

Abstract
Plant virus nanoparticles can be used as drug carriers, imaging reagents, vaccine carriers, and immune adjuvants in the formulation of intratumoral in situ cancer vaccines. One example is the cowpea mosaic virus (CPMV), a nonenveloped virus with a bipartite positive-strand RNA genome with each RNA packaged separately into identical protein capsids. Based on differences in their densities, the components carrying RNA-1 (6 kb) denoted as the bottom (B) component or carrying RNA-2 (3.5 kb) denoted as the middle (M) component can be separated from each other and from a top (T) component, which is devoid of any RNA. Previous preclinical mouse studies and canine cancer trials used mixed populations of CPMV (containing B, M, and T components), so it is unclear whether the particle types differ in their efficacies. It is known that the CPMV RNA genome contributes to immunostimulation by activation of TLR7. To determine whether the two RNA genomes that have different sizes and unrelated sequences cause different immune stimulation, we compared the therapeutic efficacies of B and M components and unfractionated CPMV in vitro and in mouse cancer models. We found that separated B and M particles behaved similarly to the mixed CPMV, activating innate immune cells to induce the secretion of pro-inflammatory cytokines such as IFNα, IFNγ, IL-6, and IL-12, while inhibiting immunosuppressive cytokines such as TGF-β and IL-10. In murine models of melanoma and colon cancer, the mixed and separated CPMV particles all significantly reduced tumor growth and prolonged survival with no significant difference. This shows that the specific RNA genomes similarly stimulate the immune system even though B particles have 40% more RNA than M particles; each CPMV particle type can be used as an effective adjuvant against cancer with the same efficacy as native mixed CPMV. From a translational point of view, the use of either B or M component vs the mixed CPMV formulation offers the advantage that separated B or M alone is noninfectious toward plants and thus provides agronomic safety.
Keywords: cowpea mosaic virus, genomic RNA-1, genomic RNA-2, in situ vaccine, cancer immunotherapy, intratumoral immunotherapy
Graphical Abstract
INTRODUCTION
Cancer immunotherapy aims to strengthen the immune response against tumors and elicits long-term immune memory to prevent recurrence.1–3 Although immunotherapy has achieved promising clinical outcomes, only a small subset of patients shows a response to treatment and the efficacy differs according to the tumor type.4,5 Immunotherapy can also trigger severe adverse effects in some patients, including autoimmune reactions, cytokine release syndrome, and vascular leak syndrome.6–8 To enhance the efficacy and reduce the side effects of cancer immunotherapy, cancer vaccine candidates have been developed using various nanomaterial formulations, including polymers, lipids, metals, and viruses, the latter offering key advantages of versatility and biocompatibility.9,10
Plant viruses are particularly suitable for cancer immunotherapy because they possess intrinsic immunostimulatory properties and are noninfectious in mammals.11,12 Plant viruses act as adjuvants by activating pattern recognition receptors (PRPs) that counter the immunosuppression in the tumor micro-environment (TME) by increasing the production of pro-inflammatory cytokines such as type I and II interferons (IFNα, IFNγ), interleukin-12 (IL-12), and tumor necrosis factor alpha (TNFα).13,14
Cowpea mosaic virus (CPMV) is a nonenveloped plant virus that has been investigated as a vaccine adjuvant for cancer immunotherapy. We have previously demonstrated the efficacy of CPMV as an in situ/intratumoral cancer vaccine in melanoma, colon cancer, breast cancer, glioma, and ovarian cancer.13,15–19 Efficacy has also been demonstrated in canine patients with melanoma and inflammatory mammary cancer.20,21 CPMV transforms immunosuppressive “cold” tumors into immunogenic “hot” tumors with high levels of innate immune cell infiltration and antigen presentation priming systemic and durable anti-tumor adaptive immunity.14,22 Mechanistically, CPMV is taken up by phagocytes and signals through toll-like receptors (TLRs) 2, 4, and 7,14 which promotes the secretion of pro-inflammatory cytokines into the TME, including IFNα, IFNγ, IL-6, and IL-12, while reducing secretion of immunosuppressive cytokines such as TGFβ and IL-10.17,18 CPMV acts on the innate immune system, which then induces systemic adaptive anti-tumor immunity; therefore, localized intratumoral (i.t.) treatment (i.e., in situ vaccination) induces systemic efficacy with observed abscopal effect and long-lasting immune memory.22 While potent as a solo therapy, CPMV in situ vaccination has also been combined with chemotherapy23 and radiation,24 increasing the number of antigen-specific effector T cells against resistant cancers and making the tumors more immunogenic, thus synergistically eliciting anti-tumor effects.
CPMV is uniquely potent and outperforms other plant viruses as an in situ vaccine.16 Studies pointed to the importance of the RNA cargo; RNA-free “empty” virus-like particles of CPMV are less potent than those of native CPMV.25 This is attributed to TLR-7 signaling from encapsidated viral single-strand RNA, leading to type I interferon secretion.14 CPMV has an icosahedral capsid that consists of 60 identical copies of a large (L, 42 kDa) and small (S, 24 kDa) coat protein. Its two bipartite positive-sense RNA genomes (RNA-1 and RNA-2) are packaged in separate capsids with identical protein compositions.26,27 This results in a mixed population of bottom (B) components containing RNA-1 (6 kb) and middle (M) components containing RNA-2 (3.5 kb). RNA-free top (T) components also form during infection in plants; however, these are not isolated using our purification protocol, which ends with an isopycnic sucrose gradient to collect the B and M components.25 While studies have pointed to the importance of the RNA cargo, it is unclear whether the two strands of genomic RNA have similar or distinct immunomodulatory effects.
In this study, we compared the effects of B component (RNA-1), M component (RNA-2), and unfractionated CPMV particles in murine dermal melanoma and colon cancer models. Our results provide insight into the immunomodulatory mechanism of CPMV and whether particle separation would be of value during manufacturing of CPMV for cancer therapy.
MATERIALS AND METHODS
Separation of CPMV into Its B and M Components.
CPMV was propagated in cowpea plants by mechanical inoculation and purified using previously published methods.28 In brief, CPMV was purified with a 1:1 chloroform:butanol extraction, PEG precipitation, and ultracentrifugation in a 10–40% sucrose density gradient. The B and M components were separated by Nycodenz gradient ultracentrifugation using a 30–60% (w/v) Nycodenz solution was prepared in 10 mM sodium phosphate buffer (pH 7.0) and filled into a SW40Ti tube.29 The CPMV suspension (0.1 mg/μL) was layered onto the gradient and centrifuged at 26,000g for 24 h at 4 °C. CPMV particles were collected under white light and concentrated by ultracentrifugation at 42,000g for 2 h at 4 °C. The concentration of collected CPMV components was determined using ultra-violet–visible (UV–vis) spectroscopy (NanoDrop) and calculated using the Beer–Lambert equation, A = ε × d × c, where A is the absorbance at 260 nm, ε is the extinction coefficient of CPMV (8.1 mL mg−1 cm−1), and d is the path length (0.1 cm−1). The ratio of B and M components was calculated by dividing the concentration of each component by collected components.
Agarose Gel Electrophoresis of CPMV Particles and Purified RNA.
CPMV particles (10 μg in 1× Gel Loading Purple dye) were loaded onto a 1.2% (w/v) agarose gel stained with GelRed nucleic acid and run at 100 V for 35 min in Tris–acetate EDTA (TAE) buffer. The gel was visualized under UV light using an AlphaImager system (ProteinSimple) after staining with Coomassie Brilliant Blue. Encapsidated RNA was extracted using the Quick-RNA Miniprep kit (Zymo Research). RNA purity was determined by measuring the absorbance at 260 and 280 nm and calculating the ratio (A260/280). RNAs extracted from mixed and separated CPMV components were analyzed by 1.2% agarose gel electrophoresis as above.
Denaturing Gel Electrophoresis.
Mixed and separated CPMV components (10 μg) in 1× LDS loading dye (Thermo Fisher Scientific) were denatured at 95 °C for 5 min before fractionation with sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE, 4–12% NuPAGE, Invitrogen) in 1× (3-(N-morpholino)propanesulfonic acid) (MOPS) buffer. The gel was stained with GelCode Blue Safe protein stain and visualized under white light and imaged on the AlphaImager System.
Transmission Electron Microscopy (TEM).
Mixed and separated CPMV components were dispersed in water at a concentration of 0.1 mg/mL and were pipetted onto a 400-mesh hexagonal copper grid on a Formvar support film (Ted Pella) followed by staining with 2% uranyl acetate for 2 min. TEM images were acquired using a JEM-1400Plus microscope (JEOL).
Dynamic Light Scattering (DLS).
For the analysis of size and surface charge, CPMV particles and components were dispersed in 0.1 M potassium phosphate (KP) buffer (pH 7.0) at concentrations of 0.1 mg/mL (for size analysis) and 1 mg/mL (for surface charge analysis). The size and zeta potential were measured using a Zetasizer Nano ZSP/Zen5600 (Malvern Panalytical).
Size Exclusion Chromatography (SEC).
CPMV particles were loaded onto a Superose 6 Increase column mounted on an Äkta Explorer chromatography system (Cytiva) at a flow rate of 0.5 mL/min in 0.1 M KP buffer (pH 7.0). The absorbance was recorded at 260 and 280 nm.
Cell Culture.
RAW-Blue cells (InvivoGen) were grown in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS), 1% (v/v) penicillin–streptomycin, and 100 μg/mL Normocin and Zeocin. B16F10 melanoma cells and CT26 colon cancer cells were obtained from the ATCC and were grown in DMEM and RPMI 1640, respectively, supplemented with 10% (v/v) FBS and 1% (v/v) penicillin–streptomycin. All cells were maintained at 37 °C in a humidified 5% CO2 incubator.
RAW-Blue Cell Assay.
RAW-Blue cells (1 × 105 cells/200 μL) were seeded in 96-well flat-bottom plates, and CPMV particles were added at a dose of 1 μg per well. The cells were incubated for 24 h at 37 °C in a 5% CO2. We then mixed 20 μL of the supernatant with 180 μL of QUANTI-Blue solution (InvivoGen) and incubated the mixture for 6 h at room temperature. The level of secreted embryonic alkaline phosphatase (SEAP) was measured at 630 nm.
Animal Studies.
All mouse studies were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California, San Diego. Female C57BL/6 J and BALB/c mice were purchased from The Jackson Laboratory. BALB/c mice (n = 7, 6–8 weeks old) were intraperitoneally (i.p.) implanted with 5 × 105 CT26 cells in 200 μL of phosphate-buffered saline (PBS). Mice were randomly assigned to four groups: (1) PBS control, (2) CPMV mix, (3) CPMV B-type, and (4) CPMV M-type. The treatments were administered i.p. three times at weekly intervals, starting on day 3 at a dose of 100 μg in 200 μL of PBS. Tumor growth was monitored by measuring abdominal circumference and body weight. For melanoma studies, 2.0 × 105 B16F10 cells in 20 μL of PBS were intradermally (i.d.) inoculated into the left flank of C57BL/6 J mice (n = 5, 6–8 weeks old). CPMV treatments were injected intratumorally (i.t.) at a dose of 100 μg in 20 μL of PBS three times per week, starting on day 7 after tumor challenge. Tumor volume was calculated using the following equation: volume = (length × width2)/2. Tumor volume and survival rates were monitored over time, and mice were euthanized once the tumor volume exceeded 1500 mm3.
Cytokine Evaluation.
Peritoneal cavity washes were collected 7 days after the third CPMV treatment, and an enzyme-linked immunosorbent assay (ELISA) was used to determine the quantity of IL-6, IL-10, IL-12, TNFα, TGFβ, and IFNγ in the supernatant (Thermo Fisher Scientific). For ex vivo cytokine secretion assays, peritoneal cavity cells were collected from tumor-bearing mice treated with PBS, 7 days after the third injection. Peritoneal cavity cells (3 × 106 cells) were seeded in 24-well plates and stimulated with 10 μg of CPMV. The supernatant was collected after incubation for 24 h. For splenocyte assays, spleens from C57BL/6 or TLR-7−/− mice were harvested and 5 × 105 splenocytes were cultured with either CPMV mix (12 μg/mL), CPMV M-type (12 μg/mL), CPMV B-type (12 μg/mL), or CPMV M-type (6 μg/mL) + CPMV B-type (6 μg/mL) in 200 μL of complete RPMI 1640 for 24 h in 96-well plates, similar to previous studies.14 The conditioned medium was analyzed by ELISA to detect IL-6 or IFNα.
Statistical Analysis.
All data were analyzed using GraphPad Prism v7.0 and are presented as means ± standard deviations (SD). ELISA data were determined by one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test. Tumor volumes were compared using an using two-way ANOVA, and survival rates were compared using a log-rank (Mantel–Cox) test. A p value below 0.05 was considered statistically significant.
RESULTS AND DISCUSSION
Preparation and Characterization of Mixed and Separated CPMV Components.
CPMV B and M components were separated by Nycodenz gradient centrifugation (Figure 1), and their properties were compared to those of the native mixed CPMV sample (Figure 2A). The B-type particles accounted for 58 ± 7.32% of the mixture by mass, and the M-type particles accounted for 42 ± 7.32% of the mixture, indicating roughly equal numbers of capsids containing RNA-1 or −2. The separation of CPMV particle types was confirmed by agarose gel electrophoresis of the RNA extracted from mixed and separated particles (Figure 2B). The mixed particles yielded two RNA bands at 3.5 kb (RNA-2) and 6 kb (RNA-1), whereas the separated particles yielded a single band each, confirming separation of the components. To confirm that the isolated components remained intact, mixed and separated CPMV particles were analyzed by agarose gel electrophoresis (Figure 2C). The separated CPMV particles were intact, as nucleic acids (RNA stain) and CPMV capsids (protein stain) co-migrate under electrophoretic separation. The three preparations were denatured and fractionated by SDS-PAGE. All samples produced identical protein bands of 42 and 24 kDa, representing the L and S coat protein subunits, respectively, and confirming that the two distinct RNAs are encapsidated in identical protein capsids (Figure 2D). TEM images revealed the monodispersity and icosahedral structure of all three particle preparations (Figure 2E). DLS showed that all three preparations have a mean particle diameter of ~30 nm (Figure 2F), and the zeta potentials indicated that the surface charge of B-type particles (−14.6 mV) and M-type particles (−14.69 mV) were similar to native mixed CPMV (−15.5 mV) (Figure 2G). SEC confirmed the structural integrity of all three preparations, with the same elution volume of 11.5 mL and an absorbance ratio (A260/280) of ~1.8, indicating intact capsid proteins with encapsidated RNAs (Figure 2H).
Figure 1.
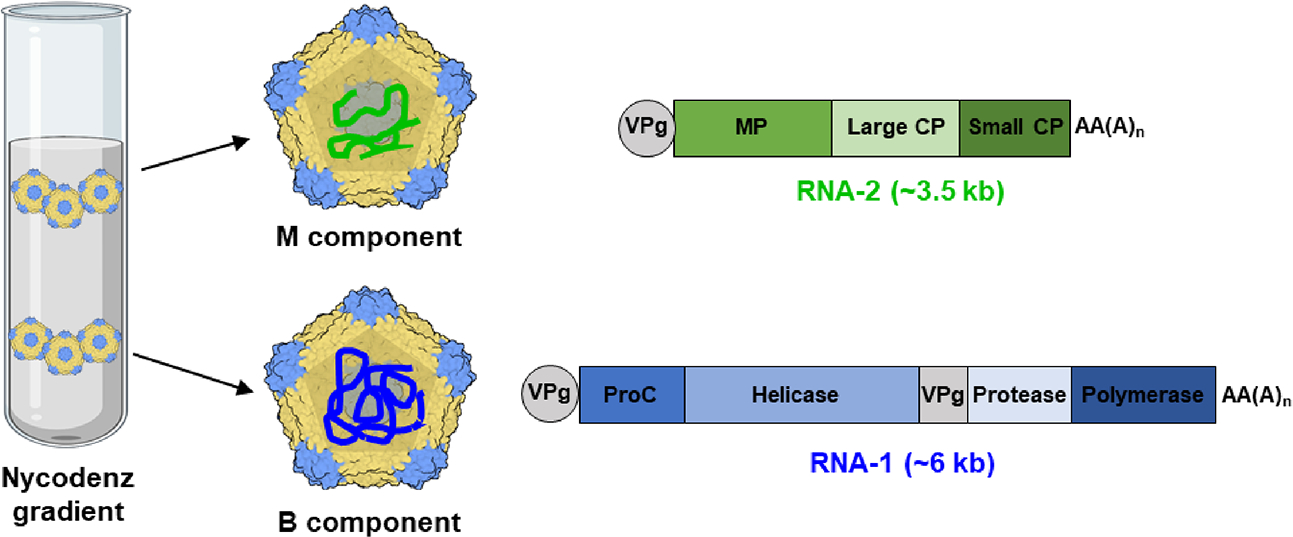
Schematic diagram of the two CPMV components. The separation of CPMV into B and M components by Nycodenz gradient centrifugation reveals identical particles carrying distinct RNAs. The small coat protein (CP) is shown in blue, and the large CP is shown in yellow. VPg—genome-linked protein, MP—movement protein, CP—coat protein, ProC—proteinase cofactor, AA(A)n—PolyA tail.
Figure 2.
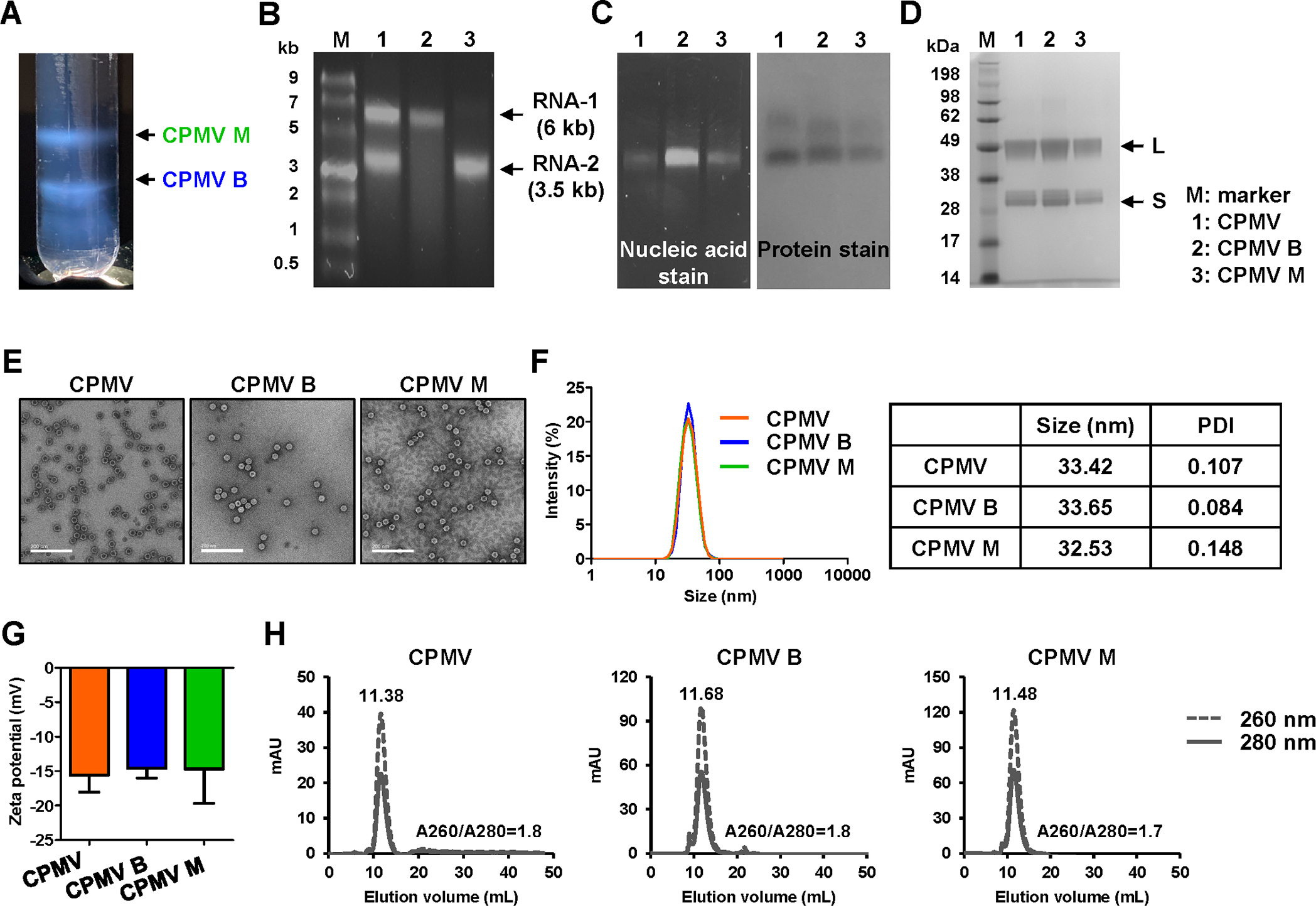
Characterization of mixed and separated CPMV components. (A) Nycodenz gradient after ultracentrifugation at 26,000g for 24 h (visualized under white light). The upper band consists of M components containing RNA-2. The lower band consists of B components containing RNA-1. (B) GelRed agarose gel electrophoresis of nucleic acids extracted from mixed and separated CPMV. Gels were stained with nucleic acid stain, and the image was acquired under UV light. M = ssRNA ladder marker. (C) GelRed agarose gel electrophoresis of mixed and separated CPMV particles. Gels containing nucleic acid stain were imaged under UV light (left, RNA detection) and then stained with protein stain (Coomassie staining) and imaged under white light (right, protein detection). (D) 4–12% Nu-PAGE gel of CPMV particles stained with GelCode Blue Safe protein stain. M = SeeBlue Plus2 molecular weight marker. (E) TEM images of CPMV particles. Scale bar = 200 nm. (F) Size distribution and (G) surface charge of CPMV particles dispersed in 0.1 M KP buffer (pH 7.0). (H) SEC profiles of CPMV particles.
Immunogenicity of Mixed and Separated CPMV Components.
The immunogenicity of the CPMV particles was confirmed using RAW-Blue cells, which are derived from the murine macrophage cell line RAW264.7 and express numerous PRRs. As expected, the mixed CPMV preparation induced a potent immunostimulatory effect activating NF-κB and eliciting SEAP production. The level of SEAP increased 4.17-fold in response to the mixed particles, 4.6-fold in response to the B component, and 3.4-fold in response to the M component, indicating that the mixed and separated particles were similar in immune cell activation (Figure 3A).
Figure 3.
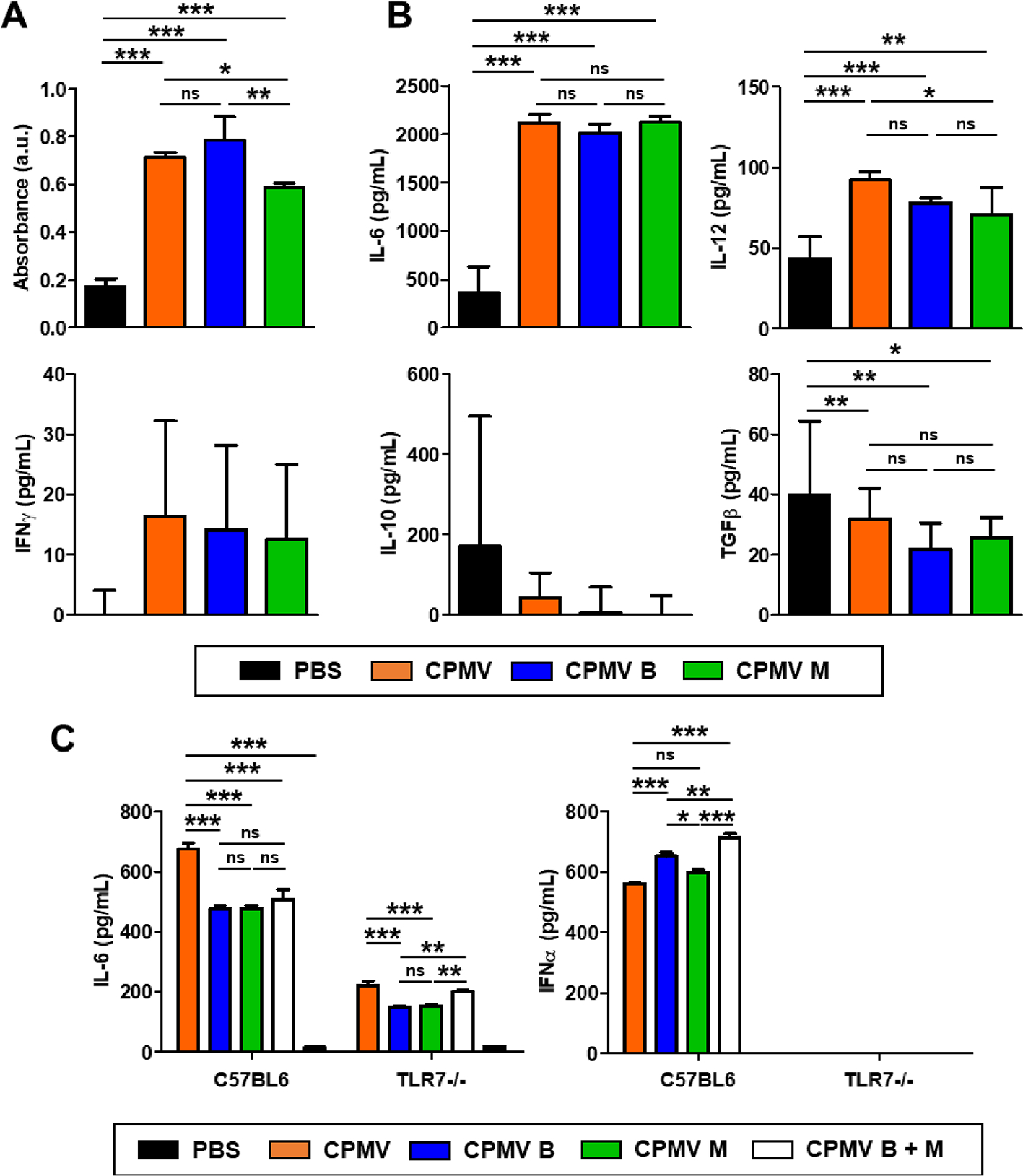
Immunogenicity of mixed and separated CPMV components is similar in a reporter line and tumor-associated peritoneal cells ex vivo. (A) Activation of RAW-Blue cells after incubation with 1 μg of CPMV for 24 h. The results in each treatment group were compared by one-way ANOVA with Tukey’s multiple comparison test. (B) Cytokine expression ex vivo of immune cells harvested from peritoneal cavity washes in PBS-treated CT26 tumor-bearing mice on day 18. Cells were treated with 10 μg of CPMV particles for 24 h, and cytokine levels in the supernatant were determined by ELISA. (C) IL-6 and IFNα production in the splenocytes of each mouse strain following exposure to CPMV particles. The ELISA results in each treatment group were compared by one-way ANOVA with Tukey’s multiple comparison test (***p < 0.001, **p < 0.01, *p < 0.05, ns = not significant).
The mechanism was confirmed by quantifying cytokine levels in peritoneal cavity washes collected from mice bearing CT26-derived tumors. Generally immune suppressive peritoneal cavity cells from the TME were treated with mixed or separated CPMV before measuring the levels of IL-6, IL-12, IFNγ, IL-10, and TGFβ by ELISA (Figure 3B). Like the mixed CPMV preparation, the B and M components increased the abundance of pro-inflammatory cytokines (IL-6, IL-12, and IFNγ), which improve antigen priming, recruit immune cells to the TME, and enhance their cytolytic activity.30 Similarly, both particle types reduced the abundance of immunosuppressive cytokines (IL-10 and TGFβ) to the same level as CPMV. There were no statistical differences between the mixed and separated particles.
To gain further insight into the activation of TLR signaling by CPMV and the separated particles, the stimulation of IL-6 and IFNα was assessed in splenocytes isolated from C57BL/6 and TLR-7−/− mice (Figure 3C). The production of IL-6 was induced to a similar extent by all three preparations. Intact CPMV particles containing viral RNA activate TLRs-2, −4, and −7 with the CPMV capsid activating TLR-2 and −4 and the viral RNA activating TLR-7.14 Therefore, release of IL-6 by all three preparations was partially suppressed in TLR-7−/− mice compared to wild-type C57BL/6 controls, but not completely eliminated due to TLR-2 and −4 activation. The reduction in activation was similar between the CPMV mixed and separated solutions. TLR-7 signaling predominantly regulates the production of type I interferons.31 Therefore, we measured the levels of IFNα to confirm the stimulation of immunity via TLR-7 (Figure 3C). We found that all three preparations induced IFNα production in wild-type mice but none did in the TLR-7−/− mutant.
We concluded that IL-6 and IFNα were similarly stimulated in splenocytes treated with mixed or separated CPMV particles and that all three preparations were similarly immunostimulatory. This similar IFN α production by all three preparations demonstrates that TLR-7 responds quantitatively similarly to both B and M components, despite no sequence similarity of the carried RNA and more RNA encapsidated per particle in B than M components.
Therapeutic Efficacy of Mixed and Separated CPMV Particles In Vivo.
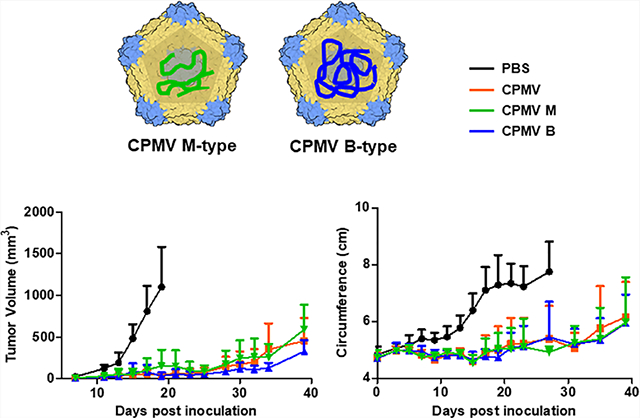
The in vivo therapeutic efficacy of the CPMV particles was investigated using CT26 colon cancer and B16F10 melanoma models. In the colon cancer model, mice were challenged i.p. with 500,000 CT26 cells and treated with each of the three different CPMV preparations or a PBS control three times at weekly intervals, starting on day 3 post tumor inoculation (Figure 4A). As expected, the mixed CPMV treatment significantly reduced the abdominal circumference of tumor-bearing mice (which increases due to tumor load and ascites) and prolonged their survival. On day 27, the average tumor volume in the PBS control group was 7.75 ± 1.06 cm, compared to 5.38 ± 1.16 cm in the CPMV treatment group, a 67% decrease (Figure 4B,C). The B and M components showed similar efficacy, reducing tumor growth on day 27 by 70.32 and 63.35%, respectively. The mice treated with mixed CPMV, B, and M components demonstrated survival rates of 57.14, 57.14, and 71.4%, respectively, at day 40, and all had a significant (**p < 0.01) improvement over the PBS control group (Figure 4D). Tumor growth in the PBS control group was aggressive, with a median overall survival of 23 days. There was no significant body weight loss among the three treatment groups, indicating safety of the treatment (Figure 4E).
Figure 4.
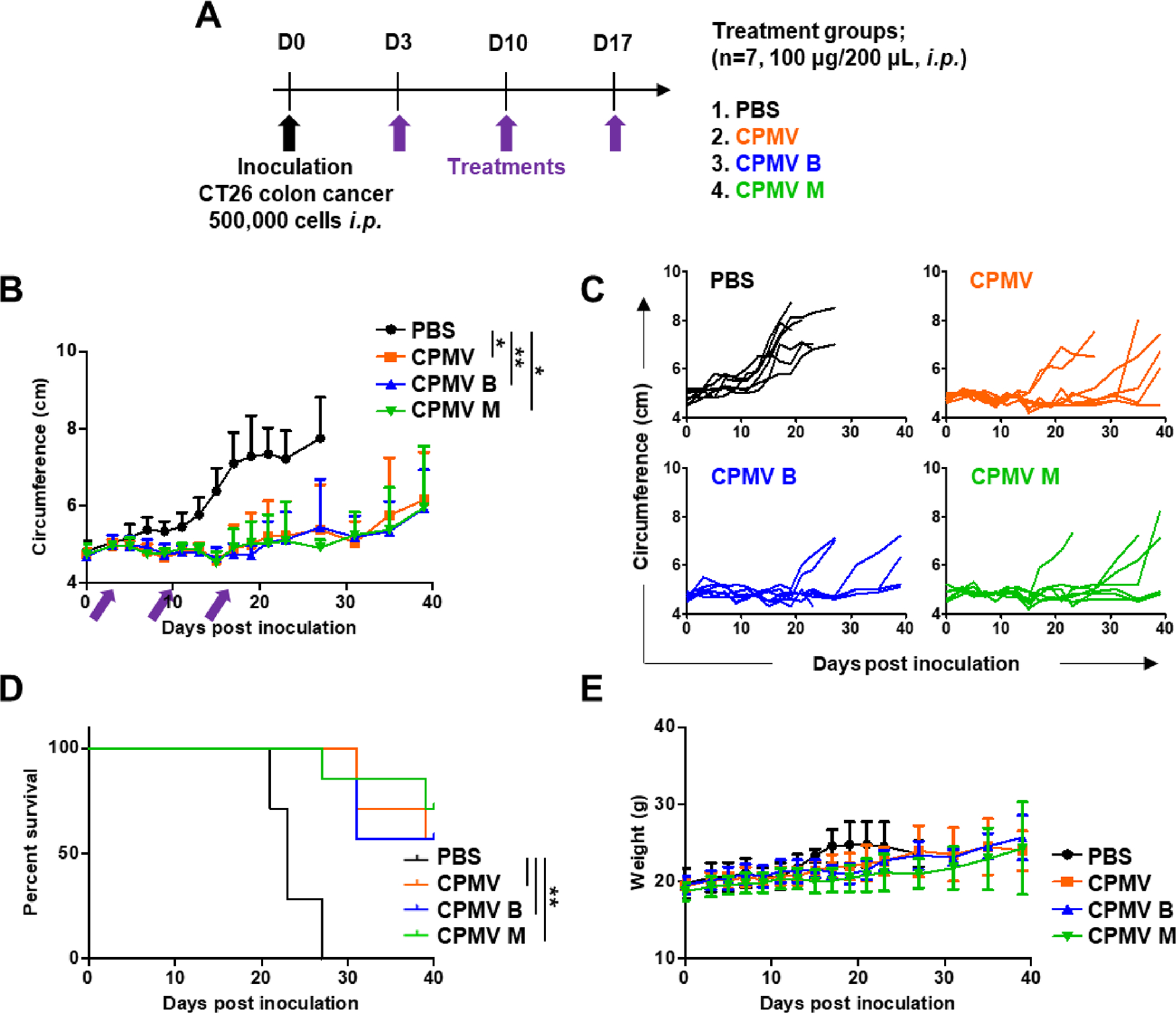
Similar therapeutic effects of mixed and separated CPMV particles in vivo in a mouse model of CT26 colon cancer. (A) Schematic timeline showing the establishment of CT26 colon cancer and subsequent treatments. (B) Change in abdominal circumference. Arrows indicate the dates of injection. (C) Tumor growth kinetics in each treatment group. (D) Survival curves of mice treated with CPMV particles. (E) Change in body weight over time. Tumor volumes were compared using two-way ANOVA, and survival rates were compared using a log-rank (Mantel–Cox) test (**p < 0.01, *p < 0.05).
Similar results were achieved in the B16F10 melanoma mouse model (Figure 5). When dermal tumors reached a volume of ~30 mm3 (day 7), mice were randomly assigned to one of the three CPMV treatment groups or to the PBS control (Figure 5A). The three CPMV treatments significantly delayed tumor growth compared to the PBS control (**p < 0.01). The day-40 tumor volumes of mice treated with mixed CPMV, B, and M components were 454 ± 272.5, 326 ± 139.2, and 590 ± 297.8 mm3, respectively (Figure 5B,C). The survival increased significantly in response to the three CPMV treatments compared to PBS (***p < 0.001), and there was no significant difference between the CPMV groups. More than 50% of the animals in the three treatment groups survived until the end of the experiment at 40 days (Figure 5D).
Figure 5.
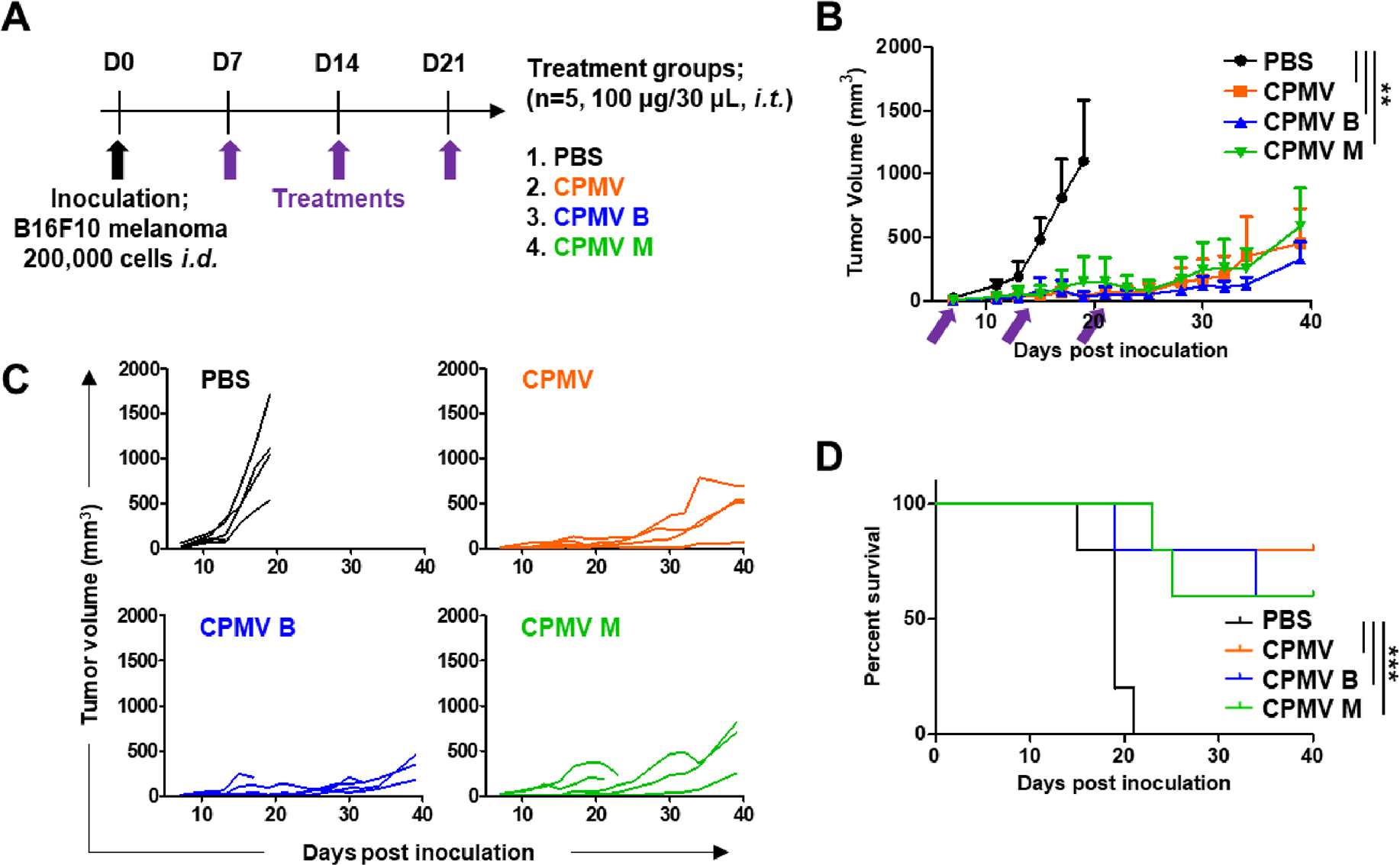
Similar therapeutic effects of mixed and separated CPMV particles in vivo in a mouse model of B16F10 melanoma. (A) Schematic timeline showing the establishment of B16F10 melanoma challenge and the subsequent treatments. (B) Tumor growth curves in mice treated with CPMV particles. Arrows indicate the dates of injection. (C) Tumor growth kinetics in each treatment group. (D) Survival curves of mice treated with CPMV particles. Tumor volumes were compared using an using two-way ANOVA, and survival rates were compared using a log-rank (Mantel–Cox) test (***p < 0.001, **p < 0.01).
To further compare the in vivo immunogenicity of the three CPMV fractions in animal models, mice were i.p. inoculated with CT26 cells and treated with the mixed or separated CPMV particles, as shown in Figure 4A. On day 24, peritoneal cavity washes were collected for analysis of cytokine levels by ELISA (Figure 6). As confirmed in the ex vivo experiments, all three CPMV treatments caused elevated production of IL-6, IL-12, and IFNγ and depleted IL-10 and TGFβ similarly between themselves and significantly compared to the PBS control. We therefore observed no significant differences in immunotherapeutic efficacy between the three CPMV treatment groups and note that all three treatments resulted in significant improvements compared to PBS controls.
Figure 6.
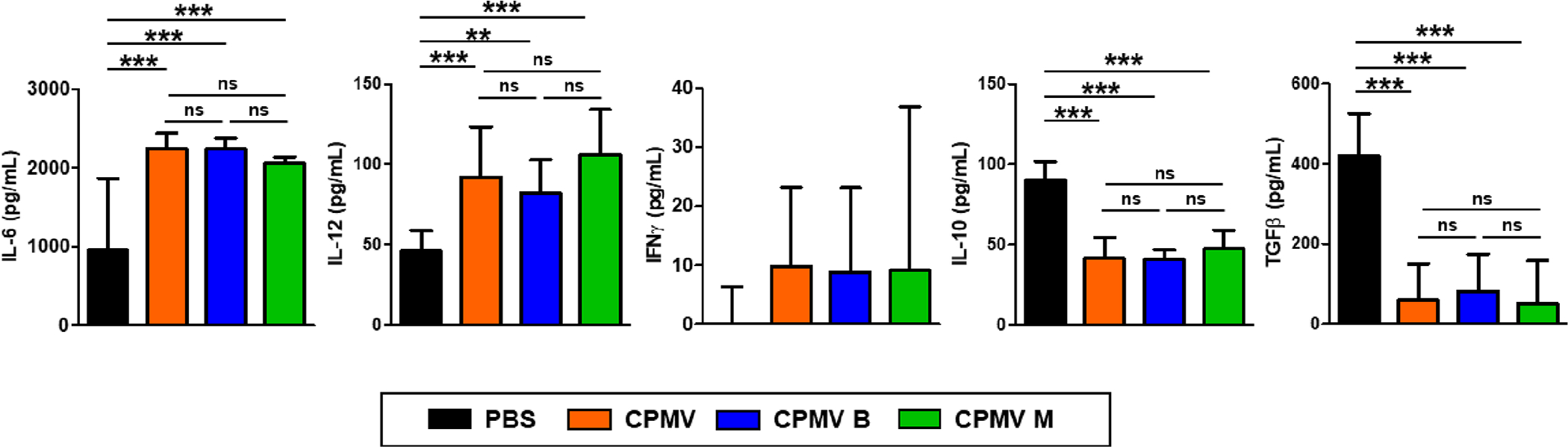
Similar modification of cytokine levels in vivo in a mouse model of CT26 colon cancer. Peritoneal cavity washes were harvested and cytokine levels were quantified by ELISA as in Figure 4A. The results were compared by one-way ANOVA (***p < 0.001, **p < 0.01, ns = not significant).
Although CPMV does not infect animal cells, it has demonstrated potency as a cancer immunotherapy due to its ability to stimulate a strong and durable antitumor response against the primary treated tumor that leads to systemic antitumor efficacy and abscopal response.16,18,32,33 Previous mechanistic studies show that TLR activation, specifically TLR-2, −4, and −7, mediate CPMV in situ vaccination efficacy.14 TLR-7 activation is induced by the RNA of CPMV; therefore, CPMV without RNA demonstrates a significant drop-off in efficacy compared to native CPMV.25 The importance of the RNA is also highlighted by studies in which inactivated CPMV was studied: When UV light or chemicals such as formalin were used to inactivate CPMV by RNA and protein crosslinking, in situ vaccine efficacy was lost or reduced, and this was consistent with a loss in TLR-7 signaling.25,34,35 These studies indicate the significance of genomic RNA in CPMV immunotherapy.
CPMV has a bipartite genome; thereby, it has identical particles carrying distinct RNAs (RNA-1 and RNA-2) and both RNAs must be in a single plant cell to mediate productive infection. In past in situ vaccine experiments, CPMV was always utilized as the harvested mixture of different nanoparticles carrying RNA-1 and RNA-2—the efficacy of each CPMV component had never been explicitly tested. Here, we investigated whether CPMV with encapsidated RNA-1 (B-type) and RNA-2 (M-type) have different immune stimulatory properties or therapeutic efficacy in cancer treatment. This study reveals that (i) both CPMV B-type and M-type attenuate tumor growth and improve survival compared to PBS similarly to mixed CPMV, (ii) CPMV components similarly immunomodulate the antitumor response through expression of pro-inflammatory and immunosuppressive cytokines, and (iii) CPMV components demonstrate similar TLR-associated immune recognition and knockout of TLR-7 dampens efficacy similarly between CPMV mixed, B, or M. Thus, the study shows that the immunotherapeutic effects of CPMV encapsidating RNA-1 or RNA-2 have similar immune-mediated anti-cancer effects. Questions remain as to what area of CPMV RNA bind to TLR-7, and more detailed mechanistic and structure-based experiments investigate that in the future. However, the lack of a difference indicates that binding to TLR-7 is specific to certain regions of the CPMV viral RNA that are present in both RNA-1 and RNA-2 and is not directly proportional to RNA mass, as the longer RNA-1 (6 vs 3.5 kb) did not induce greater immunotherapeutic responses than RNA-2. This may be similar to how unmethylated CpG oligodeoxynucleotides activate TLR-9, but this must be experimentally determined.36 This is the first study comparing immunotherapeutic differences between CPMV particles containing RNA-1 and RNA-2. Our findings show that treatments with any CPMV mixed, B, or M are equally effective immunotherapies in cancer treatment and that isolation and utilization of CPMV B or M will not further improve therapeutic efficacy. Mechanistically, this study contributes to the further understanding of the CPMV in situ vaccine, and from a translational development perspective, this study adds value in lead candidate selection and development of a manufacturing process. Mixed CPMV is infectious toward crops, but separated B or M components are not. Therefore, one could propose to separate the components and use the B or M component as the lead drug candidate. While potency is matched to that of mixed CPMV, separated B or M components offer agronomic safety, because they are noninfectious toward plants.
CONCLUSIONS
CPMV is a promising vaccine component due to its immunostimulatory potency, making it an effective therapeutic strategy for cancer. The aim of this study was to compare the therapeutic efficacy of CPMV particles containing different RNAs for cancer in situ vaccination. Both particle types induced an immune response similar in magnitude and mechanism compared to the mixed CPMV particles. In animal models of melanoma and colon cancer, both particle types showed similar therapeutic efficacy to mixed CPMV, significantly suppressing tumor growth and enhancing the survival rate. We did not observe a statistically significant difference between the native CPMV mixture and the separated RNA-1 and RNA-2 carrying B and M components in any of the efficacy and mechanistic studies. The individual particle types can therefore be utilized as effective immune adjuvants with the same efficacy as native CPMV mixtures. An advantage of using the separated components vs mixed CPMV is that the separated B and M components alone are not infectious to crops and thus provide agronomic safety.
ACKNOWLEDGMENTS
This work was supported in part by NIH grants R01 CA274640 and R01 CA224605 R01 CA253615, a Galvanizing Engineering in Medicine (GEM) Grant through UCSD’s ACTRI and IEM, a grant through CDMRP W81XWH2010742, the Shaughnessy Family Fund for Nano-ImmunoEngineering (nanoIE) at UCSD, and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, 2022R1A6A3A03066056.
Footnotes
The authors declare the following competing financial interest(s): Dr. Steinmetz and Dr. Fiering are co-founders of, have equity in, are paid consultants for, Mosaic ImmunoEngineering Inc. Dr. Steinmetz serves as a board member, and Acting Chief Scientific Officer for Mosaic. The other authors declare no potential COI.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.molpharmaceut.3c00214
Contributor Information
Eunkyeong Jung, Department of Nanoengineering, University of California San Diego, La Jolla, California 92093, United States.
Young Hun Chung, Department of Bioengineering and Moores Cancer Center, University of California San Diego, La Jolla, California 92093, United States.
Chenkai Mao, Department of Microbiology and Immunology, Dartmouth Geisel School of Medicine, Hanover, New Hampshire 03755, United States.
Steven N. Fiering, Department of Microbiology and Immunology and Dartmouth Cancer Center, Dartmouth Geisel School of Medicine, Hanover, New Hampshire 03755, United States
Nicole F. Steinmetz, Department of Nanoengineering, Department of Bioengineering, Department of Radiology, Moores Cancer Center, Institute for Materials Design and Discovery, and Center for Engineering in Cancer, Institute for Engineering in Medicine, University of California San Diego, La Jolla, California 92093, United States Center for Nano-ImmunoEngineering, University of California San Diego, La Jolla, California 92093, United States.
REFERENCES
- (1).Schreiber RD; Old LJ; Smyth MJ Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [DOI] [PubMed] [Google Scholar]
- (2).Vesely MD; Kershaw MH; Schreiber RD; Smyth MJ Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [DOI] [PubMed] [Google Scholar]
- (3).Inthagard J; Edwards J; Roseweir AK Immunotherapy: enhancing the efficacy of this promising therapeutic in multiple cancers. Clin. Sci. 2019, 133, 181–193. [DOI] [PubMed] [Google Scholar]
- (4).Arnedos M; Soria JC; Andre F; Tursz T Personalized treatments of cancer patients: a reality in daily practice, a costly dream or a shared vision of the future from the oncology community? Cancer Treat. Rev 2014, 40, 1192–1198. [DOI] [PubMed] [Google Scholar]
- (5).Li X; Shao C; Shi Y; Han W Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J. Hematol. Oncol. 2018, 11, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rotz SJ; Leino D; Szabo S; Mangino JL; Turpin BK; Pressey JG Severe cytokine release syndrome in a patient receiving PD-1-directed therapy. Pediatr. Blood Cancer 2017, 64, 12. [DOI] [PubMed] [Google Scholar]
- (7).Percik R; Nethanel A; Liel Y Capillary-leak syndrome: an unrecognized early immune adverse effect of checkpoint-inhibitors treatment. Immunotherapy 2021, 13, 653–659. [DOI] [PubMed] [Google Scholar]
- (8).Kethireddy N; Thomas S; Bindal P; Shukla P; Hegde U Multiple autoimmune side effects of immune checkpoint inhibitors in a patient with metastatic melanoma receiving pembrolizumab. J. Oncol. Pharm. Pract. 2021, 27, 207–211. [DOI] [PubMed] [Google Scholar]
- (9).Kumari P; Ghosh B; Biswas S Nanocarriers for cancer-targeted drug delivery. J. Drug Targeting 2016, 24, 179–191. [DOI] [PubMed] [Google Scholar]
- (10).Irvine DJ; Dane EL Enhancing cancer immunotherapy with nanomedicine. Nat. Rev. Immunol. 2020, 20, 321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chung YH; Cai H; Steinmetz NF Viral nanoparticles for drug delivery, imaging, immunotherapy, and theranostic applications. Adv. Drug Delivery Rev 2020, 156, 214–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mohsen MO; Bachmann MF Virus-like particle vaccinology, from bench to bedside. Cell. Mol. Immunol. 2022, 19, 993–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Murray AA; Wang C; Fiering S; Steinmetz NF In Situ Vaccination with Cowpea vs Tobacco Mosaic Virus against Melanoma. Mol. Pharmaceutics 2018, 15, 3700–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mao C; Beiss V; Fields J; Steinmetz NF; Fiering S Cowpea mosaic virus stimulates antitumor immunity through recognition by multiple MYD88-dependent toll-like receptors. Biomaterials 2021, 275, No. 120914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lizotte PH; Wen AM; Sheen MR; Fields J; Rojanasopondist P; Steinmetz NF; Fiering S In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat. Nanotechnol. 2016, 11, 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shukla S; Wang C; Beiss V; Cai H; Washington T II; Murray AA; Gong X; Zhao Z; Masarapu H; Zlotnick A; Fiering S; Steinmetz NF The unique potency of Cowpea mosaic virus (CPMV) in situ cancer vaccine. Biomater. Sci. 2020, 8, 5489–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wang C; Fiering SN; Steinmetz NF Cowpea Mosaic Virus Promotes Anti-Tumor Activity and Immune Memory in a Mouse Ovarian Tumor Model. Adv. Ther. 2019, 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kerstetter-Fogle A; Shukla S; Wang C; Beiss V; Harris PLR; Sloan AE; Steinmetz NF Plant Virus-Like Particle In Situ Vaccine for Intracranial Glioma Immunotherapy. Cancers 2019, 11, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Shukla S; Myers JT; Woods SE; Gong X; Czapar AE; Commandeur U; Huang AY; Levine AD; Steinmetz NF Plant viral nanoparticles-based HER2 vaccine: Immune response influenced by differential transport, localization and cellular interactions of particulate carriers. Biomaterials 2017, 121, 15–27. [DOI] [PubMed] [Google Scholar]
- (20).Hoopes PJ; Wagner RJ; Duval K; Kang K; Gladstone DJ; Moodie KL; Crary-Burney M; Ariaspulido H; Veliz FA; Steinmetz NF; Fiering SN Treatment of Canine Oral Melanoma with Nanotechnology-Based Immunotherapy and Radiation. Mol. Pharmaceutics 2018, 15, 3717–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Alonso-Miguel D; Valdivia G; Guerrera D; Perez-Alenza MD; Pantelyushin S; Alonso-Diez A; Beiss V; Fiering S; Steinmetz NF; Suarez-Redondo M; Vom Berg J; Pena L; Arias-Pulido H Neoadjuvant in situ vaccination with cowpea mosaic virus as a novel therapy against canine inflammatory mammary cancer. J. Immunother. Cancer 2022, 10, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Mao C; Beiss V; Ho GW; Fields J; Steinmetz NF; Fiering S In situ vaccination with cowpea mosaic virus elicits systemic antitumor immunity and potentiates immune checkpoint blockade. J. Immunother. Cancer 2022, 10, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cai H; Wang C; Shukla S; Steinmetz NF Cowpea Mosaic Virus Immunotherapy Combined with Cyclophosphamide Reduces Breast Cancer Tumor Burden and Inhibits Lung Metastasis. Adv. Sci. 2019, 6, 1802281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Patel R; Czapar AE; Fiering S; Oleinick NL; Steinmetz NF Radiation Therapy Combined with Cowpea Mosaic Virus Nano-particle in Situ Vaccination Initiates Immune-Mediated Tumor Regression. ACS Omega 2018, 3, 3702–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang C; Beiss V; Steinmetz NF Cowpea Mosaic Virus Nanoparticles and Empty Virus-Like Particles Show Distinct but Overlapping Immunostimulatory Properties. J. Virol. 2019, 93, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lin T; Chen Z; Usha R; Stauffacher CV; Dai JB; Schmidt T; Johnson JE The refined crystal structure of cowpea mosaic virus at 2.8 A resolution. Virology 1999, 265, 20–34. [DOI] [PubMed] [Google Scholar]
- (27).Kruse I; Peyret H; Saxena P; Lomonossoff GP Encapsidation of Viral RNA in Picornavirales: Studies on Cowpea Mosaic Virus Demonstrate Dependence on Viral Replication. J. Virol. 2019, 93, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Leong HS; Steinmetz NF; Ablack A; Destito G; Zijlstra A; Stuhlmann H; Manchester M; Lewis JD Intravital imaging of embryonic and tumor neovasculature using viral nanoparticles. Nat. Protoc. 2010, 5, 1406–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Steinmetz NF; Evans DJ; Lomonossoff GP Chemical introduction of reactive thiols into a viral nanoscaffold: a method that avoids virus aggregation. ChemBioChem 2007, 8, 1131–1136. [DOI] [PubMed] [Google Scholar]
- (30).Berraondo P; Sanmamed MF; Ochoa MC; Etxeberria I; Aznar MA; Perez-Gracia JL; Rodriguez-Ruiz ME; Ponz-Sarvise M; Castanon E; Melero I Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sakata K; Nakayamada S; Miyazaki Y; Kubo S; Ishii A; Nakano K; Tanaka Y Up-Regulation of TLR7-Mediated IFN-alpha Production by Plasmacytoid Dendritic Cells in Patients With Systemic Lupus Erythematosus. Front. Immunol. 2018, 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Chung YH; Park J; Cai H; Steinmetz NF S100A9-Targeted Cowpea Mosaic Virus as a Prophylactic and Therapeutic Immunotherapy against Metastatic Breast Cancer and Melanoma. Adv. Sci 2021, 8, No. e2101796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Koellhoffer EC; Steinmetz NF Cowpea Mosaic Virus and Natural Killer Cell Agonism for In Situ Cancer Vaccination. Nano Lett. 2022, 22, 5348–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Jung E; Mao C; Bhatia M; Koellhoffer EC; Fiering SN; Steinmetz NF Inactivated Cowpea Mosaic Virus for In Situ Vaccination: Differential Efficacy of Formalin vs UV-Inactivated Formulations. Mol. Pharmaceutics 2023, 20, 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chariou PL; Beiss V; Ma Y; Steinmetz NF In situ vaccine application of inactivated CPMV nanoparticles for cancer immunotherapy. Mater Adv 2021, 2, 1644–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Krieg AM; Yi AK; Matson S; Waldschmidt TJ; Bishop GA; Teasdale R; Koretzky GA; Klinman DM CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995, 374, 546–549. [DOI] [PubMed] [Google Scholar]