

Abstract
Early‐onset Alzheimer's disease (AD) is highly heritable, yet only 10% of cases are associated with known pathogenic mutations. For early‐onset AD patients without an identified autosomal dominant cause, we hypothesized that their early‐onset disease reflects further enrichment of the common risk‐conferring single nucleotide polymorphisms associated with late‐onset AD.
We applied a previously validated polygenic hazard score for late‐onset AD to 193 consecutive patients diagnosed at our tertiary dementia referral center with symptomatic early‐onset AD. For comparison, we included 179 participants with late‐onset AD and 70 healthy controls. Polygenic hazard scores were similar in early‐ versus late‐onset AD. The polygenic hazard score was not associated with age‐of‐onset or disease biomarkers within early‐onset AD. Early‐onset AD does not represent an extreme enrichment of the common single nucleotide polymorphisms associated with late‐onset AD. Further exploration of novel genetic risk factors of this highly heritable disease is warranted.
Highlights
There is a unique genetic architecture of early‐ versus late‐onset Alzheimer's disease (AD).
Late‐onset AD polygenic risk is not an explanation for early‐onset AD.
Polygenic risk of late‐onset AD does not predict early‐onset AD biology.
Unique genetic architecture of early‐ versus late‐onset AD parallels AD heterogeneity.
Keywords: age of onset, biomarkers, early‐onset Alzheimer's disease, late‐onset Alzheimer's disease, polygenic risk
1. INTRODUCTION
While Alzheimer's disease (AD) is highly heritable, only 31% is accounted for by known AD genetic variants. 1 The first discovered genetic links to AD, namely pathogenic mutations in amyloid precursor protein (APP) 2 or presenilin 1 or 2 (PSEN1/2), explain < 1% of total cases of AD. 2 , 3 , 4 Apolipoprotein E (APOE) ε4, a more common risk‐conferring variant with a large effect size, only accounts for 27% of AD heritability. 5 Partially in an attempt to explain this “missing heritability,” polygenic risk scores were developed that aggregate small effect sizes of common risk‐ or protection‐conferring single nucleotide polymorphisms (SNPs). These polygenic risk scores offer predictive power of case–control status, dementia age of onset, levels of AD biomarkers, and AD neuropathological burden. 6 , 7 , 8
While polygenic risk scores have provided new insights into late‐onset AD, the genetic basis of early‐onset AD is less well known. Early‐onset AD, which is generally defined as symptoms attributable to AD starting prior to age 65, is thought to be 90% to 100% genetic and is even more heritable than late‐onset AD. 9 But 90% of early‐onset AD remains genetically unexplained. 9 One theory is that early‐onset AD represents an extreme form of the polygenic risk of late‐onset AD. Supporting this idea is a previous report by Cruchaga et al., 10 in which patients with sporadic early‐onset AD surpassed sporadic late‐onset AD participants in the degree of enrichment of late‐onset AD polygenic risk. In the current study, we sought to extend this line of evidence by using another polygenic risk score that predicts age of onset of late‐onset AD. In particular, we explore the “polygenic hazard score” (PHS), a weighted average of 33 risk‐conferring or protective SNPs that in late‐onset AD predicts disease status, age of onset, and AD biomarkers/neuropathology. 7 , 11 We chose this particular polygenic risk score as it is able to predict earlier‐onset cases of late‐onset AD; among APOE ε3/ε3 homozygotes there is a 10‐year spread between the upper and lower tenth percentiles in age of onset. 7 We hypothesized that participants with early‐onset AD, defined here as mild cognitive impairment or dementia due to AD diagnosed before age 65, would have higher enrichment of late‐onset AD polygenic risk (as reflected by the PHS) than the late‐onset AD group.
2. METHODS
2.1. Standard protocol approvals, registrations, and patient consents
Early‐onset AD and control participants or their legal authorized representative provided written informed consent at the University of California, San Francisco (UCSF) Alzheimer's Disease Research Center (ADRC). The de‐identified clinical and genetic data for late‐onset AD participants were obtained via the National Alzheimer's Coordinating Center (NACC) and the Alzheimer's Disease Sequencing Project (ADSP), respectively. Written consent for the NACC and ADSP was obtained by the participating institutions. This study was approved by the UCSF, the Lawrence Berkeley National Laboratory, and University of Minnesota Institutional Review Boards.
2.2. Participants
One hundred ninety‐three consecutive patients at the UCSF ADRC were clinically diagnosed with symptomatic early‐onset AD (age of diagnosis ≤ 65 years of age) in accordance with National Institute on Aging–Alzheimer's Association (NIA‐AA) criteria 12 after a comprehensive clinical evaluation followed by consensus review. A subset of 82 patients had biomarker confirmation of AD pathophysiology via cerebrospinal fluid (CSF) biomarkers or Pittsburgh compound B (PiB) amyloid positron emission tomography (PET). Patients presenting with logopenic variant of primary progressive aphasia and posterior cortical atrophy were included with diagnoses established according to published criteria. 13 , 14 Participants with APP, PSEN1, or PSEN2 pathogenic variants were excluded. Clinical diagnosis was made blinded to genetic results.
Seventy healthy control participants were studied as part of the Hillblom Healthy Aging Network at UCSF and were free of any objective cognitive impairment, structural brain lesion, history of neurological disease, and were psychiatric medication‐free. The full set of 179 late‐onset AD participants without any co‐primary or secondary contributing pathologies who had clinical data from the NACC and accompanying genetic data from ADSP were obtained. The age of symptom onset for the early‐ and late‐onset AD participants was established according to the clinician's best judgement using participant and informant report. For all diagnostic groups (controls, early‐ and late‐onset AD) participants with non‐European ancestry on genetic testing were also excluded.
2.3. Lumbar puncture and CSF testing
Cerebrospinal fluid collection was performed in a subset of the early‐onset AD participants (N = 29) according to previously delineated methods. 15 In brief, CSF was collected in a polypropylene tube and stored at −80°C within 1 hour of lumbar puncture collection and sent to University of Pennsylvania. Amyloid beta (Aβ)42, total tau (t‐tau), and tau phosphorylated at threonine 181 (p‐tau) were measured using the INNO‐BIA AlzBio3 platform (Innogenetics). 16
2.4. Amyloid PET imaging
A subset of participants (N = 68) underwent 11C‐PiB amyloid PET imaging at the Lawrence Berkeley National Laboratory according to previously published protocols. 17 In brief, PET imaging was performed on a Siemens/Biograph PET/computed tomography (CT) in 3D acquisition mode. Attenuation correction using a low‐dose CT/transmission scan occurred prior to each PET scan. 11C‐PiB data were collected 50 to 70 minutes after tracer injection. Cerebellar gray matter was used as the reference region. Magnetization‐prepared rapid acquisition gradient echo sequence was processed using Statistical Parametric Mapping version 8 (SPM8, https://www.fil.ion.ucl.ac.uk/spm/) software for reference region definition and spatial warping. Amyloid positivity at 11C‐PiB was assessed both visually by an expert reader (GDR, RLJ) and via quantification using a neocortical composite score 11C‐PiB standardized uptake value ratio (SUVR) score > 1.21. 17
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using Google Scholar and PubMed. Early‐onset Alzheimer's disease (AD) is a highly heritable condition, but only 10% of cases are explained by known mutations. Exploring this “missing heritability,” one prior study by Cruchaga et al. suggested that early‐onset AD is explained by extreme enrichment of the polygenic risk of late‐onset AD. However, numerous other studies suggest the contrary: rather than polygenic, early‐onset AD may be better explained by undiscovered autosomal recessive or oligogenic mechanisms.
Interpretation: Our findings suggest that early‐onset AD is not explained by further enrichment of late‐onset AD polygenic risk. This finding points toward recessive or oligogenic modes of inheritance for early‐onset AD
Future directions: Uncovering the genetic basis of early‐onset AD will likely benefit from searching for unique novel genetic/epigenetic variants compared to late‐onset AD.
2.5. Polygenic hazard score
The PHS, composed of a weighted score of 33 risk‐ or protection‐conferring SNPs, was calculated for each participant as previously described by Desikan et al. 7 In brief, the creation of the PHS is based on data from multiple large AD genetic consortium studies. First, 17,008 clinically diagnosed AD cases and 37,154 European or European‐ancestry controls who were part of the International Genomics of Alzheimer's Project (IGAP) Stage 1 had genotyped or imputed data at 7,055,881 SNPs. One thousand eight hundred fifty‐four SNPs were identified that were associated with increased risk for AD that met the significance threshold of P < 10−5. Next, a “final” list of AD‐associated SNPs was created using the Alzheimer's Disease Genetics Consortium (ADGC) Phase 1 case–control dataset of 6409 AD dementia and 9386 healthy controls (excluding Alzheimer's Disease Center and Alzheimer's Disease Neuroimaging Initiative [ADNI] samples). Participants with an autosomal dominant (APP, PSEN1, or PSEN2) mutation, who had AD symptom onset prior to age 60, or who were of non‐European ancestry were excluded. Diagnosis of clinical AD was established using criteria for definite, probable, and possible AD. 18 A stepwise regression framework, which used a Cox proportional hazard model, identified the top AD‐associated SNPs while controlling for the effects of sex, APOE variants, and the top five genetic principal components (to control for the effects of population stratification). Age of AD onset and age of last clinical visit were incorporated to select SNPs with effects on age of onset. In each step of the stepwise procedure, the algorithm selected the one SNP from the pool that most improved model prediction (e.g., minimizing the Martingale residuals); additional SNP inclusion that did not further minimize the residuals resulted in halting of the SNP selection process. This process produced the final 31 SNPs in addition to the two SNPs that define the APOE alleles that compose the PHS. Weights of each SNP composing the PHS are enumerated by Desikan et al. 7 In an attempt to reproduce the findings of the previous report by Cruchaga et al., which stems from the IGAP stage 1 and 2 (Lambert et al. 5 ), we used their same methodology for computation of the Cruchaga et al. polygenic risk and compared it across controls, early‐, and late‐onset AD using logistic regression controlling for sex. 10 Both the Desikan et al. PHS and the Cruchaga et al. polygenic risk score are based on the IGAP, 5 , 7 , 10 in which selected variants pass Hardy–Weinberg equilibrium. SNPs included in the Desikan et al. PHS and Cruchaga et al. polygenic risk score are enumerated in Tables S5 and S6 in supporting information.
2.6. Sequencing, data alignment, and quality control
We used the same sequencing, data alignment, and quality control methodology as the one used in a previous study. 19 For early‐onset AD participants and healthy controls whole‐genome sequencing was performed at HudsonAlpha Institute for Biotechnology (Huntsville, AL) on a HiSeq X platform. Mean depth was 34x with an average of 92% of bases covered at 20x. Sequencing libraries at HudsonAlpha were prepared by Covaris shearing, end repair, adaptor ligation, and polymerization chain reaction (PCR) using standard protocols. Library concentrations were normalized using KAPA quantitative PCR prior to sequencing.
All sequencing reads from both sequencing centers were aligned to the hg19 reference genome with Burrows–Wheeler aligner. 20 Binary alignment maps were sorted, and duplicates were marked using Sambamba 0.5.4. 21 Variants were called across all samples in a single batch through the use of GATK 3.8 using the ‐newQual flag to minimize false negative singleton calls. The variant cell format (VCF) was quality filtered with a genotype‐level requirement for 95% of sites to have a minimum genotype quality (GQ) of 20 and read depth of 10 (applied using VCFtools 0.1.1521), and a variant level filter of variant quality score log‐odds (VQSLOD) > −3. Only samples from the largest cluster (European ancestry) were retained to minimize potential confounding population effects. While no participants were excluded under the following criteria, we verified that no participants carried a pathogenic variant in APP, PSEN1/2, C9ORF72, GRN, or MAPT. Additionally, Goleft indexcov 0.1.17 was used for sex checks and samples failing sex checks were excluded. Finally, individuals were not related within at least four degrees, which is the outer detection limit for relatedness for the software used to assess relatedness (KING version 2.2).
Genotyping for the late‐onset AD cohort was performed at ADSP as previously described. 22 ADSP samples underwent whole exome sequencing at one of three National Human Genome Research Institute–funded large‐scale sequencing centers at Baylor University, the Broad Institute, or Washington University. Whole exome capture was performed using either the Illumina Rapid Capture Exome kit or VCRome v2.1 kit (NimbleGen), and paired‐end reads were generated using an Illumina HiSeq 2000. Sequence reads were aligned to the GRCh37 reference genome using the Burrows–Wheeler aligner, 20 and variants were jointly called across the entire cohort using Atlas V2 software (Baylor) or GATK (Broad). Variants underwent pipeline‐specific quality control prior to merging the variants that were concordant between the two sets of variants. The ADSP also performed initial quality control checks on sample information, phenotypes, and genotype data to ensure that these data were of high quality and suitable for downstream analysis. Joint called variants with GQ scores > 30 and read depth scores > 20.
2.7. Statistical analyses
We measured the relationship between PHS and disease status (early‐onset AD, late‐onset AD, and healthy controls) using two‐sided Student t tests and logistic regression controlling for sex. Age was not incorporated as a covariate as our goal was to compare PHS scores between diagnostic groups that are already divided according to age. Comparison of categorical variables between diagnostic groups was performed via chi‐square analysis. The relationship between PHS and age of onset, disease biomarkers (CSF Aβ42, p‐tau, and t‐tau) and amyloid PET SUVR was quantified using multiple linear regression controlling for sex. Regression co‐efficients are reported via unstandardized (e.g., raw) beta (B).
2.8. Data availability
Anonymized data not published within this article will be made available by request from any qualified investigator. Genomic data for UCSF participants is available under controlled access via the Synapse AD Knowledge portal accession number syn25686500. ADSP data are available at National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site under project NG00067.
3. RESULTS
3.1. All participants
Table 1 includes demographic and clinical features of the 193 participants clinically diagnosed with early‐onset AD, 70 control participants, and 179 participants with late‐onset AD. A higher (more risk conferring) PHS was associated with a diagnosis of early‐onset AD compared to healthy controls (odds ratio [OR]: 2.2; 95% confidence interval [CI]: 1.6–3.1). As expected, this relationship is similarly present in late‐onset AD versus healthy controls (OR: 2.6; 95% CI: 1.8–3.7). However, higher PHS was not associated with a diagnosis of early‐onset AD compared to late‐onset AD (OR: 0.87; 95% CI: 0.71–1.1). Examining only the biomarker‐confirmed cases of early‐onset AD, we again found no difference in PHS predicting early‐ versus late‐onset AD (OR: 0.80; 95% CI: 0.62–1.0; Table S1 in supporting information).
TABLE 1.
Demographics, age of onset, APOE, and polygenic hazard score across diagnostic groups.
| Diagnostic group | Age of symptom onset (years) | Age at first visit (years) | Sex, % female | APOE ε4 allele frequency | PHS | PHS odds ratio (95% CI) predicting diagnostic group versus controls | PHS odds ratio (95% CI) predicting diagnostic group versus late‐onset AD |
|---|---|---|---|---|---|---|---|
|
Early‐onset AD ‐Entire group N = 193 |
55 (5.3) |
59 (5.6) |
54% |
0.35 |
0.60 (1.0) |
2.2 (1.6–3.1) |
0.87 (0.71–1.1) |
|
‐Multi‐domain N = 111 |
55 (5.2) | 59 (5.6) | 50% | 0.40 | 0.69 (1.0) | 2.4 (1.6–3.4) | 0.96 (0.75–1.2) |
|
‐Logopenic variant PPA N = 47 |
55 (5.3) | 60 (6.1) | 62% | 0.28 | 0.53 (0.91) | 2.3 (1.4–3.6) | 0.80 (0.56–1.1) |
|
‐Posterior cortical atrophy N = 35 |
55 (5.3) | 59 (5.0) | 54% | 0.27 | 0.43 (0.95) | 1.9 (1.2–3.2) | 0.72 (0.49–1.1) |
|
Late‐onset AD N = 179 |
76 (6.1) | 80 (7.0) | 57% | 0.44 | 0.73 (1.0) | 2.6 (1.8–3.7) | N/A |
|
Controls N = 70 |
N/A | 70 (6.4) | 63% | 0.13 | −0.08 (0.82) | N/A | 0.38 (0.27–0.54) |
Note. Unless otherwise noted, all values are mean ± (standard deviation). Logistic regression co‐varies for sex.
Abbreviations: AD, Alzheimer's disease; APOE, apolipoprotein E; CI, confidence interval; PHS, polygenic hazard score; PPA, primary progressive aphasia.
There was no correlation between PHS and age of onset within the early‐onset AD participants (B: −0.013; 95% CI: −0.77–0.74). We also stratified the analysis by age of onset (participants 60–65, 55–60, and ≤ 55 years old) and found no associations between PHS and age of onset or disease biomarkers in these age brackets of early‐onset AD (data not shown). As a positive control, in late‐onset AD the PHS was associated with age of onset (B: −1.1; 95% CI: −2.1 to −0.23), with higher PHS predicting younger age of onset. Figure 1 visually depicts the regression of age of onset by PHS divided by cohort. In attempt to control for recall bias in participant‐ or informant‐reported age of onset, we also examined the correlation between PHS and age at first visit for the early‐onset AD group. We again found no relationship (B: 0.011; 95% CI: − 0.71–0.92).
FIGURE 1.
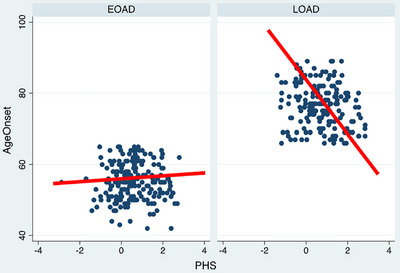
Regression of age of onset versus polygenic hazard score in early‐onset versus late‐onset Alzheimer's disease. EOAD, early‐onset Alzheimer's disease; LOAD, late‐onset Alzheimer's disease; PHS, polygenic hazard score (Desikan et al. 7 )
We stratified the analysis by early‐onset AD variant (e.g. multi‐domain [N = 110], logopenic variant of primary progressive aphasia [N = 48], or posterior cortical atrophy [N = 35]); however, we did not find any significant PHS difference between early‐onset AD variants and late‐onset AD (Table 1) nor did we find any correlation between PHS and age of onset or disease biomarkers, with the exception of higher PHS predicting higher p‐tau 181 in the posterior cortical atrophy subgroup. However, this latter finding did not survive Bonferroni correction (Table S2 in supporting information).
Finally, we assessed the association of APOE ε4 on age of onset within the early‐onset AD group and found no correlation even when controlling for sex and APOE ε2 allele (APOE ε4 heterozygous B: 0.46; 95% CI −1.0–2.3; APOE ε4/ε4 homozygous B: 0.53; 95% CI: −3.2–1.6).
3.2. Analysis of PHS independent of APOE ε2 and APOE ε4
To examine the effects of PHS independent of APOE ε4 or APOE ε2, we performed the same analyses in APOE ε3/ε3 homozygotes. Table 2 includes demographic and clinical features of the 77 participants clinically diagnosed with early‐onset AD, 43 control participants, and 54 participants with late‐onset AD who are all APOE ε3/ε3 homozygotes. While the OR predicting early‐ versus late‐onset AD was above 1 (OR: 1.1; 95% CI: 0.61–1.9), this was not statistically significant. We performed the same analyses in biomarker‐confirmed early‐onset AD and found similar results (OR: 1.1; 95% CI: 0.56–2.2; Table S3 in supporting information). We also compared a modified PHS that excludes APOE between all early‐ versus late‐onset AD participants, regardless of their APOE genotype. We similarly found a > 1 OR that was not statistically significantly associated with early‐ versus late‐onset AD (OR: 1.4; 95% CI: 0.76–1.7).
TABLE 2.
Participants homozygous for APOE ε3; demographics, age of onset, and polygenic hazard score across diagnostic groups.
|
Diagnostic group (all APOE ε3/ε3 homozygotes) |
Age of symptom onset (years) | Age at first visit (years) | Sex, % female | PHS | PHS odds ratio (95% CI) predicting diagnostic group versus controls | PHS odds ratio (95% CI) predicting diagnostic group versus late‐onset AD |
|---|---|---|---|---|---|---|
|
Early‐onset AD ‐Entire group N = 77 |
55 (5.4) |
59 (5.5) |
49% |
−0.12 (0.60) |
2.0 (1.0–3.9) |
1.1 (0.61–1.9) |
|
‐Multidomain N = 37 |
54 (5.0) | 58 (4.8) | 46% | −0.18 (0.60) | 1.6 (0.72–3.6) | 0.92 (0.46–1.8) |
|
‐Logopenic variant PPA N = 23 |
56 (6.2) | 61 (6.4) | 48% | −0.045 (0.54) | 2.8 (1.0–7.7) | 1.4 (0.61–3.1) |
|
‐Posterior cortical atrophy N = 17 |
55 (5.2) | 59 (5.1) | 59% | −0.097 (0.70) | 1.9 (0.74–5.0) | 1.1 (0.50–2.7) |
|
Late‐onset AD N = 54 |
77 (6.6) | 80 (6.6) | 57% | −0.15 (0.64) | 1.7 (0.84–3.3) | N/A |
|
Controls N = 43 |
N/A | 69 (6.6) | 70% | −0.33 (0.58) | N/A | 0.60 (0.30–1.2) |
Note. Unless otherwise noted, all values are mean ± (standard deviation). Logistic regression co‐varies for sex.
Abbreviations: AD, Alzheimer's disease; APOE, apolipoprotein E; CI, confidence interval; PHS, polygenic hazard score; PPA, primary progressive aphasia.
Examining the relation between PHS and AD biomarkers in the APOE ε3/ε3 homozygotes similarly did not demonstrate any significant relationships, except for PHS and amyloid PET (paradoxically lower amyloid PET with higher/more “risk‐conferring” PHS) and CSF Aβ42 (in the predicted direction of lower CSF Aβ42 with higher PHS) in the logopenic primary progressive aphasia group, which did not survive Bonferroni correction (Table S4).
When assessing the PHS's ability to predict age of onset, paradoxically a higher PHS within the early‐onset AD APOE ε3/ε3 homozygotes trended toward an older age of onset, although this did not meet statistical significance (B: 1.7; 95% CI −0.32–3.7).
3.3. Replicating Cruchaga et al.’s polygenic risk score
Table 3 demonstrates the association of the Cruchaga et al. polygenic risk scores between controls, early‐, and late‐onset AD. While the OR was > 1 in comparing early‐ versus‐late onset AD participants, this was not statistically significant (OR: 1.2; 95% CI: 0.78–1.8). Comparing PRS in APOE ε3/ε3 homozygotes (Table 4) there was again a non‐statistically significant association of higher enrichment in early‐ versus late‐onset AD (OR: 1.5; 95% CI: 0.71–2.9). Comparing the full group of early‐ versus late‐onset AD participants with a modified Cruchaga et al. polygenic risk score that excludes APOE, there was another non‐statistically significant association of higher enrichment in early‐ versus late‐onset AD (OR: 1.2; 95% CI: 0.76–1.8). To mirror the lack of known family history in the sporadic early‐ and sporadic late‐onset AD participants in the Cruchaga et al. study, we excluded 32 early‐ and 106 late‐onset participants with a known first‐degree relative with cognitive impairment or dementia and then compared the remaining early‐ versus late‐onset AD participants. Our N being significantly reduced, and with CIs stretching from zero to the hundreds of trillions, no trends could be discerned (full Cruchaga et al. polygenic risk OR: 2.8; 95% CI: 0.00015–53000/APOE‐independent Cruchaga et al. polygenic risk OR: 180000; 95% CI: 0.000043 – 7.5 × 10^14). Note that due to missing or unavailable data elements, we were not able to ascertain the pattern of family history or sibling status in most participants without a known family history. Finally, we examined the relationship between the Cruchaga et al. polygenic risk and age of onset within the early‐onset AD participants, and found no association with either the full or APOE independent polygenic risk score (B: −7.0; 95% CI: −32–18/B: −30; 95% CI: −89–28).
TABLE 3.
Cruchaga et al., 10 polygenic risk score: Demographics, age of onset, APOE, and polygenic risk score across diagnostic groups.
| Diagnostic group | Age of symptom onset (years) | Age at first visit (years) | Sex, % female | APOE ε4 allele frequency | Cruchaga et al., 10 PRS | PRS odds ratio (95% CI) predicting diagnostic group versus controls | PRS odds ratio (95% CI) predicting diagnostic group versus late‐onset AD |
|---|---|---|---|---|---|---|---|
|
Early‐onset AD ‐Entire group N = 193 |
55 (5.3) |
59 (5.6) |
54% |
0.35 |
0.030 (0.030) |
1.5 (0.80–2.7) |
1.2 (0.78–1.8) |
|
‐Multi‐domain N = 111 |
55 (5.2) | 59 (5.6) | 50% | 0.40 | 0.033 (0.031) | 1.7 (0.85–3.3) | 1.3 (0.82–2.1) |
|
‐Logopenic variant PPA N = 47 |
55 (5.3) | 60 (6.1) | 62% | 0.28 | 0.028 (0.027) | 1.1 (0.48–2.6) | 0.9 (0.44–1.6) |
|
‐Posterior cortical atrophy N = 35 |
55 (5.3) | 59 (5.0) | 54% | 0.27 | 0.021 (0.027) | 1.2 (0.49–2.8) | 1.3 (0.60–2.6) |
|
Late‐onset AD N = 178 a |
76.7 (6.1) | 80 (6.8) | 57% | 0.45 | 0.036 (0.031) | 1.1 (0.60–2.2) | N/A |
|
Controls N = 70 |
N/A | 70 (6.4) | 63% | 0.13 | 0.0064 (0.022) | N/A | 0.88 (0.46–1.7) |
Note. Unless otherwise noted, all values are mean ± (standard deviation). Logistic regression co‐varies for sex.
Abbreviations: AD, Alzheimer's disease; APOE, apolipoprotein E; CI, confidence interval; PHS, polygenic hazard score; PPA, primary progressive aphasia; PRS, Cruchaga et al. polygenic risk score.
One late‐onset AD participant from the prior PHS group was not available at the time of data analysis (e.g., N = 178 instead of 179).
TABLE 4.
Cruchaga et al., 10 polygenic risk score in participants homozygous for APOE ε3; demographics, age of onset, APOE, and polygenic risk score across diagnostic groups.
|
Diagnostic group (all APOE ε3/ε3 homozygotes) |
Age of symptom onset (years) | Age at first visit (years) | Sex, % female | Cruchaga et al., 10 PRS | PRS odds ratio (95% CI) predicting diagnostic group versus controls | PRS odds ratio (95% CI) predicting diagnostic group versus late‐onset AD |
|---|---|---|---|---|---|---|
|
Early‐onset AD ‐Entire group N = 77 |
55 (5.4) |
59 (5.5) |
49% |
0.0032 (0.0098) |
2.6 (1.2–6.0) |
1.5 (0.71–2.9) |
|
‐Multidomain N = 37 |
54 (5.0) | 58 (4.8) | 46% | 0.0019 (0.0091) | 2.9 (1.2–7.5) | 1.7 (0.71–3.9) |
|
‐Logopenic variant PPA N = 23 |
56 (6.2) | 61 (6.4) | 48% | 0.0075 (0.010) | 3.7 (1.1–13) | 1.6 (0.58–4.4) |
|
‐Posterior cortical atrophy N = 17 |
55 (5.2) | 59 (5.1) | 59% | 0.00068 (0.0098) | 1.7 (0.52–5.4) | 1.0 (0.34–3.2) |
|
Late‐onset AD N = 53 a |
77 (6.6) | 80 (7.3) | 58% | 0.0027 (0.086) | 1.7 (0.70–4.0) | N/A |
|
Controls N = 43 |
N/A | 69 (6.6) | 70% | −0.0013 (0.0097) | N/A | 0.59 (0.25–1.4) |
Note. Unless otherwise noted, all values are mean ± (standard deviation). Logistic regression co‐varies for sex.
Abbreviations: AD, Alzheimer's disease; APOE, apolipoprotein E; CI, confidence interval; PHS, polygenic hazard score; PPA, primary progressive aphasia; PRS, Cruchaga et al. polygenic risk score.
One late‐onset AD participant from the prior PHS group was not available at the time of data analysis (e.g., N = 53 instead of 54).
4. DISCUSSION
Early‐onset AD does not appear to be associated with an extreme burden of the polygenic risk associated with late‐onset AD. Participants with early‐onset AD had a similarly risk‐conferring polygenic risk, as reflected by the PHS and the Cruchaga et al. polygenic risk score, as late‐onset AD. Furthermore, unlike late‐onset AD, the PHS in early‐onset AD did not predict age of onset or AD biomarkers. In a similar fashion, while APOE ε4 allele burden reduces the age of onset of AD, it does not further reduce the age of onset for patients who are already in the early‐onset AD category. While excluding APOE from polygenic risk calculations appeared to tilt the balance of polygenic enrichment toward early‐onset AD, these were non‐statistically significant.
The findings presented here contrast with those from a previous study by Cruchaga et al., 10 which suggested extreme enrichment of late‐onset AD polygenic risk in sporadic early‐onset AD. Several methodological differences could account for the discrepancy between the current study and Cruchaga et al. First, the Cruchaga et al. study compared sporadic early‐ versus sporadic late‐onset AD. Sporadic in the Cruchaga et al. study was defined as the lack of any documented family history (for sporadic early‐onset AD) or without two affected siblings with another affective relative (for sporadic late‐onset AD). In the present study, known Mendelian variants were excluded from the analysis, but 1:6 early‐onset participants had a known first‐degree relative (parent, sibling, or child) with cognitive impairment or dementia and were included in the analysis. In attempt to compare only sporadic participants, we ran a subanalysis on participants without a known first‐degree relative, but our N was too small to make any conclusions. One possibility for the discordance between the Cruchaga et al. study and this study is that polygenic modes of inheritance may underlie sporadic early‐onset AD, whereas highly penetrant oligogenic or recessive inheritance may underlie early‐onset AD with positive family histories. A recent report supports this hypothesis; participants with a strong family history of dementia had a higher likelihood of harboring rare pathogenic variants with large effect sizes. 23 A second methodological difference is that the age of onset of the two studies’ early‐onset AD groups are different; the present study's mean age of AD symptom onset was 5 years younger than the prior report. Furthermore, for inclusion as an early‐onset AD participant, we used an age of diagnosis < 65 whereas the Cruchaga et al. study used an age of reported symptom onset < 65. Finally, our study has a high proportion of atypical cases: 46% of our participants had a non‐medial temporal lobe predominant subtype of AD, whereas it is unclear if these atypical cases were included in the Cruchaga et al. report. Our study thus focuses on an early‐onset AD group that is younger with a high proportion of atypical cases, which may impact the genetic similarity between our current report and the Cruchaga et al. study.
The current report's conclusions are in line with another genome‐wide association study of posterior cortical atrophy, a visuospatial variant of AD that occurs more commonly in early‐onset AD. In the report by Schott et al., 24 participants with posterior cortical atrophy variant of AD had a mean age of onset of 58 years and had a partial but not full overlap in polygenic risk compared to late‐onset AD. Furthermore, participants with the posterior cortical atrophy variant of AD had novel risk variants not previously associated with late‐onset AD. Together with the results of the present study and prior work suggesting an oligogenic or recessive inheritance pattern for early‐onset AD, 9 , 25 it is likely there are unique common and rare genetic contributions to early‐onset AD risk.
Our study's results also mirror increasing evidence that APOE ε4 does not lower the age of onset within early‐onset AD. While APOE ε4 reduces the age of onset of AD in a dose‐dependent fashion, it does so only up to a point; APOE ε4/ε4 homozygotes show an average age of symptom onset between 60 and 69 years. 26 , 27 Below age 60, however, early‐onset AD patients are equally likely to be APOE ε3/ε3 homozygotes. 26 , 28 Elsewhere, a recent study observed no effect of APOE ε4 on age of onset in men with early‐onset AD, but a decreased age of onset for females with early‐onset AD lacking an APOE ε4 allele. 29 That APOE ε4 plays less of a role in the earliest onset cases of AD further argues against the hypothesis that early‐onset AD is merely explained by an enrichment of late‐onset AD common genetic risk variants.
The differences in predictive power of the PHS and APOE ε4 in early‐ versus late‐onset AD is mirrored by differences in disease risk factors and clinicopathological features between the two forms of AD. Compared to late‐onset AD, patients with early‐onset AD show faster clinical decline 30 and greater neuropsychiatric symptoms. 31 While late‐onset AD and APOE ε4 are associated with medial temporal lobe disease, early‐onset AD patients exhibit greater impairment in brain regions outside of the medial temporal lobe, especially for patients without an APOE ε4 allele. 27 Disease risk factors may also be different between the two disease subtypes; patients with the logopenic variant of primary progressive aphasia or the posterior cortical atrophy variant of AD, which occur more commonly in early‐onset cases of AD, 32 have a higher prevalence of left‐handedness, 33 , 34 and childhood learning disabilities. 33 , 35 Neuropathologically, early‐onset AD patients have more severe neurofibrillary tangle burden and synaptic loss than late‐onset AD. 36 , 37 This age‐related heterogeneity of AD may reflect differences in underlying genetic risk.
While the current study argues against early‐onset AD representing an extreme enrichment of common late‐onset AD genetic risk variants, rare pathogenic variants in the same genes implicated in late‐onset AD (e.g. SORL1, ABCA7) have been found in early‐onset AD. 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 Conversely, mutations in genes previously associated with early‐onset AD (e.g., APP, PSEN2) also rarely cause late‐onset AD. 46 , 47 Thus, even though the location and functional effect of variants may differ across early‐ versus late‐onset AD, the affected genes may still substantially overlap.
This study has several important limitations. First, our relatively small and heterogeneous patient group of 193 may be underpowered to detect further enrichment of late‐onset AD polygenic risk in early‐onset AD. However, despite the relatively small sample size, clear differences were observed in early‐ and late‐onset AD versus controls. The lack of observable difference in PHS between early‐ versus late‐onset AD means that if such a difference truly exists, the difference is small. A second limitation is that only 82 of the 193 early‐onset AD participants had biomarker confirmation, meaning that an unknown number of the non‐biomarker confirmed cases may have had a non‐AD alternative diagnosis. We note, however, that examining only biomarker‐confirmed cases of early‐onset AD did not demonstrate any differences in polygenic risk compared to the entire early‐onset AD group. A third limitation is that our ability to compare polygenic risk to age of onset is reliant on participant or informant reporting, which is subject to variations in recall bias. We note again, however, that the late‐onset AD‐positive control group similarly relied on informant report and we found a statistically significant relationship between PHS and age of onset. Fourth, the early‐onset AD group was recruited mainly from the San Francisco Bay Area through referrals from local clinics to the UCSF ADRC, a source of ascertainment bias that may give rise to a non‐representative sample. Finally, we restricted our analysis to participants of European ancestry, which parallels the methodology of the PHS. Given important differences in the risk of APOE depending on genetic ancestry, along with important differences in linkage disequilibrium and non‐APOE AD risk factors in diverse cohorts, 48 , 49 , 50 , 51 this study may not generalize across racial groups and underlines a pressing need to improve recruitment of diverse cohorts to national AD research efforts.
5. CONCLUSION
In summary, our results are not consistent with the model that early‐onset AD risk is mainly conferred by extreme enrichment of common late‐onset AD associated risk variants. Further exploration is warranted to uncover the genetic basis of early‐onset AD.
CONFLICT OF INTEREST STATEMENT
Dr W.M. reports consulting fees from Genentech/Roche and research support from the NIH, the American Academy of Neurology, American Brain Foundation, Alzheimer's Association, Wallin Foundation, and Fesler‐Lampert Foundation. Dr N.C. reports receiving consulting fees from Caraway Therapeutics and research support from the NIH. Mr J.T. reports no disclosures. Dr E.G. reports no disclosures. Dr I.B. reports no disclosures. Dr L.B. reports no disclosures. Ms A.A. reports no disclosures. Dr D.S. reports no disclosures. Dr R.L.J. reports no disclosures. Dr L.I. reports no disclosures. Ms K.C. reports no disclosures. Ms L.E. reports no disclosures. Ms A.S. reports no disclosures. Ms H.G. reports no disclosures. Dr I.A. reports no disclosures. Dr Z.M. reports no disclosures. Dr M.G.‐T. reports no disclosures. Dr B.M. reports no disclosures. Dr R.D. is posthumously a co‐author. Dr J.K. reports royalties from Pearson, Inc. and being on an advisory board for Biogen. Dr G.R. consulting fees from Eli Lilly, GE Healthcare, Genentech, Johnson & Johnson, and Roche and research support from the NIH, Alzheimer's Association, American College of Radiology, Rainwater Charitable Foundation, Avid Radiopharmaceuticals, GE Healthcare, Genentech, and Life Molecular Imaging. Dr J.Y. reports no disclosures. The authors have no conflicts of interest listed in the supporting information.
CONSENT STATEMENT
All participants provided informed consent.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors would like to acknowledge William Jagust, Mustfa Janabi, and Kristen Norton for acquisition of PET scans. The authors thank the Genomic Services Lab at HudsonAlpha for DNA isolations, library generation, quality control, and sequencing.The NACC database is funded by NIA/NIH Grant U24 AG072122. NACC data are contributed by the NIA‐funded ADCs: P50 AG005131 (PI James Brewer, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005134 (PI Bradley Hyman, MD, PhD), P50 AG005136 (PI Thomas Grabowski, MD), P50 AG005138 (PI Mary Sano, PhD), P50 AG005142 (PI Helena Chui, MD), P50 AG005146 (PI Marilyn Albert, PhD), P50 AG005681 (PI John Morris, MD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG008051 (PI Thomas Wisniewski, MD), P50 AG008702 (PI Scott Small, MD), P30 AG010124 (PI John Trojanowski, MD, PhD), P30 AG010129 (PI Charles DeCarli, MD), P30 AG010133 (PI Andrew Saykin, PsyD), P30 AG010161 (PI David Bennett, MD), P30 AG012300 (PI Roger Rosenberg, MD), P30 AG013846 (PI Neil Kowall, MD), P30 AG013854 (PI Robert Vassar, PhD), P50 AG016573 (PI Frank LaFerla, PhD), P50 AG016574 (PI Ronald Petersen, MD, PhD), P30 AG019610 (PI Eric Reiman, MD), P30‐AG062422 (PI Gil Rabinovici, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P30 AG028383 (PI Linda Van Eldik, PhD), P50 AG033514 (PI Sanjay Asthana, MD, FRCP), P30 AG035982 (PI Russell Swerdlow, MD), P50 AG047266 (PI Todd Golde, MD, PhD), P50 AG047270 (PI Stephen Strittmatter, MD, PhD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG049638 (PI Suzanne Craft, PhD), P30 AG053760 (PI Henry Paulson, MD, PhD), P30 AG066546 (PI Sudha Seshadri, MD), P20 AG068024 (PI Erik Roberson, MD, PhD), P20 AG068053 (PI Marwan Sabbagh, MD), P20 AG068077 (PI Gary Rosenberg, MD), P20 AG068082 (PI Angela Jefferson, PhD), P30 AG072958 (PI Heather Whitson, MD), P30 AG072959 (PI James Leverenz, MD). Data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer's Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24‐AG041689), funded by the National Institute on Aging. ADSP data are available at NIAGADS under project NG00067. The Alzheimer's Disease Sequencing Project (ADSP) is comprised of two Alzheimer's disease (AD) genetics consortia and three National Human Genome Research Institute (NHGRI) funded Large Scale Sequencing and Analysis Centers (LSAC). The two AD genetics consortia are the Alzheimer's Disease Genetics Consortium (ADGC) funded by NIA (U01 AG032984), and the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) funded by NIA (R01 AG033193), the National Heart, Lung, and Blood Institute (NHLBI), other National Institute of Health (NIH) institutes, and other foreign governmental and non‐governmental organizations. The Discovery Phase analysis of sequence data is supported through UF1AG047133 (to Drs Schellenberg, Farrer, Pericak‐Vance, Mayeux, and Haines); U01AG049505 to Dr Seshadri; U01AG049506 to Dr Boerwinkle; U01AG049507 to Dr Wijsman; and U01AG049508 to Dr Goate and the Discovery Extension Phase analysis is supported through U01AG052411 to Dr. Goate, U01AG052410 to Dr Pericak‐Vance, and U01 AG052409 to Drs Seshadri and Fornage. Sequencing for the Follow Up Study (FUS) is supported through U01AG057659 (to Drs PericakVance, Mayeux, and Vardarajan) and U01AG062943 (to Drs Pericak‐Vance and Mayeux). Data generation and harmonization in the Follow‐up Phase is supported by U54AG052427 (to Drs Schellenberg and Wang). The FUS Phase analysis of sequence data is supported through U01AG058589 (to Drs Destefano, Boerwinkle, De Jager, Fornage, Seshadri, and Wijsman), U01AG058654 (to Drs Haines, Bush, Farrer, Martin, and Pericak‐Vance), U01AG058635 (to Dr Goate), RF1AG058066 (to Drs Haines, Pericak‐Vance, and Scott), RF1AG057519 (to Drs Farrer and Jun), R01AG048927 (to Dr Farrer), and RF1AG054074 (to Drs Pericak‐Vance and Beecham). The ADGC cohorts include: Adult Changes in Thought (ACT; UO1 AG006781, UO1 HG004610, UO1 HG006375, U01 HG008657), the Alzheimer's Disease Centers (ADC; P30 AG019610, P30 AG013846, P50 AG008702, P50 AG025688, P50 AG047266, P30 AG010133, P50 ag005146, P50 AG005134, P50 AG016574, P50 AG005138, P30 AG008051, P30 AG013854, P30 AG008017, P30 AG010161, P50 AG047366, P30 AG010129, P50 AG016573, P50 AG016570, P50 AG005131, P50 AG023501, P30 AG035982, P30 AG028383, P30 AG010124, P50 AG005133, P50 AG005142, P30 AG012300, P50 AG005136, P50 AG033514, P50 AG005681, and P50 AG047270), the Chicago Health and Aging Project (CHAP; R01 AG11101, RC4 AG039085, K23 AG030944), Indianapolis Ibadan (R01 AG009956, P30 AG010133), the Memory and Aging Project (MAP; R01 AG17917), Mayo Clinic (MAYO; R01 AG032990, U01 AG046139, R01 NS080820, RF1 AG051504, P50 AG016574), Mayo Parkinson's Disease controls (NS039764, NS071674, 5RC2HG005605), University of Miami (R01 AG027944, R01 AG028786, R01 AG019085, IIRG09133827, A2011048), the Multi‐Institutional Research in Alzheimer's Genetic Epidemiology Study (MIRAGE; R01 AG09029, R01 AG025259), the National Cell Repository for Alzheimer's Disease (NCRAD; U24 AG21886), the National Institute on Aging Late Onset Alzheimer's Disease Family Study (NIA‐ LOAD; R01 AG041797), the Religious Orders Study (ROS; P30 AG10161, R01 AG15819), the Texas Alzheimer's Research and Care Consortium (TARCC; funded by the Darrell K Royal Texas Alzheimer's Initiative), Vanderbilt University/Case Western Reserve University (VAN/CWRU; R01 AG019757, R01 AG021547, R01 AG027944, R01 AG028786, P01 NS026630, and Alzheimer's Association), the Washington Heights‐Inwood Columbia Aging Project (WHICAP; RF1 AG054023), the University of Washington Families (VA Research Merit Grant, NIA: P50AG005136, R01AG041797, NINDS: R01NS069719), the Columbia University Hispanic Estudio Familiar de Influencia Genetica de Alzheimer (EFIGA; RF1 AG015473), the University of Toronto (UT; funded by Wellcome Trust, Medical Research Council, Canadian Institutes of Health Research), and Genetic Differences (GD; R01 AG007584). The CHARGE cohorts are supported in part by National Heart, Lung, and Blood Institute (NHLBI) infrastructure grant HL105756 (Psaty), RC2HL102419 (Boerwinkle), and the neurology working group is supported by the National Institute on Aging (NIA) R01 grant AG033193.
The CHARGE cohorts participating in the ADSP include the following: Austrian Stroke Prevention Study (ASPS), ASPS‐Family study, and the Prospective Dementia Registry‐Austria (ASPS/PRODEM‐Aus), the Atherosclerosis Risk in Communities (ARIC) Study, the Cardiovascular Health Study (CHS), the Erasmus Rucphen Family Study (ERF), the Framingham Heart Study (FHS), and the Rotterdam Study (RS). ASPS is funded by the Austrian Science Fond (FWF) grant number P20545‐P05 and P13180 and the Medical University of Graz. The ASPS‐Fam is funded by the Austrian Science Fund (FWF) project I904, the EU Joint Programme—Neurodegenerative Disease Research (JPND) in frame of the BRIDGET project (Austria, Ministry of Science), and the Medical University of Graz and the Steiermärkische Krankenanstalten Gesellschaft. PRODEM‐Austria is supported by the Austrian Research Promotion agency (FFG; Project No. 827462), and by the Austrian National Bank (Anniversary Fund, project 15435). ARIC research is carried out as a collaborative study supported by NHLBI contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C). Neurocognitive data in ARIC is collected by U01 2U01HL096812, 2U01HL096814, 2U01HL096899, 2U01HL096902, 2U01HL096917 from the NIH (NHLBI, NINDS, NIA, and NIDCD), and with previous brain MRI examinations funded by R01‐HL70825 from the NHLBI. CHS research was supported by contracts HHSN268201200036C, HHSN268200800007C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, and grants U01HL080295 and U01HL130114 from the NHLBI with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by R01AG023629, R01AG15928, and R01AG20098 from the NIA. FHS research is supported by NHLBI contracts N01‐HC‐25195 and HHSN268201500001I. This study was also supported by additional grants from the NIA (R01s AG054076, AG049607 and AG033040) and NINDS (R01 NS017950). The ERF study as a part of EUROSPAN (European Special Populations Research Network) was supported by European Commission FP6 STRP grant number 018947 (LSHG‐CT‐2006‐01947) and also received funding from the European Community's Seventh Framework Programme (FP7/2007‐2013)/grant agreement HEALTH‐F4‐ 2007‐201413 by the European Commission under the program “Quality of Life and Management of the Living Resources” of 5th Framework Programme (no. QLG2‐CT‐2002‐ 01254). High‐throughput analysis of the ERF data was supported by a joint grant from the Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO‐RFBR 047.017.043). The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, the Netherlands Organization for Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the municipality of Rotterdam. Genetic data sets are also supported by the Netherlands Organization of Scientific Research NWO Investments (175.010.2005.011, 911‐03‐012), the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Research Institute for Diseases in the Elderly (014‐93‐015; RIDE2), and the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) Netherlands Consortium for Healthy Aging (NCHA), project 050‐060‐810. All studies are grateful to their participants, faculty, and staff. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the U.S. Department of Health and Human Services. The FUS cohorts include: the Alzheimer's Disease Centers (ADC; P30 AG019610, P30 AG013846, P50 AG008702, P50 AG025688, P50 AG047266, P30 AG010133, P50 AG005146, P50 AG005134, P50 AG016574, P50 AG005138, P30 AG008051, P30 AG013854, P30 AG008017, P30 AG010161, P50 AG047366, P30 AG010129, P50 AG016573, P50 AG016570, P50 AG005131, P50 AG023501, P30 AG035982, P30 AG028383, P30 AG010124, P50 AG005133, P50 AG005142, P30 AG012300, P50 AG005136, P50 AG033514, P50 AG005681, and P50 AG047270), Alzheimer's Disease Neuroimaging Initiative (ADNI; U19AG024904), Amish Protective Variant Study (RF1AG058066), Cache County Study (R01AG11380, R01AG031272, R01AG21136, RF1AG054052), Case Western Reserve University Brain Bank (CWRUBB; P50AG008012), Case Western Reserve University Rapid Decline (CWRURD; RF1AG058267, NU38CK000480), Cuban American Alzheimer's Disease Initiative (CuAADI; 3U01AG052410), Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA; 5R37AG015473, RF1AG015473, R56AG051876), Genetic and Environmental Risk Factors for Alzheimer Disease Among African Americans Study (GenerAAtions; 2R01AG09029, R01AG025259, 2R01AG048927), Gwangju Alzheimer and Related Dementias Study (GARD; U01AG062602), Hussman Institute for Human Genomics Brain Bank (HIHGBB; R01AG027944, Alzheimer's Association “Identification of Rare Variants in Alzheimer Disease”), Ibadan Study of Aging (IBADAN; 5R01AG009956), Mexican Health and Aging Study (MHAS; R01AG018016), Multi‐Institutional Research in Alzheimer's Genetic Epidemiology (MIRAGE; 2R01AG09029, R01AG025259, 2R01AG048927), Northern Manhattan Study (NOMAS; R01NS29993), Peru Alzheimer's Disease Initiative (PeADI; RF1AG054074), Puerto Rican 1066 (PR1066; Wellcome Trust [GR066133/GR080002], European Research Council [340755]), Puerto Rican Alzheimer Disease Initiative (PRADI; RF1AG054074), Reasons for Geographic and Racial Differences in Stroke (REGARDS; U01NS041588), Research in African American Alzheimer Disease Initiative (REAAADI; U01AG052410), Rush Alzheimer's Disease Center (ROSMAP; P30AG10161, R01AG15819, R01AG17919), University of Miami Brain Endowment Bank (MBB), and University of Miami/Case Western/North Carolina A&T African American (UM/CASE/NCAT; U01AG052410, R01AG028786). The four LSACs are: the Human Genome Sequencing Center at the Baylor College of Medicine (U54 HG003273), the Broad Institute Genome Center (U54HG003067), The American Genome Center at the Uniformed Services University of the Health Sciences (U01AG057659), and the Washington University Genome Institute (U54HG003079). Biological samples and associated phenotypic data used in primary data analyses were stored at Study Investigators institutions, and at the National Cell Repository for Alzheimer's Disease (NCRAD, U24AG021886) at Indiana University funded by NIA. Associated Phenotypic Data used in primary and secondary data analyses were provided by Study Investigators, the NIA funded Alzheimer's Disease Centers (ADCs), and the National Alzheimer's Coordinating Center (NACC, U01AG016976) and the National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site (NIAGADS, U24AG041689) at the University of Pennsylvania, funded by NIA. This research was supported in part by the Intramural Research Program of the National Institutes of health, National Library of Medicine. Contributors to the Genetic Analysis Data included Study Investigators on projects that were individually funded by NIA, and other NIH institutes, and by private U.S. organizations, or foreign governmental or nongovernmental organizations. National Institutes of Health R01‐AG080806 (W.G.M), P30‐AG062422 (B.L.M., G.D.R.), R00‐AG068271 (J.N.C.), R01‐AG062588 (J.S.Y.), R35‐AG072362 (G.D.R.), R21NS120629 (G.D.R.), K99‐AG065501 (R.L.J.), K01‐AG070376‐03 (I.J.B.), R01‐NS050915 (M.G.T), R01‐P0551783 (M.G.T.), K24‐DC015544 (M.G.T.), K23‐AG048291 (Z.A.M.), the Robert W. Katzman Fellowship Training Grant through the American Academy of Neurology in conjunction with the American Brain Foundation and Alzheimer's Association, The Wallin Neuroscience Discovery Fund, and the Fesler‐Lampert Chair in Aging Studies (W.G.M), Alzheimer's Association Research Fellowship 16‐443577 (R.L.J.) and AARGD‐22‐969900 (I.J.B) the Rainwater Charitable Foundation (G.D.R. and J.S.Y.), Department of Defense (W81XWH‐19‐1‐0709 to R.S.D. and J.S.Y.), and the Mary Oakley Foundation (J.S.Y.). Role of the funding source: the funding sources played no role in the study design, collection, analysis, or interpretation of the study.
Mantyh WG, Cochran JN, Taylor JW, et al. Early‐onset Alzheimer's disease explained by polygenic risk of late‐onset disease? Alzheimer's Dement. 2023;15:e12482. 10.1002/dad2.12482
Statistical analyses performed by: William G. Mantyh (academic), Isabel E. Allen (academic)
REFERENCES
- 1. Ridge PG, Hoyt KB, Boehme K, et al. Assessment of the genetic variance of late‐onset Alzheimer's disease. Neurobiol Aging. 2016;41:200.e13‐200.e20. doi: 10.1016/J.NEUROBIOLAGING.2016.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goate A, Chartier‐Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349(6311):704‐706. doi: 10.1038/349704a0. 1991 3496311. [DOI] [PubMed] [Google Scholar]
- 3. Sherrington R, Leresque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early‐onset familial Alzheimer's disease. Nature. 1995;375(June):754‐760. [DOI] [PubMed] [Google Scholar]
- 4. Rogaev EI, Sherrlngton R, Rogaeva EA, et al. Familial Alzheimer's disease in kindreds with missense mutations In a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995;376(August):775‐778. [DOI] [PubMed] [Google Scholar]
- 5. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45(12):1452‐1458. doi: 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tan CH, Hyman BT, Tan JJX, et al. Polygenic hazard scores in preclinical Alzheimer disease. Ann Neurol. 2017;82(3):484‐488. doi: 10.1002/ana.25029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Desikan RS, Fan CC, Wang Y, et al. Genetic assessment of age‐associated Alzheimer disease risk: development and validation of a polygenic hazard score. Brayne C, ed. PLOS Med. 2017;14(3):e1002258. doi: 10.1371/journal.pmed.1002258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Escott‐Price V, Sims R, Bannister C, et al. Common polygenic variation enhances risk prediction for Alzheimer's disease. Brain. 2015;138(12):3673‐3684. doi: 10.1093/brain/awv268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wingo TS, Lah JJ, Levey AI, Cutler DJ. Autosomal recessive causes likely in early‐onset Alzheimer disease. Arch Neurol. 2012;69(1):59. doi: 10.1001/archneurol.2011.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cruchaga C, Del‐Aguila JL, Saef B, et al. Polygenic risk score of sporadic late‐onset Alzheimer's disease reveals a shared architecture with the familial and early‐onset forms. Alzheimer's Dement . 2018;14(2):205‐214. doi: 10.1016/j.jalz.2017.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tan CH, Fan CC, Mormino EC, et al. Polygenic hazard score: an enrichment marker for Alzheimer's associated amyloid and tau deposition. Acta Neuropathol. 2018;135(1):85‐93. doi: 10.1007/s00401-017-1789-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263‐269. doi: 10.1016/j.jalz.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gorno‐Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006‐1014. doi: 10.1212/WNL.0b013e31821103e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schott JM, Lehmann M, Primativo S, et al. Consensus classification of posterior cortical atrophy. Alzheimer's Dement. 2017;13(8):870‐884. doi: 10.1016/j.jalz.2017.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. La Joie R, Bejanin A, Fagan AM, et al. Associations between [18F]AV1451 tau PET and CSF measures of tau pathology in a clinical sample. Neurology. 2018;90(4):e282‐e290. doi: 10.1212/WNL.0000000000004860. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65(4):403‐413. doi: 10.1002/ana.21610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Villeneuve S, Rabinovici GD, Cohn‐Sheehy BI, et al. Existing Pittsburgh Compound‐B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain. 2015;138(7):2020‐2033. doi: 10.1093/brain/awv112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease. Neurology. 1984;34(7):939‐939. doi: 10.1212/WNL.34.7.939 [DOI] [PubMed] [Google Scholar]
- 19. Cochran JN, Geier EG, Bonham LW, et al. Non‐coding and loss‐of‐function coding variants in TET2 are associated with multiple neurodegenerative diseases. Am J Hum Genet. 2020;106(5):632‐645. doi: 10.1016/j.ajhg.2020.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25:1754‐1760. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31(12):2032‐2034. doi: 10.1093/bioinformatics/btv098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sirkis DW, Bonham LW, Aparicio RE, et al. Rare TREM2 variants associated with Alzheimer's disease display reduced cell surface expression. Acta Neuropathol Commun. 2016;4(1):98. doi: 10.1186/s40478-016-0367-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wright CA, Taylor JW, Cochran M, et al. Contributions of rare and common variation to early‐onset and atypical dementia risk. Cold Spring Harb Mol Case Stud. 2023;9(3):a006271. doi: 10.1101/mcs.a006271. Published online 2023:mcs.a006271.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schott JM, Crutch SJ, Carrasquillo MM, et al. Genetic risk factors for the posterior cortical atrophy variant of Alzheimer's disease. Alzheimer's Dement. 2016;12(8):862‐871. doi: 10.1016/J.JALZ.2016.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cochran JN, McKinley EC, Cochran M, et al. Genome sequencing for early‐onset or atypical dementia: high diagnostic yield and frequent observation of multiple contributory alleles. Cold Spring Harb Mol Case Stud. 2019;5(6):1‐18. doi: 10.1101/mcs.a003491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davidson Y, Gibbons L, Pritchard A, et al. Apolipoprotein E ε4 allele frequency and age at onset of Alzheimer's disease. Dement Geriatr Cogn Disord. 2006;23(1):60‐66. doi: 10.1159/000097038 [DOI] [PubMed] [Google Scholar]
- 27. Smirnov DS, Galasko D, Hiniker A, Edland S, Salmon DP. Age‐of‐onset and APOE ‐related heterogeneity in pathologically confirmed sporadic Alzheimer disease. Neurology. Published online 2021:doi: 10.1212/wnl.0000000000011772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van der Flier WM, Pijnenburg YAL, Fox NC, Scheltens P. Early‐onset versus late‐onset Alzheimer's disease: the case of the missing APOE ɛ4 allele. Lancet Neurol. 2011;10(3):280‐288. doi: 10.1016/S1474-4422(10)70306-9. Published online 2011. [DOI] [PubMed] [Google Scholar]
- 29. Polsinelli AJ, Lane KA, Manchella MK, Logan PE, Gao S, Apostolova LG. APOE ε4 is associated with earlier symptom onset in LOAD but later symptom onset in EOAD. Alzheimers Dement. 2023;19(5):2212‐2217. doi: 10.1002/ALZ.12955. Published online February 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seltzer B, Sherwin I. Comparison of clinical features in early‐ and late‐onset primary degenerative dementia. Arch Neurol. 1983;40:143‐146. [DOI] [PubMed] [Google Scholar]
- 31. Falgàs N, Allen IE, Spina S, et al. The severity of neuropsychiatric symptoms is higher in early‐onset than late‐onset Alzheimer's disease. Eur J Neurol. 2021:1‐11. doi: 10.1111/ene.15203. October. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mendez MF, Lee AS, Joshi A, Shapira JS. Nonamnestic presentations of early‐onset Alzheimer's disease. Am J Alzheimers Dis Other Demen. 2012;27(6):413‐420. doi: 10.1177/1533317512454711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miller ZA, Mandelli ML, Rankin KP, et al. Handedness and language learning disability differentially distribute in progressive aphasia variants. Brain. 2013;136:3461‐3473. doi: 10.1093/brain/awt242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miller ZA, Spina S, Pakvasa M, et al. Cortical developmental abnormalities in logopenic variant primary progressive aphasia with dyslexia. Brain Commun. 2019;1(1):1‐8. doi: 10.1093/braincomms/fcz027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miller ZA, Rosenberg L, Santos‐Santos MA, et al. Prevalence of mathematical and visuospatial learning disabilities in patients with posterior cortical atrophy. JAMA Neurol. 2018;75(6):728. doi: 10.1001/jamaneurol.2018.0395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nochlin D, Van Belle G, Bird D, Sumi SM. Comparison of the severity of neuropathologic changes in familial and sporadic Alzheimer's disease. Alzheimer Dis Assoc Disord. 1993;7(4):212‐222. Accessed February 10, 2022. https://europepmc.org/article/med/8305189 [PubMed] [Google Scholar]
- 37. Spina S, La Joie R, Petersen C, et al. Comorbid neuropathological diagnoses in early versus late‐onset Alzheimer's disease. Brain. 2021;144(7):2186‐2198. doi: 10.1093/brain/awab099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin‐related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39(2):168‐177. doi: 10.1038/ng1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reitz C, Wang L, Lin C, et al. Variants in the ATP‐binding cassette and the risk of late‐onset Alzheimer disease. Jama. 2013;309(14):1483‐1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pottier C, Hannequin D, Coutant S, et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early‐onset Alzheimer disease. Mol Psychiatry. 2012;17(9):875‐879. doi: 10.1038/mp.2012.15 [DOI] [PubMed] [Google Scholar]
- 41. Verheijen J, Van den Bossche T, van der Zee J, et al. A comprehensive study of the genetic impact of rare variants in SORL1 in European early‐onset Alzheimer's disease. Acta Neuropathol. 2016;132(2):213‐224. doi: 10.1007/s00401-016-1566-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cuyvers E, De Roeck A, Van den Bossche T, et al. Mutations in ABCA7 in a Belgian cohort of Alzheimer's disease patients: a targeted resequencing study. Lancet Neurol. 2015;14(8):814‐822. doi: 10.1016/S1474-4422(15)00133-7 [DOI] [PubMed] [Google Scholar]
- 43. De Roeck A, Van den Bossche T, van der Zee J, et al. Deleterious ABCA7 mutations and transcript rescue mechanisms in early onset Alzheimer's disease. Acta Neuropathol. 2017;134(3):475‐487. doi: 10.1007/s00401-017-1714-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Steinberg S, Stefansson H, Jonsson T, et al. Loss‐of‐function variants in ABCA7 confer risk of Alzheimer's disease. Nat Genet. 2015;47(5):445‐447. doi: 10.1038/ng.3246 [DOI] [PubMed] [Google Scholar]
- 45. Sirkis DW, Bonham LW, Johnson TP, La Joie R, Yokoyama JS. Dissecting the clinical heterogeneity of early‐onset Alzheimer's disease. Mol Psychiatry. 2022;27(6):2674‐2688. doi: 10.1038/s41380-022-01531-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sherrington R, Froelich S, Sorbi S, et al. Alzheimer's disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Genet. 1996;5(7):985‐988. doi: 10.1093/HMG/5.7.985 [DOI] [PubMed] [Google Scholar]
- 47. Theuns J, Brouwers N, Engelborghs S, et al. Promoter mutations that increase amyloid precursor‐protein expression are associated with Alzheimer disease. Am J Hum Genet. 2006;78(6):936‐946. doi: 10.1086/504044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Osuntokun BO, Sahota A, Ogunniyi AO, et al. Lack of an association between apolipoprotein E ?4 and Alzheimer's disease in elderly nigerians. Ann Neurol. 1995;38(3):463‐465. doi: 10.1002/ana.410380319 [DOI] [PubMed] [Google Scholar]
- 49. Deters KD, Napolioni V, Sperling RA, et al. Amyloid PET imaging in self‐identified non‐hispanic black participants of the anti‐amyloid in asymptomatic Alzheimer's disease (A4) study. Neurology. 2021;96(11):e1491‐e1500. doi: 10.1212/WNL.0000000000011599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Blue EE, Horimoto ARVR, Mukherjee S, Wijsman EM, Thornton TA. Local ancestry at APOE modifies Alzheimer's disease risk in Caribbean Hispanics. Alzheimer's Dement. 2019;15(12):1524‐1532. doi: 10.1016/j.jalz.2019.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hoogmartens J, Cacace R, Van Broeckhoven C. Insight into the genetic etiology of alzheimer's disease: a comprehensive review of the role of rare variants. Alzheimer's Dement Diagnosis . Assess Dis Monit. 2021;13(1):1‐14. doi: 10.1002/dad2.12155 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Data Availability Statement
Anonymized data not published within this article will be made available by request from any qualified investigator. Genomic data for UCSF participants is available under controlled access via the Synapse AD Knowledge portal accession number syn25686500. ADSP data are available at National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site under project NG00067.