

Abstract
Branched metal chalcogenide nanostructures with well-defined composition and configuration are appealing photocatalysts for solar-driven organic transformations. However, precise design and controlled synthesis of such nanostructures still remain a great challenge. Herein, we report the construction of a variety of highly symmetrical metal sulfides and heterostructured icosapods based on them, in which twenty branches were radially grown in spatially ordered arrangement, with a high degree of structure homogeneity. Impressively, the as-obtained CdS–PdxS icosapods manifest a significantly improved photocatalytic activity for the selective oxidation of biomass-relevant alcohols into corresponding aldehydes coupled with H2 evolution under visible-light irradiation (>420 nm), and the apparent quantum yield of the benzyl alcohol reforming can be achieved as high as 31.4% at 420 nm. The photoreforming process over the CdS–PdxS icosapods is found to be directly triggered by the photogenerated electrons and holes without participation of radicals. The enhanced photocatalytic performance is attributed to the fast charge separation and abundant active sites originating from the well-defined configuration and spatial organization of the components in the branched heterostructures.
Highly symmetrical branched semiconductor-based heterostructured icosapods are successfully prepared by consecutive cation exchange and exhibit enhanced photocatalytic activity for photoreforming biomass-relevant alcohols.
Introduction
Exploiting solar-energy-driven organic transformations via artificial photosynthesis, e.g., selective oxidation of biomass-relevant alcohols, reduction of nitroaromatics and oxidation coupling of amines, for the production of value-added chemicals has attracted widespread attention in recent years, as it provides an alternative to replace fossil fuels, thus achieving the goal of carbon neutrality.1–10 Developing an efficient artificial photocatalyst with outstanding performance is the key toward this goal. Among various photocatalytic materials, colloidal metal chalcogenide semiconductor-based nanomaterials have shown great priorities toward these photoreforming reactions due to their large molar extinction coefficients, adjustable band edge positions and rapid carrier migration capabilities.11–18 For example, CdS nanostructures modified with a Ni cocatalyst could efficiently transform alcohols into corresponding aldehydes/ketones or selectively oxidize 5-hydroxymethylfurfural (HMF) to tunable products under visible light irradiation.19,20 However, it is still challenging to achieve desired photocatalytic performance with high conversion and excellent selectivity.21–25 Firstly, conventional architectures, such as core@shell, Janus, and dumbbell nanostructures, often suffer from fast charge recombination, poor surface passivation and slow oxidation-reduction kinetics, ultimately impeding their photocatalytic conversion rate.26–28 Secondly, many patchy semiconductor-cocatalyst systems and hierarchical heterostructures already exhibit promising results, but the randomly arranged components and unclear interfaces in these structures make it difficult to reveal structure–property correlation.29–35 Hence, delicate engineering of favorable metal chalcogenide-based photocatalysts with controlled morphology and configuration as well as a well-defined interface would be a powerful strategy to enhance their performance but it has not yet been realized.
Highly symmetrical branched metal chalcogenide nanostructures, featuring well-defined morphology and spatial organization, have been widely studied and showed versatile applications in photocatalysis, energy storage and conversion.36–40 Compared with conventional colloidal metal chalcogenides, they exhibit distinct advantages originating from their unique architecture, i.e., high-density branches with adjustable length would be beneficial for light absorption/scattering and charge separation/transport. Moreover, large specific surface area of such branches would provide abundant active sites, and accessible and interconnected networks in these nanostructures can promote the transport of guest organic species to accessible reaction sites. Therefore, it can be expected that highly symmetrical branched nanostructures could be an optimal architecture for improving activity toward photocatalytic organic transformations.41–45 However, prototypical branched metal chalcogenide nanostructures are mainly tetrapods and octapods, and other types of highly branched nanostructures with more complicated symmetries have been rarely reported.46,47
Recently, we have reported the successful synthesis of a new type of highly symmetrical branched nanostructure, Ag–CdS icosapods, in which twenty CdS nanorods were radially grown on the [111] facets of multi-twinned Ag icosahedra.48 In this work, highly symmetrical branched nanostructures with tunable compositions have been controllably synthesized via consecutive cation exchange by using Ag–CdS icosapods as templates (Scheme 1). First, Cu2S icosapods were obtained by the cation exchange of the as-prepared Ag–CdS icosapods with cuprous ions, accompanied by the removal of Ag cores to form hollow cavities. Subsequently, a series of highly monodisperse and uniform metal sulfide icosapods (MS, M = Cd, Zn, Mn, Co, and Ni) can be obtained by using Cu2S icosapods as templates for secondary cation exchange. Furthermore, PdxS icosapods and CdS–PdxS heterostructured icosapods with amorphous PdxS tips can also be produced. Interestingly, the as-prepared CdS–PdxS icosapods show excellent performance for visible light-induced biomass-relevant alcohol reformation (Sel. >94.8%) integrated with H2 evolution compared with blank CdS icosapods and their other counterparts. The apparent quantum yield (AQY) of the benzyl alcohol (BA) reforming on CdS–PdxS icosapods is measured to be as high as 31.4% at 420 nm. In-situ electron paramagnetic resonance (EPR) and reactive species trapping experiments indicate that the photoreforming process over the CdS–PdxS icosapods is directly triggered by the electrons and holes without involving radicals. The significantly improved photocatalytic activity can be attributed to effective charge separation and transport capability, which are verified by the photoelectrochemical and ultrafast transient absorption spectroscopy measurements.
Scheme 1. Schematic illustration for the preparation of a variety of icosapods with tunable compositions by consecutive cation exchange.

Results and discussion
In a typical synthesis, Ag–CdS icosapods with an arm length of ∼45 nm were well prepared according to our recent report with a slight modification (see the ESI for experimental details and Fig. S1†).48 To prepare the Cu2S icosapods, the as-obtained Ag–CdS icosapods were then dropped into a solution of CuI in acetonitrile/methanol (v/v, 4 : 1) at room temperature with ultrasonication. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images show that the morphology and size of the obtained products are well preserved with respect to the Ag–CdS icosapods, except that obvious cavities appeared in the center of the icosapods (Fig. 1a and b). The X-ray diffraction (XRD) pattern reveals that all diffraction peaks can be well matched with chalcocite Cu2S, while the diffraction peaks ascribed to fcc-Ag in pristine Ag–CdS icosapods are absent (Fig. 1c). The measured lattice fringe of 0.34 nm can be assigned to the (002) plane of chalcocite Cu2S (Fig. 1b, inset). The Ag cores may be oxidized or diffused outward to form hollow cavities during the cation exchange, which was also confirmed by scanning electron microscopy-energy dispersive X-ray spectrometry (SEM-EDX). As shown in Fig. 1d and Fig. S2a,† the Ag cannot be distinguished, and only Cu and S exist in the product with an atomic ratio of ∼1.85, which is close to the stoichiometric value for Cu2S. Furthermore, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and the corresponding energy dispersive spectroscopy (EDS) elemental mappings show that the Cu and S are homogeneously distributed over the branches of the whole icosapods (Fig. 1e). The X-ray photoelectron spectroscopy (XPS) survey spectrum displays the existence of Cu and S elements in the Cu2S icosapods (Fig. S3a†). In the high-resolution Cu 2p spectrum, the two fitted strong signals peaking at 952.7 and 932.9 eV can be ascribed to Cu+ 2p1/2 and Cu+ 2p3/2 of Cu2S, whereas the other peaks located at around 954.4 and 934.4 eV can be assigned to Cu2+ 2p1/2 and Cu2+ 2p3/2 of the remnant copper oxide by oxidization, respectively (Fig. 1f). For the S 2p spectrum, two fitted peaks located at 163.0 and 161.8 eV can be ascribed to S2− 2p1/2 and S2− 2p3/2 of Cu2S, respectively (Fig. S3b†).49,50 All in all, the Cu2S icosapods have been successfully prepared with high yield by cation exchange with the assistance of ultrasonication.
Fig. 1. (a) TEM and (b) HRTEM images of Cu2S icosapods. The inset of b displays the HRTEM image taken from the white dashed square. (c) XRD pattern of the Cu2S icosapods. The standard diffraction peaks for chalcocite Cu2S (PDF#26-1116) are used as references. (d) SEM-EDX profile and the corresponding elemental contents (inset) of the Cu2S icosapods. (e) HAADF-STEM image and the corresponding EDS elemental mappings of the Cu2S icosapods. (f) High-resolution XPS spectrum of Cu 2p for the Cu2S icosapods.

It is well known that Cu2−xS (0 < x< 1) nanocrystals with different morphologies are widely chosen as the host materials for the cation exchange reaction because of their fast Cu+ diffusion and solvation capabilities with the introduction of alkyl phosphine according to the hard-soft acid-base theory.51–54 Therefore, a variety of icosapods with tunable compositions can be easily obtained by the cation exchange of the Cu2S icosapods with different metal ions (i.e., Cd2+, Zn2+, Mn2+, Co2+, and Ni2+). TEM and SEM images illustrate the corresponding morphologies of the as-prepared five types of products, which intactly inherit the overall configuration of the Cu2S icosapods (Fig. S4a1–e1† and Fig. S2b–f†). The XRD patterns confirm that the Cu2S icosapods have been successfully transformed into the corresponding icosapods with wurtzite structures (CdS, ZnS, and MnS) and zinc blende structures (Co9S8 and Ni3S4), respectively (Fig. S4a4–e4†). The HRTEM images and fast Fourier transforms (FFTs) reveal the highly crystalline structure of the as-prepared icosapods (Fig. S4a2–e2†). The measured lattice fringes of 0.34 and 0.31 nm are consistent with the (002) planes of wurtzite CdS and ZnS, respectively (Fig. S4a2 and b2†). The FFT patterns show that the longitudinal directions of MnS, Co9S8, and Ni3S4 arms in icosapods are parallel to [0001]wz, [111]zb and [111]zb directions, respectively (Fig. S4c2–e2†). The HAADF-STEM images and corresponding EDS elemental mappings were recorded where the corresponding metals and S were homogeneously overlapped in the finally yielded icosapods (Fig. S4a3–e3†). The XPS spectra of these five types of icosapods are shown in Fig. S5–S9,† verifying the successful preparation of the corresponding metal sulfide icosapods.
Furthermore, the as-obtained CdS icosapods can also be completely or partially cation exchanged with Pd2+, by adjusting the feeding amount of the precursor, to get PdxS and CdS–PdxS icosapods, respectively. After the cation exchange reaction, the overall morphology of the PdxS and CdS–PdxS icosapods still remains the same (Fig. 2a and d) and shows a high degree of shape uniformity (Fig. S10†), but the crystal structure changes accordingly. The XRD pattern of the PdxS icosapods only displays a broad peak at around 40°, indicating the amorphous structure of the as-obtained PdxS icosapods (Fig. 2f, blue line). The HRTEM image of the PdxS branches shows the disordered arrangement without clear lattice fringes (Fig. 2b), and the selected area electron diffraction (SAED) pattern only displays diffusing rings, further confirming its amorphous structure (Fig. 2b, inset). The overview SEM-EDX profile reveals that the atomic ratio of Pd and S in PdxS icosapods is about 4.9 (Fig. S11a†). The HAADF-STEM image and the corresponding EDS elemental mappings of PdxS icosapods verify that Pd and S distribute homogeneously over the icosapods (Fig. 2c). The XPS spectrum of the PdxS icosapods indicates the existence of Pd0 and Pd2+ (Fig. S11c†).55–57 While the CdS icosapods undergo partial cation exchange with Pd2+, only the tip areas of each arm have transformed into PdxS and the main trunk of the CdS arm still remains (Fig. 2d and e). The FFT pattern of the selected area (Fig. 2e, white dashed square) demonstrates that only one set of diffraction spots appeared for a single arm, while the FFT pattern from the tip area of the arm shows no obvious diffraction spots, indicating the amorphous structure of the PdxS tip. Moreover, the XRD pattern of the CdS–PdxS icosapods is in accordance with the hexagonal wurtzite CdS and no cubic Pd or PdxS crystal structure can be detected (Fig. 2f, red line). The HAADF-STEM image of CdS–PdxS icosapods illustrates clear contrast between the arms and the tips, and the corresponding EDS elemental mappings further confirm that the Pd and Cd elements are confined in the tip and trunk areas, respectively, while the S element distributes homogeneously over the whole icosapods (Fig. 2g and h). The atomic ratio of Pd in CdS–PdxS icosapods is 5.3% according to SEM-EDX (Fig. S12a†). The XPS spectra of CdS–PdxS icosapods are given in Fig. S12b–e,† revealing the similar valence states of each element compared with the aforementioned PdxS and CdS icosapods (Fig. S11† and S5†). As seen in Fig. 2i, the CdS and CdS–PdxS icosapods exhibit similar band edge absorption at around 485 nm in the UV-vis extinction spectrum.
Fig. 2. TEM and HRTEM images of (a and b) PdxS and (d and e) CdS–PdxS icosapods, respectively. The inset of b shows the SEAD image of PdxS icosapods. The insets of (e) display FFT images taken from the selected squares. (c) HAADF-STEM image and the corresponding EDS elemental mappings of the amorphous PdxS icosapods. (f) XRD patterns of the PdxS and CdS–PdxS icosapods. (g) HAADF-STEM image and (h) the corresponding EDS elemental mappings of CdS–PdxS icosapods. (i) UV-vis extinction spectra of the CdS and CdS–PdxS icosapods.

The CdS–PdxS icosapods are expected to show excellent performance for photocatalytic organic transformations. Firstly, the icosapod structures are mainly composed of CdS, which possesses a suitable band gap (∼2.4 eV) for strong visible light absorption. Secondly, the components of CdS and PdxS form lots of interfaces, which would accelerate the separation efficiency and transport of photogenerated carriers. Finally, due to the large specific surface area of the branched nanostructures and the low-coordinated Pd in amorphous PdxS tips, the oxidation and reduction reactions induced by holes and electrons could proceed smoothly and effectively. As a proof-of-concept demonstration, upgrading oxidation of biomass-relevant alcohols into corresponding aldehydes coupled with H2 evolution under visible-light irradiation (λ > 420 nm) was investigated by using the CdS–PdxS icosapods as the photocatalyst. The H2 and corresponding liquid products were identified by gas chromatography spectrometry (GC) and gas chromatograph-mass spectrometry (GC-MS), respectively (Fig. S13–S20†). The UV-vis extinction spectra of different photocatalysts in Fig. 3a are shown in Fig. S21.† The dehydrogenation of BA into benzaldehyde (BZD) was initially carried out as a model photoreaction in an acetonitrile solution containing 0.1 mmol of BA and 10 mg of the photocatalysts, which was irradiated with a xenon lamp under a nitrogen atmosphere. As displayed in Fig. 3a, both Pd and PdxS nanoparticles don't show any activity for the dehydrogenation reaction after 2 h of illumination. Interestingly, the CdS–PdxS icosapods show a superior photoactivity compared with blank CdS icosapods and a physical mixture of CdS icosapods and PdxS nanoparticles with the same Pd content (Fig. 3a). The conversion and selectivity of BA on CdS–PdxS icosapods are up to 98.1% and 99.1%, respectively, which are 31.6 and 2.6 times higher than those on single CdS icosapods (Table 1, entry 1). Although the conversion of BA on the physical mixture is relatively moderate, many undesired products appeared and the selectivity is only 39.5%. Moreover, one-dimensional CdS–PdxS nanorods (NRs, Fig. S22†), which also possess a similar Pd atomic ratio, only show quite low conversion (43.1%) and good selectivity (94.0%), reflecting the distinct architecture superiority of the heterostructures. In this case, BA can be almost completely converted by CdS–PdxS NRs when the reaction time extends to 4 h, while the selectivity decreased to 90.0%. The interfacial electronic structures of the CdS–PdxS icosapods and nanorod photocatalysts have been measured by ultraviolet photoelectron spectroscopy (Fig. S23†). An apparent Schottky junction is formed in both CdS–PdxS samples due to very close Fermi levels of PdxS with respect to Pd NPs. In addition, the as-prepared CdS–PdxS icosapods and nanorod photocatalysts not only meet the reduction potential required for the conversion of H+ to H2, but also meet the oxidation potential of selective oxidation of BA to BAD. To evaluate the recyclability and durability of the CdS–PdxS icosapod photocatalysts, a recycling experiment has been performed, indicating a marginal loss in the photocatalytic performance even after four cycles (Fig. 3b). Impressively, the optimized H2 evolution and AQY (calculated based on H2) of the BA reforming on CdS–PdxS icosapods are measured to be 16.01 mmol g−1 h−1 and 31.4% at 420 nm, respectively, which is a competitive photocatalytic system for selective organic transformation integrated with H2 evolution (Table S1†).
Fig. 3. (a) Photocatalytic oxidation performance of BA over Pd nanoparticles, PdxS nanoparticles, CdS–PdxS NRs, CdS icosapods, physical mixture of CdS icosapods and PdxS nanoparticles, and CdS–PdxS icosapods under visible-light irradiation (λ > 420 nm). (b) Photocatalytic recycling tests for the CdS–PdxS icosapods. (c) Quenching experiments with different reactive species photocatalyzed by the CdS–PdxS icosapods. (d) In situ EPR spectra in N2 saturated CH3CN solution with the addition of DMPO.

Photocatalytic selective oxidation of biomass-relevant alcohols over CdS–PdxS icosapods under visible-light irradiation (λ > 420 nm).
| Entry | Substrate | Product | Conversion (%) | Selectivity (%) | H2 (μmol) | Reaction time (h) |
|---|---|---|---|---|---|---|
| 1 |
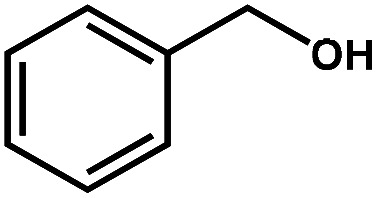
|
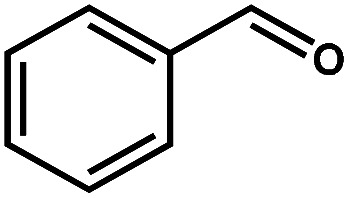
|
98.1 | 99.1 | 95.9 | 2 |
| 2 |
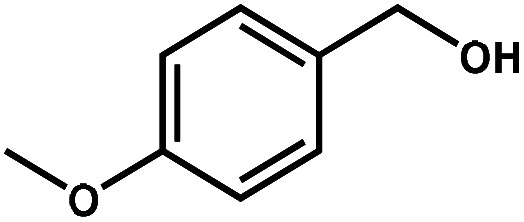
|
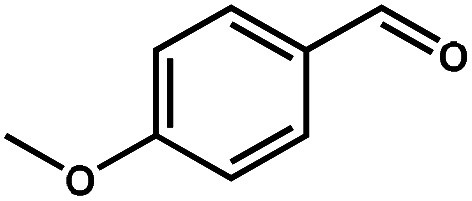
|
98.6 | 99.7 | 96.5 | 2 |
| 3 |
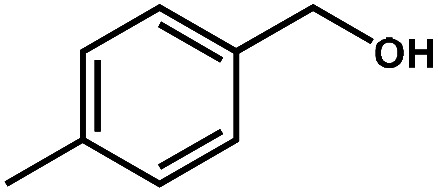
|
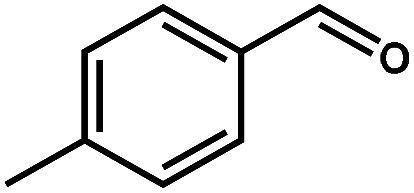
|
98.3 | 99.8 | 96.3 | 2 |
| 4 |
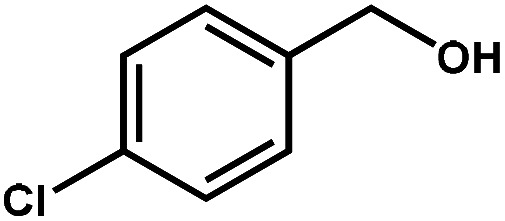
|
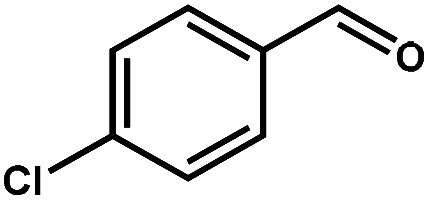
|
97.6 | 99.7 | 95.0 | 2.5 |
| 5 |
|
|
98.8 | 99.3 | 96.4 | 4 |
| 6 |
|
|
47.8 | 94.8 | 42.9 | 8 |
| 7 |
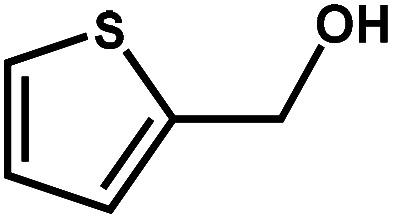
|
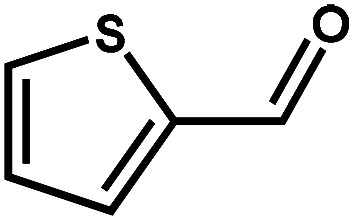
|
89.7 | 99.3 | 85.8 | 6 |
| 8 |
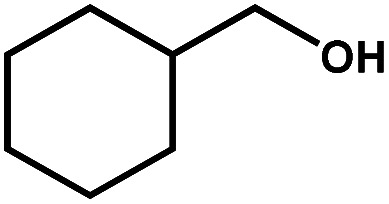
|
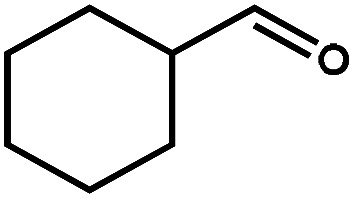
|
23.9 | 90.4 | 22.2 | 24 |
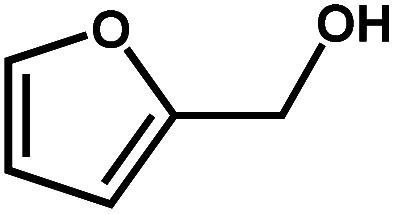
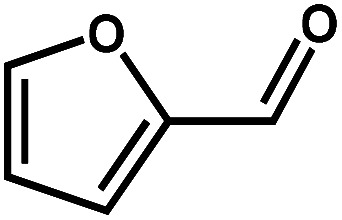
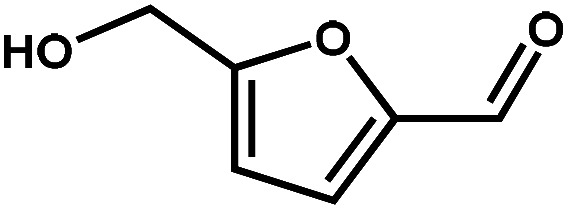
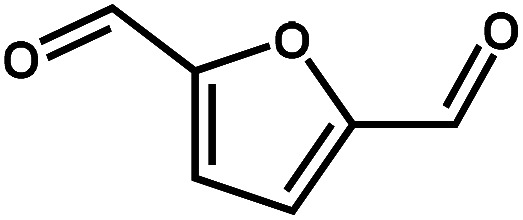
Next, the substrate scope of the CdS–PdxS icosapod system with regard to photocatalytic selective oxidation of many types of biomass-relevant alcohols has also been investigated. It is found that all aromatic and aromatic heterocyclic alcohols were oxidized smoothly to the corresponding aldehydes with excellent selectivity (>94.8%, Table 1, entries 1–7). However, in the case of alicyclic alcohols, although their conversions are low, the selectivity of dehydrogenative products is still high (Table 1, entry 8). Notably, the molar ratio of all oxidation products (aldehydes) and the reduction product (H2) is calculated to be nearly 1, implying a stoichiometric dehydrogenation reaction (Table 1, entries 1–8). The influence of the electronic effect of functional groups at the para-position on the photocatalytic oxidation of substituted benzylic alcohol by CdS–PdxS icosapods has also been observed. By taking unsubstituted BA as the reference, the aldehyde production rate is enhanced by introduction of electron-donating groups, e.g. –OCH3 and –CH3 (entries 2 and 3), while the installation of an electron-withdrawing group (–Cl, entry 4) can apparently suppress the reaction rate and the illumination time needs to be extended to 2.5 h to make the conversion up to 97%. It is worth mentioning that furfuryl alcohol (FA) and HMF, two kinds of most crucial biomass-derived platform chemicals, also show exciting conversion and selectivity into corresponding furfural and 2,5-diformylfuran over the photocatalyst (Table 1, entries 5 and 6). Specifically, under dark conditions, no transformation occurs for FA and HMF over the CdS–PdxS icosapods (Fig. S24†). However, the furfural is obtained with a conversion and selectivity of 98.8% and 99.3% after only 4 h of light irradiation, while the single materials of CdS icosapods are measured to be 1.5% and 13.2%, respectively. As for HMF, the formation of overoxidized products, such as 5-hydroxymethyl-2-furancarboxylic acid, 5-formyl-2-furancarboxylic acid, and 2, 5-furandicarboxylic acid, cannot be observed in our case over the CdS–PdxS icosapods within 8 h of illumination.14,20 The conversion and selectivity are up to 47.8% and 94.8%, which are 59.8 and 2.6 times higher than those of CdS icosapods, respectively. Compared to the reported studies, the as-prepared CdS–PdxS icosapods exhibit excellent photoreforming performance over such biomass-derived intermediates (Table S1†).
To unveil the possible role of the reactive species and radical intermediates in the photoredox-catalyzed system, trapping experiments have been executed by taking BA transformation as an example (Fig. 3c). Because the selective oxidation of BA into BZD was carried out so quickly, the photocatalytic reactions implemented in an hour were monitored in the trapping experiments. As seen in Fig. 3c, the addition of tetrachloromethane (CCl4) as an electron scavenger completely ceased the H2 evolution, while the conversion efficiency of BA oxidation remained almost unchanged over CdS–PdxS icosapods. However, when triethylamine (TEA) was introduced for irreversible consumption of holes, the formation of BZD retarded apparently and the H2 evolution kept up production. It is widely reported that the ˙CH(OH)Ph radical, which can be induced by the interaction between BA and h+, is often considered the key intermediate for selective photo-oxidation of alcohol into aldehyde.58,59 Therefore, butylated hydroxytoluene (BHT), a commonly used carbon-centered free radical trapping reagent, was added to verify whether ˙CH(OH)Ph radicals are involved in the photocatalytic process.59 Clearly, both the conversion of BA and H2 evolution remain, suggesting the simultaneous participation of electrons and holes for BZD and H2 production instead of ˙CH(OH)Ph radicals. Furthermore, the carbon-centered free radicals have been further explored by the in situ EPR measurements with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the spin-trapping regent to gain more information on the radical intermediate mechanism of the reaction (Fig. 3d). Under the dark and light irradiation conditions, no obvious signal appeared over the solution, manifesting no carbon-centered free radicals involved in the photoreaction. Otherwise, the ˙CH(OH)Ph radical is gradually evolved over CdS icosapods with illumination and eventually trapped by DMPO, as evidenced by the characteristic signal peaks belonging to the DMPO-CH(OH)Ph adduct.58,59
Based on the above results, the photoreforming of biomass-relevant alcohols can be proceeded by direct participation of the photo-generated electrons and holes in the CdS–PdxS icosapods. To deeply explore the origin of striking photocatalytic activity of the CdS–PdxS icosapods, the separation and transport dynamics of photogenerated charges were systematically surveyed. First, the photoelectrochemical (PEC) experiment reveals that the transient photocurrent response for CdS–PdxS icosapods is much higher with respect to that of the single component of CdS icosapods, indicating the importance of construction of heterostructures to enhance the photocatalytic activity (Fig. S25†). Moreover, ultrafast transient absorption spectroscopy (TA) was used to uncover the photo-excited charge dynamics of the CdS–PdxS and CdS icosapods under 400 nm laser pumping. The fluence dependence of the time-resolved transient absorption measurement indicates that the feature is derived from one-photon excitation (Fig. S26†). As shown in Fig. 4a and b, the CdS–PdxS and CdS icosapods show similar 1Σ exciton bleach signals at around 480 nm, originating from the state filling of the conduction band 1σe electron level for CdS.60 The corresponding 1Σ exciton bleach recovery kinetics within 4.8 ns for both icosapods clearly demonstrate that the kinetics recovery process becomes significantly faster over the CdS–PdxS icosapods, indicating the enhanced depopulation rate of conduction band electrons in the icosapods (Fig. S27† and 4c). As reported in the literature, the CdS nanorods with a small amount of PdxS tips would favor electron transfer in heterostructures.55,56 Similarly, the photogenerated electrons would immediately transfer from the CdS trunk to the amorphous PdxS tip in the CdS–PdxS icosapods, instead of the direct recombination of charge carriers or possible charge trapping by the defects for the CdS icosapods.61–63 In this way, spatially separated photogenerated electrons and holes in CdS–PdxS icosapods offer more opportunities to separately participate in the corresponding photocatalytic reactions. Therefore, a possible photocatalytic mechanism for selective oxidation of biomass-relevant alcohols into corresponding aldehydes coupled with H2 evolution over the CdS–PdxS icosapods is depicted in Fig. 4d. Initially, the photoinduced carriers are generated in CdS under light illumination. Then, the photogenerated electrons promptly transfer to the domains of PdxS tips, while the holes still localize at CdS trunks. The protons from–OH bonds of the biomass-relevant alcohols, which pre-adsorb on the PdxS tips of the CdS–PdxS icosapods, can be reduced by photoinduced electrons and thus form a Pd–H hydride and an alkoxide anion. Subsequently, the alkoxide anion is immediately photooxidized into corresponding aldehyde products by the photoinduced holes in the CdS trunks and releases another Pd–H hydride simultaneously. Finally, two Pd–H hydrides generate H2 in the PdxS tips (Fig. 4d).
Fig. 4. TA spectra of the (a) CdS–PdxS and (b) CdS icosapods at different delay times with 400 nm excitation, respectively. (c) Comparison of 1Σ exciton bleach recovery kinetics of the CdS–PdxS (at 484 nm) and CdS icosapods (at 480 nm) when excited at 400 nm. (d) Proposed possible photocatalytic mechanism for selective oxidation of biomass-relevant alcohols into corresponding aldehydes integrated with H2 evolution over the CdS–PdxS icosapods under visible-light irradiation.

Conclusion
In summary, a series of highly branched metal sulfides and heterostructured icosapods based on them with delicate architecture and tunable compositions have been successfully produced by consecutive cation exchange. All obtained icosapods exhibit high symmetry and integral structure homogeneity. As a proof of concept, the CdS–PdxS icosapods displays remarkably enhanced photocatalytic activity for selective oxidation of biomass-relevant alcohols, including aromatic and aromatic heterocyclic alcohols, into corresponding aldehydes (Sel. >94.8%) coupled with H2 evolution. Impressively, the AQY of the benzyl alcohol reforming on the CdS–PdxS icosapods can be achieved as high as 31.4% at 420 nm. The photoreforming of biomass-relevant alcohols is found to be followed by direct participation of the photo-generated electrons and holes in the CdS–PdxS icosapods, rather than the involvement of radicals. The superior photocatalytic performance of the CdS–PdxS icosapods results from the enhanced charge separation capability and the fast electron migration in the heterostructured icosapods. We believe that this work will inspire further study on the controllable construction of heterostructures with well-defined architectures and explore their photocatalytic performance for organic molecule activation and transformation.
Data availability
The data that support the findings of this study are available in the main text and the ESI.†
Author contributions
X.-J. W., F. C. and M. D. supervised the project. X.-J. W. and D. X. conceived the idea. D. X. carried out the synthesis of the materials. D. X., Y. M., L. Z., and H. Z. performed HRTEM measurements. D. X. collected HAADF and EDS elemental mapping data. D. X., C.-L. T., and F.-Y. G. contributed to UV-vis and XRD characterization. X.-J. W., F. C. and D. X. wrote the manuscript. All authors discussed the experimental results.
Conflicts of interest
The authors declare no conflict of interest.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (22271142, 21871129 and 21902079), Natural Science Foundation of Jiangsu Province of China (BK20190724), and the “Innovation & Entrepreneurship Talents Plan” of Jiangsu Province.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02493h
Notes and references
- Li H. Qin F. Yang Z. Cui X. Wang J. Zhang L. J. Am. Chem. Soc. 2017;139:3513–3521. doi: 10.1021/jacs.6b12850. [DOI] [PubMed] [Google Scholar]
- Li Y.-H. Zhang F. Chen Y. Li J.-Y. Xu Y.-J. Green Chem. 2020;22:163–169. doi: 10.1039/C9GC03332G. [DOI] [Google Scholar]
- Zhou P. Jiang L. Wang F. Deng K. Lv K. Zhang Z. Sci. Adv. 2017;3:1601945–1601955. doi: 10.1126/sciadv.1601945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur M. Nagaraja C. M. ACS Sustainable Chem. Eng. 2017;5:4293–4303. doi: 10.1021/acssuschemeng.7b00325. [DOI] [Google Scholar]
- Liu H. Xu C. Li D. Jiang H.-L. Angew. Chem., Int. Ed. 2018;57:5379–5383. doi: 10.1002/anie.201800320. [DOI] [PubMed] [Google Scholar]
- Han C. Li Y. H. Li J. Y. Qi M. Y. Tang Z. R. Xu Y.-J. Angew. Chem., Int. Ed. 2021;60:7962–7970. doi: 10.1002/anie.202015756. [DOI] [PubMed] [Google Scholar]
- Zhang W. Fernández-Fueyo E. Ni Y. van Schie M. Gacs J. Renirie R. Wever R. Mutti F. G. Rother D. Alcalde M. Hollmann F. Nat. Catal. 2017;1:55–62. doi: 10.1038/s41929-017-0001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo N. Montini T. Zhang J. Fornasiero P. Fonda E. Hou T. Nie W. Lu J. Liu J. Heggen M. Nat. Energy. 2019;4:575–584. doi: 10.1038/s41560-019-0403-5. [DOI] [Google Scholar]
- Li D. Zhao Y. Miao Y. Zhou C. Zhang L. P. Wu L.-Z. Zhang T. Adv. Mater. 2022;34:2207793–2207799. doi: 10.1002/adma.202207793. [DOI] [PubMed] [Google Scholar]
- Qi M. Y. Conte M. Anpo M. Tang Z. R. Xu Y.-J. Chem. Rev. 2021;121:13051–13085. doi: 10.1021/acs.chemrev.1c00197. [DOI] [PubMed] [Google Scholar]
- Zhao L.-M. Meng Q.-Y. Fan X.-B. Ye C. Li X.-B. Chen B. Ramamurthy V. Tung C.-H. Wu L.-Z. Angew. Chem., Int. Ed. 2017;56:3020–3024. doi: 10.1002/anie.201700243. [DOI] [PubMed] [Google Scholar]
- Jiang D. Chen X. Zhang Z. Zhang L. Wang Y. Sun Z. Irfan R. M. Du P. J. Catal. 2018;357:147–153. doi: 10.1016/j.jcat.2017.10.019. [DOI] [Google Scholar]
- Sun X. Luo X. Zhang X. Xie J. Jin S. Wang H. Zheng X. Wu X. Xie Y. J. Am. Chem. Soc. 2019;141:3797–3801. doi: 10.1021/jacs.8b13051. [DOI] [PubMed] [Google Scholar]
- Xia T. Gong W. Chen Y. Duan M. Ma J. Cui X. Dai Y. Gao C. Xiong Y. Angew. Chem., Int. Ed. 2022;61:202204225–202204230. doi: 10.1002/anie.202204225. [DOI] [PubMed] [Google Scholar]
- Jensen S. C. Homan S. B. Weiss E. A. J. Am. Chem. Soc. 2016;138:1591–1600. doi: 10.1021/jacs.5b11353. [DOI] [PubMed] [Google Scholar]
- Poulose A. C. Zoppellaro G. Konidakis I. Serpetzoglou E. Stratakis E. Tomanec O. Beller M. Bakandritsos A. boril R. Z. Nat. Nanotechnol. 2022;17:485–492. doi: 10.1038/s41565-022-01087-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMeglio J. L. Bartlett B. M. Chem. Mater. 2017;29:7579–7586. doi: 10.1021/acs.chemmater.7b02899. [DOI] [Google Scholar]
- Raza F. Park J. H. Lee H.-R. Kim H.-I. Jeon S.-J. Kim J.-H. ACS Catal. 2016;6:2754–2759. doi: 10.1021/acscatal.5b02798. [DOI] [Google Scholar]
- Chai Z. Zeng T.-T. Li Q. Lu L.-Q. Xiao W.-J. Xu D. J. Am. Chem. Soc. 2016;138:10128–11013. doi: 10.1021/jacs.6b06860. [DOI] [PubMed] [Google Scholar]
- Han G. Jin Y.-H. Burgess R. A. Dickenson N. E. Cao X.-M. Sun Y. J. Am. Chem. Soc. 2017;139:15584–15587. doi: 10.1021/jacs.7b08657. [DOI] [PubMed] [Google Scholar]
- Marzo L. Pagire S. K. Reiser O. König B. Angew. Chem., Int. Ed. 2018;57:10034–10072. doi: 10.1002/anie.201709766. [DOI] [PubMed] [Google Scholar]
- Li X.-B. Tung C.-H. Wu L.-Z. Nat. Rev. Chem. 2018;2:160–173. doi: 10.1038/s41570-018-0024-8. [DOI] [Google Scholar]
- Li J.-Y. Li Y.-H. Qi M.-Y. Lin Q. Tang Z.-R. Xu Y.-J. ACS Catal. 2020;10:6262–6280. doi: 10.1021/acscatal.0c01567. [DOI] [Google Scholar]
- Xiong L. Tang J. Adv. Energy Mater. 2021;11:2003216–2003234. doi: 10.1002/aenm.202003216. [DOI] [Google Scholar]
- Sun K. Lv Q.-Y. Chen X.-L. Qu L.-B. Yu B. Green Chem. 2021;23:232–248. doi: 10.1039/D0GC03447A. [DOI] [Google Scholar]
- Li Y. Zhang C. Zhuang T. T. Lin Y. Tian J. Qi X. Y. Li X. Wang R. Wu L. Liu G. Q. Ma T. He Z. Sun H. B. Fan F. Zhu H. Yu S.-H. J. Am. Chem. Soc. 2021;143:7013–7020. doi: 10.1021/jacs.1c01514. [DOI] [PubMed] [Google Scholar]
- Xin Z. K. Gao Y. J. Gao Y. Song H. W. Zhao J. Fan F. Xia A. D. Li X. B. Tung C.-H. Wu L.-Z. Adv. Mater. 2022;34:2106662–2106670. doi: 10.1002/adma.202106662. [DOI] [PubMed] [Google Scholar]
- Li X.-B. Xin Z.-K. Xia S.-G. Gao X.-Y. Tung C.-H. Wu L.-Z. Chem. Soc. Rev. 2020;49:9028–9056. doi: 10.1039/D0CS00930J. [DOI] [PubMed] [Google Scholar]
- Li F. Wang Y. Du J. hu Y. Xu C. Sun L. Appl. Catal., B. 2018;225:258–263. doi: 10.1016/j.apcatb.2017.11.072. [DOI] [Google Scholar]
- Han G. Yan T. Zhang W. Zhang Y. C. Lee D. Y. Cao Z. Sun Y. ACS Catal. 2019;9:11341–11349. doi: 10.1021/acscatal.9b02842. [DOI] [Google Scholar]
- Hao H. Zhang L. Wang W. Qiao S. Liu X. ACS Sustainable Chem. Eng. 2019;7:10501–10508. doi: 10.1021/acssuschemeng.9b01017. [DOI] [Google Scholar]
- Raza F. Yim D. Park J. H. Kim H.-I. Jeon S.-J. Kim J.-H. J. Am. Chem. Soc. 2017;139:14767–14774. doi: 10.1021/jacs.7b08619. [DOI] [PubMed] [Google Scholar]
- Huang Y. Liu C. Li M. Li H. Li Y. Su R. Zhang B. ACS Catal. 2020;10:3904–3910. doi: 10.1021/acscatal.0c00282. [DOI] [Google Scholar]
- Liu S. Yang M. Tang Z. Xu Y. Nanoscale. 2014;6:7193–7198. doi: 10.1039/C4NR01227E. [DOI] [PubMed] [Google Scholar]
- Han C. Chen Z. Zhang N. Colmenares J. Xu Y. Adv. Funct. Mater. 2015;25:221–229. doi: 10.1002/adfm.201402443. [DOI] [Google Scholar]
- Sung Y. Lim J. Koh J. H. Hill L. J. Min B. K. Pyun J. Char K. CrystEngComm. 2015;17:8423–8427. doi: 10.1039/C5CE01502B. [DOI] [Google Scholar]
- Karakus M. Sung Y. Wang H. I. Mics Z. Char K. Bonn M. Cánovas E. J. Phys. Chem. C. 2017;121:13070–13077. doi: 10.1021/acs.jpcc.7b03639. [DOI] [Google Scholar]
- Tong S. W. Mishra N. Su C. L. Nalla V. Wu W. Ji W. Zhang J. Chan Y. Loh K. P. Adv. Funct. Mater. 2014;24:1904–1910. doi: 10.1002/adfm.201303010. [DOI] [Google Scholar]
- Lee H. Lim J. Song J. Heo H. An K. Kim J. Lee S. Char K. Song H. J. Lee C. Nanotechnology. 2019;30:065401–065410. doi: 10.1088/1361-6528/aaf158. [DOI] [PubMed] [Google Scholar]
- Li X. Zhang Y. Zhai L. Tao C.-L. Xu D. Mu Z. Ding M. Wu X.-J. Angew. Chem., Int. Ed. 2021;60:3475–3480. doi: 10.1002/anie.202012537. [DOI] [PubMed] [Google Scholar]
- Bierman M. J. Jin S. Energy Environ. Sci. 2009;2:1050–1059. doi: 10.1039/B912095E. [DOI] [Google Scholar]
- Cheng C. Fan H. J. Nano Today. 2012;7:327–343. doi: 10.1016/j.nantod.2012.06.002. [DOI] [Google Scholar]
- Li X. Yu J. Jaroniec M. Chem. Soc. Rev. 2016;45:2603–2636. doi: 10.1039/C5CS00838G. [DOI] [PubMed] [Google Scholar]
- Mishra N. Vasavi Dutt V. G. Arciniegas M. P. Chem. Mater. 2019;31:9216–9242. doi: 10.1021/acs.chemmater.8b05363. [DOI] [Google Scholar]
- Nawaz A. Goudarzi S. Asghari M. A. Pichiah S. Selopal G. S. Rosei F. Wang Z. M. Zarrin H. ACS Appl. Nano Mater. 2021;4:11323–11352. doi: 10.1021/acsanm.1c01014. [DOI] [Google Scholar]
- Fiore A. Mastria R. Lupo M. G. Lanzani G. Giannini C. Carlino E. Morello G. De Giorgi M. Li Y. Cingolani R. Manna L. J. Am. Chem. Soc. 2009;131:2274–2282. doi: 10.1021/ja807874e. [DOI] [PubMed] [Google Scholar]
- Deka S. Miszta K. Dorfs D. Genovese A. Bertoni G. Manna L. Nano Lett. 2010;10:3770–3776. doi: 10.1021/nl102539a. [DOI] [PubMed] [Google Scholar]
- Zhai L. Gebre S. T. Chen B. Xu D. Chen J. Li Z. Liu Y. Yang H. Ling C. Ge Y. Zhai W. Chen C. Ma L. Zhang Q. Li X. Yan Y. Huang X. Li L. Guan Z. Tao C.-L. Huang Z. Wang H. Liang J. Zhu Y. Lee C.-S. Wang P. Zhang C. Gu L. Du Y. Lian T. Zhang H. Wu X.-J. Nat. Commun. 2023;14:2538–2547. doi: 10.1038/s41467-023-38237-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W. Y. Chakrabortty S. Guchhait A. Wong G. Y. Z. Dalapati G. K. Lin M. Chan Y. Chem. Mater. 2016;28:9132–9913. doi: 10.1021/acs.chemmater.6b04330. [DOI] [Google Scholar]
- Peng Q. Zhang S. Yang H. Sheng B. Xu R. Wang Q. Yu Y. ACS Nano. 2020;14:6024–6033. doi: 10.1021/acsnano.0c01681. [DOI] [PubMed] [Google Scholar]
- Son D. H. Hughes S. M. Yin Y. Paul Alivisatos A. Science. 2004;306:1009–1012. doi: 10.1126/science.1103755. [DOI] [PubMed] [Google Scholar]
- Fenton J. L. Steimle B. C. Schaak R. E. Science. 2018;360:513–517. doi: 10.1126/science.aar5597. [DOI] [PubMed] [Google Scholar]
- Steimle B. C. Fenton J. L. Schaak R. E. Science. 2020;367:418–442. doi: 10.1126/science.aaz1172. [DOI] [PubMed] [Google Scholar]
- Li Z. Saruyama M. Asaka T. Tatetsu Y. Teranishi T. Science. 2021;373:332–337. doi: 10.1126/science.abh2741. [DOI] [PubMed] [Google Scholar]
- Shemesh Y. Macdonald J. E. Menagen G. Banin U. Angew. Chem., Int. Ed. 2011;50:1185–1221. doi: 10.1002/anie.201006407. [DOI] [PubMed] [Google Scholar]
- Long L. L. Zhang A. Y. Huang Y. X. Zhang X. Yu H. Q. J. Mater. Chem. A. 2015;3:4301–4306. doi: 10.1039/C4TA05818F. [DOI] [Google Scholar]
- Du C. Li P. Yang F. Cheng G. Chen S. Luo W. ACS Appl. Mater. Interfaces. 2018;10:753–761. doi: 10.1021/acsami.7b16359. [DOI] [PubMed] [Google Scholar]
- Li Y. H. Qi M. Y. Tang Z. R. Xu Y.-J. J. Phys. Chem. C. 2022;126:1872–1880. doi: 10.1021/acs.jpcc.1c10911. [DOI] [Google Scholar]
- Zhu Z. Huang H. Liu L. Chen F. Tian N. Zhang Y. Yu H. Angew. Chem., Int. Ed. 2022;61:e202203519. doi: 10.1002/anie.202203519. [DOI] [PubMed] [Google Scholar]
- Wu K. Rodríguez-Córdoba W. E. Yang Y. Lian T. Nano Lett. 2013;13:5255–5263. doi: 10.1021/nl402730m. [DOI] [PubMed] [Google Scholar]
- Wu K. Du Y. Tang H. Chen Z. Lian T. J. Am. Chem. Soc. 2015;137:10224–10230. doi: 10.1021/jacs.5b04564. [DOI] [PubMed] [Google Scholar]
- Wu K. Zhu H. Liu Z. Rodriguez-Cordoba W. Lian T. J. Am. Chem. Soc. 2012;134:10337–10340. doi: 10.1021/ja303306u. [DOI] [PubMed] [Google Scholar]
- Utterback J. K. Grennell A. N. Wilker M. B. Pearce O. M. Eaves J. D. Dukovic G. Nat. Chem. 2016;8:1061–1066. doi: 10.1038/nchem.2566. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the main text and the ESI.†