

Abstract
Background:
Differentiating neuromyelitis optica spectrum disorder (NMOSD) from its mimics is crucial to avoid misdiagnosis, especially in the absence of aquaporin-4-IgG. While multiple sclerosis (MS) and myelin oligodendrocyte glycoprotein-IgG associated disease (MOGAD) represent major and well-defined differential diagnoses, non-demyelinating NMOSD mimics remain poorly characterized.
Methods:
We conducted a systematic review on Pubmed/Medline to identify reports of patients with non-demyelinating disorders that mimicked or were misdiagnosed as NMOSD. Three novel cases seen at the authors’ institutions were also included. The characteristics of NMOSD mimics were analysed and red flags associated with misdiagnosis identified.
Results:
A total of sixty-eight patients were included; 35 (52%) were female. Median age at symptoms onset was 44 years (range, 1–78). Fifty-six (82%) patients did not fulfil the 2015 NMOSD diagnostic criteria. The clinical syndromes misinterpreted for NMOSD were myelopathy (41%), myelopathy+optic neuropathy (41%), optic neuropathy (6%), or other (12%). Alternative etiologies included genetic/metabolic disorders, neoplasms, infections, vascular disorders, spondylosis, and other immune-mediated disorders. Common red flags associated with misdiagnosis were lack of CSF pleocytosis (57%), lack of response to immunotherapy (55%), progressive disease course (54%), and lack of MRI gadolinium enhancement (31%). Aquaporin-4-IgG positivity was detected in five patients by enzyme-linked immunosorbent assay (n=2), cell-based assay (n=2: serum, 1; CSF, 1), and non-specified assay (n=1).
Conclusions:
The spectrum of NMOSD mimics is broad. Misdiagnosis frequently results from incorrect application of diagnostic criteria, in patients with multiple identifiable red flags. False aquaporin-4-IgG positivity, generally from nonspecific testing assays, may rarely contribute to misdiagnosis.
Keywords: Neuromyelitis Optica spectrum disorder, differential diagnosis, mimickers, misdiagnosis, false positivity
Introduction
Aquaporin-4 (AQP4)-IgG-positive neuromyelitis optica spectrum disorder (NMOSD) is a distinct demyelinating disorder of the central nervous system (CNS) characterized by acute/subacute attacks of optic neuritis (typically recurrent and/or bilateral), transverse myelitis (typically extending over ≥ 3 continuous vertebral body segments on MRI) and, less commonly, brain or brainstem dysfunction (e.g., area postrema syndrome). Identification of AQP4-IgG in serum is highly specific and confirms the diagnosis of NMOSD, with important therapeutic implications. In some patients with suspected NMOSD, however, AQP4-IgG may be negative or AQP4-IgG serostatus may be unknown (referred to hereafter as “seronegative”), making the diagnosis challenging. Diagnostic criteria for seronegative NMOSD were published in 2015, which allow for the diagnosis of NMOSD to be made without AQP4-IgG if strict clinical-MRI requirements are met and alternative disease etiologies are excluded.1 In this context, awareness of alternative disorders that can mimic NMOSD becomes fundamental to avoid inappropriate treatment, morbidity associate with diagnostic delays, and unnecessary healthcare costs.
Previous studies have suggested that patients with NMOSD are often misdiagnosed as having MS or other disorders,2–5 but only few studies, other than case reports, have investigated what disorders are mistaken for NMOSD.6 While multiple sclerosis (MS) and MOG-IgG-associate disease (MOGAD) represent the main differential diagnosis for NMOSD in clinical practice, and several studies have highlighted clinical-MRI differences between these three entities,7–9 non-demyelinating disorders that can mimic NMOSD are rare and likely under recognized, which may lead to them being incorrectly diagnosed as seronegative NMOSD. We herein describe the main characteristics of non-demyelinating mimics of NMOSD reported in the literature, and highlight red flags that may help prevent misdiagnosis.
Materials and methods
A PubMed/Medline search was performed on January 9, 2023 using the following query: “(“NMO” or “NMOSD” or “neuromyelitis” or “devic” or “aquaporin*” or “aqp*”) AND (“mimic*” or “resembl*” or “masquerad*” or “manifest*” or “differential” or “misdiagnos*” or “misinterpret*”)”. Relevant articles were cross-referenced, and missing studies incorporated. Two investigators (PZ, AD) reviewed all studies for inclusion and discrepancies were resolved by consensus with a third neurologist (ES). We included patients with alternative neurologic disorders reported as NMOSD mimics or misdiagnosed as NMOSD during the diagnostic work-up. We excluded cases of MS or MOGAD misdiagnosed as NMOSD because these represent entities that are already well-established as differential diagnoses in clinical practice.5, 7–9 We also excluded cases with uncertain or poorly defined final diagnoses, as well as articles predating the discovery of AQP4-IgG.10 Three additional cases seen at the authors institutions were also included.
Data collection and application of NMOSD diagnostic criteria
Relevant demographic and clinical data were independently extracted for included cases by three authors (PZ, AD, SC) in an electronic database. For each included case, the 2015 diagnostic criteria for seronegative NMOSD were applied, and potential red flags associated with NMOSD misdiagnosis identified. The following red flags potentially associated with NMOSD misdiagnosis were pre-defined by the authors by consensus on the base of existing literature and clinical experience:1 (1) Lack of CSF pleocytosis, (2) lack of response to immunosuppressive therapy, (3) progressive disease course over months, (4) lack of gadolinium enhancement on brain or spinal cord MRI, (5) presence of cerebrospinal fluid (CSF)-restricted oligoclonal bands (≥ 2), and (6) fever. Collected data were reviewed by ES in case of disagreement or uncertainty.
Results
A total of 2404 articles underwent screening for eligibility. Sixty-five patients were included from 51 articles (Supplementary Figure and Table) in addition to three patients seen at the authors’ institutions, for a total of 68 patients. Fifty-six (82%) patients did not fulfil diagnostic criteria for NMOSD at the time of misdiagnosis. The median age was 44 years (range, 1–78), 35 (52%) were female, and 11 (17%) were children. Representative MRI images and a brief description of the three additional cases reported by the authors are shown in Figures 1–3 and associated figure legends.
Figure 1 -. Spinal cord infarction misdiagnosed as seronegative NMOSD.
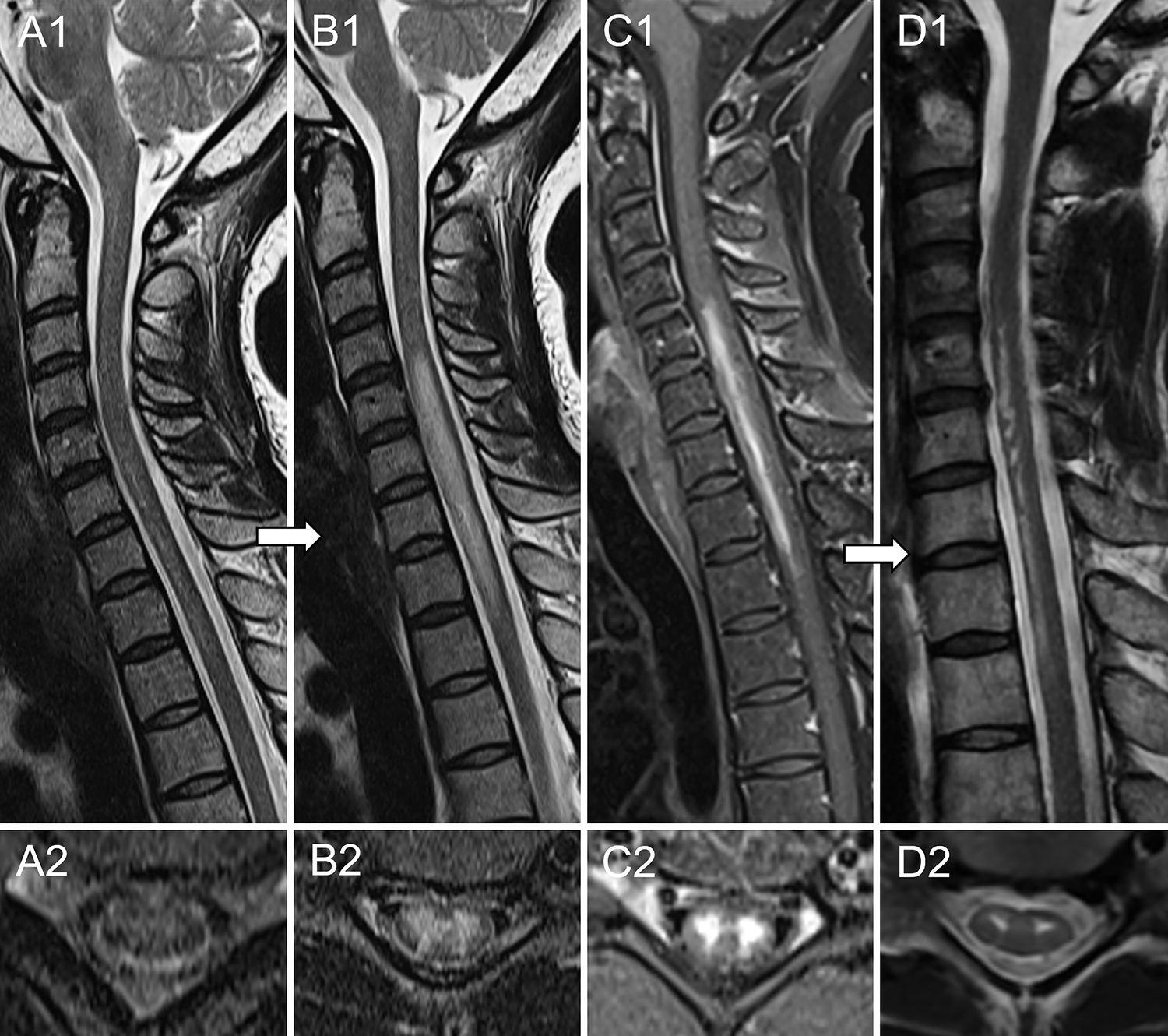
A 41-year-old African American woman with medical history of Sjögren syndrome was admitted to the emergency department for sudden onset of chest tightness. The following day she reported severe back pain and, over the course of 2–3 hours, developed acute paraplegia accompanied by numbness in lower extremities, severe weakness of upper extremities and urinary retention requiring catheterization. Spinal cord MRI obtained five hours later was unremarkable (A). CSF analysis showed 7 white blood cells and absence of oligoclonal bands; infectious and immunological evaluations were negative, except for positive anti-nuclear antibodies. Aquaporin-4-IgG and myelin oligodendrocyte glycoprotein-IgG were negative in both serum and CSF by live cell-based assay. Repeat MRI 6 days later revealed a longitudinal extensive T2-hyperintense lesion in the cervico-thoracic spinal cord (B) with associated enhancement on post-gadolinium T1-weighted sequences (C). A working diagnosis of seronegative NMOSD was made and she was treated with intravenous immune-globulins, slow taper prednisone and rituximab induction with only mild improvement of symptoms. The patient underwent a repeat MRI 2.5 months later, which showed persistence of the T2-hyperintense lesion restricted to the ventral spinal cord, with a “snake eyes” appearance on axial images (D). The patient was later referred to the Mayo Clinic (Rochester, Minnesota) where a final diagnosis of spinal cord infarction was made and rituximab was discontinued. Despite investigations no cause for the spinal cord infarct was found.
Figure 3 -. Intramedullary spinal cord abscess misdiagnosed as seronegative NMOSD.
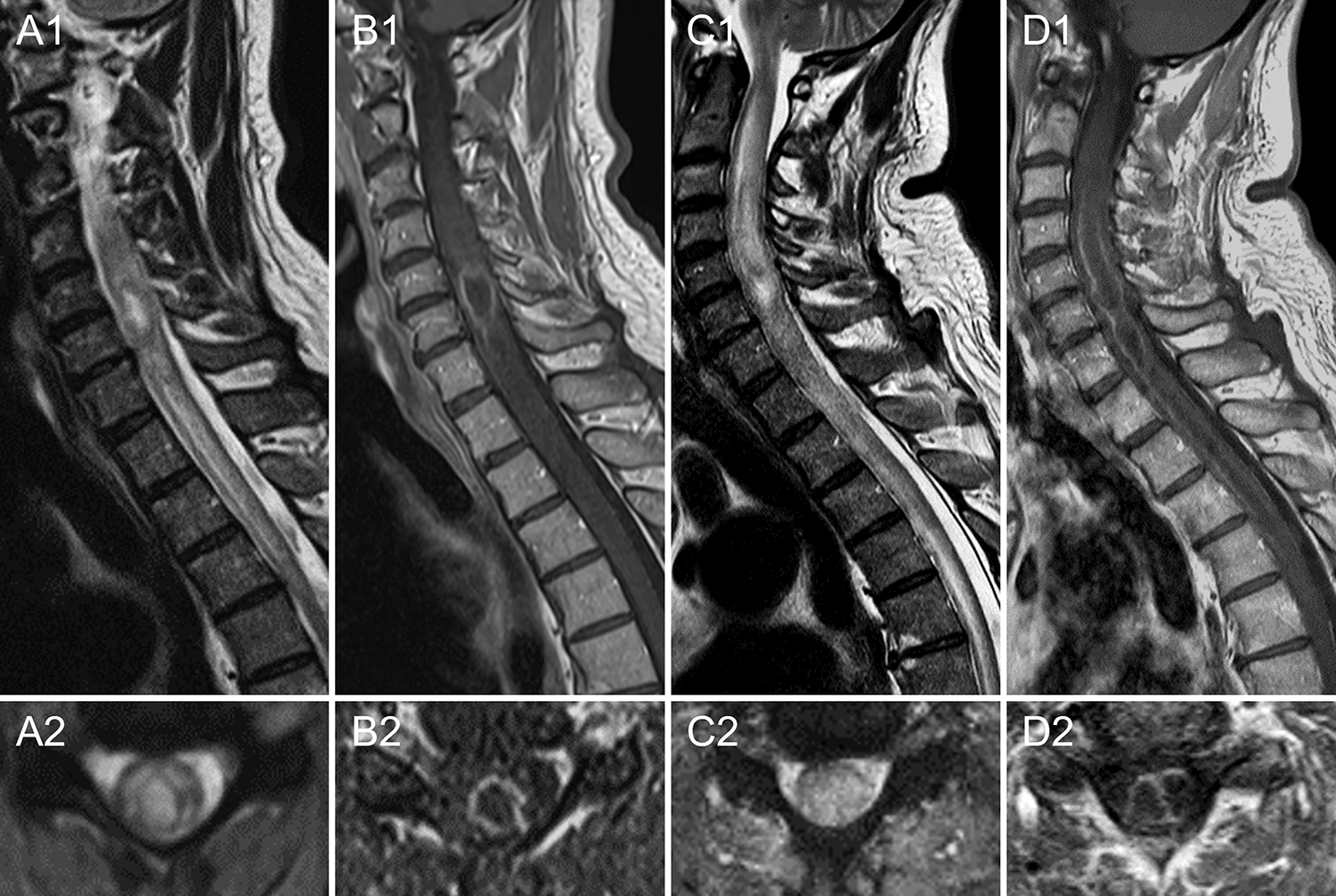
A 54-year-old Caucasian man developed severe neck and shoulder pain, followed by progressive weakness of the left limbs and sensory loss in both hands over 3 days. His medical history was unremarkable, except for several minor dentistry procedures over the prior months. Spinal cord MRI showed a longitudinally extensive T2-hyperintense lesion from C3 to C7 with a central core of higher T2-hyperintensity (A) surrounded by an irregular ring of enhancement on post-gadolinium T1 sequences (B). CSF examination revealed 85 white blood cells and protein elevation (90 g/dl). Extensive infectious screening on both serum and CSF (including blood and CSF culture) was unremarkable. Testing for aquaporin-4-IgG and myelin oligodendrocyte glycoprotein-IgG was negative on serum and CSF by live cell-based assay. He was started on intravenous methylprednisolone (1 g daily for 5 days) with no improvement. A new MRI showed extension of the lesion to the cervicomedullary junction and the thoracic spinal cord to the T4 level (C). A longitudinal extension of the ring enhancement was also observed after gadolinium administration, which appeared bilobate on axial images at cervical level (D). A working diagnosis of seronegative NMOSD was made and plasma exchange was started. Given further clinical deterioration after two plasma exchange sessions (the patient became tetraplegic), a spinal cord biopsy was performed. Cultures from intralesional material came back positive for Haemophilus Influenzae, and antibiotic therapy was started with slow improvement. No fever or increased serum procalcitonin levels were observed before the spinal cord biopsy.
Clinical features and alternative diagnosis
The clinical characteristics of included patients are summarized in Table 1. The most common neurological syndromes misdiagnosed as NMOSD were isolated myelopathy (n=29, 43%; longitudinally extensive in 24), myelopathy associated with optic neuropathy in 26 (38%), and isolated optic neuropathy (n=5, 7%; bilateral in 3). The final alternative diagnoses are listed in Table 2 and included genetic/metabolic diseases (n=20), neoplasms (n=11), infectious diseases (n=11), systemic autoimmune diseases (n=9), vascular diseases (n=8), spondylotic myelopathy (n=5), and other neurologic autoimmune diseases (n=4). Genetic/metabolic disorders were the most common alternative etiologies in both children (age <18 years) and adults, accountin for 55% (6/11) and 24% (13/55), respectively. Spondylotic myelopathy, vascular myelopathies, and systemic autoimmune diseases were exclusively reported in adults.
Table 1 –
Clinical and demographic features of 68 included patients
|
Median age, years
|
44 (range, 1–78) |
|
| |
|
Children (age <18 years)
|
11 (17%) |
|
| |
|
Female sex
|
35 (52%) |
|
| |
| Neurological syndrome misinterpreted as NMOSD | |
| Isolated myelopathy | 29 (43%) |
| Myelopathy + optic neuropathy | 26 (38%) |
| Isolated optic neuropathy | 5 (7%), bilateral in 3 (60%) |
| Isolated area postrema syndrome | 2 (3%) |
| Combinations | 6 (9%) |
|
| |
| Disease course | |
| Progressive | 36/67 (54%) |
| Monophasic | 17/67 (25%) |
| Relapsing | 14/67 (21%) |
|
| |
| MRI findings | |
| Spinal Cord MRI T2 abnormalities | 56/63 (89%) |
| Longitudinally extensive for ≥ 3 VBS | 45/56 (80%) |
| Enhancing spinal cord lesions | 38/55 (69%) |
| Brain T2 abnormalities | 36/59 (61%) |
| Optic nerve T2 abnormalities | 16/52 (31%) |
| Enhancing brain/optic nerve lesions | 15/32 (47%) |
|
| |
| CSF findings | |
| Pleocytosis (>5 WBC) | 20/46 (43%) |
| Median cell count | 40 (range, 6–1800) |
| Protein elevation (>45 mg/dl) | 25/42 (60%) |
| CSF-restricted oligoclonal bands (≥ 2) | 9/29 (31%) |
|
| |
| Treatment | |
|
| |
| Received immunotherapy | 58/66 (88%) |
| Improvement after immunotherapy | 25/56 (45%) |
| Median number of treatments per patient | 2 (range, 1–5) |
| Corticosteroids | 56/58 (97%) |
| Plasma exchange | 16/58 (28%) |
| Rituximab | 7/58 (12%) |
| Intravenous immunoglobulins | 5/58 (9%) |
| Azathioprine | 4/58 (7%) |
| Cyclophosphamide | 4/58 (7%) |
| Mycophenolate Mofetil | 4/58 (7%) |
| DMTs | 1/58 (2%) |
| Infliximab | 1/58 (2%) |
|
| |
|
Seronegative NMOSD criteria met
|
12 (18%) |
Abbreviations – CSF = cerebrospinal fluid; DMTs = disease modifying treatments for multiple sclerosis; MRI = magnetic resonance imaging; NMOSD = neuromyelitis optica spectrum disorder; VBS = vertebral body segments; WBC = white blood cells
Table 2 –
Alternative diagnoses misinterpreted as NMOSD
| Genetic/metabolic diseases (n=20) | Biotinidase deficiency (n= 9) |
| OPA1 gene mutation (n=3) | |
| Leber hereditary optic neuropathy (n=3) | |
| Copper deficiency (n=2) | |
| B12 deficiency (n=1) | |
| MTFMT deficiency (n=1) | |
| Alexander disease (n=1) | |
|
| |
| Neoplasms (n=11) | Primary CNS lymphoma (n=3) |
| Primary spinal cord glioblastoma multiforme (n=1) | |
| Primary CNS histiocytic sarcoma (n=1) | |
| Primary intracranial germinoma (n=1) | |
| Primary spinal cord oligoastrocytoma (n=1) | |
| Anaplastic astrocytoma (n=1) | |
| B-cell lymphoma (n=1) | |
| Optic nerve sheath meningioma (n=1) | |
| High grade glioma (n=1) | |
|
| |
| Infections (n=11) | Tuberculous myelopathy associated with optic neuropathy (n=2) |
| Subacute sclerosing panencephalitis (n=1) | |
| Amoebic encephalitis (n=1) | |
| Varicella Zoster virus myelitis (n=1) | |
| Syphilitic optic neuropathy (n=1) | |
| Spinal toxoplasmosis (n=1) | |
| Chikungunya associated myelitis (n=1) | |
| HIV optic neuropathy (n=1) | |
| C. Bantiana infectious myelitis (n=1) | |
| Intramedullary spinal cord abscess (n=1) | |
|
| |
| Systemic autoimmune diseases (n=9) | Neurosarcoidosis (n=3) |
| Sjogren syndrome (n=2) | |
| Neuro-Bechet (n=2) | |
| SLE (n=1) | |
| Undifferentiated connective tissue disease (n=1) | |
|
| |
| Vascular diseases (n=8) | Dural arteriovenous fistula (n=7) |
| Spinal cord infarction (n=1) | |
|
| |
| Compressive myelopathy (n=5) | Spondylotic myelopathy (n=5) |
|
| |
| Neurologic autoimmune diseases (n=4) | CRMP5-IgG-associated myelitis (n=2) |
| Clippers (n=1) | |
| GFAP astrocytopathy (n=1) | |
Abbreviations – CNS = central nervous system; CRMP5 = collapsing response mediated protein 5; GFAP = glial fibrillary acidic protein; HIV = human immunodeficiency virus; MTFMT = mitochondrial methionyl-tRNA formyltransferase; SLE = systemic lupus erythematosus;
AQP4-IgG positivity and fulfilment of NMOSD diagnostic criteria
Test for AQP4-IgG was performed in 52/58 (90%) with positive result in 5/52 (10%). Of the five patients with AQP4-IgG positivity, two tested positive by enzyme-linked immunosorbent assay (ELISA) and one of them subsequently tested negative by cell-based assay (CBA); one patient had an isolated CSF positivity by a non-specified CBA (and concomitant weak positivity for MOG-IgG in both serum and CSF); one patient was positive in serum by live CBA; and one had a transient serum positivity using a non-specified assay.
Twelve (18%) patients fulfilled the diagnostic criteria for seronegative NMOSD. Their alternative diagnoses were: biotinidase deficiency, 5 (42%); spinal cord neoplasms, 3 (25%); myelitis in the context of Sjögren syndrome, 2 (17%); myelitis associated with glial fibrillary acidic protein (GFAP)-IgG, 1 (8%); and tuberculosis, 1 (8%).
Red flags associated with NMOSD misdiagnosis
The most common red flags associated with NMOSD misdiagnosis were lack of CSF pleocytosis (26/46, 57%), lack of response to immunosuppressive therapy (31/56, 55%), progressive disease course over months (36/67, 54%), and lack of gadolinium enhancement on brain or spinal cord MRI (21/67, 31%). CSF-restricted oligoclonal bands (≥ 2) were reported in 9/29 (31%), mostly in patients with genetic and neoplastic disorders. Fever was reported in 8/68 (12%) patients. The flow diagram in Figure 4 summarizes the recommended diagnostic work-up to diagnose NMOSD, including common red flags to exclude when a diagnosis of seronegative NMOSD is considered, and associated alternative diagnoses.
Figure 4 -. Suggested diagnostic work-up in patients with suspected NMOSD.
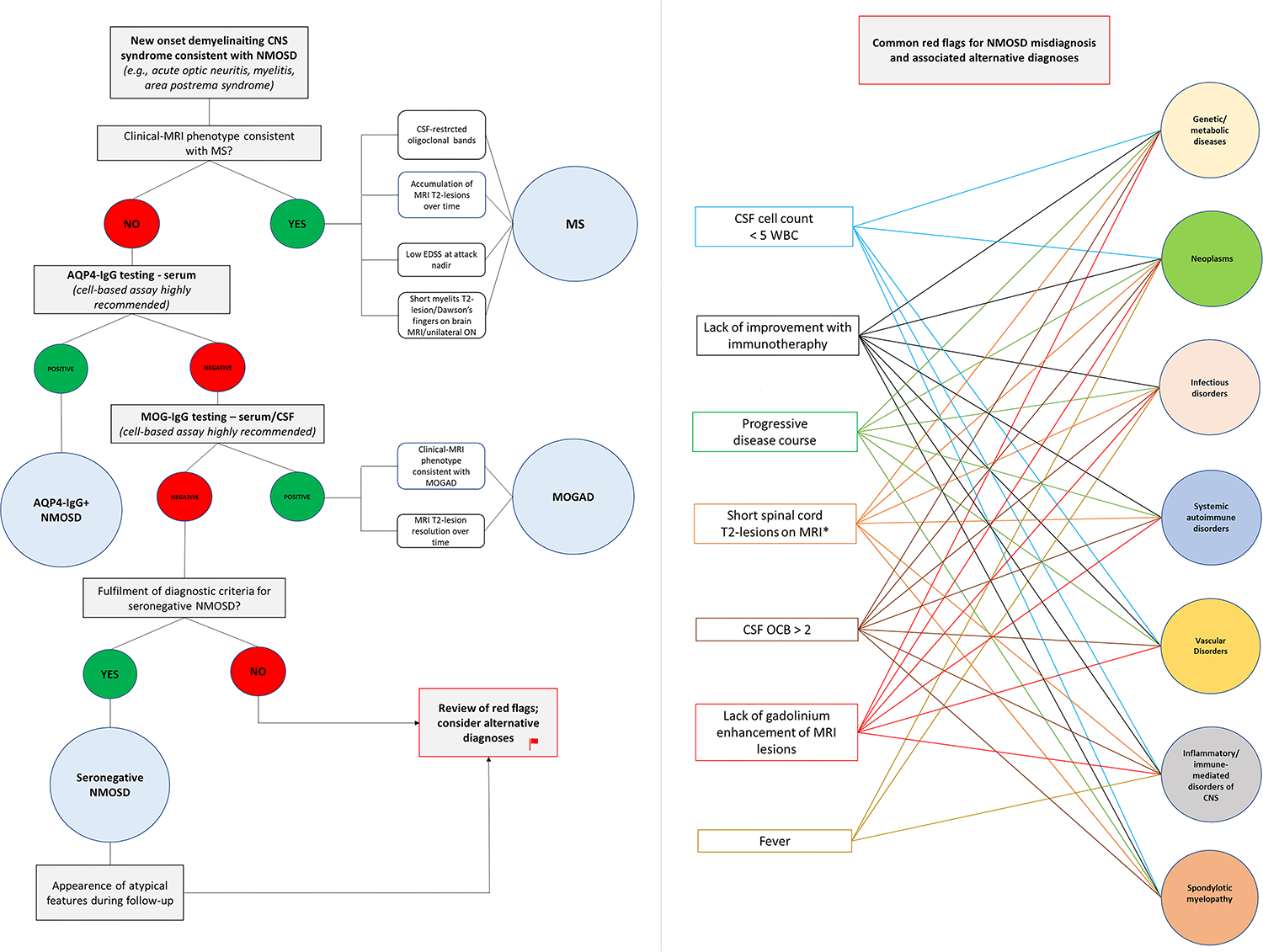
The left half of the figure shows the recommended diagnostic work-up in patients with new-onset demyelinating CNS syndromes compatible with NMOSD (e.g., optic neuritis, myelitis). Multiple sclerosis is the most common demyelinating CNS disorder and should be the first to considered in case of suggestive features (e.g., short myelitis lesions on MRI, CSF-restricted oligoclonal bands). If the clinical-MRI phenotype is not consistent with MS, testing for aquaporin-4-IgG and myelin oligodendrocyte glycoprotein-IgG by cell-based assay is recommended. In case autoantibody testing gives a negative result and a diagnosis of seronegative NMOSD is considered, a careful evaluation for common red-flags is important to avoid misdiagnosis. The right half of the figure shows the most common red-flags encountered in patients with NMOSD misdiagnosis and associated alternative etiologies reported in the literature. Once common red-flags have been excluded, a reasonable diagnosis of seronegative NMOSD can be made. In these patients, however, the lack of response to immunotherapy is another red flag that should prompt reconsideration of alternative diagnoses.
Discussion
The etiologic spectrum of NMOSD mimics is broad. Misdiagnosis frequently occurs due to incorrect application of the diagnostic criteria for seronegative NMOSD, in patients in whom one or more red flags suggestive of alternative diagnoses can be identified. False AQP4-IgG positivity may rarely contribute to misdiagnosis, especially when suboptimal methodologies for AQP4-IgG detection are used. Awareness of NMOSD mimics and associated red flags may help prevent misdiagnosis, treatment delays and inappropriate treatments that may result in severe clinical worsening.
NMOSD is a rare condition, with an incidence of approximately 0.5–7.3/million person-years.11 Its relative frequency among CNS demyelinating disorders is approximately 50 times lower compared to MS, and similar to that of MOGAD.12, 13 Due to its characteristic clinical-MRI phenotype, however, NMOSD is often regarded as the first diagnostic suspicion in patients presenting with longitudinally extensive myelopathy and/or bilateral optic neuropathy, as also observed in our study. MOGAD frequently has overlapping manifestations with NMOSD and represents a major differential diagnosis. MS typically presents with different clinical-MRI features in adults (e.g., less severe clinical attacks, short myelitis lesions on MRI) and is less likely to be misdiagnosed as NMOSD.5, 7–9, 14 However, MS patients may sometimes show preferential involvement of optic nerves and spinal cord, or multiple contiguous myelitis lesions that can resemble a longitudinally extensive lesion on MRI, that can mimic NMOSD.15 The distinction may also be challenging in children with MS, in whom longitudinally extensive myelitis is more common than adults.9 Once MS and MOGAD have been ruled out, we highlight a wide spectrum of rarer, non-demyelinating alternative diagnoses that are liable to be overlooked by clinicians (Table 2). These mimics are different from those that are generally observed in cases of autoimmune encephalitis (e.g., functional neurologic disorder, neurodegenerative disorders) and multiple sclerosis (e.g., migraine, fibromyalgia) misdiagnosis, which are more commonly encountered in clinical practice.16–18
In our study, 82% of patients in whom NMOSD was misdiagnosed did not meet diagnostic criteria. Most of these patients presented with only one of the core clinical features required for diagnosis (typically an isolated longitudinally extensive myelopathy) and/or had major red flags that precluded fulfilment of the criteria (Figure 4). Lack of CSF pleocytosis was the most common red flag in this study but is not alone sufficient to rule out a diagnosis of NMOSD, as a non-inflammatory CSF can be observed in up to 57% of patients.19 Less commonly observed in NMOSD are lack of response to acute immunotherapy (especially when plasma exchange is used in addition to corticosteroids) and a progressive disease course, which were also both found to be red flags associated with misdiagnosis.20, 21 A progressive disease course, in particular, is extremely rare in NMOSD but is common in patients with spinal cord sarcoidosis,6 spondylotic myelopathy,22 neoplastic and genetic/metabolic etiologies.23 On the contrary, a hyperacute onset within hours (often preceded by back pain or intermittent neurological symptoms during the previous week) is typical of spinal cord infarction (Figure 1).24 Gadolinium lesion enhancement during acute attacks is seen in >90% of patients with NMOSD and its absence should prompt considering other etiologies.5 Persistent enhancement over >3 months is also unusual in NMOSD but can be seen in neurosarcoidosis or neoplasms (Figure 2).25 Lastly, fever and constitutional symptoms may rarely be seen in patients with NMOSD but should prompt extensive screening for infectious etiologies (including bacteria, mycobacteria, fungi, and viruses). Intramedullary abscess is extremely rare and typically presents with an elongated, irregular ring of peripheral enhancement on spine MRI, similar to that frequently seen in NMOSD (Figure 3).26 Notably, fever is often absent in patients with intramedullary abscess and lumbar puncture may be unrevealing. Fever may also trigger worsening in certain genetic/metabolic disorders.27
Figure 2 -. Optic nerve sheath meningioma misdiagnosed as seronegative NMOSD.
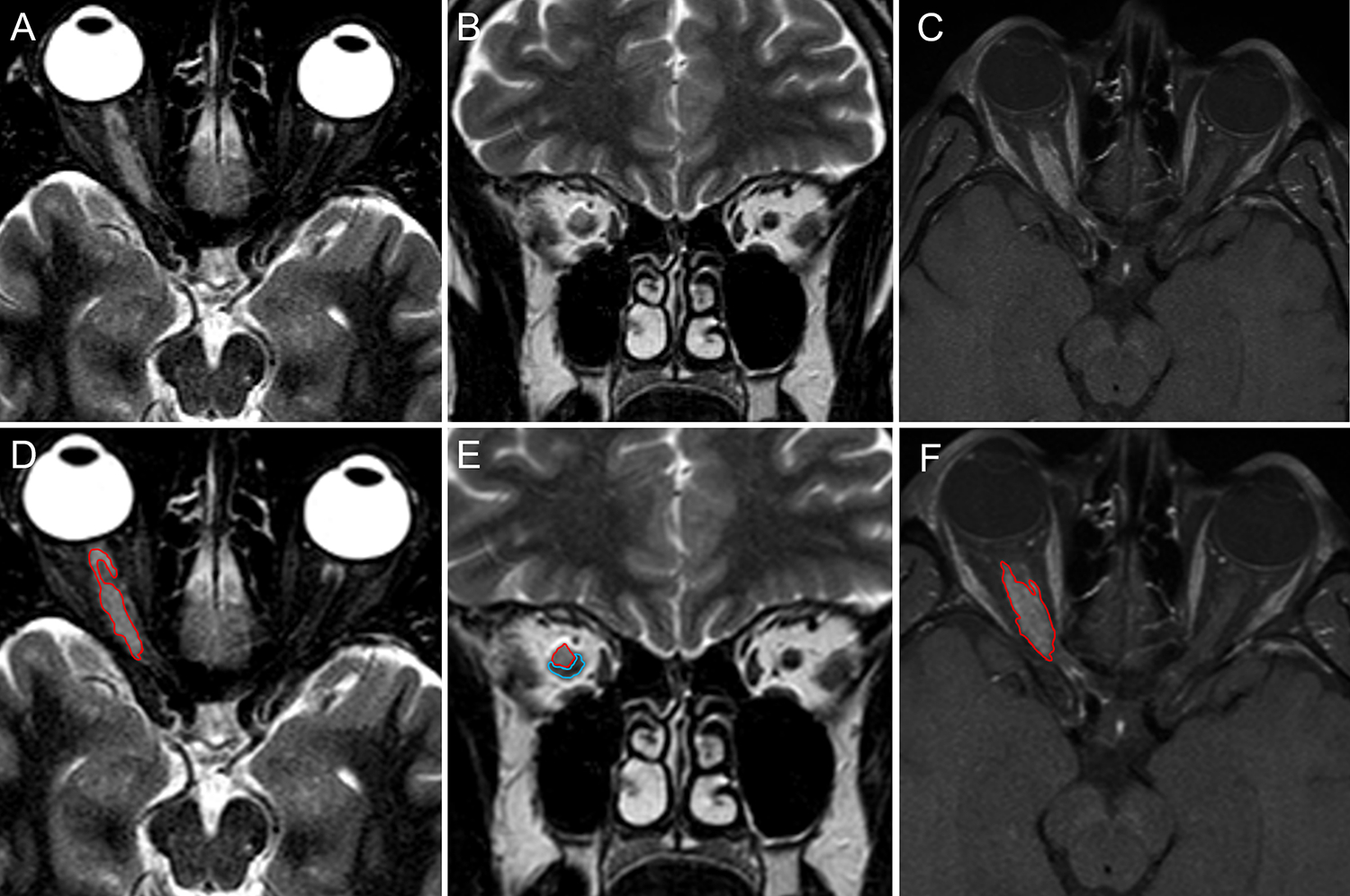
A 52-years old Caucasian woman developed blurred vision in the right eye over one week. She denied headache, diplopia, or other neurologic symptoms. Brain MRI showed marked swelling of the right optic nerve with associated gadolinium enhancement (not shown). A diagnosis of optic neuritis was made and she was treated with intravenous methylprednisolone (1 g daily for 5 days), with resolution of the visual symptoms. In the following two years, she experienced several recurrences of symptoms in the same eye interpreted as recurrent optic neuritis and treated with intravenous methylprednisolone pulses and oral prednisone taper. Repeat MRIs over the two years showed persistent enhancement of the right optic nerve, substantially unmodified compared to the first exam. Despite some temporary improvement with corticosteroid therapy, she experienced progressive vision impairment. NMOSD was eventually suspected and the patient was referred to the Neurology Unit of the University-Hospital of Sassari. Orbital MRI showed a discrete tissue mass surrounding the right optic nerve that was hyperintense on both axial (A) and coronal (B) T2-images and enhanced diffusely after gadolinium administration on fat-suppression sequences (C). The margins of the mass and adjacent optic nerve are highlighted in red and blue, respectively (D-F). A final diagnosis of probable optic nerve sheath meningioma was made based on clinical and MRI findings.
A minority of patients (18%) fulfilled diagnostic criteria for seronegative NMOSD despite having an alternative diagnosis. Biotinidase deficiency is a rare but well-described mimic of NMOSD, and should be suspected in children or young adults who do not respond to immunosuppressive therapy, or lack indirect evidence of inflammation on MRI (gadolinium enhancement) and/or in the CSF (pleocytosis).28, 29 Among other neurologic autoimmune disorders, collapsin response mediator protein-5 (CRMP5)-IgG associated optic neuritis and/or myelitis represent common mimics and may reveal the presence of an underlying cancer.30 GFAP astrocytopathy is a recently-described entity that manifests with meningo-encephalo-myelitis or limited forms thereof, and may resemble NMOSD.31 The disease course, however, is typically progressive and presentation with isolated myelitis and optic neuritis is rare.32 Lastly, CNS lymphomas are insidious mimics of NMOSD and should be suspected in case of concomitant constitutional symptoms (e.g., unintentional weight loss), persistent gadolinium enhancement on MRI and steroid-dependent course. CSF cytometry/cytological examination may be diagnostic but has low sensitivity and biopsy is often required.33
Longitudinally extensive myelitis or other CNS syndromes that can mimic NMOSD have been reported in association with different systemic immune-mediated disorders. However, these manifestations are rare and for some of these diseases the pathophysiological mechanisms underlying CNS involvement remain incompletely understood (e.g., myelitis in Sjögren’s syndrome). Since different autoimmune disorders can often coexist, testing for AQP4-IgG is always recommended when neurologic manifestation consistent with NMOSD are seen in patients with systemic autoimmune diseases.34
Five patients in this study tested positive for AQP4-IgG. Although AQP4-IgG testing by cell-based assay (CBA) is highly specific, false positive results are recognized to occur with ELISA.35, 36 Nevertheless, two patients in this study with a final diagnosis of spinal cord neoplasm tested positive for AQP4-IgG by CBA, one in serum by live CBA and one in CSF with a non-specified CBA. This highlights the rare possibility of false AQP4-IgG positivity in serum even with CBA, while reliable data on isolated CSF positivity and its clinical significance are lacking.37 Notably, both patients presented with an isolated progressive myelopathy, a clinical phenotype rarely seen in NMOSD.
Limitations of this study include the relatively small sample size and design biased towards inclusion of rare or atypical cases. It is often difficult to distinguish between cases to whom an incorrect diagnosis of NMOSD was assigned and cases that were reported as mimicking NMOSD but not clearly misdiagnosed as NMOSD, which might result in heterogeneity of included cases. However, for all included cases in this study NMOSD represented a major diagnostic suspicion during the diagnostic work-up that resulted in administration of immunotherapy in 88% of cases. We were not able to assess the impact of misdiagnosis and its duration on final outcome, but this varies widely based on the specific alternative etiology. As an example, a diagnostic delay of only few days in case of intramedullary spinal cord abscess (Figure 3) may have more severe clinical consequences compared to a longer diagnostic delay of two years in case of optic nerve sheath meningioma (Figure 2). Lastly, we were not able to assess the frequency of NMOSD misdiagnosis in clinical practice for which a different study design will be preferred.
Conclusion
NMOSD misdiagnosis frequently occurs due to incorrect application of the seronegative NMOSD diagnostic criteria. Although seronegative NMOSD is extremely rare when positivity for AQP4-IgG and MOG-IgG has been excluded with reliable assays, clinicians may often be concerned about making a rapid diagnosis and start with immunotherapy even if the criteria are only partially fulfilled (e.g., isolated longitudinally extensive myelopathy). However, it remains unclear whether seronegative NMOSD represent a single disease or a heterogeneous group of disorder, and data regarding treatment response in these patients are still scarce. While future studies will help clarify the nature of seronegative NMOSD, its diagnosis warrants extra caution and extensive evaluation for alternative etiologies in order to prevent misdiagnosis.
Supplementary Material
Footnotes
Conflict of Interest Statement
Drs. Zara, Dinoto, Carta, Floris, and Turilli have nothing to disclose. Adrian Budhram reports that he holds the London Health Sciences Centre and London Health Sciences Foundation Chair in Neural Antibody Testing for Neuro-Inflammatory Diseases and receives support from the Opportunities Fund of the Academic Health Sciences Centre Alternative Funding Plan of the Academic Medical Organization of Southwestern Ontario (AMOSO). Sergio Ferrari received speaker honoraria from Lundbeck and support for scientific meeting by Shire, Merck, Euroimmun. Paolo Solla has received speaker honoraria from Bayer and Zambon. Sara Mariotto has received speaker honoraria from Biogen, Novartis, and Sanofy. Eoin P. Flanagan has served on advisory boards for Alexion, Genentech, Horizon Therapeutics and UCB. He has received research support from UCB. He has received speaker honoraria from Pharmacy Times. He received royalties from UpToDate. Dr Flanagan was a site primary investigator in a randomized clinical trial on Inebilizumab in neuromyelitis optica spectrum disorder run by Medimmune/Viela-Bio/Horizon Therapeutics. Dr Flanagan has received funding from the NIH (R01NS113828). Dr Flanagan is a member of the medical advisory board of the MOG project. Dr Flanagan is an editorial board member of the Journal of the Neurological Sciences and Neuroimmunology Reports. A patent has been submitted on DACH1-IgG as a biomarker of paraneoplastic autoimmunity. A Sebastian Lopez Chiriboga has served on scientific advisory boards for Genentech and Horizon. Elia Sechi has received speaker honoraria and support for attending scientific meetings from Alexion. He serves as an editorial board member for BMC Neurology and Frontiers in Neurology. Dr. Sechi is a member of the medical advisory board of the MOG project.
References
- 1.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carnero Contentti E, Lopez PA, Criniti J, et al. Frequency of NMOSD misdiagnosis in a cohort from Latin America: Impact and evaluation of different contributors. Mult Scler 2023;29:277–286. [DOI] [PubMed] [Google Scholar]
- 3.Smith AD, Moog TM, Burgess KW, McCreary M, Okuda DT. Factors associated with the misdiagnosis of neuromyelitis optica spectrum disorder. Mult Scler Relat Disord 2023;70:104498. [DOI] [PubMed] [Google Scholar]
- 4.Dubey D, Pittock SJ, Krecke KN, et al. Clinical, Radiologic, and Prognostic Features of Myelitis Associated With Myelin Oligodendrocyte Glycoprotein Autoantibody. JAMA Neurol 2019;76:301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sechi E, Krecke KN, Messina SA, et al. Comparison of MRI Lesion Evolution in Different Central Nervous System Demyelinating Disorders. Neurology 2021;97:e1097–e1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flanagan EP, Kaufmann TJ, Krecke KN, et al. Discriminating long myelitis of neuromyelitis optica from sarcoidosis. Ann Neurol 2016;79:437–447. [DOI] [PubMed] [Google Scholar]
- 7.Banks SA, Morris PP, Chen JJ, et al. Brainstem and cerebellar involvement in MOG-IgG-associated disorder versus aquaporin-4-IgG and MS. J Neurol Neurosurg Psychiatry 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cacciaguerra L, Morris P, Tobin WO, et al. Tumefactive Demyelination in MOG Ab-Associated Disease, Multiple Sclerosis, and AQP-4-IgG-Positive Neuromyelitis Optica Spectrum Disorder. Neurology 2023;100:e1418–e1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fadda G, Flanagan EP, Cacciaguerra L, et al. Myelitis features and outcomes in CNS demyelinating disorders: Comparison between multiple sclerosis, MOGAD, and AQP4-IgG-positive NMOSD. Front Neurol 2022;13:1011579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 11.Papp V, Magyari M, Aktas O, et al. Worldwide Incidence and Prevalence of Neuromyelitis Optica: A Systematic Review. Neurology 2021;96:59–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flanagan EP, Cabre P, Weinshenker BG, et al. Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum. Ann Neurol 2016;79:775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sechi E, Cacciaguerra L, Chen JJ, et al. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD): A Review of Clinical and MRI Features, Diagnosis, and Management. Front Neurol 2022;13:885218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filippi M, Preziosa P, Banwell BL, et al. Assessment of lesions on magnetic resonance imaging in multiple sclerosis: practical guidelines. Brain 2019;142:1858–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asnafi S, Morris PP, Sechi E, et al. The frequency of longitudinally extensive transverse myelitis in MS: A population-based study. Mult Scler Relat Disord 2020;37:101487. [DOI] [PubMed] [Google Scholar]
- 16.Solomon AJ, Klein EP, Bourdette D. “Undiagnosing” multiple sclerosis: the challenge of misdiagnosis in MS. Neurology 2012;78:1986–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flanagan EP, Geschwind MD, Lopez-Chiriboga AS, et al. Autoimmune Encephalitis Misdiagnosis in Adults. JAMA Neurol 2023;80:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dinoto A, Zara P, Mariotto S, et al. Autoimmune encephalitis misdiagnosis and mimics. J Neuroimmunol 2023;378:578071. [DOI] [PubMed] [Google Scholar]
- 19.Jarius S, Pellkofer H, Siebert N, et al. Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies. Part 1: Results from 163 lumbar punctures in 100 adult patients. J Neuroinflammation 2020;17:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wingerchuk DM, Pittock SJ, Lucchinetti CF, Lennon VA, Weinshenker BG. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology 2007;68:603–605. [DOI] [PubMed] [Google Scholar]
- 21.Bonnan M, Cabre P. Plasma exchange in severe attacks of neuromyelitis optica. Mult Scler Int 2012;2012:787630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flanagan EP, Krecke KN, Marsh RW, Giannini C, Keegan BM, Weinshenker BG. Specific pattern of gadolinium enhancement in spondylotic myelopathy. Ann Neurol 2014;76:54–65. [DOI] [PubMed] [Google Scholar]
- 23.Sechi E, Flanagan EP. Evaluation and Management of Acute Myelopathy. Semin Neurol 2021;41:511–529. [DOI] [PubMed] [Google Scholar]
- 24.Zalewski NL, Rabinstein AA, Krecke KN, et al. Characteristics of Spontaneous Spinal Cord Infarction and Proposed Diagnostic Criteria. JAMA Neurol 2019;76:56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cacciaguerra L, Sechi E, Rocca MA, Filippi M, Pittock SJ, Flanagan EP. Neuroimaging features in inflammatory myelopathies: A review. Front Neurol 2022;13:993645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zalewski NL, Morris PP, Weinshenker BG, et al. Ring-enhancing spinal cord lesions in neuromyelitis optica spectrum disorders. J Neurol Neurosurg Psychiatry 2017;88:218–225. [DOI] [PubMed] [Google Scholar]
- 27.Sechi E, Addis A, Fadda G, Minafra L, Bravata V, Sechi G. Teaching NeuroImages: Subacute encephalopathy in a young woman with THTR2 gene mutation. Neurology 2015;85:e108–109. [DOI] [PubMed] [Google Scholar]
- 28.Bottin L, Prud’hon S, Guey S, et al. Biotinidase deficiency mimicking neuromyelitis optica: Initially exhibiting symptoms in adulthood. Mult Scler 2015;21:1604–1607. [DOI] [PubMed] [Google Scholar]
- 29.Santoro JD, Paulsen KC. Biotinidase Deficiency as a Mimic of Neuromyelitis Optica Spectrum Disorder in Childhood. JAMA Neurol 2020;78:118–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flanagan EP, McKeon A, Lennon VA, et al. Paraneoplastic isolated myelopathy: clinical course and neuroimaging clues. Neurology 2011;76:2089–2095. [DOI] [PubMed] [Google Scholar]
- 31.Flanagan EP, Hinson SR, Lennon VA, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: Analysis of 102 patients. Ann Neurol 2017;81:298–309. [DOI] [PubMed] [Google Scholar]
- 32.Sechi E, Morris PP, McKeon A, et al. Glial fibrillary acidic protein IgG related myelitis: characterisation and comparison with aquaporin-4-IgG myelitis. J Neurol Neurosurg Psychiatry 2019;90:488–490. [DOI] [PubMed] [Google Scholar]
- 33.Flanagan EP, O’Neill BP, Porter AB, Lanzino G, Haberman TM, Keegan BM. Primary intramedullary spinal cord lymphoma. Neurology 2011;77:784–791. [DOI] [PubMed] [Google Scholar]
- 34.Guerra H, Pittock SJ, Moder KG, Fryer JP, Gadoth A, Flanagan EP. Frequency of Aquaporin-4 Immunoglobulin G in Longitudinally Extensive Transverse Myelitis With Antiphospholipid Antibodies. Mayo Clin Proc 2018;93:1299–1304. [DOI] [PubMed] [Google Scholar]
- 35.Redenbaugh V, Montalvo M, Sechi E, et al. Diagnostic value of aquaporin-4-IgG live cell based assay in neuromyelitis optica spectrum disorders. Mult Scler J Exp Transl Clin 2021;7:20552173211052656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams JP, Abbatemarco JR, Galli JJ, et al. Aquaporin-4 Autoantibody Detection by ELISA: A Retrospective Characterization of a Commonly Used Assay. Mult Scler Int 2021;2021:8692328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Majed M, Fryer JP, McKeon A, Lennon VA, Pittock SJ. Clinical utility of testing AQP4-IgG in CSF: Guidance for physicians. Neurol Neuroimmunol Neuroinflamm 2016;3:e231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.