a model whereby regulation of MurA(Lmo) stability is mediated by the level of ReoM(Lmo) phosphorylation by the PrkA(Lmo) Ser/Thr protein kinase (Wamp et al., 2022, Wamp et al., 2020). According to this model, unphosphorylated ReoM(Lmo) may act as an adaptor, along with ReoY(Lmo) and MurZ(Lmo), to direct MurA(Lmo) degradation by the ClpCP(Lmo) protease. Phosphorylation of ReoM(Lmo) by PrkA(Lmo) in response to PG signals and stress are postulated to increase MurA(Lmo) amount and increase PG precursor synthesis for PG synthases in response to beta-lactam antibiotics. In support of this model, overexpression of murA(Lmo), but not murZ(Lmo), suppressed ΔgpsB(Lmo), and amino acid changes in MurA(Lmo) were identified that uncouple ReoM(Lmo)-mediated degradation by ClpCP(Lmo) (Wamp et al., 2022). Moreover, reoM(Lmo), reoY(Lmo), and clpC(Lmo) mutations suppress the conditional lethality of ΔgpsB as well as the lethality of ΔprkA in one genetic background of L. monocytogenes (Wamp et al., 2022, Wamp et al., 2020). Notably, Kelliher and colleagues confirmed this general model by isolating suppressors in this set of genes that decrease sensitivity of ΔprkA(Lmo) to β-lactam antibiotics and relieve infection-linked phenotypes (Kelliher et al., 2021). However, a link between general protein phosphorylation by the PrkA(Lmo) Ser/Thr protein kinase and GpsB function was not reported in L. monocytogenes, and it was speculated that lack of GpsB(Lmo) leads to misregulation of Class A PBP function that somehow signals to the PrkA(Lmo) kinase (Wamp et al., 2020).
In this paper, we expand our previous study of ΔgpsB suppression in S. pneumoniae D39. We report that most ΔgpsB(Spn) and ΔstkP suppressors are duplications of regions containing murZ(Spn) or murA(Spn). We show that these duplications range from ≈20 genes to >150 genes and are anchored by different repeat sequences flanking murZ(Spn) or murA(Spn), attesting to remarkable genetic plasticity in the pneumococcal chromosome (Slager et al., 2018). Consistent with the isolation of these duplication suppressors, we show that overexpression of murZ(Spn) or murA(Spn) suppressed ΔgpsB(Spn) or ΔstkP lethality. In addition, lack of the pneumococcal KhpAB RNA-binding protein resulted in overproduction of MurZ(Spn), which accounts for suppression of ΔgpsB(Spn) by ΔkhpA(Spn) or khpB(Spn). Yet, determinations of growth, morphology, and sensitivity to fosfomycin indicated that MurZ(Spn) is predominant to MurA(Spn), although their cellular amounts are approximately equal.
In addition, we isolated mutations containing amino-acid changes in a region of MurZ(Spn) away from its catalytic site that suppressed ΔgpsB(Spn) (without restoring Ser/Thr protein phosphorylation) or ΔstkP. Other amino acid changes in this region of MurZ(Spn) acted as suppressors, including one present in laboratory strains R6 and Rx1. An isolated stop-codon mutation near the end of ireB(Spn) and a constructed ΔireB(Spn) deletion also suppressed ΔgpsB(Spn) or ΔstkP. However, genetic suppression and western blotting experiments indicated that MurZ(Spn) and MurA(Spn) are not degraded by the ClpCP(Spn) protease. Tn-seq and depletion experiments further showed that StkP is essential in D39 strains and that the primary morphology phenotype caused by lack of StkP is a defect in division septation, resulting in longer, but not wider, cells. Altogether, these findings support the conclusion that GpsB(Spn) and StkP are essential in exponentially growing S. pneumoniae D39 cells, because Ser/Thr phosphorylation by StkP is required for the regulation of MurZ(Spn) and MurA(Spn) activity, but not their amounts.
2 |. RESULTS
2.1 |. Chromosome duplications containing murZ or murA are present in ΔgpsB or ΔstkP suppressor strains of S. pneumoniae D39
Previously, we reported five spontaneous missense mutations in phpP (Thr/Ser protein phosphatase) (Table 1, lines 2 and 5–8) and two mutants containing large chromosomal duplications/deletions (Table 1, lines 3–4) that suppress the essentiality of ΔgpsB in unencapsulated S. pneumoniae D39 (Rued et al., 2017). However, we did not determine the basis for ΔgpsB suppression or how the duplications/deletions formed in these mutants. To this end, we screened 20 additional ΔgpsB spontaneous suppressors from independent transformations by sequencing for phpP mutations or by PCR for the Δ(spd_1029’-spd_1037’)-region deletion present in the sup gpsB-2 and sup gpsB-3 duplication/deletion mutants (Rued et al., 2017). Fifteen of 20 suppressors contained Δ(spd_1032’-spd_1036’)-region deletions, indicative of adjacent duplications (Table 1, line 13). Whole-genome sequencing of the remaining 5 suppressors indicated that sup gpsB-8 contains an ≈163 kb (149 genes) duplication of Ω[spd_0889’-spd_1037’] (Fig. 2A, S1B, and S2B; Table 1, line 9), sup gpsB-9 and sup gpsB-10 contain an ≈18 kb (21 genes) duplication or quadruplication, respectively, of Ω[spd_0966’ to spd_0986’] (Fig. 2A and S1C; Table 1, lines 10–11), sup gpsB-11 contains a murZ(D280Y) missense mutation as well as two other mutations (Table 1, line 12 and footnote), and sup gpsB-27 contains a nonsense mutation ireB(Q84(STOP)), truncating IreB by 4 amino acids, as well as a (7→6) slippage mutation in an intergenic region (Table 1, line 14 and footnote). Genetic separation showed that murZ(D280Y) or ireB(Q84(STOP)) was necessary and sufficient for ΔgpsB suppression (Table 2, lines 6 and 12). Consistent with involvement of MurZ in ΔgpsB suppression, the duplicated regions of sup gpsB-2–3 and sup gpsB-8-10 contain murZ (spd_0967) (Fig. 2A, 3, and S1B–S1C).
Table 1.
Analysis of spontaneous ΔgpsB suppressor mutations that arose in unencapsulated S. pneumoniae Δcps D39a
| ΔgpsB suppressor designation | Strain number | Genotype | Doubling time (min)b | Growth yield (OD620)b | StkP-dependent phosphorylation phenotypec | |
|---|---|---|---|---|---|---|
| 1 | WT parent | IU1945 | 38 ± 2 | 1.00 ± 0.02 | WT | |
| 2 | sup gpsB-1 d | IU6442 | phpP(G229D) | 43 ± 4 | 1.01 ± 0.01 | similar to WT |
| 3 | sup gpsB-2 d | IU5845 | Δ[spd_1026’-spd_1037’] (≈6.3 kb, 12 genes) Ω[spd_0889’-spd_1026’] (≈150 kb, 137 genes) |
39 ± 4 | 0.8 ± 0 | reduced |
| 4 | sup gpsB-3 d | IU6441 | Δ[spd_1029’-spd_1037’] (≈8 kb, 9 genes) Ω[spd_0889’-spd_1024’] (≈148 kb, 135 genes) |
38 ± 3 | 0.88 ± 0.03 | reduced |
| 5 | sup gpsB-4 d | IU9262 | phpP(L148S) | nde | nde | nde |
| 6 | sup gpsB-5 d | IU6444 | phpP(G117D) | 41 | 0.99 | similar to WT |
| 7 | sup gpsB-6 d | IU7736 | phpP(T163P) | 45 | 1.11 | similar to WT |
| 8 | sup gpsB-7 d | IU11955 | phpP(R125P) | 38 ± 1 | 1.02 ± 0.01 | similar to WT |
| 9 | sup gpsB-8 f | IU11954 | Ω[spd_0889’-spd_1037’] (≈163kb, 149 genes) | 63 ± 6 | 0.38 ± 0.05 | reduced |
| 10 | sup gpsB-9 f,g | IU11846 | Ω[spd_0966’-spd_0986’] (≈18kb, 21 genes) tandem repeat of region | 69 ± 9 | 0.49 ± 0.16 | reduced |
| 11 | sup gpsB-10 f | IU11918 | Ω[spd_0966’-spd_0986’] (≈18kb, 21 genes) quadruplicate of reads | 47 ± 2 | 0.66 ± 0.06 | reduced |
| 12 | sup gpsB-11 h | IU11914 | murZ(D280Y) | 52 ± 3 | 0.73 ± 0.09 | reduced |
| 13 | sup gpsB-12 to -26 i | Detected Δ[spd_1032’-spd_1036’], indicative of adjacent duplication | nde | nde | nde | |
| 14 | sup gpsB-27 j | IU7735 | ireB(Q84(STOP)) | 43 ± 1. | 0.71 ± 0.04 | reduced |
Transformations were performed as described in Experimental procedures. All isolates were obtained from IU1945 (D39 Δcps), except for sup gpsB-6 and sup gpsB-4, which were isolated from IU1824 (D39 Δcps rpsL1) and Rx1, respectively. Control transformations with a Δpbp1b::aad9 amplicon gave >500 colonies in 24 h, whereas ΔgpsB<>aad9 transformations gave <10 colonies in 48 h. Mutations in the sup1–3 and sup8–11 suppressors were located by whole-genome sequencing (see Experimental procedures).
Doubling times and maximal growth yields obtained within 8 h of growth in BHI broth were determined as described in Experimental procedures. Values (means ± SEM) were obtained from 2 or more independent biological experiments except for sup-5 and sup-6. Representative growth curves are shown in Fig. S4.
Detection of proteins phosphorylated at Thr residues was performed by Western blotting using ɑ-pThr antibody as described in Experimental procedures. See Results and Fig. S6 for details.
sup gpsB-1 to sup gpsB-7 are reported in (Rued et al., 2017).
nd, not determined. The parent strain of sup4 was Rx1.
Chromosomal duplication is depicted in Fig. 1. murZ(spd_0967) is within the duplicated region.
Additional mutation detected with whole genome sequence of IU11846 includes a T deletion at intergenic spd_1376/spd_1377.
murZ(D280Y) mutation resulted from a GAC to TAC codon change. Additional mutation detected by whole genome sequence of IU11914 includes a T deletion in spd_1348 at 347/465 bp, and a G→A at intergenic spoJ/dnaA.
PCR primers specific for spd_1032 or spd_1036 (Table S1) were used to detect the deletion of spd_1032 or spd_1036 region.
In IU7735 (D39 rpsL1 Δcps ΔgpsB<>aad9), codon change that leads to ireB(Q84(STOP)) is CAA→TAA at chromosomal position 184,601. An additional spontaneous mutation identified in IU7735 by Illumina whole-genome sequencing includes a (A) 7→6 deletion at chromosome position 998,228, at an intergenic site between eutD and spd_0987.
Figure 2. Chromosomal duplications containing murZ or murA are present in ΔgpsB or ΔstkP suppressor strains of S. pneumoniae D39.
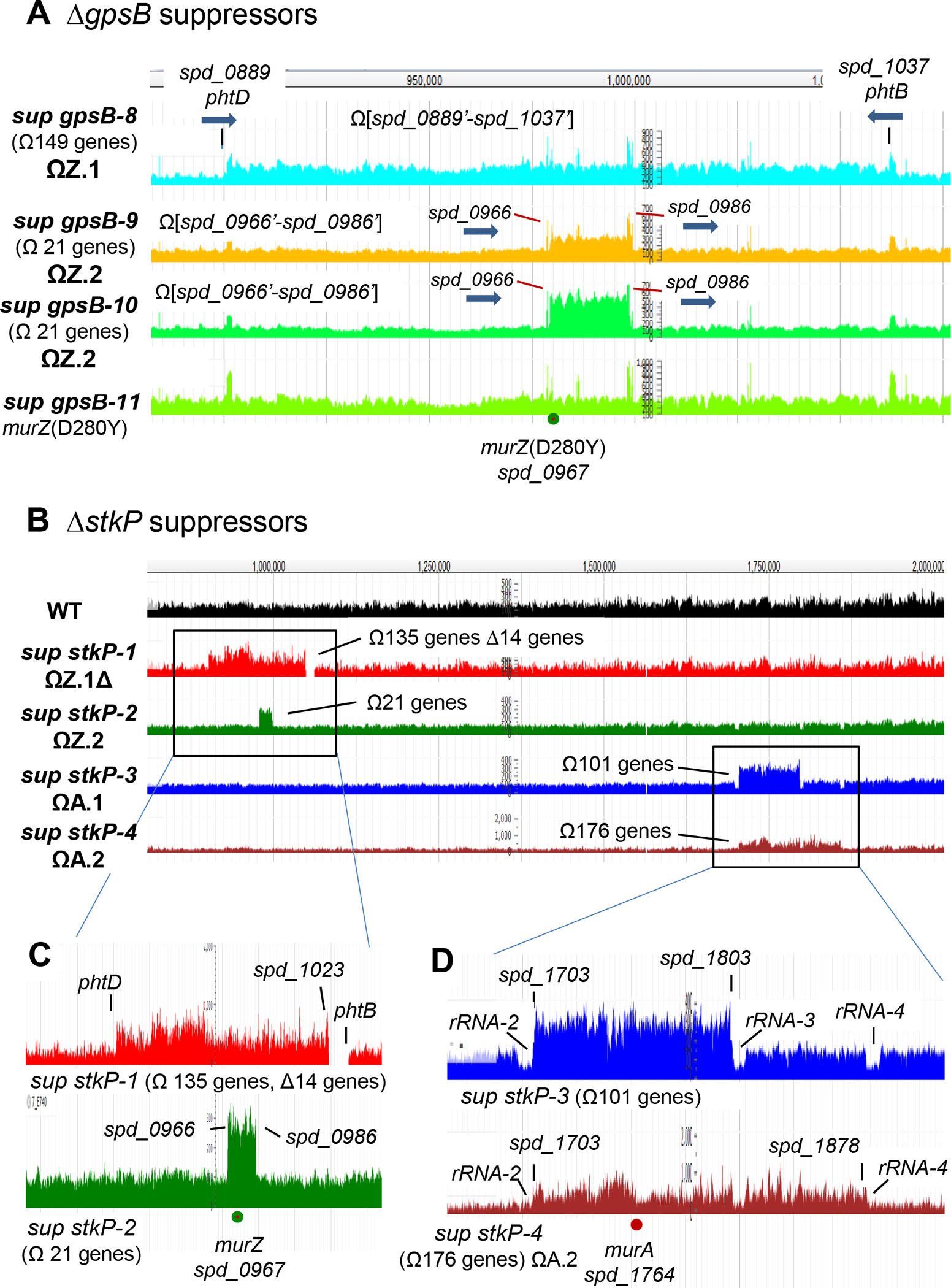
(A) Snapshot of genome browser output of ΔgpsB sup strains from genome coordinates 870 to 1100 kb. Three new ΔgpsB suppressor strains contain chromosomal duplication or quadruplication of multiple genes, all of which include murZ. Sup gpsB-8 contains a ≈163 kb duplication of chromosomal region from spd_0889’ to spd_1037’, while sup gpsB-9 and sup gpsB-10 contain a duplication or quadruplication, respectively, of the chromosomal region from spd_0966’ to spd_0986’. sup gpsB-10 has a murZ(D280Y) mutation and no chromosomal duplication. Black lines point to the flanking regions of the duplication found in sup gpsB-8, which are 1324-bp inverted repeats present in phtD (spd_0889) and phtB (spd_1037), encoding 2 of the 3 pneumococcal histidine triad proteins. The red lines point to the flanking regions (spd_0966 and spd_0986) of duplication or quadruplication found in sup gpsB-9, and -10, respectively. spd_0966 and spd_0986 are pseudogenes containing IS1167 degenerate transposase sequences. Thick blue arrows show the gene orientations of phtB, phtD, spd_0966, and spd_0986. (B) Snapshot of genome browser output of ΔstkP sup strains from genome coordinates 750 to 2,000 kb. (C) Sup stkP-1 contains a duplication/deletion between phtD and phtB, and sup stkP-2 contains a duplication between spd_0966 and spd_0986. (D) Large duplications found in sup stkP-3 and -4 are flanked by tRNA + rRNA clusters rRNA/rRNA3 and rRNA/rRNA4 respectively. Sup stkP-3 showed a decrease in sequence reads of the four rRNA-1–4 operons (rRNA-1, rRNA-2, rRNA-3, and rRNA-4) compared to the surrounding region. It is possible that either rRNA-2 or rRNA-3, or both rRNA-2 and rRNA-3, are deleted in this strain, but because of the sequence identity of the rRNA operons, deletion of one or two operons manifest as a decrease of reads for all four operons.
Table 2.
Suppression of ΔgpsB or ΔstkP mutation in S. pneumoniae Δcps D39a
| Recipient strains | Zn | Number of and appearance of colonies 20 to 22 h after transformation b | |
|---|---|---|---|
| ΔgpsB<>aad9 | ΔstkP::Pc-erm | ||
| 1. WT (IU1824)c | - | 0d | >500 fainte |
| + | 0 | >500 faint | |
| 2. gpsB+//PZn-gpsB+(IU15877)c | - | 0 | >500 faint |
| + | >500 WTf | >500 faint | |
| 3. stkP+//PZn-stkP+(IU14974)c | - | 0 | >500 faint |
| + | 0 | >500 WTf | |
| 4. murZ+//PZn-murZ+(IU13393)c | - | 0 | >500 faint |
| + | >500 small | >500 WTf | |
| 5. murA+//PZn-murA+(IU13395)c | - | 0 | >500 faint |
| + | >500 WTf | >500 WTf | |
| 6. murZ(D280Y) (IU13438) | - | >500 small | >500 WTf |
| 7. murZ(I265V, R6 allele) (IU14210) | - | >500, smallg | >500 WTf |
| 8. murZ(E259A) (IU17627) | - | >500 small | >500 WTf |
| 9. ΔkhpA (IU9036) | - | >500, small | >500, WTf |
| 10. ΔkhpB (IU10592) | - | >500, small | >500, WTf |
| 11. ΔclpP (IU17138)h | - | 0 | >500 faint |
| 12. ireB(Q84(STOP)) (IU13606) | - | >500 small | >500 WT |
| 13. ΔireB (markerless) (IU13604) | - | >500 small | >500 WT |
Recipient strains in D39 Δcps rpsL1 (IU1824) background and amplicons were obtained as described in Table S1. Transformations and visualization of colonies normalized to 1 mL of transformation mixture were performed as described in Experimental procedures. All transformation experiments were performed with Δpbp1b amplicons containing the same antibiotic selections as the positive control for detection of colonies and colony size comparison. The volumes of transformation mixture plated were adjusted to provide ≈150–500 colonies for the Δpbp1b control amplicon. Transformations with control Δpbp1b amplicons yielded >500 colonies per 1 mL of transformation mixture. Transformants were confirmed by PCR reactions. Each transformation experiment was performed 2 or more times independently with similar results.
Unless indicated, transformed colonies were generally uniform in size and of similar size as the recipient strain transformed with a control Δpbp1b amplicon.
0.4 mM (Zn2+/(1/10)Mn2+) (IU15877, IU14974, and IU13395) or 0.2 mM (Zn2+/(1/10)Mn2+) (IU13393) were added to transformation mixes and in subsequent steps to induce expression of gpsB, stkP, murZ or murA under control of the PZn zinc-inducible promoter in the ectopic bgaA site. 1/10 concentrations of Mn2+ was added to eliminate toxicity caused by addition of Zn2+ (Jacobsen et al., 2011, Martin et al., 2017, Tsui et al., 2016). The wild-type parent strain (IU1824) was transformed with the same Zn-containing condition to control for possible effects of Zn2+ on transformation efficiency.
Occasional suppressor mutants were present.
Typically only faint colonies appeared on TSAII-BA plates in the first 20 h after transformation (Fig. S3). However, upon re-streaking, these mutants show heterogeneous colony sizes.
Colonies are described as WT when the colony size and appearance are similar to the recipient strain transformed with the control Δpbp1b amplicon.
Colonies remained very small, but uniform-sized upon re-streaking on antibiotic selection plates. This strain was stored as IU14234 and verified to be ΔgpsB.
Similar results were obtained with ΔclpC (IU12462), ΔclpL (IU17136), and ΔclpE (IU17134) strains as with the ΔclpP strain after transformation with ΔgpsB<>aad9 and ΔstkP::Pc-erm amplicons.
Figure 3. Locations of repeated sequences that anchor chromosomal duplications in S. pneumoniae D39.
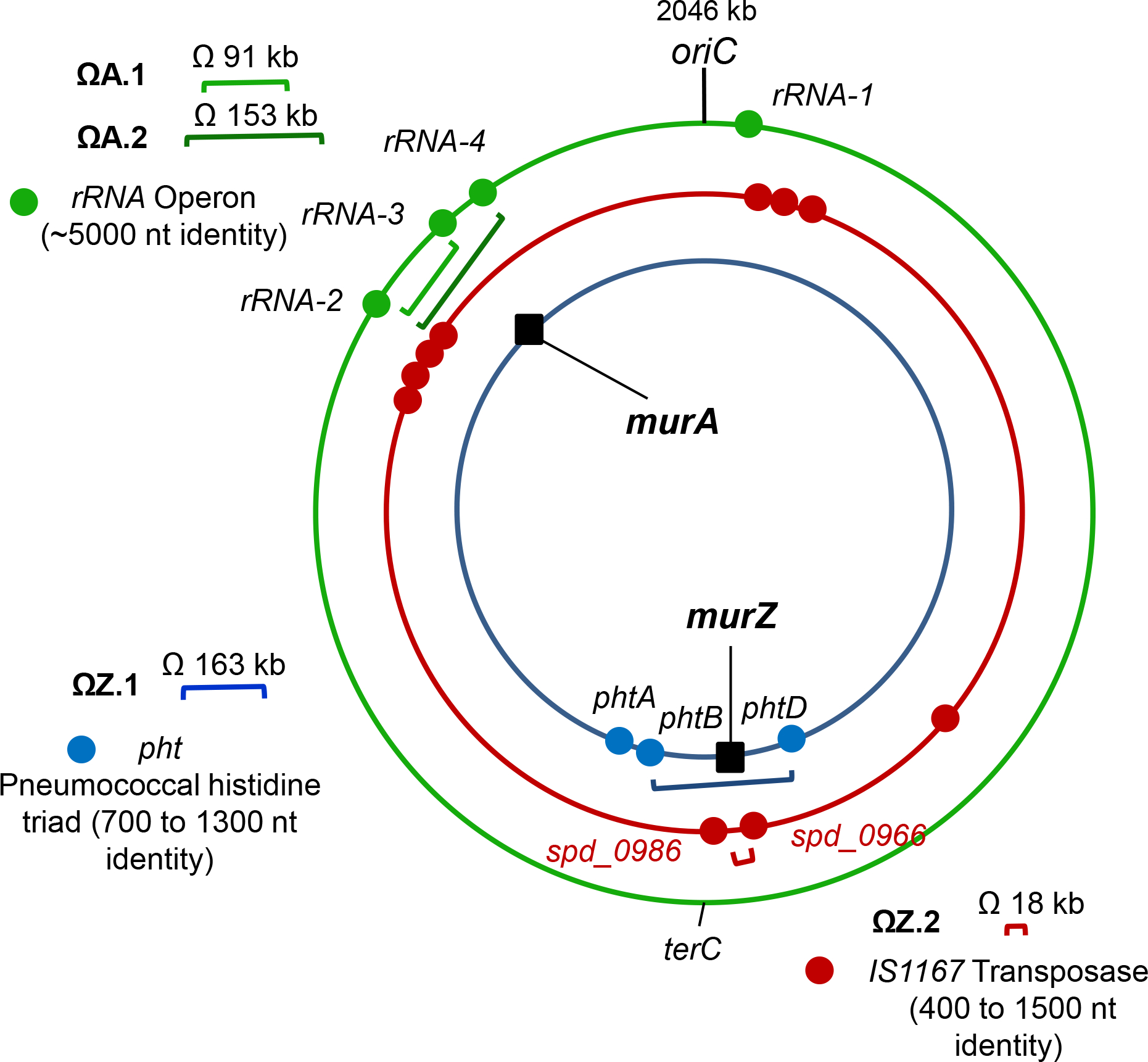
Blue, red, and green dots are locations of pht genes, IS1167 transposase, and tRNA/rRNA gene clusters, respectively. Duplications ΩZ.1 and ΩZ.2 result in duplication of murZ and surrounding genes, while ΩA.1 and ΩA.2 result in murA duplication. ΩZ.1 is present in sup gpsB-8. ΩZ.2 is present in sup gpsB-9, sup gpsB-10, and sup stkP-2. ΩA.1 is present in sup stkP-3 while ΩA.2 is present in sup stkP-4.
Since GpsB plays a role in activation of pneumococcal StkP Ser/Thr kinase activity (Fleurie et al., 2014, Rued et al., 2017), we also isolated and characterized suppressor mutations of D39 unencapsulated (Δcps) and encapsulated (cps)+ strains transformed with a ΔstkP or Δ[phpP-stkP] amplicon (Tables 2 and 3). Transformants typically appeared as faint, indistinct colonies on TSAII-BA plates after 20 h (Fig. S3A; Table 2). Re-streaking these ΔstkP and Δ[phpP-stkP] transformants resulted in heterogeneously sized, faster growing colonies, indicative of suppressor accumulation (Rued et al., 2017). We interrogated six of these re-streaked transformants for the presence of suppressor mutations (Table 3). Gene sequencing showed that none contained mutations in murZ, but one Δ[phpP-stkP] suppressor contained a 14-bp duplication within the ribosome-binding site (RBS) of ireB(Spn) (Table 3, line 9). This RBS-mutation will be described further in a report in preparation. Only one (1/6) of the transformants contained a Δ(spd_1032’-spd_1036’)-region deletion (Table 3, line 8), indicative of an adjacent phtD-phtB duplication (Fig. 2B). The genomes of the four remaining ΔstkP or Δ[phpP-stkP] transformants were sequenced (Table 3), and all were found to contain chromosomal duplications containing murZ or murA (Fig. 2B and S1B–S1D). sup stkP-1 has a duplication containing murZ and unexpectedly, a deletion similar to that of sup gpsB-2, except for the deletion junction (Fig. S1B). The deletion in sup stkP-1 accumulated during propagation of the initial ΔstkP isolate, which lacks the deletion accordingly to PCR assays. A similar duplication/deletion was reported previously in a D39 ΔstkP mutant (Ulrych et al., 2021). sup stkP-2 contains a 21-gene duplication containing murZ, similar to that of sup gpsB-9 (Fig. S1C). Notably, sup stkP-3 and sup stkP-4 contain duplications of Ω[spd_1703’-spd_1803’] and Ω[spd_1703’-spd_1878’], respectively, which contain murA (spd_1764) (Fig. 2B and S1D). Together, these results implicate overproduction of MurZ or MurA and point mutations in murZ and ireB in ΔgpsB and ΔstkP suppression.
Table 3.
Analysis of spontaneous ΔstkP suppressor mutations that arose in unencapsulated (Δcps) and encapsulated D39 S. pneumoniae D39a
| ΔstkP suppressor designation | Strain number | Genotype | Doubling time (min)b | Growth yield (OD620)b | |
|---|---|---|---|---|---|
| 1 | WT parent of sup stkP-1 | IU1824 | D39 Δcps rpsL1 | 36 ± 2 | 1.0 ± 0 |
| 2 | sup stkP-1 | IU16883 | D39 Δcps rpsL1 ΔstkP::Pc-erm Δ[spd_1024’-spd_1037’] (≈13.4 kb, 14 genes) Ω[spd_0889’-spd_1023] (≈147 kb, 135 genes) Amplification of murZ |
49 ± 2 | 1.0 ± 0 |
| 3 | WT parent of sup stkP-2, -3, -5, -6 | IU1945 | D39 Δcps | 31 ± 0.1 | 0.9 ± 0.0 |
| 4 | sup stkP-2 | E740c | D39 Δcps Δ[phpP-stkP]::Pc-erm
Ω[spd_0966’-spd_0986’] (≈18 kb, 21 genes) Amplification of murZ |
39 ± 1 | 0.9 ± 0.1 |
| 5 | sup stkP-3 | IU11912d | D39 Δcps ΔstkP::Pc-cat Ω[spd_1703-spd_1803] (≈91.3 kb, 101 genes) Amplification of murA |
51 ± 2 | 0.6 ± 0.1 |
| 6 | WT parent of sup stkP-4 | IU1690 | D39 cps+ | 44 ± 4 | 0.9 ± 0.1 |
| 7 | sup stkP-4 | IU11456 | D39 ΔstkP::Pc-erm Ω[spd_1703-spd_1878] (≈153 kb, 176 genes) Amplification of murA |
57 ± 1 (n=2) | 0.7 (n=1) |
| 8 | sup stkP-5 | E739 | D39 Δcps Δ[phpP-stkP]::Pc-erm Δ[spd_1032’-spd_1036’] detected, indicative of adjacent duplication |
41 (n=1) | 1.0 (n=1) |
| 9 | sup stkP-6 | K740 | D39 Δcps Δ[phpP-stkP]::Pc-erm 14 bp-duplication detected in the RBS of ireB(Spn); duplication status not determined |
45 (n=1) | 1.1 (n=1) |
WT D39 and its derivatives (D39 Δcps, or D39 Δcps rpsL1) were transformed with a ΔstkP::Pc-erm, ΔstkP::Pc-cat, or Δ[phpP-stkP]::Pc-erm amplicon as described in Experimental procedures. Typically, only faint colonies appeared on TSAII-BA plates in the first 20 h after transformation (Fig. S3). However, upon re-streaking, these mutants show heterogeneous colony sizes. The larger colonies were stored and analyzed by whole-genome sequencing.
Doubling times and maximal growth yields obtained within 8 h of growth in BHI broth were determined as described in Experimental procedures. Values (means ± SEM) were obtained from 2 independent biological experiments, except for sup4. Representative growth curves are shown in Fig. S4C, S4D, and S20A.
Additional mutation detected with whole genome sequence of E740 includes a sun(A324D, GCT → GAT). sun encodes rRNA small subunit methyltransferase B.
Additional mutation detected with whole genome sequence of IU11912 includes spd_0921(K420M, AAG → ATG). spd_0921 encodes a site-specific recombinase family protein.
2.2 |. Repeats in phtD and phtB, degenerate IS elements spd_0966 and spd_0986, or tRNA/rRNA gene clusters contribute to pneumococcal genomic plasticity
To understand their formation, we deduced the flanking sequences of the duplications that suppress ΔgpsB and ΔstkP (Fig. 2). The duplications were grouped into four patterns: ΩZ.1 or ΩZ.2 for duplication of the murZ region and ΩA.1 or ΩA.2 for duplication of the murA region (Fig. 3). The flanking sequences of ΩZ.1 duplications are intact and hybrid inverted repeat elements of phtD and phtB, while ΩZ.2 duplications are bordered by intact direct repeats of degenerate IS elements spd_0966 and spd_0986. The flanking sequences of ΩA.1 or ΩA.2 duplications consist of intact direct repeats of tRNA/rRNA gene clusters (Fig. 2, 3, and S1).
ΩZ.1 duplications (Fig. 3 and S1B; sup gpsB-2, -3, and -8, and sup stkP-1) are bordered by intact or hybrid (phtB’/phtD’) inverted repeat elements of phtD (spd_0889) and phtB (spd_1037) generated by homologous recombination (Fig. 2A, S1B, and S2B; where apostrophes indicate hybrid genes). phtD and phtB encode 2 of the 3 histidine triad proteins in S. pneumoniae D39 and have identical 1,324-bp sequences at their 3’-ends (Table S2). During chromosome replication when there are two copies of the genes between phtD and phtB, the large phtD and phtB inverted repeats can recombine to invert the order of intervening genes. Evidence for inversion during duplication formation is presented below for sup gpsB-3 (Fig. S2C–F).
However, the inverted phtD and phtB sequences cannot foster direct homologous recombination to form a duplication. Consequently, phtD and phtB must also contain short direct repeats or other elements that enhance short-junction (SJ) duplication (Reams & Roth, 2015) that keeps the duplication boundaries within phtD and phtB (Fig. 2A, 2B, S1B, and S2B). Indeed, there are small direct repeats of 8 and 9 bp and shorter clusters of directly repeated bps within inverted phtD and phtB that could promote SJ duplication. Of the 4 ΩZ.1 duplications, only sup gpsB-8 contains intact duplicated regions, which may be aligned in the same or an inverted orientation. The other three ΩZ.1 duplications contain slightly different deletions of duplication junctions (labeled ΩZ.1Δ; Fig. S1B and S2C). Similar remodeling by deletion of duplication junctions often occurs (Reams & Roth, 2015). Interestingly, all ΩZ.1 duplications create a second copy of the terC chromosomal replication terminus, including the difSL recombination site and xerC recombinase gene (star, Fig. S1A), that mediate chromosome dimer resolution in Streptococci/Lactococci (Le Bourgeois et al., 2007). In ΩZ.1 duplications, the two copies of difSL and xerS are oppositely oriented (Fig. S1B).
ΩZ.2 duplications are bordered by direct repeats of pseudogenes spd_0966 and spd_0986, which contain IS1167 degenerate transposase sequences (Fig. 2B, 3, and S1C; sup gpsB-9 and -10, and sup stkP-2). spd_0966 (1,492 bp) shows 91% identity with spd_0986 (1,477 bp), including 240-bp of identical sequence at their ends (Table S3). The duplications are likely joined by a spd_0986’/spd_0966’ hybrid element formed by homologous recombination (Fig. S1C). Similarly, ΩA.1 (sup stkP-3) and ΩA.2 (sup stkP-4) duplications are bordered by direct repeats; in this case, of rRNA operons that have homologous/heterologous DNA stretching over >5,000 bp (Fig. 2B, 2D, 3, and S1D; Table S4). sup stkP-3 is flanked by direct repeats of the ≈6 kb rRNA-2 and rRNA-3 operons, which are 99.9% identical and contain genes for 9 tRNAs, a 5S rRNA, a 23 S rRNA, a tRNA, a 16S rRNA, and a tRNA (Table S4). The internal junction in sup stkP-3 is likely a rRNA-3’/rRNA-2’ hybrid element (S1D). sup stkP-4 is flanked by direct repeats of rRNA-2 and rRNA-4, with a hybrid rRNA-4’/rRNA-2’ element in the internal junction (S1D). The ≈5.2 kb rRNA-4 operon contains the same (100% identity) tRNA, 5S RNA, 23S RNA, tRNA, 16S rRNA, and tRNA genes as the distal portion of rRNA-2 (Table S4). Together these results show that repeats of phtD and phtB, degenerate IS transposase genes, and tRNA/rRNA gene clusters act as endpoints for duplications of regions ranging from ≈18 kb (21 genes) to >150 kb (176 genes) in the S. pneumoniae D39 chromosome.
2.3 |. Deletions in ΩZ.1Δ duplications may enhance fitness of ΔgpsB mutants
To provide a model for formation ΩZ.1Δ duplication/deletions (Fig. 2B and S1B), we assumed that the first event was formation of an intact ΩZ.1 inverted duplication between spd_0889’ (phtD’) and spd_1037’ (phtB’), such as sup gpsB-8 (Fig. 2A, S1B, and S2B). The next event would be deletion from spd_1029’ on one side of the duplication junction to spd_1024’ on the other side (Fig. S2C). Notably, the endpoints of internal deletions of the duplication junction are slightly different for sup gpsB-3, sup stkP-1, and sup gpsB-2 (Fig. S1B and S2C). We obtained results consistent with this model by PCR analysis of sup gpsB-3 compared to WT (Fig. S2C–S2F). Primer pairs P1/P3, P1/P4, P2/P3, and P2/P4 yielded PCR products of the expected sizes for the arrangement shown for sup gpsB-3, but not WT, consistent with formation of an inverted duplication followed by deletion of the rearrangement junction (Fig. S2C).
Different deletions of the spd_1032’ to spd_1036’ region were present in most (17/26) ΔgpsB suppressors (Table 1, row 13). However, Δ(spd_1029-spd_1037) by itself had no effect on growth in BHI broth (data not shown). We therefore checked whether ΔgpsB suppressor strains that have long (135–137 gene) duplications and short (9–12 gene) deletions, such as sup gpsB-3 and sup gpsB-2, had an apparent fitness advantage over ΔgpsB suppressor strains that contain (21–149 gene) duplications, but lack duplication-junction deletions, such as sup gpsB-8 or sup gpsB-9 (Fig. 2A, S1B, and S1C; Appendix A, Tab A). Consistent with this idea, the sup gpsB-2 and -3 strains grew similarly to WT in BHI broth with higher growth rates and yields than the sup gpsB-8 and -9 strains (Table 1, lines 3–4 and 9–10; Fig. S4A). Of particular interest, although both sup gpsB-9 (Table 1, line 10) and sup stkP-2 (Table 3, line 4) contain a duplication of spd_0966’ to spd_0986’, the growth rates and yields of sup gpsB-9 were much lower than those of sup stkP-2. These results indicate a difference between suppression of ΔgpsB and ΔstkP that was also detected in other experiments described below.
2.4 |. Overexpression of murZ or murA or the presence of murZ(D280Y) suppresses ΔgpsB lethality independently of StkP-mediated Ser/Thr protein phosphorylation
The mutants described above implicated overexpression of pneumococcal murZ or murA or mutation in murZ in the suppression of pneumococcal ΔgpsB or ΔstkP (Tables 1 and 3). To test this idea further, we constructed merodiploid strains that overexpress murZ or murA under the control of a Zn2+-inducible promoter from an ectopic site. Overexpression of murZ, optimally with 0.2 mM Zn inducer (0.2 mM ZnCl2 + 0.02 mM MnSO4; 0.2 mM (Zn2+/(1/10)Mn2+), or MurA, optimally with 0.4 mM Zn inducer (0.4 mM (Zn2+/(1/10)Mn2+), suppressed ΔgpsB in transformation assays (Table 2, lines 4–5; Table S5A, lines 35–39). As a control, overexpression of catalytically inactive murZ(C116S) or murA(C120S) did not suppress ΔgpsB (Table S5A, lines 15–16, 19–20), indicating a requirement for catalytic activity. Western blot controls indicated that cellular amounts of the catalytically deficient proteins were the same as WT (Fig. S15B).
Suppression of ΔgpsB by murZ or murA overexpression was confirmed by growth of ΔgpsB murZ+//PZn-murZ+ and ΔgpsB murA+//PZn-murA+ merodiploid strains in BHI broth containing a range of Zn inducer concentrations (Fig. 4). Decreased ectopic expression of murZ or murA in a ΔgpsB mutant led to the formation of large, elongated cells that lysed (Fig. 4B, 4D, and S5; no Zn), as reported previously for ΔgpsB mutants (Cleverley et al., 2019, Land et al., 2013, Rued et al., 2017). Surprisingly, suppression of ΔgpsB was maximal when MurZ was overproduced by addition of Zn(0.2) inducer, which led to an ≈3.6-fold increase in cellular MurZ amount (Fig. 5C); but, this level of MurZ Induction did not fully restore WT growth or cell morphology to ΔgpsB cells (Fig. 4A, 4B, and S5A). In fact, induction of MurZ above this level led to decreased growth rate and yield in the ΔgpsB background (Fig. 4A). In contrast, increasing MurA cellular amount to ≈10-fold above WT suppressed ΔgpsB and largely restored WT growth and morphology to ΔgpsB cells (Fig. 4C, 4D, 5D, and S5B). Besides overexpression of murZ or murA, we tested whether overexpression of 21 other genes involved in pneumococcal division or peptidoglycan synthesis suppressed ΔgpsB (Table S6). Overexpression of these genes did not suppress ΔgpsB in transformation assays, while each ectopic construct complemented its corresponding deletion mutation (data not shown). We conclude that moderate (≈4-fold) overproduction of MurZ or MurA is sufficient to restore growth to a ΔgpsB mutant, but higher overproduction of MurZ, but not MurA, is deleterious for growth of ΔgpsB mutants in BHI broth.
Figure 4. murZ(D280Y) and overexpression of murZ or murA partially suppress ΔgpsB growth and morphology phenotypes.
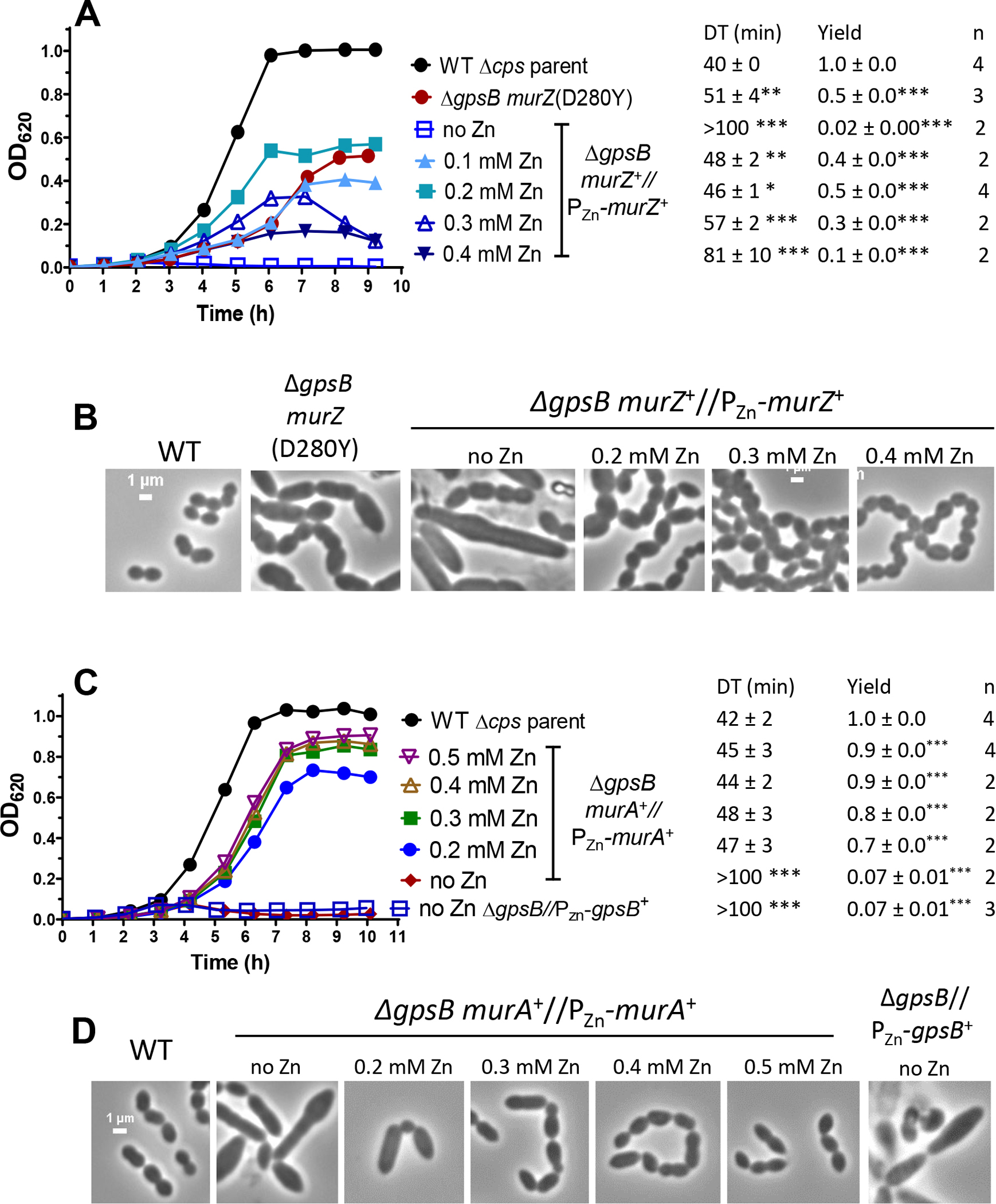
(A and B) Parent D39 Δcps rpsL1 strain (IU1824), murZ(D280Y) ΔgpsB strain (IU13509), and ΔgpsB murZ+//PZn-murZ+ (IU15860) strain were grown overnight in BHI broth with no (IU1824, IU13509) or 0.2 mM (Zn2+/(1/10)Mn2+) (IU15860), respectively. Overnight cultures were diluted to OD620 ≈0.003 in the morning in BHI broth for IU1824 and IU13509 and in BHI broth containing Zn2+/(1/10)Mn2+ for IU15860 as indicated. (A) Left, representative growth curves. Right, averages ± SEMs of doubling times (DT) and maximal growth yields (OD620) during 9 h of growth. n denotes number of independent growths. ***, p< 0.001 when compared to WT strain with one-way ANOVA analysis (GraphPad Prism, Dunnett’s test). DTs and growth yields without asterisks were statistically insignificant compared to values obtained from WT. (B) Representative phase-contrast images taken between at 3 to 3.5 h for IU1824, and between 3.5 to 4.5 h for IU13509 and IU15860. Scale bar = 1 μm. (C and D) Parent D39 Δcps rpsL1 strain (IU1824), ΔgpsB murA+//PZn-murA+ (IU15862), and ΔgpsB//PZn-gpsB+ (IU16370) were grown overnight in BHI broth with no (IU1824) or 0.5 mM (Zn2+/(1/10)Mn2+) (IU15862 and IU16370). Overnight cultures were diluted to OD620 ≈0.003 in the morning in BHI broth for IU1824 and IU16370 and in BHI broth containing (Zn2+/(1/10)Mn2+) as indicated for IU15862. Representative growth curves are shown along with averaged DT and growth yields. (D) Representative phase-contrast images taken at 3 h for IU1824 and IU16370 and between 4 to 4.5 h for IU15862. Box-and-whisker plots of cell dimensions of these strains are shown in Fig. S5.
Figure 5. Quantitative western blot assays showing nearly equivalent cellular amounts of MurZ-L-FLAG3 (-F3), MurZ(D280Y)-L-F3, and MurA-L-F3, overproduction levels of MurZ-L-FLAG3 and MurA-L-FLAG3, and lack of change when the other homolog or ClpC is deleted.
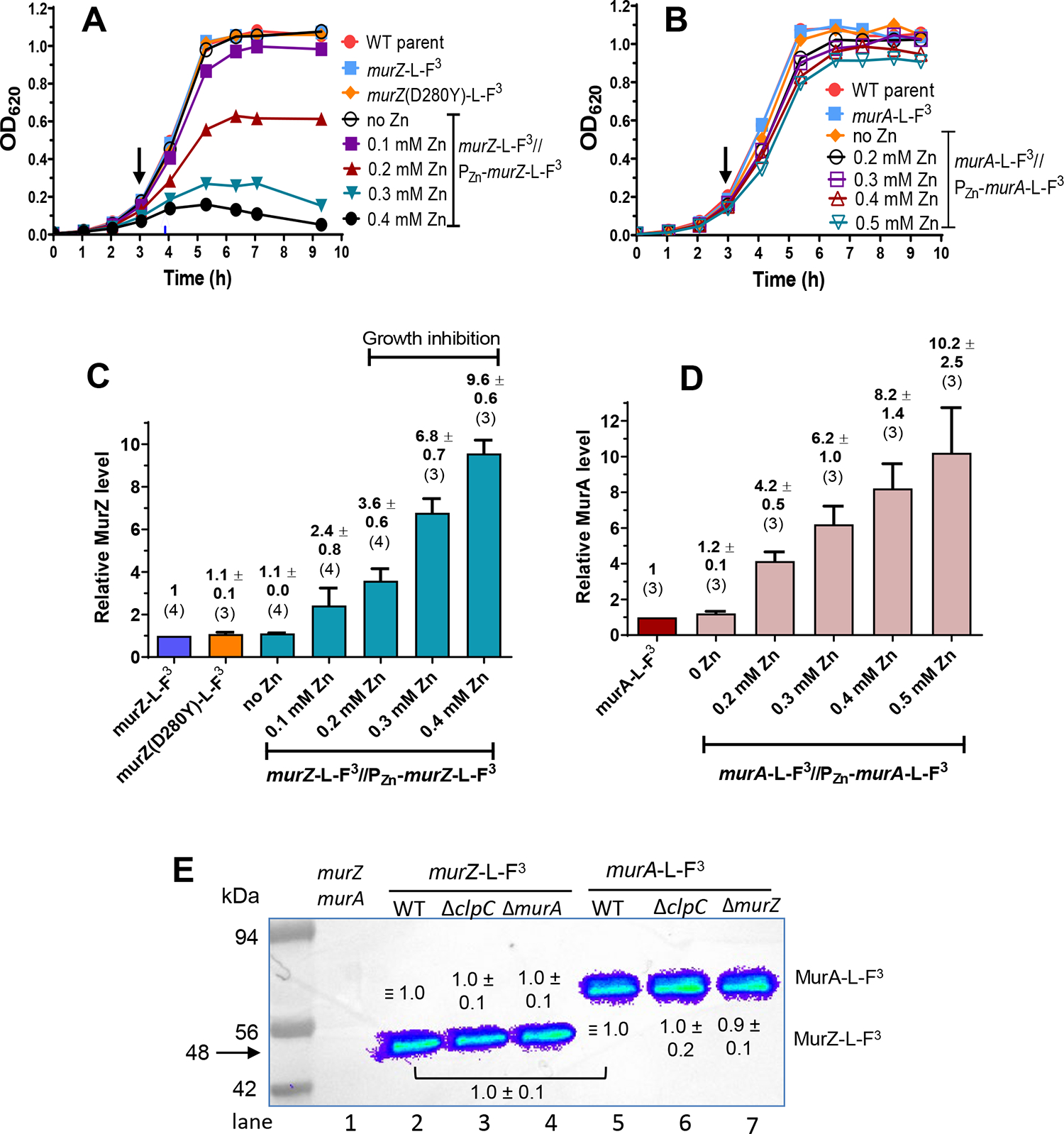
Strains tested in (A) and (C) were non-FLAG (F) - tagged murZ WT (IU1824), murZ-L-F3 (IU13502), murZ(D280Y)-L-F3 (IU13600), and murZ-L-F3//PZn-murZ-L-F3 (IU13772). Strains tested in (B) and (D) were non-F-tagged murA WT (IU1824), murA-L-F3 (IU14028), and murA-L-F3//PZn-murA-L-F3 (IU15983). Strains were grown overnight in BHI broth with no additional (Zn2+/(1/10)Mn2+), and diluted to OD620 ≈0.005 in the morning in BHI with no additional (Zn2+/(1/10)Mn2+), or in BHI broth containing 0.1, 0.2, 0.3 or 0.4 mM (Zn2+/(1/10)Mn2+) for IU13772, or in BHI broth containing 0.2, 0.3, 0.4 or 0.5 mM (Zn2+/(1/10)Mn2+) for IU15983. Black arrows point to the time (≈3 h) when samples were collected, except for IU13772 grown in the presence of 0.3 or 0.4 mM (Zn2+(1/10)Mn2+), where samples were collected at 3.6 h (blue arrow). (C) and (D) Quantitative western blotting using anti-FLAG antibody was performed as described in Experimental procedures. Calculated averages and SEMs of relative MurZ-L-F3 or MurA-L-F3 protein amounts were obtained from three or more independent experiments using anti-FLAG antibody. The numbers above each bar are averages ± SEM obtained for the number of independent biological replicates indicated in parentheses. Representative western blots are presented in Fig. S8. (E) Representative western blot showing similar cellular amounts of MurZ-L-F3 in ΔclpC or ΔmurA strains as in WT, similar cellular amounts of MurA-L-F3 in ΔclpC or ΔmurZ strains as in WT, and similar cellular amounts of WT MurZ-L-F3 and WT MurA-L-F3. Lane 1, Wild-type (IU1824); lane 2, murZ-L-F3 (IU13502); lane 3, murZ-L-F3 ΔclpC (IU14082); lane 4, murZ-L-F3 ΔmurA (IU14084); lane 5, murA-L-F3 (IU14028); lane 6, murA-L-F3 ΔclpC (IU14086); lane 7, murA-L-F3 ΔmurZ (IU14088). Numbers above MurZ-L-F3 or below MurA-L-F3 bands are calculated protein amounts (mean ± SEM) relative to murZ-L-F3 (lane 2) or murA-L-F3 (lane 5) based on three independent experiments with ΔclpC strains and two independent experiments with ΔmurZ or ΔmurA strains. 0.67 μg of protein was loaded into each lane. The predicted molecular masses of both MurZ-L-F3 and MurA-L-F3 are 48 kDa; however, MurA-L-F3 (and untagged MurA(Spn) (data not shown)) migrate slower than their predicted molecular weights.
We also tested whether the murZ(D280Y) mutations identified in the genetic screen suppressed ΔgpsB. A constructed isogenic murZ(D280Y) mutation suppressed ΔgpsB in transformation assays (Table 2, lines 1 and 6). However, the growth rate and yield of the ΔgpsB murZ(D280Y) mutant were considerably reduced compared to the WT strain (Fig. 4A), and ΔgpsB murZ(D280Y) cells were extremely large and elongated compared to WT cells (Fig. 4B and S5B), similar to ΔgpsB cells depleted for MurZ that stop growing (Fig. 4A). We conclude that the murZ(D280Y) mutation only partly suppresses the defects caused by ΔgpsB.
Finally, we assayed whether overexpression of murZ or murA or the presence of murZ(D280Y) restored general Thr phosphorylation of proteins in a ΔgpsB mutant. It was previously reported that ΔgpsB greatly reduces Thr phosphorylation by the StkP Ser/Thr kinase in S. pneumoniae, leading to the model that GpsB activates StkP function (Fleurie et al., 2014, Rued et al., 2017). We showed that ΩZ.1Δ gpsB suppressors sup gpsB-2 and sup gpsB-3 did not restore Thr phosphorylation of proteins, whereas the phpP phosphatase mutation in sup gpsB-1 restored phosphorylation (Rued et al., 2017). Likewise, all new ΩZ.1 and ΩZ.1Δ duplications that suppressed ΔgpsB from this study did not restore Thr phosphorylation of proteins to WT level (Fig. S6A), while phpP mutations that suppressed ΔgpsB did restore Thr phosphorylation (Fig. S6B). Overexpression of murZ or murA or murZ(D280Y) also failed to restore Thr phosphorylation of proteins in a ΔgpsB mutant (Fig. S7A, lanes 5 and 9; Fig. S7B, lane 6). We conclude that suppression of ΔgpsB by overexpression of murZ or murA or by murZ(D280Y) occurs by a Thr phosphorylation-independent mechanism.
2.5 |. Overproduction, absence, or catalytic inactivation of MurZ(Spn), but not MurA(Spn), results in altered growth, morphology, and sensitivity to fosfomycin or penicillin
The relative contribution of MurZ and MurA in pneumococcal cells is not well understood. Purified MurZ(Spn) from strain R6 has a higher catalytic efficiency for UDP-GlcNAc substrate than MurA(Spn) (Du et al., 2000). By contrast, the MurA-family homolog is essential or catalytically predominant in other Gram-positive species (Fig. 1) (Blake et al., 2009, Kedar et al., 2008, Kock et al., 2004, Mascari et al., 2022, Rismondo et al., 2017). The growth defects of MurZ(Spn) overproduction in the ΔgpsB mutant (Fig. 4A–B) prompted us to further characterize the relative roles of MurZ and MurA in WT pneumococcal cells.
The absence of MurZ and MurA was confirmed to be synthetically lethal in S. pneumoniae D39 (Table S5B, line 2 and S5C, line 2)(Du et al., 2000). Catalytically inactive MurZ(C116S) and MurA(C120S) also were synthetically lethal with lack of MurA or MurZ, respectively (Table S5B, line 3 and S5C, line 3). Strains expressing murZ-L -FLAG3 or murA-L-FLAG3 from their native chromosomal loci were constructed (Table S1), and production levels were assayed by quantitative western blotting (Fig. 5). Strains expressing murZ-L-FLAG3 or murA-L-FLAG3 did not show phenotypic differences in growth or transformation assays compared to their WT counterparts, including synthetic lethality (Fig. 5A–B, 6A, and 6C; Table S5B, line 4 and S5C, line 5). Consistent with comparable activities, high overproduction of MurZ-L-FLAG3 inhibited growth like MurZ overproduction (Fig. 5A and 6A). MurZ-L-FLAG3 and MurA-L-FLAG3 amounts were comparable (ratio = 0.95 ± 0.06 (SEM; n= 2)) in bacteria growing exponentially in BHI broth (Fig. 5E). Immunofluorescent microscopy showed that MurZ-L-FLAG3 and MurA-L-FLAG3 were distributed throughout the cytoplasm, and not localized at division septa or equators (Fig. S9).
Figure 6. Overproduction or absence of MurZ(Spn), but not MurA(Spn), alters growth, morphology, and sensitivity to fosfomycin or penicillin.
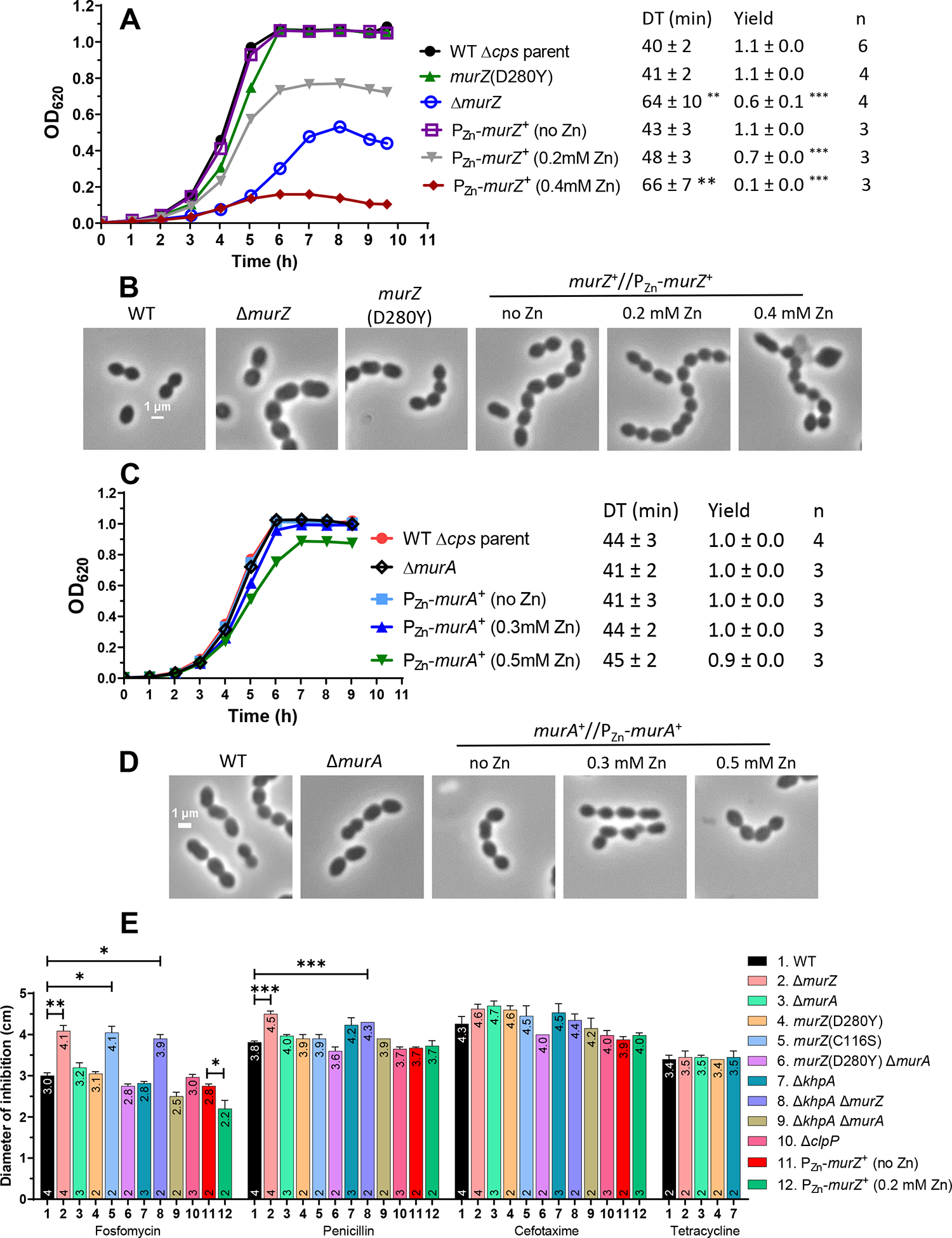
(A and B) Parent D39 Δcps rpsL1 strain (IU1824), constructed murZ(D280Y) (IU13438), ΔmurZ (IU13536), and merodiploid murZ+//PZn-murZ+ (IU13393) strains were grown overnight in BHI broth with no additional (Zn2+/(1/10)Mn2+) and diluted to OD620 ≈0.003 in the morning in BHI broth with or without (Zn2+/(1/10)Mn2+) at the concentrations indicated. (A) Representative growth curves and averaged DT and yields. ** p< 0.01; *** p < 0.001 compared to WT strain by one-way ANOVA analysis (GraphPad Prism, Dunnett’s test). (B) Representative phase-contrast images taken between 3.5 to 4 h of growth for all strains and conditions, except for IU13393 with 0.4 mM (Zn2+(1/10)Mn2+), which was taken at 5 h of growth. (C and D) Parent D39 Δcps rpsL1 strain (IU1824), ΔmurA (IU13538), and merodiploid murA+//PZn-murA+ (IU13395) strains were grown similarly to the murZ strains described above. The DTs and growth yields of all strains and conditions were not statistically different from the values obtained for the WT strain. (D) Representative phase-contrast images taken at 3 h of growth for all strains and conditions. All micrographs in (B) and (D) are at the same magnification (scale bar = 1 μm). Box-and-whisker plots of cell dimensions of murZ(D280Y) and strains overexpressing murZ or murA are in Fig. S10. (E) Disc diffusion assays were performed as described in Experimental procedures for strains: WT parent (IU1824), ΔmurZ (IU13536), ΔmurA (IU13538), murZ(D280Y) (IU13438), murZ(C116S) (IU15939), murZ(D280Y) ΔmurA (IU17748), ΔkhpA (IU9036), ΔkhpA ΔmurZ (IU13542), ΔkhpA ΔmurA (IU13546), ΔclpP (IU12462), murZ+//PZn-murZ+ (no Zn) (IU13393), and murZ+//PZn-murZ+ in 0.2 mM (Zn2+/(1/10)Mn2+). Mean diameters of zones of inhibition ± SEM are graphed from at least two independent biological replicates. Means and numbers of replicates (n) are shown at the tops and bottoms of bars, respectively. P values were obtained by the Welch t-test (GraphPad Prism). *, **, and *** denote p<0.05, p<0.01, p<0.001, respectively.
However, further experiments indicated that MurZ and MurA function was not equivalent and interchangeable in cells grown in most lots of BHI broth. ΔmurZ mutants usually grew slower, had a lower growth yield, and formed larger cells than ΔmurA mutants in exponential cultures (Fig. 6A–D, and S10A–B). While overproduction of MurZ by ≈2-fold did not change growth (Fig. 5A; Zn (0.1)), higher overproduction of MurZ by ≈4–10 fold progressively reduced growth rate and yield and led to smaller cells with increasingly defective morphologies (Fig. 5A, 6A–B, and S10A). The impaired growth patterns resulting from absence or overproduction of MurZ may be due to an increase or decrease, respectively, of UDP-GlcNAc, one of the substrates used by MurZ. UDP-GlcNAc is an important substrate for other cellular processes such as teichoic acid and nucleotide sugar synthesis (see https://www.genome.jp/kegg/pathway.html#global) (Denapaite et al., 2012). Alternately, increased MurZ amount may lead to increased metabolite flux through the PG synthesis pathway that is detrimental.
In contrast, overproduction of MurA by ≈4–10 fold did not affect cell growth or morphology (Fig. 5B, 5D, 6C–D, and S10B). Control experiments showed that the growth and size phenotypes of ΔmurZ mutants were complemented by ≈2-fold overproduction of MurZ (Fig. S11A–C, Zn(0.1) and Fig. 5C). ΔmurZ growth and morphology defects were also complemented by overproduction of MurA by ≈4–6-fold (Fig. S12A–C, Zn(0.2) and Fig. 5D)), but not fully at Zn(0.1), indicating that greater induction of MurA than MurZ was required to complement ΔmurZ. The lack of phenotypes from the absence or overproduction of MurA compared to MurZ may be linked to a lower catalytic efficiency of the MurA compared to the MurZ (Du et al., 2000).
We looked for other indications of differences in the relative roles of pneumococcal MurZ and MurA. We found that the absence of MurZ or overproduction of MurZ, but not MurA, caused similar growth defects or inhibition, respectively, in the isogenic encapsulated cps+ D39 progenitor strain as in Δcps mutants (Fig. S13). In the Δcps unencapsulated background, ΔmurZ and catalytically inactive murZ(C116S) mutants were more sensitive to fosfomycin, which covalently binds to the catalytic cysteine of MurA enzymes (Skarzynski et al., 1996), than a ΔmurA mutant in disk-diffusion assays (Fig. 6E). ΔmurZ mutants were also slightly more sensitive to the β-lactam antibiotic penicillin (Fig. 6E). Conversely, moderate overproduction of MurZ reduced sensitivity to fosfomycin compared to WT. We tested the effect of cephalosporins on Spn mutants, because deletion of murAA(Efa), but not murAB (Efa), led to increased susceptibility to cephalosporins (Vesic & Kristich, 2012). Similarly, reduced or increased expression of murA(Lmo) also led to increased or decreased sensitivity, respectively, to cephalosporins (Wamp et al., 2022). By contrast, ΔmurZ(Spn) and ΔmurA(Spn) mutants were equally sensitive as WT to the cephalosporin antibiotics cefotaxime or cefoperazone, and to tetracycline, which inhibits translation (Fig. 6E and data not shown). To investigate whether the absence of both MurA and MurZ causes the elongated-cell phenotype characteristic of GpsB depletion in the D39 background (Land et al., 2013, Rued et al., 2017), we examined the morphology of cells depleted of MurA in a ΔmurZ mutant or depleted of MurZ in a ΔmurA mutant. To the contrary, reduced amounts of MurZ and MurA inhibited growth and caused formation of rounded, heterogeneously sized cells that began to lyse (Fig. S14A–B). This result is consistent with GpsB having additional roles besides regulating MurZ and MurA function.
Mutants expressing catalytically inactive murZ(C116S)-L-FLAG3 or murZ(C116S) phenocopied ΔmurZ by showing impaired growth (Fig. S15A and S16C). By contrast, a mutant expressing catalytically inactive murA(C120S)-L-FLAG3 did not affect growth, similar to ΔmurA (Fig. S15A). Quantitative western blotting showed that murZ(C116S)-FLAG3 or murA(C120S)-FLAG3 were expressed at the same level as murZ-L-FLAG3 or murA-L-FLAG3, respectively (Fig. S15B). Consistent with its lack of catalytic activity, overproduction of MurZ(C116S) did not cause growth inhibition like WT MurZ (Fig. S15C). This result indicated that MurZ(C116S) is not dominant-negative over WT MurZ, consistent with a MurZ monomer in cells as well as in purified preparations(Du et al., 2000).
Finally, we noticed that severity of growth inhibition of ΔmurZ mutants from that shown in Fig. 6A varied with the lot of BHI powder, although cell morphology defects similar to those in Fig. 6B were detected. Therefore, we tested whether the absence of MurZ inhibited cell growth and caused defective cell morphology in C+Y medium, as occurred in animal-derived BHI broth (Fig. 6A–B). Previously, we determined the velocities of septal PG synthase components bPBP2x and FtsW and FtsZ treadmilling in WT and ΔmurZ (called ΔmurA1 there) in C+Y medium (Perez et al., 2019). The decreased velocity of bPBP2x and FtsW in the murZ mutant compared to WT provided evidence that PG synthesis drives movement of the PG synthase, rather than FtsZ treadmilling. Moreover, the MurZ and MurA substrate UDP-GlcNAc is involved in multiple pathways (Denapaite et al., 2012, Sachla & Helmann, 2021), and its amount may change in cells grown in different media and conditions. Indeed, we found that the absence of MurZ or MurA or their catalytic inactivation did not inhibit growth in C+Y medium (Fig. S16A) as in most lots of BHI medium (Fig. 6A). However, lack of MurZ or its catalytic activity resulted in longer, wider, and larger cells than WT in C+Y medium (Fig. S16B–C), similar to those in BHI broth (Fig. 6B), whereas ΔmurA and WT cells were the same size (data not shown). Altogether, we conclude that MurZ(Spn) and MurA(Spn) function is not equivalent in exponentially growing D39 cells and that in most cases, phenotypes of murZ mutants are more severe than those of murA mutants, consistent with a predominant role of MurZ in S. pneumoniae D39 cells.
2.6 |. murZ(D280Y), murZ(I265V) present in laboratory strains R6 and Rx1, and murZ(E259A) alleles suppress ΔgpsB
murZ(D280Y) was isolated as a spontaneous suppressor of ΔgpsB (Table 1, line 12), and partial ΔgpsB suppression was confirmed in a reconstructed murZ(D280Y) mutant (Table 2, line 6). Compared to WT, murZ(D280Y) ΔgpsB double mutants formed smaller colonies in transformation assays (Table 2, line 6), had reduced growth rate and yield (Fig. 4A), and formed large, aberrantly shaped cells (Fig. 4B and S5A). However, a single murZ(D280Y) mutant grew similarly to WT, formed marginally smaller (by 10%–20%) cells than WT in BHI broth, and showed the same sensitivity to fosfomycin or penicillin as WT or a murZ(D280Y) ΔmurA mutant (Fig. 6A, 6B, 6E, S10A, and S17C). Overexpression of murZ(D280Y) also inhibited growth of murZ+ or murZ(D280Y) merodiploid strains, similar to overexpression of murZ (Fig. S17). murZ(D280Y) was expressed in approximately the same amount as murZ in cells growing exponentially in BHI broth (Fig. 5C). Finally, whereas murZ(D280Y) partially suppresses ΔgpsB in transformation assays (Table 2, line 6; small colonies), it strongly suppressed ΔstkP and the requirement for Ser/Thr phosphorylation of proteins (Table 2, line 6; WT colonies; Fig. S3E, Fig. 9A–C, and S21A–B and E). Together, these results suggest that MurZ(D280Y) has comparable enzymatic activity and cellular amount as MurZ, but is not subjected to regulation that occurs in ΔgpsB or ΔstkP mutants.
Figure 9. Primary phenotypes of StkP(Spn) depletion are strongly suppressed by murZ(D280Y).
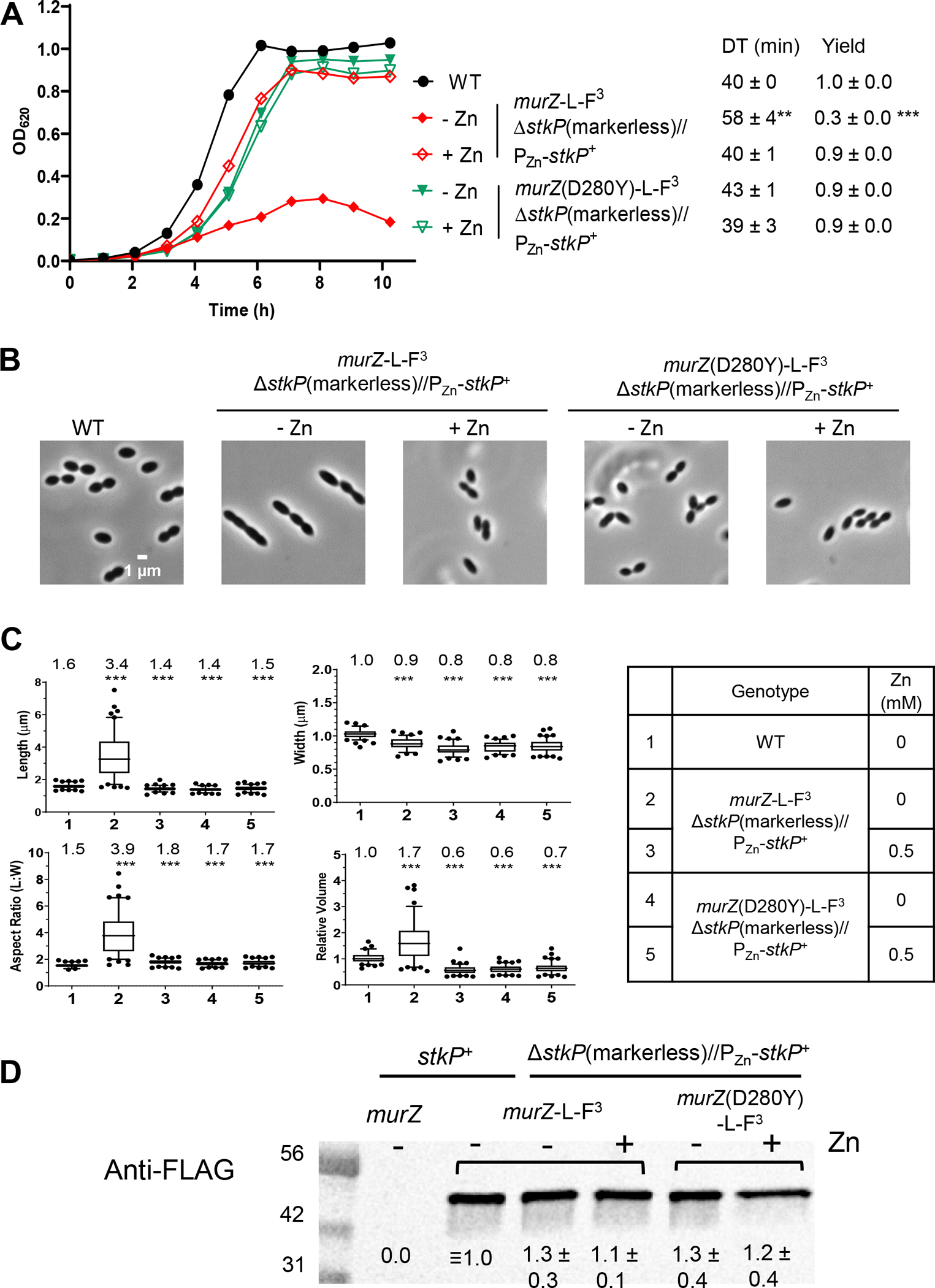
Parent D39 Δcps rpsL1 strain (IU1824), and merodiploid ΔstkP markerless//PZn-stkP+ strains containing murZ-L-FLAG3 (IU19081) or murZ(D280Y)-L-FLAG3 (IU19079) were grown overnight in BHI broth with no additional (Zn2+/(1/10)Mn2+) (IU1824) or with 0.5 mM (Zn2+(1/10)Mn2+) (IU19081 and IU19079) as described in Experimental procedures. Strains were diluted to OD620 ≈0.003 in the morning with fresh BHI broth containing no (Zn2+/(1/10)Mn2+) or 0.5 mM (Zn2+/(1/10)Mn2+). (A) Growth curves, DT, and maximal growth yields (OD620) during 10 h of growth. (B) Representative phase-contrast images taken at ≈3.5 h of growth. Scale bar = 1 μm. Growth curves and microscopy were performed in two independent experiments. (C) Box-and-whisker plots (whiskers, 5 and 95 percentile) of cell lengths, widths, aspect ratios, and relative cell volumes. P values were obtained by one-way ANOVA analysis (GraphPad Prism, Kruskal-Wallis test). *** p<0.001 compared to WT. (D) Representative western blot using anti-FLAG antibody of samples collected after 3.5 h of growth, where − or + indicates the absence of presence of 0.5 mM (Zn2+/(1/10)Mn2+) in the BHI broth. Western blotting was performed as described in Experimental procedures. 6 μL (≈2 μg) of protein samples were loaded in each lane. A standard curve was generated by loading 3, 6, 9 or 12 μL of IU13502 (murZ-L-FLAG3) samples (lanes not shown). Signal intensities obtained with anti-StkP antibody were normalized in each lane by using Totalstain Q-NC reagent (Azure Biosystems). Calculated protein amounts (mean ± SEM) relative to stkP+ murZ-L-F3 (IU13249) are based on two independent experiments.
The MurZ(D280Y) amino-acid change is located in Domain I on a surface distant from the active site of MurZ, which includes C116 (catalysis), N23 (conformation switching), D306 (deprotonation of substrate), and R398 (product release) (Fig. 7) (Jackson et al., 2009, Samland et al., 2001, Skarzynski et al., 1996). Compared to D39 strains (and WT serotype-4 strain TIGR4 (Tettelin et al., 2001)), R6 and Rx1 laboratory strains produce mutant MurZ(I265V) (Lanie et al., 2007), which has an amino-acid change near MurZ(D280Y) (Fig. 7). Like murZ(D280Y), murZ(I265V) moved into the Δcps D39 genetic background partially suppressed ΔgpsB and strongly suppressed ΔstkP in transformation assays (Table 2, lines 6–7; Fig. S3E–F and S18A–D), and D39 Δcps murZ(I265V) partially suppressed ΔgpsB in growth and morphology assays (Fig. S18A–B). Both murZ(D280Y) ΔgpsB and murZ(I265V) ΔgpsB double mutants formed large, elongated cells (Fig. 4B and S18B), reminiscent of strains depleted for GpsB (Land et al., 2013, Rued et al., 2017). Both murZ(I265V) and murZ(D280Y) strains grew similarly to WT (Fig. 5A, 6A, and S18A); however, murZ(D280Y) cells were marginally smaller than WT and murZ(I265V) cells under these growth conditions (Fig. S10A and S18E). Finally, ΔgpsB could not be transformed into an R6 ΔmurZ mutant, and ΔmurZ could not be transformed into an R6 ΔgpsB mutant, consistent with a requirement for the murZ(I265V) allele to suppress ΔgpsB in R6-derived strains (Tables S5A, lines 42–44, and S5B, line 7).
Figure 7. MurZ(D280Y), MurZ(E259A), and MurZ(I265V) that suppress ΔgpsB or ΔstkP are located on a face of Domain I of MurZ, away from its active site.
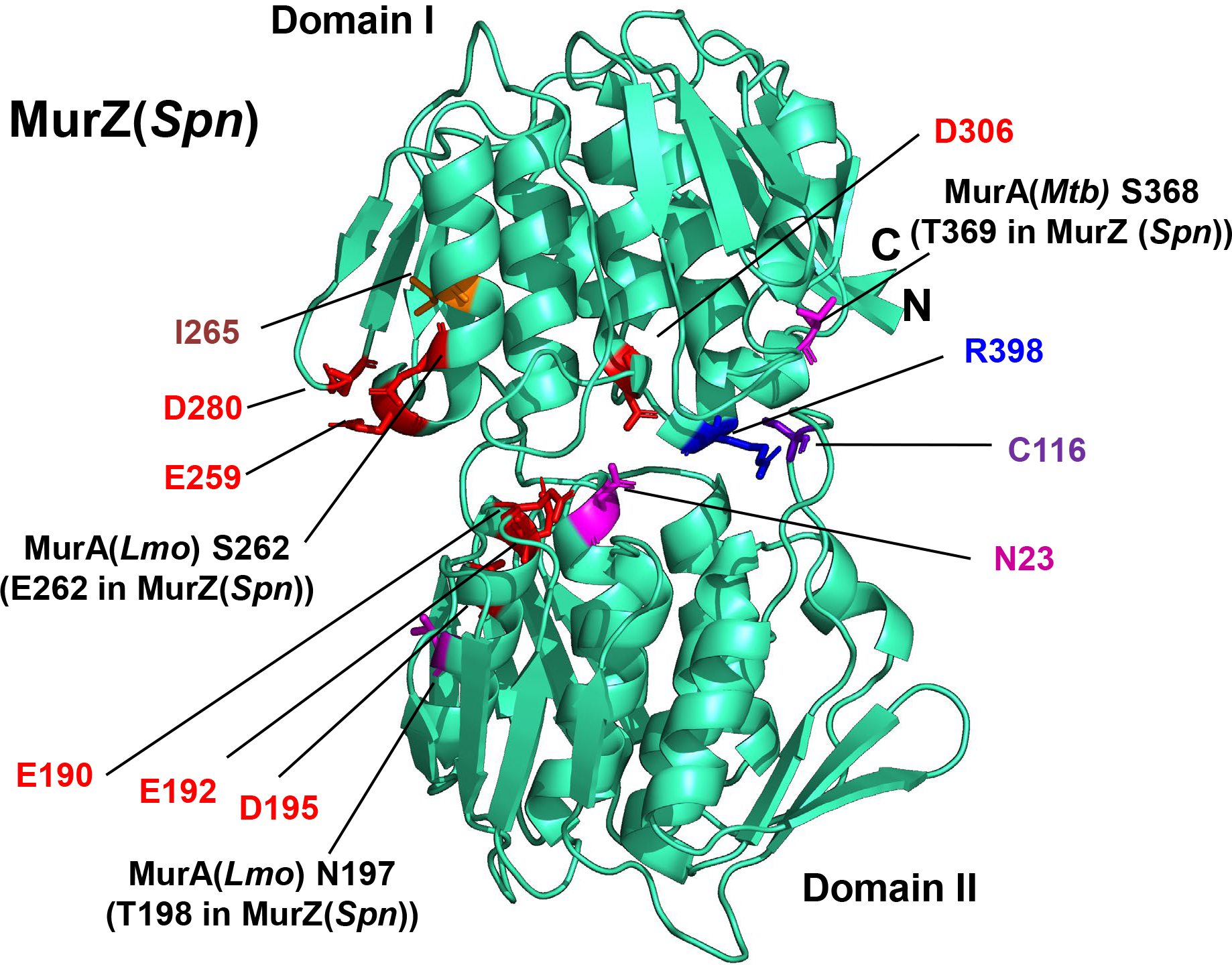
The predicted 3D-structure of MurZ(Spn) from D39 strains generated using the AlphaFold v2.0 webserver is shown in cyan, with important residues illustrated as colored sticks. Catalytic site C116, and other residues important for MurA enzymatic activity include N23 (conformation switching), D306 (initial deprotonation of the UDP substrate), and R398 (product release) (Jackson et al., 2009, Samland et al., 2001, Skarzynski et al., 1996). Although N23 and C116 are in Domain II, and D306 and R398 are in Domain I, these four residues are in close proximity on one side of the molecule. In contrast, D280, E259, and I265, for which amino acid substitutions lead to ΔgpsB suppression, are located on the opposite side Domain I compared to C116. E190, E192 and D195 are in Domain II across the cleft from D280 and do not lead to ΔgpsB suppression when substituted. Residues T198 and E262 correspond to residues MurA(Lmo) N197 and MurA(Lmo) S262 respectively. MurA(Lmo) N197D and MurA(Lmo) S262L are suppressor mutations of ΔgpsB and ΔprkA mutations in Listeria monocytogenes (Wamp et al., 2021).
Based on structure, MurZ(E259) is on the same surface as MurZ(D280Y) and MurZ(I265V) (Fig. 7). murZ(E259A) also partly suppressed ΔgpsB and strongly suppressed ΔstkP in transformation assays (Tables 2, line 8, and S5A, line 7). In contrast, analogous amino acid changes in MurA(D281Y) and MurA(E282Y) did not suppress ΔgpsB (Table S5A, lines 11–12). Finally, MurZ(E190A E192A), MurZ(E192A), and MurZ(E195A), which contain amino-acid changes in Domain II on the same side of MurZ as Domain I suppressors MurZ(D280Y), MurZ(I265V), and MurZ(E259A), failed to suppress ΔgpsB (Table S5A, lines 8–10). We conclude that the Domain I surface close to MurZ(D280) specifically mediates escape from regulation that occurs in ΔgpsB mutants.
2.7 |. MurZ and MurA are not degraded by ClpP protease in S. pneumoniae
MurA(Lmo) (the homolog of MurA(Spn); Fig. 1), accumulates to a high level (≈10-fold over WT) in ΔmurZ(Lmo) (the homolog of murZ(Spn)) or ΔclpC(Lmo) mutants of L. monocytogenes (Rismondo et al., 2017, Wamp et al., 2020). Likewise, MurAA(Bsu) (the homolog of MurA (Spn); Fig. 1) is a substrate of the ClpCP protease of B. subtilis (Kock et al., 2004), although only a marginal increase in MurAA(Bsu) amount was detected in a ΔclpC mutant in a recent study (Sun et al., 2023). Cleavage of MurA(Lmo) by the ClpCP protease is central to the model of the regulation of MurA(Lmo) cellular amount by MurZ(Lmo) and the ReoM and ReoY regulatory proteins in L. monocytogenes (Wamp et al., 2022, Wamp et al., 2020). In support of this model, ΔclpC, ΔmurZ, ΔreoM, or ΔreoY suppressed ΔgpsB or ΔprkA (lacking Ser/Thr protein kinase) in L. monocytogenes (Rismondo et al., 2017, Wamp et al., 2020, Wamp et al., 2022).
Several different results indicate that MurZ and MurA cellular amounts are not regulated by the ClpP protease and its regulatory ATPase subunits (ClpC, ClpE, or ClpL) in S. pneumoniae. We confirmed a previous report that clpP is not essential and that ΔclpP mutants do not cause strong phenotypes under non-stressed growth conditions in S. pneumoniae D39 (Robertson et al., 2003). Transformation with a ΔclpP::Pc-erm or ΔclpP::Pc-[kan-rpsL+] amplicon resulted in numerous uniform-sized ΔclpP(Spn) mutants on TSAII-BA plates, inconsistent with the accumulation of suppressor mutations in ΔclpP mutants tested for ΔgpsB or ΔstkP suppression. PCR confirmed that the WT clpP gene was not duplicated in ΔclpP mutants. Antibiotic-insertion and markerless ΔclpP mutants grew similarly to WT cells in BHI broth with no obvious cell morphology defects (data not shown). In contrast to L. monocytogenes, ΔclpP, ΔclpC, ΔclpE, or ΔclpL did not suppress ΔgpsB or ΔstkP in S. pneumoniae D39 strains (Table 2, line 11 and footnote h; Table S5A, lines 23–26; Fig. S3B). In addition, a ΔclpP mutant did not decrease sensitivity of fosfomycin, which would have been indicative of increased MurZ(Spn) or MurA(Spn) amount (Fig. 6E).
Quantitative western blot analyses further demonstrated that MurA and MurZ amounts were unchanged in pneumococcal ΔclpP, ΔclpC, ΔclpE, or ΔclpL mutants. MurA amounts were unchanged in ΔclpP or ΔclpC mutants compared to WT in blots probed with antibody against MurA(Spn) (Fig. 8A, lanes 4–6 vs lane 1). A similar result was obtained in strains that overproduced MurA (Fig. S19A, lower panel, lanes 4–5 vs lane 3). In other experiments, amounts of MurZ-L-FLAG3 and MurA-L-FLAG3 expressed from native chromosomal loci were determined in strains that did not show phenotypes different from WT (Fig. 5 and 6). The cellular amount of MurZ-FLAG3 or MurA-FLAG3 was not changed in ΔclpP, ΔclpC, ΔclpE, or ΔclpL mutants (Fig. 8B–C, lanes 3–6 vs lane 2). It could be argued that the C-terminal epitope tags interfere with degradation of MurZ-L-FLAG3 and MurA-L-FLAG3 by ClpCP. If this were true, then MurZ-L-FLAG3 or MurA-L-FLAG3 should suppress ΔgpsB. This was found not to be the case in transformation assays (Table S5A, lines 27–28). Last, N-terminal fusion of MurZ or MurA to FLAG or HT resulted in lower protein levels than C-terminal fusions (Fig. S19B–C); nevertheless, the relative amount of remaining HT-MurZ or HT-MurA detected did not change in a ΔclpP mutant (Fig. S19D). We conclude that a ClpP-protease dependent mechanism does not regulate the amounts of MurZ and MurA in S. pneumoniae D39, in contrast to MurA(Lmo) (Rismondo et al., 2017) or MurAA(Bsu) (Kock et al., 2004).
Figure 8. MurZ(Spn) and MurA(Spn) cellular amounts are unchanged in ΔclpP, ΔclpC, ΔclpL, or ΔclpE mutants lacking the ClpP protease or its ATPase subunits.
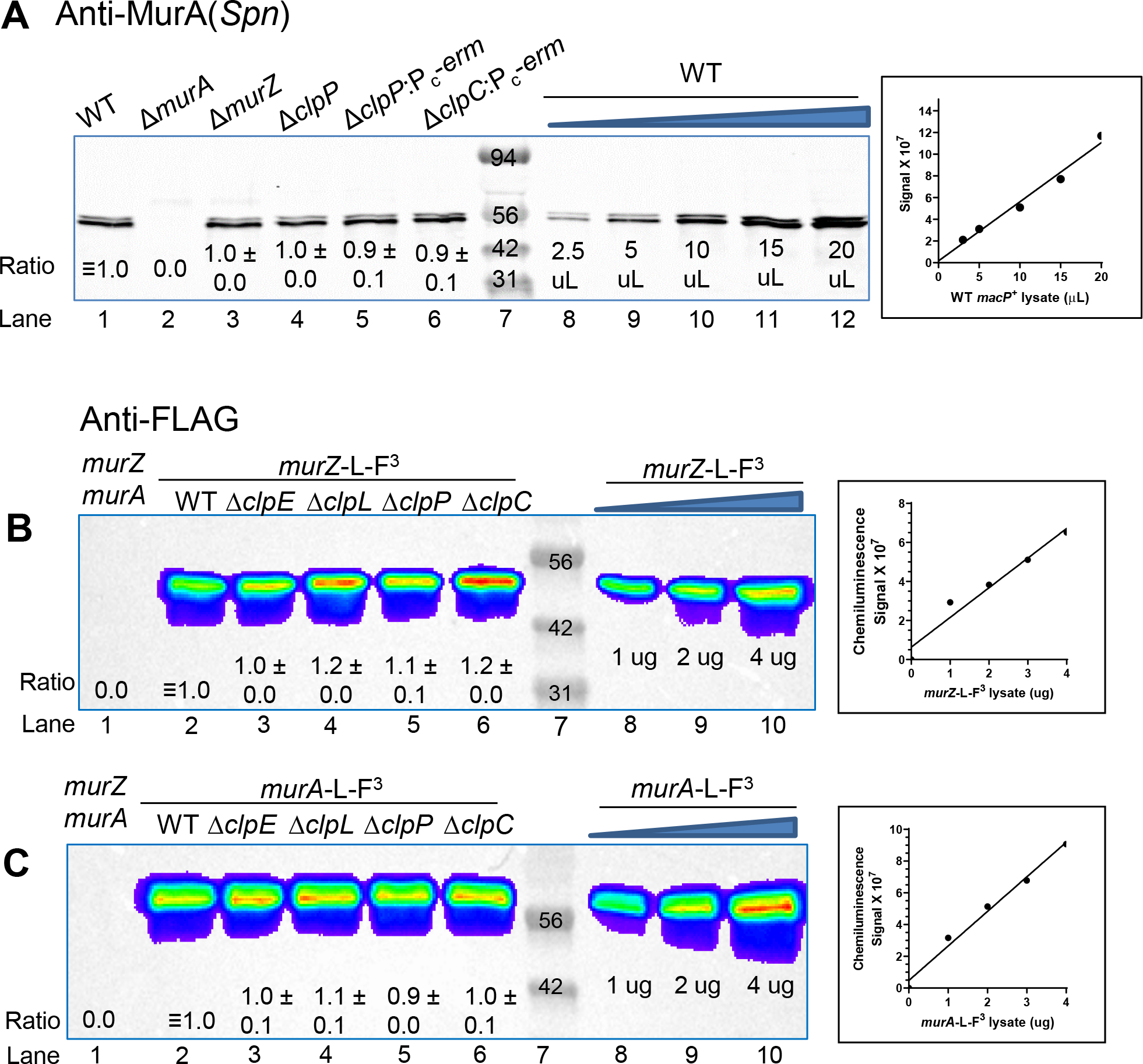
(A) Representative western blot probed with anti-MurA antibody of samples collected after 3.5 h of growth in the BHI broth. Western blotting was performed as described in Experimental procedures using Licor IR Dye800 CW secondary antibody detected with an Azure Biosystem 600. 10 μL (≈4 μg) of protein samples were loaded in each lane. Lane 1, WT (IU1824); lane 2, ΔmurA (IU13538); lane 3, ΔmurZ (IU13536); lane 4, ΔclpP markerless (IU18663); lane 5, ΔclpP::Pc-erm (IU17146); lane 6, ΔclpC::Pc-erm (IU15889). A standard curve was generated by loading 2.5, 5.0, 10, 15, or 20 μL of WT (IU1824) samples (lanes 8–12). Calculated protein amounts (mean ± SEM) relative to WT (IU1824) are based on two independent experiments. Signals obtained with anti-MurA antibody were normalized with total protein stain in each lane using Totalstain Q-NC (Azure Scientific). (B) and (C) Representative western blot using anti-FLAG antibody of samples obtained from WT parent (IU1824), murZ-L-F3 (IU13502), murZ-L-F3 ΔclpE (IU17150), murZ-L-F3 ΔclpL (IU17152), murZ-L-F3 ΔclpP (IU17154), and murZ-L-F3 ΔclpC (IU14082). (B) Western blot of samples obtained from WT parent (IU1824), murA-L-F3 (IU14028), murA-L-F3 ΔclpE (IU17158), murA-L-F3 ΔclpL (IU17160), murA-L-F3 ΔclpP (IU17162), and murA-L-F3 ΔclpC (IU14086). 3 μg of each protein was loaded onto lanes 1–6, and 1, 2, or 4 μg of either murZ-L-F3 (A) or murA-L-F3 (B) lysates were loaded in lanes 8–10 to generate standard curves for quantitation. Plots of μg of lysate obtained from IU13502 or IU14028 loaded vs chemiluminescence signal intensities are shown to the right of the blots. Calculated protein amounts (mean ± SEM) relative to murZ-L-F3 (lane 2) or murA-L-F3 (lane 2) based on two independent experiments are shown.
Finally, quantitative western blotting showed that the cellular amount of MurZ-FLAG3 or MurA-FLAG3 was not changed by ΔmurA or ΔmurZ, respectively (Fig. 5E, lane 4 vs lane 2; lane 7 vs lane 5). Consistent with this result, cellular MurA amount detected by anti-MurA(Spn) was not changed by ΔmurZ (Fig. 8A, lane 3 vs lane 1). These results indicate that in contrast to MurA(Lmo) and MurZ(Lmo) (Rismondo et al., 2017), MurZ(Spn) and MurA(Spn) cellular amounts are not interrelated.
2.8 |. murZ(D280Y) and overexpression of murZ or murA strongly suppress primary morphology phenotypes of StkP(Spn) depletion
ΔstkP mutants have been extensively characterized in R6 and Rx1 laboratory strains that contain murZ(I265V), which suppresses ΔstkP (Table 2, line 7; Fig. S3F and S18C–D) (Beilharz et al., 2012, Echenique et al., 2004, Fleurie et al., 2012, Novakova et al., 2010, Pinas et al., 2018, Saskova et al., 2007, Ulrych et al., 2016, Zucchini et al., 2018). ΔstkP mutants have also been isolated in D39 and TIGR4 strains (Beilharz et al., 2012, Giefing et al., 2010, Herbert et al., 2015, Kant et al., 2023), where chromosomal duplications and other suppressors may have arisen. In our experiments, transformants of a ΔstkP::Pc-erm amplicon into D39 Δcps strains resulted in extremely faint colonies that when re-streaked, produced colonies of variable sizes containing suppressor mutations (Table 3; Fig. S3) (Rued et al., 2017). The faint-colony phenotype of ΔstkP transformants was complemented by ectopic expression of stkP+ (Fig. S3C–D).
To resolve whether ΔstkP is essential in D39 strains, we compared Tn-seq analysis of the unencapsulated WT to a ΔkhpB mutant that suppresses the requirement for stkP (below; Table 2, lines 9–10). Viable insertions in stkP were obtained in the WT strain only in the C-terminal 144 amino acid region that contains the third and fourth extracellular PASTA domains (P3 and P4) (Fig. 10A), indicating that the intracellular, transmembrane domain, and the first two PASTA domains (P1 and P2) are essential for exponential growth in BHI broth in 5% CO2. The first TA insertion occurs in the WT strain at the TAT(Y515) codon, creating a TAA stop codon (Fig. 10A), and no insertions were detected upstream of TTA(L512) codon, indicating that StkP(M1-L512) are essential under the growth conditions tested. Moreover, the same WT Tn-seq insertion profile was obtained for encapsulated D39 strain IU1781 as for unencapsulated strain IU1824 growing in BHI broth or for IU1824 growing in C+Y, pH 6.9 medium in 5% CO2 (Fig. 10A–C; data not shown).
Figure 10. Tn-seq demonstrates the essentiality of StkP(Spn) and GpsB(Spn) is suppressed by ΔkhpB in cells growing exponentially in BHI broth in 5% CO2.
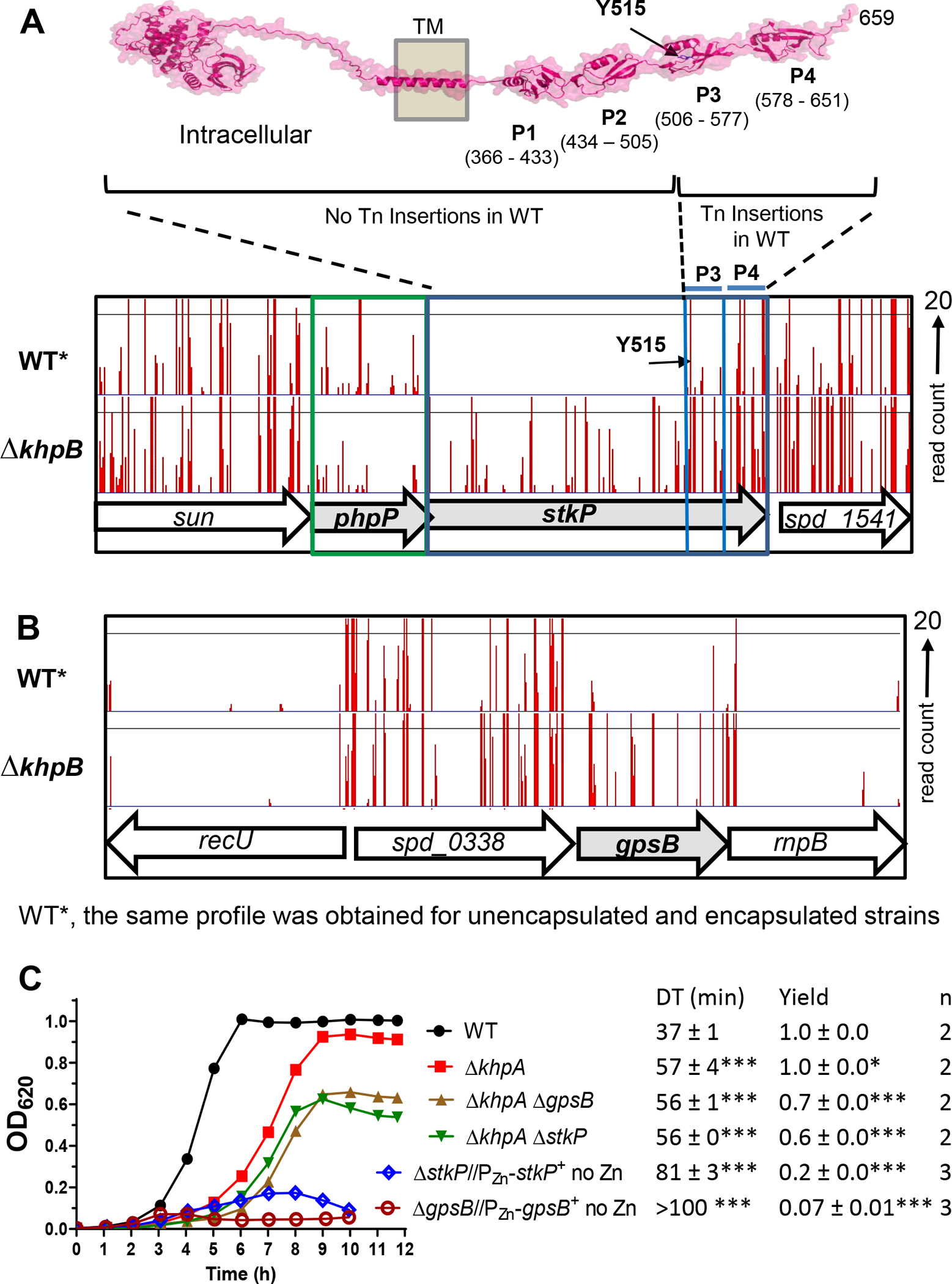
(A) Top: Predicted 3D structure of StkP(Spn) generated using the AlphaFold v2.0 webserver. P1, P2, P3 and P4 with indicated amino acid numbers are predicted extracellular PASTA domains. Bottom: Mini-Mariner Malgellan6 Tn-Seq transposon insertion profile for the genome region covering sun, phpP, stkP, and spd_1541 in the genomes of the unencapsulated WT parent (D39 Δcps rpsL1, IU1824) or ΔkhpB (IU10592) strain growing exponentially in BHI broth in 5% CO2. The same WT Tn-seq insertion profile was obtained for encapsulated D39 strain IU1781 grown in BHI broth or IU1824 grown in C+Y, pH 6.9 medium in 5% CO2 (data not shown). In vitro transposition reactions containing purified genomic DNA, Magellan6 plasmid DNA, and purified MarC9 mariner transposase, transformation, harvesting of transposon-inserted mutants, growth of pooled insertion libraries exponentially in BHI broth or C+Y, pH 6.9 medium, NextSeq 75 high-output sequencing, and analysis were performed as described in Experimental procedures based on (Lamanna et al., 2022). Sortable data for the profile shown are contained in Appendix A, Tabs C and D. Tn-insertions were recovered for the WT strains in the regions encoding P3 and P4, but not in other regions of stkP. The first TA insertion occurs in the WT strain at a TAT (Y515) codon, where the Tn insertion creates a TAA stop codon, while there is no insertion at the upstream TTA (L512) codon, indicating that StkP(M1-L512) is essential for viability. (B) Tn-Seq transposon insertion profiles for the genome region covering recU, spd_0338, gpsB, and rnpB of in the genomes of the WT parent (D39 Δcps rpsL1, IU1824) or ΔkhpB (IU10592) strain. (C) Representative growth curves of the WT parent (IU1824), ΔkhpA (IU9036), ΔkhpA ΔgpsB (IU16196) and ΔkhpA ΔstkP (IU16910) strains. Similar growth results were obtained with ΔkhpB (IU10592), ΔkhpB ΔgpsB (IU12977), and ΔkhpB ΔstkP (IU16912) strains compared to the strains of ΔkhpA background. The growths of merodiploid strains ΔgpsB//PZn-gpsB+ (IU16370) and ΔstkP::Pc-erm//PZn-stkP+ (IU16933) grown under conditions that result in depletion of GpsB or StkP were shown for comparison.
We next performed StkP depletion experiments that minimized suppressor accumulation to determine the primary phenotypes caused by lack of StkP. In these experiments, we constructed a merodiploid strain with a non-polar markerless ΔstkP at its native site and a zinc-regulatable copy of stkP+ at an ectopic site (Fig. 9). Depletion of StkP caused cessation of growth followed by a decrease in OD620 and substantial increases in the length, aspect ratio, and relative volume, but not width (Fig. 9A–C and S20A–B; no Zn inducer). Markerless ΔstkP was nearly completely complemented by an ectopic copy of stkP+ (Fig. 9 and S20; 0.5 mM Zn inducer). Quantitative western blots showed that no StkP was detectable after ≈3–4 h of depletion, and ectopic induction of StkP occurred to ≈50% of the WT level (Fig. S20D). In transformation assays, we used a ΔstkP::Pc-erm allele for selection (Table 2). The morphology of markerless ΔstkP and ΔstkP::Pc-erm cells were slightly different upon StkP deletion (Fig. S20B–C), and unlike markerless ΔstkP, ΔstkP::Pc-erm was not fully complemented back to WT by ectopic stkP expression (Fig. 20A–D). This lack of full complementation, which was not studied further here, may have been caused by retro-polarity of the insertion construct on expression of upstream phpP (phosphatase) or polarity of the constitutive Pc promoter on expression of downstream genes, such as spd_1541 (unknown membrane protein). But together, we conclude that the primary phenotype caused by the absence of StkP is a defect in septum formation in dividing cells, manifested by longer, but not wider, cells compared to WT (Fig. S20B–C).
Importantly, cellular MurZ amount was unchanged by depletion of StkP from its WT level (Fig. 9D and S20D). This result is consistent with the interpretation that StkP does not regulate MurZ amount, but rather modulates MurZ activity indirectly by an alternative mechanism. Cellular MurZ(D280Y) amount was also unchanged by depletion of StkP from its WT level (Fig. 9D), when MurZ(D280Y) suppressed the requirement for StkP (Table 2, line 6; Fig. 9A–C). In addition, murZ(I265V), murZ(E259A), and overexpression of murZ or murA suppressed ΔstkP::Pc-erm in transformation assays (Table 2, lines 4–5 (+Zn inducer) and 7–8; Fig. S3E–J) and in growth and morphology assays (Fig. S21). In contrast, ΔclpP, ΔclpC, ΔclpE, and ΔclpL did not suppress ΔstkP::Pc-erm in transformation assays (Table 2, line 11 and footnote h; Fig. S3B). We conclude that mutations that suppressed ΔgpsB also suppressed ΔstkP. Based on transformant colony size, the suppression of ΔstkP was generally complete compared to the partial suppression of ΔgpsB (Table 2; Fig. S3). The stronger suppression by murZ(D280Y) of ΔstkP compared to ΔgpsB is supported by the growth curves shown in Fig. 4A vs Fig. 9A.
2.9 |. ΔkhpA or ΔkhpB suppress ΔgpsB by increasing MurZ amount
KhpA and KhpB (EloR/Jag) are KH-domain proteins that form an RNA-binding heterodimer (Stamsas et al., 2017, Ulrych et al., 2016, Zheng et al., 2017, Winther et al., 2019). We previously reported that ΔkhpA or ΔkhpB suppressed the lethal phenotypes of Δpbp2b, ΔrodA, ΔmreCD, or ΔrodZ elongasome mutants by increasing FtsA amount (Lamanna et al., 2022, Zheng et al., 2017) (Fig. 11A). We also reported that ΔkhpA or ΔkhpB suppressed the lethal phenotypes of ΔgpsB (Zheng et al., 2017). Tn-seq, transformation, and growth assays confirmed that ΔkhpA or ΔkhpB suppressed ΔgpsB or ΔstkP (Table 2, lines 9–10; Table S5A, lines 30–31; Fig. 10C and S3K–L).
Figure 11. KhpA/B negatively and post-transcriptionally regulates MurZ(Spn), but not MurA(Spn), cellular amounts.
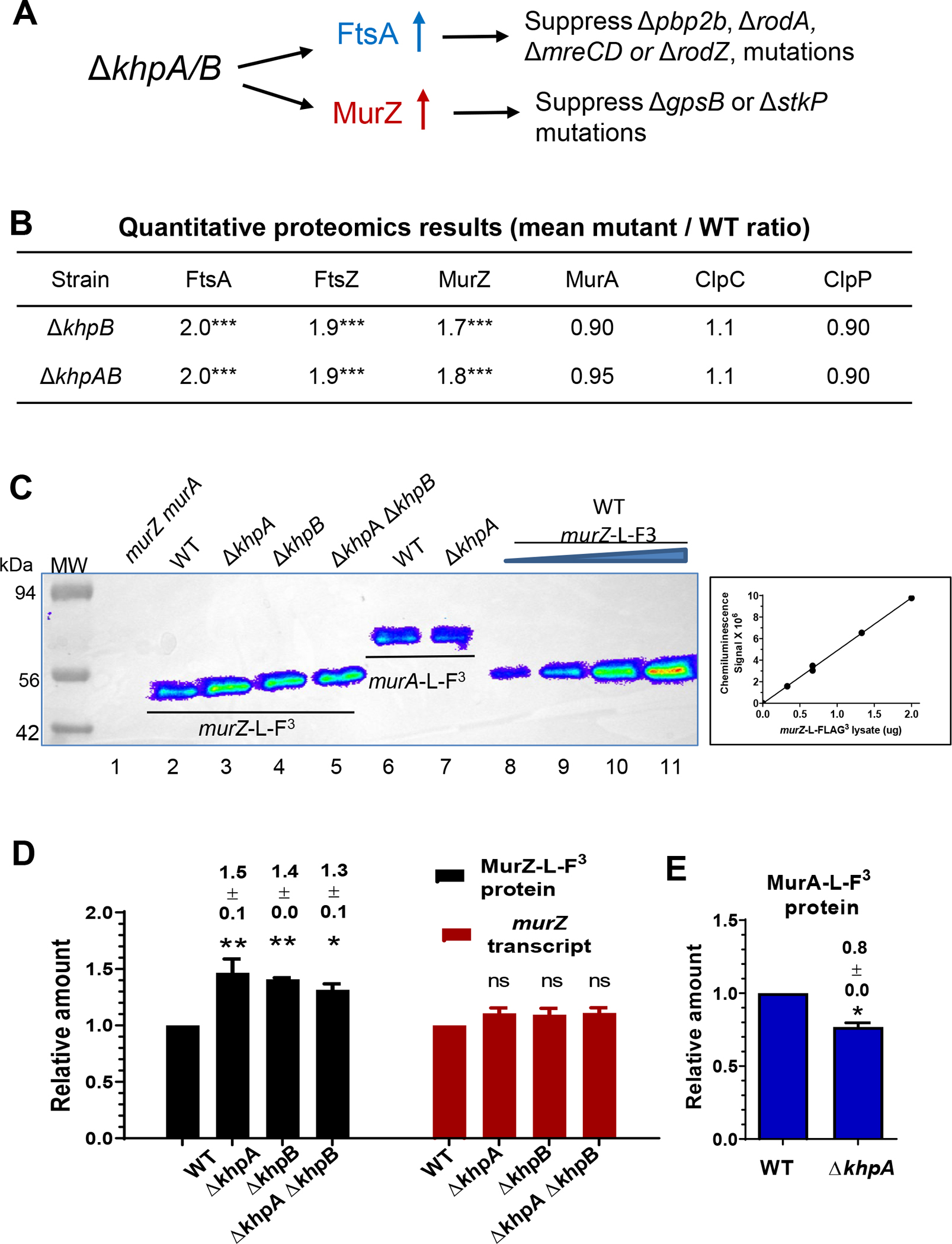
(A) Summary of suppression patterns of ΔgpsB, Δpbp2b, ΔrodA, and ΔmreCD by ΔkhpA/B mutation. The absence of KhpA and/or KhpB increases the cellular amount of FtsA, which bypasses the requirement for essential PBP2b, RodA, RodZ, and MreCD (Lamanna et al., 2022, Zheng et al., 2017). The absence of KhpA/B also moderately increases cellular MurZ amount as shown below, which bypasses the requirement for essential GpsB and StkP as described in the text and Fig. 12. (B) Quantitative proteomic results showing relative amounts of FtsA, FtsZ, MurZ, MurA, ClpC, and ClpP in ΔkhpA ΔkhpB (IU10596) or ΔkhpB (IU10592) strains compared to wild-type (IU1824). *** p < 0.001. Proteomics was performed as described in Experimental procedures, and data are contained in Appendix A, Tab E. (C) Representative Western blots using anti-FLAG antibody to determine the cellular amounts of MurZ-L-FLAG3 and MurA-L-FLAG3 in cells growing exponentially in BHI broth. Lane 1, WT parent (IU1824); lane 2, murZ-L-F3 (IU13502); lane 3, murZ-L-F3 ΔkhpA (IU13545); lane 4, murZ-L-F3 ΔkhpB (IU14014); lane 5, murZ-L-F3 ΔkhpA ΔkhpB (IU14016); lane 6, murA-L-F3 (IU14028); lane 7, murA-L-F3 ΔkhpA (IU14030). 0.67 μg of total protein from each strain were loaded in lanes 1–7. For lanes 8 to 11, 0.33, 0.67, 1.33, and 2 μg, respectively, of murZ-L-FLAG3 (IU13502) lysates were loaded to generate the standard curve at right, which showed proportionality between protein amounts and signal intensities over the range of signal intensities obtained. (C) Relative average (± SEM) of cellular amounts of MurZ-L-F3 or murZ transcripts in mutants compared to WT from 3 independent experiments. P values were obtained relative to WT by one-way ANOVA analysis (Dunnett’s multiple comparison test, GraphPad Prism). * P<0.05; ** P<0.01; ns: not significantly different. (E) Relative average (± SEM) cellular amount of MurA-L-F3 protein in a ΔkhpA mutant compared to WT from 3 independent experiments. P value was obtained relative to WT by one sample t-test (GraphPad Prism). * p<0.05.
Transformation assays strongly implicated MurZ, but not MurA, in ΔkhpA suppression of ΔgpsB. A ΔkhpA single or ΔkhpA ΔmurA mutant could be transformed by ΔgpsB, whereas a ΔkhpA ΔmurZ mutant could not be transformed by ΔgpsB (Table S5, lines 30, 33–34). Consistent with this result, ΔmurA, but not ΔmurZ, could be transformed into a ΔkhpA ΔgpsB suppressed strain (Table S5B, line 5; S5C, line 6). Thus, MurZ is required for ΔkhpA suppression of ΔgpsB.
Consistent with these genetic results, quantitative proteomic analysis detected a ≈1.8-fold (p<0.001) increase in the amount of MurZ, but not MurA, in ΔkhpB or ΔkhpA ΔkhpB mutants compared to WT (Fig. 11B). As controls, the proteomic analysis also confirmed the previous results from quantitative western blotting that FtsA and FtsZ amounts increased ≈2-fold (p< 0.001) in ΔkhpB and ΔkhpA ΔkhpB mutants compared to WT (Fig. 11B; Appendix A, Tab E). Consistent with the proteomic results, quantitative western blotting indicated that MurZ-L-F3 amount increased ≈1.4-fold in ΔkhpA, ΔkhpB, or ΔkhpA ΔkhpB mutants (Fig. 11C–D), whereas MurA-L-F3 amount decreased slightly in a ΔkhpA mutant (Fig. 11E). qRT-PCR showed that the increase in MurZ protein amount was not paralleled by an increase in relative murZ transcript amount in ΔkhpA, ΔkhpB, or ΔkhpA ΔkhpB mutants (Fig. 11D), suggestive of post-transcriptional regulation of murZ expression, including possible indirect effects of a KhpAB-regulated protease or regulator that targets MurZ. Finally, phosphorylation of KhpB by StkP did not play a role in regulating murZ expression in cells growing exponentially in BHI broth, since a khpB(T89A) phosphoablative mutation or khpB(T89D) or khpB(T89E) phosphomimetic mutations did not suppress ΔgpsB (Table S5A, line 32–34). Together, these results suggest that the absence of the KhpAB RNA-binding protein results in a modest (≈2-fold) increase in MurZ(Spn), which is sufficient to suppress ΔgpsB and ΔstkP (Tables 1 and 3, murZ duplications; Fig. 5C, 0.1 mM Zn inducer), but not enough to significantly reduce fosfomycin sensitivity (Fig. 6E).
3 |. DISCUSSION
A large majority (25/32) of suppressors of essential ΔgpsB or ΔstkP in S. pneumoniae D39 contained chromosomal duplications that increase the gene dosage of murZ or murA (Tables 1 and 3). These duplications range from ≈21 to ≈176 genes (Fig. 2 and S1), and suppressors of ΔgpsB also suppress ΔstkP, and vice versa (Table 2). This pattern attests to the extraordinary plasticity of the pneumococcal chromosome, as observed in other studies (Baylay et al., 2015, Cowley et al., 2018, Johnston et al., 2013, Robertson et al., 2003, Zheng et al., 2017). In this case, the dosage of numerous genes adjoining murZ or murA is doubled, and in some cases quadrupled, resulting in overexpression of murZ or murA and many other essential and nonessential gene products with various functions (Fig. 2; Appendix A, Tabs A1 and A2). Smaller duplications containing murZ (3/25) were anchored by direct repeats of degenerate IS elements (Fig. 3 and S1C), and large duplications containing murA (2/25) were anchored by direct repeats of tRNA/rRNA gene clusters (Fig. 2 and S1D). Deletions of duplication junctions were not detected in these two classes of duplications, which likely arose by recombination between the long homologous direct repeats of the degenerate IS elements or tRNA/rRNA genes during chromosome replication (Reams & Roth, 2015).
In contrast, the majority (20/25) of large duplications containing murZ were anchored by inverted repeats of the redundant phtD and phtB genes, which encode histidine triad proteins (Fig. 2–3, and S1B). Inverted repeats of redundant copies of genes lead to inversion of the gene order between the repeated genes (Reams & Roth, 2015), which occurred between phtD and phtB in isolates D39W and D39V of the D39 progenitor strain (Slager et al., 2018). However, the results presented here indicate that even though inverted, phtD and phtB can also anchor large duplications of about ≈150 genes surrounding murZ. To do this, phtD and phtB must contain short direct repeats or other elements that enhance short-junction (SJ) duplication (Reams & Roth, 2015). Indeed, there are small direct repeats of 8 and 9 bp and shorter clusters of directly repeated base pairs within inverted phtD and phtB that could promote SJ duplication.
Moreover, few large duplications of the murZ region (e.g., sup gpsB-8) retained an intact duplication junction (Fig. 2A, S1B, and S2B), whereas most of these duplications contained a short deletion of ≈10 genes that removed the junction region (Fig. 2B, S1B, and S2C). Remodeling of chromosome duplications by junction deletion is common and likely arises by a short-junction mechanism involving short, direct repeats or other elements (Reams & Roth, 2015). PCR experiments supported the idea that the deletion/insertion in sup gpsB-3 arose by a duplication of the phtD-phtB region, an inversion within one of the duplicated regions, and last, a short deletion of the duplication junction (Fig. S2C). In ΔgpsB mutants, junction deletion is correlated with faster growth compared to long duplication without the deletion (Tables 1; Fig. S4A). Together, these results indicate that the region between inverted phtD and phtB can readily be duplicated, providing an extra copy of murZ that suppresses ΔgpsB or ΔstkP. This capacity for duplication also raises the potential that the copy of numerous other genes in this region (Appendix A, Tab A1) can be increased in response to other stress conditions.
Besides these murZ and murA duplications, ΔgpsB was suppressed by five separate mutations in phpP, which encodes the lone Ser/Thr phosphatase in S. pneumoniae, by murZ(D280Y), and by ireB(Q84(STOP), which truncated the homolog of the IreB(Efa) and ReoM(Lmo) by four amino acids (Tables 1 and S5; Fig. S4) (Rued et al., 2017). Most of the mutations that partly suppressed ΔgpsB also almost fully suppressed ΔstkP (Table 2; Fig S3). We did not find suppressor mutations that decrease teichoic acid decoration, analogous to those in L. monocytogenes (Rismondo et al., 2017), because pneumococcal decorations contain GalNAc, which is also an essential component of the teichoic acid core structure (Denapaite et al., 2012). As reported previously, phpP suppressor mutations restore StkP-dependent protein phosphorylation and strongly suppress ΔgpsB, whereas the duplication suppressors do not (Rued et al., 2017). The new phpP and duplication suppressors reported here fit this pattern, and the murZ(D280Y) suppressor also did not restore phosphorylation (Fig. S6–S7). The presence of murZ or murA in all duplication suppressors suggested that overexpression of murZ or murA provides a mechanism for phosphorylation-independent suppression of ΔgpsB and ΔstkP. Consistent with this hypothesis, ectopic overexpression of murZ or murA was sufficient to partially suppress ΔgpsB and strongly suppress ΔstkP (Fig. 4 and S21). Likewise, suppression of ΔgpsB by ΔkhpAB, which lacks a regulator that binds to RNA (Zheng et al., 2017, Winther et al., 2019), depended on MurZ production and was correlated with MurZ, but not MurA overproduction (Table S5; Fig. 11). In addition, results presented here further confirm that ΔkhpAB increases the cellular amount of FtsA in exponentially growing pneumococcal cells (Fig. 11A–B), which leads to suppression of peptidoglycan elongasome mutations (Lamanna et al., 2022, Zheng et al., 2017).
Isolated MurZ(D280Y), constructed MurZ(E259A), and the MurZ(I265V) allele in R6 and Rx1 laboratory strains suppressed ΔgpsB and ΔstkP (Tables 2 and S5). Notably, the amino acid changes in MurZ(D280Y), MurZ(E259A), and MurZ(I265V) are distant from the catalytic region of MurZ (Fig. 7). murZ(D280Y) was expressed at the WT murZ level (Fig. 5C), and ΔmurZ or ΔmurA did not change cellular MurA or MurZ amount, respectively (Fig. 5E). These results indicate a third mechanism of suppression, distinct from loss of PhpP activity or murZ or murA overexpression. Taken together, these results fit and extend our previous model that GpsB is required for StkP-catalyzed protein phosphorylation, as well as for regulation of peptidoglycan synthesis in exponentially growing cells of S. pneumoniae (Rued et al., 2017). These new data tie the requirement for StkP-dependent protein phosphorylation to regulation of MurZ and MurA activity, but not amount (Fig. 5C and 9D). According to this updated model, protein phosphorylation drops in the absence of GpsB, which limits MurZ and MurA activity, without changing their amounts. This limitation can be overcome by decreasing PhpP-mediated protein dephosphorylation, by increasing the cellular amounts of MurZ (by ≈2-fold) or MurA (by ≈2–4-fold) by gene duplication or loss of KhpAB, or by altering the interaction of MurZ and MurA with a phosphorylation-dependent regulatory protein. This interaction could potentially be with a phosphorylated positive regulator that activates MurZ and MurA activity or with an unphosphorylated negative regulator that inhibits MurZ and MurA activity (Fig. 12).
Figure 12. Summary model for regulation of MurZ and MurA enzymatic activities by StkP-mediated phosphorylation in S. pneumoniae D39.
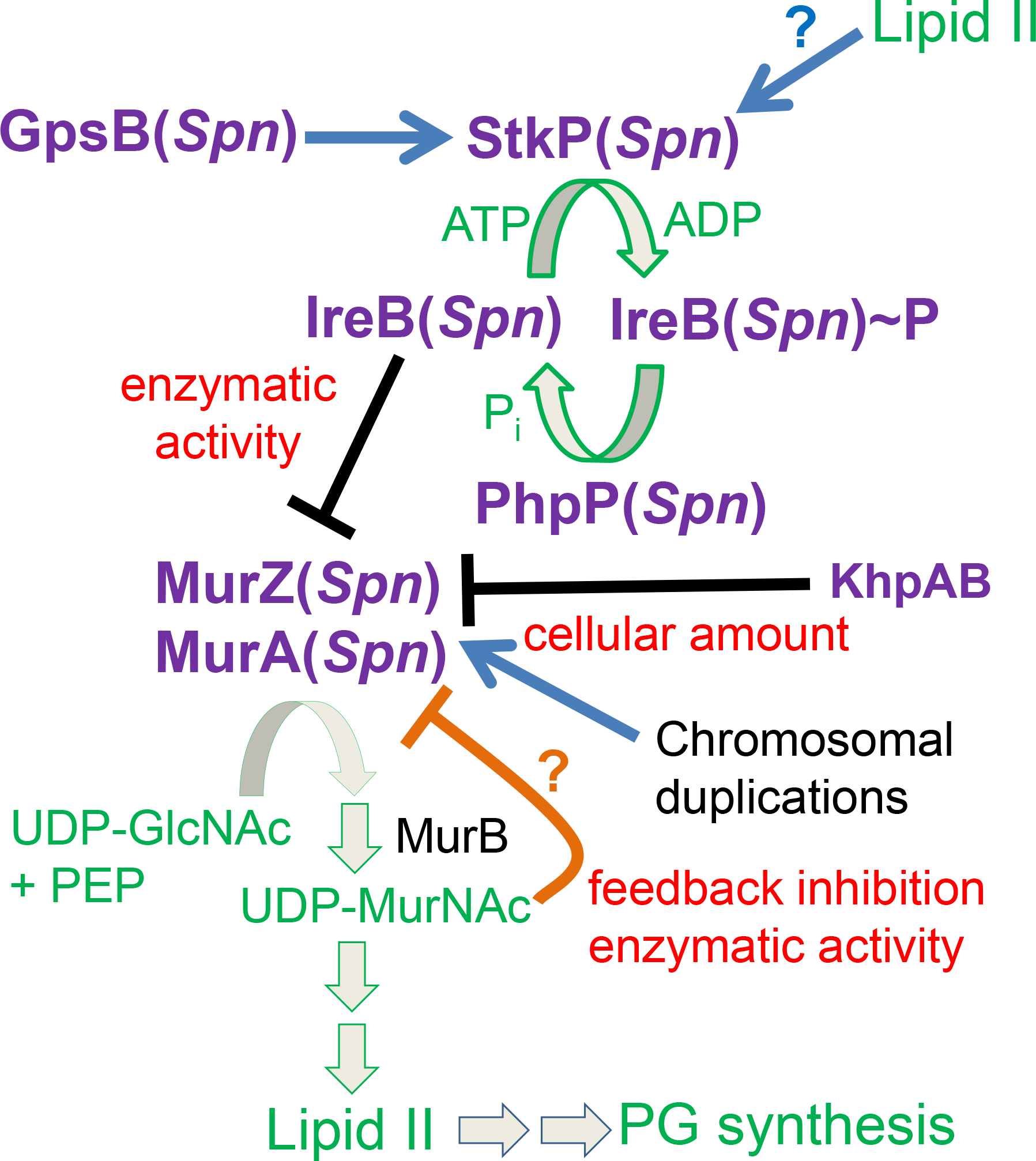
GpsB(Spn) and possibly other ligands, such as Lipid II, stimulate the phosphorylation of a negative regulator of MurZ(Spn) and MurA(Spn) enzymatic activity, but not their cellular amounts, in the first committed step of Lipid II synthesis for PG synthesis. By genetic criteria presented here, the negative regulator is unphosphorylated IreB(Spn). Phosphorylated IreB(Spn)~P does not bind to MurZ(Spn) or MurA(Spn), resulting in full enzymatic activity in pneumococcal cells growing exponentially in rich media. The absence of GpsB(Spn) significantly reduces phosphorylation of IreB(Spn) leading to inhibition of MurZ(Spn) and MurA(Spn) enzymatic activities and no growth. This inhibition can be relieved by inactivation of the cognate PhpP protein phosphatase, which allows residual phosphorylation to IreB(Spn)~P. The absence of the StkP protein kinase and the need for protein phosphorylation in pneumococcal cells growing exponentially in rich media can be suppressed by inactivation or absence of the IreB(Spn) negative regulator, by amino-acid changes in a regulatory domain of MurZ(Spn), which is enzymatically predominant over MurA(Spn), or by overexpression of murZ(Spn) or murA(Spn) in spontaneous chromosomal duplications. Moderate MurZ(Spn) overproduction sufficient to suppress the absence of StkP also occurs in the absence of the KhpAB RNA-binding protein, which also negatively regulates FtsA amount. This pathway provides a positive feedback loop, such that cells growing rapidly in rich media produce Lipid II, which may activate StkP(Spn) to fully phosphorylate IreB(Spn) and maximize MurZ(Spn) and MurA(Spn) enzymatic activities for the production of even more Lipid II for PG synthesis. Evidence for the direct interaction between unphosphorylated IreB(Spn) and MurZ(Spn) will be presented elsewhere (Merrin Joseph, unpublished result). Structures predicted by AlphaFold v2.0 also suggest that MurZ(Spn) and MurA(Spn) enzymatic activity is subject to negative pathway feedback inhibition by binding of UDP-MurNAc (UDP-N-acetylmuramic acid) near the catalytic sites of the enzymes (Mizyed et al., 2005, Schonbrunn et al., 2000). See text for additional details.
The isolation of the ireB(Spn)(Q84(STOP)) suppressor implicates IreB(Spn) as this regulator, and ΔireB suppressed ΔgpsB or ΔstkP, consistent with negative regulation (Table 2, line 13). Recent phosphoproteomic analyses show that MurZ or MurA are not phosphorylated by StkP in exponentially growing S. pneumoniae D39 cells, whereas IreB(Spn) is a prominent phosphorylated protein (Ulrych et al., 2021). According to the negative regulation model, the amino acid changes in MurZ(D280Y), MurZ(E259A), and MurZ(I265V) in Domain 1 of MurZ (Fig. 7) weaken an inhibitory interaction between MurZ and unphosphorylated IreB(Spn), thereby suppressing the absence of GpsB or StkP (Fig. 12). Details of the interaction between MurZ(Spn) and IreB(Spn) will be published elsewhere (Merrin Joseph, unpublished results). Moreover, the observation that suppression of ΔgpsB is partial, except in phpP suppressors, compared to full suppression of ΔstkP (Table 2; Fig. 4 and S21) is consistent with GpsB having additional regulatory roles in peptidoglycan synthesis (Cleverley et al., 2019, Hammond et al., 2022, Minton et al., 2022, Rued et al., 2017), besides activating StkP. Importantly, these suppression patterns indicate that regulation of MurZ and MurA is the sole essential requirement for StkP-dependent protein phosphorylation in unstressed D39 S. pneumoniae cells growing exponentially in BHI broth. Strikingly, this conclusion is similar to that drawn for L. monocytogenes, where the main function of PrkA-mediated signaling is control of MurA stability during standard laboratory growth conditions (Wamp et al., 2022), although the mechanisms of MurZ/A control are different in the two bacteria.
Experiments performed parallel to this study and published recently by Wamp and colleagues revealed similar ΔgpsB suppression phenotypes in L. monocytogenes, with some major differences (Wamp et al., 2020). The conditional, temperature-sensitive ΔgpsB mutation of L. monocytogenes was suppressed by PrpC(Lmo) protein phosphatase mutations, by overproduction of MurA(Lmo) (the homolog of MurA(Spn) (Fig. 1)), and by MurA(Lmo)(S262L), which is at a similar position to MurZ(Spn)(D280Y) in Domain 1 (Fig. 7) (Wamp et al., 2020, Wamp et al., 2022). Importantly, several lines of evidence presented here show that the mechanism of MurA homolog regulation is different in L. monocytogenes and S. pneumoniae. MurA(Lmo) stability is regulated by PrkA Thr phosphorylation of ReoM(Lmo), which is the homolog of IreB(Efa) and IreB(Spn) (Wamp et al., 2020, Wamp et al., 2022, Kelliher et al., 2021). Unphosphorylated ReoM acts with MurZ(Lmo) and ReoY(Lmo) as adaptors for degradation of MurA(Lmo) by the ClpCP(Lmo) protease (Rismondo et al., 2017, Wamp et al., 2022, Wamp et al., 2020). Hence, ReoM(Lmo) phosphorylation to ReoM(Lmo)~P makes MurA(Lmo) available for peptidoglycan synthesis, including by a special RodA3:PBP3 synthase that contributes to the intrinsic cephalosporin resistance of L. monocytogenes (Wamp et al., 2022). This mechanism causes ΔmurZ(Lmo) mutants to accumulate MurA(Lmo), which is essential in L. monocytogenes (Rismondo et al., 2017).
In contrast, MurZ(Spn) and MurA(Spn) share a synthetic lethal relationship (Table S5; Fig. S12) (Du et al., 2000), and a ΔmurZ(Spn) or ΔmurA(Spn) mutation does not result in increased cellular amounts of MurA(Spn) or MurZ(Spn), respectively (Fig. 5E). Several pieces of data in this study demonstrate that MurZ is predominant to MurA in pneumococcal cells. This is the reverse relationship to other Gram-positive bacteria, including L. monocytogenes, E. faecalis, and B. subtilis, where the MurA-family homolog is often essential or predominant to the MurZ-family homolog, which is dispensable and regulatory in the case of MurZ (Lmo) (Fig. 1) (Wamp et al., 2020). The predominance of MurZ(Spn) over MurA(Spn) was indicated by the growth and morphology defects of ΔmurZ mutants in unencapsulated and encapsulated D39 strains grown in BHI broth or C+Y medium (Fig. 6, S10, S13, and S16) and by the increased sensitivity to fosfomycin of ΔmurZ, but not ΔmurA, mutants (Fig. 6E). In addition, lower overproduction of MurZ (≈2-fold) than MurA (≈4-fold) was required to suppress ΔgpsB (Fig. 5), and overproduction of MurZ beyond 2-fold in cells grown in BHI broth led to growth inhibition (Fig. 5–6) that was not observed in C+Y medium (Fig. S10A). This predominance of MurZ compared to MurA in pneumococcal cells is consistent with the greater kinetic efficiency of purified MurZ(Spn) compared to MurA(Spn) reported earlier by Du and colleagues (Du et al., 2000). These combined results show that the relative physiological roles of MurZ(Spn) and MurA(Spn) are substantially different from those of MurA(Lmo) and MurZ(Lmo). Likewise, MurZ(Spn) and its MurZ-family homolog MurAB(Bsu) play very different physiological roles. Remarkably, MurAB(Bsu) was discovered to be required for efficient spore engulfment during sporulation of B. subtilis (Chan et al., 2022). S. pneumoniae does not sporulate.
Other evidence strongly argues for a different mechanism of MurA and MurZ regulation by ReoM/IreB homologs in S. pneumoniae compared to L. monocytogenes, B. subtilis, and E. faecalis. S. pneumoniae lacks homologs of the ReoY accessory factor required for MurA degradation by ClpCP in L. monocytogenes (Wamp et al., 2020, Wamp et al., 2022). In addition, MurA is essential in L. monocytogenes (Rismondo et al., 2017) and likely supplies Lipid II precursor to an additional RodA3:PBPB3 synthase that imparts resistance to cephalosporins (Wamp et al., 2022). Homologs of ReoY and RodA3:PBPB3 are also present in E. faecalis (Wamp et al., 2022), while S. pneumoniae lacks homologs of these proteins. Most importantly, suppression of ΔgpsB(Spn) is not dependent on ClpP(Spn) (Table 2) or ClpP-associated ATPases, including ClpC(Spn) (Table S5), and MurZ(Spn) and MurA(Spn) cellular amounts remain unchanged in a ΔclpP, ΔclpC, ΔclpE, or ΔclpL mutants (Fig. 5E and 8). In addition, cellular MurZ amount was unaffected by depletion of StkP from its WT level (Fig. 9D and S20D). Together, these results support a model in which StkP-mediated protein phosphorylation does not change the amounts of MurZ(Spn) or MurA(Spn), but rather, regulates their enzymatic activities. Interestingly, amino-acid changes in Domain I of MurZ(Spn) (I265V, D280Y, and E259A) and MurA(Lmo) (S262L) likely suppress ΔgpsB by decreasing interactions with unphosphorylated IreB(Spn) and ReoM(Lmo), respectively (Wamp et al., 2022, Wamp et al., 2020). However, amino-acid changes in Domain II of MurZ(Spn) (E190A, E192A, and E195A) did not suppress ΔgpsB (Table S5; Fig. 7), whereas MurA(Lmo)(N197D) did (Wamp et al., 2022), consistent with different mechanisms in S. pneumoniae and L. monocytogenes.
This paper also demonstrates that the StkP Ser/Thr protein kinase is essential, except for its two distal PASTA domains (P3 and P4), in S. pneumoniae D39 progenitor strains growing exponentially in BHI broth or C+Y, pH 6.9 medium (Fig. 10). PASTA domains have been shown to bind Lipid II in Ser/Thr protein kinases of other Gram-positive bacteria (Hardt et al., 2017, Kaur et al., 2019, Sun et al., 2023). We also show that the primary phenotype of StkP depletion is the formation of longer, but not wider, non-growing cells (Fig. 9), indicative of a septation defect that may be triggered by decreased cellular Lipid II amount (Fig. 12). Essentiality of stkP(Spn) has been controversial for two reasons addressed here. First, ΔstkP mutants readily accumulate gene duplications of murZ or murA that compensate for the lack of IreB phosphorylation (Table 3), as do other spontaneous mutations in murZ or ireB (Table 2). Consequently, ΔstkP mutants in D39 strains form unusual-looking, faint colonies with tiny centers containing suppressor mutants (Fig. S3) (Rued et al., 2017). Along this line, it was previously noted that the morphology of D39 ΔstkP mutant cells seemed to change upon passage (Beilharz et al., 2012). Chromosomal duplications do not result in bp changes and are not indicated in standard whole-genome sequencing reports. The recent conclusion that ΔstkP is not essential in D39 likely stems from a duplication of the phtD-phtB region (Fig. 2), as indicated by increased transcript amounts in RNA-seq (Kant et al., 2023). Suppression of ΔstkP by chromosomal duplications complicates interpretations of mutant phenotypes.
Second, the R6- and Rx1-derived laboratory strains in which previous experiments were performed carry a murZ(I265V) mutation (Lanie et al., 2007) that suppresses the requirement for ΔgpsB or ΔstkP (Table 2; Fig. S18). murZ(I265V) changes an amino acid in domain I of MurZ, near the murZ(D280Y) and murZ(E259A) suppressors (Fig. 7). Thus, ΔgpsB and ΔstkP appeared not to be essential in studies using laboratory strains that contain murZ(I265V). Compared to the D39 progenitor strain, these R6- and Rx1-derived laboratory strains contain dozens of additional mutations, besides murZ(I265V) (Cuppone et al., 2021, Lanie et al., 2007, Santoro et al., 2019). Mutational variations may account for why the level of ΔgpsB and ΔstkP suppression by murZ(I265V) varies in different R6 and Rx1 isolates (Beilharz et al., 2012, Rued et al., 2017).
Overall, this study reveals two different evolutionary strategies for the regulation of MurA function in different Gram-positive bacteria. In all cases, MurA function is linked to StkP-dependent protein phosphorylation in exponentially growing cells (Fig. 12) (Wamp et al., 2020, Wamp et al., 2022). Recent biochemical studies by Minton and colleagues and by Doubravová and colleagues demonstrate that purified GpsB directly stimulates the activity of the Ser/Thr protein kinases from E. faecalis and S. pneumoniae, respectively (Doubravová, unpublished result) (Minton et al., 2022). In L. monocytogenes, and likely E. faecalis, unphosphorylated ReoM/IreB interacts with the MurA-family enzyme, along with adaptors MurZ and ReoY to present MurA to ClpCP protease for degradation, thereby inhibiting peptidoglycan synthesis and growth (Wamp et al., 2022, Wamp et al., 2020). In S. pneumoniae, reduced phosphorylation, likely of IreB(Spn) (Merrin Joseph, unpublished results), does not change the cellular amounts of MurZ and MurA, but decreases their enzymatic activity. This inhibition of MurZ and MurA activity by unphosphorylated IreB is likely not complete, leading to the residual slow growth and elongated cell phenotype of ΔstkP strains (Fig. 9A, S3, S20, and S21), compared to the absence of growth and cell death caused by depletion/deletion of MurA and MurZ (Fig. S14). Thus, binding between MurA homologs and unphosphorylated ReoM/IreB appears to be evolutionary conserved, but S. pneumoniae did not evolve or retain the adaptor/ClpCP degradation pathway of MurA regulation present in L. monocytogenes and E. faecalis (Wamp et al., 2022, Wamp et al., 2020). It remains to be determined how the relative function and regulation of MurZ(Spn) and MurA(Spn) change in S. pneumoniae cells subjected to stress conditions, which alters protein phosphorylation by StkP (Ulrych et al., 2021), besides during the exponential growth conditions used here.
Finally, there is precedent for phosphorylated proteins modulating MurA activity directly. In Mycobacterium tuberculosis, MurA(Mtb) is inactive until it binds to phosphorylated CwlM(Mtb) regulator, which increases MurA(Mtb) enzymatic activity by 20–40-fold (Boutte et al., 2016). Homologs of CwlM(Mtb) are absent from S. pneumoniae, L. monocytogenes, E. faecalis, and B. subtilis (data not shown) (Boutte et al., 2016). A MurA(Mtb)(S368P) amino-acid change suppressed the lethal phenotype of a phosphoablative change to CwlM(Mtb)(T374A), which is unable to be phosphorylated and activate WT MurA(Mtb) (Boutte et al., 2016). Notably, MurA(Mtb)(S368P) is on the opposite side of Domain I of MurA near the active site region (Fig. 7), compared to amino-acid changes in MurA homologs, such as MurZ(Spn)(D280Y) and MurA(Lmo)(S262L), that likely decrease binding to unphosphorylated homologs of IreB/ReoM. The separate location of amino-acid changes that result in suppression is consistent with the different mechanisms of positive activation of MurA(Mtb) activity by phosphorylated CwlM(Mtb) (Boutte et al., 2016) compared to negative inhibition of MurZ(Spn) activity by an unphosphorylated regulator, such as IreB(Spn).
4 |. Experimental procedures
4.1 |. Bacterial strains and growth conditions
Strains used in this study are listed in Table S1. Strains were derived from unencapsulated strains IU1824 (D39 Δcps rpsL1) and IU1945 (D39 Δcps), which were derived from the encapsulated serotype-2 D39W progenitor strain IU1690 (Lanie et al., 2007, Slager et al., 2018). Other strains were derived from unencapsulated laboratory strain R6 (Hoskins et al., 2001). A small number of drift mutations that have accumulated in IU1824 and IU1945 compared to IU1690 were determined by whole-genome sequencing and are listed in Appendix A, Tab B. Strains containing antibiotic markers were constructed by transformation of CSP1-induced competent pneumococcal cells with linear DNA amplicons synthesized by overlapping fusion PCR (Ramos-Montanez et al., 2008, Tsui et al., 2016, Tsui et al., 2014). Strains containing markerless alleles in native chromosomal loci were constructed using allele replacement via the Pc-[kan-rpsL+] (Janus cassette) (Sung et al., 2001). Primers used to synthesize different amplicons are listed in Table S1. Bacteria were grown on plates containing trypticase soy agar II (modified; Becton-Dickinson), and 5% (vol/vol) defibrinated sheep blood (TSAII-BA). Plates were incubated at 37°C in an atmosphere of 5% CO2. TSAII-BA plates for selections contained antibiotics at concentrations described previously (Tsui et al., 2016, Tsui et al., 2014). Bacteria were cultured statically in Becton-Dickinson brain heart infusion (BHI) broth at 37°C in an atmosphere of 5% CO2, and growth was monitored by OD620 as described before (Tsui et al., 2016). Mutant constructs were confirmed by PCR and DNA sequencing of chromosomal regions corresponding to the amplicon region used for transformation. Ectopic expression of various genes was achieved with a PZn zinc-inducible promoter in the ectopic bgaA site. 0.2 to 0.5 mM (Zn2+/(1/10)Mn2+) was added to TSAII-BA plates or BHI broth for inducing conditions. Mn2+ was added with Zn2+ to prevent zinc toxicity (Jacobsen et al., 2011, Tsui et al., 2016, Rued et al., 2017).
In all experiments, cells were inoculated from frozen glycerol stocks into BHI broth, serially diluted, and incubated 12–15 h statically at 37°C in an atmosphere of 5% CO2. Parallel cultures were set up for each strain and condition for generation of growth curves and collections of samples for Western blot or microscopy. For culturing merodiploid strains that require Zn2+ for overexpressing murZ, murA, gpsB, or stkP from a Zn-dependent promoter (PZn) placed at an ectopic bgaA site (Tsui et al., 2016), 0.2 to 0.5 mM (Zn2+/(1/10)Mn2+) were added to BHI broth in the overnight cultures. BHI was supplemented with 0.2 mM (Zn2+/(1/10)Mn2+) for overnight growth of IU15860 (ΔgpsB murZ+//PZn-murZ+) and IU16897 (ΔstkP murZ+//PZn-murZ+), with 0.5 mM (Zn2+/(1/10)Mn2+) for growth of IU15862 (ΔgpsB murA+//PZn-murA+) and IU16933 (ΔstkP//PZn-stkP+), and with 0.4 mM (Zn2+/(1/10)Mn2+) for growth of IU16915 (ΔstkP murA+//PZn-murA+). The next day, cultures at OD620 ≈0.1–0.4 were diluted to OD620 ≈0.003 in BHI broth with no additional (Zn2+/(1/10)Mn2+) or the amounts of (Zn2+/(1/10)Mn2+) indicated for each experiment. Doubling time determination was performed by first examining the growth curves on a log scale to determine the time points when growth was in exponential phase. Doubling times were determined with GraphPad Prism exponential growth equation using only data points that exhibit exponential growth. Maximal growth yields were determined by the highest OD620 values obtained within 9 h of growth. Doubling times and maximal growth yields were compared to WT strain with one-way ANOVA analysis (GraphPad Prism, Dunnett’s test). Cultures were sampled for microscopy or western analysis at OD620 ≈0.1–0.2 (early to mid-exponential phase).
4.2 |. Transformation assays
Transformations were performed as previously described (Rued et al., 2017, Tsui et al., 2016). ΔgpsB<>aad9, ΔmurZ::Pc-erm, ΔmurA::Pc-erm, ΔstkP::Pc-erm amplicons, and positive control Δpbp1b::Pc-aad9 or Δpbp1b::Pc-erm amplicon were synthesized by PCR using the primers and templates listed in Table S1, and contain ≈1 kb of flanking chromosomal DNA. All transformation experiments were performed with no added DNA as the negative control, and with respective Δpbp1b amplicons containing the same antibiotic selections as the positive control for competence efficiency and colony size comparison. The volumes of transformation mixture plated (50 to 300 μL) were adjusted to provide ≈150 to 300 colonies with the Δpbp1b amplicons. Transformations with control Δpbp1b amplicons with unencapsulated or encapsulated strains typically yielded >500, or ≈300 colonies per 1 mL of transformation mixture. Transformants were confirmed by PCR reactions. Each transformation experiment was performed 2 or more times. The sizes of colonies indicated in Table 2 were relative to colonies transformed with the same recipient strain with a control Δpbp1b amplicon. For transformations in 0.2 mM or 0.4 mM (Zn2+/(1/10)Mn2+), ZnCl2 and MnSO4 stock solutions were added to transformation mixes and soft agar for plating and spread onto blood plates containing (Zn2+/(1/10)Mn2+) to induce gene expression under control of the PZn zinc-inducible promoter in the ectopic bgaA site (Jacobsen et al., 2011, Rued et al., 2017). For ΔstkP transformation experiments, a volume (≈100 to 150 μL) of transformation mix so that ≈100 colonies appeared on each plate. We ensured that there were similar numbers of the ΔstkP and positive control transformants, and that all the colonies appeared similar on each plate. Pictures of colony morphologies of strains transformed with ΔstkP::Pc-erm and the control Δpbp1b::Pc-erm amplicon were taken from transformation plates after 20 h of incubation at 37°C post-transformation, with illumination source from under the plates.
4.3 |. Whole-genome DNA sequencing
Whole-genome sequencing was used to identify suppressor mutations and to verify the genomes of constructed mutants. Strains listed in Table 1 containing suppressor mutations that allowed growth of a ΔgpsB mutant were isolated as described previously (Rued et al., 2017, Tsui et al., 2016). For strains IU11954, IU11846 and IU11918, genomic DNA preparation, DNA library construction, Illumina MiSeq or NextSeq DNA sequencing, and bioinformatics analyses were performed as described previously (Rued et al., 2017, Tsui et al., 2016). For strains IU16883, E740, IU11912 and IU11456, the NEXTFLEX Rapid DNA-Seq 2.0 kit (catalog number 5188–03) was used in place of the Nextflex Rapid DNA-Seq kit (catalog number 5144–02) used for IU11954, IU11846 and IU11918. Reads were adapter trimmed and quality filtered using Trimmomatic ver. 0.38 (http://www.usadellab.org/cms/?page=trimmomatic), with the cutoff threshold for average base quality score set at 20 over a window of 3 bases. Reads shorter than 20 bases post-trimming were excluded. More than 95% of the sequenced reads passed quality filters. Cleaned reads were mapped to Streptococcus pneumoniae D39 genome sequence (CP000410.2) using bowtie2 version 2.3.2. More than 97.5% of the cleaned reads mapped to the genome. Variants in the libraries with each group against the D39 reference were called and compared using Breseq version 0.35.1 (Deatherage & Barrick, 2014) https://barricklab.org/twiki/bin/view/Lab/ToolsBacterialGenomeResequencing). Several spontaneous drift mutations (Appendix A, Tab B) that do not cause detectable phenotypes in the IU1824 (D39 Δcps rpsL1) and IU1945 (D39 Δcps) unencapsulated parent strains (Table S1) (Lanie et al., 2007) were eliminated manually as new variants. The number of reads of each base was also mapped to the D39 reference genome by using the JBrowse program (Skinner et al., 2009, Westesson et al., 2013) to detect regions containing chromosomal duplications or large deletions (Rued et al., 2017). Sequencing data obtained with sup gpsB-8 (accession # SRR24310104), sup gpsB-9 (SRR24310106), sup gpsB-10 (SRR24310105), sup stkP-1 (SRR24310110), sup stkP-2 (SRR24310109), sup stkP-3 (SRR24310108), and sup stkP-4 (SRR24310107) are deposited in NCBI as a BioProject. Associated SRA metadata are available at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA962082
4.4 |. Cell length and width measurements
Cell lengths and widths of strain growing exponentially in BHI broth were measured as previously described (Tsui et al., 2016). For gpsB+ strains, only ovoid-shape predivisional cells were measured. For analysis that include ΔgpsB strain, all separated cells, including cells that were constricted or narrower at midcell, were measured. Unless indicated in the figure legends, more than 100 cells from at least 2 independent experiments were measured and plotted with box and whiskers plot (5 to 95 percentile whiskers). P values were obtained by one-way ANOVA analysis by using the nonparametric Kruskal-Wallis test in GraphPad Prism program.
4.5 |. RNA preparation and qRT-PCR
RNA preparation and qRT-PCR were performed as previously described (Tsui et al., 2016, Zheng et al., 2017). Primers used for qRT-PCR are listed in Table S1.
4.6 |. Quantitative western blotting
Cell lysate preparations using SEDS lysis buffer (0.1% deoxycholate (vol/vol), 150 mM NaCl, 0.2% SDS (vol/vol), 15 mM EDTA pH 8.0) and western blotting was performed as previously described (Cleverley et al., 2019, Lamanna et al., 2022). Briefly, bacteria were grown exponentially in 5 ml BHI broth to an OD620 ≈ 0.15–0.2. Frozen pellets collected from 1.8 mL of cultures at OD620 ≈0.16 were suspended in 80 μL of SEDS lysis buffer. The volume of SEDS buffer was adjusted proportional to the OD620 values. Protein assays were performed with the lysates and the μg amounts of protein lysates loaded on each lane were listed in the figure legends of each blot. The sources of antibodies used for western blotting are as below. Primary antibodies used are anti-HaloTag monoclonal antibody (Promega, G921A, 1:1000), and polyclonal rabbit antibodies: anti- FLAG (Sigma, F7425, 1:2000); anti-HA (Invitrogen, 71–5500, 1:1000); α-pThr antibody (Cell Signaling, #9381) (Rued et al., 2017), anti-StkP (1:10,000) (Beilharz et al., 2012, Rued et al., 2017), and anti-MurA(Spn) (1:7,000) (see below for antibody information). Secondary antibodies used were anti-mouse IgG conjugated to horseradish peroxidase (Invitrogen, SZ-100, 1:3300), anti-rabbit IgG conjugated to horseradish peroxidase (GE healthcare NA93AV, 1:10,000), or Licor IR Dye800 CW goat anti-rabbit (926–32,211, 1:14,000). Chemiluminescence signals obtained with secondary HRP-conjugated antibodies were detected using IVIS imaging system (Fig. 5, 8, 11, S6, S7, S8, S15), or Azure biosystem 600 (Fig. S19C) as described previously (Lamanna et al., 2022). IR signals obtained with Licor IR Dye800 CW secondary antibody was detected with Azure biosystem 600 (Fig. 9, S19A, S19B S20).
The relative expression levels of murZ and murA were measured with murZ-L-FLAG3 or murA-L-FLAG3 expressed from their native chromosomal locus (Fig. 5, 8, 9, 11, S8, S15). To ensure linearity of western signal values vs protein amounts, a range of protein samples of IU13502 (murZ-L-FLAG3) or IU14028 (murA-L-FLAG3) were loaded on the same gel as the experimental samples to provide a standard curve of μg protein amounts versus signal intensities. These plots were performed for each western quantitation experiment (see Fig. 5, 8, 9, 11, S8, S15, S20), and were used to calculate the relative protein amounts in each sample lane by extrapolation. To avoid intensity values beyond the linear range, lower μg amounts of proteins from the induced murZ-L-FLAG3 or murA-L-FLAG3 overexpression strains (IU13772 or IU15983, respectively) were loaded per lane in order for the intensity signals of these samples to stay within the linear range (Fig. S8). For Fig. 9D, 6 μL (≈2 μg) of protein samples were loaded in each sample lane for comparison. A standard curve was generated by loading 3, 6, 9 or 12 μL of IU13502 (murZ-L-FLAG3) samples (lanes not shown). For Fig. S20D, 10 μL (≈3 μg) of protein samples were loaded in each sample lane for comparison. A standard curve was generated by loading 5, 7.5, 10 or 15 μL of WT samples. Signal intensities obtained with the anti-Flag or anti-StkP antibody were normalized with total protein stain in each lane using Totalstain Q-NC reagent from Azure biosystems in these two experiments.
4.7 |. 2D-immunofluorescence microscopy (2D-IFM)
2D-IFM was performed to examine the localization pattern of MurZ and MurA as described in (Land et al., 2013) using a primary anti-FLAG antibody (Sigma, F7425, 1:100 dilution) and secondary Alexa Fluor 488 goat anti-rabbit IgG (Life Technologies, Z1034, 1:100 dilution) with strains IU13502 (murZ-L-FLAG3) and IU14028 (murA-L-FLAG3). Nucleoid DNA was labeled with mounting media SlowFade gold antifade reagent with DAPI (Life Technologies, S36936).
4.8 |. Antibiotic disk-diffusion assay
Strains were inoculated in 3 mL BHI broth from frozen glycerol stocks and grown at 37°C until early exponential phase (OD620 ≈0.09–0.15). Cells were then diluted to OD620 ≈0.009 in 1 mL BHI, and 50 μL of diluted culture was then mixed into 3 mL nutrient-broth soft agar [0.8% (w/v) nutrient broth and 0.7% (w/v) Bacto Agar (Difco)] and poured onto TSAII-BA plates. After 15 min, antibiotic Sensi-Disc™ (Becton Dickinson Pty Ltd., Fosfomycin; cat# 231709, Cefotaxime; cat# 231606, Tetracycline; cat# 230998, penicillin; cat# 230918, Cefoperazone; cat# 231612 (data not shown)), were placed at the middle of plates that were incubated 37°C for 16 h prior to measurement of zone of inhibition. Images of plates were taken using the Azure imaging system, and diameters of the zones of inhibition were measured using the Java program AntibiogramJ (Alonso et al., 2017).
4.9 |. 3D structure and residue alignment
The MurZ structure from S. pneumoniae D39 was generated using AlphaFold v2.0 (Jumper et al., 2021) on the Carbonate Research supercomputer at Indiana University, and images were generated using PyMOL (Schrödinger, LLC). For amino acid sequence comparisons, amino acid sequences of MurZ and MurA from S. pneumoniae D39 and MurA from E. coli K12 were obtained from the protein PubMed database (https://www.ncbi.nlm.nih.gov/protein/) and aligned using the Clustal Omega web server to determine locations of the catalytic Cys, and other residues demonstrated to be important for MurA function in other bacterial species.
4.10 |. Proteomic analysis
Triplicate 30-mL cultures of wild-type (IU1824), ΔkhpA ΔkhpB (IU10596) and ΔkhpB (IU10592) strains were grown in BHI broth to an OD620 ≈0.1–0.15. Cultures were then collected by centrifugation at 16,000 x g for 5 min at 4°C. Cell pellets were resuspended in 1 mL of cold PBS, centrifuged at 16,100 x g for 5 min at 4°C, and resuspended in 1 mL of lysis buffer (8 M Urea, 100 mM ammonium bicarbonate (pH 7.8), 0.5 % sodium deoxycholate, and protease inhibitor (1 mini tablet (Pierce™ A32955) per 10 mL). Resuspended cells were transferred to lysing matrix B tubes and lysed in a FastPrep homogenizer (MP Biomedicals) at a rate of 6 m/s for 40 s three times. Samples were centrifuged at 16,100 x g for 5 min at 4°C. 700 μL supernatant was transferred to a new 1.5-mL tube and concentrated using Amicon Ultra 1 mL 10K membrane filters (Millipore, catalog number: UFC501096) to ≈40 μL by centrifuging at room temperature at 14,000 x g for ≈45 min. Samples were washed in the spin filter by adding 200 μL of wash buffer (8 M Urea, 100 mM ammonium bicarbonate (pH 7.8), 0.1% sodium deoxycholate) in the spin filter and centrifuged at room temperature at 14,000 x g for ≈1 h until ≈40 μL remains in the column. 3 x volumes (≈120 μL) of 100 mM ammonium bicarbonate were added to the samples to produce a final urea concentration of 2M. Samples were concentrated by centrifugation in the spin column to ≈40 μL, which were transferred to fresh 1.5 mL microfuge tubes. Spin filters were rinsed twice with 200 μL of 25 mM ammonium bicarbonate and added to the sample tubes. The protein concentration was quantified by a Bio-Rad DC protein assay (catalog number: 5000111) using BSA in 0.2M urea and 25 mM ammonium bicarbonate (pH 7.8) as standards. Typical protein yields were 270 to 430 μg per 30-mL culture. 100 μg of protein were dried in SpeedVac concentrator for ≈15 h followed by in-solution protein digestion.
Samples were denatured in 8 M urea, 100 mM ammonium bicarbonate solution, then incubated for 45 min at 56°C with 10 mM dithiothreitol (DTT) to reduce cysteine residues. The free cysteine residue side chains were then alkylated with 40 mM iodoacetamide for 1 h in the dark at room temperature. The solution was diluted to 1 M urea and 1:100 (wt/wt) ratio of trypsin was added and the samples were digested at 37°C for 16 h. Peptides were desalted by Zip-tip.
LC-MS/MS Analysis was performed by injection of peptides into an Easy-nLC HPLC system coupled to an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific, Bremen, Germany). Peptide samples were loaded onto a 75 μm x 2 cm Acclaim PepMap 100 C18 trap column (Thermo Scientific) in 0.1% formic acid. The peptides were separated using a 75 μm x 25 cm Acclaim PepMap C18 analytical column using an acetonitrile-based gradient (Solvent A: 0% acetonitrile, 0.1% formic acid; Solvent B: 80% acetonitrile, 0.1% formic acid) at a flow rate of 300 nL/min. Peptides were separated using a 120 min gradient. The initial solvent was 2% B. This was ramped to 4% B over 30 sec. The gradient then ramped up to 32% B over 114 min, then up to 100% B over 30 sec and held there for the remaining five min. The electrospray ionization was carried out with a nanoESI source at a 260°C capillary temperature and 1.8 kV spray voltage. The mass spectrometer was operated in data-dependent acquisition mode with mass range 400 to 1600 m/z. The precursor ions were selected for tandem mass (MS/MS) analysis in the Orbitrap with 3 sec cycle time using HCD at 35% collision energy. Intensity threshold was set at 1e4. The dynamic exclusion was set with a repeat count of 1 and exclusion duration of 30 s.
The resulting data were searched against a Streptococcus pneumoniae D39 database (Uniprot UP000001452 with 1,915 entries, downloaded on 02/2020) using MaxQuant version 1.6. Carbamidomethylation of cysteine residues was set as a fixed modification. Protein N-terminal acetylation and oxidation of methionine were set as variable modifications. Trypsin digestion specificity with two missed cleavage was allowed. The first and main search peptide tolerances were set to 20 and 4.5 ppm, respectively.
Perseus Version 2.0.3.0 was used for statistical analysis of the data (Aguilan et al., 2020, Turapov et al., 2018). The fractional abundance of each protein is calculated relative to the total lysate (protein area/total lysate area) and used to estimate the fold-change. Statistical data analyzation was done in Perseus by applying the following workflow: (a) log2 data transformation and imputation based on normal distribution to eliminate division by zero, (b) removing proteins only identified in one of replicates, (c) calculating the mean of replicates, and (d) performing a t-test to determine proteins that were statistically different between wild-type and mutant. Average values reported in this study were calculated based on 5 replicates of wild-type and 3 replicates of mutant strains. Pairwise Pearson correlation coefficients among replicates of the same strain were ≥ 0.986 for all three strains. Data from the proteomic analysis is contained in Appendix A, Tab E.
4.11 |. Tn-seq transposon library generation and insertion sequencing
Tn-seq transposon library generation and insertion sequencing of WT D39 Δcps rpsL1 (IU1824) and isogenic ΔkhpB (IU10592) are as reported in (Lamanna et al., 2022). Tn-seq primary data for the region between sun (spd_1544) and spd_1541, which are upstream and downstream of phpP(spd_1543)-stkP(spd_1542), respectively, are contained in Appendix A, Tabs C and D, including run summaries, number of reads per TA site in each gene, and count ratios for each gene in the indicated mutants compared with WT. P values for comparisons of the number of reads per TA site in each gene were calculated by the nonparametric Mann-Whitney test using GraphPad Prism (9.2.0).
4.12 |. Purification of MurA(Spn) and generation of anti-MurA(Spn) polyclonal antibody
E. coli strains for protein expression were derived from strain BL21(DE3) (catalog number C2527H; NEB). Standard methods were used for transformation of E. coli and isolation of plasmid DNA (Sambrook et al., 1989). The plasmid for expressing the recombinant MurA(Spn) was prepared by first amplifying murA from S. pneumoniae D39 genomic DNA using primer pair AJP435/AJP436 (Table S1). pHis-parallel1 plasmid was amplified from BL21(DE3) pHis-parallel1 (IU6814) (Rued et al., 2019) using primer pair AJP431/AJP432 (Table S1). PCR products were ligated by Gibson assembly, and ligated plasmid was then transformed into E. coli α-select gold efficiency (Bioline, Bio-85027). Protein expression plasmid was obtained using the Qiaprep Spin Miniprep Kit (Qiagen, 27106) and transformed into BL21(DE3) (NEB, C2527H) for protein expression. MurA protein was purified as previously described (Du et al., 2000) with the following modifications. Cell cultures were grown at 37°C in LB broth supplemented with 100 μg/mL ampicillin to an OD600 = 0.6–0.8 before IPTG induction (0.5 mM). Cultures were harvested by centrifugation at 8,000 x g for 10 min at 4°C, lysed in a French Press at 18,000 psi, and centrifuged at 12,000 x g for 90 min at 4°C. Supernates were filtered through a 0.45 μm filter and loaded onto a 5 mL HisTrap HP column (Cytiva), from which bound protein was eluted with 1.0 to 250 mM imidazole gradient. The His6-tag was cleaved off of His6-MurA with His-tagged TEV protease (1 mg TEV for 20 mg of protein) (Rued et al., 2019) at 4°C during overnight dialysis against a buffer of 100 mM Tris-HCl, pH 8, 300 mM NaCl, 10 mM imidazole, 1.0 mM DTT. The proteolysis reaction products were passed over a 5 mL HisTrap HP column (Cytiva) to remove TEV and uncleaved protein. MurA that did not bind to the second Ni-NTA column was concentrated and loaded onto a Superdex G200 column (GE Healthcare) equilibrated with 100 mM Tris-HCl, pH 8, 100 mM NaCl for size exclusion chromatography. Column fractions were analyzed for purity by SDS-PAGE, concentrated, and small aliquots were fast-frozen in liquid nitrogen for storage at −80°C. The mass of purified MurA protein was verified using a Synapt G2-S mass spectrometer. Purified MurA was sent to Thermo Fisher Scientific for custom polyclonal antibody generation in rabbits.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ziyun April Ye and Bobby Walker for technical assistance in strain construction, Jonathon Trinidad and Aleš Ulrych for advice on interpretation of proteomic data, Doug Rusch and Ram Podicheti for bioinformatic assistance of whole-genome sequences, Ulf Gerth and Chris Kristich for polyclonal antibodies against MurAA(Bsu) and MurAA(Efa), respectively, and Kevin Bruce and other members of the Winkler lab for discussions about this work. This work was supported by NIH Grant R35GM131767 (to MEW), grants 18–07748S (to LD) and 19–03269S (to PB) from the Czech Science Foundation, grant LTAUSA18112 (to LD and PB) from the Ministry of Education, Youth, and Sports of the Czech Republic, and by institutional research funds from the CIBIO Department of the University of Trento (to OM). Work done on the Carbonate Research supercomputer was supported in part by the Lilly Endowment, Inc., through its support of the Indiana University Pervasive Technology Institute.
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interests.
ETHICS STATEMENT
This work did not include animal or human experimental subjects requiring formal approval or consent. Antibodies used in this study are available commercially, were published previously, or were prepared by companies approved by the Indiana University Bloomington Institutional Animal Care and Use Committee.
DATA AVAILABILITY STATEMENT
All data that support the findings of this study are reported with indicated statistical analyses and numbers of biological repeats in the main text, Supplemental Information, and Appendix A. Primary data from experiments are available from the corresponding authors upon reasonable request.
REFERENCES
- Aguilan JT, Kulej K, and Sidoli S (2020) Guide for protein fold change and p-value calculation for non-experts in proteomics. Mol Omics 16: 573–582. [DOI] [PubMed] [Google Scholar]
- Alonso CA, Dominguez C, Heras J, Mata E, Pascual V, Torres C, and Zarazaga M (2017) Antibiogramj: A tool for analysing images from disk diffusion tests. Comput Methods Programs Biomed 143: 159–169. [DOI] [PubMed] [Google Scholar]
- Baylay AJ, Ivens A, and Piddock LJ (2015) A novel gene amplification causes upregulation of the PatAB ABC transporter and fluoroquinolone resistance in Streptococcus pneumoniae. Antimicrob Agents Chemother 59: 3098–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilharz K, Novakova L, Fadda D, Branny P, Massidda O, and Veening JW (2012) Control of cell division in Streptococcus pneumoniae by the conserved Ser/Thr protein kinase StkP. Proc Natl Acad Sci U S A 109: E905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake KL, O’Neill AJ, Mengin-Lecreulx D, Henderson PJ, Bostock JM, Dunsmore CJ, Simmons KJ, Fishwick CW, Leeds JA, and Chopra I (2009) The nature of Staphylococcus aureus MurA and MurZ and approaches for detection of peptidoglycan biosynthesis inhibitors. Mol Microbiol 72: 335–343. [DOI] [PubMed] [Google Scholar]
- Booth S, and Lewis RJ (2019) Structural basis for the coordination of cell division with the synthesis of the bacterial cell envelope. Protein Sci 28: 2042–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutte CC, Baer CE, Papavinasasundaram K, Liu W, Chase MR, Meniche X, Fortune SM, Sassetti CM, Ioerger TR, and Rubin EJ (2016) A cytoplasmic peptidoglycan amidase homologue controls mycobacterial cell wall synthesis. Elife 5: e14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs NS, Bruce KE, Naskar S, Winkler ME, and Roper DI (2021) The pneumococcal divisome: dynamic control of Streptococcus pneumoniae cell division. Front Microbiol 12: e737396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown ED, Vivas EI, Walsh CT, and Kolter R (1995) MurA (MurZ), the enzyme that catalyzes the first committed step in peptidoglycan biosynthesis, is essential in Escherichia coli. J Bacteriol 177: 4194–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K, and Bradford PA (2016) β-Lactams and β-lactamase inhibitors: an overview. Cold Spring Harbor Perspect Med 6: a025247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC, (2019) Antibiotic resistance threats in the United States, 2019. Atlanta, GA;U.S. Department of Health and Human Services, CDC. Available from: http://www.cdc.gov/drugresistance/Biggest-Threats.html. [Google Scholar]
- Chan H, Taib N, Gilmore MC, Mohamed AMT, Hanna K, Luhur J, Nguyen H, Hafiz E, Cava F, Gribaldo S, Rudner D, and Rodrigues CDA (2022) Genetic screens identify additional genes implicated in envelope remodeling during the engulfment stage of Bacillus subtilis sporulation. mBio 13: e0173222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessen D, Emmins R, Hamoen LW, Daniel RA, Errington J, and Edwards DH (2008) Control of the cell elongation-division cycle by shuttling of PBP1 protein in Bacillus subtilis. Mol Microbiol 68: 1029–1046. [DOI] [PubMed] [Google Scholar]
- Cleverley RM, Rutter ZJ, Rismondo J, Corona F, Tsui HT, Alatawi FA, Daniel RA, Halbedel S, Massidda O, Winkler ME, and Lewis RJ (2019) The cell cycle regulator GpsB functions as cytosolic adaptor for multiple cell wall enzymes. Nat Commun 10: 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley LA, Petersen FC, Junges R, Jimson DJM, Morrison DA, and Hanage WP (2018) Evolution via recombination: cell-to-cell contact facilitates larger recombination events in Streptococcus pneumoniae. PLoS Genet 14: e1007410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MJ, Loman N, Bogaert D, and O’Grady J (2020) Co-infections: potentially lethal and unexplored in COVID-19. The Lancet Microbe 1: e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuppone AM, Colombini L, Fox V, Pinzauti D, Santoro F, Pozzi G, and Iannelli F (2021) Complete genome sequence of Streptococcus pneumoniae strain Rx1, a Hex mismatch repair-deficient standard transformation recipient. Microbiol Resour Announc 10: e0079921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deatherage DE, and Barrick JE (2014) Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol 1151: 165–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denapaite D, Brückner R, Hakenbeck R, and Vollmer W (2012) Biosynthesis of teichoic acids in Streptococcus pneumoniae and closely related species: lessons from genomes. Microb Drug Resist 18: 344–358. [DOI] [PubMed] [Google Scholar]
- Dias R, Felix D, Canica M, and Trombe MC (2009) The highly conserved serine threonine kinase StkP of Streptococcus pneumoniae contributes to penicillin susceptibility independently from genes encoding penicillin-binding proteins. BMC Microbiol 9: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Brown JR, Sylvester DR, Huang J, Chalker AF, So CY, Holmes DJ, Payne DJ, and Wallis NG (2000) Two active forms of UDP-N-acetylglucosamine enolpyruvyl transferase in gram-positive bacteria. J Bacteriol 182: 4146–4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echenique J, Kadioglu A, Romao S, Andrew PW, and Trombe MC (2004) Protein serine/threonine kinase StkP positively controls virulence and competence in Streptococcus pneumoniae. Infect Immun 72: 2434–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan AJF, Errington J, and Vollmer W (2020) Regulation of peptidoglycan synthesis and remodelling. Nat Rev Microbiol 18: 446–460. [DOI] [PubMed] [Google Scholar]
- Eswara PJ, Brzozowski RS, Viola MG, Graham G, Spanoudis C, Trebino C, Jha J, Aubee JI, Thompson KM, Camberg JL, and Ramamurthi KS (2018) An essential Staphylococcus aureus cell division protein directly regulates FtsZ dynamics. Elife 7: 38856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AK, Manuse S, Flores-Kim J, Garcia PS, Mercy C, Grangeasse C, Bernhardt TG, and Rudner DZ (2018) Phosphorylation-dependent activation of the cell wall synthase PBP2a in Streptococcus pneumoniae by MacP. Proc Natl Acad Sci U S A 115: 2812–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleurie A, Cluzel C, Guiral S, Freton C, Galisson F, Zanella-Cleon I, Di Guilmi AM, and Grangeasse C (2012) Mutational dissection of the S/T-kinase StkP reveals crucial roles in cell division of Streptococcus pneumoniae. Mol Microbiol 83: 746–758. [DOI] [PubMed] [Google Scholar]
- Fleurie A, Manuse S, Zhao C, Campo N, Cluzel C, Lavergne JP, Freton C, Combet C, Guiral S, Soufi B, Macek B, Kuru E, VanNieuwenhze MS, Brun YV, Di Guilmi AM, Claverys JP, Galinier A, and Grangeasse C (2014) Interplay of the serine/threonine-kinase StkP and the paralogs DivIVA and GpsB in pneumococcal cell elongation and division. PLoS Genet 10: e1004275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garde S, Chodisetti PK, and Reddy M (2021) Peptidoglycan: structure, synthesis, and regulation. EcoSal Plus 9: ESP-0010–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giefing C, Jelencsics KE, Gelbmann D, Senn BM, and Nagy E (2010) The pneumococcal eukaryotic-type serine/threonine protein kinase StkP co-localizes with the cell division apparatus and interacts with FtsZ in vitro. Microbiol 156: 1697–1707. [DOI] [PubMed] [Google Scholar]
- Grangeasse C (2016) Rewiring the pneumococcal cell cycle with serine/threonine- and tyrosine-kinases. Trends Microbiol 24: 713–724. [DOI] [PubMed] [Google Scholar]
- Halbedel S, and Lewis RJ (2019) Structural basis for interaction of DivIVA/GpsB proteins with their ligands. Mol Microbiol 111: 1404–1415. [DOI] [PubMed] [Google Scholar]
- Hammond LR, Sacco MD, Khan SJ, Spanoudis C, Hough-Neidig A, Chen Y, and Eswara PJ (2022) GpsB coordinates cell division and cell surface decoration by wall teichoic acids in Staphylococcus aureus. Microbiol Spect 10: e01413–01422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond LR, White ML, and Eswara PJ (2019) ¡vIVA la DivIVA! J Bacteriol 201: e00245–00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardt P, Engels I, Rausch M, Gajdiss M, Ulm H, Sass P, Ohlsen K, Sahl HG, Bierbaum G, Schneider T, and Grein F (2017) The cell wall precursor lipid II acts as a molecular signal for the Ser/Thr kinase PknB of Staphylococcus aureus. Int J Med Microbiol 307: 1–10. [DOI] [PubMed] [Google Scholar]
- Herbert JA, Mitchell AM, and Mitchell TJ (2015) A serine-threonine kinase (StkP) regulates expression of the pneumococcal pilus and modulates bacterial adherence to human epithelial and endothelial cells in vitro. Plos One 10: e0127212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschfeld C, Gomez-Mejia A, Bartel J, Hentschker C, Rohde M, Maass S, Hammerschmidt S, and Becher D (2019) Proteomic investigation uncovers potential targets and target sites of pneumococcal serine-threonine kinase StkP and phosphatase PhpP. Front Microbiol 10: 3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holeckova N, Doubravova L, Massidda O, Molle V, Buriankova K, Benada O, Kofronova O, Ulrych A, and Branny P (2014) LocZ is a new cell division protein involved in proper septum placement in Streptococcus pneumoniae. mBio 6: e01700–01714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins J, Alborn WE Jr., Arnold J, Blaszczak LC, Burgett S, DeHoff BS, Estrem ST, Fritz L, Fu DJ, Fuller W, Geringer C, Gilmour R, Glass JS, Khoja H, Kraft AR, Lagace RE, LeBlanc DJ, Lee LN, Lefkowitz EJ, Lu J, Matsushima P, McAhren SM, McHenney M, McLeaster K, Mundy CW, Nicas TI, Norris FH, O’Gara M, Peery RB, Robertson GT, Rockey P, Sun PM, Winkler ME, Yang Y, Young-Bellido M, Zhao G, Zook CA, Baltz RH, Jaskunas SR, Rosteck PR Jr., Skatrud PL, and Glass JI (2001) Genome of the bacterium Streptococcus pneumoniae strain R6. J Bacteriol 183: 5709–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummels KR, Berry SP, Li Z, Taguchi A, Min JK, Walker S, Marks DS, and Bernhardt TG (2023) Coordination of bacterial cell wall and outer membrane biosynthesis. Nature 615: 300–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SG, Zhang F, Chindemi P, Junop MS, and Berti PJ (2009) Evidence of kinetic control of ligand binding and staged product release in MurA (enolpyruvyl UDP-GlcNAc synthase)-catalyzed reactions. Biochem 48: 11715–11723. [DOI] [PubMed] [Google Scholar]
- Jacobsen FE, Kazmierczak KM, Lisher JP, Winkler ME, and Giedroc DP (2011) Interplay between manganese and zinc homeostasis in the human pathogen Streptococcus pneumoniae. Metallomics 3: 38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston C, Caymaris S, Zomer A, Bootsma HJ, Prudhomme M, Granadel C, Hermans PW, Polard P, Martin B, and Claverys JP (2013) Natural genetic transformation generates a population of merodiploids in Streptococcus pneumoniae. PLoS Genet 9: e1003819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, and Hassabis D (2021) Applying and improving AlphaFold at CASP14. Proteins 89: 1711–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kant S, Sun Y, and Pancholi V (2023) StkP- and PhpP-mediated posttranslational modifications modulate the S. pneumoniae metabolism, polysaccharide capsule, and virulence. Infect Immun: e0029622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur P, Rausch M, Malakar B, Watson U, Damle NP, Chawla Y, Srinivasan S, Sharma K, Schneider T, Jhingan GD, Saini D, Mohanty D, Grein F, and Nandicoori VK (2019) LipidII interaction with specific residues of Mycobacterium tuberculosis PknB extracytoplasmic domain governs its optimal activation. Nat Commun 10: 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedar GC, Brown-Driver V, Reyes DR, Hilgers MT, Stidham MA, Shaw KJ, Finn J, and Haselbeck RJ (2008) Comparison of the essential cellular functions of the two murA genes of Bacillus anthracis. Antimicrob Agents Chemother 52: 2009–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelliher JL, Grunenwald CM, Abrahams RR, Daanen ME, Lew CI, Rose WE, and Sauer JD (2021) PASTA kinase-dependent control of peptidoglycan synthesis via ReoM is required for cell wall stress responses, cytosolic survival, and virulence in Listeria monocytogenes. PLoS Pathog 17: e1009881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kock H, Gerth U, and Hecker M (2004) MurAA, catalysing the first committed step in peptidoglycan biosynthesis, is a target of Clp-dependent proteolysis in Bacillus subtilis. Mol Microbiol 51: 1087–1102. [DOI] [PubMed] [Google Scholar]
- Kumar S, Mollo A, Kahne D, and Ruiz N (2022) The bacterial cell wall: from Lipid II flipping to polymerization. Chem Rev 122: 8884–8910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamanna MM, Manzoor I, Joseph M, Ye ZA, Benedet M, Zanardi A, Ren Z, Wang X, Massidda O, Tsui HT, and Winkler ME (2022) Roles of RodZ and class A PBP1b in the assembly and regulation of the peripheral peptidoglycan elongasome in ovoid-shaped cells of Streptococcus pneumoniae D39. Mol Microbiol 118: 336–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land AD, Tsui HC, Kocaoglu O, Vella SA, Shaw SL, Keen SK, Sham LT, Carlson EE, and Winkler ME (2013) Requirement of essential Pbp2x and GpsB for septal ring closure in Streptococcus pneumoniae D39. Mol Microbiol 90: 939–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanie JA, Ng WL, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ, Tettelin H, Glass JI, and Winkler ME (2007) Genome sequence of Avery’s virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J Bacteriol 189: 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bourgeois P, Bugarel M, Campo N, Daveran-Mingot ML, Labonté J, Lanfranchi D, Lautier T, Pagès C, and Ritzenthaler P (2007) The unconventional Xer recombination machinery of Streptococci/Lactococci. PLoS Genet 3: e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuse S, Fleurie A, Zucchini L, Lesterlin C, and Grangeasse C (2016) Role of eukaryotic-like serine/threonine kinases in bacterial cell division and morphogenesis. FEMS Microbiol Rev 40: 41–56. [DOI] [PubMed] [Google Scholar]
- Martin JE, Edmonds KA, Bruce KE, Campanello GC, Eijkelkamp BA, Brazel EB, McDevitt CA, Winkler ME, and Giedroc DP (2017) The zinc efflux activator SczA protects Streptococcus pneumoniae serotype 2 D39 from intracellular zinc toxicity. Mol Microbiol 104: 636–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascari CA, Djorić D, Little JL, and Kristich CJ (2022) Use of an interspecies chimeric receptor for inducible gene expression reveals that metabolic flux through the peptidoglycan biosynthesis pathway is an important driver of cephalosporin resistance in Enterococcus faecalis. J Bacteriol 204: e0060221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massidda O, Novakova L, and Vollmer W (2013) From models to pathogens: how much have we learned about Streptococcus pneumoniae cell division? Environ Microbiol 15: 3133–3157. [DOI] [PubMed] [Google Scholar]
- Minton NE, Djorić D, Little J, and Kristich CJ (2022) Gpsb promotes pasta kinase signaling and cephalosporin resistance in Enterococcus faecalis. J Bacteriol 204: e0030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizyed S, Oddone A, Byczynski B, Hughes DW, and Berti PJ (2005) UDP-N-acetylmuramic acid (UDP-MurNAc) is a potent inhibitor of MurA (enolpyruvyl-UDP-GlcNAc synthase). Biochem 44: 4011–4017. [DOI] [PubMed] [Google Scholar]
- Mobegi FM, Cremers AJ, de Jonge MI, Bentley SD, van Hijum SA, and Zomer A (2017) Deciphering the distance to antibiotic resistance for the pneumococcus using genome sequencing data. Sci Rep 7: 42808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakova L, Bezouskova S, Pompach P, Spidlova P, Saskova L, Weiser J, and Branny P (2010) Identification of multiple substrates of the StkP Ser/Thr protein kinase in Streptococcus pneumoniae. J Bacteriol 192: 3629–3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakova L, Saskova L, Pallova P, Janecek J, Novotna J, Ulrych A, Echenique J, Trombe MC, and Branny P (2005) Characterization of a eukaryotic type serine/threonine protein kinase and protein phosphatase of Streptococcus pneumoniae and identification of kinase substrates. FEBS J 272: 1243–1254. [DOI] [PubMed] [Google Scholar]
- Perez AJ, Cesbron Y, Shaw SL, Bazan Villicana J, Tsui HT, Boersma MJ, Ye ZA, Tovpeko Y, Dekker C, Holden S, and Winkler ME (2019) Movement dynamics of divisome proteins and PBP2x:FtsW in cells of Streptococcus pneumoniae. Proc Natl Acad Sci U S A 116: 3211–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinas GE, Reinoso-Vizcaino NM, Yandar Barahona NY, Cortes PR, Duran R, Badapanda C, Rathore A, Bichara DR, Cian MB, Olivero NB, Perez DR, and Echenique J (2018) Crosstalk between the serine/threonine kinase StkP and the response regulator ComE controls the stress response and intracellular survival of Streptococcus pneumoniae. PLoS Pathog 14: e1007118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompeo F, Foulquier E, Serrano B, Grangeasse C, and Galinier A (2015) Phosphorylation of the cell division protein GpsB regulates PrkC kinase activity through a negative feedback loop in Bacillus subtilis. Mol Microbiol 97: 139–150. [DOI] [PubMed] [Google Scholar]
- Ramos-Montanez S, Tsui HC, Wayne KJ, Morris JL, Peters LE, Zhang F, Kazmierczak KM, Sham LT, and Winkler ME (2008) Polymorphism and regulation of the spxB (pyruvate oxidase) virulence factor gene by a CBS-HotDog domain protein (SpxR) in serotype 2 Streptococcus pneumoniae. Mol Microbiol 67: 729–746. [DOI] [PubMed] [Google Scholar]
- Reams AB, and Roth JR (2015) Mechanisms of gene duplication and amplification. Cold Spring Harb Perspect Biol 7: a016592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rismondo J, Bender JK, and Halbedel S (2017) Suppressor Mutations Linking gpsB with the first committed step of peptidoglycan biosynthesis in Listeria monocytogenes. J Bacteriol 199: e00393–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rismondo J, Cleverley RM, Lane HV, Grosshennig S, Steglich A, Moller L, Mannala GK, Hain T, Lewis RJ, and Halbedel S (2016) Structure of the bacterial cell division determinant GpsB and its interaction with penicillin-binding proteins. Mol Microbiol 99: 978–998. [DOI] [PubMed] [Google Scholar]
- Robertson GT, Ng WL, Gilmour R, and Winkler ME (2003) Essentiality of clpX, but not clpP, clpL, clpC, or clpE, in Streptococcus pneumoniae R6. J Bacteriol 185: 2961–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohs PDA, and Bernhardt TG (2021) Growth and Division of the peptidoglycan matrix. Ann Rev Microbiol 75: 315–336. [DOI] [PubMed] [Google Scholar]
- Rued BE, Zheng JJ, Mura A, Tsui HT, Boersma MJ, Mazny JL, Corona F, Perez AJ, Fadda D, Doubravova L, Buriankova K, Branny P, Massidda O, and Winkler ME (2017) Suppression and synthetic-lethal genetic relationships of ΔgpsB mutations indicate that GpsB mediates protein phosphorylation and penicillin-binding protein interactions in Streptococcus pneumoniae D39. Mol Microbiol 103: 931–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco MD, Hammond LR, Noor RE, Bhattacharya D, Madsen JJ, Zhang X, Butler SG, Kemp MT, Jaskolka-Brown AC, Khan SJ, Gelis I, Eswara PJ, and Chen Y (2022) Staphylococcus aureus FtsZ and PBP4 bind to the conformationally dynamic N-terminal domain of GpsB. bioRxiv: 2022.2010.2025.513704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachla AJ, and Helmann JD (2021) Resource sharing between central metabolism and cell envelope synthesis. Curr Opin Microbiol 60: 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samland AK, Etezady-Esfarjani T, Amrhein N, and Macheroux P (2001) Asparagine 23 and aspartate 305 are essential residues in the active site of UDP-N-acetylglucosamine enolpyruvyl transferase from Enterobacter cloacae. Biochem 40: 1550–1559. [DOI] [PubMed] [Google Scholar]
- Santoro F, Iannelli F, and Pozzi G (2019) Genomics and genetics of Streptococcus pneumoniae. Microbiol Spectr 7: GPP3–0025-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saskova L, Novakova L, Basler M, and Branny P (2007) Eukaryotic-type serine/threonine protein kinase StkP is a global regulator of gene expression in Streptococcus pneumoniae. J Bacteriol 189: 4168–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonbrunn E, Eschenburg S, Luger K, Kabsch W, and Amrhein N (2000) Structural basis for the interaction of the fluorescence probe 8-anilino-1-naphthalene sulfonate (ANS) with the antibiotic target MurA. Proc Natl Acad Sci USA 97: 6345–6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender V, Hentrich K, and Henriques-Normark B (2021) Virus-induced changes of the respiratory tract environment promote secondary infections with Streptococcus pneumoniae. Front Cell Infect Microbiol 11: 643326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarzynski T, Mistry A, Wonacott A, Hutchinson SE, Kelly VA, and Duncan K (1996) Structure of UDP-N-acetylglucosamine enolpyruvyl transferase, an enzyme essential for the synthesis of bacterial peptidoglycan, complexed with substrate UDP-N-acetylglucosamine and the drug fosfomycin. Structure 4: 1465–1474. [DOI] [PubMed] [Google Scholar]
- Skinner ME, Uzilov AV, Stein LD, Mungall CJ, and Holmes IH (2009) JBrowse: a next-generation genome browser. Genome Res 19: 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slager J, Aprianto R, and Veening JW (2018) Deep genome annotation of the opportunistic human pathogen Streptococcus pneumoniae D39. Nuc Acids Res 46: 9971–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamsas GA, Straume D, Ruud Winther A, Kjos M, Frantzen CA, and Havarstein LS (2017) Identification of EloR (Spr1851) as a regulator of cell elongation in Streptococcus pneumoniae. Mol Microbiol 105: 954–967. [DOI] [PubMed] [Google Scholar]
- Sun X, Ge F, Xiao CL, Yin XF, Ge R, Zhang LH, and He QY (2010) Phosphoproteomic analysis reveals the multiple roles of phosphorylation in pathogenic bacterium Streptococcus pneumoniae. J Proteome Res 9: 275–282. [DOI] [PubMed] [Google Scholar]
- Sun Y, Hürlimann S, and Garner E (2023) Growth rate is modulated by monitoring cell wall precursors in Bacillus subtilis. Nat Microbiol 8: 469–480. [DOI] [PubMed] [Google Scholar]
- Sung CK, Li H, Claverys JP, and Morrison DA (2001) An rpsL cassette, Janus, for gene replacement through negative selection in Streptococcus pneumoniae. Appl Environ Microbiol 67: 5190–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi A, Page JE, Tsui HT, Winkler ME, and Walker S (2021) Biochemical reconstitution defines new functions for membrane-bound glycosidases in assembly of the bacterial cell wall. Proc Natl Acad Sci U S A 118: e2103740118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares JR, de Souza RF, Meira GL, and Gueiros-Filho FJ (2008) Cytological characterization of YpsB, a novel component of the Bacillus subtilis divisome. Bacteriol 190: 7096–7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, Durkin AS, Gwinn M, Kolonay JF, Nelson WC, Peterson JD, Umayam LA, White O, Salzberg SL, Lewis MR, Radune D, Holtzapple E, Khouri H, Wolf AM, Utterback TR, Hansen CL, McDonald LA, Feldblyum TV, Angiuoli S, Dickinson T, Hickey EK, Holt IE, Loftus BJ, Yang F, Smith HO, Venter JC, Dougherty BA, Morrison DA, Hollingshead SK, and Fraser CM (2001) Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293: 498–506. [DOI] [PubMed] [Google Scholar]
- Tsui HC, Zheng JJ, Magallon AN, Ryan JD, Yunck R, Rued BE, Bernhardt TG, and Winkler ME (2016) Suppression of a deletion mutation in the gene encoding essential PBP2b reveals a new lytic transglycosylase involved in peripheral peptidoglycan synthesis in Streptococcus pneumoniae D39. Mol Microbiol 100: 1039–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui HT, Boersma MJ, Vella SA, Kocaoglu O, Kuru E, Peceny JK, Carlson EE, VanNieuwenhze MS, Brun YV, Shaw SL, and Winkler ME (2014) Pbp2x localizes separately from Pbp2b and other peptidoglycan synthesis proteins during later stages of cell division of Streptococcus pneumoniae D39. Mol Microbiol 94: 21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turapov O, Forti F, Kadhim B, Ghisotti D, Sassine J, Straatman-Iwanowska A, Bottrill AR, Moynihan PJ, Wallis R, Barthe P, Cohen-Gonsaud M, Ajuh P, Vollmer W, and Mukamolova GV (2018) Two faces of CwlM, an essential PknB substrate, in Mycobacterium tuberculosis. Cell Rep 25: 57–67 e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrych A, Fabrik I, Kupčík R, Vajrychová M, Doubravová L, and Branny P (2021) Cell wall stress stimulates the activity of the protein kinase StkP of Streptococcus pneumoniae, leading to multiple phosphorylation. J Mol Biol 433: 167319. [DOI] [PubMed] [Google Scholar]
- Ulrych A, Holeckova N, Goldova J, Doubravova L, Benada O, Kofronova O, Halada P, and Branny P (2016) Characterization of pneumococcal Ser/Thr protein phosphatase phpP mutant and identification of a novel PhpP substrate, putative RNA binding protein Jag. BMC Microbiol 16: 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesic D, and Kristich CJ (2012) MurAA is required for intrinsic cephalosporin resistance of Enterococcus faecalis. Antimicrob Agents Chemother 56: 2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer W, Massidda O, and Tomasz A (2019) The cell wall of Streptococcus pneumoniae. Microbiol Spectr 7: GPP3–0018-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamp S, Rothe P, Stern D, Holland G, Döhling J, and Halbedel S (2022) MurA escape mutations uncouple peptidoglycan biosynthesis from PrkA signaling. PLoS Pathog 18: e1010406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamp S, Rutter ZJ, Rismondo J, Jennings CE, Möller L, Lewis RJ, and Halbedel S (2020) PrkA controls peptidoglycan biosynthesis through the essential phosphorylation of ReoM. Elife 9: e56048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser JN, Ferreira DM, and Paton JC (2018) Streptococcus pneumoniae: transmission, colonization and invasion. Nat Rev Microbiol 16: 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westesson O, Skinner M, and Holmes I (2013) Visualizing next-generation sequencing data with JBrowse. Brief Bioinform 14: 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2017) List of bacteria for which new antibiotics are urgently needed. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/news/releases/2017/bacteria-antibiotics-needed/en. [Google Scholar]
- Winther AR, Kjos M, Stamsas GA, Havarstein LS, and Straume D (2019) Prevention of EloR/KhpA heterodimerization by introduction of site-specific amino acid substitutions renders the essential elongasome protein PBP2b redundant in Streptococcus pneumoniae. Sci Rep 9: 3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JJ, Perez AJ, Tsui HT, Massidda O, and Winkler ME (2017) Absence of the KhpA and KhpB (JAG/EloR) RNA-binding proteins suppresses the requirement for PBP2b by overproduction of FtsA in Streptococcus pneumoniae D39. Mol Microbiol 106: 793–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Cai Y, Liu Y, An H, Deng K, Ashraf MA, Zou L, and Wang J (2022) Breaking down the cell wall: Still an attractive antibacterial strategy. Front Microbiol 13: 952633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchini L, Mercy C, Garcia PS, Cluzel C, Gueguen-Chaignon V, Galisson F, Freton C, Guiral S, Brochier-Armanet C, Gouet P, and Grangeasse C (2018) PASTA repeats of the protein kinase StkP interconnect cell constriction and separation of Streptococcus pneumoniae. Nat Microbiol 3: 197–209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data that support the findings of this study are reported with indicated statistical analyses and numbers of biological repeats in the main text, Supplemental Information, and Appendix A. Primary data from experiments are available from the corresponding authors upon reasonable request.